
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
1
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
Wszystkie skały magmowe i metamorficzne odsłaniające się na powierzchni ziemi powstały w warun-
kach wyższych ciśnień i temperatur niż te panujące na powierzchni. W dużym stopniu zbudowane są z
minerałów, które są w warunkach hipergenicznych termodynamicznie nietrwałe (są metatrwałe) i powin-
ny ulec przemianom w produkty trwałe przy 1 atmosferze i około 25
o
C. Jednym z istotniejszych proce-
sów hipergenicznych dających wyraz tej nietrwałości jest wietrzenie. Wietrzenie można zdefiniować jako
mechaniczne rozdrabnianie i chemiczne zastępowanie lub usuwanie (np. przez rozpuszczanie) minerałów
i substancji mineralnych wystawionych na powierzchni ziemi na działanie powietrza, wody, czynników
klimatycznych i żywych organizmów. Czynnikami wietrzenia fizycznego są np. nasłonecznienie, zamróz,
wzrost wytrącających się kryształów, niszcząca działalność mechaniczna roślin i zwierząt, i inne czynniki
opisane w podręcznikach geologii fizycznej (geologii dynamicznej). Prowadzą one do rozdrobnienia czyli
dezintegracji granularnej. Czynnikami wietrzenia chemicznego są procesy rozpuszczania, hydrolizy, utle-
niania/redukcji, i inne reakcje zachodzące (zazwyczaj) na granicy ciało stałe – roztwór wodny, często z
udziałem mikroorganizmów. Woda jest głównym medium pośredniczącym w większości procesów wie-
trzenia i dlatego geochemia wód nieodłącznie wiąże się z geochemią większości procesów hipergenicz-
nych. Wszystkie te procesy odbywają się na granicy minerał – roztwór i zależą zarówno od właściwości
fizycznych i chemicznych ciała stałego jak i cieczy. Poznanie geochemii tych zjawisk miało olbrzymi
wpływ na rozwój nowoczesnej inżynierii mineralnej, technologii oczyszczania wody i ścieków oraz spo-
sobów walki ze skutkami zanieczyszczeń środowiska. Jednocześnie wiedza na temat geochemii procesów
zachodzących w glebach przyczynia się znacząco do postępów w nowoczesnym rolnictwie i walce z gło-
dem na świecie.
7.1. Rozpuszczanie
Minerały rozpuszczalne w wodzie (to znaczy głównie te, które zawierają wiązanie jonowe) w trakcie
rozpuszczania przechodzą całkowicie do roztworu dysocjując na wolne jony. Tak się dzieje np. z solami
(np. halit NaCl, sylwin KCl), które w wyniku rozpuszczania po prostu przestają istnieć. Jest to rozpusz-
czanie KONGRUENTNE („całkowite”).
NaCl <=> Na
+
+ Cl
-
KCl <=> K
+
+ Cl
-
krystaliczne
roztwór
ciało stałe
wodny
Warto zwrócić uwagę na dwie cechy takich reakcji rozpuszczania kongruentnego. Po pierwsze zazwy-
czaj nie zależą one zbytnio od pH roztworu, a po drugie prowadzą do odtransportowania drogą wodną
substancji mineralnych z lądu do morza.
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
2
Reakcje miedzy minerałami a kwaśnymi czynnikami wietrzenia nazywa się reakcjami hydrolizy kwa-
śnej. Najistotniejszym czynnikiem hydrolizy kwaśnej są węglany. Dwutlenek węgla CO
2(gaz)
z powietrza
na kontakcie z wodą rozpuszcza się w niej, w wyniku czego powstaje słaby kwas węglowy nadający czy-
stej wodzie odczyn lekko kwaśny (pH około 5,6).
CO
2(gaz)
+ H
2
O
(ciecz)
<=> H
2
CO
3(roztwór)
H
2
CO
3
<=> H
+
+ HCO
3
-
Hydroliza kwaśna kalcytu:
CaCO
3(c.stałe)
+ H
2
CO
3(roztwór)
<=> Ca
2+
(roztwór)
+ 2HCO
3
-
(roztwór)
Hydroliza kwaśna forsterytu (oliwinu magnezowego):
Mg
2
SiO
4(c.stałe)
+ 4H
2
CO
3(roztwór)
<=> 2Mg
2+
(roztwór)
+ 4HCO
3
-
(roztwór)
+ H
4
SiO
4(roztwór)
Przytoczone powyżej przykłady są również reakcjami całkowitego kongruentnego rozpuszczania (tak jak
w innych reakcjach z udziałem minerałów krzemianowych, wyrażenie H
4
SiO
4(roztwór)
reprezentuje
wszystkie formy krzemu Si rozpuszczonego w wodzie).
Powszechność występowania na powierzchni ziemi wtórnych minerałów wietrzeniowych spowodowa-
na jest w dużym stopniu zachodzeniem reakcji „niecałkowitego” rozpuszczania NIEKONGRUENT-
NEGO. W tym wypadku w wyniku rozpuszczania minerału w roztworze wodnym część składników
uwalnianych jest w formie jonowej do roztworu a część wytrąca się w postaci minerału wtórnego. Po-
wszechnie znanym wtórnym minerałem wietrzeniowym powstającym na przykład podczas wietrzenia
chemicznego skał krystalicznych (granitów, gnejsów) jest kaolinit. Niekongruentna reakcja rozpuszczania
skaleni na drodze kwaśnej hydrolizy z udziałem wszechobecnych węglanów może wyglądać na przykład
tak:
CaAl
2
Si
2
O
8(c.stałe)
+ H
2
CO
3(roztwór)
+ H
2
O
<=> Al
2
Si
2
O
5
(OH)
4(c.stałe)
+ Ca
2+
(roztwór)
+ 2HCO
3
-
(roztwór)
+ 4H
4
SiO
4(roztwór)
anortyt
2NaAlSi
3
O
8(c.stałe)
+ H
2
CO
3(roztwór)
+ 9H
2
O
<=> Al
2
Si
2
O
5
(OH)
4(c.stałe)
+ 2Na
+
(roztwór)
+ 2HCO
3
-
(roztwór)
+ 4H
4
SiO
4(roztwór)
albit
kaolinit
Reakcje rozpuszczania mogą być z powodzeniem modelowane komputerowo. Jednym z programów
używanych do obliczeń równowag chemicznych pomiędzy roztworami wodnymi a minerałami przy
uwzględnieniu udziału dwutlenku węgla i tlenu z powietrza jest program PHREEQC (Parkhurst 1995).
Obecność jonów hydro-
niowych obniża pH roz-
tworu powodując zakwa-
szenie
Jon wodorowęglanowy
wchodzi w reakcje z wie-
trzejącymi minerałami

