
Konspekt wykładu „Chemia Organiczna” dla studentów Wydziału Inżynierii Chemicznej i Procesowej
WĘGLOWODORY AROMATYCZNE
5 godz.
1.
Nomenklatura
2.
Właściwości fizyczne homologów benzenu
2.1.
Gęstość i rozpuszczalność w wodzie - lżejsze od wody i bardzo trudno w niej
rozpuszczalne.
2.2.
Toksyczność - bardzo toksyczne, wdychanie par jest bardzo niebezpieczne.
2.3.
Palność - pary benzenu są ciężkie i bardzo łatwo palne, stanowią poważną groźbę
pożaru, palą się świecącym, silnie kopcącym płomieniem.
2.4.
Temperatura wrzenia a masa cząsteczkowa - temperatura wrzenia alkilobenzenów
wzrasta wraz ze wzrostem masy cząsteczkowej, ale prawie wcale nie zależy od
położenia podstawników w pierścieniu.
2.5.
Temperatura topnienia a symetria podstawienia pierścienia - pochodne o budowie
symetrycznej topią się zazwyczaj w wyższej temperaturze niż ich niesymetryczne
izomery.
3.
Przemysłowe metody otrzymywania – na przykładzie suchej destylacji węgla
kamiennego.
1
Sucha destylacja węgla kamiennego
– ogrzewanie w 1000-1300
°
C bez dostępu powietrza
gaz świetlny:
H
2
(52% obj.), CH
4
(32% obj.),
CO, CO
2
, N
2
, CH
3
-CH
3
smoła węglowa:
węglowodory aromatyczne,
aminy, fenole
woda
amoniakalna
ekstrakcja ługiem
węglowodory aromatyczne, aminy
fenole
ekstrakcja kwasem mineralnym
węglowodory aromatyczne, m. in.: benzen, toluen,
ksyleny, naftalen i jego pochodne alkilowe
aminy
Konspekt wykładu „Chemia Organiczna” dla studentów Wydziału Inżynierii Chemicznej i Procesowej
4.
Struktury Kekulego a właściwości chemiczne odróżniające benzen od alkenów
4.1.
Reakcja z wodnym roztworem KMnO
4
4.2.
Reakcja z bromem w niepolarnym rozpuszczalniku organicznym
4.3.
Reakcja z bromem w obecności FeBr
3
5.
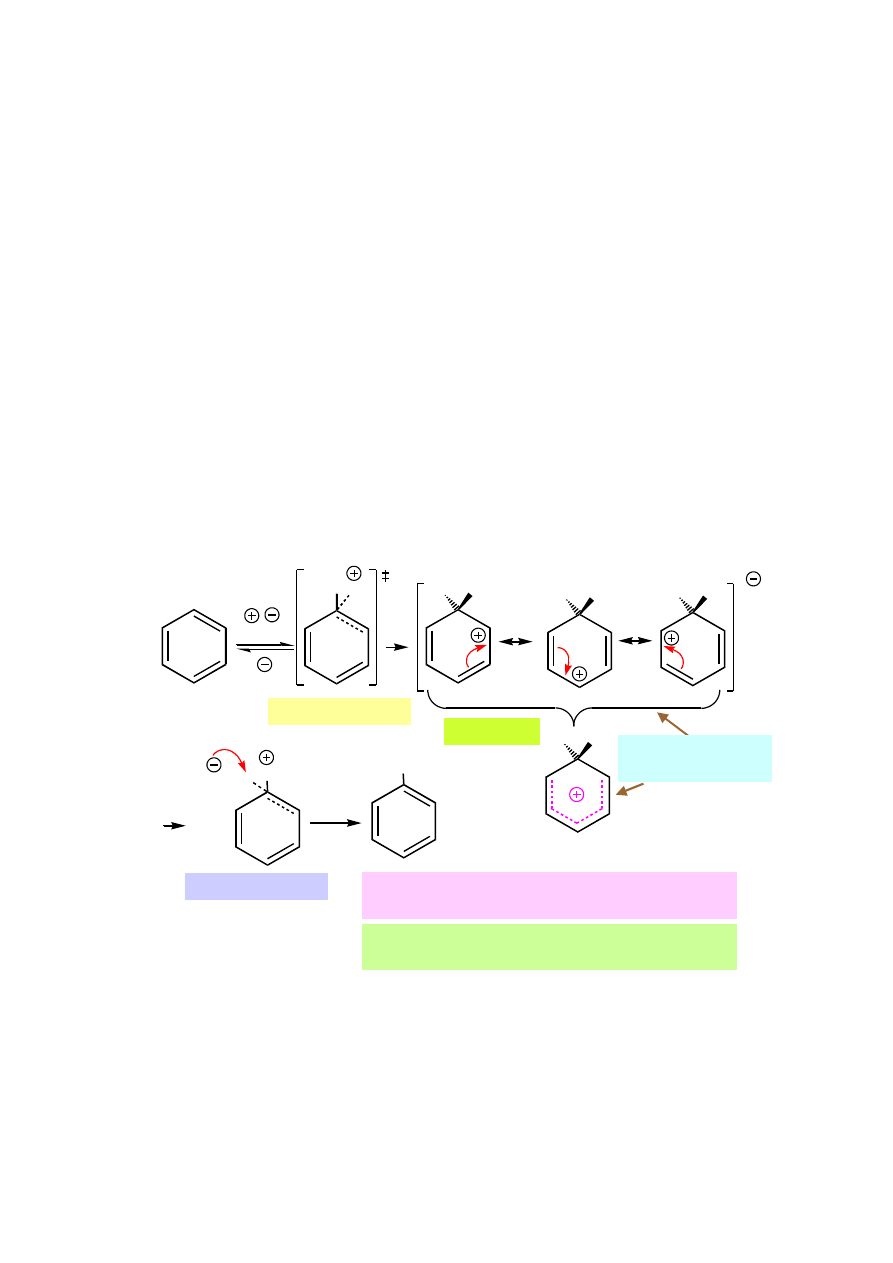
Mezomeryczna budowa cząsteczki benzenu
Rzeczywisty rozkład elektronów
π
przedstawiają dwie równoważne struktury
mezomeryczne; żadna ze struktur samodzielnie nie odzwierciedla
