
PODSTAWOWE MOLEKUŁY W ŻYWYM
PODSTAWOWE MOLEKUŁY W ŻYWYM
ORGANIZMIE
ORGANIZMIE

Dna moczanowa
Dna moczanowa
(artretyzm, skaza
(artretyzm, skaza
moczanowa, - przewlekła choroba związana
moczanowa, - przewlekła choroba związana
z zaburzeniem metabolizmu kwasu
z zaburzeniem metabolizmu kwasu
moczowego powstającego z puryn.
moczowego powstającego z puryn.
Wyróżniamy dnę pierwotną i wtórną.
Wyróżniamy dnę pierwotną i wtórną.
W 90% przypadków dny pierwotnej,
W 90% przypadków dny pierwotnej,
przyczyną choroby jest upośledzone
przyczyną choroby jest upośledzone
wydalanie kwasu moczowego do światła
wydalanie kwasu moczowego do światła
cewek nerkowych za sprawą obniżenia
cewek nerkowych za sprawą obniżenia
filtracji lub sekrecji kwasu moczowego lub
filtracji lub sekrecji kwasu moczowego lub
zwiększenia jego resorpcji.
zwiększenia jego resorpcji.
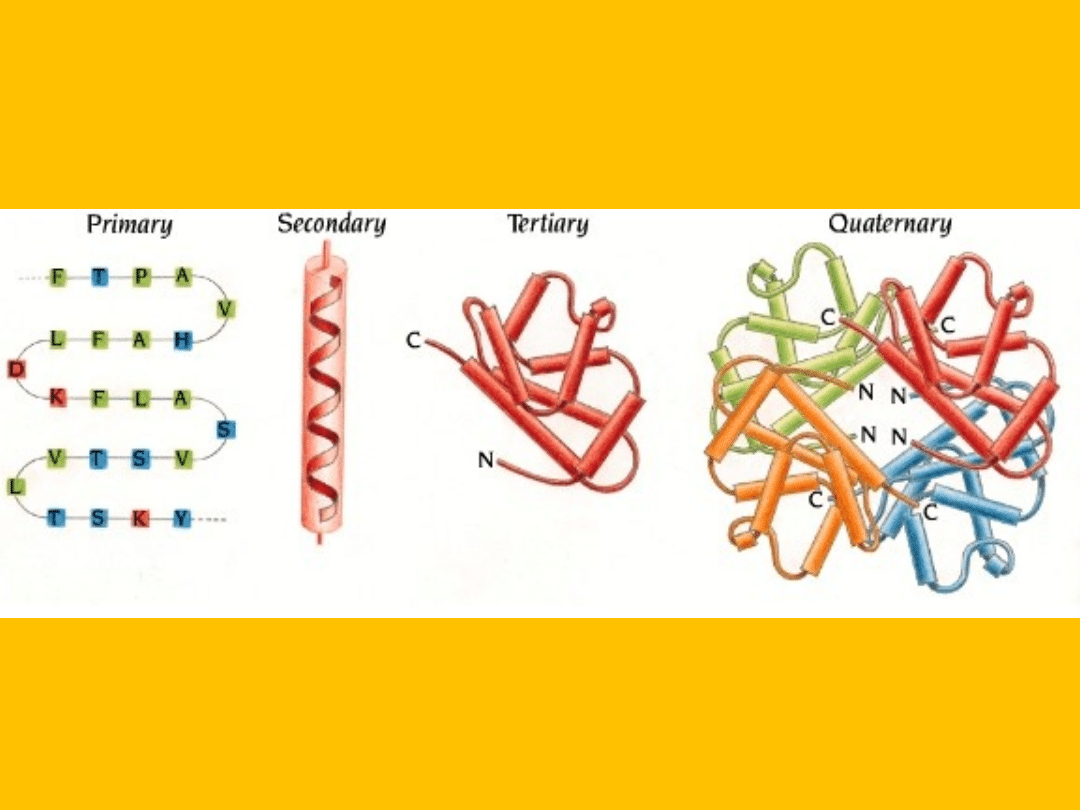
W budowie białek wyróżnia się cztery poziomy ich struktury,
(struktura I-IV rzędowa),
czyli cztery zasadnicze poziomy organizacji łańcucha polipeptydowego.

Funkcje białek:
1. KATALIZA ENZYMATYCZNA
Enzymy (E) - prawie wszystkie są białkami (odkrycie katalitycznej aktywności
RNA pozwala nam wyobrazić sobie świat, istniejący wcześnie w ewolucji życia,
przed pajawieniem się DNA i białka).
- dotychczas w pełni scharakteryzowano kilka tysięcy E,
- b. silne katalizatory, przyspieszają V reakcji przynajmniej milion razy,
2. TRANSPORT I MAGAZYNOWANIE
wielu małych cząsteczek i jonów zachodzi z udziałem białek,
np. a)
Hb - przenosi tlen - w krwinkach czerwonych,
Mioglobina - przenosi tlen - w mięśniach,
b)
transferyna - przenosi Fe - w osoczu krwi,
c)
ferrytyna – w kompleksie z Fe - w wątrobie, przechowywanie Fe.
3. RUCH UPORZĄDKOWANY
Białkowe układy kurczliwe;
- skurcz mięśnia.
- poruszanie się plemników za pomocą wici,
- przemieszczanie się chromosomów podczas mitozy, etc.
4. FUNKCJE MECHANICZNO-STRUKTURALNE
np. kolagen (białko fibrylarne) - elastyczność mięśni i tkanki kostnej

Funkcje białek c.d. :
5. OCHRONA IMMUNOLOGICZNA
Przeciwciała - białka o dużej swoistości; odgrywają istotną rolę w rozróżnianiu
między
tym co własne i obce (wirusy, bakterie, itd.) dla danego organizmu.
6. WYTWARZANIE I PRZEKAZYWANIE IMPULSÓW NERWOWYCH
Reakcje komórek nerwowych na specyficzne bodźce przebiegają z udziałem
białek receptorowych (np. rodopsyna - białko fotoreceptorowe, występujące w
komórkach pręcikowych siatkówki).
7. KONTROLA WZROSTU I RÓŻNICOWANIA
Białka kontrolują przepływ energii i materii w komórkach.
Kontrola odpowiedniej kolejności ekspresji informacji genetycznej jest
zasadniczym warunkiem uporządkowanego wzrostu i różnicowania komórek.
Istotnym elementem kontroli są: - u bakterii - białka represorowe
- w organizmach wyższych - białkowe czynniki
wzrostu
W organizmach wielokomórkowych hormony koordynują aktywność różnych
komórek.
- wiele hormonów to białka (np. insulina, hormon tyreotropowy)

