1
Oprac. dr Ewa Więckowska-Bryłka,
KCh WNoŻ SGGW
Wykład VII. Koloidy. Metody spektroskopowe
UKŁADY DYSPERSYJNE
Mieszaniny otrzymane przez rozproszenie (zdyspergowanie) dowolnej
substancji (zw. fazą zdyspergowaną) w jakimkolwiek ośrodku (zw. ośrodkiem
rozpraszającym lub dyspersyjnym)
rozdrobnienie koloidowe
(układ koloidowy,
zol)
1nm ≤ 2r
< 100 nm
Układ lub stan koloidowy
Specyficzny stan rozdrobnienia substancji w danym rozpuszczalniku
Podstawą do zaklasyfikowania danego układu są:
właściwości fizyczne i chemiczne
1
Cechy charakterystyczne układów dyspersyjnych:
Doświadczenie lub
zjawisko
Roztwór
Układ koloidowy
Zawiesina
Obserwacja cząstek fazy
rozproszonej w
mikroskopie
niewidzialne
niewidzialne
widzialne
Obserwacja w
ultramikroskopie
niewidzialne
widzialne
widzialne
Ruchy Browna
nie występują
występują
nie występują
Efekt Tyndalla
nie występuje
występuje
występuje
Dializa
zachodzi
nie zachodzi
nie zachodzi
Dyfuzja
zachodzi
zachodzi
nie zachodzi
Przechodzenie fazy
rozdrobnionej przez
bibułę filtracyjną
przechodzi
przechodzi
nie przechodzi
Przechodzenie przez
ultrasączek
przechodzi
nie przechodzi
nie przechodzi
2
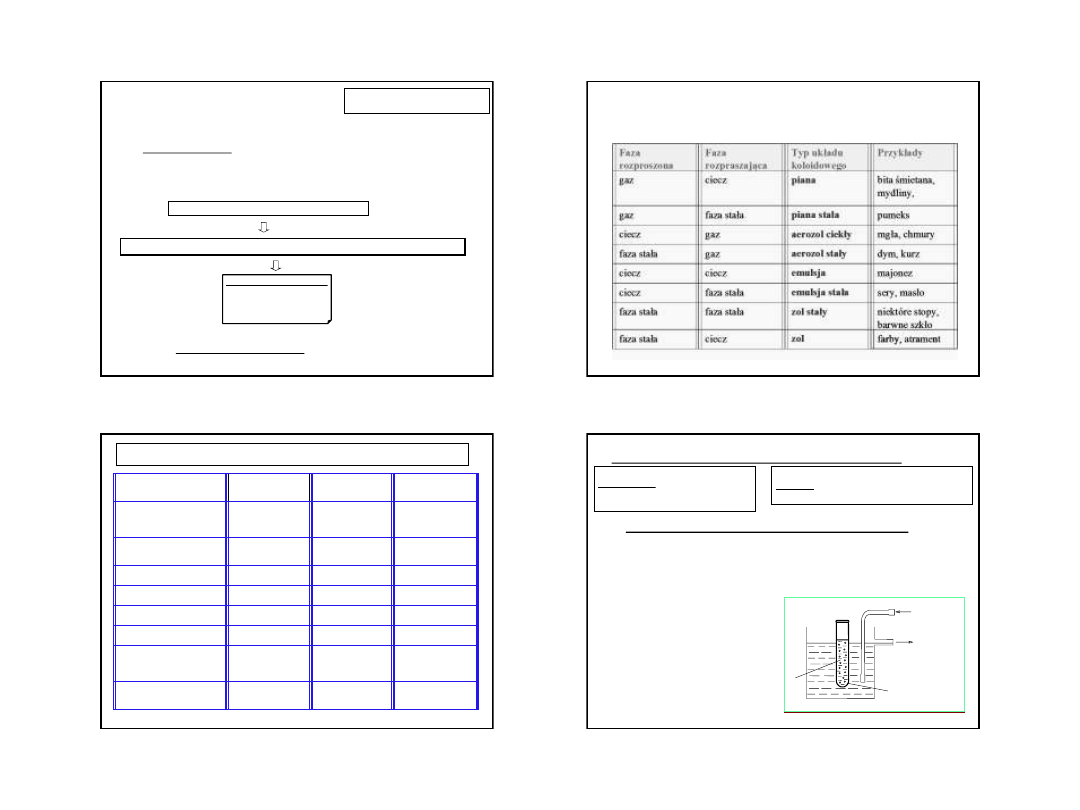
Typy układów koloidowych w zależności od stanu skupienia fazy
rozproszonej (faza zdyspergowana) i fazy rozpraszającej (ośrodek
rozpraszający)
3
Metody otrzymywania układów koloidowych:
Dyspersyjne -
Rozdrabnianie jednej fazy w celu
otrzymania
z
niej
cząstek
o
wymiarach koloidowych
Kondensacyjne, polimeryzacyjne-
Łączenie mniejszych cząstek fazy w
większe o rozmiarach koloidowych
Metody oczyszczania układów koloidowych:
zwykle od rozpuszczonych substancji krystalicznych i elektrolitów
cząstki koloidów przenikają przez sączki z bibuły filtracyjnej
są zatrzymywane przez naturalne (roślinne i zwierzęce) i sztuczne
(kolodium, celofan) błony półprzepuszczalne
dializa – wykorzystuje dyfuzję
innych składników przez błony
półprzepuszczalne. Jony lub
rozpuszczone małe cząsteczki
przechodzą przez błonę z zolu do
wody
d o p r o w a d z e n i e
w o d y
o d p r o w a d z e n i e
w o d y
b ł o n a d i a l i z u j ą c a
z o l
d o p r o w a d z e n i e
w o d y
o d p r o w a d z e n i e
w o d y
b ł o n a d i a l i z u j ą c a
z o l
4
2
Właściwości kinetyczne układów koloidowych
x
r
N
3
t
T
R
=
x
A
2
Δx – średnia wartość długości przesunięcia w czasie Δt na
podstawie dużej liczby pomiarów dla pojedynczej cząstki
koloidowej, r – promień cząstki koloidowej, η –lepkość
ośrodka, R – stała gazowa, T – temperatura, K, N
A
– liczba
Avogadra,
2
A
x)
(
η
π
3N
Δt
RT
r
Pomiar wielkości cząstek ważny w produkcji: artykułów spożywczych,
kosmetyków, farmaceutyków, papieru, cementu, farb i barwników, przeróbki
kopalin
Sedymentacja – opadanie cząstek koloidowych na dno naczynia pod wpływem
siły ciężkości; prędkość opadania cząstek koloidowych:
9
g
)
d
-
(d
r
2
=
v
o
2
d – gęstość ośrodka rozpraszającego, d
o
– gęstość
cząstki koloidowej, g – przyspieszenie ziemskie,
–
lepkość
Ruchy Browna – cząstki w bezustannym,
chaotycznym ruchu
5
•
rozpraszanie światła na cząstkach o wielkości porównywalnej z długością fali
światła. Dzięki rozpraszaniu światła w dzień niebo jest niebieskie, a o świcie i
zmierzchu czerwone. Dzięki rozpraszaniu widzimy chmury i dym.
