
Rodzaje
aberracji
chromosomowyc
h
Aberracje
chromosomowe
Strukturalne
Liczbowe

Zaburzenia liczbowe
• Poliploidalne:
- triploidie
- tetraploidie
• Aneuploidalne
autosomalne:
- monosomie
- trisomie
• Aneuploidalne
chromosomów płciowych

Trisomia 21
• wywołuje zespół Downa
• ok. 1/800 żywych urodzeń!
• cechy: upośledzenie umysłowe, wady serca,
utrata słuchu, charakterystyczne rysy twarzy
• ryzyko urodzenia dziecka z zespołem Downa
wzrasta wraz z wiekiem matki
• mozaicyzm występuje u 1-3%

Trisomia 18
• wywołuje zespół Edwardsa
• 1/6000 żywych urodzeń
• tylko 10% przeżywa do 12. miesiąca
• cechy: liczne wady rozwojowe, poważne
upośledzenie umysłowe, charakterystyczne rysy
twarzy
• istnieje wyraźny związek pomiędzy
występowaniem choroby a wiekiem matki

Trisomia 13
• wywołuje zespół Patau
• ok. 1/10 000 urodzeń
• cechy: rozszczep wargi, małe, nieprawidłowo
rozwinięte oczy, wady rozwojowe ośrodkowego
układu nerwowego
• 90% noworodków nie przeżywa jednego roku

Monosomia chromosomu
X
45,X
• zespół Turnera
• 1/2500-5000 żywych noworodków płci żeńskiej
• 99% płodów z tym kariotypem ulega poronieniu,
• cechy: zredukowane rozmiary ciała, nie rozwijają się
drugorzędowe cechy płciowe, większość bezpłodna
• ok. 30-40% to mozaiki (45,X/46,XX, rzadziej
45,X/46,XY)
• posiadanie w niektórych komórkach chromosomu Y
predysponuje do nowotworów złośliwych
(gonadoblastoma) w pasmach gonadowych

Trisomia 47, XXY
• zespół KIinefeltera
• 1/1000 noworodków płci męskiej
• cechy: zwiększone rozmiary ciała, u ok. 1/3występuje
rozwój gruczołów piersiowych, większość bezpłodna
• częstość występowania choroby wzrasta u
potomstwa matek starszych
• zdarzają się także przypadki o fenotypie mężczyzny i
kariotypie 48,XXXY lub 49,XXXXY, nieprawidłowości
somatyczne wzrastają z każdym dodatkowym X

Trisomia chromosomu
X
47, XXX
• ok. 1/1000 kobiet
• nie niesie ze sobą bardzo poważnych konsekwencji
zdrowotnych, ale większość kobiet bezpłodna,
niewielki stopień obniżenia IQ
• większa część przypadków wynika z
nieprawidłowości u matki i zwiększa się wraz z
wiekiem
• zdarzają się kobiety z czterema, pięcioma, a nawet
większa liczbą X, każdemu dodatkowemu
towarzyszy zwiększony stopień upośledzenia
umysłowego i nieprawidłowości fizycznych