FAŁDOWANIE BIAŁKA
Skład AA wpływa na strukturę przestrzenną.
Łańcuch polipeptydowy uzyskuje strukturę natywnego białka.
Fałdowanie białek przebiega w procesie postępującej stabilizacji
form pośrednich, do osiągnięcia konformacji najbardziej stabilnej
energetycznie:
U M N
U - stan rozpleciony (unfolded)
M - struktura częściowo statystycznego kłębka (zwartej
struktury)
N - stan natywny
Dla 100 AA liczba możliwych struktur wynosi 5x10
47
w czasie
1,6x10
27
lat [Paradoks Levinthala]. Jednak czas rzeczywisty
fałdowania jest bardzo krótki, gdyż zostają zachowane stany
częściowo poprawne.

Kierowanie białek i ich przemiany
Za wyjaśnienie mechanizmu kierowania białek w komorce Gunter
Blobel otrzymał nagrodę Nobla w 1999r.
Transport nowo zsyntetyzowanych białek do różnych przedziałów
komórkowych, w tym także do błon, określa się jako:
kierowanie białek lub
adresowanie białek do ich miejsc docelowych
Fałdowanie się większości białek w komórce jest wspomagane
przez enzymy
np. disulfidoizomeraza (izomeraza dwusiarczkowa)
izomeraza peptydyloprolinowa (przyspiesza izomeryzację cis-,
trans-wiązań tworzonych przez Pro).
Obecnie znamy ponad 100 różnych motywów fałdowania się (splotów)
białek.
Projektowanie białek
Skomplikowane programy komputerowe umożliwiają szerokie
wykorzystanie szybko wzrastającej ilości danych na temat sekwencji
i struktury, zebranych w odpowiednich bazach danych.

WYMIANA DOMEN STRUKTURALNYCH
JAKO MECHANIZM OLIGOMERYZACJI BIAŁEK
ang. Domain Swapping (DS)
odkrył Eisenberg (1994) w
strukturze toksyny błonicy.
- def. DS – zastąpienie elementu struktury białkowej („domeny”)
identyczną domeną innej cząsteczki i odtworzenia pierwotnej
struktury przestrzennej.
· - Jeżeli wymiana domen jest wzajemna, to powstaje dimer lub
oligomer przeplecionych cząsteczek białka.
np. -amyloid (ch. Alzheimera)
lizozym
fibrynogen
insulina
apolipoproteina
białka prionowe
Mutacje powodują tworzenie agregatów i choroby
związane z patologiami
konformacyjnymi białek
A

Patologie konformacyjne białek
Jaki jest związek wymiany Domeny Strukturalnej z procesem
amyloidogenezy?
Ludzka cystatyna C, małe białko monomeryczne, o dużym znaczeniu
jako inhibitor
proteaz cysteinowych, jest istotnym składnikiem złogów amyloidowych
tworzących się
w mózgu w podeszłym wieku.
Angiopatia mózgowa (endemiczna choroba na Islandii) wiąże się z
dziedziczną mutacją punktową L68Q w sekwencji tego białka.
Choroba objawia się masową amyloidozą naczyń krwionośnych mózgu,
gwałtownymi wylewami krwi do mózgu i śmiercią w młodym wieku.
Białka choroby Alzheimera

BIAŁKA OPIEKUŃCZE, CZAPERONY (ang.
chaperones)
- niezbędne w każdym żywym organizmie,
- funkcja:
1. pomoc w poprawnym zwijaniu białek,
2. w ochronie białek przed czynnikami stresowymi
3. uczestniczą w:
- transporcie białek między organellami komórkowymi,
- w degradacji niepożądanych białek.
Wynikiem działania chaperonów nie jest przeprowadzenie
określonej reakcji, a
jedynie zmiana struktury przestrzennej substratu.
RODZINY BIAŁEK OPIEKUŃCZYCH (CZAPERONÓW), tzw.
Hsp - BIAŁKA
SZOKU TERMICZNEGO (ang. heat shock protein)

Wiele groźnych chorób ma swoją przyczynę w źle zwiniętych
cząsteczkach białkowych;
- syndrom Marfana punktowa mutacja (R1137P) w
fibrylinie.
- Encefalopatia gąbczasta, tzw. „choroba szalonych
krów”- zmiany strukturalne
białka prionowego.
- ch. Alzheimera - tworzenie amyloidowych płytek przez
peptyd -amyloidowy.
Do tej samej grupy chorób, których przyczyną jest
chorobotwórcza struktura (ang.
toxic folds) należą choroby;
- Hantingtona
- Parkinsona

BUDOWA I ROLA BIAŁKA tau
· białko cytoplazmatyczne,
· funkcje:
polimeryzacja tubuliny,
stabilizacja mikrotubul.
TAUOPATIE
- zespoły otępienne związane z nieprawidłową
przemianą białka tau
ch. Alzheimera (AD),
otępienie czołowo-skroniowe (FTP),
zespół Downa,
ch. Parkinsona (FTPD-17),
porażenie ponadjądrowe (PSP),
zwyrodnienie korowo-podstawne (CBD)
Tauopatie te mogą mieć podłoże w
nieprawidłowej fosforylacji
tau w komórkach nerwowych mózgu.

