
wykład 2
BIOCHEMIA
9.X.2000
TEMAT:BIOCHEMIA KWASÓW NUKLEINOWYCH
Dzisiaj na temat metodologii badania badania genomu, ograniczymy się do metod stosowanych
w spółczesnej diagnostyce. przypominam ,że ostatnio zakończyliśmy na etapie transkrypcji a
teraz przejdziemy do translacji.Zreplikowaliśmy DNA i poddaliśmy go transkrypcji i podaliśmy
go obróbce posttranskrypcyjnej prowadzocej metodą splicingu powstania transkryptu i dziś nasz
traskrypt opuścił jądro znalazł się w cytoplaźmie ,wszedł na rybosomy -zaczyna się pierwsza ...
tym pośrednikiem jest aminoacylo-tRNA, czyli tRNA z połączonym z nim aminokwasem.To są
cząsteczki niewielkie liczące ok. 80 nukleotydów, które cechują się zarówno występowaniem
miejsc dwuniciowych z obecnymi sparowaniami, oraz 4 sekwencji jednoniciowych-nieważne jest
ich funkcja, ważne że występują tutaj zmodyfikowane zasad:dihydrourydyna,w pętli T Psi C
występuje rybotymidyna,chodz wiadomo, że tymina nie występuje w RNA i modyfikacje o
których wspominałem występują na drodze modyfikacji posttranskrypcyjnej
tRNA.NAjisotniejsze z punktu widzenia procesu, którym się zajmujemy jest pętala
antykodonowa,która asocjuje z odpowiednim kodonem znajdującym się w mRNA.Na 3' końcu
cząsteczki tRNA następuje przyłączenie aminokwasu.Aminokwas przyłączony jest do grupy
hydroksylowej adeniny swoją grupą karboksylową i ta struktura nosi nazwę aminoacylo-
tRNA.W takiej formie aminokas zdąża na rybosom aby w konsekwencji procesu translacji
tworzyć łańcuch polipeptydowy.Ale to nie jedyna funkcja tego tRNA, posiada tych funkcji
znacznie więcej oprócz rybosomalnej produkcji białka .U organizmów, które dysponują ścianą
komórkową,a więc u roślin oaz organizmów prokariotycznych tRNA bierze udział w syntezie
ściany komórkowej .U organizmów, które wytwarzają chlorofil jest zangażowany w biosyntezę
chlorofilu.Dalej bierze udział w syntezie fosfoguanozyny.Bierze udział w transporcie
aminokwasów .U bakterii gramujemnych bierze udział w syntezie glikopolisachorydów,które są
endotoksynami bakterii. Bierze udział w degradacji białek oraz ostnią funkcją -niezwykle
istotną- jest primerem odwrotnej transkryptazy (polimeraza DNA RNA-zależna, więc musi
mieć jakiś primer, tym primerem są cząsteczki tRNA) I wreszcie za pomocą zmutowanych
cząsteczek tRNA spowodowanych mutacjami w genach kodujących tRNA możliwe jest
wystąpienie mutacji supresorowej tzn.tRNA mimo napotkania kodonu typu non-sens ,który ma
oznaczać koniec translacji nie terminuje tylko przyłącza częst błędny aminokwas.Takie białko
powstałe w wyniku przyłączenia do miejsca kodonu nonsensownego zmutowanego tRNA -
tzw.tRNA supresorowego -takie białko nosi nazwę białka supresorowego.Mimo, iż sekwencja
aminokwasowa może być odmienna to efekt dla komórki jest lepszy niż w momencie gdy to
białko wogóle by nie powstało, często to białko nawet po zamianie aminokwasu pełni tą samą
funkcję, albo funkcja jego jest upośledzona , ale to lepsze niż by tego białka nie było.Dla naszych
dalszych rozważań ważne są trzy punkty kontaktowe w cząsteczce,mianowicie miejsce A,miejsce
P oraz miejsce E.Do miejsca oznaczonego jako A przyłącza się aminoacylo-tRNA z wyjątkiem
pierwszego.W miejscu P znajduje się peptydylo-tRNA czyli tRNA z przyłączonym narastającym
peptydem.I wreszcie miejsce E to jest miejsce, które zajmuje peptydylo-tRNA przed wyjściem z
rybosomu.
TRANSLACJA
Inicjacja procesu translacji rozpoczyna się od zajęcia przez pierwszy aminoacylo-tRNA,którym
w przypadku organizmów eukariotycznych jest metionylo-tRNA,czyli tRNA z przyłączoną
metioniną a w przypadku organizmów prokariotycznych metionina ulega modyfikacji i powstaje
formylometionina i formylometionino-tRNA,ale kodon rozpoznawania jest ten sam, ponieważ
kodonu dla formylometioniny nie ma.I ten pierwszy inicjujący proces translacji aminoacylo-
tRNA zajmuje miejsce P, a nie miejsce A.Po zajęciu miejsca P do miejsca A przyłącza się
kolejny aminoacylo-tRNA.Do akcji wkracza enzym-transferaza peptydylowa-jest to enzym,
który syntetyzuje wiązanie peptydowe.I wiązanie peptydowe -przypominam,że połączone są

grupą karboksylową aminokwasy z tRNA -wiązanie jest stworzone pomiędzy grupą
karboksylową tego narastającego peptydu, a w przypadku pierwszego aminoacylo-tRNA między
metionylo-tRNA a grupą aminową przyłączonego w pozycję A aminoacylo-tRNA.Z tego
wynika, że narastanie peptydu następuje od końca N w kierunku końca C,czyli zawsze dawcą
grupy karboksylowej jest peptydylo-tRNA ( a w przypadku pierwszego metionylo-tRNA ) a
dawcą grupy aminowej aminokwas, który znajduje się w miejscu A przyłączonego tRNA .Tak
więc w naszym konkretnym przypadku po zadziałaniu transferazy peptydylowej powstał nam
dipeptyd, zbudowany z metioniny i jakiegoś drugiego aminokwasu, który okupuje miejsce A
rybosomu.Do akcji wkracza translokaza .Translokaza przesuwa nasze w tym przypadku
dipeptydylo-tRNA w miejsce P, miejsce A ulega zwolnieniu i tu znowu przyłącza się jakiś
aminoacylo-tRNA do miejsca A, no i następuje na podobnej zasadzie wygenerowanie trzeciego
aminokwasu, czyli drugiego wiązania peptydowego i mamy już tripeptyd. W tym czasie
zwolniony tRNA przechodzi do miejsca P i w miarę jak rybosom się przesuwa, następuje
opuszczenie rybosomu przez tRNA. I tak wszystko trwa długo, aż pojawi się kodon nonsensowny
i wówczas czynniki, które terminują translację, odpowiednie grupy białek enzymatycznych i
nieenzymatycznych, doprowadzają do odłączenia się powstałego peptydu od rybosomu . No i
teraz wiedząc jak ten proces przebiega przyjrzymy się substancjom, które ten proces modulują.
Inhibitory translacji
I- Inhibitory inicjacji
-Trimetoprim
Pamiętając, że pierwszym aminoacylo-tRNA jest formylometionylo-tRNA, a on powstaje przez
przyłączenie grupy formylowej do metionylo-tRNA. Ostatno już z tym związkiem się
spotkaliśmy - trimetoprim, jest inhibitorem reduktazy kwasu foliowego, czyli trimetoprim
uniemożliwia powstanie FA
4
, grupa formylowa jako grupa jednowęglowa jest przenoszona przez
tetrahydrofo- lian, czyli nie może powstać FA
4
, przez to nie może powstać formylometionylo-
tRNA i u bakterii, bo tylko u nich ten proces się od tego związku zaczyna, nie może rozpocząć
się translacja.
II-Inhibitory elongacji
-antybiotyki aminoglikozydowe
Jest to bardzo szeroko stosowana grupa antybiotyków przeciwbakteryjnych, dobrze znane nazwy:
>Streptomycyna
>Gentamycyna
>Neomycyna
>Kanamycyna
>Tebramycyna
One działają na rybosom bakteryjny, i mają dwa działania : hamowanie inicjacji translacji (
wprawdzie powstaje formylometionylo-tRNA, ale nie może się przyłączyć do rybosomu, bo do
miejsca P nie może się przyłączyć formylometionylo-tRNA) a druga - hamowanie procesu
elongacji ( a to w ten sposób, że zaburzają prawidłowe przyłączanie kolejnych aminoacylo-tRNA
do rybosomu lub powodują błędny odczyt ).
