Postępy Biochemii 59 (3) 2013
257
Jerzy Kotlinowski
Józef Dulak
Alicja Józkowicz
Zakład Biotechnologii Medycznej, Wydział
Biochemii, Biofizyki i Biotechnologii, Uniwer-
sytet Jagielloński, Kraków
Zakład Biotechnologii Medycznej, Wydział
Biochemii,
Biofizyki
i
Biotechnologii,
Uniwersytet Jagielloński, ul. Gronostajowa 7,
30-387 Kraków; tel.: (12) 66 46 411, e-mail alicja.
jozkowicz@uj.edu.pl
Artykuł otrzymano 2 lutego 2013 r.
Artykuł zaakceptowano 9 kwietnia 2013 r.
Słowa kluczowe: cukrzyca, komórki progeni-
torowe śródbłonka, powikłania cukrzycowe
Wykaz skrótów: T2DM (ang. type 2 diabetes
mellitus) — cukrzyca typu 2; EPC (ang. endothe-
lial progenitor cells) — komórki progenitorowe
śródbłonka
Cukrzyca typu 2 upośledza funkcjonowanie
komórek progenitorowych śródbłonka
StreSzczenie
c
ukrzyca typu 2 (t2DM, ang.
type 2 diabetes mellitus) jest chorobą związaną z defektem
działania insuliny prowadzącym do szeregu zmian w metabolizmie komórek. Połącze-
nie insulinooporności i hiperglikemii, obecne u pacjentów z T2DM, jest przyczyną zabu-
rzeń w funkcjonowaniu zarówno dojrzałych, jak i progenitorowych komórek śródbłonka
(EPC, ang.
endothelial progenitor cells). EPC charakteryzowane są jako jednojądrzaste ko-
mórki eksprymujące równocześnie markery śródbłonkowe oraz progenitorowe. Do opisu
EPC wykorzystuje się też ich właściwości angiogenne: tworzenie kapilar
in vitro, tworzenie
wtórnych kolonii komórek śródbłonka, produkcję czynników proangiogennych czy udział
w naprawie naczyń
in vivo. Komórki progenitorowe pochodzące ze szpiku kostnego biorą
udział w regeneracji naczyń krwionośnych. Wykazano jednak, że ich funkcjonowanie jest
upośledzone u osób chorych na cukrzycę. W artykule scharakteryzowano wpływ T2DM na
funkcjonowanie EPC, uwzględniając ponadto powikłania cukrzycowe i wpływ stosowanych
powszechnie leków regulujących poziom glukozy. Zmniejszona liczba i pogorszone funk-
cjonowanie EPC u osób chorych na cukrzycę może być jednym z czynników prowadzących
do zwiększenia ryzyka chorób sercowo-naczyniowych oraz powstawania mikro- i makroan-
giopatii.
WProWaDZEniE
Cukrzyca (DM, łac. Diabetes mellitus) jest schorzeniem towarzyszącym ludz-
kości od pokoleń. Pierwsze informacje o tej chorobie pochodzą z XVI wieku
p.n.e. i dotyczą klasycznych objawów, czyli nadmiernego wydzielania moczu
i jego słodkiego smaku. W starożytnej Grecji Areteusz z Kapadocji — jak się
współcześnie uważa, po raz pierwszy — wprowadził termin diabetes, opisujący
poliurię. Drugi człon łacińskiej nazwy — mellitus, oznaczający po łacinie miód —
został wprowadzony dopiero w 1675 roku przez angielskiego lekarza Thomasa
Willisa [1].
Przez wiele lat nie wiedziano ani jak leczyć cukrzycę, ani jaka jest jej pato-
geneza. Stosowane terapie nie przynosiły zamierzonych rezultatów, choć poja-
wiały się również właściwe przesłanki wiążące otyłość z rozwojem cukrzycy i
zalecające pacjentom aktywność fizyczną [1]. Duży wkład w odkrycie patologii
cukrzycy mieli Joseph von Mering i Oskar Minkowski. W 1889 roku wywołali
oni cukrzycę u psów, którym usunęli operacyjnie trzustkę [2]. O krok dalej po-
sunęli się w swoim eksperymencie Frederick Banting i Charles Best w 1921 roku,
którzy podobnie jak poprzednicy poddali psy pankreatektomii prowadzącej do
cukrzycy, następnie jednak podawali im ekstrakt z trzustek zdrowych zwierząt,
co spowodowało ustąpienie symptomów choroby. Jak przypuszczano, ekstrakt
ten zawierał czynną substancję, której nazwę — insulina — (łac. insula, wyspa)
zaproponowano kilka lat wcześniej [3]. Dalsze badania insuliny potoczyły się
dużo szybciej i doprowadziły do jej oczyszczenia z trzustek bydlęcych oraz te-
rapeutycznego zastosowania hormonu w formie iniekcji u osób chorych na cu-
krzycę już w 1922 roku [1].
Współczesna definicja cukrzycy przyjęta przez Światową Organizację Zdro-
wia (WHO, ang. World Health Organization) podaje, że jest to choroba metabo-
liczna związana z defektem wydzielania lub działania insuliny. W wyniku tych
zmian, dochodzi do przewlekłej hiperglikemii odpowiedzialnej za zaburzenia
czynności oraz uszkodzenie nerek, oczu, nerwów czy naczyń krwionośnych.
Należy podkreślić, że hiperglikemia jest zjawiskiem pierwotnym, prowadzącym
do rozwoju powikłań w cukrzycy, które u osób nieleczonych mogą doprowa-
dzić do śmierci [4]. W ustanowionej przez WHO klasyfikacji tej choroby wy-
różnia się 4 typy cukrzycy: cukrzycę typu 1 (T1DM, ang. type 1 diabetes mellitus),
cukrzycę typu 2 (T2DM, ang. type 2 diabetes mellitus), cukrzycę o znanej etiologii
i cukrzycę ciężarnych [1,4]. Przyczyną występowania T1DM jest niszczenie ko-
mórek β trzustki w procesie autoimmunologicznym, prowadzącym do braku

258
www.postepybiochemii.pl
endogennej insuliny. Natomiast do rozwoju T2DM docho-
dzi na skutek insulinooporności, mimo produkowania tego
hormonu przez komórki β. Do grupy cukrzyc o znanej etio-
logii należy najwięcej form tej choroby. Zalicza się do niej
m. in. cukrzycę spowodowaną chorobami genetycznymi,
zakażeniami oraz wpływem substancji chemicznych. Jako
przyczynę rozwoju tej choroby podaje się również endo-
krynopatie i choroby wydzielniczej części trzustki. Ostat-
ni, 4 typ cukrzycy opisuje się w przypadku każdego stanu
hiperglikemicznego pojawiającego się u zdrowych dotąd
kobiet będących w ciąży. Należy podkreślić, że T2DM jest
najczęstszą postacią schorzenia, diagnozowaną u 90-95%
wszystkich pacjentów cukrzycowych [1,4].
Ze względu na duży wzrost liczby zachorowań oraz
skomplikowaną terapię, eksperci WHO uznali cukrzycę za
epidemię XXI wieku. Szacują oni, że obecnie na świecie cho-
ruje ponad 220 milionów osób, a liczba ta ma się podwoić
przed rokiem 2030 [4]. Podobne wyniki otrzymali Shaw i
wsp., którzy liczbę chorych na świecie w 2010 roku oceni-
li na 285 milionów [5]. Tylko w 2004 roku bezpośrednio z
powodu hiperglikemii lub w związku z rozwojem powi-
kłań cukrzycowych zmarło około 3,4 miliona osób. Pacjenci
chorzy na cukrzycę są również bardziej podatni na rozwój
przewlekłych powikłań, takich jak trudno gojące się rany,
uszkodzenia nerek czy neuropatie, które znacząco obniżają
jakość życia [5].
objaWy i DiagnosTyKa CuKrZyCy
Prawidłowe rozpoznanie cukrzycy i wczesne rozpoczę-
cie terapii jest utrudnione przez brak swoistych i jedno-
znacznych objawów. Ocenia się, że nawet 50% chorych na
cukrzycę jest nieświadomych choroby [1]. Sytuację dodat-
kowo utrudnia fakt, że wiele przypadków T2DM przebie-
ga praktycznie bezobjawowo, dlatego w rozpoznaniu tej
choroby bardzo istotne znacznie ma wykrycie czynników
ryzyka i badanie przesiewowe. Występowanie nadciśnienia
tętniczego, otyłości czy cukrzycy w rodzinie chorego po-
winno być wskazaniem do badań w kierunku cukrzycy. Jak
wskazują statystyki, np. otyłość, zwłaszcza typu brzuszne-
go, występuje aż u 85% pacjentów z T2DM [1].
Standardowym badaniem wykorzystywanym w diagno-
styce cukrzycy jest pomiar glikemii [1,6]. Oznaczenia stęże-
nia glukozy wykonuje się w osoczu krwi żylnej metodami
enzymatycznymi zarówno na czczo jak i po obciążeniu
glukozą (tzw. test tolerancji glukozy). Z kolei doustny test
tolerancji glukozy (OGTT, ang. oral glucose tolerance test) jest
najczęściej wykonywanym testem czynnościowym. Polega
na oznaczeniu glikemii na czczo oraz na sprawdzeniu jak
szybko glukoza jest pobierana z krwi przez komórki doce-
lowe. Standardowo w teście tym określa się glikemię przed
i dwie godziny po wypiciu przez pacjenta szklanki wody
zawierającej 75 g glukozy [6]. Natomiast w przypadku osób
cierpiących na cukrzycę, do oceny skuteczności zastosowa-
nej terapii wykorzystuje się pomiar stężenia glikowanej he-
moglobiny (HbA1c) w krwi [6]. Ze względu na średni czas
życia erytrocytów wynoszący 120 dni, HbA1c odzwiercie-
dla trzymiesięczną historię choroby. Opisane testy pozwa-
lają na przeprowadzenie diagnozy cukrzycy, zgodnie z al-
gorytmem przedstawionym na rycinie 1.
