
Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
1/113
Neurobiologia
wybrane zagadnienia „neuroscience”
Dariusz Adamek
Zakład Neuropatologii, Inst. Neurologii, CM UJ
Materiały do wykładów z neurobiologii dla studentów Wydziału Lekarskiego i Stomatologii CM UJ
Materiały stanowią własność autora (w rozumieniu praw autorskich) i mogą być wykorzystywane jedynie
jako pomoc i podstawa do przygotowania się do zaliczenia przez studentów zajęć fakultatywnych z
zakresu neurobiologii, którym zostały udostępnione nieodpłatnie.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
2/113
Neurobiologia
(wybrane zagadnienia „neuroscience”)
Podstawy hierachiczności i budowy SN
•Z perspektywy ewolucji system nerwowy ZWIĘKSZA SZANSE NA PRZEśYCIE I REPRODUKCJĘ
•Schemat budowy i funkcjonowania komórki nerwowej jest podobny u wszystkich zwierząt począwszy od
jamochłonów !
•Ogólny „plan” budowy CSN u wszystkich kręgowców jest TAKI SAM.
Camillo Golgi: teoria retikularna SN (sieciowa, „syncytialna”)
Ramon y Cajal: doktryna neuronalna (każdy neuron jest niezależną komórką)
Charles Sherrington: postulat synapsy od greckiego synaptein = łączyć (1897). 50 lat później zobaczono
synapsy w ME. On też zdefiniował pojęcie odruchu. („The integrative action of nervous system” 1906).
Kompleks synaptyczny przypuszczalnie wywodzi się z desmosomów nabłonków.
Hierarchia systemów nerwowych
Najprostszy: hydra (stułbia) –
1) pierwsze czuciowe dwubiegunowe neurony (biegun recepcyjny i biegun transmisyjno-efektorowy).
Wyspecjalizowane komórki czuciowe pozwalają m.in. na pojawienie się różnych typów czucia.
Układ „od-do” (FUNKCJONALNA BIEGUNOWOŚĆ o fundamentalnym znaczeniu!) obowiązuje w
czuciowych neuronach także u człowieka.
2) pierwsze neurony ruchowe (m) (pośredniczące między n.czuciowym i efektorem). Razem: Od neuronu
czuciowego (jego dendrytów) poprzez (drugi) neuron ruchowy do efektora daje najprostszy łuk odruchowy.
(Wypustki między neuronami ruchowymi Cajal nazwał „amakrynowymi” .Nota bene kk. amakrynowe
występują w siatkówce i opuszce węchowej.)
Są też początki „centralizacji” dystrybucji neuronów (np. wokół otworu gębowego).
Cajal: teoria biegunowości czynnościowej. Konwergencja (motoneuron otrzymuje sygnały z wielu neuronów
czuciowych) i dywergencja.
Cnidaria: począwszy od stułbi mają SN
Robaki płaskie:
Symetria dwustronna; zróżnicowanie (umownie) „grzbiet-brzuch” oraz „głowa-ogon” (cefalizacja)
Pojawia się interneuron (w tym amakrynowy = „niepolarny”). Interneuron może działać jako „włącznik-
wyłącznik” oraz jako pacemaker. Interneuron „lokalny” i „projecting”. Najprostsze układy z cechami
centralizacji i cefalizacji.
Dendryty u bezkręgowców rozgałęziają się odchodząc od aksonów! (u kręgowców to aksony mogą odchodzić
od dendrytów)
Ludzki układ nerwowy jelita („enteralny”) ma cechy „prymitywne”.
Pierścienice i stawonogi:
segmentacja i brzuszna lokalizacja (podwójnego) sznura nerwowego (ventral nerve cord)
Kręgowce: Wszystkie mają ten sam plan układu nerwowego i ten sam plan rozwoju embrionalnego.
Rdzeń umiejscowiony jest grzbietowo od struny grzbietowej (notochord).
Początek – płytka nerwowa (wyosobniona grupa komórek w formie „łyżeczki” w grzbietowej części ektodermy)
Cechy REGIONALIZACJI komórek płytki
„Neurulacja”
1)Pęcherzyki oczne, infundibulum, otic rhombomere
2)Zagłębianie się płytki nerwowej tworzy „fałdy nerwowe” (n.folds) i dalej cewę nerwową (n.tube) z
neuroporami (tworzy ją pojedyncza warstwa komórek tzw. „neuroepitelium”)
3)
Fazy: „trzech pęcherzyków” i „pięciu pęcherzyków”
4) Proliferacja neuroepitelium i tworzenie tzw. strefy płaszcza (mantle layer) w której następuje różnicowanie się
neuronów i ich migracja oraz segregacja w warstwy i jądra.
W tyłomózgowiu i rdzeniu kręgowym pierwsze różnicują się MOTONEURONY.
(u płodu pojawiają się nieskoordynowane ruchy zanim wytworzą się łuki odruchowe)
•Tzw „układy farmakologiczne” (oparte o ten sam neurotransmiter i podległe działaniu tych samych leków) nie
pokrywają się z układami anatomicznymi

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
3/113
•Podobnie jest w przypadku układów charakteryzujących się ekspresją tych samych genów „gene expressinig
system”
•Jest błędem przypuszczenie a priori, że przetwarzanie informacji w SN podlega również hierarchiczności.
Rola astrocytów:
Regulacja zewnątrzkomórkowego stężenia K+
usuwanie neuroprzekaźnika ze szczeliny synaptycznej
„uszczelnianie” synaps
zaopatrzenie neuronów w glukozę
współtworzenie bariery krew-mózg
tworzenie membrana gliae limitans externa
Przepływ informacji w systemie nerwowym
cz. I: geneza potencjału błonowego i potencjału czynno
ś
ciowego.
Przewodnictwo nerwowe.
Neurotoksyny biogenne
„Elektryka układu nerwowego”
RÓWNANIE NERNSTA
Równanie Nernsta (Walter Nernst 1888)
(równanie potencjału równowagi dla określonego jonu) t.j. równowagi między gradientem stężeń i
„gradientem” elektrycznym: na przykładzie potasu
(po prawej wersja uproszczona dla temp. 37 st. C
)
R= stała gazowa (8,315 J K-1 mol -1);
F = stała Faraday’a (96485 Culombów/mol)
T=temp.Kelvina; z = wartościowość jonu
Culomb = 6x1018 ładunków;
V = J/C (wolt = Joul / Coulomb)
Ponadto ln(x) = Log10(x) / Log10e = Log10(x) / 0,434;
L Avogadry = 6 x 1023 mol-1
K+ o = stężenie pozakom K+,
K+i = stężenie wewnkom. K+
Dla [K+ zew]/[K+ wew] = 4/155 = 0,026
Log10 0,026 = -1,58 a zatem 61,5 x (–1,58) = -97,7 mV
EK = -97,7mV (potencjał równowagi dla K)
Potencjał EK równoważy potencjał dyfuzji przy różnicy stężeń z przykładu poprzedniego (155 i 5 mmol/l)
Gdy uwzględnimy inne jony (Równanie Goldman-Hodgkin-Katz) wyjaśnia się dlaczego potencjał
spoczynkowy różni się od potencjału równowagi dla potasu (jak również od pozostałych jonów!)
Różne populacje komórek w CSN mają różne wartości potencjału spoczynkowego. Mogą wartości zależeć
od pory dnia, np. neurony wzgórza są hyperspolaryzowane w nocy zmniejszając w ten sposób impulsację
dokorową.
×
×
=
+
+
i
O
K
K
K
zF
RT
E
]
[
]
[
ln
303
,
2
z
V(wolt)
C
z
J
C
z
mol
mol
K
K
J
mol
C
z
mol
K
K
J
=
=
∗
=
÷
Równanie Goldman-Hodgkin-Katz:
Wyznacza potencjał spoczynkowy komórki uwzgledniając
najważniejsze jony i ich relatywne (w stosunku do potasu)
przepuszczalności
×
=
+
+
i
O
K
K
K
Log
mV
E
]
[
]
[
10
5
,
61
+
+
+
+
×
=
−
+
+
−
+
+
O
Cl
i
Na
i
K
i
Cl
O
Na
O
K
m
Cl
p
Na
p
K
p
Cl
p
Na
p
K
p
F
RT
V
]
[
]
[
]
[
]
[
]
[
]
[
ln

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
4/113
Względne przepuszczalności dla jonów
P
P
P
P
KKKK
::::
PPPP
Na
Na
Na
Na
::::
PPPP
Cl
Cl
Cl
Cl
pozostaja w nastepującej relacji:
PPPP
KKKK
::::
PPPP
Na
Na
Na
Na
::::
PPPP
Cl
Cl
Cl
Cl
=
=
=
= 1:0,02:
1:0,02:
1:0,02:
1:0,02:0,45
0,45
0,45
0,45
Indeksy „o” oraz „i” przy nawiasach kwadratowych oznaczają odpowiednio, że
Indeksy „o” oraz „i” przy nawiasach kwadratowych oznaczają odpowiednio, że
Indeksy „o” oraz „i” przy nawiasach kwadratowych oznaczają odpowiednio, że
Indeksy „o” oraz „i” przy nawiasach kwadratowych oznaczają odpowiednio, że
stężenie danego jonu odnosi się do przestrzeni pozakomórkowej („O”
stężenie danego jonu odnosi się do przestrzeni pozakomórkowej („O”
stężenie danego jonu odnosi się do przestrzeni pozakomórkowej („O”
stężenie danego jonu odnosi się do przestrzeni pozakomórkowej („O”---- outside) i
outside) i
outside) i
outside) i
wewnatrzkomórkowej (”i” inside)
wewnatrzkomórkowej (”i” inside)
wewnatrzkomórkowej (”i” inside)
wewnatrzkomórkowej (”i” inside)
POTAS
•70 kg
•28L wewnątrzkomórkowej
–140 mEq/L
•14L zewnątrzkomórkowej
–4,2mEq/L
–Około 98 proc. całkowitych zasobów wewnątrzustrojowych potasu znajduje się w przestrzeni
wewnątrzkomórkowej, a ok. 2 proc. w przestrzeni zewnątrzkomórkowej.
–Prawidłowe stężenie potasu w surowicy wynosi 3,5–5,0 mEq/l. Gdy stężenie potasu w surowicy krwi spada
poniżej 3,5 mEq/l, rozpoznaje się hipokaliemię.
–Zasoby ustrojowe potasu wynoszą ok. 50 mEq/kg (czyli u osoby ważącej 70 kg – ok. 3 500 mEq).
•Hodkin i Katz w 1949 r eksperymentem (na aksonie kałamarnicy) ze zmianą stężenia pozakomórkowego K ustalili, że
potencjał równowagi zachowuje się „prawie” zgodnie z równaniem Nernsta, a zatem doszli do wniosku, że błona
komórkowa jest znacznie bardziej przepuszczalna dla K niż dla innych jonów i że to właśnie potas najbardziej wpływa na
zachowanie potencjału błonowego
Pompy jonowe i białka transporterowe
„pompy jonowe”:
3Na
+
-2K
+
-ATPaza
(„elektrogeniczna”)
Ca
++
-Mg
+
-ATPaza
Wymieniacz
e
(głównie „kosztem” Na
+
a zatem pośrednio korzystające z energii gradientu
elektrochemicznego wynikającego z różnicy Na wytworzonej przez Na/K ATP-azę):
Wymieniacz 1Ca
++
- 1Na
+
Wymieniacz 1Cl
-
- 1Na
+
/HCO
3
-
Wymieniacz 1H
+
- 1Na
+
Wymieniacze Na
+
/transportery neurotransmiterów
•Wymieniacz 1Ca++- 1Na+
•Wymieniacz 1Cl- - 1Na+/HCO3-
•Wymieniacz 1H+- 1Na+
•Wymieniacze Na+/transportery neurotransmiterów
Kanały jonowe
•Kanał jonowy to rodzaj „poru” w błonie komórkowej, kontrolowanego przez otwierające się i
zamykające „bramki”
Ruch określonych jonów poprzez błonę komórkową jest formą prądu elektrycznego zależnego od:
•„siły napędowej” będącej różnicą potencjału (spoczynkowego lub po prostu rzeczywistego aktualnego
potencjału np. wyznaczonego dowolnie w technice „voltage-clamp”) Em i potencjału równowagi dla
określonego jonu np. dla potasu Ek
•(np. dla prądu potasowego: Em – Ek) Jeśli Em = Ek prądu nie ma.
•Przewodnictwo błony dla określonego jonu (de facto jest to odwrotność oporu elektrycznego zgodnie z prawem
Ohma tj. i = V/R).
•Dla potasu prąd określi równanie:
•Ik= gk (Em – Ek)
•gdzie Ik – oznacza prąd jonowy potasu; gk – oznacza przewodnictwo dla potasu (g = 1 Siemen gdy 1 volt
powoduje przepływ 1 ampera)
•Przewodnictwo (conductance) przelicza się na powierzchnię błony w cm2

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
5/113
Technika „voltage-clamp”
•Techniki tej używali w latach 50-tych Hodgkin i Huxley wyjaśniając dzięki niej mechanizm m.in. potencjału
czynnościowego jako zjawiska, które można wytłumaczyć i opisać poprzez zmieniające się w czasie
właściwości przewodności błony komórkowej dla poszczególnych jonów
•Technika patch-clamp (Erwin Neher, Bert Sakmann 1976 Max Planck Inst. Goettingen) umożliwiła badanie
przepływów jonowych (prądów) dla indywidualnych kanałów.
•Ostatecznie udowodniła istnienie kanałów jonowoselektywnych a jednocześnie potwierdziła wcześniejsze
postulaty odnośnie istnienia takich kanałów proponowane przez Hodgkina i Huxley’a
•„Makroskopowy” prąd jest sumą mikroskopowych prądów pojedynczych (napięciowozależnych) kanałów
•„Makroskopowe” prądy potasowe – dozewnątrz (outward) również są zsumowanymi prądami kanałów
potasowych
•Zarówno kanał potasowy jak i sodowy muszą posiadać „voltage sensor” – strukturę „wyczuwającą” napięcie
Eksperymenty z patch-clamp wykazały podobieństwa i różnice pomiędzy różnymi kanałami jonowymi
Podobieństwa
kanałów K i Na:
•jonoselektywność, zależność prawdopodobieństwa otwarcia od napięcia, zamykanie kanałów Na i K przez
hyperpolaryzację
•oraz
różnice
•w kinetyce otwarcia (szybkość, czas otwarcia) w fakcie, że depolaryzacja w kanale Na prowadzi oprócz
otwarcia także do jego inaktywacji ale nie w przypadku kanału potasowego
••Wykryto również takie napięciowozależne kanały Na które nie są inaktywowane depolaryzacją i prowadzące
do długotrwających Pcz – (blokowane przez lidokainę, benzokainę)
Tetrodotoksyna (TTX) wytwarzana przez ryby typu puffer fish
blokuje napięciowozależne kanały sodowe
tym samym blokując przewodnictwo nerwowe
Techniki patch-clamp otworzyły drogę do poznania bardzo wielu kanałów jonowych i ich własności
Typy kanałów jonowych:
1) napięciowo-zależne
2) aktywowane ligandem
3) aktywowane fizyczną zmianą kształtu (rozciąganiem)
4) zależne od temperatury
5) Są też kanały zależne od pH- „ acid sensing ion channels (ASICs)
Aktywowane ligandem kanały jonowe nie są zazwyczaj tak wysokoselektywne dla określonych jonów jak
kanały napięciowo-zależne
Oprócz błony cytoplazmatycznej obecne są w błonach organelli wewnątrzkomórkowych
Kanały K aktywowane wapniem, kanały aktywowane cyklicznymi nukleotydami oraz ASICs mają
wewnątrzkomórkowe domeny
detekcji ligandu (Kanały aktywowane cykl. nukleotydami graja rolę m.in.
w węchu i recepcji światła)!
Liczne geny kodują różne kanały jonowe i tak:
Kanały jonowe napięciowo-zależne selektywne dla: Na (10 genów), Ca (16 genów, szczeg. ważne kanały
konieczne dla uwolnienia pęcherzyków synaptycznych), K (prawie 100 genów, prawdopodobnie
najważniesza rola to generowanie potencjału spoczynkowego).
Równanie prądu jonowego obowiązuje dla pojedynczego kanału (np. potasowego)
is= gs (Em – Ek)
„przeciętny” kanał jonowy posiada przewodność średnią ok. 20 pS (pikosiemensów czyli 10-12S)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
6/113
Jeśli gradient napięcia wynosi 50mV a przewodność 20pS, to prąd przechodzący przez pojedynczy kanał
wynosi 10-12A (ampera) czyli ok. 6 mln jednowartościowych jonów/sek. (1A = ok. 6 x 1018 ładunków
elektrycznych / sek)
Kanały jonowe (na przykładzie kanałów potasowych)
1.Wszystkie kanały potasowe potrafią
rozróżnić między K+ i Na+ (dwoma najbardziej rozpowszechnionymi metalami alkalicznymi w
przyrodzie). Miejsce w kanale (w przypadku kanału dla K+ kanał jest tetramerem), które ze względu na
największe przewężenie jest „bramkarzem” nazywane jest „filtrem selektywności”. Jest ono
najprawdopodobniej wspólne dla całej rodziny kanałów potasowych.
•Przez kanał potasowy przechodzą tylko
nieuwodnione
jony K+
Kanały potasowe są najbardziej zróżnicowane
(np. czasem inaktywacji od milisekund do minut).
Wyróżnia się wiele szczególnych podtypów (podgrup kanałów potasowych) np:
-
„Voltage-Gated” Czyli Kanały Napięciowozależne
-
„Inward Rectifiers” Czyli „Dośrodkowe Prostowniki”
-
Kanały potasowe z „opóźnionym” działaniem „delayed rectifier channels” (najważniejsze w fazie
repolaryzacji potencjału czynnościowego)
-
Kanały potasowe wapniowo-zależne
-
„Maxi K+” 200-300 pS, blokowane przez charybdotoksynę (ze skorpiona)
-
Kanały o pośrednim przewodnictwie (18-80 pS) aktywowane obrzękiem komórek
-
„Małe” kanały potasowe 10-14 pS, blokowane przez toksynę pszczół – apaminę
-
Kanały typu „A-current” blokowane przez 4-aminopirydynę
-
Kanały typu IsK z pojedynczą domeną transbłonową
Potasowe kanały tzw. „dośrodkowe/dokomórkowe-prostujące” (inward rectifying potasssium channels =
K-ir) to szczególna podgrupa kanałów potasowych.
Potasowe „dowewnątrz prostujące” (inwardly rectifying) kanały jonowe
Ich cechą jest osłabione przewodnictwo w warunkach depolaryzacji i podwyższone w warunkach
hyperpolaryzacji
.
To podgrupa kanałów potasowych, których wspólnym mianownikiem jest zdolność do
wytwarzania większego dokomórkowego napływu jonów (influx) niż wypływu (efflux). W tym typie
kanałów potasowych oprócz selektywności dla potasu (niezależnej dla kierunku „do” i „od” komórki,
występuje blokowanie kierunku „od” czyli wypływu jonów potasowych w warunkach gdy potencjał
błonowy jest bardziej dodatni niż potencjał spoczynkowy (depolaryzacja).
UWAGA! Pomimo nazwy („inwardly rectifying”) w praktyce niemal zawsze przepuszczają jony potasowe
na zewnątrz (kierunek zależy od wartości potencjału błonowego w relacji do potencjału równowagi dla
potasu czyli –80mV).
Blokowanie wypływu jonów jest skutkiem działania wewnątrzkomórkowego
magnezu (Mg2+) oraz polyamin (spermina, putrescyna, spermidyna). Poznano 7 podrodzin potasowych
kanałów prostujących, (K-ir) które różnią się m.in. Stopniem „prostowania”. K-ir grają istotną rolę w
kontroli i regulacji potencjału spoczynkowego oraz wartości potencjału progowego.
„Dośrodkowe prostujące kanały potasowe” pozwalają na dłuższe odpowiedzi depolaryzacyjne np. w sercu (gdzie
potencjał czynnościowy trwa 100-600msec), oraz grają rolę w tzw „fertilization potential” w komórkach
jajowych (który trwa minuty). Zapobiegają utracie K+ w czasie przedłużonej depolaryzacji i mogą pozwalać
na re-entry K+ T-tubul w mięśniach.
Przykłady chorób spowodowanych nieprawidłowością działania kanałów jonowych („channelopathies”)
1.Mukowiscydoza:
zmutowany kanał chlorkowy
(epithelium chloride channel Cystic Fibrosis
Transmembrane Conductance Regulator – CFTR)
2.Zesp. Bartter’a : Alkaloza, hipokaliemia, hyperaldosteronizm, hyperreninemia, bez nadciśnienia (brak
odpowiedzi na aldosteron), zab. wzrastania, słabość mięśniowa, zaparcia, wielomocz.:
mutacja
napięciowozależnego kanału chlorkowego.
3.Myotonia congenita: Inna mutacja tego samego kanału
(napięciowozależnego kanału chlorkowego)
.
4.Cholera: biegunka spowodowana działaniem toksyny bakteryjnej, która pobudza cAMP w nabłonku
jelita i w następstwie
pobudza kanał CFTR
i w rezultacie wywołuje sekrecję chloru do światła jelita. (w
mukowiscydozie toksyna nie działa!).
5.Rodzinna hypoglikemia z hyperinsulinizmem;
mutacja ATP-zależnego kanału potasowego
w komórkach
beta trzustki, która powoduje ,że kanał stymuluje wydzielanie insuliny
6.Zesp. Liddle’a: wrodzone nadciśnienie:
nadaktywność kanału sodowego w nabłonkach
.
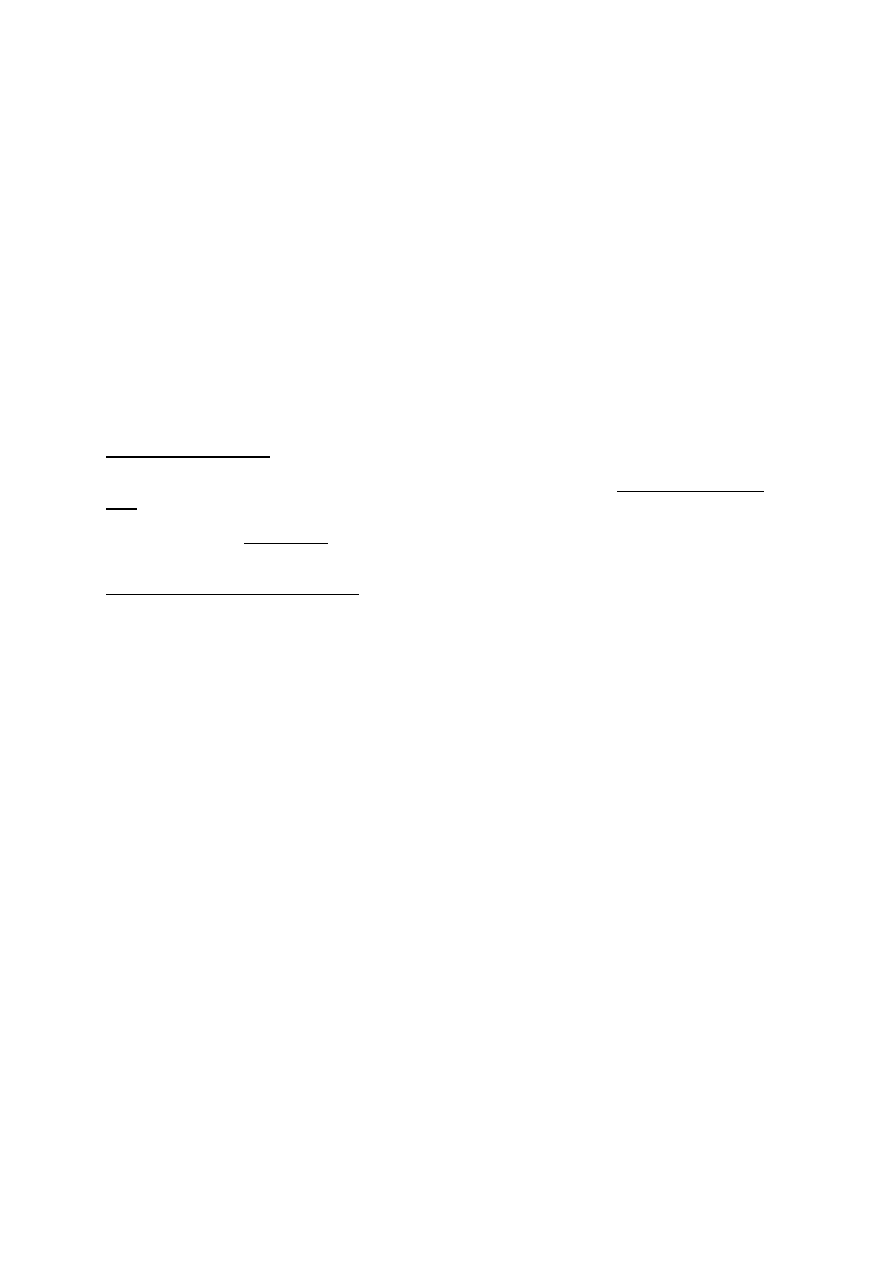
Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
7/113
7.Zesp. Lamberta-Eatona (miasteniczny):
przeciwciała przeciw kanałom Ca
Przykłady schorzeń mózgu nzwiązanych z nieprawidłowymi kanałami jonowymi („KANAŁOPATII”
mózgowych)
Nie ma jednoznacznego wytłumaczenia dlaczego określona mutacja kanału jonowego powoduje dane
objawy
Istnieją różne typy mutacji w danym kanale jonowym różniące się także fenotypowo (objawami).
Zaburzenia napięciowo-zależnych kanałów wapniowych (Ca):
1.Familial hemiplegic migraine (FHM) = (rodzinna migrena z hemiplegią)
2.Episodic ataxia type 2 (EA2)
3.X-linked congenital stationary night blindness (CSNB)
2.
Defekt kanałów sodowych (Na):
1.Generalized epilepsy with febrile seizures (GEPS)
Defekt kanałów potasowych (K)
1.Benign familial neonatal convulsions (BFNC) – objawy drgawek zanikają po okresie noworodkowym
Potas może zabijać…
„Mikstura” do wykonywania wyroku śmierci w niektórych stanach USA (tzw. lethal injection – za
źródłem internetowym): thiopental sodium 6 g, pancuronium bromide 150 mg, potassium chloride 360
mEq
•KCl podawany iv ponad 20 miliekwiwalentów/h jest toksyczny. (nie powinno się przekraczać 80
mEq/dobę a ponadto maksymalnie 40mEq/godz) Podawanie uśmiercające jest szybkie a jego skutkiem
jest zaburzenie czynności elektrycznej serca (indukcja cardiac arrest). •Przy założeniu, że objętość
przestrzeni pozakomórkowej wynosi 14 litrów
w (bardzo bardzo) dużym uproszczeniu można spróbować obliczyć potencjał równowagi dla potasu po
podaniu 360 mEq (tzn dawki podawanej w „lethal injection”) 360/14 litrów (prz.-pozakom.)= 25,7 mEq/L .
W rzeczywistości lokalnie wokół naczyń, zanim dojdzie do pełnego wyrównania będzie znacznie wyższe
stężenie, (w samej krwi wyniosłoby ok. 72mEq/1).
Stąd zgodnie z równaniem Nernsta potencjał równowagi dla K zewnątrzkomórkowego = 25,7 mEq/L i
wewnątrzkomórkowego = 140 mEq wyniesie Vrównowagi = -45,3 mV a potencjał spoczynkowy wg równania
Goldmana-HK -48,8mV (Ampułka 20 ml 15% KCl zawiera 2mEq/ml czyli w 20 ml 40 mEq K+ )
Potencjał czynnościowy (Pcz):
•Zmiana stężenia pozakomórkowego sodu prowadzi do wyraźnej zmiany amplitudy Pcz ale „prawie” nie ma
wpływu na potencjał spoczynkowy
•Wniosek Hodgkina i Katza : w czasie Pcz następuje gwałtowny wzrost przepuszczalności dla sodu
•Pcz w różnych neuronach ma różny kształt („waveform”) ale wnioski z eksperymentów na kałamarnicy
zasadniczo obowiązują wszędzie
•Różne kształty Pcz są związane z dodatkowymi prądami (kanałami) jonowymi (Ca-, Kir)
(omówienie Pcz znajduje się w skrypcie Prof.Konturka dlatego tu nie rozwijam tego zagadnienia)
(omówienie Pcz znajduje się w skrypcie Prof.Konturka dlatego tu nie rozwijam tego zagadnienia)
(omówienie Pcz znajduje się w skrypcie Prof.Konturka dlatego tu nie rozwijam tego zagadnienia)
(omówienie Pcz znajduje się w skrypcie Prof.Konturka dlatego tu nie rozwijam tego zagadnienia)
„Kablowe” właściwości neuronów i ich wypustek
Błona komórkowa jako kondensator q = CV
q –ładunek ; C – pojemność (mF/cm2); V – napięcie (mV)
Obliczmy: ile ładunków elektrycznych musi być zgromadzone na/przy błonie o powierzchni 1 mm2 ( jeśli
C = 1 mF/cm2 a V = 100mV
Ilość ładunków/ 1 mm2 = 1 mF/cm2 x 0,1V
(cm2= 10000x10000 mm2 = 108 mm2 )
= 1/108 mF/mm2 x 0,1V
= 1/109 mFV /mm2 (FV= Culomb bo q=CV)
= 1/109 mCulomb /mm2 = 1/1015 Culomb /mm2 (1Culomb=6,24 x 1018 ładunków el.)
= (6,24 x 1018)/ 1015 ładunków /mm2 = 6,24 x 103 /mm2 =
6240 /mm2
Elektrotonus, potencjały elektrotoniczne
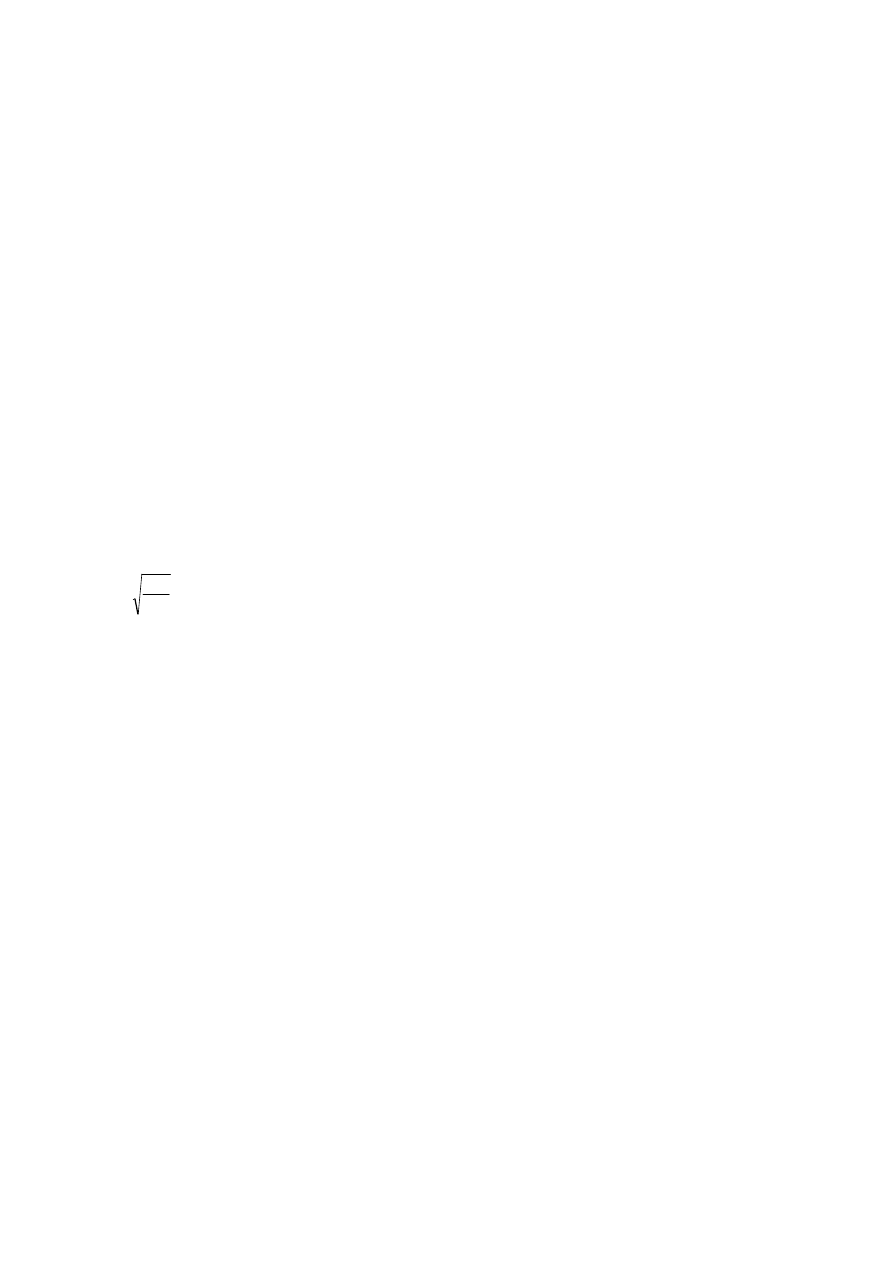
Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
8/113
Potencjały podprogowe (nie wywołujące potencjału czynnościowego) rozprzestrzeniają się biernie
(elektrotonicznie) i ulegają osłabieniu wraz z odległością od miejsca ich powstania (np. w błonie
postsynaptycznej) i wraz z upływem czasu.
Własności elektrotoniczne neuronów nazywane są też „właściwościami kablowymi”
Ich poznanie pozwala na zrozumienie przede wszystkim funkcjonowania dendrytów (aksony przewodzą
potencjały czynnościowe, nie ulegające dekrementowi). Służy temu formułowanie tzw. obwodów
ekwiwalentnych, która to czynność wymaga przyjęcia (niestety) wielu upraszczających założeń np..
1 - wyodrębniony segment wypustki jest walcowaty (ta sama średnica)
2 – potencjał elektrotoniczny pojawia się w postaci czymkolwiek spowodowanej zmiany „potencjału
spoczynkowego” (o ile w ogóle coś takiego istnieje) a jego wartość w „punkcie zero” wynosi V =
Vzmieniony - Espocz (także Espocz bywa pomijane).
3 – Prąd elektrotoniczny jest zgodny z prawem Ohma: E (potencjał) = IR
4 – Opór (R) ma składową „osiową” (inaczej „wewnętrzną”) oraz oporność błonową
5 - Pomijana jest właściwość „kondensatorowa” błon komórkowych
„Kablowe” właściwości neuronu
Równanie kablowe opisuje relację (zmian) napięcia wzdłuż (modelowej) wypustki neuronu w stosunku do
odległości i czasu, a jego rozwiązanie:
Vx = V0 e
–x/l V
t = V0 e
–t/t
λ
oznacza tzw. „stałą długości” (length constant,
space constant, characteristic length) i jest to odległość od punktu „0”, w której napięcie zmniejszy się do
37% wartości pierwotnej (w punkcie „0”).
Zatem jeśli x= l to Vx=0,37V0 a więc w odległości l napięcie wyniesie 0,37 napięcia początkowego
Stała długości w relacji do cech neurytu:
a – promień neurytu (0,02 mm - 25 mm) 1000x!Rm =oporność charakterystyczna błony (103 – 105
Wcm2)Rw =oporność wewnętrzna (50 – 200 Wcm)
„Długość elektrotoniczna”
L
„kabla” o długości
x
(np. w milimetrach) wynosi
L= x/ l
(dla wielu neuronów 0,3 –1,5)
•Vt = V0 e
–t/t
•t - „stała czasu” t= RmCm
•gdzie Cm to pojemność charakterystyczna błony która jest dość stała (0,75 m Fcm-2) a Rm oporność
charakterystyczna błony (103 – 105 Wcm2) (F-farad = 1Culomb/1Volt)
•Szybkość przewodzenia elektrotonicznego theta
θθθθ
= 2 (l/ t
))))
Aksony zmielinizowane tej samej średnicy przewodzą impulsy 100x szybciej niż niezmielinizowane (ale
przy średnicy poniżej 1 [mm] niezmielinizowane są „szybsze”).
Empirycznie stwierdzono, że szybkość propagacji potencjału czynnościowego w aksonie zmielinizowanym
w metrach/sek. jest równa ich średnicy w mikronach (mikrometrach) pomnożonej przez 6.
Vimpulsu [w m/s] = śr. włókna [w mm] x 6
(tzw. czynnik Hursh’a)
Najgrubsze aksony u ssaków o śr. 20 [mm] przewodzą 120 m/s
Cienkie, o śr. 1 [mm] przewodzą 5 – 10 m/s.
Zróżnicowanie długości i grubości aksonów i szybkości przewodnictwa Pcz
•Ok 1 mm w inhibitory interneurons do 1 metra lub więcej
•Średnica najgrubszego axonu (squid giant axon) bliska 1 mm a w niezmielinizowanych aksonach korowych
ssaków (od 0.08 do 0.4 µm)
•Axonalne opóźnienie zależy zasadniczo od prędkości Pcz (między 0.1 m/s w niezmielinizowanych axonach i
100 m/s w dużych zmielinizowanych) – znaczenie dla funkcji integracyjnych (np.. W lokalizacji źródła
w
m
R
aR
2
=
λ

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
9/113
dźwięku – zob. wykład na temat słuchu), zależy też od zgrubień aksonalnych – „varicosities” i podziałów
(spowalniają) oraz od powtarzanej stymulacji (również spowalnia) i wpływu określonych kanałów jonowych
•Zjawisko (objaw?) „conduction failure” w aksonie – czyli zatrzymanie propagacji Pcz. występuje w punktach
rozgałęzień aksonu oraz w zgrubieniach i w „wejściu” do somy neuronu oraz na skutek powtarzanych stymulacji
(akumulacja K pozaaksonalnie i stąd depolaryzacja). Może być spowodowane blokowaniem Na/K ATP-azy
przez OUABAINĘ (depolaryzacja) oraz przez hyperpolaryzację.
Możliwość „cofki” (pojęcie wzięte z hydrogeologii) Pcz oraz interakcje „efatyczne”
•U bezkręgowców stwierdzono „cofanie” się Pcz („odbicie” - reflection) który wcześniej ulegał prawie
wygaśnięciu w miejscach rozgałęzień aksonu. U ssaków stwierdzono to zjawisko w dendrytach.
•„Cofka” Pcz powstająca w rozgałęzieniach jest tłumaczona występującym tam opóźnieniem przewodnictwa
które „przeczekuje” okres refrakcji.
•Pcz w aksonie powoduje zmianę wrażliwości (ekscytatyczności) aksonu sąsiedniego (zwykle początkowo
obniżenie potem podwyższenie). Prowadzić to może do synchronizacji przebiegów Pcz w pęczku aksonów. Są
to tzw. wpływy (interakcje) efatyczne.
W komórkach Purkinjego włókna pnące powodują silne EPSPs (pobudzające potencjały postsynaptyczne)
w całym drzewie dendrytycznym (po 100 000 kolców), których rezultatem jest prawdopodobnie
synchroniczne wyładowanie w postaci „wapniowych potencjałów czynnościowych” przenoszących się do
wzgórka aksonalnego i powodujących modulację „normalnych” (Na+/K+) aksonalnych Pcz.
Natomiast w dendrytach obserwuje się
wapniowe potencjały czynnościowe
wywołane impulsacje włókien
pnących. Te potencjały modyfikują „sodowo-potasowe” potencjały czynnościowe wzgórka aksonalnego.
Rola dendrytów
•PIERWSZA ZASADA: NEURON (JEŚLI MA) MA TYLKO JEDEN AKSON (ALE DLACZEGO ???)
•ZASADA (KOLEJNA): CZĘSTOTLIWOŚCIOWEGO KODOWANIA WYSYŁANEJ INFORMACJI
–INTEGRACJA NASTĘPUJĄCA W DENDRYTACH WPŁYWA NA „OUTPUT”
•Ale badania ostatnich lat udowodniły, że nawet neurony z aksonem mogą używać dendrytów jako środka
„komunikacji wychodzącej” (OUTPUT). Jest to udowodnione zwłaszcza u bezkręgowców (np. detekcja
ruchu u much mięsnych blowfly)
NAWET ODLEGŁE DENDRYTY MOGĄ ZNACZNIE EFEKTYWNIEJ WPŁYWAĆ NA „AXONAL
OUTPUT”
•Dendryty stanowią zasadniczy i najważniejszy obszar przetwarzania informacji w neuronie
•Obecnie wiemy, że miejsce inicjacji potencjału czynnościowego może się zmieniać w kierunku nawet
dystalnych dendrytów.
•Najnowsze badania wskazują, że kolce dendrytyczne pełnią funkcje „microintegrative units” (operacje
logiczne AND, OR AND-NOT i operacje „liczenia”), mogą też pełnić funkcje sekwestratorów jonów
wapniowych (toksycznych dla komórki). Dlatego neuron może być porównany do „mikrochipa” w którym
znajdują się dziesiątki tysięcy logicznych i kalkulacyjnych układów.
•Kolce dendrytyczne zapewne maja też udział w mechanizmach pamięci
Dendryty nie są tylko rozległą siecią receptorową !
W świecie bezkręgowców ogromna część neuronów nie ma aksonów!
U kręgowców są praktycznie tylko dwa typy takich bezaksonowych komórek:
komórki amakrynowe siatkówki
węchowe komórki ziarniste
Najwięcej informacji o neurotransmisji i innych procesach związanych z przetwarzaniem informacji
uzyskano z badań nad neuronami z długimi aksonami.
•Badania Stuarta i Sackmanna (1994) wykazały m.in., że potencjał czynnościowy może propagować
wstecznie do drzewa dendrytycznego. „Wsteczny” Pcz może mieć na celu m.in.. „resetowanie” potencjału
błonowego.
Prawdopodobnie wzmaga on też reaktywność synaps. Co więcej, stwierdzono, że
wsteczny Pcz
powoduje w korze mózgowej oddziaływanie GABAergicznych dendrytów interneuronów na zakończenia
aksonalne komórek piramidalnych
a z kolei glutamatergiczne dendryty komórek piramidalnych oddziałują na
zakończenia aksonalne interneuronów. Te połączenia i związki mogą mieć znaczenie w patogenezie drgawek!

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
10/113
Przepływ informacji w systemie nerwowym
cz. II:
Neurotransmisja cz. A – neurotransmitery
Thomas Elliot (XIX/XX) zaobserwował skurcz nieunerwionych mięśni gładkich pod wpływem epinefryny.
Otto Loewi – dowody na chemiczną neurotransmisję i na rolę w niej acetylocholiny (Ach) na podstawie
eksperymentów z sercem żaby
•Otto Lewi 1926 eksperyment na sercu żaby : odkrycie „vagus substance” (którą później okazała się
acetylocholina)
Rodzaje sygnalizacji międzykomórkowej
(nie tylko synaptyczne i nie tylko między neuronami i ich komórkami „docelowymi”)
Humoralna
Parakrynna
Autokrynna
Efatyczna (poprzez przestrzeń-pozakomórkową)
Synaptyczna: elektryczna (m-komórkowe prądy jonowe bezpośrednio poprzez gap junction)
Synaptyczna: chemiczna
Neurotransmiter („klasyczne” kryteria)
1.Substancja musi być syntetyzowana (obecna) w neuronie
2.Musi być obecna w zakończeniach synaptycznych i uwalniana po stymulacji (depolaryzacji) a następnie
musi wywoływać reakcję komórki efektorowej (musi być izolowana i identyfikowalna chemiczne lub
farmakologicznie)
3.Neurotransmiter musi wywoływać te same zmiany w komórce postsynaptycznej jak stymulacja neuronu
presynaptycznego
4.Powinien istnieć swoisty receptor w komórce postsynaptycznej, co oznacza w praktyce, że podając
substancję egzogennie można wywołać podobny efekt jak stymulując synapsę, ponadto powinno być
możliwe dawkozależne blokowanie przez kompetytywnego antagonistę. Również blokowanie syntezy
neurotransmitera powinno blokować efekty stymulacji presynaptycznej
5.Powinien być mechanizm aktywnego usuwania uwolnionego neurotransmitera (enzymatyczny
rozkład/transport)
6.Uwalnianie neurotransmitera niemal zawsze (niektórzy formułują Ca-zależne uwalnianie jako
jednoznacznie konieczne kryterium) łączy się z napływem Ca++ do zakończenia (musi być wapń w
przestrzeni pozakomórkowej)
Składowe procesu neurotransmisji
1.Synteza neurotransmitera
2.Magazynowanie n-t w zakończniach presynaptycznych
(„klasyczne” n-t gromadzą się w mniejszych
pęcherzykach -50nm, peptydowe w większych –100 z gęstym rdzeniem). Ponieważ w większości synteza n-
t jest w cytozolu istnieje aktywny mechanizm „ładowania” pęcherzyków („vesicular transporter protein”)
3.Uwalnianie n-t do szczeliny synaptycznej
zwykle z udziałem wapnia, może być „konstytutywne” (bez
stymulacji np. czynniki wzrostu) lub stymulowane
4.Wiązanie i rozpoznawanie n-t przez receptor komórki docelowej
(receptory jonotropowe i
metabotropowe), receptory mogą być na tym samym neuronie (tzw. autoreceptory – mech.regulacyjne)
5.Aktywne zakończenie działania n-t (dla klasycznych n-t)
enzymatyczna degradacja i/lub wychwyt - jest
też „pasywne” zakończenie działania n-t, poprzez dyfuzję)
Neurotransmitery drobnomolekularne:
„jasne pęcherzyki” 40-60 nm, ( katecholaminy mają pęcherzyki dense-core)
synteza w strefie synaps,
enzymy syntetyzujące transportowane poprzez slow axonal transport (0.5 – 5 mm/doba)
Neurotransmitery peptydowe (neuropeptydy):
„Ciemnordzeniowe” pęcherzyki 90-250 nm
synteza w ciele neuronu (ew. modyfikacja prekursorów w strefie synaps)
transport do synaps poprzez „fast axonal transport” (400mm/doba)
(kinezyna – motoryczne białko zużywające ATP)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
11/113
Neurotransmitery („klasyczne”)
Acetylocholina, Glutaminian, Glicyna, kw.gammaaminomasłowy (GABA), adrenalina,
noradrenalina, dopamina, serotonina (5-HT)
•
Do „klasycznych” neurotransmiterów niektórzy zaliczają również:
–ATP i inne puryny
–Neuropeptydy
•Do „nieklasycznych” (niekonwencjonalnych) neurotransmiterów zaliczane są:
–Endokanabinoidy
–Tlenek azotu (NO)
Neurotransmitery katecholaminowe
•DOPAMINA
•NOREPINEFRYNA
•EPINEFRYNA
N-T katecholaminowe
DOPAMINA
Hydroksylaza tyrozyny (TH) jest kluczowym enzymem w produkcji amin katecholowych
Neurony albo zwiększają jego syntezę albo aktywność przez fosforylację.
Aktywność TH jest regulawana także poprzez kofaktor BH4 czyli
tetrahydrobiopterynę
.
Mutacja GTP-cyklohydrolazy (syntetyzującego BH4) prowadzi do tzw. dystonii wrażliwej na DOPA
(DOPA-responsive d.)
Dekarboksylaza DOPA (3,4 dihydroxyphenylalaniny) gra również rolę w syntezie 5-HT, jest
„finalnym” enzymem w neuronach dopaminergicznych,
- działa „superszybko”.
•Fosforylacja Hydroksylazy tyrozyny zmieniając jej konformację zwiększa jej aktywność a zatem i
syntezę katecholamin
•Hydroksylaza tyrozyny jest substratem dla wielu różnych kinaz będących elementami różnych
szlaków sygnalizacyjnych które działają poprzez cAMP, Ca++ lub DAG
•Są to takie kinazy jak PKA, CaMKII,, MAPK, PKC
Neurony dopaminergiczne znajduja się w:
substancja czarna
Ventral Tegmental Area
L-DOPA jest kluczowym związkiem w leczeniu ch.Parkinsona, (w której jest niedobór
dopaminy w prążkowiu) ponieważ w przeciwieństwie do dopaminy przenika przez BBB.
Musi być podawana wraz z inhibitorem obwodowych dekarboksylaz !
Niestety L-DOPA podawana przewlekle hamuje aktywność endogennej dekarboksylazy
DOPA w mózgu !!! (hamuje w ten sposób przejście w dopaminę).
„Załadunek i wyładunek” katecholamin
Pęcherzyki chronią N-T przed degradacją
Rola Vesicle Monoamine Transporter (VMAT):
VMAT (poznano 2 typy) nie jest wysoko specyficzny, wymaga Mg2+; hamuje go
rezerpina
(b.stary lek
w nadciśnieniu i psychozach – rozrywa pęcherzyki i uwalnia monoaminowy n-t tj zarówno katecholamin
jak i 5-HT)
VMAT może też grać rolę w sekwestracji toksyn
Uwalnianie n-t:
egzocytoza (jest Ca2+ zależna)
oraz inne procesy (np. odwrócenie działania transporterów)
Rola autoreceptorów w regulacji
hamują uwalnianie n-t i prawdopodobnie syntezę.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
12/113
Inaktywacja katecholamin:
Enzymatyczna:
(kiedyś sądzono, że najważniejsza, obecnie uważa się że gra rolę gł tylko
we krwi)
MAO (monoaminooksydaza) na zewn. Bł. mitochondrialnej
COMT (catechol-O-metylotransferaza)
Wychwyt n-t przez neurony
(najważniejszy sposób inaktywacji katecholamin w mózgu
!!!). Sklonowano 2 transportery, oba spokrewnione z transporterami 5-HT i EAAT). Są one różne od
transporterów pęcherzykowych i nie są zależne od Mg2+ i nie są hamowane prze rezerpinę. Pomimo
nazw, żaden z nich nie jest specyficzny dla DA czy NE.
DAT =
Transporter dopaminy
(jest raczej
poza
synaptyczny !! Stąd być może katecholaminy działaja nie tylko synaptycznie ale i „parakrynnie”;
faktycznie b.duże ilości dyfundują poza synapsy)
NE-T
Transporter norepinefryny
(trójcykliczne antydepresanty np.
doxepina, imipramina
blokują ten transporter)
Psychostymulujący efekt
kokainy i amfetaminy
polega na blokowaniu wychwytu katecholamin (oraz w
przypadku kokainy także 5-HT) przez blokowanie transporterów (kokaina szczeg. blokuje DAT).
Amfetamina bardziej niż blokująco, działa poprzez odwracanie działania transportera !
Histamina
•Oprócz katecholamin również należy do grupy „amin biogennych”
•Główny rejon: nucleus tuberomammillaris hypothalami
•Promuje aktywność mózgu („arousal”), wspomaga uwagę
–Leki antyhistaminowe używane m.in. jako antyuczuleniowe powodują senność i są np. przeciwwskazane przy
prowadzeniu pojazdów!
•Histamina m.in. odgrywa rolę w kontroli układu przedsionkowego
•Przypuszczalnie histamina może też regulować przepływ mózgowy krwi
•Nie zidentyfikowano transportera histaminy
•Rozkład: metylotransferaza histaminy i MAO
•Wszystkie znane receptory histaminy należą do metabotropowych (sprzężonych z białkiem
G)
Serotonina (5-HT)
•Wraz z histaminą i katecholaminami należy do „amin biogennych”
•Główny ośrodek serotoninergiczny: n.raphe
•Bierze udział w regulacji snu i czuwania
•Aktywacja receptorów serotoniny powoduje m.in. uczucie sytości
Mózg zawiera jedynie 1% zasobów(5-HT)
Szczególną uwagę zwrócono na 5-HT z powodu jej podobieństwa do LSD i w związku z teoriami, że 5-HT
gra rolę w schizofrenii i depresji.
Hydroksylaza tryptofanu limituje produkcję 5-HT.
Tryptofan dostaje się do mózgu a zwiększenie podaży tryptofanu zwiększa syntezę 5-HT
5-HT nie przechodzi do mózgu ale przechodzi 5-HydroxyTryptofan (5-HTP)
Rezerpina
zmniejsza również 5-HT w pęcherzykach synapt.
Jest 15 różnych receptorów 5-HT !
W szyszynce 5-HT jest wyjściowym substratem dla produkcji
melatoniny
SER-T : Transporter serotoniny odpowiedzialny za jej inaktywację poprzez wychwyt jest transporterem o
wysokim powinowactwie (jest też spokrewniony z DA-T i NE-T).
Selektywnym inhibitorem SER-T jest lek antydepresyjny
fluoxetyna (Prozac). Pośrednio wskazuje to także na rolę serotoniny w procesach psychicznych
.
Aminy biogenne w chorobach psychicznych
•Pomimo relatywnie niewielkiej ilości neuronów aminergicznych w obrębie CSN ich rola w regulacji stanów
psychicznych jest bardzo duża
•Rezerpina blokująca wychwyt noradrenaliny i obniżenie ciśnienia krwi a jednocześnie powodująca stany
depresyjne zwróciła uwagę na rolę amin biogennych w mechanizmach zjawisk psychicznych (np. nastrój). Była
też pierwszym lekiem antypsychotycznym

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
13/113
•Obecnie wiemy, że aktywacja układu dopaminergicznego odgrywa rolę w psychozach i większość leków
antypsychotycznych chloropromazyna, haloperidol, benzperidol) w ten lub inny sposób hamuje układ
dopaminergiczny (np. blokując receptory dopaminergiczne).
•Aminy odgrywają też rolę w mechanizmach stanów lękowych – inhibitory MAO były wykorzystywane jako
leki przeciwlękowe
•Stany depresyjne : leki przeciwko depresji takie jak inhibitory MAO (fenelezyna), trójcykliczne antydepresanty
(desipramina – blokująca wychwyt noradrenaliny), blokery wychwytu serotoniny (Prozac), trazodon – wszystkie
ingerują w przewodnictwo aminergiczne
•Amfetamina – stymulująca uwalnianie noradrenaliny z zakończeń nerwowych powoduje stany (tzw. „high”),
które można uznać za „odwrotność” depresyjnego działania rezerpiny!
GABA (
γγγγ
-amino butyric acid, kwas
γγγγ
-aminomasłowy)
główny hamujący N-T mózgu (odkryty w 1950)
GABA jest częścią metabolizmu glukozy (w przeciwieństwie do katecholamin)
GAD
= glutamic acid decarboxylase jest enzymem krytycznym dla tworzenia GABA i jest tylko w
neuronach GABA-ergicznych (ma dwie izoformy o różnych własnościach)
Zastanawia przeciwieństwo Glu i GABA przy tym samym szlaku metabolicznym i związku z cyklem
Krebsa
GABA-T = transaminaza GABA-ketoglutaranu tworzy z a-ketoglutaranu kw.glutaminowy, który w
neuronie jest dekarboksylowany przez dekarboksylazę kw.glutaminowego (
GAD
) do GABA.
GAD jest wyłącznie w neuronach GABA-ergicznych
GAD wymaga kofaktora w postaci fosforanu pirydoksalu (pochodnego vit B
6
). Niedobór vit B
6
prowadzi do niedoboru GABA (drgawki u dzieci niekiedy śmiertelne)
Ten sam enzym GABA-T (obecny w mitochondriach ale też w synaptosomach) inaktywuje GABA !!!
(do succinic semialdehyde = semialdehyd bursztynylowy)
UWAGA!
Z pobudzającego N-T (kw.glutaminowy) powstaje hamujący (GABA).!
Glicyna
•Hamujący NT obecny głównie w rdzeniu
•Powstaje z seryny (mitochondrialna hydroksymetylotransferaza)
•Ładowana do pęcherzyków przez ten sam transporter co transporter dla GABA
•Receptory (wyłącznie) jonotropowe – kanały dla Cl (blokowanie przez alkaloid strychninę)
•Usuwana z przestrzeni pozakomórkowej przez transportery glicynowe (ich mutacja prowadzi do
hyperglicynemii
– choroby wrodzonej i śmiertelnej z sennością, opóźnieniem rozwoju umysłowego, drgawkami)
Glutaminian i asparaginian
Glutaminian i asparaginian – metabolity i neurotransmitery (żaden z nich nie przekracza
bariery krew-mózg!)
Glutaminian to najważniejszy pobudzający NT
Powstaje na drodze różnych przemian chemicznych ale głównie z alfa-ketoglutaranu (z cyklu
Krebsa) oraz z glutaminy.
Po uwolnieniu z błony presynaptycznej jest wychwytywany przez astrocyty za pomocą
specjalnych białek transporterowych. W astrocytach ulega przemianie do glutaminy i która
powraca do neuronu gdzie przekształcana jest ponownie w glutaminian. Nadmierne
uwalnianie glutaminianu lub niefektywny wychwyt prowadzą do tzw. ekscytotoksycznego
uszkodzenia tkanki mózgu i odgrywa rolę w wielu chorobach OUN (zob. Dodatek).
Glutaminian działa poprzez receptory jonotropowe i metabotropowe (zob. dalej)
Acetylocholina (pierwszy poznany n-t)
Acetylocholina –: synteza najprostsza ze wszystkich n-t (1 etap)
ChAT – acetylotransferaza choliny -
marker neuronów cholinerg.
Ac-CoA pochodzi z pirogronianu i poprzez pirogronian wiąże syntezę Ach z metabolizmem glukozy,
Jego transport z mitochondriów limituje syntezę Ach ?
Albo cholina ? (próby leczenia ch. Alzheimera lecytyną – prekursorem choliny)
Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
14/113
Acetylocholine esterase (działa pozakomórkowo i sama może być neurotransmiterem ?
Transporter choliny nie jest elementem inaktywacji ACh
Receptory cholinergiczne
Receptory nikotynowe (jonotropowe); 40 typów; w złączu N-M
Receptory muskarynowe (metabotropowe), gruczoły. M-oka
Inaktywacja Ach poprzez enzym acetylocholinoesteraza (AChE) hydrolizująca Ach.
Substancje blokujące Ach-esterazę prowadzą do akumulacji Ach i depolaryzacyjnej
inaktywacji mięśni są to:
sarin („nerve gas”)
zw. fosforoorganiczne (insektycydy np. parathion),
Kompetycyjne blokery służą do zwiotczenia w anestezji (sukcynylocholina - prototypowy
zwiotczający lek depolaryzujący) i jako leki w miastenii
Cholinergiczne neurony „czuwania”
•Cholinergiczne neurony aktywującego układu siatkowatego na
pograniczu mostu i śródmózgowia odgrywają kluczową rolę w regulacji aktywności mózgu (czuwanie kontra
sen)
•Inne nurotransmitery (odpowiednie neurony) również biorące udział w kontroli aktywności „sen-czuwanie” to
serotonina (n.raphe) oraz noradrenalina (l.coeruleus) i histamina (n. tuberomamillaris hypothalami) a także
neuropeptyd orexyna (obszar podwzgórza około jądra n. tuberomammillaris)
zob. dodatek II „sen i czuwanie”
Puryny: ATP, AMP, adenozyna
•ATP jest obecne niemal we wszystkich pęcherzykach synaptycznych („co-
transmiter”?)
•Pozakomórkowe podawanie puryn może wywołać odpowiedzi elektryczne neuronów (lata 20-ste!)
•ATP działa pobudzająco w motoneuronach rdzenia w zwojach autonomicznych i czuciowych, w hipokampie
•Puryny odgrywają rolę w przewodzeniu bólu i mechanorecepcji jednak w większości ich funkcja jest nieznana
•Kofeina i teofilina blokują receptory purynergiczne (ale dlaczego działają pobudzająco?)
•Receptory purynergiczne (jono i metabotropowe) są w całym mózgu.
•Adenozyna nie jest obecna w pęcherzykach a zatem jest „nieklasyczna”
•Usuwanie puryn: enzymy (apiraza, ecto-5-nukleotydaza, białka transporterowe nukleozydów)
Neuropeptydy
N-T peptydowe n-t
podobnie jak klasyczne są identyczne pomiędzy gatunkami,
magazynowane są w pęcherzykach i uwalniane w sposób zależny od Ca
2+
jednak inna jest biosynteza i
inaktywacja.
Peptydy syntetyzowane są w ciele komórki początkowo jako prohormon (potem aktywowany peptydazą) i
transportowane w pęcherzykach na obwód. Pęcherzyki są większe i z tzw. dense core.
Hipoteza Henry Dale’a 1neuron=1n-t
Obecnie wiadomo, że neuron może dysponować 2 lub więcej neurotransmiterami
•Peptydy aktywują receptory w niskich stężeniach (mili i mikromolowych)
•Szczególnie istotona rola w jelicie i pozazwojowych neuronach sympatycznych
•Neuropeptyd Y odgrywa rolę w regulacji zachowań pokarmowych (sytość, otyłość).
•Neuropeptydy
powstają jako tzw. pre-propeptydy, następnie przetwarzane do propeptydów (proteoliza) i ładowane do
pęcherzyków gdzie zachodzą ostateczne przemiany (powstają definitywne formy neuropeptydu oraz ich
modyfikacje – np. fosforylacja, glikozylacja)
•Zwykle w pęcherzyku są różne pochodne peptydy (i uwalniane są razem)
•Katabolizm neuropeptydów: peptydazy
•Ważniejsze poznane role neuropeptydów:
•Substancja P oraz opioidy grają rolę w percepcji bólu
•Melanocyte Stimulating Hormone, ACTH, beta-endorfina – reakcje na stress
Peptydowe N-T nie mają specyficznego wychwytu i są inaktywowane przez dyfuzję i enzymatyczny
rozkład (który nie jest swoisty dla określonego peptydu ale np. dla dipeptydu). Produkty „dezaktywacji”
mogą też być aktywne.
Substancja P
•Substancja P:
działanie hipotensyjne
; 11-aminokwasów (odkryta 60 lat temu jako „
P
owder
extract” mózgu i jelita)
•tzw. brain/gut peptide (hipokamp, neocortex, jelito, aferentne włókna C z dróg czucia bólu i temperatury)
•W rdzeniu antagonistycznie do substancji P działają opioidy
•Szereg innych neuropeptydów jest kodowanych przez ten sam gen co gen substancji P (neurokinina A,
neuropeptyd K i neuropeptyd gamma)
Opioidy
•Opioidy – peptydy działające na te same receptory, na które działa opium (morfina)
•Dzielą się na 3 grupy (pochodzące z 3 osobnych odpowiednio genów): endorfiny, enkefaliny, dynorfiny

