
E-skrzat
Notatki studentów kierunku lekarskiego z zakresu
Biologii Medycznej
Autor oryginału
Wielka Przedwieczna
Opracowanie wersji elektronicznej
Mały Współczesny
Wydanie pierwsze
Gdańsk 2013

Autor poniższej pracy ograniczył się do przepisania dostępnych
notatek z Biologii Medycznej o roboczej nazwie „Skrzat” do
wersji cyfrowej. Tekst niemal w ogóle nie był modyfikowany ani
analizowany pod kątem merytorycznym, mogą pojawić się
literówki. Na wypadek gdyby znalazła się kiedyś osoba chętna
udoskonalić poniższe notatki informuję, że plik .docx można
pobrać ze strony
https://www.box.com/eskrzat
„Wspólnota nie jest sumą interesów, lecz sumą ofiar”
Budowa i rola kwasów nukleinowych
Ludzki genom:
- 46 chromosomów
- 22 pary autosomów
- dwa chromosomy płci
- 2 metry DNA
- 30000-40000 genów
* rośliny mają więcej genów!
* gen BRCA1 jest medialny
Struktura DNA
- Rosalind Franklin
- James Watson i Francis Crick (B-DNA)
DNA budowa nukleotydu:
- reszta fosforanowa

- cukier deoksyryboza lub ryboza w RNA
- jedna z 4 zasad azotowych
C i T – zasady pirymidynowe
A i G – zasady purynowe
* komplementarność zasad w DNA zaobserwował Erwin Chargaff
adenina = tymina (2 wiązania wodorowe)
guanina ≡ cytozyna (3 wiązania wodorowe)
* każda nić ma końce 5’ i 3’
5’ – do 5 węgla jest przyłączona deoksyryboza
3’ – grupa OH potrzebna do wytworzenia wiązań fosfodiestrowych
* DNA jest podwójną helisą
* prymaza – wytwarza kodon START
* tworzenie DNA – wytworzenie wiązań fosfodiestrowych i wiązań wodorowych
* A-DNA – w komórkach odwodnionych
* siły Van der Waalsa odpowiadają za ostateczne uformowanie podwójnej helisy
* DNA pełni jedną funkcję – nośnik informacji genetycznej
RNA (dwie funkcje)
- jednoniciowy
- ryboza zamiast deoksyrybozy
- uracyl zamiast tyminy
- nośnik informacji
- tRNA, rRNA, mRNA -> biorą udział w procesie translacji
- struktura: pętla, szpilka do włosów
Rodzaje RNA
a) RNA kodujące
- mRNA – RNA informacyjne, transkrypt genów
b) RNA niekodujące
- rRNA – RNA rybosomowe, składnik rybosomów
- tRNA – RNA transportujące
* RNA cięte przez nukleazy to RNA interferencyjne
Synteza DNA
- DNA i RNA powstają na matrycowym DNA
- synteza DNA wymaga obecności startera
- wydłużenie od 5’ do 3’
- rozplecenie podwójnej helisy przed replikacją
- replikacja jest procesem semikonserwatywnym
Replikacja – białka kompleksu
- helikaza – rozrywa helisę
- białka SSB – obejmują odcinki jednoniciowe
- prymaza – synteza odcinka starterowe (iRNA) potrzebuje odcinka jednoniciowego
- polimeraza DNA – potrzebuje odcinka startowego i jednoniciowego, rozpoczyna proces
- egzonukleaza – usuwa odcinek starterowy
- ligaza – wytwarza wiązania fosfodiestrowe między dwoma nukleotydami jednej nici
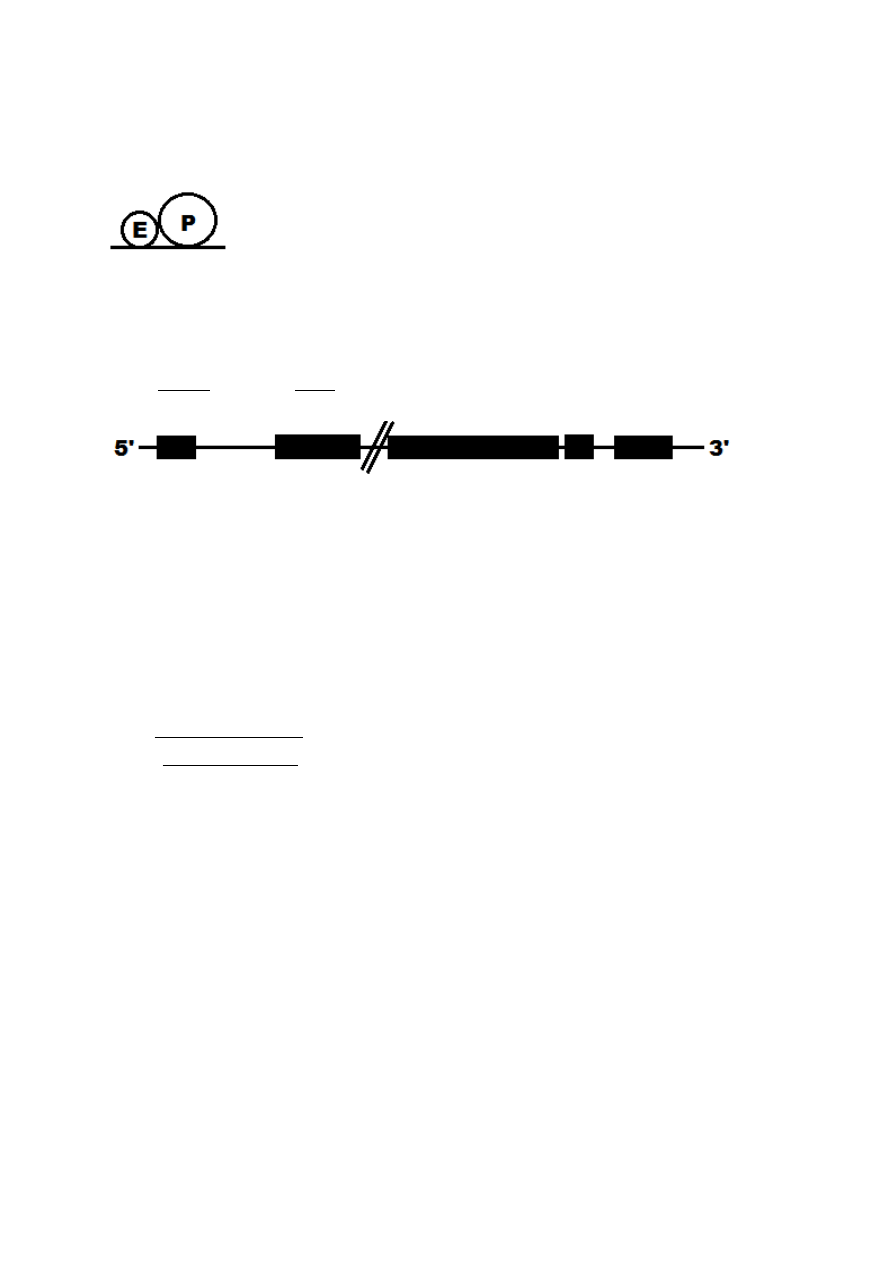
*polimeraza DNA syntetyzuje nową nić oraz sprawdza poprawność wbudowanych
nukleotydów, usuwając nieprawidłowe
fragmenty Okazaki
usuwa wstawia
gdy stykają się to jedno usuwa (E) i od razu produkuje (P) nowe nukleotydy
* gen koduje białko oprócz tRNA i rRNA!
DNA –
transkrypcja
-> mRNA –
translacja
-> białko
miejsce inicjacji
sygnał
sygnał
transkrypcji
start
stop
promotor
sekwencja odp.
właściwa sekwencja
terminator
za wiązanie rybosomu
kodująca
Struktura genu
* eksony kodują, a introny nie (tylko u Eukariota)
* transkrypcja – przebiega w jądrze komórkowym, jeden gen koduje jedno białko
* translacja – przebiega w cytosolu przy udziale rybosomów
Transkrypcja
inicjacja, elongacja, terminacja
3’ ---
NIĆ DNA MATRYCOWA
---> 5’
5’ ----
NIĆ DNA KODUJĄCA
----> 3’
Obróbka potranskrypcyjna
- do 5’ przyczepia się czapeczka
- do 3’ przyczepia się ogonek polyA
- składanie genów (wycinanie intronów)

zastosowanie: przeciwciała – dużo różnych antygenów; to samo DNA może produkować
wiele różnych białek
Rybosomy
60% rRNA i 40% białka
A – aminoacylo
P – tworzenie łańcuchów, wiązań
E – exit
* 3 kodony STOP (UAA, UAG, UGA), 1 kodon START (AUG)
* do kodonu STOP przyłącza się czynnik uwalniający
Genetyka
-> Genotyp – wszystkie geny organizmu
-> Fenotyp – cechy zewnętrzne, powstaje po ekspresji
-> Allel – odmiana genu
-> Locus – miejsce genu w chromosomie
-> Homozygota – osobnik posiadający te same allele w danym locus
-> Linia czysta – osobnik o fenotypie dominującym w pokoleniu F
1
, F
Prawa Mendla
- usuwał z kwiatów pręciki
„W czasie tworzenia gamet każda para alleli ulega segregacji, a gameta przenosi tylko
jeden allel danego genu.”
- krzyżówka jednogenowa
1:2:1
- krzyżówka dwugenowa
9:3:3:1
„Geny należące do różnych par alleli dziedziczą się niezależnie i są przekazywane do
gamet oddzielnie, na zasadzie segregacji losowej.”
* Dominacja całkowita – w fenotypie heterozygoty ujawnia się cecha dominująca
* Dominacja niecałkowita – w fenotypie heterozygoty ujawnia się cecha pośrednia
* Kodominacja – w fenotypie heterozygoty ujawniają się obydwa allele, np. grupy krwi AB
* Dziedziczenie ograniczone płcią – geny dziedziczone z płcią
Dominacja częściowa
- rozszczepienie genotypów 1:2:1
- rozszczepienie fenotypów 1:2:1
Współdziałanie genów nieallelicznych -> geny uzupełniające się
np.
enzym A
enzym B
prekursor
bezbarwny leukozwiązek
antocyjan
np.
A – gen warunkujący syntezę enzymu A
a – allel genu A, warunkujący brak syntezy enzymu A
B – gen warunkujący syntezę enzymu B
b – allel genu B, warunkujący brak syntezy enzymu B
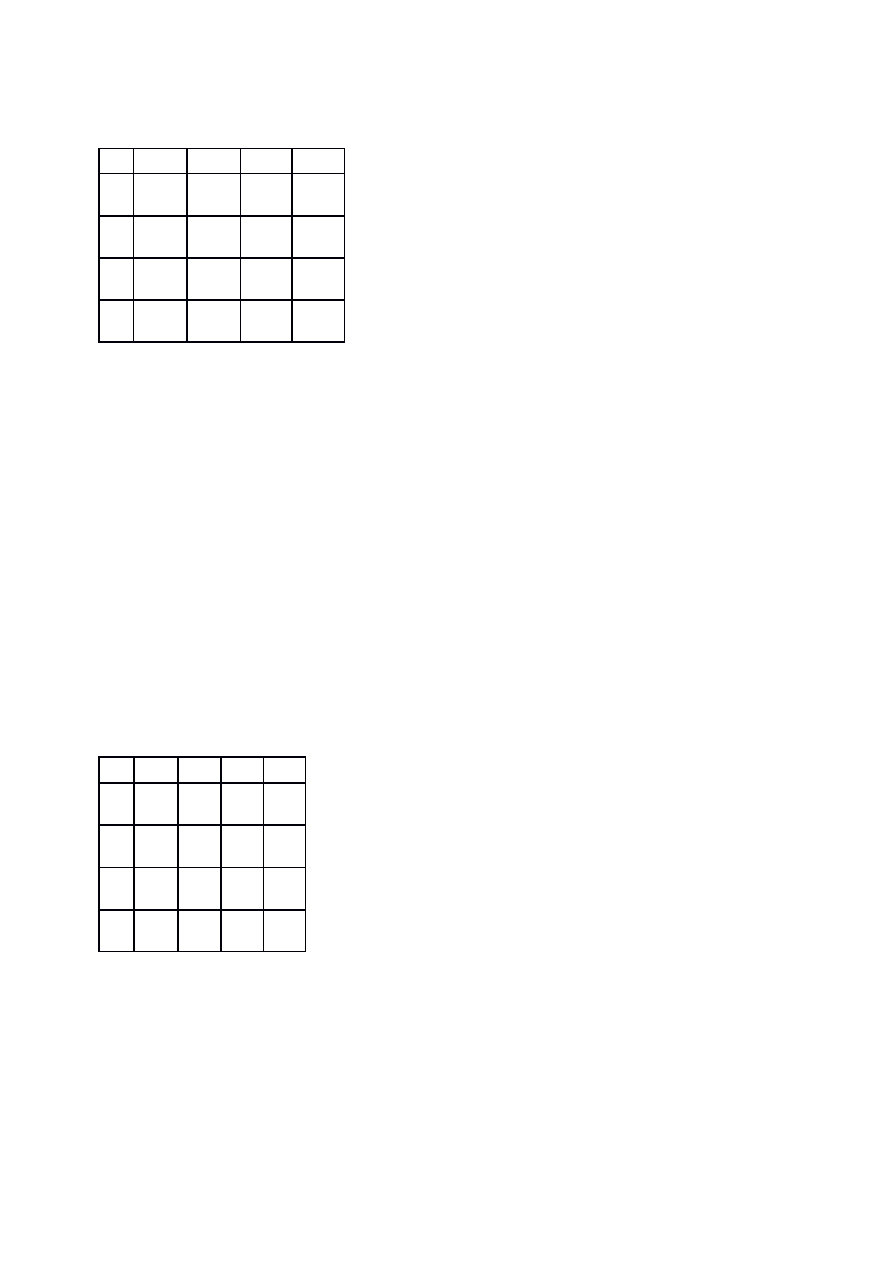
F
2
AB
Ab
aB
ab
A
B
AA
BB
AA
Bb
AaB
B
AaB
b
A
b
AA
Bb
AAb
b
AaB
b
Aab
b
a
B
AaB
B
AaB
b
AaB
B
aaB
b
a
b
AaB
b
Aab
b
aaB
b
aab
b
10:6 stosunek fenotypowy
np. albinizm – przykład genów uzupełniających
* test komplementacji – sprawdza czy dane mutacje zaszły w tym samym locus
Epistaza – współdziałanie genów nieallelicznych polegające na oddziaływaniu jednego genu
na fenotypowe przejawianie się drugiego genu nieallelicznego w ten sposób, że ujawnia się
fenotypowo tylko jeden z tych genów
- gen epistatyczny – gen, który hamuje fenotypowe przejawianie się innych genów
- gen hipostatyczny – gen, którego fenotypowe przejawianie się zostaje zahamowane
Epistaza dominująca
np.
C – gen podstawowego ubarwienia
c – allel genu C, warunkuje brak ubarwienia
I – gen inhibitora, hamujący powstawanie ciemnego ubarwienia
i – allel genu I, nie hamujący ekspresji genu C
F
2
IC
Ic
iC
ic
I
C
IIC
C
IIC
c
IiC
C
IiC
c
Ic
IIC
c
IIc
c
IiC
c
Iic
c
iC
IiC
C
IiC
c
iiC
C
iiC
c
ic
IiC
c
Iicc iiC
c
iic
c
13:3 duże C lub ii
np. krew Lewis (locus układu jest sprzężone z cechą wydzielania Se)
Genotyp Se
Genotyp Lewis
Fenotyp

