
1
dr Paweł Golik
dr Ana Stanković
mgr Mateusz Baca
mgr Martyna Molak
Analiza naturalnych populacji – ekologia molekularna. Analiza DNA w
archeologii. Genetyka w Kryminalistyce.
Na ostatnich ćwiczeniach z cyklu „Genetyka z inżynierią genetyczną” chcielibyśmy
przedstawić pojęcie markera genetycznego i opowiedzieć o jego wykorzystywaniu w
różnych dyscyplinach: genetyce populacji (ludzi zwierząt i roślin), genetyce ochronnej,
archeologii i kryminalistyce.
Marker genetyczny
Markerem genetycznym nazywamy każdą sekwencję nukleotydową, której
polimorfizm (zmienność pomiędzy osobnikami lub grupami taksonomicznymi) pozwala
ocenić różnorodność genetyczną.
Rodzaje markerów genetycznych
SNPs (ang. Single Nucleotide Polimorphisms) – mutacje punktowe (substytucje: tranzycje i
transwersje) w sekwencji nukleotydowej. Zaletą ich analizy jest ich ogromna liczba w
genomie - u człowieka kilka milionów. Wykrywanie SNPs opiera się na analizie hybrydyzacji
z oligonukleotydami (krótkie zsyntetyzowane chemicznie jednoniciowe cząsteczki DNA
(poniżej 50 pz), które będą hybrydyzowały jedynie w sytuacji całkowitej komplementarności
z badaną sekwencją DNA).
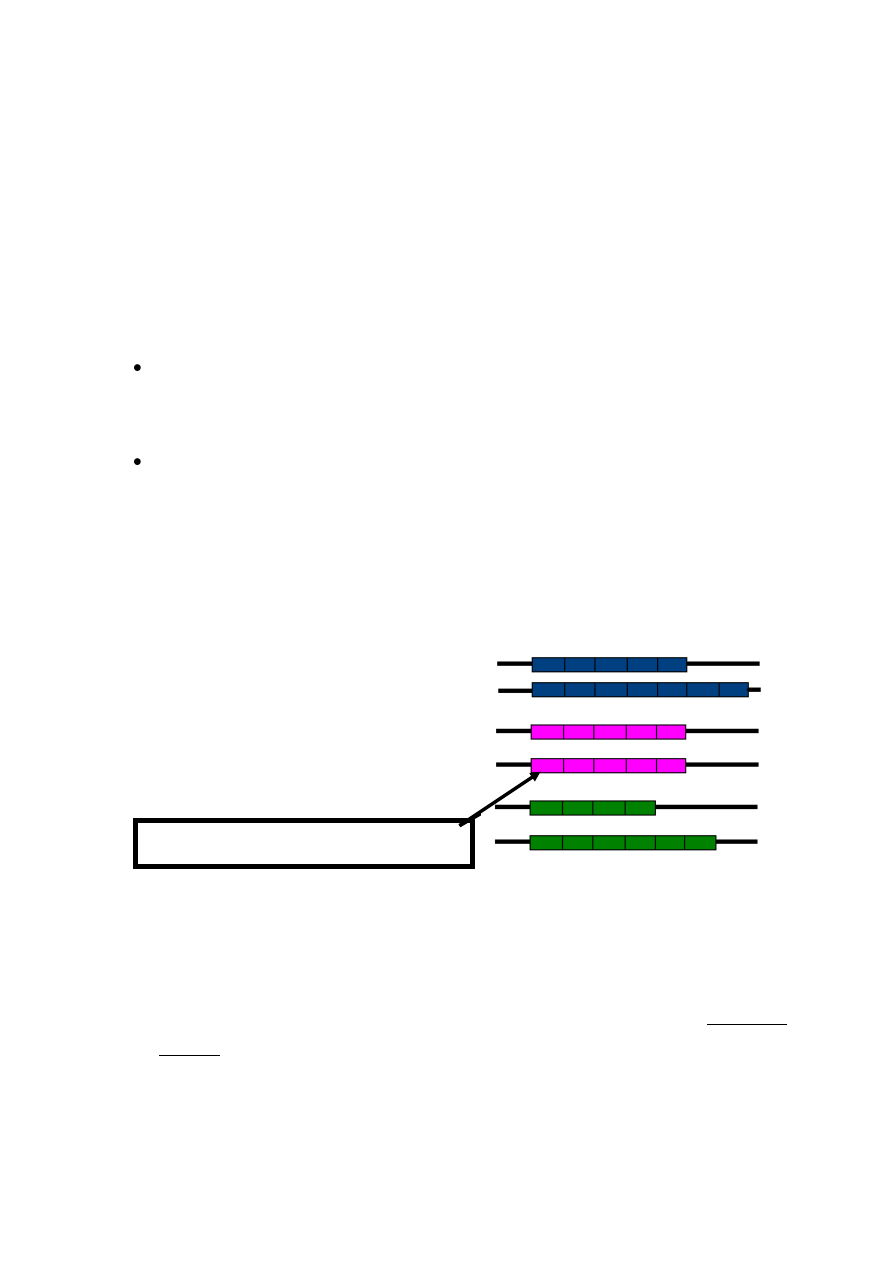
2
SSLP (ang. simple sequence length polimorphism) są szeregami powtórzeń sekwencji - a
więc różnymi allelami zawierającymi różną liczbę jednostek powtarzalnych. Podstawą
zmienności są indele. [Indel – delecja lub insercja. Aby ustalić homologię dwóch sekwencji
nukleotydowych wykonujemy ich przyrównanie (uliniowanie), czyli tzw. ang. alingment.
Podczas przyrównania sekwencji nie można odróżnić od siebie delecji od insercji. Nadaje im
się zatem wspólną nazwę indele].
Minisatelity-VNTR (ang. variable number of tandem repeats) – złożone są z
jednostek powtarzających się, złożonych z kilkudziesięciu nukleotydów (10-100 pz).
Mikrosatelity – STR, msDNA (ang. simple tandem repeats), czyli proste powtórzenia
tandemowe, w których powtarzalny motyw złożony jest z 2 do 6 pz (np. CAAG, TGA