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
15/113
•Uważa się, że ich wydzielanie jest podstawą efektu znieczulającego akupunktury
•Opioidy grają rolę w zachowaniach seksualnych oraz agresyjno-submisywnych, być może także w schizofrenii i
autyzmie
neurotensyna
Prohormon - 170 aminokwasów;
Dense-core vesicles
Uwalnianie Ca2+ zależne;
Inaktywacja przez metaloendopeptydazy
NT może być internalizowana po związaniu z receptorem metabotropowym.
Może być wstecznie transportowana (np. do śródmózgowia)
NT kolokalizuje zwł z dopaminą.
NT jest też w jelicie cienkim
Orexyny A i B
Neuropeptydy biorące udział w regulacji aktywności ukłądu nerwowego, snu-czuwania
oraz przyjmowania pokarmu i prawdopodobnie emocji
Orexyna-A (33 aminokwasy) jest identyczna u ludzi, myszy, szczurów, krowy, świni,
orexyna-B (28 aminokw.) ludzi różni się 2 aminokw. od gryzoni
Znane są dwa receptory oreksyn:
OX
1
R (orphan G-protein coupled receptor) – 10x bardziej swoisty (affinity) dla Orexyny A niż dla Or-B
OX
2
R – nieselektywny dla obu oreksyn
Neurony orexyno-dodatnie
z LHA (=Lateral hypothalamic area – „centrum odżywienia”) i obszarów
okołosklepieniowych mają b.liczne połączenia z korą, opuszką węchową, hipokampem, c.migdałowatymi,
przegrodą, diagonal band of Broca, bed nucleus of stria terminalis, wzgórzem, resztą podwzgórza,
śródmózgowiem, pniem, rdzeniem kręgowym.
Immunoreaktywność dla oreksyn stwierdzono też w układzie nerwowym jelita i trzustce a mRNA również
w jądrach (!?). Receptory z grubsza powielają dystrybucję neuropeptydu
Rola oreksyn
Utrzymywanie stanu czuwania (wakefulness)
Orexin-Knock-out myszy zachowują się jak narkoleptycy
Badania u ludzi również wskazują na rolę orexyn w narkolepsji (u narkoleptyków nie ma orexyn w CSF)
Dokomorowe wstrzyknięce oreksyn wywołuje stan podwyższonej aktywności lokomotorycznej, różnych
czynności np. czyszczenia się (grooming) i spożywania jedzenia.
Obecność receptorów oreksyn w substancji czarnej może sugerować, że oreksyny modulują
dopaminergiczne działanie w procesach związanych z aktywnością, emocjami. (Antagoniści dopaminy
dawkozależnie blokują indukowane oreksyną zachowanie nadruchliwe i czyszczące)
Stymulacja jedzenia przez oreksyny jest słabsza od NPY ale o dłuższym działaniu.
Oreksyny powodują wzrost zużycia tlenu, spadek poziomu prolaktyny i GH oraz wzrost kortykosteronu w
osoczu.
Wpływają na układ autonomiczny m.in. Powodując wzrost ciśnienia krwi i częstości akcji serca.
Oreksynowe neurony są aktywowane przez hypoglikemię
Neuroregulacja odżywienia
1)Lateral
hypothalamic area (LHA) – centrum odżywienia (feeding center)
1)Uszkodzenie LHA prowadzi do hypofagii, wzrostu metabolizmu i obniżenia stopnia czuwania (arousal)
2)Antagonistyczne układy stymulacji i supresji apetytu (orexigenic vs. Anorectic) orexis = gr. apetyt
3)Układ orexygenny: neuropeptyd Y (NPY), melanin concentrating hormone (MCH – produkowany w
LHA),
1)MCH dawkozależnie podwyższa spożywanie pokarmu, genetyczne uszkodzenie MCH prowadzi do
hypofagii
2)orexyny A i B
(wytwarzane w LHA).
4)Układ anorexygenny: alfa-melanocyte stimulating hormone (a-MSH), leptyny,
Endokanabinoidy (EK)
•Uważane są za nieklasyczne neurotransmitery bo nie są magazynowane w
pęcherzykach a często biorą udział w sygnalizacji „wstecznej” (uwalniane są z komórki postsynaptycznej i
następnie dyfundują do komórek)
•Znane są 2 substancje (endokanabinoidy):
anandamid
oraz
2-arachidonylglicerol (2-AG
), które reagują z
receptorem egzogennego aktywnego składnika marihuany – delta
9
-tetrahydrokanabinolem
•Substancje te o charakterze tłuszczów powstają z lipidów błon komórkowych
•Endokanabinoidy (EK) są wychwytywane przez aktywny transport i hydrolizowane (hydrolaza kwasów
tłuszczowych - FAAH)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
16/113
•Głównym receptorem EK jest receptor CB1 – jest to receptor typu metabotropowego (GPCR)
spokrewniony z metabotropowymi receptorami Ach.
•Receptory dla EK są szczególnie liczne m.in. w substantia nigra oraz skorupie (caudate putamen).
•EK synetyzowane i uwalniane pod wpływem Ca
++
hamują wstecznie wydzielanie neurotransmiterów (np.
GABA) w synapsach
•Wsteczne hamowanie uwalniania GABA przez EK
Inne neurotransmitery niekonwencjonalne:
NO i CO (i Hydrogen sulfide?)
N-T = jakakolwiek substancja, która zapewnia przepływ
informacji z neuronu do neuronu (miesza się z pojęciem hormonu bo brak „celu” i czasowych
właściwości neurotransmisji, a co z rolą gleju?).
Azotany, nitrogliceryna znane od dziesiątków lat jako
rozszerzające naczynia. Dopiero w latach 80tych zidentyfikowano NO (endothelial-derived relaxing
factor,
NO jest stymulowany także przez Glu w mózgu prowadząc do wazodilatacji!).
NO: Lista wątpliwości jest długa:
NO nie jest magazynowany, nie ma receptora, nie ma mechanizmu
inaktywacji,
NO stymulując cyklazę guanylową powoduje wzrost cGMP
(podobnie jak Glu, który w ciągu sekund 3x
wzmaga aktywność NOS poprzez ciąg: NMDA:Ca2+:wiązanie kalmoduliny:aktywacja nNOS
(neuronalna nNOS w 1% neuronów);
„Klasyczna” droga NO:
Ach w endotelium stymuluje szlak IP
3
prowadząc do wzrostu Ca2+.
Ca2+ aktywuje NOS. NO dyfunduje z endotelium do mięśniówki gładkiej naczynia gdzie
aktywuje cyklazę
guanylową i produkcję cGMP
. cGMP aktywuje GMP-zależne kinazy proteineowe co prowadzi do
rozluźnienia mięśnia.
NO trwa tylko kilka sekund w płynach ustrojowych
inaktywowany przez nadtlenki i tworzenie kompleksów z hemem Np. w oksyhemoglobinie
Rola NOS i cyklazy guanylowej
NO :
relaksacja mi
ęś
niówki gładkiej w obwodowych
naczyniach, oraz mi
ęś
niówki w jelicie, zabijanie obcych komórek w makrofagach.
Konstytutywna NOS w neuronach i w endoteliach
Indukowana NOS (szczególnie w makrofagach stymulowana przez cytokiny) iNOS jest
zwi
ą
zana z kalmodulin
ą
i działa w warunkach niepodwy
ż
szonego poziomu Ca2+
nNOS jest silnie aktywna w komórkach ziarnistych mó
ż
d
ż
ku i dlatego NO aktywuj
ą
c
cyklaz
ę
guanylow
ą
w kk.Purkinjego i powoduj
ą
c wzrost cGMP indukuje Long-Term Depression.
NO jest głównym stymulatorem cGMP
NO mo
ż
e równie
ż
działa
ć
niezale
ż
nie od cGMP (np. uwalnianie N-T)
Nitrogliceryna i nitroprusydek sodu s
ą
dawcami NO i wzmagaj
ą
c cGMP rozlu
ź
niaj
ą
mi
ęś
niówk
ę
naczy
ń
prowadz
ą
c do obni
ż
enia ci
ś
nienia krwi.
2 typy cyklazy guanylowej: cytozolowa i błonowa
(błonowa cyklaza guanylowa jest receptorem dla neuropeptydów takich jak atrial natriuretic
peptide i brain natriuretic peptide)
•
CO
: powstaje przy degradacji hemu (oxygenaza hemu-2 w myenteric plexus) i razem z NO
bierze udział np. w neurotransmisji w jelicie (zwiększają relaksację, a u knock-outowych myszy
relaksacja jest skrócona ).
Czynniki wzrostu jako niekonwencjonalne N-T
Mogą wpływać na neurony presynaptyczne,
kontrolują rozwój, różnicowanie i utrzymywanie neuronów. Ich ekspresja jest stymulowana lub
hamowana przez aktywność i inne N-T
(np. Glu i Ach podwyższa ekspresję BDNF i NGF a
GABA obniża.)
BDNF (brain derived neurotrophic factor) – może być magazynowany w pęcherzykach
NGF (nerve growth factor)
Neurotrofina-3 (NT-3)
Ich uwalnianie jest:
1)konstytutywne
2)stymulowane depolaryzacją (aktywnością neuronu). Jest to uwalnianie niezależne od
zewnątrzkomórkowego Ca2+ (jak w klasycznych N-T) ale od zapasów wewnątrzkomórkowego
Ca2+.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
17/113
NEUROTRASNSMISJA cz B
SYNAPSY – CZĘŚĆ PRESYNAPTYCZNA
Synapsa chemiczna pozwala wielokrotnie wzmacniać sygnał
oraz umożliwia wieloczynnikową regulację
transmisji i w związku z tym procesy adaptacyjne i inne np. powodujące krótko i długotrwałe zmiany
przewodnictwa („PLASTYCZNOŚĆ SYNAPTYCZNA”).
Potencjał czynnościowy uwalnia N-T który łącząc się z receptorem powoduje w zależności od typu kanału
jonowego pobudzający („excitatory postsynaptic potential” EPSP) lub hamujący („inhibitory” IPSP)
postsynaptyczny potencjał (depolaryzacja przesuwa potencjał błonowy w kierunku progu potencjału
czynnościowego lub odwrotnie - hyperpolaryzacja).
Z pojedynczego pęcherzyka („magazynu”) uwalnia się ok. 5000 molekuł N-T powodując powstanie tzw.
MINI-EPSP lub MINI-IPSP (tzw. „minis”, które można rejestrować poprzez mikroelektrodę)
Gradient protonowy (dzięki ATPazowej pompie protonowej) jest dostarczycielem energii do transportu
N-T do wewnątrz pęcherzyków (realizowanemu przez transportery N-T).
Są znane 4 typy transporterów przenoszących N-T do pęcherzyków:
dla Ach,
dla [katecholamin/serotoniny],
dla glutaminianu,
dla [GABA/glicyny].
Mają one pokrewieństwo z bakteryjnymi transporterami odpowiedzialnymi za odporność przeciw lekom.
Są różne od transporterów błonowych wychwytujących N-T z przestrzeni pozakomórkowej.
Uwalnianie neurotransmitera
Opóźnienie („synaptic delay”) między PCz i uwolnieniem N-T wynosi mniej niż 0,2 msek. (a w złączu n-
mięśń. do depolaryzacji postsynaptycznej upływa ok.. 0,5msek).
To raczej wyklucza jakiekolwiek systemy aktywacji enzymów oraz udziału np. hydrolizy ATP
(wykluczono to też przez usunięcie-chelatowanie Mg2+, którego brak uniemożliwia skorzystanie z a ATP
przez jakikolwiek enzym).
Uważa się natomiast, że istnieją „gotowe do fuzji” kompleksy pęcherzyków-błony synaptycznej, dla
których Ca jest jedynie „trigerem” -wyzwalaczem nagłych zmian konformacyjnych prowadzących do
otwarcia pęcherzyka. (tworzenie tych kompleksów nazywa się „dokowaniem” i odbezpieczniem (priming).
Energia potrzebna do fuzji nie może pochodzić z ATP. (ale w ogóle jest potrzebna)
Prawdopodobnie istnieje wspólny mechanizm dla wszelkich procesów fuzji błon (np..
Endosomy+lizsomy, ER-Golgi.)
Potencjał czynnościowy powoduje otwarcie kanałów wapniowych
Napięciowo-zależne kanały wapniowe są bardzo selektywne.
Na każdą strefę aktywną przypada 100 kanałów wapniowych (w złączu nerwowo-mięśniowym jest ok.
1000 stref aktywnych - active zones),
Na każdy zakotwiczony pęcherzyk przypada 10 kanałów wapniowych w odległości do 50nm;
Stężenie Ca2+ zdolne do aktywowania egzocytozy pęcherzyków gwałtownie spada nieco dalej od kanałów
wapniowych (buforowanie Ca przez cytozol)
Uważa się, że rolę sensora Ca2+ prowadzącego do uwolnienia N-T z pęcherzyka gra
synaptotagmina
Co się dzieje gdy potencjał czynnościowy dociera do złącza nerwowo-mięśniowego?
Potencjał czynnościowy (PCz) powoduje otwarcie kanałów wapniowych (Ca2+ wewnątrzkomórkowe jest
zaledwie rzędu 100nM, po otwarciu kanałów skacze do 100mM lub więcej tuż przy kanale).
Dwuwartościowe kationy takie jak Co2+ lub Mn2+ blokują transmisję.
Zespół miasteniczny Lamberta-Eatona
: (głównie paraneoplastyczny) jest spowodowany przeciwciałami
przeciwko presynaptycznym napięciowo-zależnym kanałom wapniowym
Białka pęcherzyków synaptycznych
Skład i budowa pęcherzyków zostały dość dobrze poznane
Nie zależą od rodzaju transmitera

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
18/113
Białka lepiej poznane i/lub ważniejsze z nich to:
1.ATP-azowy transporter protonów .- Zakwasza światło pęcherzyków (gradient umożliwiający ładowanie
N-T)
2.Transportery pęcherzykowe dla poszczególnych N-T (umożliwiają
wypuszczenie
protonów „w zamian”
za N-T)
3.Synaptic vesicle protein (SV2): funkcja nieznana
4.Synaptofizyna
: funkcja nieznana (ale wykorzystywana w diagn. histopatologicznej)
5.Synaptotagmin:
łączy się z błonowymi białkami kompleksu SNARE (syntaxin) i prawdopodobnie
pełni rolę
„sensora” Ca2+
(a calmodulina może pełnić rolę modulacyjną).6.Rab-3 (GTP-binding
protein): łączy się z błonowym białkiem rabphilin i innymi białkami jak RIM1 (składnik aktywnej strefy),
7.Synapsin: prawdopodobnie łączy pęcherzyk z aktyną cytoszkieletu; fosforylacja synapsyny umożliwia
ich uwolnienie
SNARE complex = (SNAP- REceptor complex)
kompleks 3 białek: (1)Vesicle-associated membrane protein (VAMP=synaptobrevin) (2) syntaxin (3)
synaptosomal associated protein (SNAP-25 kDa).
Synapsy bez SNARE nie uwalniają N-T.
„SNAREs” oznacza całą klasę „membrane-trafficking” protein.
Blokujące uwalnianie N-T toksyny Gr+ pałeczek beztlenowców Clostridium: botulinowe i tężcowa są
enzymami proteolitycznymi tnącymi komponenty SNARE
N-Ethylmaleimide sensitive factor (NSF) jest ATP-azą, która prawdopodobnie dostarcza energii
koniecznej do odnawiania pęcherzyków.
„Kiss and run” raczej gra mniejszą rolę niż endocytoza w „recyklingu” pęcherzyków.
Białko
dynamina
„odcina” opłaszczony
klatryną (clathrin)
pęcherzyk od błony komórkowej
(mutanty shibire drozofili w odpowiedniej temperaturze są gwałtownie zparaliżowane na skutek
wrażliwości zmutowanej dynaminy)
„Biologia kwantowa” neurotransmiterów
„Quantal size” = jednostkowa odpowiedź na uwolnienie 1 kwantu N-T (w postaci amplitudy sygnału
elektrycznego w komórce postynaptycznej)
„Quantal content” = średnia ilość kwantów uwalnianych przez pojedynczy impuls
Złącze nerwowo-mięśniowe:
Pojedynczy potencjał czynnościowy motoneuronu uwalnia nawet 300 „kwantów” N-T
Każdy receptor posiada przewodnictwo o wartości 25 pS i otwiera się na 1,5 ms. (przepuszczając 35 000
jonów dodatnich).
Otwarcie pojedynczego pęcherzyka (ok. 5000 molekuł n-t, związując ok. 2000 receptorów) powoduje
napływ ok. 70 mln jonów (2000 x 35000) w receptorowych kanałach dając pojedynczy „MINI”
= kilka mV
Synapsa glutamatergiczna:
1 potencjał czynnościowy uwalnia 5-10 kwantów N-T, każdy z nich aktywuje ok. 30 kanałów a 1 quantum
powoduje EPSP=1mV (zdecydowanie za mało do wywołania potencjału czynnościowego)
„Kwantowe” zagadnienia związane z neurotransmisją:
„Model standardowy” Katz’a (ok.1950) pasuje głównie do zł.ner-mies:
Model standardowy Katz’a („kwantowego przekaźnictwa”)
1)Potencjał czynnościciowy podnosi prawdopodobieństwo egzocytozy N-T i uwolnienia „kwantu” N-T
2)Kwant N-T daje w przybliżeniu ten sam efekt elektryczny w kom.postsynaptycznej („quantal size” = Q )
3)Kwanty N-T mogą sumować się liniowo dając wielokrotność „quantal size”)
4)Średnia ilość uwolnionych kwantów „m” („quantal content”) N-T m jest określona równaniem: m = n p
gdzie
n=ilość dostępnych kwantów;
p=prawdopodobieństwo uwolnienia kwantu
5)Przeciętna odpowiedź na bodziec jest iloczynem (Q m) (= Qnp)
6)Względne prawdopodobieństwo obserwacji 0,1,2,...,n kwantów jest dane rozkładem dwumianowym z
parametrami n oraz p
7)„Quantal size” = jednostkowa odpowiedź na uwolnienie 1 kwantu N-T (w postaci amplitudy sygnału
elektrycznego w komórce postynaptycznej)
„Quantal content” = średnia ilość kwantów uwalnianych przez pojedynczy impuls

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
19/113
„Model standardowy” Katz’a jest wykorzystywany w badaniach nad neuromodulacją i wpływem
potencjalnych leczniczych substancji (wskazuje na miejsce uchwytu („target”) potencjalnego
neuromodulatora (potencjalnego leku)
Wpływ na częstość i amplitudę mini-EPP:
np.
bloker receptora postsynaptycznego albo substancja wpływająca na „ładowanie”
pęcherzyków
nie zmieni częstości „mini-EPP” ale zmieni ich amplitudę oraz amplitudę stymulowanych EPPs.
Natomiast bloker lub modulator kanałów wapniowych
zmieni prawdopodobieństwo uwolnienia N-T z pęcherzyka (a w związku z tym tzw. „quantal content”
m=pn (p=prawdopodobieństwo uwolnienia, n=liczba dostępnych kwantów), czyli przeciętną ilość
uwolnionych pęcherzyków przez pojedynczy potencjał czynnościowy.
W obrębie CNS model standardowy często nie pasuje bo m.in.
Pęcherzyki mogą zawierać różne ilości N-T
Prawdopodobnie nie zawsze opróżniana jest cała zawartość pęcherzyka
Jest bardzo trudna rejestracja EPP i wyznaczenie wartości „miniEPP”
Synapsy konwergują na neuronach
Istnieją różne izoformy kanałów Ca2+ a dodatkowo fosforylacja zmienia ich właściwości. Być może w
niektórych przypadkach uwolnienie pęcherzyka hamuje uwolnienie innych pęcherzyków.
Nie tyle ilość dostępnych pęcherzyków ale
ilość receptorów decyduje o EPP
(np. dla Glu o EPP decyduje ilość i cechy receptorów. Istnieją też „ciche synapsy”)
Transmisja synaptyczna:
zmiany przepuszczalności błony postsynaptycznej w czasie aktywności
synapsy (neurotransmisji)
Technika „patch-clamp”
Erwin Neher i Bert Sakman, którzy rozwin
ę
li j
ą
w latach 70-tych otrzymali Nagrod
ę
Nobla w
1991 r
Pozwala na zmierzenie pr
ą
du płyn
ą
cego przez pojedynczy kanał jonowy
A) Eksperyment typu „outside-out” – pipeta zawiera roztwór o składzie podobnym do
cytoplazmy. Na zewn
ą
trz błony z kanałem roztwór jest podobny do zewn
ą
trzkomórkowego.
Pozakomórkowy płyn stanowi „uziemienie” układu elektrycznego w którym wzmacniacz
utrzymuje stałe napi
ę
cie przezbłonowe. Rejestrowane jest nat
ęż
enie pr
ą
du przechodz
ą
cego
przez kanał.
Mierzone jest nat
ęż
enie pr
ą
du.
Natomiast wolta
ż
jest stabilizowany na dowolnie wybranej warto
ś
ci
B) Przepływ pr
ą
du płyn
ą
cego przez pojedynczy kanał jonowy po podaniu „od zewn
ą
trz”
acetylocholiny (Ach)
w sposób ci
ą
gły
.
Pr
ą
d NIE PŁYNIE STALE LECZ W POSTACI „IMPULSÓW”.
Ilustruje to odpowied
ź
receptora w postaci „wszystko-albo-nic”.
Zwi
ę
kszenie st
ęż
enia Ach nie powoduje zmiany nat
ęż
enia pr
ą
du lecz wzrost
PRAWDOPODOBIE
Ń
STWA otwarcia kanału!
Pr
ą
d przepływaj
ą
cy przez 1 kanał jest rz
ę
du 10-12 amperów (pA)
Efekt postsynaptyczny jest wynikiem sumowania potencjałów z wielu kanałów jonowych.
Czas otwarcia jest ró
ż
ny ale „amplituda” (nat
ęż
enie pr
ą
du) zawsze ta sama.
Technika patch-clamp:
rezultaty eksperymentów z podawaniem N-T przy zmienianych i
ró
ż
nych warto
ś
ciach (-40mV, -20mV, 0mV, +20mV) stabilizowanego napi
ę
cia w
mikroelektrodzie (w relacji do na zewn
ą
trz błony komórkowej ze „złapanym” kanałem
receptorowo-zale
ż
nym
WNIOSKI: Po zwi
ą
zaniu z NT cz
ę
stotliwo
ść
i
ś
redni czas otwarcia kanału s
ą
niezale
ż
ne od
napi
ę
cia jednak kierunek i amplituda pr
ą
du zale
ż
y od napi
ę
cia. Kierunek pr
ą
du „d
ąż
y” do
osi
ą
gni
ę
cia równowagi zgodnie z równaniem Goldmana-Hodgkina-Katza.
Badania prądów w złączu nerwowo-mięśniowym:
•EPP – end plate potential; EPC – end plate current•
EPC jest proporcjonalny do ilości otwartych kanałów
•W normalnym mięśniu dośrodkowy (inward) EPC depolaryzuje błonę
•Powstająca zmiana potencjału błonowego nazywana jest EPP (potencjałem płytki końcowej)
Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
20/113
Badano prądy postsynaptyczne (end-plate currents - EPC) w zależności od postsynaptycznego napięcia
błonowego.
•Badania te (wraz z manipulacją stężeń jonów) pomogły ustalić przepływ jakich jonów tworzy te prądy
(małżeństwo Akira i Noriko Takeuchi 1960)
•Kierunek i wielkość prądu EPC zależą od zastosowanego (voltage-clamped) postsynaptycznego napięcia
błonowego
•Przy -110 mV prąd (ładunki +) jest dośrodkowy, przy napięciu 0 mV prąd jest zerowy (EPC=0) a powyżej 0mV
prąd zaczyna się odwracać na dozewnątrz (outward)
•Potencjał 0mV nazywamy dlatego „reverse potential” (potencjał odwrócenia)
EPC jest proporcjonalny do różnicy między danym napięciem (oznaczanym jako Vm) i potencjałem odwrócenia
(Erev)
•Ponadto EPC jest proporcjonalny do przewodnictwa (conductance = odwrotność oporu) błony aktywowanej
acetylocholiną (oznacznaej jako gACh)
•Stąd EPC = gACh (Vm – Erev)
•Potencjał odwrócenia „leży” pomiędzy potencjałam równowagi dla jonów ECl, ENa
•Obniżenie zewnątrzkomórkowego Na powoduje przesunięcie „reversal potential” w stronę wartości ujemnych
•Podwyższenie zewnątrzkomórkowego stężenia jonów K powoduje przesunięcie „reversal potential” w stronę
wartości dodatnich
•Dla typowego potencjału spoczynkowego mieśnia (-90 mV) w EPC dominuje prąd dośrodkowy jonów Na
(dlatego efekt netto prądów obu jonów Na i K jest też dośrodkowy)
•Prądy dla potencjału powyżej „potencjału odwrócenia” hyperpolaryzują komórkę postsynaptyczną a dla
potencjału poniżej „potencjału odwrócenia” depolaryzują komórkę.
•W przypadku komórki mięśniowej praktycznie każdy EPP wywołuje Pcz
•W przypadku komórek nerwowych rezultat zależy od „sumacji” potencjałów postsynaptycznych (PSP) w tym
pobudzających i hamujących
•Glutaminian zwykle również powoduje otwarcie kanałów przepuszczalnych zarówno dla Na jak i K dlatego
ogólny opis zależności jest podobny jak w złączu nerwowo-mięśniowym
•Różnica między EPSP i IPSP
•EPSP (pobudzający) charakteryzuje się tym, że jego potencjał odwrócenia (Erev) jest bardziej dodatni niż próg
pobudliwości (threshold) komórki
•IPSP (hamujący) charakteryzuje się tym, że jego potencjał odwrócenia jest bardziej ujemny niż potencjał
progowy.
•Ważna uwaga: EPSP jest depolaryzujący ale IPSP może być hyperpolaryzujący ale nie musi (w pewnych
warunkach może być hyperpolaryzujący! Wystarczy aby jego Erev był poniżej progu pobudzenia czyli
powstania potencjału czynnościowego)
Oprócz szybkich EPSP zaobserwowano,
ż
e istniej
ą
tak
ż
e wolne EPSP.
Szybkie EPSP wywołane s
ą
otwarciem kanału jonowego bramkowanego ligandem
(neurotransmiterem).
Wolne EPSP opieraja si
ę
na znacznie bardziej skomplikowanym mechanizmie receptorowymi
(receptory te nazwano metabotropowymi).
Czasowe i przestrzenne sumowanie PSP
W przykładzie obok, sumowanie PSP w neuronie ruchowym umożliwia wygenerowanie p.cz. pomimo, że
pojedynczy p.cz. w każdym z presynaptycznych neuronów czuciowych wywołuje EPSP równy 1 mV, a do
osiągnięcia progu pobudzenia potrzeba przynajmniej ok. 20 mV lub więcej.
W sumowaniu czasowym
nie ma dokładnego „sumowania” ponieważ nawet jeśli przewodnictwo pozostaje
takie samo w kolejnych otwarciach kanału, jednak zgodnie z prawem Ohma natężenie prądu jonowego
jest oprócz przewodniości kanału (g) proporcjonalne do różnicy potencjału błonowego is= gs (Em – Ek) (a
ta nieco zmniejsza się na skutek wypływu jonów w czasie trwania kolejnego otwarcia kanału)
W sumowaniu czasowym istotna rolę gra znana nam „stała przestrzenna” l(space constant = length
constant)
Vx = V0 e –x/
l
Sumowanie czasowe
pozwala na integracj
ę
kolejnych potencjałów postsynaptycznych w danej
synapsie natomiast
sumowanie przestrzenne
pozwala na integracj
ę
postsynaptycznych potencjałów z ró
ż
nych
okolic neuronu

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
21/113
NEUROTRANSMISJA cz. C
Receptory neurotransmiterów
Receptory dzielą się na:
JONOTROPOWE
(tworzące i po związaniu z ligandem otwierające kanały jonowe, dużych rozmiarów,
zbudowane z podjednostek).
Ich pobudzenie wywołuje szybko potencjał postsynaptyczny (PSP), który jest jednak
krótkotrwały („fast-PSP”- typowo ok. 20ms)
METABOTROPOWE
(działające poprzez aktywację GTP-wiążących białek – tzw. G-protein; stąd
zwane G-protein coupled receptors GPCRs; są utworzone przez pojedynczy polipeptyd). Ich pobudzenie
wywołuje długo trwający (slow-PSP)
NEUROTRANSMISJA jest częścią szerszego pojęcia – „sygnalizacji międzykomórkowej”
W ramach
sygnalizacji międzykomórkowej
recepcja sygnału odbywa się za pośrednictwem:
RECEPTORÓW JONOTROPOWYCH
(tworzących „szybką komunikację” (w milisekundach) w
ramach układu nerwowego oraz pomiędzy układem nerwowym i innymi narządami).
RECEPTORÓW METABOTROPOWYCH – (=GPCRs)
(najliczniejszych, działających znacznie dłużej –
nawet do wielu godzin, i w znacznej mierze spełniających funkcje regulacyjne)
RECEPTOROWYCH KINAZ TYROZYNOWYCH
(Stanowiących receptory dla czynników wzrostu,
czynników troficznych regulujących długotrwałe zmiany decydujące o wzroście, różnicowaniu i „losie”
komórek)
Receptory jonotropowe
Badania genetyczno-molekularne oraz strukturalne oparte o prX umożliwiły dokładne poznanie budowy i
funkcji poszczególnych fragmentów podjednostek
Dwie rodziny pochodzące z 2 różnych genetycznych „przodków”
1- receptory: nikotynowy Ach -dla kationów
(pozostałe iR są wariantami budowy nACh),
GABAA (g-amino butyric acid), -dla anionów
glicynowy, -dla anionów 5-HT3 (jedna z
podklas receptora dla 5-HT), -dla kationów
2- (jonotropowe) receptory glutamatergiczne
Nikotynowy receptor Ach (nACh)
(model struktury jonotropowych receptorów)
Receptor nikotynowy ACh (nACh) jest receptorem
1.w złączu nerwowo-mięśniowym
2.W synapsach pomiędzy przedzwojowymi i pozazwojowymi neuronami obu części układu
autonomicznego (parasympatycznego i sympatycznego)
3.W mózgu
Blokerem kanału jonowego w nACh jest hexametonium, antagonistą miejsca łączącego a ACh jest
trimetafan
Poznany najlepiej m. in. z powodu dostępności (electric organ Torpedo ray, wyspecjalizowany mięsień
generujący napięcie nawet do 500V)
α
αα
α
-bungarotoksyna z wysoką swoistością wiąże się z nACh co pozwala na superczyste ekstrakty receptora
w chromatografii
Nikotyna jest wzorcowym agonistą nACh
Kanał receptorowy jest umiarkowanie selektywny: przepuszczalny dla Na+, K+, i nieco słabiej dla Ca2+,
które przechodzą zgodnie z gradientem elektrochemicznym.
Każdy receptor ma dwa miejsca wiążące Ach
Otwarcie kanału następuje na skutek zmian konformacyjnych białek receptora po przyłączeniu Ach
Desensytyzacja jest w nACh mięśniowym wolna, znacznie wolniejsza niż w mózgu (stała 50-100ms)
Miastenia gravis
: jedna z najlepiej poznanych chorób autoimmunologicznych jest spowodowana
autoagresją przeciw receptorom Ach w mięśniach
Neuronalny nAChR
Neuronalny nAChR zbudowany jest jedynie z podjednostek
α
αα
α
i
ββββ

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
22/113
Zidentyfikowano wiele wariantów budowy podjednostki a (9) oraz b (4) (niektóre specyficzne dla
niektórych zwierząt). Możliwa jest kombinacja nawet tysięcy różnych „wersji” receptora z różnymi
właściwościami !
Np.
Przewodnością
(może być od 5 do 50 pS.)
Przepuszczalnością dla Ca2+
(zwykle większą niż dla rec. mięśniowych)
Czasem desensytyzacji
Neuronalny nAChR prawdopodobnie jest odpowiedzialny za psychofizyczne efekty uzależnienia od
nikotyny.
Receptor serotoninowy – (podklasa 5-HT3)
jest receptorem jonotropowym (większość receptorów 5-HT jest metabotropowa)
Receptor 5-HT3 występuje w obwodowych zakończeniach nerwów czuciowych i w CNS
Receptor serotoninowy 5-HT3 jest przepuszczalny dla K+ i Na+ (nieprzepuszczalny dla Ca2+ i innych
dwuwartościowych jonów, pomimo podobnej szerokości otworu jak w nACh)
Ma budowę podobną do nACh ale składa się z 5 kopii tej samej podjednostki (zbliżonej do podtypu a7 z
nACh)
Antagoniści receptora 5-HT3 są używani jako
leki przeciwwymiotne (ONDASETRON, GRANISETRON – blokują 5-HT3 receptory m.in. W dnie
kom.IV i obwodowo w zak.nerwu X),
antypsychotyczne i anksjolityki.
Receptory GABAA- główne receptory hamujące w CSN
GABAA to najczęściej występujący hamujący receptor GABA
Tworzą go podjednostki (pentamer tak jak r nACh) nazwane dgba e oraz r (głównie w
siatkówce); dla każdej z nich znane są różne podtypy.
W podjednostce a znajduje się miejsce wiążące ligand
Kanał jonowy receptora GABAA jest
selektywny dla Cl-,
co powoduje hyperpolaryzację po
otwarciu kanału (stąd hamowanie i IPSP)
GABAC jest głównie w siatkówce (podobnie jak GABAA jest związany z kanałem jonowym
dla Cl-.)
(GABAB jest metabotropowy !)
Receptory GABAA- to główne receptory hamujące w CSN
Receptor GABAA może być modulowany przez wiązanie różnych substancji np.
barbituranów (luminal) i benzodiazepin (diazepam).
Obie grupy leków potęgują wiązanie
GABA (i podwyższają hamowanie).
Odwrotnie działają substancje powodujące drgawki picrotoxin (blokuje kanał) i
bicucullin, która zmniejsza wiązanie GABA.
Także
penicylina
hamuje receptor blokując otwór dla jonów
(UWAGA! drgawki należą do
działań niepożądanych penicyliny!).
Również progesteron, kortykosteron, testosteron potęgują działanie receptora
Receptory siatkówkowe GABA nie są wrażliwe na bicuculinę (choć są wrażliwe na
pikrotoksynę), nie są wrażliwe na barbiturany i benzodiazepiny
Jonotropowe receptory glutamatergiczne – iGluR najliczniejsze receptory
pobudzające w CSN
Składają się z 4 podjednostek (tetramery). Podjednostki są znacznie większe od podjednostek AChR
W latach 70-tych zaobserwowano (Watkins i wsp.), że egzogenny związek NMDA
kwas N-metylo-D-
asparaginowy
swoiście stymuluje (agonista) część iGluR. W latach 80 i 90tych ujawniono i
scharakteryzowano budowę oraz właściwości poszczególnych podgrup iGluR.
Ich nazwy wywodzą się od nazw specyficznych agonistów iGluR, którzy różnicują te receptory na szereg
podgrup o odmiennych właściwościach. Są to zatem receptory typu:
1)
NMDA (
kwas N-metylo-D-asparaginowy)
2)
„non-NMDA”
takie jak:
AMPA
kwas
α
-Amino-3-hydroksy-5-metylo-4-izoksazolopropionowy
Kainianowy
Glutamatergiczny receptor typu NMDA
1
Otwarcie kanału 2-10ms, EPSC >>100ms, 50pS

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
23/113
Wybitne przewodnictwo dla jon
ó
w Ca
2+
i
Na
+
; najwi
ę
ksza g
ę
sto
ść
w
sektorze CA1 hipokampa;
pe
ł
ni
ą
fizjologiczn
ę
rol
ę
w
plastyczno
ś
ci i
uczeniu si
ę
i
patologiczn
ą
w
ekscytotoksyczno
ś
ci
,
najliczniejszy. NR2(A-D) same nie tworz
ą
kanałów,
Wymaga depolaryzacji aby uwolni
ć
si
ę
od
blokady magnezowej! –Cecha ta powoduje,
ż
e receptor NMDA jest „detektorem
jednoczesno
ś
ci” zdarze
ń
(„bramka logiczna „AND”).
Antagoni
ś
ci: MK-801
#
AP5
4
Mg
2+
Receptory NMDA bardzo istotne w rozwoju mózgu, uczeniu, pamięci oraz w patologii
NMDA jest selektywnym agonistą jednak jest co najmniej 10x słabszy niż L-glutaminian
Mają budowę podobną jak receptory non-NMDA, ( podjednostki NR1, NR2A, NR2B, NR2C, NR2D,
NR3A, NR3B) z TM2 nieprzechodzącym przez błonę i który odpowiedzialny jest za przepuszczalność dla
Ca2+
Ich cechy charakterystyczne:
1)Napięciowo zależne blokowanie przez Mg2+ (błona musi być zdepolaryzowana aby uwolnić się od
blokowania przez Mg2+!)
2)Glicyna związana z tzw. „miejscem glicynowym” konieczna dla efektywnego otwarcia kanału
3)Przewodzą Ca2+
(potencjalnie patologiczne znaczenie prowadzące do tzw. ekscytotoksyczności)
Zagadkowa modulacja przez sperminę wzmagającą aktywację NMDA
Glutamatergiczny receptor typu AMPA
2
(podjednostki: GluR1, GluR2, GluR3,
GluR4)
B.szybkie
otwarcie kanału <1ms, krótki pr
ą
d EPSC ok. 10ms. Szybkie przeka
ź
nictwo
synaptyczne, m.in czucie b
ó
lu, zwiazane z
kana
ł
ami dla Na
+
, K
+
i
Ca
2+
,
(z GluR2 słaba
przepuszczalno
ść
dla Ca
2+
) antagon
ś
ci: NBQX
5
, CNQX
6
Różnorodność właściwości iGluR (non-NMDA) nie pochodzi tylko z różnych „kombinacji”
podjednosteknp. Istnieją „splice variants” nazywane flip i flop (flop wykazuje silniejszą desensytyzację a
stąd mniejsze prądy jonowe)
Szybkie i wolne potencjały postsynaptyczne (PSP)
A)Szybkie PSP (Poprzez kanały jonowe)
B)Wolne PSP (metabotropowe) działaj
ą
przez po
ś
redników (np. cAMP) Potencjał powstaje
wolniej (wymaga syntezy cAMP) ale trwa wielokrotnie dłu
ż
ej (Nawet gdy fosfodiesteraza cAMP
zako
ń
czy działanie wtórnego przeka
ź
nika to ufosforylowany kanał potasowy dalej przepuszcza
jony. Trwa to a
ż
fosfataza proteinowa zdefosforyluje kanał)
Jeszcze dłu
ż
sze działanie mo
ż
e by
ć
wtedy, gdy aktywowane s
ą
geny i modulowany metabolizm
Receptor glicynowy – główny receptor hamujący w
rdzeniu kręgowym
i pniu mózgu
Przepuszczalny dla Cl-
Pentamer zbudowany z podjednostek a oraz bTrzy glicyny muszą być przyłączone aby otworzyć kanał
Strychnina
(alkaloid z gat. roślin strączkowych) jest antagonistą receptora glicynowego
Jonotropowe receptory purynowe
(„purynergiczne”)
Są również opisane
jonotropowe receptory purynowe
P2x1-7 (oraz P2z?) choć większość z receptorów
purynowych jest metabotropowa.
Ligandem jest ATP lub adenozyna lub inne analogi nukleotydów
Są one uwalniane w niektórych synapsach w sposób kwantowy (wraz z katecholaminami i Ach).
Mogą przepuszczać zarówno aniony jak i kationy
Receptory jonotropowe (podsumowanie):
są odpowiedzialne za szybkie pobudzające i hamujące potencjały synaptyczne
Przeciętnie prąd
przechodzący przez pojedynczy kanał wynosi 10-12A (ampera) czyli ok. 6 mln
jednowartościowych jonów/sek.
Zwiększenie stężenia N-T nie zmienia przewodnictwa kanału (ok.20pS) ale zmienia
prawdopodobieństwo jego otwarcia.
Otwarcie kanału ma charakter procesu typu „wszystko-albo-nic”
Gdy bierzemy pod uwagę nie pojedynczy kanał ale ich liczny zbiór zwiększone
prawdopodobieństwo otwarcia kanału przekłada się na zwiększoną ilość otwartych
kanałów, których efekty się sumują.
Najważniejsze receptory jonotropowe: podsumowanie
Nikotynowe receptory
acetylocholinowe

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
24/113
Receptory GABAAi GABAC
Receptory serotoninergiczne 5-HT3
Receptory glicynowe
Jonotropowe receptory purynergiczne P2x oraz P2z Jonootropowe receptory
glutamatergiczne (NMDA, AMPA)
Receptory metabotropowe
wła
ś
ciwsza nazwa –
receptory sprz
ęż
one z białkiem G
(G-protein coupled receptors = GPCRs) :
Receptory „regulacyjne” - „brama” do biochemicznego i metabolicznego wn
ę
trza komórki
G-protein coupled receptors (GPCR = „metabotropowe”)Zidentyfikowano ponad 1000 !
Receptorów metabotropowych
Bardziej wskazana nazwa to GPCR, ponieważ bardzo często poprzez aktywację białka G
modulują kanały jonowe
a nie bezpośrednio „wpływają na metabolizm”
Aktywacja białka G (GTP-binding protein) oznacza wymianę GDP w GTP
Tak zaktywowane białko
zmienia aktywność enzymów oraz kanałów jonowych.
Powstają
też „second messengers”m.in. Aktywujący kinazy proteinowe.
Typowo działanie GPCR musi być wolniejsze niż w przypadku rec. jonotropowych ale też
jest zarazem znacznie dłuższe (w efektach).
Większość małych N-T ma zarówno jono jaki metabotropowe receptory.
Na podstawie struktury GPCR dzieli się na 3 podrodziny:
1.Rodopsyny i receptorów adrenergicznych.
2.Sekretyny i vazoaktywnego intest. peptydu (VIP)
3.Metabotropowego receptora glutaminianergicznego
Najważniejsze receptory metabotropowe
Muskarynowe
receptory acetylocholinowe
Receptory adrenergiczne
Receptory dopaminergiczne
Receptory GABAB
Metabotropowe receptory serotoninergiczne
Metabotropowe receptory purynergiczne
Metabotropowe receptory glutamatergiczne
Receptory neuropeptydów (wszystkie są GPCRs)
Ogólny model receptora metabotropowego
GPCR składa się z pojedynczego polipeptydu.
Receptor zawiera 7 transbłonowych helikalnych segmentów. Każda z 7 domen
transbłonowych zawiera ok. 24 aminokwasów.
GPCRs mogą istnieć jako pojedyncze jednostki a także tworzyć homo i heterooligomery
Miejsce wiążące N-T znajduje się w dużej części GPCR w środku receptora jednak
nie
dotyczy to mGluR i GABAB oraz Rec. Neuropeptydowych
.
Zamiany pojedynczego aminokwasu może niekiedy zmniejszać siłę wiązania liganda 1000
a nawet 10000x
Związanie agonisty stabilizuje konformację aktywną i przesuwa równowagę w kierunku
formy aktywnej izomeru (aktywującej białko G)
Układ sygnalizacji poprzez receptory sprz
ęż
one z białkiem G
„składa si
ę
” z :
•1) Receptora
•2) Białka G po wewn
ę
trznej stronie błony cytoplazmatycznej, które mo
ż
e by
ć
stymulowane
przez zaktywowany receptor
•3) Efektorowego enzymu „wytwarzaj
ą
cego” (zmieniaj
ą
cego st
ęż
enie) II-rz
ę
dowego
przeka
ź
nika lub efektorowego kanału jonowego (w odpowiedzi na aktywacj
ę
białka G)
Dwie drogi sygnalizacji poprzez receptory sprzężone z białkiem G:
Dwie drogi sygnalizacji poprzez receptory sprzężone z białkiem G:
Dwie drogi sygnalizacji poprzez receptory sprzężone z białkiem G:
Dwie drogi sygnalizacji poprzez receptory sprzężone z białkiem G:
jjjjedna „w kierunku” aktywacji
edna „w kierunku” aktywacji
edna „w kierunku” aktywacji
edna „w kierunku” aktywacji kinazy
kinazy
kinazy
kinazy PKA,
PKA,
PKA,
PKA,

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
25/113
po złączeniu z ligandem aktywacja cyklazy adenylowej
po złączeniu z ligandem aktywacja cyklazy adenylowej
po złączeniu z ligandem aktywacja cyklazy adenylowej
po złączeniu z ligandem aktywacja cyklazy adenylowej –––– wytworzenie cAMP,
wytworzenie cAMP,
wytworzenie cAMP,
wytworzenie cAMP,
aktywacja kinazy białkowej PKA
aktywacja kinazy białkowej PKA
aktywacja kinazy białkowej PKA
aktywacja kinazy białkowej PKA
druga
druga
druga
druga „w kierunku” altywacji
„w kierunku” altywacji
„w kierunku” altywacji
„w kierunku” altywacji kinazy
kinazy
kinazy
kinazy PKC
PKC
PKC
PKC
Po złączeniu z ligandem aktywacja fosfolipazy C
Po złączeniu z ligandem aktywacja fosfolipazy C
Po złączeniu z ligandem aktywacja fosfolipazy C
Po złączeniu z ligandem aktywacja fosfolipazy C ––––powstanie diacyloglicerolu i
powstanie diacyloglicerolu i
powstanie diacyloglicerolu i
powstanie diacyloglicerolu i
trójfosfoinozytolu, bezpośrednia aktywacja kinazy białkowej C (PKC) przez
trójfosfoinozytolu, bezpośrednia aktywacja kinazy białkowej C (PKC) przez
trójfosfoinozytolu, bezpośrednia aktywacja kinazy białkowej C (PKC) przez
trójfosfoinozytolu, bezpośrednia aktywacja kinazy białkowej C (PKC) przez
diacyloglicerol oraz pośrednia poprzez uruchomienie jonów wapn
diacyloglicerol oraz pośrednia poprzez uruchomienie jonów wapn
diacyloglicerol oraz pośrednia poprzez uruchomienie jonów wapn
diacyloglicerol oraz pośrednia poprzez uruchomienie jonów wapnia (przez
ia (przez
ia (przez
ia (przez
trójfosfolinozytol) z retikulum endoplazmatycznego
trójfosfolinozytol) z retikulum endoplazmatycznego
trójfosfolinozytol) z retikulum endoplazmatycznego
trójfosfolinozytol) z retikulum endoplazmatycznego
Jest znanych ponad 600 receptorów (w tym dla światła!), które są sprzężone z jednym lub więcej z
27
rodzajów białka G (podjednostki alfa).
Z kolei białka G regulują jeden lub więcej z dwudziestu kilku różnych kanałów i enzymów.
Białko G musi „wykryć” aktywację receptora!
Aktywacja enzymu
np. cyklazy adenylowej produkującej cAMP lub fosfolipazy C generującej
diacyloglicerol oraz IP3 – (IP3 uwalnia Ca2+ z magazynów śródkomórkowych) stanowi czynnik
wzmacniający
sygnał.
cAMP, Ca2+, diacyloglicerol
aktywują kinazy proteinowe
dla wielu różnych substratów (enzymy, kanały,
białka strukturalne, czynniki transkrypcyjne).
cAMP, cGMP, kwas arachidonowy i Ca2+ mogą też bezpośrednio otwierać i modulować kanały jonowe.
Białka G mogą także bezpośrednio sprzęgać się z kanałmi jonowymi (bez pośrednictwa „second
messnger”)
Cechy sygnalizacji poprzez receptory sprz
ęż
one z białkiem G (metabotropowe)
Konkretny typ
receptora może łączyć się na ogół (w większości) tylko z jednym rodzajem białka G
Neuron ma tylko określony podzbiór GPCR i białek G
Amplifikacja sygnału w układzie receptora typu GPCR:
1- zaktywowany receptor może aktywować wiele „sztuk” białek G
2- każda cyklaza adenylowa może zsyntetyzować wiele cAMP
3- każda kinaza proteinowa może ufosforylować wiele kopii swojego substratu
Proces jest z początku wolniejszy (w porównaniu np. z receptorem jonotropowym) ale trwa dłużej.
„Orkiestracja” odpowiedzi komórkowej
Ten sam wtórny przekaźnik może
jednocześnie
aktywować liczne i różne szlaki metaboliczne i aktywować
transkrypcję genów (tzw. „orchestrated response”.)
Białka G maj
ą
trymerow
ą
(trójzło
ż
on
ą
) budow
ę
z podjednostek
a
b
g
(
a
podjednostka ma cechy GTPazy).
Zwi
ą
zanie z receptorem uwalnia GDP (redukcja powinowactwa do GDP) a poniewa
ż
w komórce
jest przewaga GTP, miejsce wi
ążą
ce nukleotyd zostaje zaj
ę
te przez GTP i jednocze
ś
nie
nast
ę
puje dysocjacja białka G na
a
-GTP oraz (
gb
).
Wymiana GTP-GDP jest powolna dlatego
„stan-on” jest zwykle bardzo niski.
Ponadto
a
-GTP odł
ą
cza si
ę
od receptora (redukcja powinowactwa).
•Oprócz heterotrymerycznych białek G (złożonych z 3 podjednostek) sa także monomeryczne białka G (tzw.
small G-protein), które także biorą udział w przekaźnictwie sygnałów. Należy do nich (pierwsze odkryte z tej
grupy) białko
„ras”
(od wirusa powodującego mięsaka szczurzego - rat sarcoma) przekazujące sygnał z receptora
sprzężonego z kinazą i wpływające na różnicowanie i proliferację komórek
Ź
ródła specyficzno
ś
ci sygnalizacji poprzez receptory sprz
ęż
one z białkiem G
Znanych jest 27
r
óż
nych gen
ó
w podjednostki
a
oraz 5
b
i 13
g
Ale wszystkie teoretyczne kombinacje podtyp
ó
w
gba
s
ą
mo
ż
liwe.
Tradycyjnie u
ż
ywa si
ę
nazw z liter
ą
G i indeksem dolnym
Np.
Gs (
a
s) – stymuluje
cyklaz
ę
adenylow
ą
; Gi – hamuje
cyklaz
ę
adenylow
ą
Gp –stymuluje
fosfolipaz
ę
Go – inne efekty
Dimery [
gb
] równie
ż
stymuluj
ą
ró
ż
ne enzymy i kanały.
Specyficzno
ść
sygnalizacji przez okre
ś
lony receptor osi
ą
gana jest poprzez fakt,
ż
e tylko
ograniczona liczba podtypów receptora oraz białek G i efektorów jest „na wyposa
ż
eniu”
poszczególnych neuronów.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
26/113
Ponadto istnieje
„
kompartmentacja
”
efektorów (np.. Ten sam receptor mo
ż
e regulowa
ć
kanały
Ca2+ w zako
ń
czeniu nerwowym a fosfolipaz
ę
C w dendrycie).
Szlak sygnalizacji cyklazy adenylowej i PKA
Poziom cAMP jest regulowany poprzez
przeciwstawne enzymy:
cyklazy adenylowe
(tworz
ą
ce cAMP) i fosfodiesterazy (PDEs)
degraduj
ą
ce cAMP.
Cyklazy adenylowe tworz
ą
du
żą
rodzin
ę
, (wyró
ż
nia si
ę
grupy izoform I-IX).
Niektóre cyklazy adenylowe
(zwł tzw. grupy A)
mog
ą
by
ć
„
detektorami koincydencji” poprzez
sprz
ę
ganie sygnału prowadz
ą
cego do wzrostu cAMP z sygnałem (od innego neurotransmitera)
wzrostu Ca2+.
N-T które u
ż
ywaj
ą
cAMP jak wtórnego przeka
ź
nika poprzez aktywacj
ę
lub hamowanie cyklazy
to m.in. : epinefryna, norepinefryna, dopamina, serotonina, VIP, somatostatyna.
cAMP-zale
ż
na kinaza białkowa (PKA) neurotransmisja poprzez aktywacj
ę
adenylocyklazy i cAMP, fosforyluje reszty Ser/Tre
-Regulacja ekspresji genów
poprzez [cAMP response element-building protein] = CREB
-Regulacja syntezy katecholamin (poprzez hydrolaz
ę
tyrozyny)
-Regulacja MAP-2 (microtubule associated protein)
-Regulacja przewodnictwa błonowego (kanały K+)
-Regulacja czuło
ś
ci receptora AMPA
-PKA jest kotwiczona do ró
ż
nych miejsc przy pomocy tzw białek kotwicz
ą
cych (anchoring
proteins AKAPs np. do AMPAR)
Specyfika działania receptorów metabotropowych
W momencie połączenia z białkiem G
wzmaga się powinowactwo agonisty do receptora a zarazem do samego białka G i to
pozytywne sprzężenie
wydłuża działanie agonisty !
Białka G są różne co oznacza także, że ich różne podtypy aktywizują różne enzymy a stąd różne szlaki
metaboliczne.
Poszczególne izomery receptora łączą się z innymi rodzajami białka G i stymulują
fosfolipazę C
albo
fosfolipazę C i jednocześnie fosfolipazę A2.
Komórki regulują wrażliwość na agonistę poprzez zmianę ilości receptora !
Innym istotnym mechanizmem regulacyjnym jest
desensytyzacja.
Desensytyzacja
chroni system sygnalizacji przed saturacją. Jej efektem i zarazem wykładnikiem jest
konieczność zwiększenia ilości agonisty aby wywołać mierzalne skutki aktywacji (np. ilość cAMP).
-(szybka desensytyzacja) przez fosforylację receptora
- („wolna” desensytyzacja) przez endocytozę receptora
Szlak aktywacji kinazy PKC (poprzez fosfolipaz
ę
C, trójfosfoinozytol,
diacyloglicerol i jony wapnia)
(np. w muskarynowym receptorze Ach)
IP3 mobilizuje wapń działając poprzez specyficzne receptory.
Wapń jest zarówno elektrogennym jonem jak też wtórnym przekaźnikiem.
Wapń wolno dyfunduje (wiązany przez proteiny) dlatego jako przekaźnik działa lokalnie !
Najważniejszym mediatorem dla Ca2+ jest
kalmodulina
, która po przyłączeniu Ca2+ zmienia
konformację zwiększając powinowactwo do i aktywując ponad 20 enzymów m.in. różne kinazy.
Fosfatydylocholina jest również źródłem DAG
poprzez PKD ponadto aktywacja PLA2 prowadzi do powstania kw.arachidonowego
•Śródkomórkowe receptory-kanały wapniowe uwalniają wapń z zasobów wewnątrzkomórkowych (Ca jest
tu „trzeciorzędowym” przekaźnikiem):
•1. Receptor IP3•2. Receptor ryanodinowy* (m.in. w mięśniach - aktywowany depolaryzacją)
–(*ryanodine – roslinny alkaloid z tropikalnej rośliny Ryania speciosa używany w mieszankach jako
insektycyd)
PKC (kinaza proteinowa C) (główny cel systemu sygnałowego PI)
-PKC to
kolektywna nazwa dla
rodziny kinaz
ł
ą
cz
ą
cych si
ę
z sygnalizacj
ą
przez PI i fosforyluj
ą
cych reszty Ser/Thr
-PKC jest aktywowana przez diacyloglicerol (DAG) i Ca2+.
-W nowotworach PKC jest stymulowana nietypowo przez estry forbolu (promotory
nowotworowe), które symuluj
ą
działanie DAG. Estry forbolu prowadz
ą
do długotrwałej
stymulacji (godziny i dni) PKC i z niejasnych przyczyn wywołuj
ą
degradacj
ę
PKC
-Kinazy PKC s
ą
monomerami

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
27/113
-Podobnie jak inne kinazy, PKC jest „kotwiczona” do ró
ż
nych miejsc przez tzw. anchoring
proteins, które kotwicz
ą
równie
ż
inne kinazy (np.. A kinase anchoring protein - AKAP79)
-Do jej substratów nale
żą
liczne białka regulacyjne cyklu i wzrostu komórki oraz m.in. kanałów
jonowych i receptorów takich jak NMDA i AMPA
Metabotropowe receptory dla poszczególnych NT
Receptory muskarynowe ACh
Muskaryna (alkaloid obecny w grzybach amanita muscaria) jest
agonistą metabotropowego mACh. Najbardziej znanym i stosowanym antagonistą mACh jest
atropina (oraz ipratropium)
mACh pełnią dominującą rolę w cholinergicznej neurotransmisji w
mózgu. Poza mózgiem są receptorami wszystkich komórek efektorowych unerwianych przez pozazwojowe
neurony układu PARASYMPATYCZNEGO
(także niektórych układu sympatycznego unerwianych przez
cholinergiczne neurony).
Są zarówno
pre jak i postsynaptyczne
a ich głównym mechanizmem jest
działanie poprzez zmiany
kanałów jonowych.
Kanał dla K+ w sercu
, który gwałtownie wzmaga przepuszczalność w odpowiedzi na
pojawienie się acetylocholiny (uwolnionej z n.X)
był pierwszym kanałem jonowym, dla
którego udowodniono modulację poprzez metabotropowy muskarynowy receptor dla
ACh.
Presynaptyczny mACh reguluje uwalnianie Ach (działa tu zazwyczaj hamująco, co prowadzi do
sprzężenia zwrotnego).
Najbardziej znanym i stosowanym antagonistą mACh jest
atropina
Rodzina obejmuje 5 członków m1-m5:
m1, m3, m5 wiążą się z białkami G aktywującymi fosfolipazęC (PLC)
m2, m4 wiążą się z białkami G które hamuja cyklazę adenylową oraz regulującymi bezpośrednio
kanały dla K+ i Ca2+
M.in. Acetylocholina poprzez receptory muskarynowe może wpływać na syntezę NO (następny slajd)
•M1 – jelito i gruczoły jelita
•M2 – serce, naczynia krwionośne
•M3 – mięśniówka gładka i różne gruczoły
Receptory adrenergiczne
Receptory dla noradrenaliny, adrenaliny (obie wiążą się do tego
receptora) - Dzielą się na trzy klasy
a1, a2, b
Każda klasa a1, a2, ma 3 podklasy, również
receptory klasy b mają 3 subklasy: b1, b2, b3.
Receptory b aktywują cyklazę adenylową a receptory a hamują cyklazę adenylową.
W mózgu głównymi receptorami adrenergicznymi są a2 i b1
Adrenergiczne receptory a i b są typowymi
receptorami neuronów postganglionicznych
autonomicznego układu sympatycznego.
Inne różnice w dystrybucji receptorów adrenergicznych:
a1 : mięśniówka większości naczyń krwion, mięsień rozszerzający źrenicę, mięśnie
pilomotoryczne
a2 : CSN, płytki krwi, zakończenia obwodowych nerwów adrenergicznych
b1 : serce, CSN
b2 : drogi oddechowe, macica, mięśnie części naczyń
Skutki stymulacji adenergicznej zależą od rodzaju receptora.
Receptory adrenergiczne – punkt uchwytu wielu leków
Receptory b aktywują cyklazę adenylową a
receptory a hamują cyklazę adenylową.
Agonistami receptorów
aR
są adrenalina i noradrenalina -
Adrenalina działa tak samo na aR i bR
Noradrenalina działa silniej na aR (ale działa też na bR)
Antagonistą receptorów aR jest fentolamina (słabo wiąże też b)
Agonistą receptorów b jest isoproterenol (izoprenalina)
Wybiórczy agonista receptorów b2 – fenoterol, salbutamol
Antagonistą receptorów b jest propranolol (selekt. bloker b1 są metoprolol, atenolol)
Inne różnice w dystrybucji receptorów adrenergicznych:
a1 : mięśniówka większości naczyń krwion, mięsień rozszerzający źrenicę, mięśnie pilomotoryczne
a2 : CSN, płytki krwi, zakończenia obwodowych nerwów adrenergicznych
b1 : serce, CSN
b2 : drogi oddechowe, macica, mięśnie części naczyń

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
28/113
Receptory dopaminergiczne
80% receptorów dopaminergicznych w mózgu znajduje się w obrębie
corpus striatum, które otrzymuje główna impulsację z
substantia nigra.
Poza tym receptory
dopaminergiczne znajdują się w korze mózgowej.
Wyróżnia się 5 podtypów receptorów dopaminergicznych (DA1-DA5)
D1 i D5 aktywuja cyklazę adenylową
a
D2
, D3 i D4 hamują cyklazę adenylową.
Receptory dopaminergiczne wiążą (ale mało swoiście) różne leki jak bromokryptyna, haloperidol,
klozapina
Receptory GABAB
Obecne są w całym CSN, niekiedy kolokalizując z jonotropowymi receptorami
GABA (GABAA).
Mają pokrewieństwo do mGluR.
Są zarówno pre jak i postsynaptyczne.
Receptory postsynaptyczne
GABAB: działanie
hamujące
- wywołują tzw. „slow inhibitory postsynaptic
potential” (powolna hyperpolaryzacja poprzez aktywację przewodnictwa potasowego K+)
Receptory presynaptyczne
GABAB są elementem mechanizmu autorecepcyjnego hamującego uwalnianie
N-T poprzez aktywację kanałów K+ i hamowanie prądu wapniowego
Mogą niekiedy modulować kanały K+ bez pośrednictwa białka G.
Agonistą
GABAB jest lek baclofen.
Metabotropowe receptory serotoninowe: 5-HT(1,2,4,5)
(Uwaga! 5-HT3 jest jonotropowy)W mózgu: jądro szwu (n.raphae) w pniu mózgowym
Dzielą się na podtypy 5-HT1 – 5-HT5 (5-HT3 jest jonotropowym)
Receptory są sprzężone z cyklazą adenylową (pobudzając ją lub hamując)
5-HT bierze udział w modulacji rytmów
dobowych,
jedzenia, podwyższa ciśnienie krwi.
Metabotropowe receptory purynergiczne
Wiążą ATP, inne analogi nykleotydów i adenozynę
(adenozyna w przeciwieństwie do ATP nie jest obecna w pęcherzykach synaptycznych dlatego jest
„nieklasycznym” N-T, adenozyna akumuluje się, w stanach nadmiernego zużycia ATP i niewystarczającej
regeneracji ATP, jednocześnie adenozyna przenika przez błony komórkowe i dlatego łatwo
rozprzestrzenia się, stąd może nieść sygnał komunikujący o metabolicznym statusie neuronów do
komórek „sąsiedztwa”.)
Receptory adenozynowe oznacza się nazwami: A1, A2a, A2b, A3
Receptory ATP oznacza się literą: P2(x,y,z,t,u) (ale z nich P2x i P2z są jonotropowe)
Aktywacja receptora A1 (licznego w mózgu) hamuje cyklazę adenylową i powoduje wzrost fosfolipazy C.
Glutamatergiczne receptory metabotropowe
Receptory zwi
ą
zane nie z kanałem jonowym,
ale z białkiem G,
fosfolipaz
ą
C
i systemem wtórnych przeka
ź
ników (trifosfoinozytol IP3 ,
diacyloglicerol DAG), obecne s
ą
we wszystkich strukturach mózgu; wyst
ę
puj
ą
te
ż
pozamózgowo w anatomicznym ukł.nerwowym, w sercu, jelitach, ko
ś
ciach; moduluj
ą
aktywno
ść
neuronów i reguluj
ą
uwalnianie GLUW przeciwie
ń
stwie do innych
metabotropowych receptorów miejsce wi
ążą
ce agonist
ę
znajduje si
ę
nie „wewn
ą
trz” w obr
ę
bie
rejonu transbłonowego ale
na zewn
ą
trz błony
komórkowej.
Ponadto mGluR tworz
ą
dimery (inne receptory metabotropowe s
ą
monomerami).
Dziel
ą
si
ę
na grupy:
Grupa I
Zwi
ą
zane z aktywacj
ą
fosfolipazy C (wzrost IP
3
i DAG),
działaj
ą
aktywuj
ą
co
Receptory grupy I
s
ą
zlokalizowane postsynaptycznie (obwodowe cz
ęś
ci synaps),
Grupa II
Zwi
ą
zane z zahamowaniem cyklazy adenylowej (spadek poziomu cAMP),
działaj
ą
hamuj
ą
co,
receptory z grupy II i III s
ą
głównie zlokalizowane presynaptycznie (udział w regulacji syntezy
i hamowaniu uwalniania glutaminianu?)
Aktywacja receptorów prowadzi tak
ż
e do zahamowania kanałów Ca
2+
Grupa III Mechanizm działania podobny do grupy II, zlokalizowane bli
ż
ej centrum synapsy ni
ż
recept.gr.II,
Receptory peptydowe (dla neuropeptydów)
Bardzo (bardzo...) liczna rodzina – ale żadne nie są
kanałami jonowymi
Albo są typu „metabotropowego” – (GPCRs) albo są sprzęgnięte z kinazą tyrozynową