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
3
7.2. Wietrzenie glinokrzemianowych minerałów skałotwórczych
Przedstawione powyżej reakcje rozpuszczania skaleni nie wyczerpują wszystkich sposobów, na jakie
mogą zachodzić procesy wietrzenia minerałów skałotwórczych z grupy krzemianów i glinokrzemianów.
Podatność pospolitych minerałów skałotwórczych z grupy krzemianów i glinokrzemianów na wietrzenie
jest różna. Zapamiętanie wzajemnych zależności znakomicie ułatwia skojarzenie kolejności podatności na
wietrzenie z serią Bowena krystalizacji frakcjonalnej magmy: kolejność jest odwrotna (Rys. ...). Ta
zbieżność nie jest przypadkowa. Można ją przynajmniej częściowo wytłumaczyć różnicą temperatur po-
między warunkami powstania minerału a warunkami panującymi w strefie hipergenicznej. Minerały kry-
stalizujące z magmy wcześnie (oliwin, anortyt) są minerałami wysokotemperaturowymi. Powstają one w
wyższych temperaturach niż minerały krystalizujące z magmy pod koniec (miki, K-skaleń, kwarc). Więk-
sza różnica temperatur wskazuje na to, że w warunkach powierzchniowych oliwin i anortyt są minerałami
bardziej nietrwałymi, można by powiedzieć „bardziej metatrwałymi”. Ponadto, mniej trwałymi są też
minerały o większym udziale wiązań jonowych w strukturze jak np. oliwin – krzemian wyspowy (Fe,
Mg)
2
SiO
4
o silnie jonowym wiązaniu pomiędzy kationami Fe
2+
i Mg
2+
a tetraedrami krzemianowymi
SiO
4
. Woda, główny czynnik wietrzenia chemicznego, rozpuszcza znacznie lepiej minerały z wiązaniami
jonowymi.
Rys. ... Sekwencja wzrastającej odporności na wietrzenie przypomina serię Bowena krystalizacji ze stopu
magmowego. W nawiasach jest szacunkowa trwałość 1-mm kostki minerału wyrażona w latach.
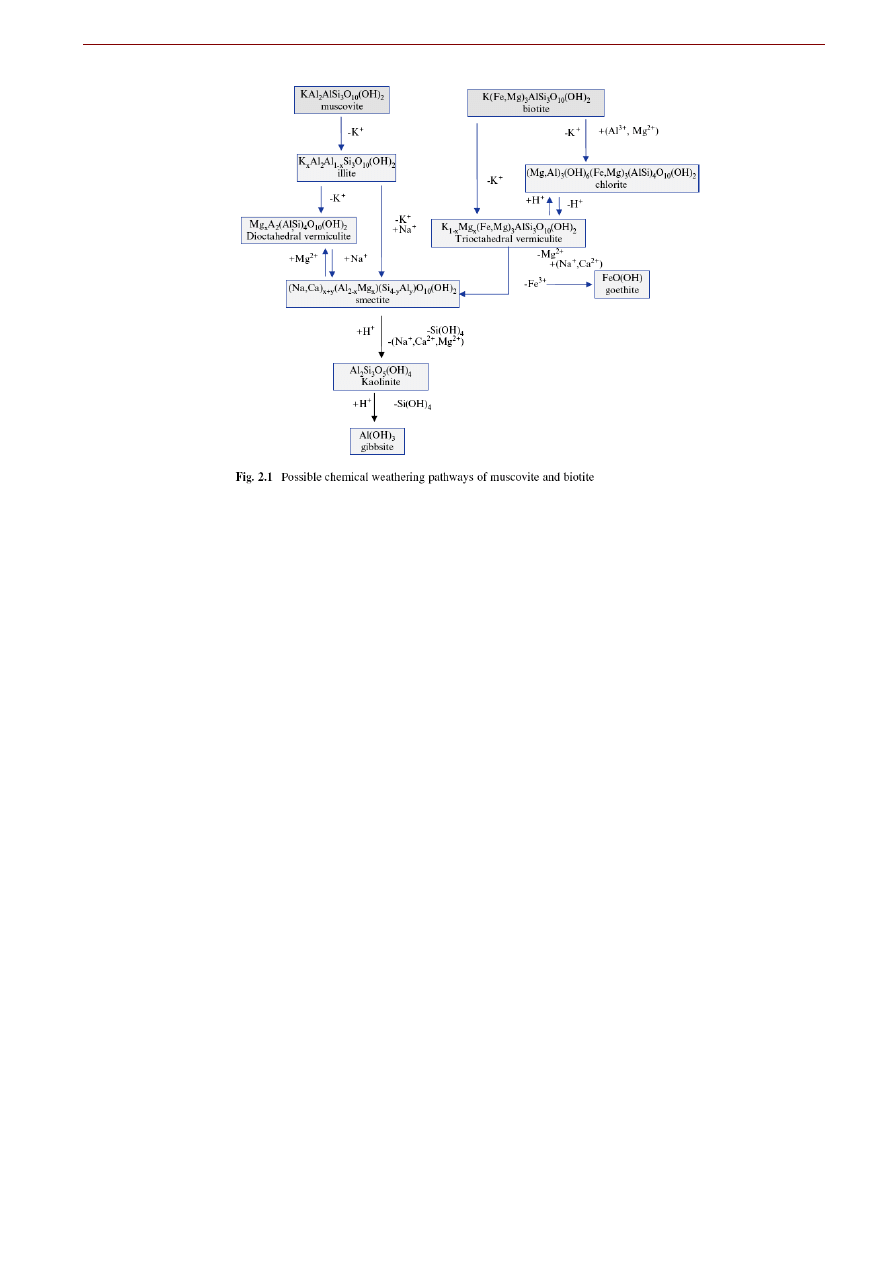
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
4
7.3. Diagramy trwałości minerałów
Bardzo istotnym czynnikiem wietrzenia jest pH roztworów będących na kontakcie z wietrzejącymi mi-
nerałami. Zarówno rozpuszczalność minerałów pierwotnych (ulegających wietrzeniu) jak i minerałów
wtórnych (produktów wietrzenia) zależy od pH. Determinuje to zarówno minimalne jak i maksymalne
stężenia składników (jonów) napotykane zazwyczaj w wodach naturalnych. Minimalne, bo stężenia pier-
wiastków są co najmniej takie jakie powstaną z uwolnienia składników podczas rozpuszczania minerałów
pierwotnych. Maksymalne, bo ograniczona rozpuszczalność minerałów wtórnych „zmusza” nadmiar
uwolnionych pierwiastków do wytrącenia się w postaci minerałów wtórnych.
Trwałość minerałów w środowisku, wynikająca z ich rozpuszczalności w różnych warunkach pH może
być przedstawiona w postaci diagramów trwałości minerałów. Jest pewien standard rysowania tego typu
wykresów, z którym warto się zapoznać, aby w przyszłości łatwiej i lepiej rozumieć publikowane dane.
Na osi X umieszcza się zazwyczaj pH. Na osi Y przedstawione są stężenia wybranych składników uwal-
nianych podczas rozpuszczania minerału. Dla umożliwienia porównania wykresów ze sobą i z chemicz-
nym zapisem reakcji rozpuszczania stężenia muszą być wyrażone w jednostkach molowych (np. w mo-
lach na litr). Jednak stężenia molowe pierwiastków w wodach naturalnych są zazwyczaj bardzo niewiel-
kie. W efekcie otrzymuje się bardzo małe liczby, którymi trudno się operuje. Dlatego na osi Y umieszcza
się zazwyczaj logarytm dziesiętny stężeń (lub logarytm dziesiętny aktywności, czyli skorygowanych stę-
żeń). Na przykład zapis log[Al]
całkowity
oznacza, że przedstawiane są w formie logarytmicznej aktywności
glinu „całkowitego”, czyli suma stężeń wszystkich jonów i związków zawierających glin obecnych w
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
5
roztworze. Linie na wykresie są liniami równowagi minerał–roztwór i dzielą obszar na dwie części: po-
wyżej każdej linii jest ciało stałe a poniżej roztwór. Wartości stężeń rosną na wykresie w górę. Wynika z
tego, że czym wyżej linia na wykresie tym większe maksymalne stężenie pierwiastka w roztworze w
obecności danego minerału (wyrażone na osi Y) a więc większa rozpuszczalność tego minerału.
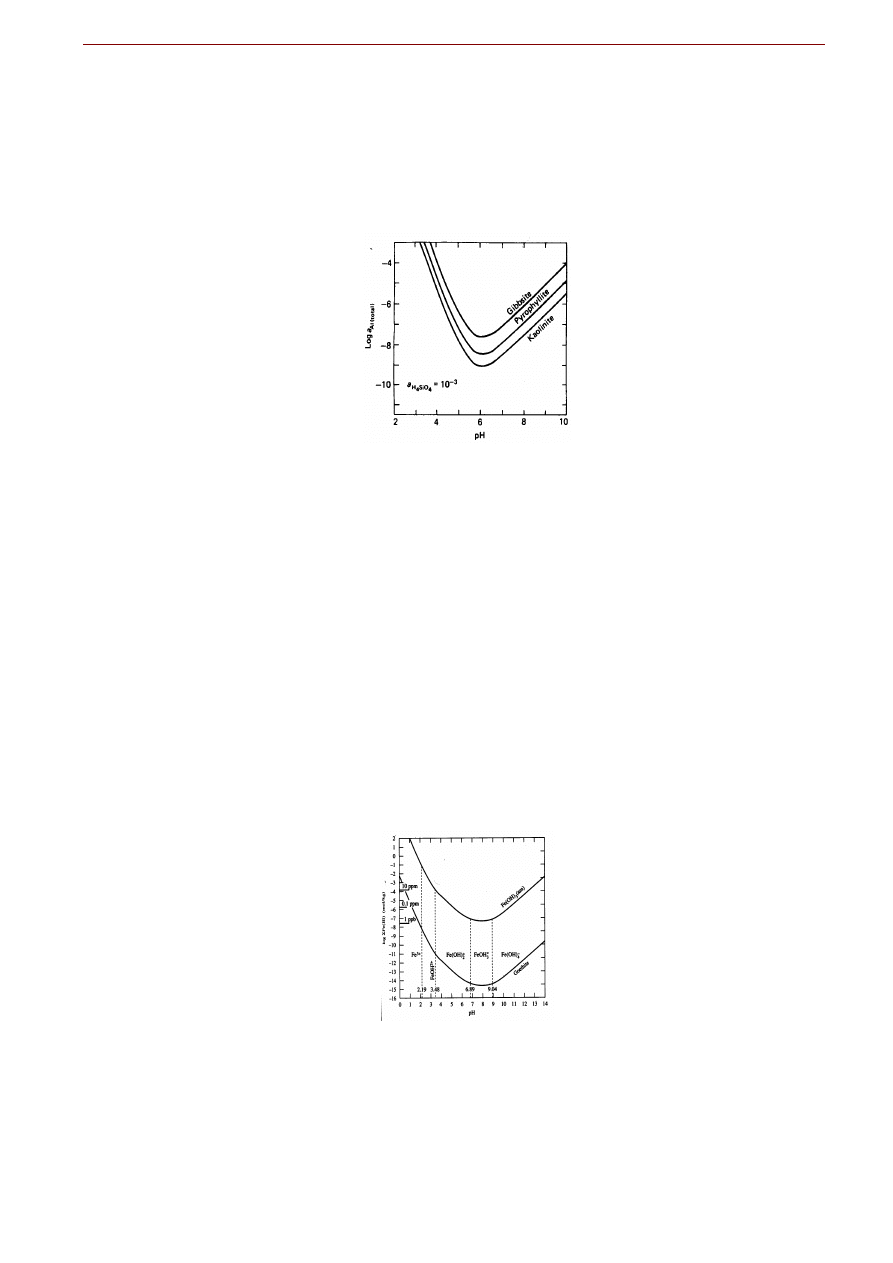
Rys. ... Diagram trwałości przedstawiający zależność rozpuszczalności wybranych glinokrzemianów war-
stwowych od pH przy ustalonym stężeniu Si wyrażonym przez H
4
SiO
4
=0,001 mol/L.
Figura … przedstawia diagramy trwałości dla trzech minerałów zawierających Al: kaolinitu, pirofyllitu
i gibbsytu. Ponieważ niektóre z nich zawierają również krzem, wykresy skonstruowano dla stałego, przy-
jętego realistycznie stężenia Si w roztworze. Pamiętając, że czym wyżej linia na wykresie tym wyższe
stężenie Al
całk
(a więc większa rozpuszczalność minerału w równowadze z roztworem) można poczynić
dwie istotne obserwacje na powyższym rysunku. Po pierwsze dla całego zakresu pH rozpuszczalność
kaolinitu jest zawsze najmniejsza a gibbsytu największa (przynajmniej tak jest dla podanego, stałego stę-
żenia H
4
SiO
4
w roztworze). Po drugie, rozpuszczalność wszystkich trzech minerałów zmienia się ze
zmianą pH osiągając minimum w okolicach pH=6, a rosnąc w bardziej kwaśnych i w alkalicznych roz-
tworach.
Rys. ... Diagram trwałości obrazujący
zależność
rozpuszczalności od pH dla wybranych faz
wtórnych żelaza Fe
3+
.
Figura … przedstawia diagramy trwałości dla związków żelaza: amorficznego wodorotlenku
Fe(OH)
3(am)
oraz goethyt FeOOH. Podobnie jak poprzednio można zaobserwować, że goethyt jest znacz-
nie mniej rozpuszczalny od amorficznego wodorotlenku żelaza. Podobnie jak dla glinu, najniższych stę-
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
6
żeń Fe
3+
w roztworze należy się spodziewać dla wód naturalnych o pH zbliżonym do neutralnego, dla
którego obydwie substancje mają najniższą rozpuszczalność. Stężenia wyższe spowodują, że roztwór
staje się przesycony (współczynnik nasycenia SI>0) i nadmiar żelaza powinien wytrącić się w postaci
wtórnych minerałów, na przykład Fe(OH)
3(am)
lub goethytu. Stężenia Fe mogą osiągnąć nieco wyższe
wartości w wodach o kwaśnym i alkalicznym odczynie, bo rozpuszczalność faz stałych Fe wtedy wzrasta.
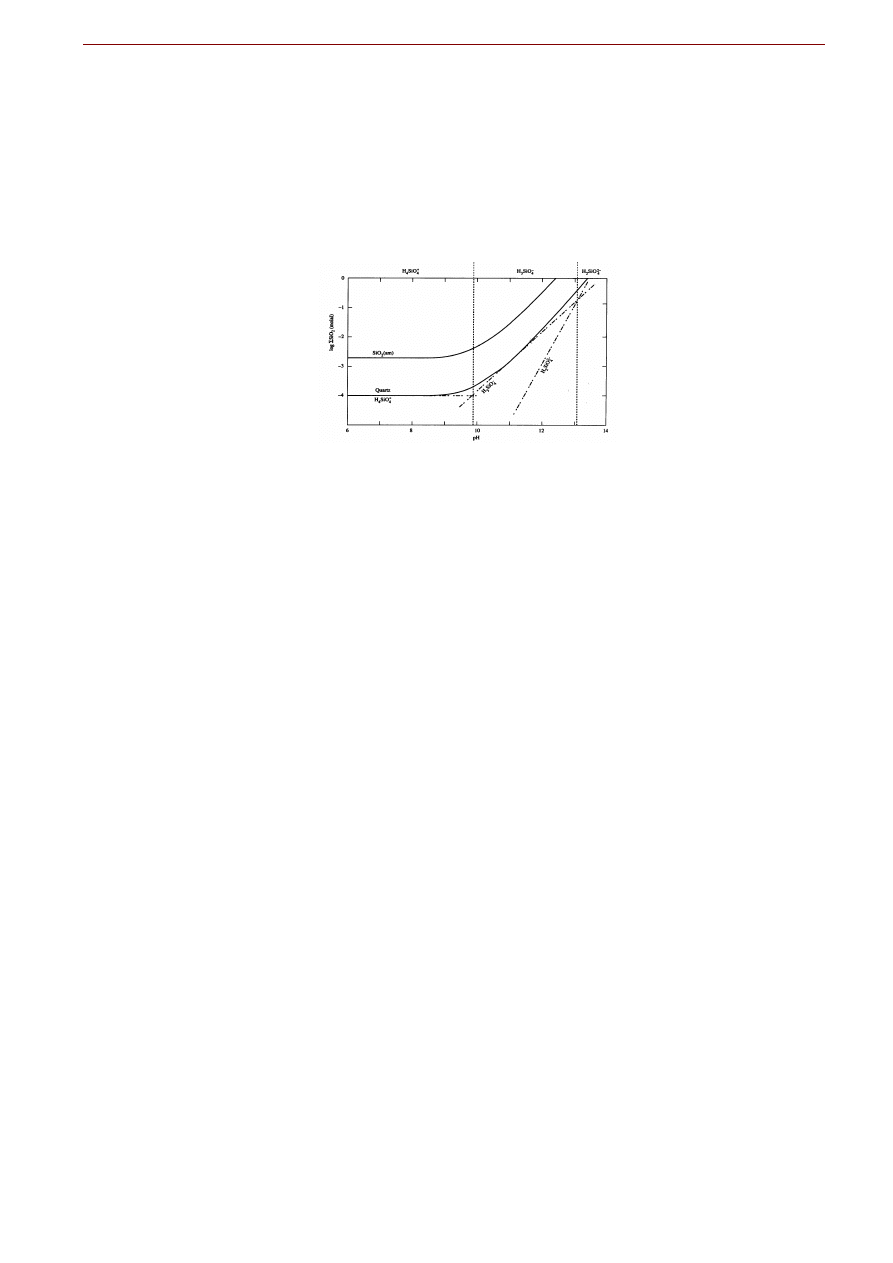
Rys. ... Diagram trwałości
pokazujący
porównanie zmienności rozpuszczalności kwarcu i
amorficznej krzemionki SiO
2(am)
w zależności od pH (w temp. 25
o
C i przy ciśnieniu 1
atm.).
Diagram trwałości dla substancji SiO
2
w postaci powszechnie występującego kwarcu lub w postaci tzw.
amorficznej krzemionki SiO
2(am)
przedstawiony na Fig. … wygląda inaczej od poprzednich. Wynika to z
odmiennej zależności rozpuszczalności od pH. Po pierwsze, w przeciwieństwie do poprzednich wykre-
sów dla wybranych minerałów Al i Fe, nie obserwujemy U-kształtnego wykresu z minimum przy pH w
okolicach obojętnego. Zarówno kwarc jak i krzemionka amorficzna mają niską rozpuszczalność w kwa-
śnych i obojętnych roztworach, natomiast ich rozpuszczalność wzrasta w roztworach alkalicznych. Po-
dobnie jednak jak w przypadku związków żelaza, amorficzna forma krzemionki ma wyższą rozpuszczal-
ność niż forma krystaliczna. Ta ostatnia prawidłowość jest regułą i obserwuje się ją również dla wielu
innych substancji, które mogą występować zarówno w postaci amorficznej jak i krystalicznej.
7.4. Krystalizacja z roztworu
Krystalizacja homogeniczna
Krystalizacja homogeniczna ma miejsce gdy nukleacja (czyli tworzenie się zarodków krystalizacji) i
wzrost kryształów zachodzą w całej objętości roztworu. Zazwyczaj jej rezultatem są euherdalne (auto-
morficzne) kryształy z dobrze wykształconymi zakończeniami z obydwu stron. Zdjęcie na fig. … przed-
stawia obraz z mikroskopu elektronowego (powiększenie 3000x) kryształów apatytu ołowiowego
Pb
5
(PO
4
)
3
Cl (minerał piromorfit) powstałego na drodze krystalizacji homogenicznej z roztworu wodnego.
Widać dobrze wykształcone heksagonalne słupki zakończone bipiramidą, często zbliźniaczone.