Zespół 47,XYY
• ok. 1/1000 mężczyzn
• nie wywołuje poważnych problemów fizycznych,
obniżony stopień IQ
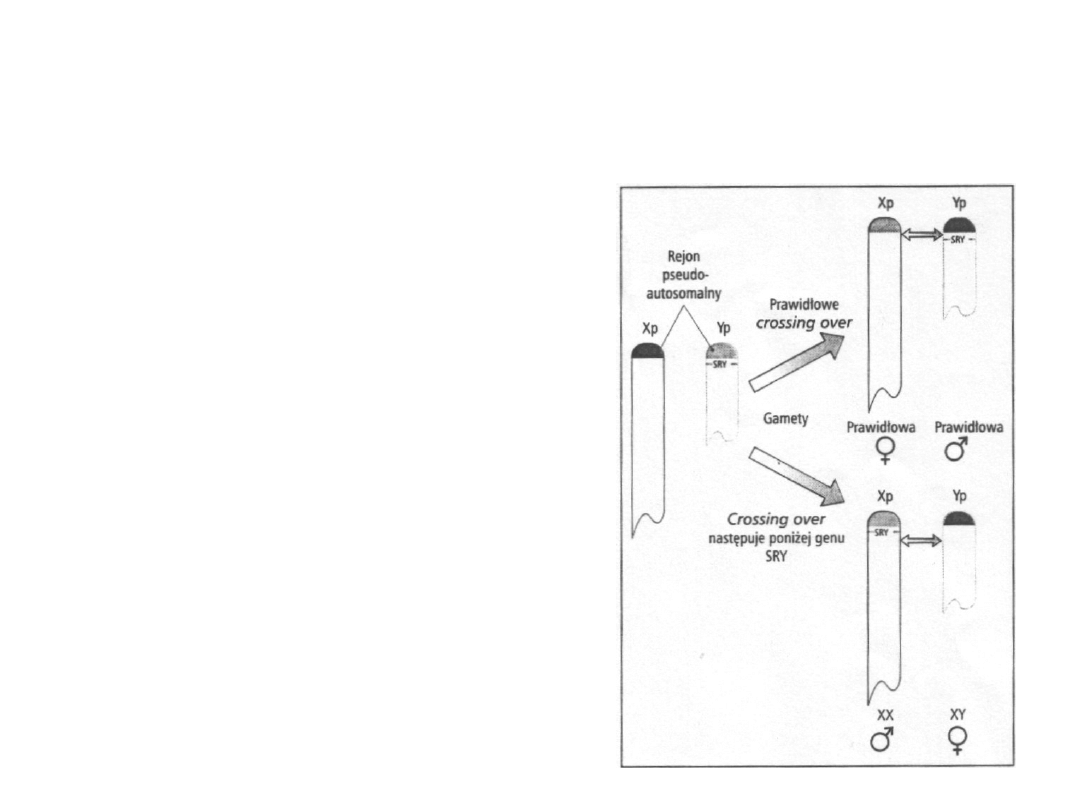
Mężczyźni XX, kobiety XY i genetyczne
podstawy określania płci
• podczas prawidłowej mejozy u
mężczyzn proces c. o. następuje
pomiędzy końcem krótkiego
ramienia chromosomu Y i
końcem krótkiego ramienia
chromosomu X, pod tym rejonem
na chromosomie Y położony jest
gen SRY
• jeśli proces c. o. zajdzie po
centromerowej stronie genu SRY,
to zostanie on przeniesiony na
chromosom X – potomek
otrzymujący taki chromosom X
będzie mężczyzną XX, natomiast
potomek otrzymujący
chromosom Y pozbawiony tego
genu będzie kobietą XY
Aberracje
strukturalne
• Niezrównoważone – rearanżacja powoduje dodanie lub
utratę materiału chromosomowego
• Zrównoważone – rearanżacja nie powoduje dodania lub
utraty materiału chromosomowego
• Zmiany struktur mogą być spowodowane:
- ustawieniem się chromosomów homologicznych
nieprawidłowo w linii podczas mejozy
- nie naprawionymi lub źle naprawionymi pęknięciami
chromosomów podczas mejozy lub mitozy
- prawdopodobieństwo pęknięcia może się zwiększyć w
przypadku obecności pewnych szkodliwych czynników
zwanych klastogenami (promieniowanie jonizujące, pewne
zakażenia wirusowe, niektóre środki chemiczne)
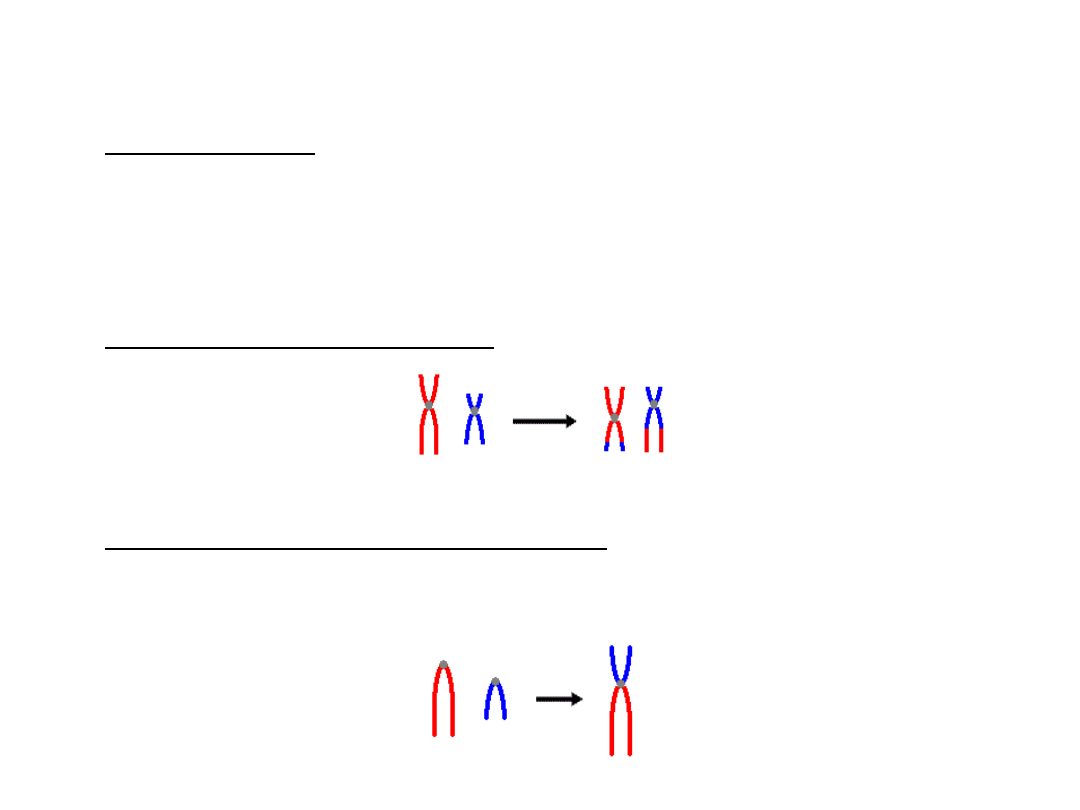
Translokacje
• Translokacja – wymiana materiału
genetycznego pomiędzy niehomologicznymi
chromosomami
• przynajmniej 1/500 osób jest nosicielem
translokacji zrównoważonej
• Translokacje wzajemne są powodowane przez
dwa pęknięcia na różnych chromosomach, z
następującą wymianą materiału
• Translokacje robertsonowskie to takie, gdzie
dwa krótkie ramiona dwóch niehomologicznych
chromosomów zostają utracone, a ramiona
długie łączą się w centromerze, tworząc
pojedynczy chromosom