Degradowane białka znakowane są UBIKWITYNĄ (76 AA),
która przyłącza się ( nawet do 100 cząsteczek) do białka i
wędruje do
PROTEASOMU – kompleks E (26S) („komórkowa maszyna
do utylizacji śmieci”) uruchamiany przez UBIKWITYNĘ,
która „pasuje jak klucz do zamka”) powstające peptydy,
AA są wykorzystywane do budowy nowych białek, a
UBIKWITYNA przechodzi do cytoplazmy.
MEDYCYNA
Brak UBIKWITYNY lub zaburzenia w jej funkcjonowaniu
ma związek
z wieloma chorobami:
· stany zapalne,
· nowotwory,
· schorzenia układu nerwowego;
1. ch. Alzheimera,
2. ch. Parkinsona
· mózg – tworzenie złogów w komórkach
nerwowych
Degradacja
białek
bez
naznaczenia
UBIKWITYNĄ
prowadziłaby do całkowitego chaosu i śmierci komórek
oraz całego organizmu.
PERSPEKTYWY dalszych badań:
czego nie wiemy? jak ubikwityna rozpoznaje „chore”
białka ?
dlaczego jednak czasem białko naznaczone ubikwityną
przeżywa?

BILANS AZOTOWY USTROJU
BILANS AZOTOWY USTROJU
różnica pomiędzy ilością azotu
różnica pomiędzy ilością azotu
obecnego w spożytym w ciągu
obecnego w spożytym w ciągu
doby pokarmie a ilością tego
doby pokarmie a ilością tego
pierwiastka w związkach
pierwiastka w związkach
wydalonych w tym samym czasie
wydalonych w tym samym czasie
w moczu i kale
w moczu i kale

BILANS AZOTOWY USTROJU
BILANS AZOTOWY USTROJU
1. Zerowy (wyrównany) – stan
1. Zerowy (wyrównany) – stan
równowagi azotowej, w którym ilość
równowagi azotowej, w którym ilość
azotu pobrana w pokarmie jest
azotu pobrana w pokarmie jest
równa ilości azotu wydalonego
równa ilości azotu wydalonego
występuje u ludzi dorosłych,
występuje u ludzi dorosłych,
zdrowych, prawidłowo odżywionych
zdrowych, prawidłowo odżywionych

BILANS AZOTOWY USTROJU
BILANS AZOTOWY USTROJU
2. Ujemny – stan, w którym ilość azotu
2. Ujemny – stan, w którym ilość azotu
pobrana w pokarmie jest mniejsza od
pobrana w pokarmie jest mniejsza od
ilości azotu wydalonego
ilości azotu wydalonego

PRZYCZYNY UJEMNEGO BILANSU
PRZYCZYNY UJEMNEGO BILANSU
AZOTOWEGO
AZOTOWEGO
głód
głód
brak lub niedobór białka w diecie
brak lub niedobór białka w diecie
spożywanie białka niepełnowartościowego
spożywanie białka niepełnowartościowego
zaburzenia trawienia białka
zaburzenia trawienia białka
zaburzenia wchłaniania peptydów i aminokwasów
zaburzenia wchłaniania peptydów i aminokwasów
zaburzenia wykorzystania produktów trawienia
zaburzenia wykorzystania produktów trawienia
białka
białka
przyspieszony rozpad białek ustrojowych
przyspieszony rozpad białek ustrojowych
utrata białka z moczem i kałem
utrata białka z moczem i kałem

BILANS AZOTOWY USTROJU
BILANS AZOTOWY USTROJU
3. Dodatni – stan, w którym ilość azotu
3. Dodatni – stan, w którym ilość azotu
pobrana w pokarmie jest większa od
pobrana w pokarmie jest większa od
ilości azotu wydalonego
ilości azotu wydalonego

WYSTĘPOWANIE DODATNIEGO
WYSTĘPOWANIE DODATNIEGO
BILANSU AZOTOWEGO
BILANSU AZOTOWEGO
zdrowe noworodki, niemowlęta i dzieci
zdrowe noworodki, niemowlęta i dzieci
rekonwalescencja po chorobie lub zabiegu
rekonwalescencja po chorobie lub zabiegu
operacyjnym
operacyjnym
kobiety ciężarne i karmiące
kobiety ciężarne i karmiące

AMINOKWASY NIEZBĘDNE
AMINOKWASY NIEZBĘDNE
I NIENIEZBĘDNE
I NIENIEZBĘDNE
Aminokwasy niezbędne: muszą być
Aminokwasy niezbędne: muszą być
dostarczone ustrojowi w pokarmie,
dostarczone ustrojowi w pokarmie,
aby mógł być zachowany odpowiedni
aby mógł być zachowany odpowiedni
bilans azotowy
bilans azotowy

AMINOKWASY NIEZBĘDNE
AMINOKWASY NIEZBĘDNE
I NIENIEZBĘDNE
I NIENIEZBĘDNE
Aminokwasy niezbędne
Aminokwasy niezbędne
Aminokwasy
Aminokwasy
nieniezbędne
nieniezbędne
fenyloalanina
fenyloalanina
izoleucyna
izoleucyna
leucyna
leucyna
lizyna
lizyna
metionina
metionina
treonina
treonina
tryptofan
tryptofan
walina
walina
arginina
arginina
histydyna
histydyna
alanina
alanina
asparagina
asparagina
asparaginian
asparaginian
cysteina
cysteina
glicyna
glicyna
glutamina
glutamina
glutaminian
glutaminian
prolina
prolina
seryna
seryna
tyrozyna
tyrozyna

WARTOŚĆ BIOLOGICZNA BIAŁKA
WARTOŚĆ BIOLOGICZNA BIAŁKA
Białka pełnowartościowe – zawierają
Białka pełnowartościowe – zawierają
wszystkie aminokwasy niezbędne w
wszystkie aminokwasy niezbędne w
ilości
ilości
i proporcjach zapewniających
i proporcjach zapewniających
właściwe pokrycie zapotrzebowania
właściwe pokrycie zapotrzebowania
jaja, mięso, mleko, sery
jaja, mięso, mleko, sery

WARTOŚĆ BIOLOGICZNA BIAŁKA
WARTOŚĆ BIOLOGICZNA BIAŁKA
Białka częściowo niepełnowartościowe
Białka częściowo niepełnowartościowe
–
–
zawierają wszystkie niezbędne
zawierają wszystkie niezbędne
aminokwasy, lecz co najmniej jeden
aminokwasy, lecz co najmniej jeden
w ilościach niewystarczających do
w ilościach niewystarczających do
pokrycia zapotrzebowania
pokrycia zapotrzebowania
kasze, mąki
kasze, mąki