-Tetracykliny
>Doxycyklina, czy Vibramycyna ( jak kto woli )
Ona hamuje przyłączanie aminoacylo-tRNA do rybosomu, czyli nie może wydłużać się łańcuch
polipeptydowy. Natomiast nie zaburza procesu inicjacji translacji.
- Chloranfenikol
- Cykloheksymid
Są inhiobitorami transferazy peptydylowej. Przyczym chloranfenikol działa na rybosom

bakteryjny, a cykloheksymid działa na rybosom eukariotyczny.Chloranfenikol jest bardzo
toksycznym antybiotykiem, rzadko obecnie stosowanym, a jeśli się go stosuje to głównie w
salmonellozach, czyli durze brzusznym oraz w schigellozach, czyli czerwonce bakteryjnej,
rzadko stosowany ze względu na toksyczne działanie w stosunku do szpiku kostnego. Natomiast
cykloheksymid nie znalazł zastosowania terapeutycznego, natomiast znalazł on zastosowanie
jako pewnego rodzaju narzędzie w biochemii komórki. To nie jest cytostatyk.
-Antybiotyki makrolitowe
>Erytromycyna
>Dadercyna
-Toksyna błonicza
Są inhibitorami translokazy, czyli tego enzymu, który umożliwia przesunięcie peptydylo-tRNA z
miejsca A do miejsca P. Antybiotyki makrolitowe działają na rybosom bakteryjny, natomiast
toksyna błonicza działa na rybosom eukariotyczny. Błonica to jest choroba bakteryjna,
wywoływana przez maczugowca błonicy, obecnie choroba nie występuje dzięki szczepieniom
ochronnym, które podaje się już bardzo małym niemowlętom, natomiast dawniej, rozwuj
maczugowca odbywał się w gardle dając objawy anginy, czy zapalenia gardła, natomiast
wydzielana przez niego toksyna uszkadzała głównie mięsień sercowy, na szczęście obecnie nie
występuje.
III-Inhibitory terminacji
-Puromycyna
Jest analogiem tyrozynylo-tRNA, czyli udaje takie tRNA, do którego przyłączona jest tyrozyna.
No i jak się pojawia kodon dla tyrozyny, to podjeżdża sobie puromycyna, w to miejsce się
włącza, ale ona nie jest w stanie utworzyć wiązania peptydowego, czyli na tym etapie biosynteza
białka staje.
No i zsyntetyzowaliśmy łańcuch polipeptydowy, który następnym razem będziemy modyikować
posttranslacyjnie, aby uzyskać w pełni funkcjonalne białka. Poza bardzo małymi peptydami,
każde inne białko ulega modyfikacjom, zarówno modyfikacjom wewnątrzkomórkowym jak i
zewnątrzkomórkowym.
Metody badania genomu człowieka
Jak wspominałem, będziemy się zajmować tylko metodami łatwymi do zrozumienia, szeroko
stosowanymi w praktyce, takimi, którymi my się również w zakładzie zajmujemy, takimi,
które pokażemy w czasie ćwiczeń laboratoryjnych.
Genom człowieka jest trudny do badania, dlaczego: ze względu na jego wielkość
Escherichia coli 4500 genów
Drożdże
6500 genów
Muszka owocowa 8000 genów
Człowiek 80 000 genów
Dlatego badania, które mają być ladamoment ukończone, na poznanie pełnej sekwencji
genomu ludzkiego, trwały od wielu, wielu lat i były obarczone pewnymi trudnościami, ze
wzgłędu na jego cech i ze względu na trudność w pozyskiwaniu.
Cech genomu człowieka :
-gęstość genów ( czyli ilość genów przypadająca na jednostkę długości DNA, a tą jednostką
jest ilość par zasad - pz.)
- geny w mitochondrium upakowane są dość gęsto , bo sam genom jest stosunkowo
nieduży - 10,45 kb ( tys. zasad )
- natomiast w jądrze geny są stosunkowo rozsiane 140 - 45 kb
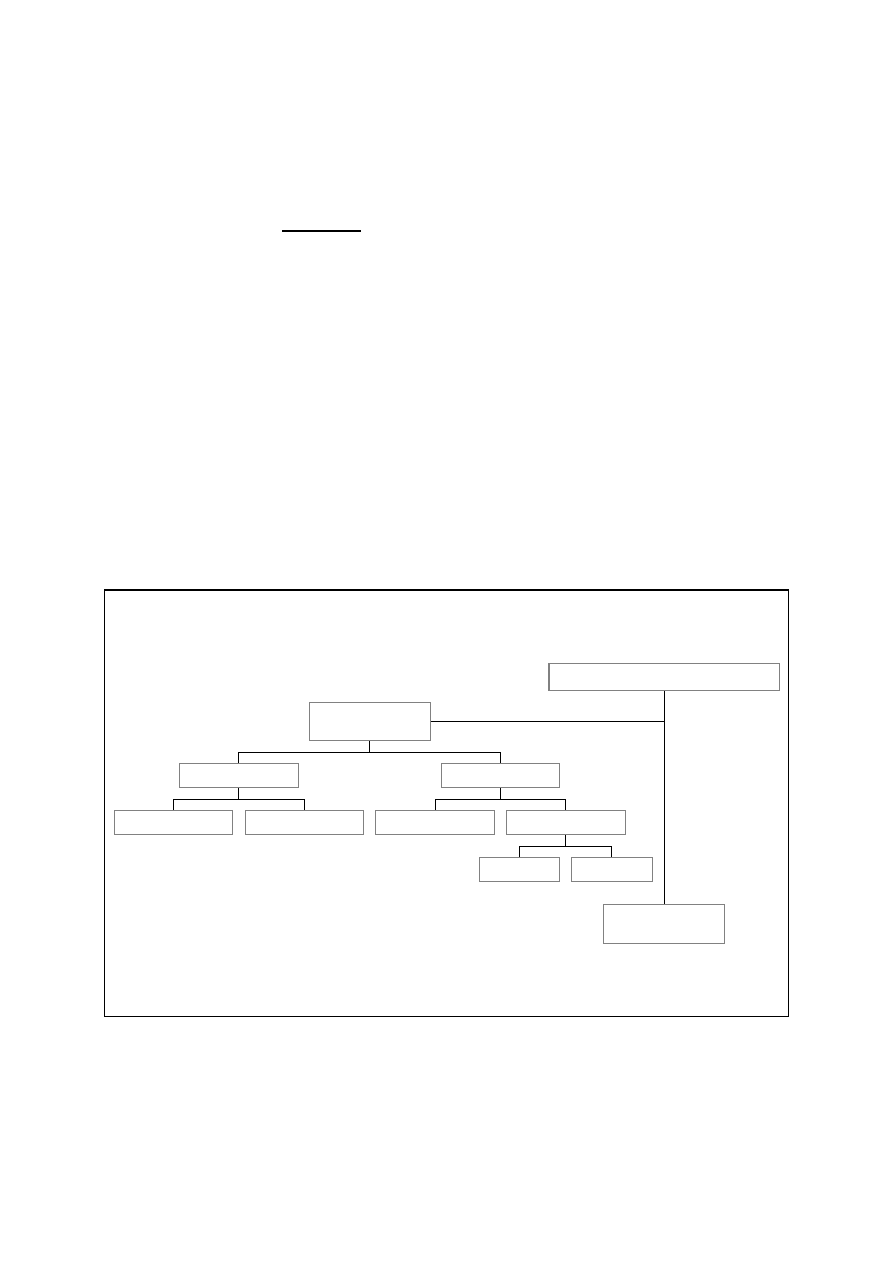
-wielkość genu ( długość genu ) 10 - 15 kb
-odstępy między genowe, czyli tzw. spacer 25 - 30 kb
Geny człowieka są bardzo zróżnicowanepod względem ilość eksonów. Są geny bardzo proste
mające jeden ekson, często są to geny kodujące np. tRNA. Natomiast największy gen genomu
człowieka posiada 79 eksonów i jest to gen, którego nazwę warto zapamiętać, to jest gen, który
koduje białko o nazwie dystrofina . Natomiast gen jest oznaczony literami DMD, co znaczy
dystrofia mięśniowa Duchanne'a, ponieważ mutacje w obrębie tego genu prowadzą do pewnej
uwarunkowanej gentycznie choroby mięśni - dystrofia mięśniowa Duchanne'a.