Chromosom Philadelphia
• translokacja wzajemna pomiędzy chromosomem 9 i 22 ( t(9;22)
(q34;q11) )
• translokacja powoduje powstanie genu fuzyjnego bcr-abl
(fragment bcr znajdujący się na chromosomie 22 w rejonie q11
zostaje połączony z genem abl znajdującym się na chromosomie 9
w rejonie q34
Chromosom Philadelphia
występuje w ponad 95%
przypadkach przewlekłych
białaczek szpikowych.
Spotyka się go również w
ostrych białaczkach
limfoblastycznych - (25-30%
u dorosłych, <10% u dzieci),
niekiedy
również w ostrych
białaczkach szpikowych

Gen abl
• jest protoonkogenem
• koduje białko z rodziny kinaz tyrozynowych, które jest
odpowiedzialne za różnicowanie, podział, adhezję i odpowiedź
na uszkodzenie komórek
• ekspresja tego genu, a zatem synteza białka podlega ścisłej
regulacji
• po połączeniu z fragmentem bcr, powstały gen znajduje się
ciągle w pozycji włączonej (staje się onkogenem) i wymyka się
spod kontroli komórki
• produkowane nowe białko o ciężarze 210 kDa lub 185 kDa
przyczynia się do wzrostu częstotliwości podziałów
komórkowych, dodatkowo blokując naprawę DNA, powoduje
szybkie gromadzenie się mutacji w nowych pokoleniach
komórek
• wzmożona produkcja kinazy bcr-abl upośledza zdolność
komórek do apoptozy

