
Patofizjologia ośrodkowego
układu nerwowego
©Jacek M. Witkowski
2006
Wykład 15
© JMW 2006 & EKST
RANET AMG
2
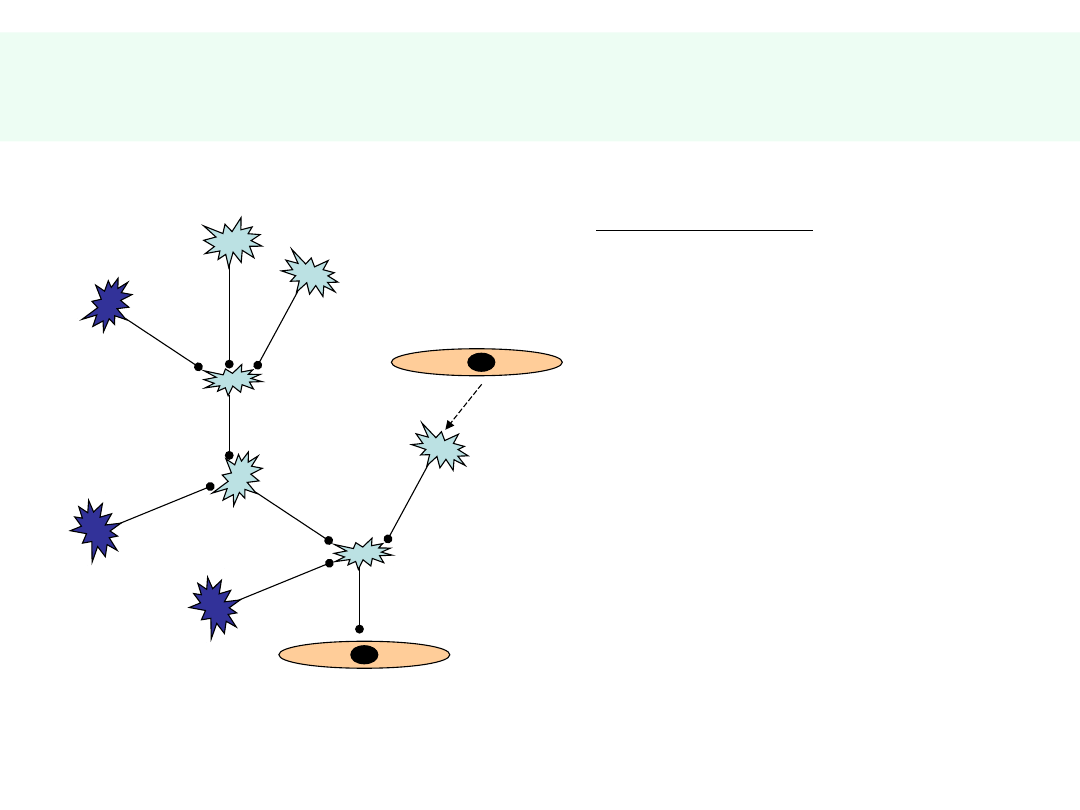
Układ nerwowy
KOMUNIKACJA
CZUCIE
RUCH
ODRUCHY
CZYN. WEGETATYWNE
CZYN. PSYCHICZNE
•
PAMIĘĆ
•
INTELIGENCJA
•
MYŚLENIE LOGICZNE
•
MOWA
•
UCZUCIA
•
STRUKTURA OSOBOWOŚCI
_
_
_
© JMW 2006 & EKST
RANET AMG
3
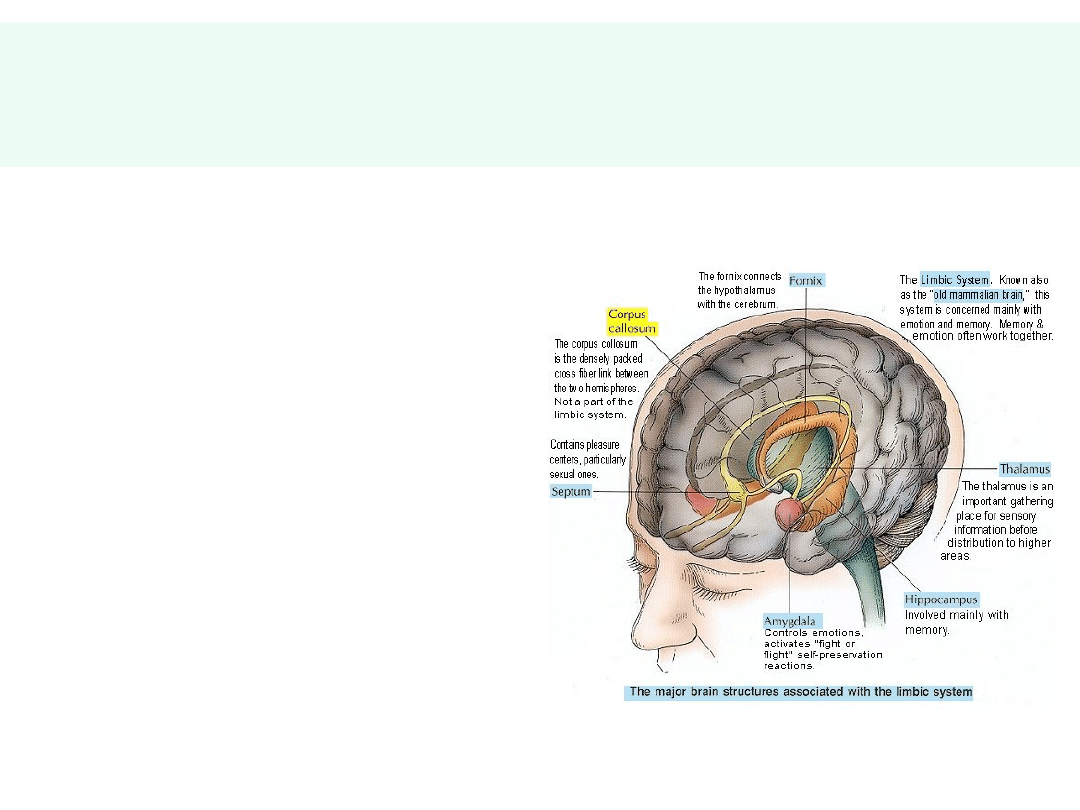
Anatomiczne podłoże czynności
umysłowych
• Neocortex:
• płaty czołowe (nastrój,
planowanie, ustalanie i
dążenie do celu,
priorytyzacja)
• płaty ciemieniowe (czytanie,
matematyka)
• płaty skroniowe (mowa,
pamięć)
• Układ limbiczny:
• Zakręt obręczy (emocje
uświadamiane)
• Ciała migdałowate (emocje
— gniew, lęk,
• uczenie się, pamięć)
• Hipokamp (pamięć
długotrwała)
© JMW 2006 & EKST
RANET AMG
4
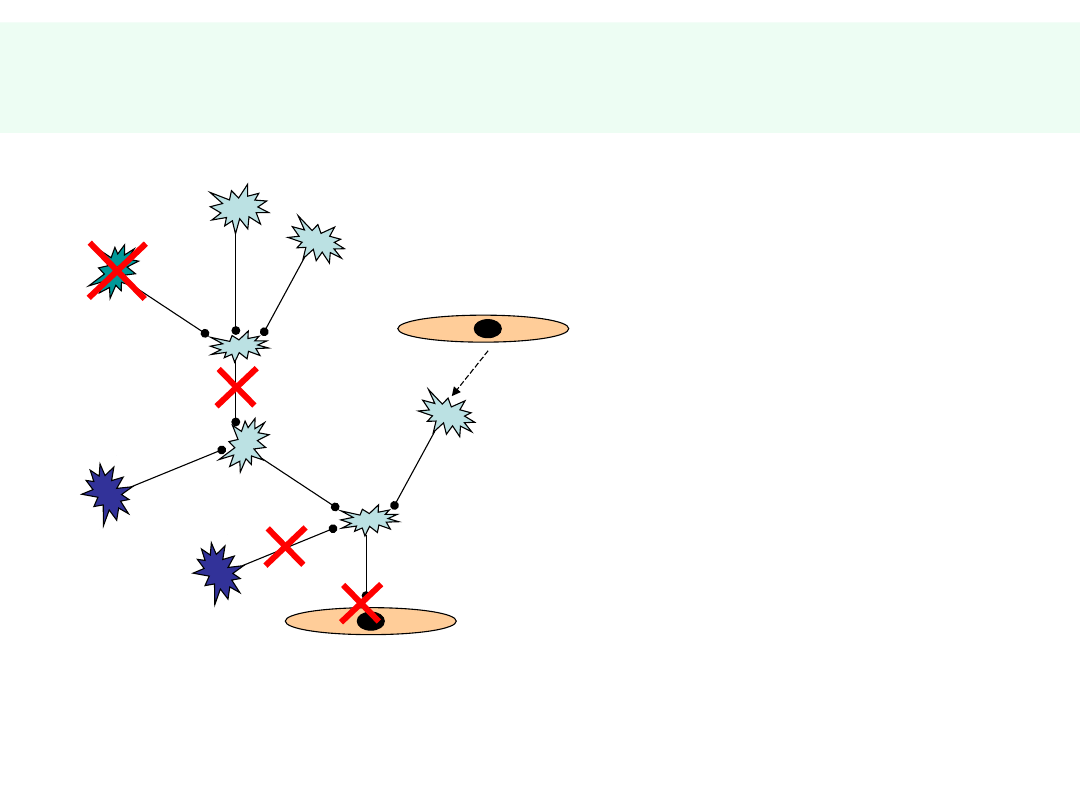
Możliwe zaburzenia
ANATOMICZNE:
•
WRODZONE
• URAZY
METABOLICZNE:
WRODZONE
EGZOGENNE
• TOKSYNY
• LEKI
• NARKOTYKI
INFEKCJE
np. prionowe
NACZYNIOWE
miażdżyca
niedokrwienie
zawał
wylew
AUTOIMMUNIZACJA
stwardnienie rozsiane
miastenia
DEGENERACYJNE:
STARZENIE
CH. ALZHEIMERA
_
+
+
_
© JMW 2006 & EKST
RANET AMG
5
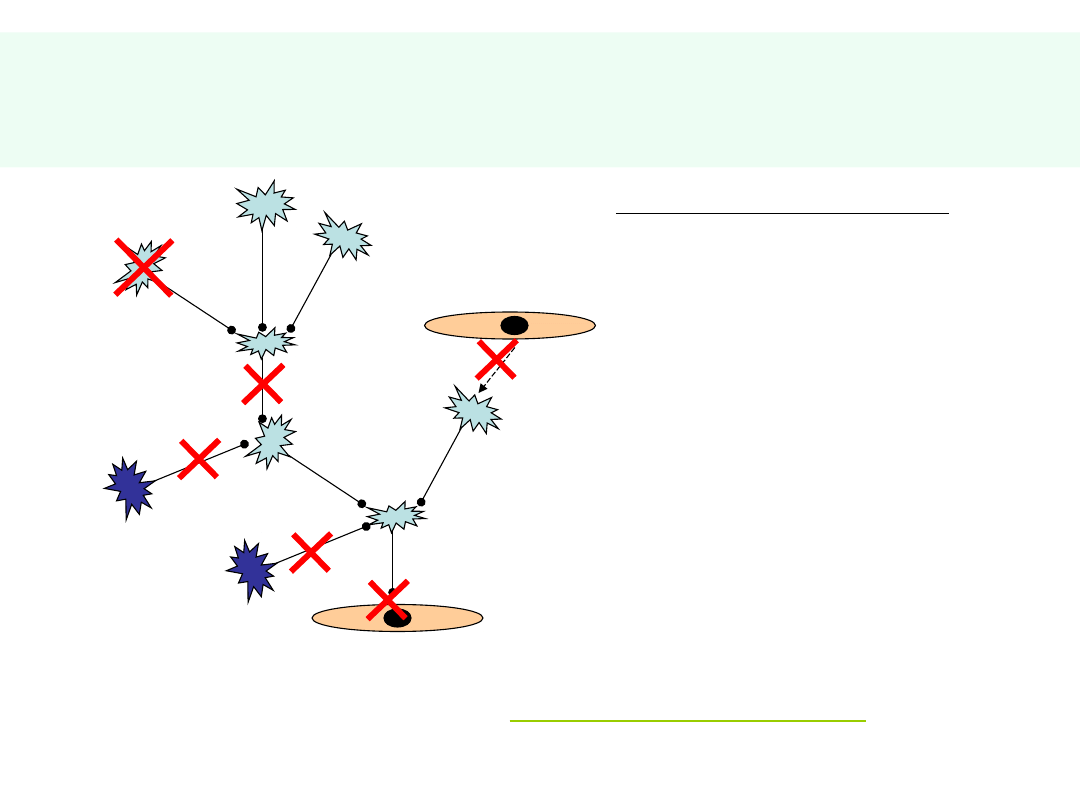
Skutki…
ZABURZENIOM ULEGAJĄ:
RUCH +/-
CZUCIE +/-
ODRUCHY +/-
CZYN. WEGETATYWNE +/-
CZYN. PSYCHICZNE
•
PAMIĘĆ
•
INTELIGENCJA
•
MYŚLENIE LOGICZNE
•
MOWA
•
UCZUCIA
•
STRUKTURA OSOBOWOŚCI
W KONFIGURACJI ZALEŻNEJ OD RODZAJU I LOKALIZACJI USZKODZENIA
_
_
CHOROBY PRIONOWE
GĄBCZASTE ZWYRODNIENIA TKANKI
MÓZGOWEJ
_
+
+
_
© JMW 2006 & EKST
RANET AMG
7
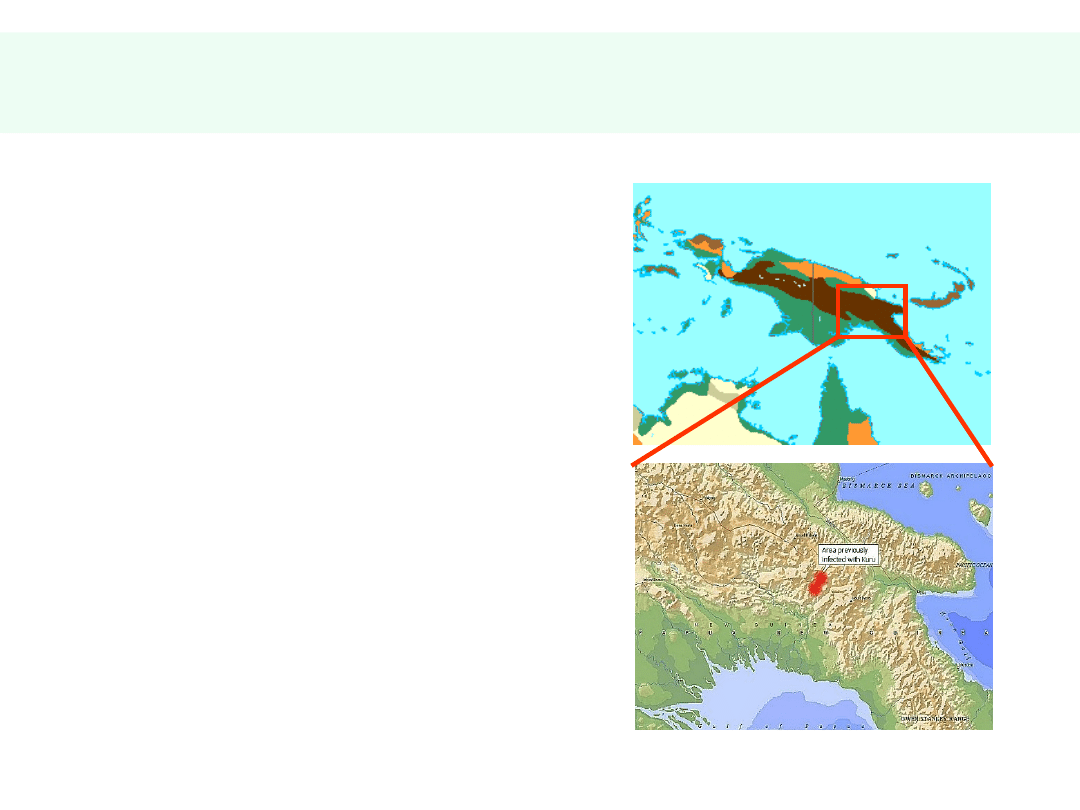
Choroby prionowe człowieka
• Kuru („śmieszne drgawki”,
„śmieszna śmierć” w języku ludu
Fore z Nowej Gwinei)
• „Klasyczna” forma choroby
Creutzfeld-Jakoba (CJD)
• „nowy wariant” choroby
Creutzfeld-Jakoba (vCJD)
• Zespół Gerstmanna –
Strausslera – Scheinkera
(GSS)
• Fatalna rodzinna bezsenność
(FFI)
• Zespół Alpersa (AS, dzieci)