Zjawisko Tyndalla -
strumień światła przechodzący przez układ koloidowy
ulega ugięciu i częściowo rozproszeniu na cząstkach koloidowych; z boku –
jasna opalizująca smuga w postaci stożka
6
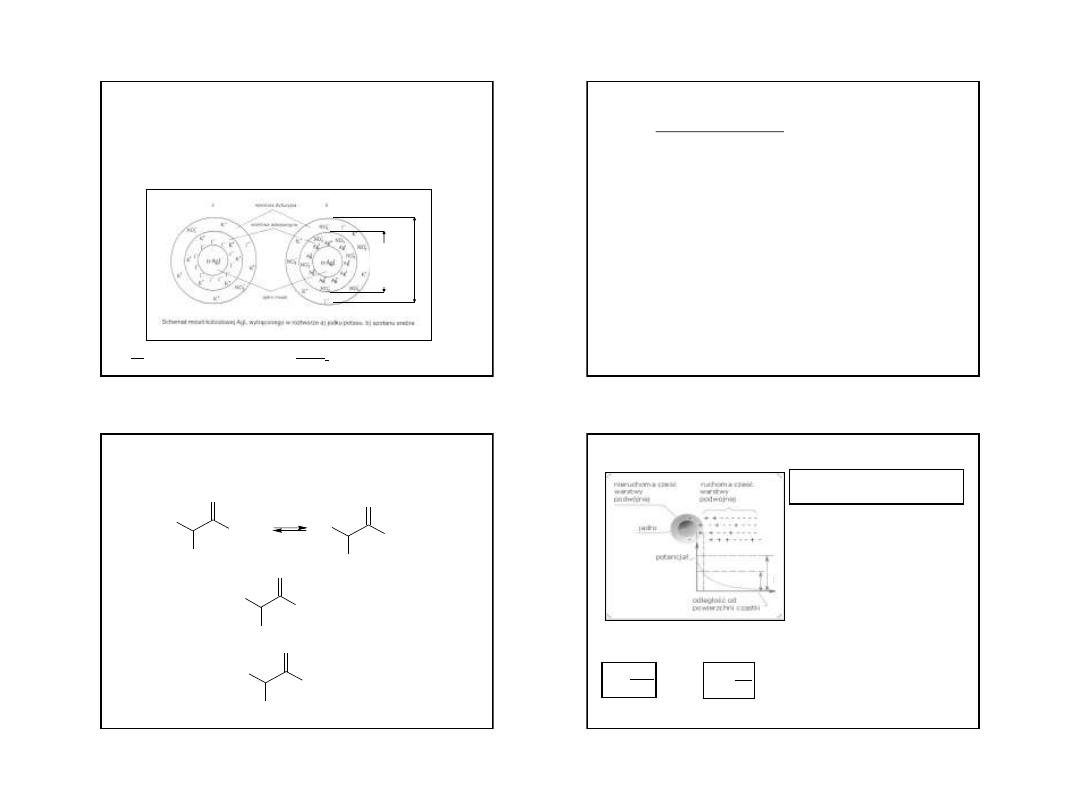
Właściwości elektrokinetyczne układów koloidowych:
Ze względu na zdolność oddziaływania cząstek koloidowych z cząsteczkami
ośrodka rozpraszającego:
Koloidy
Liofobowe
•
cząstki koloidowe trudno ulegają
solwatacji cząsteczkami fazy
rozpraszającej
•
m. in. hydrofobowe (H
2
O jako
rozpuszczalnik)
•
rozdrobnione złoto w wodzie
Liofilowe
•
cząstki koloidowe łatwo ulegają
solwatacji
•
m. in. hydrofilowe (H
2
O jako
rozpuszczalnik)
•
m. in. koloidy białkowe
7
8
Właściwości
Koloidy liofobowe
Koloidy liofilowe
Otrzymywanie
Metodami dyspersji lub
kondensacji
Można otrzymać przez
rozpuszczenie
Struktura cząstek
Przeważnie zespoły cząstek
Często makrocząsteczki
Stężenie fazy
rozproszonej
Na ogół nieznaczne
Może być duże
Ruchy Browna
Wyraźne
Często bardzo niewyraźne
Efekt Tyndalla
Wyraźny
Niewyraźny
Barwa układu
Często zabarwione
Najczęściej bezbarwne
Ładunek elektryczny
Cząstki zawsze naładowane Ładunek może nie występować
Lepkość
Zbliżona do fazy
rozpraszającej
Większa niż fazy rozpraszającej
Tworzenie piany
Nie tworzą piany
Łatwo tworzą pianę
Pęcznienie
Nie pęcznieją
Pęczniejąc zwiększają objętość
Tworzenie galaret
Nie tworzą
Łatwo tworzą
Wrażliwość na działanie
elektrolitu
Koagulacja pod wpływem
małych stężeń elektrolitu
Mało wrażliwe, pod wpływem
dużych stężeń następuje
wysalanie lub koacerwacja
Wrażliwość na działanie
środków dehydratujących
Nieznaczna i dopiero przy
dużych stężeniach
Przy dużych stężeniach znaczna
Charakter koagulacji
Nieodwracalna
Odwracalna
Napięcie powierzchniowe
Zbliżone do fazy
rozpraszającej
Mniejsze od fazy rozpraszającej
Właściwości koloidów liofobowych i liofilowych
3
Micela koloidowa – cząstki zwykle obdarzone ładunkiem elektrycznym
wskutek:
•
adsorpcji jonów elektrolitu z układu
•
procesu dysocjacji elektrolitycznej koloidu cząsteczkowego
•
procesu dysocjacji cząsteczek tworzących cząstkę koloidową
Pozostałość – ośrodek międzymicelarny