WARTOŚĆ BIOLOGICZNA BIAŁKA
WARTOŚĆ BIOLOGICZNA BIAŁKA
Białka niepełnowartościowe
Białka niepełnowartościowe
– zawierają
– zawierają
aminokwasy niezbędne w ilościach
aminokwasy niezbędne w ilościach
niewystarczających do pokrycia
niewystarczających do pokrycia
zapotrzebowania lub wykazują brak
zapotrzebowania lub wykazują brak
co najmniej jednego aminokwasu
co najmniej jednego aminokwasu
niezbędnego
niezbędnego
żelatyna (brak tryptofanu)
żelatyna (brak tryptofanu)

Fenyloketonuria
Fenyloketonuria
Fenyloketonurię po raz pierwszy opisano
Fenyloketonurię po raz pierwszy opisano
w 1934 roku. Norweski lekarz i biolog
w 1934 roku. Norweski lekarz i biolog
A.Fölling poszukiwał przyczyny
A.Fölling poszukiwał przyczyny
upośledzenia umysłowego dwójki
upośledzenia umysłowego dwójki
rodzeństwa. Zaobserwował, że może to
rodzeństwa. Zaobserwował, że może to
mieć związek z przemianą fenyloalaniny.
mieć związek z przemianą fenyloalaniny.
Pięć lat później w 1939 r. inny naukowiec
Pięć lat później w 1939 r. inny naukowiec
C.A Jervis wykazał że fenyloketonuria
C.A Jervis wykazał że fenyloketonuria
jest chorobą uwarunkowaną genetycznie.
jest chorobą uwarunkowaną genetycznie.
Dopiero po kolejnych 14 latach w 1953
Dopiero po kolejnych 14 latach w 1953
roku ten sam autor udowodnił, że
roku ten sam autor udowodnił, że
przyczyną choroby jest utrata zdolności
przyczyną choroby jest utrata zdolności
przekształcania fenyloalaniny do tyrozyny.
przekształcania fenyloalaniny do tyrozyny.

PKU to skrót od nazwy
PKU to skrót od nazwy
Phenyloketonuria, czyli nazwy -
Phenyloketonuria, czyli nazwy -
Fenyloketonuria pisanej w języku
Fenyloketonuria pisanej w języku
angielskim
angielskim
-
-
choroba metaboliczna,
choroba metaboliczna,
-przewlekła,
-przewlekła,
-rozpoznanie u noworodków jest możliwe
-rozpoznanie u noworodków jest możliwe
dzięki sprawnemu działaniu systemu
dzięki sprawnemu działaniu systemu
noworodkowych badan
noworodkowych badan
przesiewowych,
przesiewowych,
-Organizm osoby chorej na
-Organizm osoby chorej na
fenyloketonurię nie jest zdolny do
fenyloketonurię nie jest zdolny do
prawidłowego metabolizmu jednego ze
prawidłowego metabolizmu jednego ze
składników diety - fenyloalaniny.
składników diety - fenyloalaniny.
Aminokwas
Aminokwas
fenyloalanina znajduje
fenyloalanina znajduje
się w pokarmach
się w pokarmach
zawierających dużo
zawierających dużo
białka, takich jak:
białka, takich jak:
mięso, jaja, ryby,
mięso, jaja, ryby,
mleko, ser oraz (w
mleko, ser oraz (w
mniejszych ilościach) w
mniejszych ilościach) w
produktach zbożowych,
produktach zbożowych,
warzywach i owocach.
warzywach i owocach.
Z fenyloalaniny
Z fenyloalaniny
powstaje inny
powstaje inny
aminokwas – tyrozyna
aminokwas – tyrozyna
Nadmierne stężenie
Nadmierne stężenie
fenyloalaniny we krwi
fenyloalaniny we krwi
oraz produktów jej
oraz produktów jej
rozkładu jest przyczyną
rozkładu jest przyczyną
upośledzenia rozwoju
upośledzenia rozwoju
fizycznego i
fizycznego i
intelektualnego.
intelektualnego.

Bez właściwego i
Bez właściwego i
szybkiego leczenia
szybkiego leczenia
(które w tym przypadku
(które w tym przypadku
polega na odpowiedniej
polega na odpowiedniej
diecie) gromadzenie się
diecie) gromadzenie się
znacznych ilości
znacznych ilości
fenyloalaniny
fenyloalaniny
doprowadza do
doprowadza do
postępującego
postępującego
uszkodzenia mózgu,
uszkodzenia mózgu,
szczególnie we
szczególnie we
wczesnym okresie
wczesnym okresie
życia, kiedy ten narząd
życia, kiedy ten narząd
intensywnie się rozwija.
intensywnie się rozwija.
Jeśli w ciągu pierwszych
Jeśli w ciągu pierwszych
tygodni życia wykryje
tygodni życia wykryje
się chorobę i rozpocznie
się chorobę i rozpocznie
leczenie, rozwój dziecka
leczenie, rozwój dziecka
postępuje bez szwanku.
postępuje bez szwanku.
Przeoczenie
Przeoczenie
fenyloketonurii w okresie
fenyloketonurii w okresie
pierwszych kilku tygodni
pierwszych kilku tygodni
życia powoduje
życia powoduje
upośledzenie umysłowe
upośledzenie umysłowe
(zazwyczaj umiarkowane
(zazwyczaj umiarkowane
bądź głębokie) oraz
bądź głębokie) oraz
objawy neurologiczne:
objawy neurologiczne:
drgawki, zwiększone
drgawki, zwiększone
napięcie i drżenia
napięcie i drżenia
mięśniowe, a także
mięśniowe, a także
zaburzenia zachowania.
zaburzenia zachowania.
Jedynym sposobem
Jedynym sposobem
zapobiegania takim
zapobiegania takim
zmianom jest długotrwałe
zmianom jest długotrwałe
stosowanie diety - aż do
stosowanie diety - aż do
momentu ukończenia
momentu ukończenia
intensywnego rozwoju
intensywnego rozwoju
mózgu, czyli wieku 12-14
mózgu, czyli wieku 12-14
lat.
lat.