© JMW 2006 & EKST
RANET AMG
8
Kamienie milowe:
• 1920 – H.G. Creutzfeld – pierwszy opis
zespołu zmian, określanych później jako CJD
• 1966 – Carleton Gajdusek – kuru i CJD to
choroby zakaźne
, powodowane przez
„powolny wirus”
• 1982 – Stanley Prusiner – odkrycie
prionów
• 1997 – Stanley Prusiner – nagroda Nobla

© JMW 2006 & EKST
RANET AMG
9
SKALA PROBLEMU
(częstość zachorowań na świecie):
• „Klasyczna” forma choroby Creutzfeld-Jakoba (CJD):
1:milion
• „nowy wariant” choroby Creutzfeld-Jakoba (vCJD):
1:30milionów (do roku 2002: 138 przypadków, 128 w Wlk. Brytanii
(około 15 przypadków/rok)
Prognoza epidemiologiczna: od kilkuset do >150.000 zachorowań
ocenia się, że 1:10.000 zmarłych jest zakażony CJD
• Kuru – wysoka częstość wśród rytualnych kanibali do lat 60-tych XX
wieku, obecnie zanik choroby (zanik obyczaju jedzenia mózgów
zmarłych członków rodziny)
• Zespół Gerstmanna – Strausslera – Scheinkera (GSS)
• Fatalna rodzinna bezsenność (FFI)
• Zespół Alpersa (AS, dzieci)
GSS, FFI, AS – częstości 100 razy niższe od CJD

© JMW 2006 & EKST
RANET AMG
10
Infekcje prionowe – herezja w
mikrobiologii
• Priony – WYŁĄCZNIE BIAŁKOWY
czynnik infekcyjny (
białko PrP
sc
,
pochodne PrP
c
)
• BRAK kwasów nukleinowych
w
czynniku zakaźnym

© JMW 2006 & EKST
RANET AMG
11
Jak dostać choroby
prionowej?
• Nabyta infekcja
• Rozwój sporadyczny (spontaniczna
mutacja PrP)
ALBO
• Transmisja dziedziczna mutacji PrP
(Mendlowska) – cecha autosomalna,
dominująca (10-15% przypadków CJD,
większość GSS, FFI, AS)

© JMW 2006 & EKST
RANET AMG
12
Czynniki ryzyka w chorobach
prionowych
•
Kanibalizm
(obecnie raczej nie występuje)
•
Genetyczne
: homozygotyzm Met/Met polimorficznego
kodonu 129 w genie PrP białka PrP
c
zwiększa podatność na
infekcję i/lub konwersję
–
Met/Met > Met/Val > Val/Val,
–
w Polsce 45% populacji Met/Met
•
Jatrogenne
:
–
Przeszczep materiału zakażonego:
• Rogówka
• Opona twarda
• Wątroba
• Szpik kostny
• Krew
–
Podawanie hormonu wzrostu z zakażonych przysadek (obecnie
rzadkie ze względu na produkcję rekombinowanego hormonu)
–
Soczewki kontaktowe wielokrotnego użycia
•
Spożywanie zakażonego mięsa
i podrobów
???