lub CA). Typowe loci mikrosatelitarne zawierają 10-30 (maksymalnie 50) powtórzeń
motywu i osiągają długość 100 do 400 pz. Zlokalizowane są najczęściej w regionach
niekodujących genomu.
Allele mikrosatelitów (warianty długości jednego locus) są kodominujące i
dziedziczą się w sposób mendlowski.
Ze względu na ich budowę, w mikrosatelitach często zachodzą mutacje.
Powtórzenia tandemowe powodują, że polimeraza DNA „ślizga się” (ang. polimerase
slippage), dodając lub opuszczając najczęściej pojedyncze powtórzenie, wydłużając
lub skracając tym samym sekwencję mikrosatelitarną. Oblicza się, że częstość
występowania mutacji w tych markerach u ssaków wynosi ok. 10
-2
– 10
-4
/na
pokolenie. Jest ona zmienna u różnych grup organizmów i wyższa u zwierząt niż u
Powtarzający się motyw np. ATCG
osobnik1
osobnik2
osobnik3
Rys 1. Budowa loci
mikrosatelitarnych

3
roślin. Im więcej powtórzeń znajduje się w danym allelu mikrosatelitarnym, tym
większe prawdopodobieństwo jego mutacji. Stąd po pewnym czasie dla danego locus
(odcinka DNA) występować będzie wiele alleli. Mikrosatelity, w związku z szybkim
tempem mutacji, są bardzo dobrym markerem do badań populacyjnych.
Zastosowanie badań genetycznych w ekologii
Od około 20 lat techniki genetyczne są wykorzystywane w badaniach ekologicznych,
przede wszystkim do ustalania polimorfizmu genetycznego populacji. Czynniki i procesy
ekologiczne determinują strukturę genetyczną. W latach 70. i 80. XX wieku badano przede
wszystkim polimorfizm allozymów. W latach 90. nastąpił gwałtowny rozwój zastosowania
techniki PCR i sekwencjonowania. Obecnie w badaniach populacyjnych wykorzystuje się
najczęściej niekodujące sekwencje DNA, podlegające minimalnie lub wcale presji selekcyjnej
(doborowi naturalnemu). Sekwencje te są z reguły wysoce polimorficzne. Należą do nich w
jądrze sekwencje mikrosatelitarne (msDNA). Z sekwencji mitochondrialnych w badaniach
głównie filogeograficznych i populacyjnych wykorzystuje się cytochrom b (będący wysoce
polimorficznym genem) oraz pętlę D.