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
29/113
Rola wapnia w neurotransmisji
Ca2+ jest przeka
ź
nikiem „orkiestruj
ą
cym”
M.in. Calmodulina do aktywacji cz
ęś
ci enzymów wymaga podwy
ż
szonego Ca2+
Ca2+-Kalmodulinozale
ż
na kinaza białkowa: Wielofunkcyjna kinaza białkowa
(CaMKII)
dekoduje wszelkie sygnały które podwy
ż
szaj
ą
poziom Ca2+
CaMKII fosforyluje resztyn
Ser/Tre i jest złożona prawdopodobnie z 14 podjednostek z których każda posiada domenę katalizującą i
regulatorową
CaMKII fosforyluje hydrolaz
ę
tyrozyny, MAP-2, synapsin, kanały wapniowe, receptory
glutaminianowe, Ca2+-ATPase, czynniki transkrypcyjne,
-Kinaza CaMII jest aktywowana wapniem niezale
ż
nie od jego
ź
ródła
-Jest szczególnie obfita w neuronach (nawet 2% wszystkich protein w hipokampie tj. 50x wi
ę
cej
ni
ż
w innych tkankach )
-Składa si
ę
z 12 podjednostek, „kotwiczona” przez m.in..AKAP79 (A kinase anchoring protein);
podobnie jak PKC i PKA fosforyluje reszty Ser/Thr
-
-Autofosforylacja
jest jedn
ą
z najistotniejszych cech CaMII. Powoduje 400x wzrost
powinowactwa do kalmoduliny i w efekcie aktywno
ść
CaMII trwa wiele sekund po spadku
poziomu wapnia !
Korzy
ś
ci zwi
ą
zane z sygnalizacj
ą
poprzez receptory sprz
ęż
one z białkiem G (w porównaniu z
szybk
ą
transmisj
ą
)
1.Amplifikacja sygnału
1.Nawet rzędu wielu tysięcy razy poprzez aktywację enzymów
2.Duży zakres czasowy
- Stosunkowo szybkie działanie poprzez modyfikację kanałów jonowych
- Wydłużone działanie gdy sygnał przenoszony jest na przekaźniki wtórne (od kilkuset msek w
sygnalizacji węchu z cAMP i IP3) do sekund lub minut
3.Duży zakres przestrzenny
Częściowo na skutek wydłużonego działania modulacja może dotyczyć odległych w stosunku do
receptora procesów komórkowych (IP3 , DAG, mogą wpływać m.in. na ekspresję genów)
4.„Cross talk”
- Składniki transdukcji sygnału i ich enzymatyczne efektory (np. kinazy) oddziałuja wzajemnie na
siebie
5.Skoordynowana modulacja („orkiestracja” różnych procesów)
PKA, PKC, CaMKII s
ą
tzw. „ kinazami kognitywnymi”
-PKA, PKC, CaMKII podlegaj
ą
trwałym zmianom aktywno
ś
ci nie ust
ę
puj
ą
cym nawet po zanikni
ę
ciu stymuluj
ą
cego je sygnału
(wtórnego przeka
ź
nika)
-Kinazy te moduluj
ą
aktywno
ść
synaptyczn
ą
-
Po długiej stymulacji np. serotoninergicznej aktywne pojednostki C dostaj
ą
si
ę
do j
ą
dra gdzie stymuluj
ą
syntez
ę
genów proteinaz (fosforylacja CREB) dla
podjednostki R co powoduje trwałe wydłu
ż
enie aktywno
ś
ci PKA
Fosfatazy proteinowe wykonuj
ą
„robot
ę
” przeciwn
ą
do kinaz
Enzymy (odwrotnie ni
ż
kinazy ale
w tych samych resztach) defosforyluj
ą
(hydrolizuj
ą
estrowe wi
ą
zanie) ufosforylowane reszty
Ser/Thr albo Tyr (albo wszystkich wymienionych)
•Fosfatazy podobnie jak kinazy kontroluj
ą
procesy metaboliczne, neurotransmisji, ekspresji
genów, plastyczno
ś
ci, wzrostu etc.
•Spo
ś
ród fosfataz tylko kalcineuryna (PP-2B) odpowiada bezpo
ś
rednio na wtórny przeka
ź
nik
(wzrost Ca2+).
•Kalcineuryna (PP-2B) jest zale
ż
na od Ca2+kalmoduliny i jest obfita w mózgu ale zakres
działalno
ś
ci PP-2B jest w znacznej cz
ęś
ci odmienny od równie
ż
Ca2+kalmodulinozale
ż
nej
kinazy CaMKII.
Dlatego sygnał wapniowy nie powoduje bezowocnego fosfo i
defosforylowania.!!!
•Fosfatazy fosfotyrozynowe („anty-kinazy” tyrozynowe) stanowi
ą
osobn
ą
klas
ę
fosfataz,
niektóre z nich defosforyluj
ą
równie
ż
Ser/Thr; mog
ą
by
ć
zarówno zakotwiczone w błonie
komórkowej (maj
ą
wtedy domeny pozakomórkowe umo
ż
liwiajace „odbieranie sygnałów” z
zewn
ą
trz) jak te
ż
rozpuszczone w cytozolu.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
30/113
Kinazy i fosfatazy proteinowe oraz ich substraty stanowi
ą
rodzaj zintegrowanej sieci.
Pomi
ę
dzy kinazami i fosfatazami trwa rodzaj dialogu („cross-talk”)
Modulacja funkcji neuronu przez kinazy i fosfatazy
Kinazy i fosfatazy modyfikuj
ą
1/5 wszystkich
białek
Kinazy katalizuj
ą
transfer
g
fosforanowej grupy ATP na grupy –OH reszt Ser, Thr (treonina) lub
Tyr w specyficznych lokalizacjach białek. Wi
ę
kszo
ść
fosforyluje albo Ser/Thr albo Tyr.
Fosfatazy hydrolizuj
ą
grupy fosforylowe z Ser/Thr lub Tyr lub ze wszystkich.
Fosfatazy i kinazy proteinowe maj
ą
wzajemnie odwrotne działanie.
Skutki fosforylacji
Zmiana aktywności katalitycznej enzymów
Zmiana powinowactwa enzymów do substratów lub kofaktorów
Modyfikuje interakcje między fosfoproteinami i DNA, fosfolipidami, innymi proteinami
Reguluje desensytyzacje receptorów ich wiązanie z innymi molekułami
Zmiana charakterystyk kanałów jonowych
Modyfikacja kinaz może redukować ich wymagania odnośnie wtórnego przekaźnika (znaczenie w
plastyczności neuronalnej)
Sygnalizacyjne układy wewn
ą
trzkomórkowe PODSUMOWANIE
Sygnalizacja poprzez receptory
sprz
ęż
one z kanałami jonowymi bramkowanymi ligandem
Sygnalizacja poprzez receptory sprz
ęż
one z kinazami tyrozynowymi (gł. dla czynników wzrostu
i troficznych)
Sygnalizacja poprzez receptory sprz
ęż
one z białkiem G (najliczniejsze odmiany receptorów)
Sygnalizacja poprzez receptory wewn
ą
trzkomórkowe
Modulacja funkcji neuronu poprzez kinazy proteinowe i fosfatazy
Sygnalizacja wł
ą
czaj
ą
ca i reguluj
ą
ca ekspresj
ę
genów

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
31/113
Neurotransmisja i geny, ekscytotoksycznosc
A.
Na styku neurotransmisji i ekspresji genów
Sygnalizacja wpływaj
ą
ca na ekspresj
ę
genów
•Sygnalizacja m-komórkowa nie tylko ogranicza si
ę
do regulacji funkcji białek istniej
ą
cych w
danym momencie w komórce, ale równie
ż
mo
ż
e prowadzi
ć
do regulacji syntezy białek poprzez
ekspresj
ę
odpowiednich genów.
•Wydaje si
ę
,
ż
e fosforylacja (i defosforylacja) białek oraz regulacja ich ekspresji (genetyczna)
s
ą
najwa
ż
niejszymi czynnikami le
żą
cymi u podstaw plastyczno
ś
ci neuronalnej
•Zmiany ekspresji genów prowadza do długotrwałych zmian funkcji neuronów
Sygnalizacja wpływaj
ą
ca na ekspresj
ę
genów
•Aktywatory transkrypcji (np..CREB =cAMP response element-building protein)
•mog
ą
mie
ć
poło
ż
enie odległe od podstawowego aparatu transkrypcyjnego
•Aktywator transkrypcji CREB
po fosforylacji
(np. przez szlaki PKA lub ras i inne) tworzy
poł
ą
czenie z aparatem transkrypcyjnym przez po
ś
rednictwo białka CBP (CREB-binding
protein). CBP jest form
ą
tzw. białka adapterowego o cechach acetylotransferazy histonów
(„luzuj
ą
cej” ich poł
ą
czenie z DNA; inne białka adapterowe maja funkcj
ę
deacetylazy histonów
hamuj
ą
cej transkrypcj
ę
; s
ą
te
ż
metyltransferazy i demetylazy histonowe o podobnych
efektach)
Białko CREB nale
ż
y do licznej rodziny spokrewnionych białek.
Białek spokrewnionych z tzw. „activating transcription factors” (ATFs) oraz „CRE-
modulators” (CREMs)
•Rozsiane sekwencje regulatorowe genu rozpoznawane s
ą
przez z reguły multimeryczne
czynniki transkrypcyjne (tworz
ą
ce kompleksy np. poprzez tzw. „leucine-zipper”)
•Czynniki transkrypcyjne odległe (na nici DNA w relacji do basal promoter) kontaktuj
ą
si
ę
z
„basal promoter” poprzez po
ś
rednicz
ą
ce białka adaptorowe (np. CBP) , które cz
ę
sto pełni
ą
funkcje enzymatyczne modyfikuj
ą
ce histony
•Czynniki transkrypcyjne mog
ą
by
ć
:
–Takie jak np. CREB -trwale zwi
ą
zane z regulacyjnymi cis-elementami DNA (tzw. cAMP
response elements = CRE obecnymi w wielu genach)
CREB jest aktywowany przez aktywn
ą
podjednostk
ę
PKA
, która w tym celu musi wnikn
ąć
do j
ą
dra.
CREB jest aktywowane je
ś
li jest ufosforylowane na Ser-133 Tylko ufosforylowane CREB
rekrutuje CBP (acetylotransferaza histonów)
•Inne czynniki transkrypcyjne (poza CREB):
–Takie jak STATs „signal tranducers and activators” (np. dla cytokin), fosforylowane przez
receptorowe kinazy Trk aby dosta
ć
si
ę
do j
ą
dra i ł
ą
czy
ć
z DNA
–Takie jak np. NF
k
B jest wyj
ś
ciowo w cytoplazmie zwi
ą
zany z I
k
B. I
k
B gdy ulegnie fosforylacji
uwalnia NF
k
B i umo
ż
liwia jego wej
ś
cie do j
ą
dra
Sygnalizacja wpływaj
ą
ca na ekspresj
ę
genów
•CREB mo
ż
e by
ć
ufosforylowane nie tylko przez
aktywna podjednostk
ę
PKA ale te
ż
inne kinazy: CaMII, CaMIV oraz kinaz
ę
RSK2
Je
ś
li
ż
adna z tych kinaz z osobna nie daje wystarczaj
ą
co silnego sygnału wymagana jest
KONWERGENCJA wszystkich lub wielu z nich (wymagany jest
sygnał koincydencji
)
Rola fosforylacji CREB w innych resztach ni
ż
Ser-133 nie jest poznana ale nie wykluczone,
ż
e
mo
ż
e by
ć
wykorzystywana do bardziej precyzyjniejszej regulacji ??.
•Geny aktywowane przez CREB:
–c-fos
–BDNF
–Hydroksylaza tyrozyny
–Neuropeptydy (np. enkefalina, CRH)
• CREB odgrywa rolę (poprzez aktywowane geny) w długotrwałej plastyczności synaptycznej, uczeniu, pamięci)
Geny aktywowane transkrypcyjnie w wyniku pobudzenia synaptycznego, przez leki, lub
czynniki wzrostu mo
ż
na podzieli
ć
na dwie grupy (podział nie jest do ko
ń
ca jednoznaczny):
Cellular immediate-early genes (IEGs)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
32/113
Late-response genes
-Cellular immediate-early genes (IEGs)
-Aktywowane gwałtownie szybko, w ci
ą
gu minut, a tak
ż
e przej
ś
ciowo i nie wymagaj
ą
ce
uprzedniej syntezy nowych białek. „Klasycznie” s
ą
to geny dla czynników transkypcyjnych.
(np. gen c-fos dla białka c-Fos)
-Tych genów (białek przez nie kodowanych) zacz
ę
to u
ż
ywa
ć
jako
markerów (synaptycznej)
aktywno
ś
ci neuronalnej !!
-Białkowe produkty IEGs funkcjonuj
ą
ce jako czynniki tranksrypcyjne wi
ążą
si
ę
z elementami
cis-regulatorowymi genów odpowiedzi pó
ź
nej (late-response genes) aby je aktywowa
ć
lub
blokowa
ć
. Zatem IEGs mo
ż
na nazwa
ć
„trzeciorz
ę
dowymi przeka
ź
nikami”
-Late-response genes
-Geny indukowane (lub hamowane) wolniej (w ci
ą
gu godzin) i wymagaj
ą
ce syntezy nowych
białek (czynników transkrypcyjnych)
C-Fos – marker aktywności neuronów
•c-Fos nazwano w zwi
ą
zku z homologi
ą
do białka
wirusa (FBR osteogenic sarcoma virus).
•c-Fos jest produktem genu c-fos (jednego z ogromnej rodziny czynników transkrypcyjnych
posiadaj
ą
cych tzw. Leucine zipper pełni
ą
cy rol
ę
w ich dimeryzacji z innymi białkami zwł. z
rodziny Jun, które równie
ż
razem z Fos s
ą
tzw. immediately-early genes)
•Białka c-Fos ł
ą
cz
ą
c si
ę
ze specyficznymi sekwencjami DNA moduluj
ą
ekspresj
ę
genów
(„ekspresjonowanych” pó
ź
niej)
•Poniewa
ż
wiele neuronów wykazuje ekspresj
ę
c-Fos tylko po stymulacji synaptycznej białko
c-Fos lub jego mRNA mo
ż
e by
ć
u
ż
ywany jako
marker aktywno
ś
ci synaptycznej np.
immunohistochemicznie
•Dzi
ę
ki temu mo
ż
na ocenia
ć
które neurony były aktywne po stymulacji np. okre
ś
lonym lekiem
czy innym bod
ź
cem.
UWAGA
-Wiele białek neuronalnych jest produkowana w wyniku bezpo
ś
redniej indukcji bez u
ż
ywania
„genów natychmiastowo-wczesnych” (IEGs).
Np. geny koduj
ą
ce neuropeptydy (proenkefalina, prodynorfina, niektóre czynniki
neurotroficzne) s
ą
aktywowane w odpowiedzi na depolaryzacj
ę
, lub cAMP poprzez fosforylacj
ę
konstytutywnie ekspresjonowanego CREB
Induktory ekspresji genów w układzie nerwowym:
-Podział na czynniki wzrostu, troficzne,
cytokiny jest cz
ę
sto arbitralny natomiast sygnałowe mechanizmy
wewn
ą
trzkomórkowe
mog
ą
by
ć
u
ż
yteczne w podziale
zewn
ą
trzkomórkowych
ró
ż
nych czynników sygnalizacyjnych
-Czynniki działaj
ą
ce poprzez receptorow
ą
kinaz
ę
tyrozynow
ą
-Czynniki wzrostu, brain derived neurotrophic factor – (BDNF), neurotrofina-3 (NT-3), epidermal
growth factor (EGF), fibroblast growth f. (FGF)
-Czynniki działaj
ą
ce poprzez niereceptorowe kinazy tyrozynowe
: Cytokiny: leukemia inhibitory
factor (LIF),
-Ciliary neurotrophic factor (CNTF), Interleukina-6 (IL-6)
-Receptory tych („non-Trk zale
ż
nych”) czynników zawieraj
ą
podjednostk
ę
przekazuj
ą
c
ą
sygnał
gp-130 i współdziałaj
ą
z niereceptorowymi kinazami z rodziny „Janus kinase” (Jak) takimi jak
Jak1, Jak2, Tyk2. Kinazy te fosforyluj
ą
jedno lub wi
ę
cej białek typu STAT. Dimery STAT
przechodz
ą
do j
ą
dra gdzie rozpoznaj
ą
„cytokine response elements” Ró
ż
ne białka z grupy
STATs swoi
ś
cie („preferencyjnie”) aktywowane s
ą
przez ró
ż
ne receptory cytokinowe.
-Hormony steroidowe
(gluko i mineralokort., estrogen, testosteron) przenikaj
ą
do wn
ę
trza
komórki i ich receptorami s
ą
same czynniki transkrypcyjne, które po aktywacji przez hormon
przechodz
ą
do j
ą
dra i tam rozpoznaj
ą
„cognate response elements” wzmagaj
ą
c lub hamuj
ą
c
odpowiednie geny

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
33/113
Energetyka kr
ąż
enie obrazowanie
„Energetyka” mózgu•
W jaki sposób potrzeby funkcjonalne aktywności mózgu są ściśle związane z aktywnością metaboliczno-
energetyczną
•Rola kooperacji neuronalno-glejowej
•Glukoza jest głównym „paliwem” dla „siłowni” komórek mózgowych
•Waga mózgu to 2% ciała* ale zużycie glukozy sięga 25% ! (zużycie tlenu nieznacznie mniej)
•W mózgu glukoza niemal w 100% jest utleniana do CO2 i wody poprzez glikolizę, cykla Krebsa i oksydatywną
fosforylację, które to procesy „wyciskają” z mola glukozy 36 moli ATP (38-2)
Tzw współczynnik mózg/ciało:
U człowieka = 0.02 (zużycie tlenu 25%)
Zużycie glukozy w mózgu
Różnica poziomu glukozy między krwią tętniczą i żylną gluc(A-V) wynosi
0,55 mmol/l (= 0,55
µµµµ
mol/ml)
(Ide K. I wsp.
J Appl Physiol 87: 1604-1608, 1999;
Przepływ krwi wynosi
0,55 ml/g/min
Zatem zużycie glukozy na min na gram wynosi:
0,55 mmol/ml x 0,55 ml/g/min =
0,3025 mmol/g/min
(czyli ok. 30 mmol/100g/min)
Jeśli mózg waży 1200g (a doba = 1440 min) to na dobę mózg zużywa
0,3028 mmol/g/min x 1200 x 1440 = 522720 mmol (= 0,52 mola)
1 mol = 180 g a zatem odpowiada to ok.
94 g glukozy/mózg/doba
(standardowa dieta dorosłego wynosi 2500 kcal tj równoważnik 625 g glukozy)
Mózg dorosłego mężczyzny – ok. 1350 g, kobiety – 1200g
C6H12O6
+
6O2
6CO2
+
6H2O
+ energia (z 0,5 mola glukozy jest 3 mole CO
2
)
3mole x 25,4 litra/mol gazu = 76,2 litra CO2 (w temp. 36 st C) Tyle dwutlenku węgla powstanie
Wyznaczanie lokalnego zużycia glukozy:
2-DG (deoksygukoza) łatwo przenika przez BBB (ten sam transporter jak dla glukozy) i tak samo jest
fosforylowana przez heksokinazę ale dalej już nie włącza się w szlak glikolizy i jest akumulowana w
komórkach. Tzw.
local cerebral metabolic rates
glukozy (LCMRglu) może być u zwierząt wyznaczany
autoradiograficznie po uprzednim podaniu dożylnym (18F-labeled 2-DG)
Wyznaczone
u szczurów
za pomocą autoradiografii stopnie zużycia glukozy (poprzez 2-DG) wahają się od
50 do 150 mmol/100g/1min
.
Na to samo, poprzez użycie (18F-labeled 2-DG) pozwala
przyżyciowo
u ludzi i zwierząt technika PET,
Glukoza jest poprzez
glikolizę
oraz cykl Krebsa podstawowym „paliwem” dla mózgu
Jednak w warunkach ketogennych takich jak:
cukrzyca, głodzenie i karmienie piersią
ciała ketonowe również mogą dostarczać energii
Fosfofruktokinaza
–
główny enzym limituj
ą
cy glikoliz
ę
jest hamowana przez ATP !
Spadek ATP stymuluje enzym i jednocze
ś
nie glikoliz
ę
Metabolizm glukozy w mózgu jest podobny jak w innych narządach i obejmuje trzy podstawowe szlaki:
Glikoliza
Cykl kwasów trójkarboksylowych
GLIKOLIZA: szlak od glukozy do pirogronianu daje netto 2 ATP z 1 glukozy
Gdy glikoliza „produkuje” nadmiar pirogronianu i nie jest on efektywnie utleniany w cyklu Krebsa
(konsumpcja tlenu niewspółmierna do utylizacji glukozy) wtedy powstaje nadmiar kwasu mlekowego. Jest
to sytuacja w czasie aktywacji kory i jest podobna do mięśni w czasie wysiłku.
Dehydrogenaza kwasu mlekowego (LDH) zamienia mleczan w pirogronian
Wrodzona kwasica mleczanowa (zaburzenia przemiany węglowodanów) występuje w:
•Niedobór
karboksylazy kwasu pirogronowego
•Niedobór kompleksu dehydrogenazy kwasu pirogronowego (PDHC)
•Niedobór enzymów łańcucha oddechowego (szczególnie kompleksu I i IV)
•Pojęcie kwasicy mleczanowej łączy encefalopatie mitochondrialne i niemitochondrialne zaburzenia
metaboliczne przemiany glukozy - zmiany neuropatologiczne w tych zespołach (congenital lactic acidosis,
Leigh’s s., Kearns-Sayre s., MELAS, MERRF) wykazują znaczne podobieństwa (zob. dalej)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
34/113
Całkowite spalanie glukozy w warunkach aerobowych
C6H12O6
+
6O2
daje 6CO2
+
6H2O
+ energia
W warunkach aerobowych dehydrogenaza pirogronianu dekarboksyluje pirogronian do
Acetylo-CoA, który nast
ę
pnie ł
ą
czy si
ę
ze szczawiooctanem tworz
ą
c cytrynian rozpoczynaj
ą
cy
cykl Krebsa.
Oksydatywna fosforylacja daje 18 ATP z jednego mleczanu (pirogronianiu)
Zamiana mleczanu w pirogronian nie wymaga ATP i dlatego
mleczan jest
dogodniejszym „paliwem” dla neuronów niż glukoza.
Również przemiana pirogronianu
w Glutaminian nie wymaga zużycia tlenu.
ATP i NADPH
są podstawowymi nośnikami energii dla mózgu
BILANS spalania glukozy
C6H12O6
+
6O2
daje w wyniku 6CO2
+
6H2O + energia
Przy zało
ż
eniu pełnego „spalania” 1 cz
ą
steczki glukozy otrzymujemy:
36 ATP
(z 36 ATP tylko 2 ATP z glikolizy, reszta z oksydatywnej fosforylacji)
Zużycie glukozy w mózgu:
ok. 30 mikromola/100g/min
Przy zało
ż
eniu pełnego „spalania”
Na 6 moli tlenu przypada 1 mol glukozy
I rzeczywi
ś
cie dla mózgu współczynnik poboru (zu
ż
ycia) O2/glukoza wynosi ok.
6/1
.
Wysiłek powoduje spadek tego współczynnika a jeszcze bardziej współczynnika poboru dla
w
ę
glowodanów O2/(glukoza +1/2mleczanu) bo dodatkowo jest wykorzystywany mleczan z krwi.
Ponadto je
ś
li dobrze obliczy
ć
to współczynnik poboru tlen/glukoza jest nieco ni
ż
szy ni
ż
6/1
(zob. „afera”)
Współczynnik poboru (zużycia) O2/glukoza wynosi ok.6/1. Wysiłek powoduje spadek tego współczynnika
a jeszcze bardziej współczynnika poboru dla węglowodanów O2/(glukoza +1/2mleczanu) bo dodatkowo
jest wykorzystywany mleczan z krwi.
Zu
ż
ycie tlenu przez mózg wynosi:
160
m
ikromol tlenu/100g tkanki mózgu/1min
(niektóre wyliczenia mog
ą
da
ć
nieznacznie odmienne wyniki) np.
Przepływ krwi w mózgu (CBF) u dorosłego wynosi
55
- 57ml/100g tkanki/1min
Ró
ż
nica zawarto
ś
ci tlenu mi
ę
dzy krwi
ą
t
ę
tnicz
ą
i
ż
yln
ą
(z opuszki
ż
yły szyjnej) wynosi:
[AO2-VO2] = 3,1mikromol/ml
Zatem zu
ż
ycie tlenu wyniesie
[AO2-VO2] X przepływ[AO2-VO2] 3,1
m
ikromol/ml x
0,55
ml/g/min = 1,705
m
ikromol/g/min]
1,705
m
ikromol/g/min = 170,5
m
ikromol/100g/min (to jest nieco wi
ę
cej ni
ż
160
m
ikromol)
Nie mniej niezale
ż
nie od sposobów obliczania w przybli
ż
eniu mózg zu
ż
ywa ok. 20% całego tlenu
zu
ż
ywanego przez cały organizm
i w przybli
ż
eniu tyle samo produkuje CO2 (tzw współczynnik
oddechowy = 1)
ALE UWAGA !!!
Mózg zużywa glukozy:
30 mikromol glukozy / 100g tkanki / 1min
W przypadku współczynnika oddechowego = 1 obliczenie stechiometryczne wskazuje, że
6mmol O2 potrzeba na utlenienie 1mmol glukozy. A zatem jeśli 160
mikro
moli tlenu
podzielimy przez 6 otrzymamy 26,7 (a więc wyraźnie mniej niż obliczone
eksperymentalnie przy pomocy pomiaru glukozy w krwi tętniczej tętnic szyjnych i żylnej
w opuszce żyły szyjnej) zużycie glukozy 30
mikro
mol/100g/min !)
Na co więc idzie te „ekstra” 3,3 mikromola glukozy? (wyjaśnienie „afery cukrowej”)
1)Część (stosunkowo niewielka co prawda) glukozy jest „spalana” tylko do kwasu mlekowego (a zatem
bez konsumpcji tlenu)
2)Nieco glukozy jest „magazynowane” w postaci glikogenu
3)glukoza jest konieczna dla konstrukcji makromolekuł takich jak: glikolipidy, glikoproteiny
4)Glukoza jest konieczna do wytwarzania 3 kluczowych neurotransmiterów : Glu, GABA, ACh.
Metabolizm energetyczny jest w mózgu wyraźnie porozdzielany („compartmentacja”):
b-oksydacja wolnych kwasów tłuszczowych odbywa się
tylko w astrocytach

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
35/113
Oprócz glukozy jedynym potencjalnym pełnym „zamiennikiem” glukozy jest
mannoza
, która przenika
przez BBB i poprzez 2 reakcje jest przekształcana do fruktozo-6-fosforanu (elementu glikolizy). Jednak
mannoza nie jest normalnie obecna we krwi.
Najnowsze obserwacje wskazują, że mleczan wbrew poprzednim mniemaniom może przedostawać się
przez BBB.
Zarówno mleczan jak i pirogronian mogą być preferowanymi substratami energetycznymi dla aktywnych
neuronów
Oprócz glukozy
ciała ketonowe
(acetooctan, D-3-hydroksymleczan) stanowią istotne
„paliwo” w przypadku karmionych piersią osesków
(mleko ma 55% tłuszczu w porównaniu z 30-35% w diecie po okresie karmienia piersią,
więc jest to także rodzaj adaptacji do diety).
Ciałą ketonowe są też zarówno dostarczycielami energii jak i
prekursorami dla
lipogenezy
, bardzo istotnej w okresie formowania się mieliny!
Ale w wieku niemowlęcym nawet glukoza może być metabolizowana do substratów dla
lipogenezy (mielinizacja!).
Także wolne kwasy tłuszczowe poprzez acetyl-CoA mogą służyć do produkcji ATP
Również
głodzenie (i cukrzyca)
powoduje wzrost użycia ketonów, których poziomy są
podwyższone z powodu katabolizmu lipidów jako dostarczycieli energii.
Oprócz ATP drugim ważnym związkiem energetycznym jest NADPH (zredukowany
nicotinamide adenine dinucleotide phosphate) wytwarzany z glukozy w cyklu
pentozowym
Cykl pentozowy: produkuje NADPH
Jeśli potrzeba silnych związków redukujących np. do wytwarzania kwasów tłuszczowych
z acetylo-CoA lub produkcji mieliny spadek NADPH aktywuje cykl pentozowy.
NADPH jest też konieczny do usuwania ROS
NADPH (produkowane w cyklu pentozowym) jest konieczny dla usuwania ROS (powstałych np. w
oksydatywnej fosforylacji, w wyniku działania enzymów jak: hydroksylaza tyrozyny, NOS,
lipooksygenaza, cyklooksygenaza)
Zesp. Wernickego-Korskakoff’a
– patogeneza zwi
ą
zana z elementem cyklu pentozowego
(transketolaza)
Zesp W-K dotyczy alkoholików; objawy: zab. pami
ę
ci, zab.chodu i mi
ęś
ni okulomotorycznych.
Zesp W-K wynika ze zmniejszonej aktywno
ś
ci transketolazy i braku vit B1 (pirofosforan tiaminy
jest kofaktorem transketolazy)
U osób wra
ż
liwych tiamina 10x słabiej ł
ą
czy si
ę
z enzymem i je
ś
li s
ą
alkoholikami lub
przewlekle niedo
ż
ywionymi powstaje zespół Korsakoff’a
Mleczan i pirogronian
pozwalają na utrzymanie aktywności neuronalnej w izolowanym
skrawku mózgu (pozbawionym krążenia !). To one są preferowanymi substratami dla
produkcji energii w neuronach !
Jednak ich przechodzenie przez BBB choć w świetle najnowszych badań możliwe jest
prawdopodobnie niewielkie?
(Wykryto też transportery dla kwasów monokarboksylowych w kapilarach mózgu!)
Mózg w znacznej mierze „pracuje lokalnie”. Logiczne jest zatem, że posiada mechanizmy
lokalnie regulujące dopływ substratów energetycznych, a więc
regulujące przepływ krwi
!
Zobacz DODATEK
Badania przy pomocy PET (Zobacz DODATEK) z jednoczesnym obserwowaniem Local Cerebral
Metabolic Rate dla tlenu (
konsumpcja tlenu
), oraz dla
glukozy
a także
przepływu
krwi pozwoliły na ocenę
ich wzajemnych relacji. Stwierdzono, że w ludzkim mózgu
te trzy parametry metaboliczne są ze sobą
związane
(czyli wzrastają jednocześnie i proporcjonalnie w czasie aktywacji mózgu) ale w różnych
okolicach stopień korelacji może być różny!
Mózg w sytuacji zwiększonego zapotrzebowania na energię używa przede wszystkim
glikolizy a później oksydatywnej fosforylacji.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
36/113
Na co mózg zu
ż
ywa energi
ę
?
Najbardziej konsumującym energię procesem w mózgu
(ok. 50% całej
energii z PODSTAWOWEJ oksydacji glukozy, czyli w stanie nieaktywnym neuronów)
jest
utrzymanie
gradientów jonowych
pomiędzy błonami cytoplazmatycznymi.
Energię (ATP) zużywają pompy a szczególnie Na+K+ATPase
Attwell i Laughin (2001) obliczyli
wydatek energetyczny na transmisje synaptyczną
:
Koszty wychwytu i recyklingu Glu wynoszą 2,67 ATP/molekułę Glu.
Ponieważ pęcherzyk zawiera 4000 Glu zatem
koszt recyklingu Glu na 1 pęcherzyk wynosi
blisko ok. 1,1 x 104 ATP (11000)
Odnowienie gradientów jonowych po impulsie Glu i aktywacji NMDAR i non-NMDAR
wynosi ok. 1,4 x 105 ATP (140000 ATP) / pęcherzyk.
Suma zużycia energii w przeliczeniu na pęcherzyk synaptyczny: 1,1 x 104 + 1,4 x 105 =
1,51 x 105 ATP (151000)
Podstawowa (spoczynkowa) konsumpcja
energii
dla neuronu
wynosi 3,4 x 108 ATP/komórkę/sek
dla komórki glejowej
1 x 108 ATP/komórkę/sek
Wydatek na „aktywność”
2,8 x 109 ATP/neuron/sek
Ok. 87% energii idzie na Glu-mediowaną neurotransmisję a 13% na utrzymanie
potencjału spoczynkowego
Astrocyty i metabolizm mózgu
Potrzeby energetyczne zale
żą
od typu, wielko
ś
ci (tak
ż
e
długo
ś
ci aksonu) i obci
ąż
enia prac
ą
neuronu.
Energi
ę
dostarczaja tak
ż
e komórki gleju i endotelia.
np. stosunek ilo
ś
ci astrocytów do neuronów w przybli
ż
eniu wynosi 1:1 ale im wi
ę
kszy mózg
stosunek ten jest wy
ż
szy na korzy
ść
astrocytów. Astrocyty otaczaj
ą
c endotelia „stópkami
ss
ą
cymi” stanowi
ą
pierwsz
ą
„stacj
ę
” przeładunkow
ą
(i przetwórcz
ą
) dla glukozy. Jednocze
ś
nie
szczelnie otaczaj
ą
c synapsy i wychwytuj
ą
c N-T
s
ą
najlepszymi kandydatami do roli czujników
aktywno
ś
ci neuronalnej.
Podstawowy
poziom zużycia glukozy obliczony w hodowli mieszanej astrocytów i neuronów (Magistretti i
Pellerin 1999) wynosi:
Dla astrocytów 20 nmol/mg/min
Dla neuronów 2 nmol/mg/min
Te wartości są zbliżone do uzyskanych in vivo metodami autoradiograficznymi z 2-DG w korze mózgu
(10-20 nmol/mg/min)
Zatem …astrocyty zużywają w warunkach podstawowych znacznie więcej glukozy niż neurony !!!
Ale aktywowane astrocyty przy pomocy glutaminianu (in vivo Glutaminian aktywuje astrocyty w czasie
aktywności neuronalnej) zwiększają zużycie glukozy w sposób zależny od stężenia Glutaminianu i
zjawisko to zależy nie od receptorów Glutaminianu ale od transporterów (EAAT, grają rolę m.in. w
patogenezie ALS)
Transport Glutaminianu do astrocytów jest sprzężony z wprowadzaniem jonów Na+ (2-3 Na+ na 1
Glu)(nb. wraz z Glu do astrocytów wchodzi też woda)
Podwyższenie stężenia Na+ w astrocytach stymuluje pompę jonową (Na+K+-ATPazę). Powoduje to
spadek ATP co z kolei stymuluje enzymy glikolizy: fosfofruktokinazę i heksokinazę.
Innymi słowy: „wpuszczanie Na+ wraz z Glu do astrocyta powoduje wzrost Na+ co stymuluje
Na+K+ATPazę a ta z kolei stymuluje glikolizę.
Aktywacja NA+K+ATPazy wywołuje stymulację glikolizy?
Wykazano, że ATP jest negatywnym regulatorem fosfofruktokinazy a konsumpcja
(spadek ATP) wywołuje wzrost aktywności tego enzymu (który limituje glikolizę).
Również następuje wzrost aktywności heksokinazy (fosforylującej glukozę i 2-DG)
Na 1 Glu i 3 Na+ wchodzące do astrocyta przypada wejście 1 glukozy, produkcja 2 ATP i 2 mleczanów w
procesie glikolizy. Z tych 2 ATP jeden „idzie” na pompę Na+K+ATPazową, drugi na syntezę Gln
W ten sposób metabolizm glukozy związany jest z aktywnością neuronalną.
Glutaminian pobudza astrocyty do zwiększenia metabolizmu glukozy (czyli wtedy gdy
neurony „bardziej pracują” uwalniając Glutaminian, astrocyty zwiększają metabolizm...)
Glutaminian stymuluje glikolizę w astrocytach a także stymuluje wychwyt glukozy i fosforylację

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
37/113
pobór 1 Glutaminian z 3 Na+
1 glukoza wchodzi do astrocyta
glikoliza daje 2 ATP i 2 mleczany
mleczany przechodz
ą
do neuronu
w neuronie daj
ą
po 18 ATP
Dominuj
ą
ca przemiana Glutaminianu w astrocycie to amidacja wymagaj
ą
ca syntazy glutaminy
(GS) i ATP UWAGA! To jest te
ż
usuwanie amoniaku!
Glutamina jest oboj
ę
tna dla neurotransmisji dlatego „bezpiecznie” przenika do neuronu
Glutaminaza w mitochondriach neuronu przekształaca glutamin
ę
(Gln) w glutaminian (Glu)
(powstaje te
ż
w tej reakcji NH
4
+
)
Neuron może uzupełnić pulę Glutaminianu przez wykorzystując mleczan
Wiemy ju
ż
,
ż
e
wzmo
ż
ona aktywno
ść
neuronalna powoduje stymulacj
ę
poboru glukozy i glikolizy w
astrocytach ALE CO Z ENERGI
Ą
DLA NEURONÓW ?
Wiadomo,
ż
e neuronom w hodowli „wystarcza” mleczan i pirogronian.
Z hodowli astrocytów uwalnia si
ę
głównie mleczan, a pirogronian 10x mniej, natomiast inne
produkty glikolizy w jeszcze mniejszych wr
ę
cz
ś
ladowych ilo
ś
ciach (alfa-ketoglutaran,
cytrynian, maleate).
Wykazano,
ż
e aktywacja neuronów powoduje wzrost uwalniania mleczanu z astrocytów oraz
wzrost jego poboru przez neurony.
Krew najprawdopodobniej nie jest
ż
ródłem mleczanu dla mózgu.
Tak
ż
e spektroskopia MRJ potwierdza wzrost mleczanu w strefach aktywacji neuronów (np. w
korze wzrokowej).
MLECZAN JEST BARDZO „WYGODNYM” PALIWEM DLA NEURONÓW
Spalanie oksydatywne mleczanu wytwarza 18 ATP. Ponadto przemiana mleczanu w
pirogronian (enzym LDH lactate dehydrogenase)
nie wymaga ATP
(jak to jest w przypadku
wst
ę
pnego etapu glikolizy.
Trzeba te
ż
pami
ę
ta
ć
,
ż
e mleczan i pirogronian s
ą
substratami dla Glu ! (st
ą
d wspomniana cz
ęść
przyczyn „rozprz
ę
gni
ę
cia” konsumpcji glukozy i tlenu)
ROLA GLIKOGENU
Glikogen (akumulowany głównie w astrocytach, kk. ependymy i niektórych du
ż
ych neuronach)
jest najwi
ę
kszym energetycznym rezerwuarem dla mózgu.
W mózgu jednak w porównaniu z innymi narz
ą
dami (w
ą
troba, mi
ęś
nie) jest go 100x mniej ni
ż
w
w
ą
trobie i 10 mniej ni
ż
w mi
ęś
niach.
Jest zatem raczej „buforem” metabolicznym ni
ż
„zapasem paliwa”.
Jednak stwierdzono,
ż
e wymiana glikogenu w mózgu jest bardzo szybka i skoordynowana z
aktywno
ś
ci
ą
synaptyczn
ą
.
Magistretti i wsp. w 1993 r stwierdzili,
ż
e
w ogólnym znieczuleniu (atenuacja aktywno
ś
ci
synaptycznej) gwałtownie wzrasta poziom glikogenu w mózgu
.
Natomiast w hodowli samych astrocytów anestetyki nie daj
ą
tego efektu.
A zatem jest to efekt mediowany przez neurony (zahamowanie ich aktywno
ś
ci wzmaga zasoby
glikogenu.
Potwierdza to równie
ż
fakt,
ż
e w obszarach gdzie zniszczone s
ą
neurony i powstaje glioza, jest
ona bogata (reaktywne astrocyty) w glikogen („zahamowanie” neuronalne z powodu braku
neuronów ???).
Stwierdzono,
ż
e niektóre szlaki nerwowe stymuluj
ą
pobór energii z zapasów glikogenu (np..
stymulacja wibrysów u szczurów obni
ż
ała zapasy glikogenu w w tzw. polu „baryłkowym” kory
somatosensorycznej.
Astrocyt i neuron tworz
ą
rodzaj wspólnej „jednostki metabolicznej”
Metabolizm glukozy jest regulowany czasowo, przestrzennie i funkcjonalnie i (jak si
ę
wydaje)
ś
ci
ś
le odzwierciedla aktywno
ść
neuronaln
ą
!
Ale miejscem gdzie wzrasta metabolizm glukozy nie jest „ciało neuronu” ale raczej NEUROPIL,
gdzie s
ą
zlokalizowane pre i postsynaptyczne struktury otoczone
ś
ci
ś
le wypustkami
astrocytów.
W odpowiedzi na wyrzut glutaminianu zwi
ą
zany z aktywno
ś
ci
ą
neuronaln
ą
astrocyty uwalniaj
ą
metabolit glukozy – mleczan, który jest potrzebny neuronom jako paliwo.
Glukoza dostarcza te
ż
„w
ę
glowego szkieletu” dla odnawiania puli neurotransmitera
(Glutaminianu) a kluczow
ą
rol
ę
pełni astrocyt m.in. poniewa
ż
wył
ą
cznie on posiada

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
38/113
karboksylaz
ę
pirogronianu,
która przekształcaj
ą
c pirogronian do szczawiooctanu (tzw. reakcja
anaplerotyczna) „otwiera drog
ę
” do cyklu Krebsa.
Wskazuje to na wyspecjalizowanie funkcji
syntezy glutaminianu i innych aminokwasów w tych komórkach.
Inny selektywny dla astrocytów enzym:
syntetaza glutaminy
zamienia glutaminian na glutamin
ę
i dopiero glutamina przechodzi do neuronów gdzie (dzi
ę
ki glutaminazie) jest przetwarzana w
glutaminian (dzi
ę
ki enzymowi
glutaminazie
w mitochondriach która hydorolizuje glutamin
ę
do
glutaminianu z wytworzeniem NH
4
+
).
Glukoza dostarcza „węglowego kręgosłupa” dla glutaminianu natomiast azot dostarczany
jest przez leucynę i dzięki transaminazie leucyny (LT)
Grupa aminowa przenoszona jest na a-ketoglutaran i w rezultacie powstaje glutaminian
oraz a-ketoizocapronian (a-KIC)
UWAGA!
Dehydrogenaza
a
-ketoglutaranu -
enzym kluczowy dla losów
a
-ketoglutaranu katalizuj
ą
cy jego
konwersj
ę
do sukcynylo-CoA ma nisk
ą
aktywno
ść
u wielu chorych na Alzheimera !?
Encefalopatia w
ą
trobowa- przykład jak zaburzenie metabolizmu astrocytów
wpływa na funkcje całego mózgu
•Astrocyty łączą z neuronami ścisłe więzy
metaboliczne
•Zaburzenie funkcji astrocytów w encefalopatii wątrobowej (selektywnie dotyczące tych
komórek) powoduje szereg objawów neurologicznych i psychiatrycznych. Świadczy to
pośrednio jak bardzo zależne są neurony od astrocytów.
•Amoniak w nadmiarze wnikając do astrocytów zmusza je do detoksyfikacji (syntaza
glutaminy) na co spożytkowują zarówno na bieżąco produkowaną energię jak i zapasy
energetyczne (glikogen). Astrocyty ulegają degeneracji widocznej w mikroskopie. Stają
się one niewydolne w prawidłowym zaopatrywaniu neuronów w metabolity

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
39/113
Neuro-synapto-geneza, plastyczność
Podstawy embriogenezy układu nerwowego
Sk
ą
d si
ę
bior
ą
„dojrzałe” komórki układu nerwowego
oraz ich funkcjonalne zwi
ą
zki?
•Komórki „rodz
ą
si
ę
” i dojrzewaj
ą
poddane zarówno cechom własnym (ekspresja genów
w okre
ś
lonej
sekwencji czasowej
) jak i wpływom sygnałów zewn
ę
trznych
•Ostateczne „dojrzewanie” i „umocowanie” w społeczno
ś
ci układu nerwowego jest równie
ż
zwi
ą
zane z
migracj
ą
, dzi
ę
ki której komórki wygenerowane w ró
ż
nym czasie i z ró
ż
nymi wła
ś
ciwo
ś
ciami wchodz
ą
we wzajemne kontakty tworz
ą
c układy zdolne do kooperacji.
•Kooperacja kształtuje si
ę
poprzez wymian
ę
informacji głównie w postaci aktywno
ś
ci elektrycznej
(potencjały czynno
ś
ciowe) i neurochemicznej (transmisja synaptyczna)
Podstawy ró
ż
nicowania i rozwoju układu nerwowego
Eksperymenty Mangold’a i Spemann’a z lat 30-tych wskazały na istnienie „tkanki
organizuj
ą
cej” powoduj
ą
cej indukcj
ę
płytki i w konsekwencji układu nerwowego z ektodermy.
Liczne badania ujawniły,
ż
e ektoderma nie tyle potrzebuje indukcji aby utworzy
ć
struktury
nerwowe ale b
ę
d
ą
c „uprzednio zaprogramowana” do ró
ż
nicowania si
ę
w neuroektoderm
ę
potrzebuje jedynie raczej tylko niewielkiego bod
ź
ca.
W ko
ń
cu XX wieku ujawniono „molekularnych graczy” bior
ą
cych udział w ró
ż
nicowaniu si
ę
układu nerwowego. Obraz tych zale
ż
no
ś
ci był zaskakuj
ą
cy.
Komórki ektodermy w warunkach „normalnych” pod działaniem protein zwanych Bone
Morphogenetic Proteins (BMPs) staj
ą
si
ę
naskórkiem, natomiast BLOKOWANIE BMPs
prowadzi do „naturalnie zaprogramowanej („preprogrammed”) drogi w kierunku
neuroektodermalnym.
Podstawy ró
ż
nicowania i rozwoju układu nerwowego
(tzw.
„default model”
potwierdzony
cho
ć
nie do ko
ń
ca
m.in. w knock-out
zwierz
ę
tach)
„Nerwowy organizator” działa poprzez uwalnianie (noggin, chordin, follistatin,
cerberus, nr3) -
czynników blokuj
ą
cych białka BMP
Jednak knock-out-owe zarodki pozbawione np. noggin rozwijaj
ą
układ nerwowy cho
ć
znacznie
zredukowany..
Zapewne wi
ę
c istniej
ą
jeszcze inne czynniki blokuj
ą
ce BMP i indukuj
ą
ce układ nerwowy ??
W morfogenezie układu nerwowego graj
ą
rol
ę
równie
ż
cadherins (ok. 80 typów molekuł
adhezyjnych ł
ą
cz
ą
cych komórki posiadaj
ą
ce te same typy molekuł a wewn
ą
trzkomórkowo
zł
ą
czone ze szkieletem aktynowym*)
*Mutacje czynników reguluj
ą
cych polimeryzacje aktyny prowadz
ą
do zaburze
ń
rozwoju CSN
Ró
ż
nicowanie si
ę
komórek
Ró
ż
nicowanie si
ę
wzdłu
ż
ró
ż
nych osi opiera si
ę
o GRADIENTY ró
ż
nych molekuł
sygnalizacyjnych i detekcj
ę
tych gradientów przez komórki.
1.Neurony wykazuj
ą
ogromne ró
ż
nice fenotypowe (np. por. pr
ę
ciki siatkówki i kk Purkinjego)
2. „Zewn
ę
trzni” i „wewn
ę
trzni” (wen
ą
trzneuronalni) „kontrolerzy” ró
ż
nicowania neuronów
(=„
determinanty
”)
3. Zewn
ę
trzni „kontrolerzy” to czynniki hormonalne i czynniki wzrostu oraz czynniki
„parakrynne” (z „s
ą
siedztwa”). Działaj
ą
poprzez receptory i system wtórnych przeka
ź
ników
4. Wewn
ę
trzni kontrolerzy ró
ż
nicowania - zwłaszcza u bezkr
ę
gowców dominuje „determinative
development” neuralnych progenitorów (determinacja wewn
ę
trzna), w przeciwie
ń
stwie do nich
u kr
ę
gowców dominuje „regulowany” sposób ró
ż
nicowania.
5. „Cartesian coordinate” genes (decyduj
ą
o prawidłowym schemacie ró
ż
nicowania wzdłu
ż
osi
AP)
Determinanty wewn
ę
trzne s
ą
molekułami wytwarzanymi w komórce i przekazywanymi w
okre
ś
lony niezmienny sposób do komórek potomnych
Ró
ż
nicowanie si
ę
komórek nerwowych na przykładzie rdzenia
„floor plate” jest wtórnym
ź
ródłem „sonic hedgehog” = SHH (po strunie grzbietowej) a sama jest te
ż
indukowana przez
SHH

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
40/113
W zale
ż
no
ś
ci od st
ęż
enia SHH s
ą
wł
ą
czane lub wył
ą
czane geny szeregu tzw. homeodomain
protein (czynniki transkrypcyjne)
Tzw białka Klasy I (homeodomain protein np. Pax-6) s
ą
zablokowane
dla okre
ś
lonych st
ęż
e
ń
SHH
Białka klasy II (np. Nkx-2.2) s
ą
aktywowane
dla okre
ś
lonych st
ęż
e
ń
SHH.
Granice domen neuronalnych „zaostrza” krzy
ż
owe blokowanie si
ę
pomi
ę
dzy obiema klasami
genów (analogicznie u drozofili działa „o
ś
” ró
ż
nicowania boczno-przy
ś
rodkowa)
Podstawy ró
ż
nicowania DV (brzuszno-grzbietowego)
Decyduj
ą
sygnały ze struny grzbietowej (gradient Sonic hedgehog) (oraz BMP4 i BMP7 od
strony grzbietowej z ektodermy)
•Gdy tylko powstan
ą
szcz
ą
tkowe struktury OUN zaczyna si
ę
proces generacji ró
ż
nych populacji
komórek, który (poza szczególnymi wyj
ą
tkami) ko
ń
czy si
ę
przed urodzeniem. Potem komórki
prekursorowe zanikaj
ą
.
•Strefa okołokomorowa jest głównym obszarem generacji komórek prekursorowych (oblicza si
ę
,
ż
e
nawet 250 000 komórek powstaje w tej strefie w ka
ż
dej minucie w okresie najwi
ę
kszej fali generacji)
•W układach „warstwowych” mózgu, (kora mózgu, mó
ż
d
ż
ku, wzgórki górne) ka
ż
da warstwa jest
generowana w okre
ś
lonym czasie
•Komórki strefy okołokomorowej znakowane radioaktywn
ą
tymin
ą
w ró
ż
nych okresach
ż
ycia zarodka
zasiedlaj
ą
ró
ż
ne warstwy kory – najwcze
ś
niej jest to tymczasowa tzw. strefa subplate (z której niektóre
prze
ż
ywaj
ą
w obr
ę
bie istoty białej) oraz kom. Cajala-Retziusa (warstwa I)
Ró
ż
nicowanie kk glejowych
nast
ę
puje pó
ź
niej
ni
ż
powstawanie pierwszych „neuroblastów”.
PDGF i NT-3 wydzielane przez astrocyty stymulują proliferację prekursorów oligodendrocytów
Ró
ż
nicowanie si
ę
komórek kory mózgowej
Asymetryczne podziały komórek okołokomorowej
strefy germinalnej powoduj
ą
tworzenie si
ę
neuronów z przeznaczeniem do zasiedlenia kory
(poprzez migracj
ę
wzdłu
ż
komórek gleju promienistego)
Najwcze
ś
niej wygenerowane komórki zasiedlaj
ą
gł
ę
bsze warstwy a najpó
ź
niej wygenerowane
zasiedlaj
ą
powierzchowne
Z biegiem czasu progenitory tracą multipotencjalność w zasiedlaniu kory o czym świadczą eksperymenty
z przeszczepianiem
Migracja neuronów obwodowego SN do mózgu
Komórki LHRH (luteinizing hormone releasing
hormone) podwzgórza pochodz
ą
z układu w
ę
chowego.
S
ą
to zatem komórki (neurony) mózgu pochodz
ą
ce spoza niego!
Zaburzenie tej migracji prowadzi do tzw. zespołu Kallman’a (anosmia, hypogonadyzm,
niepłodno
ść
)
Neurogeneza i migracjaKomórki grzebieni nerwowych migruja w odległe miejsca tworz
ą
c
obwodowy układ nerwowy (w tym zwoje układu sympatycznego i parasympatyczngo,
paraganglia, zwoje czuciowe w tym tak
ż
e cz
ęś
ciowo nerwów czaszkowych, melanocyty,
komórki chromafinowe nadnerczy a tak
ż
e chrz
ę
stne elementy ko
ś
ci czaszki !). •W migracji
wa
ż
n
ą
rol
ę
odgrywa interakcja ze składnikami macierzy pozakomórkowej (ECM) takimi jak
fibronektyna, laminina , kolagen
•W poł
ą
czeniu z nimi po
ś
redniczy
receptor integrynowy
na migruj
ą
cych kk.grzebieni nerwowych
•(podanie przeciwciał przeciw temu receptorowi hamuje migracj
ę
)
Jak „
ś
ledzimy” komórki migruj
ą
ce?•1. wszczepianie komórek znakowanych 3H-tymidyn
ą
do
„nieoznakowanego” gospodarza (1963)
•2. tworzenie „chimer” poprzez wszczepianie fragmentu cewy i grzebieni z innego gatunku do innego
gatunku (gł ptaka np.. Przepiórki do zarodka kurcz
ę
cia które ró
ż
ni
ą
si
ę
obrazem chromatyny j
ą
drowej)
LeDouarin 1982
•J.w. ale komórki przepiórek s
ą
znakowane specyficznymi przeciwciałami (dla przepiórek a nie dla
kurcz
ą
t)
•Choroba Hirschprunga
jest przykładem defektu migracji kk. grzebieni n. z odcinka „bł
ę
dnego”.
•Prawdopodobnie nieprawidłowe ró
ż
nicowanie si
ę
lub uszkodzenie kom. grzebieni nerwowych
i ich pochodnych w obr
ę
bie twarzoczaszki le
żą
u podstaw zespołu Parry-Romberga
•Ró
ż
nicowanie si
ę
komórekLos komórek nerwowych zale
ż
y (równie
ż
) od tkanek docelowego
unerwienia, które wytwarzaj
ą
czynniki troficzne dla neuronów i ich aksonówNp. Zmiana typu

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
41/113
transmitera w neuronach układu sympatycznego unerwiaj
ą
cych gruczoły potowe z
noradrenaliny na acetylocholin
ę
pod wpływem dojrzewania gruczołów potowych (szczur)
Jak rosn
ą
aksony i jak znajduj
ą
drog
ę
do celu
Aksonogeneza i synaptogeneza oraz tworzenie obwodów neuronalnych
„Uzbrajanie” terenu pod budow
ę
jest niezale
ż
ne od tego co tam ma by
ć
zbudowane.
Podobnie aksony docieraj
ą
w „strefy działek budowlanych” gdy jeszcze niewiele tam si
ę
dzieje”
Ramon y Cajal po raz pierwszy u
ż
ył nazwy „sto
ż
ek wzrostu” aksonu i przypuszczał,
ż
e
wykonuje ruchy ameboidalne.
Sto
ż
ek wzrostu tworzy liczne cienki „mikrokolce” (filipodia) promieniuj
ą
ce w ró
ż
ne strony.
Wła
ś
ciwe poł
ą
czenie aksonu z jego docelow
ą
komórk
ą
(struktur
ą
) realizowane jest w sposób w
którym mo
ż
na wyró
ż
ni
ć
2 fazy. (Goodman i Shatz 1993)
Wzrost aksonu („kierunek”) jest cz
ęś
ciowo zaprogramowany
Doświadczenie z 1980r W zarodku kurczęcia segmenty T7-LS3 zostały odwrócone w fazie S15-16 (przed
wypuszczaniem aksonów) a mimo to aksony motoneuronów „kierują się” do tych samych mięśni.
Są więc jakby zaprogramowane do swych celów.
W latach 60-tych XX w Roger Sperry wysun
ą
ł hipotez
ę
„chemoaffinity” w oparciu o
eksperymenty na nerwie i układzie wzrokowym
ż
ab i ryb. Sperry zaobserwował,
ż
e nawet po
odci
ę
ciu i „przekr
ę
ceniu” oka u
ż
aby aksony komórek zwojowych siatkówki docierały do
prawdiłowego miejsca w tectum. (zob dalej o tych eksperymentach)
Wzrost aksonu kierowany jest równie
ż
przez „wskazówki” zewn
ę
trzne, które mo
ż
na podzieli
ć
na działaj
ą
ce na du
ż
e dystanse (Long-range cues) i na krótkie (short-range cues). W ka
ż
dej
klasie tych czynników s
ą
zarówno „przyci
ą
gaj
ą
ce” jak i „odpychaj
ą
ce”. Jednak te podziały w
praktyce s
ą
wybitnie „zamazane”.
Wszystkie te czynniki działa
ć
mog
ą
jednocze
ś
nie w ka
ż
dym z odcinków-etapów wzrostu
aksonu (czyli zarówno „popychanie jak i przyci
ą
ganie”).
Badania lat 90-tych umo
ż
liwiły identyfikacj
ę
4 rodzin molekuł sygnalizacyjnych bior
ą
cych
udział w nakierowywaniu aksonów:
S
ą
to :
SEMAFORYNY,
NETRYNY,
SLITS ,
EFRYNY
Ponadto inne molekuły najprawdopodobniej równie
ż
bior
ą
udział w kierowaniu wzrostem
aksonów
S
ą
to najprawdopodobniej:
CAMs (cell adhesion molecule)
składniki pozakomórkowej macierzy,
cadherins,
fosfatazy transbłonowe.
Wyznaczanie drogi dla aksonu to wspólna gra czynnika zewn
ę
trznego i stopnia nasilenia
ekspresji receptora dla tego czynnika (czynników)
Np..
Efryny
i ich receptory w poł
ą
czeniach siatkówka-tectum, oraz białka
SLIT
i ich receptory
Robo
w neuronach komisuralnych rdzenia
[Czynnik(f)] x [receptor(dla-f)] = efekt
SEMAFORYNY:
W wi
ę
kszo
ś
ci „odpychaj
ą
”
aksony ale jednoznaczna aktywno
ść
wielu z nich nie jest jeszcze
ustalona!!!. (mog
ą
zapewne równie
ż
„przyci
ą
ga
ć
” dendryty w korze co zale
ż
y od poziomu
cGMP w komórce).
7 klas u kr
ę
gowców (ogółem ok.20 białek)
Z wyj
ą
tkiem klasy 3 receptorami semaforyn s
ą
spokrewnione z nimi białka nazwane
pleksynami
Klasa 3 obejmuje semaforyny wydzielane, pozostałe s
ą
białkami transbłonowymi (dział
ą
j
ą
krótkodystansowo). Receptorami dla klasy 3 s
ą
neuropiliny
(które wi
ążą
si
ę
równie
ż
z
pleksynami)
Semaforyna 3A została odkryta w 1993 r jako substancja „odpychaj
ą
ca” w hodowli aksonów
czuciowych. Wpływa nie tylko na wzrost aksonu ale równie
ż
dendrytów.
Polleux i wsp. w 1998r stwierdzili istotna rol
ę
semaforyny 3A w pierwotnym ró
ż
nicowaniu si
ę
komórki piramidalnej kory (tj w tworzeniu aksonu, który zmierza „wgł
ą
b” i dendrytu, który
kieruje si
ę
ku pia mater). Ich eksperymenty wykazały,
ż
e
strefa brze
ż
na (marginal zone) pod