© JMW 2006 & EKST
RANET AMG
13
Cechy chorób prionowych
• Rozwój choroby – od
6 do 40 lat
od
kontaktu z czynnikiem zakaźnym, średnio 10-
12 lat („powolny wirus”)
• Jedynie rozwój vCJD szybki; śmierć w ciągu
miesięcy
(mediana 13)
od kontaktu
• CJD - śmierć w ciągu
poniżej roku od pierwszych
objawów
• Wiek chorych – powyżej 50 (mediana 68)
lat, z wyjątkiem:
• vCJD –
średnio ok. 28 lat, najmłodszy chory 12 lat
• Zespół Alpersa – początek w
niemowlęctwie

© JMW 2006 & EKST
RANET AMG
14
Objawy chorób prionowych
• Otępienie
(CJD, vCJD)
• Depresja
• Zaburzenia pamięci
• Zaburzenia chodu i in. ruchów celowych
• Śmierć w wyniku infekcji, najczęściej
zapalenia płuc
• Bezsenność
– skrócenie fazy REM i
NREM (FFI)
• Ataksja
- zaburzenia koordynacji
mięśniowej (GSS, CJD)
• Drgawki
(kuru, zespół Alpersa)

© JMW 2006 & EKST
RANET AMG
15
Choroby prionowe – zespół
GSS
• Rodzinna (genetyczna) i sporadyczna
postać choroby
• Typowo występuje w 4-5 dekadzie życia
• Charakterystycznym objawem jest
ataksja móżdżkowa (zaburzenia
ruchowe)
• Otępienie mniej powszechne i późne
• Wolny (wieloletni) postęp choroby

© JMW 2006 & EKST
RANET AMG
16
Choroby prionowe – zespół
Alpersa
• Opóźnienie rozwoju fizycznego
• Postępujące opóźnienie umysłowe
• Hipotonia mięśniowa (obniżone napięcie –
wiotkość), lub hipertonia aż do stanu spastycznego
• Otępienie
• Drgawki kloniczne i/lub padaczkowe
• Zanik nerwu wzrokowego
• Marskość wątroby
Udział zaburzeń mitochondrialnych („choroba
mitochondrialna”)

© JMW 2006 & EKST
RANET AMG
17
Patomorfologia chorób
prionowych
• Gąbczaste
zwyrodnienie tkanki
mózgowej:
– Kora mózgowa (CJD,
vCJD
, z. Alpersa)
– Ośrodki
podkorowe, zwoje
podstawne (CJD,
vCJD
, z. Alpersa)
– Wzgórze (FFI,
vCJD
)
– Móżdżek (GSS, CJD,
vCJD
)
UWAGA:
BRAK CECH ZAPALENIA!
APOPTOZA?!
Ubytki tkanki
Złogi amyloidu
Glioza astrocytowa
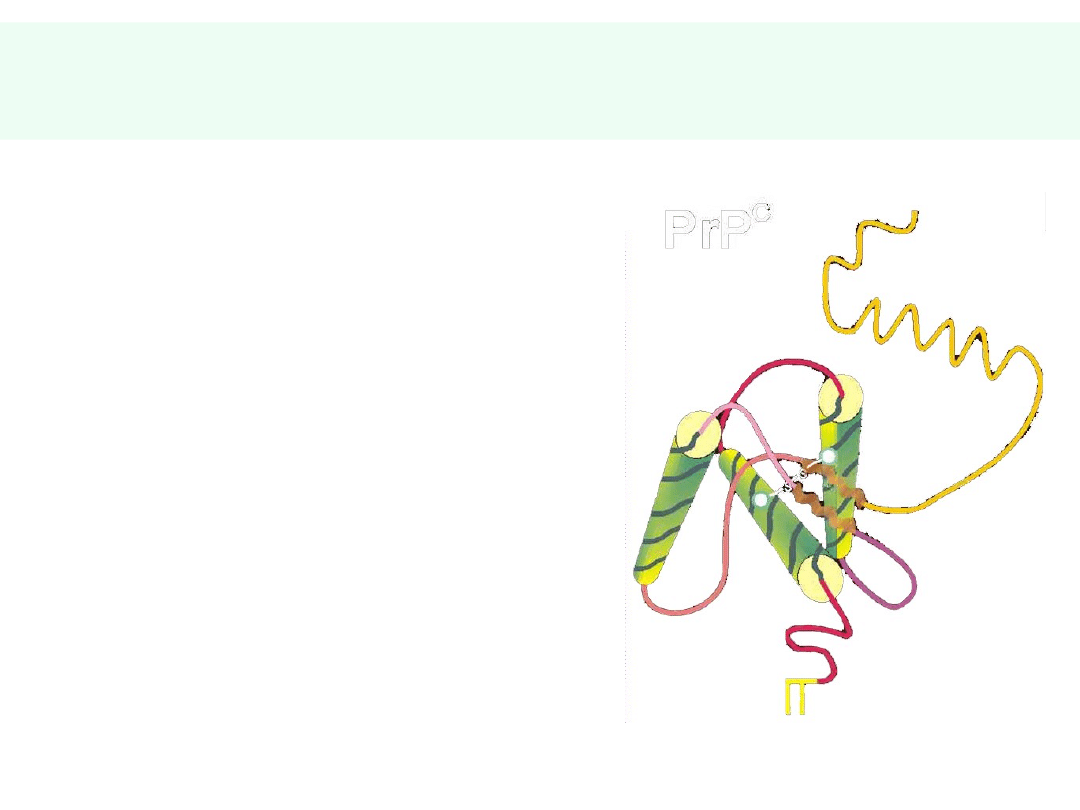
© JMW 2006 & EKST
RANET AMG
18
Właściwości białka PrP
c
• Białko macierzyste:
PrP
c
33-35, (254
aminokwasy), produkt
genu PrP na
chromosomie 20.
• EKSPRESJA: komórki
nerwowe, limfocyty,
szpik kostny nabłonki
i inne.
• Białko
PrP
c
33-35
związane z błoną
komórkową przez
fosfatydyloinozytol (PI)
© JMW 2006 & EKST
RANET AMG
19
Do czego służy komórkom
PrP
c
?
• Wiązanie jonów Cu
2+
– rola w procesach
oksydoredukcji
– detoksykacja
– „zmiatanie” wolnych
rodników
– materiał „genetyczny”
starszy od RNA i DNA
(drożdże, Paramecium)?
– regulacja rytmu dobowego
i snu?
BARDZO WYSOKA (8095%) HOMOLOGIA MIĘDZYGATUNKOWA

© JMW 2006 & EKST
RANET AMG
20
Powstawanie zakaźnego białka
PrP
sc
• PrP 33-35
(zmutowane?)
podlega
zmianom posttranslacyjnym w
endosomach prowadzącym do
przemiany w
białko prionowe PrP
sc
27-30
:
1.glikozylacja
2.wytworzenie wiązania S=S pomiędzy dwoma
resztami Cys
3.usunięcie N-terminalnego peptydu sygnałowego i
pierwszych 57 aminokwasów
4.zastąpienie C-terminalnego segmentu
hydrofobowego przez fosfatydyloinozytol (PI).
• PrP
sc
- sialoglikoproteina (27-30 kDa),
145 aminokwasów
• PrP
sc
posiada znaczną zdolność do
polimeryzacji z wytworzeniem złogów
AMYLOIDU