rzeczywistości.
Swobodny przepływ elektronów
π
sprawia, że są one zdelokalizowane, tzn. ich
dystrybucja jest identyczna w obrębie każdego wiązania między atomami
węgla.
6.
Reakcje benzenu i jego pochodnych: chlorowanie, bromowanie, nitrowanie,
sulfonowanie, alkilowanie Friedla-Craftsa, alkilowanie przy użyciu alkoholi lub
alkenów, acylowanie Friedla-Craftsa, formylowanie Gattermana-Kocha
Wszystkie te reakcje przebiegają według jednego mechanizmu substytucji
elektrofilowej (S
E
) i polegają na podstawieniu atomu wodoru z pierścienia
benzenowego innym atomem lub grupą atomów.
Reakcje przebiegają zwykle w temperaturze 0-50 °C, ale warunki ich
prowadzenia mogą być łagodniejsze lub ostrzejsze ze względu na obecność
innych podstawników już obecnych w pierścieniu.
7.
Mechanizm reakcji substytucji elektrofilowej (S
E
)
7.1.
Mechanizm chlorowania i bromowania
7.2.
Mechanizm nitrowania:
Reakcja benzenu ze stężonym kwasem azotowym jest bardzo niebezpieczna,
ponieważ tlenki azotu pochodzące z rozkładu kwasu azotowego mogą
spowodować eksplozję.
2
E
H
X
−
HX
E
H
E
H
E
H
E
H
E
H
E
E
X
−
X
orto
para
orto
X
kompleks
σ
stan przejściowy 2
delokalizacja
ładunku dodatniego
E
+
- elektrofil; cząsteczka obdarzona całkowitym
lub częściowym ładunkiem dodatnim
stan przejściowy 1
X
−
- nukleofil; cząsteczka obdarzona całkowitym
lub częściowym ładunkiem ujemnym

Konspekt wykładu „Chemia Organiczna” dla studentów Wydziału Inżynierii Chemicznej i Procesowej
Do nitrowania benzenu stosuje się mieszaninę stężonych kwasów: azotowego i
siarkowego (tzw. mieszaninę nitrującą). Dodatek stężonego kwasu siarkowego
sprawia, że reakcja zachodzi szybciej i w niższej temperaturze.
Do nitrowania mało reaktywnych substratów (np. nitrobenzenu, benzoesanu
metylu, benzaldehydu lub bromobenzenu) stosuje się 100% HNO
3
(d=1.5 g/cm
3
) (tzw. bezwodny HNO
3
) i stężony H
2
SO
4
.