Warto pamiętać, że do opisu stanów hiperglikemicznych
stosuje się kilka terminów: stan przedcukrzycowy (ang. pre-
diabetes), który rozpoznaje się na podstawie wykrycia nie-
prawidłowej glikemii na czczo (IFG, ang. impaired fasting
glucose) i/lub upośledzonej tolerancji glukozy (IGT, ang.
impaired glucose tolerance). Diagnozuje się go na podstawie
wartości IFG i IGT wynoszących odpowiednio 100–125 mg/
dL i 140–199 mg/dL. U osób z rozpoznanym stanem przed-
cukrzycowym zaleca się powtarzanie badań co 2 lata [6]. Z
kolei cukrzycę stwierdza się w przypadku spełnienia przez
pacjentów jednego z poniższych kryteriów:
1. występowania typowych objawów klinicznych w po-
łączeniu z wartością glikemii o dowolnej porze dnia ≥ 200
mg/dL,
2. dwukrotnego pomiaru glikemii na czczo ≥ 126 mg/dL,
3. glikemii ≥ 200 mg/dL 120 minut po doustnym obciąże-
niu 75 g glukozy (test OGTT).
ETiologia i roZWój CuKrZyCy TyPu 2
Czynnikami ryzyka zwiększającymi prawdopodobień-
stwo rozwoju T2DM są zarówno uwarunkowania genetycz-
ne, jak i czynniki środowiskowe. Do najważniejszych czyn-
ników środowiskowych zalicza się otyłość typu brzusznego
i małą aktywność fizyczną, jednak do rozwoju choroby do-
chodzi tylko u części osób. U pozostałych, sprawność wy-
dzielnicza komórek β jest wystarczająco duża, by utrzymu-
jąca się przez długi czas hiperinsulinemia kompensowała
hiperglikemię. Osoby te charakteryzuje również zbliżony
do prawidłowego poziom glukozy w krwi, brak insulino-
oporności oraz rozrost podskórnej tkanki tłuszczowej, dzię-
ki czemu nie dochodzi do magazynowania tłuszczów w
wątrobie, sercu, mięśniach i trzustce [7,8].
Wydaje się jednak, że wpływ czynników środowisko-
wych promujących rozwój cukrzycy zaczyna się już w trak-
cie życia płodowego. Badania populacyjne wykazały, że u
rycina 1. Algorytm rozpoznawania cukrzycy, na podstawie [1], zmienione.
Postępy Biochemii 59 (3) 2013
259
dzieci, których matka w trakcie ciąży miała cukrzycę, wcze-
śniej dochodzi do rozwoju T2DM [9]. Przykładem może być
też analiza częstości zachorowań w rodzinach, w których
matki rodziły dzieci zarówno przed, jak i po zachorowaniu
na T2DM. Stwierdzono, że dzieci urodzone zanim doszło
do rozwoju T2DM u matki rzadziej zapadały na tą chorobę
[10]. Mimo istotnego wpływu historii rodzinnej na rozwój
cukrzycy stwierdzono, że tylko u około 10% pacjentów roz-
wój choroby można wytłumaczyć znanymi dziś mutacjami.
Jedno z ostatnich badań populacyjnych przeprowadzonych
w Szwecji w 2010 roku wykazało zwiększone ryzyko wystą-
pienia cukrzycy u dzieci, których rodzeństwo również cho-
rowało na cukrzycę. Ryzyko nie było natomiast związane ze
stanem zdrowia rodziców. Wynik ten sugeruje recesywny
wzór dziedziczenia u osób narażonych na podobne czynni-
ki środowiskowe [11].
Badania porównawcze genomów pacjentów cukrzyco-
wych pozwoliły na wykrycie wielu potencjalnie istotnych
genów, których mutacje mogą wpływać na rozwój choroby.
Są to przede wszystkim geny związane z upośledzeniem
funkcjonowania komórek β, insulinoopornością oraz oty-
łością [12]. Najważniejsza znana mutacja promująca rozwój
T2DM zlokalizowana jest w intronie 3 genu kodującego
czynnik transkrypcyjny TCF7L2 (ang. transcription factor
7-like 2) [13]. TCF7L2 jest składnikiem kompleksu trans-
krypcyjnego β-katenina/TCF i reguluje przekaz sygnału w
szlaku Wnt. Jego funkcja w cukrzycy polega na stymulacji
proliferacji komórek β trzustki oraz zwiększaniu produkcji
glukagonopodobnego peptydu-1 (GLP-1, ang. glucagon-like
peptide-1) [13].
Rozwój T2DM przebiega wieloetapowo. Zbyt wysokie,
toksyczne dla organów wewnętrznych stężenie glukozy w
krwi upośledza szereg procesów m. in. działanie insuliny
[14]. Pojawiająca się w pierwszej fazie T2DM insulinoopor-
ność prowadzi do zaburzeń we wchłanianiu i metabolizmie
glukozy przez komórki mięśniowe, adipocyty i hepatocy-
ty. W odpowiedzi na gorsze działanie insuliny, organizm
zwiększa jej produkcję, kompensując w ten sposób przez
pewien czas hiperglikemię. Jest to jednak działanie krót-
kotrwałe, ponieważ nadmierna eksploatacja komórek β
trzustki prowadzi do zmniejszenia ich masy i stopniowego
spadku wydzielania insuliny, aż do całkowitego zaprzesta-
nia funkcji wydzielniczych. Występująca w końcowej fazie
cukrzycy apoptoza komórek β prowadzi do całkowitego
braku endogennej insuliny, nasilenia hiperglikemii i ko-
nieczności podawania egzogenngo hormonu [14].
WPłyW hiPErgliKEMii na KoMórKi śróDbłonKa
Podwyższony poziom glukozy w krwi zaburza funkcjo-
nowanie hepatocytów, adipocytów, komórek mięśniowych
oraz komórek śródbłonka, zarówno dojrzałych (EC, ang.
endothelial cells), jak i progenitorowych (EPC, ang. endothe-
lial progenitor cells) [14,15]. Wpływ na komórki śródbłonka
wydaje się najistotniejszy, ponieważ związany jest z uszko-
dzeniami naczyń krwionośnych.
Glukoza jest podstawowym źródłem energii wszystkich
komórek organizmu człowieka. W warunkach prawidło-
wych jest przekształcana do acetylo-CoA, który bierze na-
stępnie udział w wytwarzaniu i magazynowaniu energii
w postaci ATP. Produktem ubocznym tych przemian jest
zawsze powstawanie niewielkiej ilości reaktywnych form
tlenu (ROS, ang. reactive oxygen species). W hiperglikemii,
oprócz prawidłowej syntezy ATP dochodzi do zwiększonej
produkcji ROS, niespecyficznej glikacji białek (AGE, ang.
advanced glycation endproducts), aktywacji kinazy białkowej
C oraz indukcji szlaku poliolowego i heksozoaminowego
[16].
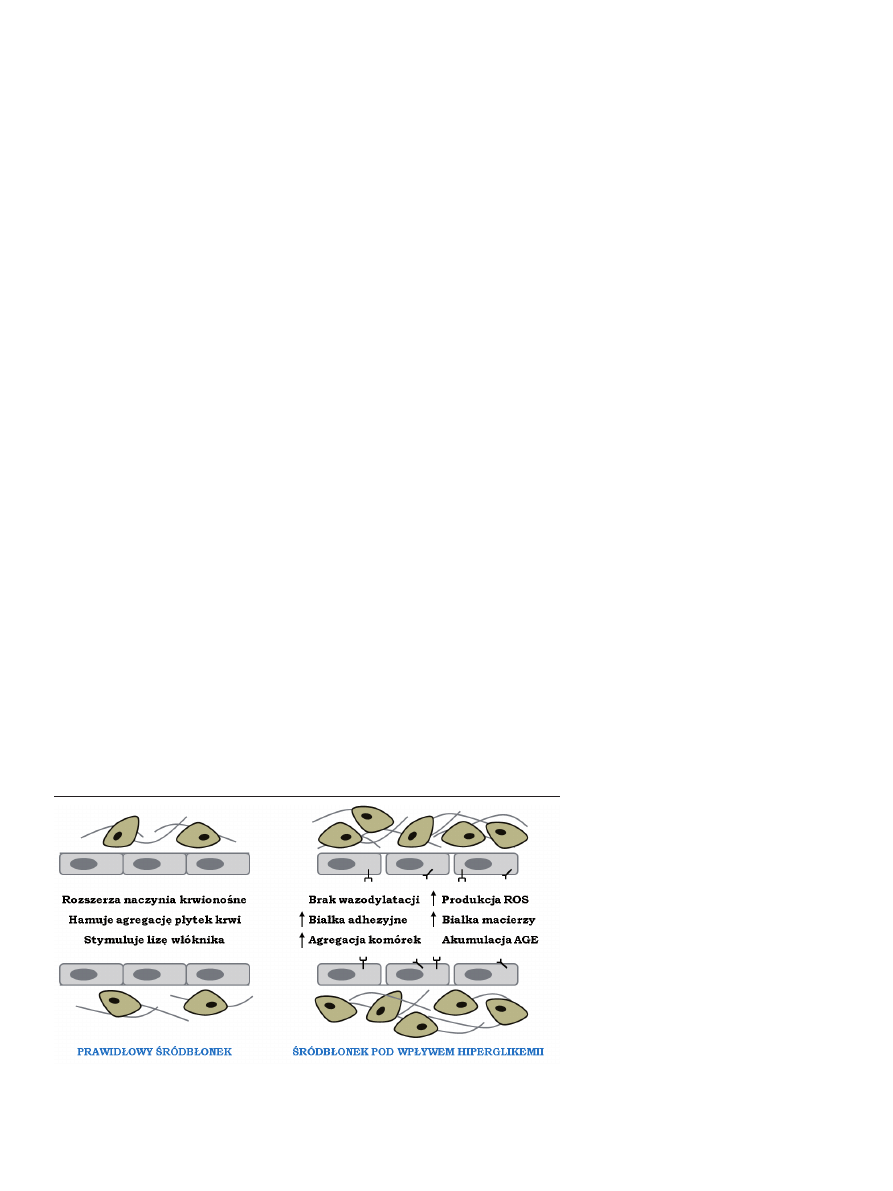
Produkcja ROS i AGE uważana jest za kluczowy czyn-
nik uszkadzający naczynia krwionośne i przyczyniający się
do rozwoju mikro- i makroangiopatii, głównych powikłań
cukrzycy. Wykazano, że wiązanie AGE z receptorem
wywołuje indukcję oksydaz NADPH, wzrost produkcji
ROS, a w konsekwencji prowadzi do uszkodzeń w DNA [17].