© JMW 2006 & EKST
RANET AMG
21
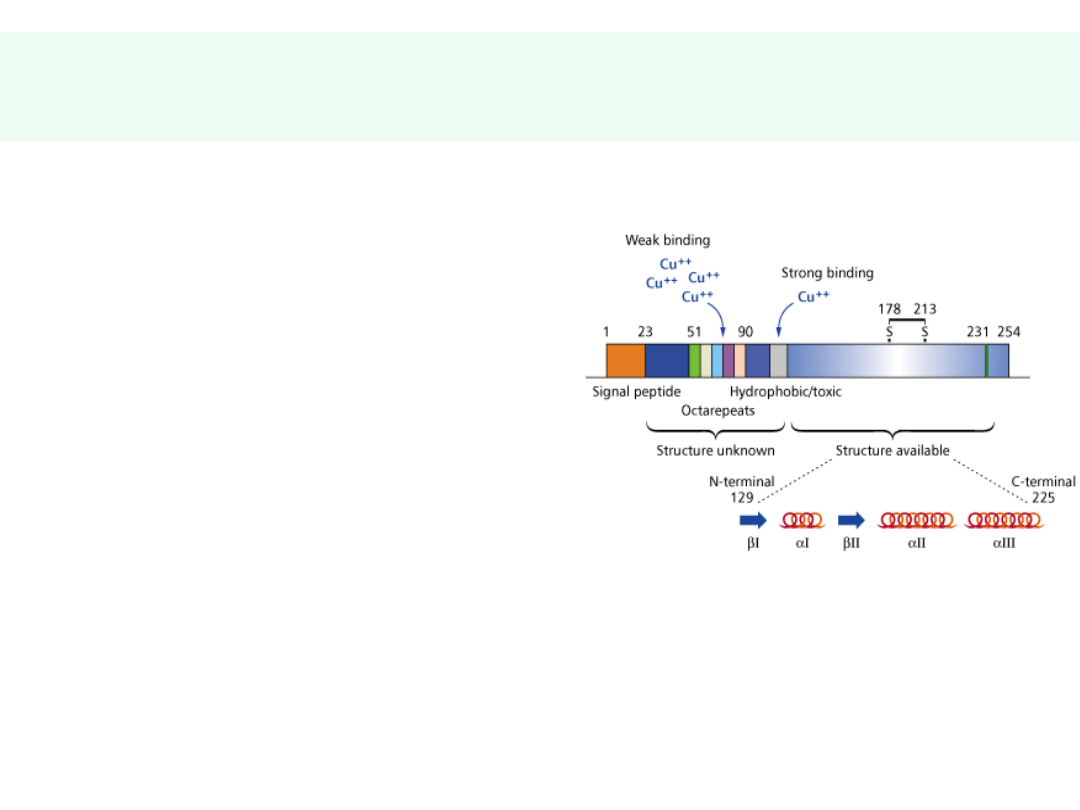
Genetyka chorób prionowych –
charakterystyczne mutacje PrP
• CJD
- częste D178 N, V210L, E200K
• GSS
– D178 N, A117V oraz P102L
• FFI
– równoczesna obecność polimorfizmu Met-
Met w pozycji 129 oraz mutacji D178 N
Mutacje zwiększają szybkość i/lub częstość
konwersji PrP
c
PrP
sc
LUB
• są warunkiem podatności na infekcję
Mutacje mogą decydować o lokalizacji uszkodzeń
© JMW 2006 & EKST
RANET AMG
22
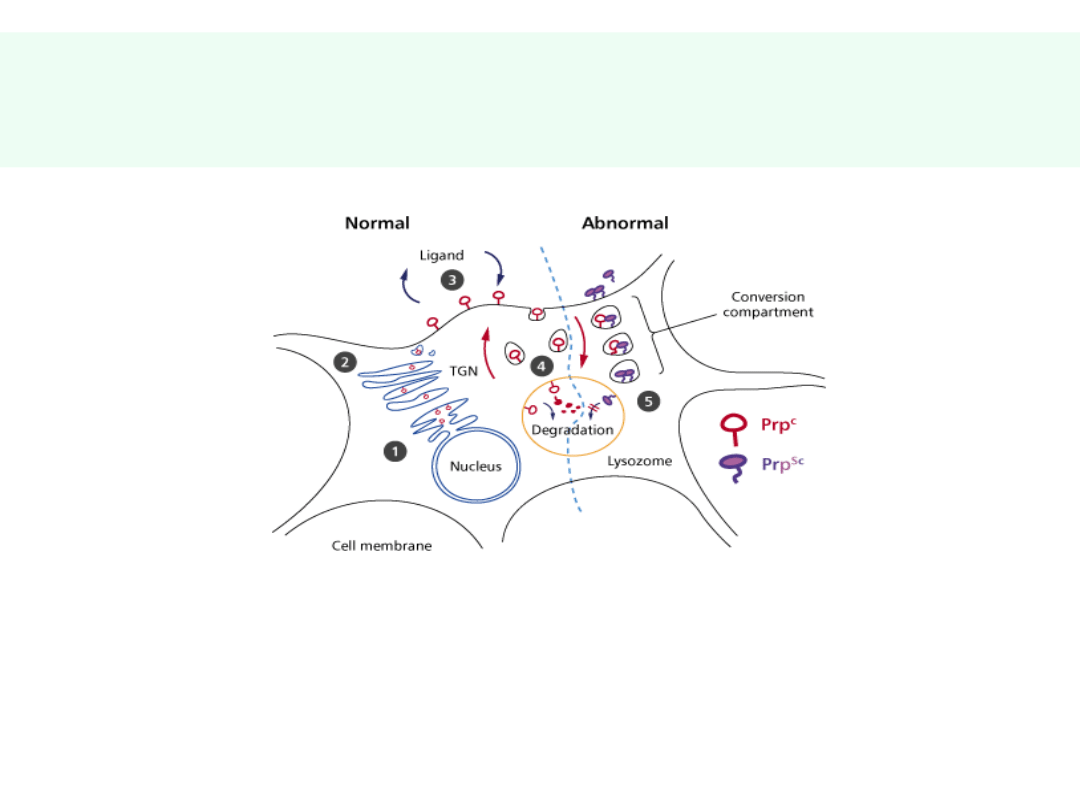
Konwersja PrP
c
PrP
sc
(1)
PrP
sc
jest odporne na proteazy,
wypełnia i uszkadza lizosomy – autoliza?
uszkadza mitochondria - apoptoza
© JMW 2006 & EKST
RANET AMG
23
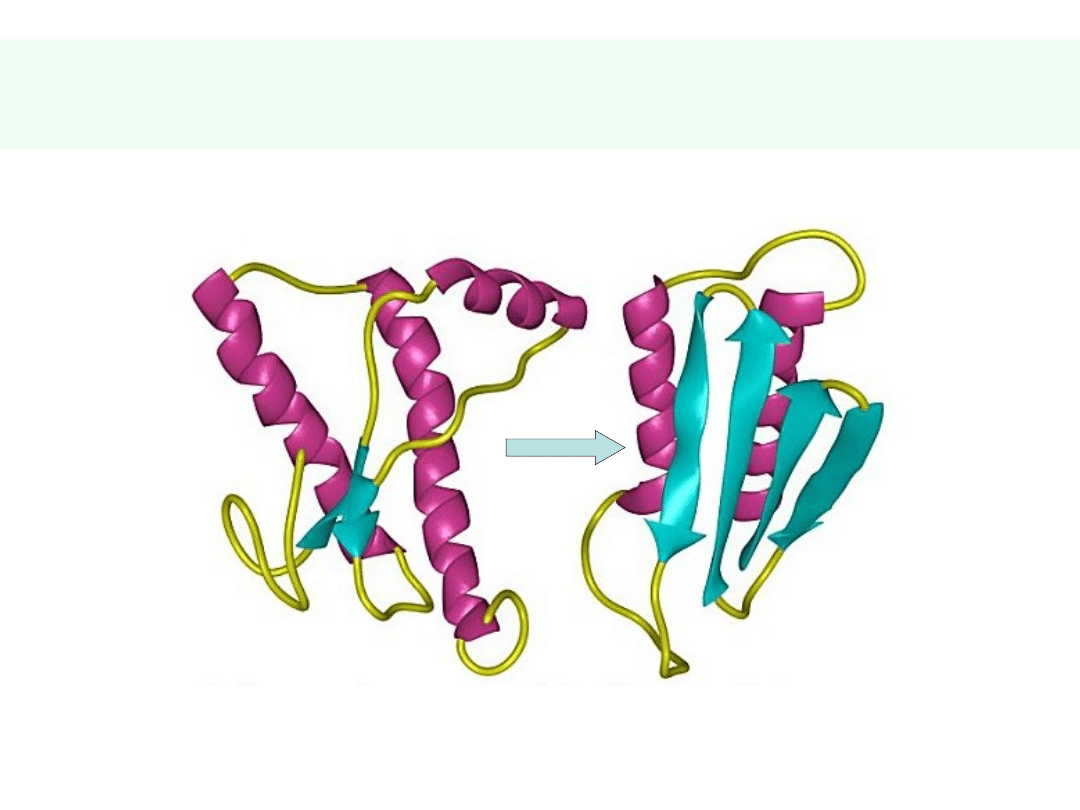
Konwersja PrP
c
PrP
sc
(2)
Mutacja 178
Asp
(D) 178
Asn
(N) w spontanicznym CJD
Forma NORMALNA (PrP
c
33-35) Forma ZAKAŹNA (PrP
sc
27-30)
© JMW 2006 & EKST
RANET AMG
24
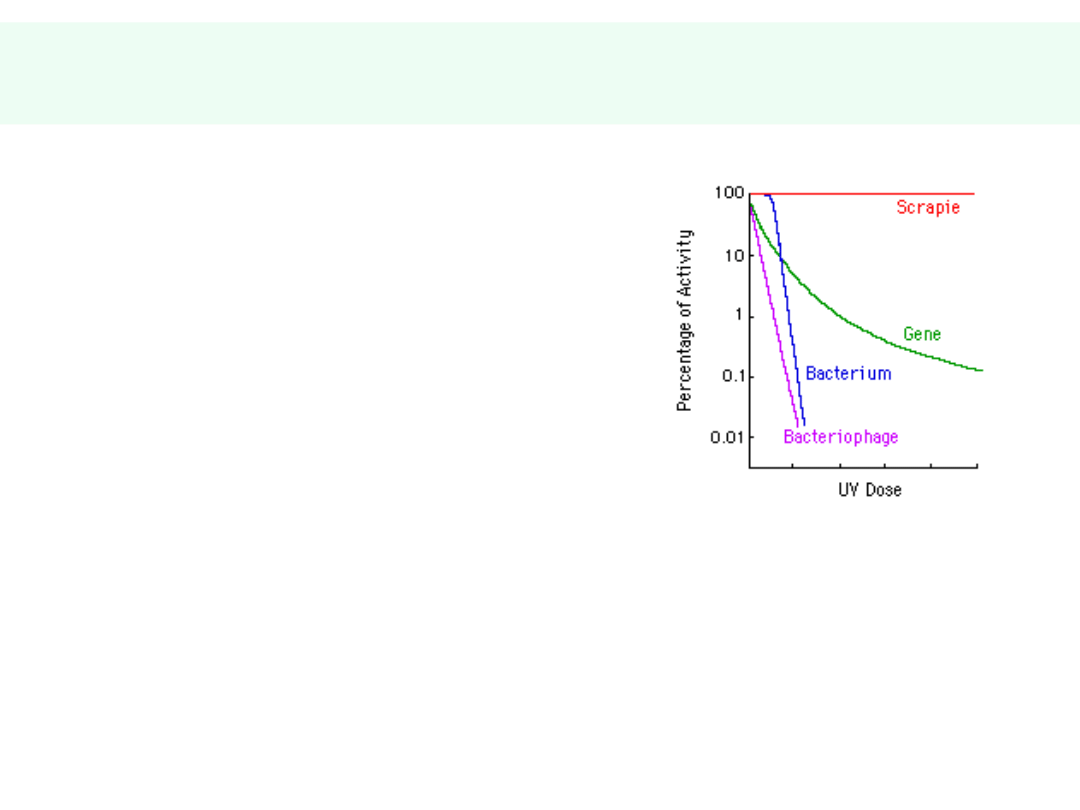
Białko prionów PrP
sc
jest odporne
na:
• Proteolizę (proteazy
wewnątrzkomórkowe i
pozakomórkowe)
• Promieniowanie UV, γ
• Środki dezynfekcyjne: alkohol,
chloraminę, formaldehyd
• Silne kwasy
• Silne zasady
• Autoklawowanie
• Wysoką temperaturę -
zakaźność po
spopieleniu w temperaturze 600
o
C
= 15%, zanik zakaźności dopiero po
wyżarzeniu w temperaturze 1000
o
C
Potencjalnie:
konieczność używania wyłącznie
jednorazowych
narzędzi chirurgicznych, zwłaszcza w
neurochirurgii,
oftalmologii i laryngologii