Pętla D – (ang. displacement loop, D-loop) zwana też regionem kontrolnym, (ang.
control region) jest najbardziej zmiennym odcinkiem genomu mitochodrialnego. Ma on
długość około 1000 pz. Cechą tego markera genetycznego jest jego neutralność selekcyjna. W
obrębie pętli D mieszczą się trzy hiperzmienne regiony (ang. hipervariable region) HVR1,
Replikacja
Poślizg
Nowy cykl replikacji
+ 1 motyw
- 1 motyw
Rys 3. Przykład poślizgu polimerazy podczas replikacji
(http://www.scielo.br/img/revistas/gmb/v29n2/a18fig02.gif)

4
HVR2 oraz rzadziej wykorzystywany w analizach HVR3, w których mutacje powstają
znacznie częściej aniżeli w pozostałej części regionu kontrolnego. Funkcją D-loop jest
rozpoczęcie replikacji genomu mitochondrialnego. Tutaj mieści się region ori.
Informacje uzyskane w oparciu o analizę sekwencji mtDNA i msDNA wykorzystuje
się w badaniach nad migracjami gatunków, określaniu efektywnej wielkości populacji, jej
heterozygotyczności, pokrewieństwa osobników, dystansu genetycznego między populacjami,
przejścia populacji przez wąskie gardło itd. Pamiętać należy, że mitochondrialny DNA
dziedziczy się wyłącznie w linii matczynej, w związku z czym jego analiza dostarcza
informacji dotyczących jedynie części populacji (samic lub kobiet). W zależności od
charakteru badań wykorzystuje się jeden lub obydwa wyżej opisane markery molekularne.
Genetyka ochronna (ang. Conservation genetics).
Zgodnie z powszechnie akceptowanymi zasadami ekologii na świecie, restytucję bądź
ochronę gatunków powinna poprzedzać staranna analiza genetyczna, której wyniki pozwalają
ocenić, czy zakres polimorfizmu genetycznego introdukowanych lub zachowywanych
populacji jest wystarczający dla powodzenia projektów ochronnych. Zastosowanie metod
genetycznych pozwala poznać różnorodność (polimorfizm genetyczny) populacji, jak i
określić przepływ genów między populacjami. Możliwe jest także ustalenie stopnia i
obecności hybrydyzacji pomiędzy gatunkami autochtonicznymi a pokrewnymi,
introdukowanymi przez człowieka. Wiedza ta jest niezbędna dla prawidłowej odbudowy
populacji gatunku, który na danym obszarze kiedyś występował lub umożliwienie naturalnego
jej odrodzenia się. Wyżej opisane działania zyskały nazwę conservation genetics – w wolnym
tłumaczeniu „genetyka ochronna” (Hedrik, 2001). Genetyka ochronna to połączenie ekologii,
genetyki populacyjnej, modelowania matematycznego, taksonomii i mikroewolucjonizmu.
Poznanie dokładnie struktury ekologicznej i genetycznej pomaga w aktywnej ochronie
gatunków.

5
Genetyka w kryminalistyce.
Identyfikacja osób i określanie pokrewieństwa.
W ostatnich latach, rozwój technik izolacji DNA oraz namnażania i analizy wielu loci
mikrosatelitarnych jednocześnie, dostarczyły niezawodnego narzędzia do ustalania
identyczności lub stopnia pokrewieństwa osobników. Możliwości te wykorzystywane są
dzisiaj w wielu zagadnieniach medycyny sądowej (ang. forensic science) takich jak określanie
związku pomiędzy śladami biologicznymi, identyfikacja ofiar katastrof, ustalanie ojcostwa
lub pokrewieństwa dla celów spadkowych czy imigracyjnych.
Identyfikacja osób – podstawy teoretyczne:
Aby stwierdzić, że przykładowo, plama krwi znaleziona na miejscu morderstwa
pochodzi od konkretnego podejrzanego, trzeba porównać profile mikrosatelitarne uzyskane z
krwi oraz od podejrzanego. Aby uzyskane wyniki były wiarygodne i porównywalne np.
pomiędzy bazami danych w różnych krajach, na potrzeby takich organizacji jak FBI w USA
czy ISFS (ang. International Society of Forensic Science) w Europie stworzono zestawy
autosomalnych loci mikrosatelitarnych (położonych na chromosomach autosomalnych)
wykorzystywane do celów identyfikacyjnych. W USA zestaw ten nazwano CODIS (ang.