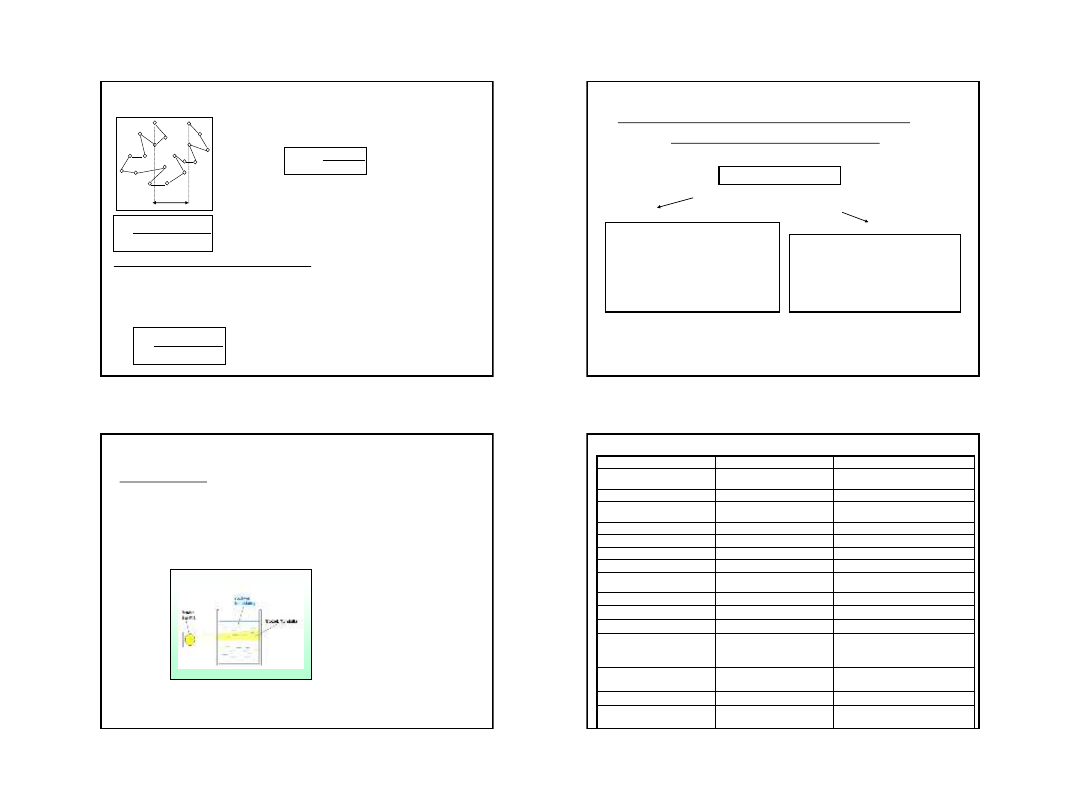
Micela koloidu liofobowego:
m
ic
el
a
gr
a
n
u
la
a) KI + AgNO
3
AgI + KNO
3
b) AgNO
3
+ KI AgI + KNO
3
9
Koloidy liofilowe – cząstki mogą być obdarzone ładunkiem, np. cząstki białek
aminokwasy
O
NH
2
R
OH
O
NH
3
+
R
O
-
jon
obojnaczy
– w środowisku H
+
:
O
NH
3
+
R
OH
jądro miceli otoczone
warstwą rozmytą
zawierającą jony OH
–
– w środowisku OH
–
O
NH
2
R
O
-
jądro miceli otoczone
warstwą rozmytą
zawierającą jony H
+
10
•
stan, w którym ładunek cząstek koloidowych sprowadzi się do zera
przez adsorpcję jonów przeciwnego znaku (zarówno w przypadku
koloidu liofobowego, jak i liofilowego)
•
liofobowy [mAgI]nI
–
(ładunek ujemny)
Punkt izoelektryczny koloidu
dodatek Ag
+
(np. AgNO
3
) zobojętnia I
–
, nadmiar Ag
+
–
przeładowanie cząstek i powstanie układu [mAgI]nAg
+
•
stan tuż przed „przeładowaniem” – nietrwały zw. punktem
izoelektrycznym
•
koloid liofobowy – ulega koagulacji, liofilowy – pozostaje zolem
•
w przypadku amfoterycznych koloidów np. białek – punkt
izoelektryczny występuje przy wartości pH zw. wartością pH punktu
izoelektrycznego
11
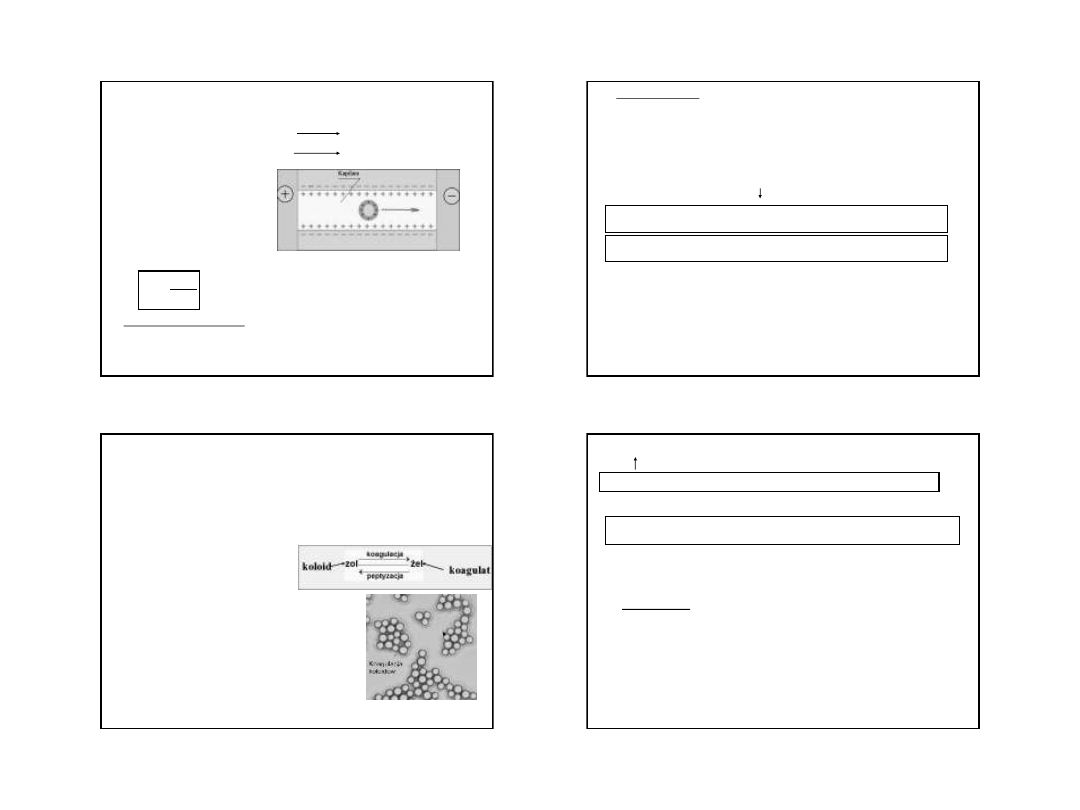
Potencjał elektrokinetyczny ξ
Na granicy warstwy adsorpcyjnej i
dyfuzyjnej różnica potencjałów zw.
potencjałem ξ (czyt. dzeta)
Elektroosmoza – przyłożenie pola
elektrycznego
powoduje
ruch
warstwy
dyfuzyjnej
jonów
i
cząsteczek rozpuszczalnika. Jest to
jednokierunkowy ruch ośrodka
dyspersyjnego
przez
błonę
półprzepuszczalną
Ruchliwość elektroosmotyczna czyli prędkość przesuwania się warstwy rozpuszczalnika
podczas elektroosmozy:
η
ξ
ε
E
=
v
ε
E
η
v
ξ
stąd:
E – natężenie pola elektrycznego, równe
ilorazowi U/l (napięcia przez odległość
między elektrodami podłączonymi do źródła
prądu), ε – stała dielektryczna ośrodka,
η – lepkość ośrodka.
E
12
4
Elektroforeza – wędrówka cząstek koloidowych pod wpływem pola
elektrycznego względem nieruchomego ośrodka rozpraszającego
Cząstki (–) wędrują do anody
anaforeza
Cząstki (+) wędrują do katody
kataforeza
Elektroforeza w
kapilarze
Ruchliwość elektroforetyczna
prędkość przesuwania się miceli koloidowych w
polu elektrycznym o natężeniu E
Zastosowania elektroforezy:
—
rozdzielanie i analiza białek, kwasów itp.; odpowiedni dobór
stężenia i pH ośrodka – składniki mieszaniny wędrują z różnymi
prędkościami i mogą być rozdzielone
η
ξ
ε
E
=
v
13
Trwałość układów koloidowych
spowodowana ładunkiem na powierzchni cząstki koloidowej
solwatacją (wytworzenie wokół cząstki koloidowej otoczki złożonej z cząsteczek
rozpuszczalnika), zwłaszcza w przypadku koloidów liofilowych
Zole – nietrwałe wskutek ruchów Browna i przechodzą w żele
Koagulacja – proces łączenia się mniejszych cząstek koloidu w większe, przejście zolu w
żel
Objawy koagulacji:
•
zmiana barwy,
•
zmętnienie lub wytrącenie osadu,
•
zmniejszenie
wartości
potencjału
elektrokinetycznego
•
zmniejszenie prędkości elektroforezy itp.
Koagulację przyspieszają:
•
podwyższenie temperatury,
•
bodźce mechaniczne (wstrząsanie układu),
•
przepływ prądu elektrycznego,
•
działanie światła,
•
dodatek nieelektrolitów,
•
dodatek elektrolitu (zwłaszcza w przypadku
koloidów hydrofobowych)
14
Wpływ elektrolitu:
wzrost mocy jonowej roztworu,
szybki zanik warstwy dyfuzyjnej
większa możliwość zbliżenia się dwóch miceli.
całkowity zanik warstwy dyfuzyjnej oznacza wartość potencjału
elektrokinetycznego równą zeru (punkt izoelektryczny).
Próg koagulacji – minimalna liczba milimoli danego elektrolitu niezbędna do
skoagulowania 1 dm
3
układu koloidowego
zależy od rodzaju dodanego elektrolitu, a zwłaszcza od wartościowości
dodawanych jonów
Im wyższa jest wartościowość jonu o znaku przeciwnym do ładunku cząstek
koloidowych, tym niższy jest próg koagulacji
Szeregi liotropowe Hofmeistera:
•
dla jednowartościowych kationów litowców zdolność do wywołania
koagulacji maleje w szeregu: Cs
+
< Rb
+
< K
+
< Na
+
< Li
+
.
•
podobnie w przypadku anionów:
2
4
SO
< anion kwasu cytrynowego < CH
3
COO
–
< Br
–
<
3
NO
I
–
< Br
–
< Cl
–
.
oraz:
15
Wysalanie
– koagulacja koloidów liofilowych pod wpływem
dużych ilości elektrolitu
elektrolit usuwa cząsteczki rozpuszczalnika stanowiące otoczkę solwatacyjną
Zniszczenie otoczki solwatacyjnej:
pod wpływem innych środków dehydratujących, np. alkoholu, acetonu
w przypadku białek – denaturacja (proces nieodwracalny).