© JMW 2006 & EKST
RANET AMG
25
Infekcja i patogeneza
odzwierzęca:
•
PRIONY zawarte w pokarmie są absorbowane w
kępkach Peyera (MALT)
•
Komórki układu MALT (limfocyty, makrofagi,
DC?) zawierające PRIONY wędrują do narządów
limfatycznych (węzły chłonne, śledziona,
migdałki)
•
Replikacja PRIONÓW w narządach
limfatycznych
•
Wsteczny transport PRIONÓW wzdłuż
aksonów komórek unerwiających narządy
limfatyczne do rdzenia kręgowego i mózgu
DOWODY INFEKCYJNOŚCI PRIONÓW:
•
Myszy SCID są oporne na infekcję PRIONAMI
(rola układu odpornościowego)
•
Myszy PrP null (nokaut) nie mogą być
zainfekowane
•
Karmienie zwierząt drapieżnych tkanką
mózgową/mięsem zwierząt chorych na BSE
rozwój TSE
•
Podanie oczyszczonego białka prionowego
(BSE, CJD) myszom lub chomikom rozwój TSE
(międzygatunkowa transmisja choroby)
W LATACH 80-TYCH XX WIEKU ZJEDZONO OKOŁO 750
TYSIĘCY SZTUK WOŁOWINY ZAINFEKOWANYCH
PRIONEM BSE

© JMW 2006 & EKST
RANET AMG
26
Choroby prionowe zwierząt
(i odzwierzęce?)
• Choroba wściekłych krów (BSE)
vCJD?
• Scrapie (owce i kozy)
• Zakaźna encefalopatia zwierząt
futerkowych
• Chroniczna choroba
wyniszczeniowa saren i jeleni
• Zakaźna encefalopatia
kotowatych
• Drób? Ryby?
TSE (transmissible spongiform encephalitis) –
zakaźne gąbczaste zwyrodnienie mózgu

© JMW 2006 & EKST
RANET AMG
27
Postulaty Kocha – warunek uznania
etiologii choroby zakaźnej – są tylko
częściowo spełnione w przypadku
prionów
• Mikroorganizm musi być stale izolowany z materiału,
pochodzącego od chorych
• Mikroorganizm hodowany in vitro musi zachować
patogenność w stosunku do podatnego gatunku zwierząt,
wywołując u nich te same objawy chorobowe (warunek
niespełniony, ale materiał z chorych zwierząt zaraża inne
zwierzęta)
• Mikroorganizm musi być możliwy do izolacji z tak zakażonych
zwierząt
SEKWENCJA AMINOKWASÓW PRIONÓW
IZOLOWANYCH OD CHORYCH na vCJD
ODPOWIADAŁA SEKWENCJI PRIONÓW BSE
© JMW 2006 & EKST
RANET AMG
28
Krowa trojańska?

© JMW 2006 & EKST
RANET AMG
29
Choroba Alzheimera i inne
otępienia
Dr Alois Alzheimer (1864-
1915, profesor psychiatrii
we Wrocławiu, później
profesor patologii w
Monachium)

© JMW 2006 & EKST
RANET AMG
30
Choroby otępienne
• PROGRESYWNE ZABURZENIA I
ZNACZNA UTRATA WYŻSZYCH
CZYNNOŚCI UMYSŁOWYCH:
• pamięci
• inteligencji
• zdolności logicznego myślenia
• zdolności do kontaktów społecznych
• przeżywania uczuć (emocji)
• struktury osobowości

© JMW 2006 & EKST
RANET AMG
31
Postaci chorób otępiennych
• Podłoże organiczne:
• Choroba Alzheimera (70%)
• Choroba Parkinsona
• Choroba ciałek Levy’ego
• Podłoże naczyniowe:
• Otępienie w wyniku mnogich udarów
• Naczyniowe otępienie podkorowe (choroba Binswangera)
• Otępienie związane z uszkodzeniem okolicy czołowej
• lub czołowo-skroniowej:
• Choroba Picka (10%)
• Choroba (pląsawica) Huntingtona (monogenowa)
• Otępienie na tle infekcji:
• Kiła
• AIDS
• Choroba Creutzfeldt-Jakoba
UWAGA:
Podeszły wiek jest czynnikiem ryzyka, ale nie
bezwzględnym! Nie istnieje jednostka chorobowa „otępienie
starcze”!

© JMW 2006 & EKST
RANET AMG
32
Choroba Alzheimera
• 10 milionów przypadków na świecie, 200 tysięcy w Polsce
(50% wszystkich przypadków otępień)
• OBECNIE NIEULECZALNA
• Postępująca (rozwój wieloletni, 5 - 20 lat)
• Neurodegeneratywna – postępujące niszczenie neuronów
• Forma spontaniczna zwykle po 65 roku życia
• Forma rodzinna — po40 rż. (1% wszystkich
• przypadków)
• CZYNNIKI RYZYKA
• Podeszły wiek (65 lat: 1/100, 85 lat: 25~
50
/100)
• Rodzinne występowanie choroby
• Czynniki genetyczne
• Zespół Downa
• Urazy głowy
• Płeć żeńska
• Niski poziom wykształcenia

© JMW 2006 & EKST
RANET AMG
33
Objawy choroby Alzheimera - 1
• Wczesne:
• Utrata pamięci
• Zaburzenia uwagi
• Wydłużenie reakcji
• Niezrozumienie (np.
utrata wątku w trakcie
rozmowy, czytania
książki)
• Zaburzenia mowy (np.
powtarzanie się)
• Zaburzenia oceny sytuacji
• Depresja