Combined DNA Identification System) i zawiera on 13 loci mikrostaelitarnych (CSF1PO,
FGA, TH01, TPOX, VWA, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539,
D18S51, D21S11) oraz fragment genu amelogeniny pozwalający na określenie płci. W
Europie funkcjonuje kilka systemów zawierających podobne zestawy, obejmujące tzw. ang.
European Core Loci set (FGA, TH01, VWA, D2S1338, D3S1358, D8S1179, D16S539,
D18S51, D19S433, D21S11 oraz fragment genu amelogeniny)
*
.
Analiza zestawu loci mikrosatelitarnych, w wyniku której otrzymuje się profil
mikrosatelitarny danej osoby nazywa się DNA fingerprinting (genetycznym odciskiem
palca).
Jeżeli obydwa uzyskane profile są jednakowe (tzn. posiadają identyczne allele we
wszystkich loci), mamy podstawy by sądzić, że pochodzą one od jednej osoby. Jednak
możliwe jest, że 2 lub więcej niespokrewnionych osób w populacji posiada taki sam profil
*
znajomość nazw loci wykorzystywanych w opisanych zestawach nie jest wymagana

6
genetyczny zupełnie przypadkowo. Prawdopodobieństwo takiego zdarzenia określane jest
jako random match probability (mp, prawdopodobieństwo przypadkowej zgodności). Takie
prawdopodobieństwo obliczane jest za każdym razem dla danego profilu genetycznego na
podstawie danych populacyjnych z danego regionu. Prawdopodobieństwo przypadkowego
pojawienia się danego profilu u dwóch niespokrewnionych osób w populacji zależy do ilości
wykorzystanych loci oraz ich zmienności w populacji. Dla zestawu CODIS wynosi ono
średnio 5 x 10
-15
a dla dostępnych komercyjnie zestawów sięga nawet 7,2 x 10
-19
. Innymi
słowy oznacza to, że konkretny profil genetyczny wystąpi raz na miliony miliardów osób, a
jest to liczba znacznie większa niż kiedykolwiek żyło ludzi na Ziemi. Zapewnia to prawie
100% pewność, że zgodne (identyczne) profile należą do jednej osoby. Jednak tak, jak z
całkowitą pewnością można stwierdzić, że profile nie należą do tej samej osoby (nie są
zgodne), tak prawdopodobieństwo, że profile pochodzą od jednej osoby nigdy nie osiąga
100%.
W kryminalistyce często wykorzystuje się mikrosatelity zlokalizowane na
chromosomie Y. Takie analizy wykonuje się szczególnie w przypadku przestępstw na tle
seksualnym, kiedy uzyskana próbka jest mieszaniną materiału pochodzącego od kobiety i od
mężczyzny i nie udaje się uzyskać czystego profilu mikrosatelitów autosomalnych sprawcy.
Specyficzna amplifikacja mikrosatelitów zlokalizowanych na chromosomie Y pozwala na
uzyskanie profilu, który można porównać z profilami podejrzanych. Wadą markerów na
chromosomie Y jest to, że dziedziczą się niezmienione z ojca na syna, przez co nie można
odróżnić krewnych w linii męskiej. Z tego powodu z reguły traktuje się je jako tzw. markery
wykluczające. Oznacza to, że można jedynie wykluczyć podejrzanego, kiedy profile są
niezgodne.
Określanie pokrewieństwa
Jeżeli dwie osoby są ze sobą spokrewnione, to posiadają pewną część jednakowych
alleli. Allele jednakowe u dwóch osób, kiedy ich identyczność wynika z pokrewieństwa (czyli
odziedziczenia tego samego allelu po wspólnym przodku), określa się jako IBD (ang.
Identity-by-descent, identyczne poprzez pochodzenie). Przeciwieństwem takiej sytuacji jest
występowanie identycznych alleli u dwóch osób, kiedy to allele te nie pochodzą od jednego
przodka/chromosomu. O takich allelach mówimy, że są IBS (ang. Identity-by-state).
7
Posiadając profile genetyczne dwóch osób, można określić na podstwie frekwencji
poszczególnych alleli w populacji, jaka część ich alleli jest wspólna ze względu na
pochodzenie (IBD) i na tej podstawie określić stopień pokrewieństwa pomiędzy tymi
osobami.