Następujące typy zaburzeń
Następujące typy zaburzeń
neurologicznych:
neurologicznych:
-wzmożone lub zmniejszone
-wzmożone lub zmniejszone
napięcie mięśniowe;
napięcie mięśniowe;
-drżenia spoczynkowe;
-drżenia spoczynkowe;
-ruchy pląsawiczne lub
-ruchy pląsawiczne lub
atetotyczne;
atetotyczne;
-zaburzenia chodu i
-zaburzenia chodu i
niezborność ruchów;
niezborność ruchów;
-padaczka oporna na leczenie;
-padaczka oporna na leczenie;
-atrofia nerwów wzrokowych;
-atrofia nerwów wzrokowych;
Na uwagę zasługują :
Na uwagę zasługują :
-niekontrolowane wybuchy
-niekontrolowane wybuchy
złości,
złości,
-agresja wobec osób trzecich,
-agresja wobec osób trzecich,
-napady nadpobudliwości i
-napady nadpobudliwości i
autoagresji , z ciężkimi
autoagresji , z ciężkimi
okaleczeniami.
okaleczeniami.
W fenyloketonurii,
W fenyloketonurii,
podobnie jak w innych
podobnie jak w innych
wadach metabolizmu,
wadach metabolizmu,
określenie "leczenie"
określenie "leczenie"
jest nieco mylące. Nie
jest nieco mylące. Nie
podajemy bowiem
podajemy bowiem
żadnych leków, nie
żadnych leków, nie
stosujemy zabiegów
stosujemy zabiegów
chirurgicznych.
chirurgicznych.
Postępowanie w tej
Postępowanie w tej
chorobie polega na
chorobie polega na
utrzymywaniu
utrzymywaniu
odpowiedniej diety i
odpowiedniej diety i
dokonywaniu
dokonywaniu
pomiarów stężenia
pomiarów stężenia
fenyloalaniny we krwi.
fenyloalaniny we krwi.

DIETA CHORYCH NA FENYLOKERONURIĘ:
DIETA CHORYCH NA FENYLOKERONURIĘ:
Podstawą diety są specjalne preparaty białkozastępcze
Podstawą diety są specjalne preparaty białkozastępcze
o bardzo małej zawartości fenyloalaniny (zapisywane na
o bardzo małej zawartości fenyloalaniny (zapisywane na
receptę) pozwalające na prawidłowy wzrost i rozwój
receptę) pozwalające na prawidłowy wzrost i rozwój
dziecka oraz jarzyny, owoce i tłuszcze. Dietę wylicza i
dziecka oraz jarzyny, owoce i tłuszcze. Dietę wylicza i
układa lekarz pediatra lub dietetyk
układa lekarz pediatra lub dietetyk
.
.
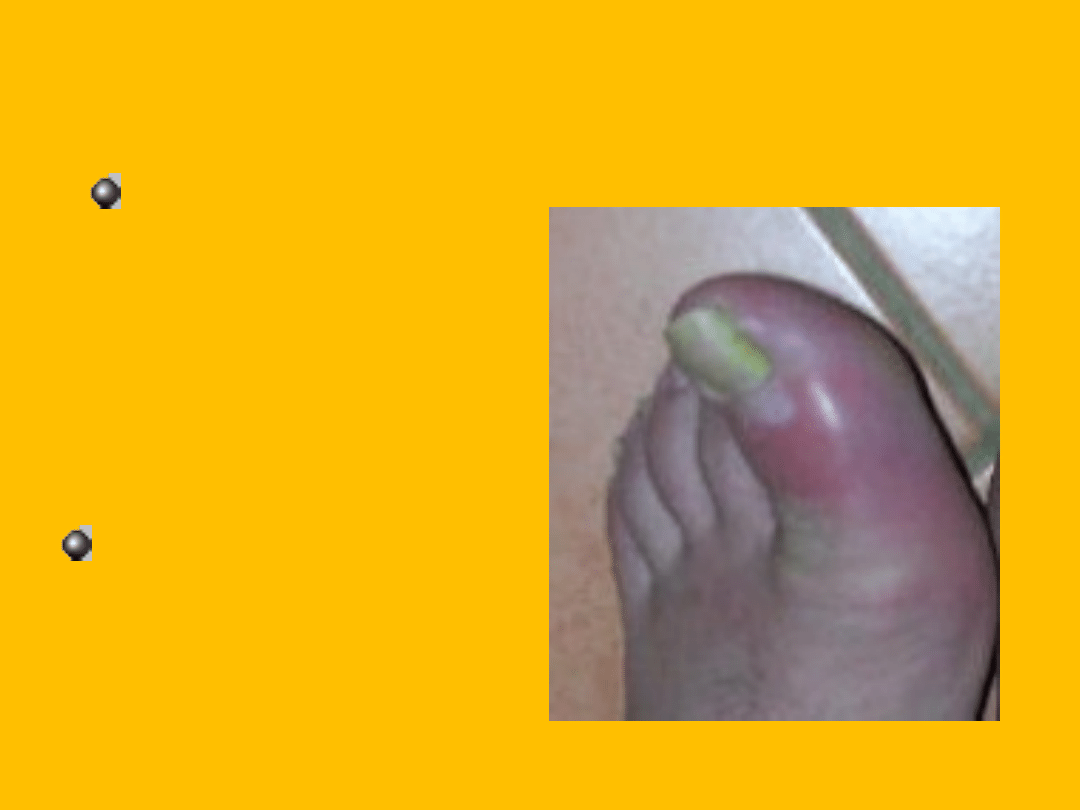
Alkaptonuria
Alkaptonuria
rzadka choroba
rzadka choroba
metaboliczna
metaboliczna
uwarunkowana
uwarunkowana
genetycznie.
genetycznie.
Alkaptonuria podlega
Alkaptonuria podlega
dziedziczeniu
dziedziczeniu
autosomalnemu
autosomalnemu
recesywnemu.
recesywnemu.
Jednym z objawów
Jednym z objawów
może być
może być
artretyzm(
artretyzm(
zdjęcie
zdjęcie
obok)
obok)