Koacerwacja – proces powstawania agregatów w postaci kropelek cieczy wskutek dodania
elektrolitu do koloidu liofilowego
Koagulacja wzajemna – zobojętnienie ładunku cząstek koloidu liofobowego po dodaniu
innego koloidu liofobowego o cząstkach przeciwnie naładowanych
Peptyzacja
jeżeli do świeżo skoagulowanego koloidu (żelu) dodać odpowiedniego elektrolitu
ulegającego silnej adsorpcji na powierzchni cząstek, to żel może ponownie przejść w zol
np. podczas rozcieńczania wodą destylowaną białka kurzego skoagulowanego pod
wpływem Na
2
SO
4
.
proces przeciwny do koagulacji czyli przejścia żelu w zol, a więc ponownego
rozdrobnienia (koloidy odwracalne i koagulacja odwracalna)
Dodatek soli metali ciężkich, np. Cu
2+
, Pb
2+
, czy kwasu nieorganicznego (np.
HCl) do białka kurzego lub jego koagulacja pod wpływem temperatury
powoduje otrzymanie koloidu nieodwracalnego.
16

5
Żele
układy koloidowe, w których ośrodkiem rozpraszającym jest
ciecz, a fazą rozproszoną koloid liofilowy
układ w pewnym sensie sztywny i nie wykazuje płynności.
stan zbliżony do stanu stałego (np.
roztwory żelatyny, krochmalu i mydła lub
naturalnego
kauczuku
w
benzenie,
przechodzą samorzutnie ze stanu ciekłego
do zbliżonego do stałego)
otrzymuje się je przez sporządzenie układu
koloidowego na gorąco, a następnie
schłodzenie (np. żelatyna do otrzymywania
galaret, galaretek owocowych, dżemów)
cząsteczki rozpuszczalnika w żelu wypełniają luźną sieć
przestrzenną złożoną z cząstek koloidu liofilowego
Pęcznienie żeli – wysuszone żele mają zdolność wchłaniania cieczy
(rozpuszczalnika)
17
mieszaniny złożone z przynajmniej dwóch nie mieszających się ze sobą
cieczy, z których jedna jest rozproszona, w postaci drobnych kropelek, w
drugiej stanowiącej ośrodek rozpraszający
W celu utrwalenia emulsji dodaje się tzw. emulgatorów:
Emulsje
na ogół układy nietrwałe (krople cieczy
dążą do łączenia się ze sobą, gdyż duże
mają mniejszą energię powierzchniową)
każda z tych cieczy, zależnie od
warunków, może być zarówno
ośrodkiem rozpraszającym jak i
składnikiem rozproszonym, np.
emulsja wody w oleju lub odwrotnie –
emulsja oleju w wodzie
emulgatorami mogą być np. elektrolity, których jony adsorbują się na
powierzchni kropelek cieczy powodując ich jednoimienne naładowanie, co z
kolei nie sprzyja zlewaniu się kropelek cieczy w krople
w przemyśle, w celu utrwalenia emulsji przeprowadza się homogenizację, np.
mleka, kremów, margaryny (krople tłuszczu są rozbijane na tak małe fragmenty,
że oddzielają się od mleka znacznie trudniej).
Emulsje spotykane w życiu codziennym to np. mleko, mleczne soki roślin, tłuszcze
zwierzęce, masło, majonez (w majonezie emulgatorem jest albumina jaj,
utrwalająca emulsję oleju w wodzie).
18
•
układy, w którym fazą rozproszoną jest gaz, a
rozpraszającą – ciecz
•
można je otrzymać wstrząsając ciecz z
nierozpuszczalnym w niej gazem
•
wymiary zawieszonych w cieczy pęcherzyków
gazów mogą znacznie przewyższać rozmiary
cząstek koloidowych
•
mają wiele cech zolów koloidowych
•
można je utrwalać przez dodatek substancji
powierzchniowo czynnych, obniżających napięcie
powierzchniowe cieczy (mydło, saponiny, niektóre
·
białka) tzw. substancji pianotwórczych
•
tworzenie piany korzystne w procesie prania ręcznego,
ale np. w procesie barwienia tkanin, dodaje się
specjalnych środków niszczących pianę.
Piany
19
Analiza instrumentalna
metody spektrofotometryczne
metody elektrochemiczne
metody radiometryczne
metody chromatograficzne
Metody spektroskopowe – zespół metod w chemii fizycznej i fizyce zajmujących się
badaniem przejawów oddziaływania między promieniowaniem elektromagnetycznym a
materią (czyli zbiorem atomów, cząsteczek, itd.) oraz badaniem na tej podstawie
budowy i właściwości atomów, cząsteczek i jąder atomowych.
Klasyfikacja metod spektroskopowych ze względu na charakter oddziaływania
promieniowania elektromagnetycznego z materią:
spektroskopię absorpcyjną
emisyjną
rozpraszania
Klasyfikacja metod spektroskopowych ze względu na rodzaj badanego układu (cząstek
materii):
spektroskopia atomowa
spektroskopia molekularna lub cząsteczkowa
spektroskopia jądrowa
spektroskopia kryształów
20
6
Klasyfikacja metod spektroskopowych ze względu na rodzaj wzbudzeń
(przejścia między poziomami elektronowymi, oscylacyjnymi, rotacyjnymi,
jądrowymi) i formę energii molekuł:
spektroskopia elektronowa
spektroskopia oscylacyjna
spektroskopia rotacyjna
spektroskopia elektronowego rezonansu paramagnetycznego (EPR)
spektroskopia jądrowego rezonansu magnetycznego (NMR)
drganie pola elektrycznego, któremu towarzyszy drganie pola magnetycznego
długość fali
(odcinek drogi promieniowania, na którym mieści się jeden okres
drgania pola czyli jedno drganie), [cm],
częstość drgań pola na sekundę
, [s
–1
],
Promieniowanie elektromagnetyczne:
liczba falowa
czyli częstość drgań pola na centymetr, [cm
–1
],
c
c – prędkość promieniowania w próżni,
3
.