-wielkość eksonu wynosi około 70 pz. i z reguły zależy od wielkości genu, czyli małe geny mają
małe eksony, duże geny mają duże eksony. Największym znanym eksonem u człowieka jest
ekson 26-sty, występujący w genie kodującym białko ApoB . ApoB, to jest apolipoproteina,
czyli biał- ko wchodzące w skład lipoprotein osocza i wielkość tego eksonu wynosi 7,6kb.
Również wielkość intronu zależy od wielkości genu. Największy intron stwierdza się w genie
kodującym dystrofine. Jego długość wynosi 30kb, a skoro już jesteśmy już przy genie DMD, jego
długość wynosi 2,5 mln par zasad, czyli jest genem ogromnym.Tutaj mamy pokazany gen
insuliny, wielkość genu 1400 pz.zaledwie 3 egzony. Na przeciwstawnym biegunie mamy gen
dystrofii,czyli ten DMD, którego wielkość wynosi 2,5 mln.pz i który posiada 79 egzonów,czyli to
pokazuje jak ogromną różnorodność posiada genom człowieka. Jak zorganizowany jest genom
człowieka? Genom człowieka można podzielić na dwi główne frakcje,najważniejsza to jest
genom jądrowy,którym się za chwilę szczegółowo zajmiemy ,a druga frakcja to jest genom
mitochondrialny-o którym mówiłem ostatnio.Spośród 80 000 genów zaledwie 37-przypominam-
występuje w genomie mitochondrialnym.
Patrząc na genom jądrowy to znacznie mniejszą część stanowią fragmenty DNA, które kodują (
szacuje się, że ok 30% to jest DNA kodujące a ok. 70% materiału genetycznego tworzy tzw.
Eksony ( 10% )
Introny ( 90% )
Geny ( 30% )
Unikatowe ( 80% )
Zgrupowane
Rozproszone
Powtarzalne ( 20% )
Pozageny ( 70% )
Genom jądrowy
Genom pozajądrowy
( mitochondrium )
Organizacja genomu w komórce

pozagenowy DNA ).Teraz przyjrzyjmy się najpierw genom .Sekwencje kodujące zaledwie 10% z
tych 30%, natomiast w większości są to sekwencje niekodujące, które przez nasz użytek możmy
je ogólnie nazwać intronami.Natomiast pozagenowy DNA tworzy dwie zasadnicze frakcje a
mianowicie tzw. sekwencje unikatowe, których jest ok. 80% a 20% stanowią tzw. sekwencje
powtórzone.Zkolei sekwencje powtórzone dzielą się jeszcze na dwie frakcje mianowicie
sekwencje zgrupowane i sekwencje rozproszone.Warto pamietać o tym przypomniałem
poprzednio, że geny człowieka mają charakter nieciagły,sekwencje egzonowe są poprzedzielane
sekwencjami intronowymi poza kilkoma wyjątkowymi mianowicie geny ciągłe spotykamy w
mitochondrium ,geny ciągłe kodują histony.Wiekszość małych genów kodujacych cząsteczki
tRNA również mają charakter ciągły, dalej geny niektórych receptorów hormonalnych.
Wymienia się , że należy do nich receptor dopaminergiczny typu I ,receptor dopaminergiczny
typu V -o nich już więcej na biochemi państwo ode mnie nie usłyszą, natomiast trzeci tu
wymieniony receptor dla bradykininy typu II będzie jak zły sen się często pojawiał. No i np.
takim genem jest gen produkujący intrferon alfa,o którym więcej powiem przy omawianiu
cytokin ukł. immunologicznego.Teraz zajmiemy się sekwencjami, które mają charakter
sekwencji powtórzonych .Pierwsza grupa są to duże bloki należące do sekwencji zgrupowanych,
są to tzw. satelity, czyli satelitarny DNA .Taka jednostka, która ulega powtórzeniu liczy od 200-
5 000 pz i one praktycznie nie mają znaczenia w diagnostyce molekularnej, którą się zajmujemy i
do nich nie będziemy wracać ,służą m.in. do identyfikacji chromosomów.Natomiast nas
interesują powtórzenia rozproszone, którymi zainteresowanie datuje się na ok. ostatnie 10 lat , są
to tzw. mini- i mikro-satelity.Co to są minisatelity? One pojawiaja się z częstością 2-400 ,
jenostka, która się powtarza to jest od 7 do 100 pz. i one mają znaczenie w diagnostyce
molekularnej,natomiast najbardziej ciekawą strukturą są mikrosatelity, które obejmują jednostki
powtarzające się od 1-6 pz ,najintensywniej zgłębiane jest występowanie powtórzeń 3 i 4
nukleotydowych .Jaka jest funkcja mikrosatelit to nikt naprawdę nie wie.One występują poza
genami , czyli nie kodują białka, natomiast razem z genem mogą ulec transkrypcji i przypuszcza
się, że jedną z ich funkcji może być stabilizacja transkryptu przed działaniem enzymów
nukleolitycznych, które mogły by ten transkrypt zdegradować podczas jego drogi do rybosomu.
Metodologią służącą do badania mikrosatelitów jest technika zwana STR ( short tandem repeat)
co znaczy krótki powtórzenia tandemowe.W skutek wystąpienia jakieś mutacji w wyniku analizy
pojawiających się mikrosatelit wzorzec występujacy u danego człowieka może ulec zmianie i
badając dzidziczenie genu razem z układem sprzężeń z tymi mikrosatelitami można wnosić o
defekcie genu bez bezpośredniego badania tego genu-wyłącznie na podstawie rozkładu sekwencji
mikrosatelitarnych,które u pewnych osób w wyniku mutacji mogą zaniknąć.Poprostu w wyniku
mutacji nie pojawi się ta jednostka powtórzona -to jest oczywiste.Szczególnie jednostką
chorobową,w ktorej bada się polimorfizm sekwencji mikrosatelitarnych jest rak jelita grubego
rzwijający się na podłożu rodzinnej polipowatości.Są osoby, które mają pecha i które w wyniku
pewnego genu mają całe jelito grube usiane tysiącami polipów i jest to stan przed rakowy,
ponieważ polipy ulegają transformacji nowotworowej i ryzyko wystąpienia takiego nowotworu
można ocenić na podstawie analizy genotypu a konkretnie występowania krótkich powtórzeń
tandemowych. Teraz przejdziemy do badania tego DNA, który koduje.Od zawsze wielkką
trudnością w badaniu DNA było to, że było go w komórce mało-zbyt mało aby użyć typowe
metody chemiczne do analizy .I najbardziej starymi historycznie metodami, chociaż dalej
używanymi jest badanie DNA nieamplifikowanego, czyli tyle ile uzyskaliśmy w komórce tyle
badamy.Skoro rutynowe techniki badawcze tutaj zawodzą to musimy znaleść wzmacniacz,ktory
wykrywa daną cząsteczke DNA i tutaj musimy uciec się do hybrydyzacji i do struktury zwaną
sondą molekularną .Sonda molekularna jest znakowana,zjwisko które pokazałem nosi nazwę
hybrydyzacji , czyli sonda molekularna kiedy napotka komplementarny fragment DNA to na
takiej samej zasadzie jak tu występuje chociaż by w procesie replikacji następuje
komplementarne połączenie z danym naszym DNA.Dawniej znacznikiem były głównie izotopy

promieniotwórcze i metodą radioautografii należało przyłożyć kliszę fotograficzną i sprawdzić
czy dany gen czy fragment genu został wykryty czy teą nie. Obecie używane znaczniki są to
znaczniki fluorescencyjne , także można bez używania pr.jonizującego identyfikować proces
hybrydyzacji Znaczny postęp rozwoju biologii molekularnej był możliwy dzięki zastosowaniu
technik, które amplifikują DNA.Ampllfikować DNA możemy in vivo i taka metoda nosi nazwę
klonowania ,do tego celu naszymi niewolnikami są bakterie, które tresujemy w taki sposób,żeby
z własnym chromosomem bakteryjnym replikowały w kolejnych cyklach podziałowych
wprowadzony przez nas DNA w formie wektora plazmidowego.Metoda ta znalazła zastosowanie
głównie w biotechnologii i w produkowaniu enzymów czy hormonów.Oszukujemy bakterię i ona
poprostu odtwarza nasze DNA.Natomiast calkowity przełom nastąpił w wyniku wprowadzenia
do badań techniki amplifikacji genu in vitro ,która powszechnie znana jest metodą PCR-reakcja
łańcuchowa polimerazy ( polimerase chain reaction )Ale najpierw zajmiemy się tymi metodami,
które dotyczyły materiału niezamplifikowanego.Pierwszy etap,gdzie następuje hybrydyzacja z
sondą molekularną. Dawniej stosowane były metody hybrydyzacji w roztworach , ale były
uciążliwe,więc opracowano metodę przenoszenia kwasów nukleinowych na nośniki stałe.Tym
stałym nosnikiem jest nitroceluloza lub nylon .Możliwe było immobilizowanie materiału
genetycznego i działanie sondą molekularną w fazie stałej i obecnie te metody mają największe
zastosowanie.No i wreszcie pieśń ostatnich lat to jest technika FISH ( fluorencens insitu
hybrydysation ) czyli hybrydyzacja fluorescencyjna in situ,oznacza to, że celem
przeprowadzenia reakcji hybrydyzacji, nie musimy wogóle izolować materiału genetycznego z
komórki, tylko mając preparat cytologiczny na szkiełku podstawowym, działamy sondą
molekularną, ktora na poziomie choromosomu wykrywa nam istniejący allel lub sekwencję
poszukiwaną, informuje nas o tym mówiąc trywialnie świecąc
Ale teraz szczegółowo zajmiemy się hybrydyzacją immobilizowanego DNA. Technika nosi
nazwę blottingu.Blotting jest to trasfer DNA na filtr ( nitrocelulozowy albo nylonowy ) i tu nie
mogą być państwu obce trzy pojęcia .Mianowicie pierwsze to jest Northern blotting -
materialem przez nas poszukiwanym,diagnozowanym jest RNA i RNA jest przenoszone na
odpowiedni filtr i następuje hybrydyzacja z sondą maolekularną którą jest DNA znakowany.