sese
le
a
le
a,
le
a
le
le (a+b-)
Sese, SeSe
le
a
le
a,
le
a
le
le (a-b+)
Epistaza recesywna
C – gen odpowiedzialny za syntezę melaniny
c – allel genu C, warunkujący brak syntezy melaniny
B – gen warunkujący rozmieszczenie barwnika u podstawy
b – allel genu B, warunkujący brak melaniny w podstawie
Genotyp F
2
9:3:4
Grupy krwi AB0
- A i B to antygeny obecne na powierzchni krwinek
- epistaza recesywna
gen 0 grupa krwi 0
glikoproteina H (HH, Hh)
gen A grupa krwi A; obecny antygen A (N-
acetylogalaktozamina)
gen B grupa krwi B; obecny antygen B (D-galaktoza)
brak glikoproteiny H (hh) dowolna para genów A, B, 0 grupa krwi 0
* gen epistatyczny hamuje ekspresję genów fenomen bombajski!
sprzężenie – łączne przekazywanie genów, które są zlokalizowane w tym samym
chromosomie lub cząsteczce kwasu nukleinowego
Rodzaje sprzężeń:
* całkowite – brak rekombinantów w potomstwie krzyżówki testowej; rozszczepienie
fenotypowe w
stosunku 1:1
- układ zgodności tkankowej HLA
- łańcuch beta HbA i delta HbA
2
* częściowe – procent rekombinantów mniejszy niż procent potomstwa o fenotypach
rodzicielskich
- grupa krwi Lutheran (Lu)
- cecha wydzielania antygenów grupowych krwi AB (Se)
- grupa krwi 0 i zespół paznokieć rzepka
- eliptocytoza i czynnik Rh
- grupa krwi Duffy i dziedziczna katarakta
Penetracja - % częstości z jaką przejawia się gen dominujący lub recesywny np.
retinoblastoma (siatkówczak) penetracja ok. 90%
Ekspresywność – stopień fenotypowego przejawiania się genu, np. choroba Recklinghausena
(nerwiakowłóknikowatość) plamki na ciele – więcej melaniny
* geny letalne – geny, które w przypadku ujawnienia się w fenotypie powodują niezdolność
do życia i śmierć organizmu
Podział genów letalnych ze względu na stopień penetracji:
- letalne powodują śmierć 90-100% nosicieli
- semiletalne powodują śmierć 50-90% nosicieli
- subwitalne powodują śmierć 10-50% nosicieli

np. kot Manks, mysz Cuenot
u człowieka:
a) choroba Taya-Sachsa, typ II
- choroba spichrzeniowa
- mutacja w genie kodującym betaheksozaminidazę A – enzym odpowiedzialny za
hydrolizę
gangliozydów
- fenotyp: zaburzenia rozwoju umysłowego i fizycznego prowadzące do śmierci
organizmu
np. u Żydów aszkenazyjskich
b) Osteogenesis imperfecta, typ II
- dziedziczenie autosomalne dominujące
- mutacja powoduje wytwarzanie nieprawidłowego kolagenu
- fenotyp: ciężkie złamania, deformacja, zgon w 2 miesiącu życia
Budowa chromosomów i chromatyny
Chromosomy:
- szczególny składnik komórki
- każdy gatunek posiada charakterystyczną liczbę
- skondensowana chromatyna
- człowiek posiada 23 pary chromosomów
- każdy chromosom zawiera jedną cząsteczkę DNA uorganizowaną na kilku poziomach
upakowania
* Geny i sekwencje związane z genami (30%)
- eksony genów 10%
- DNA niekodujący 90% (pseudogeny, fragmenty genów, introny, sekwencje nie
ulegające
translacji)
* DNA pozagenowy (70%)
- sekwencje powtórzone 20% (powtórzenia zgrupowane lub rozproszone)
- sekwencje unikatowe 80%
Szczególne postacie genów
- geny zachodzące na siebie – występują u bakteriofagów, u człowieka w mitochondriach
- geny w genach – 1 gen znajduje się wewnątrz intronu innego genu
Sekwencje kodujące:
- pojedyncze geny – kodujące określone białka
- geny zduplikowane – powstały w skutek duplikacji
- geny występujące w licznych kopiach – kodujące histony, tRNA, rRNA
- pseudogeny – sekwencje przypominające funkcjonalne geny, na skutek mutacji niezdolne do
produkcji białka
- fragmenty genów – fragmenty sekwencji kodujących
Chromatyna

- komórki eukariotyczne mają problem z
„upakowaniem” DNA
- długość DNA E. coli to około 1.2mm,
natomiast człowieka 2m
Białka dzielimy na
a) histonowe
- histon H1 – łącznikowy
- histony H2A, H2B, H3, H4 – tworzące oktamer
* DNA histonów nie zawiera intronów!
b) niehistonowe
- enzymatyczne (polimerazy DNA, RNA, Poli(A), ligazy, nukleazy, topoizomerazy,
proteinazy,
metylotransferazy, acetylotransferazy)
- strukturalne (topoizomeraza II)
- regulatorowe (czynniki transkrypcji)
* polimeraza DNA – rozpoczyna replikację
* polimeraza RNA – rozpoczyna transkrypcję
* Poli(A) – dołącza ogonek poliadenylowy
* ligaza – składa rozcięte fragmenty DNA w procesie replikacji
* nukleazy – wycinają nukleotydy
* topoizomerazy – przecinają i skręcają lub rozkręcają DNA
* proteinazy – tną białka
* metylotransferazy – przenoszą grupy –CH3 (metylowe) na DNA
* acetylotransferazy – modyfikują histony
* topoizomeraza II – pełni funkcję szkieletową
Nukleosom
- histony (H2A, H2B, H3, H4)
- DNA
- zbudowany z oktameru histonów (po dwa histony każdego rodzaju) oraz DNA o dł. 146 par
zasad
- histon H1 znajduje się na zewnątrz nukleosomu (pełni funkcję stabilizującą)
- włókno o dł. 30 nanometrów = solenoid
Pętle chromatynowe
- tworzone przez solenoid
- zmniejszenie kondensacji ułatwia replikację
enzym telomeraza – odbudowuje
telomery, fizjologicznie czynny w gametach
* u człowieka obecne chromosomy: metacentryczne, submetacentryczne i akrocentryczne –
brak ch. telocentrycznych!
* akrocentyczne u myszy
Rodzaje chromatyny

a) euchromatyna – zdekondensowana postać chromatyny, aktywna transkrypcyjnie (replikuje
we wczesnej fazie S)
b) heterochromatyna – skondensowana postać chromatyny, nieaktywna transkrypcyjnie
(replikuje w późnej fazie S)
- konstytutywna
- fakultatywna
- interkalarna
* prążki G
- jasne – euchromatyna (bogate w zasady GC)
- ciemne – heterochromosomy (bogate w zasady AT)
* prążki C – wybarwienie heterochromatyny
* barwienie przy użyciu AgNOR
Wrodzone wady rozwojowe i choroby uwarunkowane genetycznie
człowieka
* genetyka – gr. genesis (pochodzenie)
* Grzegorz Mendel – twórca podstaw genetyki
* 1900r. – rozpoczęcie badań nad genetyką
* 1910r. – teoria dziedziczenia Morgana
* do 1930r. – wszystkie badania
* 1953r. – odkrycie DNA
* polidaktylia – wielopalczastość
* achondroplazja – karłowatość
* delecja interstycjalna ch. XV (achondroplazja)
* fokomelia – bardzo krótkie kończyny
* zespół Marfana (dot. tkanki łącznej, duża zmienność fenotypowa)
* dolichocefalia (długa głowa)
* ekrodaktylia – ręce niczym szczypce homara
* zespół Patau ( trisomia 13. chromosomu; cyklop)
* zespół Klinefeltera (mężczyzna XXY)
* zespół Downa (trisomia 21. chromosomu)
* choroba von Recklinghausena (liczne guzki na ciele)
* progeria – przedwczesne starzenie się
* kiła wrodzona
* padaczka
* dystrofia mięśniowa Duchenne (zanik mięśni)
* jednostronny rozszczep wargi
* kariotyp – kompletny zestaw chromosomów danego gatunku lub jednego osobnika
* chromosomy człowieka podzielono na 7 grup autosomów oznaczonych literami od A do G
oraz na chromosomy płciowe
* kryteria klasyfikacji chromosomów:
- względna wielkość
- kształt zależny od położenia centromeru
* zależnie od położenia centromeru wyróżnia się chromosomy:

a) metacentryczne – centromer położony centralnie (A – długie, F – krótkie)
b) submetacentryczne – B, C, E
c) akrocentryczne – D – długie, G – krótkie
d) telocentryczne – u człowieka nie występują
* kariogram – obraz zespołu chromosomów pojedynczej, dowolnie wybranej komórki,
uporządkowanych w sposób systematyczny, uwzględniający ich wielkość i położenie
centromerów
* w celu określenia kariotypu jednej osoby trzeba ułożyć kilka kariagramów
* mozaicyzm – występowanie u jednej osoby co najmniej dwóch populacji komórek (linii
komórkowych) z odmiennym kariotypem
Rodzaje tkanek pobieranych do badań cytogenetycznych
- limfocyty T krwi obwodowej
- fibroblasty (gdy kariotyp limfocytów nie odpowiada obrazowi klinicznemu chorego, np. w
mozaikowatości)
- komórki płynu owodniowego (amniocyty), kom. trofoblastu – badania prenatalne, krew
płodowa
- komórki nowotworowe
- komórki guzów litych (z płynów wysiękowych, tkanki guza)
- komórki nowotworowe szpiku (biopsja lub komórki uzyskane z krwi obwodowej)
Rutynowa metoda uzyskiwania chromosomów z limfocytów krwi obwodowej:
- dodanie heparyny do pobranej krwi
- dodanie krwi do podłoża hodowlanego z dodatkiem miogenu (fitohemaglutynina)
- inkubacja w 37*C przez 72h
- zatrzymanie komórek na etapie metafazy przy udziale kolchicyny
- proces utrwalania (hipotonik, utrwalacz)
- przygotowanie preparatów mikroskopowych
- barwienie preparatów
- analiza mikroskopowa
Techniki barwienia stosowane do otrzymywania wzoru prążków na chromosomach:
1) G – odczynnik giemzy po łagodnej proteolizie
wzór prążków: ciemne – heterochromatyna, jasne – euchromatyna
2) R – odczynnik giemzy po denaturacji cieplnej
wzór prążków: ciemne – euchromatyna, jasne – heterochromatyna
3) Q – odczynnik akrydynowy np. atebryna
wzór prążków: prążki intensywnie świecące – heterochromatyna, prążki słabo
świecące - euchromatyna
4) C – odczynnik giemzy po denaturacji wodorotlenkiem baru
wzór prążków: barwi się tylko heterochromatyna okołocentromerowa
Wzór opisu chromosomów
6q21.1
p – krótkie ramię, q – długie ramię
chromosom
ramię region prążek podprążek