Translokacja
robertsonowska
chromosomów 14 i 21
• w zależności od segregacji w gametach matki,
potomstwo może mieć:
- trisomię 21 (zespół Downa)
- kariotyp prawidłowy
- translokację zrównoważoną z prawidłowym
fenotypem
- monosomię 21
- płody z trisomią 14 i monosomią 14 nie
przeżywają do porodu
(należy zwrócić uwagę, że te trisomie i
monosomie są genetycznie identyczne z tymi,
które powstały w wyniku nondusjunkcji,
ponieważ tylko długie ramiona tych
chromosomów zawierają istotny materiał
genetyczny)
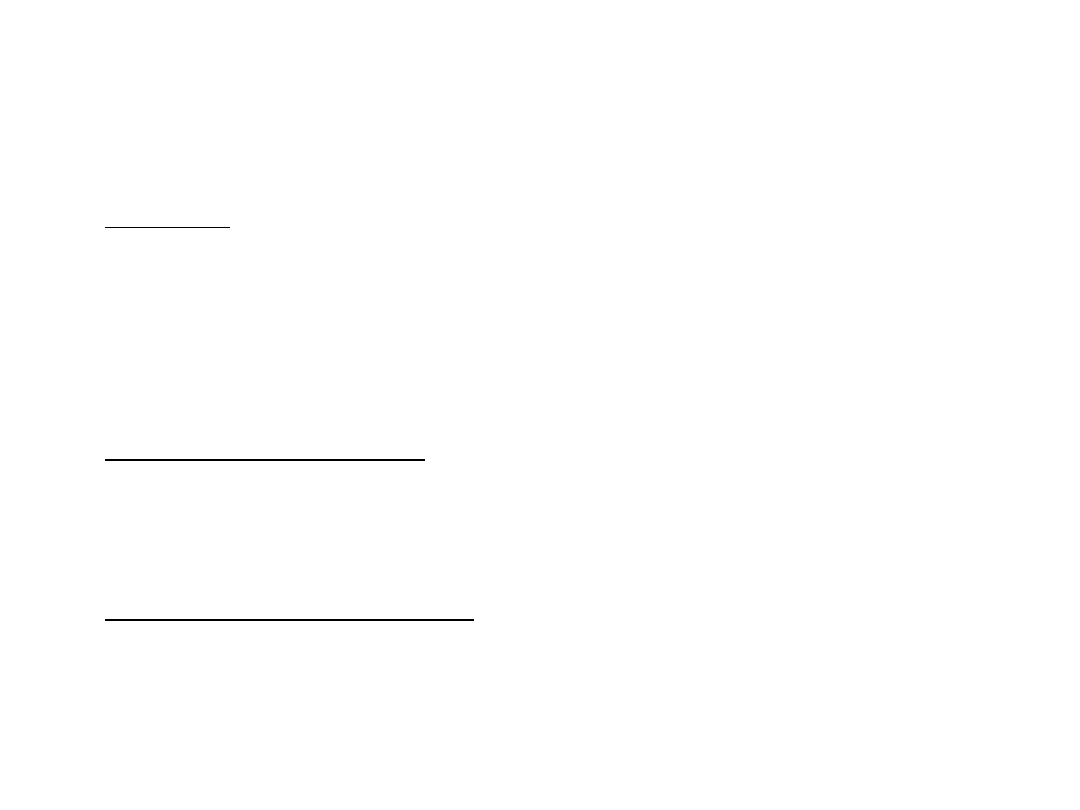
Delecje
• Delecje powstają na skutek pęknięcia chromosomu i
następującej utraty materiału genetycznego; dotyczą
zwykle dość dużej liczby genów i wywołują
charakterystyczne zespoły chorobowe; są możliwe do
zobaczenia pod mikroskopem
• Delecja terminalna powstaje, kiedy dochodzi do
pojedynczego pęknięcia chromosomu i utraty materiału z
końca chromosomu
• Delecja interstycjalna jest efektem dwóch pęknięć
chromosomu i utraty materiału pomiędzy nimi

Zespół cri-du-chat
• Delecja krótkiego ramienia 5p
chromosomu
• cechy kliniczne są związane z regionami
chromosomu
• piskliwy płacz dziecka w okresie
niemowlęcym występuje w przypadku
utraty proksymalnego 5p.15.3
• pozostałe charakterystyczne cechy
zespołu występują przy utracie małego
regionu w obrębie centralnego 15p,12.2
• zespół cechuje się upośledzeniem
umysłowym (średnie IQ około 35), małą
głową i dość charakterystycznym
wyrazem twarzy
• częstość występowania dużych wad
rozwojowych jest zmienna
• chorzy rzadko dożywają pełnoletności

Mikrodelecje
• Mikrodelecje to podtypy delecji chromosomów, które
można zaobserwować tylko w barwionych chromosomach
lub przy zastosowaniu metod genetyki molekularnej
• generalnie obejmują delecje całej serii sąsiadujących genów

Zespoły mikrodelecji
Zespół
Cechy kliniczne
Delecje
chromosomo
we
Pradera-Willego
upośledzenie umysłowe, niski wzrost,
otyłość, hipotonia, charakterystyczne
rysy twarzy, małe stopy
15q11-13
Langera-
Giediona
charakterystyczne rysy twarzy, rzadkie
włosy, egzostoza, zmienne upośledzenie
umysłowe
8q24
Millera-Diekera
brak zakrętów mózgowych,
charakterystyczne rysy twarzy
17p13.3
DiGeorge’a
charakterystyczne rysy twarzy rozszczep
podniebienia, wada serca
22q11
Smitha-
Magenisa
upośledzenie umysłowe,
hiperaktywność, cechy dysmorficzne,
autoagresje
17p11.2
Williamsa
zaburzenia rozwojowe,
charakterystyczne rysy twarzy,
nadzastawkowe zwężenie aorty
7q1
Brak tęczówki/
guz Wilmsa
upośledzenie umysłowe, brak tęczówki,
predyspozycja do wystąpienia guza
Wilmsa, defekty narządów płciowych
11p13

Disomia jednorodzicielska
• jedno z rodziców przekazuje potomstwu dwie kopie danego chromosomu, a drugie
żadnej
• izodisomia występuje, kiedy jedno z rodziców przekazuje dwie kopie
homologicznego chromosomu
• heterodisomia występuje w przypadku przekazania przez rodzica jednej kopii
każdego z homologów
• skutkiem izodisomii jednorodzicielskiej może być homozygotyczność zmutowanych
genów znajdujących się w objętych nią chromosomach (ujawnienie choroby
autosomalnej recesywnej)
• disomia jednorodzicielska chromosomu 15 wywołuje zespół Angelmana (opóźnienie
umysłowe, zaburzenia ruchowe, śmiech i wesołość) i Pradera-Williego (opóźnienie
umysłowe, niski wzrost, oczy w kształcie migdałów, chorobliwy apetyt)
Zespół Pradera-Williego
zespół
Angelmana
Piętno genomowe w Zespole
Pradera-Williego i Angelmana
•
gen związany z AS (gen UBE3A
kodujący ligazę ubikwityny) ulega w
mózgu ekspresji wyłącznie z
matczynego allelu
•
ekspresja genów związanych z
regionem PWS ulega ekspresji z
ojcowskiego allelu

