
Niedoczynność przytarczyc
• Częstość występowania – nieznana (po
strumektomii 1-10%)
• Objawy: tężyczka, mrowienie, drętwenie
kończyn, zaćma patologiczna,
zwapnienia jąder podstawy mózgu
• Niedostateczne wydzielanie PTH lub
oporność na PTH
• Badania biochemiczne: hipokalcemia,
hiperfosfatemia, hipokalciuria

Nadczynność przytarczyc
•Częstość występowania – 25-28/100000/rok
•Trzykrotnie częściej u kobiet
•Hiperkalcemia, hipofosfatemia, hiperkalcuria,
wysokie wydzielanie parathormonu
•Ubytek masy kostnej, resorpcja podokostnowa,
torbiele kostne, kamica nerkowa
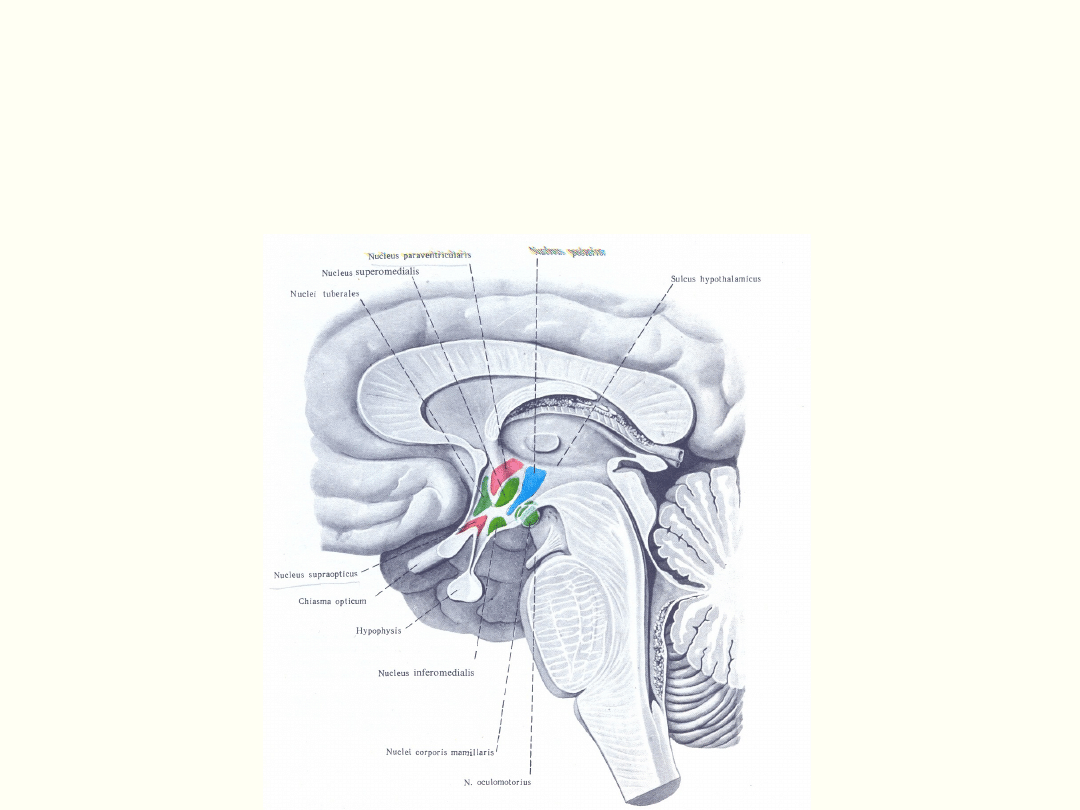
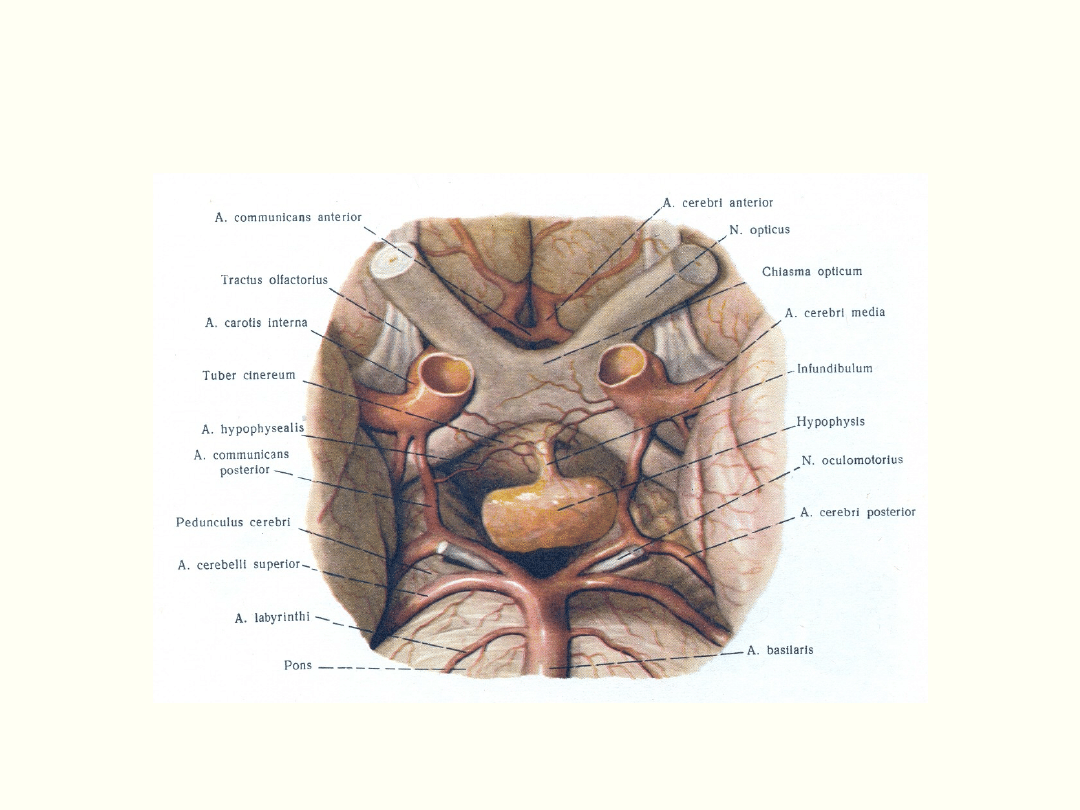
Podwzgórze i przysadka
mózgowa
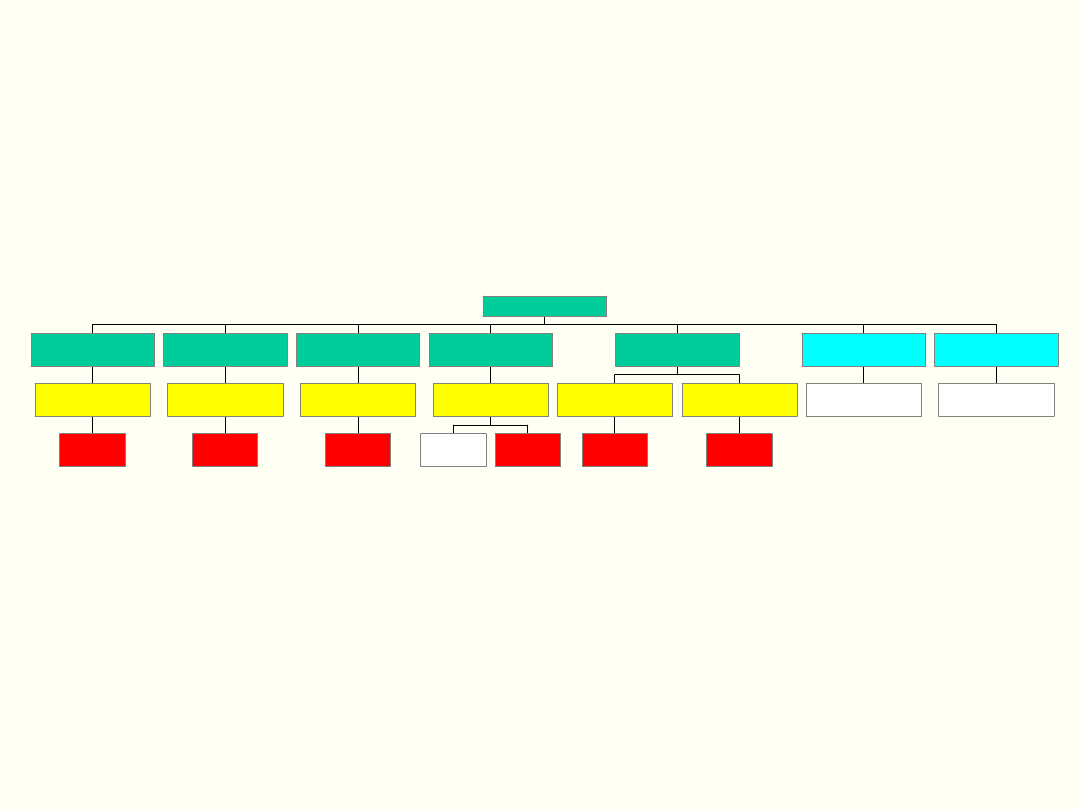
T a r c z y c a
T 3 i T 4
T S H
T R H
I G F - 1
G H
G H - R H
N a d n e r c z a
C o r tiz o l
A C T H
C R F
S u tk i
J a jn ik i
P r o la k ty n a
P I F
D o p a m in a - h a m u je P R L
J a jn ik i/ J ą d r a
F S H
J a jn ik i/ J ą d r a
L H
G N - R H
K a n a lik i d y s t a ln e n e r e k
M ię ś n ie g ł n a c z y ń
W a z o p r e s y n a
M ię ś n ió w k a m a c ic y
P r z e w o d y m le k o w e
O k s y t o c y n a
P o d w z g ó r z e