Do nitrowania średnio reaktywnych substratów (np. benzenu, naftalenu lub
toluenu) stosuje się 65% HNO
3
(d=1.4 g/cm
3
) (stężony HNO
3
, tzw. azeotrop) i
stężony H
2
SO
4
.
Do nitrowania reaktywnych substratów (np. 1,2-dimetoksybenzenu lub fenolu)
stosuje się 40% HNO
3
.
7.3.
Mechanizm sulfonowania
Najczęściej stosuje się stężony (>95%) kwas siarkowy lub dymiący kwas
siarkowy (tzw. oleum) czyli roztwór SO
3
w 100% kwasie siarkowym.
Sulfonowanie jest reakcją odwracalną; w odpowiednich warunkach można
przeprowadzić desulfonowanie, czyli usunąć grupę sulfonową z pierścienia.
Sekwencja reakcji sulfonowanie-desulfonowanie jest często wykorzystywana
w syntezach złożonych pochodnych benzenu.
7.4.
Mechanizm alkilowania Friedla-Craftsa
ilość AlCl
3
(katalizatora) - 0.1 – 0.2 mol na 1 mol halogenku alkilowego.
Reaktywność halogenków alkilowych w reakcji alkilowania Friedla-Craftsa:
jodki < bromki < chlorki; fluorki są niereaktywne w reakcji alkilowania
Friedla-Craftsa - nie stosuje się ich.
7.4.a.
Ograniczenia reakcji alkilowania Friedla-Craftsa
Izomeryzacja (przegrupowanie) karbokationu na przykładzie alkilowania
benzenu 1-chlorobutanem lub 2-metylo-1-chorobutanem.
Polialkilowanie - ze względu na elektronodonorowy charakter grup
alkilowych, wprowadzenie pierwszego podstawnika do pierścienia
aromatycznego zwiększa, w porównaniu z benzenem, jego podatność na dalsze
podstawienie elektrofilowe.
Powstawaniu polipodstawionych produktów sprzyja wysoka
temperatura i wydłużony czas reakcji.
Polialkilowanie można ograniczyć, stosując znaczny nadmiar
benzenu w stosunku do halogenku alkilowego.
Reaktywność pierścienia aromatycznego spowodowana obecnością różnych
podstawników:
Alkilowaniu Friedla-Craftsa ulegają tylko takie pochodne benzenu,
które posiadają podstawniki: alkilowe (-R), alkoksylowe (-OR), lub
atom fluorowca (-Cl, -Br, -F).
Pochodne benzenu posiadające następujące podstawniki nie ulegają
alkilowaniu Friedla-Craftsa: -NO
2
(nitrozwiązki); -SO
3
H (kwasy
sulfonowe); -CN (nitryle); -C(O)R (ketony); -C(O)H (aldehydy);
-C(O)OR (estry); -CO
2
H (kwasy karboksylowe); -NH
2
, -NHR, -NR
2
(aminy);-OH (fenole).
7.5.
Mechanizm alkilowania przy użyciu alkoholi lub alkenów
Reakcja katalizowana kwasami mineralnymi: H
2
SO
4
, H
3
PO
4
lub HF.
7.6.
Mechanizm acylowania Friedla-Craftsa
7.6.a.
Generowanie elektrofila (kationu acyliowego) z chlorku kwasu karboksylowego;
ze względu na tworzenie się kompleksu produktu z AlCl
3
, w praktyce stosuje się
1.1 mol AlCl
3
na 1 mol chlorku kwasowego.
3

Konspekt wykładu „Chemia Organiczna” dla studentów Wydziału Inżynierii Chemicznej i Procesowej
7.6.b.
Generowanie elektrofila (kationu acyliowego) z bezwodnika kwasu
karboksylowego; w praktyce stosuje się 2.1 mol AlCl
3
na 1 mol bezwodnika
kwasowego.
7.6.c.
Różnice między reakcją acylowania Friedla-Craftsa, a reakcją alkilowania Friedla-
Craftsa
Produkty reakcji acylowania Friedla-Craftsa (ketony) są niepodatne na dalsze
acylowanie, dlatego nie powstają produkty poliacylowania.
Kationy acyliowe nie ulegają przegrupowaniu; dlatego można tę reakcję
wykorzystać do syntezy alkilobebzenów niemożliwych do otrzymania w
reakcji alkilowania Friedla-Craftsa; przykład – otrzymywanie propylobenzenu.
7.7.