Skoro mamy mRNA to łatwo zgadnąć, że mamy transkrypt a chcemy się dowiedzieć czego to jest
transkrypt? Doszło do ekspresji genu zadajmy sobie pytanie jakiego genu ?Takie jest
zastosowanie techniki Northern blotting.Technika Southern blotting to jest technika,w której
immobilizujemy na filtrze nitrocelulozowym lub nylonowym DNA i hybrydyzujemy go również
sondą DNA , czyli to badanie nie prowadzimy nie na transkrypcie tylko na genie DNA.Jest to
technika szeroko stosowana w identyfikacji osobniczej .No i wreszcie z tymi dwoma pojęciami
trudno pominąć Western blotting, Western blotting nie dotyczy DNA, jest to technika
polegająca na przeniesieniu na podłoże stałe rozdzielonych elektroforetycznie białek .Ale na
podstawie masy cząsteczkowej trudno nam odpowiedzieć na pytanie co to za białko? Takim
jedynym faktycznym sposobem powiedzenia to jest to białko to jest zadziałanie przeciwciałem
monoklonalnym wykrywającym daną sekwencję aminokwasową daną determinantę antygenową i
wtedy białko nasze jest wykrywane przeciwciałem przeciwko białku ( nie ma tu żadnej
hybrydyzacji z sondą molekularną ani kwasów nukleinowych ) I tu mamy naszą technikę
Southern blotting , mamy wysoko cząsteczkowy DNA, który wyizowaliśmy np. z krwi
obwodowej , następnie trawimy je endonukleazami i dokonujemy rozdziału elektroforetycznego,
następnie przenosimy przy użyciu specjalnego urządzenia do trasferu na nitroceluloze albo na
nylon i działamy sondą molekularną w takim piecu hybrydyzacyjnym -tak nazywa się to
urządzenie, ulega to mieleniu przez kilka do kilkunastu godzin i następnie uzyskujemy wzór
autoradiograficzny.Prążki powstałe są to prążki gdzie nastąpiła hybrydyzacja,czyli jeśli mamy
jakiś marker ,który wykrywamy i mamy dla nigo komplementarną sondę molekularną tylko
wtedy kiedy on występuje sonda go rozpoznaje.Jeśli nastąpiła mutacja to sekwencja
nukleotydowa jest inna ,wówczas sonda molekularna z takim fragmentem się nie łączy i prążka

radiograficznego tu nie widzimy.Do dziś technika Southern blotting używana jest bardzo często
w analizie osobniczej jako tzw.DNA fingerprinting ,czyli badanie typu odciskpalca.DNA ulega
trawieniu enzymami często tnącymi, a ponieważ każdy z nas różni się od drugiej osoby
sekwencjami genotypu to każdy ma bardzo charakterystyczny układ prążków.A tutaj mamy
technikę FISH,mamy preparat cytologiczny to nic innego jak chromosomy,podajemy sondę
znakowaną fluorescencyjnym barwnikiem,pozwalamy na hybrydyzację z chromosomem,nie
doprowadzmy do izolowania materiału genetycznego,czyli jaka szybkość potężna tego typu
badania, a następnie w mikroskopie fluorescencyjnym patrzymy czy gdzieś nam świeci, no i
widzimy że tutaj świeci to jakiś marker genetyczny został wykryty.
AMPLIFIKACJA DNA
Pierwszą metodą klonowaniem się nie zajmujemy, celem klonowania jest uzyskanie
zamplifikownego całego fragmentu genu, ponieważ w przeciwieństwie do techniki PCR
fragmenty, które amplifikujemy magą być znacznie większe ,mogą być nawet do 2 000 000
pz,czyli cały gen może ulec amplifikowaniu .Natomiast ograniczeniem techniki PCR jest
wielkość amplifikowanego fragmentu DNA, która nie przekracza 5 000 pz.( 5 kb) No i jest tu
pokazane że musimy nasz DNA wstwić w plzmid, następnie plazmid wstawić w bakterię,
bakteria podczas kolejnych podziałów odtwarza nasz plazmid z zawartym tam genem i następnie
wyciągamy to z bakterii uśmiercając ją, ale on jest takim swoistym perpetum mobile i po
oczyszczeniu wyprodukowanego białka mamy gotową insulinę, albo hormon wzrostu.Natomiast
w laboratorium i to dzisiaj zobaczycie wykonujemy badanie zwane amplifikacją genu in vitro ,
czyli technikę PCR. Do PCR-u nie potrzebujemy żadnych wielkich skomplikowanych urządzeń,
całe urządzenie jest wielkości tego rzutnika i nosi nazwę termocyklera albo termoblocku i w tym
naszym układzie in vitro odtwarzamy dokładnie to co zachodzi podczas replikacji DNA w
komórce, czyli sprawa jest genialnie prosta ,a ponieważ jest genialnie prosta w 19993 k.Bullis
otrzymał nagrodę Nobla.Technika PCR: najpierw musimy wyizolować DNA ( wystarczy DNA z
jednej komórki ) w przeciwieństwie do technik hybrydyzacyjnych możemy miec od 50-100 razy
mniej DNA żeby to badanie wykonać...i dokładnie samą tylko za pośrednictwem
enzymów.Natomiast my do tego celu wykorzystujemy temperaturę.Następnie do naszego
wyizolowanego DNA -musimy wiedzieć co chcemy amplifikować, czyli musimy znać
sekwencję nukleotydów w analizowanym fragmencie -mianowicie jeśli mamy dwie nici DNA
odpowiednio spolaryzowane -tak będzie powstawała nowa nić od końca 5'-to ona musi od
któregoś miejsca zacząć i my musimy wprowadzić rodzj primera , to są kawałki składające się z
kilkunastu kawałków no do 30 nukleotydów, ale po to aby się one nam w tym miejscu
przyłączyły do matrycy ...czyli ograniczającą amplifikowany DNA . Ona musi mieć jak łatwo
zgadnąć wolną grupę 3' , bo inaczej nie mogła by tu działać polimeraza DNA ,czyli powoli
znajdujemy co do tej zupy musimy wrzucić .Musimy wrzucić matrycowy DNA ktory uległ
denaturacji, następnie wrzucamy startery,jak łatwo zgadnąć musimy mieć dwa, bo zrówno
sekwencja na jednej flance jak i na drugiej jest odmienna,czyli mamy dwa startery. Następnie
musimy mieć enzym, który to wszystko będzie robił, czyli polimerazę DNA no i wresz-cie
musimy mieć cegiełki, z których to wszystko jest zbudowane, czyli musimy mieć trójfoforany
dezoksyrybonukleotydów ( dNTP ); Odpowiedni bufor; polimeraza; startery; matryca; -to jest
przepis na naszą zupę, a naszym garnkiem są probówki. No i w kolejnych etapach po związaniu
starterów następuje zwielokronienie naszego analizowanego DNA, który możemy techniką
elektrofortyczną wykryć no a bardziej wyrafinowanym sposobem jest stosowanie użądzenia,
które nam mierzy ile zamplifikowaliśmy DNA. Reakcja przebiega w następujących etapach:
-denaturacja, która zachodzi w 95
o
C, dla konkretnej reakcji jest ona konkretnie dobierana,ale z
reguły jest to ta temperatura, kiedy nici się od siebie oddzielają
-następnie temperaturę obniżamy, do jakiej za chwilę powiem, aby dołączać startery, i to łączenie
starterów nosi nazwę annealing (czytaj aniling). Każdy DNA ma określoną temperaturę
topnienia, którą oznaczamy T
m
i temperatura annealingu jest dobierana dla każdego układu

eksperymentalnie lub na podstawie pewnych kalkulacji i wynosi o 5
0
mniej w stosunku doT
m
.