Określanie pokrewieństwa jest wykorzystywane przy testowaniu ojcostwa, sprawach
imigracyjnych czy identyfikacji zmarłych. Wykorzystuje się je także w badaniach nad
restytucją gatunków. Przykładowo ważne jest określenie stopnia pokrewieństwa narybku,
którym prowadzi się zarybienia. Ponadto zaplanować można dokładny schemat krzyżowań
osobników przeznaczonych do rozrodu, by uniknąć chowu wsobnego.
Przypomnieć sobie:
Jądrowy i mitochondrialny DNA (charakterystyka ogólna, szczególnie człowieka),
prawo Hardy’ego-Weinberga
Literatura:
„Genomy” T.A. Brown,
PWN (2009) str. 598-622 (rozd. 19)
lub
PWN (2001) str. 400-421 (rozd. 15)
„Genetyka molekularna” P. Węgleński i in., PWN (2006) str. 430-474 (rozd. 11) –
szczególnie zwrócić uwagę na koncepcję „Pożegnania z Afryką” (ang. „out of
Africa”, rozd. 11.3.8)
Literatura uzupełniająca – mini i mikrosatelity: „Genomy” T.A. Brown – PWN (2001) –
str. 20-22, 136-137 lub (2009) str. 68-72, 217-218.

8
MATERIAŁ DODATKOWY:
Na podstawie zmian mutacyjnych (substytucji oraz indeli) - przede wszystkim w
obrębie HVR1, utworzono haplogrupy, czyli grupy sekwencji (haplotypów) o pewnym
wspólnym wzorze nukleotydów (np. u ludzi: haplogrupy A,B,C,D itd.). Wewnątrz każdej
haplogrupy istnieją haplotypy, zawierające pewne wspólne podstawienia nukeotydowe,
specyficzne dla danej haplogrupy. Haplotyp jest właściwym określeniem w stosunku do
mtDNA (nie allel!). Wszystkie geny genomu mtDNA są dziedziczone wspólnie, zatem
odmiany sekwencji są haplotypami. Ponadto na podstawie danych tysięcy sekwencji
nukleotydowych ludzi stworzono mapę migracji ludzkości na świecie. Podobne badania
przeprowadza się na innych gatunkach rekonstruując ich historię i ewolucję. Mogą to być
badania filogeograficzne (np. rozprzestrzenianie się populacji po zlodowaceniach) oraz
filogenetyczne (czyli rekonstrukcja pokrewieństwa różnych taksonów).
Wykorzystanie loci mikrosatelitarnych w badaniach populacyjnych
Mikrosatelity znajdują zastosowanie w badaniach mechanizmów ewolucyjnych, które
zachodzą w stosunkowo krótkim czasie. Za ich pomocą analizuje się dystans genetyczny
pomiędzy populacjami. Ponadto bada się procesy demograficzne, takie jak gwałtowne
wahania liczebności.
Mogą one być spowodowane efektem wąskiego gardła (ang. bottleneck) lub efektem
założyciela (ang. founder effect), czyli kolonizacją małą liczbą osobników. Na takie populacje
silnie działa dryf genetyczny (losowe utrwalanie się – fiksacja (ang. fixation) lub eliminacja
części alleli oraz zmiana ich frekwencji) ze względu na odtwarzanie populacji z małej liczby
przypadkowych osobników.
Z kolei czynniki behawioralne, do których należy dobór płciowy, terytorializm,
decydują o tym, które genotypy (określone zestawy alleli danych genów w osobniku) będą
uczestniczyły w rozrodzie, a tym samym zostaną przekazane do kolejnych pokoleń. Na tym
poziomie rozgrywa się tzw. selekcja określonych fenotypów, które jednak mają swoje
odzwierciedlenie w konkretnym genotypie. Poligamia również jest źródłem ograniczenia
polimorfizmu danej populacji, szczególnie mało liczebnej.