Mechanizm formylowania Gattermana-Kocha - otrzymywanie aldehydów
aromatycznych w reakcji benzenu i jego pochodnych alkilowych (-R) oraz
alkoksylowych (-OR) z równomolową mieszaniną CO/HCl. Reakcja nie nadaje się
do formylowania fenoli (-OH) i amin aromatycznych (-NH
2
,
-NHR, - NR
2
). Reakcja jest szeroko stosowana przemyśle.
7.7.a.
Otrzymywanie równomolowej mieszaniny CO/HCl z kwasu chlorosulfonowego i
kwasu mrówkowego.
7.7.b.
Generowanie kationu formylowego z chlorku formylu.
7.8.
Podsumowanie wiadomości o S
E
8.
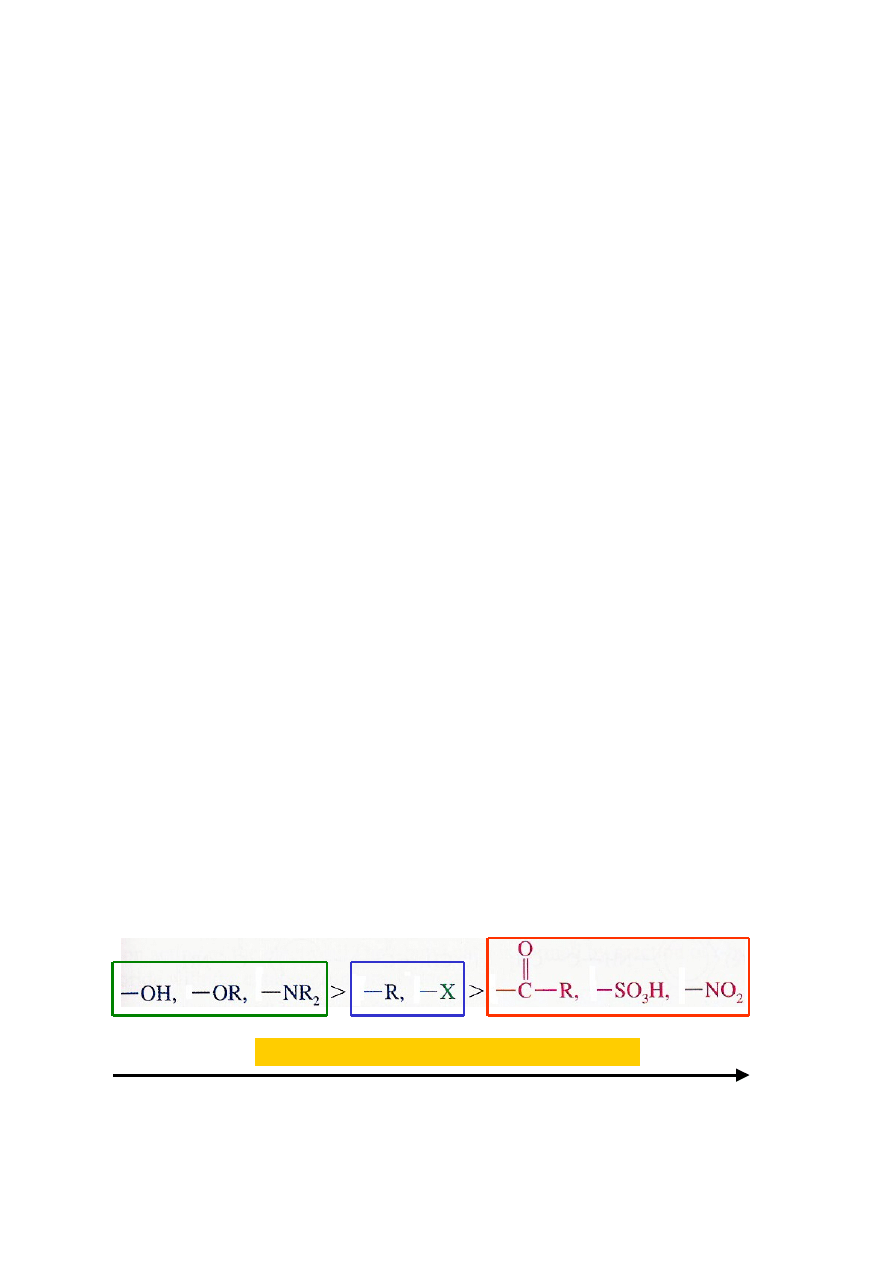
Wpływ podstawników obecnych w pierścieniu aromatycznym na szybkość
reakcji substytucji elektrofilowej (S
E
)
Szybkość zachodzenia reakcji podstawienia elektrofilowego zależy od rodzaju
podstawnika obecnego w rozpatrywanym związku aromatycznym. Może ona
być większa lub mniejsza od szybkości danej reakcji z udziałem benzenu;
porównanie szybkości reakcji w szeregu: fenol > toluen > benzen >
chlorobenzen > nitrobenzen.