Objawy
Charakterystycznym objawem alkaptnurii jest
zabarwianie się moczu i woskowiny pod wpływem
powietrza na czerwono lub czarno (w zależności od
posiłku zjedzonego parę godzin wcześniej) pod
wpływem tworzonego kwasu homogentyzynowego.
Łatwo zauważalne jest to na pieluchach niemowląt. U
dorosłych (zazwyczaj po 40. roku życia) rozwija się
stopniowo artretyzm (zwłaszcza w okolicy kręgosłupa).
Przyczyną artretyzmu jest długotrwałe nagromadzanie
kwasu homogentyzynowego w kościach i chrząstkach
stawowych.
Leczenie
Profilaktyka jest niemożliwa, leczenie objawowe.
Zmniejszenie ilości przyjmowanej tyrozyny i
fenyloalaniny do minimum spowalnia rozwój choroby.
Fenyloalanina jest aminokwasem egzogennym i jej
przyjmowanie w pokarmie jest niezbędne do przeżycia.

Acyduria mewalonianowa.
Acyduria mewalonianowa.
Acyduria mewalonianowa (kwasica
Acyduria mewalonianowa (kwasica
mewalonianowa, ang. mevalonic
mewalonianowa, ang. mevalonic
aciduria, MVA) – choroba metaboliczna
aciduria, MVA) – choroba metaboliczna
uwarunkowana genetycznie.
uwarunkowana genetycznie.
Spowodowana jest niedoborem
Spowodowana jest niedoborem
enzymu kinazy mewalonianowej.
enzymu kinazy mewalonianowej.
Acyduria
Acyduria
mewalonianowa
mewalonianowa
choroba metaboliczna
choroba metaboliczna
uwarunkowana
uwarunkowana
genetycznie.
genetycznie.
Spowodowana jest
Spowodowana jest
niedoborem enzymu
niedoborem enzymu
kinazy
kinazy
Objawia się opóźnieniem
Objawia się opóźnieniem
umysłowym, postępującą
umysłowym, postępującą
ataksją, przełomami
ataksją, przełomami
gorączkowymi z
gorączkowymi z
biegunką i wymiotami,
biegunką i wymiotami,
wysypką na skórze,
wysypką na skórze,
hepatosplenomegalią,
hepatosplenomegalią,
cechami dysmorficznymi
cechami dysmorficznymi
twarzy, nieprawidłowym
twarzy, nieprawidłowym
rozwojem i wzrostem.
rozwojem i wzrostem.

Tyrozynoza
Tyrozynoza
Blok metaboliczny – brak hydrolazy
Blok metaboliczny – brak hydrolazy
fumaryloacetooctanowej i hydrolazy
fumaryloacetooctanowej i hydrolazy
maleiloacetooctanowej – u niemowląt w postaci
maleiloacetooctanowej – u niemowląt w postaci
ostrej występuje biegunka i wymioty
ostrej występuje biegunka i wymioty
przypominające kapustę- nieprawidłowy rozwój
przypominające kapustę- nieprawidłowy rozwój
i wzrost Nieleczona tyrozynoza prowadzi do
i wzrost Nieleczona tyrozynoza prowadzi do
śmierci w ciągu 6-8 miesięcy z powodu
śmierci w ciągu 6-8 miesięcy z powodu
niewydolności wątroby. W chorobie tej zwiększa
niewydolności wątroby. W chorobie tej zwiększa
się stężenie tyrozyny w osoczu i innych
się stężenie tyrozyny w osoczu i innych
aminokwasów- metioniny. Terapia polega na
aminokwasów- metioniny. Terapia polega na
braku w diecie tyrozyny, fenyloalaniny i
braku w diecie tyrozyny, fenyloalaniny i
metioniny.
metioniny.

Albinizm
Albinizm
Brak oksydazy fenolowej
Brak oksydazy fenolowej
przekształcającej tyrozynę w melaninę-
przekształcającej tyrozynę w melaninę-
Brak tego enzymu manifestuje się jasną
Brak tego enzymu manifestuje się jasną
karnacją- włosy białe od urodzenia.
karnacją- włosy białe od urodzenia.

Choroba Hartnupów
Choroba Hartnupów
Jest to dziedziczne zaburzenie matabolizmu
Jest to dziedziczne zaburzenie matabolizmu
tryptofanu, charakteryzujące się wysypką skórną
tryptofanu, charakteryzujące się wysypką skórną
przypominającą pelagrę, nawracającą ataksją
przypominającą pelagrę, nawracającą ataksją
móżdżkową i upośledzeniem umysłowym.Mocz
móżdżkową i upośledzeniem umysłowym.Mocz
zawiera zwiększone ilości indolooctanu oraz
zawiera zwiększone ilości indolooctanu oraz
tryptofanu
tryptofanu
Choroba Hartnupów (ang. Hartnup disease,
Choroba Hartnupów (ang. Hartnup disease,
Hartnup disorder, HND) – dziedziczone
Hartnup disorder, HND) – dziedziczone
autosomalnie recesywnie zaburzenie transportu
autosomalnie recesywnie zaburzenie transportu
aminokwasów, głównie tryptofanu, w świetle
aminokwasów, głównie tryptofanu, w świetle
jelita i kanalikach nerkowych. Przyczyną choroby
jelita i kanalikach nerkowych. Przyczyną choroby
jest mutacja w genie SLC6A19 w locus 5p15.
jest mutacja w genie SLC6A19 w locus 5p15.

