
Aberracje
chromosomowe
Liczbowe
Aneuploidia
-dodanie lub
utrata jednego z
dwóch
chromosomów
Poliploidia –
dodanie całego
haploidalnego
garnituru
chromosomów
Strukturalne –
przyczyna:
przemieszczenie
materiału genet.
W obrębie
chromosomu lub
między
chromosomami

Nieprawidłowości chromosomów
podejrzewamy gdy:
Pary małżeńskie z bezpłodnością
Poronienia samoistne i martwe
urodzenia
Żywe urodzenia z wadami
wrodzonymi

Aberracje liczbowe
Trisomia:
Trisomia 21- Zespół Downa
Trisomia 13 – Zespół Pataua
Trisomia 18 – Zespół Edwardsa
Zespół Klinefertera 47, XXY
Trisomia 47, XYY
Trisomia 47, XXX
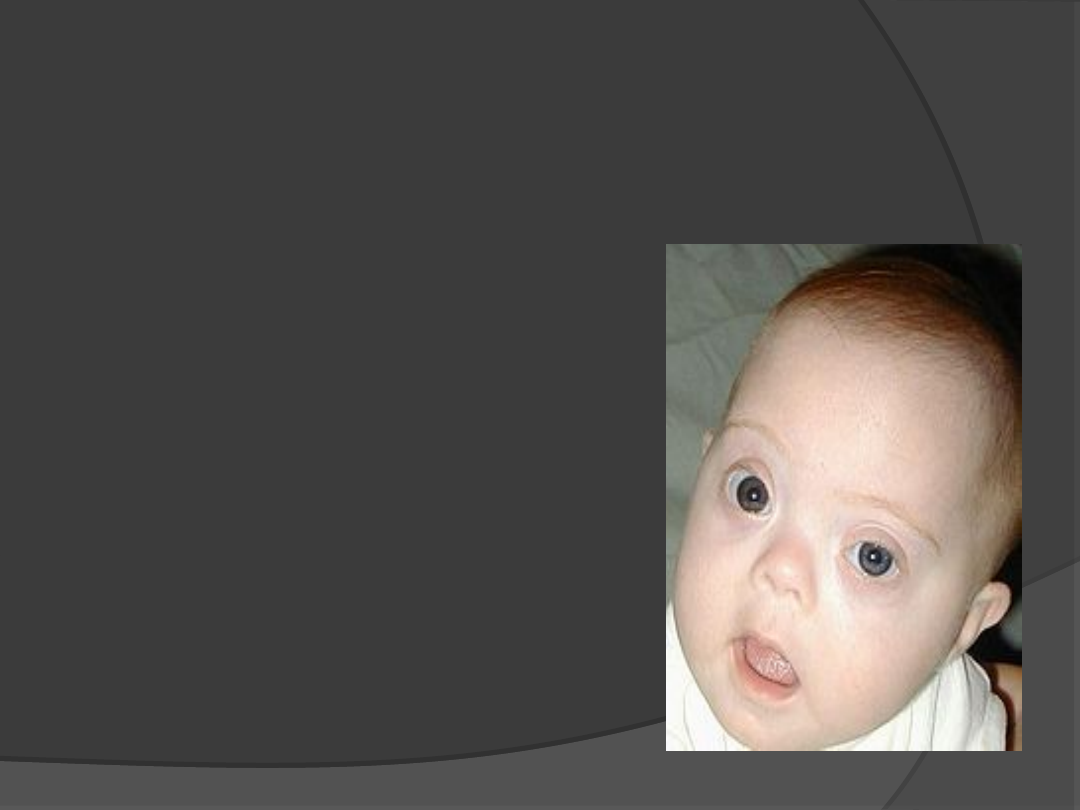
Trisomia 21- Zespół
Downa
1: 680 na żywo
urodzonych
Cechy:
Płaska nasada nosa
Plamki Brushfielda
Płaska potylica
Wrodzone wady serca
Opóźniony rozwój umysłowy
Niski wzrost
Przedstarcza demencja typu
Alzheimera
50-60 lat przeżywają
Bruzda poprzeczna na dłoni
Trisomia 21 – Zespół
Downa