Stwierdzono również, że wywołana przez hiperglikemię i
ROS apoptoza EC zależy od szlaku NFκB, c-Jun i aktywacji
kaspaz [18]. Upośledzenie funkcji śródbłonka w warunkach
hiperglikemii wynika też z hamowania szlaku kinazy fosfa-
tydyloinozytolu-3 (PI3K, ang. phosphoinositide-3-OH kinase)
przez AGE. W prawidłowych warunkach aktywacja PI3K
poprawia żywotność EC oraz zwiększa ich zdolność do mi-
gracji i produkcji tlenku azotu (NO, ang. nitric oxide) przez
śródbłonkową syntazę NO (eNOS, ang.
endothelial nitric oxide synthase) [19]. W
hiperglikemii dochodzi do rozprzęgnię-
cia eNOS, czego efektem jest zmniej-
szenie syntezy NO, nasilenie produkcji
anionorodnika ponadtlenkowego (O
2
-
∙)
i w konsekwencji powstawanie bardzo
toksycznego nadtlenoazotynu (ONOO-)
[19].
U chorych na cukrzycę dysfunkcja
śródbłonka związana jest ponadto z
nasiloną syntezą cytokin prozapalnych,
adhezyn i czynników wzrostowych.
Prowadzi to do nadmiernej adhezji lim-
focytów, płytek krwi i monocytów, co
indukuje reakcję zapalną i może zwięk-
szać ryzyko rozwoju miażdżycy [16].
Dodatkowym czynnikiem ułatwiającym
rycina 2. Prawidłowy śródbłonek naczyń krwionośnych i mechanizmy odpowiedzialne za jego uszkodzenie
w hiperglikemii, na podstawie [40], zmienione.

260
www.postepybiochemii.pl
infiltrację monocytów jest zwiększona przepuszczalność
śródbłonka w warunkach hiperglikemii, spowodowana
osłabieniem połączeń międzykomórkowych [20]. Wyka-
zano też, że AGE wywołując agregację białek macierzy są
przyczyną spadku elastyczności naczyń i hamowania akty-
wacji metaloproteinaz (Ryc. 2) [21].
Wywołane przez hiperglikemię i ROS zmiany w funkcjo-
nowaniu EC oraz zaburzony skład i struktura macierzy ze-
wnątrzkomórkowej prowadzą do dysfunkcji naczyń krwio-
nośnych, zmniejszenia ich światła i upośledzenia przepływu
krwi. Wszystkie te zmiany wywołują powikłania cukrzyco-
we, które u osób nieleczonych mogą doprowadzić nawet do
śmierci [21]. Dlatego kluczowym zagadnieniem jest moż-
liwość poprawy funkcjonowania naczyń krwionośnych u
osób z cukrzycą, a jedną z możliwych strategii terapeutycz-
nych jest wykorzystanie komórek progenitorowych.
oDKryCiE KoMórEK ProgEniToroWyCh
śróDbłonKa
Przez wiele lat uważano, że tworzenie de novo naczyń
krwionośnych z niezróżnicowanych komórek prekurso-
rowych zachodzi jedynie w trakcie rozwoju płodowego.
Sądzono, że u osób dorosłych proces ten nie występuje, a
regeneracja i tworzenie nowych naczyń przebiega na dro-
dze migracji i różnicowania in situ dojrzałych komórek
śródbłonka. Ten powszechnie zaakceptowany pogląd uległ
zmianie po odkryciu przez Asaharę i współpracowników z
zespołu prof. Jeffrey’a Isnera, w krwi dorosłych osób komó-
rek progenitorowych śródbłonka zdolnych do proliferacji,
migracji i inkorporowania do istniejących naczyń [22].
Praca opublikowana w 1997 roku w Science, po raz
pierwszy udokumentowała obecność w ludzkiej krwi ob-
wodowej komórek frakcji leukocytarnej, zdolnej do różni-
cowania in vitro do komórek śródbłonka. Do izolacji EPC
wykorzystano antygeny CD34 i VEGFR-2 (KDR) ulegające
ekspresji zarówno w komórkach śródbłonkowych jak i w
hematopoetycznych komórkach macierzystych (HSC, ang.
hematopoietic stem cells). Pozyskane od dorosłych ochotni-
ków komórki CD34(+)/VEGFR-2(+) hodowano in vitro i już
po kilku dniach stwierdzono syntezę charakterystycznych
markerów śródbłonka, takich jak: CD31, E-selektyna, Tie-2
czy eNOS [22]. W tej samej pracy opisano też doświadcze-
nie, polegające na izolacji EPC z transgenicznych myszy wy-
kazujących ekspresję genu reporterowego β-galaktozydazy
i podaniu ich zwierzętom z niedotlenioną kończyną. Wy-
kazano obecność komórek β-gal(+)/CD31(+) w regenerują-
cych naczyniach krwionośnych, wskazując tym samym na
ich udział w waskulogenezie [22]. Kolejne doświadczenia
wykonywane przez ten sam zespół potwierdzały udział ko-
mórek EPC uwalnianych ze szpiku kostnego w tworzeniu
naczyń krwionośnych zarówno w stanach fizjologicznych
(przerost błony śluzowej macicy, ukrwienie ciałka żółtego,
gojenie ran) jak i patologicznych (wzrost nowotworu, za-
wał serca czy niedokrwienie kończyny tylnej). W badaniach
przeprowadzano rekonstytucję szpiku kostnego u napro-
mieniowanych myszy, przeszczepiając im komórki wyka-
zujące ekspresję β-galaktozydazy pod kontrolą promotorów
charakterystycznych dla komórek śródbłonka, czyli Tie-2
lub VEGFR-2. Po wykonaniu rekonstytucji szpiku kostnego
badacze stwierdzali obecność znakowanych komórek w no-
wopowstałych naczyniach [23,24]. Eksperymenty wykona-
ne przez Asaharę i wsp. dały początek wielu pracom, które
pozwoliły na lepsze poznanie funkcjonowania EPC.
Mianem EPC określa się dziś jednojądrzaste komórki
eksprymujące równocześnie markery śródbłonkowe i pro-
genitorowe [25]. EPC wykazują ponadto zdolność wiązania
lektyny, białek macierzy zewnątrzkomórkowej (np. fibro-
nektyny) oraz pochłaniania acetylowanych lipoprotein o
niskiej gęstości (AcLDL, ang. acetylated low density lipopro-
tein). Mimo wielu lat badań nie udało się jednak do dzisiaj
zidentyfikować antygenu ulegającego ekspresji jedynie na
EPC, który jednoznacznie definiowałby tylko tę populację
komórek. Wśród markerów obecnych na powierzchni ludz-
kich EPC wyróżnia się CD31, CD34, VE-kadherynę, CD146,
CXCR4, VEGFR-2, Tie-2 czy vWF. Synteza CD133 i c-kit,
markerów progenitorowych, zanika na powierzchni ludz-
kich EPC w trakcie ich różnicowania do EC [25]. Charak-
terystyka mysich EPC w dużej mierze jest podobna. Warto
jednak pamiętać, że komórki mysie wiążą lektynę BS1 Grif-
fonia simplicifolia (ludzkie — Ulex europaeus) i eksprymują
Sca-1. Podobnie jak w przypadku komórek ludzkich, róż-
nicowanie mysich EPC wiąże się z utratą Sca-1 i c-kit [25].
Do analizy EPC wykorzystuje się również właściwości
angiogenne tych komórek, takie jak zdolność do tworzenia
kapilar in vitro, tworzenie wtórnych kolonii EC, produkcję
czynników proangiogennych czy udział w naprawie naczyń
in vivo. Należy podkreślić, że analiza testów funkcjonalnych
jest bardzo istotna, ponieważ pozwala na ocenę rzeczywi-
stej funkcji EPC i ich ewentualnej przydatności w terapii.