Jeśli temperatura jest zbyt niska następuje nieswoiste przyłączanie różnych nukleotydów, a nie
tych, na których by nam zależało.
-po przyłączeniu starterów następuje podwyższenie temperatury i w tej temperaturze zaczyna
działać polimeraza DNA, która wydłuża łańcuch.
No i reakcja zaczyna przebiegać od początku, podwyższenie tem. , denaturacja, itd ok. 25-35 razy
co zajmuje ok. 3 h . Następnie urządzenie można zostawić na noc , schładza nasz preparat do tem.
4
0
C. Ta temperatura denaturacji ( T
m
) zależy od :
-długości łańcucha, im łańcuch dłuższy, tym trzeba przyłożyć więcej energii żeby go
zdenaturować
-skład zasad, czyli odsetkowego % par G-C, które są złączone 3 wiązaniami wodorowymi, a para
A-T dwoma, czyli trzeba więcej energii do denaturacji
-środowiska chemicznego. Głównym czynnikiem stabilizującym są jony sodu, natomiast
głównym czynnikiem destabilizującym są czynniki denaturujące, czyli najczęściej używany jest
mocznik
Jakie jest główne ograniczenie tej metody:
-no oprócz trgo, że musimy znać sekwencje genu, a właściwie flankujące, a tego w klonowaniu
nie potrzebujemy znać
-drugim ograniczeniem jest wielkość, jak powiedziałem nie może przekraczać 5 tys. zasad
-trzecim ograniczeniem jest to, że ta polimeraza popełnia błędy. Po 20 obrotach reakcji PCR, w
40% zawartych tam zamplifikowanych odcinków DNA są błędy. Dlaczego tam są błędy. Otórz
polimerazy DNA poza dwoma tu wymienionymi, które używane są w PCR, nie posiadają
aktywności 3' - 5' nukleazy, czyli nie mają tego fragmentu, który w polimerazie bakteryjnej, czy
w somatycznych komórkach eukariota, sprawdza, czy nukleotyd został wbudowany w sposób
prawidłowy. Ale ponieważ w pozostałych cząsteczka DNA wbudowanie nieprawidłowego
nukleotydu następuje w sposób losowy, a nie zawsze w tym samym miejscu - nieprawidłowego,
dlatego w dalszej analizie sekwencyjnej nie ma to większego znaczenia. Ale proszę o tych
pomyłkach pamiętać.Najbardziej historyczną polimerazą w reakcji PCR był fragment Klenova
poli- merazy DNA I , ale ten enzym ma to ograniczenie, że jest enzymem termolabilnym. A
ponieważ reakcje przeprowadzamy w zmiennej temperaturze, dochodzącej do 95
0
C, to z czasem
dochodziło do denaturacji enzymu i należało wlewać kolejne jego porcje, co znacznie podrażało
jego badanie. Więc przypomniano sobie, że są organizmy, które lubią wysoką temperaturę,
żyjące w ciepłych źródłach, geizerach. I wykryto Thermus aquaticus, która cechuje się
występowaniem polimerazy termicznie stabilnej i stąd używanie w PCR polimerazy Taq . Inne
polimeraza używane są rzadko, ze względu na to, że są kosztowne, np. Vent i Pfu, które mają
aktywność 3' 5' nukleazową, czyli spraw- dzają, czy prawidłowo włączyły nukleotyd do
wydłużanej nici. Natomiast korzyścią ze stosowania polimerazy Bst jest możliwość amplifikacji
bardzo długich fragmentów dochodzących do 7 tys. par zasad. Czyli jeśli wybieramy jakiś
fragment genu, który musi ulec amplifikacji i jest długi, wtedy uciekamy się do takiej polimerazy.
A co robimy, jeśli nie mamy pieniędzy na polimerazę Bst, a materiał, który mamy analizować
dalej jest długi, to wtedy amplifikujemy poszczególne fragmenty genu, ale tak wybieramy , żeby
poszczególne fragmenty zachodziły na siebie, jeśli mamy amplifikować bardzo duży gen do
badań, oczywiście nie do techniki inżynierii genetycznej, bo do tego używamy klonowania.Jakie
jest zastosowanie techniki PCR:
- wykrywanie mutacji i tym się zajmiemy przedewszystkim.
Metody używane w wykrywaniu mutacji dzielimy na :
> metody Screening-owe, które mają odpowiedzieć na pytanie : czy w analizowanym genie
wystąpiła mutacja ?. W przypadku pozytywnej odpowiedzi na to pytanie zadajemy drugie : Na
czym ta mutacja polega ? Czyli ją identifikujemy, a jeśli już ją zidentyfikujemy i okazuje się , że
ona często współistnieje, ta konkretna mutacja, ze znaną chorobą genetyczną to ustalamy dalszą
metodologie dla wyszukiwania taniego i szybkiego tej konkretnej mutacji w całej populacji.
Wtedy jak już wiemy na czym polega, nie rozpoznajemy u każdego pacjenta, czyli nie
rozpoczynamy od screeningu, tylko wprost wykonujemy metodę na wykrywanie tej konkretnej
mutacji.
Metody, które mają zastosowanie w diagnostyce mutacji :
1.PCR - HD
2.PCR - SSCP
3.PCR - DGGE
To są metody, które państwa obowiązują i jeszcze dwie, na temat których nic nie powiem, bo to
jest dla ultra zainteresowanych. I wreszcie obowiązuje państwa metodologia Sekwencjonowania
DNA.
1. PCR - HD czyli PCR heterodupleks
Po reakcji PCR, doprowadzamy do denaturacji produktu PCR. A następnie doprowadzamy do
renaturacji. Nici łączą się ze sobą na chybił - trafił w sposób losowy. Co się dzieje, jeżeli osoba
jest heterozygotą, czyli w jednym genie wystąpiła mutacja, to wówczas możemy mieć takie
możliwości
-albo połączą się dwie nici prawidłowe
-albo połączą się dwie nici zmutowane
-albo połączy nam się jedna nić prawidłowa i jedna nić zmutowana - to nazywamy
hetrodupleksem
Następnie poddajemy naszą mieszanine kombinacji - elektroforezie. W elektroforezie obserwuje-
my, że heterodupleksy są najbardziej leniwe i najwolniej migrują w polu elektrycznym.
Ograniczenia metody :
-jeśli fragmenty mają poniżej 200pz to skuteczność metody ulega obniżeniu, przy długości
fragmentów do 200pz skuteczność metody wynosi 95 %
-wykrywa mutacje punktowe, dlaczego jest to takie ważne. Gdybyśmy w przypadku mutacji
punktowych wyizolowali DNA i badali wyłącznie techniką elektroforezy, to zmiany w masie
cząsteczkowej analizowanego preparatu są tak małe, że nie jesteśmy wykryć zmian w ruchliwości
elektroforetycznej.
-nie określa nam, w którym miejscu wystąpiła mutacja, bo odpowiada na pytanie czy wystąpiła .