Prolaktinoma
• Gruczolak z komórek laktotropowych
przysadki mózgowej
• Hipogonadyzm hipogonadotropowy
• Mlekotok
• Brak miesiączki/zaburzenia płodności
• U mężczyzn impotencja
• Zapadalność 6/100 000 osób rocznie

Akromegalia
Charakterystyka kliniczna:
Akromegalia charakteryzuje się powiększeniem
twarzoczaszki, rąk i stóp, obrzmienier(l i rozrostem
tkanek miękkich, kości oraz narządów wewnętrznych
w - następstwie nadmiernego wydzielania hormonu
wzrostu (GH) przez gruczolak przysadki wywodzący
się z komórek somatotropowych.
Chorobowość w akromegalii wynosi 50-70/mln.
populacji, natomiast zachorowalność, czyli liczba
nowo rozpoznanych przypadków w ciągu roku wynosi
około 4/1 mln. populacji. Chorobę rozpoznaje się
dwukrotnie częściej u kobiet.

Akromegalia
• Akromegalia i gigantyzm są niemal zawsze skutkiem gruczolaka
przysadki wydzielającego hormon wzrostu. Gruczolaki tego typu
stanowią około 1/5 wszystkich gruczolaków przysadki (i są
drugie co do częstości występowania po prolactinoma).
• Znacznie rzadszą przyczyną akromegalii jest ektopowe
wydzielanie GRH, występujące u niektórych pacjentów z
rakowiakiem lub wyspiakiem trzustki. Ektopowe wydzielanie
hormonu wzrostu spotyka się bardzo rzadko.
• W akromegalii obserwuje się charakterystyczne zaburzenia
regulacji wydzielania GH. Wzrasta liczba i wielkość wyrzutów
tego hormonu, przy jednoczesnym braku typowego wyrzutu
nocnego.
• Zmienia się też odpowiedź wydzielania GH na czynniki
pobudzające bądź hamujące. Większość objawów przewlekłej
nadprodukcji GH jest związana z syntezą zwiększonych ilości
somatomedyny C (IGF-I), co prowadzi do charakterystycznego
wzrostu kości, chrząstek i tkanek miękkich oraz do
powiększenia narządów wewnętrznych.
• Insulinooporność i obniżona tolerancja glukozy są bezpośrednim
skutkiem nadmiaru GH .

Akromegalia - objawy
• Rozrost tkanek miękkich z powiększeniem dłoni i stóp oraz
pogrubieniem rysów twarzy.
• Zwiększona potliwość, tycie, łojotok oraz uczucie
zmęczenia.
• W momencie postawienia diagnozy (zwykle 5-10 lat od
początku choroby) zmiany układu chrzęstnego i kostnego -
głównie czaszki -pogrubienie sklepienia czaszki,
powiększenie zatok czołowych, powiększenie nosa oraz wzrost
żuchwy i szerokie rozstawienie zębów.
• Zwiększenie wymiarów dłoni i stóp - konieczność zwiększenia
rozmiarów
• Przerost chrząstek powoduje bóle stawowe, zwyrodnienie
stawów kola
nowych, biodrowych i kręgosłupa.
• U niemal połowy pacjentów pojawia się fotofobia niejasnego
pochodzenia.
• W akromegalii dochodzi także do powiększenia narządów we
wnętrznych, ale klinicznie uchwytne jest zwykle wole i
powiększenie ślinianek.
• Kardiomegalia wynika raczej z nadciśnienia tętniczego.
• Rzadko rozwija się klinicznie jawna cukrzyca.
• U około połowy pacjentów występują objawy hipogonadyzmu.