Grupy chromosomów
- A (1-3)
- B (4-5)
- C (6-12)
- D (13-15)
- E (16-18)
- F (19-20)
- G (21-22)
Chromosomy płci nie przynależą do żadnej grupy!
* prążki C – wybarwienie heterochromatyny konstytutywnej
* polimorfizm:
- ilość chromatyny w chromosomach 1, 9, 16, Y
- satelity chromosomu 21
- odwrócenie 9 (inwersja)
Zasady zapisu kariotypu
- prawidłowy:
46, XY lub 46, XX
- mozaikowy:
mos 47, XY; +21/46, XY
ch. dodatkowy
Mutacje genowe
* mutacja – nagła, skokowa zmiana, która zaszła w materiale genetycznym
* polimorfizm – zmiana w materiale genetycznym, która dotyczy ponad 1% populacji
* konsekwencje mutacji:
- mutacje w regionach kodujących mogą powodować powstanie białka o zmienionej funkcji
- mutacje o sekwencjach regulatorowych mogą prowadzić do zmiany ekspresji genu lub do
błędnego składania mRNA
Podział mutacji
- mutacje punktowe – zmiana/uszkodzenie dotyczy od jednej do kilkudziesięciu par zasad
- mutacje genowe – pojedynczy gen
- mutacje chromosomowe – struktura chromosomu
- mutacje genomowe – zaburzenia liczbowe chromosomów
Mutacje
genowe i punktowe
chromosomowe
transwersje
strukturalne
liczbowe
tranzycie

inwersje
delecje euploidalne(2n, 3n)
duplikacje
insercje
aneuploidalne(2n+1)
translokacje
duplikacje
* mutacje spontaniczne – są wynikiem błędu w procesie replikacji, bardzo rzadkie
Dokładność procesu replikacji zawdzięczamy zdolności korektorskiej jednej z podjednostek
polimerazy DNA. Usunięcie tej podjednostki powoduje 1000-krotne podwyższenie poziomu
mutacji spontanicznych
* mutacje indukowane – są wynikiem działania czynników mutagennych:
- mutagenów fizycznych
- mutagenów chemicznych
- mutagenów biologicznych
Choroby:
- ataksja-teleangiektazja
- zespół Blooma (trudno gojące się rany)
- Xeroderma pigmentosum (nadwrażliwość na światło)
* mutacje somatyczne – powstają w komórkach somatycznych; prowadzą do powstania
organizmów mozaikowych – nie dziedziczą się; mogą prowadzić do powstawania
nowotworów
* mutacje generatywne – powstają w gametach, mogą być przekazywane potomstwu,
pozostają bez wpływu na fenotyp, w którym wystąpiły
Rodzaje mutacji punktowych
- tranzycje – zamiana purynę na purynę lub pirymidynę na pirymidynę
- transwersje – zamiana puryny na pirymidynę lub odwrotnie
Mutacje powodujące przesunięcie ramki odczytu
- insercja – wstawienie (jednej lub kilku par zasad) do łańcucha DNA
- delecja – wypadnięcie (jednej lub kilku par zasad) z łańcucha DNA
Efekty mutacji typu tranzycji i trans wersji
1. Nie ma zmiany sekwencji aminokwasów (mutacja milcząca)
GAA
CAA
kwas glutaminowy kwas glutaminowy
2. Zmiana jednego aminokwasu na inny (mutacja zmiany sensu)
GAA
GGA
kwas glutaminowy
glicyna

3. Terminacja translacji (mutacja nonsensowna)
GAA
UAA
kwas glutaminowy
kodon stop
Zmiana ramki odczytu
Na mRNA sekwencja genu jest zakodowana w sposób „trójkowy”, czyli 3 pary zasad tworzą
KODON
Mutacje
proste
powrotne
prawdziwe
supresorowe
* mutacja powrotna – powoduje odzyskanie cechy zmienionej lub utraconej w trakcie
pierwotnej mutacji
* mutacja powrotna prawdziwa – przywraca pierwotny układ nukleotydów w zmienionych w
czasie pierwszej mutacji
CCT
TCT
CCT
* mutacja powrotna supresorowa – powoduje częściowe lub całkowite przywrócenie fenotypu
CCT
TCT
TCG
GGA
AGA
AGC
Molekularne mechanizmy powstawania mutacji spontanicznych u człowieka
1. Tranzycje i transwersje
- formy tautomeryczne zasad
- deaminacja 5-metylocytozyny
- błędne włączanie zasad w czasie replikacji
- depurynacja i depirymidynacja
2. Delecje i insercje
- nieprawidłowe parowanie zasad w czasie replikacji
- ekspansja repetytywnych sekwencji trójneklotydowych
* formy tautomeryczne
C – w formie aminowej
C-T
C-A
A-C
forma rzadka, nieprawidłowa
G-T
3. Modyfikacje chemiczne zasad w DNA
deaminacja cytozyny – w wyniku tego procesu powstaje uracyl, jest on zasadą
niewystępującą w DNA dlatego jest usuwany na drodze glikozylacji. Wycięcie uracylu
powoduje powstanie miejsca AP (apurynowego) , które jest rozpoznawane przez

endonukleazy. Enzymy te wycinają miejsce AP wraz z sąsiednimi parami zasad, a następnie
polimeraza DNA uzupełnia brakującą lukę.
metylacja cytozyny do 5-metylocytozyny – obecność samej 5-metylocytozyny nie
powoduje mutacji punktowych, bo łączy się ona prawidłowo z guaniną. Metylocytozyna
podlega jednak samorzutnej deaminacji do tyminy. Efektem tej zmiany jest mutacja
punktowa, zamiast parowania z guaniną
deaminacja
C
U
metylacja
5-metylocytozyna
deaminacja
T
* mylne wstawienie zasady w czasie procesu replikacji
* ślizganie się polimerazy – może mieć miejsce gdy w DNA występują długie powtórzenia
tych samych par zasad
Dziedziczenie mitochondrialne
1. każde mitochondrium zawiera pewną liczbę kopii kolistego genomu. Niektóre enzymy mit.
są kodowane wyłącznie przez geny zlokalizowane w mitDNA
2. Prawie całe mitDNA jest pochodzenia matczynego
3. Różne tkanki zawierają różna ilość mitochondriów
Choroby:
- KSS – paraliż mięśni oka, choroby serca, utrata słuchu
- LHON – ślepota, wynikająca z uszkodzenia nerwu wzrokowego
Mutacje dynamiczne
Ekspansja trójnukleotydowych sekwencji repetytywnych prowadzi do powstania
niektórych chorób (zespół łamliwego chromosomu X, dystrofia mięśniowa Kennedy’ego,
choroba Huntingtona)
- premutacja – występowanie liczby powtórzeń trójnukleotydowych powyżej normy, ale
poniżej wartości progowej, powodującej występowanie objawów
- antycypacja – występowanie coraz cięższych objawów i wcześniejszego początku choroby
w następujących po sobie pokoleniach, co wiąże się z ilością powtórzeń
* Choroby powstające w wyniku dziedziczenia:
- cukrzyca
- otyłość
- schizofrenia
- rozszczepy podniebienia
- zwichnięcie stawów biodrowych
- wady cewy nerwowej
- nadciśnienie

* Czynniki środowiskowe:
- dieta
- palenie papierosów
- alkohol
- leki
- inne choroby
- stres
- wiek
* konkordancja - % osób posiadających daną cechę
* Czynniki genetyczne o dużym komponencie:
- temperament (stabilność – niestabilność, ekstrawersja – introwersja)
- inteligencja (sprawność umysłowa, logiczne myślenie, wysokie umiejętności operacji
myślowych)
- talent (rodzinne występowanie)
- orientacja przestrzenna
* ryzyko empiryczne ponownego pojawienia się choroby w rodzinie
- dla dziedziczenia mendlowskiego (jeden gen) możemy obliczyć to ryzyko znając genotyp
rodziców
- dla dziedziczenia wieloczynnikowego…???
ryzyko empiryczne – jest to obserwowana częstość występowania choroby w określonej
populacji; ryzyko empiryczne może wzrosnąć dla określonej choroby w zależności:
- stopnia ciężkości choroby
- liczby chorych członków rodziny
- stopnia pokrewieństwa z chorą osobą
- płci
np. rozszczepienie wargi
* zmienność skokowa (alternatywna) – fenotypy występują w dwóch nienakładających się
klasach, np. pierwiosnek chiński: kolor czerwony
* zmienność ciągła – np. biały -> brązowy
Aberracje
Aberracje strukturalne
* aberracja chromosomowa – nieprawidłowość chromosomowa widoczna w mikroskopie
świetlnym; powstają w komórkach somatycznych
- aberracje liczbowe
- aberracje strukturalne
del – delecja
der – pochodne
inv – inwersja
dup – duplikacja
p – ramię krótkie
t – translokacja

r – chromosom pierścieniowaty
* Aberracje:
- symetryczne
- asymetryczne
* chromosomy minutkowe???
Aberacje chromosomowe
- delecje
- duplikacje
- inwersje
- translokacje
np. delecja interstycjalna – tracony fragment ze środka
Rodzaje abberacji strukturalnych
pierwotne
wtórne
chromatydowe
chromosomowe
chromosomowe
(koniec fazy S
(faza G1, początek S)
(bez względu na
pochodzenie
lub faza G2)
mutacji pierwotnej)
Pęknięcia
w jednym miejscu bez ponownego
w dwóch miejscach z nowym
połączenia wolnych fragmentów
połączeniem wolnych końców
delecja terminalna
wymiany
asymetryczne
symetryczne
(powstaje fragment acentryczny)
interchromosomowe
intrach.
interch.
intrach.
dicentryk +
chromosom
translokacja
inwersje
fragment centryczny
pierścieniowy
wzajemna
delecja
interstycjalna
Symetryczna1. Intra
inwersja pericentryczna
inwersja paracentryczna
A
B
C
D
E
F

A
B
C
D
E
F
A
B
C
D
E
F
A
B
C
E
D
F
(schematycznie przedstawione
chromsomy; czarne pola to
centromery)
2. Inter

translokacja wzajemna
Asymetryczne
1. Intra
a)
b)
Delecja:
A B C D E F G H I
A B C
H I
D E F G
A B C H I
- terminalna
np. del (9)(q32)
- interstycjalna
np. del (5)(q13q32)
Duplikacja:
- w wyniku duplikacji chromosom może posiadać kopie niektórych genów
- powstają często w wyniku nierównego crossing-over
- mogą być proste lub odwrócone
odwrócona np:
inv dup (4)(p16p14)
Inwersja:
- 2 miejsca pęknięć
- pericentryczna – angażuje centromer; może prowadzić do zmiany kształtu chromosomu (p i
q)
- paracentryczna – poza centromerem; nie prowadzi do zmiany kształtu chromosomu (p i p
lub q i q)
zapis
zaczynamy
od
telomeru
Zmiany:
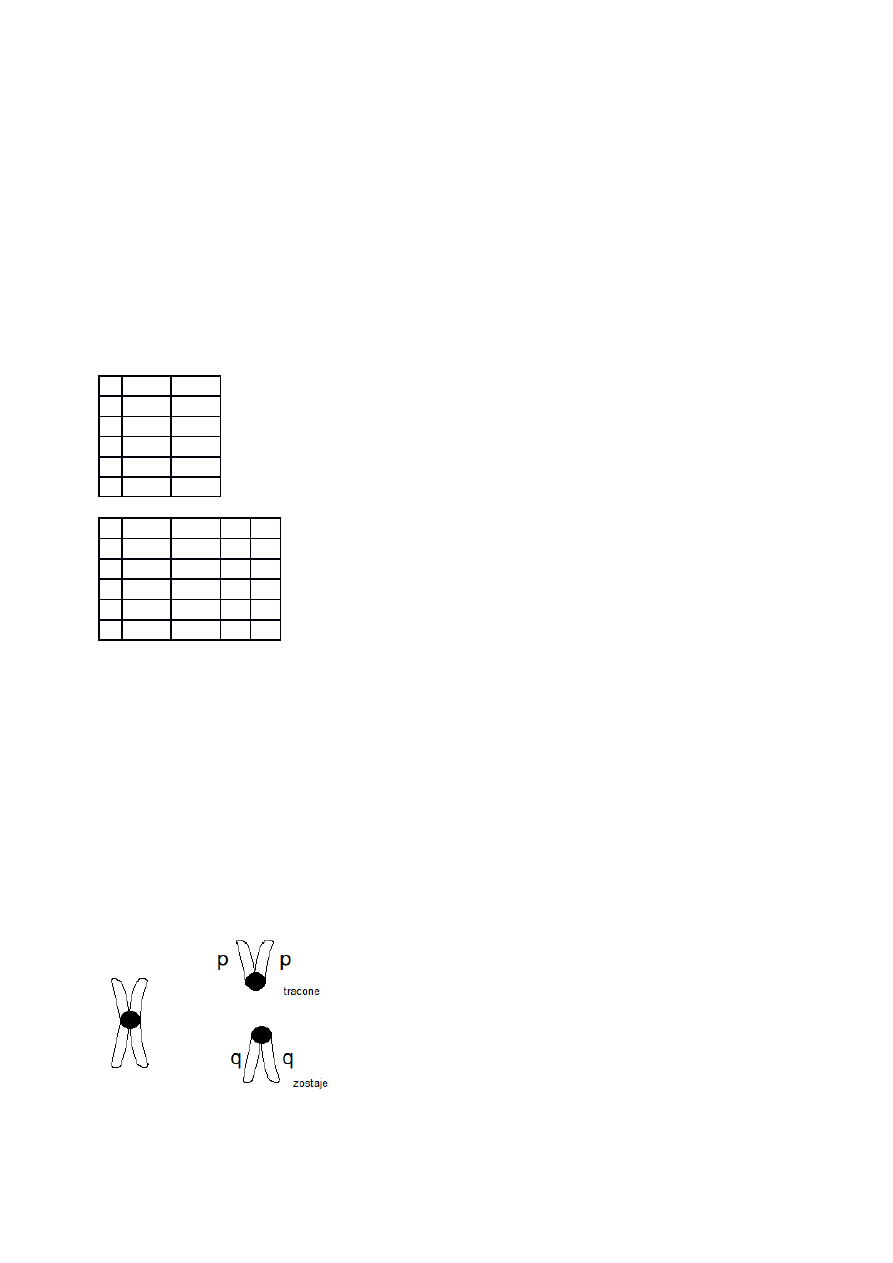
- niezrównoważone – prowadzą do zmiany fenotypu
- zrównoważone – nie prowadzą do zmiany fenotypu
Translokacje:
- wzajemne – wymiana między dwoma ch.
- niewzajemne – wymiana między dwoma ch., ale tylko jeden przyjmuje
np. 46,XX,t(7;17)(p13;q21) -
zapis poprawny z pełnym kariotypem
Translokacja robertsonowska – fuzja centryczna – ulegają wyłącznie chromosomy
akrocentryczne
np. der(14;21)(q10;q10)
(gdy łączą się 2 centromery powstaje 1)
G:
1
14
21
2
14
14/21
3
14/21
21
4
14/21
5
21
6
14
F
1
:
1
14
21
14
21
2
14
14/21
14
21
3
14/21
21
14
21
4
14/21
14
21
5
21
14
21
6
14
14
21
trisomie aktywne letalnie: 13, 18, 21
monosomia21 – 2, 14 – 1
monosomia21 – 1, 14 – 2
* wszystkie monosomie autosomów są letalne
* 1/3 ryzyko powstania letalnej zygoty
* 1/6 ryzyko urodzenia się dziecka z zespołem Downa
* kariotyp osoby z translokacją robertsonowską
45,XY, der(14;21)(q10;q10)
* kariotyp osoby z translokacyjnym zespołem Downa
46,XY, der(14;21)(q10;q10),+21
Izochromosom:
i(X)(q10)
również w zespole Turnera
Chromosom pierścieniowy:
r(18)(p11,3q23)
podprążek
Aberracje liczbowe
* euploidie (polipolidie) – cały zestaw chromosomów jest powielony