2.PCR - SSCP czyli single strended comformation polimorfism. Polimorfizm fragmentów jedno-
niciowych.To samo mamy zamplifikowany produkt, poddajemy go denaturacji, czyli uzyskujemy
fragmenty jednoniciowe a następnie fragmenty jednoniciowe rozdzielamy elektroforetycznie, w
warunkach żelu niedenaturującego. Dlaczego jest to takie istotne ? Nasze fragmenty jednoniciwe
w warunkach niedenaturujących mogą wykazywać pewne wewnętrzne sparowania. Przyjmą taką
ciekawą konformację. Jeżeli nastąpi tu w którymś miejscu mutacja, ten konformer przyjmie inny
kształt niż wszystkie pozostałe, w związku z czym w warunkach żelu niedenaturującego jego
ruch-liwość elektroforetyczna będzie zależała od dwuch czynników : jego masy cząsteczkowej i
od jego konformacji przestrzennej. Jeżeli zastosowalibyśmy żel denaturujący np. z dodatkiem
mocznika to wówczas nastąpi zarwanie tych wiązań wewnętrznych i jedynie ruchliwość
elektroforetyczna będzie zależała od masy cząsteczkowej, a ta w przypadku mutacji punktowych
ulega bardzo niewielkim zmianom. Jeśli ulega wypadnięciu duża część naszego genu to wpływa
znacznie na zmianę masy cząsteczkowej, to do żadnej z tych metod nie musimy się uciekać tylko
dokonujemy zwykłej elektroforezy, bo wtedy różnica w ruchliwości jest widoczna "gołym
okiem" i z taką najprostszą techniką również się na ćwiczeniach zapoznacie : jest to badanie
polimorfizmu genu konwertującego, czyli AC. Mniejwięcej właściwości podobne do HD:
-analiza fragmentu do 200pz
-wykrywa mutacje punktowe
-nie wykrywa miejsca mutacji

3. PCR - DGGE czyli Denaturating gradient gel elktroforesis, czyli elektroforeza w gradiencie
czynnika denaturującego. Fragmenty dwuniciowe rozdzielamy elektroforetycznie, a w żelu
wytwarzamy gradient albo chemiczny albo temperaturowy. Nasz fragment dwuniciowy wędruje
tak długo dopuki nie trafi na takie stężenie czynnika denaturującego, który sprawia, że nici
denaturują, czyli się oddzielają i wtedy fragment dalej już nie wędruje. Jeśli nastąpiła mutacja
punktowa powoduje to, że miejsce wędrowania następuje do innego punktu, niż się to dzieje w
przypadku gdy mutacji nie ma .Jest trudniejsza do wykonania, ale ma taką samą wartość jak
wcześniej opisane.
I tu jeszcze nasze pozostałe dwie metody
4.PCR - CCM, czyli chemical cleaning of mishmashes, czyli chemiczne trawienie miejsc zmuto-
wanych. kosztowna i nie powszechnie używana, a jej zaletą są dwie rzeczy, po pierwsze można
analizować znacznie większe fragmenty do 1000pz, i można się zorientować, w którym miejscu
nastąpiła mutacja. Jeżeli nastąpiła w jakimś miejscu mutacja, chemicznie można to miejsce
odpowiednio modyfikować, i albo chemicznie, albo enzymatycznie, w tym miejscu nasz
powstały DNA strawić . I lokalizując w elektroforezie, wiedząc jaka jest ruchliwość
elektroforetyczna frag-mentów można dowieść, w którym miejscu nastąpiła mutacja, w
następstwie której doszło do strawienia DNA w tym miejscu naszego analizowanego genu.
5.PTT, czyli Protein truncation test . Niezbyt popularna ze względu na cenę i to, że jest jeszcze
w fazie eksperymentu. Jest to metoda, która wykrywa białka niekompletnie zsyntetyzowane, w
skutek wystąpienia w DNA mutacji nonsensownej. Metoda jest intelektualnie interesująca,
bowiem nasz wyizolowany DNA, przepuszcza się w komórce w układzie in vitro przez
syntetyczny układ transkrypcyjno-translacyjny. W wyniku tego uzyskujemy w układzie in vitro -
białko . I analizując to białko pod względem jego masy cząsteczkowej, determinant
antygenowych możemy stwierdzić, czy zaburzenia w produkcji białka są wynikiem oddziaływań
supresorowych ( mutacja nonsen-sowna ) czy też nie. Metoda kosztowna mało stosowana
No i wreszcie jak już odkryjemy, że gdzieś nastąpiła mutacja tylko jeszcze nie wiemy jaka, to
kolejną rzeczą jaką musimy ustalić to jest, w którym miejscu nastąpiła.Czyli DNA musimy
poddać sekwencjonowaniu . Isnieją dwie metody :
1.Metoda chemiczna Maxama - Gilberta - rzadko stosowana,
2.Metoda Sangera zwana inaczej Dideoxy . Dlaczego się tak nazywa. Do naszego układu
reagu-jącego wprowadzamy takie oszukańcze nukleotydy. To są nukleotydy, z których usuwamy
grupę hydroksylową z 3 atomu dezoksyrybozy , a wiemy, że polimeraza musi mieć wolną grupę
3' OH dla wytworzenia wiązania fosfodiestrowego. Czyli po wprowadzeniu takiego fałszywego
nukleotydu, następuje terminacja replikacji. Proces prowadzimy w sekwenatorach, urządzenia do
sekwencjonowania DNA właśnie tak się nazywają. I z reguły aparaty te pracują w takim
systemie, że na 4 ścieżkach, mamy cztery niezależne elektroforezy. A przedtem zanim do tych
ścieżek dojdzie, mamy cztery niezależne PCR robione. W każdym z tych czterech PCR-ów inny
nukleo-tyd jest zmodyfikowany. W reakcji zwanej A - wprowadzona jest adenina dideoxy (ddA )
na inną ścieżkę ddC i dalej tymina ( ddT ) i guanina ( ddG) . W momenci, w którym polimeraza
chce wbudować taki nukleotyd następny nie jest już wbudowywany, ponieważ nie ma grupy 3' i
repli-kacja staje. W wyniku elektroforetycznego rozdziału te znakowane, zmodyfikowane
nukleotydy mogą być znakowane barwnikami fluorescencyjnymi i tak najczęściej jest, detektor
laserowy ustawia nam sekwencje pokolei dołączanych nukleotydów, którą odczytujemy za
pomocą software-u np. wydrukowane na drukarce. I teraz możemy nasz odczyt porównać z tzw.
bibliote- ką genów, czyli z genami prawidłowymi szczepu dzikiego.
No i teraz tak . Wiemy na czym polega mutacja i mamy 1000 osób do przebadania na obecność
tej mutacji i tu używamy już innych technik. I znowu kilka metod:
1.PCR-RFLP, czyli PCR sprzężony z polimorfizmem długości framentów restrykcyjnych

2.PCR-ASO
3.PCR-ARMS
4.OLA
1 i 2 mają największe znaczenie
1.PCR-RFLP. Kożysta z enzymów restrykcyjnych tzw. endonukleaz restrykcyjnych, które
pozyskiwane są z komórek bakteryjnych i one występują u bakterii. W chwili obecnej znanych
jest kilka tysięcy endonukleaz, ale używanych jest zaledwie kilkaset. Dlaczego ? Ponieważ
niektóre, rzadko występujące sekwencje, są trawione przez tak unikalne enzymy, że koszt ich
zastosowania, wychodowania odpowiedniego szczepu bakterii, wielokrotnie przekracza sens
analizy genetycznej. I to jest jedyne, aczkolwiek istotne ograniczenie. Praktycznie każda
sekwencja DNA ma swoją endonukleazę, która ją trawi. Dlaczego one nie trawią własnego DNA.
Ponieważ ich DNA jest zmodyfikowane przy użyciu enzymów zwanych metylazami . A
normalna funkcja, bo ktoś zapyta po co one właściwie są w tych komórkach bakteryjnych, no
przecież nie po to, żebyśmy się nimi bawili w zakresie biologii molekularnej. One chronią DNA
komórki bakteryjnej przed atakiem bakteriofagów. Dany bakteriofag, który ma szczególną
predylekcję do infekowania komórki bakteryjnej, komórka bakteryjna ma na niego oręż w postaci
endonukleazy restrykcyjnej, jak tam się pojawia to w łeb dostaje taką endonukleazą, bo jego
DNA zostaje obrucone w niwecz. One nie trawią DNA jakkolwiek. One trawią w tzw. miejscach
palindromowych, czyli o podwójnej odwróconej symetrii i co ważne trawią obydwie nici . Nazwa
każdej z nich utworzona jest od nazwy gatunku bakterii, w którym dana endonukleaza została
znaleziona Eco R1 - Escherichia coli szczep R enzym pierwszy. Obok są zawsze podawane
sekwencje, które są rozpoznawane i trawione, i jak wiemy, w którym miejscu nastąpiła mutacja,
następnie bierzemy katalog, patrzymy za ile pieniędzy można taką endonukleazę przysłać.