Duplikacje
• mogą występować u potomstwa osób będących
nosicielami translokacji wzajemnej
• mogą zachodzić podczas rekombinacji homologicznej
w mejozie, w wyniku nierównomiernego procesu
crossing over
• generalnie wywołują mniej poważne konsekwencje
niż delecje
• Choroba Charcota-Marie’a-Tootha
• duplikacja chromosomu 17p11.2
• neuropatia, zaburzenia mielinizacji i regeneracji
aksonu, zanik włókien nerwowych, zanik mięśni
• gen PMP22 (peripheral myelin gene
)
w rejonie
chromosomu 17p11.2
Chromosomy pierścieniowe
• często ulegają utracie, co powoduje monosomię
chromosomową w niektórych komórkach
• zostały opisane przynajmniej w jednym przypadku dla
każdego autosomu człowieka
• chromosomy pierścieniowe 13 i 14 wywołują zespół
związany z upośledzeniem umysłowym
• zespół pierścieniowego chromosomu 20 powodujący
padaczkę i upośledzenie umysłowe
• powstają gdy na obu końcach chromosomów dochodzi
do delecji, a następnie końce te łączą się ze sobą
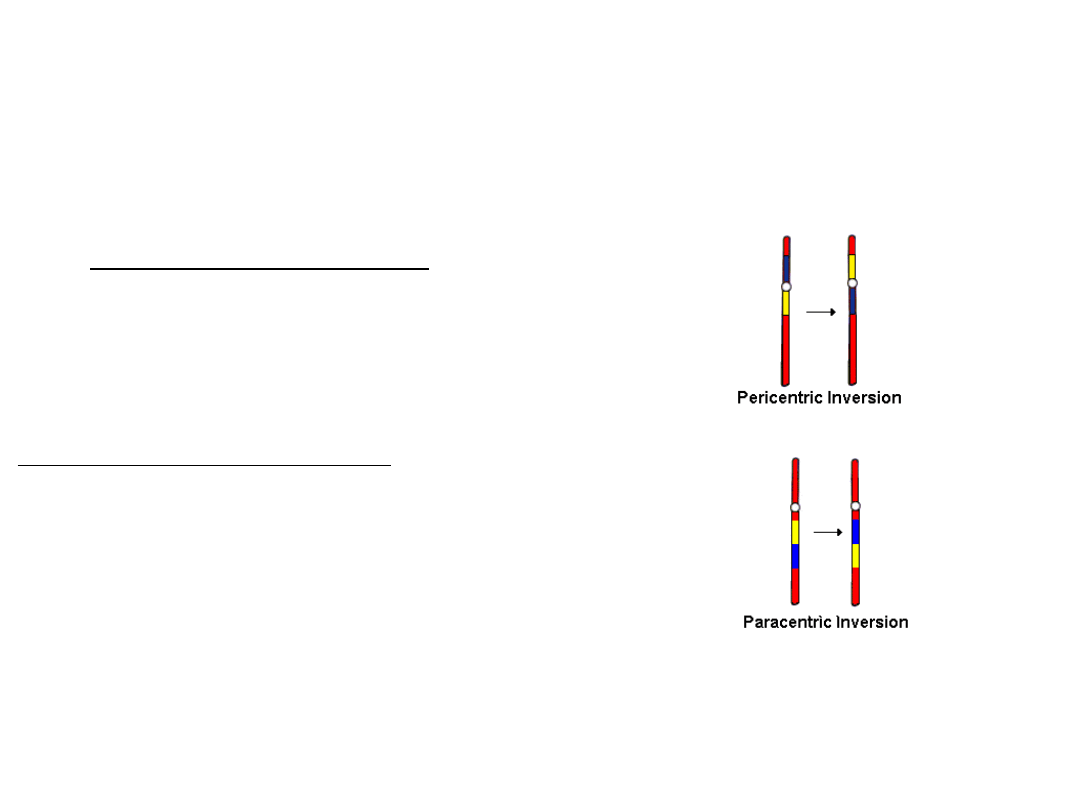
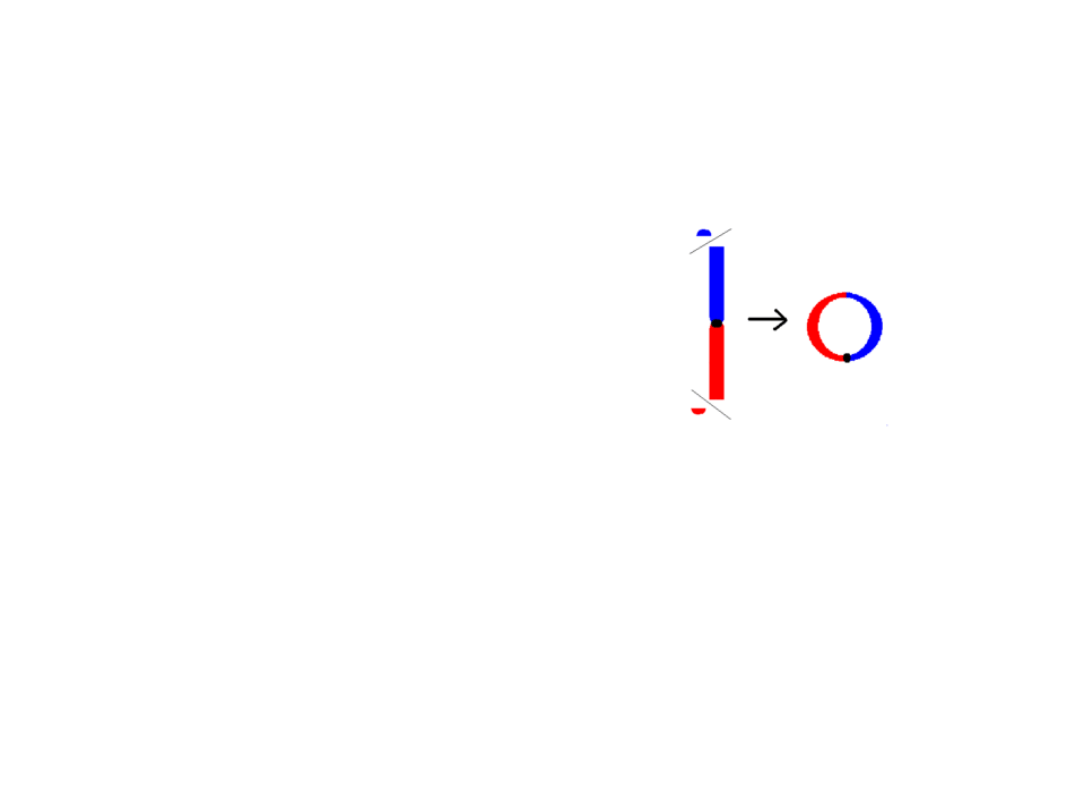
Inwersje
• są one rzadko przyczyną choroby u
nosiciela inwersji, ale inwersja która
przerywa gen czynnika krzepliwości
VIII, jest przyczyną poważnej
hemofilii A
• są rezultatem dwóch pęknięć, po których następuje reinsercja brakującego
fragmentu w oryginalnym miejscu, ale w odwrotnej kolejności
• inwersja pericentryczna obejmuje centromer
•inwersja paracentryczna nie obejmuje centromeru
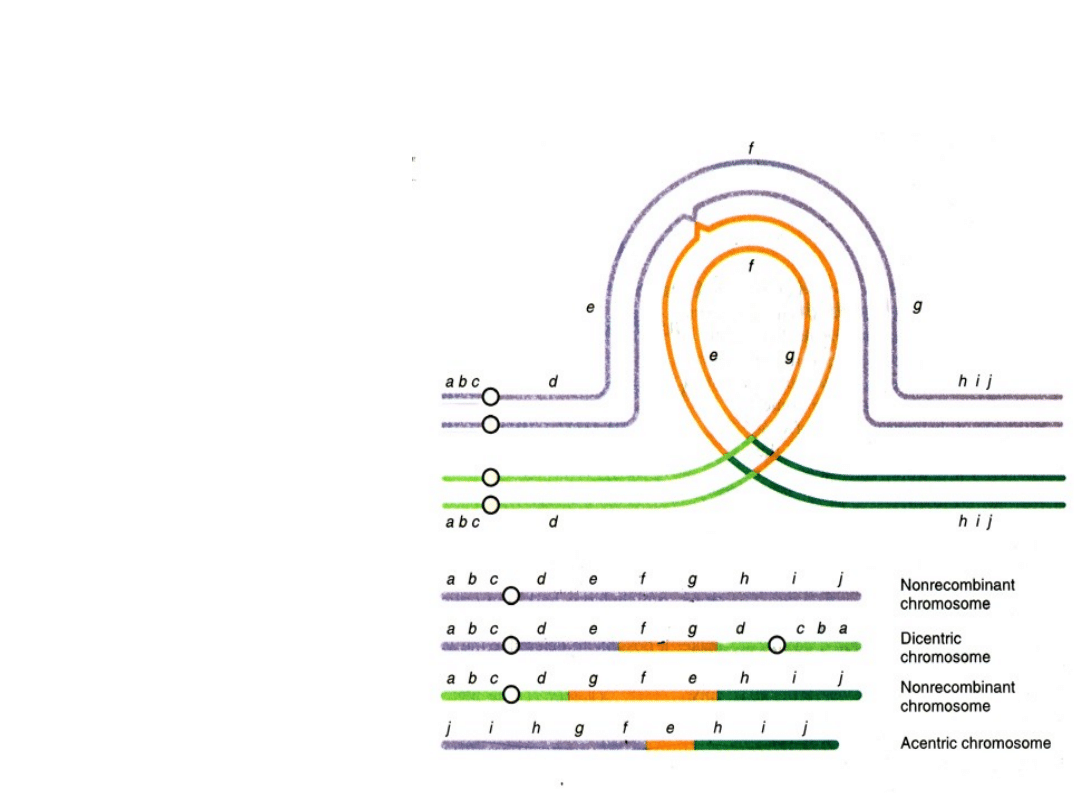
Rodzice z inwersjami i ich dzieci
• inwersje mogą wpływać na proces mejozy,
wywołując aberracje chromosomowe u potomstwa
nosicieli
• aby chromosom z inwersją ustawił się w