* aneuploidzie – występuje jeden dodatkowy chromosom
Euploidie
- autopoliploidie – zwielokrotniona liczba chromosomów pochodzi z tego samego gatunku, a
często od tego samego osobnika np. pszenica
- allopoliploidie – powstają z mieszańców pomiędzy blisko spokrewnionymi gatunkami
* Poliploidy o nieparzystej liczbie chromosomów są bezpłodne
Poliploidie
- u człowieka letalna
- triploidia – powstaje zazwyczaj w wyniku zapłodnienia oocytu przez 2 plemniki lub przez
połączenie dwóch gamet, z których jedna jest diploidalna
- tetraploidia – skutek braku pierwszego podziału zygoty, co powoduje podwojenie liczby
chromosomów bezpośrednio po zapłodnieniu
np. diploidalne truskawki, pomidory
Przykłady zapisów kariotypów w aberracjach liczbowych
Euploidie
triploidia (3n)
tetraploidia (4n)
69, XXX
92, XXYY
69, XXY
92, XXXX
69, XYY
Aneuploidie
- należą do najczęściej występujących aberracji chromosomowych
- najczęstszą postacią jest trisomia – obecność dodatkowego chromosomu, tzn. trzech zamiast
dwóch kopii danego ch.
- brak jednego chromosomu w diploidalnym zestawie określamy jako monosomia – aberracja
letalna
- powstają w wyniku nondysjunkcji – nie rozdzielenia się chromosomów homologicznych lub
chromatyd siostrzanych ch. w czasie podziału komórki lub na skutek opóźnienia ruchu ch.
podczas anafazy
np.
monosomia (2n -1)
podwójna monosomia (2n -1 -1)
45,X
44,XY,-13,-14
45,XX,-13
44,X,-X,-18
nullisomia (2n -2)
trisomia (2n +1)
44,XX,-15,-15
47,XY,+21
44,XY,-18-18
47,XX,+18
podwójna trisomia (2n +1 +1)
tetrasomia (2n +2)
48,XX,+13,+21
48,XY,+21+21
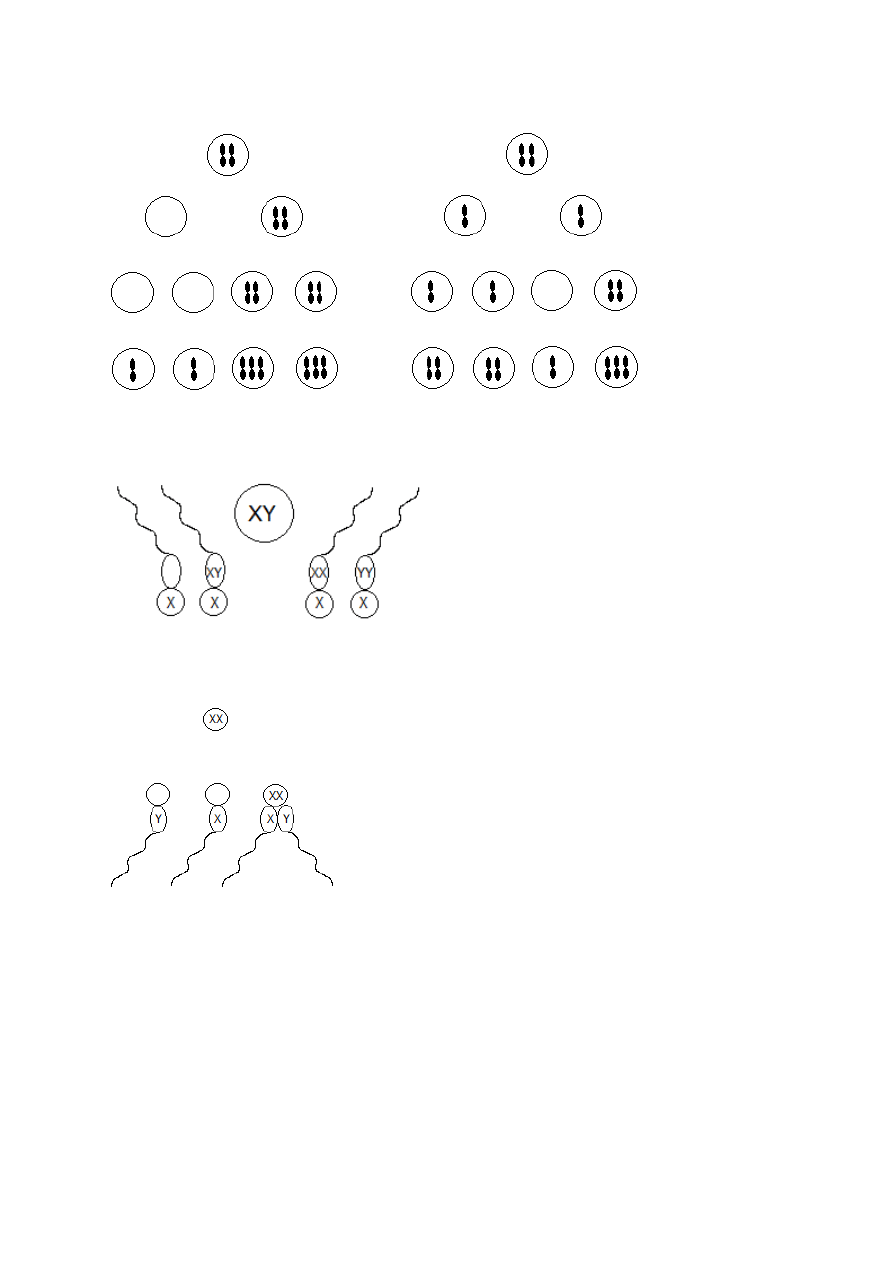
Nondysjunkcja
Nondysjunkcja spermatogenezy
mejoza I
mejoza II
XO XXY XXX XYY
Turner Klinefelter
(zw. poziom agresji)
Nondysjunkcja oogenezy
YO
XO
XXX
XXY
Turner
Klinefelter
Do powstania aberracji liczbowych przyczynia się:
a) euploidie
- c mitoza
- c mejoza
- endomitoza
- fuzja komórek somatycznych
- dispermia
- połączenie prawidłowej gamety z diploidalną
b) aneuploidie

- nondysjunkcja w mitozie
- nondysjunkcja w mejozie
- opóźnienie w anafazie
Czynniki mutagenne
Mutageny
1) fizyczne (promieniowanie)
2) chemiczne (różne związki chemiczne)
3) biologiczne (wirusy)
Ad.1 Fizyczne
- promieniowanie jonizujące (X, α, β, γ, neutrony, promieniowanie kosmiczne) – jest
wysokoenergetyczne, które przenikając przez środowisko powoduje jonizację napotkanych
atomów. Najbardziej wrażliwe są tkanki o komórkach intensywnie dzielących się
Powoduje powstanie wolnych rodników
jeśli wolny rodnik przyłączy się do zasady – osłabienie wiązania, wypadnięcie
nukleotydu
jeśli przyłączy się do gr. fosforanowej – pęka nić DNA; jeżeli stanie się tak po obydwu
stronach helisy może pęknąć cały chromosom
- promieniowanie UV – dłuższe od jonizującego, emituje fale pochłaniane przez DNA
(~260nm), większa długość fali = mniejsza energia, a tym samym mniejsza zdolność
przenikania przez tkanki. Nie powoduje jonizacji, ale wzbudza atomy. Powstają dimery
pirymidynowe (T-T), powstają wiązania DNA z białkami, rzadziej: pękanie pojedynczej nici
Ad.2 Chemiczne
- analogi zasad
- związki alkilujące
- związki nitrozowe
- barwniki akrydynowe
- nitropireny
analogi zasad
* działanie na zasadzie hamowania kom petycyjnego
* antagoniści składników kwasów nukleinowych lub ich syntezy
- 5-bromouracyl – analog tyminy, wiąże się z guaniną zamiast z adeniną
- 2-aminopuryna – analog adeniny, wiąże się z cytozyną zamiast z tyminą
- 6-merkaptopuryna – hamuje syntezę puryn (AML)
- 5-fluorouracyl – rak żołądka, jamy nosowo-gardłowej, pęcherza moczowego, jajników
- ametopteryna – metotreksat – antagonista kwasu foliowego (leczenie ALL u dzieci), działa
w fazie S
- cytarabina – arabinozyd cytozyny, białaczka limfatyczna
związki nitrozowe (HNO
2
)
Powodują deaminację DNA i RNA zmieniając:
- cytozynę w uracyl
- guaninę w ksantynę (para z cytozyną)
- adeninę w hipoksantynę (para z cytozyną)

związki alkilujące
Powodują przyłączenie dodatkowych rodników alkilowych do DNA (najczęściej do guaniny
związki jednofunkcyjne). Następuje wycinanie zmodyfikowanym zasad, przez endonukleazy
lub specyficzne glikozydazy DNA – miejsce purynowe lub pirymidynowe lub trans wersje
(A-C, T-G, C-T)
alkilacja grupy fosforanowej – pękanie łańcucha DNA (zw. jednofunkcyjne)
alkilacja zasad – wiązanie krzyżowe w obrębie jednej lub dwóch nici (zw.
dwufunkcyjne)
związki
jednofunkcyjne
dwufunkcyjne
- sulfonian dwumetylowy
- iperyt azotowy (g. musztardowy)
- sulfonian etylom etylowy
- busulfan
- cyklofosfamid
* bardzo małe dawki powodują wzmożenie podziałów komórkowych
* hamują wzrost komórek, podziały
* zaburzenia mitozy w komórkach szybko rosnących (np. cebulki włosów)
* działanie cytostatyczne głównie tkanki chłonnej (białaczka przewlekła, ziarnica złośliwa,
szpiczak mnogi)
* np. CYKLOFOSFAMID (Endoxan) – pochodna iperytu azotowego
* konieczny metabolizm w wątrobie przez system oksydaz
* raki: żołądka, jajnkia, prostaty, oskrzeli
barwniki akrydynowe
- bromek etydyny
- oranż akrydynowy
- proflawina
- akryflawina
- quinakryna
Interkalują w DNA zniekształcając matrycę – wypadnięcie lub dostawienie zasady podczas
replikacji
Leki o działaniu mutagennym – leki przeciwnowotworowe
* mechanizm działania:
- hamowanie replikacji
- destrukcja DNA
- hamowanie syntezy białek
- niszczenie struktur komórkowych
* leki stosuje się na różnych etapach:
- syntezy dezoksyrybonukleotydów-5-fluorouracylowych
- niszczenie wrzeciona kariokinetycznego (kolchicyna)
- gotowego DNA-cisplatyna (wiązanie krzyżowe)
Cl
H
3
N
Pt
2+
cisplatyna
Cl
H
3
N

* swoiste dla cyklu komórkowego (słabiej na fazę G0):
- leki alkilujące (cyklofosfamid, busulfan)
- antybiotyki (adriamycyna, bleomycyna)
* swoiste dla fazy cyklu komórkowego:
- antymetabolity (metotreksat – antagonista kwasu foliowego, odpowiedzialnego za
segregację chromosomów) – faza S
- alkaloidy (winkrystyna, winblastyna) – mitoza
- inhibitory topoizomerazy II (etopozyd) – hamuje działanie topoizomerazy, niezbędnej do
poreplikacyjnego rozdziału DNA i kondensacji chromosomów w mitozie
antybiotyki
- aktynomycyna – hamuje transkrypcję
- bleomycyna – rozcina łańcuchy DNA
- doksorubicyna – antybiotyki antracyklinowe – interkalacja
alkaloidy
- kolchicyna
- winkrystyna
- winblastyna
- adrianomycyna
* stosuje się także hormony i antyhormony – jeśli nowotwory są hormonozależne
(nowotwory gonad i piersi, tarczycy, trzustki)
* promieniowanie jonizujące (191 Au, 32 P, 131 I)
inne leki
* leki immunosupresyjne (np. metotreksat)
- leczenie chorób autoagresji, zapobieganie odrzutom przeszczepów, choroba hemolityczna
noworodków
- działanie immunosupresyjne poprzez wpływ na rozmnażanie i różnicowanie komórek
immunologicznie kompetentnych
* środki odkażające
- etakrydyna (rivanol) – związek interkalujący, stosuje się 0,1-0,5% roztwór; działa na
bakterie, odkażająco na skórę i błony śluzowe
Metody badania mutagenności
Prokaryota
1) test mutagenności (test Amesa) – test rewersji mutacji
Bakterie „his-” miesza się z homogenatem z wątroby szczura; wylewa się na podłoże
minimalne (nie zawierające histydyny) i dodaje badanego związku. Jeśli jest mutagenny,
przeżyją tylko te, które „nauczyły się” syntetyzować histydynę (his+), czyli rewertanty
(mutacja powrotna)
2) test indukcji faga λ
Dwa szczepy bakteryjne, jeden z lizogenicznym fagiem, drugi normalny. Hodowla na
stałym podłożu, dodaje się badanego związku – indukuje faga (uszkodzenie DNA i

przestawienie na cykl lityczny). Wirusy atakują zdrowe komórki. Ilość łysinek świadczy o
stopniu mutagenności
Eukaryota
1) test plamkowy
A – gen agouti (barwnik u nasady i góry włosa)
B – gen ciemnej barwy
S – jednolite barwienie
aa/bb/SS
AA/BB/SS
a – jednolite wybarwienie
b – brązowa barwa
Aa/Bb/SS
Aa/bb/SS
s – łaciatość
agouti czarne agouti z
brązową
łatką na czarnym
tle
* związek jest mutagenem, jeżeli na 500 myszek urodzą się 4 z brązową łatą
2) test kometowy (test degradacji DNA)
- czynnik mutagenny
- zawieszenie w kropli agarozy
- szkiełko
- obróbka zaczyna się w roztworze do lizy (niszczenie błon komórkowych)
- elektroforeza DNA
- wybarwienie fluorochromem
Addukty
Reagują z cząsteczką DNA, pochodne węglowodorów aromatycznych. Nie są
mutagenne w takiej postaci, w jakiej je wdychamy, dopiero po szeregu obróbek
metabolicznych łączą się z zasadą azotową.
Konsekwencje:
- zahamowanie lub obniżenie szybkości replikacji DNA
- niewłaściwe parowanie zasad podczas replikacji
- generowanie miejsc apurynowych
Chromosomopatie
- skutki zmian aberracji chromosomowych
- zespoły chorobowe spowodowane aberracjami zarówno strukturalnymi jak i liczbowymi
Aneuploidie:
- trisomie – tylko trzy rodzaje dają szansę na narodziny – chromosomu 13, 18 i 21
- monosomie – letalne w przypadku autosomów i chromosomu Y (brak chromosomu X)
* Aberracje liczbowe autosomalne
Trisomie:
1) chromosom 21 – zespół Downa
2) chromosom 18 – zespół Edwardsa
3) chromosom 13 – zespół Patau