Endonukleazy albo są w roztworze albo w formie liofilizowanej. No i każda ma swoją
specyfikację : w jakiej tem. trawii, itp. W wyniku tra-wienia endonukleazami powstają tzw.
fragmenty restrykcyjne , które rozdzielane elektrofore-tycznie dają obraz charakterystyczny dla
danego genu. Inny obraz da DNA zmutowany a inny zdrowy, bo w wyniku działania mutagenu
jedne miejsca restrykcyjne mogą zaniknąć a inne się pojawić. Jeśli wiemy na czym mutacja
polega to możemy wybrać taką endonukleazę, która właśnie miejsce zmutowane będzie nam
cieła. A w innym przypadku wybieramy taką, która w wyniku mutacji straci swoją sekwencję
palindromową. Miejsce cięcia endonukleazą może przechodzić albo przez oś symetrii takiej
sekwencji palindromowej i powstają wówczas końce tępe no i wreszcie kiedy to jest w pewnym
oddaleniu od tej osi symetrii to powstają końce lepkie , czyli kohezyjne. Zarówno do rearanżacji
genów możemy używać końców tępych jak i lepkich. Te tępe najpierw musimy specjalnie
zmodyfikować, ale najłatwiej postąpić jest z końcami lepkimi, bo wtedy z innym genem można to
przylepić i zmontować.
No i sprawa jest bardzo prosta wyizolowany allel po amplifikacji ( PCR-RFLP) poddajemy
trawieniu endonukleaz, jeśli endo. miejsca nierozpoznał to znaczy, że jest wszystko w porządku
uzyskujemy jednolity obraz prążków .
2.PCR-ASO - allel specyfic oligonukleotyde. Mamy dwa allele, jeden dziki i drugi zmutowany.
Nasze podejście może być dwojakie możemy hybrydyzować: nasz zamplifikowany materiał albo
sondą molekularną, czyli oligonukleotydem specyficznym dla allelu albo dzikiego, albo allelu
zmutowanego. Tak dobieramy długość allelu, że w przypadku gdy trafi na miejsce o niekomple-
mentarnej sekwencji wówczas nie hybrydyzuje. Szczególnie dobre są takie miejsca, które
wypadają w środku naszego oligonukleotydu, bo wywołują sytuację w której następuje szypkie
oddysocjowanie naszego oligonukleotydu i on nie wykrywa naszego miejsca zmutowanego. No
jeśli miejsce zostanie rozpoznane prawidłow to jest jakiś marker radioaktywny lub fluororescen-
cyjny i w ten sposób stwierdzamy, czy miejsce zmutowane występuje czy nie.
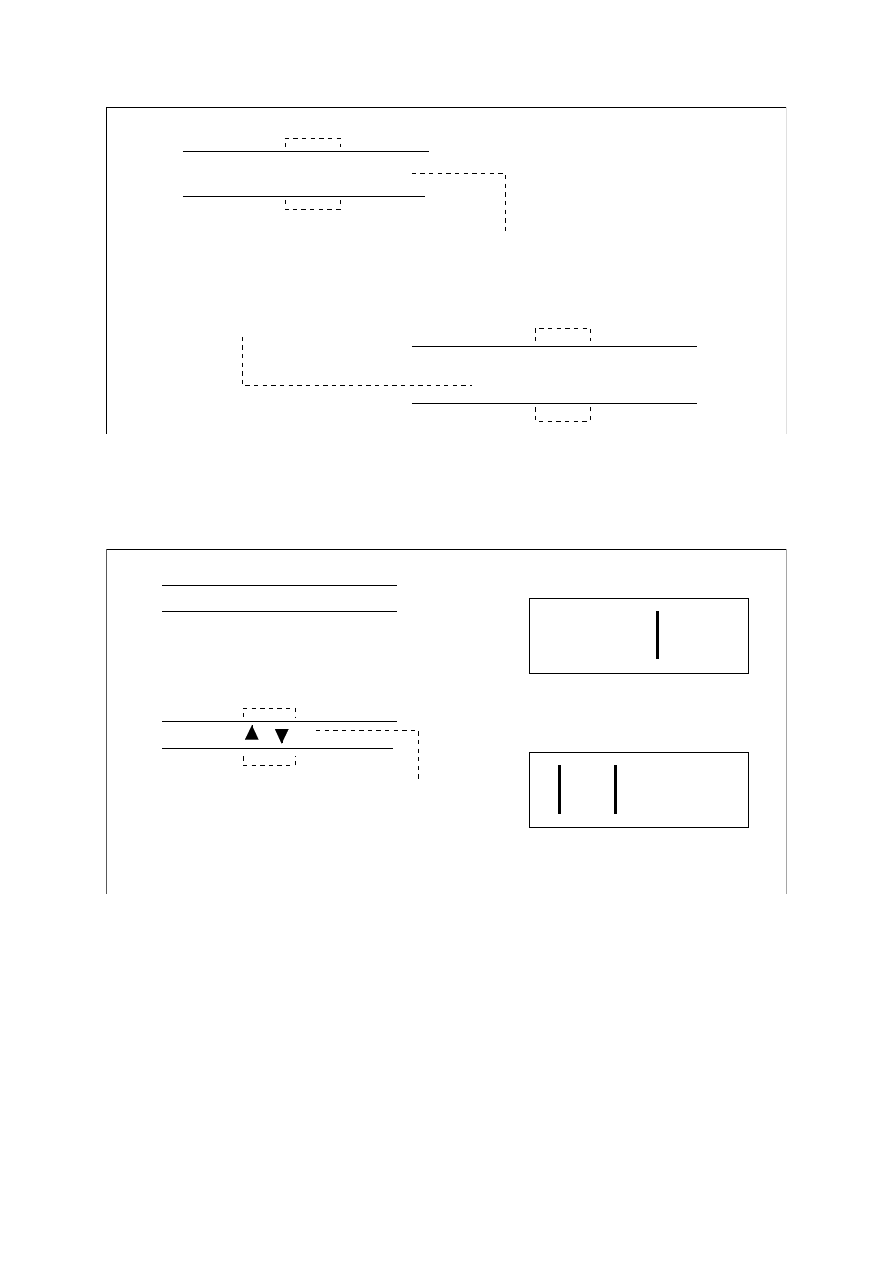
3.ARMS- amplification refractory mutation system, czyli mutacja prowadzi do niemożności wy-
TAGACGTA
ATCTGCAT
Stara prawidłowa nić DNA
( pozbawiona palindromu - restryktaza ją mija )
ATCGGCAT
TAGCCGTA
Sekwencja po mutacji punktowej
( powstał palindrom, który restryktaza rozpoznaje )
1. Sytuacja, w której restryktaza nie przcieła nici
cieńcie w sekwencji
palindromowej
2.Nastąpiło przecięcie nici.
200 zasad
200 zasad
70 zasad 130 zasad
130 zasad 70 zasad
Elektroforeza
1.Sytuacja prawidłowa
2.Sytuacja patologiczna
70 130 200
200

konania PCR-u.Wobraźmy sobie, że tu mamy miejsce prawidłowe i następuje prawidłowy
przebieg PCR-u. Natomiast jeśli nastąpiła mutacja, to wówczas stosowany dla danego allelu
specyficzny starter tego miejsca nie rozpoznaje, związjku z tym nie ma amplifikacji. Czyli
wybieramy miejsce znajdujące się w miejscu wiązania starteru, czyli w sekwencjach
flankujących. Czyli tak dobieramy startery by wykrywały tylko jeden z alleli. Więc w tej sytuacji
naszym oligonukleotydem jest primer . I rozpoznaje tylko jeden z dwóch alleli. Rozpoznał
prawidłowy zachodzi reakcja PCR, nie rozpoznał prawidłowego
PCR
nie zachodzi .
4.OLA- czyli oligonukleotyde ligation assay , czyli ligacja oligonukleotydów. Wybieramy dwa
fragmenty DNA i nazwijmy je umownie sondami, które muszą idealnie komplementować poszu-
kiwane przez nas miejsce - punktową mutację. One są sprzężone z odpowiednim markerem, np.
enzymatycznym i w tym układzie musimy mieć enzym ligazę, jeśli wszystko następuje jak
trzeba, następuje połączenie, czyli ligacja tych dwóch fragmentów oligonukleotydowych i
następuje sygnał enzymatyczny i generacja jakiegoś produktu i sygnał - reakcja zaszła - wszystko
jest OK. Jeśli jest mutacja punktowa nie następuje ligacja półfragmentów i sygnału nie ma.
Cele:
- wykorzystanie technik identyfikacji pozwala np. na postawienie pacjentowi szybko diagnozy w
ciągu kilku godzin, a normalnie by wychodować szczep bakterii z tej pobranej próbki potrzeba 6
tygodni
-synteza genów - klonowanie, PCR synteza genów do celów przemysłu spożywczego, farmaceu-
tycznego . To jest produkcja białek (enzymów ), hormonów i synteza nowych genów, którymi
możemy zastąpić geny zdefektowane, albo zdefektowane w sposób uwarunkowany genetycznie,
dziedzicznie albo zmutowanych sporadycznie i to nosi nazwę terapii genowej .