10
8
m/s
1
21
Natura promieniowania elektromagnetycznego:
wiązka promieniowania jest zbiorem porcji energii (natura
korpuskularna), czyli kwantów energii, biegnących w kierunku
rozchodzenia się promieniowania
wielkość
pojedynczego kwantu, zwanego fotonem, określa
zależność Plancka:
c
1
h
h
lub
c
h
gdzie: h – stała Plancka, 6,63 · 10
–34
J·s.
kwanty energii (czyli fotony) są tym większe, im większa jest częstość
drgań fali elektromagnetycznej, czyli im mniejsza jest jej długość
zależność Plancka wiąże charakter falowy i korpuskularny
promieniowania.
22
Widmo promieniowania elektromagnetycznego:
zbiór fal elektromagnetycznych o różnej długości
promieniowanie widzialne przez oko ludzkie (światło) obejmuje niewielki
zakres promieniowania elektromagnetycznego, którego długość fal wynosi
od około 400 nm (fiolet) do 760 nm (czerwień).
10
3 .10
11
3 .10
12
3 .10
3 .10
13
3 .10
14
3 .10
15
3 .1016
3 .10
17
18
3 .10
1
cm
[
]
10
fale
radiow e
mikrofale
prom .
w idz.
nadfiolet
prom .
rentgenow skie
10
8
7
10
10
6
10
5
10
4
10
3
2
10
1
10
-1
10
-2
10
-3
10
-4
10
-5
10
-6
10
-7
-8
10
prom .
23
2 . 10
-
22
2 . 10
-
20
2 . 10
-
19
2 . 10
-
21
2 . 10
-
15
2 . 10
-
18
2 . 10
-
17
2 . 10
-
16
2 . 10
-
1.2 . 10
-2
1.2 . 10-1
1.2
1.2 . 10
1
1.2 . 10
2
1.2 . 10
3
4
1.2 . 10
1.2 . 10
1.2 . 10
1.2 . 10
1.2 . 10
1.2 . 10
1.2 . 10
1.2 . 10
1.2 . 10
1.2 . 10
5
1.2 . 10
6
podczerw ień
.
J
]
foton
-1
ajnsztajn
[kJ . m ol -1]
[H z]
_
-1]
[cm
23
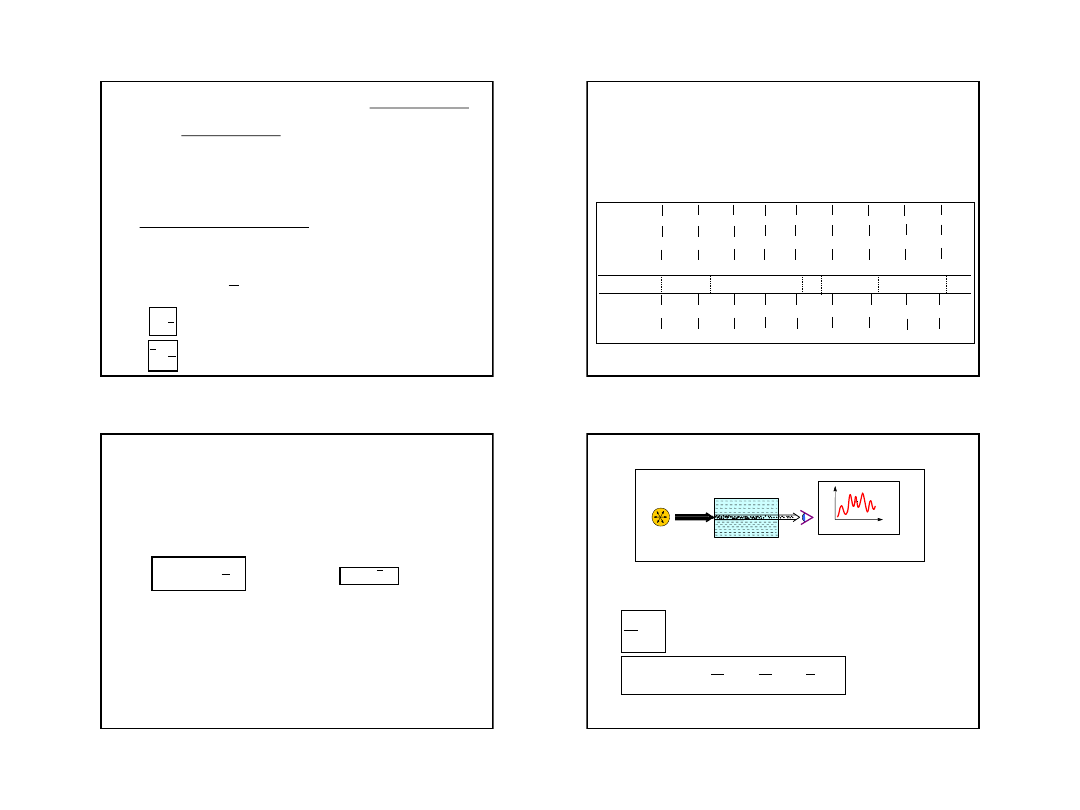
Absorpcja promieniowania przechodzącego przez roztwór:
I
0
– natężenie promieniowania przed przejściem przez roztwór,
I – natężenie promieniowania po przejściu przez roztwór.
T
I
I
0
T – transmitancja (przepuszczalność), np. 70 %
promieniowania przechodzi przez próbkę T = 0,70.
A
T
1
log
I
I
log
I
I
log
T
log
0
0
A – absorbancja, wartość mierzona absorbcji
I
o
I
d e t e k t o r
r e j e s t r a t o r
ź r ó d ł o
p r o m i e n i o w a n i a
A
24
7
Widmo absorpcyjne: krzywa absorpcji, poszczególne składowe noszą nazwę pasm
Interpretacja widma:
pozwala na identyfikację jakościową
(występowanie
charakterystycznych
maksimów
odpowiadających
poszczególnym atomom, cząsteczkom lub
ugrupowaniom atomów w cząsteczkach)
pozwala na analizę ilościową próbki
ustalenie struktury związków
badanie mechanizmów i kinetyki reakcji
A
Do zastosowań analitycznych
należy wybrać tzw. analityczną
długość fali
max.