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
42/113
opon
ą
wydziela semaforyn
ę
3A, co powoduje „uciekanie” aksonów w przeciwnym kierunku a
jednocze
ś
nie działa jako „atraktant” dla dendrytów („apikalnych
NETRYNY:
sekrecyjne białka
– (u kr
ę
gowców zidentyfikowano ok. 6 białek). Pierwsze odkryto w
rdzeniu kr
ę
gowym; maja
działanie przyci
ą
gaj
ą
ce aksony
, ale netryna-1 działa jednocze
ś
nie „od
tyłu odpychaj
ą
co” (ich homologi odkryto te
ż
w Caenorhabditis elegans i Drosophila m.)
Receptorami dla netryn s
ą
białka z rodziny DCC (wszystkie s
ą
pokrewne zarówno u kr
ę
gowców
jak i bezkr
ę
gowców). Netryny działaj
ą
na du
ż
e odległo
ś
ci ale równie
ż
i na małe.
SLITS: s
ą
to du
ż
e
sekrecyjne
(wydzielane) proteiny; u ssaków poznano 3 typy maj
ą
ce
działanie
odpychaj
ą
ce
aksony w przodomózgowiu; (s
ą
te
ż
o drozofili i Caenorhabditis e.) ; efekt
odpychaj
ą
cy slits jest „mediowany” przez receptory, którymi s
ą
białka z rodziny
Robo
(które
wraz z DCC i UNC5 nale
żą
do nadrodziny immunoglobulin). Slits sa tak
ż
e regulatorami
(pozytywnymi) rozgał
ę
ziania aksonów i dendrytów)
EFRYNY: s
ą
rodzina molekuł powierzchniowych komórek, dziel
ą
si
ę
na dwie grupy: A i B,
równie
ż
ich receptory dziel
ą
si
ę
na klasy A i B. Ich role stwierdzono m.in. W tworzeniu
precyzyjnych topograficznych projekcji ł
ą
cz
ą
cych komórki zwojowe siatkówki z ciałem
kolankowatym bocznym u kr
ę
gowców.
Molekuły kieruj
ą
ce wpływaj
ą
na sto
ż
ki wzrostu aksonów.
Wypuszczanie licznych filipodiów w ró
ż
nych kierunkach jest „naturaln
ą
” (wewn
ę
trzn
ą
) cech
ą
prawidłowego sto
ż
ka. Gdy jeden z filipodiów uzyskuje stabilny kontakt i zwi
ą
zek z okre
ś
lona struktur
ą
nast
ę
puje retrakcja tych filipodiów, które nie uzyskały „kontaktu”
Molekuły „sygnalizacyjne” wpływaj
ą
na proces polimeryzacji aktyny, który wspomaga „wysuwanie si
ę
”
filipodiów.
Aktyna de i repolimeryzuje si
ę
przesuwaj
ą
c czoło aksonu.
Za aktyną „następuje” polimeryzacja mikrotubul.
Semaforyny, netryny, slits, efryny wpływają na proces de i polimeryzacji aktyny w stożku wzrostu.
SEMAFORYNY, NETRYNY, SLITS ,EFRYNY aktywuj
ą
c swoje receptory po
ś
rednio lub
bezpo
ś
rednio wpływaj
ą
na aktywno
ść
GTP-az z rodziny Rho
(np.. Rac lub RhoA), które
odgrywaj
ą
kluczow
ą
rol
ę
jako regulatory polimeryzacji aktyny.
Aksony ł
ą
cz
ą
ce odległe cele w organizmie wykształcaj
ą
si
ę
jak najwcze
ś
niej, póki zarodek jest
niedu
ż
y. Niekiedy docieraj
ą
do miejsc przeznaczenia wcze
ś
niej ni
ż
narz
ą
d (struktura) z któr
ą
maj
ą
si
ę
poł
ą
czy
ć
osi
ą
gn
ę
ła wła
ś
ciwy stopie
ń
dojrzało
ś
ci (i niemo
ż
liwe jest powstanie
funkcjonalnych poł
ą
cze
ń
).
Aksony wykazuj
ą
niekiedy objawy „powstrzymywania si
ę
” i oczekiwania na dojrzewanie ich
celu. Np. takie zjawisko obserwuje si
ę
w tworzeniu drogi w
ę
chowej. Aksony oczekuj
ą
na
opuszk
ę
w
ę
chow
ą
kilka dni aby odpowiednio si
ę
wykształciła i wtedy nast
ę
puje „inwazja”
aksonów z nabłonka w
ę
chowego do opuszki w
ę
chowej.
Wykazano,
ż
e w przypadku nieprawidłowego receptora dla semaforyn (neuropilin-1) aksony
w
ę
chowe nie czekaj
ą
na uformowanie opuszki w
ę
chowej i „przerastaj
ą
” w kierunku CSN nie
tworz
ą
c odpowiednich poł
ą
cze
ń
z opuszk
ą
(Renzi i wsp 2000).
U kr
ę
gowców interneurony komisuralne w rozwijaj
ą
cym si
ę
rdzeniu wysyłaj
ą
aksony
„zwabiane” do brzusznej cz
ęś
ci rdzenia przez wytwarzane tam netryny (ich receptorami s
ą
białka z rodziny DCC obecne na aksonach) ale aksony te nie ko
ń
cz
ą
si
ę
w tej strefie poniewa
ż
przekraczaj
ą
lini
ę
ś
rodkow
ą
i „zakr
ę
caj
ą
” o 900 i zmierzaj
ą
do innych poziomów rdzenia.
Po
wkroczeniu do linii środkowej
następuje silna ekspresja receptora Robo
(receptora dla Slits) w aksonach a
jednocześnie utrata reakcji na netrynę.
Komórki strefy środkowej produkują oprócz netryny również semaforyny i białka typu slit. Teraz okolica
środkowa rdzenia staje się „odpychająca” dla aksonów. Ponadto receptor Robo wiąże się z DCC
„wyciszając” reaktywność aksonów na netrynę.
Selekcja docelowych narz
ą
dów które maj
ą
by
ć
unerwionePoznanie mechanizmów dzi
ę
ki
którym neurony rozpoznaj
ą
i unerwiaj
ą
ich docelowe narz
ą
dy jest jednym z głównych zada
ń
neurobiologii.
Istotne etapy – zagadnienia tego procesu
1)„przecieranie
ś
cie
ż
ki” przez wzrastaj
ą
cy akson
2)„delayed interstitial axon branching”

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
43/113
1)Badania wzrostu aksonów neuronów projekcyjnych V-warstwy kory wskazuj
ą
na
powstawanie odgał
ę
zie
ń
do j
ą
der mostu ju
ż
po przej
ś
ciu sto
ż
ka wzrostu aksonu (istnienie
chemoatraktantów)
2)Liczne przykłady wskazuj
ą
na „
ś
ródmi
ąż
szowe rozgał
ę
zianie si
ę
” aksonów jako na
zasadniczy pierwotny mechanizm „selekcji celu” unerwiania
3)Proces eliminacji „niefunkcjonalnych” kolaterali aksonalnych (ale niekoniecznie neuronów)
np. w corpus callosum („wiosenne przycinanie drzewek...”). „Przycinane” kolaterale lub cz
ęś
ci
aksonu nie s
ą
„bł
ę
dami” ale raczej planowym zaprogramowanym procesem!
Przeszczepiane neurony V warstwy wysyłaj
ą
aksony zgodnie z nowym miejscem do którego
zostały przeszczepione (np. z kory wzrokowej V do ruchowej M)
(szczur)
Eksperyment Sperry’ego z przeci
ę
ciem nerwu wzrokowego
ż
aby i przekr
ę
ceniem oka o 180O i
umo
ż
liwieniem ponownego wrostu aksonów z komórek zwojowych siatkówki do tectum
opticum –
ś
aba zachowywała si
ę
jakby widziała „
ś
wiat przekr
ę
cony” i nie uzyskano
ż
adnego efektu
treningu.
WNIOSEK: regeneruj
ą
ce aksony tworzyły identyczny wzorzec poł
ą
cze
ń
w tectum.
Istniej
ą
gradienty molekuł „kieruj
ą
cych” oraz odpowiednie wzorce receptorów aksonalnych
Modelowy układ eksperymentalny Bonhoffer’a:
Aksony skroniowych cz
ęś
ci siatkówki s
ą
„odpychane” przez ephriny (A2, A5) i pod
ąż
aj
ą
drog
ą
wzdłu
ż
pasków z tectum pozbawionych ephryn
Trajektoria wzrostu aksonu dzieli si
ę
na krótkie odcinki (przypuszczalnie po ok. kilkaset
mikrometrów)
Zale
ż
no
ść
„troficzna” neuronu i komórki docelowej
•Odci
ę
cie zawi
ą
zków nogi w zarodku kurzym
powoduje brak wykształcenia motoneuronów dla nieistniej
ą
cej nogi
•W
ż
yciu zarodkowym kr
ę
gowców powstaje nawet 3 razy wi
ę
cej neuronów postmitotycznych ni
ż
prze
ż
ywa ostatecznie – te które nie tworz
ą
prawidłowych poł
ą
cze
ń
s
ą
eliminowane („si
ę
eliminuj
ą
”)
Kompetytywne mechanizmy redukcji polineuronalno
ś
ci – „przegrywaj
ą
cy” akson „odpada”
•Eksperymenty z usuwaniem lub dodawanie zawi
ą
zków nogi pokazuj
ą
,
ż
e liczba neuronów nie jest
„zaprogramowana” (np.. Genetycznie) ale,
ż
e mo
ż
e by
ć
(i jest) modyfikowana przez interakcj
ę
z
innymi („docelowymi”?) komórkami
•„Nadmiarowe” (w przypadku usuni
ę
cia zawi
ą
zka ko
ń
czyny) neurony gin
ą
poniewa
ż
nie otrzymały
troficznego „wsparcia” ze strony komórek docelowych (w tym przypadku mi
ęś
niowych).
•Mo
ż
na st
ą
d wysnu
ć
wniosek,
ż
e neurony musz
ą
„walczy
ć
” o co
ś
(czynnik troficzny), co zapewne nie
wyst
ę
puje w nadmiarze i st
ą
d mo
ż
na mówi
ć
o „kompetytywnych interakcjach” w tworzeniu poł
ą
cze
ń
neuronów.
•Prawdopodobnie te kompetytywne interakcje mog
ą
tłumaczy
ć
mechanizm redukcji „nadmiarowego”
unerwienia np. włókien mi
ęś
niowych (komórka mi
ęś
niowa pierwotnie unerwiana jest przez wiele
neuronów), które u ssaków jest redukowane tuz po urodzeniu. W efekcie dana komórka mi
ęś
niowa
jest unerwiona ostatecznie tylko przez jeden neuron (jakkolwiek jeden neuron mo
ż
e unerwia
ć
wiele
komórek mi
ęś
niowych)
•Jest to nie tyle eliminacja synaps ile raczej redukcja poł
ą
cze
ń
aksonalnych od ró
ż
nych neuronów:
•Blokowanie przewodnictwa nerwowego przeciwdziała tej redukcji „polineuronalno
ś
ci” unerwienia
•Nie znamy jednak mechanizmów w jaki sposób funkcjonalne („elektryczne”) poł
ą
czenie neuronu
stymuluje wytwarzanie czynnika troficznego
Neurony s
ą
zale
ż
ne od utworzenia prawidłowego kontaktu synaptycznego z komórk
ą
(komórkami) docelowymi.
„Czynniki troficzne” (neurotroficzne) wytwarzane przez komórki docelowe s
ą
konieczne dla utworzenia
i utrzymania kontaktu oraz dla prze
ż
ycia neuronu!
Czynniki neurotroficzne decyduj
ą
te
ż
o stopniu rozwoju drzewa dendrytycznego oraz o
konwergencji i
dywergencji
poł
ą
cze
ń
.
•Neurotrofiny: czynniki wytwarzane przez neurony i nieneuronalne komórki docelowe –
–odpowiedzialne s
ą
za 3 typy odpowiedzi: prze
ż
ycie/
ś
mier
ć
komórki, stabilizacja/eliminacja synaps,
wzrost/retrakcja wypustek komórek nerwowych
•Rodzina neurotrofin:
–NGF (nerve growth factor): odkryty w latach 50-tych, obfity w
ś
liniankach myszy

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
44/113
–BDNF (brain derived neurotrophic factor): odkryty w latach 80-tych,
–Neurotrophin-3 (NT-3) (lata 90-te)
–Neurotrophin-4/5 (NT-4/5)
NGF (nerve growth factor):•NGF odgrywa rol
ę
troficzn
ą
w przypadku narz
ą
dów docelowych
unerwianych przez neurony układu sympatycznego oraz cz
ęś
ci zwojowych neuronów czuciowych –
prawdopodobnie nie gra roli w OUN
•NGF stał si
ę
modelem czynnika neurotroficznego: a) W w/w neuronach brak sygnału NGF (ze strony
komórki docelowej) prowadzi do
ś
mierci neuronu, b) nadmiar NGF prowadzi do prze
ż
ycia zwi
ę
kszonej
ilo
ś
ci neuronów (zale
ż
nych od NGF), c) aksony wykazuj
ą
obecno
ść
receptora dla NGF, d) komórki
docelowe wytwarzaj
ą
NGF.
•NGF nie jest molekuł
ą
ś
ci
ś
le bior
ą
c sekrecyjn
ą
(?) (a zatem zapewne w warunkach fizjologicznych
nie ma działania chemotropowego) i oddziałuje jedynie na aksony wchodz
ą
ce w kontakt z komórkami
docelowymi
•Eksperymenty demonstruj
ą
ce działanie NGF de facto jako czynnika chemotropowego –
dyfunduj
ą
cego w medium hodowli komórkowej (nast
ę
pny slajd) prawdopodobnie nie oddaj
ą
całkowicie
rzeczywistej roli NGF w fizjologii
Po zł
ą
czeniu z NFG nast
ę
puje wzajemna fosforylacja obu podjednostek receptora TrkA
•Neurotrofiny działaj
ą
poprzez receptory typu kinazy tyrozynowej (Poszczególne neurotrofiny ró
ż
ni
ą
si
ę
mi
ę
dzy sob
ą
punktem działania oraz podtypem receptora Trk
–NGF - TrkA
–BDNF - TrkB
–Neurotrophin-3 (NT-3) - TrkC
–Neurotrophin-4/5 (NT-4/5) – TrkB
Czy aktywno
ść
neuronalna odgrywa rol
ę
w formowaniu si
ę
„mapy” poł
ą
cze
ń
siatkówkowo-
tektalnych?
Wydaje si
ę
,
ż
e raczej tak ale nie PIERWSZORZ
Ę
DN
Ą
!
Liczne eksperymenty z blokowaniem przewodnictwa nerwowego w okresie formowania si
ę
tych poł
ą
cze
ń
nie miały wi
ę
kszego wpływu na tworzenie ich charakterystycznych układów!
Np. przeszczepianie oczu aksolotla do traszki kalifornijskiej (endogennie produkuj
ą
cej
tetrodotoksyn
ę
TTX blokuj
ą
c
ą
kanały sodowe) dało prawidłowy układ poł
ą
cze
ń
pomimo
„uciszenia” aksonów RGC (komórek zwojowych siatkówki) przez TTX.
Podobnie nie zaobserwowano wpływu blokerów NMDA na tworzenie poł
ą
cze
ń
.
Jak do celu (wła
ś
ciwego mi
ęś
nia) trafiaja aksony motoneuronów rdzenia
Aksony
nakierowywane s
ą
przez ró
ż
ne układy molekuł „sygnalizacyjnych”.
Przeszczepione motoneurony były w stanie prawidłowo dotrze
ć
do odpowiednich mi
ęś
ni.
Sygnały „nakierowuj
ą
ce” NIE POCHODZ
Ą
OD MI
ĘŚ
NI!
Bardzo wa
ż
n
ą
rol
ę
najprawdopodobniej pełni
ą
komórki mezenchymalne w strefach gdzie
tworz
ą
si
ę
sploty nerwowe.
Istotn
ą
rol
ę
odgrywa ephrin-A (receptory Eph-A4 sa na aksonach)
Synaptogeneza – zł
ą
cze nerwowo-mi
ęś
niowe
Najlepiej poznana w tym zł
ą
czu m.in. dzi
ę
ki
łatwo
ś
ci eksperymentów z manipulacj
ą
i regeneracj
ą
tego zł
ą
cza, obfito
ś
ci
ą
analogicznych
struktur organu elektrycznego morskiej płaszczki Torpedo
Od kontaktu aksonu z miotub
ą
do utworzenia funkcjonalnej synapsy wystarczy by
ć
mo
ż
e
nawet mniej ni
ż
godziny
W pełni dojrzałe zł
ą
cze tworzy si
ę
(u ssaków) dopiero po kilku tygodniach.
Motoneurony INDUKUJ
Ą
ró
ż
nicowanie struktur postsynaptycznych (na podstawie eksp. Z
hodowl
ą
)
Synaptyczna basal lamina (wyspecjalizowana cz
ęść
basal lamina) zawiera elementy
sygnalizacyjne zarówno do tworzenia
struktur
pre jak i postsynaptycznych
Indukcja zmian postsynaptycznych poprzez układ sygnalizacyjny, którego jednymi z
najistotniejszych elementów s
ą
białka:
AGRIN i MuSK
Agrin
indukuje fosforylacj
ę
MuSK

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
45/113
AGRIN jest wytwarzana w motoneuronach.
MuSK jest mi
ęś
niowym receptorem o wła
ś
ciwo
ś
ciach kinazy tyrozynowej
Myszy bez agrin jak i bez MuSK nie wytwarzaj
ą
synaps i gin
ą
natychmiast po urodzeniu (nie
ruszaj
ą
si
ę
i nie oddychaj
ą
)
U takich myszy aksony dochodz
ą
do włókien mi
ęś
niowych ale bezcelowo przerastaj
ą
mi
ę
sie
ń
nie tworz
ą
c synaps !
Musi istnie
ć
system zwrotny od mi
ęś
nia do aksonu „powiadamiaj
ą
cy” o utworzeniu
funkcjonalnego kontaktu i zatrzymuj
ą
cy wzrost aksonu oraz powoduj
ą
cy utworzenie struktur
presynaptycznych.
Agrin
– białko motoneuronów (ok. 200-kDa bardzo wiele izoform) kluczowe w indukcji struktur
postsynaptycznych
Szereg dowodów (m.in. blokowanie p-ciałem) wskazuje
ż
e
sekrecja agryny z zako
ń
czenia
aksonu powoduje skupianie si
ę
receptorów AChR oraz szeregu innych białek (AChE, Rapsyn,
mi
ęś
niowa Neuregulin NRG-1 i jej receptorów oraz białek ErbB)
(hipoteza alternatywna): S
ą
jednak dowody,
ż
e nawet bez Agrin miotuby agreguj
ą
AChR i
prawdopodobnie Agrin konieczna jest raczej do UTRZYMANIA ni
ż
samej indukcji klasteryzacji
AChR
Agrin podawana zewn
ę
trznie nie powoduje wzrostu ekspresji AChR
Agrin
prawdopodobnie reguluje ekspresj
ę
NRG-1 i jego receptorów i „doprecyzowuje” budow
ę
synapsy.
Synaptogeneza – przed inerwacj
ą
…
Najnowsze badania wskazuj
ą
,
ż
e ograniczone do miejsca
przyszłej synapsy i zlokalizowane tworzenie potrzebnych białek (w tym receptora) we włóknie
mi
ęś
niowym nie wymaga obecno
ś
ci aksonów!
(„mięśnie same wiedzą gdzie chcą mieć
przyłączenie kabla...)
i prawdopodobnie same „przygotowują” to miejsce.”)
„Mięsień gorączkowo przygotowuje się na nadejście Pana Aksona…”
Synaptogeneza – po inerwacji
(„zjawił się nerwowo zachowujący się Pan Akson wraz towarzyszącą mu Agryną”)
Agryna utrzymuje
stymulacj
ę
syntezy AChR a aktywno
ść
elektryczna aksonu najprawdopodobnie „wycisza”
ekspresj
ę
AChR w całej reszcie włókna mi
ęś
niowego.
Unerwienie (poprzez agrynę) zawęża strefę receptorów (AChR) i blokuje syntezę receptorów w innych
miejscach
Niewiele wiadomo natomiast o mechanizmach formowania si
ę
postsynaptycznej cz
ęś
ci aparatu
synaptycznego w CSN, np. jak neurony nakierowuj
ą
odpowiednie receptory do błon
postsynaptycznych
„Uzbrajanie” terenu pod budow
ę
jest niezale
ż
ne od tego co tam ma by
ć
zbudowane. Podobnie
aksony docieraj
ą
w „strefy działek budowlanych” gdy jeszcze niewiele tam si
ę
dzieje”
Plan „budynku” zawiera miejsca „przył
ą
czenia kabla aksonalnego” i zgodnie z „planem” te
miejsca s
ą
tworzone w budowanym obiekcie.
Podobnie strefy przyszłych synaps tworz
ą
si
ę
bez udziału aksonów.
Gdy dochodzi do wła
ś
ciwego „podł
ą
czenia” nast
ę
puj
ą
procesy „dostrojenia” aparatury
odbiorczej i nauki wła
ś
ciwego u
ż
ytkowania „internetu”...
Zmiany progresywne w embriogenezie układu nerwowego •Proliferacja komórek
•Migracja
•Ró
ż
nicowanie
•Rozrost aksonów i dendrytów
•Synaptogeneza
•Plastyczno
ść
synaptyczna
Regresywne zmiany w embriogenezie układu nerwowego•Eliminacja aksonów
•Eliminacja synaps
•Eliminacja neuronów
Eliminacja synaps
Tzw. „input elimination” – eliminacja aksonów unerwiaj
ą
cych komórki
docelowe
Nieznane jest nasilenie procesu eliminacji synaps w rozwijaj
ą
cym si
ę
CSN u zarodka
REDUKCJA DYWERGENCJI AKSONALNEJ

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
46/113
Przykłady eliminacji pourodzeniowej synaps u ssaków:
1)Kora wzrokowa (w. IV) z dwuocznego na jednooczne unerwienie (małpa, kot, fretka)
2)Ciało kolankowate boczne: z 2-ocznego na 1-oczne oraz z ponad 20 aksonów siatkówkowych
(>20/1) na 1-2/1 (Cheng i Regehr 2000) w 1 miesi
ą
cu! (ale pozostałe aksony s
ą
znacznie
skuteczniejsze)
3)KK Purkinjego z ponad 3 wł.pn
ą
cych/1kom.Purk. na 1wł/1kom.Purk. włókno pn
ą
ce (climbing
f.) [(z 3/1) na 1/1)]
4)Zwoje sympatyczne szyjne z ok. 14 przedzwojowych aksonów na ok. 7
(ale WZRASTA
LICZBA SYNAPS !)
5)Zł
ą
cze nerwowo-mi
ęś
niowe: z 2-6 aksonów na włókno do 1 aksonu z jednego motoneuronu
(na włókno).
Eliminacja synaps: REDUKCJA DYWERGENCJI AKSONALNEJ
Raczej
nie jest to „naprawa bł
ę
dów”
ale „wyostrzanie specyficzno
ś
ci” i dokładno
ś
ci „map
topograficznych poł
ą
cze
ń
”
Zjawisko eliminacji synaps istnieje niezale
ż
nie (?) od „przycinania” rozgał
ę
zie
ń
aksonów.
Eliminacja synaps
likwiduje
„nadmiarowo
ść
” neuronów (redundancy)*
(unerwienia) i np. w
przypadku mi
ęś
ni pozwala na precyzyjniejsz
ą
regulacj
ę
siły (napi
ę
cia) poprzez rekrutacj
ę
dodatkowych jednostek motorycznych.
Bezkr
ę
gowce nie wykazuj
ą
„nadmiarowo
ś
ci” unerwienia i co za tym idzie procesu eliminacji
synaps. W rezultacie ich aktywno
ść
jest znacznie bardziej zaprogramowana (zakodowana)
wewn
ę
trznie w przeciwie
ń
stwie do ssaków które wymagaj
ą
„zewn
ę
trznych” bod
ź
ców (po
prostu „nauki”) – por. natychmiastowy lot przepoczwarzonej wa
ż
ki i długi proces uczenia si
ę
chodzenia, (tak
ż
e do pewnego stopnia latania ptaków ...)
Synapsy s
ą
eliminowane w systemie „kompetytywnym” (?) (znaczenie słowa „kompetytywny”
jest bardzo szerokie, oznacza tu po prostu,
ż
e z „kilku” zostaje jeden)
Nie ma jak si
ę
wydaje
ż
adnych reguł ani stereotypowo
ś
ci w wynikach tej „rywalizacji” (efekt nie
jest np. genetycznie zaprogramowany).
Eliminacja synaps w korze wzrokowej podobnie jak w zł
ą
czach nerwowo-mi
ęś
niowych jest
mechanizmem kompetytywnym realizowanym poprzez aktywno
ść
oka (siatkówki) i
motoneuronów.
Proponowane jest istnienie „sygnałów karnych” (oraz „sygnałów ochronnych”) wewn
ą
trz
komórek docelowych (unerwianych) prowadz
ą
cych do eliminacji synaps pobudzanych
niesynchronicznie. Eliminacja synaps prowadzi do retrakcji aksonu lub jego rozgał
ę
zienia.
Kompetytywna eliminacja synaps wyst
ę
puje w całej drodze wzrokowej.
Efektem eliminacji synaps jest powstawanie map retinotopowych na poziomie tectum opticum i
w korze u kr
ę
gowców odwzorowuj
ą
cych obrazy. Neurony kory wzrokowej aktywowane ka
ż
dym
okiem tworz
ą
charakterystyczny
paskowaty wzór „dominacji ocznej”
(pasek wyznacza neurony aktywowane lewym lub prawym okiem)
(A)Kolumny
dominacji ocznej
u małpy widoczne po podaniu do jednego oka znakowanej 3H proliny
transportowanej do kory wzrokowej wzdłuż aksonów (jasne paski z oka nastrzyknietego) -
paski są takiej
samej szerokości
(B) Po zablokowaniu (przez zszycie powiek) jednego oka od 2 tyg. po urodzeniu przez 18 miesięcy
następuje obkurczenie się kolumn wraz z ekspansją kolumn z drugiego oka. –
paski są różnej szerokości
1) Istnieje
„okres krytyczny”
w którym zablokowanie oka zmienia szeroko
ść
kolumn dominacji.
2) Je
ś
li zablokujemy równie
ż
oko z niezaszytymi powiekami przez wstrzykni
ę
cie
tetrodotoksyny (blokada całkowita aktywno
ś
ci siatkówki wł
ą
cznie z blokad
ą
aktywno
ś
ci
spontanicznej, nie wymagaj
ą
cej stymulacji
ś
wiatłem) reakcja kory „przesunie si
ę
” w kierunku
oka z zaszytymi powiekami ale w którym zachowana jest spontaniczna aktywno
ść
neuronalna !
3) Równie
ż
je
ś
li zaszyjemy oba oczy powstan
ą
kolumny dominacji ale
gdy do obu podamy TTX
(tetrodotoxin np. z traszki kalifornijskiej lub puffer fish)
wtedy kolumny nie powstan
ą
.
4) Nawet bez stymulacji
ś
wietlnej niedojrzał
ą
siatkówka emituje spontaniczn
ą
aktywno
ść
w
sposób, który pozwala na tworzenie „mapy” poł
ą
cze
ń
z kor
ą
wzrokow
ą
(pobudzenia kk
zwojowych wyst
ę
puj
ą
w postaci „fali” rozprzestrzeniaj
ą
cej si
ę
w siatkówce) a „fale” z obu oczu
praktycznie nigdy nie s
ą
synchroniczne.
Dalsze doprecyzowanie poł
ą
cze
ń
projekcyjnych drogi wzrokowej nast
ę
puje pod wpływem
bod
ź
ców wzrokowych.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
47/113
Mapy wzrokowe mo
ż
na wytworzy
ć
w obszarach słuchowych je
ś
li skierowa
ć
tam drog
ę
wzrokow
ą
!
W gryzoni istnieje dokładna mapa korowa reprezentuj
ą
ca poszczególne „wibrysy” czyli „w
ą
sy”
w postaci tzw. barrels (jednostek funkcjonalnych w korze mózgowej).
Postembrionalna modyfikacja układów neuronalnych i synaps –
„okresy krytyczne”
Plastyczno
ść
„dojrzałych” synaps i obwodów neuronalnych
Krótkotrwała plastyczno
ść
synaptyczna
Facylitacja
(ułatwienie): osi
ą
ga szczyt w ok. 1 sek i potem szybko zanika. Wystarczy jeden
potencjał i nast
ę
pny jest wi
ę
kszy (je
ś
li jest w ci
ą
gu ok. sek.).
Augmentacja
: wolniejsze w czasie nasilaj
ą
ce si
ę
powi
ę
kszanie PSP
Potencjacja
(wzmocnienie po długiej „t
ęż
cowej” stymulacji ): w niektórych synapsach trwaj
ą
ce
wiele minut podwy
ż
szenie PSP.
Wszystkie w/w zjawiska tłumaczy si
ę
kumuluj
ą
cym si
ę
wzrostem st
ęż
enia Ca2+
Depresja
: w niektórych synapsach (przeciwie
ń
stwo facylitacji)
Depresja mo
ż
e by
ć
skutkiem spadku ilo
ś
ci p
ę
cherzyków, deaktywacji miejsca uwalniania N-T
lub istnienia hamuj
ą
cych autoreceptorów. Ponadto mo
ż
e wynika
ć
z tzw.
desensytyzacji
receptora
Mechanizmy LTP (Długotrwałe wzmocnienie synaptyczne) LTD (długotrwałe
osłabienie synaptyczne)
Długotrwałe wzmocnienie synaptyczne = (Long-term potentiation = LTP)Bliss i Lomo 1973
LTP jest to trwały wzrost potencjału postsynaptycznego (EPSP), który mo
ż
e by
ć
indukowany w
sposób szybki poprzez gwałtowne wyładowanie potencjałów czynno
ś
ciowych (spike activity)
we włóknach aferentnych (presynaptycznych). Przypuszcza si
ę
,
ż
e LTP jest elementem
mechanizmów pami
ę
ci i uczenia si
ę
Postulat Donalda Hebba: „Cells that fire together, wire together”
Warunki dla LTP:
1)Napływ Ca2+
2)Jednoczesna depolaryzacja
Najwa
ż
niejszym mechanizmem LTP jest prawdopodobnie wzrost liczby AMPA-R
Współdziałanie –(Cooperativity)
Specyficzno
ść
ze wzgl
ę
du na „wej
ś
cie” - (Input specificity)
– LTP dotyczy tylko synapsy stymulowanej
Asocjacyjno
ść
(Associativity)
– gdy jednoczasowo stymulowane obie synapsy LTP powstaje w obu z nich –
prawdopodobnie jest to podło
ż
em
warunkowania
asocjatywnego (klasycznego)
•Koincydencja (w „okienku” czasowym do 100 ms) depolaryzacji neuronu CA1 i pojedynczej stymulacji
kolaterali Schaffera wystarczy do wytworzenia LTP (przypomnie
ć
postulat Donalda Hebb’a)
•LTD oraz LTP mog
ą
si
ę
wzajemnie znosi
ć
(likwidowa
ć
)
LTD: Long-term depression
Powstaje w wyniku długotrwałej stymulacji o niskiej
frekwencji (np. 1Hz przez 10min.)
Zadaniem LTD mo
ż
e by
ć
likwidacja LTP
Mo
ż
e by
ć
mediowana przez NMDA oraz inne receptory (np. metabotropowe mGluR)
Przykład:
•LTD na synapsie k.Purkinjego z włóknem równoległym (paralel fiber)
LTD w mó
ż
d
ż
ku
•W przeciwie
ń
stwie do LTD w hipokampie nie bierze udziału receptor NMDA
(nieobecny w kk.Purkinjego) ale efekt (internalizacja AMPAR) jest ten sam.
Indukcja LTD wymaga jednoczesnego napływu Ca2+ do kom.Pur. z cf (przez VD-Ca-Kanały)
oraz aktywacji mGluR1 na synapsach pf z komórkami Purkinjego
PKC fosforyluje AMPAR i powoduje zmniejszenie wra
ż
liwo
ś
ci AMPAR
LTD na komórkach Purkinjego wywołuje dysinhibicj
ę
poniewa
ż
w efekcie „hamowane s
ą
hamuj
ą
ce” komórki Purkinjego

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
48/113
Czucie somatyczne
Układy czucia somatosensorycznego:
dwie „zasady”:
modalno
ść
(„typy” czucia a
ż
do kory czuciowej s
ą
reprezentowane
przez osobne szlaki)
i
somatotopia
•Typy receptorów czucia somatycznego:
–Mechanoreceptory
–Receptory bólu (nocyceptory)
–Receptory ciepła (termoreceptory)
•Receptory czucia somatycznego z uwagi na morfologi
ę
dziel
ą
si
ę
na:
–Wolne
•Wolne zako
ń
czenia nerwowe (ból, temperatura)
–Otorebkowane
•Niezale
ż
nie od typu wszystkie receptory czucia somatycznego funkcjonuj
ą
na podobnej zasadzie:
bodziec zmienia przepuszczalno
ść
jonow
ą
receptora co prowadzi do powstania „potencjału
receptora”. Potencjał receptora mo
ż
e prowadzi
ć
do potencjału czynno
ś
ciowego
•„Jako
ść
” czucia („rodzaj”) zale
ż
y od typu pobudzanego receptora i umiejscowienia jego o
ś
rodka w
CSN
•„Siła” bod
ź
ca jest kodowana w postaci cz
ę
stotliwo
ś
ci potencjałów czynno
ś
ciowych powstaj
ą
cych w
receptorze
••Zale
ż
no
ść
siła/cz
ę
sto
ść
P.cz. nie jest prosta i ró
ż
na jest w ró
ż
nych receptorach.
•Z uwagi na typ zale
ż
no
ś
ci dzielimy receptory (a zarazem tworz
ą
ce je neurony) na
–szybko adaptuj
ą
ce si
ę
(„fazowe”)
•Rejestruj
ą
dynamiczne zmiany bod
ź
ca (jego zmienno
ść
)
–wolno adaptuj
ą
ce si
ę
(„toniczne”)
•Rejestruj
ą
statyczne cechy bod
ź
ca (jego trwało
ść
)
•Podział typów włókien z lat 20-30tych XXw!
•A (najwi
ę
ksze i najszybsze) – B (po
ś
rednie) i C najmniejsze i najwolniejsze
–S
ą
te
ż
podgrupy :
α
,
β
,
δ
•Z kolei aferentne włókna mi
ęś
niowe dziela si
ę
na I (najszybsze), II, III (po
ś
rednie) i IV (najwolniejsze)
–S
ą
te
ż
dodatkowe podgrupy (a, b)
•To co odczuwamy zale
ż
y nie tylko od tego które i jak silnie s
ą
pobudzane receptory ale od czynników
regulowanych z poziomu CSN (nie zauwa
ż
amy ubrania, ew. tylko gdy chcemy lub gdy to jest
potrzebne)
•Aktywne („badawcze”) dotykanie prawdopodobnie daje inne (nieco) odpowiedzi neuronalne
o
ś
rodkowego neuronu czuciowego ni
ż
w przypadku „biernej” stymulacji
•Pola recepcyjne zmieniaj
ą
si
ę
w czasie działania bod
ź
ca
•Wniosek:
•W przeciwie
ń
stwie do klasycznych („statycznych”) pogl
ą
dów na temat pól recepcyjnych obecnie
wiemy,
ż
e maj
ą
one charakter dynamiczny.
•Mechanoreceptory odpowiedzialne za „propriocepcj
ę
” (głównie pozycja ko
ń
czyn i ciała)
–Wrzeciona mi
ęś
niowe
–Narz
ą
dy Golgiego
–Receptory stawowe
––Ponadto:
–(pozycja głowy jest ustalana poprzez organ przedsionkowy)
–(jeszcze inne mechanoreceptory b
ę
d
ą
ce cz
ęś
ci
ą
trzewnego układu ruchowego sa obecne w sercu i
naczyniach – detekcja rozci
ą
gni
ę
cia naczy
ń
)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
49/113
•Czuciowa somatosensoryczna kora drugiego rz
ę
du (SII) otrzymuje konwerguj
ą
ce sygnały z kory SI i
wysyła aksony do hipokampa i c.migdałowatego
•UWAGA! Kora czuciowa wysyła bardzo liczne aksony (wi
ę
cej ni
ż
ich „otrzymuje”!) w kierunku
wzgórza, pnia i rdzenia (zapewne słu
ż
y to modyfikacji odczuwania bod
ź
ców poprzez wpływ na o
ś
rodki
ni
ż
sze dróg czucia)
•Reorganizacja pól recepcyjnych czucia somatycznego, wzroku, słuchu (oraz ruchowych) na skutek
zablokowania lub dozna
ń
czuciowych lub (odwrotnie) nadmiernej stymulacji
Po amputacji palca 3 s
ą
siaduj
ą
ce pola recepcyjne „przejmuj
ą
” obszar amputowanego palca
•Silne u
ż
ywanie palców 2 i 3 powoduje poszerzenie pól recepcyjnych dla tych palców
•Na skutek ataku padaczki powstaj
ą
ce zmiany zwiazane z plastyczno
ś
ci
ą
synaptyczn
ą
(np..LTP)
mog
ą
trwale jeszcze bardziej zaburza
ć
obwody neuronalne
•Tzw kindling
polega na powtarzanej słabej stymulacji c.migdałowatych u szczura nie prowadz
ą
cej
pocz
ą
tkowo do drgawek a po pewnym czasie doprowadza do sytuacji,
ż
e ten sam słaby impuls
wywołuje uogólnione drgawki. Efekt ten jest trwały nawet przez ponad rok.
•
•CZUCIE BÓLU
•Receptorami bólu (nocyceptorami) oraz temperatury (termoreceptory) s
ą
wolne (u
ż
ywa si
ę
te
ż
okre
ś
lenia „nagie” ang. bare) zako
ń
czenia nerwowe (neuronów zwojów korzonków grzbietowych i
zwoju n.V, a tak
ż
e n. VII, n.IX, n.X). Wolne zako
ń
czenia umo
ż
liwiaj
ą
dost
ę
p substancjom
chemicznym, które równie
ż
wywołuj
ą
uczucia bólowe. Neurotransmiterem aferentów bólowych jest
Glutaminian i subst.P.
••Termoreceptory: Wolne zako
ń
czenia nerwowe b
ę
d
ą
ce niebólowymi receptorami temperatury
–Receptory wra
ż
liwe na zimno (przewodzenie „szybkie” włóknami A
δ
)
–Receptory wra
ż
liwe na ciepło (przewodzenie „wolne” włóknami C)
•Termoreceptory wskazuj
ą
głównie zmiany temperatury a słabo warto
ś
ci bezwzgl
ę
dne (przykład z
trzymaniem jednej r
ę
ki w zimnej a drugiej w ciepłej wodzie i nast
ę
pnie wło
ż
enie obu r
ą
k do tej samej
„letniej” wody)
••Nocyceptory znajduj
ą
si
ę
w skórze oraz w narz
ą
dach wewn
ę
trznych
••Dwie szybko
ś
ci przewodzenia bólu:
–20m/sek (włókna A
δ
; tzw. „pierwszy ból” – o charakterze ostrym
–2m/sek (włókna C, tzw.”drugi ból”- doznania polimodalne o charakterze t
ę
pym, piek
ą
cym)
••Trzy typy receptorów bólu (nocyceptorów) w skórze
–A
δ
mechanosensytywne (tak
ż
e A
δ
mechanotermiczne)
–Polimodalne (poprzez włókna C) – bod
ź
ce mechaniczne, termiczne i chemiczne
•Pola recepcyjne „bólowych” neuronów korowych s
ą
relatywnie du
ż
e
Doznanie bólu nie jest „ilo
ś
ciowym” wariantem innych dozna
ń
mechanicznych i termicznych
ale zupełnie osobn
ą
jako
ś
ci
ą
(modalno
ś
ci
ą
) zwi
ą
zan
ą
z osobnymi włóknami i drogami.
•Wolne zako
ń
czenia nerwowe (b
ę
d
ą
ce receptorami bólowymi w znaczeniu „neurofizjologicznym”)
posiadaj
ą
receptory (w znaczeniu „molekularnym”) wra
ż
liwe na ró
ż
ne substancje chemiczne.
•Najsilniejszym sygnałem bod
ź
ca bólowego charakteryzuje si
ę
polipeptyd bradykinina
•Dobrze poznano grup
ę
receptorów b
ę
d
ą
cych kanałami jonowymi dla Na i Ca (gdy s
ą
aktywne
wpuszczaj
ą
te jony co prowadzi do generacji P.cz.) nazwanych:
–vanilloid receptor VR-1 (= transient receptor potential TRPV-1) obecny we włóknach A
δ
i C;
aktywowany przez capsaicyn
ę
(z papryki chili) i temperatur
ę
450. (nic dziwnego,
ż
e papryk
ę
odczuwamy jak co
ś
gor
ą
cego…!) Prawdopodobnie podobnie jak w przypadku endogennych opiatów
istniej
ą
endogenne substancje – „endovanilloidy” odgrywaj
ą
ce rol
ę
w reakcji bólowej w odpowiedzi na
uszkodzenie tkanki. Receptor VR-1 jest równie
ż
aktywowany przez kwas (H+) i anandamid
(endokanabinoid).
–vanilloid-like receptor VRL-1 (=TRPV-2) obecny we włóknach A
δ
; aktywowany przez temperatur
ę
520 i niewra
ż
liwy na capsaicyn
ę
A co z zimnem ?
•Podgrupa receptorów typu Transient Receptor Potential (TRP) b
ę
d
ą
cych kanałami jonowymi
aktywowanymi przez temperatur
ę
(thermoTRP) odgrywa istotna rol
ę
w recepcji temperatury oraz bólu.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
50/113
•Cztery z nich (TRPV1-4), aktywowane s
ą
ciepłem (
ąż
do indukcji bólu) natomiast receptory TRPM8 i
TRPA1 aktywowane s
ą
„przyjemnym chłodem” jak równie
ż
„bolesnym zimnem”
•ThermoTRP s
ą
te
ż
chemosensytywne (ró
ż
ne substancje ro
ś
linne i endogenne mediatory zapalne).
•TRPM8, „minty-cool ion channel”
•TRP s
ą
obecne w neuronach zwojów korzonków grzbietowych (myszy).
•Receptor „mentolowo-chłodny” CMR1 (od CoolMintR1) = (TRPM8), aktywowany jest progow
ą
temperatur
ą
~28°C, a aktywowane pr
ą
dy wzrastaj
ą
przy spadku temperatury a
ż
do 8°C obejmuj
ą
c
zarówno nieszkodliwe jak i nieprzyjemne-uszkadzaj
ą
ce temperatury.
•Receptory TRPA1 (aktywowane tak
ż
e substancjami takimi jak allyl isothiocyanate (w musztardzie),
cinnamaldehyde (cynamon) wyst
ę
puj
ą
razem z receptorami „ciepła” TRPV1 na tych samych
zako
ń
czeniach nerwowych.
•Natomiast receptory „chłodu-i-mentolu” TRPM8 wyst
ę
puj
ą
na oddzielnych zako
ń
czeniach
nerwowych.
Kapsaicyna wywołuje pieczenie skóry ale stosowana przewlekle powoduje desensytyzacj
ę
zako
ń
cze
ń
nerwowych i blokad
ę
uwalniania substancji P i VIP i w efekcie działa przeciwbólowo.
•Zarówno zwykłe obserwacje jak i badania eksperymentalne wskazuj
ą
na ogromna rol
ę
czynników
„pozaobiektywnych” na odczuwanie bólu.
•Uszkodzenie czuciowej ciemieniowej kory somatosensorycznej nie znosi (odczucia) bólu chocia
ż
upo
ś
ledza inne mechano-sensoryczne „modalno
ś
ci” czuciowe (po drugiej stronie ciała).
Ból wywołuje aktywacj
ę
bardzo wielu o
ś
rodków.
Wraz ze wzrostem nasilenia bólu wi
ę
cej okolic mózgu staje si
ę
aktywnych.
•Mo
ż
na zatem wyró
ż
ni
ć
dwie komponenty dozna
ń
bólowych.
•Dwie „komponenty” dozna
ń
bólowych:
•1) czuciowo-dyskryminatywna
–Koduj
ą
ca lokalizacj
ę
, intensywno
ść
i jako
ść
bod
ź
ca
–„realizowana” przez „klasyczne” drogi i o
ś
rodki bólu
•2) afektywno (emocjonalno)-motywacyjna
–Decyduj
ą
ca o stopniu „nieprzyjemno
ś
ci” bólu i aspekcie emocjonalnym
–„realizowana” przez osobne szlaki obejmuj
ą
ce kor
ę
(przedni zakr
ę
t obr
ę
czy, wyspa i pie
ń
mózgu
(n.parabrachialis)
–Do j
ą
der parabrachialnych docieraja aksony z neuronów blaszki I Rexeda rdzenia natomiast
pozostałe aksony drogi bólu-temperatury pochodz
ą
z neuronów blaszki.V rdzenia (nastepuje to
rozdzielenie dwóch aspektów doznania bólowego: dyskryminatywnego i emocjonalnego)
–N.parabrachialis wysyła aksony do c.migdałowatego i podwzgórza („o
ś
rodki” emocji i motywacji) oraz
do substancji szarej okołowodoci
ą
gowej, która odgrywa rol
ę
w kontroli aktywno
ś
ci szlaków bólu
Substancja szara okołowodociagowa:
O
ś
rodek kontroli aktywno
ś
ci dróg bólowych
•Hyperalgezja: nadwra
ż
liwo
ść
na ból w okolicy, w której wyst
ę
puje uszkodzenie tkanki (np. wzrost
wra
ż
liwo
ś
ci na temperatur
ę
w strefie oparzenia słonecznego)
•Wyst
ę
puje w takim miejscu obwodowe uwra
ż
liwienie na temperatur
ę
i ból spowodowane
oddziaływaniem na receptory bólu i temperatury czynników obecnych w „zupie zapalnej” (produkty
uszkodzenia tkanki takie jak H+, metabolity lipidów, prostaglandyny, nukleotydy, bradykinina,
histamina, serotonina, NGF wszystkie oddziałuj
ą
na wolne zako
ń
czenia nerwowe i ich receptorowe
kanały jonowe wzmagaj
ą
c ich odpowiedzi).
•Ponadto zako
ń
czenia nerwowe wydzielaj
ą
calcitonine-gene-related peptide (CGRP) i substancj
ę
P
oraz ATP oddziałuj
ą
ce na naczynia (poszerzenie) i na mastocyty oraz neutrofile.
•Prostaglandyny obni
ż
aj
ą
próg pobudliwo
ś
ci zako
ń
czenia nerwowego (st
ą
d m.in. efekt przeciwbólowy
niesterydowych leków przeciwzapalnych które hamuj
ą
COX), jednocze
ś
nie w podobny sposób
oddziałuj
ą
na neurony rogów tylnych rdzenia.
•O
ś
rodkowe mechanizmy nadwra
ż
liwo
ś
ci na bod
ź
ce bólowe i termiczne:
•Wzrost pobudliwo
ś
ci neuronów rogów tylnych na skutek uprzedniej aktywno
ś
ci aferentnych nerwów
„bólowo-termicznych”

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
51/113
•Wzrost pobudliwo
ś
ci „uogólnia si
ę
” na inne (niebólowe) bod
ź
ce z mechanoreceptorów i w efekcie
uprzedniego silnego bod
ź
ca bólowego nast
ę
pne (w normalnych warunkach) niebólowe dra
ż
nienie
powoduje ból – co nazywamy ALLODYNI
Ą
.
•Uszkodzenie tkanki dotycz
ą
ce równie
ż
zako
ń
cze
ń
nerwowych prowadzi do ich przetrwałej
nadwra
ż
liwo
ś
ci i do tzw. BÓLU NEUROPATYCZNEGO (skrajnie dokuczliwego i bardzo opornego na
leczenie)
•Współczesne pogl
ą
dy na ból zwracaj
ą
uwag
ę
na fakt,
ż
e ból jest zjawiskiem zło
ż
onym i nie jest
ograniczony do prostej stymulacji receptorów „bólowych”. Mo
ż
na powiedzie
ć
,
ż
e raczej „boli mózg” ni
ż
konkretny narz
ą
d…?
•Neuronalna modulacja bólu
–Zst
ę
puj
ą
ce drogi do korzeni tylnych rdzenia (oraz n.tr.spinalis n.V)
•Teoria „bramkowa” bólu (Ronald Melzack, Patrick Wall 1965)- zakłada,
ż
e ból podlega modulacji
zarówno na poziomie rdzenia (np. aktywacja mechanoreceptorów przez „pocieranie” łagodzi ostry ból)
jak i pod wpływem impulsów pochodz
ą
cych z mózgu
•Transmisja informacji z pierwotnych aferentów do wtórnych neuronów w rdzeniu i wy
ż
ej, podlega
modulacji („bramkowaniu”). W rdzeniu miejscem „bramkowania” jest substantia gelatinosa
•Typy bramkowania:
1. Lokalne – (rdzeniowe, odcinkowe)
2. Rozlane – drogi zst
ę
puj
ą
ce z pnia mózgu.
Toria bramkowania zakłada,
ż
e ból jest funkcj
ą
równowagi mi
ę
dzy informacj
ą
dochodz
ą
c
ą
do rdzenia
przez du
ż
e i małe włókna nerwowe. Je
ś
li przewa
ż
a impulsacja z du
ż
ych nie ma bólu, je
ś
li z małych
(C) jest ból.
••Ró
ż
ne metody np. elektrostymulacji jako
ś
rodka przeciwbólowego oparte s
ą
ne tej teorii.
•Efekt PLACEBO
–Mo
ż
e by
ć
„dawkozale
ż
ny”!
–mo
ż
e by
ć
likwidowany przez nalokson! (nie jest czysto „psychiczny”)
–Wskazuje to te
ż
,
ż
e nie chodzi tu o likwidowanie „wyobra
ż
onego” (udawanego?) bólu.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
52/113
SŁUCH
Kodowanie siły (amplitudy d
ź
wi
ę
ku) w nerwie słuchowych.
Wraz ze wzrostem amplitudy d
ź
wi
ę
ku, wzrasta cz
ę
stotliwo
ść
potencjałów czynno
ś
ciowych we
włóknie nerwowym nerwu słuchowego (dla charakterystycznej cz
ę
stotliwo
ś
ci) a
ż
do saturacji.
Włókna maj
ą
ró
ż
n
ą
cz
ę
stotliwo
ść
spontanicznej aktywno
ś
ci.
"low spontaneous rates” saturacja wolna- koduj
ą
sił
ę
dla gło
ś
niejszych d
ź
wi
ę
ków
„High spontaneous rate” saturacja szybka – koduj
ą
dla cichszych d
ź
wi
ę
ków.
Kazde włókno w nerwie słuchowym posiada swoja tzw chrakaterystyczn
ą
cz
ę
stotliwo
ść
(characteristic
frequency – CF) – jest to taka cz
ę
stotliwo
ść
drga
ń
fali d
ź
wi
ę
kowej dla której potrzebna jest minimalna
energia aby stymulowa
ć
potencjały czynno
ś
ciowe ww włóknie . Z reguły jest to ta sama lub zblizona
cz
ę
stotliwo
ść
do cz
ę
stotliwo
ś
ci rezonansowej tej cz
ęś
ci błony podstawnej narz
ą
du Cortiego od której
odchodzi dane włókno nerwu słuchowego
.
Pitagorejskie pojmowanie muzyki (i
ś
wiata)
•Odkrycie matematycznych zwi
ą
zków mi
ę
dzy tonami muzycznymi oraz d
ź
wi
ę
ku jako cyklicznej
oscylacji
•Interwały muzyczne definiowane jako relacje długo
ś
ci struny i zarazem cz
ę
stotliwo
ś
ci drgania
(fundamentalnego): oktawa – 2:1, kwinta – 3:2, kwarta – 4:3
•Tony o relatywnych cz
ę
stotliwo
ś
ciach 1:2 postrzegane s
ą
jako podobne = „takie same” (oktawy) ALE
DLACZEGO ???
•Dlaczego niektóre interwały postrzegamy jako dysonanse (zwykle budz
ą
ce uczucie „napi
ę
cia”) a
niektóre jako konsonanse („przyjemne”)?
••Schwartz, Howe i Purves (2003 J Neurosci 32:7160-7168) uwa
ż
aj
ą
,
ż
e natura tonalno
ś
ci jest
pochodn
ą
głównych (dominuj
ą
cych) cz
ę
stotliwo
ś
ci wyst
ę
puj
ą
cych w mowie człowieka a muzyka jest
by
ć
mo
ż
e „ubocznym” efektem doskonalenia recepcji mowy (w tym jej emocjonalnego jak te
ż
czysto
j
ę
zykowego-informatycznego znaczenia)
•Wytrawny muzyk mo
ż
e rozró
ż
ni
ć
ró
ż
nic
ę
1 Hz pomi
ę
dzy tonem 1000 Hz i 1001 Hz
•Potrafimy rozró
ż
ni
ć
brzmienie poszczególnych instrumentów w orkestrze a dyrygent potrafi wskaza
ć
drobne bł
ę
dy intonacji
•Zmysł słuchu jest przykładem mechanorecepcji której zadaniem jest detekcja i postrzeganie
(rozumienie) d
ź
wi
ę
ków oraz rozpoznawanie ich
ź
ródła (kierunku sk
ą
d dochodz
ą
)
•Podobnie jak w przypadku organu przedsionkowego (oraz linii bocznej ryb) detektorami s
ą
wyspecjalizowane komórki nabłonkowe (neuronalne?)– tzw. komórki włoskowate
Organ słuchu ma wspólne cechy funkcjonalne i ewolucyjne podobie
ń
stwa z innymi systemami
mechanorecepcyjnymi takimi jak organ przedsionkowy (równowagi) i układ linii bocznej u ryb
poniewa
ż
wszystkie te narz
ą
dy u
ż
ywaj
ą
tego samego typu komórek recepcyjnych :
tzw.
Komórek włoskowatych
i we wszystkich tych narz
ą
dach bodziec odbierany powoduje
odkształcenie rz
ę
sek komórek włoskowatych (stereociliów) jednak w odró
ż
nieniu od
pozostałych narz
ą
dów mechanorecepcyjnych w narz
ą
dzie słuchu komórki recepcyjne s
ą
wra
ż
liwe na d
ź
wi
ę
k. U zwierz
ą
t „naziemnych” receptorowe komórki włoskowate pozostały w
ś
rodowisku „wodnym” (kanały
ś
limaka).
•D
ź
wi
ę
k jest no
ś
nikiem bardzo wielu cz
ę
sto kluczowych dla prze
ż
ycia osobnika informacji
•Z uwagi na jego cechy fizyczne (interferencja, dyfrakcja, refrakcja) i brak „pierwotnej mapy
przestrzennej” wydobycie (odkodowanie) informacji zawartej w fali d
ź
wi
ę
kowej wymaga bardzo
zło
ż
onych mechanizmów.
•Realizowane s
ą
one na wielu pi
ę
trach OUN – dlatego „droga” słuchowa jest najbardziej
skomplikowanym szlakiem ze wszystkich szlaków czuciowych.
•D
ź
wi
ę
k to
podłu
ż
na
oscylacja (fala) ci
ś
nienia powietrza o cz
ę
stotliwo
ś
ci si
ę
gaj
ą
cych wielu
tysi
ę
cy Hz.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
53/113
v = f
λ
Intensywno
ść
d
ź
wi
ę
ku = I okre
ś
lona jest przez W (moc) podzielon
ą
przez powierzchni
ę
A :I =
W/A
Je
ś
li przyjmiemy punktow
ą
emisj
ę
d
ź
wi
ę
ku (fala rozchodzi si
ę
w postaci kulistej wtedy
powierzchni
ę
w relacji do promienia opisuje równanie:
A = 4
π
r 2
Podstawiaj
ą
c do powy
ż
szego wzoru
I = W/4
π
r 2 st
ą
d wiemy,
ż
e
Intensywno
ść
d
ź
wi
ę
ku zmniejsza si
ę
z kwadratem odległo
ś
ci
Z kolei (przy braku absorbcji) amplituda ci
ś
nienia fal d
ź
wi
ę
kowych (proporcjonalna do
pierwiastka kw. z intensywno
ś
ci) zmniejsza si
ę
odwrotnie proporcjonalnie do promienia
(odległo
ś
ci) czyli do 1/r.
Czuło
ść
si
ę
ga od 0 dB (komar z odl. 3m)
do 120 (próg bólu, gwizd lokomotywy z odl. 1m))
•Słuch człowieka wra
ż
liwy jest na ogromny zakres cz
ę
stotliwo
ś
ci: od ok. 20Hz do 20 000 Hz
(najwra
ż
liwszy w zakresie 1-4 kHz)* Wi
ę
kszo
ść
energii d
ź
wi
ę
kowej zawartej w ludzkiej mowie
jest przenoszona w zakresie cz
ę
stotliwo
ś
ci od 0,25 kHz do 3 kHz.
•Potencjały receptorowe komórek włoskowatych s
ą
przekształcane i kodowane w potencjały
czynno
ś
ciowe nerwu słuchowego. W mózgu poprzez m.in. Ró
ż
nic
ę
w dotarciu d
ź
wi
ę
ku do
jednego i drugiego ucha (rz
ę
du 30-10
m
s !!) nast
ę
puje lokalizowanie kierunku, z którego
dochodzi d
ź
wi
ę
k.
•* U nietoperzy próg zaczyna si
ę
od 20 kHz a ko
ń
czy na 200 kHz
•Słuch jest najbardziej wra
ż
liwy w zakresie cz
ę
stotliwo
ś
ci około 3 kHz (takie cz
ę
stotliwo
ś
ci
równie
ż
działaj
ą
silnie uszkadzaj
ą
co!)
Amplituda ci
ś
nienia d
ź
wi
ę
ku wyra
ż
ana jest w skali logarytmicznej (decybele dB) z uwagi m.in.
Na ogromn
ą
skal
ę
słyszalnych ró
ż
nic ci
ś
nienia (od 0 dB do uszkadzaj
ą
cych i wywołuj
ą
cych ból
120 dB). Przy progu słyszalno
ś
ci (0dB) ruch drobin powietrza wynosi zaledwie ok. 0,01
nanometra !!! (trylionowa cz
ęść
Wata / m2)
Na całkowicie „bezd
ź
wi
ę
cznej” planecie oznaczałoby to,
ż
e
ź
ródło d
ź
wi
ę
ku mocy 1 Wata i
cz
ę
stotliwo
ś
ci 3 kHz mogłoby by
ć
słyszalne z odległo
ś
ci 450 km !
Ucho reaguje logarytmicznie na zmiany siły d
ź
wi
ę
ku.
Obwodowy narz
ą
d słuchu składa si
ę
z ucha zewn
ę
trznego,
ś
rodkowego i wewn
ę
trznego.
Ucho zewn
ę
trzne: mał
ż
owina i zewn. kanał słuchowy.
pozwala poprzez wpływ na przepływaj
ą
c
ą
fal
ę
d
ź
wi
ę
kow
ą
na lokalizacj
ę
ź
ródła nawet
przy słyszeniu jednousznym - szczeg. lokalizacj
ę
wysoko
ś
ci z której dochodzi d
ź
wi
ę
k.
(konstrukcja ucha zewn
ę
trznego powoduje,
ż
e składowe wysokiej cz
ę
stotliwo
ś
ci d
ź
wi
ę
ków s
ą
lepiej przewodzone je
ś
li ich
ź
ródło jest wy
ż
ej)
Tzw. nie-liniowo
ść
zachowania ucha wewn
ę
trznego
1. Podwojenie siły fali oddziałuj
ą
cej na błone podstawn
ą
narz
ą
du Cortiego nie daje podwojenia
wyładowa
ń
w nerwie słuchowym (“output” jest mniej ni
ż
podwojony – tzw saturating non-
linearity).
2. Dodanie drugiego tonu ró
ż
ni
ą
cego si
ę
cz
ę
stotliwo
ś
ci
ą
obni
ż
a odpowied
ź
na pierwszy ton -
tzw Two-tone suppression)
3. Zagranie dwóch tonów np. 1000 i 1200 Hz spowoduje odczucie trzeciego tonu (w tym
przypadku o cz
ę
stotliwo
ś
ci 800 Hz) -tzw Cubic Difference Tone.
WZMOCNIENIE FALI GŁOSOWEJ W UCHU
Ś
RODKOWYM
Ucho
ś
rodkowe: pozwala na silne wzmocnienie fali d
ź
wi
ę
kowej poprzez system kostek
(młoteczek, kowadełko, strzemi
ą
czko) i przeniesienie fali ze
ś
rodowiska powietrznego na
wodne
ś
limaka (normalnie d
ź
wi
ę
k raczej odbija si
ę
od powierzchni wody). Wzmocnienie
d
ź
wi
ę
ku nast
ę
puje m.in. poniewa
ż
błona b
ę
benkowa ma powierzchni
ę
35 razy wi
ę
ksz
ą
ni
ż
okienko owalne, do którego przylega strzemi
ą
czko (daje to w przybli
ż
eniu ten sam
współczynnik wzrostu ci
ś
nienia.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
54/113
Zysk ci
ś
nienia wynosi ok. 25-30 dB w zakresie po
ś
rednich cz
ę
stotliwo
ś
ci (ponad to co byłoby
gdyby fala d
ź
wi
ę
kowa powietrza bezpo
ś
rednio docierała do okienka owalnego
ś
limaka).
TYPY GŁUCHOTY
Głuchota przewodz
ą
ca (conductive hearing loss)zazwyczaj polega na procesie tzw.
„otosklerozy”, który powoduje zrastanie strzemi
ą
czka z ko
ś
ci
ą
wokół okienka owalnego
(leczenie: - „stapedectomia”)
Głuchota czuciowo-nerwowa (sensorineurial hearing loss) jest spowodowana uszkodzeniem
nerwu słuchowego lub komórek włosowatych ucha wewn
ę
trznego. (Test Webera z
kamertonem)
•W
ś
limaku nast
ę
puje dekompozycja d
ź
wi
ę
ku na sinusoidalne komponenty z zachowaniem
cz
ę
stotliwo
ś
ci, amplitudy i fazy
•Tonotopia : ró
ż
nice wra
ż
liwo
ś
ci na poszczególne cz
ę
stotliwo
ś
ci w zale
ż
no
ś
ci od miejsca wzdłu
ż
przebiegu
ś
limaka. Zale
ż
no
ść
ta („separacja” cz
ę
stotliwo
ś
ci) jest utrzymywana w przebiegu
centralnych szlaków słuchowych w mózgu
Kluczowe centralne neuronalne elementy uczestnicz
ą
ce w analizie słuchu
•(Zwój spiralny
ś
limaka – cz
ęść
obwodowa)
•J
ą
dro
ś
limaka (grzbietowe, przedniobrzuszne i tylnobrzuszne), z którego sygnał rozdziela si
ę
w
kierunku szeregu struktur mózgowia
•Superior olivary complex (interakcja z druga stron
ą
umo
ż
liwiaj
ą
ca lokalizacj
ę
d
ź
wi
ę
ku)
•J
ą
dro wst
ę
gi bocznej (n. lemniscus lateralis)
•Wzgórki dolne
ś
ródmózgowia (kontakt z układem motorycznym!)
•Wzgórze (medial geniculate complex)
•Kora mózgowa
Ś
limak
jest wła
ś
ciwym organem czuciowym układu słuchowego.
•Koncepcja Bekesy zakłada jak gdyby istnienie licznych rezonatorów (dla ró
ż
nych cz
ę
stotliwo
ś
ci)
wzdłu
ż
ś
limaka („filtruj
ą
cych” poszczególne cz
ę
stotliwo
ś
ci)
MECHANORECEPTORY: Wewn
ę
trzne i zewn
ę
trzne KOMÓRKI WŁOSKOWATE
Wewn
ę
trze komórki włosowate stanowi
ą
zasadniczy mechanoreceptor odbiorczy fal
d
ź
wi
ę
kowych.
Synapsa na zł
ą
czu komórki włosowatej i aksonu obwodowego neuronu
zwoju spiralnego
ś
limaka - transmiter: najprawdopodobniej Glu
Synapsa ta
adaptuje
si
ę
(ubytek neurotransmitera) i jest te
ż
odpowiedzialna za tzw.
maskowanie
recepcji d
ź
wi
ę
ku
•Czy komórka włosowata mo
ż
e by
ć
uznana za rodzaj neuronu?
–Za: Tworzy synaps
ę
(a tak
ż
e jest postsynaptyczna)
–Przeciw: Nie wytwarza jednak potencja
łów czynno
ś
ciowych •
Zmiany potencjału receptorowego w kierunku hyperpolaryzacji s
ą
znacznie mniejsze ni
ż
w kierunku
depolaryzacji
Wychylenie tylko w jednej osi powoduje zmiany potencjału receptora
Wysokie st
ęż
enie potasu w endolimfie na skutek jego wydzielania przez stria vascularis. (kwas
etakrynowy jest ototoksyczny poniewa
ż
uszkadza wydzielaj
ą
ce potas komórki stria vascularis)
•Oscylacyjne zmiany potencjału receptorowego potrafi
ą
dokładnie powiela
ć
przebieg pobudzenia
mechanoreceptora przez fal
ę
d
ź
wi
ę
kow
ą
a
ż
do cz
ę
stotliwo
ś
ci 3kHz (u człowieka)
•„Stała czasowa” receptora (RC time constant) powoduje zmniejszanie amplitudy waha
ń
potencjału
receptorowego wraz ze wzrostem cz
ę
stotliw
ś
ci
•Przy stymulacji powy
ż
ej 3 kHz zanika komponenta zmienna (a.c.) ale dalej utrzymuje si
ę
komponenta
depolaryzacyjna (d.c.) potencjału receptorowego („rektyfikacja”) co pozwala na dalsze uwalnianie
neurotransmitera.
ROLA ZEWN
Ę
TRZNYCH KOMÓREK WŁOSOWATYCH
Teoria Bekesy’ego nie wystarcza do wyja
ś
nienia mechanizmu wysoce selektywnego rozkładu
zło
ż
onej fali d
ź
wi
ę
kowej na składowe o ró
ż
nych cz
ę
stotliwo
ś
ciach