Choroba z moczem o
Choroba z moczem o
zapachu syropu klonowego
zapachu syropu klonowego
Jest to dziedziczne zaburzenie
Jest to dziedziczne zaburzenie
matabolizmu waliny, izoleucyny i
matabolizmu waliny, izoleucyny i
leucyny .Spowodowane jest brakiem lub
leucyny .Spowodowane jest brakiem lub
zmniejszeniem aktywności
zmniejszeniem aktywności
dekarboksylazy ketokwasów. Prowadzi to
dekarboksylazy ketokwasów. Prowadzi to
do wyraźnego zwiększenia stężenia
do wyraźnego zwiększenia stężenia
waliny, izoleucyny i leucyny we krwi i
waliny, izoleucyny i leucyny we krwi i
moczu. Mocz osób cierpiących ma zapach
moczu. Mocz osób cierpiących ma zapach
syropu klonowego. Niezasastosowanie
syropu klonowego. Niezasastosowanie
diety ubogiej w walinę , izoleucynę i
diety ubogiej w walinę , izoleucynę i
leucynę prowadzi do wystąpienia
leucynę prowadzi do wystąpienia
niedorozwoju umysłowego i fizycznego.
niedorozwoju umysłowego i fizycznego.

ENZYMY PROTEOLITYCZNE
ENZYMY PROTEOLITYCZNE
1. Endopeptydazy
1. Endopeptydazy
działają na cząsteczkę białka lub polipeptydu
działają na cząsteczkę białka lub polipeptydu
rozdrabniając ją na mniejsze fragmenty
rozdrabniając ją na mniejsze fragmenty
polipeptydowe i oligopeptydowe
polipeptydowe i oligopeptydowe
2. Egzopeptydazy
2. Egzopeptydazy
działają na oligopeptydy odszczepiając jedynie
działają na oligopeptydy odszczepiając jedynie
skrajne aminokwasy
skrajne aminokwasy

ENZYMY PROTEOLITYCZNE
ENZYMY PROTEOLITYCZNE
Endopeptydazy
Endopeptydazy
(hydrolazy peptydylo-peptydowe)
(hydrolazy peptydylo-peptydowe)
pepsyna
pepsyna
enteropeptydaza (enterokinaza)
enteropeptydaza (enterokinaza)
trypsyna
trypsyna
chymotrypsyna
chymotrypsyna
elastaza
elastaza

ENZYMY PROTEOLITYCZNE
ENZYMY PROTEOLITYCZNE
Egzopeptydazy
Egzopeptydazy
aminopeptydazy
aminopeptydazy
karboksypeptydazy A i B
karboksypeptydazy A i B
dipeptydazy
dipeptydazy

PROTEOLIZA W ŻOŁĄDKU
PROTEOLIZA W ŻOŁĄDKU
Pepsyna I, II, III
Pepsyna I, II, III
-
-
powstaje z pepsynogenu (wydzielany przez komórki
powstaje z pepsynogenu (wydzielany przez komórki
główne dna żołądka)
główne dna żołądka)
- aktywacja przez jony wodorowe (ograniczona
- aktywacja przez jony wodorowe (ograniczona
proteoliza) i autokatalitycznie
proteoliza) i autokatalitycznie
- powoduje rozpad ok. 10% wiązań peptydowych
- powoduje rozpad ok. 10% wiązań peptydowych
- optimum działania: pH 1 – 3
- optimum działania: pH 1 – 3
- specyficzność działania: wiązania peptydowe
- specyficzność działania: wiązania peptydowe
utworzone przez aminokwasy aromatyczne,
utworzone przez aminokwasy aromatyczne,
dikarboksylowe i pomiędzy leucyną i glutaminianem
dikarboksylowe i pomiędzy leucyną i glutaminianem
- produkty działania: mieszanina białka
- produkty działania: mieszanina białka
niestrawionego
niestrawionego
(ok. 85%) i polipeptydów (m.cz.
(ok. 85%) i polipeptydów (m.cz.
600 – 3000)
600 – 3000)

ROLA SOKU ŻOŁĄDKOWEGO
ROLA SOKU ŻOŁĄDKOWEGO
1.
1.
Dentauracja, uwodnienie i
Dentauracja, uwodnienie i
pęcznienie białek
pęcznienie białek
2.
2.
Aktywacja pepsynogenu
Aktywacja pepsynogenu
3.
3.
Optimum dla działania pepsyny
Optimum dla działania pepsyny
4.
4.
Działanie bakteriobójcze
Działanie bakteriobójcze

PROTEOLIZA W JELITACH
PROTEOLIZA W JELITACH
Dwunastnica:
Dwunastnica:
50 – 60% białka ulega strawieniu i wchłonięciu
50 – 60% białka ulega strawieniu i wchłonięciu
Jelito czcze i kręte:
Jelito czcze i kręte:
30 – 40% białka ulega strawieniu i wchłonięciu
30 – 40% białka ulega strawieniu i wchłonięciu
Jelito grube:
Jelito grube:
ok. 10% białka ulega strawieniu i wchłonięciu
ok. 10% białka ulega strawieniu i wchłonięciu

PROTEOLIZA W JELITACH
PROTEOLIZA W JELITACH
Trypsyna
Trypsyna
-
-
powstaje z trypsynogenu pod wpływem
powstaje z trypsynogenu pod wpływem
enterokinazy
enterokinazy
i autokatalitycznie
i autokatalitycznie
- optimum działania: pH 7 – 9
- optimum działania: pH 7 – 9
- specyficzność działania: wiązania peptydowe
- specyficzność działania: wiązania peptydowe
utworzone przez aminokwasy zasadowe,
utworzone przez aminokwasy zasadowe,
uwalnia
uwalnia
peptydy z argininą lub lizyną jako
peptydy z argininą lub lizyną jako
aminokwasami
aminokwasami
terminalnymi
terminalnymi

PROTEOLIZA W JELITACH
PROTEOLIZA W JELITACH
Chymotrypsyna
Chymotrypsyna
-
-
powstaje z chymotrypsynogenu pod wpływem
powstaje z chymotrypsynogenu pod wpływem
trypsyny
trypsyny
i autokatalitycznie
i autokatalitycznie
- optimum działania: pH 8
- optimum działania: pH 8
- specyficzność działania: wiązania peptydowe
- specyficzność działania: wiązania peptydowe
utworzone przez aminokwasy aromatyczne,
utworzone przez aminokwasy aromatyczne,
uwalnia
uwalnia
peptydy z fenyloalaniną, tyrozyną i
peptydy z fenyloalaniną, tyrozyną i
tryptofanem jako
tryptofanem jako
aminokwasami
aminokwasami
terminalnymi
terminalnymi