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
7
Krystalizacja heterogeniczna
Krystalizacja heterogeniczna ma miejsce, gdy nukleacja i wzrost kryształów zachodzą na powierzchni
obecnych ciał stałych. Zazwyczaj jej rezultatem są subherdalne kryształy przyczepione do powierzchni.
Zdjęcie na fig. … przedstawia obraz z mikroskopu elektronowego (powiększenie 15 000x) kryształów
tego samego apatytu ołowiowego Pb
5
(PO
4
)
3
Cl (piromorfitu) co poprzednio, tym razem powstałego na
drodze krystalizacji heterogenicznej z roztworu wodnego na powierzchni ziaren chlorapatytu
Ca
5
(PO
4
)
3
Cl, które były obecne w zawiesinie. Widać dobrze wykształcone heksagonalne słupki przycze-
pione jednym końcem do powierzchni, na której powstały.
Rys. ... Przykład morfologoii kryształów powstałych w wyniku krystalizacji homogenicznej z roztworu (po
lewej) i krystalizacji heterogenicznej w obecności innego minerału w zawiesinie (po prawej).
Nukleacja (zarodkowanie) nowych kryształów prowadzi czasem do wzrostu bardzo wielu bardzo drob-
nych kryształków a kiedy indziej znów przeciwnie – powstaje niewielka ilość, ale za to dorodnych krysz-
tałów. To zjawisko jest obserwowane zarówno przy krystalizacji z roztworów jak i przy krystalizacji sty-
gnącego stopu. W magmowych skałach głębinowych (np. gabro) dostrzegamy kryształy gołym okiem
(np. struktura grubokrystaliczna) a w ich odpowiednikach wylewnych czy żyłowych o takim samym
składzie chemicznym kryształy można rozróżnić dopiero mikroskopowo (np. struktura skrytokrystaliczna
lub afanitowa). W przypadku intruzji żyłowych obserwujemy wykształcenie bardzo drobnych kryształ-
ków przy kontakcie a znacznie większe kryształy powstają w centrum żyły. Intuicyjnie wyczuwamy tu
prawidłowość. Aby wykrystalizował stop magmowy musi zostać ostudzony poniżej temperatury topnie-
nia. Podczas erupcji magmy różnica temperatur pomiędzy stopem a otoczeniem jest wielka i następuje
silne przechłodzenie stopu. Nukleacja następuje szybko i wszędzie a wzrost kryształów nie może za tym
nadążyć. W efekcie powstaje wiele małych kryształków. Natomiast w przypadku intruzji w głębi ziemi
gradient temperatury jest znacznie mniejszy, powstawanie zarodków jest stopniowe i istnieje dość czasu i
dość niezakrzepniętego jeszcze stopu dookoła powstających kryształów, aby mogły spokojnie urosnąć.

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
8
Podobnie jest w przypadku krystalizacji z roztworu tyle, że w tym wypadku rolę przechłodzenia stopu
jako czynnika stymulującego krystalizację pełni przesycenie roztworu wyrażone ilościowo poprzez
współczynnik SI >0. Ponieważ znacznie łatwiej jest prowadzić eksperymenty i obserwować krystalizację
z roztworu niż ze stopu znacznie więcej wiemy na ten temat. Zaobserwowano, że nie każdy powstający
zarodek zamienia się w rosnący kryształ. Wręcz przeciwnie, większość zarodków kryształów wypadają-
cych z roztworu w czasie nukleacji ulegnie z powrotem rozpuszczeniu. Jako mechanizm tego procesu
można sobie wyobrazić lokalny spadek stężenia jonów w roztworze w wyniku ich konsumpcji przez po-
wstające zarodki i kryształy: jeśli nowo powstały zarodek znajdzie się w otoczeniu innych powstających
zarodków i wzrastających kryształów taki spadek stężeń spowoduje lokalne obniżenie współczynnika
nasycenia roztworu SI do wartości poniżej nasycenia a więc spowoduje rozpuszczanie. Tylko zarodki
wystarczająco duże, aby swoją „strefą wpływów” (a więc powierzchnią styku z roztworem) przekraczały
te lokalne fluktuacje stopnia przesycenia, są w stanie przetrwać walkę o byt i kontynuować wzrost krysz-
tału, którego trwałość wzrasta z rozmiarami.
Z wykładu trzeciego wiemy, że istnieją związki termodynamiczne pomiędzy współczynnikiem nasyce-
nia SI a energią swobodną Gibbsa. A więc trwałość zarodków krystalizacji i powstających z niego krysz-
tałów można wyrazić poprzez energię swobodną Gibbsa pamiętając o ogólnej zasadzie, że niższa ΔG
oznacza większą trwałość a przebieg procesów samorzutnych zmierza w kierunku obniżania tej energii.
Na przykład dla maleńkiego sześciennego kryształka o krawędzi długości r na energię swobodna Gibbsa
składa się człon zależny od powierzchni zewnętrznej (odzwierciedlający reaktywność niewysyconych
wiązań chemicznych, wnoszący dodatni przyczynek) oraz człon zależny od objętości kryształka (od-
zwierciedlający spójność połączonych wiązaniami atomów we wnętrzu kryształka, wnoszący ujemny
przyczynek do wzoru):
ΔG = 6r
2
ΔG
s
– r
3
ΔG
v
Zależność ΔG od wielkości zarodka przedstawia Fig. …a. A więc jeśli powstanie zarodek o rozmiarach
mniejszych od promienia krytycznego r
k
ulegnie samorzutnie rozpuszczeniu. Natomiast, jeśli powstanie
zarodek o rozmiarach przekraczających r
k
będzie on ulegał samorzutnie dalszemu wzrostowi. Dla więk-
szych stopni przesycenia roztworu mamy mniejsze wartości promienia krytycznego a więc podczas spon-
tanicznego zarodkowania większa liczba powstałych zarodków będzie przekraczała r
k
i dalszy proces
krystalizacji będzie jednoczesnym wzrostem większej ilości kryształów niż w przypadku niewielkiego
przesycenia roztworu (Fig. …b).