Szczególne zastosowanie znajdują badania polimorfizmu mikrosatelitów u gatunków
zagrożonych wymarciem, które często są populacjami izolowanymi o ograniczonym
polimorfizmie i przepływie genów. Poznanie dokładnie ich struktury ekologicznej i
genetycznej pomaga w aktywnej ochronie tych gatunków. Migracyjność osobników

9
przeciwdziała wsobności (ang. inbreeding) i sprzyja wymianie materiału genetycznego (ang.
outbreeding). Wykorzystując sekwencje mikrosatelitarne, możliwe jest wykrycie migrantów
lub ich potomstwa w danej populacji oraz wskazanie, z jakiej populacji prawdopodobnie
pochodziły.
Z czynników środowiskowych, które silnie oddziałują na strukturę genetyczną
wymienić można stopień izolacji, którego wpływ skutkuje ograniczeniem polimorfizmu
populacji, jeśli jest ona mało liczebna, w związku z ograniczonym przepływem genów. Na
taką izolowaną populację będą silniej oddziaływać czynniki losowe, takie jak np. dryf
genetyczny. Nasilone kojarzenie wsobne w tych populacjach może prowadzić do załamania
się populacji (ang. inbreeding depression). Możliwe jest określenie efektywniej wielkości
populacji (N
E
), która jest wyznacznikiem kondycji genetycznej danej populacji. Jej wartość
zwykle drastycznie maleje w wyniku wspomnianych wyżej efektów wąskiego gardła,
założyciela czy występowania wsobności. Czasami jednak kojarzenie wsobne może mieć
działanie korzystne dla populacji (ang. puring selection). Jako że w takiej populacji większość
osobników to homozygoty pod względem wielu genów, w wyniku selekcji pozostaną jedynie
osobniki najlepiej przystosowane do lokalnych warunków środowiska. Większe jest też
prawdopodobieństwo wyeliminowania alleli genów powodujących choroby genetyczne.
Krzyżowanie się w takiej sytuacji osobników z dwóch populacji przystosowanych do
odmiennych warunków środowiska, może mieć skutek w gorszym dostosowaniu potomstwa
poprzez rozrywanie korzystnych kombinacji alleli różnych genów (ang. outbreeding
depression). Z drugiej strony, taka pozbawiona polimorfizmu populacja jest mało odporna na
zmiany warunków środowiska, np. pojawienie się nowego czynnika chorobotwórczego. Poza
tym łatwiej dochodzi do ujawniania się recesywnych chorób genetycznych.
Analiza mikrosatelitów
Obecnie mikrosatelity analizuje się za pomocą elektroforezy kapilarnej (ang. capilary
electrophoresis) w sekwenatorze. W tym celu jeden z 2 starterów znakuje się barwnikiem
fluorescencyjnym. Ustalanie długości produktów PCR polega na rejestrze czasu, jaki upływa
od rozpoczęcia elektroforezy w żelu, do momentu otrzymania sygnału świetlnego,
pochodzącego od fluorescencyjnie znakowanych nukleotydów. Za pomocą standardu, oblicza
się długość analizowanych fragmentów DNA (czas migracji produktu w żelu jest
proporcjonalny do jego długości). Istnieje wiele programów, dzięki którym można ustalić
długości alleli (np. Peak Scanner). Przed analiza statystyczną, należy wykluczyć obecność:

10
tzw. ang. allelic dropout – ADO, obecność tylko 1 allelu (z 2) mającego źródło w
jego preferencyjnym namnażaniu się, najczęściej krótszego (dotyczy to zwykle
alleli powyżej 300 pz),
fałszywych alleli (ang. false alleles, FA) powstających w wyniku błędów analizy
artefaktów elektroforetycznych (ang. electrophoresis artefacts, EA) powstających
między innymi na skutek błędów sekwenatora,
alleli zerowych (ang. null alleles, NA) – braku odczytu istniejącego allelu
mającego źródło w wyniku zmiany sekwencji w miejscu przyłączania się startera,
na skutek mutacji.
Wykonuje się też analizę ewentualnego sprzężenia loci mikrosatelitarnych ze sobą
(ang. linked loci) czyli nielosowej dystrybucji alleli (ang. linkage disequilibrium). Może być
ona zaburzona, jeśli dwa loci leżą blisko siebie na jednym chromosomie. W przypadku
obliczeń wykonanych na loci sprzężonych ze sobą można otrzymać fałszywe wyniki.
Wykonuje się analizy używając sprzężonych loci, ale wykorzystuje się wówczas inne
algorytmy. Wymienione artefakty (poza FA) oraz sprzężenie doprowadzają do
przeszacowania liczby homozygot (ang. false homozygote, FH).