Trisomia 18 – Zespół
Edwardsa
1: 3500 na żywo urodzonych
Większość ciąż to spontaniczne
poronienia
Na USG płodu widzimy: wady serca i
przepuklinę przeponową
Cechy:
Mała twarz
Wydatna kość potylicy
Palce zachodzą na siebie
Wady narządów wewnętrznych
Upośledzenie umysłowe
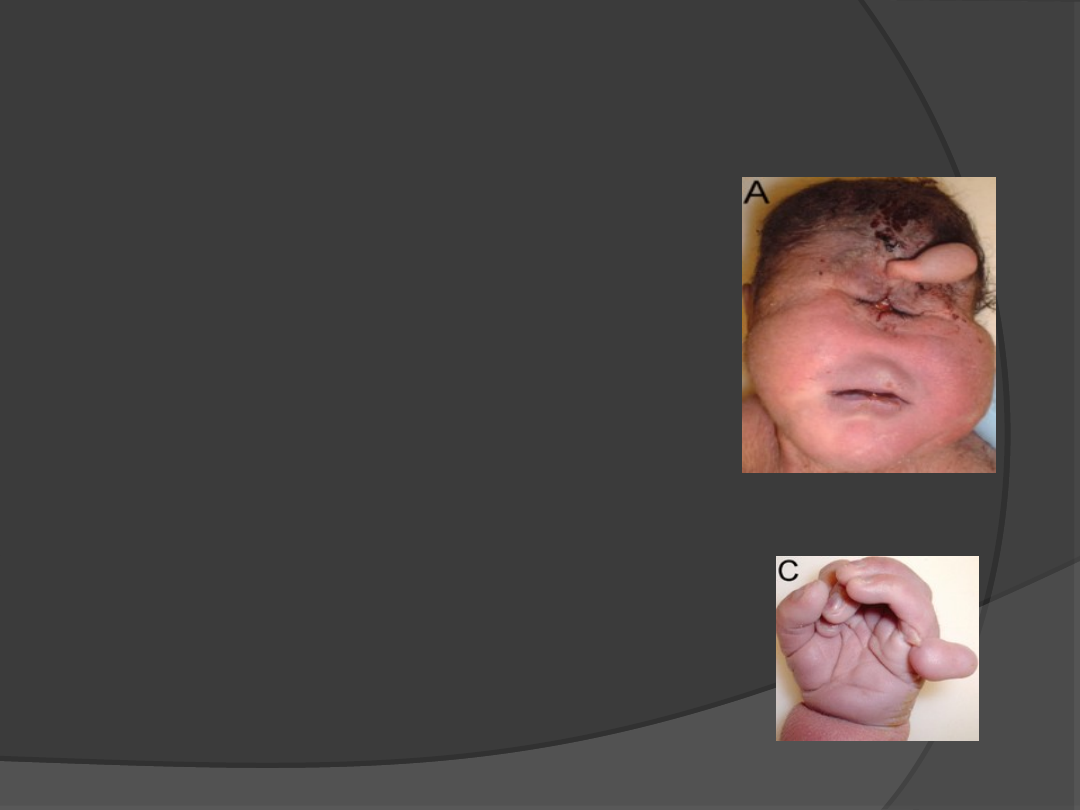
Trisomia 13 – Zespól Pataua
1: 5000
Cechy:
Nieprawidłowość w linii
środkowej mózgu np.
ubytki skóry na czaszce
Dodatkowe palce
Letalne wrodzone wady
serca
Upośledzenie głębokie
Przepuklina pępowinowa