Fenotypowa charakterystyka EPC jest natomiast wciąż
tematem ożywionych dyskusji i licznych kontrowersji. Nie
ma również powszechnie uznawanej, wystandaryzowanej
metody ich izolacji i hodowli co dodatkowo utrudnia inter-
pretację uzyskanych wyników [26]. Dokładne porównanie
i podsumowanie stosowanych metod przedstawili ostatnio
Richardson i Yoder, potwierdzając jednocześnie, że używa-
ny obecnie termin EPC nie definiuje ściśle jednej populacji
komórek [26]. Dodatkowo, charakterystykę EPC utrudnia
ekspresja niektórych antygenów zarówno na powierzchni
EPC, EC jak i HSC. Wykorzystanie różnych protokołów
izolacji, odmiennych warunków hodowli, czy wreszcie cha-
rakteryzowanie EPC w oparciu o niejednakowe markery i
testy funkcjonalne, utrudnia porównywanie wyników uzy-
skanych przez różne grupy badawcze. Jednak, co wydaje
się dziś bezsporne, komórki progenitorowe biorą udział w
regeneracji naczyń krwionośnych [25,27].
EPC i KonDyCja naCZyń KrWionośnyCh
Komórki progenitorowe śródbłonka rezydują w niszach
szpiku kostnego, skąd w odpowiedzi na uszkodzenie lub
niedotlenienie tkanek są mobilizowane i włączane w proce-
sy regeneracyjne [24]. Mobilizacja wiąże się z całą kaskadą
zdarzeń obejmujących uwolnienie EPC do krwi, dotarcie do
uszkodzonej tkanki oraz stymulację powstawania nowych i
naprawę już istniejących naczyń [28]. Jednymi z najważniej-
szych mediatorów odpowiedzialnych za mobilizację EPC
są czynniki VEGF-A i czynnik-1α pochodzenia stromalne-

Postępy Biochemii 59 (3) 2013
261
go (SDF-1α, ang. stromal cell-derived factor-1α). Wiążą się one
do receptorów VEGFR-2 (VEGF-A) i CXCR4 (SDF-1α) na
powierzchni komórek podścieliska, indukując fosforylację
kinaz PI3K/AKT i aktywując syntazę tlenku azotu. Wzrost
produkcji NO aktywuje metaloproteinazę 9 macierzy ze-
wnątrzkomórkowej (MMP-9, ang. matrix metalloproteinase
9), która uwalnia czynnik komórek macierzystych (KitL,
ang. kit ligand) obecny w błonie komórek podścieliska. Roz-
puszczalna forma KitL (sKitL), po związaniu z receptorem
na powierzchni EPC, indukuje ich mobilizację. W kolejnym
etapie, naprowadzane przez gradient chemokin EPC docie-
rają do uszkodzonych naczyń i biorą udział w ich naprawie
(Ryc. 3) [28].
Po wniknięciu do uszkodzonych lub ischemicznych tka-
nek EPC stymulują powstawanie naczyń krwionośnych.
Uważa się, że biorą udział w waskulogenezie (tworzeniu de
novo nowych naczyń), różnicując w dojrzałe komórki śród-
błonkowe. Mogą też bezpośrednio inkorporować do uszko-
dzonych, ale już istniejących struktur, wypełniając luki w
warstwie śródbłonka [22]. Z powodu rozbieżnych wyników
dotyczących udziału EPC w utworzonych naczyniach (od
0% do 80% komórek śródbłonkowych) trudno jednak jed-
noznacznie stwierdzić, jakie jest nasilenie i znaczenie tego
procesu [29-31].
Coraz częściej zwraca się natomiast uwagę na istotną rolę
działania parakrynnego. EPC po dotarciu do uszkodzonej
tkanki wydzielają czynniki wzrostowe, aktywując zarówno
EC jak i pericyty. Do proangiogennych cytokin produko-
wanych w wysokich stężeniach przez EPC zalicza się IL-8,
czynnik wzrostu hepatocytów (HGF, ang. hepatocyte growth
factor), VEGF-A, płytkowy czynnik wzrostu-BB (PDGF-BB,
ang. platelet-derived growth factor-BB), zasadowy czynnik
wzrostu fibroblastów (bFGF, ang. basic fibroblast growth fac-
tor) oraz białko chemotaktyczne-1 dla monocytów (MCP-
1, ang. monocyte chemoactractant protein-1) [32]. Znaczenie
produkowanych przez EPC czynników wzrostowych wy-
kazali Di Santo i wsp. analizując pożywki kondycjonowa-
ne zebrane znad EPC (EPC CM). Media te indukowały in
vitro wzrost kapilar w teście krążków aortalnych na matri-
gelu oraz hamowały apoptozę EC [33]. Co najważniejsze,
domięśniowe podanie EPC CM do niedotlenionej kończy-
ny szczura przyśpieszało regenerację przepływu, poprzez
zwiększenie liczby dojrzałych kapilar. Autorzy pracy zaob-
serwowali taki sam efekt terapeutyczny po podaniu komó-
rek i kondycjonowanych mediów, co potwierdza kluczowe
znaczenie parakrynnej aktywności EPC [33].
Proangiogenny potencjał komórek progenitorowych
śródbłonka i ich duże znaczenie dla prawidłowej kondycji
naczyń krwionośnych nie budzi dziś wątpliwości, mimo
trudności z jednoznacznym wskazaniem najważniejszego
mechanizmu ich działania oraz braku ścisłej definicji tych
komórek. Wykazano, że funkcjonowanie EPC jest zaburzo-
ne w stanach patologicznych. Stwierdzono m. in. odwrotną
korelację pomiędzy liczbą i zdolnością EPC do migracji, a
czynnikami ryzyka choroby niedokrwiennej serca [34]. Wy-
kazano też istnienie zależności pomiędzy liczebnością EPC
w krwi i skalą Framingham, oceniającą ryzyko wystąpienia
zawału serca lub śmierci spowodowanej chorobą niedo-
krwienną serca w perspektywie 10 lat [34]. U pacjentów z
cukrzycą wydaje się natomiast, że nasilenie niekorzystnych
zmian w naczyniach krwionośnych wiąże się ze zmniejszo-
ną liczbą oraz osłabionym potencjałem klonogennym EPC
[34].
KonDyCja KoMórEK EPC u PaCjEnTóW
ChoryCh na CuKrZyCę
Pacjenci chorzy na cukrzycę 3–4 razy częściej zapadają na
choroby układu krążenia niż osoby z prawidłową glikemią.
Stwierdzono, że najważniejszą przyczyną tych komplikacji
jest dysfunkcja śródbłonka. Wydaje się, że nasilenie nieko-
rzystnych zmian w naczyniach wynika również ze zmniej-
szonej liczby i osłabionego potencjału klonogennego EPC
[35].
Analizy laboratoryjne przeprowadzone na materiale
pobranym od ludzi wykazały niekorzystny wpływ hiper-
glikemii na aktywność EPC. Hodowanie komórek jedno-
jądrzastych (MNC, ang. mononuclear cells) izolowanych z
krwi zdrowych osób w warunkach podwyższonego stę-
żenia glukozy zmniejszało liczbę uzyskanych EPC. Ko-
mórki wykazywały też wolniejszą migrację, zmniejszo-
ną produkcję NO oraz spadek aktywności MMP-9 [36].
Wpływ hiperglikemii na EPC uzyskane od zdrowych
ochotników i hodowane in vitro był porównywany z
wpływem stymulacji czynnikiem-α martwicy nowotwo-
rów (TNF-α, ang. tumor necrosis factor-α). Stwierdzono,
że w obu przypadkach dochodzi do aktywacji kinazy
p38 [37]. Wydaje się, że zarówno indukcja p38 wynika-
jąca z fosforylacji reszt treoniny 180 i tyrozyny 182 jak
i upośledzenie aktywności szlaku kinazy Akt ogrywają
istotną rolę w hamowaniu proliferacji i starzeniu się EPC
u pacjentów z cukrzycą [38]. Komórki EPC izolowane
z krwi chorych i hodowane in vitro wykazują obniżoną
o prawie 50% proliferację i znacznie mniej wydajnie in-
korporują do kapilar. Mimo prawidłowej adhezji do ko-
lagenu, fibrynogenu i spoczynkowych EC, ich zdolność
do wiązania się z aktywowanym śródbłonkiem jest za-
burzona [39]. Tymczasem wiązanie EPC z uszkodzonym
śródbłonkiem ma kluczowe znaczenie, gdyż warunkuje
możliwość naprawy naczyń [39]. Cukrzyca wpływa też
na działanie parakrynne ludzkich EPC. Kondycjonowane
pożywki znad takich komórek mniej wydajnie stymulują
tworzenie kapilar przez dojrzałe komórki śródbłonkowe.
Osłabienie aktywności cukrzycowych EPC utrzymuje się
nawet po kilkudniowej hodowli w warunkach normogli-
kemii [40].
rycina 3. Schemat mobilizacji komórek progenitorowych śródbłonka ze szpiku
kostnego.

262
www.postepybiochemii.pl
Badania in vivo potwierdziły niekorzystny wpływ hiper-
glikemii na liczbę i funkcjonowanie EPC u ludzi, sugerowa-
ny przez doświadczenia in vitro. Wykazano między innymi
odwrotną korelację pomiędzy stężeniem glukozy a liczbą
EPC (komórek o fenotypie CD34(+)/KDR(+)) w krwi obwo-
dowej pacjentów z T2DM. Spadek u osób chorych wynosił
około 40%, co znaczyło, że liczba EPC zmniejszała się z oko-
ło 70 do około 40 na milion komórek [41]. Wykazano też,
że liczba EPC w krwi osób zdrowych i z cukrzycą zdiagno-
zowaną po raz pierwszy (charakteryzującą się poziomem
HbA1c poniżej 7,5%) nie różni się. Kluczową rolę hipergli-
kemii w pogorszeniu funkcjonowania EPC dokumentują
również prace opisujące zwiększoną zdolność jednojądrza-
stych komórek krwi obwodowej do różnicowania in vitro w
kierunku EPC u pacjentów z lepszą kontrolą poziomu glu-
kozy [42,43].