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
55/113
U ssaków mechanizm amplifikacji wra
ż
liwo
ś
ci na d
ź
wi
ę
ki jest zwi
ą
zany z wła
ś
ciwo
ś
ciami
„elektromotorycznymi”
zewn
ę
trznych komórek włosowatych
ś
limaka
(zmiana długo
ś
ci w
wyniku zmian napi
ę
cia elektrycznego) w czym gra rol
ę
białko zwane PRESTIN.
Myszy knock-outowe pozbawione genu prestyny maj
ą
podwy
ż
szony próg wra
ż
liwo
ś
ci (a
heterozygotyczne dla prestyny maj
ą
po
ś
redni próg). Uszkodzenie zewn
ę
trznych kom.
włosowatych osłabia czuło
ść
narz
ą
du Cortiego zarówno na „sił
ę
” jak i na cz
ę
stotliwo
ść
drga
ń
.
Rola CHLORU
Prestyna reaguje na wewn
ą
trzkomórkowe zmiany Cl-.
Zewn
ę
trzne komórki włoskowate (Outer hair cells) s
ą
„nabytkiem” ssaków i pozwalaj
ą
na 100x
(40dB) podwy
ż
szenie wra
ż
liwo
ś
ci na d
ź
wi
ę
ki wysokiej cz
ę
stotliwo
ś
ci
W szczególno
ś
ci ZKW zwi
ę
kszaj
ą
rozró
ż
nianie poszczególnych składowych cz
ę
stotliwo
ś
ci co
ma ogromne znaczenie w rozpoznawaniu mowy.
ROLA ZEWN
Ę
TRZNYCH KOMÓREK WŁOSOWATYCH (ZKW) c.d.
Komórki te (ZKW) pod wpływem dra
ż
nienia mechanicznego (odkształcenie włosków)
wytwarzaj
ą
potencjał receptorowy (podobnie jak komórki włosowate wewn
ę
trzne) który
jednocze
ś
nie oddziałuje na ich długo
ść
!
Oscylacje ZKW wzmacniaj
ą
drgania błony podstawnej co prowadzi do efektu „wzmacniacza
ś
limakowego” a ponadto powoduje tzw.
otoacustic emissions
- emisje otoakustyczne (David
Kemp 1978) propagowane wstecznie do błony b
ę
benkowej. Emisje otoakustyczne mog
ą
by
ć
samoistne (u 1/3 ludzi) lub wywoływane (w kilka do kilkudziesi
ę
ciu milisekund po krótkim
d
ź
wi
ę
ku).
Uwa
ż
ano je za „echo
ś
limakowe” ale ich energia jest wi
ę
ksza ni
ż
d
ź
wi
ę
ku stymulujacego
Emisje otoakustyczne s
ą
wykorzystywane w diagnostyce skriningowej słuchu (zanikaj
ą
przy
utracie ponad 30 dB) oraz słuchu u noworodków.
S
ą
te
ż
najprawdopodobniej przyczyn
ą
powstawania tzw. tonów Tartiniego.
Nie s
ą
natgomiast przyczyn
ą
„dzwonienia” w uszach (tinnitus), którego przyczyna tkwi
najprawdopodobniej w OUN.
Tony kombinacyjne („tony Tartiniego”):
„Dodatkowe” tony („distortion products”, „intermodulation products”) pojawiaj
ą
ce si
ę
na skutek tzw.
nieliniowo
ś
ci
ucha wewn
ę
trznego w czasie wspólnego brzmienia dwóch
ró
ż
nych (konsonansowych?) tonów o cz
ę
stotliwo
ś
ciach f1 and f2 i których cz
ę
stotliwo
ść
jest
sum
ą
lub ró
ż
nic
ą
cz
ę
stotliwo
ś
ci dwóch tonów „wywołuj
ą
cych”
f= m f1 ± n f2
(m, n - liczby całkowite)
W szczeg. f=2f1 - f2 jest całkiem słyszalny
Tony kombinacyjne („tony Tartiniego”):
Zazwyczaj słyszalne s
ą
tony ró
ż
nicowe (najlepiej 2f1-f2)
(sumacyjne s
ą
prawdopodobnie maskowane?)
Tony Shepard’a/Risset’a
Shepard wymy
ś
lił w 1964 roku układ emituj
ą
cy d
ź
wi
ę
k składaj
ą
cy si
ę
z samych
„harmonicznych” składowych (oktawy), z powodu czego ucho nie jest w stanie dokładnie
okre
ś
li
ć
wysoko
ś
ci d
ź
wi
ę
ku („fundamentalnej” składowej) i w efekcie d
ź
wi
ę
k je
ś
li powtarzany
odbierany jest jako pseudo wzrastaj
ą
cy a je
ś
li jest stały ma si
ę
wra
ż
enie „syrenowego
glissanda” o pseudowzrastaj
ą
cej wysoko
ś
ci d
ź
wi
ę
ku (Risset).
Unerwienie eferentne n.Cortiego
pochodzi z kompleksu j
ą
der górnej oliwki
(medial Olivary Complex OC unerwia oba ucha)
Lateral OC prawie wył
ą
cznie to
ż
stronne)
Aferentne unerwienie n.Cortiego
przez obwodowe aksony (niektórzy nazywaj
ą
je dendrytami)
dwóch typów neuronów w zwoju spiralnym
ś
limaka (aksony centralne tworz
ą
nerw słuchowy).
Typ I neuronu – unerwia komórki włosowate wewn
ę
trzne (wł. zmielinizowane) w relacji 1:1
komórki włosowate wewn
ę
trzne (KWW)
Typ II neuronu - unerwia komórki włosowate zewn
ę
trzne (KWZ) (wł.bezmielinowe – wolne
przewodzenie) w relacji 1:5-100
Centralne aksony neuronów Typu I stanowi
ą
95% (ok.. 30 000 u człowieka) włókien
n.słuchowego! i to one stanowi
ą
główny przekaz informacji do mózgu. (5% aksony neuronów
Typu II)
Rola aferentnego unerwienia KWZ (zewn
ę
trznych) nie jest poznana (same komórki WZ
odgrywaj
ą
rol
ę
„wzmacniacza” ale w relacji do KWW)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
56/113
NERW SŁUCHOWY
Selektywna wra
ż
liwo
ść
poszczególnych włókien nerwowych otrzymuj
ą
cych impulsacj
ę
z
wewn
ę
trznych komórek włosowatych, oceniana w postaci tzw. krzywej strojenia (tuning curve)
Tuning curve tip (cz
ę
stotliwo
ść
charakterystyczna włókna, dla której włókno jest najbardziej
wra
ż
liwe) jest najprawdopodobniej wynikiem działania zewn
ę
trznych komórek włosowatych
silnie wzmacniaj
ą
cych selektywno
ść
cz
ę
stotliwo
ś
ciow
ą
włókien nerwu słuchowego
•
Phase-locking – odwzorowanie fazy drgania
•Odwzorowanie fazy drgania – phase locking (=fazy potencjału receptorowego) w cz
ę
sto
ś
ci
„spikes” (potencjałów czynno
ś
ciowych) w nerwie słuchowym (odst
ę
py czasu pomi
ę
dzy
„spikes” s
ą
równe okresowi drga
ń
fali (lub jej całkowitym wielokrotno
ś
ciom)
•Phase-locking jest wierne dla ni
ż
szych cz
ę
stotliwo
ś
ci (<3kHz). Powy
ż
ej 3kHz kapacytacja
komórek włosowatych powoduje zanikanie phase-locking.
Znaczenie neuronów górnej oliwki
(tzw. olivocochlear neurons OC-neurons) w adaptacji do silnych d
ź
wi
ę
ków oraz „wyostrzaniu”
słuchu (nast
ę
pny slajd)
Funkcja relacji cz
ę
sto
ś
ci „wyładowa
ń
” do siły d
ź
wi
ę
ku (sound pressure level) we włóknie
nerwu słuchowego (Rate-level function) w zale
ż
no
ś
ci od obecno
ś
ci lub braku jednoczesnej
stymulacji neuronów górnej oliwki (tzw. olivocochlear neurons OC-neurons)
Aktywno
ść
OC-neuronów powoduje „przestrojenie” włókien nerwu słuchowego do d
ź
wi
ę
ku o
wy
ż
szym nasileniu
Ponadto
stymulacja OC obni
ż
a odpowied
ź
na szum ale podwy
ż
sza na bodziec
(Jest to tzw.
antimasking czyli w gruncie rzeczy „wyostrzanie” słuchu )
Zasada „tonotopii” na całej długo
ś
ci dróg słuchowych
Główne morfologiczne typy neuronów j
ą
der
ś
limakowych oraz odpowiadaj
ą
ce im
charakterystyczne histogramy ich wyładowa
ń
(potencjałów czynno
ś
ciowych) nast
ę
puj
ą
cych
po stymulacji d
ź
wi
ę
kiem (post-stimulus time = PST)
Zró
ż
nicowanie wskazuje na ró
ż
nice funkcji pomi
ę
dzy komórkami w analizie ró
ż
nych aspektów
perceptu d
ź
wi
ę
kowego
Najwi
ę
ksze synapsy w mózgu zwi
ą
zane s
ą
z przewodzeniem słuchu
„Spherical bushy cells” daj
ą
najlepsze „powielenie” impulsacji z nerwu słuchowego dzi
ę
ki
olbrzymim poł
ą
czeniom synaptycznym – tzw. „endbulbs of Held” . Równie
ż
inne olbrzymie
synapsy (tzw. Kielichy – calyces) s
ą
obecnie w drodze słuchowej (na neuronach medial
nucleus of trapezoid body)
•
Eferentne unerwienie ucha:
–komórki włosowate: otrzymuj
ą
eferentne unerwienie z neuronów j
ą
der oliwki górnej OC (omówiono
poprzednio)
– mi
ęś
nie ucha wewn
ę
trznego unerwione ruchowo:
•(m.stapedius – n.VII, m.tensor tympani – n.V)
•Mi
ęś
nie te s
ą
unerwione przez motoneurony w relacji 1:1 (jak mi
ęś
nie okoruchowe –
b.precyzyjnie)
•Skurcz mi
ęś
ni powoduje zmniejszenie przewodzenia w zakresie niskich cz
ę
stotliwo
ś
ci (<1kHz)
o 25 dB (inaczej ni
ż
kom. OC)
•Efektem jest ochrona narz
ą
du słuchu oraz lepsze rozumienie mowy. Z kolei ich skurcz w
czasie mówienia mo
ż
e hamowa
ć
odpowiedzi na d
ź
wi
ę
ki własne osobnika (skurcz w czasie
mówienia).
–Unerwienie sympatyczne (autonomiczne) naczy
ń
krwiono
ś
nych ucha. (mało poznane - osobi
ś
cie
uwa
ż
am,
ż
e mo
ż
e mie
ć
znaczenie w „strojeniu” słuchu w sytuacjach emocjonalnych, stresowych itp..
??)
•
Rozpoznawanie kierunku
ź
ródła d
ź
wi
ę
ku
–1. mi
ę
dzyuszna ró
ż
nica czasu (Interaural Time Difference)
•Dla ni
ż
szych cz
ę
stotliwo
ś
ci dzi
ę
ki odwzorowaniu fazy drga
ń
ró
ż
nica fazy drgania
pomi
ę
dzy jednym i
drugim uchem mo
ż
e by
ć
wykorzystana do rozpoznawania kierunku
ź
ródła d
ź
wi
ę
ku jednak dla
wy
ż
szych cz
ę
stotliwo
ś
ci musz
ą
by
ć
u
ż
yte inne metody
–2. mi
ę
dzyuszna ró
ż
nica nat
ęż
enia (Interaural Level Difference)
•Wykorzystywana dla wysokich cz
ę
stotliwo
ś
ci
Dla (minimalnie rozpoznawanej) ró
ż
nicy kierunku
ź
ródła
d
ź
wi
ę
ku wynosz
ą
cej ok. 1 stopnia odpowiednie ró
ż
nice wynosz
ą
:

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
57/113
różnica czasu = 10 mikrosekund
różnica natężenia = 1 dB
•Wzgórki dolne –
•Miejsce integracji informacji z j
ą
der
ś
limakowych jak i górnej oliwki (dane o kierunku)
••prawdopodobnie miejsce tworzenia „mapy przestrzennej d
ź
wi
ę
ku” (podobnie jak w
przypadku wzgórków górnych tworz
ą
cych map
ę
wzrokow
ą
)
•Ciało kolankowate przy
ś
rodkowe (medial geniculate)
•Przynajmniej cz
ęś
ciowo zorganizowane tonotopowo
•Ma poł
ą
czenia z c.migdałowatym (wykorzystywane w warunkowaniu reakcji strachu na bod
ź
ce
d
ź
wi
ę
kowe)
•Otrzymuje bardzo silne poł
ą
czenia ze strony kory słuchowej !
•Proste sygnały (w tym szum)
aktywuj
ą
pierwotn
ą
kor
ę
słuchow
ą
lecz nie kor
ę
wtórn
ą
, natomiast mowa aktywuje kor
ę
wtórn
ą
•Cz
ęść
kory (Pole AI) posiada neurony wra
ż
liwe na ró
ż
nice czasowe i intensywno
ś
ci d
ź
wi
ę
ku
(pozwalaj
ą
ce na lokalizacje
ź
ródła)
•Ciało migdałowate nadaje znaczenie emocjonalne bod
ź
com czuciowym (w tym słuchowym).
Neuroproteinopatie neurodegeneracje
W poszukiwaniu jednolitej teorii neurodegeneracji.
Rola patologicznych białek w patogenezie schorzeń zwyrodnieniowych ośrodkowego
układu nerwowego
Stanley B. Prusiner laureat Nagrody Nobla za odkrycie prionów twierdzi, że schorzenia
neurodegeneracyjne są chorobami wywołanymi przez nieprawidłowe mechanizmy przetwarzania
(„processing”) białek.
Mechanizmy te obejmują
:
nieprawidłowe składanie białek w struktury przestrzenne (Misfoilding)
nieprawidłowe modyfikacje potranslacyjne
nieprawidłowe rozcinanie protein (cleavage)
nieprawidłowe składanie białek (splicing)
nieprawidłowa ekspresja
zmniejszone fizjologiczne procesy niszczenia białek
Prusiner i inni zwracają uwagę na to, że wszystkie schorzenia neurodegeneracyjne mają postacie
sporadyczne (zwykle najczęstsze spośród przypadków danego typu choroby) oraz rodzinne (genetyczne) w
większości schorzeń rzadkie ( z wyjątkiem HD i ataksji rdzeniowo-móżdżkowych, w których występują
praktycznie wyłącznie przypadki genetycznie uwarunkowane - rodzinne)
Wspólnym mianownikiem neurodegeneracji są depozyty różnych białek ale powstaje pytanie: czy te
agregacje są per se patologiczne (czy są pierwotną i rzeczywistą przyczyną neurodegeneracji) czy też są
przejawem sekwestracji białek, których usuwanie z komórki jest niewydolne (np. przez mechanizmy
związane z proteasomem 26S) ?
Nie stwierdzono jednoznacznych korelacji między ilością depozytów i ciężkością choroby.
Agregacaje tworzą często bardzo różne mieszaniny białek.
Agregacje występują u osób bezobjawowych
Za przyczynową rolą depozytów białkowych przemawiają badania genetyczne.
Przypadki rodzinne neurodegeneracji
Mutacje w genach kodujących agregujące białka są genetycznie związane z rodzinnymi postaciami chorób
neurodegeneracyjnych (AD, ALS, TSE) powodują wcześniejsze pojawienie się objawów choroby i cięższy
przebieg niż w przypadkach sporadycznych.
Modele zwierzęce
Transgeniczne myszy, które wykazują nadekspresję zmutowanego ludzkiego APP wytwarzają typowe

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
58/113
zmiany dla AD (morphologicznie depozyty amyloidowe i „kliniczne” objawy zaburzeń kognitywnych)
[Games i wsp 1995].
Podobnie jest w przypadku transgenicznych myszy z ludzkim dzikim typem genu
α
αα
α
-synucleiny, które
wykazują kliniczno-patologiczne zmiany (nie wszystkie) typowe dla PD (ciałka Lewy’ego, ubytek
dopaminergicznych zakończeń w zwojach podstawy i zaburzenia ruchowe „typu pozapiramidowego”)
[Masliah i wsp. 2000].
Transgeniczne myszy zawierające exon 1 ludzkiego białka huntingtin z ekspansją nukleotydów 115-156
CAG wytwarzają śródjądrowe inkluzje neuronalne zawierające huntingtin i ubiquitin oraz później
zaburzenia ruchowe typu HD a także utratę wagi. [Mangiarini i wsp. 1996]
Nadekspresja zmutowanego ludzkiego genu PrP u myszy powoduje zmiany gąbczaste typowe dla
sporadycznej TSE. [Hsiao i wsp. 1990]. Niekiedy same insercje ludzkiego genu do genomu myszy
prowadzą do objawów klinicznopatologicznych i agregacji „misfolded” protein!
Transgeniczne myszy z ludzkim SOD1 z objawami „ALS-like”.
Niektóre formy TSE, AD i ataksji u zwierząt i ludzi nie wykazują wykrywalnych agregacji pomimo
uszkodzenia mózgu i objawów chorobowych stąd m.in. wynika, że degeneracje mogą być spowodowane
samym nieprawidłowym białkiem, które niekoniecznie musi tworzyć depozyty...
Wstrzyknięcie homogenatu bogatego w „misfolded proteins” przyspiesza chorobę w modelu zwierzęcym
(modele AD i innych amyloidoz) Amyloid-A (z SAA) [Lundmark i wsp. 2002],
Udowodniono in vitro, że pierwotnie rozpuszczalne białka takie jak: amylina (IAPP), lizozym po dodaniu
doustnym niewielkiej ilości ich form włókienkowych rozpoczynają proces agregacji.!!!).
Spośród „chorób konformacyjnych” tylko TSE w sposób przekonywujący wykazują właściwości
„zakaźne” (transmisja)
Dlatego można wyróżnić:
1) amyloidozy pasażowalne
2) amyloidozy niepasażowalne
Objawy kliniczne i zmiany neuropatologiczne różnych neurodegeneracji mogą nakładać się
Proteinopatie CSN (wg typów białek szczególnie uwikłanych w patogenezę)
agregacje włókienek białka A
ββββ
(amyloidozy)
agregacje białka MAP-tau (tauopatie)
agregacje białkowe w chorobach wywołanych niestabilnością
powtarzalnych tripletów
nukleotydowych
agregacje prionowe (CJD i inne TSE)
agregacje
α
αα
α
-synukleiny i podjednostek neurofilamentów w
ciałach Lewy’ego (synukleinopatie:
ch.Parkinsona, Demencja+LB, multiple system atrophy)
agregacje innych różnych białek (depozyty np. w SZB, w zesp. Shy-Drager, aktyna w c.Hirano)
Amyloidozy = choroby konformacyjne białek
(Glenner GG – 1980 NEJM 52:148 -
ββββ
-fibrylozy)
ββββ
-fałdowa struktura pozwalająca na liczne wiązania wodorowe pomiędzy fibrylami białka co sprawia, że
struktura jest stabilna.
Białka mogące tworzyć amyloid: (ponad 20 białek)
łańcuchy lekkie przeciwciał (AL),
surowicze białko amyloidu A (SAA –protein),
białka „endokrynne”,
białko amyloidowe A
ββββ
W „stanie rozciągniętym” (wprzeciwieństwie do helikalnego) Paulinga grupy NH i CO, (które tworzą
wiązania wodorowe) „wystają” pod kątem prostym w stosunku do osi pasma białka. Jeśli dwa takie
pasma w „stanie rozciągniętym” są ułożone jeden wzdłuż drugiego tworzą się wiązania wodorowe

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
59/113
pomiędzy pasmami. W zależności od kierunku ułożenia „pozycje” oznaczane są jako równoległe lub
„antyrównoległe”. „Antyrównoległe” ustawienie jest bardziej stabilne bo mostki wiązań wodorowych są
lepiej „spasowane”.
(amyloidoza uogólniona, odczynowa)
serum amyloid associated : SAA (20mg/L) wytwarzane w wątrobie, po stymulacji zapalnej
(makrofagi, IL-6, IL-1)
→
→
→
→
>1g/L
(białko AA = 76 resztowy N-koncowy odc. SAA)
(amyloidoza uogólniona, pierwotna)
szpiczak i inne proliferacje kk-B
(białko AL)
(inne amyloidozy uogólnione)
tow.hemodializie (
ββββ
2-mikroglobulina – składnik MHC class - I)
rodzinne amyloidozy (transthyretyna – transport tyroksyny i retinolu)
(amyloidozy zlokalizowane)
a. Mózgowe (np. AD, amyloid precursor p. APP - A
ββββ
),
endokrynne (typ II cukrzycy – amylina)
Amyloidozy mózgowe :
APP .(Amyloid-
ββββ
protein odkryte przez Glennera i Wonga w 1984 – początek „nowej ery” w badaniach
nad Ch.Alzheimera)
Ch.Alzheimera, wrodzony krwotok mózgowy z amyloidozą-holenderski: HCHWA-D, zesp.Downa,
mózgowa angiopatia amyloidowa
PrP (CJD, GSS, Kuru, FFI)
Cystatyna C (zmutowany inhibitor proteinazy cysteinowej) HCHWA-1 (typ islandzki)
transtyretyna (wariant) .
Rodzinna amyloidoza mózgowa – typ węgierski
ponadto mutacje transtyretyny prowadzą do:
wrodzone neuropatie (rodzinne polineuropatie amyloidowe) amyloid gromadzi się w nerwach
prowadząc do zespołów typu HSAN hereditary sensory and autonomic neuropathy
PROTEIN MISFOLDING: Rola chaperonów w zachowaniu
funkcjonalności struktury
Z wyjątkiem białka tau głównie zawierającego alfa helisy w pozostałych białkach odcinki bogate w
ββββ
-
fałdy uważane są za uwikłane w neurodegenerację.
Pasma
ββββ
-fałdowe biegną prostopadle do długiej osi włókien.
Nie wiadomo czy misfolding wywołuje agregację czy też oligomeryzacja indukuje zmiany konformacyjne.
Protofibryle: bogate w struktury
ββββ
sheet , nierozgałęzione, szer. 3-6 nm, dług. do 100 nm, wydłużają się
przez „coalescence” i mają fizykochemiczne cechy amyloidu. Pozostają też w dynamicznej równowadze z
oligomerami A
ββββ
i są prekursorami włókien amyloidowych.
Mechanizmy „spaczonego składania” i agregacji białek
W A
ββββ
, PrP, alfa-synukleinie odpowiedzialne są miejsca hydrofobowe
W HD i innych chorobach „tripletów” CAG agregacja jest związana wiązaniami wodorowymi (glutamina
ma aminową grupę dostarczającą spolaryzowane boczne łańcuchy i tendencją do tworzenia wiązań

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
60/113
wodorowych z wodą).
Liczne hipotezy próbujące wyjaśnić (względnie) selektywne zajęcie różnych struktur OUN w AD, HD, PD,
ALS.
Wśród proponowanych mechanizmów „spaczonego składania” i agregacji białek rozważa się
rolę:
jonów metali,
patologicznych białek chaperonowych,
pH,
stresu oksydatywnego,
nadmiernego nagromadzenia makromolekuł
Mechanizm „nukleacji” (seeding/nucleation) przypominający krystalizację
Oligomery białkowe stanowią jądra krystalizacji.
Mechanizmy neurodegeneracji w „proteinopatiach”
1. Utrata funkcji (rozważana w ALS, HD i TSE)
In vitro PrP, SOD1, A
ββββ
wykazują aktywność SOD (stąd utrata funkcji może promować ROS-y)
2. Uzyskiwanie właściwości (neuro)toksycznych „Gain of toxic activity” (przez „źle
poskładane” białka)
To jest najbardziej szeroko akceptowana hipoteza.
Badania in vitro: agregacje „źle poskładanych” białek powodują apoptozę, podobnie jest w przypadku
oligomerów ze strukturami typu
ββββ
-sheet w białkach nie kojarzonych z jakąkolwiek chorobą (być może
jakichkolwiek białek!) [Bucciantini i wsp. 2002].
A) aktywacja apoptozy poprzez sygnalizację pozakomórkową
np. poprzez wieloligandowy receptor RAGE (Receptor for Advanced Glycation End products)
B) zabieranie istotnych białek komórkowych do kompleksów amyloidowych
np. składniki cytoszkieletowych białek, proteasomu, czynników transcrypcyjnych, chaperonów są obecne
w agregacjach
α
αα
α
-synucleiny i huntingtiny.
C) Formacja kanałów jonowych lub porów śródbłonowych
D) Indukcja stresu oxydatywnego: wolne rodn., oxydacja lipidów, Ca2+
Być może ważniejsze jest samo „misfolding” i oligomeryzacja niż agregacja? [Hartley i wsp. 1999,
Goldberg i Lansbury Jr 2000]
Agregacja i sekwestracja może być mechanizmem obronnym przeciwdziałającym toksycznym efektom
„źle poskładanych” białek poprzez ich izolację [Watase i wsp 2000].
3. Hipoteza patogenezy proteinopatii poprzez wywoływanie odczynu zapalnego przez
„źle poskładane” białka
ZA: astroglioza i aktywacja mikrogleju w sąsiedztwie złogów. Podwyższenie poziomu różnych
czynników zapalnych w mózgu takich jak: cytokiny, chemokiny, czynniki wzrostu. In vitro – „żle
poskładane” białka indukują wydzielanie białek zapalnych przez mikroglej i astroglej. Niektóre dane
wskazują na pozytywne działanie w AD leków przeciwzapalnych.
PRZECIW: Reakcja zapalna może mieć charakter obronny (zahamowanie aktywacji składnika
C3 komplementu wzmagało neurodegenerację w transgenicznych zwierzących modelach AD)[Wyss-
Coray i Mucke 2002].
Choroby neurodegeneracyjne z otępieniem (demencją):
(podział na „korowe” i „podkorowe” nieco umowny)
•
KOROWE
•
Ch.Alzheimera,
•
Otępienie czołowo-skroniowe (Fronto-temporal dementia)
•
Ch. Picka,

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
61/113
•
Choroba rozsianych ciałek Lewy’ego (DLBD),
•
Zwyrodnienie korowo-podstawne (corticobasal ganglionic degeneration-CBGD)
•
Chr.17-linked d.,
•
Postępująca glejoza podkorowa (Prog.subc.gliosis)
PODKOROWE
•
Ch.Parkinsona,
•
Postępujące zwyrodnienie nadjądrowe (Progressive supranuclear palsy = Ch.Steele’a-
Richardsona-Olszewskiego) ,
•
ALS-plus, (ALS+demencja)
•
Neurodegeneration with brain iron accumulation type-I =NBIA-1 = (gen kinazy pantotenianu vit
B5) = (dawniej zwana “Hallervorden-Spatz disease” )
ch.Huntingtona
Choroba Alzheimera:
Główna przyczyn otępienia
Nieprawdłowa przemiana prekursora amyloidu (beta-amyloid precursor protein czyli APP) w amyloid
beta protein czyli A
ββββ
.;
Patologia: „blaszki starcze” NFT, kongofilna angiopatia, zwyr.ziarnisto-wodniczkowe
UWAGA : blaszki starcze mogą występować u osób bez cech otepienia dlatego nie sam fakt wystąpienia
blaszek ale ich ilość (szacowana wg kryteriów tzw. CERAD) jest podstawą do neuropatologicznego
rozpoznania (w gruncie rzeczy – jedynie potwierdzenia rozpoznania klinicznego) choroby Alzheimera.
Funkcja APP nieznana, knock-out-owe myszy wykazują jedynie niewielkie zaburzenia motoryczne i
niespecyficzną gliozę.
Ostatnio sugeruje się rolę APP jako:
1)
Receptora dla kinezyny-1 (szybki transport aksonalny)
2)
„
ββββ
-stub” z C-końcem APP pełni rolę wnikającej do jądra molekuły sygnalizacyjnej (analogia z
Notch intracellular domain w embriogenezie)
α
αα
α
-sekretaza: proces realizowany najprawdopodobniej przez kilka enzymów i specyficzny nie dla
określonej sekwencji aminokwasów w łańcuchu ale dla „dystansu” ok. 16 reszt licząc od błony
komórkowej.
„Droga amyloidogenna” endoproteolizy APP
Procesy enzymatyczne :
ββββ
-sekretaza i
γγγγ
-sekretaza (cięcie w domenie śródbłonowej) tworzą „drogę
amyloidogenną” (
ββββ
-sheet)
Być może proces
γγγγ
-sekretazy musi poprzedzać dodatkowo hipotetyczna
εεεε
-sekretaza
Obydwa procesy
ββββ
-sekretazy i
γγγγ
-sekretazy zachodzą niemal jednocześnie po INTERNALIZACJI APP
Podobne procesy dotyczą białka Notch i ErbB4
ββββ
-sekretazę zidentyfikowano jako enzym o nazwie:
ββββ
-site APP-cleaving enzyme (BACE)
Jest to potencjalnie obiecujący terapeutycznie „target”
(BACE-knock-out-owe myszy są bezobjawowe a zatem przypuszczalnie jest małe prawdopodobieństwo, że
blokowanie enzymu może grozić bardzo poważnymi działaniami ubocznymi)
Akumulacja zwłaszcza amyloidogennych form A
ββββ
otwiera HIPOTETYCZNĄ tzw. „KASKADĘ
AMYLOIDOWĄ”
UWAGA ! Nie każdy peptyd A
ββββ
jest amyloidogenny!
Peptyd A
ββββ
kończący się na reszcie 40 (A
ββββ
40) stanowi 90% wydzielanych peptydów A
ββββ
i jest prawidłowym
i nieamyloidogennym produktem komórkowym - jakkolwiek nieznana jest jego rola.
Reszta peptydów A
ββββ
to amyloidogenne A
ββββ
42 i A
ββββ
43.
Przypuszczalnie mogą one pełnić funkcję ośrodków nukleacji i tworzenia amyloidu także z udziałem
A
ββββ
40.
Za tworzenie różnych wariantów białka A
ββββ
(A
ββββ
42 i A
ββββ
43) odpowiedzialne są RÓśNE farmakologicznie
γγγγ
-
sekretazy.
Mutacje APP oraz białek presenilin 1 i 2 (PS1, PS2) w rodzinnej AD
prowadzą do nasilonej produkcji fibrylo (amyloido)-gennego A
ββββ
42
Zdecydowana większość zidentyfikowanych mutacji prowadzących do rodzinnej AD dotyczy presenilin!

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
62/113
Mutacje presenilin PS1 i PS2, które prowadzą do rodzinnej choroby Alzheimera (FAD) powodując
nadprodukcję amyloidogennego A
ββββ
42
•
Dokładna rola presenilin i nikastryny w procesie
γγγγ
-sekretazy nie jest całkowicie jasna
•
Szereg obserwacji wskazuje na własności proteazowe presenilin jednak m.in. odmienna
dystrybucja wewnątrzkomórkowa presenilin w stosunku do miejsc gdzie zarówno APP jak i
Notch1 są rozszczepiane w domenie wewnątrzbłonowej (tzw. „spatial paradox”) i inne dane
czynią identyfikację
γγγγ
-sekretaza = presenilina niepewną.
•
γγγγ
-sekretazowa czynność jest prawdopodobnie realizowana przez więcej niż jeden enzym
•
Preseniliny są raczej „kofaktorami”
γγγγ
-sekretazy
Białko tau:
gen na chr. 17
białko towarzyszące mikrotubulom (MAP-tau), obecne w aksonach oraz w astrocytach i
oligodendrocytach.
W Ch. Alzheimera występuje w postaci nadmiernie ufosforylowanej (wzmożona aktywność kinaz? lub
osłabiona fosforylaz?) i tworzy charakterystyczne zwyrodnienia czyli NFT (neurofibrillary tangles),
Złogi białka tau nie wykazują przewagi
ββββ
-fałdowej lecz
α
αα
α
-helikalne [Sadqi i wsp. 2002].
Udział MAP-tau w AD jest raczej wtórny.
Genetyka ch. Alzheimera
(rodzinne postacie AD – dziedziczone autosomalnie dominująco)
1. Mutacje genu APP (chr.21),
efektem jest nadprodukcja l/lub przyspieszona agregacja A
ββββ
trisomia 21 (efekt „dawki genu),
Hered.cerebr.hemorrhage-with-amyloidosis (HCHWA)
2. Mutacje preseniliny (PS1 – chr.14, PS2 – chr.1)
białka (wraz z nikastryną) pełnia istotną rolę w rozszczepianiu różnych transbłonowych protein
związanych z transdukcją sygnałów (np. Notch, ErbB4), także APP. (PS1 jest być może
gamma sekretazą? Zob dyskusje powyżej)
Kilkadziesiąt mutacji (zamiana poj.aminokwasów)
związanych z rodzinną AD i wczesnymi
objawami
(nadprodukcja A
ββββ
)
Rola apolipoproteiny E w zagrożeniu ch. Alzheimera
Polimorfizm apolipoproteiny E (chr.19)
(białko łączy się z A
ββββ
; ułatwia agregację?)
Zwiększone 8x ryzyko AD (late-onset AD >65 l.) w przypadku genotypu homozygotycznego E4/E4 (dwa
identyczne allele dla izoformy E4), i 4x gdy tylko jeden z alleli jest typu E4. Natomiast ryzyko jest
zmniejszone w przypadku gdy nie ma E4 a zwłaszcza w przypadku układu E2/E2
izoforma
εεεε
4 może stanowić „patologiczny chaperon” dla A
ββββ
(prowadzący do tworzenia konformacyjnych
struktur amyloidowych)
Alan Roses wykazał w 1994, że interakcja
εεεε
4 z białkiem tau może być odpowiedzialna za szybsze
tworzenie paired helical filaments (PHF), natomiast
εεεε
2 i
εεεε
3 sekwestrują białko tau i opóźniają tworzenie
PHF.
KRYTERIA ROZPOZNAWANIA DEFINITYWNEGO CHOROBY ALZHEIMERA
Wytyczne CERAD
KRYTERIA ROZPOZNAWANIA HISTOPATOLOGICZNEGO CHOROBY ALZHEIMERA WG
CERAD OPARTE SĄ NA ILOŚCIOWEJ OCENIE WYSTĘPOWANIA BLASZEK STARCZYCH
(ZŁOGÓW W KTÓRYCH GŁÓWNYM BIAŁKIEM JEST BETA-AMYLOID)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
63/113
Synukleinopatie:
1
CH.PARKINSONA,
2
DEMENCJA+LB,
3
ZANIK WIELOUKŁADOWY (multiple system atrophy)
(4?)
(Neurodegeneration with brain iron accumulation type-I =NBIA-1 = gen kinazy
pantotenianu vit B5)
alfa-synukleina: 140 aminokwasów; zakończenia presynaptyczne, funkcja nieznana
•
α
αα
α
-synukleina gra rolę w sekwestracji dopaminy w pęcherzykach synaptycznych,
•
mutatcje
α
αα
α
-synukleiny (nie-amyloidowego komponentu blaszek starczych w AD) powodują
wzrost tendencji do tworzenia form protofibrilli, co prowadzi do nieprawidłowej formacji i/lub
przepuszczalności pęcherzyków z dopaminą i do neurotoksyczności
•
α
αα
α
-synukleina może wytwarzać kanały („pory”) dziurawiące błonę komórkową
•
α
αα
α
-synukleina jest obecna w c.Lewy’ego razem z ubikwityną, podjednostkami proteasomu, heat
shock proteins, neurofilamentami
•
po 70 r.ż. 1,5% populacji ma zmiany typu PD („senile” condensation of
α
αα
α
-synuclein and hence
tendency to form oligomers with
ββββ
-pleated sheets)
Choroba Parkinsona
•
Sporadyczna w 95%, genetycznie uwarunkowana w <5%
•
Klin.: sztywność, hypokineza, spowolnienie, drżenie stałe, „liczenie pieniędzy”, otępienie-późno,
początek ok.58 r.z. (początek w rodzinnej chorobie Parkinsona jest znacznie wcześniej)
•
Degeneracja neuronów w s.nigra prowadzi do redukcji dopaminy w striatum (leczenie L-dopą,
prekursorem dopaminy)
•
czasami mutacje alfa-synucleiny (non-amyloid proteinaceous component of senile plaques in AD)
•
Alfa-synuclein jest obecna w ciałkach Lewy’ego (LB)
•
Ciałka.Lewy’ego w s.nigra (staje się blada) i w korowych neuronach
•
W 5-7% autopsji są parkinson-type zmiany bez klinicznych symptomów („preclinical phase”?),
•
Po 70 r.ż 1,5% populacji ma zmiany typu PD
•
Diffuse Lewy Body Dementia (Otępienie z rozsianymi korowymi LB)
•
Degeneracja neuronów w s.nigra prowadzi do redukcji dopaminy w striatum (leczenie L-dopą,
prekursorem dopaminy)
•
Modele eksperymentalne: „toksyczne” i „genetyczne”
•
Objawy, które w różnym stopniu imitują kliniczne i patologiczne cechy choroby Parkinsona
można wywołać m.in. Podając zwierzętom: MPTP oraz rotenon (środek insektobójczy blokujący
mitochondrialny kompleks I co powoduje wzrost ROS i uszkodzenie komórek)
•
Transgeniczna Drosophila m. Z ludzkim dzikim lub „typowo” (A53T; A30P) zmutowanym genem
α
αα
α
-synukleiny (drozofila nie posiada białka
α
αα
α
-synukleiny ). W 30-60 dniu wykazuje selektywna
deplecję DA-neuronów i zab motoryczne (brak ujemnej reakcji geotaktycznej). Zaburzenia te
likwiduje podanie lewodopy, bromokryptyny lub pergolidu.
Choroba Parkinsona - genetyka
•
Mutacje łączące się z rodzinną PD – geny (niektóre):
–
Locus PARK1: kodujący
α
αα
α
-synuclein (autosomalna dominująca early-onset PD)
–
Locus PARK2: kodujący parkin (=E3 ubiquitin ligase, bierze udział w „ubiquitin-
proteasome pathway” UPP; autosom reces, juvenile-onset PD)
–
Locus PARK5: kodujący UCHL1 (ubiquitin carboxy-terminal hydrolase L1,
odpwiedzialna za recycling molekuł ubiquitin w UPP; autosom.dom. z niepełną
penetracją)
–
Gen białka DJ1 (białko DJ1 - locus PARK7 – (autosomalnie recesywna PD)
Stwardnienie boczne zanikowe (ALS)
•
Etiologia nieznana – liczne teorie patognetyczne
•
Czas trwania ok.. 2 lat
•
Choroba sporadyczna (90%) lub dziedziczna (rodzinna)
•
W 5-10% choroba dziedziczna (rodzinna – Familal amyotrophic lateral sclerosis – FALS) –
zwykle początek objawów 10 wcześniej niż w sporadycznym SBZ

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
64/113
•
Mutacja genu z locusem na chr. 18 jest prawdopodobnie najczęstszą wśród rodzinnej choroby
•
Jak dotąd wśród konkretnie rozpoznanych mutacji określonego genu najczęstszą jest mutacja
genu Cu/Zn SOD1 (25% wszystkich FALS)
•
Delecja homozygotyczna genów SMN2 dodatkowo pogarsza rokowanie (czynnik modyfikujący)
•
Ubytek „górnych” i „dolnych” motoneuronów
•
Degeneracja mieliny w drogach piramidowych
•
Wtręty (ciałka Buniny, wtręty „skeinowe”-nitkowate, wtręty szkliste, ubikwityno-pozytywne)
•
Sferoidy aksonalne, chromatoliza, glioza,
Choroby prionowe
Choroby z patogenezą prionową
•
ZWIERZĘTA
•
Scrapie:owce, kozy
•
TME (pasażowalna encefalopatia norek)
•
CWD (przewlekłą wyniszczająca choroba jeleni): jeleń, łoś
•
BSE (gąbczasta encefalopatia bydła)
•
Inne EG: koty, antylopy, pumy, tygrysy (ZOO)
•
LUDZIE (podział wg. obrazu klinicznego)
•
CJD: Choroba Creutzfeldta-Jacoba
•
GSS: Gerstmann-Sträussler-Scheinker syndrome
•
FFI: Śmiertelna rodzinna bezsenność
•
Kuru
•
Postępujące zglejowacenie podkorowe ?
PRIONY
"PROTEINACEOUS INFECTIOUS PARTICLES”
Białko PrPc (cellular), jest prawidłowym białkiem kodowanym przez pojedynczy exon z pojedynczej kopii
genu na krótkim ramieniu chr. 20 (ok. 210 aminokw.)
Prion, oznaczany jako PrPsc (scrapie) jest zmodyfikowaną formą prawidłowego białka komórkowego
PrPc białko błonowe (synaptyczne?) neuronów złączone z glikoinozytolowym fosfolipidowym
zakotwiczeniem
PrPc jest wrażliwe na proteazę natomiast PrPsc jest oporne na proteazę
PrPsc wprowadzone do komórki powoduje trwałe chemiczne i/lub konformacyjne zmiany PrPc
prowadzące do powstania „kopii” PrPsc
•
Jednym z największych i szokujących obserwacji były obserwacje „zakażenia” małp przez materiał
pochodzący z rodzinnych (genetycznych) przypadków CJD !! (w 1981 przeniesienie fCJD na naczelne)
•
Oprócz PrPc podobne własności „samoreplikacji-konformacyjnej” mają białka drożdży Ure2p i Sup35.
Białko PrPc jest szczególnie silnie ekspresjonowane w neuronach gdzie prawdopodobnie może pełnić rolę
w magazynowaniu lub „sygnalizacji” miedzi (Cu) [Brown Qin, Herms i inni Nature 1997, 390:684-7]
Zmiany konformacyjne białka priona mogą występować bez zmian istotnych modyfikacji chemicznych
- oporne na procedury modyfikujące kwasy nukleinowe
- Gen (PRNP) u człowieka na krótkim ramieniu chr. 20; pojedynczy exon (3)
- PrPc 27-30 kD (prekursor 33-35kD) 254 aminokwasy.
Po odcięciu N- i C-końcowych sekwencji
„dojrzały” prion (reszty od 23 do 231) ma ok. 210 aminokwasów
„Własne” allele genu PrP oraz układ odpornościowy konieczny do efektywnego zakażenia*
*myszy z zesp. SCID oraz myszy „null” (PrP o/o) (Büeler et al., 1993)
czyli pozbawione alleli dla PrP są oporne na infekcję.
Konwersja PrPC do PrPSc obejmuje redukcję struktur alfa helix i wzrost „beta fałdowych” struktur w
obrębie protein prionowych (Pan et al., 1993)
Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
65/113
Wraz z wiekiem prawdopodobnie obniża się bariera energetyczna pomiędzy stanem energii „low-level”
dla prawidłowej konformacji PrP i „high-level” dla patologicznegpo konformera
Neuropatologia chorób „prionowych”
•
Ogólnie choroby prionowe u ludzi i zwierząt można z grubsza podzielić na takie w których:
•
1. (większość chorób) są zmiany gąbczaste i akumulacje PrP ale bez (lub z nielicznymi) plakami
amyloidowych
•
2. (tylko przypadki GSS) Liczne plaki amyloidowe PrP (białko jest silnie „okrojone”) i różnie
nasilone zgąbczenie
•
3. (nowy „wariant” CJD - vCJD) Silne zgąbczenie, liczne akumulacje PrP oraz liczne plaki PrP.
(W vCJD nie ma mutacji PRNP.)
Choroba Creutzfeldta-Jakoba
Opisana w 1920-1921 jako postępująca demencja z przeżyciem poniżej 1 roku z miokloniami a później z
piramidowymi i pozapiramidowymi objawami
Sporadyczna CJD
1/1000 000/rok; 55-70 r.ż.; przebieg śr. 7 mieś.
1. Możliwa (postępujące otępienie, czas trwania poniżej 2 lata, bez typowych zmian EEG i objawy jak
niżej)
2. Prawdopodobna [postępujące otępienie, typowy EEG i min. 2 z objawów: a) mioklonie, b) zab. widzenia
lub móżdżkowe, c) zab. piramid-pozapiramidowe, d) mutyzm akinetyczny]
3. Definitywna (typowy obraz neuropatologiczny i/lub stwierdzenie złogów PrP immunohistochemicznie)
Rozpoznanie neuropatologiczne CJD:
Prawdopodobna [typowe objawy kliniczne oraz zmiany gąbczaste bez innych niejsnych interpretacyjnie
zmian morfologicznych]
Definitywna : wymagane spełnienie jednego z dodatkowych (oprócz powyższych) kryteriów:
- obecność amyloidowych złogów (plak typu Kuru) PrP
- obecność PrPSc
- wykazanie transmisji choroby na zwierzęta
- obecność patogenetycznej mutacji PRNP
Kliniczna diagnostyka CJD
•
EEG w 60% (1-2 cykli/s uogólnione trójfazowe periodyczne zespoły ostrej fali)
•
CSF: białko pow.0.4g/L, białka: p130,p13, 14-3-3,
•
CT/NMR: norma lub atrofia, spektroskopia NAA bez rezultatów
Konieczność weryfikacji neuropatologicznej
Różnicowanie neuropatologiczne CJD
wymaga różnicowania (przynajmniej w teorii) wszelkich typów otępień.
•
Zmiany gąbczaste spotyka się w licznych schorzeniach takich jak ch. Alzheimera, ch. Picka, ch. Ciałek
Lewy’ego, otępienie typu czołowego, zespół ALS-plus,
•
„status spongiosus” (nieswoista wakuolizacja w przebiegu różnych schorzeń, oraz artefaktyczna)
•
Glioza i zaniki neuronalne są często spotykane w niemal wszystkich neurodegeneracjach i innych
schorzeniach OUN
Neuropatologia CJD
•
Wyróżnia się wiele podtypów z uwagi na dystrybucję zmian.
•
Podstawowymi zmianami są:
–
zgąbczenie, (Masters i Richardson określają „stan gąbczasty – st.spongiosus” z
większymi wakuolami do 100 mikrometrów oraz „zwyrodnienie gąbczaste – spongiform
degeneration” w którym wakuole są małe 5-25 mikrometrów)
–
zanik neuronów,
–
astroglioza,
–
niekiedy blaszki amyloidowe , nazywane też tzw. blaszkami-Kuru (w 5-10%)
•
Zmiany mogą się bardzo różnić w poszczególnych przypadkach stopniem zasilenia i dystrybucją.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
66/113
•
Zmiany w istocie białej uważa się za wtórne (chociaż tzw. vacuolar myelopathy może też należeć
do zmian pierwotnych)
Neuropatologia nowego wariantu: nvCJD (vCJD)
•
Do końca 2001 zachorowało 106 osób w Wlk.Brytanii.
•
Śr. Wiek 28 lat, średni czas choroby – 13 mieś., obj.psychiczne
•
śaden z chorych nie miał mutacji PRNP
•
Wszyscy zbadani (76) mieli Met/Met w kodonie 129
•
Charakterystyczne są bardzo liczne PAS+ amyloidowe blaszki prionowe oraz masywne
prymitywne PrP depozyty które są PAS-ujemne. Blaszki najliczniejsze są w móżdżku i w płatach
potylicznych. (W przeciwieństwie do GSS gdzie też sa bardzo liczne blaszki, PrP w vCJD jest
oporne na proteazy oraz choroba nie jest dziedziczna.)
•
Jest też duże nasilenie zmian gąbczastych (zwłaszcza w j.podstawy i wzgórzu).
•
We wzgórzu jest szczególnie silna astroglioza.
•
Wiele blaszek jest szczególnie specyficznych – tzw. „florid”
•
Typowe jest też powstawanie „klasterów” blaszek
•
PrP stwierdza się poza CSN (migdałki)
•
Status kodonu 129 PRNP a fenotyp choroby
sporadycznej
•
*UWAGA! Status kodonu 129 białka priona ma kluczowe znaczenie dla fenotypu choroby
prionowej zarówno sporadycznej oraz jatrogennej a także rodzinnej. (zob dalej slajd „genetyka
CJD”)
•
Na podstawie analizy 300 przypadków SPORADYCZNEJ CJD z USA, i Europy wyróżniono
kilka osobnych molekularno-neuropatologiczno-klinicznych grup z uwagi na:
–
status kodonu* 129 (MM, MV, VV) oraz rezultat cięcia proteinazą K (typ 1 = 21 kDa i
typ 2 = 19 kDa). W rezultacie wyróżniono grupy:
•
MM1 lub MV1 (70% przyp „typowej CJD”)
•
VV2 (16% CJD z ataksja i późną demencją bez typowych zmian EEG i mioklonii)
•
MV2 (9% ataksja i relatywnie powolna demencja)
•
MM2 (4% trudna diagnostycznie, bezsenność: Sporadic Fatal Insomnia -SFI,)
•
VV1 (1% rozległa wakuolizacja bez zmian w EEG i bez ataksji)
•
Ogólnie w sporadycznej CJD 90% chorych jest homozygotyczna dla albo Met albo Val
•
Natomiast 100% przypadków wariantu (vCJD) była homozygotyczna dla Met.
Genetyka CJD
•
W 15% CJD jest rodzinna i dotyczy wtedy młodszego wieku; (w 85% CJD jest sporadyczna);
•
W rodzinnej CJD
najbardziej typowe mutacje PRNP to:
–
a) kodon 200 (zamiast kw. Glu jest lizyna Glu=>Lys)
–
b) kodon 178 (zamiast kw. Asparaginowego jest asparagina Asp => Asn) – Ma też
znaczenie status kodonu 129 (zob. niżej)
–
c) dodatkowe insercje powtarzalnych oktapeptydów, których w normalnym PRNP jest 5.
–
Ad b) w przypadku mutacji w kodonie 178 o fenotypie decyduje kodon 129- jeśli jest w
nim Val to jest CJD, jeśli jest w nim Met to jest FFI,
•
W sporadycznej CJD NIE MA MUTACJI natomiast podwyższone ryzyko dla sporadycznej i
jatrogennej CJD jest u homozygot w kodonie 129 PRNP, ponadto ryzyko wzrasta z wiekiem.
•
1%>> przypadkowe zakażenie (hormon wzrostu, przeszczepy),
Genetyka śmiertelnej rodzinnej bezsenności FFI
Mutacja kodonu 178 identyczna jak w części przypadków rodzinnej CJD Asp (acid)=> Asn (asparagine).
(Mutacje wykryto w 1992 a w 1995 chorobę przeniesiono na gryzonie.)
Obraz kliniczny zależy od kodonu 129, Jeśli (oprócz mutacji w kodonie 178), w kodonie 129 jest
homozygotyczność z metioniną (Met/Met) to przypadek wykazuje cechy odpowiadające FFI, a jeśli jest
Val/Val to jest to CJD.
Opisano też przypadek sporadycznej FI bez mutacji genu PRNP (ale również z zajęciem wzgórza) i
przeniesiono chorobę na myszy uzyskując taki sam obraz neuropatologiczny jak w FFI. [Mastrianni i wsp.
NEngJMed 1999, 340, 1630-8]

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
67/113
Genetyka GSS
•
Niemal wyłącznie przypadki rodzinne, dominująco somatycznie z typową mutacją kodonu 102
(zamiast proliny jest leucyna pro=>leu P102L) będącą pierwszą opisaną mutacją w chorobie prionowej
a także m.in. w potomstwie oryginalnego przypadku rodziny opisanej przez Gerstmanna.
•
Na non-human primates przeniesiono w 1981r. Zmutowane białko jest względnie wrażliwe na
proteinazy w przeciwieństwie do pozostałych chorób prionowych.
•
Inne rzadsze mutacje w GSS to zamiany aminokwasów w kodonach 105, 117, 198, 217, oraz stop w
kodonie 145 (skrócone białko PrP i atypowy wariant GSS przypominający chorobę Alzheimera)
Porównanie „klasycznej” CJD
i (nowego) „wariantu” of CJD
•
KLASYCZNA:
•
wiek: VII dekada
•
Czas trwania: 7ms.
•
WARIANT:
•
Wiek: III dekada.
•
Czas trwania: 1-2 lat
•
Obj. kliniczne: zab. osobowości; późne otępienie; brak typowych zmian w EEG
•
Neuropathologia: liczne Kuru-like plaki i tzw. „florid-plaques” (z wakuolami)
Neuropatologia Kuru
•
Kuru (choroba najprawdopodobniej zaczęła się między 1900 i 1920 rokiem od pojedynczego
sporadycznego przypadku CJD, następnie zjedzonego...;
•
kobiety i dzieci częściej chorowały niż mężczyźni, (którzy zjadali wyłącznie mięso)
•
zmiany w móżdżku, zanik neuronów, „torpedowate” aksony komórek Purkinjego, glioza
Bergmanna, zmiany gąbczaste, zmiany w korze mózgu niewielkie i głównie w obszarach
„przyśrodkowych” (również w skorupie, j,ogoniastym).; liczne amyloidowe blaszki o promienistej
budowie zwłaszcza w w.ziarnistej, PAS+ („kuru-plaques”) !! W 1966 udowodniono zakaźność
homogenatów mózgu z ch. Kuru (w 1968 także dla sporadycznej CJD, a w 1981 dla fCJD)
Neuropatologia niektórych pozostałych chorób prionowych
•
GSS: wielordzeniowe, PAS+, multicentryczne blaszki amyloidowe, w móżdżku blaszki dominują
w warstwie drobinowej kory (w przeciwieństwie do CJD)
–
Niektóre przypadki GSS (niektóre typy mutacji) wykazują obecność silniejszych niż dla
danego wieku zmian neurofibrylarnych (NFT). NFT sa podobnie jak w Alzheimerze
zbudowane z ufosforylowanego białka tau. Ponadto w mutacji Q217R stwierdza się złogi
białka amyloidowego A
ββββ
na obrzeżach plak PrP.
–
W mutacji GSS (Y145Stop - silnie skrócone białko PrP) stwierdzono obfite złogi PrP w i
wokół naczyń w parenchymie mózgu i oponach, natomiast bez zgąbczenia.
•
FFI oraz sporadyczna FI: charakterystyczne „wybiórcze” zajęcie wzgórza z zanikiem neuronów
jednak bez zgąbczenia.
Choroby wywołane niestabilnością powtarzalnych
tripletów nukleotydowych
w większości ekspansja CAG (kodon glutaminy)
(CTG leu) Dystrofia miotoniczna
(GAA glu) Ataksja Friedreicha
Choroby wywołanych niestabilnością powtarzalnych tripletów nukleotydowych (w 100% genetyczne)
Choroba Huntingtona (ekspansja CAG w genie huntingtyny, chr.4)
Choroby z niestabilnością TN w obrębie sekwencji podlegającej translacji
Ch. Kennedy’ego
Ataksja rdzeniowo-móżdżkowa-
dentatorubropallidoluysial atrophy

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
68/113
Choroby j.w. w obrębie sekwencji niekodującej (zab. reg. transkrypcji)
Dystrofia miotoniczna (CTG leu)
Ataksja Friedreicha (GAA glu)
Ch.Huntingtona
:
Autosom.domin 4-7/100 000. (gen HD huntingtin, 4p16.3, kr. ekspansja CAG (glutamina- schorzenie
„poliglutaminowe”)
norma 9-37 w HD nawet ponad 100
Początek w III & IV dek. (im bardziej liczne triplety tym wcześniejszy początek)
Objawy:
hyperkinezje
(ruchy mimowolne, chorea, atetoza),
dementia;
Typ hyperkinetyczny (częstszy),
Typ akinetyczny-ze sztywnością (rzadki, zwykle z młodzieńczym początkiem, dziedziczony od ojca,
szczególnie agresywny)
Późny początek – (pow. 49) – dziedz. od matki
PATOLOGIA
Ubytek głównie medium spiny cells (GABA neurons), (póżniej także „aspiny”,
Astrogliosis;
Inkluzje (huntingtin) wewnątrzjądrowe szczeg w neuronach kory
Patologiczne neuryty w korze (ubiq+)
Inne (poza Ch.Huntingtona) choroby wywołane niestabilnością
powtarzalnych tripletów nukleotydowych
Choroby z niestabilnością TN w obrębie sekwencji podlegającej translacji
Ch. Kennedy’ego
(eksp. CAG, receptor androgenowy)
–genetyka: X-liked; klin.: pocz.w III dek.;
zesp. dolnego motoneuronu, (bulbospinal), niepłodność, ginekomastia, czuciowa neuropatia
neuropatologicznie: wtręty jądrowe białka AR wykrywane p-ciałem
przeciwko AR (lub p-
ubikwitynie)
Choroby wywołanych niestabilnością powtarzalnych tripletów nukleotydowych - Choroby j.w. w obrębie
sekwencji niekodującej (zab. reg. transkrypcji)
Ataksja Friedreicha :
autosm.reces. GAA (Glu) w genie frataxin; nawet ponad 1000 powtórzeń kodonu
(zaburzenie transportu żelaza – gromadzenie żelaza w mitochondraich - stres oksydatywny)
w 60% kardiomiopatia, w 10% cukrzyca
Tauopatie
Choroby z patologicznym białkiem tau („tauopatie”)
Zanik wieloukładowy - Multiple system atrophy
Pojęcie wprowadzone przez Grahama i Oppenheimera w 1969 obejmuje n/w zesp., - wszystkie z
obecnością depozytów białek (ubikwit., tau, tubulina,
α
αα
α
B-crystalin,
α
αα
α
-synuclein) w cytoplazmie kom.
glejowych (oligodendrocyty) oraz w cytoplazmie i jądrach kom. nerwowych.
1)
Zespół Shy-Dragera : b.rzadki; początek V-VI dekada; przewaga mężczyzn; początkowo objawy
wegetatywne (niedocisnienie ortostat., impotencja, zmniejszone wydzielanie potu, osłabiona
tolerancja ciepła, bezdech senny) później inne objawy (parkinsonizm, zesp.móżdżkowe, porażenie
gałkoruchowe, obj.piramidowe, porażenia zwieraczy, porażenia opuszkowe); zgon w ciągu kilku
lat (ponad 50% w ciągu 6 lat). Neuropatologicznie (oprócz wtrętów): zaniki neuronalne w

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
69/113
różnych okolicach mózgu, móżdżku, pnia (i.czarna, m.sinawe), zanik neuronów zwł. Pośrednio-
bocznych strefach i.szarej rdzenia kręgowego (może być nieobecny); astroglioza, wtręty w
oligodendrocytach
2)
Degeneratio striato-nigralis (zwyrodnienie prążkowia i istoty czarnej)
3)
Atrophia olivo-ponto-cerebellaris (zanik oliwkowo-mostowo-móżdżkowy)
Otępienia nie-Alzheimerowskie:
Otępienie czołowo-skroniowe (i warianty-odmiany)
Choroba Picka
Otepienie czołowo-skroniowe lub otępienie bez wyróżniającej patologii
Postepująca afazja
Otepienie semantyczne
Otępienie z ALS
Otępienie mezolimbiczne
Zwyrodnienie korowo-podstawne
Postępujące porażenie nadjądrowe (Ch. Steele’a-Richardsona-Olszewskiego)
Postępująca podkorowa glioza
Otępienie z ziarnami srebrochłonnymi
Otępienie z włosowatopodobnymi srebrochłonnymi wtrętami
Otępienie czołowo-skroniowe (FTD)
Choroba rodzinna w ok. ½ przypadków (17q21-22)
Klinicznie: otępienie i głębokie zmiany osobowości i zachowania.
Początkowo brak dbałości o higienę osobistą, nieostrożna (reckless) jazda samochodem, „odhamowanie”
zachowania, nadmierne jedzenie, alkoholizm, wczesny zaniki świadomości społecznej, świadomości osoby,
zachowania perseweracyjne i rytualne, zanik sprawności językowej. Zachowanie orientacji i praksji.
Przebieg powolny, w późniejszym okresie dochodzą obj.pozapiramidowe
Choroba neuropatologicznie i klinicznie bywa rozpatrywana w łączności z chorobą Picka i tzw. dementia
lacking distinctive neuropathology (DLDN) a ponadto niektórzy (Brun i wsp, Giannakopulos i wsp.) do
„grupy FTD” dołączają zespół ALS-plus.
Choroba Picka
Bardzo rzadka; charakterystyczny zanik płata czołowego i skroniowego i zaoszczędzenie tylnych 2/3
zakrętu skroniowego górnego.
Zaniki neuronów zwł. III warstwy kory, glejoza, achromatyczne neurony (tzw. komórki Picka) oraz ciała
Picka („kule srebrochłonne”)
Ciała Picka są dodatnie dla ubikwityny oraz MAP-tau i chromograniny. Występują w nich „paired-
helical-filaments” oraz proste tubule
Komórki i ciała Picka najliczniejsze w zakręcie zębatym i w korze czołowej. Są też zmiany typu
zwyrodnienia ziarnisto-wodniczkowego.
Zwyrodnienie korowo-podstawne (CBD = corticobasal ganglionic degeneration =cortico-nigral
degeneration)
Opisane w 1967, dotąd opisano ok. 100 przypadków;
Przebieg: trudności wykonywania złożonych czynności pojawiają się asymetrycznie w jednej kończynie a
później w następnych („pozycja dystoniczna” kończyny), trudności chodzenia, sztywność
pozapiramidowa, mioklonie, zespół „obcej kończyny” (alien limb), dysartria, porażenie nadjądrowe
ruchów oczu; otępienie późno po 4-5 latach choroby.
Neuropatologia (kryteria NINDS): obrzmiałe achromatyczne neurony w III, V i VI warstwach kory
dodatnie dla MAP-tau i neurofilamentów. Wtręty „korowo-podstawne” w s.nigra (ubikwityno+ i tau+);
zmiany mieszają się często z PSP oraz ze zmianami alzheimerowskimi i parkinsonowskimi.
Postępujące porażenie nadjądrowe
Ch. Steele’a-Richardsona-Olszewskiego
Choroba zdefiniowana w 1964 r., zwykle sporadyczna choć obserwowano przypadki rodzinne (ale te
raczej uważa się za warianty tzw. FTDP-17). Chorobę rozpoznaje się posługując się kryteriami NINDS
(PSP możliwe, prawdopodobne, definitywne).