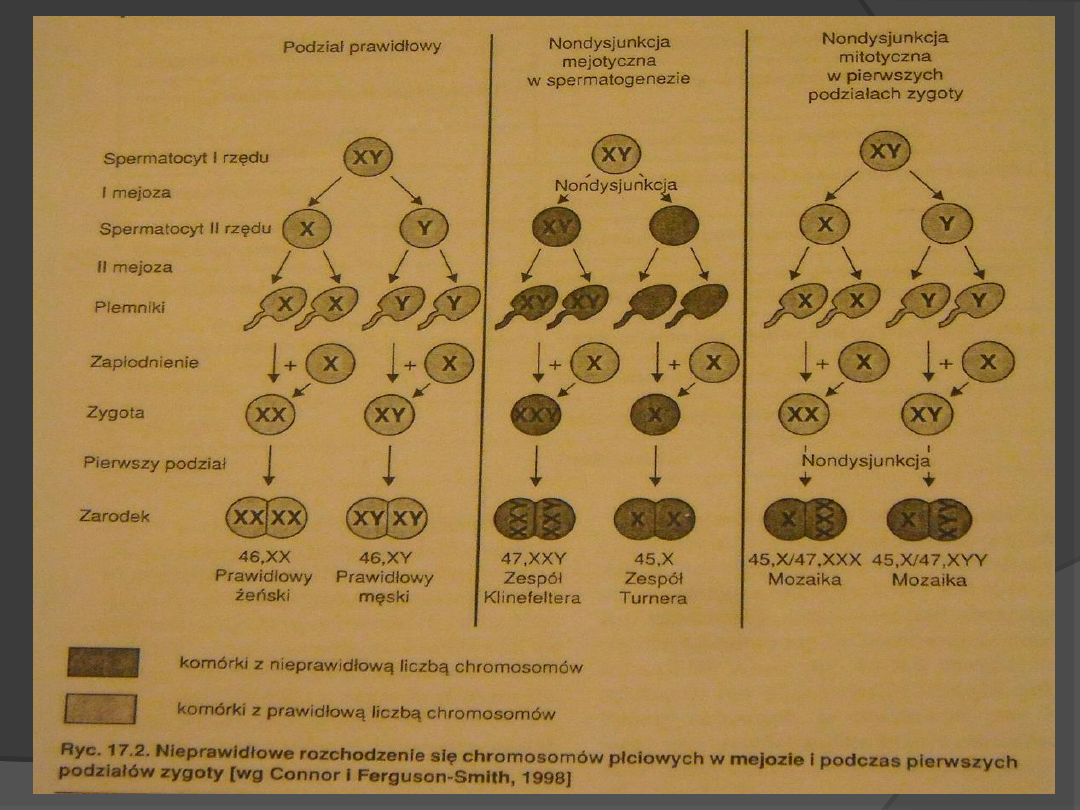
Czynniki patogenne:
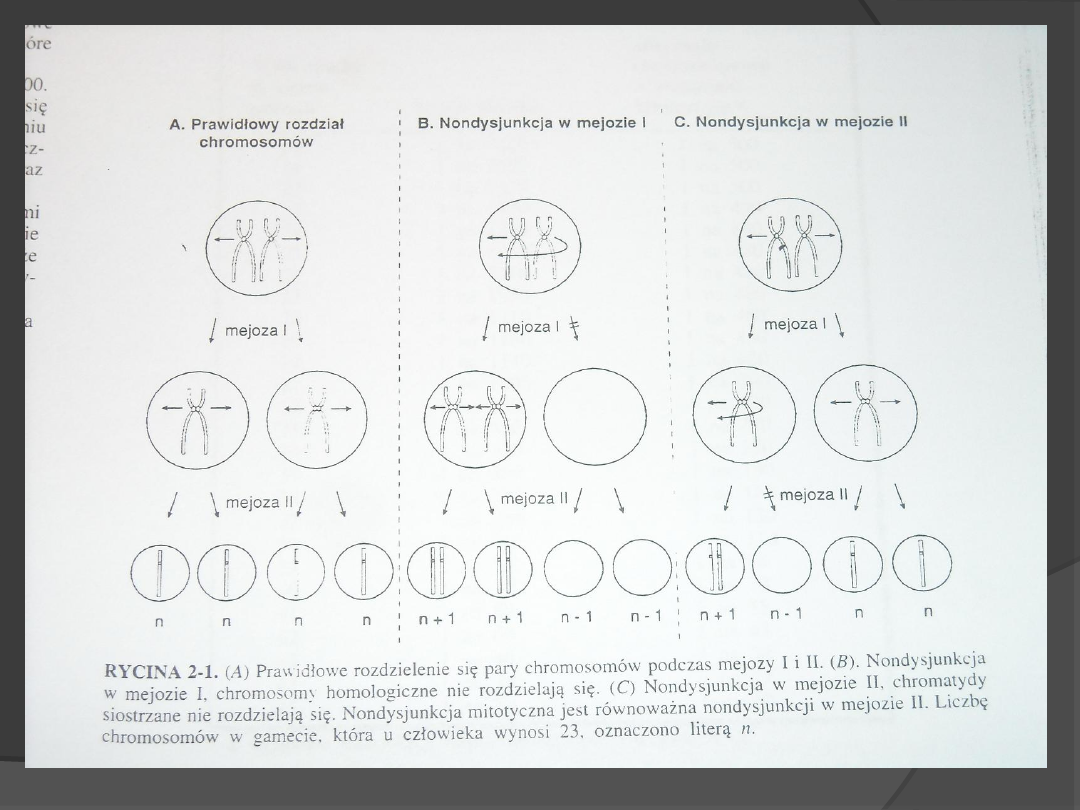
Nondysjunkcja- nierozdzielenie
chromosomów lub chromatyd