Ad.1 – zespół Downa, trisomia ch. 21
- 1/700 urodzeń
- wzrasta z wiekiem matki
- wykrywany w badaniach prenatalnych USG; torbiel pęcherzykowa okolicy szyjnej, obrzęk,
wady serca, zwężenie lub zrośnięcie dwunastnicy
Fenotyp
- upośledzenie umysłowe
- zmarszczka nakątna
- dysplastyczne uszy
- wystający duży język
- małpia bruzda
Kariotyp
- prosta trisomia 47,XY,+21 (96%)
- zespoły translokacyjne (3%)
46,XX,der(14;21)(q10;q10),+21
46,XX,der(21;21)(q10;10),+21
- kariotyp nosiciela translokacji zrównoważonej
45,XX,der(14;21)(q10;q10)
45,XY,der(21;21)(q10;q10)
- kariotypy mozaikowe (1%)
mos46,XX/47,XX,+21
Ryzyko urodzenia dziecka z zespołem Downa w przypadku nosicielstwa translokacji
robertsonowskiej wynosi:
- 10-15% gdy nosicielem jest kobieta
- 2-5% gdy nosicielem jest mężczyżna
Ad.2 – zespół Edwardsa, trisomia ch. 18
- 1/3000
- przewaga płci żeńskiej
- wysoka umieralność we wczesnym okresie niemowlęcym (30%), poronienia (95%)
Kariotyp
47,XX,+18
mos46,XX/47,XX,+18
Fenotyp
- retrognatia, wydatna potylica
- zniekształcone, nisko osadzone uszy
- stopy o łukowato wygiętych podeszwach
Ad.3 – zespół Patau, trisomia ch. 13
- 1/5000
- większość płodów ulega poronieniu
- upośledzenie umysłowe
Kariotyp
47,XX,+13
Fenotyp
- nisko położone oczy
- rozszczepienie wargi

- dysplastyczne uszy
- zmarszczka nakątna
Aberracje strukturalne
1) zespół Cri du chat (częściowa monosomia ch. 5)
- 1/50 000
- ok. 75% chorych umiera w pierwszym miesiącu życia
- przeważają dziewczynki
Kariotyp
46,XX,del(5)(p13)
46,XX,del(5)(p14)
46,XX,del(5)(p15)
- 10-15% to potomstwo nosicieli translokacji zrównoważonej
Fenotyp
- płacz dzieci przypomina miauczenie kota
- małogłowie
- nisko osadzone uszy
- małożuchwie
- hiperteloryzm
- opóźnienie umysłowe
Zespoły chorobowe człowieka spowodowane aberracjami liczbowymi
1) zespół Turnera, monosomia chromosomu X (45,XX)
- 1/5000 u dziewczynek
- fenotyp żeński
- 99% poronień
Fenotyp
- płetwiasta szyja
- niski wzrost
- szeroka klatka piersiowa i gruczoły sutkowe
- nisko schodząca linia włosów na karku
- koślawość łokci
- niedorozwój cech płciowych
- pasma łącznotkankowe w jajnikach
- wcześnie rozwijająca się osteoporoza
- brak upośledzenia umysłowego
Kariotyp
- monosomia chromosomu X: 45,X
- aberracje strukturalne chromosomu X: 46,X,del(X)(p11)
46,X,del(X)(q13)
46,X,i(X)(q10)
46,X,idąc(X)(p11)
46,X,r(X)(p22;q28)
- kariotypy mozaikowe
mos46,XX/45,X

mos46,XY/45,X
mos45,X/46,X,der(X)+(X;X)(q28;q28)
2) zespół Klinefeltera (dodatkowy ch. X)
- 1/1000 urodzeń chłopców
- fenotyp męski
- wysoki wzrost
- małe jądra
- rozwój piersi
- bezpłodność
- brak upośledzenia
- słaby rozwój drugorzędowych cech płciowych
Kariotyp
47,XXY
mos46,XY/47,XXY
3) 47,XXX
- 1/1000 urodzeń dziewczynek
- fenotyp żeński
- czasami wtórny brak miesiączki i przedwczesna menopauza
- 75% osób płodnych
- łatwiej zapadają na choroby psychiczne
4) 47,XYY
- 1/1000 urodzeń chłopców
- fenotyp męski
- wysoki wzrost i prawidłowa budowa ciała
- płodni
- nie są to mężczyźni o większej agresji
Euploidie
- płody ulegają poronieniu (poliploidie)
triploidia
69,XXY
tetraploidia
92,XXYY
Techniki molekularne
1953r. – opisano model DNA
*Materiałem do badań genetycznych jest DNA i RNA
* źródło – każda kom. jądrzasta
- rutynowo – krew (limfocyty)
- w diagnostyce prenatalnej – kom. płynu owodniowego trofoblastu
- w medycynie sądowej – każdy ślad biologiczny
- w diagnostyce nowotworów – fragment guza, biopsje, szpik kostny
Izolacja kwasów nukleinowych
- wszystkie kom. jądrowe zawierają DNA i RNA
- większość kom. RNA stanowi rRNA, tylko 3-5% to mRNA
1) liza komórek – zniszczenie błony kom. związkami chem. lub enzymami

2) trawienie białek (pro kinaza k)
3) oczyszczanie
4) precypitacja
5) suszenie
6) rozpuszczenie
7) sprawdzenie ilości i jakości
I etap – liza komórek
II etap – oczyszczanie kwasów (nast. jest precypitacja)
III etap – zawieszanie DNA i sprawdzanie jakości
stosunek fali 260/280
Elektroforeza
- DNA naładowane ujemnie, bo ma resztę kwasu fosforowego
- im mniejszy fragment tym szybciej migruje
* Rodzaje żeli stosowanych w elektroforezie:
- agarozowy – stosowany do rozdziału fragmentów DNA wielkości od 100pz do
20000pz
- poliakrylamidowy – stosowany do rozdziału fragmentów DNA wielkości od 10pz do
500pz
* Barwienie żeli:
- bromek etydyny – mutagen, interkalacja !!!
- SYBR green
- srebro
Technika PCR – replikacja in vitro (1983r. Nagroda Nobla)
- helikaza
- temperatura denaturacji 93-96*C /stosowane białka SSB/
- nukleotydy (deoksyrybonukleotydy)
- startery – primaza
- matryca
- enzym – polimeraza DNA + magnez + bufor
* bakteria Thermus aquaticus – jej polimeraza jest używana w reakcji PCR
PCR:
- denaturacja DNA
- przyłączenie startera 5’-3’ (do końca 3’)
- wydłużenie startera, synteza
denaturacja, hybrydyzacja, elongacja
polimeraza Taf
72*C – optymalna temperatura działania polimerazy Taf
Kopie DNA
1.
2
2.
4

3.
8
4.
16
5.
32
6.
64
po 3 cyklu powstają nowe fragmenty „bez ogona”
Rodzaje technik PCR
- long PCR
- ASA-PCR
- multiplex PCR
- RT-PCR
- in situ PCR
- Real time PCR (qPCR)
1) Long PCR – diagnostyka dużych delecji genu LDLR –zły cholesterol o niskiej gęstości – w
hipercholesterolemii rodzinnej (FH)
- powielamy fragmenty kilku tysięcy nukleotydów (w przeciwieństwie do zwykłego,
który ma
kilkaset)
- wykrywamy delecje
hipercholesterolemia – choroba autosomalna
2) ASA-PCR – diagnostyka mutacji R35OOQ w genie APOB w hipercholesterolemii
rodzinnej (FH)
(gen apoliproteiny typu B)
- szukamy konkretnej mutacji
3) Multiplex PCR – diagnostyka dystrofii mięśniowej Duchenna (DMD) – brak eksonu 45
- gen dystrofiny (Xp21,2)
- 2/3Mb (79 eksonów)
- kilka par starterów
4) RT-PCR – technika, która angażuje mRNA; PCR z odwrotną transkrypcją (na wirusach);
identyfikacja genu fuzyjnego BCR/ABL w przewlekłej białaczce szpikowej (CML)
- mRNA ma na końcu poliA; izolacja;
- starter oligoDT
- tworzy się cDNA
- translokacja 9,22
* enzymy restrykcyjne – specyficznie tną DNA
- dotychczas poznano ok. 200, np:
* EcoR1
* Hpa I
* Hind III
- najczęściej rozpoznają palindromowi sekwencje 4-8pz
- po cięciu powstają tępe lub lepkie końce
- używane do wykrywania mutacji i klonowania
VNTR – Variable Number Tandem Repeats
- wysoce polimorficzne allele; zmienne ilości powtórzeń danej sekwencji

- wykorzystywane w medycynie sądowej
- zespół łamliwego chromosomu X
DNA fingerprinting
- np. wykluczanie ojcostwa
5) RFLP-PCR – enzymy restrykcyjne używane do detekcji (wykrywania) mutacji; rozpoznają
one sekwencje nukleotydów; tnie sekwencje zmutowaną a dzikiej(???) nie tnie
w heterozygocie są 3 prążki
w homozygocie są 2 prążki
* Sekwencjonowanie – odczytanie sekwencji nukleotydów DNA
Metoda dideoksy – Sangera
- na tym miejscu się zatrzyma na dideoksynukleotydzie
potrzebujemy:
- 1 startera
- matryca: produkt PCR
- polimeraza
- 4 nukleotydy
dodajemy dideoksynukleotydów wysyconych odpowiednim barwnikiem (jak dideoks. się
pojawia to brak replikacji; wynikiem jest denaturacja) po dołączeniu dideoks. zawsze stop
syntezy
FISH – fluorescencyjna hybrydyzacja in situ
- fragment DNA komplementarny
- znakowany fluorochromem
sondy:
- specyficzna
- centromerowa
- telomerowa
- malująca (translokacje)
- można wykrywać:
- trisomie
- delecje
* Zespół Pradera-Williego
- amplifikacja HER-2/NEU
- identyfikacja translokacji t(9;22) w CML
CGH – porównawcza hybrydyzacja genowa
- znakowanie DNA fluorochromem
DNA badany
DNA kontrolny
- precypitacja mieszaniny DNA badanego i referencyjnego z cot-1 DNA
- denaturacja
- hybrydyzacja
- płukanie
- detekcja

Determinacja i różnicowanie płci
Płeć – zespół cech genetycznych, anatomicznych, histologicznych, fizjologicznych,
różniących osobniki męskie od żeńskich
- istnieją gatunki hermafrodytyczne – pasożyty oraz takie, które mogą zmieniać płeć
pod
wpływem środowiska
- większość gatunków posiada zdolność kształtowania płci
- stosunek ilościowy samców do samic wynosi 1:1 w większości organizmów
Determinacja płci – procesy różnicujące rozwój organizmów w kierunku org. żeńskich lub
męskich
* Rola heterochromosomów w determinacji płci:
- płeć heterogametyczna
- samce heterogametyczne
- samce homogametyczne
* Typy determinacji płci:
- syngamia – płeć determinowana w momencie zapłodnienia (człowiek, muszka
owocowa)
- progamia – płeć determinowana przed zapłodnieniem (ptaki, motyle Abraxas)
- metagamia – płeć determinowana po zapłodnieniu przez czynniki środowiskowe (B.
viridis)
* Determinacja płci u Drosophilia melanogaster
- jest determinowana poprzez stosunek chromosomów X do autosomów
- gdy stosunek ten jest równy 1 powstaje samica (XXAA)
0.5 powstaje samiec (XAA)
- w wyniku nondysjunkcji chromosomów płci mogą powstać różne Aneuploidie
Kryteria klasyfikacji płci u człowieka:
- chromosomalna – uwarunkowana obecnością ch. Y
- chromatynowa – obecność chromatyny płciowej
- gonadalna – budowa histologiczna gonady
- zew. narządy płciowe
- wew. narządy płciowe
- hormonalna
- psychiczna
- metrykalna (urzędowa)
Chromosomy płci:
46,XX – kariotyp żeński
46,XY – kariotyp męski
Chromosom X – zidentyfikowano ponad 1000 genów
Chromosom Y – zidentyfikowano ok. 330 genów
* przewody Mullera – prekursory jajowodów, macicy, dolnej części pochwy
* przewody Wolffa – prekursory nasieniowodów, pęcherzyków nasiennych, gruczołu
krokowego