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
9
Fig. … . a) Zależność trwałości zarodków krystalizacji od ich rozmiarów dla różnych stopni przesycenia roz-
tworu. Promień krytyczny r
k
przypada na moment przegięcia na krzywej zmian ΔG. Na lewo od r
k
znajduje
się obszar nietrwałości zarodków krystalizacji: ΔG jest > 0 a trwałość zarodka rośnie (czyli ΔG spada) w mia-
rę jego rozpuszczania (w lewo na osi X). Dopiero zarodek, którego rozmiary są większe od r
k
zaczyna osiągać
korzyści energetyczne z powiększania rozmiarów i jego trwałość zwiększa się w miarę wzrostu (w prawo na
osi X). b) Kolejne krzywe pokazują, że czym większy stopień przesycenia roztworu wyrażony wartością SI
tym mniejsza średnica krytyczna r
k
: powstaje więcej małych kryształków bo większa jest szansa pojawienia
się trwałych zarodków („przeskoczenia” bariery energetycznej z pola nietrwałości do pola trwałości). W roz-
tworach o niskim stopniu przesycenia tylko sporadycznie zdarza się utworzenie na tyle wielkich zarodków, że
przetrwają one i będą kontynuowały wzrost pojedynczych kryształów.
Proces krystalizacji się kończy, gdy system osiągnie stan równowagi. W stanie równowagi krystaliczny
osad otoczony jest roztworem o takim stężeniu składników, że osiągnięty jest stan nasycenia. Jest to stan
równowagi dynamicznej: procesy rozpuszczania i krystalizacji zachodzą cały czas ale z jednakową pręd-
kością w związku z czym nie obserwuje się żadnych zmian w stężeniach składników roztworu. Obserwu-
jemy jednak często zmiany w obrębie świeżo powstałego osadu, które ogólnie określa się mianem „sta-
rzenia osadu”. Zaraz po wypadnięciu z roztworu osad zazwyczaj jest mieszaniną drobniejszych i nieco
większych kryształków. Ponieważ mniejsze kryształki są nieznacznie mniej trwałe niż te większe, więc w
miarę trwania równowagi dynamicznej często zdarza się, że rozpuszczeniu ulegnie mały kryształek a jed-
noczesna krystalizacja zajdzie na powierzchni większego kryształka. W efekcie po kilku dniach czy mie-
siącach osad ulega przekrystalizowaniu i drobniutkie kryształki znikają a w ich miejsce rozwijają się
kryształki większe. Zjawisko to, obserwowane zarówno w przyrodzie jak i wykorzystywane w technolo-
gii i syntezie chemicznej znane jest pod angielską nazwą „Oswald ripening” (Fig. …).
Fig. … . Obraz ze skaningowego mikroskopu elektronowego próbek krystalicznego osadu pobieranych kolej-
no po 2 min, 48 godzinach i po roku od syntezy.
a
b
dominuje
człon 6r
2
ΔG
s
równania
dominuje
człon – r
3
ΔG
v
równania

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
10
7.5. Procesy utleniania i redukcji
Przypomnienie podstawowych pojęć z chemii.
Utlenienie to podniesienie stopnia utlenienia pierwiastka związane z oddaniem elektronów, np. utlenia-
nie żelaza:
Fe
o
– 2e -> Fe
2+
Fe
o
– 3e -> Fe
3+
Fe
2+
– 1e -> Fe
3+
Przeciwnie, redukcja to obniżenie stopnia utlenienia związane z przyjęciem elektronów, np.:
Fe
3+
+ 1e -> Fe
2+
S
o
+ 1e -> S
-
C
+4
+ 4e -> C
o
Procesy redoks zachodzą zawsze parami: aby pierwiastek uległ redukcji inny pierwiastek musi ulec utle-
nieniu dostarczając odpowiedniej liczby elektronów. Dlatego aby uzgodnić reakcję redoks trzeba zrobić
bilans wymienianych elektronów.
…
Wymiana elektronów podczas reakcji oznacza „przepływ prądu elektrycznego” a powstające napięcie
(siła elektromotoryczna) jest mierzone względem reakcji utleniania wodoru 1/2H
2
– 1e -> H
+
na przykład:
Zn + 2H
+
-> Zn
2+
+ H
2
E
o
= -0,76 Volt
Dla eksperymentu przeprowadzonego w 25
o
C, przy ciśnieniu 1 bar i dla aktywności jonów H
+
i Zn
2+
w
roztworze równych jedności E
o
jest potencjałem standardowym. Potencjały standardowe pierwiastków o
znaczeniu geologicznym zebrane są w tabeli … w postaci tzw. szeregu elektrochemicznego. Wszystkie
reakcje połówkowe mają formę zredukowaną po lewej stronie. Umownie, substancje będące silniejszymi
reduktorami od standardowej elektrody wodorowej mają przypisany ujemny znak potencjału. U góry ta-
beli znajdują się silne reduktory a u dołu silne utleniacze. Pierwiastek będący w tabeli wyżej może samo-
rzutnie wyprzeć z roztworu jon pierwiastka znajdującego się w tabeli niżej (tak są zrobione baterie i
ogniwa elektryczne: samorzutna reakcja redoks powoduje wytwarzanie prądu elektrycznego). Jakościowo
można przewidzieć, że jeśli forma zredukowana pierwiastka spotka się z formą utlenioną innego, położo-
nego niżej w tabeli, to zajdzie samorzutna reakcja pomiędzy nimi. Na przykład:
MnO
2
+ 4H
+
+ 2Fe
2+
=> Mn
2+
+ 2H
2
O + 2Fe
3+
Reakcja połówkowa dla żelaza
Fe
2+
<=> Fe
3+
+ 1e
E
o
= +0,77 V
dla której formą zredukowana jest Fe
2+
jest położona wyżej w tabeli niż reakcja
Mn
2+
+ 2H
2
O <=> MnO
2
+ 4H
+
+ 2e
E
o
= +1,23 V
a potencjał reakcji jest równy różnicy potencjałów dla reakcji połówkowych

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
11
+0,77 – (+1,23) = –0,46 V.
Ujemny znak potencjału (siły elektromotorycznej) reakcji redoks wskazuje na samorzutność przebiegu
procesu w prawo, ponieważ E jest proporcjonalna do energii swobodnej Gibbsa reakcji ΔG
r
:
ΔG
r
o
= nFE
o
gdzie n to ilość elektronów wymienionych w myśl zapisu reakcji a F to stała Faradaya F=96485 culom-
bów (mnożenie kulombów z voltami daje energię w Joulach, tak jak to jest wymagane dla ΔG). Otrzymu-
jemy więc dla tak zapisanej reakcji ujemne wartości ΔG (ΔG<0) a to, jak pamiętamy z rozważań wykładu
trzeciego o termodynamice, oznacza samorzutny przebieg reakcji w prawo. I co dalej? Reakcja będzie
przebiegać aż do osiągnięcia stanu równowagi. Użyte przez nas do obliczeń wartości potencjału standar-
dowego z tabeli są podane dla takich stężeń jonów w roztworze, ze ich aktywności wynoszą 1. A to na
pewno nie sa stężenia równowagowe powyższej reakcji. A więc reakcja ta będzie przebiegała w prawo
powodując zmianę stężeń wszystkich jonów w roztworze tak, aby uzyskać stężenia równowagowe. Wte-
dy ΔG osiągnie wartość zero, a więc i potencjał E uzyska wartość zero.
Okazuje się więc, że E jest uzależnione od stężeń reagentów podobnie jak ΔG. Analogia jest kompletna
nawet w zapisie matematycznym:
ΔG
r
= ΔG
r
o
+ RT lnQ
E = E
o
+(RT lnQ)/nF
gdzie Q to wyrażenie kryjące aktywności produktów i substratów reakcji. W naszym przykładzie:
Q = (a
Mn2+
)
.
(a
Fe3+
)
2
/(a
H+
)
4.
(a
Fe2+
)
2
W stanie równowagi Q staje się identyczne ze stała równowagi reakcji K.
Potencjał utleniająco-redukcyjny Eh
Powyższe obliczenia wskazują na to, że pomiar potencjału redoks w roztworze może nam pozwolić
określić czy system jest w równowadze czy nie a jeśli nie, to w którym kierunku potencjalnie zachodzą
właśnie reakcje złapane na gorącym uczynku. Takiej możliwości nie daje nam ΔG. A pomiar elektryczny
w roztworze wydaje się względnie łatwy. I rzeczywiście, podobnie jak elektrody pH do pomiaru kwaso-
wości roztworu, zostały skonstruowane elektrody Eh do pomiaru potencjału utleniająco-redukcyjnego
(zwanego w żargonie potencjałem redoks). Pomiar potencjału Eh ma również podobny sens do pomiaru
odczynu pH: podaje nam gotowość badanego środowiska do potencjalnego oddania elektronów lub po-
brania elektronów gdyby pojawił się w nim utleniacz lub reduktor. Podobnie pomiar pH podaje nam
zdolność środowiska do oddania lub pobrania jonów hydroniowych H
+
(gdyby pojawiła się zasada goto-
wa je pobrać lub kwas gotowy je oddać).
Sam pomiar Eh jest równie prosty jak pomiar pH: wystarczy wetknąć odpowiednią elektrodę do roz-
tworu i odczytać z miernika wynik w voltach. Jeśli na przykład mamy w zlewce rozpuszczone sole żelaza
możemy się dowiedzieć, która forma jonowa przeważa w roztworze, zredukowana Fe
2+
czy utleniona
Fe
3+
. Jeśli pomiar Eh wskazuje +0,6 volt, porównujemy ten odczyt z potencjałem standardowym dla re-