Trudno dziś jednak jednoznacznie stwierdzić, czy wystę-
powanie mikro- i makroangiopatii w cukrzycy spowodo-
wane jest zaburzeniem funkcji EPC, czy też na skutek roz-
woju powikłań, dochodzi do zmian w biologii EPC. Anali-
zy kliniczne sugerują jedynie, że występowanie angiopatii
u osób chorych na cukrzycę, zmniejsza liczbę EPC w krwi
obwodowej, a stopień zaawansowania choroby naczyń ob-
wodowych jest odwrotnie proporcjonalny do liczby tych
komórek [41].
ZaburZonE funKCjonoWaniE KoMórEK
EPC a PoWiKłania CuKrZyCoWE
Powikłania przewlekłe powstają na skutek wieloletniej
ekspozycji komórek na podwyższone stężenie glukozy i
niemożności uniknięcia silnych wahań glikemii w trakcie
długotrwałej choroby. Stopniowe nagromadzanie uszko-
dzeń w układzie krwionośnym prowadzi bardzo często,
nawet u osób leczonych, do rozwoju dysfunkcji śródbłonka.
Rozwój powikłań przewlekłych jest największym zagroże-
niem dla pacjentów cukrzycowych. Oszacowano, że w 2000
roku przyczyniły się one do śmierci 2,9 miliona osób na ca-
łym świecie [4,5].
Najczęściej diagnozowanym przewlekłym powikłaniem
jest neuropatia cukrzycowa, występująca aż u 50% cho-
rych. Jest ona bezpośrednim skutkiem hiperglikemii, któ-
ra w połączeniu ze stresem oksydacyjnym i zaburzeniami
produkcji czynników wzrostowych prowadzi nie tylko do
demielinizacji, ale również do zaniku nerwów [1]. Badania
wpływu neuropatii cukrzycowej na właściwości EPC w
modelu szczurzym wykazały, że upośledzona mobilizacja
tych komórek związana jest z mniejszą liczbą zakończeń
nerwowych w szpiku kostnym. Obok zaniku nerwów au-
torzy pracy wykazali wzrost liczby EPC w szpiku, przy jed-
noczesnym spadku ich liczebności w krwi obwodowej [43].
Podobne wyniki uzyskano analizując nerwy kulszowe u
myszy z cukrzycą wywołaną iniekcją streptozotocyny. Wy-
kazano u nich pogorszony przepływ krwi, mniejszą liczbę
kapilar oraz słabsze przewodzenie w nerwach czuciowych
i ruchowych. Obserwowane zmiany ustąpiły po domię-
śniowym podaniu EPC, które preferencyjnie lokowały się
w naczyniach odżywiających nerwy i w okolicy dojrzałych
komórek śródbłonka. Proangiogenne i proneurogenne wła-
ściwości EPC przyczyniły się do nasilenia proliferacji komó-
rek Schwanna i EC [44].
Neuropatia, wraz ze zmniejszonym przepływem krwi
w naczyniach obwodowych, dysfunkcją śródbłonka oraz
upośledzoną angio- i arteriogenezą stanowi najważniejszy
czynnik prowadzący do powstawania trudno gojących się
ran, czyli takich, które goją się dłużej niż 8 tygodni, nie goją
się wcale, lub po wygojeniu pojawiają się powtórnie [45].
Skrajnym przypadkiem są owrzodzenia stóp występujące
u kilku procent pacjentów, które u około 1% chorych koń-
czą się amputacją [46]. W 2004 roku w USA 60% amputacji
kończyn dolnych niezwiązanych z urazem mechanicznym
było spowodowanych występowaniem cukrzycy z powi-
kłaniami [47]. Przedłużenie gojenia ran lub całkowite za-
hamowanie tego procesu w cukrzycy wynika z rozwijającej
się neuropatii i waskulopatii lub może być spowodowane
zakażeniami. Wydaje się, że zmniejszenie liczebności i upo-
śledzenie funkcji EPC w cukrzycy, może przyczyniać się do
zaburzeń gojenia ran, gdyż hiperglikemia wpływa zarów-
no na zdolności EPC do bezpośredniej naprawy naczyń,
jak również na wydzielanie czynników wzrostowych [48].
Jak wykazali Barcelos i wsp., podanie pożywek zebranych
znad komórek CD133(+) (CD133(+) CM) przyśpiesza goje-
nie owrzodzeń u myszy cukrzycowych. Proangiogenny po-
tencjał CD133(+) CM potwierdzono również in vitro: media
te stymulowały migrację i tworzenie tubul przez dojrzałe
komórki śródbłonka, a efekt był hamowany w obecności
przeciwciał wiążących VEGF-A lub IL-8 [48].
Zaangażowanie EPC w gojenie ran przebiega wieloeta-
powo, obejmując przekaz sygnału do szpiku, uwalnianie
EPC do krwi, migrację komórek do miejsca zranienia i ak-
tywne włączanie się w proces odbudowy. Zakłócenie które-
gokolwiek z tych etapów może opóźnić lub całkowicie za-
burzyć regenerację tkanek [28]. Wykazano, że upośledzone
uwalnianie komórek progenitorowych ze szpiku kostnego
w cukrzycy w odpowiedzi na m. in. VEGF-A i SDF-1α spo-
wodowane jest przede wszystkim zmniejszoną aktywnością
eNOS i wynikającym z tego osłabieniem przekazu sygnału
od sKitL [39,40]. SDF-1α jest jedną z najważniejszych che-
mokin odpowiedzialnych za mobilizację komórek progeni-
torowych. EPC migrują zgodnie z gradientem tego białka,
którego stężenie w prawidłowych warunkach powinno być
najwyższe w miejscu uszkodzenia naczyń [28]. U myszy
cukrzycowych stwierdzono jednak niższą ekspresję SDF-1α
w zranionej tkance. Dopiero odtworzenie naturalnie wystę-
pującego gradientu poprzez lokalne podanie rekombino-
wanego białka indukowało mobilizację EPC i przyśpieszało
gojenie ran [28]. Podobne wyniki otrzymano analizując trzy
odmienne modele myszy cukrzycowych (db/db, NOD i
myszy nastrzyknięte streptozotocyną). We wszystkich mo-
delach zaobserwowano spowolnienie gojenia ran, co wią-
zało się z mniejszą liczebnością EPC w miejscu zranienia.
Tak jak w przypadku SDF-1α, miejscowe podanie PDGF-BB
przyspieszało gojenie i stymulowało powstawanie nowych
naczyń [49]. Wyniki te sugerują, że stymulacja komórek EPC
może pomóc w terapii przewlekłych ran oraz owrzodzeń.
Terapia komórkowa z wykorzystaniem EPC może być
bardzo obiecująca, choć nie można wykluczyć niepożą-
danych skutków ubocznych tej metody. Potencjalnie EPC

Postępy Biochemii 59 (3) 2013
263
mogą bowiem przyczyniać się do wzrostu unaczynienia
nowotworów czy rozwoju proliferacyjnej formy retinopatii
cukrzycowej. Badania potwierdziły, że EPC mogą uczest-
niczyć w patologicznej neowaskularyzacji [50], przy czym
wydaje się, że proces ten zależy od mobilizacji EPC induko-
wanej przez SDF-1. Wykazano, że w ciele szklistym myszy
będących modelem proliferacyjnej retinopatii cukrzycowej,
stężenie SDF-1 jest wysokie, a blokowanie aktywności tego
białka in vivo zapobiega neowaskularyzacji [51].
Niekontrolowany wzrost naczyń krwionośnych w przy-
padku retinopatii cukrzycowej jest przeciwieństwem upo-
śledzonej angiogenezy opisywanej zarówno u pacjentów
T1DM jak i T2DM [38,39]. Jedna z hipotez tłumaczących ten
paradoks zakłada, że w odpowiedzi na lokalną mobilizację
EPC w niedotlenionej siatkówce dochodzi do wzrostu ka-
pilar, ale ich dalsze funkcjonowanie jest upośledzone. Hi-
poteza ta została sformułowana na podstawie wyników,
które otrzymali Asnaghi i wsp., analizując EPC krążące w
krwi obwodowej osób zdrowych, cierpiących na T1DM bez
powikłań oraz T1DM współistniejącą z retinopatią cukrzy-
cową. Potencjał klonogenny EPC u pacjentów z cukrzycą
i retinopatią był większy niż u pozostałych grup. Autorzy
potwierdzili również osłabienie potencjału klonogennego
cukrzycowych EPC (od pacjentów z T1DM bez retinopa-
tii) w porównaniu do komórek osób zdrowych [52]. Także
Fadini i wsp. analizując komórki CD34(+)/KDR(+) w krwi
obwodowej pacjentów z T2DM stwierdzili, że występowa-
nie powikłań koreluje z liczebnością EPC. W porównaniu
do osób z cukrzycą bez komplikacji, pacjenci z cukrzycą i
z chorobą naczyń obwodowych mieli mniej EPC. Współ-
istnienie cukrzycy i retinopatii było natomiast związane ze
zwiększoną liczbą tych komórek [53].