Diagnostyka akromegalii
• Oznaczenie hormonu wzrostu i IGF- I
• Test hamowania glukozą. U zdrowych osób podanie
doustne 100 g glukozy powoduje po godzinie spadek
stężenia GR poniżej 2 ng/ml; w akromegalii stężenie to
niemal zawsze przekracza tę wartość.
• W rzadkich przypadkach także stwierdzenie pobudzenia
wydzielania GR przez TRH lub hamowania - przez
dopaminę lub bromokryptynę.
• Ze względu na częste współistnienie hiperprolaktynemii
u pacjentów z akromegalią należy oznaczyń Prolaktynę
• MRI ew. TK
• W przypadku braku zmian w obrębie siodła lub sugestii
rozlanej hiperplazji przysadki należy rozważyć
możliwość ektopowego wydzielania GRH lub, ewentualnie,
GR.

• Leczenie neurochirurgiczne -skuteczność do
80%.
• Za kryterium wyleczenia zwykle przyjmuje się
trwałe utrzymywanie poziomów GR poniżej 5
ng/mI.
• W przypadku nieskuteczności leczenia
chirurgicznego należy rozważyć zastosowanie
konwencjonalnej radioterapii w dawce 4500-
5000radów. (liczne powikłania)
• Analogi somatostatyny - m.in. Sandostatyna LAR
(Octreotide) lub nowszy - Lanreotyd.
• Bromokryptyna - skuteczność jedynie u 10-20%
pacjentów z akromegalią.
Diagnostyka i leczenie
akromegalii

Choroba i zespół Cushinga
• Choroba Cushinga charakteryzuje się zaczerwienieniem
i zaokrągleniem twarzy, czerwonymi rozstępami w
skórze, otłuszczeniem karku i tułowia, zanikami mięśni,
głównie kończyn, nadciśnieniem tętniczym, cukrzycą i
osteoporozą w następstwie hiperkortyzolemii
wywołanej nadmiernym wydzielaniem ACTH przez
gruczolak przysadki wywodzący się z komórek
kortykotropowych .
• Gruczolak przysadki wydzielający ACTH - choroba
Cushinga - jest najczęstszą przyczyną hiperkortyzolemii
prowadzącej do zespołu Cushinga. Zapadalność roczna
wynosi 0,1-1/100 000. Chorują głównie dorosłe kobiety
(8 x częściej niż mężczyźni).

• W 1932 r. Harvey Cushing przedstawił obraz kliniczny chorych, u
których stwierdził w badaniu sekcyjnym zasadochłonnego gruczolaka
przedniego płata przysadki.
• Następnie opisano podobny zespół objawów klinicznych w
przypadkach guzów nad nerczy, a zakres chorób nazwanych zespołem
Cushinga znacznie się rozszerzył. Postęp w medycynie, a szczególnie w
badaniach biochemicznych i technikach ob:t;azowych, w tym próby
wykrywania guzów ektopowo wydzielających ACTH/CRH lub ich
przerzu tów za pomocą scyntygrafii z użyciem znakowanych analogów
somatostatyny, znacznie ułatwiły rozpoznawanie całego spektrum
patologii
związanych
hiperkortyzolemią.
W
ostatnich
latach
zastosowano syntetyczny (lub bydlęcy) CRH do testu stymulującego z
jednoczesnym cewnikowaniem zatok skalistych dolnych z oznaczaniem
ACTH. Mimo stałego postępu ustalenie etiologicznego rozpoznania
zespołu Cushinga często wymaga wielokrotnego powtarzania badań,
jest trudne i nie zawsze pewne.
• Po stwierdzeniu klinicznych objawów sugerujących hiperkortyzolemię,
postępowanie diagnostyczne obejmuje:
• - potwierdzenie rozpoznania hiperkortyzolemii,
• - ustalenie etiologii i lokalizacja zmiany odpowiedzialnej za powstanie
zespołu Cushinga.