© JMW 2006 & EKST
RANET AMG
34
Objawy choroby Alzheimera - 2
• Późniejsze:
• Nasilenie objawów wczesnych
• Uproszczenie języka
• Zaburzenia oceny wzrokowo-
przestrzennej
• Zobojętnienie i inne zaburzenia
osobowości
• Pomieszanie (konfuzja) — utrata
związku z otaczającym światem
• Utrata zdolności poznawczych
• Zanik rytmu biologicznego
• Utrata zdolności do
podejmowania podstawowych
czynności (jedzenie, ubieranie,
higiena osobista)
• Utrata kontroli nad
czynnościami fizjologicznymi
• Infekcje - ŚMIERĆ
© JMW 2006 & EKST
RANET AMG
35
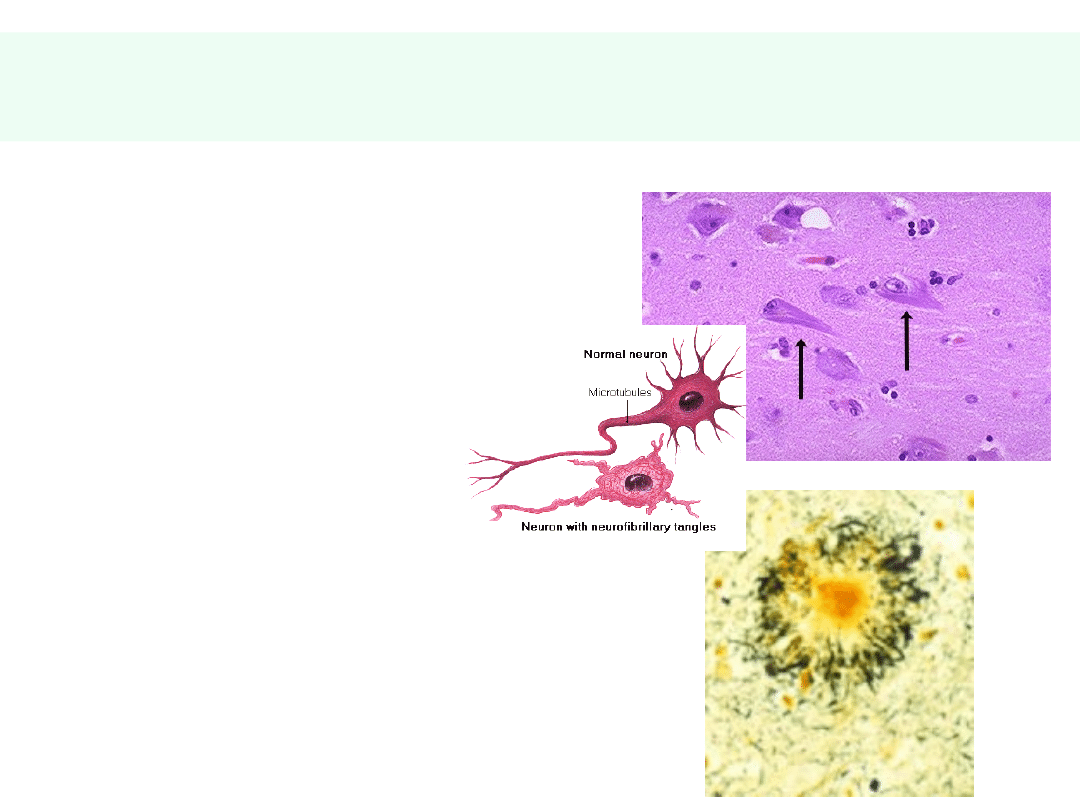
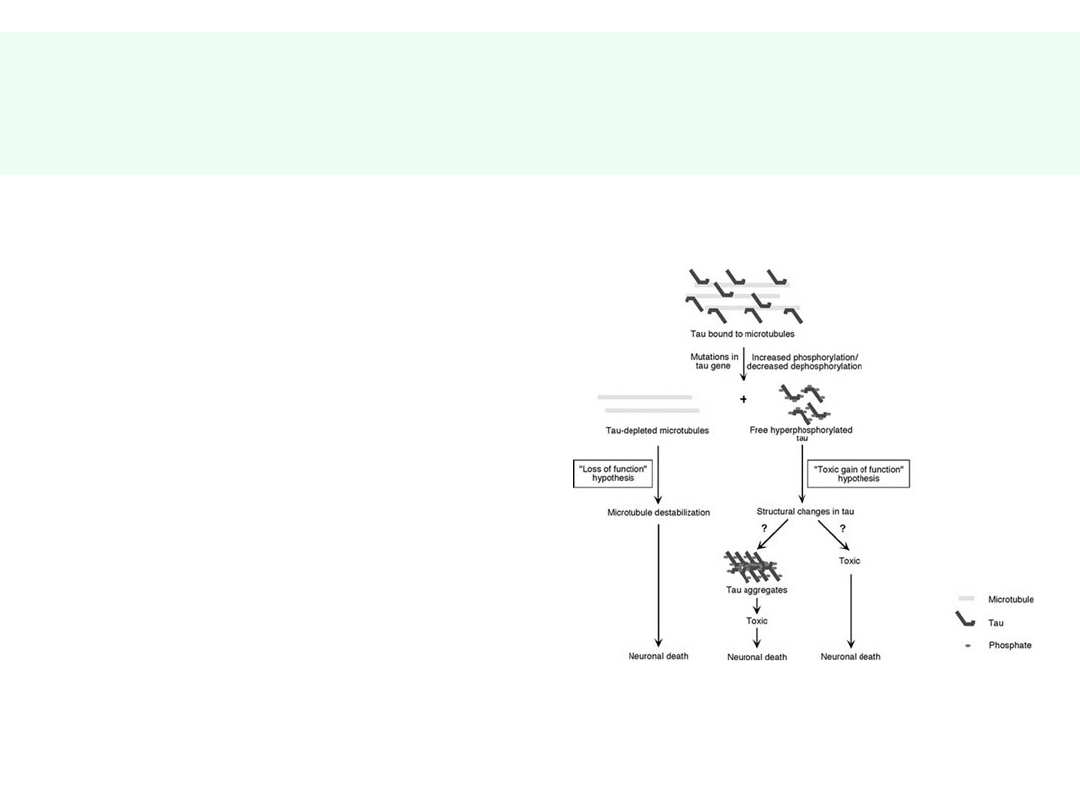
Patomorfologia AD
• SPLOTY
NEUROFIBRYLLARNE
• Białko tau
• BLASZKI
AMYLOIDOWE:
• Beta-amyloid (fragment
białka APP)
• Apolipoproteina E (ApoE)
• Mikroglej
© JMW 2006 & EKST
RANET AMG
36
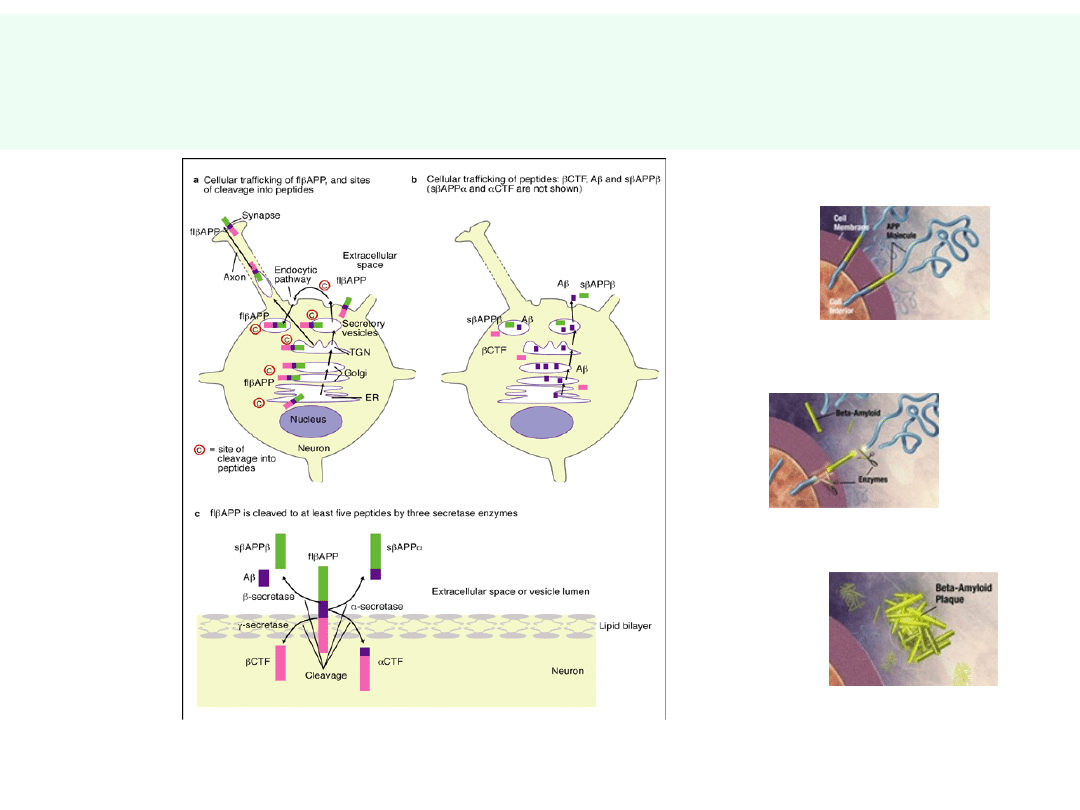
Obróbka APP – powstawanie
β-amyloidu
Nieprawidłowa obróbka proteolityczna APP —
powstanie β-amyloidu (p40,p42) — forma
agregująca,
nierozpuszczalna
© JMW 2006 & EKST
RANET AMG
37
Niszczenie neuronów w chorobie
Alzheimera
• Podłoże toksyczno-
zapalne:
• Toksyczny (bezpośredni)
wpływ amyloidu i/lub tau
• Gromadzenie mikrogleju
• Sekrecja cytokin, wolnych
rodników, NO
• Efekt cytotoksyczny
• Apoptoza
• Rola odporności
swoistej?
• AD choroba zaburzeń
fałdowania białka?
© JMW 2006 & EKST
RANET AMG
38
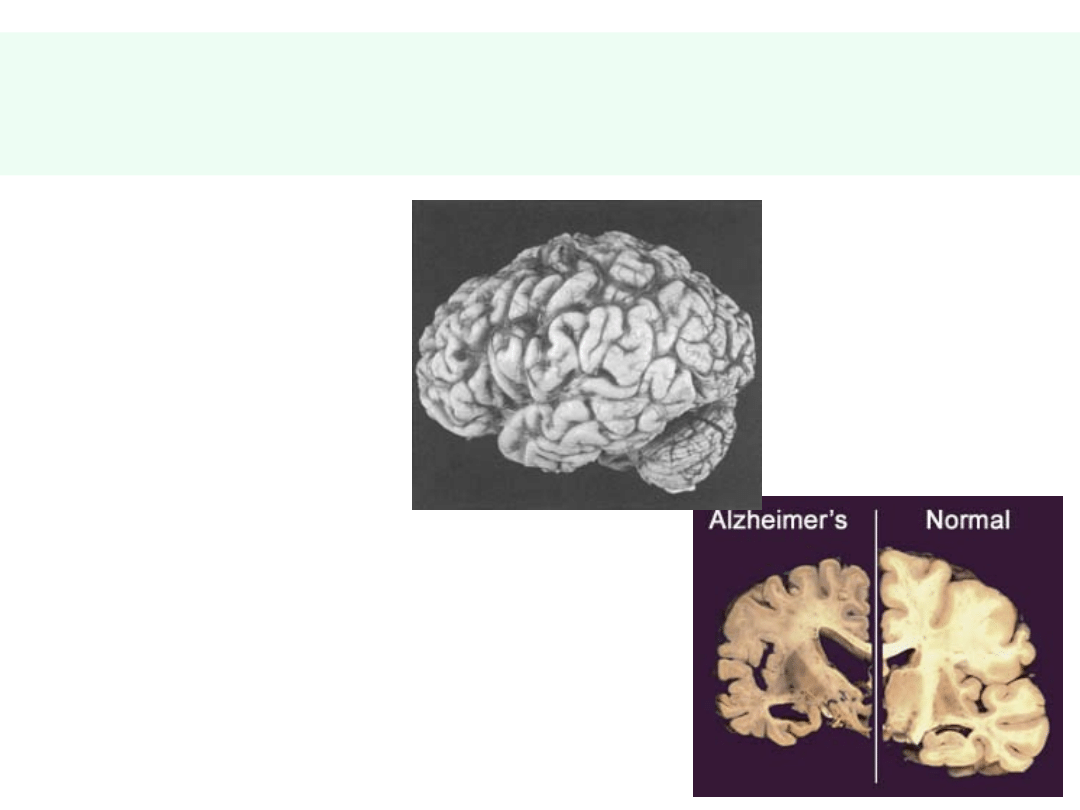
Konsekwencje zaniku neuronów w
AD
• Poszerzenie
bruzd
• Obniżenie
zakrętów
• Powiększenie
komór mózgu
© JMW 2006 & EKST
RANET AMG
39
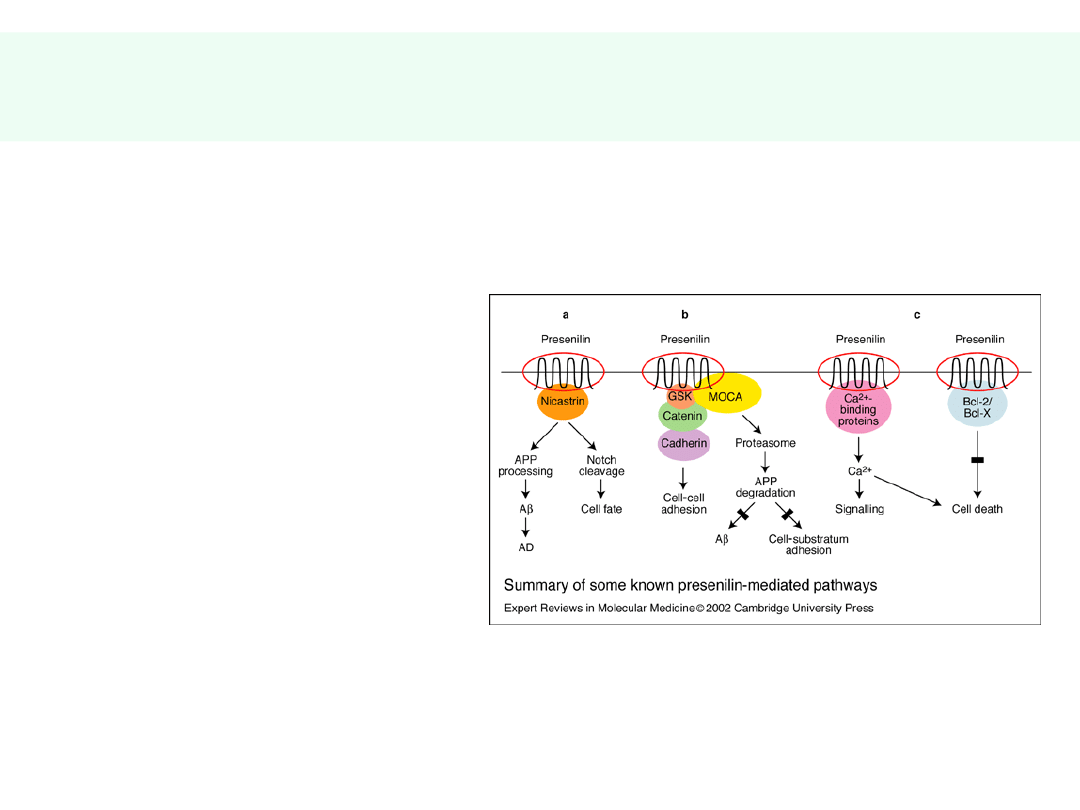
Genetyka choroby Alzheimera -
1
• Choroba nie zależy od
zaburzeń w pojedynczym
genie
• Rodzinna choroba
Alzheimera — mutacje
genów w chromosomach: 1,
14, 21
• Presenilina-1 (PS-1) —
mutacje prowadzą do
wzrostu produkcji i
odkładania AMYLOIDU
• PS-1 jest składnikiem
kompleksu enzymatycznego
tnącego APP (gamma —
sekretazy)