MożliWośCi PoPraWy funKCjonoWania EPC
Ze względu na potencjalne znaczenie terapeutyczne EPC,
podejmowane są liczne próby poprawy ich właściwości. Ba-
dania przeprowadzone u zdrowych ochotników sugerują,
że nawet jednorazowy wysiłek fizyczny stymuluje uwalnia-
nie EPC. Rehman i wsp. opisali prawie czterokrotny wzrost
ich liczby w krwi osób poddanych ćwiczeniom w stosunku
do grupy kontrolnej [54]. Podobny efekt zaobserwowano
w przypadku regularnych ćwiczeń trwających 3 miesiące.
Przeprowadzone doświadczenia wykazały, że długotrwała
gimnastyka zwiększa zarówno potencjał klonogenny EPC,
jak i ich zdolność do migracji. Autorzy udokumentowa-
li również pogorszenie funkcjonowania EPC u osób star-
szych, przy czym trend ten został częściowo odwrócony w
wyniku fizykoterapii [55].
Ćwiczenia fizyczne wpływają korzystnie na komórki
EPC nie tylko u osób zdrowych, ale także u cierpiących na
choroby układu krążenia. Poprawę liczebności, potencjału
migracyjnego i klonogennego EPC wykazano np. u pacjentów
z chorobą tętnic obwodowych poddanych regularnemu
treningowi. Efektem treningu było zwiększenie dystansu,
jaki chorzy mogli przejść bez wystąpienia dolegliwości
bólowych [56]. Ćwiczenia fizyczne miały również korzystny
wpływ na EPC u chorych z przewlekłą niewydolnością
serca, chorobą niedokrwienną serca i cukrzycą typu 2 [57,58].
Również nasze badania wykazały wzrost liczby krążących
EPC u pacjentów z miażdżycą tętnic obwodowych już w
trzy godziny po jednorazowym wysiłku na bieżni, choć
efekt taki nie był obserwowany po jednorazowym wysiłku
u pacjentów trenujących regularnie przez trzy miesiące [59].
Poprawę funkcjonowania EPC obserwuje się również
po zastosowaniu odpowiednio dobranej farmakoterapii.
Badania in vitro pokazały, że insulina stymuluje tworzenie
kapilar oraz zwiększa potencjał klonogenny EPC. Związane
jest to z aktywacją szlaku receptora dla insulinopodobnego
czynnika wzrostu-1 (IGF-1, ang. insulin-like growth factor) i
kinaz MAP (Erk1/2 i p38) [60]. Insulina wpływa także na
mobilizację komórek CD34(+)/CD133(+) u pacjentów z
cukrzycą. Wykazano, że kilkutygodniowa insulinoterapia
doprowadziła do obniżenia poziomu glikowanej hemoglo-
biny oraz do znaczącego wzrostu liczby komórek EPC w
krwi. Wyniki opublikowane ostatnio przez Fadini i wsp. po-
twierdzają, że normalizacja glikemii u pacjentów z T2DM i
makroangiopatią poddanych insulinoterapii związana jest z
poprawą kondycji naczyń krwionośnych. Towarzyszy temu
obniżenie produkcji prozapalnych białek adhezyjnych: mię-
dzykomórkowej cząsteczki adhezyjnej-1 (ICAM-1, ang. in-
tercellular adhesion molecule-1), naczyniowej cząsteczki ad-
hezyjnej-1 (VCAM-1, ang. vascular cell adhesion molecule-1),
E-selektyny oraz zwiększenie liczby krążących EPC [61].
U chorych na cukrzycę wykazano także korzystny
wpływ na EPC agonistów receptora aktywowanego przez
proliferatory peroksysomów-gamma (PPARγ, ang. peroxi-
some proliferator-activated receptor gamma). PPARγ to czyn-
nik transkrypcyjny z nadrodziny receptorów jądrowych,
który w wyniku aktywacji tworzy heterodimer i wiąże się
do sekwencji PPRE (PPAR, ang. responsive element) na pro-
motorach genów, bezpośrednio aktywując ich transkrypcję.
Działa głównie na geny zaangażowane w regulację meta-
bolizmu lipidów i glukozy. Jego ekspresja jest najwyższa w
białej tkance tłuszczowej, a indukcja PPARγ jest warunkiem
dojrzewania adipocytów [62]. Wzrost aktywności PPARγ
zwiększa także liczbę EPC w krwi oraz poprawia ich wła-
ściwości in vitro [42,63,64]. Syntetycznymi agonistami
PPARγ, które stosowano u pacjentów z insulinoopornością
są związki z grupy tiazolidinedionów (TZD, ang. thiazolidi-
nediones) takie jak rosiglitazon i pioglitazon.
Pierwsze wyniki dotyczące wpływu TZD na funkcjono-
wanie EPC u pacjentów z T2DM opublikowali Pistrosch i
wsp. w 2005 roku. W pracy opisano wyniki trzymiesięcznej
terapii rosiglitazonem u osób z cukrzycą, w wyniku której
zaobserwowano normalizację upośledzonej migracji EPC
oraz wzrost ich liczby. Korzystne efekty utrzymywały się
nawet przez 9 tygodni od zakończenia terapii [42]. Podobne
wyniki uzyskano w przypadku dodatkowego podawania
pioglitazonu pacjentom leczonym metforminą, przy czym
w grupie leczonej dodatkowo TZD obserwowano normali-
zację parametrów biochemicznych, takich jak poziom trigli-
cerydów, lipoprotein o bardzo małej gęstości (VLDL, ang.
very low density lipoproteins) i białka C-reaktywnego. Zarów-
no bezpośrednie jak i pośrednie działanie pioglitazonu na
EPC doprowadziło do zwiększenia ich liczby, poprawy mi-
gracji oraz nasilenia adhezji do fibronektyny i kolagenu. Za-
obserwowano również normalizację proliferacji i potencjału
klonogenennego EPC u osób poddanych terapii [65].

264
www.postepybiochemii.pl
Efekty aktywatorów PPARγ nie zależą wyłącznie od
wpływu na insulinooporność, gdyż korzystne działanie
pioglitazonu na EPC wykazano również u pacjentów z pra-
widłową tolerancją glukozy cierpiących na chorobę niedo-
krwienną serca. Trwająca 30 dni terapia zwiększała liczbę
komórek CD34(+)/KDR(+), nasilała migrację zależną od
SDF-1 i wzmacniała potencjał klonogenny in vitro [64]. Ak-
tywacja PPARγ znosiła również negatywny wpływ AGE –
stymulacja EPC rosiglitazonem zmniejszała zahamowanie
proliferacji, migracji, aktywacji Akt i fosforylacji eNOS wy-
woływane przez AGE [64].
Szczegółowe badania analizujące wpływ TZD na funk-
cjonowanie EPC pozwoliły na poznanie niektórych mecha-
nizmów odpowiedzialnych za ten proces. Do szlaków zwią-
zanych z aktywacją PPARγ przez TZD i mających korzystny
wpływ na cukrzycowe EPC zalicza się m. in. aktywację telo-
merazy w komórkach śródbłonka. Krótkotrwałe, kilkudnio-
we podawanie myszom pioglitazonu nie tylko zwiększało
liczbę EPC, ale przede wszystkim indukowało wzrost za-
wartości czynnika wiążącego telomer, typ 2 (TERF-2, ang te-
lomer repeat-binding factor 2) oraz obniżało apoptozę tych ko-
mórek [63]. Z kolei u myszy, którym podawano pioglitazon
przez miesiąc stwierdzono w komórkach aortalnych in vivo
indukcję aktywności telomerazy, wzrost syntezy TERF-2
oraz spadek zawartości białek będących markerami starze-
nia się komórek: p16 i p53. Analogiczne wyniki uzyskano
stymulując in vitro ludzkie komórki EPC, a efekty były za-
leżne od aktywacji kinazy Akt [66]. Wykazano również, że
inkubacja in vitro ludzkich EPC w obecności pioglitazonu,
podobnie jak w modelach zwierzęcych, nasila aktywność
telomerazy [67].
Nie jest natomiast jasne, na ile aktywatory PPARγ dzia-
łają bezpośrednio poprzez aktywację PPARγ, a na ile ich
aktywność jest plejotropowa i wynika z wpływu na inne
ścieżki sygnałowe. Dokładne poznanie mechanizmów dzia-
łania jest utrudnione przez brak odpowiedniego modelu
zwierzęcego, myszy pozbawionych PPARγ. Brak PPARγ
jest bowiem letalny, przy czym jednym z obserwowanych
zaburzeń jest upośledzenie unaczynienia łożyska [68], co
sugeruje bezpośrednie znaczenie PPARγ dla angiogene-
zy. Nie ma również żadnych danych wskazujących na ile
zmniejszenie zawartości lub aktywności PPARγ np. u pa-
cjentów z zespołem niewrażliwości na ligandy PPARγ, u
pacjentów z hiperhomocysteinemią lub w niektórych mo-
delach cukrzycy może wpływać na właściwości angiogenne
dojrzałych i progenitorowych komórek śródbłonka.
PoDsuMoWaniE
Zmniejszona liczba i upośledzenie funkcji EPC u osób
chorych na cukrzycę może być jednym z czynników pro-
wadzących do zwiększenia ryzyka chorób sercowo-na-
czyniowych oraz powstawania mikro- i makroangiopatii.