Etiologia hiperkortyzolemii
• 1. Zaburzenia podwzgórzowo-przysadkowe (choroba Cushinga).
• 2. Zaburzenia pierwotnie nadnerczowe (zespół Cushinga), w
tym: rak, gruczolakorak, gruczolak kory nadnerczy,
drobnoguzkowa dysplazja kory nadnerczy, pierwotna pig
mentacyjna guzkowa dysplazja kory nadnerczy bez zespołu
Carneya lub z towarzyszącym zespołem Carneya.
• 3. Zaburzenia kory nadnerczy bez supresji ACTH (guzkowa
dysplazja kory nadnerczy).
• 4. Zespół ektopicznego wytwarzania ACTH i CRH, ACTH lub CRH.
• 5. Rozsiany nowotwór wielogruczołowy MEN I - guz kory
nadnerczy wytwarzający kortyzol lub, o wiele rzadziej, guz
przysadki wytwarzający ACTH.
• 6. Przerost kory nadnerczy wywołany przez GIP.
• 7. Zespół Cushinga w pierwszym roku życia.
• 8. Zespół rodzinnej oporności na glikokortykoidy.
• 9. Hiperkortyzolemia jatrogenna.
• 10. Rzekomy zespół Cushinga występujący w stanach depresji,
jadłowstręcie psychicznym, alkoholizmie.

Objawy hiperortyzolemii
• Objawami występującymi w różnej częstości są:
• - nadmierny przyrost masy ciała z charakterystycznym
rozkładem tkanki tłuszczowej na twarzy, tułowiu;
•
- zmiany skórne: ścieńczenie skóry, sinoczerwone
rozstępy, zmiana zabarwienia skóry na ciemniejszą, trądzik,
meszek, hirsutyzm, wybroczyny, wylewy podskórne, obrzęki,
• plamy soczewicowate (lentigines);
• - podwyższone ciśnienie tętnicze krwi;
• - zaburzenia psychiczne: depresja, senność, chwiejność
emocjonalna, nadmierna drażliwość; - łatwe męczenie się,
słabość fizyczna;
• - bóle kręgosłupa, zaburzenia uwapnienia kości;
• - bóle głowy, bóle brzucha;
• - nawracające infekcje;

Objawy hiperkortyzolemii u
dzieci
• U dzieci objawem występujacym w 100% jest zmiejszenie
szybkości wzrastania prowadzące do niskorosłości,
opóźniony wiek kostny
• wczesne pojawienie się owłosienia płciowego
• androgenizacja w formie przerostu łechtaczki u dziewcząt,
makrogenitosomii u chłopców bez powiększenia objętości
jąder
• objawy zależne od nadmiaru androgenów nadnerczowych
• opóźnienie występowania rozwoju gruczołów piersiowych
• opóźnianie się wystąpienia menarche lub zaburzenia
miesiączkowania

Badania w hiperkortyzolemii
• podwyższenie stężenia hemoglobiny i liczby
erytrocytów, leukocytoza obojętnochłonnaz
eozynopenią;
• nieprawidłowa tolerancja węglowodanów w teście z
doustnym obciążeniem glukozą lubjawna cukrzyca;
• zaburzenia w gospodarce elektrolitowej i kwasowo-
zasadowej (alkaloza hipokalerniczna);
• podwyższone markery resorpcji kostnej (wzmożone
wydalanie wapnia z moczem);
• cechy osteoporozy w badaniach RTG kości czaszki,
kręgosłupa;- podwyższenie poziomu cholesterolu i
triglicerydów we krwi;
• badanie okulistyczne w przypadku guzów okolicy siodła
może wykazać ograniczenie pola widzenia, osłabienie
ostrości wzroku, zanik nerwów wzrokowych, obrzęk tarcz
nerwów wzrokowych.

• 1. Oznaczenie ACTH w osoczu (RIA) w próbkach rannych i nocnych po
2-3 próbki.
• Wartości prawidłowe dla próbek rannych: < l 0-80 pg/ml « 2,2-17,6
pmol/L).
• Pobieranie próbek krwi do oznaczania ACTH powinno być wykonane z
przestrzeganiem wszystkich zasad obowiązujących w tym oznaczaniu
(probówki schłodzone, próbki krwi przenoszone do laboratorium w
lodzie i odwirowane natychmiast w wirówce z chłodzeniem).
• W chorobie Cushinga wartości ACTH mogą być nieco podwyższone lub
w normie.
• W zespole Cushinga mogą być w dolnej granicy normy, poniżej lub
nieoznaczalne.
• W zespole ektopicznego ACTH/CRH są w górnej granicy normy lub
wysokie.
• Nie we wszystkich przypadkach wartość stężenia ACTH różnicuje
wymienione postacie hiperkortyzolemii.
• Badanie ACTH ma niską wartość w różnicowaniu pomiędzy chorobą
Cushinga a zespołem ektopii.
Badania w hiperkortyzolemii