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
12
akcji Fe
2+
-> Fe
3+
+ 1e w tabeli z szeregiem elektrochemicznym pierwiastków, który wynosi +0,77 volt.
Zmierzony potencjał jest niższy, a więc zgodnie z umową bardziej redukcyjny: w środowisku będzie
przeważać zredukowana forma Fe
2+
.
Diagramy pH – Eh
Pomimo, że pomiar Eh jest tak szybki i prosty bardzo szybko okazało się, że rzeczywiste praktyczne
wykorzystanie tego pomiaru do interpretacji geochemicznych warunków utleniająco-redukcyjnych w
glebach, wodach naturalnych czy ściekach jest znacznie ograniczone. Główną przyczyną ograniczeń jest
fakt istnienia skomplikowanych wieloskładnikowych systemów redoks w środowiskach naturalnych. A
więc podczas pomiaru w zlewce w laboratorium, jak to było podane w przykładzie powyżej, możemy
domyślić się jakiej reakcji redoks przypisać odczytany wynik. Natomiast niehomogeniczne, wieloskład-
nikowe i często pod silnym wpływem mikroorganizmów środowiska naturalne nie poddają się jedno-
znacznej interpretacji. Dodatkowym czynnikiem jest różna prędkość w dochodzeniu do równowagi elek-
trody redoks względem różnych reakcji. A więc pomiar dokonany w terenie sam w sobie obarczony jest
zazwyczaj trudnym do określenia błędem. Pomimo tych ograniczeń, prowadząc pomiary lub opróbowanie
wód, gleb czy ścieków, jeśli wykonywane są pomiary pH i innych parametrów należy zawsze wykonać
pomiar Eh.
W literaturze często pojawiają się diagramy pH – Eh sporządzane dla pierwiastków istotnych środowi-
skowo a zdolnych do występowania na różnych stopniach utlenienia, pozwalające przynajmniej w przy-
bliżeniu określić ich zakres występowania w formie mineralnej lub jonowej w roztworach. …
Przykłady procesów utleniania i redukcji z udziałem żelaza i siarki
AMD
Wiele minerałów ulegających wietrzeniu chemicznemu zawiera żelazo. A żelazo zachowuje się w pro-
cesach wietrzenia nieco odmiennie od innych pospolitych pierwiastków. Może ono w warunkach hiper-
genicznych występować albo na II albo na III stopniu utlenienia (w roztworze w formie jonu Fe
2+
albo w
formie jonu Fe
3+
). Podczas wietrzenia biotytu, augitu czy hornblendy zostaje uwolnione do roztworu że-
lazo w formie zredukowanej Fe
2+
. (Fig. …) W obecności wszędobylskiego tlenu atmosferycznego, w
normalnych wodach powierzchniowych o pH z zakresu 5 do 8, ulega ono łatwo utlenieniu do Fe
3+
. Po-
nieważ minerały utlenionego żelaza mają w tym zakresie pH bardzo małą rozpuszczalność, wytrącają się
z roztworu tworząc barwne osady hematytu Fe
2
O
3
(wiśniowo-czerwony), goethytu i lepidokrokitu Fe-
OOH (żółte i pomarańczowo-brązowe). Podobny ogólnie mechanizm dotyczy innych pierwiastków łatwo
zmieniających stopień utlenienia np. siarki, manganu, węgla zawartego w materii organicznej, a także
pierwiastków stanowiących skażenia środowiska jak chrom czy arsen. Poważny udział w procesach utle-
niania (i redukcji) tych pierwiastków mają mikroorganizmy. Reakcje utleniania i redukcji zachodzące w

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
13
strefie hipergenicznej pod wpływem tlenu atmosferycznego i mikroorganizmów są m.in. przyczyną po-
wstawania niepożądanych gleb glejowych czy kwaśnych wód kopalnianych (AMD – Acid Mine Draina-
ge).
7.6. Elementy geochemii powierzchni mineralnej i kontaktu minerał-roztwór
Większość procesów geochemicznych w naturze, a w strefie hipergenicznej w szczególności, zachodzi
na kontakcie powierzchni mineralnej z innymi fazami. Dlatego zagadnieniom oddziaływania fizycznego i
chemicznego powierzchni ciał stałych i roztworów poświęca się bardzo wiele uwagi zarówno w bada-
niach przyrodniczych jak i w technologii chemicznej i inżynierii mineralnej. Związane z tym są po-
wszechnie używane angielskie określenia, które nie znajdują jeszcze w polskim języku równie trafnych
odpowiedników. Dziedzina badająca relacje pomiędzy powierzchnią mineralną a roztworami nosi miano
„mineral-water interaction”. Sama granica faz (powierzchnia styku) pomiędzy ciałem stałym a roztwo-
rem lub gazem nazywana jest „interface”, czyli strefa pomiędzy fazami. Na ile ta „strefa” jest odrębna,
że zasługuje na uwagę?
Fizycznie, powierzchnia minerału to najbardziej zewnętrzna warstwa atomów (jonów). Można ja po-
traktować jak jeden wielki „defekt kryształu”, związany m.in. z zaburzeniem struktury krystalicznej i
obecnością nieskompensowanych wiązań chemicznych. Czy te niewykorzystane wiązania chemiczne
„wiszą” sobie w przestrzeni poza kryształem? Wiązania jonowe to po prostu elektrostatyczne oddziały-
wania ładunków, które rozchodzą się we wszystkie strony. A więc na powierzchni minerału należy rze-
czywiście spodziewać się, że takie niezrównoważone oddziaływania elektrostatyczne będą przy po-
wierzchni minerału rozchodzić się w przestrzeń. Ponadto, najbardziej zewnętrzna warstwa jonów będzie
silniej przyciągana do wnętrza minerału, przez co odległości międzyjonowe w tej warstwie są nieco
mniejsze: zaburzona jest w tej warstwie struktura krystaliczna. Jeszcze silniejsze zaburzenia obserwuje
się w przypadku występowania wiązań atomowych, które są wiązaniami skierowanymi w określonym
kierunku w przestrzeni. Taka przebudowa struktury kryształu spowodowana występowaniem nieskom-
pensowanych wiązań na granicy kryształu nosi nazwę relaksacji i rekonstrukcji powierzchni. Relaksacja
to po prostu zmniejszenie odległości międzyatomowych. Rekonstrukcja, to bardziej gruntowna zmiana
uporządkowania, gdzie zmiana długości wiązań połączona jest ze zmianą kątów pomiędzy nimi a więc z
zaburzeniem periodyczności sieci krystalicznej. Pojedynczy kryształ może wymagać różnej rekonstrukcji
powierzchni na różnych krystalograficznie ścianach. To szczególnie silnie zaznacza się w przypadku gli-
nokrzemianów warstwowych takich jak kaolinit, smektyty czy miki i chloryty. W minerałach tych po-
wierzchnia równoległa do warstw jest niejako naturalną powierzchnią strukturalną i wymaga niewielkiej
przebudowy. Świeżo odłupane blaszki muskowit są atomowo gładkie i używane często jako podstawa do
sporządzania preparatów w mikroskopii sił atomowych. Natomiast powierzchnie prostopadłe do warstw

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
14
wykazują obecność wielu nieskompensowanych wiązań i w badaniach właściwości sorpcyjnych można
wykazać ich odrębne zachowanie w porównaniu z resztą powierzchni minerału.
Sam kryształ, a więc i jego powierzchnię można sobie wyobrazić jako zbudowane z cegiełek (atomów,
jonów), które w trakcie wzrostu są dobudowywane z roztworu, w trakcie rozpuszczania są odrywane, a w
innych przypadkach wchodzą kolejno w reakcje lub inne oddziaływania ze światem zewnętrznym (Fig.
…). Atomy czy jony zajmujące różne pozycje na powierzchni charakteryzują się różna reaktywnością a
więc charakter powierzchni może wpłynąć na zachowanie ziaren mineralnych w środowisku. Ten sam
minerał wykształcony w postaci różnych kryształów czy też obecny w formie pokruszonych ziaren Może
wykazywać zgoła odmienne właściwości w oddziaływaniach z identycznymi roztworami. Dlatego wiedza
o ogólnych własnościach minerałów jest często niewystarczająca dla scharakteryzowania przebiegu pro-
cesów i w konkretnych przypadkach musi być uzupełniona wiedzą na temat geometrii i geochemii po-
wierzchni mineralnych.
Fig. … . Schematyczny model różnych form morfologicznych w skali atomowej na powierzchni kryształu: ta-
rasy, stopnie, kolanka, nisze, defekty krystaliczne.
Jedną z oczywistych konsekwencji specyficznych właściwości powierzchni mineralnych jest wykazy-
wanie ładunku powierzchniowego. Wsypanie do roztworu o znanym pH sproszkowanego minerału po-
woduje zmianę pH. W eksperymentach elektroforezy obserwuje się ruch drobnych ziaren minerałów w
zawiesinie pod wpływem zewnętrznego pola elektrycznego: cząstki o dodatnim ładunku na powierzchni
przemieszczają się w kierunku ujemnej elektrody a cząstki o ładunku ujemnym – do dodatniej (Fig. …).
Ładunek σ na powierzchni ziaren mineralnych w zawiesinie jest częściowo wynikiem właściwości same-
go minerału (tw. trwały ładunek σ
min
) a częściowo wynika z reakcji ze składnikami roztworu (σ
rcj
). Spo-
śród składników roztworu zawsze wpływ na ładunek powierzchniowy wykazują jony hydroniowe H
+
lub
jony OH
-
, przez co całkowity ładunek σ zależy od pH roztworu. Zmieniając odczyn pH zawiesiny i jed-
nocześnie śledząc poruszanie się cząstek mineralnych w eksperymencie elektroforezy można dla każdego
minerału znaleźć taką wartość pH, przy której ziarenka przestają się poruszać: oznacza to, że ładunek na
ich powierzchni został przez jony hydroniowe H
+
lub jony OH
-
zobojętniony do zera. Taka wartość pH
nazywa się „pH punktu zerowego ładunku” (ang. pH point of zero charge pH
pzc
). Dla kwarcu czy skaleni