© JMW 2006 & EKST
RANET AMG
40
Genetyka choroby Alzheimera -
2
• Apolipoproteina E (ApoE) (składnik blaszek towarzyszący
amyloidowi)
• 3 allele — ApoE 2, 3, 4
• homozygoty ApoE 2 — protekcja
• homozygoty i heterozygoty ApoE4 — większe ryzyko, wcześniejszy rozwój
• Chorzy na AD częsta hipercholesterolemia
© JMW 2006 & EKST
RANET AMG
41
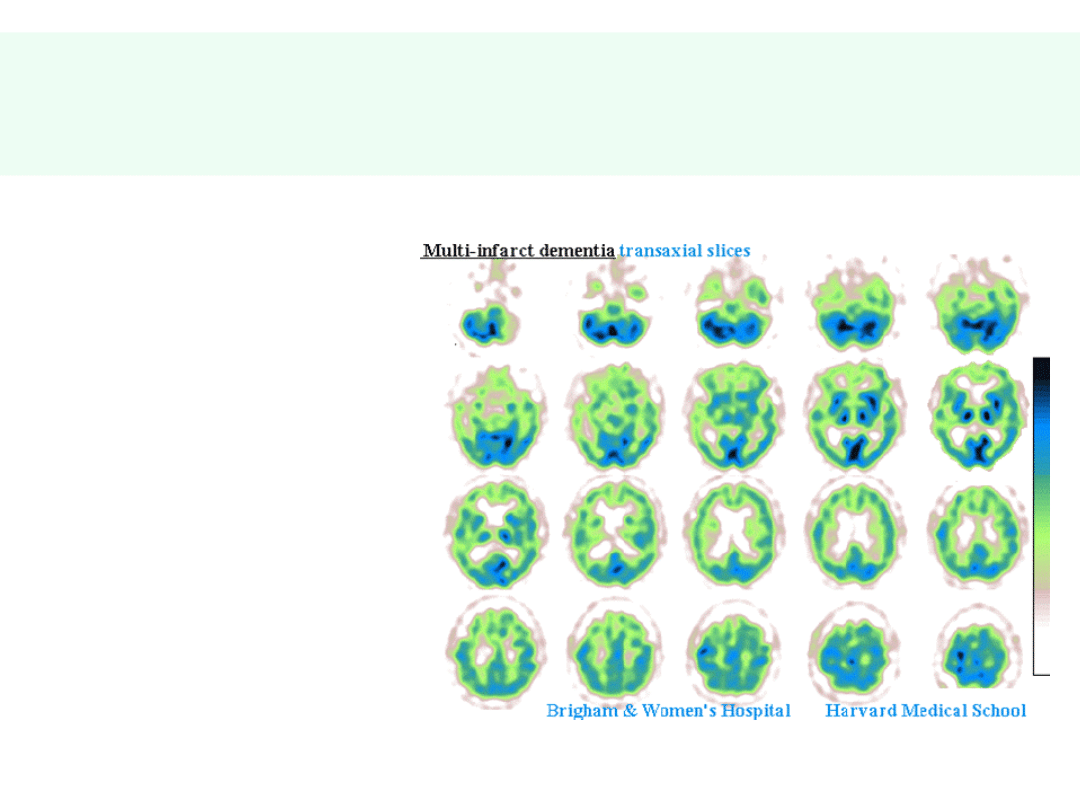
Otępienie w wyniku mnogich
udarów
• dowolna
lokalizacja
zaburzenia
naczyniowego
= dowolność
objawów
psycho- i
neurologicznyc
h
© JMW 2006 & EKST
RANET AMG
42
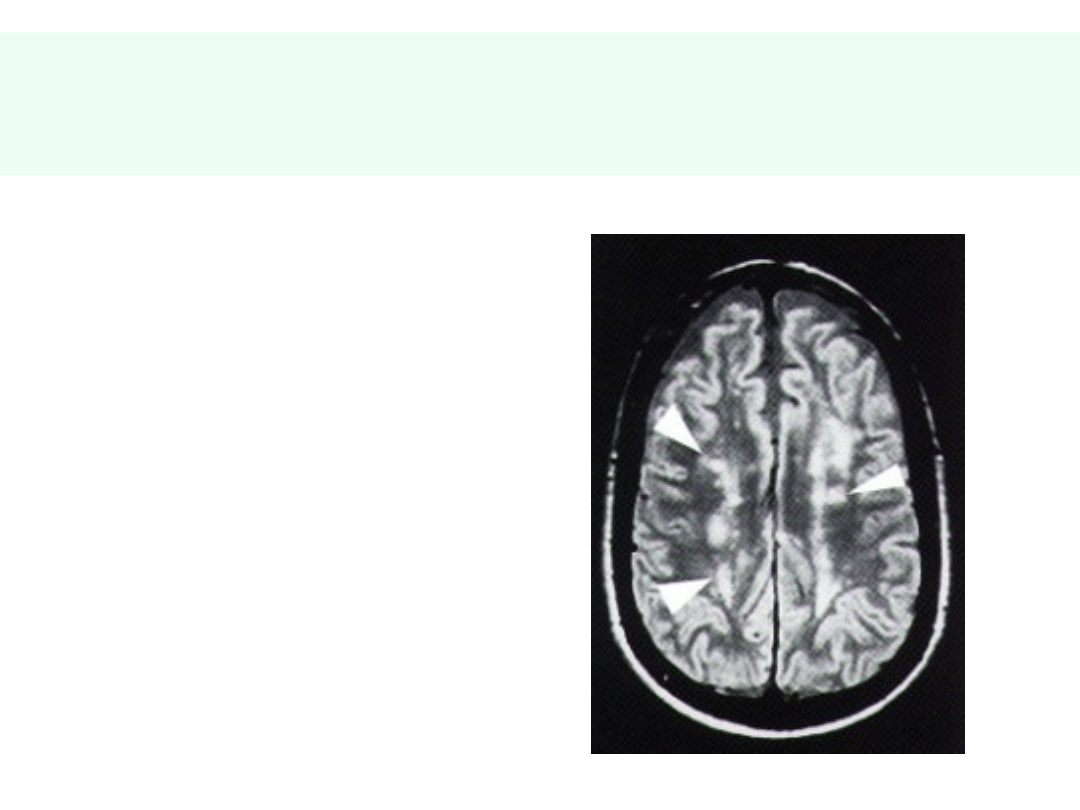
Naczyniowe otępienie podkorowe
(ch. Binswangera)
• Spowolnienie,
ospałość
• Skoki
emocjonalne
• Specyficzne
uszkodzenia
naczyń
podkorowej istoty
białej