8.1.
Podstawniki deaktywujące: F, Cl, Br, -NO
2
, -SO
3
H, -CN, -C(O)R, -C(O)H,
-C(O)OR, -CO
2
H, -C(O)NH
2;
reakcja z udziałem związków zawierających takie
podstawniki zachodzi wolniej niż ta sama reakcja z udziałem benzenu.
4
(kation formylowy)
CO/HCl, AlCl
3
FORMYLOWANIE
GATTERMANA-KOCHA
(kation acyliowy)
chlorek [RC(O)Cl] lub
bezwodnik kwasowy
[(RCO)
2
O)], AlCl
3
ACYLOWANIE
FRIEDLA-CRAFTSA
R
+
(karbokation)
alken lub alkohol,
kwas mineralny
ALKILOWANIE
R
+
(karbokation)
halogenek alkilowy
(RX), AlCl
3
ALKILOWANIE
FRIEDLA-CRAFTSA
SO
3
H
2
SO
4
st. lub oleum
SULFONOWANIE
NO
2
+
(kation nitroniowy)
HNO
3
st., H
2
SO
4
st.
NITROWANIE
Br
+
Br
2
, Fe
BROMOWANIE
Cl
+
Cl
2
, Fe
CHLOROWANIE
ELEKTROFIL
REAGENTY
REAKCJA
(kation formylowy)
CO/HCl, AlCl
3
FORMYLOWANIE
GATTERMANA-KOCHA
(kation acyliowy)
chlorek [RC(O)Cl] lub
bezwodnik kwasowy
[(RCO)
2
O)], AlCl
3
ACYLOWANIE
FRIEDLA-CRAFTSA
R
+
(karbokation)
alken lub alkohol,
kwas mineralny
ALKILOWANIE
R
+
(karbokation)
halogenek alkilowy
(RX), AlCl
3
ALKILOWANIE
FRIEDLA-CRAFTSA
SO
3
H
2
SO
4
st. lub oleum
SULFONOWANIE
NO
2
+
(kation nitroniowy)
HNO
3
st., H
2
SO
4
st.
NITROWANIE
Br
+
Br
2
, Fe
BROMOWANIE
Cl
+
Cl
2
, Fe
CHLOROWANIE
ELEKTROFIL
REAGENTY
REAKCJA
O
C
R
≡
−
⊕
O
C
H
≡
−
⊕
Konspekt wykładu „Chemia Organiczna” dla studentów Wydziału Inżynierii Chemicznej i Procesowej
8.2.
Podstawniki aktywujące: -NH
2
, -NHR, -NR
2
, -OH, -OR, -NHC(O)R, R (alkil);
reakcja z udziałem związków zawierających takie podstawniki zachodzi szybciej
niż ta sama reakcja z udziałem benzenu.
9.
Wpływ podstawników obecnych w pierścieniu aromatycznym na
regioselektywność reakcji substytucji elektrofilowej (S
E
)
9.1.
Grup alkilowych (na przykładzie nitrowania toluenu): otrzymuje się mieszaninę
para- i orto-podstawionych alkilobenzenów, ponieważ podczas ataku elektrofila na
pozycje para lub orto mogą powstać 3° karbokationy (trwalsze od karbokationów
2° powstających podczas ataku elektrofila na pozycję meta).
9.2.
Posiadających wolne pary elektronowe na atomie związanym z pierścieniem
aromatycznym (na przykładzie bromowania fenolu): otrzymuje się mieszaninę
para- i orto-podstawionych produktów, ze względu na możliwość dodatkowej
delokalizacji ładunku dodatniego podczas ataku elektrofila na pozycje para i orto.
9.2.a.
Inne podstawniki kierujące w pozycje orto i para: F, Cl, Br, -NH
2
, -NHR, -NR
2
,
-OH, -OR, -NHC(O)R.
9.3.
Posiadających cząstkowy ładunek dodatni na atomie związanym z pierścieniem
aromatycznym (na przykładzie nitrowania nitrobenzenu): otrzymuje się meta-
podstawione produkty, ponieważ tylko ładunek dodatni nie jest zlokalizowany na
atomie węgla związanym z podstawnikiem posiadającym cząstkowy ładunek
dodatni.