Test z Dexametazonem
• Test hamowania małą i dużą dawką deksametazonu.
• Badanie wyjściowe: dwie kolejne DZM w celu oznaczenia 17 -OHCS,
WKM, kreatyniny.
• Po wyjściowych zbiórkach moczu rozpoczyna się I i II dzień testu. 8
dawek deksametazonu po 0,5 mg co 6 godzin, zaczynając o godz 8°°.
Rano w dniu I (przed pierwszym podaniem deksametazonu) i w dniu III
pobiera się krew na oznaczenie stężenia kortyzolu i androgenów
nadnerczowych. W II dobie prowadzi się DZM w celu oznaczenia 17-
0HCS, WKM i kreatyniny.
• W dniu III i IV pacjent otrzymuje deksametazon po 2 mg co 6 godzin;
pierwsze podanie dużej dawki następuje po pobraniu krwi w dniu III o
godz. 8°°.
• W dniu V, 6 godzin po ostatniej dawce 2 mg deksametazonu, czyli o
godzinie 8°° pobiera się krew na oznaczenie poziomu korty zolu i
androgenów nadnerczowych.
• DZM prowadzi się w IV i V dobie na badania, jak przed testem i w II
dobie.

17 -OHCS w moczu:
• u zdrowych wartość 17 -OHCS obniża się poniżej 50% wartości
wyjściowej po małej dawce (DZM w II dobie).
•Brak hamowania małą dawką deksametazonu potwierdza stan
hiperkortyzolemii.
• Obniżenie 17 -OHCS do poniżej 64% wartości wyjściowej po dużej
dawce przemawia za przysadkowym pochodzeniem hiperkortyzolemii,
•Brak hamowania do wartości poniżej 64% średniej z dwóch DZM
wyjściowych wskazuje na zespół Cushinga pochodzenia nadnerczowego
lub na ektopię.
•WKM: obniżenie ilości wydalonego kortyzolu w moczu do poniżej 90%
wartości wyjściowej po dużej dawce przemawia za chorobą Cushinga.
•W hiperkortyzolemii pochodzenia nadnerczowego lub w zespole
ektopicznego ACTH nie następuje obniżenie wydalanego WKM.
Kortyzol we krwi: u zdrowych poziom kortyzolu w osoczu obniża się do
< 50 nmolll (<1,8 ~g/dl) po drugim dniu podawania małej dawki. U
pacjentów z chorobą Cushinga następuje obniżenie stężenia kortyzolu
do poniżej 50% wartości wyjściowej po dużej dawce. Jeżeli spadek
nastąpi już po małej dawce, test można przerwać.Niedostateczne
zmniejszenie wydalania 17-0HCS, WKM i poziomu kortyzolu we krwi po
dużej dawce deksametazonu przemawia za pierwotnie nadnerczowym
zespołem lub ektopowym wydzielaniem ACTH.

Leczenie hiperkortyzolemii
Choroba Cushinga
• Leczenie jest chirurgiczne. Selektywnie usuwa się gruczolaka przysadki
dojściem przez zatokę klinową lub stosuje częściową hipofizektomię.
Alternatywą w stosunku do reoperacji jest radioterapia z towarzyszącym
leczeniem lekami blokującymi steroidogenezę nadnerczową, szczegól
nie jeśli podczas pierwszej operacji stwierdzono naciekanie opony
twardej lub zatoki jamistej. Obustronna adrenalektomia może być
konieczna u pacjentów z chorobą Cushinga, u których nie uzyskano
wyleczenia.
Zespół Cushinga
• Leczenie chirurgiczne: usunięcie jednego lub obu nadnerczy. W
przypadkach raka obowiązuje leczenie mitotanem po wykonanej
adrenalektomii lub stosowanie chemioterapii.
Zespół ektopowego wydzielania ACTH/CRH
• Leczenie chirurgiczne - wycięcie guza. Jeżeli lokalizacja guza nie została
ustalona lub zabieg był nieskuteczny, stosuje się leczenie zachowawcze
lekami hamującymi wytwarzanie kortyzolu.
• Leczenie farmakologiczne: Ketokonazol (3x 1),
Aminoglutetymid,

Guz chromochłonny
• Guz chromochłonny (pheochromocytoma) charakteryzuje się
napadowym nadciśnieniem z zaburzeniami rytmu, bladością
skóry, potami i bólami głowy spowodowanymi zwiększonym
wydzielaniem katecholamin (noradrenaliny, adrenaliny,
dopaminy) najczęściej przez guz rdzenia nadnerczy.
• Guz chromochłonny występuje u około 0,1% chorych na
nadciśnienie tętnicze i w 90% jest guzem sporadycznym,
zlokalizowanym w rdzeniu nadnerczy; 20% pozornie
sporadycznych guzów ma podłoże genetyczne, z czym wiąże
się większa częstość guzów mnogich, o lokalizacji
pozanadnerczowej (paraganglioma) i złośliwych.