© JMW 2006 & EKST
RANET AMG
43
Choroba Picka
• Objawy związane z
lokalizacją (czołowo -
skroniowa):
• Zaburzenia osobowości
• Zmiany w zachowaniu
(utrata zahamowań)
• Zaburzenia mowy i
języka (wulgaryzacja)
• Możliwe podłoże
genetyczne
© JMW 2006 & EKST
RANET AMG
44
Choroba Parkinsona
• Zanik w obrębie
substantia nigra
• Zaburzenia
koordynacji ruchowej
• Ruchy zamiarowe
(trudność rozpoczęcia
ruchu celowego)
• Zaburzenia mowy
• Drżenia
• Późne otępienie,
nasilające się pod
wpływem terapii
znoszącej powyższe
objawy
_
+
+
_
© JMW 2006 & EKST
RANET AMG
45
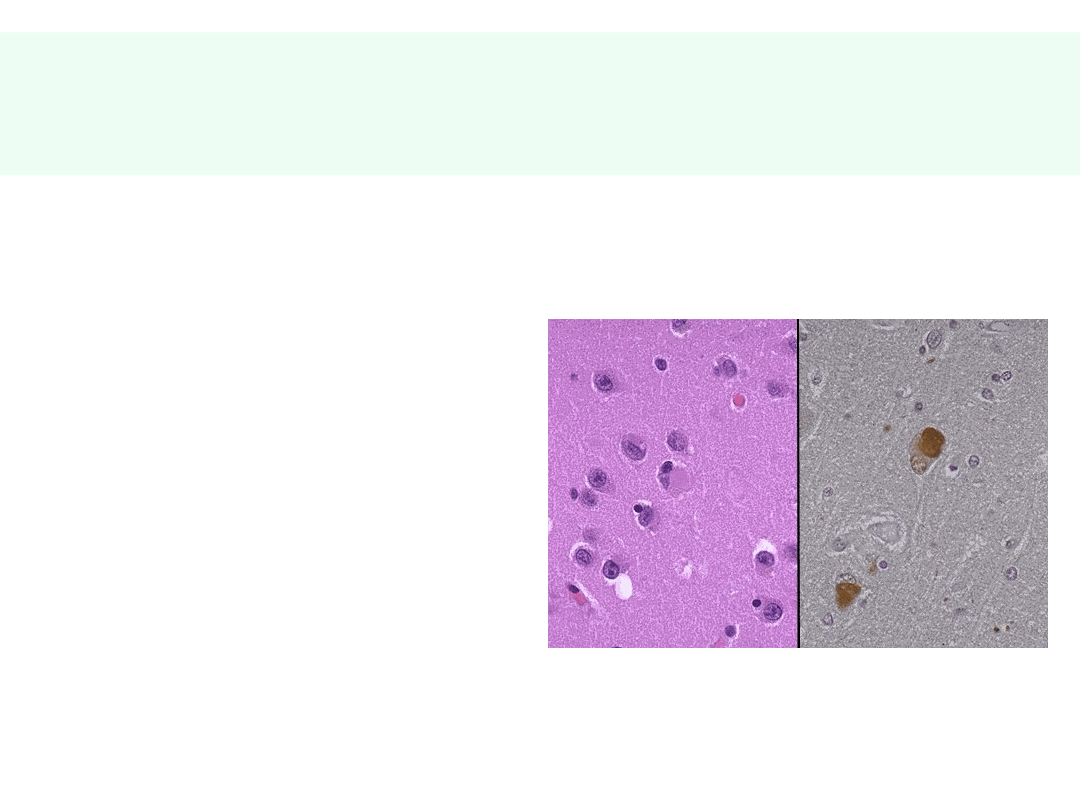
Choroba ciałek Levy’ego
• Gromadzenie
wewnątrzkomórkowych inkluzji z:
• (neuro)melaniny
• synukleiny
• objawy ch. Alzheimera (wariant?)
• zaburzenia pamięci
krótkotrwałej
• zaburzenia zachowania,
uwagi
• zaburzenia mowy
• objawy ch. Parkinsona —
• zaburzenia koordynacji
ruchowej, drżenia
• złożone halucynacje wzrokowe
• znaczna fluktuacja nasilenia
objawów
© JMW 2006 & EKST
RANET AMG
46
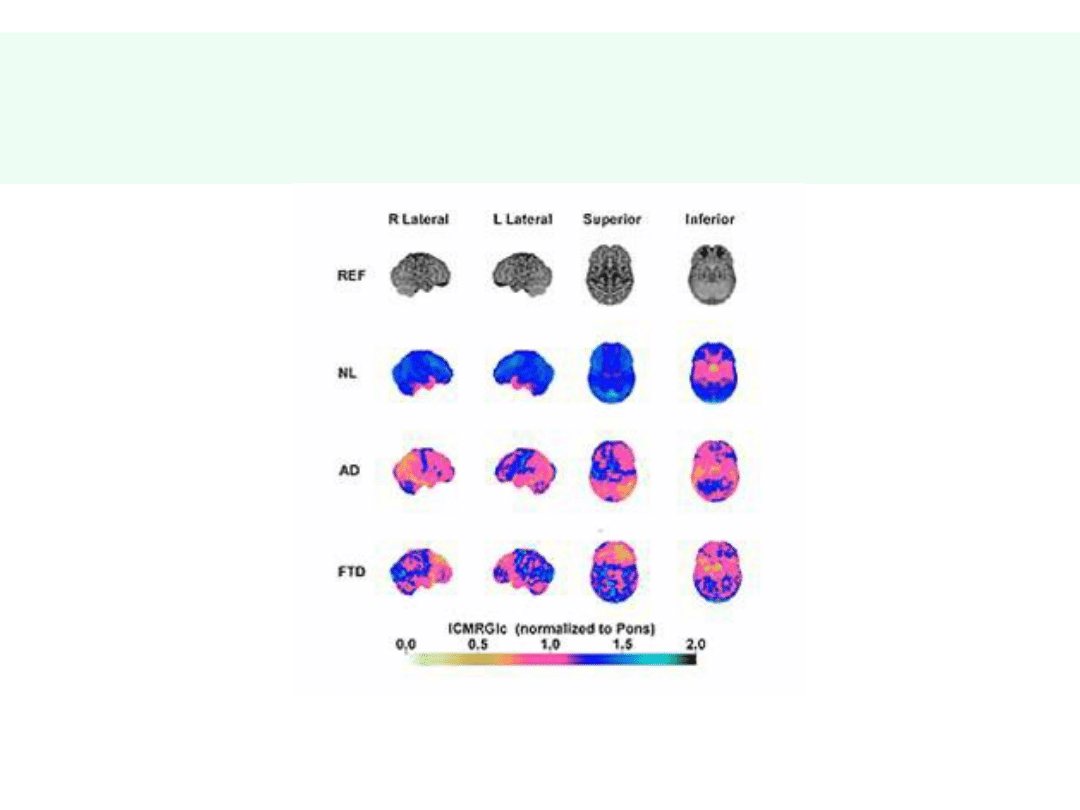
Metabolizm glukozy w mózgu w
chorobach otępiennych
•
NL – kontrola
•
AD – choroba Alzheimera
•
FTD – otępienie czołowo-skroniowe

© JMW 2006 & EKST
RANET AMG
47
Zaburzenia fałdowania i agregacji białek –
wspólne podłoże chorób
neurodegeneracyjnych?
• Choroba Alzheimera - białko β-amyloidu tworzące tzw.
„blaszki starcze”
• Choroba Parkinsona - α-synukleina tworząca ciałka
Lewy’ego
• Choroba Huntingtona – huntingtyna tworząca wtręty
śródjądrowe
• Choroba Creutzfeldta-Jakoba i inne pionowe- agregacje
białka prionowego
Rodzaj choroby (zespół objawów) zależy od rodzaju uszkodzonego
(zmutowanego) białka, skutków mutacji oraz lokalizacji złogów.

© JMW 2006 & EKST
RANET AMG
48
KONIEC
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
Wyszukiwarka
Podobne podstrony:
W18 Patofizjologia OUN choroba Alzheimera i inne otępienia, choroby prionowe
choroba alzheimera i inne zespoły otępienne wieku podeszłego
W8 Choroba Alzheimera i inne zespoły otępienne
Patofizjologia, WYKłAD - Choroby OUN, WYKŁAD
15 Zaburzenia neurologiczne w chorobach wewnetrznych
15 G08 H06 Choroby zakaźne wersja IHiT
15 Zaburzenia neurologiczne w chorobach wewętrznych 1 lightid 16253 ppt
13 Patofizjologia wybrane choroby infekcyjne 2011id 14478 ppt
Patofizjologia Zatrucia i Choroby Krwi, Wykłady
wyklady 2009, oun, Choroby OUN
Szkol Choroby Prionowe
15 Patofizjologia narządu ruchu nie potrzebny
Ćwiczenie 15, Patofizjologia, Ćwiczenia 13-15 (wydalniczy, nerwowy, nowotwory, toksykologia, rytmy b
Choroby prionowe, biologia, wykłady
więcej podobnych podstron