idealnym porządku ze swoim prawidłowym
homologiem podczas profazy I, musi
uformować pętlę
•crossing over w ramach tej pętli może prowadzić do
wystąpienia duplikacji i delecji w chromosomach
komórek potomnych
•crossing over w ramach tej
pętli może prowadzić do
wystąpienia duplikacji i delecji
w chromosomach komórek
potomnych
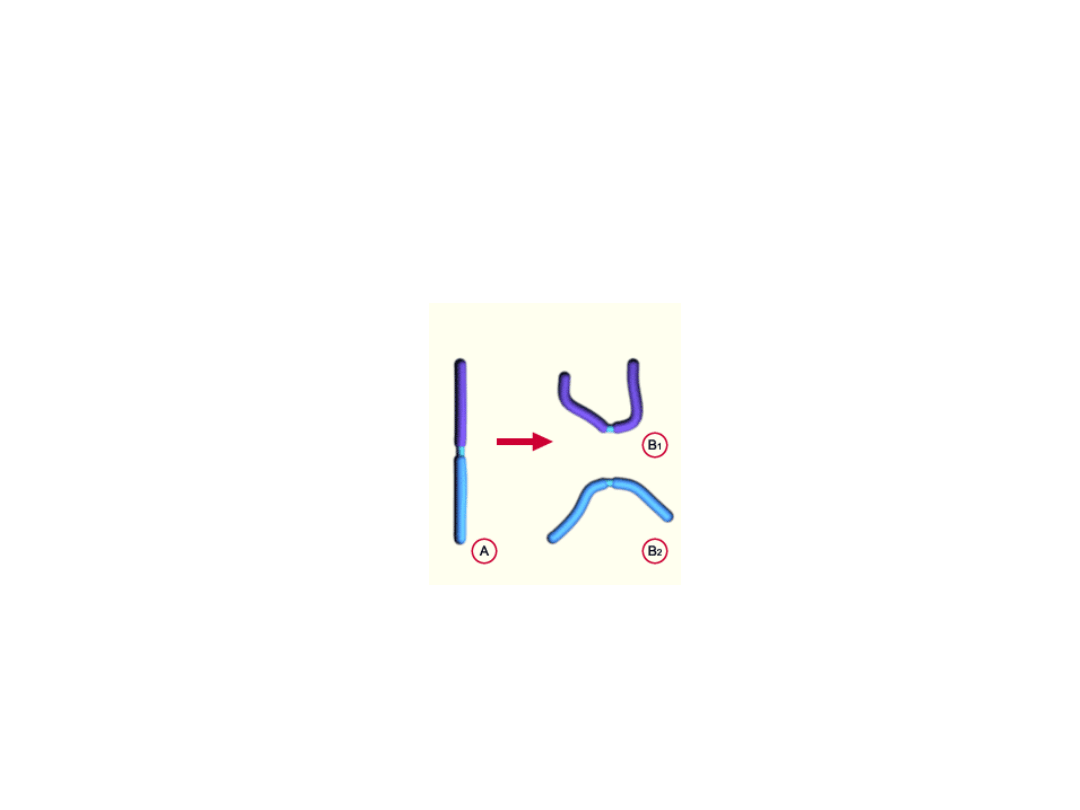
Izochromosomy
• powstają w rezultacie podziału wzdłuż osi prostopadłej do zwykłej osi
podziału chromosomu
• posiadają dwie kopie jednego ramienia i żadnej kopii drugiego
ramienia
• Izochromosomy większości chromosomów są śmiertelne
• większość izochromosomów obserwowanych u żywych noworodków dotyczy
chromosomu X, a dzieci z chromosomem Xq (46,X,i[Xq]) zwykle wykazują
cechy zespołu Turnera
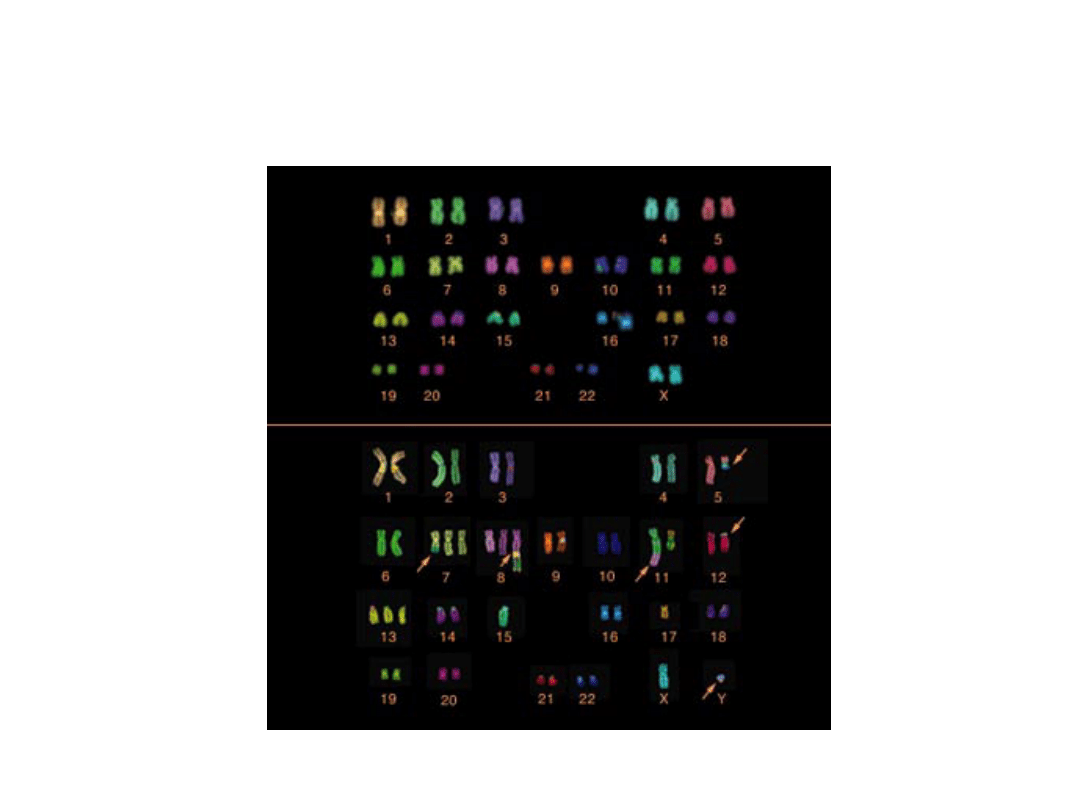
Aberracje w nowotworach