No i odmianą techniki PCR jest technika RT-PCR można ją z filozofii działania przyrównać do
Northern Blottingu. Tam również mieliśmy RNA, które identyfikowaliśmy za pomocą sondy
molekularnej znakowanej, którym DNA, natomiast tu mamy mRNA, który amplifikujemy, czyli
mamy transkrypt, który amplifikujemy, żeby się dowiedzieć skąd się wziął ten transkrypt. W tym
celu pierwszym etapem jak łatwo zgadnąć jest odwrotna transkryptaza, a następnie uzyskany w
ten sposób DNA amplifikujemy typowo jak to robiliśmy w reakcji PCR.
GENY W ONKOGENEZIE
Geny, które biorą udział w onkogenezie można podzielić na 3 kategorie :
1. Onkogeny, czyli są to takie geny, które stymulują proliferację komórek, można je przyrównać
do pedału gazu w samochodzie. Jak się na taki pedał gazu naciśnie , albo w tym wypadku
zaktywuje taki protoonkogen - komórka w sposób niepochamowany zaczyna proliferować.
2. Antyonkogeny, czyli drugi rodzaj genów, nazywane także genami supresorowymi. I one przy-
pominają hamulec w samochodzie. Normalnie ten hamulec jest zaciągnięty . Komórka rusza,
czyli samochód rusza np. z górki i ten hamulec ulegnie uszkodzeniu
3. Mutatorowe . Geny mutatorowe normalnie biorą udział w naprawie DNA, dbają o
integralność genomu. Jeśli jedziemy to też są swego rodzaju hamulcem jeden to np. hamulec
ręczny a drugi nożny . Jeśli ten hamulec ulegnie uszkodzeniu następuje gromadzenie błędów w
informacji gene-tycznej zawartej w komórce i w konsekwencji prowadzi to do rozwoju
niektórych chorób nowo-tworowych
Zainteresowanie onkogenami spowodowane jest tym, że już w latach 20-tych, stwierdzono, że
niektóre wirusy mogą wywoływać u zwierząt doświadczalnych nowotwory. Takim najpierwej
od-krytym genem był gen Src, który wywołuje mięsaka Russa ( ? ). Tylko retrowirusy, które
posiada-ją tego rodzaju geny, zwane onkogenami, są w stanie wywołać transformację
nowotworową komórki . Natomiast wirusy, które nie posiadają tego rodzaju onkogenów,
transformacji nie wy- wołują np. HIV powoduje niszczenie limfocytów L
H
, ale transformacji
nowotworowej nie wywo-łuje. Warunkiem, żeby retrowirus wywołał transformację jest

posiadanie onkogenu. Ale jak się przyjrzano bliżej tym wirusom onkogennym, że wirusy te
posiadają geny jako żywo przypominają-ce te występujące u człowieka. Powstała więc
koncepcja, że pradziadek albo prababcia odwiedza-jąc komórkę zwierzęcia, na drogę sobie
zabrała taki bagaż w postaci genu prawidłowego, który następnie w wyniku mutacji uległ
pewnym zmianom. Onkogen nie jest do niczego potrzebny wirusowi . Wirus posiada w swoim
genomie geny kodujące jego białka, czyli białka kapsydu, kwasów nukleinowych potrzebne do
replikacji - odwrotna transkryptaza, natomiast onkogen to ten niepotrzebny bagaż. No i z takich
wirusów DNA, które nie posiadają onkogenów, bo onko-geny posiadają jedynie wirusy RNA
retrowirusy, mamy takie, które są szczególnie ważne dla człowieka. I to są dwa :
>HPV 6, który wywołuje raka szyjki macicy
>no i mamy wirus Ebschteina-Barra. Wirus ten wywołuje mononukleozę zakaźną w naszej strfie
klimatycznej. Ale jest on również postrzegany jako czynnik etiologiczny wywołujący raka noso-
gardzieli. Natomiast u dzieci w Afryce równikowej ten sam wirus wywołuje złośliwego
chłoniaka, czyli nowotwór ukł. chłonnego tzw. chłoniaka Bertita (?). Wszystko zależy w jaki
sposób asocjuje z genomem gospodarza. Natomiast najważniejsze retrowirusy tzw. wirusy ostro
trasformujące to są właśnie wirusy onkogenne posiadające onkogeny. I każdy z wymienionych
tutaj onkogenów wirusowych ma swój odpowiednik w komórce gospodarza. U każdego z nas
takie onkogeny występują.
Terminologia:
Jeżeli to co występuje u wirusa nazywamy onkogenem to to co występuje w formie prawidłowej
można nazwać protoonkogennem.
Ale można i mówić inaczej to co występuje w komórce jest onkogennem, a to co powoduje
rozwój nowotworu onkogen zaktywowany.
Te geny, które znajdują się u wirusów poprzedzone są literką ''v'' ( v- )czyli virus, natomiast te
on-kogeny, które występują u nas poprzedzone są literką ''c'' (c-) czyli cellular , czyli
komórkowy.
Co to są za geny ? Są to geny zaangażowane w różne etapy różnicowania, proliferacji komórki i
odpowiedzi komórki na różne sygnały, czyli nie są to jakieś byle jakie geny. Nie są to np. geny
kodujące enzymy glikolizy, nie są to enzymy beta-oksydacji zakodowane, ale transfer sygnału jak
to się mądrze mówi transdukcja sygnału i proliferacji komórki.
Są to na przykład geny :
- kodujące czynniki wzrostu, które regulują proliferację komórek ( Sis )
- kodujące receptory powierzchniowe dla czynników wzrostu . Bo to, że jest czynnik wzrostu to
jest nic. To tak jakby mieć klucz i będziemy potrzebować jeszcze zamek - receptor, bo inaczej to
nie działa (ERBB - gen receptora powierzchniowego )
- kodujące składniki systemu transdukcji ( RAS )
- kodujące czynniki transkrypcyjne, czyli białka oddziaływujące z DNA ( MYC , JUN )
- geny zaagażowane w kodowanie cyklin oraz CDK, czyli Cyklin-depend kinases , kinazy zależne
od cyklin (i tutaj mamy onkogen PRADT 1 ).No i co one takiego ciekawego robią ? Taka jest
choroba -to są mięsaki lub białaczki
-wirusowy onkogen Hau-cis,a on koduje płytkopochodny czynnik wzrostu PDGF łańcuch b
-onkogen Ras -wywołuje u szczurów mięsaka , czuli nowotwór tkanek miękkich , u człowieka
koduje jedno z białek Ras
-onkogen Abl , który u myszy wywołuje białaczkę, u człowieka koduje kinazę tyrozynoswoistą
-onkogen Fos-wirusowy, który wywołuje mięsaka kości, u człowieka zangażowany w
kodowanie kinazy tyrozynoswoistej i czynników transkrypcyjnych
Gdyby to prześledzić na schemacie komórki są to cząstki sygnałowe zaangażowane w receptory
dla cząstek sygnałowych ...

Wyszukiwarka
Podobne podstrony:
BIOCHEMIA KWASOW NUKLEINOWYCH egzamin
BIOCHEMIA KWASOW NUKLEINOWYCHegz
dobroszycki,biochemia L, Izolacja kwasów nukleinowych z komórek?kterii
Skład kwasów nukleinowych, Kosmetologia, Notatki i wyłady, Biochemia
Skrypt - Rozdzial 2 Izolowanie i amplifikacja kwasów nukleinowych CALOSC, materiały medycyna SUM, bi
Hybrydyzacja kwasów nukleinowych, III rok, Genetyka kliniczna
sprawozdanie 3, Właściwości białek i kwasów nukleinowych
Kartkówka Budowa i rola kwasów nukleinowych
hybrydyzacja kwasów nukleinowych
BIAŁEK I KWASÓW NUKLEINOWYCH REAKCJE CHARAKTERYSTYCZNE
analiza kwasow nukleinowych
budowa i rola kwasów nukleinowych
Techniki izolacji kwasów nukleinowych
Lekcja I Skladniki i struktura kwasow nukleinowych (powtorzenie podstawowych informacji
PORÓWNANIE KWASÓW NUKLEINOWYCH
Budowa i funkcje kwasów nukleinowych
więcej podobnych podstron