siostrzanych podczas podziału
komórkowego
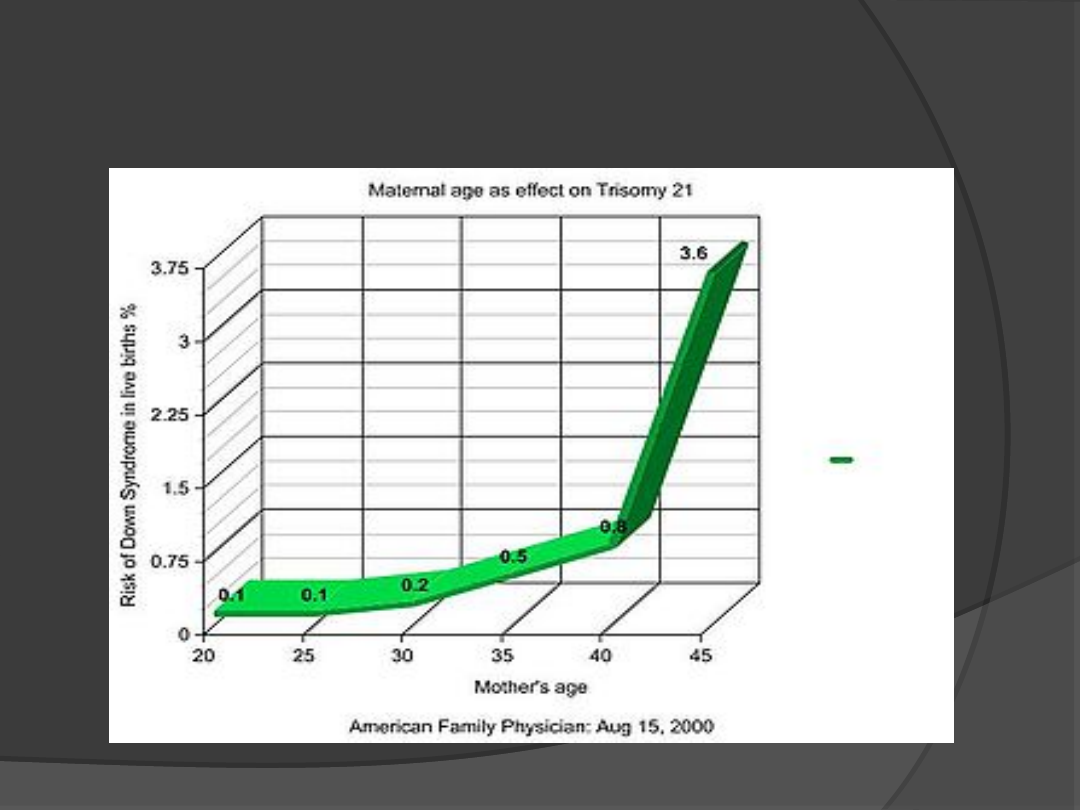
Wiek matki
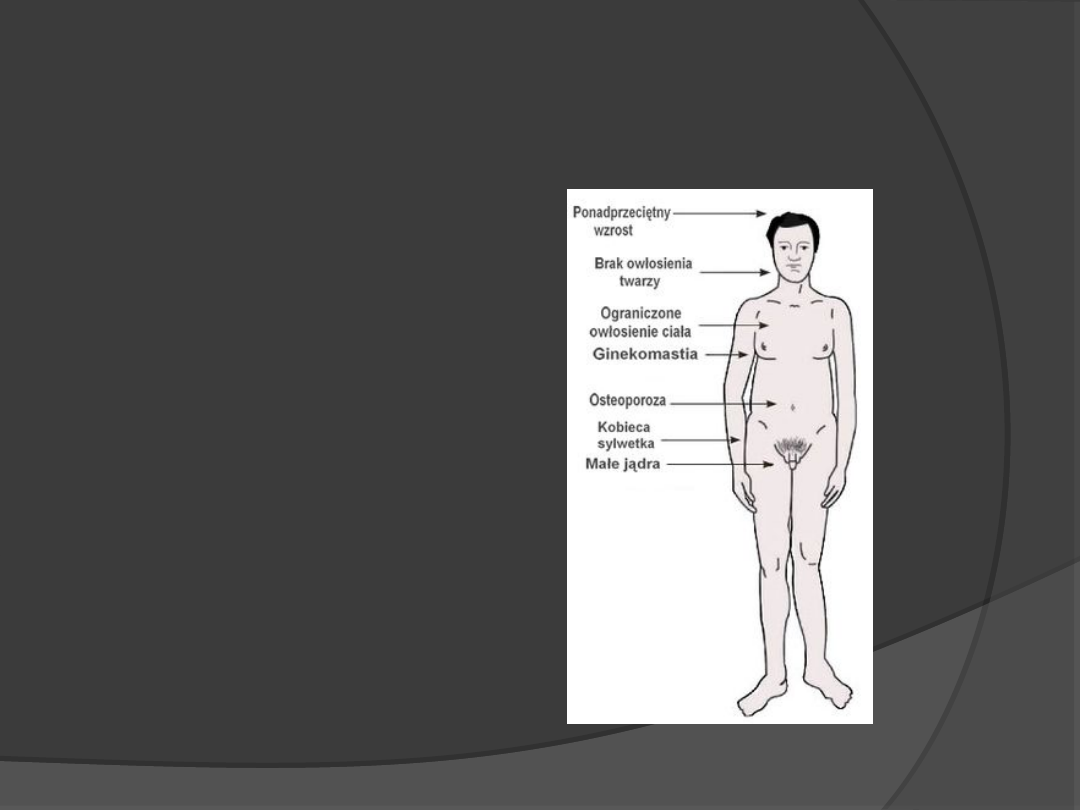
Zespół Klinefertera
47,XXY
1:800 chłopców
Cechy:
Łagodne opóźnienie
Małe, miękkie jądra
Przerost sutków
Większe ryzyko
schizofrenii
Bezpłodność
Zwłóknienie kanalików
nasiennych
Zmniejszona produkcja
testosteronu