Literatura
• Bruce R. Korf; „Genetyka człowieka. Rozwiązywanie
problemów medycznych"; PWN, Warszawa 2000
• L. B. Jorde, J. C. Carey, M. J. Bamshad, R. L. White;
„Genetyka medyczna”; Wydawnictwo Czelej Sp. z o.o. 2000
• Michael Connor, Malcolm Ferguson-Smith; „Podstawy
genetyki medycznej”; Wydawnictwo Lekarskie PZWL 1998
• http://web.feccbologna.it/abstract_home.htm
• http://www.cvmbs.colostate.edu/bms/bowen.htm
• http://www.sciencemuseum.org.uk/on-line/genes
• http://www.cafamily.org.uk/inherita.html
• http://www.ibis-birthdefects.org/index.htm
• http://learn.genetics.utah.edu/
• http://www.wikipedia.org

Dziękuję za uwagę
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
Wyszukiwarka
Podobne podstrony:
Rodzaje aberracji chromosomowych pop
genetyka, RODZAJE ABERRACJI CHROMOSOMOWYCH WWL
Rodzaje aberracji chromosomowych
Rodzaje aberracji chromosomowych 2
Rodzaje aberracji chromosomowych
Rodzaje aberracji chromosomowych pop
Aberracje chromosomowe i mutacje
ABERRACJE CHROMOSOMOWE, BIOLOGIA MEDYCZNA
aberracje chromosomowe prezentacja
2012.11.05 Zespoły aberracji chromosomowych, Lekarski I rok ŚUM, biologia
Aberracja chromosomowa referat, biologia, biologia medyczna
131 Aberracje chromosomow po napromieniowaniu
więcej podobnych podstron