Wyznaczamy ją na podstawie
zależności
A = f (
)
max
odpowiada największa
absorbancja (a jednocześnie
najmniejsza T)
A
[nm]
c
1
c
2
c
3
<
<
c
2
c
3
c
1
max
25
Absorbancja A (wielkość
bezwymiarowa) jest
proporcjonalna do stężenia
molowego roztworu c i grubości
warstwy absorbującej l (w cm).
A
b
s
o
rb
a
n
c
ja
Stężenie wzorca [mg/L]
0.1
0
0.5
0.1
1.0
1.5
2.0
0.5
1.0
1.5
Liniowa zależność absorbancji
od stężenia wzorca
Warunki oznaczenia:
technika płomieniowa
długość fali - 285.2 nm
szczelina - 0.7 mm
c
l
ε
I
I
log
A
0
•
rozpuszczalnik
nie
absorbuje
promieniowania,
•
brak jakichkolwiek oddziaływań między
cząsteczkami substancji absorbującej
czy też między cząsteczkami tej
substancji i rozpuszczalnika
– współczynnik proporcjonalności,
tzw. molowy współczynnik absorbancji
(molowy współczynnik pochłaniania),
[dm
3
.
mol
–1.
cm
–1
].
Prawo Lamberta-Beera jest
spełnione gdy:
Prawo Lamberta-Beera:
Wykres
wzorcowy
-
po
zmierzeniu wartości A dla
roztworu o nieznanym c
x
,
wyznacza się jego stężenie z
wykresu
26
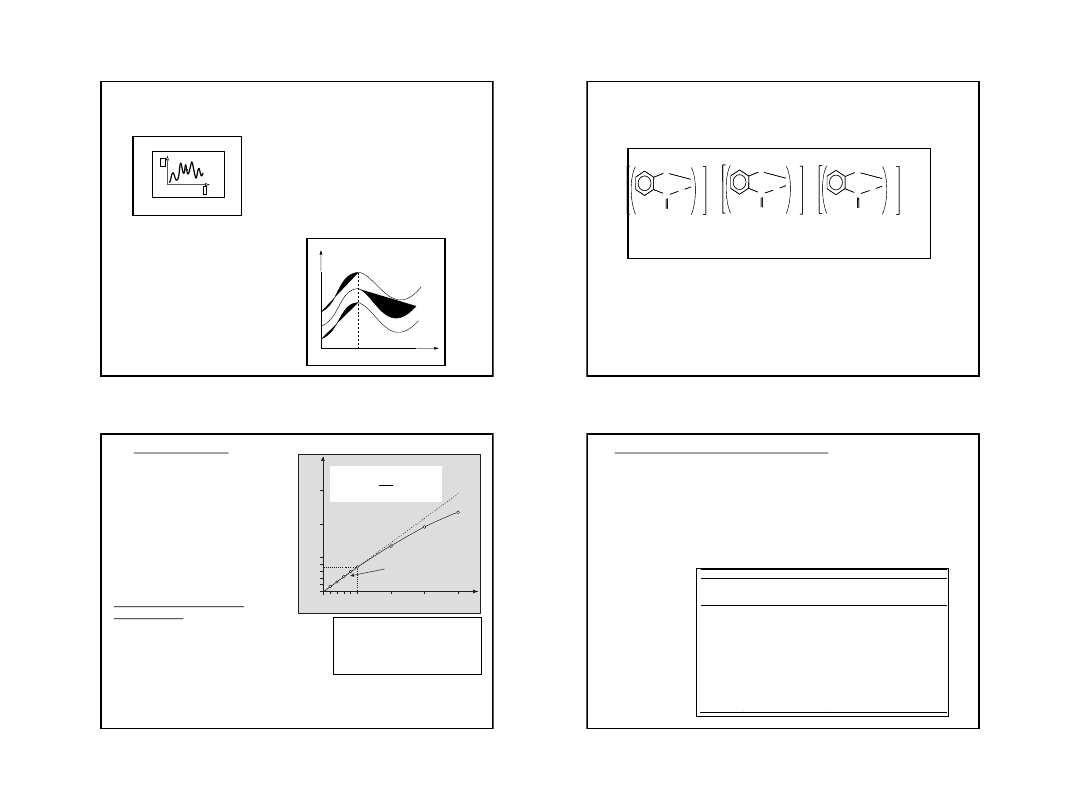
Oznaczanie stężenia jonów żelaza (III) metodą kolorymetryczną
przeprowadzenie jonów żelaza (III) w związek barwny za pomocą kwasu
salicylowego
kationy Fe
3+
tworzą z kwasem salicylowym (w zależności od pH roztworu)
trzy rodzaje jonów:
3
O
C
O
O
F e
3 _
t r i s a l i c y l a n
ż e l a z a ( I I I )
m o n o s a l i c y l a n
ż e l a z a ( I I I )
+
O
C
O
O
F e
d i s a l i c y l a n
ż e l a z a ( I I I )
2
O
C
O
O
F e
_
3
O
C
O
O
F e
3 _
t r i s a l i c y l a n
ż e l a z a ( I I I )
m o n o s a l i c y l a n
ż e l a z a ( I I I )
+
O
C
O
O
F e
d i s a l i c y l a n
ż e l a z a ( I I I )
2
O
C
O
O
F e
_
w środowisku kwaśnym przy pH < 2 w roztworze badanym występuje jedynie
monosalicylan żelaza (III) o barwie fioletowej
dodając roztworu kwasu salicylowego do roztworów o różnej zawartości jonów Fe (III)
otrzymuje się roztwory wzorcowe o różnym stężeniu związku kompleksowego (o różnej
intensywności zabarwienia), a więc o różnej wartości A
po określeniu
max
i ustawieniu jej na przyrządzie, wykonuje się pomiary A dla serii
roztworów wzorcowych
sporządza się wykres zależności A = f(c)
umożliwia on określenie ilości kationów Fe
3+
w analizowanym roztworze po zmierzeniu
jego absorbancji.