Trisomia 47, XYY
1:800 chłopców
Cechy:
Wysoki
Problemy z dojrzałością społeczną
Szybki wzrost ciała
Powikłania trądziku
Płodni, zdrowe potomstwo
Bark tendencji do zachowań agresywnych i
kryminalnych

Trisomia 47, XXX
1:800 dziewczynek
Cechy:
Brak objawów klinicznych
Płodne, zdrowe potomstwo
Kościozrost kości łokciowej i promieniowej
Skąpe miesiączkowanie
Wczesna menopauza
Większe ryzyko zaburzeń psychicznych

Aberracje liczbowe
Monosomia-
obecność jednego z dwóch
chromosomów danej pary w kariotypie
Monosomia autosomalna – letalna
Monosomia chromosomu X, 45X

Monosomia chromosomu X – Zespół
Turnera 45, X
1:2500 dziewczynek
Cechy:
Niski wzrost
Krępa budowa, puklerzowa
klatka piersiowa
Płetwiasta szyja
Wady narządów
wewnętrznych np. objaw
Archibala,objaw Kosowicza
Bark miesiączki, niepłodność
Trudności w nauce
zwłaszcza matematyki
Wady nerek
Obrzęki limfatyczne
grzbietu, rąk, stóp u
noworodka

Aberracje liczbowe:
Aneuploidia mozaikowa
Przykłady:
Mozaikowata trisomia chromosomu 8
Mozaikowa trisomia chromosomów 13, 18, 21
Mozaikowatość chromosomu płci
Nierozpoznawalna mozaikowatość niewielkiego
stopnia -mikromozaikowatość
Mozaikowatość występująca wyłącznie w kosmówce

Aberracje liczbowe
Disomia
jednorodzicielska
– UPD -
w diploidalnej
linii komórkowej oba
chromosomy danej
pary pochodzą tylko od
jednego z rodziców
Poliploidia
Triploidia -
obecność
w komórce 69
chromosomów z
chromosomami płci
XXX, XXY, XYY
Tetraploidia – obecności
w komórce 92
chromosomów z
chromosomami płci
XXXX, XXYY

Aberracje strukturalne
Delecje:
5p Cri du chat
(5p15)
4p Zespół Wolfa –
Hrischhorna
(4p16)
Pierścieniowy
chromosom 14
(14p11, 14q32)
Mikrodelecje:
Zespół Pradera –
Willego (15q11-
13)
Zespół Millera-
Diekera (17p13.3)
Zespół
Angelmana
(15q11-q13)
Zespół DiGeorge