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
70/113
Neuropatologiczne rozpoznanie opiera się również na kryteriach NINDS (Typowy PSP, Złożony PSP,
CBD)
Typowo: wiek powyżej 40 r.ż., powolny narastający przebieg, zesp. Parkinsonowski (akinetyczny ze
sztywnością) nie reagujący na leczenie L-dopą; porażenie pionowych ruchów gałek ocznych*, porażenie
rzekomoopuszkowe zaburzenia ruchów sakkadowych i częste upadki w pierwszym roku choroby, wczesna
dysartria, wczesne otępienie.
* Rostral interstitial nucleus (vertical gaze center)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
71/113
Neuronalna kontrola ruchu
Dwa „piony” dowodzenia ruchem:
–„Osiowy-posturalny”
–„Boczny-wolicjonalny” („precyzyjny”)
Trzy typy układu motorycznego:
szkieletowy,
autonomiczny
neuroendokrynny
Rola impulsacji czuciowej:
Receptory w mi
ęś
niach
Receptory w stawach
Receptory skórne,
ś
luzówkowe, inne
Impulsacja bł
ę
dnikowa i oczna
J
ą
dra podstawy i mó
ż
d
ż
ek uczestnicz
ą
w kontroli ruchu poprzez Górny Motoneuron (GMN)
Mi
ęś
nie szkieletowe – zł
ą
cze nerwowo-mi
ęś
niowe – jednostki motoryczne
Motoneurony unerwiaj
ą
ce okre
ś
lony mi
ę
sie
ń
tworz
ą
„pul
ę
” (motor neuron
pool)
w substancji szarej rogów przednich rdzenia (w kształcie wrzecionowatego klasteru)
•Trzy podstawowe typy włókien mi
ęś
niowych w zale
ż
no
ś
ci od szybko
ś
ci wytwarzania
skurczu w odpowiedzi na pobudzenie z nerwu i na odporno
ś
c na zm
ę
czenie
(obserwacja zmiany siły skurczu na pocz
ą
tku i po wielu minutach dra
ż
nienia stał
ą
cz
ę
sto
ś
ci
ą
wyładowa
ń
):
•S (Slow twitch) = typ 1
(czerwone, obfita mioglobina i mitochondria )
•FR (fast (fatigue) resistant)
= 2a
•FF (fast „fatigable” twitch) = typ 2b
(blade, mało mitochondriów, grubsze-silniejsze, jednostki
motoryczne „obsługuj
ą
” liczniejsze włókna – wi
ę
kszy współczynnik unerwienia „innervation ratio”)
Jednostki motoryczne
–(od 3-4 włókien do 2000 – tzw. współczynnik unerwienia – Innervation Ratio)
–Mi
ęś
nie oczu IR
≈
3-10
–Mi
ęś
nie poruszaj
ą
ce palcami
≈
100
–Mi
ę
sie
ń
gastrocnemius
≈
2000
•
UWAGA!
Mi
ę
sie
ń
, który kurczy si
ę
wolniej, wolniej si
ę
rozkurcza (stabilniejsza praca)
•Regulacja siły skurczu: dwie kooperuj
ą
ce metody
•1
. wzrost cz
ę
sto
ś
ci wyładowa
ń
w
α
-motoneuronach
•2
. rekrutacja kolejnych jednostek motorycznych wg „zasady wielko
ś
ci”
(size principle
) czyli rozmiaru
motoneuronu
•Kombinacja obu metod ró
ż
na w ró
ż
nych mi
ęś
niach (np. w drobnych mi
ęś
niach r
ę
ki szybka rekrutacja
wielu jednostek a potem gradacja cz
ę
stotliwo
ś
ci wyładowa
ń
)
•AD 1.
Schemat po prawej:
zale
ż
no
ść
siły skurczu od cz
ę
stotliwo
ś
ci potencjałów cz. we włóknie nerwowym – sumowanie si
ę
efektów pojedynczych potencjałów – a
ż
do skurczu t
ęż
cowego
Odpowiada to wzrastaj
ą
cej cz
ę
sto
ś
ci wyładowa
ń
w nerwie od 8/sek do 100/sek („fused tetanus”) .
•AD 2.
„SIZE PRINCIPLE” (Henneman 1957) – zasada sekwencji rekrutacji typów jednostek
motorycznych wraz ze zwi
ę
kszaniem siły skurczu: S - pierwsze, poniewa
ż
maj
ą
ni
ż
szy potencjał
progowy.
•S (pierwsze)
→
FR
→
FF (ostatnie)
•Co przekłada si
ę
równie
ż
na rozmiar
α
-motoneuronów
•S (najmniejsze
α
-motoneurony)
→
→
FF (du
ż
e
α
-motoneurony)
•(im wi
ę
kszy neuron tym wi
ę
ksza „siła” jednostki)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
72/113
Struktura rdzenia – strefy Rexeda
•Somatopowa organizacja motoneuronów rdzenia
•Obszary przy
ś
rodkowe – mi
ęś
nie tułowia i proksymalne mi
ęś
nie ko
ń
czyn
–Interneurony tworz
ą
poł
ą
czenia niemal na wszystkich poziomach rdzenia
•Obszary boczne – mi
ęś
nie dystalne ko
ń
czyn
–Interneurony tworz
ą
poł
ą
czenia na niewielu s
ą
siaduj
ą
cych poziomach rdzenia
•Wa
ż
niejsze interneurony:
–Kk. Renshaw’a
, - tzw. interneurony rekurentne (glicyna, GABA?). Powoduj
ą
tzw. rekurentne
hamowanie (ujemne sprz
ęż
enie zwrotne) motoneuronów. S
ą
pobudzane przez te same neurony
(tak
ż
e przez motoneurony z mi
ęś
ni synergistycznych), które nast
ę
pnie hamuj
ą
. Przypuszcza si
ę
,
ż
e
ich zadaniem jest „poboczne hamowanie” innych synergistycznych motoneuronów w celu zwi
ę
kszenia
„kontrastu” mi
ę
dzy aktywnymi i nie(słabo)aktywnymi motoneuronami.
–IN - Ia
(glicyna, blaszka VII) – stymulowany przez włókna Ia z wrzecion mi
ęś
niowych hamuje
motoneurony mi
ęś
ni antagonistycznych (hamowanie recyprokalne). Z reguły sygnały stymuluj
ą
ce
dany motoneuron (np.. Z kory) jednocze
ś
nie stymuluj
ą
IaIN dla motoneuronów mi
ęś
ni
antagonistycznych. IaIN sa równie
ż
stymulowane przez lokalne CPG.
–Interneurony hamuj
ą
ce presynaptycznie
(ró
ż
ne podklasy dla włókien Ia, Ib, II (Ib z narz
ą
dów
Golgiego), czucia powierzchownego. – hamuj
ą
wydzielanie neurotransmitera w zako
ń
czeniach
włókien aferentnych ró
ż
nych dróg czuciowych (ale nie np.. Dróg korowo-rdzeniowych). Przeł
ą
czanie
stymulacji pomi
ę
dzy interneuronami dla włóken czuciowych Ia i dla Ib pozwala na mo
ż
liwo
ść
regulacji
balansu mi
ę
dzy odpowiedzi
ą
na impulsacj
ę
włókien Ia (rozci
ą
gni
ę
cie) lub włókien Ib (obci
ąż
enie).
Rdze
ń
kr
ę
gowy kontroluje funkcje motoryczne poprzez mechanizmy odruchowe
Odruch miotatyczny
•Wrzeciono mi
ęś
niowe nale
ż
y (wraz z narz
ą
dami Golgiego i receptorami stawowymi do
proprioceptorów (podrodzina mechanoreceptorów)
•Unerwienie wrzeciona mi
ęś
niowego: aksony włókien Ia tworz
ą
tzw.”pierwotne zako
ń
czenia
czuciowe”, włókna II tworz
ą
wtórne zako
ń
czenia czuciowe
Wrzeciono mi
ęś
niowe – cz
ęść
układu reguluj
ą
cego długo
ść
mi
ęś
nia
•Dwa typy intrafuzalnych włókien mi
ęś
niowych: typu „nuclea bag” i „nuclear chain”
•Pierwotne włókna aferentne Ia maj
ą
pocz
ą
tek w obu typach włókien mi
ęś
niowych
•Mniejsze tzw. wtórne aferenty typu II odchodz
ą
tylko z włókien typu „nuclear chain”
•Aferenty z włókien typu „nuclear bag” sygnalizuj
ą
pr
ę
dko
ść
rozkurczania si
ę
mi
ęś
nia i b.szybko
adaptuj
ą
si
ę
– jest to odpowied
ź
fazowa po któej nast
ę
puje „cisza” je
ś
li nie zmienia si
ę
długo
ść
mi
ęś
nia
•Oba typy aferentów (Ia i II) z włókien „ła
ń
cuchowych” wykazuja impulsacj
ę
proporcjonaln
ą
do
długo
ś
ci mi
ęś
nia
Ró
ż
nice impulsacji w biernym rozci
ą
ganiu mi
ęś
nia i w aktywnym skurczu
Rola p
ę
tli gamma (
γ
):
Wpływ p
ę
tli gamma (
γ
) na efektywno
ść
odruchu miotatycznego (jest to okrae
ś
lane mianem
„gain” czyli stopnia zysku reakcji odruchowej)
Aktywno
ść
γ
jest wy
ż
sza w trakcie ruchów wymagaj
ą
cych precyzji.
Efektywno
ść
odruchu miotatycznego regulowana jest te
ż
poprzez oddziaływanie na
alfamotoneurony.
Odruch z narz
ą
dów Golgiego reguluje napi
ę
cie mi
ęś
nia i pomaga np. w utrzymaniu kartki papieru
W
ś
ci
ę
gnie przeci
ę
tnego mi
ęś
nia znajduje si
ę
ok. 100 zako
ń
cze
ń
Golgiego. Ka
ż
de odpowiada
na skurcz okre
ś
lonej liczby włókien mi
ęś
niowych z danej jednostki motorycznej.
Odruchy wywoływane bólem – (receptory skórne):
1.Skurcz zginaczy po tej samej stronie
2.Skurcz prostowników po stronie przeciwnej (utrzymanie równowagi i postawy)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
73/113
(Ale co b
ę
dzie gdy wywoła
ć
odruch jednocze
ś
nie z obu stron ??? obustronne odruchy „znios
ą
si
ę
” ?
nie jest to jasne)
•Uwaga!
•Istniej
ą
równie
ż
tzw. transkortykalne (cz
ęś
ciowo te
ż
prawdopodobnie „subkortykalne”)
odruchy rozci
ą
gowe (long-latency stretch reflexes).
•Odruchy pozostaj
ą
pod kontrol
ą
o
ś
rodków wy
ż
szych (przykład spastyczno
ś
ci i „szoku rdzeniowego”)
Central pattern generator
O
ś
rodkowy generator wzorca motorycznego
Motoryczny układ szkieletowy (hierarchia „wojskowa”?!)
Najni
ż
ej -
a
motoneurony (MN)
(„Wy
ż
ej”?) – interneurony rdzenia i pnia
Wy
ż
ej – tzw. generatory wzorców ruchowych („motor pattern generators” –MPG (układy
interneuronów)
Najwy
ż
ej -- inicjatory wzorców ruchowych (motor pattern initiators MPIs)
„Wej
ś
cie-input” I,S,C mo
ż
liwe do ka
ż
dego „szczebla hierarchii”
MPGs i MPIs równie
ż
maj
ą
własn
ą
hierarchi
ę
•Generator wzorca ruchu (MPG = CentralPG = program motoryczny) to grupa zwi
ą
zanych
funkcjonalnie interneuronów koordynuj
ą
cych okre
ś
lon
ą
aktywno
ść
ruchow
ą
realizowana przez wiele
mi
ęś
ni (np. odruchy unikania - withrawal,
ż
ucie, oddychanie, połykanie, kaszel, kichanie, chód)
•Najlepiej poznane u homarów (lobster) i minoga morskiego (lamprey)
•CPG – s
ą
w pniu i rdzeniu
Oscylatorowa teoria pływania u morskiego minoga (lamprey)•W obr
ę
bie CPG wyst
ę
puj
ą
neurony
charakteryzuj
ą
ce si
ę
oscylacyjn
ą
aktywno
ś
ci
ą
(„pacemakers”)
•Ta sama „sie
ć
” neuronalna mo
ż
e pod wpływem ró
ż
nych bod
ź
ców zmienia
ć
charakterystyk
ę
aktywno
ś
ci
•Czy jest szansa aby wykorzysta
ć
CPG do rehabilitacji po uszkodzeniu rdzenia?
•Input czuciowy w czasie mechanicznej stymulacji mo
ż
e stymulowa
ć
CPG, zmienia te
ż
syntez
ę
GABA
w hamuj
ą
cych interneuronach
•Leki serotoninergiczne i adrenergiczne prawdopodobnie mog
ą
sprzyja
ć
ponownemu odtworzeniu
aktywno
ś
ci CPG
•Sytuacja lepsza gdy zachowana jest cz
ęść
włókien korowo-rdzeniowych
•Receptory sygnalizuj
ą
ce pozycj
ę
i ruch ko
ń
czyn (propriocepcja i kinestezja) :
–Aferenty stawów (joint afferents)
–Wrzeciona mi
ęś
niowe
–Narz
ą
dy Golgiego
–Aferenty dotykowe (tactile afferents) – w mi
ęś
niach i skórze np. wolno adaptuj
ą
ce si
ę
aferenty
Ruffiniego poprzez sygnalizacj
ę
napi
ę
cia skóry informuj
ą
o pozycji palców
Kora ruchowa i przedruchowa
•Pierwotna kora ruchowa (pole Brodmanna 4) charakteryzuje si
ę
bardzo niskim progiem dla
pobudzenia ruchu w mi
ęś
niach.
•Komórki piramidowe Betza (s
ą
to wła
ś
ciwe „górne motoneurony” kory) z pierwotnej kory (warstwy V)
bezpo
ś
rednimi i po
ś
rednimi drogami korowo-rdzeniowymi i korowo-opuszkowymi tworz
ą
poł
ą
czenia z
dolnymi neuronami ruchowymi.
Płaty czołowe:
Okolica przedczołowa:
9,10,11,12,48
Okolica
przed
ś
rodkowa:
4 (ruchowe)
6 (przedruchowe)
44 (wieczkowe)
8 (czołowe po
ś
rednie)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
74/113
Dywergencja motoneuronów pierwotnej kory ruchowej•Jeden motoneuron pierwotnej kory
ruchowej „zaopatruje” kilka mi
ęś
ni i raczej reprezentuje okre
ś
lony ruch. („muscle field” górnego MN)
Konwergencja neuronów pierwotnej kory ruchowej•„pola” kory ruchowej, z których obserwowano
pobudzanie tych samych mi
ęś
ni (EDC= extensor digitorum communis)
„Strojenie” kierunkowe UMN pierwotnej kory ruchowej
Niektóre neurony pierwotnej kory ruchowej wykazuj
ą
ró
ż
n
ą
cz
ę
stotliwo
ść
potencjałów czynno
ś
ciowych
w zale
ż
no
ś
ci od kierunku zamierzonego ruchu.
Kora przedruchowa
•Tu równie
ż
mamy do czynienia z „górnymi motoneuronami”
•Zwrotne (dwustronne) poł
ą
czenia mi
ę
dzy kor
ą
przedruchow
ą
i pierwotn
ą
kora ruchow
ą
•Kora przedruchowa komunikuje si
ę
te
ż
bezpo
ś
rednio poprzez drogi korowo-rdzeniowe, w których
włókna z kory przedruchowej stanowi
ą
ok. 30%
•Wyró
ż
niamy boczn
ą
i przy
ś
rodkow
ą
kor
ę
przedruchow
ą
Kora przedruchowa: boczna i przy
ś
rodkowa•Ogólnie w korze przedruchowej przebiega
proces
selekcji (wyboru?)
akcji
planowanie, inicjowanie i ukierunkowywanie ruchów dowolnych.ruchowej,
•Boczna kora przedruchowa
: du
ż
a asocjacja aktywno
ś
ci neuronalnej w zale
ż
no
ś
ci od kierunku
(
zamierzonego
) ruchu i znacznie wcze
ś
niej przed wykonaniem ruchu (podobnie jak w korze
pierwotnej ale tutaj jest to zwi
ą
zane z warunkowanymi zadaniami motorycznymi, np..
warunkowanymi bod
ź
cami wzrokowymi lub słuchowymi). Uszkodzenie kory przedruchowej u
człowieka mo
ż
e te
ż
prowadzi
ć
do trudno
ś
ci w wykonywaniu zada
ń
wyznaczanych werbalnie
•Przy
ś
rodkowa kora przedruchowa
: równie
ż
prowadzi „selekcj
ę
” (= wybór z dost
ę
pnych,
wykonywalnych) zada
ń
ruchowych ale raczej inicjowanych przez bod
ź
ce wewn
ę
trzne (np.
odtworzenie ruchu „z pami
ę
ci”) ni
ż
zewn
ę
trzne (u małpy z uszkodzeniem kory przedruchowej
przy
ś
rodkowej obserwuje si
ę
redukcj
ę
ruchów „spontanicznych” przy zachowaniu ruchów
stymulowanych bod
ź
cami zewn
ę
trznymi – warunkowanych). Aktywno
ść
neuronów znacznie
wyprzedza aktywno
ść
ruchow
ą
podobnie jak w korze przedruchowej bocznej.
Droga korowo-rdzeniowa/opuszkowa (inicjacja ruchów dowolnych)Droga korowo-czerwienna
Poprzez szlak czerwienno-rdzeniowy wspomaga kontrol
ę
ruchów r
ą
k
Droga korowo-opuszkowa
Droga korowo-rdzeniowa boczna (skrzy
ż
owana) -(zawiaduje mi
ęś
niami odsiebnymi r
ą
k)
Droga korowo-rdzeniowa przednia (zawiaduje mi
ęś
niami osiowymi tułowia i proksymalnymi)
Nowy pogl
ą
d na mechanizm „o
ś
rodkowego” pora
ż
enia nerwu twarzowegoObszar ruchowy w
przednim zakr
ę
cie obr
ę
czy unerwia obustronnie cz
ęść
j
ą
dra n.VII dla górnej cz
ęś
ci twarzy
Bezpo
ś
rednie (droga korowo-rdzeniowa) i po
ś
rednie poł
ą
czenie kory z
rdzeniem
Poł
ą
czenie po
ś
rednie:
Poprzez j.czerwienne do mi
ęś
ni dystalnych r
ą
k
Poprzez twór siatkowaty do mi
ęś
ni osiowych i proksymalnych (wspomaganie utrzymania
równowagi wyprzedzaj
ą
ce zamierzony ruch ).
Twór siatkowaty
Czasowa i przestrzenna koordynacja ruchu
O
ś
rodek licznych odruchów (np.połykania, kichania,
ż
ucia)
Kontrola snu i czuwania
Kontrola pracy serca
Regulacja oddychania
•Górna cz
ęść
t.siatkowatego odgrywa
rol
ę
moduluj
ą
c
ą
(kontrola stanu
ś
wiadomo
ś
ci i czuwania),
ponadto j.szwu, j miejsca sinawego, wpływaj
ą
na liczne obszary przodomózgowia).
•Dolna cz
ęść
pełni
rol
ę
„premotoryczn
ą
”
koordynuj
ą
c
ą
ruchy trzewne i somatyczne (np., oddechowe,
akcj
ę
serca,
ż
ucie, ekspresje twarzy, odruchy wymiotne, ziewania, kichania, czkawki, połykanie).
•Te liczne stereotypowe czynno
ś
ci motoryczne maj
ą
w obr
ę
bie tworu siatkowatego „dedykowane”
obwody neuronalne czyli tzw. CPG (central pattern generators).
Twór siatkowaty w kontroli postawy – antycypacja skutków ruchu i dostosowanie napi
ę
cia
mi
ęś
ni wyprzedzaj
ą
ce zamierzony ruch („anticipatory maintenance of body posture” )
•Eksperymentalne uszkodzenie włókien korowo-rdzeniowych (drogi bezpo
ś
redniej) powoduje u
naczelnych małp zanik precyzyjnych ruchów r
ę
ki z zachowaniem kontroli postawy.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
75/113
J
ą
dro czerwienneWysyła aksony do bocznych stref motoneuronów i neuronów wstawkowych odcinka
szyjnego rdzenia – (ruchy r
ą
k).
Motoneurony kontroluj
ą
ce ruchy głowy (tractus tectospinalis).
„Adresatami” neuronów j
ą
der przedsionkowych s
ą
głównie mi
ęś
nie tułowia i ksobne mi
ęś
nie
ko
ń
czyn (równowaga i kontrola poło
ż
enia ciała).
•„Spastyczno
ść
”
– (wzrost napi
ę
cia mi
ęś
niowego, wygórowanie odruchów i klonusy):
•Prawdopodobnie spowodowana przerwaniem hamuj
ą
cego oddziaływania kory na j
ą
dra
przedsionkowe i tworu siatkowatego
•Eksperymentalnie łagodzono spastyczno
ść
uszkadzaj
ą
c j
ą
dra przedsionkowe oraz korzonki tylne
nerwów rdzeniowych (osłabienie „gain” dla odruchów rozci
ą
gowych)
J
ą
dra podstawy
•J
ą
dra podstawy s
ą
formacj
ą
blokuj
ą
c
ą
niepo
żą
dan
ą
aktywno
ść
kory ruchowej
a jednocze
ś
nie przygotowuj
ą
górne motoneurony do wykonania i inicjacji ruchu („priming”).
•Aby program ruchowy mógł by
ć
zainicjowany konieczne jest
zablokowanie tonicznej aktywno
ś
ci
hamuj
ą
cej
(„dysinhibicja”)
pallidum lub
(w przypadku sakkadowych i innych ruchów oczu) inhibicja
pars reticulata s.nigra
b
ę
d
ą
cych głównymi „outputowymi” o
ś
rodkami j
ą
der podstawy.
Drogi „wej
ś
cia” do zwojów podstawy
•J.ogoniaste i skorupa s
ą
„bramami wej
ś
ciowymi” j
ą
der podstawy
•„Input” pochodzi z niemal całej kory oraz z s.czarnej (wyj
ą
tki to pierwotne kory wzrokowe i
słuchowe)
•(tzw. corticostriatal pathway”)
•J
ą
dro ogoniaste
otrzymuje sygnały z kor asocjacyjnych („multimodalnych”) oraz z tzw. czołowej kory
wzrokowej.
•Skorupa
otrzymuje sygnały z pierwotnej i wtórnej kory czucia somatycznego, z „pozapr
ąż
kowanej”
kory wzrokowej, z kory ruchowej i przedruchowej oraz słuchowej kory asocjacyjnej z płata
skroniowego.
•Równie
ż
w j
ą
drach podstawy mo
ż
na mówi
ć
o „reprezentacjach” ruchowych okre
ś
lonych
cz
ęś
ci ciała w postaci „pasm” neuronalnych (tzw. striosomów) uło
ż
onych w kierunkach
przednio-tylnych.
•Komórki kolczyste otrzymuj
ą
te
ż
impulsacj
ę
z pars compacta i z interneuronów wzgórza.
Aktywno
ść
komórek kolczystych typowo jest bardzo niska ale
pojawia si
ę
przed wykonaniem
ruchu
(w j
ą
drze ogoniastym ruchu oczu a w skorupie ruchu ciała).
[koduj
ą
raczej decyzj
ę
celu
ruchu]
Rola j
ą
der podstawy w funkcjach
niemotorycznych•Pami
ęć
proceduralna
•Ró
ż
ne funkcje niemotoryczne
•P
ę
tla „okoruchowa”
–Modulacja aktywno
ś
ci czołowego pola ocznego
•P
ę
tla przedczołowa
–Rola w zaburzeniach kognitywnych towarzysz
ą
cych np. Ch.Parkinsona
–
W zesp. Tourette, (nadmierna aktywacja przedczołowych pól zwi
ą
zanych z mow
ą
)
•P
ę
tla limbiczna
–Zaburzenia emocjonalne i motywacji
–Schiozofrenia
–? zaburzenia obsesyjno-kompulsywne (jest to wg mnie swoisty odpowiednik „pl
ą
sawicy
my
ś
lowej”? pogl
ą
d własny)
Czy „hypokognicja” to analog hypokinezy ?
– czy „my
ś
lenie” nie jest dla j
ą
der podstawy jeszcze jedn
ą
form
ą
ruchu?...
Pytania póki co bez
odpowiedzi…
Rola mó
ż
d
ż
ku w kontroli ruchu
Główne „wej
ś
cia” do mó
ż
d
ż
ku (dolne i
ś
rodkowe
konary)
Główne „wej
ś
cia” do mó
ż
d
ż
ku (dolne i
ś
rodkowe konary)
Z kory mózgu:
Kora czołowa (pierwotna i wtórna ruchowa)
Kora ciemieniowa (pierwotna i wtórna czuciowa-somatyczna, wtórna wzrokowa)
Kora obr
ę
czy (limbiczna)
Z innych okolic:
Rdze
ń
kr
ę
gowy (kolumny Clarka)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
76/113
J
ą
dra czerwienne
Wzgórki górne
J
ą
dra przedsionkowe
Twór siatkowaty
Dolne j
ą
dra oliwki
Locus coeruleus
Somatotopowa organizacja kory mó
ż
d
ż
ku w spinocerebellum
Reprezentacja somatyczna w korze mó
ż
d
ż
ku jest zgodna co do strony ciała!Uszkodzenie przedniej
cz
ęś
ci robaka (w alkoholizmie) powoduje zaburzenia chodu (ko
ń
czyny dolne)
Drogi „wyj
ś
ciowe” z mó
ż
d
ż
ku
- Kora ruchowa (poprzez brzuszno-boczn
ą
cz
ęść
wzgórza -VL
thalamus)
- J
ą
dra czerwienne
- Wzgórki górne
- Twór siatkowaty
- J
ą
dra przedsionkowe (bez po
ś
rednictwa j
ą
der gł
ę
bokich mó
ż
d
ż
ku)
Znaczenie obwodów mó
ż
d
ż
kowych w uczeniu si
ę
poprzez „korekcj
ę
bł
ę
du”
•Wg
modelu Masao Ito i wsp. „koincydencja” sygnału na synapsach włókien równoległych i sygnału
„bł
ę
du” z włókien pn
ą
cych prowadzi do LTD i jest to mechanizm „uczenia si
ę
”
•LTD na komórkach Purkinjego wywołuje dysinhibicj
ę
poniewa
ż
w efekcie „hamowane s
ą
hamuj
ą
ce” komórki Purkinjego
•Przykład cyklicznej aktywno
ś
ci kk.Purkinjego – diadochokineza
•
Przykłady zale
ż
nego od mó
ż
d
ż
ku uczenia si
ę
przez korekt
ę
bł
ę
du:
–Eksperyment z cz
ęś
ciowym „podci
ę
ciem” mi
ęś
nia odwodz
ą
cego jednego oka – adaptacja sakkad
do nowej sytuacji
–Odruch przedsionkowo-oczny (utrzymuj
ą
cy fiksacj
ę
oczu na obserwowanym obiekcie w trakcie ruchu
głowy) – adaptacja zakresu ruchu oczu po nało
ż
eniu okularów „pomniejszaj
ą
cych”
Obie nauczone „umiej
ę
tno
ś
ci” (adaptacje) znikaj
ą
lub s
ą
nieosi
ą
galne po uszkodzeniu robaka-
mó
ż
d
ż
ku
Pamięć proceduralna:
podukład „móżdżkowy”
Model uczenia si
ę
mó
ż
d
ż
kowego
(ruchowego) Marra, Albersa i Ito
U królika mó
ż
d
ż
ek zaanga
ż
owany jest w:
odruch zamykania powieki (tzw. migotki) i odruch cofania gałki ocznej (w reakcji na podmuch
powietrza w kierunku oka.)
Odruch ten mo
ż
na warunkowa
ć
np.
dzwonkiem
uruchamianych tu
ż
przed dmuchni
ę
ciem.
D
ź
wi
ę
k aktywuje synapsy mi
ę
dzy włóknami równoległymi i kk.Purkinjego.
Sygnał dmuchni
ę
cia dociera do kk Purkinjego poprzez włókna pn
ą
ce.
Koincydencja dzwonka i dmuchni
ę
cia powoduje wytwarzanie LTD na synapsach włókien
równoległych i kk Purkinjego.
Zmniejszenie pobudliwo
ś
ci komórek Purkinjego powoduje wzrost
sygnału z mó
ż
d
ż
ku do mi
ęś
ni gałki ocznej
(„hamowanie hamuj
ą
cych” kk.Purkinjego)
Dalsze trenowanie odruchu warunkowego doprowadza do szybszej odpowiedzi której czas
wyst
ą
pienia zostaje zoptymalizowany do maj
ą
cego wyst
ą
pi
ć
dmuchni
ę
cia
Objawy uszkodzenia mó
ż
d
ż
ku
•Zaburzenia koordynacji ruchowej
•Zaburzenia ruchów r
ą
k (dysdiadochokineza, dysmetria, dr
ż
enie zamiarowe)
•Oczopl
ą
s
•Zaburzenia chodu,
•Zaburzenia mowy
Mó
ż
d
ż
ek to nie tylko ruch•Wiele danych wskazuje równie
ż
,
ż
e mó
ż
d
ż
ek w ró
ż
ny sposób moduluje
procesy kognitywne, np. zwi
ą
zane z mow
ą
, kontrol
ą
afektów, pami
ę
ci
ą
robocz
ą
! W rezultacie
uszkodzenie mó
ż
d
ż
ku mo
ż
e prowadzi
ć
do osłabienia zdolno
ś
ci intelektualnych
•(czy
ż
by mo
ż
na było mówi
ć
o „dysmetrii my
ś
li”?).

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
77/113
Analogie wojskowe neuronalnej kontroli ruchu: system hierarchiczny (ale
bardzo nowoczesny)
•„Pi
ę
tra dowodzenia” - rozkazy na poziomie operacyjnym, zwi
ą
zków
taktycznych, oddziałów i pododdziałów, dru
ż
yn…
•System „dowodzenia” bardzo elastyczny („armia XXI-wieku”) –
–Mo
ż
liwo
ść
bezpo
ś
redniego „dowodzenia” niewielkimi „pododdziałami” wykonuj
ą
cymi zadania
specjalne (np. ruchy palców)
–Mniej precyzyjne dowodzenie „mas
ą
ciała” (mi
ęś
niówka „osiowa”)
–Na ka
ż
dym poziomie oddziały posiadaj
ą
du
żą
niezale
ż
no
ść
i mo
ż
liwo
ść
dopasowania do „sytuacji
pola walki” (input czuciowy modyfikuje działanie motoneuronów)
–„Armia” dowodzona jest w oparciu o doskonale wyuczone i „wbudowane” schematy współdziałania
(CPG)
Kontrola ruchów oczu
Typy ruchów oczu:
•–Stabilizuj
ą
ce wzrok
•Ruchy przedsionkowo-oczne (vestibulo-ocular m. sygnał ruchu z przedsionka)
•(Ruchy optokinetyczne – sygnał ruchu z fotoreceptorów)
–Wodz
ą
ce wzrok (gaze-shifting)
•Ruchy sakkadowe (saccades)
•Jednostajne ruchy wodz
ą
ce/
ś
ledz
ą
ce (smooth pursuit m.)
–Ruchy zbie
ż
ne (vergence m.)
Nerwy kontroluj
ą
ce ruchami oczu
N. III prowadzi włókna tak
ż
e do d
ź
wigacza powieki oraz włókna parasympatyczne z j. Westfal-
Edingera
Technika stabilizacji obrazów na siatkówce:
obraz stabilizowany gwałtownie zanika
(adaptacja siatkówki? zapewne konieczno
ść
„od
ś
wie
ż
ania”?)
Mechanizm korowy tzw. transferu mi
ę
dzyocznego czyli osłabienia percepcji w drugim oku przy
stabilizacji obrazu.
Wodzenie:
Ruch dowolny,
Bez okre
ś
lonego „celu” jest trudny do wykonania (zwykle wtedy wyst
ę
puje sakkada)
Tzw. (prawidłowy) oczopl
ą
s optokinetyczny jest cykliczn
ą
sekwencj
ą
wodzenia i szybkiej
„powrotnej” sakkady.
Sakkady:
Mog
ą
by
ć
inicjowane dowolnie ale wyst
ę
puj
ą
te
ż
odruchowo.
200ms opó
ź
nienia potrzebne na „obliczenie” zakresu ruchu
Maj
ą
„balistyczny” charakter (Tak jak w przypadku rakietowego pocisku balistycznego po
„odpaleniu” nie mo
ż
na ju
ż
zatrzyma
ć
ani „przekierowa
ć
”).
Wyst
ę
puj
ą
te
ż
w fazie REM
Kontrola ruchów sakkadowych
•Informacja neuronalna musi kodowa
ć
kierunek i amplitud
ę
(zakres) ruchu.
•Amplituda ruchu kodowana jest w cz
ę
stotliwo
ś
ci wyładowa
ń
w nerwie poruszaj
ą
cym oko (na ryc.
N.VI)
•Kierunek ruchu wyznaczaj
ą
dwa osobne o
ś
rodki w tworze siatkowatym pnia:
–Paramedian pontine reticular formation PPRF (= o
ś
rodek kontroli spojrzenia w bok, horizontal
gaze center)
––Rostral interstitial nucleus (vertical gaze center, o
ś
rodek kontroli spojrzenia w pionie)
Ruch oczu w prawo
•W j
ą
drze n.VI oprócz alfamotoneuronów s
ą
neurony „mi
ę
dzyj
ą
drowe” wysyłaj
ą
ce aksony do cz
ęś
ci
j.n.III dla mi
ęś
nia prostego przy
ś
rodkowego (poprzez p
ę
czek podłu
ż
ny przy
ś
rodkowy)
•Aktywacja prawego PPRF prowadzi do spojrzenia w prawo
Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
78/113
•Poprzez neurony wstawkowe tworu siatkowatego nast
ę
puje hamowanie mi
ęś
ni przeciwstawnych
(podobnie jak w rdzeniu)
Wpływ wzgórków górnych
Wraz z czołowym polem ocznym kodowanie kierunku (poło
ż
enia) okre
ś
lonego (interesuj
ą
cego)
punktu w polu widzenia (mapa pola widzenia).
Wzgórki górne zawieraj
ą
te
ż
mapy przestrzenne dla (kierunków) bod
ź
ców słuchowych oraz
czucia somatycznego. Pozwala to np. na nakierowywanie głowy w zale
ż
no
ś
ci od
napływaj
ą
cych bod
ź
ców
Wzgórki górne maj
ą
poł
ą
czenia z o
ś
rodkami skojarzonego spojrzenia po drugiej stronie.
Siatkówka nie jest ani konieczna ani wystarczaj
ą
ca do inicjacji sakkady.
„Zarz
ą
dzanie” - bezpo
ś
rednie ruchem oczu
- po
ś
rednie – przez wzgórki górne
Oba o
ś
rodki kontroli ruchów oczu (korowy i wzgórki) wzajemnie si
ę
uzupełniaj
ą
i cz
ęś
ciowo
mog
ą
wzajemnie kompensowa
ć
uszkodzenia.
Nie s
ą
jednak równorz
ę
dne: uszkodzenie kory powoduje niemo
ż
liwo
ść
dowolnej sakkady w kierunku
obiektu, który znikn
ą
ł z pola widzenia oraz tzw. antysakkad
ę
(niemo
ż
liwo
ść
„oderwania” wzroku od
obiektu, który stymulował sakkad
ę
). Czołowa kora oczna jest istotna dla skanowania pola widzenia i
wyławiania interesuj
ą
cych obiektów.
„
Ś
lepota zmian” („change blindness”)•Zjawisko obserwowane głównie (ale nie tylko) w czasie
ruchów sakkadowych oczu
•
Ruchy oczu w pionie
•Slow vertical saccades in motor neuron disease: correlation of
structure and function. ‘Averbuch-Heller L i wsp. Ann Neurol.'); 1998 Oct;44(4):641-8 Postmortem
examination in both patients demonstrated cell loss in the rostral interstitial nucleus of the medial
longitudinal fasciculus (riMLF)
•Study of the rostral midbrain atrophy in progressive supranuclear palsy.
Kato N i wsp J Neurol Sci. 2003 Jun 15;210(1-2):57-60. Rostral midbrain atrophy in progressive
supranuclear palsy (PSP) is detected by mid-sagittal plain magnetic resonance imaging (MRI).
The shape of the atrophy looks like
the bill of a hummingbird (hummingbird sign).
•
Ruchy oczu stabilizuj
ą
ce wzrok•Ruchy przedsionkowo-oczne
(vestibulo-ocular m. sygnał ruchu z
przedsionka)
–Dostosowuje kierunek osi optycznej oczu kompensuj
ą
c ruch głowy (wykorzystuje informacj
ę
z
bł
ę
dnika)
–Jednak po zablokowaniu dopływu
ś
wiatła ruch zanika po ok.. 30 sek
–Ponowne np. otwarcie oczu wznawia ruch korekcyjny ale jest on inicjowany na podstawie obrazu
(jest to ruch optokinetyczny (zob. ni
ż
ej)
•(
Ruchy optokinetyczne
– sygnał ruchu z fotoreceptorów)
–W warunkach normalnych współdziała z ruchem przedsionkowo-ocznym
Ruchy zbie
ż
ne (vergence m.)•Cz
ęść
odruchowej „triady” adaptuj
ą
cej wzrok do ogl
ą
dania
przedmiotów z bliska:
–1. ruch zbie
ż
ny, 2. akomodacja soczewek, 3. zw
ęż
enie
ź
renic (wi
ę
ksza gł
ę
bia ostro
ś
ci)
Zaburzenia ruchów oczu w praktyce klinicznej!•Zanik wieloukładowy (z.Shy-Drager)
•Zwyrodnienie korowo-podstawne
–(pora
ż
enie nadj
ą
drowe ruchów oczu)
•
Ch. Steele’a-Richardsona-Olszewskiego
–pora
ż
enie pionowych ruchów gałek ocznych, zaburzenia ruchów sakkadowych, pora
ż
enie
rzekomoopuszkowe i cz
ę
ste upadki w pierwszym roku choroby,
Gdzie mog
ą
znajdywa
ć
si
ę
„najwy
ż
sze” pi
ę
tra kontroli i koordynacji ruchu?
Koncepcja tzw.
„action system”,
(Rothi, Ochipa i Heilman 1991) który ma by
ć
najwy
ż
szym pi
ę
trem
kontroli i koordynacji ruchu. – („idee ruchu” ?…)
•Wg w/w autorów plany-wzorce ruchu w postaci tzw. „praxiconów” (rodzaj engramu pami
ę
ciowego)
zakodowane s
ą
głównie w lewej korze ciemieniowej.
•S
ą
to jakby przestrzenno-czasowe „idee” okre
ś
lonego wyuczonego ruchu.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
79/113
•„idee” ruchu s
ą
przesyłane do kory przedruchowej, •gdzie s
ą
„rozkodowywane” na rozkazy dla
okre
ś
lonych grup mi
ęś
niowych i •przesyłane do pierwotnej kory ruchowej a w przypadkach ruchu
ko
ń
czyny lewej przekazywane poprzez spoidło wielkie do prawej kory przedruchowej (i potem prawej
kory ruchowej)
„Najwy
ż
szy” neuron ruchowy (?) by
ć
mo
ż
e tam, gdzie uszkodzenie prowadzi
do apraksji??
•Apraksja (dyspraksja) typy wg Merritt’a
–Ruchowa-kinetyczna:
•Uszkodzenie kory przedruchowej i innych okolic asocjacyjnych, osłabiona umiej
ę
tno
ść
posługiwania
si
ę
przedmiotami
–Ideacyjna-czuciowa :
•Odpowiednik afazji czuciowej
•Brak „planu motorycznego” zło
ż
onych czynno
ś
ci przy zachowaniu spontanicznych czynno
ś
ci;
uszkodzenie tylnej cz
ęś
ci półkuli dominuj
ą
cej
–Ideo-motoryczna- kondukcyjna
•Zachowane ruchy spontaniczne ale upo
ś
ledzone ruchy wykonywane na polecenie (cz
ę
sto
spotykana, cho
ć
umykaj
ą
ca w badaniu)
•Odpowiednik afazji kondukcyjnej
•Typowo w ko
ń
czynach kontrolowanych przez półkul
ę
niedominuj
ą
c
ą
przy uszkodzeniu półkuli
dominuj
ą
cej

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
80/113
Autonomiczny Układ Nerwowy
•Układ autonomiczny (trzewny):
–Walter Gaskell i John Langley (XIX w) z Cambridge
–Walter Cannon (pocz
ą
tek XX w) z Harvardu
Sympatyczne unerwienie gruczołów potowych jest cholinergiczne
Gruczoły potowe, rdze
ń
nadnerczy i mi
ęś
nie piloerekcyjne otrzymuj
ą
niemal wył
ą
cznie
unerwienie sympatyczne
•Zwoje układu parasympatycznego le
żą
bli
ż
ej unerwianych narz
ą
dów
•Neurony zwojów parasympatycznych w porównaniu z komórkami zwojowymi zwojów sympatycznych
maj
ą
bardzo nieliczne dendryty (mniejsza konwergencja)
•Mniejsza jest te
ż
dywergencja parasympatycznych neuronów przedzwojowych
•Autonomiczny (=trzewny) układ nerwowy
–Komponenta motoryczna
•Unerwienie sympatyczne (współczulne) i parasympatyczne (przywspółczulne) narz
ą
dów
–Komponenta czuciowa
•Dostarcza informacji („input”) słu
żą
c lokalnym obwodom odruchowym reguluj
ą
cym funkcjonowanie
narz
ą
dów
•Dostarcza informacji („input”) do wy
ż
szych koordynacyjnych o
ś
rodków układu autonomicznego
•Komponenta czuciowa
autonomicznego układu nerwowego („czucie trzewne”)
–W porównaniu z neuronami czucia somatycznego neurony czucia trzewnego s
ą
mniej liczne
(prawdopodobnie co najmniej 10x) - st
ą
d znacznie mniej dokładna lokalizacja bólu trzewnego. Bardzo
niewiele z impulsacji czuciowej trzewnej dochodzi do
ś
wiadomo
ś
ci.
–J
ą
dro pasma samotnego (=JPS) (n.tr.solitarius)
rdzenia przedłu
ż
onego (głównie cz
ęść
kaudalna)
jest zasadniczym „hubem” „autonomicznej” informacji czuciowej (jego cz
ęść
rostralna nale
ż
y do
drogi czucia smaku)
–„pierwszorz
ę
dowe trzewne neurony czuciowe” (I-rz n.)
•Aferenty czuciowe j
ą
dra pasma samotnego pochodz
ą
z
–komórek zwojów korzonków grzbietowych (w których s
ą
te
ż
komórki przewodz
ą
ce czucie
somatyczne). Obwodowe aksony ko
ń
cz
ą
ce si
ę
czuciowymi zako
ń
czeniami (wra
ż
liwe na ró
ż
ne
modalno
ś
ci np. nocyceptory, rozci
ą
ganie, ci
ś
nienie) lub docieraj
ą
ce do receptorów czuciowych
(chemoreceptory) biegn
ą
razem z nerwami sympatycznymi.
–zwojów czuciowych n.X i n.IX (aksony docieraj
ą
do j
ą
dra pasma samotnego)
–„drugorz
ę
dowe trzewne neurony czuciowe”(II-rz.n.) – (ich aksony docieraj
ą
do) : a) j
ą
dra pasma
samotnego, b) brzuszno-tylnej cz
ęś
ci wzgórza c) tworu siatkowatego) d) – aksony z neuronów
regionu kanału centralnego rdzenia ł
ą
cz
ą
si
ę
z powrózkami tylnymi (nowopoznana droga
trzewnego czucia bólu!).
Do II-rz. trzewnych neuronów czuciowych nale
żą
:
•Neurony rogów tylnych
(cz
ęść
aksonów I-rz.neuronów konwerguje na neuronach czucia
somatycznego co stanowi podło
ż
e „bólu odniesionego” ponadto cz
ęść
aksonów autonomicznych I-
rz.neuronów czuciowych ko
ń
czy si
ę
w rogach bocznych rdzenia w strefach autonomicznych neuronów
przedzwojowych – stanowi
ą
cz
ęść
autonomicznych łuków odruchowych „trzewno-trzewnych”)
•Neurony w okolicy kanału centralnego rdzenia (ich aksony zob. wy
ż
ej podpunkt „d”)
•O
ś
rodki układu autonomicznego w mózgu tworz
ą
ce
„o
ś
rodkow
ą
sie
ć
autonomiczn
ą
”
odpowiedzialn
ą
m.in. za odczucia i odpowiedzi „autonomiczno-emocjnalne”:
–Tylna cz
ęść
kory wyspy (czuciowy input autonomiczny)
–Przy
ś
rodkowa kora przedczołowa (motoryczny o
ś
rodek układu autonomicznego ?)
–Podwzgórze
(centrum koordynacyjne układu autonomicznego-trzewnego) – zarz
ą
dza poprzez
–o
ś
rodki autonomiczne układu siatkowatego (centrum wielu łuków odruchowych kontroluj
ą
cych prac
ę
serca, reakcje seksualne, funkcje oddychania, oddawania moczu, reakcje wymiotne)
•Podwzgórze – obszar ł
ą
cz
ą
cy i integruj
ą
cy układ nerwowy i hormonalny – funkcje:
–Kontrola układu kr
ąż
enia

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
81/113
–Regulacja metabolizmu
–Regulacja funkcji reprodukcyjno-seksualnych
–Koordynacja odpowiedzi w warunkach zagro
ż
enia
•Biofeedback czyli próba
ś
wiadomego „trenowania” autonomicznego układu nerwowego.
••Biofeedback zalicza si
ę
do tzw. medycyny alternatywnej (komplementarnej) i polega na ci
ą
głych
pomiarach niektórych parametrów biofizycznych takich jak ci
ś
nienie krwi, temperatura skóry, t
ę
tno,
„galvanic skin response” (pomiar pocenia si
ę
), napi
ę
cie mi
ęś
ni (w EMG) i elektryczna aktywno
ść
mózgu (EEG) i prezentacji ich wyników w czasie rzeczywistym aby umo
ż
liwi
ć
ś
wiadom
ą
ich kontrol
ę
(kontrol
ę
nad aktywno
ś
ci
ą
, która tradycyjnie wydawała si
ę
podlega
ć
wył
ą
cznie mechanizmom
odruchowym bez udziału
ś
wiadomo
ś
ci).
•Parametry u
ż
ywane w biofeedbacku: EMG, galvanic skin response training, EEG, temperatura skóry

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
82/113
Emocje odczuwanie i ekspresja
•„pierwotne” emocje:
–Zło
ść
–Strach
–Przyjemno
ść
–Smutek
–Obrzydzenie
uwagi na wstepie1. Emocje (reakcje emocjonalne) to główny „nap
ę
d” – (= motywacja) aktywno
ś
ci.
2. Poprzez obserwacj
ę
reakcji emocjonalnych mo
ż
emy pozna
ć
ukryt
ą
prawd
ę
o (drugim) człowieku a
zarazem samemu si
ę
„zdradzi
ć
”.
3. Umiej
ę
tno
ś
ci prawidłowej oceny emocji oraz wpływania na emocje własne i innych decyduj
ą
o
osobniczym sukcesie społecznym.
4. Nieprawidłowe reakcje emocjonalne cechuj
ą
liczne schorzenia psychiatryczne
5. Nastrój mo
ż
na rozumie
ć
jako „przedłu
ż
ony” stan emocjonalny
•Przeciwstawne teorie emocji :
–James’a-Lange’a (koniec XIX w.): objawy generowane poprzez działanie głównie autonomicznego
układu nerwowego (np.. Uczucie kołatania serca) odczuwamy jako emocj
ę
–Cannon’a-Barda (lata 20-te XX w.): Czynnik postrzegany jako „emocjonalny” (np. widok gro
ź
nego
zwierz
ę
cia) wywołuje reakcj
ę
centralnego układu nerwowego prowadz
ą
c
ą
do odpowiednich zmian w
funkcji ró
ż
nych narz
ą
dów i reakcji ruchowych – somatycznych (np. ucieczka). Innymi słowy teoria
wskazuje,
ż
e „do
ś
wiadczenie emocjonalne” wyst
ę
puje niezale
ż
nie od reakcji somatycznej (np. po
przeci
ę
ciu rdzenia)
•U
ś
wiadomione i nieu
ś
wiadomione emocje stanowi
ą
istotny (najistotniejszy?) „nap
ę
d”
motoryczny –motywacje zarówno działa
ń
apetytywnych „po
żą
daniowych” (w celu uzyskania
po
żą
danego dobra lub stanu) jak i unikowych (maj
ą
cych na celu unikanie oddziaływa
ń
i stanów
„awersyjnych”)
•Ekspresja emocji anga
ż
uje cz
ęść
ruchowego układu somatycznego oraz trzewny układ nerwowy
(autonomiczny). Ten ostatni jest szczególnie silnie zwi
ą
zany z ekspresj
ą
i odczuciem emocji
•Dwie główne drogi (bezpo
ś
redniej) ekspresji emocji:
–Reakcje trzewnego ruchowego układu nerwowego (autonomicznego)
–Mimika twarzy
•Istotne składniki układów neuronalnych ekspresji emocji:
–Podwzgórze
i jego poł
ą
czenia z pniem mózgu (
układ siateczkowaty pnia
) oraz
neuronami
przedzwojowymi układu autonomicznego
tworz
ą
zasadnicz
ą
struktur
ę
dzi
ę
ki której nast
ę
puje
skoordynowana reakcja motoryczna ekspresji emocji. (do
ś
wiadczenia Barda z chirurgicznym
uszkodzeniem mózgu wywołuj
ą
cym „sham rage” i Hessa ze elektryczn
ą
stymulacj
ą
struktur
podwzgórza)
–Układ limbiczny (we współczesnym rozumieniu zob. dalej)-
Druga „o
ś
” emocjonalna (cz
ęś
ciowo
„konwerguj
ą
ca” na pierwszej – zob. wy
ż
ej) jest wytworzona przez tzw.
układ limbiczny
, równie
ż
ł
ą
cz
ą
cy si
ę
z pniem mózgu (twór siateczkowaty) oraz neurony przedzwojowe układu autonomicznego.
••Odczucie okre
ś
lonego stanu emocjonalnego jest
ś
ci
ś
le zwi
ą
zane nie tylko z aktywno
ś
ci
ą
„układów
emocji” w mózgu ale równie
ż
z „inputem” z narz
ą
dów wewn
ę
trznych i czucia somatycznego.
(Koncepcja James’a i Lange’a z pocz
ą
tku XXw. zakładała,
ż
e emocje to tylko odczucie wewn
ę
trznych
stanów narz
ą
dów np. kołatanie serca, pocenie, etc)
•Przykład „obustronno
ś
ci” zwi
ą
zku „emocjonalnego umysłu” i ciała:
–Wyobra
ż
enie stanu emocjonalnego
→
„obwodowe” zmiany „emocjonalne” (np. wzrost akcji serca,
pocenie si
ę
itp.) [techniki biofeedback?? Zdolno
ś
ci fakirów??]
–„Instrumentalne” (według instrukcji czysto mechanicznej) wytwarzanie emocjonalnego wyrazu twarzy
→
„obwodowe” zmiany „emocjonalne” (zgodne z mimik
ą
). [aktor mo
ż
e znacznie gł
ę
biej „wczuwa
ć
si
ę
”
w rol
ę
…?]
•Wywołanie ekspresji „totalnej agresji-w
ś
ciekło
ś
ci” („sham rage”) u zwierz
ę
cia
Usuni
ę
cie półkul mózgowych z pozostawieniem podwzgórza wywołuje „sham rage” .
Podobne rezultaty daje dodatkowe (oprócz kory mózgu) usuni
ę
cie przedniej cz
ęś
ci podwzgórza.
Je
ś
li dodatkowo i tylna cz
ęść
podwzgórza jest usuni
ę
ta nie ma efektu w postaci „sham rage”.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
83/113
•Twarz jest zasadniczym „medium” emocji •Na twarzy „maluj
ą
” si
ę
emocje…
•Osobna wolicjonalna i emocjonalna kontrola neuronalna motoryki twarzy.
•Badania Duchenne doprowadziły do stwierdzenia,
ż
e cz
ęść
motoryki twarzy podległa jest woli (tzw.
„u
ś
miech piramidowy”) a cz
ęść
nie (ekspresja emocji – „u
ś
miech Duchenne”).
•Rola układu limbicznego w ekspresji emocji
•Układ limbiczny w rozumieniu „klasycznym” (plus tzw.
≈
„obwód Papez’a” 1937):
–Układ limbiczny w „klasycznym” rozumieniu obejmuje ciała suteczkowate, sklepienie, zakr
ę
ty obr
ę
czy
i parahipokampalne wraz z hipokampem. Ponadto wg Papeza j
ą
dro przednie-grzbietowe wzgóza.
Obecnie przewa
ż
pogl
ą
d,
ż
e z wymienionych struktur „układu limbicznego” tylko zakr
ę
t obr
ę
czy
bierze istotny udział w „kontroli” emocji.
•Układ limbiczny w rozumieniu współczesnym (cz
ęść
„emocjonalna”) :
–1. Zakr
ę
ty nadoczodołowe oraz przy
ś
rodkowa kora przedczołowa
–2. Brzuszna cz
ęść
j
ą
der podstawy
–3. Przy
ś
rodkowo-grzbietowe (n.mediodorsalis) j
ą
dro wzgórza
–4. Ciało migdałowate
–Zakr
ę
t obr
ę
czy
–Cz
ęść
podwzgórza
–(1,2,3,4 tworz
ą
układ silnych poł
ą
cze
ń
) zob slajd dalej
UWAGA:
Do układu limbicznego zalicza si
ę
równie
ż
zakr
ę
t parahipokampalny z hipokampem oraz
c.suteczkowate podwzgórza i sklepienie ale nie maj
ą
zwi
ą
zku z emocjami
•Zespół Kl
ü
ver-Bucy
zaobserwowany u małp po wyci
ę
ciu obu płatów skroniowych•Małpy wykazywały
agnozj
ę
wzrokow
ą
, (cho
ć
nie były
ś
lepe), nadaktywno
ść
(szczególnie seksualn
ą
i w eksploracji
otoczenia typowo z udziałem ust), stawały si
ę
całkowicie
„
potulne
”
bez l
ę
ku przed cz
ł
owiekiem i
w
ęż
em, oboj
ę
tne na cokolwiek było z nimi robione.
•Pó
ź
niej okazało si
ę
,
ż
e wystarczy usun
ąć
ciała migdałowate•John Downer (50-te) po usuni
ę
ciu 1
c.migdałowatego małpie i po przeci
ę
ciu włókien komisuralnych oraz skrzy
ż
owania n.wzrokowych
zaobserwował,
ż
e gdy małpie pozwolono u
ż
ywa
ć
1 oka od strony usuni
ę
tego c.migdałowatego
zachowywała si
ę
„bez emocji” na widok człowieka, gdy ogl
ą
dała człowieka okiem od strony
nieuszkodzonego c.migd. wykazywała typow
ą
agresj
ę
. Agresywnie reagowała jednak na bod
ź
ce
czucia somatycznego niezale
ż
nie od strony dra
ż
nionej.
Poł
ą
czenia c.migdałowatego z przedczołowa kor
ą
daj
ą
dost
ę
p m.in. do
kognitywnych o
ś
rodków mózgu co integruje emocjonalne znaczenie bod
ź
ców
–Cz
ęść
afektywno-emocjonalnej drogi przewodzenia bólu obejmuje c.migdałowate.
•N.parabrachialis wysyła aksony do c.migdałowatego i podwzgórza („o
ś
rodki” emocji i motywacji)
oraz do substancji szarej okołowodoci
ą
gowej, która odgrywa rol
ę
w kontroli aktywno
ś
ci szlaków bólu.
Ponadto n.parabrachialis bierze te
ż
udział w kontroli oddychania (wzmo
ż
enie w strachu).
–Grupa przy
ś
rodkowa j
ą
der c.migdałowego ma poł
ą
czenia z opuszk
ą
w
ę
chow
ą
i kor
ą
w
ę
chow
ą
–Grupa podstawno-boczna ma poł
ą
czenia z kor
ą
przedczołow
ą
(zakr
ę
ty oczodołowe i
przy
ś
rodkow
ą
), asocjacyjn
ą
kor
ą
przedniej cz
ęś
ci płata skroniowego oraz z brzuszn
ą
cz
ęś
ci
ą
j
ą
der podstawy i n.mediodorsalis thalami.
–Grupa centralna ma poł
ą
czenia z podwzgórzem, pniem mózgu (m.in. z dopaminergiczn
ą
tzw
VENTRAL TEGMENTAL AREA, noradrenergicznym n.l. coeruleus, oraz z n.parabrachialis,
n.tr.solitarius)
(Stria terminalis
ł
ą
czy c.migdałowate z podwzgórzem
Ponadto poł
ą
czenie to zapewnia równie
ż
tzw.
ventral amygdalofugal pathway)
•Ciało migdałowate nadaje znaczenie emocjonalne bod
ź
com czuciowym (do
ś
wiadczenia na
szczurach i królikach).
•Badania na szczurzym modelu reakcji (na d
ź
wi
ę
k) warunkowanej strachem
•Ciało kolankowate jest niezb
ę
dne do wytworzenia odruchu warunkowanego na bodziec słuchowy
(d
ź
wi
ę
k)
•Kolejne uszkodzenia pkt 1,2,3: – dopiero uszkodzenie „3” znosi reakcj
ę
strachu (wzrost ci
ś
nienia
t
ę
tniczego i wyst
ą
pienie „freezing”)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
84/113
•Za reakcj
ę
podniesienia ci
ś
nienia odpowiedzialne s
ą
poł
ą
czenia c.migdałowatego (centralnej
grupy j
ą
der) z podwzgórzem poprzez stria terminalis
•Za reakcje „freezing” odpowiedzialne s
ą
poł
ą
czenia c.migdałowatego (centralnej grupy j
ą
der)
z tworem siatkowatym
ś
ródmózgowia (midbrain)
Rola układów monoaminergicznych w mediacji emocji
•Na bardzo wa
ż
na rol
ę
układów monoaminergicznych wskazuj
ą
m.in. „emocjonalne” efekty ró
ż
nych
substancji np..
–Yohimbina (kora drzewa z Pd Afryki) – antagonista alfa-2-receptora noradrenergicznego prowadzi w
cz
ęś
ci zwierz
ą
t do reakcji panicznych poniewa
ż
wzmaga wydzielanie noradrenaliny poprzez
zablokowanie ujemnego sprz
ęż
enia zwrotnego reguluj
ą
cego uwalnianie NE, którego istotnym
elementem jest presynaptyczny receptor alfa-2.
–Odwrotnie działa clonidyna – b
ę
d
ą
ca agonist
ą
receptora noradrenergicznego alfa-2.
–Rezerpina – obni
ż
aj
ą
ca poziom monoamin wywołuje depresj
ę
–Leki podwy
ż
szaj
ą
ce poziom monoamin s
ą
lekami przeciwdepresyjnymi
–W stanach depresji obserwuje si
ę
obni
ż
enie ilo
ś
ci metabolitów NE (3-metoxy-4-hydroksyfenyloglikol
– MHPG) i serotoniny (kwas 5-OH– indolooctowy – 5-HIAA)
–Redukcja poziomu serotoniny mo
ż
e by
ć
istotna w zachowaniach impulsywnych w tym w próbach
samobójczych.
–W depresji obserwuje si
ę
zmniejszenie aktywno
ś
ci zarówno układów noradrenergicznych jak i
serotoninergicznych
–Główne o
ś
rodki monoaminergiczne graj
ą
ce rol
ę
w emocjach:
–Locus coeruleus – Norpinefryna
–J
ą
dra szwu – serotonina
–Ventral tegmental area oraz s.nigra - dopamina
•AGRESJA
•Agresja (mediowana m.in. przez c. migdałowate) decyduje o pozycji w grupie zwierz
ą
t
(Do
ś
w. Pribram’a z lat 50-tych – po usuni
ę
ciu c. migdałowatego u dominuj
ą
cej małpy jej pozycja
spadała)
•Próby operacyjnego „leczenia” (przy okazji równie
ż
padaczki) agresji poprzez uszkodzenie
c.migdałowatego. Tzw. psychochirurgia (Egas Moniz – Nobel 1949 r.)
•W agresji obserwuje si
ę
spadek aktywno
ś
ci serotoninergicznej (podobnie jak w depresji)
•Agresja nie koreluje z pozycj
ą
w grupie (usuni
ę
cie samca dominuj
ą
cego u małp prowadzi do
przej
ę
cia dominacji przez samca o sztucznie podwy
ż
szonej aktywno
ś
ci serotoninergicznej co oznacza
samca mniej agresywnego !)
•Typy agresji:
–Agresja “efektywna” - Predatory aggression („silent-biting”)
•Cel – zdobycie po
ż
ywienia
•Cechy: brak wokalizacji, małe zaanga
ż
owanie układu sympatycznego
•System: boczne podwzgórze – medial forebrain bundle – Ventral Tegmental Area
•–Agresja afektywna - Affective aggression
•Cel – „na pokaz”
•Cechy: wokalizacje, znaczna aktywacja układu sympatycznego
•System: przy
ś
rodkowe podwzgórze – fasciculus longitudinalis dorsalis – periaqueductal gray matter
(PAG)
•Ciało migdałowate uczestniczy w nadaniu „emocjonalnej warto
ś
ciowo
ś
ci” (emotional valence)
bod
ź
com czuciowym oraz w asocjatywnym uczeniu si
ę
(opartym na warunkowaniu – jest to
prawdopodobnie równowa
ż
ne tzw. pami
ę
ci emocjonalnej)
•Asocjatywne uczenie oparte na koincydencji bod
ź
ców (wg modelu Hebb’a) zachodzi w
c.migdałowatym o czym
ś
wiadczy obecno
ść
LTP oraz blokowanie nauczania w reakcji
warunkowanego strachu poprzez podanie antagonistów receptora NMDA
•Przypadek choroby dziedzicznej (autosom.reces.) Urbach-Wiethe:
•Zwapnienia i zniszczenie c.migdałowatego – „agnozja” emocjonalna (rozpoznawanie emocji z
twarzy)
(Chora o inicjałach S.M. badana w latach 90-tych przez grup
ę
Antonio Damasio z Uniw. Iowa)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
85/113
•Zwapnienia w c.migdałowatym – „agnozja” emocjonalna – nie rozpoznawanie (w tym przypadku
negatywnych) emocji z twarzy
•UWAGA! Chorzy z chorob
ą
Urbach-Wiethe nie maj
ą
problemów z rozpoznawaniem twarzy „jako
takiej” (nie maj
ą
cech prozopagnozji) a jedynie z rozpoznawaniem emocjonalnego wyrazu strachu
na twarzy ! Prawidłowo rozpoznaj
ą
te
ż
„pozytywne emocje” na twarzy.
Asymetria „emocjonalna” półkul mózgowych
•W rozpoznawaniu i ekspresji emocji ró
ż
ny jest udział lewej i prawej półkuli mózgu.
•Półkula prawa odgrywa wi
ę
ksz
ą
rol
ę
w percepcji emocji werbalnej (emocjonalny ładunek mowy) –
uszkodzenie tylnej cz
ęś
ci prawego płata czołowego i przedniej płata ciemieniowego prowadzi do tzw.
aprozodii czyli mowy bez emocji (po stronie lewej uszkodzenie tego samego obszaru prowadzi do
afazji Broca)
•Półkula lewa – „o
ś
rodkiem” pozytywnych emocji, półkula prawa – negatywnych
•Prawa półkula lepiej rozpoznaje przejawy emocji na twarzy (np. Przy ekspozycji fotografii twarzy)
•Równie
ż
ekspresja emocji na twarzy jest szybsza po lewej stronie (a zatem „zarz
ą
dzanej” przez
praw
ą
półkul
ę
). St
ą
d sugestia,
ż
e wi
ę
kszo
ść
ludzi jest „lewotwarzowa” (analogia – cho
ć
przeciwstronna do „prawor
ę
czno
ś
ci”)
•Obserwacja twarzy (w tym ekspresji emocji) szczególnie silnie anga
ż
uje ciała migdałowate
•Aktwacja amygdali w te
ś
cie oceny „wiarygodno
ś
ci” obserwowanej twarzy.
•Aktywacja c.migdałowatych jest tym wi
ę
ksza im obserwowana przez badanego twarz na fotografii
mniej budzi zaufania (implicitely) jak te
ż
gdy zadaniem (explicit) jest ocena „wiarygodno
ś
ci”
obserwowanej twarzy