Wiadomo obecnie, że stosowanie leków regulujących po-
ziom glukozy poprawia funkcjonowanie EPC, przy czym
molekularne mechanizmy tego działania są różnorodne i
odmienne dla poszczególnych grup leków. Ich zrozumienie
jest niezbędne, aby w przyszłości można było skuteczniej
wykorzystać prowaskulogenne właściwości leków prze-
ciwcukrzycowych w praktyce klinicznej.
PiśMiEnniCTWo
1. Sieradzki J (2005) Cukrzyca i zespół metaboliczny, W: Szczeklik A
(red) Choroby wewnętrzne. Medycyna Praktyczna. Kraków, str. 1179-
1215
2. Luft R (1989) Oskar Minkowski: discovery of the pancreatic origin of
diabetes, 1889. Diabetologia 32: 399-401
3. Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA (1922) Pan-
creatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc
J 12: 141-146
4. Jagielski AK, Piesiewicz A (2011) Diabetes as the challenge to 21 cen-
tury medicine-insights from clinical and biochemical investigations.
Postepy Biochem 57: 191-199
5. Shaw JE, Sicree RA, Zimmet PZ (2010) Global estimates of the preva-
lence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 87: 4-14
6. Grzeszczak W (2011) Zalecenia kliniczne dotyczące postępowania u
chorych na cukrzycę. Diabetologia Praktyczna A1-A46
7. Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J
Clin Invest 116: 1802-1812
8. Defronzo RA (2009) Banting Lecture. From the triumvirate to the omi-
nous octet: a new paradigm for the treatment of type 2 diabetes mel-
litus. Diabetes 58: 773-795
9. Pettitt DJ, Lawrence JM, Beyer J, Hillier TA, Liese AD, Mayer-Davis B,
Loots B, Imperatore G, Liu L, Dolan LM, Linder B, Dabelea D (2008)
Association between maternal diabetes in utero and age at offspring’s
diagnosis of type 2 diabetes. Diabetes Care 31: 2126-2130
10. Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir
MM, Roumain J, Bennett PH, Knowler WC (2000) Intrauterine expo-
sure to diabetes conveys risks for type 2 diabetes and obesity: a study
of discordant sibships. Diabetes 49: 2208-2211
11. Hemminki K, Li X, Sundquist K, Sundquist J (2009) Familial risks for
type 2 diabetes in Sweden. Diabetes Care 33: 293-297
12. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP,
Zeggini E, Huth C, Aulchenko YS, Thorleifsson G et al. (2010) Twelve
type 2 diabetes susceptibility loci identified through large-scale asso-
ciation analysis. Nat Genet 42: 579-589
13. Jin T, Liu L (2008) The Wnt signaling pathway effector TCF7L2 and
type 2 diabetes mellitus. Mol Endocrinol 22: 2383-2392
14. Pawlak J, Derlacz RA (2011) The mechanism of insulin resistance in
peripheral tissues Postepy Biochem 57: 200-206
15. Fadini GP, Avogaro A (2010) Potential manipulation of endothelial
progenitor cells in diabetes and its complications. Diabetes Obes Me-
tab 12: 570-583
16. Schleicher E, Friess U (2007) Oxidative stress, AGE, and atherosclero-
sis. Kidney Int Suppl S17-26
17. Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki
T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H
(2000) High glucose level and free fatty acid stimulate reactive oxygen
species production through protein kinase C-dependent activation of
NAD(P)H oxidase in cultured vascular cells. Diabetes 49: 1939-1945
18. Ho FM, Lin WW, Chen BC, Chao CM, Yang CR, Lin LY, Lai CC, Liu
SH, Liau CS (2006) High glucose-induced apoptosis in human vascu-
lar endothelial cells is mediated through NF-kappaB and c-Jun NH2-
-terminal kinase pathway and prevented by PI3K/Akt/eNOS path-
way. Cell Signal 18: 391-399
19. Sessa WC (2005) Regulation of endothelial derived nitric oxide in he-
alth and disease. Mem Inst Oswaldo Cruz Suppl 1: 15-18
20. Scalia R, Gong Y, Berzins B, Zhao LJ, Sharma K (2007) Hyperglycemia
is a major determinant of albumin permeability in diabetic microcircu-
lation: the role of mu-calpain. Diabetes 56: 1842-1849
21. Spinetti G, Kraenkel N, Emanueli C, Madeddu P (2008) Diabetes and
vessel wall remodelling: from mechanistic insights to regenerative the-
rapies. Cardiovasc Res 78: 265-273

Postępy Biochemii 59 (3) 2013
265
22. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T,
Witzenbichler B, Schatteman G, Isner JM (1997) Isolation of putative
progenitor endothelial cells for angiogenesis. Science 275: 964-967
23. Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Ke-
arne M, Magner M, Isner JM (1999) Bone marrow origin of endothelial
progenitor cells responsible for postnatal vasculogenesis in physiolo-
gical and pathological neovascularization. Circ Res 85: 221-228
24. Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, Ma-
gner M, Isner JM, Asahara T (1999) Ischemia- and cytokine-induced
mobilization of bone marrow-derived endothelial progenitor cells for
neovascularization. Nat Med 5: 434-438
25. Rafii S, Lyden D (2003) Therapeutic stem and progenitor cell trans-
plantation for organ vascularization and regeneration. Nat Med 9:
702-712
26. Richardson MR, Yoder MC (2011) Endothelial progenitor cells: quo
vadis? J Mol Cell Cardiol 50: 266-272
27. Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R,
Temm CJ, Prchal JT, Ingram DA (2007) Redefining endothelial proge-
nitor cells via clonal analysis and hematopoietic stem/progenitor cell
principals. Blood 109: 1801-1809
28. Liu ZJ, Velazquez OC (2008) Hyperoxia, endothelial progenitor cell
mobilization, and diabetic wound healing. Antioxid Redox Signal 10:
1869-1882
29. Kopp HG, Ramos CA, Rafii S (2006) Contribution of endothelial pro-
genitors and proangiogenic hematopoietic cells to vascularization of
tumor and ischemic tissue. Curr Opin Hematol 13: 175-181
30. Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimo-
vitz-Friedman A, Fuks Z, Kolesnick R (2003) Tumor response to radio-
therapy regulated by endothelial cell apoptosis. Science 300: 1155-1159
31. Urbich C, Dimmeler S (2004) Endothelial progenitor cells functional
characterization. Trends Cardiovasc Med 14: 318-322
32. Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher
AM, Dimmeler S (2005) Soluble factors released by endothelial proge-
nitor cells promote migration of endothelial cells and cardiac resident
progenitor cells. J Mol Cell Cardiol 39: 733-742
33. Di Santo S, Yang Z, Wyler von Ballmoos M, Voelzmann J, Diehm N,
Baumgartner I, Kalka C (2009) Novel cell-free strategy for therapeutic
angiogenesis: in vitro generated conditioned medium can replace pro-
genitor cell transplantation. PLoS One 4: e5643
34. Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi
AA, Finkel T (2003) Circulating endothelial progenitor cells, vascular
function, and cardiovascular risk. N Engl J Med 348: 593-600
35. Loomans CJ, van Haperen R, Duijs JM, Verseyden C, de Crom R, Le-
enen PJ, Drexhage HA, de Boer HC, de Koning EJ, Rabelink TJ, Staal
FJ, van Zonneveld AJ (2009) Differentiation of bone marrow-derived
endothelial progenitor cells is shifted into a proinflammatory pheno-
type by hyperglycemia. Mol Med 15: 152-159
36. Seeger FH, Haendeler J, Walter DH, Rochwalsky U, Reinhold J, Urbich
C, Rössig L, Corbaz A, Chvatchko Y, Zeiher AM, Dimmeler S (2005)
p38 mitogen-activated protein kinase downregulates endothelial pro-
genitor cells. Circulation 111: 1184-1191
37. Kuki S, Imanishi T, Kobayashi K, Matsuo Y, Obana M, Akasaka T
(2006) Hyperglycemia accelerated endothelial progenitor cell sene-
scence via the activation of p38 mitogen-activated protein kinase. Circ
J 70: 1076-1081
38. Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz
GR, Levine JP, Gurtner GC (2002) Human endothelial progenitor cells
from type II diabetics exhibit impaired proliferation, adhesion, and in-
corporation into vascular structures. Circulation 106: 2781-2786
39. Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C,
de Boer HC, Verhaar MC, Braam B, Rabelink TJ, van Zonneveld AJ
(2004) Endothelial progenitor cell dysfunction: a novel concept in the
pathogenesis of vascular complications of type 1 diabetes. Diabetes 53:
195-199
40. Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, Mene-
golo M, de Kreutzenberg SV, Tiengo A, Agostini C, Avogaro A (2005)
Circulating endothelial progenitor cells are reduced in peripheral va-
scular complications of type 2 diabetes mellitus. J Am Coll Cardiol 45:
1449-1457
41. Kusuyama, T, Omura T, Nishiya D, Enomoto S, Matsumoto R, Ta-
keuchi K, Yoshikawa J, Yoshiyama M (2006) Effects of treatment for
diabetes mellitus on circulating vascular progenitor cells. J Pharmacol
Sci 102: 96-102
42. Pistrosch F, Herbrig K, Oelschlaegel U, Richter S, Passauer J, Fischer S,
Gross P (2005) PPARgamma-agonist rosiglitazone increases number
and migratory activity of cultured endothelial progenitor cells. Athe-
rosclerosis 183: 163-167
43. Busik JV, Tikhonenko M, Bhatwadekar A, Opreanu M, Yakubova N,
Caballero S, Player D, Nakagawa T, Afzal A, Kielczewski J, Sochac-
ki A, Hasty S, Li Calzi S, Kim S, Duclas SK, Segal MS, Guberski DL,
Esselman WJ, Boulton ME, Grant MB (2009) Diabetic retinopathy is
associated with bone marrow neuropathy and a depressed peripheral
clock. J Exp Med 206: 2897-2906
44. Jeong JO, Kim MO, Kim H, Lee MY, Kim SW, Ii M, Lee JU, Lee J, Choi
YJ, Cho HJ, Lee N, Silver M, Wecker A, Kim DW, Yoon YS (2009) Dual
angiogenic and neurotrophic effects of bone marrow-derived endothe-
lial progenitor cells on diabetic neuropathy. Circulation 119: 699-708
45. Baum CL, Arpey CJ (2005) Normal cutaneous wound healing: clinical
correlation with cellular and molecular events. Dermatol Surg 31: 674-
686
46. Wu SC, Driver VR, Wrobel JS, Armstrong DG (2007) Foot ulcers in the
diabetic patient, prevention and treatment. Vasc Health Risk Manag
3: 65-76
47. National diabetes fact sheet: national estimates and general infor-
mation on diabetes and prediabetes in the United States, 2011., Cen-
ters for Disease Control and Prevention. Department of Health and
Human Services, Centers for Disease Control and Prevention, 2011.