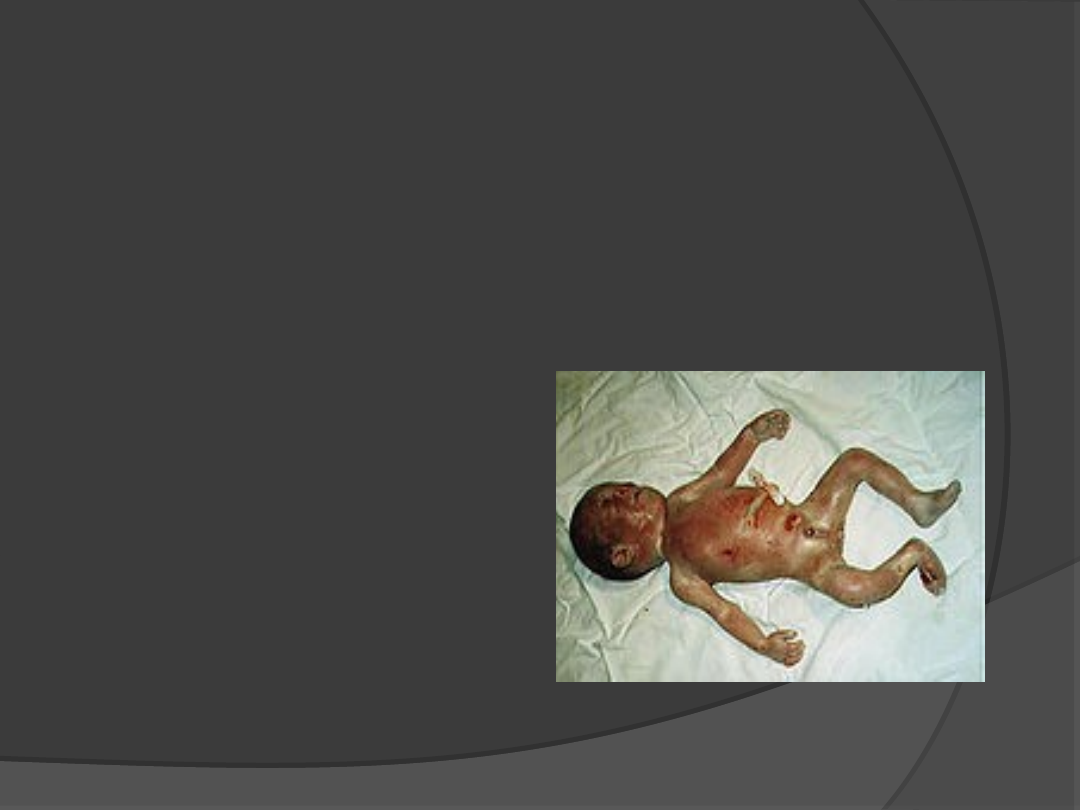
Cri du chat – Zespół kociego
krzyku
5p
Cechy:
Okrągła twarz
Płacz przypomina
miauczenie kota
Wady serca
Upośledzenie
umysłowe
Powstaje de novo
Punkt krytyczny 5p15
Zespół Wolfa -
Hirschhorna
4p (4p16)
Cechy:
Wypukłe czoło, szeroka
nasada oczu tj. hełm
greckiego wojownika
Upośledzenie
umysłowe i rozwoju
fizycznego

Zespół Pradera - Willego
1:50000 żywo
urodzonych
Cechy:
Zmniejszone napięcie
mięśniowe
Otyłość
Upośledzenie umysłowe
Zaburzenia zachowania
i okresy napadów złości
Twarz o wąskim
wymiarze
dwuskroniowym

Mikrodelecje
Zespół Millera-
Diekera
1:10000-1:50000
żywo urodzonych
Brak bruzd i
zakrętów mózgu
Gładkomózgowie i
małomózgowie
Zespół DiGeorge
Rozszczepienie
podniebienia
Rybie usta
Wady serca
Brak grasicy
Hipokalcemia

Zespół Angelmana – Zespół
szczęśliwej kukiełki
Cechy:
Charakterystyczny chód na szerokiej
podstawie z odwiedzionymi i zgiętymi w
stawach łokciowych kończynami górnymi
Brak mowy
Krótkogłowie
Napady padaczkowe
Delecja długiego ramienia chromosomu 15
(15q11-q13)

Aberracje strukturalne
Duplikacje
Obecność dodatkowego
materiału
chromosomowego.
To jest obecność dwóch
kopii danego fragmentu
chromosomu
powstających
najczęściej w wyniku
translokacji, inwersji i
powstawania
izochromosomu.
Izochromosomy –
jedno z ramion p lub q
jest podwojone
powodując brak całego
materiału drugiego
ramienia chromosomu.
W większości letalne
Do powstania dochodzi
w wyniku
nieprawidłowego
poprzecznego podziału
chromosomu w obrębie
centromeru.