Determinacja płci u ssaków zachodzi na drodze tzw. przełączenia rozwojowego – przejścia z
jednego szlaku rozwojowego na drugi
Gen SRY
- zlokalizowany na krótkim ramieniu chromosomu Y, bardzo blisko regionu
pseudoautosomalnego
- produkt genu SRY – czynnik determinujący jądro TDF, wysoce konserwatywne białko
wiążące się z DNA
Różnicowanie płci u człowieka
- obecność prawidłowego genu SRY na chromosomie Y (z rdzenia pierwotnej gonady
rozwijają się jądra)
- brak lub niefunkcjonalny gen SRY
Różnicowanie płci męskiej
SRY
Jądra
podporowe komórki Sertoliego
śródmiąższowe komórki Leydiga
hormon antymullerowski
testosteron
supresja przewodu Mullera
(kom. Leydiga produkują testosteron)
Różnicowanie płci żeńskiej
- gonada przekształca się w jajnik
- brak testosteronu powoduje zanik przewodów Wolffa
- brak hormonu antymullerowskiego – przewody Mullera kontynuują rozwój
Mutacje
1) w genie SRY
efekt: normalnym rozwój w kierunku żeńskim
2) w genie kodującym
efekt: obecność przewodów Mullera u mężczyzn
hormon antymullerowski
3) w genie odpowiedzialnym
efekt: wczesny rozwój w kierunku żeńskim,
maskulinizacja
za syntezę testosteronu
w okresie dojrzewania
4) w genie kodującym
efekt: brak sygnału dla rozwoju zewnętrznych
cech męskich
konwertazę testosteronu
- mężczyźni o kariotypie 46,XX – translokacja terminalnego regionu Yp obejmującego gen
SRY

Hermafrodytyzm
Prawdziwy
Rzekomy
(Pseudohermafrodytyzm)
osoba zawiera zarówno zawiązki
występuje jeden typ gonad, ale
cechy
jąder jak i jajników; nie występuje
płciowe zewnętrzne i wewnętrzne
są
jednoczesna produkcja gamet
charakterystyczne dla płci
przeciwnej
charakterystycznych dla obu płci
(ma gen SRY na X, bo zaszła
translokacja)
Pseudohermafrodytyzm męski
- obecność jąder i cech fenotypowych żeńskich
Zespół nadwrażliwości na androgeny
- kariotyp 46,XY
- mutacje lub delecje genu receptora dla androgenów
- żeński fenotyp
Niedobór 5α-reduktazy
- dziedziczony autosomalnie recesywnie
- wewnętrzne narządy płciowe – w pełni prawidłowe, męskie
- przeważa fenotyp żeński
Pseudohermafrodytyzm żeński
- 46,XX
- obecność jajników
- szczątkowe zewnętrzne męskie narządy płciowe
- wrodzony przerost kory nadnerczy
Inaktywacja chromosomu X
- jeden z chromosomów X w każdej ssaczej komórce żeńskiej jest inaktywowany na
wczesnym etapie embriogenezy
- inaktywacja chromosomu X jest mechanizmem równoważącym ekspresje genów
chromosomu X
Inaktywowany chromosom X w żeńskiej komórce jest widoczny w postaci grudki
chromatyny zwanej ciałkiem Barra lub „pałeczką dobosza” w komórkach granulocytów.
Liczba ciałek Barra jest zawsze o jeden mniejsza niż liczba chromosomów X w kariotypie.
Choroby jednogenowe dziedziczone w sposób autosomalny
Rodowody oznaczenia:
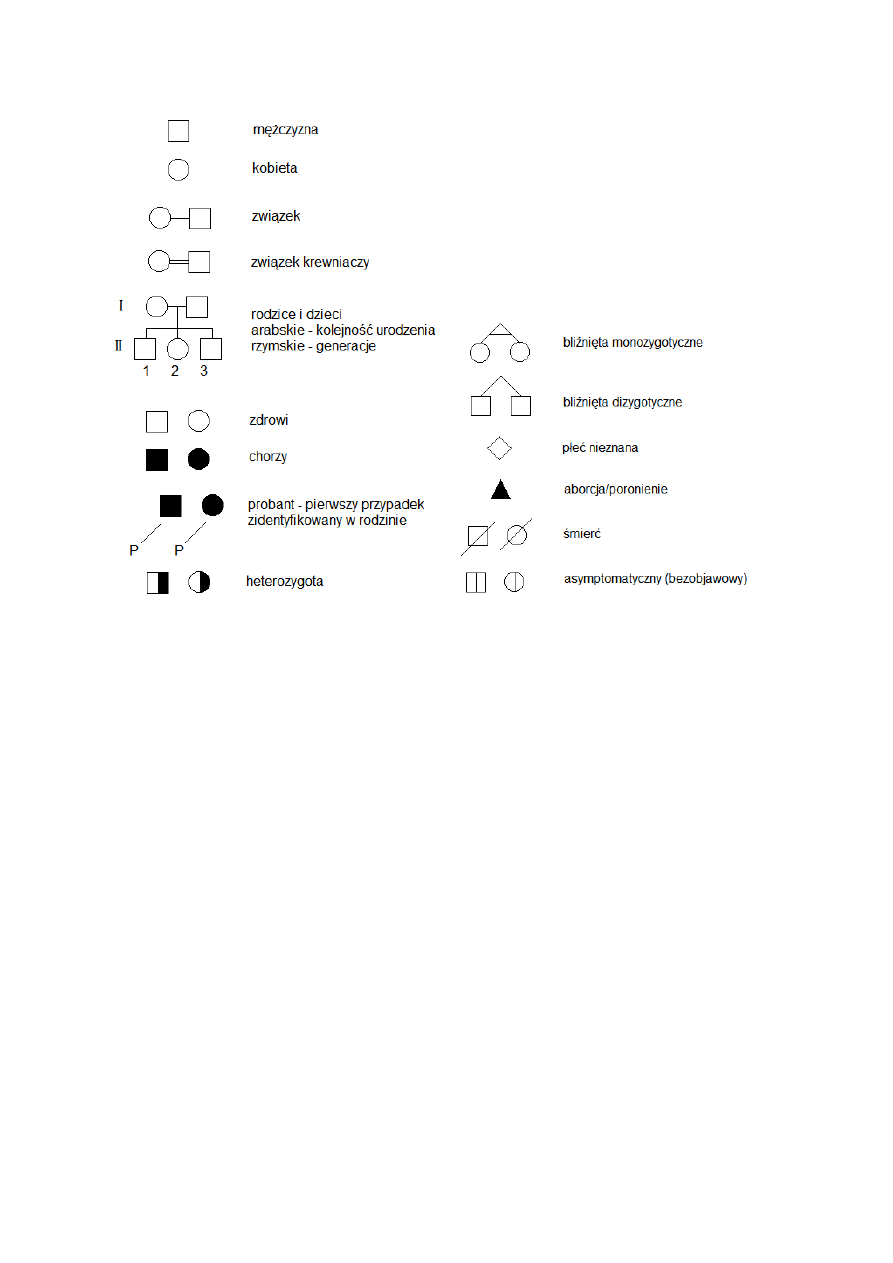
Kryteria dziedziczenia autosomalnego dominującego:
- cecha występuje w każdym pokoleniu bez przeskoków
- każde dziecko z daną cechą ma przynajmniej jednego z rodziców z tą cechą
- w potomstwie heterozygoty z homozygotą recesywną przeciętnie połowa dzieci ma daną
cechę
- osoby nie posiadające danej cechy nie przekazują jej potomstwu
- częstość występowania cechy jest jednakowa u obu płci
- niepełna penetracja mogłaby sprawić, że cecha występuje bez przeskoku
Choroby:
1) wielotorbielowatość nerek:
- AD
- 1/1000
- obraz torbieli w nerkach, wątrobie, trzustce i śledzionie
- w 7-15% przypadków przewlekłej niewydolności nerek
- mutacje genu policystyny (PKD1)
- pierwsze objawy w 3 dekadzie życia
- guzy nerek, krwiomocz, krwinkomocz, kolka nerkowa z powodu niedrożności moczowodu
zatkanego skrzepem krwi, infekcje – poważne powikłanie choroby
- urografia: powiększenie cienia nerki i deformacje układu kielichowo-miedniczkowego
2) hipercholesterolemia rodzinna:
- AD
- heterozygoty 1/500, homozygoty 1/1000000
- mutacje genu receptora LDL (LDL wiąże się z cząsteczką cholesterolu egzogennego i
transportuje ją do komórki)

- choroba polega na znacznym wzroście poziomu cholesterolu we krwi nawet do 1000mg%
(norma 200mg%). Cholesterol odkłada się w różnych miejscach w organizmie
- podwyższenie poziomu LDL od urodzenia
- zmiana ksantomatyczne głównie ścięgien Achillesa, prostowników dłoni i stóp, łokci i
kolan, w pierwszej dekadzie życia u homozygot, w drugiej u heterozygot
- przedwczesna miażdżyca tętnic (aorty, płucnej, wieńcowych)
- rąbek starczy dookoła rogówki
- odkładanie się złogów cholesterolu w ścięgnach, mózgu, płucach, wątrobie, śledzionie,
nerkach
3) polidaktylia
- penetracja 65%
- cecha dziedziczona jako cecha izolowana w sposób autosomalny dominujący
- polidaktylia przedosiowa i zaosiowa
- dodatkowy palec
3b) syndaktylia
- zrośnięcie palców
Kryteria dziedziczenia autosomalnego recesywnego:
- jeżeli cecha występuje rzadko, to rodzice i krewni z wyjątkiem rodzeństwa są zwykle
pozbawieni danej cechy
- w potomstwie z heterozygot cecha występuje przeciętnie w ¼ potomstwa
- wszystkie dzieci rodziców posiadających daną cechę też ją posiadają
- częstość występowania jest jednakowa dla obu płci
- jeżeli cecha występuje rzadko, jej częstość wzrasta u dzieci rodziców spokrewnionych
- choroby:
- ataksja teleangiektazja
- zespół Blooma
- xero derma pigmentosum
- anemia sierpowato-krwinkowa
- zwłóknienie torbielowate = mukowiscydoza
Mukowiscydoza
- AR
- częstość choroby 1/2000-1/4000
- częstość nosicieli (heterozygoty) – 1/20-1/25
- mutacje genu CFTR kodującego kanały chlorkowe, zlokalizowanego w ramieniu q
chromosomu 7
- efektem są zaburzenia funkcjonowania gruczołów wydzielania zewnętrznego, co prowadzi
do zalegania gęstego śluzu – głównie w obrębie układu oddechowego (płuca) i pokarmowego
(trzustka) oraz wysokiego poziomu chlorków w pocie
- objawy ze strony układu oddechowego (występują u ponad 90% chorych):
- gęsty i lepki śluz, który zatyka oskrzela i jest podłożem dla rozwoju bakterii
- występuje suchy i uciążliwy kaszel, niekiedy duszność (pierwsze objawy w wieku
niemowlęcym)
- nawracające zapalenie oskrzeli i płuc, trudno poddające się typowemu leczeniu,
prowadzą
do rozstrzenia oskrzeli i włóknienia płuc

- przewlekłe zapalenie zatok bocznych nosa z polipami (głównie u starszych dzieci)
- objawy ze strony układu pokarmowego (występują u ok. 75% chorych):
- gęsty i lepki śluz blokuje przewodu trzustkowe i uwalnianie enzymów trawiennych
- występują obfite, nie uformowane, cuchnące tłuszczowate stolce od wczesnego
dzieciństwa
- powiększenie objętości brzucha, niekiedy wypadanie odbytnicy
- tzw. niedrożność smółkowa jelit w okresie noworodkowym
bardziej chodzi o blokowanie śluzem jąder (???)
w normalnym genie CFTR
nukleotyd
AAT ATC ATC TTT GGT
delecja fenyloalaniny w pozycji 508
blokada w obróbce potranslacyjnej tego białka
Przemiany fenyloalaniny
fenyloketonuria
albinizm
fenyloalanina
tyrozyna
dihydroksyfenyloalanina
kwas
kwas
fenylopirogronowy
hydroksyfenylopirogronowy
melanina
kwas
homogentyzynowy
alkaptonuria
maleiloacetooctan
fumaryloacetooctan
Fenyloketonuria
- AR
- 1/10000
- defekt metabolizmu aminokwasów (fenyloalaniny)
- mutacje genu PAH zlokalizowanego na chromosomie 12q. Następstwem jest blok
metaboliczny prowadzący do niedoboru lub braku hydroksylazy fenyloalaniny, co prowadzi
do wytworzenia kwasu fenylopirogronowego z innych fenylo kwasów drogą zastępczego toru
metabolicznego
- wtórnie prowadzi do niedoboru tyrozyny
- choroba ujawnia się we wczesnym dzieciństwie
- uporczywe wymioty, pobudliwość nerwowa, mysi zapach pieluch, potu
- jasna karnacja skóry, jasne włosy, niebieskie oczy
- opóźnienie rozwoju psychoruchowego, umysłowego
- napady drgawkowe, zaburzenie chodu
Albinizm (bielactwo)
- AR