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
86/113
Pamięć uczenie kora asocjacyjna kognicja
NEUROBIOLOGICZNE PODSTAWY PAMIĘCI
Franz Josef Gall (twórca frenologii) w XIX w. uważał, że każdy z „fakultetów” mózgu („władz umysłowych”)
posiada własny rodzaj pamięci.
Psycholog William James pod koniec XIX w. wprowadził m.in. istotne rozróżnienie między
pamięcią świeżą i
pamięcią długotrwałą
(wprowadził też pojęcie „
strumienia świadomości
”).
Pierwsza połowa XX w dziedzinie badań nad pamięcią została zdominowana przez koncepcje
redukcjonistyczne, które sprowadzały pamięć do mechanizmów asocjacyjnych i oparte były także na
behawioralnym podejściu do psychologii.
W latach 60 i 70-tych ugruntowało się przekonanie, że są różne typy pamięci, które realizowane są przez różne
wyodrębnione układy w mózgu
NIE ISTNIEJE POJEDYNCZY UNIWERSALNY SYSTEM LUB MECHANIZM PAMIĘCI NATOMIAST
RÓśNE UKŁADY POSŁUGUJĄ SIĘ RÓśNYMI „MECHANIZMAMI PAMIĘCI”
Zjawiska LTP i LTD (LTD szczeg. w móżdżku) leżą u podstaw mechanizmów pamięci.
Kliniczny obraz różnych typów chorób otępiennych rzuca światło m.in. na mechanizmy pamięci i funkcji
kognitywnych mózgu.
Np. choroba Alzheimera początkowo manifestuje się głównie zaburzeniami tzw. pamięci deklaratywnej i
pamięci „świeżej” (hipokamp) natomiast choroba Huntingtona, dotyka głównie pamięci tzw. proceduralnej
(obwody korowo-prążkowiowe)
Stadia choroby Alzheimera –
Początkowe dostrzegalne zmiany dotyczą kory śródwęchowej, której neurony (IIw) tworzą główne drogi
doprowadzające hipokampa
•Amnezja retrograde (wsteczna) i anterograde (następcza)
•Przyczny:
–Zamknięcie obu tętnic tylnych mózgu (brak ukrwienia przyśrodkowej części pł. Skroniowch, zwł. hipokampa)
–Guzy obszaru środkowego (zniszczenie obustronne części przyśrodkowej wzgórza)
–Uraz, chirurgiczne wycięcie obustronne przyśrodkowego pł skroniowego (chory H.M.)
–Infekcje (HSV-encephalitis) j.w.
–Niedobór Vit. B1 (zespół Korsakoff’a) (uszkodzenie c.suteczkowatych i przyśrodkowego wzgórza)
–Leczenie elektrowstrząsami (miejsce uszkodzenia niejasne)
NAJWAśNIEJSZE „ZASADY” I MECHANIZMY PAMIĘCI I UCZENIA (Byrne 1987)
1.W mózgu istnieją liczne systemy-układy pamięciowe
2.Krótkotrwałe formy pamięci i uczenia wymagają zmian w istniejących obwodach neuronalnych
3.Te zmiany dotyczą różnych mechanizmów komórkowych w obrębie poszczególnych neuronów
4.Zmiany wewnątrzkomórkowe obejmują systemy wtórnych przekaźników
5.Pamięć i uczenie często dotyczy zmian w funkcjonowaniu kanałów błonowych (jonowych)
6.Pamięć długotrwała wymaga syntezy białek podczas gdy pamięć krótkotrwała nie.
PAMIĘĆ KRÓTKOTRWAŁA I DŁUGOTRWAŁA (William James k.XIX)
Pamięć krótkotrwała
(STM, „świeża”): ograniczona pojemność, utrzymuje się przez krótki czas, ulega
osłabieniu przez nowe zdarzenia, zaburzana przez anestetyki i oziębienie mózgu ale nie jest zaburzona w
amnezji, często utożsamiana z tzw. „pamięcią roboczą” (working m.)
Jednym z istotnych elementów tej pamięci jest tzw.
Pętla fonologiczna
(wymaga aktywności lewej
półkuli; zapewnia zrozumienie dźwięków tworzących wyrazy i zdania, dzięki czemu rozumiemy mowę) oraz
pętla wzrokowo-przestrzenna
(prawa półkula)
Pamięć długotrwała
(LTM, odległa): zachowuje „engramy” doznań przez bardzo długi czas. Jest uszkodzona w
wyniku amnezji ale odporna na anestetyki
Relacje STM i LTM nie są jasne (dwie możliwości: 1: LTM jest wynikiem tzw. Konsolidacji STM, 2: LTM jest
wynikiem selekcji STM)
Nie tylko uszkodzenie hipokampa wywołuje zaburzenia pamięci
Główna projekcja wychodząca z hipokampa
poprzez sklepienie dociera do ciał suteczkowatych, mających połączenia z
jądrami przednimi wzgórza (tractus
mammillothalamicus)
.
Kora skroniowa i c.migdałowate maja połączenia z
jądrem grzbietowo-przyśrodkowym wzgórza
.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
87/113
Obustronne uszkodzenia tych struktur wywołują różne zaburzenia pamięci ale
tylko łączne ich uszkodzenie
wywołuje ciężkie zaburzenia pamięci niekiedy zbliżone do pacjenta H.M.
W zespole Korsakoffa (amnezja wsteczna i następcza) uszkodzenie dotyczy c.suteczkowatych i jąder
grzbietowo-przyśrodkowych wzgórza na skutek niedoboru wit B1 (w alkoholizmie)
Uszkodzenie c.migdałowatego
u zwierząt prowadzi do zaniku zdolności do nauczenia warunkowej reakcji
strachu (po dźwięku następuje szok elektryczny – zwierzę reaguje złożoną reakcją układu autonomicznego,
hormonalnego i objawami behawioralnymi strachu) (zaburzenie „
pamięci emocjonalnej
”)
Cohen i Squire w 1980 wprowadzili rozróżnienie: pamięci proceduralnej i pamięci deklaratywnej uważając,
że są one realizowane przez osobne układy w mózgu.
Jeden z tych układów (można nazwać „proceduralnym”) jest odpowiedzialny za tworzenie i doskonalenie
zdolności „zręcznościowych”. Pamięć rozłączona (w całości lub częściowo rozłączona) ze świadomością
(„nieświadoma”) różne układy i sekwencje warunkowane (przykład nauki gry w piłkę, gry na fortepianie)
Drugi z układów („deklaratywny”) odpowiada za kodowanie, magazynowanie i przywoływanie faktów i
wydarzeń. Pamięć polegająca na zdolności do
przypomnienia-przywołania
zdarzeń nie pozostających w
prostej kontynuacji bieżącej świadomej obserwacji mijających chwil i zdarzeń w tych chwilach
Daniel Schacter wprowadził zasadniczo „kompatybilne” do powyższych pojęć Cohena i Squire pojęcia implicit
(p.proceduralna) i explicit (p.deklaratywna) ekspresji pamięci.
Pamięć deklaratywna („explicit”)
”” mieści się”” (jest krytycznie związana z) w przyśrodkowym obszarze płata
skroniowego oraz w centralnych obszarach międzymózgowia (diencencephalon)
Pamięć proceduralna („implicit”)
jest związana z różnymi układami mózgu wyspecjalizowanymi w różnych
funkcjach poznawczych i motorycznych
Kategorie jakościowe pamięci
•Pamięć deklaratywna (cz.przyśrodkowa pł. skroniowych
–Zdarzenia
–Słowa i ich znaczenie
–Historia
•Pamięć nie-deklaratywna (raczej niezależna od pł skroniowych (cz. Przyśrodkowej)
–Zręczności ruchowe
–Asocjacje (kojarzenia)
–Priming cues(to co uprzednio poznalismy wpływa na odbiór następnych informacji)
–Puzzle-solving skills
Kategorie czasowe pamięci
•Pamięć natychmiastowa – immediate memory: (ułamki sekund-
sekundy) „śledzenie rzeczywistości on-line”
–Bardzo duża pojemność
–Prawdopodobnie osobne rejestry dla różnych typów doznań (wzrokowe, słuchowe, dotykowe, itd.)
•Pamięć robocza - working memory: (sekundy-minuty)
–Ujawnia się np. w przebiegu poszukiwania klucza (np.pamiętanie co już zostało przeszukane)
–Powtórzyć serię liczb (standard 7-9 liczb)
•Pamięć długotrwała: (dni-lata)
•„Pamięć” filogenetyczna – (wrodzona):
•Przykład ptaków których pisklęta reagują na kształt rzeczywistego jak i „udawanego” drapieżnika
Skąd wiadomo, że kora mózgu jest odpowiedzialna za
deklaratywną
pamięć długotrwałą
•Leczenie elektrowstrząsami powoduje amnezję wsteczną (od kilku dni nawet do lat) i następczą. (U podstaw
zastosowania elektrowstrząsów był m.in. fakt, że zauważono, że spontaniczne ataki padaczki poprawiają stan u
chorych z depresją)
•Kora mózgu jest strukturą uszkadzaną głównie w tej metodzie (najprawdopodobniej w mechanizmie
ekscytotoksycznym) stąd wniosek, że długotrwała pamięć jest „realizowana” w korze mózgu.
• Potwierdzono to u szczurów w testach z labiryntem wodnym.
Zniszczenie obszaru górnego płata skroniowego powoduje utratę rozumienia słów (afazja). (Kora asocjacyjna
odpowiedzialna za łączenie określonych dźwięków ze znaczeniem leksykalnym)
•Uszkodzenie płata ciemieniowego powoduje utratę zdolności rozpoznawania przedmiotów i/lub twarzy.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
88/113
•Badania fMRI (BOLD) pokazują, że istnieją określone obszary kory aktywowane wzrokowo tymi samymi
klasami obiektów (np.. Krzesła, twarze, domy) i to zarówno obrazem jak i myślową „wizualizacją” tego samego
obrazu)
„Trenowanie” working memory•Zapamiętywanie informacji „bez znaczenia” jest ograniczone (7-9 liczb np.)
ale może być znacznie wzmocnione po treningu (nawet do 80 cyfr) poprzez tworzenie asocjacji (nadawanie
znaczeń)
•Znawca np. szachów znacznie więcej pozycji figur zapamiętuje niż „profan” gdy dotyczy konkretnego realnego
ustawienia z określonej gry a nie przypadkowego „bezsensownego”
•Przykłady „mnemonistów” („pi” do 40000 miejsc) Alexander Aitken, Arturo Toscanini
•Ogromne możliwości zapamiętywania czegokolwiek jeśli jest motywacja i zainteresowanie dyscypliną wiedzy,
sportu etc
•Zespół savanta („Idiot savant”): osoba z uszkodzeniem mózgu i głębokim ogólnym upośledzenie lecz z
niezwykłymi szczególnymi zdolnościami (szczególnie językowymi). („Rain Man” z Dustinem Hoffmanem)
Uczenie
Proces uczenia jest ściśle związany (o ile nie polega nawet) na wzmocnieniu „siły” („wagi”) synaps.
Niektóre synapsy ulegają wzmocnieniu gdy oba tworzące je neurony ulegają jednoczesnemu pobudzeniu – a
zatem wymagają zaistnienia „asocjacji” (są to tzw. Synapsy Hebba –
Donald Hebb Kanad. Psycholog 1949)
„Cells that fire together, wire together”
Synapsy te są tzw „detektorami równoczesności”
Szczególna rolę gra tzw.
Długotrwałe wzmocnienie synaptyczne
(long-term potentiation LTP, trwające wiele
godzin a nawet dni), które może być zarówno asocjacyjne jak i nieasocjacyjne.
Uczenie (się) – badania na modelach kręgowców
Rola LTP
Zjawisko LTP najczęściej jest badane w
synapsach miedzy piramidalnymi neuronami CA1 i CA3 hipokampa. Polega ono na wzroście efektywności
przewodzenia syaptycznego po „tężcowej” stymulacji krótkotrwałym bodźcem elektrycznym z wysoką (100Hz)
częstotliwością włókien aferentnych neuronów pola CA3 (włókna Schaffera). Efektem jest to, że pojedynczy
bodziec po uprzedniej „tetanizacji” wywołuje wyższy EPSP.
Również „długotrwałe osłabienie synaptyczne” (LTD) jest elementem uczenia
Bliss i Lomo w 1973 wykazali LTP w synapsach mi
ę
dzy włóknami przeszywaj
ą
cymi i neuronami
fascia dentata
Układ hipokampa
Układ połączeń hipokampa:
1.
Wejście pobudzające z kory entorinalnej (śródwęchowej) dociera tzw. Drogą przeszywającą do dendrytów
komórek ziarnistych zakrętu zębatego.
2.
Włókna komórek ziarnistych (aksony) z.zębatego projektują do komórek piramidalnych hipokampa (pole
CA3).
3.
Aksony komórek z pola CA3 docierają do CA1 (
tzw. kolaterale Schaffera tworzące synapsy na komórkach
pola CA1)
oraz do komórek podwzgórza i do przeciwstronnego hipokampa.
4.
Aksony komórek z pola CA1 tworzą synapsy z neuronami kory śródwęchowej.
W całym tym „obwodzie”
dominuje Glu chociaż są też modulujące synapsy GABA-ergiczne, cholinergiczne naradrenergiczne i
serotoninergiczne.
Aksony kom.CA3 poprzez włókna spoidłowe docierają także do hipokampa po drugiej stronie.
Włókna eferentne wychodzą też przez sklepienie (fornix) w kierunku podwzgórza i wzgórza.
Warunki dla
LTP:
1)Napływ Ca2+
2)Jednoczesna depolaryzacja i stymulacja kolaterali Schaffera bod
ź
cem o niskiej cz
ę
stotliwo
ś
ci nie
spełnia warunku wystarczaj
ą
cej depolaryzacji i uwolnienia „blokady” magnezowej (za mało otwartych
kanałów receptorowych AMPA (Ca2+)
Podawanie MK-801 (antagonisty NMDA) blokuje LTP co jest jednym z dowodów na rol
ę
NMDAR w
LTP.
Cechy LTP:
1)Współdziałanie (Cooperativity)
Prawdopodobie
ń
stwo wywołania LTP wzrasta wraz ze zwi
ę
kszeniem
liczby włókien aferentnych Schaffera stymulowanych tetanicznie
2) Specyficzno
ść
wej
ś
cia (Input specificity)
LTP pojawia si
ę
tylko w synapsach, które podlegały tetanicznej stymulacji
3) Asocjacyjno
ść
(Associativity)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
89/113
Poniewa
ż
kolaterale Schaffera docieraj
ą
do neuronów CA1 równie
ż
z drugiego hipokampa
(włókna spoidłowe) oba systemy włókien konwerguj
ą
na neuronach CA1 ale jeden ma „słabe”
oddziaływanie (nieliczne włókna) a drugi (z tej samej strony) – silne (liczne włókna). Tetaniczne
dra
ż
nienie „słabej” (w przeciwie
ń
stwie do „silnej”) drogi włókien nie wywołuje LTP. Je
ś
li jednak istnieje
„asocjacja” czasowa stymulacji obu dróg (tetaniczne dra
ż
nienie zarówno „słabej” jak i „silnej” drogi –
czyli włókien Schaffera z obu stron) pojawi si
ę
LTP na synapsach z obu stron („słabych” i „silnych”).
Warunkiem jest aby oba „wej
ś
cia” były
jednocze
ś
nie
pobudzone tetanicznie.
Najwa
ż
niejszym mechanizmem LTP jest prawdopodobnie wzrost liczby AMPA-R ale s
ą
te
ż
inne
mechanizmy LTP
Znaki zapytania dotyczące LTP:
Wątpliwości:
Nie jest do końca jasna rzeczywista funkcja LTP (czy na pewno pamięć?)
U zwierząt transgenicznych, u których nie dochodzi do LTP w hipokampie pomiędzy włóknami
przeszywającymi i kk.ziarnistymi zakrętu zębatego nie ma zaburzeń w nawigacji przestrzennej (ale być może te
synapsy nie mają związku z tym rodzajem pamięci ???)
Czy mechanizm dotyczy neuronu postsynaptycznego czy też (również) presynaptycznego (np. Wzrost ilości
pęcherzyków)?
Które z obserwowanych zjawisk można definiować jako LTP (obecnie „szeroka” definicja mówi o każdym
przypadku gdzie synapsa staje się „mocniejsza” w wyniku uprzedniej aktywności)?
Czy na pewno rezultaty eksperymentów na płatach wyciętego hipokampa wskazujące na istnienie LTP pokazują
coś, co jest faktycznie w żywym mózgu „in situ”?
Ale jednak ...
Tężcowa stymulacja jednak nie jest czymś „sztucznym” bo występuje również „naturalnie”;
Różne manipulacje ograniczające LTP powodują osłabienie uczenia
MK-801 utrudnia uczenie (co jednak nie wyklucza, że ten efekt może nie mieć związku z blokadą receptora
NMDA ale z jakimś innym mechanizmem !)
„Komórki miejsca” wyładowują zgodnie z rytmem theta EEG, (4-10Hz) widocznym przy „nauce” w labiryncie i
odzwierciedlającym aktywność hipokampa (inne komórki „milczą”).
Stymulacja „naśladująca” rytm theta pomaga w uczeniu.
Uczenie się: nabywanie zmian w zachowaniu i ich odtwarzanie pod wpływem danego doświadczenia – jest
wyrazem plastyczności mózgu.
Gra podstawową rolę w tworzeniu
pamięci proceduralnej.
Uczenie się asocjacyjne jest oparte na tworzeniu odruchu warunkowego i wymaga dwóch bodźców,
pierwszy – tzw. warunkowy
występuje tuż przed
drugim – tzw. bezwarunkowym
, który może być
Atrakcyjny-apetytywny
(pokarm) lub
awersyjny
(np. ból)
Mechanizmy uczenia się:
1.Nieasocjacyjne
(habituacja lub sensytyzacja)
1)Habituacja (osłabianie reakcji na wielokrotnie powtarzany bodziec, najprawdopodobniej towarzyszy jej
m.in. eliminacja synaps)
2)Dyshabituacja (odtworzenie uprzednio zredukowanej „zhabituowanej” odpowiedzi pod wpływem
silnego bodźca)
3)Sensytyzacja (zwiększenie reakcji na bodziec pojawiający się tuż po silnym bodźcu awersyjnym,
związana z tworzeniem nowych synaps)
2.Asocjacyjne (odruchy warunkowe)
Bodźce mogą być:
Typu NAGRODY („appetitive”) lub Typu KARY („aversive”, „fear”)
Uczenie się:
Asocjacyjne „klasyczne” (poprzez odruchy warunkowe)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
90/113
1)Bodziec bezwarunkowy – reakcja bezwarunkowa np. widok pokarmu i wydzielanie śliny. Jeżeli bodziec
bezwarunkowy jest bólowy nazywamy go awersyjnym (warunkowanie awersyjne)
2)Bodziec warunkowy (conditioned stimulus) musi poprzedzać bodziec bezwarunkowy (np. dzwonek przed
podawaniem jedzenia)
3)Wytworzona reakcja (warunkowa) np. wydzielanie śliny po dzwonku
4)Bodziec warunkowy i bezwarunkowy muszą być blisko w czasie (zasada koincydencji) a b.warunkowy musi
poprzedzać b.bezwarunkowy
5)Reakcja warunkowa ulega
stopniowemu wygaszeniu
po zaprzestaniu występowania bodźca bezwarunkowego
(także po wydłużeniu czasu pomiędzy bodźcami)
2)Asocjacyjne instrumentalne
tzw. instrumentalne odruchy warunkowe
(np. naciśnięcie dźwigni aby dostać cukierek -
bodziec wzmacniający
)
Klasyczne warunkowanie wg Pawłowa.
Obecnie zwraca się uwagę nie tyle na istnienie „czasowej ciągłości” bodźca warunkowego i bezwarunkowego
ale na fakt, że pomiędzy tymi bodźcami istnieje
związek INFORMACYJNY
!
Bodziec warunkowy w procesie warunkowania nabiera cech sygnału niosącego informację !
Obecnie wyróżniamy następujące główne typy pamięci:
PAMIĘĆ DEKLARATYWNA (opisowa)
PAMIĘĆ NIEDEKLARATYWNA
•Pamięć proceduralna (pamięć sposobów postępowania)
•Pamięć emocjonalna
•Pamięć robocza „working memory” [KORA]
•Pamięć percepcyjna „perceptual memory” [KORA]
–Priming, Puzzle solving
•Podział na typy pamięci i wzajemne relacje podtypów jest wynikiem dokonanych eksperymentów jak również
ciągle przedmiotem badań i dyskusji
Np. w podziale wg L.R. Squire na następnym slajdzie brak pamięci roboczej a pamięć emocjonalna wraz z
„skeletal responses” jest zaliczona do „klasycznego warunkowania” (nie ma też relacji do pojęć LTM i STM)
Pamięć deklaratywna
Pamięć deklaratywna („explicit”)
jak wskazują przypadki amnezji po uszkodzeniu przyśrodkowych okolic
płatów skroniowych u ludzi
pozwala na zachowywanie i świadome przypominanie faktów i wydarzeń
.
Pamięć deklaratywna pozwala też na
wyciąganie uogólnień oraz implikacji (inferrences) z zapamiętanych
faktów
, co jest nieodzowne i jednocześnie charakterystyczne dla ekspresji pamięci deklaratywnej.
Pamięć deklaratywna („explicit”)
związana z hipokampem
jest (na podst. badań na zwierzętach) szybko
nabywana i „napędzana” raczej ciekawością a nie systemem kara-nagroda i dotyczy głównie przestrzennego
poznawania (spatial cognition).
Struktury odpowiedzialne za p.deklaratywną:
pola asocjacyjne neocortex,
oraz
dla p. epizodycznej kora przedczołowa,
dla p.semantycznej zwł. przedni lewy płat skroniowy, korowe obszary sąsiadujące z hipokampem i hipokamp.
Wiedza semantyczna:
Jest to zorganizowany zasób informacji niezależny od „epizodycznych” zdarzeń (episodic
representations), które tworzyły ten zasób.
Prawdopodobnie „engramy” pamięci dotyczące podobnych pojęć mają osobne lokalizacje (przypadki
chorych dobrze przypominających owoce ale kiepsko warzywa)
Mózg tworzy zhierarchizowane obszary odrębne dla poszczególnych pojęć (np. rośliny, zwierzęta,
pojazdy, rzeki, meble etc) podobnie jak zhierarchizowana jest nasza wiedza semantyczna.
Badania wykazały, że przypominanie obiektów należących do dwóch różnych pojęć takich jak
narzędzia i zwierzęta oprócz aktywowania kory środkowego zakrętu skroniowego łączyło się z
aktywacją w
przypadku
narzędzi
– lewej kory przedruchowej
(te same obszary aktywowało
wyobrażanie manipulacji narzędziami u praworecznych), w
przypadku
zwierząt
z aktywacją kory
przyśrodkowej potylicznej
(związanej z przetwarzaniem obrazów).
Prawdopodobnie tworząc nową kategorię (pojęcie) mózg tworzy (przyporządkowuje) osobne miejsce ?

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
91/113
„COŚ” (układ, moduł, mózg, umysł???) w nas stale organizuje napływającą nieprzerwanie rzekę
informacji czyli „faktów” lub „zdarzeń”, które są zintegrowanymi i przetworzonymi zbiorami impulsacji
czuciowej
„Piksele” z obrazu na siatkówce tworzą reprezentacje np. określonych liter w korze wzrokowej, w
innych obszarach są integrowane w wyrazy, następnie w zrozumiałe zadania, pojęcia itd.
Organizacja opiera się głównie na kojarzeniu wspólnych elementów.
Organizacja tej rzeki informacji pozwala na odtwarzanie (przypominanie) faktów i ich znaczeń
Pamięć deklaratywna („explicit”)
- test dla małp naczelnych
Visual paired comparison task:
Zwierzętom prezentuje się dwa jednakowe obrazki.
Zapamiętanie ocenia się na podstawie czasu fiksacji wzroku na zmienionym obrazku (zmiana obrazu następuje
po różnym okresie czasu)
Delayed non-matching-to-sample task „dobieranie nie według wzoru”:
Zwierz
ę
ma po ró
ż
nym czasie (w tym czasie nie widzi
ż
adnych innych obiektów) rozpozna
ć
nowy
obiekt (kul
ę
). Nagrod
ą
jest smakołyk.
Zwierz
ę
ta po uszkodzeniu przy
ś
rodkowych struktur skroniowych (zwłaszcza okolic
ś
ródw
ę
chowych i
parahipokampalnych)
ź
le wykonuj
ą
test chocia
ż
np. maj
ą
nie osłabione funkcje uczenia si
ę
zr
ę
czno
ś
ci
(np. wyci
ą
gania cukierka przy pomocy zagi
ę
tego pr
ę
ta)
Pamięć deklaratywna („explicit”) u gryzoni
The Morris water maze test (labirynt wodny Morrisa):
Nieprzezroczysta woda ukrywa znajdującą się tuż pod jej powierzchnią platformę umożliwiającą mu „ratunek”
(nie musi pływać gdy na niej stanie), którą szczur uczy się odnajdywać na podstawie obiektów znajdujących się
na zewnątrz baseniku śr 1,3 m (różne przedmioty rozmieszczone w pokoju w którym są przeprowadzane testy).
Szczury z uszkodzonym hipokampem mają słabe wyniki testu, chociaż mają bardzo dobre wyniki w opanowaniu
pływania do platformy widocznej i umieszczanej w różnych miejscach kuwety.
Podanie kolchicyny niszcząc
część hipokampa po 12 tygodniach od zakończenia testów (uczenia) nie wywoływało różnic w porównaniu z
kontrolą. (wcześniejsze podawanie kolchicyny niszczyło pamięć).
Test of transitive inference
czyli badanie zdolności wnioskowania cechy przechodniości tzn. Jeśli A>B i B>C to
A>C (u dzieci wykształca się do 7 roku życia Piaget)
Gryzonie uczą się na podstawie zestawów par różnych zapachów, który z zapachów w danej parze jest
preferowany (związany z nagrodą-smakołykiem)
Tzn A>B oznacza, że w piasku z zapachem A jest zagrzebana nagroda a nie jest zagrzebana w piasku
oznaczonym zapachem B
Kolejne pary zapachowe, z jednym nagradzanym (symbol >) symbolicznie zapisujemy jako B>C, C>D, D>E.
Test polega na „wykoncypowaniu”, że w
parze B,D
preferowany jest zapach B (transitive inference). Trzeba
zauważyć, że w innych układach zarówno zapach B jak i zapach D może być i nie być nagradzany!
Szczury z
uszkodzeniem hipokampa uczą się prawidłowego wyboru w poszczególnych parach tak jak kontrolne
(„zdrowe”) natomiast nie potrafią zupełnie „kojarzyć” (dostrzegać) „przechodniości”, którą kontrolne szczury
zauważają.
Social transmission of food preference task;
Szczur testowany spotyka się z innym szczurem, który tuż wcześniej zjadł określone jedzenie. Szczur testowany
wyczuwa zapach jedzenia w oddechu szczura „demonstratora”. Następnie (natychmiast oraz po 24 godz. )
szczurowi testowanemu przedstawia się szereg różnych pożywień, wśród których znajduje się to, które jadł
„demonstrator”.
Normalny szczur preferuje to pożywienie zarówno natychmiast jak i po 24 godz.
Szczur z uszkodzonym hipokampem preferuje również ale tylko w próbie natychmiastowej, natomiast po 24
godz. nie preferuje (zapomniał...)
Tzw. „place cells” („komórki miejsca”)w hipokampie są aktywne, gdy zwierzę znajduje się w określonym
przestrzennie miejscu (mają „pole recepcyjne miejsca” kodujące cechy i relacje otoczenia konkretnego
miejsca).(testowane tzw. Labiryntami)
Inne komórki wykazują impulsację związana z określonym miejscem a nawet z wyborem dalszej drogi.
Przypuszcza się, że hipokamp (przynajmniej u szczurów) jest miejscem tworzenia „map przestrzennych” (map
poznawczych) aktualizowanych ciągle pod wpływem uczenia się („epizodycznego”)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
92/113
Hipokamp jest odpowiedzialny za bardzo szybkie lub nawet natychmiastowe kodowanie zdarzeń i tworzenie
reprezentacji epizodycznych. Ale to nie hipokamp jest trwałym miejscem deponowania reprezentacji epizodów
ale kora, ponieważ uszkodzenie hipokampa nie niszczy pamięci epizodów dawnych.
Hipokamp zatem tworzy reprezentacje epizodów i poprzez połączenia (wielokrotne) z korą umożliwia tworzenie
w korze reprezentacji pamięciowych epizodów niezależnie od siebie (hipokampa)
Pamięć proceduralna („implicit”)
Pamięć proceduralna jest najczęściej używana w codziennym
życiu. Jest to długotrwała pamięć zręczności ruchowej, percepcyjnej i asocjacyjnej.
Używamy jej wykonując zwyczajowe prace, zręczności, adaptując ruchy stosownie do informacji czuciowych.
Zazwyczaj ten rodzaj pamięci funkcjonuje
niezależnie od świadomości.
(Zwykle takie zdolności uznajemy jako
coś oczywistego)
Jednak w przypadku zaburzenia tej pamięci zmuszeni bylibyśmy szczegółowo „przemyśliwać” każdy ruch czy
czynność.
Pamięć proceduralna jest realizowana przez dwa anatomicznie i funkcjonalnie różne układy.
Jeden z głównym „ośrodkiem” w
NEOSTRIATUM
nabywanie stereotypowych zręczności
Drugi, którego głównym centrum jest
MÓśDśEK
korekcja i kompensacja zwłaszcza zmiennych warunków
obciążenia ruchu oporem lub grawitacją
Pamięć proceduralna dzieli się na dwa podtypy:
1)Nabywanie zwyczajów i zręczności, różnych stereotypowych nieświadomych zachowań (
odpowiedzialne:
neostriatum)
2)Dokonywanie specyficznych czuciowo-ruchowych adaptacji (np. na zmianę obciążenia) i doskonalenie
odruchów (np. chwytu szklanki):
odpowiedzialny: móżdżek
Pamięć proceduralna jest wynikiem uczenia się:
1)Nieasocjacyjnego (habituacja lub sensytyzacja)
2)Asocjacyjnego (poprzez odruchy warunkowe)
Pamięć proceduralna:
podukład „neostriatalny”
Nabywanie zwyczajów i zręczności, różnych stereotypowych nieświadomych zachowań
Układ neostriatun prawie nie ma bezpośrednich połączeń z pniem i rdzeniem dlatego nie ma bezpośredniego
wpływu na czynności motoryczne (tylko za pośrednictwem kory)
„Układ neostriatum”
obejmuje nabywanie „stereotypowych” (?) zwyczajów i zręczności (jazda na rowerze, gra
na pianinie) a jego centralnym „realizatorem anatomicznym” są jądra podstawy (prążkowie –
neostriatum
Packard i Knowlton 2002). Neostriatum
otrzymuje impulsację z kory i z kolei wysyła impulsy do innych jąder
podkorowych oraz do wzgórza. Te jądra z kolei wysyłają impulsację do kory ruchowej i do kory asocjacyjnej
przedczołowej.
System ten wydaje się nie łączyć bezpośrednio z „dolnymi motoneuronami” !
Pamięć proceduralna:
podukład „móżdżkowy”
Układ móżdżku:
jest podukładem pamięci proceduralnej realizującym przystosowania czuciowo-ruchowe oraz
korekcję odruchów która potrzebna jest np. w sytuacji konieczności adaptacji do zwiększonego obciążenia.
Centralną rolę w nim gra
móżdżek
.
(„objawy móżdżkowe” : dysmetria, niezborność, zab. równowagi)
Móżdżek dzięki licznym połączeniom od i do z pniem i rdzeniem (a także z korą) posiada możliwość
bezpośredniego wpływania na czynności ruchowe.
Dokonywanie specyficznych czuciowo-ruchowych adaptacji (np. na zmianę obciążenia) i doskonalenie
odruchów (np. chwytu szklanki):
Model uczenia się móżdżkowego (ruchowego) Marra, Albersa i Ito
Proces „uczenia się” w móżdżku opiera się na LTD (long-term depression) wytwarzanym w synapsach
pomiędzy włóknami równoległymi (pf) (z dróg korowo-mostowo-móżdżkowych) i komórkami
Purkinjego.
Aktywność tych synaps występuje w koincydencji z aktywnością synaps włókien pnących (cf)
pochodzących z oliwek, którymi do kk.Purkinjego docierają „sygnały błędu”.
LTD występuje na synapsach włókien równoległych.
U królika móżdżek zaangażowany jest w:
odruch zamykania powieki (tzw. migotki) i
odruch cofania gałki ocznej
(w reakcji na podmuch powietrza w kierunku oka.)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
93/113
Odruch ten można warunkować np.
dzwonkiem
uruchamianych tuż przed dmuchnięciem.
Dźwięk aktywuje synapsy między włóknami równoległymi i kk.Purkinjego.
Sygnał dmuchnięcia dociera do kk Purkinjego poprzez włókna pnące.
Koincydencja dzwonka i dmuchnięcia powoduje wytwarzanie LTD na synapsach włókien równoległych i kk
Purkinjego.
Zmniejszenie pobudliwości komórek Purkinjego powoduje wzrost sygnału z móżdżku do mięśni gałki ocznej
(„hamowanie hamujących” kk.Purkinjego)
Dalsze trenowanie odruchu warunkowego doprowadza do szybszej odpowiedzi której czas wystąpienia zostaje
zoptymalizowany do mającego wystąpić dmuchnięcia
Pamięć emocjonalna:
ciało migdałowate
Asocjacja określonych bodźców ze szczególnymi
pozytywnymi lub negatywnymi afektami oraz z reakcją m.in. układu autonomicznego
(niekiedy dla jednych „obojętne” emocjonalnie rzeczy dla innych osobników stają się źródłem
emocjonalnego pobudzenia)
Świadome odtworzenie jest zwykle trudne lub niemożliwe.
Ciało migdałowate moduluje zakres konsolidacji tworzącej się pamięci. Zdarzenia uznawane za ważne
powodują pobudzenie całego układu nerwowego co wspomaga konsolidację pamięci czyli tworzenie
trwałych jej „engramów”. Wzbudzenie mediowane przez c.migdałowate polega na aktywacji
hormonalnej (oś podwzgórze-przysadka-wydzielanie kortyzolu z nadnerczy ponadto wzmaga się
wydzielanie katecholamin).
Przykład wpływu pamięci emocjonalnej:
zdarzenie z dzieciństwa takie jak pogryzienie przez psa –
a)Silnie wzmocniona pamięć deklaratywna zdarzenia
b)Nieświadomy nabyty lęk i wstręt do psów
Glukokortykoidy
mogą bezpośrednio aktywować (przechodzą do mózgu) swoje receptory w
hipokampie i c.migdałowatym.
Tylko małe dawki glukokortykoidów wspomagają pamięć! (stres mały jest „dobry”, duży jest „zły”)
Przecięcie blaszki krańcowej (odprowadzającej włókna z c.migdałowatego) blokuje wpływ różnych
substancji (takich jak substancje działające przez receptory GABA, opioidowe, adrenergiczne) na
procesy pamięci.
M.in. Charakterystyczne jest zniesienie działania różnych substancji na procesy pamięci poprzez
podanie
propranololu
(przypuszczalnie modulacja adrenergiczna gra główna rolę w tych procesach).
Podawanie
noradrenaliny i adrenaliny
po wstępnym etapie uczenia się poprawia
przypominanie nabytych wiadomości. Przy czym tylko pośrednie dawki tych hormonów
działają pozytywnie (ani niskie ani wysokie).
Katecholaminy nie przechodzą do mózgu
i zatem prawdopodobnie działają obwodowo
poprzez interakcję z receptorami unerwianymi przez n.X i następnie aktywacje j.pasma
samotnego.
Stamtąd drogi projekcyjne pobudzają j.miejsca sinawego (dopaminergiczne), które z
kolei ma połączenia z c.migdałowatym i hipokampem i w ten sposób może dochodzić do
oddziaływania na procesy pamięci .
Elektryczna stymulacja n.X ma podobne działanie jak podawania katecholamin.
Stymulacja c. Migdałowatego stymuluje (lub hamuje ) zapamiętywanie i jest
uzależnione od sprawnych
nadnerczy!
Ciało migdałowate odgrywa rolę w emocjonalnej modulacji procesów pamięciowych
Wspomnienia wydarzeń o silnym ładunku emocjonalnym są żywsze i dłużej trwające oraz dokładniejsze
niż wspomnienia emocjonalnie obojętne.
Emocjonalnie silne wydarzenie powoduje aktywację układu sympatycznego i osi podwzgórze-przysadka-
nadnercza (wyrzut noradrenaliny i kortykosteroidów, tzw odpowiedź „flight-or-fight”), celem tych reakcji
jest nie tylko aktywacja neuro-metaboliczna ale wzmożenie procesów pamięci (dzięki temu osobnik
lepiej
uczy się
postępowania w sytuacji stresowej (zagrożenia etc)
Pamięć robocza (working memory,
„pamięć bezpośrednia”):
różne obszary kory
(Wraz z „perceptual m.” jest „realizowana” przez korę
bez udziału innych struktur mózgu.)
Pamięć robocza jest to świadoma reprezentacja poszczególnych składników bieżącego doświadczenia.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
94/113
Jest krótkotrwała (często utożsamia się pamięć roboczą z STM czyli z pamięcią krótkotrwałą, ale w tej pamięci
są również elementy LTM).
Uważa się, że pamięć robocza (bezpośrednia) pozwala na równoczesne rejestrowanie i przetwarzanie informacji
czuciowej, pamięci STM oraz „stałego dostępu” do LTM.
Przykładem może być
prowadzenie pojazdu.
input czuciowy
aktualnie obowiązujące (w danym odc.drogi) znaki drogowe
cel podróży (LTM)
„Na bieżąco” rejestruje i podtrzymuje „pośrednie” produkty myślenia oraz reprezentacje wytwarzane przez
system perceptualny (postrzegania)Dostęp do pamięci długotrwałej „składa się” z aktywowanych sekcji LTM
(pamięci długotrwałej) tzw. „chunks”„chunk” jest strukturą hierarchiczna i symbolicznąJednoczasowo ok. 7 +/-
2 „chunks” jest aktywnych
Pamięć perceptualna – „priming”
Jest to rodzaj pamięci realizowany bez udziału świadomości, który
dotyczy zdolności identyfikacji i klasyfikacji przedmiotu będącej rezultatem wcześniejszych „postrzeżeń” tego
przedmiotu lub przedmiotu „pokrewnego”.
Typowy przykład zadania angażującego pamięć perceptualną (priming) to (po uprzedniej ekspozycji sekwencji
określonych obiektów) nabycie zdolności do identyfikacji całej sekwencji tych obiektów (np.słów) na podstawie
początkowej części sekwencji.
Ten rodzaj pamięci jest zachowany u chorych z amnezją (czyli brakiem pamięci deklaratywnej) którzy maja
uszkodzoną przyśrodkową część płata skroniowego (medial temporal lobe)
Badania fMRI wykazały OBNIśENIE AKTYWNOŚCI KORY w zadaniach testujących „priming” (w
przeciwieństwie do pamięci deklaratywnej)!
Modele eksperymentalne mechanizmów pamięci
Pamięć proceduralna: badania nad Aplysia
Slimak morski Aplysia A) odruch cofania skrzela i syfonu do jamy płaszcza na dotkni
ę
cie syfonu
lub strzykni
ę
cie strugi morskiej wody (jest to przykład tzw. Odruchu wycofania)
B) Odruch cofania ogona, syfonu i skrzela po stymulacji ogona
W Aplysia można obserwować i badać mechanizmy uczenia się zarówno
nieasocjacyjnego jak i asocjacyjnego
Habituacja
Powtarzana 10x stymulacja powoduje zwi
ę
kszenie napływu jonów wapnia i tzw.
osłabienie
homosynaptyczne
(tej samej synapsy, która jest pobudzana w odruchu - jest to
zako
ń
czenie aksonu
neuronu czuciowego
). Polega na zmniejszeniu wydzielania neurotransmitera w neuronach czuciowych
syfonu.
Obserwuje si
ę
te
ż
długotrwał
ą
habituacj
ę
, której przyczyn
ą
jest
zmniejszenie liczby synaps.
Sensytyzacja
:
Seria silnych bod
ź
ców bólowych (
stymulacja bólowa ogona)
powoduje uwolnienie modulacyjnych
neurotransmiterów np. serotoniny (5-HT) ze specyficznej klasy neuronów (tzw. neuronów
facylitacyjnych), których zadaniem jest modulacja odpowiedzi neuronów czuciowych
(heterosynaptyczna facylitacja). Neurony facylitacyjne (
wstawkowe serotoninergiczne neurony
toruj
ą
ce)
tworz
ą
synapsy akso-aksonalne na zako
ń
czeniach nerwów czuciowych. W przeciwie
ń
stwie
do habituacji sensytyzacja jest
procesem heterosynaptycznym
(aktywacja innego neuronu ni
ż
neuron
czuciowy)
W
sensytyzacj
ę
krótkotrwał
ą
zaanga
ż
owane s
ą
metabotropowe receptory serotoniny sprz
ęż
one z
cAMP i kinaz
ą
białkow
ą
PKA oraz receptory sprz
ęż
one z DAG i kinaz
ą
PKC)
Kinaza fosforyluje kanał potasowy powoduj
ą
c zmniejszenie pr
ą
du potasowego a st
ą
d wydłu
ż
enie
depolaryzacji (wydłu
ż
enie potencjału czynno
ś
ciowego). Rezultatem jest wzrost napływu jonów Ca2+
do zako
ń
cze
ń
aksonalnych i przedłu
ż
one i nasilone uwalnianie neurotransmitera.
Z kolei fosforylacja przez PKC kanału wapniowego L powoduje przedłu
ż
ony napływ wapnia przez ten
kanał (i równie
ż
nasilenie uwalniania neurotransmitera z zako
ń
czenia neuronu czuciowego co
wzmaga sił
ę
odruchu).
Sensytyzacja (krótkotrwała i długotrwała)
Długotrwała sensytyzacja wymaga syntezy białek
(blokowanie syntezy ogranicza sensytyzacj
ę
do 3 godz.)
Sensytyzacja długotrwała
na modelu Aplysia
Wymaga stymulacji trwaj
ą
cej godzin
ę
lub wi
ę
cej ale jej efekty trwaj
ą
przez co najmniej 24 godz.
Czynnikiem który powoduje przej
ś
cie sensytyzacji krótkotrwałej w długotrwał
ą
jest m.in. Kinaza PKA,
która
zmienia ekspresj
ę
genów
(aktywna podjednostka PKA dostaje si
ę
do j
ą
dra komórki i fosforyluje
czynnik transkrypcyjny CREB (cAMP responsive element binding protein) aktywuj
ą
cy syntez
ę
białek,

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
95/113
w tym m.in. proteazy dla podjednostki regulatorowej PKA co wydłu
ż
a działanie PKA i m.in.
Fosforylacj
ę
kanału K+).
Ponadto aktywacja MAPK prowadzi do fosforylacji i „derepresji” CREB2 co dodatkowo stymuluje
CREB1.
Długotrwała sensytyzacja wi
ąż
e si
ę
z
a)
translacj
ą
wielu innych białek
(ponad 10 produktów genowych
zostało zidentyfikowanych) i
b)
rozgał
ę
zianiem si
ę
aksonów
oraz
c)
tworzeniem nowych synaps
.
PKA, PKC, CaMKII s
ą
tzw. „kognitywnymi kinazami”
-PKA, PKC, CaMKII podlegaj
ą
trwałym zmianom aktywno
ś
ci nie ust
ę
puj
ą
cym nawet po zanikni
ę
ciu stymuluj
ą
cego je sygnału
(wtórnego przeka
ź
nika)
-Kinazy te moduluj
ą
aktywno
ść
synaptyczn
ą
„Asocjatywna facylitacja” u Aplysia (forma uczenia się asocjacyjnego)
Odruchy „ucieczki”
(withrawal) u Aplysii mogą być użyte do klasycznego warunkowania
Odruch cofania skrzela mo
ż
na wzmocni
ć
nie tylko na drodze sensytyzacji ale równie
ż
poprzez
tzw.
Asocjatywn
ą
facylitacj
ę
(jest to forma asocjatywnego uczenia) czyli dzi
ę
ki warunkowaniu reakcji
behawioralnej.
Bod
ź
cem warunkowym (CS) jest łagodne dra
ż
nienie syfonu
Bod
ź
cem bezwarunkowym (US) jest podra
ż
nienie ogona (tylnej cz
ęś
ci nogi), który powoduje
gwałtown
ą
reakcj
ę
.
CS poprzedza US o 0,5 sek. Wielokrotne powtórzenie sekwencji CS-US powoduje powstawanie silnej
reakcji bezwarunkowej w odpowiedzi jedynie na bodziec CS (łagodny dotyk syfonu)
POSTULOWANY MECHANIZM MOLEKULARNY:
Molekularnym podło
ż
em tej zmiany jest
wzmocnienie efektywno
ś
ci przewodnictwa w synapsie mi
ę
dzy
neuronem czuciowym i ruchowym
.
Dzieje si
ę
tak dzi
ę
ki moduluj
ą
cemu wpływowi neuronów toruj
ą
cych, które w wyniku działania bod
ź
ca
bezwarunkowego (US) s
ą
zaktywowane i wyrzucaj
ą
serotonin
ę
w synapsie akso-aksonalnej przy
zako
ń
czeniu aksonalnym neuronu czuciowego.
1) W wyniku bod
ź
ca CS uwalniany jest Ca2+.
(Przerwa mi
ę
dzy CS i US musi by
ć
krótka bo wap
ń
jest szybko buforowany w cytozolu.)
2) Nast
ę
pnie serotonina uwolniona w wyniku działania US aktywuje
cyklaz
ę
adenylow
ą
w aksonie
neuronu czuciowego, która staje
si
ę
wra
ż
liwa na wap
ń
.
(Ale nie ma jasno
ś
ci dlaczego CS musi poprzedza
ć
US.)
Rola obszarów asocjacyjnych kory mózgowej
Główne połączenia kory asocjacyjnej
•„wejściowe”
–Korowo-korowe tożstronne i przeciwstronne
–Wzgórze
–C. migdałowate
–Hipokamp
–Pień mózgu (ośrodki modulacyjne)
•„wyjściowe”
–Do pozostałych obszarów kory (tak jak te „wejściowe”)
–Wzgórze
–C. migdałowate
–Prążkowie (j.ogoniaste i skorupa)
–Pień mózgu
–Rdzeń kręgowy
Główne „specjalizacje” kory asocjacyjnej
Kora ciemieniowa:
uwaga
–Contralateral neglect syndrome
po uszkodzeniu prawej kory ciemieniowej: - deficyt
postrzegania (uwagi) dotyczącego „lewej strony świata”. (pierwszy opis dr W.R.Brain 1941 w:
Brain
).
Uwaga! „niedobór” postrzegania dotyczy lewej części każdego obiektu. Prawa półkula „obsługuje” obie
strony „rzeczywistości” wewnętrznej (ciało) i zewnętrznej (otoczenie) a lewa półkula tylko prawą
stronę. Po uszkodzeniu lewej półkuli jest tylko minimalny deficyt po stronie prawej.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
96/113
•„Neurony uwagi”
•Detekcja neuronów związanych z uwagą
(„attention neurons”) – eksperymenty z implantowanymi elektrodami u małp rhesus Niektóre neurony kory
ciemieniowej są aktywne jeśli zwierzę aktywnie obserwuje „target”
•„Neurony rozpoznania”
•„Neurony rozpoznania” np. twarzy (recognition neurons, - „face cells”) w płatach skroniowych. Są
prawdopodobnie zorganizowane w kolumny, w których poszczególne komórki „odpowiadają” (rozpoznają?) na
różne cechy twarzy
•„Neurony planowania” (czołowa kora asocjacyjna)
•Badane neurony w czasie tzw. „delayed response task” są aktywne w czasie „pamiętania” (między ekspozycją
jedzenia i podniesieniem ekranu pozwalającego na odszukanie jedzenia) i przestają być aktywne gdy zwierzę
„odgaduje” gdzie było włożone jedzenie
•Kora skroniowa:
rozpoznawanie znaczenia i natury bodźców
–Prosopagnosia: nierozpoznawanie znajomych twarzy : uszkodzenie spodniej części
prawego
dolnego płata skroniowego (po stronie lewej uszkodzenie powoduje zaburzenia językowe). Np.
przypadek L.H., po urazie nie potrafiącego rozpoznawać twarzy (częściowo także zwierząt i ich
ekspresji) ale potrafiącego rozpoznawać ludzi np. po głosie lub sposobie chodzenia. Lokalizacja
„ośrodka rozpoznawania twarzy” w prawym płacie skroniowym („od spodu”) potwierdzona w fMRI. W
zależności od rozległości uszkodzenia agnozja może dotyczyć również innych obiektów (nie tylko
twarzy).
•Kora skroniowa:
rozpoznawanie znaczenia i natury bodźców
–Uszkodzenie płata skroniowego po
stronie lewej
uszkodzenie powoduje zaburzenia językowe. Cechy
osłabionego rozumienia mowy związane są z uszkodzeniem dolnej części płata skroniowego (rejon
gyrus fusiformis).
–Zaburzenia językowe dotyczą zewnętrznej części płata skroniowego lewego
•Kora czołowa:
planowanie odpowiedzi behawioralnych
–Kora czołowa jest największym obszarem asocjacyjnym, realizującym bardzo liczne funkcje, jej
zaburzenia często przejawiają się jako „zmiana charakteru” (osobowości?)
–Przypadek zmian osobowości Phineas’a Cage’a w wyniku urazu w 1848 r na budowie linii kolejowej
niszczącego płaty czołowe (zm w 1863 r). Gł niezdolność do planowania działań.
–Wisconsin Cart Sorting Task (planowanie) testuje funkcje płatów czołowych
–„obsługa” working memory
–Social restraint functions (poczucie co jest społecznie niewłaściwe)
Język-mowa
•Korowa reprezentacja języka-mowy jest osobna w stosunku do ośrodków kontrolujących
motorycznie mięśnie fonacyjne (krtani, języka, itp.) a także w stosunku do ośrodków recepcyjnych dźwięków i
wzroku.
•Jest to więc reprezentacja „komunikacji” i/lub znaczenia znaków i symboli niezależnie od sposobu przekazu
(mowa, pismo, język migowy)
•Gramatyka, syntaxa, prozodia (emocjonalne znaczenie)
•Ośrodek Broca (uszkodzenie –afazja motoryczna, rozumienie zachowane)
•Ośrodek Wernicke’go (uszkodzenie – afazja czuciowa, recepcyjna, mowa płynna)
•Afazja kondukcyjna
•Podział afazji wg Normana Geschwind’a (znalazł asymetrię w „planum temporale”)
•Lateralizacja funkcji językowych – badania Rogera Sperry z Caltech (60,70-te) u chorych ze „split-brain”
(rozcięcie ciała modzelowatego)
Ośrodek Broca (uszkodzenie –afazja motoryczna, rozumienie zachowane)
Ośrodek Wernicke’go (uszkodzenie – afazja czuciowa, recepcyjna, mowa płynna)
Lateralizacja niektórych funkcji w półkulach mózgu•Nie jest prawdą, że „mówimy lewą półkulą” ale
niewątpliwie lewa półkula odgrywa w mowie większą rolę choć i prawa ją w dużym stopniu uzupełnia.
•Badania Geschwinda:
„planum temporale”
po stronie lewej jest znacznie większa (o ok.50%), jednak tylko u
67% (2/3) ludzi (już od urodzenia!) podczas gdy „przewaga” lewej półkuli w funkcjach mowy jest obecna u 97%
ludzi.
•Badania Geschwinda: obszar tzw.
„planum temporale”
po stronie lewej jest znacznie większa (o ok.50%),
jednak tylko u 67% (2/3) ludzi (już od urodzenia!) podczas gdy „przewaga” lewej półkuli w funkcjach mowy
jest obecna u 97% ludzi.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
97/113
Język-mowa: test Juhn Wada’y
•Wstrzyknięcie amytalu sodu (krótkotrwałą anestezja) do lewej
tętnicy szyjnej powoduje krótkotrwałe „wyłączenie” półkuli i jeśli jest ona dominująca „językowo” testowany
wykazuje przejściową afazję
•
Badania neuroobrazowania funkcjonalnego z tachistoskopem i inne (Penfield) wskazują na aktywację różnych
obszarów kory w czasie mówienia
•Różne kategorie słów (ludzie, narzędzia, zwierzęta) maja różne „lokalizacje”
•„Ośrodki mowy” prawej półkuli decydują o „prozodii” mowy (ładunku emocjonalnym)
•Również u chorych głuchych od urodzenia i posługujących się językiem migowym obserwuje się
„lewopółkulowy” charakter języka znakowego i podobne cechy „afazji” w przypadku uszkodzenia lewej półkuli.
–Wniosek: reprezentacje korowe „mowy” dotyczą nie tyle języka ile ogólnego
systemu symboliczno-
komunikacyjnego
.
Tajemniczy Gyrus angularis (pole Brodmanna 39)
•V.S.Ramachandran uważa, że g.marginalis jest
odpowiedzialny za
rozumienie metafor.
S twierdził to u chorych z uszkodzeniem lewego z.kątowego którzy
okazywali się nie mieć defektów mowy natomiast nie rozumieli znaczenia metafor – rozumieli jedynie dosłowne
znaczenie przenośni (czy to jest to samo co słynny „skrót myślowy” znanego ministra?).
•Ponadto chorzy zachowywali się odmiennie niż ponad 90% populacji w teście Wofganga Kohlera polegającym
na przyporządkowaniu sztucznego słowa „Booba” kształtowi zaokrąglonemu oraz „kiki” kształtowi
kanciastemu. Ma to świadczyć o zaburzeniu zdolności u tych chorych przyporządkowywania bodźców
wzrokowych językowi.
•Ramachandran uważa, że zakręt kątowy (szczególnie duży u człowieka) może pełnic istotną rolę w asocjacji
zmysłu słuchu, wzroku i czucia somatycznego (dotyk).
•Blanke i wsp drażnieniem elektrycznym g.marginalis wywołali u chorej na padaczkę doznania typu tzw. „out-
of-body-experience” (Blanke i wsp. Stimulating illusory own-body perceptions. Nature, 419:269-270, 2002 )
DOMINACJA POŁKUL
•97% osób wykazuje dominację językową lewej półkuli mózgu (w tym większość leworęcznych!)
•9/10 osób jest praworęcznych ale i u leworęcznych większość ma językową „dominację” lewej półkuli (choć
większość osób z dominacją językową prawej półkuli to osoby leworęczne!).
•Fonemy (w angielskim 40 po ok. połowie spółgłoski i samogłoski)-sylaby-wyrazy
•Niewykluczone, że fonemy odnoszą się raczej do wyuczonego w czasie nauki czytania nawyku wymawiania
słów uważanego za poprawne a nie do „naturalnych” brzmień mowy (słyszanej)
•Noam Chomsky: język jest zbyt skomplikowany i nie może być po prostu nauczony lecz musi być
„predicated
on „universal grammar””
– „gramatyce” wykształconej w ewolucji naszego gatunku.
•Chomsky unikał neurobiologicznych analogii ale zapewne tą „uniwersalną gramatykę” realizuje ewolucyjne i
genetycznie zaprogramowana specyfika budowy i działania kory mózgu
•Prawdopodobnie analiza języka ludzkiego może dać obraz i pozwolić na zrozumienie jak pracuje mózg tworząc
niezwykłe „urządzenie” pozwalające na wytwarzanie asocjacji (najprawdopodobniej jest to fundamentalna cecha
kory mózgu)
Funkcje kognitywne
FUNKCJE „KOGNITYWNE” TO:
POSTRZEGANIE, PAMIĘĆ, ŚWIADOMOŚĆ, JĘZYK
Ale co właściwie znaczy „MYŚLENIE”,co to znaczy „MYŚLEĆ”, „BYĆ ŚWIADOMYM”,
„SAMOŚWIADOMYM”?
Czy wystarczy spojrzeć w lustro aby „zobaczyć siebie”?
Tzn. nie tylko dostrzec obraz jakiegoś-tam osobnika ale SIEBIE
Czy wystarczy spojrzeć w lustro aby „zobaczyć siebie”, to znaczy uświadomić sobie o „istnieniu siebie”, o
istnieniu czegoś absolutnie unikalnego w całym wszechświecie, unikalnego bytu, unikalnego „JA”, wobec
którego cała reszta wszechświata jest czymś zasadniczo różnym?
Jaki układ mózgu powoduje, że jesteśmy SAMOŚWIADOMI ?
Niewątpliwie wzrok i cały system jego percepcji i analizy jest najbardziej „podejrzany” o istnienie „w
nim”
korelatów świadomości
.
Ale czy ślepy od urodzenia nie ma poczucia świadomości???!!!
Czy zatem w ogóle trzeba spojrzeć w lustro aby uświadomić sobie o istnieniu siebie, aby dostrzec przepaść
między tym co nazywam „JA” i resztą, czyli „NIE-JA”, aby dostrzec (albo wymyślić) że istnieje (realnie –