Incydentaloma nadnercza
• Przypadkowo wykryty guz nadnercza - incydentaloma -
jest to nieprawidłowa masa tkankowa w obrębie
nadnercza, wykryta w czasie badania obrazowego
podjętego z innych wskazań.
• Guzy nadnerczy wykrywa się przypadkowo w 1-3%
badań metodą tomografii komputerowej i w 2-15%
badań autopsyjnych, najczęściej po 60. roku życia, około
2,5 razy częściej u kobiet niż u mężczyzn

Niedoczynność kory nadnerczy
• Niedoczynność kory nadnerczy charakteryzuje się ogólnym
osłabieniem, brakiem apetytu i chudnięciem, niedociśnienie
ortostatycznym, hiponatremią, hiperkaliemią i hipoglikemią,
na skutek pierwotnego lub wtórnego niedoboru hormonów
kory nadnerczy (glukokortykoidów, mineralokortykoidów i
androgenów nadnerczowych
• Częstość występowania:
Pierwotna nkn: 60-140/milion, zależnie od badanej
populacji. 80% na skutek procesu autoimmunologicznego
(adrenalitis)
Wtórna nkn: brak danych (szacunkowo: > 200/ milion?)

Moczówka prosta
• Diagnostyka różnicowaW postępowaniu diagnostycznym
należy przede wszystkim wykluczyć inne przyczyny poliurii.
Są one związane najczęściej z diurezą osmotyczną, tak jak w
przypadku cukrzycy. W takich przypadkach osmolalność
moczu jest zbliżona do osmolalności osocza. W
przeciwieństwie do powyższego, osmolalność moczu w
moczówce prostej lub polidypsji psychogennej jest wyraźnie
niższa od osmolalności osocza. Tak więc ciężar właściwy
moczu poniżej 1,005 (osmolalność 200 mosm/kg wody)
pozwala na wyklucze nie poliurii związanej z diurezą
osmotyczną. W badaniu podmiotowym należy zwrócić
szczególną uwagę na rytm picia i oddawania moczu.
• Podstawowym badaniem jest określenie osmolalności
pobranych w tym samym czasie próbek osocza i moczu. W
obu formach moczówki prostej mocz będzie bardziej
rozcieńczony niż osocze, przy jednocześnie podwyższonej
osmolalności osocza.

Testy w moczówce prostej
Próba zagęszczania moczu.
• Pacjent powinien zostać zważony, a następnie pozbawiony możliwości
przyjmowania płynów. Każdą oddaną porcję moczu pobiera się w celu
określenia ciężaru właściwego lub osmolalności. Okres 18 godzin jest
zwykle wystarcza jący dla potwierdzenia diagnozy. Próbę przerywa się
wcześniej, jeśli ciężar ciała pacjenta obniży się o ponad 3% (ze
względu na niebezpieczeństwo znacznego odwodnienia pacjentów z
moczówką). Staranny nadzór nad pacjentem w trakcie tej próby jest
wymagany także ze względu na fakt, że chorzy na polidypsję psychogenną
będą usilnie staraćsię o dostęp do wody. U osób zdrowych poddanych tej
próbie szybko dochodzi do ograniczenia diurezy i zagęszczenia moczu,
natomiast chorzy na moczówkę prostą nie zmiennie wydalają duże ilości
moczu o niskim ciężarze właściwym.
Test z wazopresyną
• Pozwala na zróżnicowanie obu form moczówki prostej. Podanie 5 jednostek
wazopresyny (s. c.) lub 1 ~g dezmopresyny (Adiuretin), syntetycznego
analogu wazopresyny (i. V., i.m. lub s.c.) powoduje u pacjentów z
neurogenną postacią moczówki obserwowany po godzinie wzrost
osmolalności moczu powyżej osmolalności osocza. W celu zwiększenia
czułości tego testu można go poprzedzić okresem ograniczonego
przyjmowania płynów.

Leczenie moczówki prostej
• Dezmopresyna (l-dezamino-8-D-A VP),
syntetyczny analog wazopresyny
• Adiuretin – aerozol, krople do nosa co 12
godzin
• Minirin tabletki.
• Adekwatność podawanych dawek należy
kontrolować poprzez oznaczanie osmolalności
surowicy (początkowo co 1-2 tygodnie,
następnie co 3 miesiące).