9.3.a.
Inne podstawniki kierujące w pozycję meta: -NO
2
, -SO
3
H, -CN, -C(O)R, -C(O)H,
-C(O)OR, -CO
2
H, -C(O)NH
2
.
10.
Wykorzystanie efektu kierującego podstawników w syntezie organicznej na
przykładzie otrzymywania p-bromonitrobenzenu i m-bromonitrobenzenu
11.
Efekt kierujący podstawników w pochodnych benzenu z dwoma
podstawnikami w reakcjach S
E
:
11.1.
Zgodny efekt podstawników tego samego rodzaju (na przykładzie otrzymywania
1,3-dimetylo-4-nitrobenzenu z m-ksylenu)
11.2.
Niezgodny efekt podstawników tego samego rodzaju (na przykładzie
otrzymywania mieszaniny 1,2-dimetylo-4-nitrobenzenu i 1,2-dimetylo-3-
nitrobenzen z o-ksylenu)
11.3.
Zgodny efekt podstawników różnego rodzaju (na przykładzie otrzymywania
2,4-dinitrotoluenu z p-nitrotoluenu)
11.4.
Niezgodny efekt podstawników różnego rodzaju (na przykładzie otrzymywania
mieszaniny kwasu 4-metoksy-2-nitrobenzenosulfonowego i kwasu 2-metoksy-4-
nitrobenzenosulfonowego z m-nitroanizolu)
Jeśli efekt kierujący dwóch podstawników jest niezgodny, to o kierunku
podstawienia kolejnego podstawnika decyduje efekt podstawnika o większej
„mocy” kierowania, na przykładzie reakcji substytucji p-krezolu, m-krezolu i p-
chlorotoluenu.
5
malejąca „moc” efektu kierującego podstawnika

Konspekt wykładu „Chemia Organiczna” dla studentów Wydziału Inżynierii Chemicznej i Procesowej
12.
Reakcje zachodzące w łańcuchach bocznych węglowodorów aromatycznych
12.1.
Substytucja rodnikowa w pozycji benzylowej na przykładzie bromowania
propylobenzenu w obecności promieniowania ultrafioletowego (h
ν
) lub
N-brosukcynoimidu (NBS)
12.2.
Utlenianie łańcuchów bocznych na przykładzie otrzymywania:
12.2.a.
kwasu benzoesowego z toluenu,
12.2.b.
kwasu benzoesowego z n-propylobenzenu,
12.2.c.
kwasu p-tert-butylobenzoesowego z p-tert-butylobenzenu
Atom węgla w pozycji benzylowej przekształca się w grupę karboksylową
(powstaje odpowiednia pochodna kwasu benzoesowego), a pozostała część
łańcucha ulega utlenieniu do CO
2
Łańcuchy alkilowe posiadające 4° atom węgla w pozycji benzylowej nie
utleniają się
12.3.
Addycja elektrofilowa w aryloalkenach
12.3.a.
Przyłączanie halogenowodoru (HBr, HCl) w obojętnym rozpuszczalniku
organicznym na przykładzie 1-fenylopropenu - atom halogenu przyłącza się do
benzylowego atomu węgla
12.3.b.
Przyłączanie halogenu w roztworze wodnym na przykładzie 1-fenylopropenu -
grupa hydroksylowa (OH) przyłącza się do benzylowego atomu węgla
13.
Wykorzystanie efektu kierującego podstawników w syntezie organicznej na
przykładzie otrzymywania:
p-Bromonitrobenzenu z benzenu
m-Bromonitrobenzenu z benzenu
Kwasu p-nitrobenzoesowego z toluenu
Kwasu m-nitrobenzoesowego z toluenu
m-Nitroetylobenzenu z benzenu
p-Bromopropylobenzenu z benzenu
4-(2-Bromoetylo)bromobenzenu z benzenu
6
Document Outline
- Konspekt wykładu „Chemia Organiczna” dla studentów Wydziału Inżynierii Chemicznej i Procesowej
- WĘGLOWODORY AROMATYCZNE 5 godz.
Wyszukiwarka
Podobne podstrony:
O eter W aromat W miner id 3259 Nieznany
Aromaty 13 id 69167 Nieznany (2)
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
Misc3 id 302777 Nieznany
więcej podobnych podstron