- 1/35000
- gen zlokalizowany na chromosomie 11q, blok metaboliczny powoduje niedobór lub brak
enzymu tyrozynazy
- objawy związane z niedoborem melaniny
- jasna barwa skóry, białe włosy, czerwone przebarwione tęczówki
- światłowstręt, niedowidzenie
- nadwrażliwość na słońce
Alkaptonuria (choroba niebieskich pieluch)
- AR
- 1/250000
- gen zlokalizowany na chromosomie 3q, blok metaboliczny – brak oksydazy kwasu
homogentyzynowego
- odkładanie polimerów kwasu homogentyzynowego w chrząstkach
- ciemnienie moczu na powietrzu (kw. h. jest utleniany do chinonów)
- ciemnobrązowe przebarwienie tęczówek, chrząstek nosa, ścięgien, więzadeł, chrząstek
międzykręgowych
- zesztywnienie kręgosłupa i stawów kończyn
- osteoporoza
- przeżycie normalne
Choroby dziedziczone w sposób autosomalny dominujący:
- pląsawica Huntingtona (m. dynamiczne)
- nerwiakowłókniakowatość
Zjawiska utrudniające analizę rodowodów:
- fenokopia
- genokopia – warunkowanie tej samej cechy przez dwa różne geny
- heterogenia – różny sposób dziedziczenia
- niepełna penetracja
- zróżnicowana ekspresywność – stopień nasilenia danej cechy
Choroby jednogenowe sprzężone z chromosomem X
Kryteria XDom.:
1) każde dziecko z daną cechą posiada co najmniej jednego z rodziców z tą cechą
2) mężczyzna z daną cechą przekazuje ją wyłącznie córkom
3) heterozygotyczne kobiety przekazują cechę połowie potomstwa obydwu płci
4) kobieta homozygotyczna przekazuje cechę wszystkim dzieciom obu płci
5) jeżeli cecha występuje rzadko, to występuje dwa razy częściej u kobiet niż mężczyzn (dla
zygot męskich jest często letalna)
6) przy całkowitej penetracji genu cecha występuje we wszystkich pokoleniach bez
przeskoków
Krzywica witamino-D-oporna rodzinna
- najważniejsze objawy – karłowatość kości długich, bóle, zniekształcenia kości, szczególnie
dolnych, wykrzywianie kończyn, niski wzrost, hipofosfatemia na czczo, hiperfosfatemia
- 1/20000
- sposób dziedziczenia – dominujący sprzężony z chromosomem X, mutacje w genie PHEX
- przebieg – wczesne rozpoznanie i leczenie może zmniejszyć nasilenie deformacji kostnych,
a dorośli mogą osiągnąć wzrost ponad 170cm; w starszym wieku wzmożona osteoporoza
- pierwotny defekt dotyczy uszkodzenia genu, którego ekspresja zachodzi w osteoblaście –
miejscu czynnej mineralizacji kości
Zlokalizowano gen choroby (PHEX) na chromosomie X w rejonie p22.2 oraz poznano
sekwencję i strukturę genu. Białko PHEX bierze udział w potranslacyjnej obróbce.
Kryteria dziedziczenia XRec.:
1) chory mężczyzna nie przekazuje cechy dzieciom żadnej płci, ale jego wszystkie córki są
nosicielkami
2) w potomstwie kobiety nosicielki połowa synów choruje, a połowa córek jest nosicielkami
3) chory mężczyzna ma matkę nosicielkę
4) kobiety z daną cechą moają ojca z tą cechą i matkę nosicielkę
5) częstość występowania cechy jest znacznie wyższa u mężczyzn niż u kobiet
6) cecha nie musi występować w każdym pokoleniu
Choroby XR:
- ślepota na barwę zieloną i czerwoną
- zespół łamliwego chromosomu X
- niespecyficzne upośledzenie umysłowe
- sprzężone z chromosomem X
- dystrofia mięśniowa Duchenne’a
- dystrofia mięśniowa Beckera
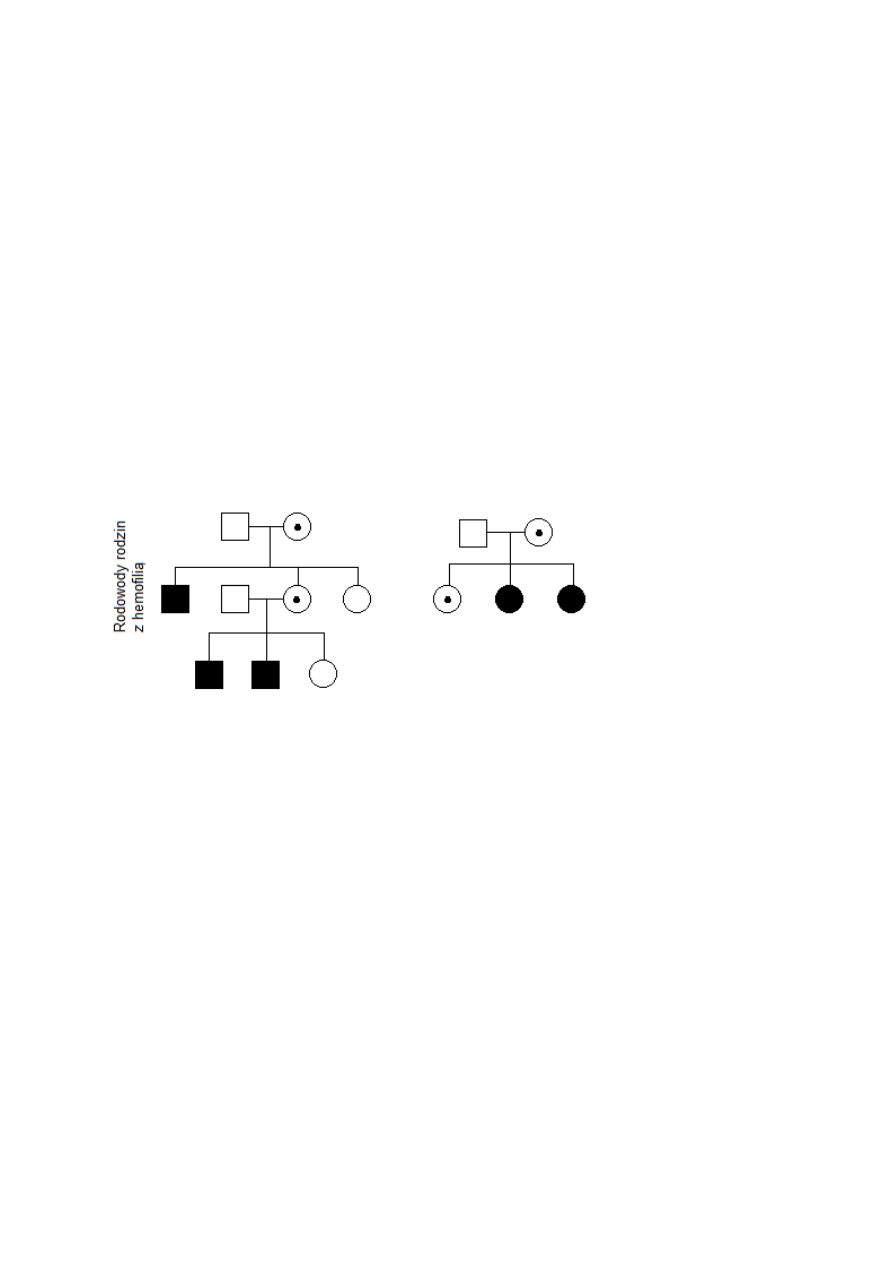
- hemofilia A
- hemofilia B
- rybia łuska – a’la łuszczyca
Podział hemofilii w zależności od aktywności czynnika VIII/IX:
- postać łagodna:
- aktywność czynnika VIII/IX 50% - nie ma objawów
- 25-50% - skłonność do krwawień po dużych zabiegach i poważnych wypadkach
- 5-25% - krwawienia po małych zabiegach i niewielkich skaleczeniach
- postać umiarkowana:
- 1-5% - ciężkie krwawienia po małych zabiegach, niekiedy samoistne wylewy
- postać ciężka:
- poniżej 1% - samoistne wylewy do stawów i mięśni

Dystrofia mięśniowa Duchenne’a (DMD):
- objawy: postępujące osłabienie mięśni proksymalnych, kończyn; przykurcze stawów;
skolioza; chód kaczkowaty; upośledzenie umysłowe; kardiomiopatia; objaw gnoma –
rzekomy przerost mięśni łydek; objaw Gawersa – wspinanie się po sobie
- występowanie: 1/3000 żywo urodzonych chłopców
- sposób dziedziczenia: recesywny sprzężony z chromosomem X; mutacja w genie dystrofiny
(DMD)
- przebieg: szybki postęp niedowładów mięśni, zachodzi w drugiej dekadzie życia, wózek
inwalidzki, od ok. 12 roku życia osłabienie mięśni gardła i oddechowych śmierć ok. 18 r.ż. z
powodu niewydolności oddechowej, zastoinowej niewydolności krążenia, zapalenie płuc lub
zachłyśnięcia
- stopniowy i nieodwracalny zanik mięśni wywołany uszkodzeniem białka dystrofiny
- dotyka tylko chłopców, kobiety są bezobjawowymi nosicielkami
Dwie postacie kliniczne:
- typ Duchenne’a (DMD) – ostry przebieg
- typ Beckera (BMD) – łagodny przebieg
W ostrej postaci pacjenci przestają chodzić samodzielnie w wieku 8-10 lat, a śmierć na skutek
niewydolności oddechowej następuje ok. 20 r.ż.
Dystrofia DMD i BMD
Podłoże molekularne: mutacje w genie DMD uszkadzające syntezę dystrofiny
- delecje (60%)
- duplikacje (5-10%)
- mutacje punktowe (30%)
Gen dystrofiny (DMD)
- wielkość – 2,5 mln pz, 75 eksonów
- wielkość mRNA – 14 tys. pz
- wielkość białka – 3368aa
Zaburzenia rozpoznawania barw – daltonizm
- achromatyzm lub monochromatyzm – całkowita ślepota na barwy, tritanomalia – nie widać
barwy niebieskiej – cechy autosomalne
- dichromatyzm – brak rozpoznawania barwy czerwonej lub zielonej
- protanopia – nie widać barwy czerwonej
- deuteranopia – nie widać barwy zielonej
Sprzężone z chromosomem X
Dziedziczenie wieloczynnikowe
- gen, środowisko
- determinowane przez wiele genów z różnych loci (każdy wywiera mały, lecz kumulujący się
efekt) wraz z czynnikami środowiskowymi, np. większe prawdopodobieństwa:
- dziecko wysokich rodziców będzie wyższe niż dziecko niskich
* geny termowrażliwe - wrażliwe na temperaturę
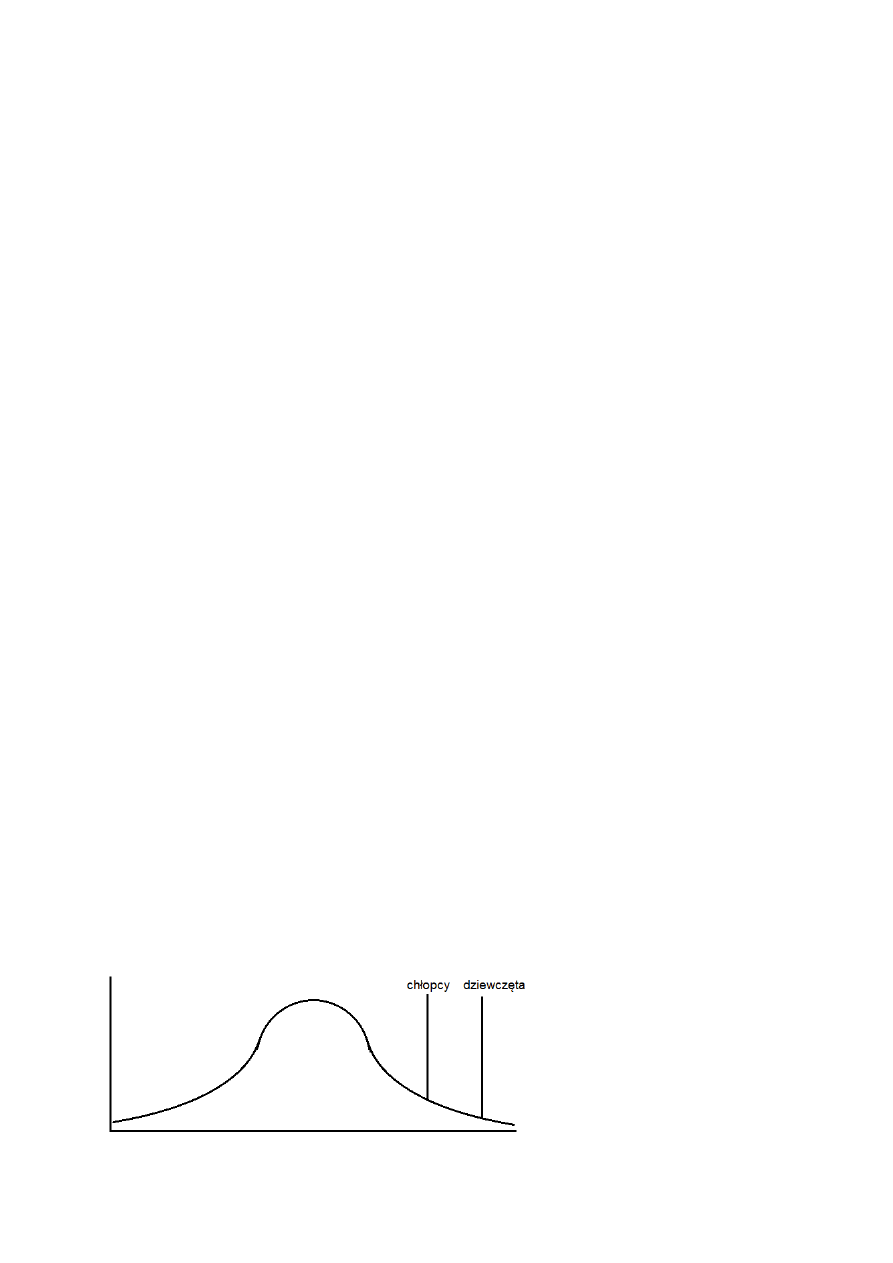
Dziedziczenie poligonowe – cechy dziedziczone wyłącznie genetycznie, przez wiele genów
znajdujących się w różnych loci, np. kolor skóry
transgresja – przekroczenie fenotypu rodzicielskiego; przekroczenie zakresu cechy
wyznaczonego przez pokolenie rodzicielskie
- może być dodatnia, gdy osobniki idą w kierunku o maksymalnym natężeniu cechy,
lub
ujemna
Cechy ilościowe i jakościowe
1) cechy ilościowe – to takie, które są mierzalne. Należą do nich: wzrost, waga, poziom
cholesterolu we krwi, inteligencja, ciśnienie krwi, objętość krwinek czerwonych
- cechy ilościowe wykazują zmienność ciągłą – istnieje nieskończona liczba pośrednich
wartości pomiędzy ekstremami (krzywa Gaussa)
- dla większości tych cech istnieje rozkład Gaussa – większość ludzi pasuje do środka
krzywej
- pierwsze badania nad dziedziczeniem wieloczynnikowym zostały przeprowadzone przez
Galtona – zauważył on „regresję do przeciętności” – potomstwo wydawało się „cofać” do
wartości pośrednich
2) cechy jakościowe – nie mogą być zmierzone – są to fenotypy wszystko-albo-nic. W
praktyce, probant z cechą jakościową jedną w rodzinie
- przykłady: depresja, cukrzyca insulino zależna, epilepsja, nadciśnienie, schizofrenia
* wartość progowa – wartość, po przekroczeniu której występują objawy chorobowe
Wieloczynnikowe czynniki dziedziczenia:
- zaangażowane kilka loci
- brak dominacji lub recesywności
- kumulacja efektów fenotypowych
- oddziaływanie czynników środowiskowych z genotypem
Choroby dziedziczące się w sposób wieloczynnikowy:
- większość dzieci dotkniętych chorobą ma zdrowych rodziców (większość geniuszy ma
przeciętnych rodziców)
- ryzyko urodzenia kolejnego chorego dziecka zwiększa się wraz z liczbą chorych dzieci w
rodzinie
- ryzyko powtórzenia się choroby u krewnych jest wyższe w przypadku ciężko chorych
probantów
- pokrewieństwo rodziców tylko nieznacznie zwiększa ryzyko urodzenia chorego dziecka
- ryzyko urodzenia chorego dziecka spada gwałtownie wraz ze spadkiem pokrewieństwa w
przeciwieństwie do chorób dziedziczonych autosomalnie dominująco o niepełnej penetracji
- ryzyko powtórzenia się choroby jest wyższe dla krewnych tych dzieci, których płeć jest
rzadziej dotykana chorobą