27
Spektroskopia w nadfiolecie i w zakresie widzialnym:
substancja pochłania ze światła białego (z zakresu widzialnego promieniowania
elektromagnetycznego) promieniowanie o określonej długości fali, czyli o
określonej barwie,
o barwie dostrzeganej okiem, decyduje mieszanina przepuszczonych i odbitych
składników barwnych światła,
barwę obserwowaną nazywamy dopełniającą do barwy absorbowanej (w
połączeniu z barwą promieniowania zaabsorbowanego tworzy ona światło białe),
selektywna absorpcja promieniowania – barwa przedmiotu (roztworu), jest jego
cechą fizyczną związaną ściśle z absorpcją promieniowania o określonym zakresie
długości fal
Absorpcja
promieniowania
widzialnego i barwy
dopełniające
Promieniowanie absorbowane
Długość
fali, nm
Barwa
promieniowania
pochłanianego
Barwa dopełniająca –
obserwowana
400 – 440
440 – 470
470 – 480
480 – 490
490 – 495
495 – 560
560 – 570
570 – 575
575 – 590
590 – 600
600 – 620
Fiolet
Błękit indygo
Błękitna
Niebieska
Niebieskozielona
Zielona
Zielonożółta
Żółta
Żółtopomarańczowa
Pomarańczowa
Pomarańczowoczerwona
Żółta
Żółta
Żółtopomarańczowa
Pomarańczowa
Czerwona
Czerwonopurpurowa i fioletowa
Purpurowofioletowa
Fioletowa i indygo
Błękitna i niebieska
Niebieska
Niebieskozielona
28
8
29
Spektrum światła białego
Większość związków kompleksowych metali bloku d jest barwna
pasma absorpcji jonów tych pierwiastków przypisuje się przejściom elektronowym w
orbitalach d, zwanych przejściami typu d
d
•
W wolnym jonie elektrony d znajdują się w polu o symetrii kulistej.
•
W kompleksie pod wpływem ligandów tworzą związki o symetrii
przeważnie tetraedrycznej lub oktaedrycznej.
o konfiguracji elektronowej [Ar]
4s
0
3d
5
jest bezbarwny, nie
obserwuje się głównego pasma
absorpcji w zakresie widzialnym.
Z
pięciu
elektronów
trzy
obsadzają pojedynczo orbitale
poziomu t
2g
, a dwa – orbitale
poziomu e
g
i nie ma możliwości
przeniesienia
elektronu
z
orbitalu t
2g
do orbitalu e
g
(brak
pustych orbitali e
g
, a stanem
podstawowym
jest
stan
o
maksymalnej
liczbie
niesparowanych
elektronów).
Jest
to
spowodowane
obecnością stosunkowo słabego
pola ligandów – cząsteczek wody
(słabe rozszczepienie poziomu d,
niezbyt duża różnica energii
między poziomami t
2g
i e
g
) i
powstaniem
kompleksu
wysokospinowego.
3
6
2
)
O
H
(
Fe
Jon
Fe
3+
t
2g
e
g
orbitale d
Fe(H
2
O)
6
3+
30
Jony CN
–
jako ligandy
silnego pola, powodują
duże
rozszczepienie
poziomu d i w stanie
podstawowym powstaje
kompleks niskospinowy
(pięć
elektronów
obsadza poziom t
2g
o
niższej energii). Wskutek
absorpcji
promieniowania
następuje
przeskok
elektronu z poziomu t
2g
do e
g
i pojawia się barwa
obserwowana zielona.
3
6
)
CN
(
Fe
Inaczej wygląda sytuacja w jonie
w roztworze wodnym
]
)
CN
(
Fe
[
K
6
3
Fe
3+
e
g
t
2g
orbitale d
Fe(CN)
6
3
_
31
Analogiczna sytuacja w monosalicylanie żelaza (III):
•
różnica energii między poziomami duża (wszystkie e na niższym poziomie)
•
po pochłonięciu światła możliwy przeskok e z orbitalu o niższej energii na
orbital o energii wyższej
•
dopełniająca barwa obserwowana – fioletowa, bo pochłaniana
= 530 nm
(barwa zółtozielona).
Metoda spektroskopowa wykorzystująca zjawisko pochłaniania czyli absorpcji
światła przez roztwory do ilościowego oznaczania substancji barwnych lub
barwiących się w wyniku reakcji oznaczanego składnika z odpowiednim
odczynnikiem, nosi nazwę analizy kolorymetrycznej (lub po prostu:
kolorymetrii).
Do pomiarów stosuje się metody wizualne lub przyrządy: kolorymetry w
zakresie widzialnym promieniowania elektromagnetycznego, spektrometry i
spektrofotometry pracujące w zakresie nadfioletu i widzialnym, wyposażone
w układy detekcyjne i urządzenia umożliwiające bezpośredni odczyt wartości
mierzonej absorpcji.
32
Wyszukiwarka
Podobne podstrony:
Pr. KSPS BZ 2014-2015 (1), bezpieczeństwo wewnętrzne materiały, Program KSPS i materia+éy do zaj¦Ö¦ç
materia y?ment dobre
Materia�y do wyk�ad�w z ha�asu
ch fiz ogniwa
ch.fiz.12, Chemia fizyczna
zobowi b9zania 2c+cz ea 9c e6+og f3lna+ +materia b3y+z+wyk b3adu BFIB2LADIBZNOPKLSRY7HBVHGTQ6WB26NNH
materia éy do ¦çw 3
pracownia specjaliz sp ch fiz zmf 2st, Praca magisterska, mikroelektrody
wyk 10 wysilek fiz
badania identyfikujące E.coli, materiały farmacja, Materiały 4 rok, epidemiologia, Higiena i epidemi
oznaczanie bakterii psychro i mezofilnych, materiały farmacja, Materiały 4 rok, epidemiologia, Higie
Ch ż Wykl1 2 2013
materialki6i9 7 8, Elektrotechnika AGH, Semestr III zimowy 2013-2014, semestr III, semestr III, Inży
HIGIENA PRACY CZYNNIKI CHEMICZNE, materiały farmacja, Materiały 4 rok, epidemiologia, Higiena i epid
Definicje- NOTATKI, AWF Katowice(materiały studenckie), IVrok; VIII semestr, PEDAGOGIKA SPECJALNA, D
więcej podobnych podstron