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
98/113
artyficjalnie?) dychotomia między „
podmiotem
-postrzegającym” i „
przedmiotem
-postrzeganym”? -
„Przedmiotem-postrzeganym”, do którego to pojęcia należy również nasze (całe?) ciało?
[„JA-ŚWIADOME” – VS – „JA-CIELESNE”]
– VS – „NIE-JA”
Gdzie „się mieści” świadomość ?
W poszukiwaniu korelatów świadomości
Neurobiolodzy poszukują tzw. „nerwowych korelatów”
świadomości (neural corrrelates of consciousness), czyli struktur lub miejsc albo specyficznych form
aktywności mózgu, które jeśli nawet nie są jednoznacznie odpowiedzialne za „świadomość” to
przynajmniej są skorelowane ze świadomością
Crick i Koch w 1995 r sformułowali hipotezę, że funkcją świadomości ciągłe wytwarzanie („on
line”/”live”) możliwie najlepszej interpretacji „sceny obejmowanej wzrokiem” (visual scene) w postaci
zwartej (compact) i udostępnianie tej informacji dla „stanów planowania” (planning stages) mózgu.
Już tylko z tego powodu, że pierwotna kora wzrokowa (V1) nie wykazuje bezpośrednich projekcji do
obszarów przedruchowych i przedczołowych (uważanych za pełniących „funkcje kognitywne”) Crick i
Koch sądzą, że pole V1 nie jest częścią „korelatów świadomości”
Wiele innych danych również przemawia
za tym, że pierwotna kora wzrokowa nie zapewnia świadomej percepcji wzrokowej.
Badania czynności
elektrycznej mózgu (EEG) w czasie snu połączone z funkcjonalnym obrazowaniem MRJ (fMRJ)
wskazują, że w czasie fazy snu REM (gdy są „obrazowe” sny) aktywność pierwotnej kory wzrokowej (V1)
jest obniżona. Wiadomo również, że u chorych z uszkodzoną korą wzrokową V1 stwierdza się „wizualne
sny”. Wniosek: badania te sugerują, że kora V1 nie jest odpowiedzialna za świadome odbieranie wrażeń
wzrokowych.
Zjawisko obuocznej rywalizacji
Eksperymenty z fMRJ w czasie przeprowadzania testów obuocznej rywalizacji również wskazują, że to
nie V1 jest aktywna
w czasie „przełączania widzenia” ale obszary brzusznej strefy kory „nieprążkowanej”
(otaczającej korę wzrokową i otrzymującej również bodźce wzrokowe) oraz obszary w płatach
ciemieniowych i przedczołowych (m.in. Związane z kontrolą uwagi przestrzeni).
Nie jest jasne w jaki sposób jakikolwiek proces fizyczny, taki jak aktywność neuronalna może być
źródłem subiektywnego zjawiska jakim jest świadomość.!
Nawet teoretyczna możliwość istnienia tego rodzaju związku przyczynowego (między czynnością fizyczną
a świadomością) jest kontrowersyjna nawet dla filozofów!!!
Dlatego poszukiwanie nerwowych korelatów świadomości jest badaniem empirycznym, które powinno
pozostawać (przynajmniej w fazie wstępnej) neutralne wobec problemów przyczynowości ale skupiać się
na identyfikacji i charakteryzowaniu wzorów aktywności, które korelują specyficznie z „doświadczeniem
świadomości” a nie z postrzeganiem lub aktywnością nieświadomą
Świadomość zapewne „rodzi się” z „koherencji” doznań zmysłowych i z „wbudowanej” (hipokamp?)
zdolności inferencji (rozumowania).
Dotykam palcem czoła i czuję jednoczesny sygnał z palca i ze skóry czoła.
Widzę ścianę i powiększające się obrysy kształtów a w chwilę potem doznaję efektów czuciowych bólu i
dotyku w zetknięciu ze ścianą. Stąd nabieram przekonania o rzeczywistości istnienia ściany. W dalszej
konsekwencji nabieram przekonania, że wszystko to co widzę zapewne realnie istnieje.
(wg mnie w tym
ciągu rozumowania jest wiele „implicitów”)
Reguły wnioskowania (
zasada przechodniości
) dostrzegamy już u szczura. Również zwierzęta
niewątpliwie doznają efektów „koherencji” doznań czuciowych różnej i tej samej „modalności” (np.
wzrok i słuch).
A więc co najmniej są na najlepszej drodze do osiągnięcia stanu świadomości, ale czy również
„samoświadomości”?
Te pytania dotyczą kluczowych aspektów pojęcia „człowieczeństwa” i (wyróżników człowieczeństwa) !!!
Sądzę, że jest jeszcze jeden istotny aspekt (wymiar, rodzaj) świadomości :
Nazwałbym go
„świadomością krytyczną”.
Jest to świadomość wątpliwości, negacja realizmu doznań
czuciowych, świadomość złudzeń, świadomość niepełności (prawdy) doznań czuciowych
•Poglądy Eugene d’Aquili i Andrew Newberg, z Filadelfii*:
–„
kognitywny imperatyw
” ludzkiego umysłu (bardzo trudno „nic nie myśleć”…)
–„
kognitywne operatory
”, które (nie przesądzając o ich lokalizacji – zapewne korowej) charakteryzują
funkcjonowanie ludzkiej kognicji:
•Operator holistyczny (dostrzegać całość)
•Operator redukcjonistyczny (dostrzegać elementy)
•Operator abstrakcji (m.in. taksonomia)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
99/113
•Operator ilościowy („matematyczny”)
•Operator przyczynowości (pytania: „dlaczego”, „z jakiego powodu”)
•Operator binarny (podziałów binarnych, dychotomii, przeciwieństw)
•Operator egzystencjalny (nadający sens i „założenie” istnienia i rzeczywistości doznań czuciowych)
•Operator emocjonalnej wartości
•
•*Eugene d’Aquili i Andrew Newberg „Why God Won’t Go Away: Brain science and the Biology of
Religious Experience”
Poszukiwanie „korelatów świadomości” jest pozornie „neutralne” ale wyobraźmy sobie co człowiek
potrafiłby zrobić gdyby (wydawało by mu się, że) posiadł taką wiedzę ???
Otóż jak każda wiedza i taka wiedza podobnie jak np. wiedza o energii rozszczepienia jąder atomu uranu
może prowadzić do destrukcji
(nazwałbym to „wiedzą letalną”).
W jaki sposób?
Np. spróbowano by sposobów (operacyjnych, farmakologicznych, genetycznych) do celowego wyłączenia
świadomości !!!
Po co?
Np. aby znieść lęk śmierci u żołnierzy ...
Aby ... ... „bardziej HUMANITARNIE (??)” zabić ...
Nie mówiąc już o tym co by można zrobić gdyby móc poznać TREŚĆ ŚWIADOMOŚCI...
Dla lekarza istotne jest aby człowiek, (umownie zwany „pacjentem”) którym lekarz się zajmuje (to szerzej
niż tylko „leczy”) nie był postrzegany jako (kolejny) metaboliczno-biochemiczno-genetyczno-
molekularno-patologiczny „PRZYPADEK” ale BYT ŚWIADOMY
To jest postulat humanistyczny !!
Nie mam nic przeciwko temu, aby podobny stosunek do pacjenta charakteryzował również
WETERYNARZA ! (nie możemy wykluczyć, że i zwierzęta mają świadomość, choć prawdopodobnie
„częściową” w stosunku do ludzkiej).
Gdy spotykamy chorego (ale niekoniecznie) – po prostu - człowieka pamiętajmy, że
nie
jest to
„humanoidny metaboliczny komputer” działający w schemacie „INPUT-PROCESSING-OUTPUT”.
POSTULAT „DOMNIEMANIA ŚWIADOMOŚCI” (analog „domniemania niewinności” w procesie
sądowo-karnym) – wynikający z moralnej normy: lepiej zaniechać wymierzenia kary niż ryzykować
skrzywdzenie niewinnego.
Neuroteologia
… czyli co
ś
w rodzaju Gagarina za monitorem tomografu MRJ…
•W obszarze które nazwałbym „zagadnieniami krańcowymi” tkwi „neuroteologia” (termin wg. Wikipedia
po raz pierwszy użyty przez Aldous Huxley’a w książce „Island” 1962)
•„NEUROTEOLOGIA”:
–Neurobiologiczne podstawy przeżyć religijnych - próby dociekań co w mózgu, koreluje („wywołuje”?) z
przeżyciami religijnymi. Jest zatem „(neuro)biologia duchowości”.–Poszukiwania „modułu Boga” w mózgu
–
–Uwagi własne:
–Wydaje się, że jak na razie „neuroteologia” jest raczej intrygującym „słowem-haczykiem” które (głównie
intuicyjnie) nieźle wskazuje na takie obszary neurobiologii i nauk pokrewnych, które dotyczą choćby w pośredni
sposób zagadnień religii. Z pewnością nie jest to jak na razie w pełni samodzielna gałąź nauki (złośliwi skłonni
są raczej zaliczać neuroteologię do pseudonauk).
–Wg mnie jeśli jest jakikolwiek sens w poszukiwaniu toposu (bardziej „przyziemnie” – lokalizacji…) ducha-
duchowości to miejscem, w którym to należy robić jest oczywiście mózg. Można jednak zadać w związku z tym
pytanie: CZY PŁÓD BEZMÓZGOWY MA „DUSZĘ”…?
– Jeśli prawdą jest, że kiedyś-tam teologowie toczyli debaty ile aniołów może zmieścić się w łebku szpilki,
czemóżby nie mogli (wspólnie z neurobiologami itp) zastanowić się np. nad tym ile neuronów wystarczy aby
„osiedlił się” (?) w nich duch-dusza …? Problem chyba nie jest z punktu widzenia religii i teologii błahy i w
gruncie rzeczy wiąże się chociażby z powyżej wspomnianym zagadnieniem duszy płodu bezmózgowego
(anencephalon) …?
–Zagadnienia te przedstawiam w ramach neurobiologii raczej jako „ciekawostkę”
próbując znaleźć właściwą dla nich miarę i traktuje te zagadnienia raczej jako zachętę
do własnych przemyśleń studentów.
Badania i poglądy „neuroteologiczne” d’Aquili i Newberga
Jeśli założyć, że obszar tylnej górnej części płata ciemieniowego lewego nazwane przez Eugene d’Aquili i
Andrew Newberga „ORIENTATION ASSOCIATION AREA” jest (po stronie lewej) miejscem

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
100/113
odpowiedzialnym za wytwarzanie poczucia „self” mającego zaznaczone granice przestrzenne i umiejscowionego
w konkretnej przestrzeni i czasie (to raczej strona prawa) a inny obszar (kora przedczołowa) jest miejscem
(m.in.) kontroli uwagi i decyzji wolicjonalnych („ATTENTION ASSOCIATION AREA”) obserwacje stopnia
aktywacji tych obszarów mogą rzucić światło na pracę mózgu w stanach związanych ze „zmienioną
świadomością” np. w stanach religijnego transu.
Eugene d’Aquili i Andrew Newberg, z Filadelfii, autorzy m.in. „The Mystical Mind: Probing the
Biology of Religious Experience” oraz „Why God Won’t Go Away: Brain science and the Biology of Religious
Experience” w latach 90-tych badali aktywność mózgu w czasie religijnych przeżyć przy pomocy
funkcjonalnego obrazowania czynności mózgu (SPECT). Badano modlące się zakonnice franciszkanki oraz
medytujących mnichów buddyjskich.
Badania ich wskazują, że w czasie religijnego przeżycia silnie aktywowane są obszary czołowe (odpowiedzialne
m.in. za koncentrację uwagi „ATTENTION ASSOCIATION AREA”) natomiast
zmniejszona aktywność
dotyczy obszarów w tylnej górnej części płatów ciemieniowych w
lewej półkuli
mózgu (odpowiedzialnych za
orientację w czasie i przestrzeni „ORIENTATION ASSOCIATION AREA”).
d’Aquili i Newberg uważają, że tzw. „stany unitarne” („jedności
z wszechbytem”)
są skutkiem „deafferentacji”
(odcięcia doznań czuciowych) ORIENTATION ASSOCIATION AREA przy jednoczesnej silnej aktywności
ATTENTION ASSOCIATION AREA. W rezultacie wytwarza się stan „awareness without self”.
Stany te są spokrewnione z reakcjami seksualnej przyjemności.
One też są „zdrowotną” metodą relaksacji i zarazem bardzo starym nabytkiem ewolucyjnym.
Autorzy ci m.in. uważają, że różne przeżycia chorych w stanie „near death” („śycie po życiu”) mają charakter
neurologiczny oraz uznają równoważność mózg i umysł (brain and mind).
Neurobiologia stanów mistycznych
wg. Eugene d’Aquili i Andrew Newberg
•Możliwość osiągania różnego stopnia stanów unitarnych („unitary continuum” – okr. d’Aquili i Newberga) i
związanej z nimi „deafferentacji” ORIENTATION ASSOCIATION AREA (OAA).
•Dzieje się to np. na skutek oddziaływania rytmicznych („ritual”) bodźców stymulujących układ limbiczny i
autonomiczny (prowadzą one do aktywacji działania hipokampa w taki sposób, że blokuje OAA).
•Stany unitarne osiągane są na drodze „biernej” (np. zachwyt nad czymś) i czynnej medytacji (gdzie d’Aquili i
Newberg również rozróżniają techniki bierne – i aktywne) następny slajd
••„Płytkie stany mistyczne-unitarne” (okr. moje na podstawie d’Aquili i Newberga) – wszystkie cechuje różnego
stopnia redukcja „ego” o poczucie „absorpcji” siebie (self) przez rodzaj szerszej rzeczywistości. Np. publiczność
„porwana” przez występ artysty
–Zachwyt nad pięknem
–Trans
–ekstaza
•Głębokie stany mistyczno-unitarne:
–„Unio Mystica”
–Krańcowy stan unitarny (Absolute Unitary State - AUB)
Neurobiologia medytacji
wg. Eugene d’Aquili i Andrew Newberg
–Bierne techniki medytacji:
wola „oczyszczenia umysłu” z myśli, emocji i percepcji
.
•Aktywacja wolicjonalnego PRAWEGO OBSZARU ASOCJACYJNEGO UWAGI, który poprzez WZGÓRZE
oddziałuje na HIPOKAMP w taki sposób, że blokuje input czuciowy m.in. do OAA (obszar asocjacyjny
orientacji). OAE staje się „deafferented”. Z „deaferentowanego” OAA impulsacja oddziałuje na UKŁAD
LIMBICZNY I PODWZGÓRZE prowadząc do emocjonalnego uspokojenia
•Całkowite odcięcie (deaferentacja) OAA w obu półkulach powoduje:
–po stronie prawej – oznacza to brak impulsacji koniecznej w tworzezniu przestrzennego-topologicznego
umiejscowienia „self” (w zwiazku z tym nastepuje utrata poczucia miejsca i czasu);
–po stronie lewej deaferentacja OAA oznacza całkowity zanik granic „self” (w praktyce zanik poczucia samego
siebie. („awareness without self”)
–W rezultacie następuje „absolute sense of unity” („nirvana”)
–
–Czynne techniki medytacji:
skupienie myśli na określonym obiekcie (zwł sakralnym) i
jego kontemplacji.
d’Aquili i Newberg podają nieco inny przebieg aktywności w mózgu niż w technikach biernych medytacji ale
rezultat jest zasadniczo ten sam: redukcja „self” i poczucie stanu unitarnego

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
101/113
Krytycy d’Aquili i Newberga podkreślaja m.in. , żę redukowanie przeżyć religijnych do „stanów
unitarnych” upraszcza zagadnienia przeżycia religijnego i religii w ogóle, że ich „teoria religii” zupełnie
pomija aspekty społeczne, w szczególności moralne i etyczne fundamenty religii…?
Niezależnie od tych wątpliwości, badania „neuroteologiczne” d’Aquili i Newberga sugerują, że przepaść
dualizmu „DUCHOWE-MATERIALNE” może zostać przynajmniej częściowo „zasypana”i prowadzą do
przełamywania poglądów, że odczucia duchowe mają charakter wyłącznie kulturowy.
z komentarza na temat prac d’Aquili i Newberga z National Catholic Reporter)
„… Niewątpliwie z
punktu widzenia religii
jedynym miejscem, gdzie Bóg mógłby zamanifestować swe
istnienie są poplątane neuronalne połączenia.
„Słowo-logos” musi zapewne w jakiś sposób stać się
„elektrochemiczne” aby zaiskrzyć pomiędzy synapsami i powędrować poprzez materialne ciała naszych
neuronalnych połączeń.”
(
„Moduł Boga”…(?)•Vilayanur Ramachandran, (Center for Brain and Cognition – Univ. California) - Brain and
Perception Laboratory
•W 1997 Ramachandran z zespołem zasugerowali na podstawie obserwacji chorych z padaczka skroniową
(temporal lobe epilepsy - TLE), którzy wykazują bardzo silne emocjonalne przeżycia religijne, odkrycie "God
module" w mózgu człowieka, który to „moduł” jest jak twierdzą, odpowiedzialny z ewolucyjny instynkt wiary w
religię. Wg Ramachandrana "God module" jest zgrupowaniem komórek w płacie skroniowym, które jeśli sa
stymulowane wydają się wywoływać religijne przeżycia.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
102/113
Uwagi na zakończenie wykładów
z neurobiologii
Neurobiologia musi w takim czy innym miejscu dotrzeć do zagadnień, które określiłbym „zagadnieniami
krańca” (krańca „materialnej neurofizjologii”).
Są to zagadnienia takie jak dychotomia „umysł-mózg” oraz problemy związane ze świadomością.
Zaznaczam, że to nie są jedyne zagadnienia typu „krańcowego” w biologii i medycynie, bo np. podobnie
„krańcowym” zagadnieniem jest np. „problem definicji życia” (co jest żywe a co jest martwe..., orzeczenie
śmierci, śmierci mózgu etc), powstania życia, ewolucji etc.
Wspólną cechą tych wszystkich „krańcowych” problemów jest ich związek z filozofią, wnioski o znaczeniu
etycznym (moralnym) jak również duży wpływ etyki-moralności na sam sposób rozwiązywania tych zagadnień,
a wreszcie interpretacja wyników badań obarczona silnym „skrzywieniem” (raczej po prostu „wpływem” co
brzmi bardziej neutralnie) religijnym (ang. „religious bias”)
DLA LEKARZA NIE SĄ TO TYLKO PROBLEMY „TEORETYCZNE”
Zawód lekarza, do którego się przygotowujecie jest „zatopiony” w tych problemach.
Przykład jeden z najprostszych:
będziecie musieli „stwierdzić zgon” i wystawić odpowiedni dokument…
A propos świadomości
Człowiek pragnący poznać rzeczywiste „ja” drugiego człowieka jest podobny do kogoś kto jest np. pozbawiony
jakiegoś zmysłu i jest zdany wyłącznie na pośrednie poznanie czym ten zmysł jest. Np. człowiek niewidomy od
urodzenia może sobie jakoś wyobrazić widzenie i świat wokół oraz siebie ale to nie to samo co naprawdę
zobaczyć !!!
A zatem usiłując poznać świadomość drugiej osoby stoimy na pozycji głuchego, któremu „na migi” tłumaczą na
czym polega świat dźwięków.
(Przykłady o domyślaniu się przez małpę bonobo myśli człowieka lub małpiej „mowy”, wyobraźni
niekoniecznie trafne – jest bardzo prawdopodobne,że są to po prostu efekty asocjatywnego uczenia)
Być może jeśli nie dowodem, to bardzo silną przesłanką przemawiającą za lub przeciw świadomym procesom
jest coś co nazwałbym roboczo
„neurobiologicznym kryterium transcendencji”
Chodzi o to czy dana istota zachowuje się tylko wyłącznie na bazie odruchowej. Czy jej zachowania można
wytłumaczyć i opisać w postaci deterministycznej „przestrzeni odruchowo-behawioralnej” (hipotetyczny rodzaj
matematycznej formuły zbioru zdarzeń i relacji między nimi). Czy też dana istota potrafi „wyzwolić się” od
mechanizmów odruchowych.
ALE... Rodzi się pytanie:
Czy sam człowiek rzeczywiście spełnia takie „neurobiologiczne kryterium transcendencji” ???
W układzie nerwowym dominuje przetwarzanie informacji
(input – processing – output) ale
mózg to nie tylko ładunki elektryczne...
Mózg jest też częściowo „narządem wydzielniczym” (nie tylko dlatego, że podwzgórze wytwarza i wydziela
neurohormony ale także ponieważ wiele innych komórek nerwowych wydziela różne substancje nie tylko
pełniące funkcje neurotransmitera
w ścisłym znaczniu
(neurohormony, czynniki wzrostu, „nieklasyczne”
neurotransmitery). Pamiętajmy też, że w związku z intensywnym spalaniem glukozy na potrzeby energetyczne
mózg w gruncie rzeczy „wydziela” dużo wody i dwutlenku węgla – stąd uzasadniony niejako jest żart o
„uderzaniu wody sodowej do głowy…”. (woda sodowa to przecież woda i dwutlenek węgla…)
Aktywność natury elektrycznej (elektromagnetycznej – uwaga n.t.TMS) choć tak spektakularnie nasuwająca
analogie z
„mózgiem elektronowym”
(to zapomniana pierwsza popularna nazwa komputerów) jest najściślej
powiązana z procesami natury (bio)chemicznej oraz z aparatem genetycznym, który także jest sui generis
systemem informatycznym (przechowuje, udostępnia, koryguje, kontroluje informację zawartą w DNA i RNA).
Można więc śmiało zaryzykować tezę, że
(jeżeli) myślenie jest funkcją mózgu
, to nie jest tylko funkcją
„galaktyk neuronów i synaps” działających na podobieństwo „gugolicznej” centrali telefonicznej ale jest w
równym stopniu funkcją metaboliczno-genetyczno-biochemiczną („googol”= 10100 E. Kasner 1878 –1955
Amer. matematyk)
Lekarz musi znać i w sposób ustawiczny pogłębiać wiedzę (farmakologiczno-patofizjologiczną-molekularno-
genetyczną etc)
To jest również wymóg deontologiczny (w kodeksie i przysiędze lekarza)
Ale koniecznie trzeba aby „w świadomości lekarza” (jego zakręt obręczy...?) była stała gotowość i stały alert do
refleksji, że nie chodzi tylko o molekuły, komórki i procesy ale, że ma kontakt z
tajemniczą głębią
samoświadomego bytu.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
103/113
Trzeba pamiętać że między skomplikowaną impulsacją neuronalną, skomplikowaną grą metaboliczno-
genetyczną neuromediatorów, receptorów, wtórnych neuromediatorów, czynników tranksypcyjnych a
rzeczywistym świadomym doznaniem „bodźca” istnieje być może niepoznawalna (do końca) przepaść.
UWAGI NA TEMAT MOWY-JĘZYKA
Można by nieskończenie opowiadać o treści i pięknie obrazu ale nic nie zastąpi obejrzenia go...
Tym niemniej „myślimy słowami” i komunikujemy się słowami (to jest jeden z wykładników „umysłu” i
człowieczeństwa) i w słowach próbujemy opisać wszystko.
UWAGA!
Słowami komunikujemy się także ZE SOBĄ
(nie tylko z innymi!).
„Słowa nasze, nawet co
ważniejsze słowo ściera się w użyciu jak ubiór co sparciał...” W.Majakowski tłum S.Pollak (Majakowski pisał
bzdury, ale jak pięknie, jak ekspresyjnie…)
Umysł na bieżąco („on-line”) stale komentuje percepcję
rzeczywistości, czyli to co postrzegają zmysły. Komentuje to w postaci SŁOWNEJ!
Jest to stały wewnętrzny dialog (czasem przechodzący w dialog „głośny” z innymi lub z samym sobą).
Ten dialog-monolog tworzy syntezę i semantyczną systematykę która jest zapamiętywana.
„Myślenie słowami” („słowa nas mówią” - lingwiści) jest wg mnie jeśli nie najważniejszym to jednym z
najważniejszych mechanizmów samo-świadomości ponieważ jest to DIALOG-Z-SOBĄ czyli procedura
zakładająca przynajmniej częściowe funkcjonowanie „pseudo-dwóch-JA”.
Procedura ta nadmiernie izolując „dwa-JA” może prowadzić do „rozdwojenia jaźni”, do „niezgody z sobą” itp.
często autodestrukcyjnych (choć niekoniecznie) syndromów.
KTO DO MNIE MÓWI ? Czy zawsze wiemy? Czy zawsze prawidłowo identyfikujemy?
•Dzięki (wyjaśniającym-komentującym) słowom odkrywamy więcej niż jest „bezpośrednio
dane” w doznaniu zmysłowym.
•Przykład muzyczny.... („fuga życia”)
Das Wohltemperierte Klavier („Komentarz” do fugi No.4 cis-moll J.S.Bacha)
(...)
Fuga czwarta cis-moll, z pierwszego tomu „Das Wohltemperierte Klavier” ...
(...)
Temat fugi i następne jego przeprowadzenia są niczym błądzenie po omacku,
Pełne smutku i lęku chodzenie „ciemną doliną”...
Zdaje się, że usłyszeć w nich też można coś, co przypomina prośbę, wołanie o pomoc?
Zarazem jakby rezygnację, zwątpienie...
I tak pośród początkowego zamętu, niepewności, zaciemnienia, zabłądzenia,
Nagle zjawia się drugi temat...(niektórzy twierdza, że jest to trzeci )
Osiem dźwięków, z których pierwsze dwa stanowią skok o kwartę czystą w górę,
Dwa następne są dokładnym powtórzeniem drugiego...
Tylko osiem dźwięków... a jakiż niesamowity efekt!
Jakby nieoczekiwana jasność pojawiła się pośród głębokiej nocy.
Jakby nagłe ukazanie się księżyca w pełni, wcześniej spowitego przez gęste chmury...
Te osiem dźwięków...
Rozbłyska jak snop światła,
Wskazówka,
Morska latarnia,
Muzyczne: „NIE LĘKAJ SIĘ, OTO JESTEM, JESTEM Z TOBĄ...”!
Nie jest odrzucone to ośmiodźwiękowe „SŁOWO”.
(NIE-LĘKAJ-SIĘ-OTO-JESTEM-JESTEM-Z-TOBĄ)
Nowy motyw, przenika i oświetla całą resztę fugi.
Oświetla, ale przecież nie oślepia i nie likwiduje ciemności!
Mrok i niepokój początkowego tematu pozostaje obecny do samego końca...
SŁOWO jest jednak przyjęte,
Na trwale zaakceptowane, ustawicznie rozważane, chronione.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
104/113
Jego rytm zaczyna bić jak serce,
Staje się sercem,
Pulsującym żarem, pewnością,
Radością ponownie przeżywanej wiosny...
(...)
(Fuga) nie jest trudna aby ją przynajmniej najskromniej „odczytać”?
Odczytać? Co?
Nuty?
Sens?
Jak bardzo szczęśliwy musi być ktoś, kto w swej fudze życia czuje, że słyszy taki właśnie
temat...
Ośmiodźwiękowe SŁOWO-LOGOS:
„NIE-LĘKAJ-SIĘ-OTO-JESTEM-JESTEM-Z-TOBĄ!”
Światło, aby móc przestać się kręcić w miejscu,
Pośród „ciemnej doliny”...
Wreszcie pójść do przodu,
Grać takt po takcie tą swoją „fugę”,
Choćby nie wiem jak była trudna i zagmatwana...
……………………………………………………………………………………………………
Słowa „dopełniają” , „pogłębiają” input czuciowy
Input czuciowy prowokuje ekspresję słowną
Przyczyna miesza się ze skutkiem
To co pierwotne z tym co wtórne
To co zasadnicze z tym co jest „epifenomenem”
Dźwięki muzyki inicjują szczególny stan emocjonalny, który jest interpretowany przez
„kognitywny operator przyczynowości” jako stan „ducha”…???
Może jednak to, co uważam za skutek jest w rzeczywistości przyczyną?
Czy "duchowość" to tylko szczególny rodzaj stanu emocjonalnego?, Jeśli tak, to może
Kartezjusz mylił się!?
To nie szyszynka ale raczej ciało migdałowate ("hub" emocjonalności) byłoby siedzibą
duchowości ("duszy"?). Jest ono jednak podwójne i stąd byc może potrafimy zachowywać się
jako dwie różne osoby ...???
Mój "kognitywny operator przyczynowości" (postulowany przez d'Aquili i Newberga)
nakazuje mi jednak w takim razie dalej poszukiwać przyczyny ... a mianowicie skąd
właściwie wziął sie "operator przyczynowości" ...
Powstaje rodzaj "pętli myslowej"...
Może jednak to co uważam za skutek ("duchowość") jest w rzeczywistości przyczyną ?
Lepiej na tym zakończyć…
sci-fi „Gedanken experiment”
A co się stanie, jeśli pewnego dnia komputer zacznie zachowywać się w taki sposób, że zaczniemy
podejrzewać go o „świadomość” ????
Co wtedy ?
No cóż, trzeba będzie się zachowywać zgodnie z procedurami przynależnymi ludziom (albo przynajmniej
zwierzętom)...
No i wtedy po prostu informatycy-elektronicy będą musieli kończyć medycynę (lub weterynarię !!!)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
105/113
Dodatek nr. 1
Bariera krew-mózg i jej zaburzenia
Komórki endotelium wytwarzające BBB wykazują bardzo wysoką opór elektryczny do
8000 Ω cm
−2
, co upodabnia je do komórek nabłonkowych.
Komórka endotelium w mózgu odpowiedzialna za wytworzenie bariery krew-mózg jest „spolaryzowana” –
posiada bowiem inny zestaw białek transporterowychw błonie od strony krwi i inny w błonie od strony
mózgu.(rycina powyżej).Aminokwasy (AA) i glukza (G) przechodzą biernie z krwi do płynu pozakomórkowego z
pomocą systemów przenośnikowych obecnych w obu częściach błony komórkowej endotelium czyli od strony
krwi i od strony mózgu (L, y
+
, G). Zależny od sodu transport aminokwasów jest obecny w błonie „abluminalnej”
(od strony mózgu). Są to transportery oznaczone jako A i B
o,+
. Wydaje się więc służyć ograniczeniu napływu
pewnych aminokwasów. Transportery dla glutaminianu (Glu) usuwaja Glu z mózgu Razem z systemem
transportowym dla glutaminy (Gln, N) systemy te biorą udział w regulacji równowagi azotu. Ponadto
transportery takie jak Na
+
/H
+
antiporter, Na channel, Na
+
/K
+
adenosine triphosphatase w obu częściach błony
komórkowej endotelium (luminal i abluminal) ułątwiają kierunek przepływu płynu od krwi do mózgu.
150ml PMR (u dorosłych)
z tego:
ok. 25 ml w komorach
ok. 125 ml w przestrz. podpaj.
(30 ml okołordzeniowo)
Wytwarzanie PMR 500ml/doba
Na+ aktywnie transportowany do CSF „pociąga” Cl- i wodę
W porównaniu z osoczem:
ciśnienie osmotyczne takie samo
Znacznie mniej białek (kilkaset x)
K, Ca, glukoza 30-50% mniej
Cl, Mg nieco wyższe niż w osoczu,
Wodogłowie
Blok odpływu lub nadprodukcja PMR lub
Zanik mózgu („hydrocephalus ex vacuo”)
Nowotwór lub guz nienowotworowy, zapalenie, włóknienie i zrosty oponowe, zwężenie wodociągu mózgu
U dzieci są dodatkowe liczne przyczyny wodogłowia takie jak malformacje.
•
Bariera krew-mózg/rdzeń
–Blokowane substancje hydrofilne
–Mogą przechodzić substancje lipofilne do ok. 500 Da (eter, alkohol, fenytoina, pentobarbital, kofeina, nikotyna
itp.)
–Istnieją liczne mechanizmy transporterowe
•Bariera krew-PMR
–Przepuszczalna (relatywnie) dla hydrofilnych makromolekuł (np. białka)
–Albuminy (200x mniejsze stężenie w PMR niż w surowicy) są używane jako markery funkcji barierowych
–Lipofilność także ułatwia przechodzenie wielu np. leków, antybiotyków, cytostatyków
•Przestrzeń międzykomórkowa w strefie kontaktującej się z PMR (na głębokość maks. paru milimetrów)
prawdopodobnie jest składem zbliżona do PMR.
•Ponad 80% białek PMR pochodzi w surowicy (albuminy pochodzą wyłącznie z surowicy)
•Pozostałe to:
–Immunoglobuliny - częściowo intratekalnie syntetyzowane przez limfocyty (stanowią 5-12% białek PMR)
–Inne białka syntetyzowane lokalnie (głównie w splotach nacz.) i dostające się do PMR:
•syntetaza D prostaglandyn (10mg/L) (sploty i opony)
•Transtyretyna (17 mg/L + 1 mg/L z surowicy) (prealbumina)
•τ-transferyna (6 mg/L)
•Cystatyna C (6 mg/L)
•–białka parenchymy OUN
Indeks albumin Ralb = AlbPMR/Albsurowica - wskaźnik bariery krew-PMR i wymiany PMR

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
106/113
•Norma: Ralb poniżej 0,007 (noworodek 0,05; 1 mieś 0,015; od 6 mieś do 20 lat max. 0,005; do 40 lat max.
0,0065; do 60 lat max. 0,008)
Indeks albumin w różnych stanach chorobowych:•
Uszkodzenie niewielkiego stopnia (do 0,01)
–SM, HIV-encephalitis, Herpes-zoster ganglionitis, polineuropatia alkoholowa, SLA
•Uszkodzenie umiarkowanego stopnia (do 0,02)
–Meningitis virusalis, polineuropatia cukrzycowa, zawał mózgu, różne stany z rozległym zanikiem kory
•Uszkodzenie dużego i bardzo dużego stopnia (ponad 0,02)
–S. Guillain-Barre, Meningopolineuritis boreliozowe, HSV-encephalitis,
–Meningitis TBC, meningitis purulenta (nawet ponad 0,1)
•Ponadto Ralb wzrasta w przypadkach bloku przepływu PMR w kanale kr. (brak wymiany PMR) np. w guzach
rdzenia, dyskopatiach i innych zmianach nawet do wartości ponad 0,1
Ocena Ralb nie pozwala na rozróżnienie między zaburzeniem bariery krew-PMR oraz blokiem przepływu i
wymiany PMR
•Próbą pokonania tej trudności jest stosowanie „wskaźnika ICAM” (ICAM index)
•ICAM-1 = intercellular adhesion molecule -1
•Ekspresja tego białka w endoteliach jest indukowana przez prozapalne cytokiny (IL-1, TNFα, IFNγ)
•Białko jest wtedy uwalniane do PMR
•ICAM-1 osiąga wartości w PMR: 44 µg/L (meningitis) 4,5 µg/L (SM), 16,2 µg/L (s. Guillain-Barre)
•ICAM index w niezapalnych chorobach wynosi do 0,7, w SM 1,0, w zapaleniach opon 1,5 – 1,7
Wykrywanie lokalnej intratekalnej syntezy immunoglobulin
•Ralb = AlbPMR/Albsurowica (poniżej 0,007)
•RIgG = IgGPMR/IgGsurowica
•Indeks IgG = RIgG / Ralb (poniżej 0,7)
W których miejscach (częściowo) nie funkcjonuje bariera Krew-Mózg
(tzw. CIRCUMVENTRICULAR ORGANS)
Obszary w któych BBB jest słaba nazywane są „narządami okołokomorowymi” "circumventricular
organs". Dzięki nim mózg może monitorować skład krwi. Są to:
Szyszynka
wydziela melatoninę i peptydy neuroaktywne. Rola w cyklu dobowym (circadian)
Tylna (nerwowa) część przysadki. Neurohypophysis (posterior pituitary)
Uwalnia neurohormony
(oxytocna i wzopresyna
Area postrema
AP
„Centrum wymiotów” : umożliwia częsciową obronę przed toksycznymi substancjami
poprzed uruchomienie odruchu wymiotnego
Narząd podsklepieniowy Subfornical organ
Gra rolę w regulacji płynów ustrojowych.
Vascular organ of the lamina terminalis
OVLT
Obszar chemoreceptorowy (detekcja różnych peptydów i
innych molekuł).
Wyniosłość pośrodkowa Median eminence
ME
Reguluje przednią część przysadki poprzez uwalnianie
neurohormonów.
Subcallosal organ SCO -
ma znaczenie w życiu płodowym.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
107/113
Dodatek nr. 2
Sen i czuwanie
Po co sen? Czym jest sen?
•Wartość i znaczenie snu można poznać pośrednio, poprzez następstwa często spotykanych zaburzeń snu (insomnia, sleep
apnea syndrome, narkolepsja).
•Pozbawienie snu prowadzi do śmierci (fatal familial insomnia). Najdłuższy okres bezsenności u młodego ochotnika bez
farmakologicznego „wspomagania” wyniósł 453 godz. ok. 19 dni., po którym to czasie po kilku dniach dłuższych niż
zwykle okresach spania powrócił do normalnego cyklu snu bez zauważalnych oznak utraty zdrowia.
•Wartość snu musi być istotna chociażby dlatego, że teoretycznie zmniejsza szanse na przeżycie zwierzęcia…?
•Sen nie jest prostym obniżeniem aktywności układu nerwowego lecz raczej czymś co (być może) można by porównać do
aktywności w supermarkecie po godzinach otwarcia lub urzędzie w godzinach gdzie nie przyjmuje się interesantów…
•Sen jest raczej serią-sekwencją ściśle kontrolowanych (głównie przez niektóre jądra pnia mózgu) stanów aktywności mózgu,
które najłatwiej prześledzić w elektroencefalografie.
•Okresowość sen-czuwanie z grubsza pokrywa się z cyklem nocy i dnia przy czym „zegar wewnętrzny” w przypadku
odcięcia bodźców z zewnątrz „ustawia się” na cykl 26 godzinny (eksperymenty z ludźmi zamkniętymi bez dostępu do
informacji o dniu lub nocy).
•Cykl dobowy (cirkadialny) dotyczy całego ciała m.in. poprzez dobowe cykliczne zmiany wielu parametrów np. poziomy
hormonów, temperatura ciała
•
Zwierzęta pozbawiane snu tracą wagę pomimo zwiększenia przyjmowania pokarmu oraz tracą regulację
temperatury ciała (wzrasta), stają się podatniejsze na infekcje.
•Szczury po kilku tygodniach deprywacji snu giną.
•Zwierzęta drapieżne sypiają długo (w noc lub dzień w zależności od typu aktywności). Zwierzęta które
stanowią pokarm drapieżników (np.zając) śpią bardzo mało w postaci niekiedy kilkuminutowych „drzemek”.
•Delfiny i foki zasypiają naprzemiennie jedną półkulą mózgu.
Drogi sygnalizacji dobowych zmian światła
•Specyficzne światłoczułe komórki zwojowe siatkówki które w przciwieństwie do czopków i pręcików ulegają
depolaryzacji pod wpływem światła i zawierają inny pigment – tzw.
melanopsin.
•Następnie pobudzenie biegnie drogą
siatkówkowo-podwzgórzową
do jądra
n.suprachiasmatic
(„centrum
rytmów okołodobowych”)
•Następną „stacją” są kolejno: A) jądro N.paraventricularis podwzgórza B) neurony przedzwojowe ukł.
symatycznego w rdzeniu szyjnym, C) zwój szyjny górny, D) szyszynka, która (gdy jest noc) wzmaga syntezę
MELATONINY
Stadia snu w EEG
Badania EEG ujawniły, że sen nie jest prostym „wyłączeniem” szeregu funkcji mózgu ale zaprogramowaną
sekwencją zmian czynności.
W ciągu pierwszej godziny stopniowo amplituda fal EEG zwiększa się a jednocześnie zmniejsza się
częstotliwość.
W fazie II występują okresowe zwiększone wyłądowania (tzw. wrzeciona senne trwające po kilka sekund)
Najgłębszy sen występuje w tzw. fazie IV charakteryzującej się falami „delta” (wolne, 0,5 – 2 Hz o wysokiej
amplitudzie – świadczące o synchronizacji aktywności elektrycznej neuronów kory).
Po fazie IV następuje faza snu REM (rapid eye movement) trwająca ok.. 10 min. W której EEG przypomina
normalną aktywność dzienną (tzw. rytm beta 15-60 Hz amplituda ok.. 30 mikroVolt)
Cykl zmian i faz snu (odpowiadających „głębokości” snu) powtarza się ale zwykle w ciągu 1 nocy faza IV
(najgłębszy sen) występuje tylko 2 razy.
Kolejne fazy REM są coraz dłuższe.
Fazom snu towarzyszą liczne zmiany funkcjonowania całego ciała
W całym okresie snu non-REM (fazy I-IV) obniża się napięcie mięśni, częstotliwość tętna, oddechu, spada
ciśnienie tętnicze, temperatura i metabolizm (najniższe wartości w fazie IV). Występują powolne ruchy gałek
ocznych.
W fazie snu REM w/w parametry wracają niemal do stanu czuwania a ponadto występują: gwałtowne ruchy
gałek ocznych, skurcze drobnych mięśni palców, erekcja oraz marzenia senne.
Uwaga! Przypadki somnabulizmu oraz mówienia przez sen nie występują w fazie REM (a zatem nie w fazie
marzeń…!). Marzenia senne występują także w fazach non-REM.
W 60% treść snów jest smutna lub przygnębiająca a tylko w 10% dotyczy seksu!
Sen –REM : „aktywny umysł w nieaktywnym ciele…”
Wybitnie obniżona aktywność ruchowa mięśni (oprócz wspomnianych mięśni oczu i palców) spowodowana jest
silną aktywnością neuronów GABA-ergicznych tworu siatkowatego mostu, które hamują dolne motoneurony
rdzenia.
Ponadto te same neurony hamują neurony czuciowe rogów tylnych rdzenia.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
108/113
Ruchy sakkadowe w fazie REM są rezultatem wysokiej aktywności ośrodków ruchu gałek ocznych –
paramedialpontine reticular formation (PPRF) oraz rostral interstitial nucleus.
Doświadczenia Horace Magoun i Giuseppe Moruzzi z 1949 wskazały na obecność aktywującego układu
siateczkowego (cholinergiczne neurony na pograniczu mostu i śródmózgowia) a doświadczenia Waltera Hessa
na (przeciwną) rolę wzgórza.
Okolice aktywne i nieaktywne w czasie snu REM.
Zwiększona aktywność c.migdałowatego tłumaczy silnie emocjonalny charakter snów w fazie REM.
Obniżona aktywność kory przedczołowej może tłumaczyć „społeczną niewłaściwość” treści marzeń w fazie
REM (kora ta bierze udział w ocenie i wyborze „prawidłowego” zachowania w warunkach czuwania).
Wszystkie 4 układy (noradrenergiczne, serotoninergiczne, histaminergiczne, i cholinergiczne) posługujące
się 4 różnymi neurotransmiterami łącznie odpowiedzialne są za stan czuwania.
•Oreksyny wydzielane przez neurony w okolicy tuberomammillary nucleus aktywują ośrodki „czuwania”
•Hamująco natomiast oddziałują GABA-ergiczne jądra ventrolateral preoptic (VLPO) podwzgórza (ich
uszkodzenie powoduje bezsenność)
•Neurony wzgórza (tzw. wzgórzowo-korowe) odgrywają kluczową rolę w synchronizacji i desynchronizacji
aktywności neuronów kory (i stąd w obrazie EEG)
•Ich aktywność w okresie czuwania ma charakter toniczny, natomiast w fazie snu oscylacyjny.
•Aktywność oscylacyjna neuronów „wzgórzowo-korowych” prowadzi też do powstawania „wrzecion
aktywności” EEG w II fazie snu. (następny slajd)
•Neurony wzgórzowo-korowe oraz neurony jąder siatkowatych wzgórza pozostają pod wpływem aktywujących i
hamujących układów (pnia mózgu)
•Oscylacyjna aktywność neuronów wzgórzowo-korowych powoduje „rozłączenie” kory mózgu od oddziaływań
zewnętrznych.
•Zaburzenia snu należą do najczęstszych dolegliwości!!!
•
Narkolepsja: nagłe „ataki snu REM” (bez faz pośrednich)
•Sleep apnea syndrom: napady bezdechu, sen jest płytki, prawie bez faz REM i IV; efekty: niedotlenienie
mózgu, brak pełnego wypoczynku po śnie.
•Bezsenność
•Kofeina działa „pobudzająco” („antysennie”) ponieważ jest antagonistą receptora adenozynowego
•Receptor adenozynowy najprawdopodobniej indukuje sen, ale nie wiadomo jak ?

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
109/113
DODATEK Nr 3:
Schorzenia, w których stwierdzono lub podejrzewa się istotną rolę mechanizmów
ekscytotoksycznych
Udar (niedokrwienie) mózgu
Otępienie naczyniopochodne (multi-infarct)
Uraz mózgu i rdzenia
Padaczka
Choroby zwyrodnieniowe
ch. Alzheimera, ch. Parkinsona, stwardnienie zanikowe boczne (ALS), pląsawica Huntingtona
Rasmussen encephalitis
Stwardnienie rozsiane
Schizofrenia
GLU w niedokrwieniu (niedotlenieniu)
Czynnikiem uwalniającym GLU może być:
utrata zasobów energetycznych przez komórki, spadek produkcji ATP w mitochondriach ?
zaburzenie działania pomp jonowych
Wzrost GLU pozakomórkowego Aktywacja receptorów
Napływ jonów Ca2+ do komórek Aktywacja syntazy NO i innych enzymów (fosfolipaza A2)
Wzrost wolnych rodników
GLU 100-500 mikromoli/L prowadzi do martwicy w hodowli neuronów
GLU 20 mikromoli/L prowadzi do ich apoptozy
Eksperymentalny bloker NMDA MK-801 (dizocilpine) zmniejsza uszkodzenie poischemiczne,
pohipoglikemiczne i pourazowe
Podobne działanie (i mechanizm) ma nimodypina
Niestety, blokery receptorów GLU mają działania uboczne (represja normalnej neurotransmisji)
GLU w padaczce
Już w latach 50-tych stwierdzono, że drgawki można wywołać podając GLU na korę mózgową psa.
Wykazano hamowanie aktywności drgawkowej zarówno przez antagonistów receptora NMDA oraz AMPA.
Korzystne jest zastosowanie kombinacji zwłaszcza substancji blokujących receptor AMPA z lekami
przeciwpadaczkowymi (fenobarbital, fenytoina, karbamazepina, walproinian).
Są nadzieje na zastosowanie inhibitorów receptoraów metabotropowych
Rasmussen encephalitis
Stwierdzono obecność przeciwciał z krwi obwodowej przeciwko podjednostce GluR3 receptora AMPA co
ma prowadzić do uszkodzenie ekscytotoksycznego neuronów kory mózgu (Rogers SW i wsp. Science 265,
648-651, 1994; He XP i wsp. Neuron, 20, 153-163, 1998)
Stwierdzono również obecność p-ciał przeciw białku synaptycznemu Munc-18 co prowadzi do
zaburzonego przewodnictwa (Yang R i wsp. Neuron, 28, 375-383, 2000)
Stwardnienie rozsiane
Niszczenie oligodendrocytów (które jak wiadomo posiadają receptory glutaminergiczne, zwł. AMPA i
kainianowe) może być spowodowane przez Glu uwalniany w dużych ilościach przez aktywowane komórki
układu odpornościowego. Podawanie NBQX (blokera AMPA) myszom z experimental autoimmune encephalitis
zmniejszało objawy oraz uszkodzenie oligodendrocytów (Pitt i wsp. Nat Med 2000 Jan;6(1):67-70 ).
Ekscytotoksyczność może zatem brać udział w autoimmunologicznej demielinizacji.
Rola kwasu glutaminowego
w nowotworach OUN
Glu stymuluje proliferacj
ę
komórek nowotworowych a efekt ten blokowany jest przez
antagonistów Glu (MK-801, memantyna)
Glejaki wytwarzaj
ą
ce i wydzielaj
ą
ce Glu rosn
ą
nawet 15x szybciej ni
ż
nie wdzielaj
ą
ce Glu
(Takano i wsp. Nature Med.. 2001, Rzeski i wsp. Proc NA Sci USA 2001)
GLU w schizofrenii
Postuluje się hypofunkcję receptorów NMDA w korze limbicznej.
MK-801 (bloker NMDA) wywoływał degenerację w tym obszarze kory.
(Teoria hypofunkcji układu glutamatergicznego)

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
110/113
„Dysinhibicja” transmisji NMDA ma prowadzić do nadmiernej stymulacji receptorów AMPA i
ekscytotoksyczności.
Fencyklidyna, ketamina – antagoniści NMDA mogą indukować psychozę podobną do schi.
Postępującej hypofunkcji receptorów NMDA przypisuje się też rolę w osłabieniu pamięci i innych procesach
starzenia mózgu.
Zauważono zwiększenie ekspresji EAAT1-2 we wzgórzach chorych na schi.
Neurotoksyny
•Toksyny działające na kanały jonowe
–Tetrodotoksyna (TTX) puffer fish – blokowanie kanału Na+
•Toksyny działające na uwalnianie
neurotransmitera
–Botulinum toxin (Bacteria) Blokuje uwalnianie acetylcholine
–Crotoxin
S. (American
Rattlesnake) redukuje uwalnianie acetylcholine
–Calciseptine (Black Mamba) Blokuje voltage-gated calcium channels
•Toksyny działające na
receptory postsynaptyczne
–α-bungarotoxin (Krait snake) Blokuje acetylcholine (nicotinic) receptor
•Toksyny działające na kanały
jonowe (pochodzą od zwierząt lub roślin):
•
Ważniejsze toksyny kanałów jonowych dla Na+ (najbardziej poznane i najczęściej stosowane
w badaniach eksperymentalnych)
–Tetrodotoksyna (TTX) puffer fish – blokowanie kanału Na+–Saxitoxin – kanał Na (Dinoflagellatae)
działanie j.w.
–α-toxin (skorpion) – spowalnia inaktywację kan. Na; wydłużenie Pcz,
–β-toxin (skorpion) – zmienia zależność napięciową
–Batrachotoxin (żaby z Pd Am) – blokuje inaktywację kan. Na
–Akonityna (Buttercups - jaskier) – działnie podobne j.w.
–Weratrydyna (lilie) - działanie podobne j.w.•
Ważniejsze toksyny działające na kanał K+ (wszystkie
znane działają blokująco)
–Dendrotoxin (osy)
–Apimin (pszczoły)
–Charybdotoxin (skorpiony)
Już w w r.1890 Brytyjczyk C.Sherrington wykazał wzrost przepływu krwi w ciemieniowych okolicach
mózgu w wyniku stymulacji czuciowej i sformułował twierdzenie, że przepływ krwi w mózgu jest
regulowany zgodnie z potrzebami metabolicznymi oraz , że rolę w tym grają jony wodorowe. Ale późnej
okazało się, że lokalne wzrosty przepływu wyprzedzają zmiany
pH
! (zob. slajdy dalej nt. regulacji
przepływu mózgowego)
Regulacja krążenia mózgowego
Prawidłowe krążenie jest krytyczne dla funkcjonowania i przeżycia mózgu!
Znaczenie stałego monitorowanie przepływu mózgowego u chorych z urazem mózgu•
Ciśnienie wewnątrzczaszkowe, intracranial pressure- ICP
Cisnienie perfuzji mózgowej - Cerebral perfusion pressure, CPPPrawidłowe ICP : 0-10mmHg
(Interwencja gdy jest powyżej 20mmHg?)
średnie ciśnienie tętnicze – mean arteriar pressure - MAP
CPP jest równe różnicy ciśnienia tętniczego i wewnątrzczaszkowego:
CPP = MAP – ICP
IPP powinno byc utrzymywane w granicach: 70-80 mmHg
Specyfika krążenia mózgowego
1)„Sprzęgnięcie” miedzy przepływem krwi i metabolizmem
2)Utrzymywanie stałego przepływu pomimo zmiennego ciśnienia
3)Wpływ gazów (O2, CO2) na przepływ
4)Czynniki neurowaskularne (unerwienie naczyń)
Już w w r.1890 Brytyjczyk C.Sherrington wykazał wzrost przepływu krwi w ciemieniowych okolicach
mózgu w wyniku stymulacji czuciowej i sformułował twierdzenie, że przepływ krwi w mózgu jest
regulowany zgodnie z potrzebami metabolicznymi oraz , że rolę w tym grają jony wodorowe. Ale okazało
się, że lokalne wzrosty przepływu wyprzedzają zmiany
pH
!
Regulacja krążenia mózgowego – czynniki metaboliczne

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
111/113
Wiadomo,
ż
e lokalny przepływ krwi w mózgu jest regulowany potrzebami metabolicznymi
(zwi
ą
zanymi z aktywno
ś
ci
ą
danej okolicy) ale jednoznaczne okre
ś
lenie, które czynniki s
ą
mediatorami tego zwi
ą
zku (aktywno
ść
– przepływ) nie jest łatwe.
Rozważano wpływ
K+
: (im większa intensywność synaptyczna, tym więcej potasu jest w przestrzeni
pozakomórkowej !) Ale astrocytarnego buforowania jonów potasu przez Ba2+ nie hamuje reaktywności
naczyń a ponadto sam K+ nie działa wystarczająco szybko w poszerzaniu naczyń
Oraz
adenozyny
(silny wazodilator, jej wzrost następuje w efekcie spadku ATP, ale jej stężenie musiałoby wzrastać
co najmniej 10x a potem nagle spadać, czego nie stwierdzono)
a także
mleczanu
Regulacja przepływu mózgowego krwi
Rozważane są dwa ogólne mechanizmy:
1)Sygnalizacja poprzez molekuły będące („ubocznym”?) rezultatem aktywności neuronalnej (czynniki
„metaboliczne”)
-wzrost K+ ?, wzrost adenozyny ?, mleczanu?, spadek pH?
-Generalnie zmiany tych metabolitów pojawiają się z opóźnieniem rzędu sekund (nieco za długo w
stosunku do reakcji przepływu)
2)Mechanizmy neurogenne wiążące się z uwalnianiem specyficznych neurotransmiterów które mogą
nawet działać „z wyprzedzeniem” (lub „równolegle” do pojawienia się aktywności w danym obszarze
mózgu)
Możliwe m.in. dlatego, że naczynia mózgowe są bogato unerwione
Specyfika krążenia mózgowego c.d.
Wiadomo,
ż
e lokalny przepływ krwi w mózgu jest regulowany potrzebami metabolicznymi
(zwi
ą
zanymi z aktywno
ś
ci
ą
danej okolicy) ale jednoznaczne okre
ś
lenie, które czynniki s
ą
mediatorami tego zwi
ą
zku (aktywno
ść
– przepływ) nie jest łatwe.
Wielkie nadzieje wi
ą
zano z
NO
NO to najbardziej „pasuj
ą
cy” kandydat na „neurowazomediatora” – zwłaszcza,
ż
e GLU
poprzez mechanizm receptorowy (NMDA) stymuluje jego syntez
ę
. Bardzo cenn
ą
cech
ą
NO jest
jego krótkotrwało
ść
.
Jednak NO nie jest jedynym takim mediatorem poniewa
ż
w niektórych eksperymentach,
w których hamowano aktywno
ść
NOS dalej obserwowano efekty sprz
ęż
enia aktywno
ś
ci i
przepływu !
Wiele danych przemawia za rolą gazów (tlenu i dwutlenku węgla) w regulacji przepływu.
Hyperkapnia
(wzrost CO2)
jest silnym wazolilatatorem. Prawdopodobnie efekty hyperkapni zależą od NO (np.
zaobserwowano redukcje przekrwienia wywołanego hyperkapnią po zahamowaniu NOS i było to
niezależne od od zmian metabolicznych) Ale dla skrajnej hyperkapni efekt wzmożenia przepływu jest
niezależny od NO.
NOS-knock-outowe myszy mają normalne reakcje na hyperkapnię! Summa summarum ... Nie wiadomo
jak to jest ...!
Wiadomo, że
hypoksja
jest czynnikiem poszerzającym naczynia ale i tu mechanizm jest nieznany!
Auto-regulacja krążenia mózgowego
Naczynia mózgowe wykazuj
ą
autoregulacj
ę
(szeroko
ś
ci w stosunku do ci
ś
nienia perfuzji) w
której uczestnicz
ą
korowe naczynia oporowe). Jest to prawdopodobnie wewn
ę
trzny
mechanizm myogenny chocia
ż
cz
ęś
ciowo mo
ż
e by
ć
zale
ż
ny od czynnika(ów) uwalnianych
przez endotelia. Nie znamy dokładnego mechanizmu autoregulacji „pary” przepływ-ci
ś
nienie.
Ustalono jednak,
ż
e stymulacja układu sympatycznego powoduje „przesuni
ę
cie” wzrostu
przepływu w kierunku wy
ż
szych ci
ś
nie
ń
(koniecznie jeszcze wy
ż
sze ci
ś
nienie aby spowodowa
ć
wzrost przepływu – jest to mechanizm ochronny dla mózgu)
Czynniki neurogenne
Naczynia mózgu są bogato unerwione zarówno przez „zewnętrzne” nerwy (autonomicznego układu) jak
też przez wypustki neuronów mózgu, np. pnia
Wykryto receptory dla licznych neurotransmiterów (NA, 5-HT, ACh, VIP, NPY, substancja P,
calcitonine-gene related peptide) w ścianie naczyń krwionośnych. (m.in. gęste unerwienie naczyń splotu

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
112/113
naczyniastego przez nerwy adrenergiczne) Mogą one tłumaczyć „neurogenny” mechanizm regulacji
przepływu.
Właśnie NPY i NA najprawdopodobniej grają rolę w „przesunięciu” wykresu wzrostu przepływu pod
wpływem stymulacji ukł sympatycznego. Neurogenna kontrola naczyń mózgowych jest zarówno
„wewnętrzna” (nerwy bezpośrenio z mózgu) jak i „zewnętrzna” (nerwy wnikające spoza czaszki).
Pierwszy (Intrinsic) system jest słabo poznany. „Zewnętrzne” unerwienie naczyń mózgowych obejmuje
zarówno nerwy sympatyczne (NA, NPY) jak i parasympatyczne (Ach, VIP, peptide histidine-methonine =
PHM, wszystkie mogą kolokalizować w tych samych neuronach)
Potencjalni kandydaci do funkcji sygnalizatorów aktywno
ś
ci neuronalnej przekazuj
ą
cych
sygnał do naczy
ń
to: noradrenalina, Ach, serotonina, oraz peptydy takie jak: VIP, Neuropeptyd
Y, calcitonine gene-related peptide (CGR) i substancja P.
Innym potencjalnym czynnikiem neurogennym mo
ż
e by
ć
„o
ś
” GLU-NO (zob slajdy dalej)
Układ parasympatyczny
(zasadniczo wazodilatacyjny) jest bardzo efektywny i może „przebić” wpływy metaboliczne !
Głównym jądrem jest jądro ślinowe górne w moście, którego wypustki wychodzą nerwem VII a następnie
łączą się ze zwojami skrzydłowopodniebiennymi (pterygopalatine) i usznymi.
Stymulacja parasympatyczna powoduje wzrost przepływu.
Wpływ parasympatyczny nie zmienia reakcji na hyperkapnię ani hypoksję i nie wpływa na autoregulację
naczyniową.
Układ sympatyczny:
Unerwienie bierze początek w
jądrach podwzgórza
następnie poprzez pośrednio-boczne słupy rdzenia
skąd neurony wysyłają wypustki do
zwoju szyjnego górnego
a stąd do ściany naczyń tętniczych, w tym
naczyń oponowych.
Na ogół jednak nerwy sympatyczne towarzyszą naczyniom wnikającym do mózgu na niewielką odległość.
Część naczyń wewnątrzmózgowych jest unerwiana przez locus coeruleus.
Pobudzenie sympatyczne wywołuje obkurczenie naczyń i spadek przepływu. Efekt jest sygnalizowany
zarówno poprzez NA (receptor alfa-1) jak też przez NPY często kolokalizujący z NA.
Skurcz dotyczy głównie większych naczyń a
szczególnie żył!
Jego efektem jest omówiony wcześniej wpływ na autoregulację (przesunięcie zmian pod wpływem wzrostu
ciśnienia)
Bardzo atrakcyjny neurogenny model regulacji przepływu krwi wymaga jednak dalszych bada
ń
.

Adamek.D. Neurobiologia - konspekty wykładów
Neurobiologia
113/113
DODATEK NR 6.
Obrazowanie funkcjonalne
METODY WIZUALIZACJI I PRZYśYCIOWEJ ILOŚCIOWEJ OCENY
LOKALNEGO PRZEPŁYWU KRWI W MÓZGU ORAZ LOKALNEGO POZIOMU
METABOLIZMU
1.MRJ: techniki „diffusion weighted imaging” i pochodne (mapy dyfuzji etc)
2.MRJ: „perfusion weighted imaging”
3.fMRJ TECHNIKA „BOLD” (blood oxygenation level dependent)
4.Zlokalizowana spektroskopia MRJ
5.SPECT (Single photon emission computed tomography)
6.PET (positron emission tomography)
fMRJ TECHNIKA „BOLD” (blood oxygenation level dependent)
OksyHb jest diamagnetykiem, deoksyHb jest paramagnetykiem. W 1982 r Thulborn wykazał szybszy zanik
sygnału FID (free induction decay) w obecno
ś
ci deoksyHb.
Na szybko
ść
rozfazowywania si
ę
spinów wpływa bardzo wiele lokalnych czynników – nazywanych
lokalnymi niehomogenno
ś
ciami pola i Hb jest tylko jednym z nich, krew stanowi ok. 6% obj
ę
to
ś
ci kory
mózgu dlatego m.in. zmiany magnetycznych jej własno
ś
ci s
ą
bardzo nikłe.
Aktywacja kory powoduje wzrost przepływu, który przewy
ż
sza pocz
ą
tkowo (3-6 sek.) siln
ą
deoksygenacj
ę
hemoglobiny.
Nast
ę
powy wzrost przepływu powoduje,
ż
e krew
ż
ylna jest mniej odtlenowana (czyli bogatsza w
oksyhemoglobin
ę
) i st
ą
d sygnał jest silniejszy!
Na wpływ Hb czuły jest przede wszystkim czas T2 Wykazał
to po raz pierwszy Seiji Ogawa w 1990 i nadał nazw
ę
technice – BOLD.
Zlokalizowana spektroskopia MRJ
Wzorcem dla stopnia przesuni
ę
cia chemicznego jest zwi
ą
zek
((CH
3
)
4
Si), w którym „osłonowy” efekt elektronów
na j
ą
dra
1
H i
13
C jest silniejszy od jakichkolwiek zwi
ą
zków biologicznych, jego pozycja oznacza 0 na tej
skali
Chemical Shift (
d
) = shift observed [(
w
-
w
ref
)/ (
w
ref
)] x 10
6
(Dlatego wyra
ż
one w ppm)
Innym j
ą
drem wykorzystywanym w spektroskopii NMR jest
31
P
Teoretycznie mo
ż
na wykorzysta
ć
tak
ż
e
13
C,
15
N,
17
O.
SPECT (Single photon emission computed tomography).
U
ż
ywany jest m.in. radioizotop technetu Tc-99m który emituje pojedynczy foton pr. gamma o
energii 140KeV i ma half-life ok. 6 godz. Foton rejestrowany jest przez tzw Gamma kamer
ę
z
kolimatorem ołowiowym, który umo
ż
liwia separacj
ę
promieniowania z ró
ż
nych punktów ciała
(tzw kamera Anger’a) dzi
ę
ki tysi
ą
com w
ą
skich równoległych kanalików w płycie ołowiowej.
Fotony absorbuj
ą
kryształy NaI emituj
ą
c fotony
ś
wiatła.
PET (positron emission tomography):
podawane s
ą
ró
ż
ne zwi
ą
zki z „podstawionym” krótkotrwałym izotopem z rozpadem
b
, przy
którym nast
ę
puje emisja pozytonu. Pozyton ulega anihilacji napotykaj
ą
c elektron a wyzwolone
fotony energii rozchodz
ą
si
ę
w przeciwnych kierunkach pod k
ą
tem 180
o
. Gammadetektory
lokalizuj
ą
miejsce anihilacji z dokładno
ś
ci
ą
do kilku milimetrów. Typowo u
ż
ywanymi izotopami
s
ą
:
15
O,
11
C,
18
F. Przykładowo:
Przepływ oceniany jest za pomoc
ą
wody znakowanej
15
O (H
2
15
O)
Utylizacja glukozy
przy pomocy
18
F-2-deoksyglukozy (
18
F-labeled 2-DG)
Zu
ż
ycie tlenu przez podawanie do oddychania
15
O
PET pozwala
przyżyciowo
na wyznaczenie tzw.
local cerebral metabolic rates
glukozy (LCMRglu) poprzez
użycie (18F-labeled 2-DG) u ludzi i zwierząt.
•Je
ś
li zbadamy t
ą
sam
ą
aktywacj
ę
poprzez podanie H215O i okre
ś
lenie przepływu mózgowego oka
ż
e
si
ę
,
ż
e
wzmo
ż
enie przepływu pokryje si
ę
z akumulacj
ą
18F-2-D
LCMRglu wyznaczony przy pomocy PET u ludzi daje rezultaty ok. o ½ niższe niż u gryzoni.
PET pozwala na obserwację „w czasie rzeczywistym” zużycia glukozy w określonych lokalizacjach w
zależności od rozmaitych bodźców (wzrokowych, czuciowych) i aktywacji różnych funkcji mózgu (w tym
uczuciowych i intelektualnych)
Wyszukiwarka
Podobne podstrony:
Zaburzenia lękowe Neurobiologia lęku Terapia lęku
sylabus neurobiologia 11 12 v 1
Zarys neurobiologii cw-03 SZABLON, psychologia I rok, BPZ
Psycholingwistyka - Rozdział 1 (Part 1), Psychologia UŚ, Semestr II, Zarys neurobiologii
Neurobiol Ontogeneza UN
neuroblastoma, Medycyna, PATOMORFOLOGIA, Noworodek
Zarys neurobiologii przykładowe pytania
Neurobit Lite
pytania z neuro, III, IV, V ROK, SEMESTR II, PODSTAWY NEUROBIOLOGII ZACHOWANAI I ETOLOGII, pytania
Zarys neurobiologii zagadnienia
elektyw neurobiologia reakcji stresowej
NEUROBIOLOGIA DEPRESJI
Ezzel Neurobiologia samobójstwa [SN]
rozwuj neurobilogi
EEG Neurobiofeedback
Neurobiologia- prof. Andrzej Głąbiński-obieralny, Biotechnologia Medyczna, Pytania na licencjat LOL
NEUROBLASTOMA
więcej podobnych podstron