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
15
pH
pzc
wynosi ok. 2, dla kaolinitu ok. 5, dla hematytu czy goethytu ok. 7 a dla tlenków i wodorotlenków
glinu wynosi ok. 9. Znajomość pH
pzc
jest istotna dla przewidywania zachowania zawiesin mineralnych w
środowisku. W roztworach o pH poniżej pH
pzc
zawiesina mineralna niesie ze sobą ładunek po-
wierzchniowy dodatni natomiast w roztworach o pH powyżej pH
pzc
cząstki mineralne mają ładunek
ujemny. Przykładowe reakcje odpowiedzialne za wpływ pH to:
reakcje protonacji, np.:
≡Si-OH + H
+
-> ≡Si-OH
2
+
≡Al-OH + H
+
-> ≡Al-OH
2
+
reakcje deprotonacji, np.:
≡Si-OH + OH
-
-> ≡Si-O
-
+ H
2
O
≡Al-OH + OH
-
-> ≡Al-O
-
+ H
2
O
Trzy kreski po lewej stronie wzorów (np. symbol ≡Si-OH) wskazują, że wzór Si-OH nie oznacza wodo-
rotlenku lecz granicę faz („interface”): grupę hydroksylową pochodzącą z wody skoordynowaną z ato-
mem Si lub Al na powierzchni mineralnej.
Tak więc znając skład mineralny zawiesiny, przez pomiar pH roztworu możemy ocenić, jaki ładunek
występuje na powierzchni ziaren. To właśnie mechanizm protonacji i deprotonacji powierzchni powoduje
również, że wprowadzenie do roztworu sproszkowanego minerału powoduje wspomnianą powyżej zmia-
nę istniejącego pH roztworu.
Opierając się na tym, co zostało powiedziane powyżej można wymienić cztery podstawowe przyczyny
obecności ładunku na powierzchni ziaren minerałów obecnych w zawiesinach wodnych lub glebach:
1. Heterowalentna substytucja izomorficzna czyli podstawienia w strukturze minerału jonów o innym
ładunku. Przykładem są spotykane dość często u minerałów ilastych podstawienia Al
+3
za Si
+4
w war-
stwie tetraedrycznej lub Mg
+2
za Al
+3
w warstwie oktaedrycznej.
2. Defekty kryształu: częstym defektem w sieci krystalicznej smektytów (np. montmorillonitu) jest brak
Al
+3
lub brak międzypakietowego Na
+
czy K
+
powodujące niedobór ładunku dodatniego a tym samym
nadmiar ładunku ujemnego ziarna mineralnego.
3. Zerwane wiązania: opisane powyżej zerwane lub niewysycone wiązania na ściankach kryształów lub
na krawędziach odruchów mineralnych dają w sumie ujemny ładunek przez nadmiar O
-2
lub OH
-
; jest to
główne źródło ładunku powierzchniowego dla kaolinitu, nie tak ważne u smektytów czy illitu.
4. Reakcje na powierzchni z utworzeniem centrów sorpcji: ładunek powierzchniowy minerałów z grupy
tlenków, wodorotlenków, fosforanów czy węglanów może powstać przez jonizację grup na powierzchni
lub przez reakcje z roztworem do utworzenia centrów sorpcji, np.
≡FeOH + OH
-
-> ≡FeO
-
+ H
2
O

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
16
Koloidy
Koloidy są układami dyspersyjnymi złożonymi z małych cząstek jednej fazy rozproszonych w innej
fazie. Zawiesiny koloidalne mogą być w postaci aerozoli (cząstki cieczy lub ciała stałego rozproszone i
zawieszone w gazie, na przykład pył atmosferyczny), w postaci emulsji (mikrobowej wielkości kropelki
jednej cieczy rozproszone w innej a ciecze wzajemnie się nie rozpuszczają, np. mleko) czy też w postaci
sol (mikrobowej wielkości cząstki stałe zawieszone w cieczy). Przykładem układu typu sol jest galaretka
z żelatyny. Układy koloidalne są metatrwałe, czyli z czasem ulegają rozdzieleniu na osobne fazy. Ze
względu na wielka łączną powierzchnię cząstek wchodzących w skład zawiesin koloidalnych, na przy-
kład zawiesin w roztworze wodnym, ich własności są wynikiem oddziaływania powierzchni cząstek z
roztworem. Oddziaływania roztworu z powierzchnią ziaren minerałów („mineral-water interaction” na
granicy faz) stanowią istotny mechanizm inżynierii procesów oczyszczania wód i ścieków przez osadza-
nie zawiesin zanieczyszczeń w osadnikach. Odpowiednio modyfikując skład roztworu przez dodatki jo-
nowe i zmianę pH można spowodować, że cząstki zawiesiny będą ulegać koagulacji i flokulacji (zbijaniu
się w agregaty) przez co szybciej ulegają sedymentacji pozostawiając klarowny roztwór. Łatwo samemu
sprawdzić to eksperymentalnie dodając kilka kropel soku z cytryny do mleka. Podobne zjawisko obser-
wuje się w przyrodzie na wielką skalę. Wystarczy wziąć globus i przyglądnąć się ujściom rzek: gdy takie
rzeki wpadają do morza w wielu wypadkach powstaje delta natomiast gdy rzeka wpada do jeziora z wodą
słodką delta zazwyczaj nie powstaje. Dlaczego tak się dzieje? Rzeki niosą ze sobą olbrzymie ilości za-
wiesiny mineralnej. Drobne ziarenka zawiesiny, których powierzchnia jest naładowana ujemnie, odpy-
chają się od siebie, co zapobiega ich koagulacji w większe agregaty a tym samym podtrzymuje zawiesinę
i zapobiega szybkiej sedymentacji. Kiedy jednak rzeka wpada do morza zmienia się drastycznie skład
roztworu otaczającego drobiny mineralne. Obecne w wodzie morskiej w dużej ilości jony adsorbują się
lub otaczają powierzchnię ziaren neutralizując ładunek ujemny (Fig. …). Zneutralizowane cząstki zawie-
siny szybko zbijają się w agregaty, które osiadają na dno przy ujściu rzeki tworząc deltę. Zjawisko takie
nie występuje, gdy rzeka wpada do słodkowodnych jezior. Dlatego w tym wypadku, jeśli powstaje mała
delta, to jedynie z powodu ustania turbulentnego nurtu rzeki.
Fig. … . Jony zawarte w wodzie morskiej częściowo adsorbują się na powierzchni minerału a częściowo ota-
czają ziarno chmurą neutralizując ujemny ładunek powierzchniowy.

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
17
7.7. Elementy geochemii sorpcji
Jednym ze zjawisk towarzyszącym oddziaływaniom jonów z roztworów z powierzchnią szkieletu ziar-
nowego gleb lub z zawiesinami mineralnymi wód jest zjawisko sorpcji: gdy do roztworu doda się sprosz-
kowane ciało stałe stężenia niektórych składników roztworu zmniejszą się. Spadek stężenia spowodowa-
ny jest z ich akumulacją na ciele stałym. Wiele mechanizmów może przyczynić się do zjawiska sorpcji.
W przypadku sorbentów mineralnych może to być adsorpcja na powierzchni, wymiana jonowa, absorpcja
w głąb struktury minerału czy też strącanie się nowych faz mineralnych na powierzchni sorbenta. Ponie-
waż oddziaływania te zachodzą na powierzchni styku fazy stałej z cieczą („interface”), ich efektywność
jest większa dla substancji o dużej powierzchni właściwej. Powierzchnia właściwa, wyrażana na przykład
w metrach kwadratowych na gram substancji, rośnie ze wzrostem rozdrobnienia: Pośród silnie aktywnych
powierzchniowo minerałów występujących na powierzchni ziemi istotną role odgrywają na przykład mi-
nerały ilaste takie jak kaolinit, smektyty (np. montmorillonit), illit, zeolity, drobnodyspersyjne tlenki i
wodorotlenki żelaza (goethyt, ferrihydryt) czy tlenki i wodorotlenki manganu. Istotną rolą jako sorbent
odgrywa też materia organiczna. Wspólną cechą wymienionych minerałów jest duża powierzchnia wła-
ściwa oraz zdolność do wchodzenia w wymienione powyżej reakcje z roztworem na drodze adsorpcji,
wymiany jonowej, absorpcji czy strącania, jak również wiele innych.
Wymiana jonowa.
Ten rodzaj sorpcji dotyczy substancji obecnych w roztworze w formie jonowej, a wiec obdarzonych
ładunkiem. Każdy jon w roztworze otoczony jest otoczka hydratacyjną cząsteczek wody, których dipole
przyciągane są siłami elektrostatycznymi. Gdy do roztworu dostaną się cząstki mineralne, które zawierają
na swej powierzchni zaadsorbowane jony, mogą one zostać wyparte i zastąpione przez jony z roztworu.
Wtedy stężenie jonów wypieranych rośnie w roztworze a stężenie jonów wypierających spada. Minerał
zachowuje się wtedy jak wymieniacz jonowy. Nie da się jednoznacznie opisać mechanizmu związania
wymienialnego jonu z powierzchnią mineralną, ponieważ jest to możliwe na wiele sposobów, które zale-
żą od specyficznych właściwości każdego sorbentu mineralnego. Względna łatwość, z jaką zachodzi ten
proces wskazuje, że siły wiążące jon z powierzchnia nie są zbyt silne. Jeden z opisanych mechanizmów
opiera się o zjawisko tzw. niespecyficznej sorpcji fizycznej opisanej poniżej i polega głównie na elektro-
statycznym oddziaływaniu niewysyconych wiązań. Badania spektroskopowe wykazały, że przyłączane do
powierzchni jony często nie tracą nawet całkowicie swej otoczki hydratacyjnej. Metale alkaliczne (Na, K)
i metale ziem alkalicznych (Ca, Mg) mają tendencję do uczestniczenia w wymianie jonowej i mogą być
wymieniane między sobą lub z innymi jonami z roztworu. Inny zgoła mechanizm, ideowo bardziej zbli-
żony do podstawień izomorficznych w strukturze kryształów związany jest z jonowymiennymi właściwo-
ściami zeolitów. Zeolity to grupa glinokrzemianów o otwartej strukturze (dużych przestrzeniach między-