Inwersja -odwrócenie odcinka
chromatyny pomiędzy dwoma
miejscami załamań w chromosomie
Pericentryczna
Złamanie i
przemieszczenie
fragmentu
chromosomu po
obu stronach
centromeru
Paracentryczna
Złamanie i
przemieszczenie
po tej samej
stronie
centromeru

Aberracje strukturalne
Miejsca łamliwe
chromosomów
Opisano 100 miejsc
Wyróżniamy
pospolite i rzadko
występujące
Miejsca łamliwe
zlokalizowane w
chromosomie X
powodują
opóźnienie rozwoju
umysłowego.
Chromosomy
markerowe
Powstają w wyniku
przegrupowań
strukturalnych
materiału
chromosomowego
To są chromosomy
dodatkowe
Pochodzenie i
mechanizm
powstawania jest
nieznany

Translokacja
robertsonowska
Powstaje w wyniku połaczenia całych ramion
długich chromosomów akrocentrycznych
tzn. chromosomów 13-14, 21-22
.
Przykłady kliniczne: 45,XX lub XY
U nosicieli translokacji chromosomu 21, 14
brak zmian
Po zapłodnieniu niezrównoważone produkty
translacji wywołują trisomię i monosomię
14, 21 powodując wczesne poronienia

Zespoły dysgenzji gonad
Dysgenzja gonad-
nieprawidłowa
budowa i czynność gonad
spowodowana aberracjami
chromosomowymi lub mutacjami
genowymi
Cechy:
Fenotyp żeński
Brak pierwotnie miesiączki
Brak II i III rzędowych cech płci

Czysta dysgenzja gonad z
kariotypem 46,XY – Zespół
Swyera
Cechy:
Fenotyp żeński
Szerokie barki, wąskie biodro
Macica mała
Wirylizacja – przerost łechtaczki
Brak miesiączki, bezpłodność
Narządy typowo żeńskie
Skąpe owłosienie pachowe i łonowe.

Dysgenzja
Czysta dysgenzja
gonad z
kariotypem 46,
XX
Fenotyp męski
Zaburzenia w budowie
ciała i gruczołów płci
Nieprawidłowa budowa
narządów płci
Bark miesiączki,
bezpłodność
Brak zahamowań
wzrostu kości
Mieszana dysgenzja
gonad z kariotypem
45X, 46XY
Obojnacze narządy płci
Przerost łechtaczki
Asymetryczny rozwój
gonad
Bark miesiączki
Macica, pochwa w różnym
stopniu rozwinięte
Bezpłodność
Słaby rozwój II i III
rzędowych cech płci
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
Wyszukiwarka
Podobne podstrony:
Rodzaje aberracji chromosomowych pop
Aberracje chromosomowe i mutacje
ABERRACJE CHROMOSOMOWE, BIOLOGIA MEDYCZNA
2012.11.05 Zespoły aberracji chromosomowych, Lekarski I rok ŚUM, biologia
Aberracja chromosomowa referat, biologia, biologia medyczna
131 Aberracje chromosomow po napromieniowaniu
PRZYKLADY ZAPISU KARIOTYPOW Z ABERRACJAMI CHROMOSOMOWY MI
Slajd z wykładu (aberracje chromosomowe)
ZAPIS KARIOTYPÓW Z ABERRACJAMI CHROMOSOMOWYMI WWL
genetyka, RODZAJE ABERRACJI CHROMOSOMOWYCH WWL
2. Aberracje chromosomowe, VI rok, Genetyka, Genetyka, Egzamin
Aberracja chromosomowa, AWF, Genetyka, Genetyka
Aberracje chromosomalne, NOTATKI gene
Rodzaje aberracji chromosomowych
ABERRACJE CHROMOSOMOWE W BIALACZKACH, VI rok Lekarski CM UMK, Genetyka CMUMK 2015 VI rok, Genetyka,
Aberracje chromosomowe, fizjoterapia, pfk
więcej podobnych podstron