Zespół Conn’a
• Hyperaldoteronizm około 0,1-2% populacji chorych z
nadciśnieniem tętniczym.
• 60-70% HP to gruczolak kory nadnercza wytwarzający
aldosteron (zespół Conna).
• Rzadziej HP spowodowany jest obustronnym rozlanym
lub guzkowym przerostem warstwy kłębkowatej kory
nadnerczy (tzw. idiopatyczny przerost kory nadnerczy).
• Zdarza się, że HP jest wywołany genetycznym
zaburzeniem syntezy aldosteronu i kortyzolu, znanym
jako aldosteronizm wrażliwy na deksametazon.
• Najrzadziej przyczyną HP jest rak kory nadnerczy, który
oprócz aldosteronu może wytwarzać również
glikokortykoidy i androgeny.

• Objawy kliniczne zależą od nadmiaru aldosteronu lub DOC i niedoboru jonów
potasowych w organizmie.
• Głównym objawem jest nadciśnienie tętnicze.
• Osłabienie mięśniowe, wielomocz (szczególnie nocny), wzmożone
pragnienie, bóle głowy.
• Rozpoznanie HP opiera się na stwierdzeniu nadciśnienia tętniczego,
hipokaliemii, alkalozy.
• Prawidłowe stężenie potasu w surowicy nie wyklucza jednoznacznie HP,
ponieważ mogą występować postacie choroby z prawidłową kaliemią.
Stężenie sodu jest najczęściej prawidłowe lub w górnej granicy normy, może
występować hipomagnezemia.
• Od 25 do 50% chorych z HP ma upośledzoną tolerancję glukozy, wynikającą
z wpływu nieprawidłowych stężeń potasu na wydzielanie insuliny.
• Dla ustalenia rozpoznania HP decydujące znaczenie ma wykazanie
zwiększonego stężenia aldosteronu we krwi i moczu, nie ulegającego
zmniejszeniu pod wpływem czynników hamujących wydzielanie tego
hormonu.
• Test obciążenia solą (4-dniowy) z oceną stężenia K i aldosteronu stosuje się
jako podstawowy test hamowania.
• Ustalenie, czy przyczyną choroby jest gruczolak, czy przerost kory
nadnerczy jest istotne ze względu na odmienny sposób leczenia. Najbardziej
powszechnym testem różnicującym jest test pionizacji, polegający na
oznaczeniu w surowicy stężenia aldoste ronu po nocnym spoczynku i pod
koniec trwającej 2 godziny pionizacji pacjenta. Zmniejszenie lub brak zmian
stężenia aldosteronu po pionizacji może przemawiać za obecnością
gruczolaka, natomiast wzrost stężenia aldosteronu może wskazywać na
idiopatyczny przerost kory nadnerczy.

• Leczenie HP spowodowanego gruczolakiem polega
na operacyjnym usunięciu guzka, które zazwyczaj
prowadzi do normalizacji ciśnienia tętniczego i
ustąpienia zaburzeń metabolicznych wynikających z
nadmiaru aldosteronu.
• Przygotowanie przedoperacyjne polega na
uzupełnieniu niedoborów potasu poprzez stosowanie
diety niskosodowej i bo gatopotasowej oraz
podawanie spironolaktonów (100-400 mg/dz.) przez
kilka tygodni przed zabiegiem.
• W idiopatycznym przeroście kory nadnerczy
leczeniem z wyboru jest przewlekle podawanie
spironolaktonów, które w razie potrzeby stosuje się
łącznie z lekami hipotensyjnymi.
Leczenie zespołu Conn’a
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
Wyszukiwarka
Podobne podstrony:
choroby przysadki stomatologia
Choroby przysadki mózgowej i nadnerczy
Choroby przysadki 2c tarczycy i nadnerczy oraz metody immunochemiczne
leki stosowane w chorobach podwzgorza i przysadki
choroby przysadki stomatologia
Choroby przysadki mózgowej i nadnerczy
Choroby przysadki
ZABURZENIA HORMONALNE U CHORYCH Z PRZEWLEKŁĄ CHOROBĄ NEREK CZĘŚĆ I PODWZGÓRZE I PRZYSADKA HIPOGONADY
Nadczynność przysadki mózgowej
1 - Podwzgórze i przysadka, MEDYCYNA, patofizjologia, Prelekcje
Hormony przysadki mózgowej antastic pl
Wpływ na skórę przysadka, tarczyca
Fw bulki, LB 5, Dla oceny czynności hormonalnej układu podwzgórze - przysadka mózgowa - jądro nie st
PRZYSADKA MÓZGOWA
więcej podobnych podstron