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
18
atomowych często w formie kanałów i rurek) umożliwiającej wymianę jonów nie tylko obecnych na ze-
wnętrznej powierzchni, ale i wewnątrz struktury kryształów.
Ze względu na skomplikowany i mieszany charakter mechanizmów wymiany jonowej, porównanie
właściwości różnych sorbentów jest w praktyce czysto empiryczne. Wielkość zjawiska zależy zarówno
od rodzaju substancji mineralnej użytej jako sorbent (na przykład montmorillonit wykazuje zazwyczaj
lepsze właściwości jonowymienne niż kaolinit) jak i od rodzaju wymienianych jonów (małe jony oraz
jony o wyższym ładunku sa silniej związane i trudniej wymienialne), nie wspominając już o temperatu-
rze, obecności innych jonów czy pH. Standardowo, zdolność wymiany jonowej (CEC – ang. kation ex-
change capacity) oznacza się przez pomiar ilości adsorbowanego i desorbowanego jonu amonowego
NH
4
+
z 1M (1 mol/L) roztworu octanu amonu CH
4
COONH
4
przy pH=7 w 25
o
C. Dobrymi wymieniacza-
mi jonowymi są…
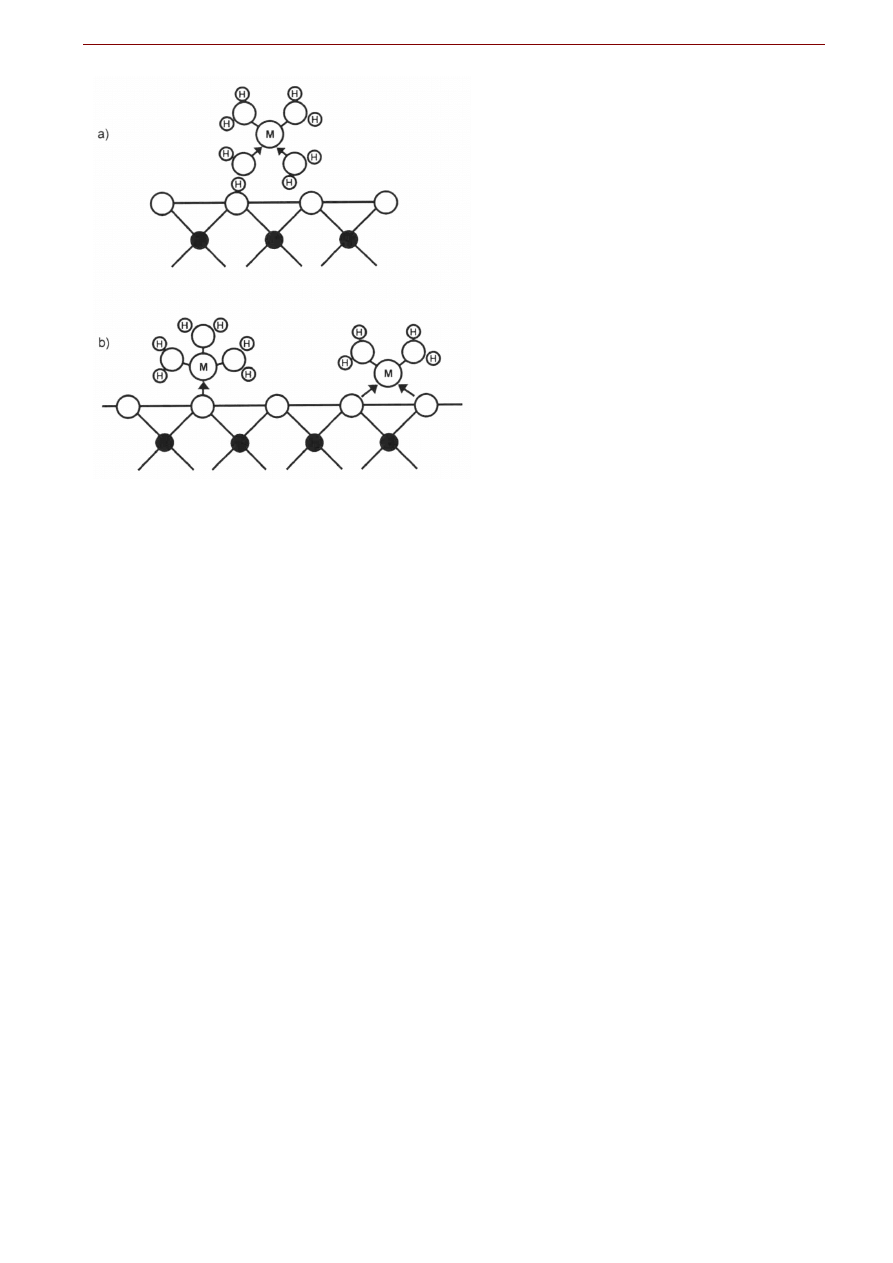
Adsorpcja
Adsorpcja określa zjawisko, w którym powierzchnia ciała stałego przyciąga i zatrzymuje warstwę jo-
nów, atomów lub molekuł z roztworu. Pośród różnych mechanizmów dwa zasługują na wspomnienie:
niespecyficzna sorpcja fizyczna i adsorpcja specyficzna czyli chemisorpcja.
Niespecyficzna sorpcja fizyczna opiera się na słabych oddziaływaniach Van der Waalsa lub elektrosta-
tycznych (jak w przypadku jednego z mechanizmów umożliwiających wymianę jonową opisanych powy-
żej). Nie powstaje wiązanie chemiczne między substancją sorbowaną a sorbentem a jony zachowują
przynajmniej częściowo swoją otoczkę hydratacyjną. Adsorpcja jest nazywana „niespecyficzna” gdyż
mechanizm nie za bardzo zależy od szczególnego układu minerał – pierwiastek sorbowany. Metale alka-
liczne i metale ziem alkalicznych mają tendencję do takiej sorpcji. Mogą być wymieniane z innymi jona-
mi z roztworu. Ten typ adsorpcji jest silnie zależny od ładunku na powierzchni i od pH roztworu. Dobry-
mi sorbentami są kaolinit, montmorillonit, zeolity, a w glebach substancja organiczna i tlenki i wodoro-
tlenki żelaza i manganu.
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
19
Adsorpcja specyficzna (chemisorpcja) opiera się na powstaniu wiązania chemicznego (przy jednocze-
snym częściowym lub całkowitym usunięciu otoczki hydratacyjnej) pomiędzy sorbowaną substancją w
roztworze a powierzchnią sorbentu. Metale przejściowe chętnie sorbują się w ten sposób. Ten typ adsorp-
cji jest słabo zależny od ładunku na powierzchni sorbentu i od pH czy siły jonowej roztworu. Ze wzgl.du
na powstawanie wiązań sorpcja ta zachodzi w konkretnych pozycjach na powierzchni minerału i silnie
zależy od specyfiki konkretnego układu sorbent – składnik roztworu.
Rozmiary sorpcji zależą od składu mineralnego sorbentu, jego ilości i frakcji ziarnowej (wielkości do-
stępnej powierzchni sorpcyjnej), stężenia sorbowanego składnika oraz od temperatury i w różnym stopniu
od pH roztworu. Proces sorpcji jest zazwyczaj dość szybki i po kilku czy kilkunastu godzinach ustala się
stan równowagi a wielkość stężeń w roztworze i ilości zaadsorbowanej substancji stabilizują się. Pomiar
eksperymentalny sorpcji przeprowadza się wytrząsając w stałej temperaturze jednakowe ilości powietrz-
nie-suchej gleby lub innego sorbentu z roztworem o znanych stężeniach składników do momentu ustale-
nia się równowagi. Eksperyment powtarza się wielokrotnie dla różnych stężeń. Po każdym eksperymen-
cie wykonuje się pomiar stężenia równowagowego, co pozwala wyliczyć ilość zaadsorbowaną z różnicy
między stężeniem początkowym a równowagowym.
Wykres przedstawiający zależność ilości zaadsorbowanego składnika przez jednostkę masy sorben-
tu od stężenia równowagowego w roztworze jest charakterystyczny dla danego układu i nazywa się
izotermą. Kształt izotermy jest czysto empiryczny i nie świadczy o mechanizmie sorpcji. Zazwyczaj jed-
nak postać izotermy można przybliżyć jakimś modelem matematycznym, który pozwala przewidzieć
wielkość sorpcji w przypadku użycia innych niż w eksperymencie wartości stężeń w roztworze. Najprost-
7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
20
szym modelem jest izoterma liniowa, ponadto często spotykane są tzw. izoterma Freundlicha (model
funkcji wykładniczej), izoterma Langmuira czy izoterma BET. Z kształtu izotermy liniowej i Freundlicha
wynika, że sorpcja może zachodzić w nieograniczony sposób na materiale sorbenta (Fig. …), co jest
oczywiście nierealne, ale dla układów rozcieńczonych, dalekich od nasycenia sorbenta substancją sorbo-
waną, z jakimi często mamy do czynienia w praktyce, taki model może być adekwatny. W modelu izo-
termy Langmuira zakłada się jednowarstwowe zapełnianie powierzchni a aparat matematyczny użyty do
interpretacji pomiarów pozawala na termodynamiczną interpretację otrzymanych wyników. Model izo-
termy BET ma zastosowanie do substancji, które ulegają sorpcji w wielu warstwach na powierzchni sor-
benta.
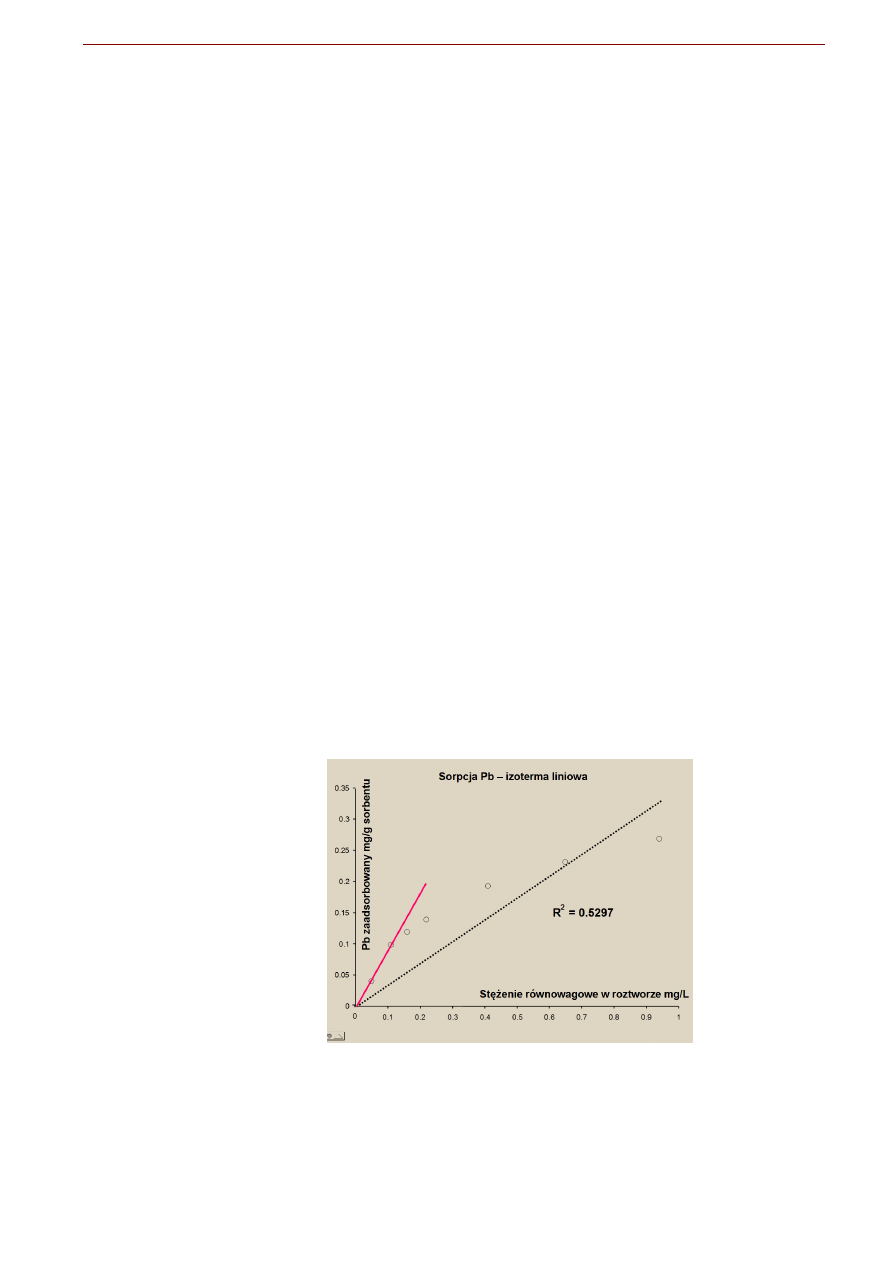
Przykład.
Przeprowadzono eksperyment adsorpcji jonów ołowiu Pb
+2
na glebie. Eksperyment polegał na wytrzą-
saniu próbek 10.18g gleby z 200 mL roztworu Pb
+2
o różnych stężeniach początkowych. Wyniki porów-
nano z przebiegiem różnych izoterm.
Stężenie początkowe Stężenie równowagowe
mg/L
mg/L
2.07
0.05
5.11
0.11
6.22
0.16
7.28
0.22
10.2
0.41
12.4
0.65
14.6
0.94
Izoterma sorpcji Pb – dopasowanie izotermy do danych eksperymentalnych. Izoterma liniowa