Równowaga Hardy’ego-Weinberga (HWE) a analizy struktury genetycznej populacji
W populacjach znajdujących się w stanie równowagi zgodnie z prawem
Hardy’ego-Weinberga nie obserwuje się wysokiej częstości mutacji oraz zjawisk selekcji,
migracji i dryfu genetycznego. Warunkiem dochodzenia do stanu tej równowagi jest także
losowość kojarzenia się osobników oraz ich wystarczająco duża liczba. Odstępstwa od HWE
świadczą o obecności procesów demograficznych, które zmieniają (zaburzają) frekwencje i
rozkład alleli.
Polimorfizm loci:
W badaniach zmienności (polimorfizmu) loci mikrosatelitarnych wykorzystuje się
następujące wskaźniki:
Liczba alleli w danym locus (Na)
Bogactwo alleliczne (R) – uwzględnia wielkość próby, przyrównując do populacji o
najmniejszej liczebności

11
Liczba alleli prywatnych (Np) – liczba alleli charakterystyczna tylko dla danej
populacji.
Heterozygotyczność o oczekiwana (H
E
) – dla frekwencji alleli danego locus w populacji
szacuje się, jaki powinien być udział heterozygot w warunkach równowagi
Hardy’ego-Weinberga.
Heterozygotyczność obserwowana (H
O
) – oblicza się rzeczywista frekwencje
heterozygot występujących w populacji.
Statystyka Wrighta
Współczynnik wsobności (ang. inbred, fixation index):
F
IS
= 1 – (H
O
/ H
E
) - wsp. inbredu – określa stopień wsobność, czyli proporcję
heterozygotyczności obserwowanej do oczekiwanej w obrębie populacji, losowość
kojarzenia się osobników (panmiksje). Wartości F
IS
< -1; 1 >:
o Nieistotne wartości F
IS
oraz F
IS
= 0 oznaczają, że populacja jest w
równowadze Hardy’ego Weinberga i brak jest struktury wewnętrznej w tej
populacji.
o Istotne statystycznie wartości F
IS
> 0 mogą wskazywać na: efekty wsobności
(ang. inbreeding), istnienie struktury wewnętrznej w populacji (subpopulacji –
efekt Wahlunda), dryf genetyczny, istnienie doboru płciowego, fizyczne
sprzężenie loci, jako że w populacji występuje nadmiar homozygot.
o
Znaczące wartości F
IS
< 0 oznaczają, iż w populacji występuje nadmiar
heterozygot, który może być wynikiem selekcji na heterozygoty albo efektem
wąskiego gardła.
Współczynnik F
ST
– wsp. utrwalenia - określa spadek heterozygotyczności w
subpopulacji w stosunku do całej populacji, na skutek np. selekcji lub dryfu
genetycznego. Jego wartości wskazują, jak intensywny jest przepływ genów
pomiędzy subpopulacjami. Mówi o dystansie genetycznym pomiędzy
subpopulacjami lub populacjami.
Wartości:
o 0 - 0,05 – małe genetyczne zróżnicowanie populacji
o 0,05 - 0,15 – średnie genetyczne zróżnicowanie populacji
o 0,15 - 0,25 – duże genetyczne zróżnicowanie populacji
o 0,25 – bardzo duże genetyczne zróżnicowanie populacji
Wyszukiwarka
Podobne podstrony:
ANALIZA DNA W ARCHEOLOGII
analiza notatki 3 id 559208 Nieznany (2)
instrukcja przeciwpozarowa gene Nieznany
Lab5 Analiza sygnalu mowy Lab5 Nieznany
analiza ilosciowa 6 id 60541 Nieznany (2)
dodatkowe1 analiza 11 12 2 sem Nieznany
4 Analiza progu rentownosci id Nieznany (2)
Analiza finansowa wskazniki cd Nieznany (2)
dodatkowe8 analiza 2011 12 id 1 Nieznany
analiza zwiazkow organiczna id Nieznany (2)
Analiza struktury id 61534 Nieznany (2)
analiza ilosciowa 2 id 60539 Nieznany
analiza sk adu aminokwasowego g Nieznany (2)
B14 analiza plu przedzialy id 7 Nieznany
Cw Analiza finansowa bankow id Nieznany
więcej podobnych podstron