Atlanta, GA: U.S.
48. Barcelos LS, Duplaa C, Kränkel N, Graiani G, Invernici G, Katare R,
Siragusa M, Meloni M, Campesi I, Monica M, Simm A, Campagnolo
P, Mangialardi G, Stevanato L, Alessandri G, Emanueli C, Madeddu P
(2009) Human CD133+ progenitor cells promote the healing of diabe-
tic ischemic ulcers by paracrine stimulation of angiogenesis and acti-
vation of Wnt signaling. Circ Res 104: 1095-1102
49. Keswani SG, Katz AB, Lim FY, Zoltick P, Radu A, Alaee D, Herlyn
M, Crombleholme TM (2004) Adenoviral mediated gene transfer of
PDGF-B enhances wound healing in type I and type II diabetic wo-
unds. Wound Repair Regen 12: 497-504
50. Grant MB, May WS, Caballero S, Brown GA, Guthrie SM, Mames RN,
Byrne BJ, Vaught T, Spoerri PE, Peck AB, Scott EW (2002) Adult hema-
topoietic stem cells provide functional hemangioblast activity during
retinal neovascularization. Nat Med 8: 607-612
51. Butler JM, Guthrie SM, Koc M, Afzal A, Caballero S, Brooks HL, Ma-
mes RN, Segal MS, Grant MB, Scott EW l (2005) SDF-1 is both neces-
sary and sufficient to promote proliferative retinopathy. J Clin Invest
115: 86-93
52. Asnaghi V, Lattanzio R, Mazzolari G, Pastore MR, Ramoni A, Ma-
estroni A, Ruggieri D, Luzi L, Brancato R, Zerbini G (2006) Increased
clonogenic potential of circulating endothelial progenitor cells in pa-
tients with type 1 diabetes and proliferative retinopathy. Diabetologia
49: 1109-1111
53. Fadini GP, Sartore S, Baesso I, Lenzi M, Agostini C, Tiengo A, Avogaro
A (2006) Endothelial progenitor cells and the diabetic paradox. Diabe-
tes Care 29: 714-716
54. Rehman J, Li J, Parvathaneni L, Karlsson G, Panchal VR, Temm CJ,
Mahenthiran J, March KL (2004) Exercise acutely increases circulating
endothelial progenitor cells and monocyte-/macrophage-derived an-
giogenic cells. J Am Coll Cardiol 43: 2314-2318
55. Hoetzer GL, Van Guilder GP, Irmiger HM, Keith RS, Stauffer BL, De-
Souza CA (2007) Aging, exercise, and endothelial progenitor cell clo-
nogenic and migratory capacity in men. J Appl Physiol 102: 847-852
56. Schlager O, Giurgea A, Schuhfried O, Seidinger D, Hammer A, Gröger
M, Fialka-Moser V, Gschwandtner M, Koppensteiner R, Steiner S
(2011) Exercise training increases endothelial progenitor cells and de-

266
www.postepybiochemii.pl
Type 2 Diabetes Mellitus impairs endothelial progenitor cells functions
Jerzy Kotlinowski, Józef Dulak, Alicja Józkowicz
Department of Medical Biotechnology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, 7 Gronostajowa St., 30-387
Krakow, Poland
e-mail: alicja.jozkowicz@uj.edu.pl
Key words: diabetes, endothelial progenitor cells, diabetic complications
absTraCT
Type 2 diabetes mellitus (T2DM) is a metabolic disease caused by insulin resistance that leads to changes in glucose metabolism. importan-
tly, both insulin resistance and hyperglycemia are present in T2DM patients as a hallmarks of metabolic syndrome. They negatively affect
functions of many cells, for example endothelial cells. Endothelial progenitor cells (EPC) is a population of mononuclear cells that expresses
endothelial and progenitor markers. EPC are also characterized by ability to form tubes on matrigel, outgrowth into mature endothelial cells,
produce proangiogenic factors or take part in the blood vessels formation. upon injury endothelial progenitor cells are mobilized from bone
marrow, home to injured site and take part in vessels formation. it was shown however, that functions of EPC in T2DM patients are impaired.
in this review we focused on the T2DM and its detrimental effects on EPC biology. taking also into account the beneficiary role of anti-diabet-
ic drugs. Decreased number and impaired functions of EPC in T2DM patients might lead to increased frequency of cardiovascular incidents
and development of micro- or macroangiopathies.
creases asymmetric dimethylarginine in peripheral arterial disease: A
randomized controlled trial. Atherosclerosis 217: 240-248
57. Sarto P, Balducci E, Balconi G, Fiordaliso F, Merlo L, Tuzzato G, Pap-
pagallo GL, Frigato N, Zanocco A, Forestieri C, Azzarello G, Mazzuc-
co A, Valenti MT, Alborino F, Noventa D, Vinante O, Pascotto P, Sarto-
re S, Dejana E, Latini R (2007) Effects of exercise training on endothelial
progenitor cells in patients with chronic heart failure. J Card Fail 13:
701-708
58. Reinhard H, Jacobsen PK, Lajer M, Pedersen N, Billestrup N, Man-
drup-Poulsen T, Parving HH, Rossing P (2010) Multifactorial treat-
ment increases endothelial progenitor cells in patients with type 2
diabetes. Diabetologia 53: 2129-2133
59. Nowak WN, Mika P, Nowobilski R, Kusinska K, Bukowska-Strakova
K, Nizankowski R, Józkowicz A, Szczeklik A, Dulak J (2012) Exercise
training in intermittent claudication: effects on antioxidant genes, in-
flammatory mediators and proangiogenic progenitor cells. Thromb
Haemost 108: 824-831
60. Humpert PM, Djuric Z, Zeuge U, Oikonomou D, Seregin Y, Laine K,
Eckstein V, Nawroth PP, Bierhaus A (2008) Insulin stimulates the clo-
nogenic potential of angiogenic endothelial progenitor cells by IGF-1
receptor-dependent signaling. Mol Med 14: 301-308
61. Fadini GP, de Kreutzenberg SV, Mariano V, Boscaro E, Bertolini F,
Mancuso P, Quarna J, Marescotti M, Agostini C, Tiengo A, Avogaro A
(2011) Optimized glycaemic control achieved with add-on basal insu-
lin therapy improves indexes of endothelial damage and regeneration
in type 2 diabetic patients with macroangiopathy: a randomized cross-
over trial comparing detemir versus glargine. Diabetes Obes Metab
13: 718-725
62. Koppen A, Kalkhoven E (2010) Brown vs white adipocytes: the PPAR-
gamma coregulator story. FEBS Lett 584: 3250-3259
63. Gensch C, Clever YP, Werner C, Hanhoun M, Böhm M, Laufs U (2007)
The PPAR-gamma agonist pioglitazone increases neoangiogenesis
and prevents apoptosis of endothelial progenitor cells. Atherosclero-
sis 192: 67-74
64. Werner C, Kamani CH, Gensch C, Böhm M, Laufs U (2007) The per-
oxisome proliferator-activated receptor-gamma agonist pioglitazone
increases number and function of endothelial progenitor cells in pa-
tients with coronary artery disease and normal glucose tolerance. Dia-
betes 56: 2609-2615
65. Wang CH, Ting MK, Verma S, Kuo LT, Yang NI, Hsieh IC, Wang SY,
Hung A, Cherng WJ (2006) Pioglitazone increases the numbers and
improves the functional capacity of endothelial progenitor cells in pa-
tients with diabetes mellitus. Am Heart J 152: 1051 e1-8
66. Werner C, Gensch C, Pöss J, Haendeler J, Böhm M, Laufs U (2011)
Pioglitazone activates aortic telomerase and prevents stress-induced
endothelial apoptosis. Atherosclerosis 216: 23-34
67. Imanishi T, Kobayashi K, Kuroi A, Ikejima H, Akasaka T (2008) Piogli-
tazone inhibits angiotensin II-induced senescence of endothelial pro-
genitor cell. Hypertens Res 31: 757-765
68. Nadra K, Quignodon L, Sardella C, Joye E, Mucciolo A, Chrast R, Des-
vergne B (2010) PPARgamma in placental angiogenesis. Endocrinol-
ogy 151: 4969-4981
Wyszukiwarka
Podobne podstrony:
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
cw med 5 id 122239 Nieznany
D20031152Lj id 130579 Nieznany
więcej podobnych podstron