Ponieważ analiza rodowodu jest niewystarczająca, konieczne są analizy zgodności u bliźniąt
jednojajowych i korelacji rodzinnej
Zgodność
Zgodność % = [X/X+Y] x 100%
X – oba bliźnięta posiadają cechę
Y – jedno z bliźniąt posiadających cechę
Jeżeli cecha jest wyłącznie uzależniona od genów monozygotyczne bliźnięta będą miały
100% zgodności, podczas gdy bliźnięta dizygotyczne mające tylko połowę genów wspólnych,
będą wykazywać wartość niższą. Jeżeli natomiast cecha jest wyłącznie środowiskowa,
zarówno bliźnięta mono- jak i dizygotyczne będą wykazywać taką samą zgodność.
Wprowadzenie do badań rodzinnych
Stopień pokrewieństwa
Przykłady
Proporcja wspólnych genów
PIERWSZY
rodzic i dziecko,
1/2
rodzeństwo
DRUGI
Dziadkowie i wnuki,
1/4
siostrzeńcy, bratankowie,
ciotki i wujowie
TRZECI
kuzyn pierwszego stopnia
1/8
cukrzyca – poligonowy model dziedziczenia
nowotwór – największa komponenta środowiskowa
Odziedziczalność
Określa, jaka część zmienności cechy zależy od wpływu genów, a jaka od środowiska.
Jest to genetyczny składnik całkowitej zmienności fenotypowej danej cechy wyrażony w
procentach
V
G
– wariancja genetyczna
V
E
– wariancja środowiskowa
Korelacja
Jest to liczbowa miara zależności cech. Współczynnik korelacji +1 oznacza korelację
całkowitą dodatnia, np. im wyższy ojciec tym wyższy syn.
Korelacja w granicach 0.7-1 jest duża, 0,3-0,7 średnia, poniżej 0,3 – bardzo słaba
* wraz ze wzrostem pokrewieństwa maleje częstość występowania choroby
Genetyka populacyjna
- badanie częstości genów populacji
Prawo Hardy’ego-Weinberga
- prawo to, określając liczbowo zależności między częstością genów a częstością genotypów
w populacji, przedstawia stan dynamicznej równowagi w tzw. populacji idealnej
- wykazali, że częstość poszczególnych genotypów w populacji rozmnażającej się płciowo,
diploidalnego gatunku osiąga równowagę po jednym pokoleniu losowego kojarzenia
populacja idealna:
- duża liczba
- brak selekcji naturalnej
- brak mutacji
- brak migracji
- panmiksja (losowe kojarzenie)
- izolacja
(p +q)
2
= 1
p + q = 1
p – częstość allelu dominującego
q – częstość allelu recesywnego
p
2
– częstość homozygot dominujących
q
2
– częstość homozygot recesywnych
2pq – częstość heterozygot
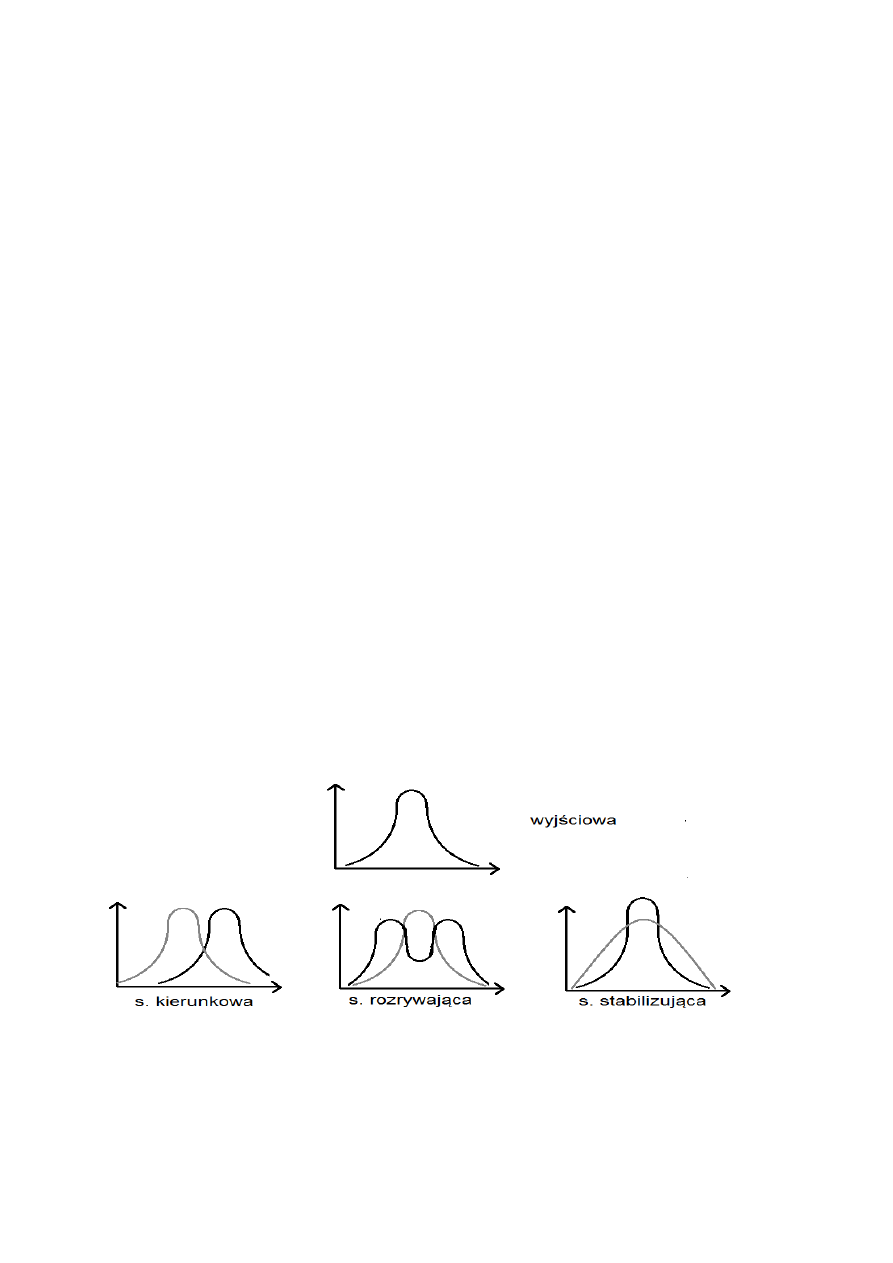
* Na naturalną populację wpływa szereg czynników:
1) mutacje
2) selekcja (dobór naturalny)
- selekcja stabilizująca
- selekcja kierunkowa
- selekcja rozrywająca
Selekcja nie oddziaływuje bezpośrednio na genotyp organizmu, lecz działa na fenotyp
- białe ćmy pod wpływem
- zięby Darwina które
- wielkość owoców
przemysłu zmieniają się
przystosowują się dziobem
- masa urodzeniowa
na ciemne
do środowiska aby przeżyć
noworodka
- samice motyli Papilio
- selekcja
preferująca
Dardanus naśladują gatunki
heterozygoty, np. anemia

motyli niejadalnych
sierpowata odporna na
malarię
* pomiędzy mutacją a selekcją istnieje równowaga
3) migracja
- prowadzi do specjacji
specjacja allopatryczna i sympatryczna
Przykładem specjacji sympatrycznej są allopoliploidy u roślin, np. liczne gatunki poziewnika,
wiele roślin uprawnych tj. pszenica
4) dryf genetyczny
- efekt wąskiego gardła (np. gepard)
- efekt założyciela (np. słoń morski)
- nielosowe kojarzenie się
Zasadę H-W można wykorzystać do określenia częstości allelu i genotypu kiedy nie
można rozróżnić homozygoty dominującej od heterozygoty (co często obserwuje się w
chorobach recesywnych)
Zad.
studenci – 28 osób
wrażliwi – 21
niewrażliwi – 7
p
2
+ 2pq + q
2
= 1
p
2
+ 2pq = = 0,75
q
2
= = 0,25 q – 0,5
p
2
+ 2*0,5p = 1
p
2
+ p = 0,75
p(p + 1) = 0,75
p + q = 1
p = 0,5
q = 0,5
Zad.
W pewnej populacji ludzi płatek ucha jest przyrośnięty (cecha recesywna) i występuje z
częstością 1/2500. Oblicz częstość hetero- i homozygot dominujących.
q
2
=
q =
p + q = 1 p =
p
2
=
2
2pq = 2 = 0,04
Hemoglobina
- białko występujące w erytrocytach
- transportuje tlen z płuc do tkanek całego ciała

- posiada strukturę czwartorzędową
2 α-globiny
2 β-globiny
- pomiędzy hem
* pseudogeny – sekwencje, które przypominają geny, ale nie pełnią ich funkcji
- gen 11 – β + 1 pseudogen
Hemoglobina:
nazwa hemoglobiny:
- embrionalny okres
ξ2ε2
gower 1
(zarodkowy)
α2ε2
gower 2
ξ2γ2
Portland 2
- płodowy okres
α2β2
HbA
α2γ2
HbF
- dorosłość
α2β2
HbA
α2δ2
HbA
2
Hemoglobinopatie
- zaburzenia struktury – zmieniona budowa globiny, bez zmiany poziomu ekspresji białka
- zaburzenia poziomu ekspresji (talasemie) – obniżony poziom syntezy jednego lub kilku
łańcuchów globinowych: efekt to zaburzenie stosunku ilościowego łańcuchów α i β
względem siebie
Substytucje aminokwasowe
β6(A3) Glu Val
Warianty strukturalne hemoglobiny
- powodujące anemię hemoglobiny
- HbS (tworzenie agregatów anemia sierpowata; krwinki sierpowate powodują
zatory)
- HbC (powoduje anemię)
- rodzaj mutacji: substytucja
- sposób dziedziczenia: autosomalny recesywny
- hemoglobiny o zmienionym powinowactwie do tlenu
- HbM (Zawsze Fe
3+
nie transportuje tlenu; sinice bezobjawowe)
- Hb Kempsey(podwyższone powinowactwo do O
2
)
- Hb Kansas (sinica); obniżone powinowactwo do O
2
- rodzaj mutacji: substytucja
- sposób dziedziczenia: autosomalny dominujący
- fenotyp talasemii
- HbE (substytucja, mutacja: obniżenie poziomu syntezy, nieprawidłowe składanie
RNA
obraz łagodnej talasemii; AR)
Talasemie
- dziedziczenie nieprawidłowości syntezy
- zmiany ekspresji globin α i β

1) α-talasemia – utrata 2 lub więcej genów kodujących α-globiny
brak α -globin jest letalny
- fenotyp:
- normalny
- bezobjawowy nosiciel
- α-talasemia łagodna – łagodna anemia, małe erytrocyty
- HbH
- hydrops letalis – cecha letalna
2) β-talasemia – wynik mutacji punktowych; wpływają na ekspresję
heterozygota – talasemia minor – łagodna anemia, hipochromazja, mikrocytoza erytrocytów
homozygota – talasemia maior – ostra anemia, nieprawidłowa erytropoeza, hipochromazja,
różny
rozmiar i kształt erytrocytów
- efekty mutacji:
- utrata funkcji (α- lub β-talasemia)
- wzmocnienie funkcji (Hb Kempsey)
- nowe właściwości (sickle cell anemia – anemia sierpowata)
- ekspresja genu w niewłaściwym czasie (np. dziedziczenie warunkujące wytwarzanie
płodowej hemoglobiny w okresie płodowym)
- ekspresja genu w niewłaściwym miejscu
Nieprawidłowości budowy błony erytrocytów:
1) sferocytoza
- autosomalna dominująca
- 1/5000
- nieprawidłowości białka cytoszkieletu
- komórki mają kształt sferyczny, są niszczone w śledzionie
- zwiększona podatność osmotyczna
- klinicznie: łagodna anemia; żółtaczka hemolityczna; przełom plastyczny, powiększenie
śledziony; prawdopodobny przyrost kości
2) eliptocytoza
- autosomalna dominująca
- 1/3000
- nieprawidłowe interakcje białek cytoszkieletu
- normalna wrażliwość osmotyczna
- u większości osób przebiega bezobjawowo
Pasożytnictwo – jest związkiem antagonistycznym, pomiędzy osobnikami populacji dwóch
różnych gatunków
Wyszukiwarka
Podobne podstrony:
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
więcej podobnych podstron