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
21
Izoterma sorpcji Pb – dopasowanie izotermy do danych eksperymentalnych.
Izoterma Freundlicha i Langmuira.
Znając z odrębnych badań dominujący mechanizm adsorpcji badanego układu można przewidzieć
kształt krzywej adsorpcji. Należy jednak pamiętać, że NIE MOŻNA WNIOSKOWAĆ O MECHANIŹ-
MIE SORPCJI W OPARCIU O KSZTAŁT IZOTERMY! Jest to częsty błąd. Krzywa adsorpcji jest bar-
dzo użyteczną krzywą EMPIRYCZNĄ.
7.8. Elementy geochemii gleb
Tam, gdzie tylko klimat na to pozwala, w wyniku współdziałania procesów wietrzenia i organizmów
żywych powstają na kontynentach gleby. Gleba jest tworem geologicznym pod wieloma względami wy-
różniającym się od innych. Przede wszystkim żyje pełnią życia: mikroorganizmy, grzyby, owady, zwie-
rzęta, rośliny stanowią o prawidłowo funkcjonującej glebie jako systemie. I nawet terminologia oddaje
traktowanie gleb jak „żywego organizmu” na różnych etapach rozwoju: mówi się o glebach młodych, o
glebach dojrzałych, a gdy organizmy umierają - gleba przekształca się w paleoglebę i jej historia się koń-
czy. Najbardziej charakterystyczną cecha morfologiczną gleb jest obecność horyzontów glebowych róż-
niących się wyglądem i składem. Fizycznie jest to układ trzech faz: szkieletu ziarnowego, roztworów
porowych i gazów, głównie powietrza obecnego w przestrzeni międzyziarnowej. Z punktu widzenia geo-
chemicznego, jest to system otwarty z jednokierunkowym transportem materii, która lokalnie ulega roz-
puszczaniu a lokalnie wytrącaniu pod wpływem przesiąkających w dół profilu wód deszczowych. Pod
względem mineralnym, głównymi składnikami szkieletu ziarnowego gleby są minerały ilaste i piasek
kwarcowy, w różnych proporcjach. Towarzyszą im zazwyczaj autogeniczne wytrącenia minerałów żelaza
i manganu, które wyraźnie odcinają się rudą lub czarną barwą, oraz w różnych ilościach minerały węgla-
nowe, pierwotne lub wtórne w zależności od składu wietrzejącej skały macierzystej. Natomiast roślinność
porastająca glebę dostarcza jej z powietrza składnika, którego początkowo w zwietrzelinie nie było: wę-
gla organicznego, który na drodze fotosyntezy jest przez rośliny asymilowany z gazowego CO
2
a w wy-
niku obumierania fragmentów roślin wchodzi do obiegu geochemicznego w profilu gleby.

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
22
A więc, choć to może wydawać się zdumiewające, bez względu na to czy na podłożu zbudowanym z
gabra, granitu czy piaskowca ogólny skład szkieletu mineralnego dojrzałej gleby jest podobny i zależy
raczej od klimatu (a więc od procesów glebotwórczych) niż od petrografii materiału wyjściowego. Dlate-
go m.in. gleb nie da się klasyfikować w oparciu o skład mineralny, gdyż główne składniki są podobne.
Petrografii podłoża też nie można ignorować, wpływa ona na domieszki poboczne i charakter gleby,
przez co np. winogrona lepiej się udają na wulkanicznych tufach paleowulkanu Tokaj niż gdzie indziej.
Jednakże to klimat determinuje ewolucję procesów geochemicznych i biologicznych formujących glebę a
nie skład zwietrzeliny podłoża i dojrzałe gleby powstałe w podobnym klimacie są pod wieloma wzglę-
dami podobne do siebie.
W terenie badanie gleby przeprowadza się wykopując prostokątną szurfę i odsłaniając na jej ścianie
dostęp do profilu (Fig. …) . W klimacie umiarkowanym zazwyczaj daje się wyróżnić czarno-brązowy
horyzont organiczny O, dalej jasny horyzont wymywania A, następnie ciemniejszy i zawierający rude i
czarne plamy związków żelaza i manganu horyzont B oraz leżący najgłębiej horyzont C, wynikający ra-
czej z charakteru geologii podłoża niż z ewolucji gleby.
Fig. … . Przykładowe profile gleb: czarnoziem, gleba bielicowa
Najbardziej charakterystyczną cechą geochemii gleb jest to, że główną przyczyną wykształcenia hory-
zontów jest ciągły i jednokierunkowy przepływ roztworów w głąb profilu powodujący wymywanie
składników z wyższych i akumulację w niższych warstwach lub odprowadzenie ich do wód podziem-
nych. Glin ulega przemieszczeniu z górnych horyzontów do horyzontu B, ale generalnie jest najmniej
wymywany do wód podziemnych i z czasem ogólna zawartość Al w glebie jako całości wzrasta. Podob-
nie żelazo i mangan choć te pierwiastki stanowią poboczne składniki gleb. Natomiast Na, K, Ca i Mg
mogą z czasem być w większości bezpowrotnie usunięte przez przesiąkające roztwory. Na przykład w
glebach leśnych klimatu umiarkowanego woda deszczowa przesiąkając przez horyzont organiczny może
stać się dość kwaśna pod wpływem kwasów organicznych (pH rzędu 3,5). Takie roztwory skutecznie
wymywają metale alkaliczne i ziem alkalicznych z horyzontu A. W kwaśnych, nieco redukcyjnych roz-
tworach glebowych łatwiej rozpuszcza się również żelazo, mangan i glin, produkty wietrzenia minerałów

7. GEOCHEMIA POWIERZCHNI ZIEMI I: strefa hipergeniczna
23
szkieletu ziarnowego horyzontu A. Ulega on więc zubożeniu w te pierwiastki co zazwyczaj objawia się
rozjaśnieniem barwy. Glin i żelazo są tylko w małej części transportowane w postaci jonowej. Ich głów-
nymi formami są związki koloidalne i rozpuszczone kompleksy metaloorganicznych. W miarę przesiąka-
nia pH roztworów wzrasta i w niższym horyzoncie powstają warunki do wytrącenia się niektórych skład-
ników. Powstają minerały ilaste, tlenki i wodorotlenki glinu, tlenki i wodorotlenki żelaza, tlenki manga-
nu, czasem minerały węglanowe (np. tak zwane kukiełki). Pozostałe składniki, w tym nadmiar Si, są od-
prowadzane z roztworami w głąb do zwierciadła wód gruntowych. W glebach wilgotnego ale bardziej
tropikalnego klimatu wymywanie jest tak silne, że nawet minerały ilaste ulegają w większości rozkłado-
wi, Si ulega wyprowadzeniu i pozostaje jedynie mieszanina tlenków żelaza i glinu – gleby laterytowe i
boksyty. Ich skład mineralny stanowią głównie hematyt (nadający glebie czerwona barwę) i goethyt oraz
gibbsyt, boehmit i diaspor, czasem z domieszką kaolinitu. Natomiast w warunkach klimatu suchego opa-
dy nie są w stanie wystarczająco nasycić gleby, aby wypłukać z nich wszystkie składniki. Część wilgoci
w miarę wysychania gleby podsiąka z większych głębokości wytrącając konkrecje kryształów węglanów
i siarczanów wapnia w postaci twardych, pokręconych i malowniczych form zwanych „caliche” (Fot. ?).
W glebach dojrzałych procesy przemian ustają, skład i położenie horyzontów się stabilizuje i jeśli klimat
nie ulega zmianie to i gleba w stanie naturalnym pozostaje niezmieniona przez setki i tysiące lat.
Wyszukiwarka
Podobne podstrony:
07 geochemia powierzchni Ziemi strefa hipergeniczna
NexusV 07 6 Strefa mroku 6 07
Konspekt strefa hipergeniczna 2
Konspekt strefa hipergeniczna 1
5 Strefa hipergeniczna id 3980 Nieznany
EŚT 07 Użytkowanie środków transportu
07 Windows
07 MOTYWACJAid 6731 ppt
Planowanie strategiczne i operac Konferencja AWF 18 X 07
Wyklad 2 TM 07 03 09
ankieta 07 08
Szkol Okres Pracodawcy 07 Koszty wypadków
Wyk 07 Osprz t Koparki
więcej podobnych podstron