PROTEOLIZA W JELITACH
PROTEOLIZA W JELITACH
Elastaza (pankreatopeptydaza)
Elastaza (pankreatopeptydaza)
-
-
powstaje z proelastazy pod wpływem trypsyny
powstaje z proelastazy pod wpływem trypsyny
- specyficzność działania: wiązania peptydowe
- specyficzność działania: wiązania peptydowe
utworzone przez aminokwasy alifatyczne,
utworzone przez aminokwasy alifatyczne,
uwalnia
uwalnia
peptydy z waliną, leucyną, seryną i
peptydy z waliną, leucyną, seryną i
alaniną jako
alaniną jako
aminokwasami terminalnymi
aminokwasami terminalnymi

PROTEOLIZA W JELITACH
PROTEOLIZA W JELITACH
Karboksypeptydaza A
Karboksypeptydaza A
- powstaje z prokarboksypeptydazy pod wpływem
- powstaje z prokarboksypeptydazy pod wpływem
trypsyny
trypsyny
- specyficzność działania: wiązania peptydowe
- specyficzność działania: wiązania peptydowe
utworzone przez aminokwasy C-terminalne,
utworzone przez aminokwasy C-terminalne,
głównie
głównie
aromatyczne i alifatyczne uwolnione
aromatyczne i alifatyczne uwolnione
przez
przez
chymotrypsynę i elastazę
chymotrypsynę i elastazę

PROTEOLIZA W JELITACH
PROTEOLIZA W JELITACH
Karboksypeptydaza B
Karboksypeptydaza B
- powstaje z prokarboksypeptydazy pod wpływem
- powstaje z prokarboksypeptydazy pod wpływem
trypsyny
trypsyny
- specyficzność działania: wiązania peptydowe
- specyficzność działania: wiązania peptydowe
utworzone przez aminokwasy C-terminalne,
utworzone przez aminokwasy C-terminalne,
głównie
głównie
zasadowe (lizynę i argininę) uwolnione
zasadowe (lizynę i argininę) uwolnione
przez
przez
trypsynę
trypsynę

PROTEOLIZA W JELITACH
PROTEOLIZA W JELITACH
Produktem działania peptydaz
Produktem działania peptydaz
jelitowych jest mieszanina krótkich
jelitowych jest mieszanina krótkich
peptydów (2 – 6 aminokwasów) i
peptydów (2 – 6 aminokwasów) i
wolnych aminokwasów
wolnych aminokwasów

PROTEOLIZA W RĄBKU
PROTEOLIZA W RĄBKU
SZCZOTECZKOWYM I ENTEROCYTACH
SZCZOTECZKOWYM I ENTEROCYTACH
AMINOPEPTYDAZY
AMINOPEPTYDAZY
DIPEPTYDAZY
DIPEPTYDAZY
-
-
dłuższe peptydy podlegają hydrolizie przez enzymy
dłuższe peptydy podlegają hydrolizie przez enzymy
rąbka
rąbka
szczoteczkowego do dipeptydów i tripeptydów
szczoteczkowego do dipeptydów i tripeptydów
- dipeptydy i tripeptydy podlegają hydrolizie przez enzymy
- dipeptydy i tripeptydy podlegają hydrolizie przez enzymy
wnętrza
wnętrza
enterocytów
enterocytów
- peptydy złożone z proliny, hydroksyproliny, argininy i
- peptydy złożone z proliny, hydroksyproliny, argininy i
lizyny są
lizyny są
hydrolizowane wyłącznie przez enzymy wnętrza
hydrolizowane wyłącznie przez enzymy wnętrza
enterocytów
enterocytów
- aminopeptydazy hydrolizują wiązania peptydowe
- aminopeptydazy hydrolizują wiązania peptydowe
utworzone
utworzone
przez
przez
aminokwasy N-terminalne
aminokwasy N-terminalne

WCHŁANIANIE AMINOKWASÓW
WCHŁANIANIE AMINOKWASÓW
-
-
transport aktywny do komórki enterocyta
transport aktywny do komórki enterocyta
- wspólny nośnik dla AA i jonów sodu
- wspólny nośnik dla AA i jonów sodu
- pompa sodowa w bocznej i podstawnej błonie
- pompa sodowa w bocznej i podstawnej błonie
komórkowej
komórkowej
- źródłem energii podtrzymującym aktywność pompy
- źródłem energii podtrzymującym aktywność pompy
sodowej jest ATP
sodowej jest ATP
- dyfuzja ułatwiona do krwi
- dyfuzja ułatwiona do krwi
- wchłanianiu ulegają L-izomery
- wchłanianiu ulegają L-izomery
- aminokwasy nie ulegają metabolizmowi w
- aminokwasy nie ulegają metabolizmowi w
enterocytach
enterocytach
z wyjątkiem glutaminianu i asparaginianu
z wyjątkiem glutaminianu i asparaginianu

BIAŁKA
BIAŁKA
Definicja
Definicja
Występowanie
Występowanie
Podział i charakterystyka
Podział i charakterystyka
Reakcje charakterystyczne na wykrywanie białek
Reakcje charakterystyczne na wykrywanie białek
:
:
-
-
K
K
santoproteinowa
santoproteinowa
-
-
B
B
iuretowa
iuretowa

DEFINICJA
DEFINICJA
Białka są to wielocząsteczkowe, skomplikowane związki
Białka są to wielocząsteczkowe, skomplikowane związki
organiczne, substancje naturalne, zbudowane z chemicznie
organiczne, substancje naturalne, zbudowane z chemicznie
powiązanych ze sobą reszt amino kwasowych zawierają
powiązanych ze sobą reszt amino kwasowych zawierają
węgiel, wodór, tlen, azot i siarkę; są podstawowymi
węgiel, wodór, tlen, azot i siarkę; są podstawowymi
składnikami struktury komórek.
składnikami struktury komórek.
Białka są syntetyzowane na podstawie DNA, ich budowa oraz
Białka są syntetyzowane na podstawie DNA, ich budowa oraz
związana z nią struktura jest uwarunkowana kolejnością
związana z nią struktura jest uwarunkowana kolejnością
zasad azotowych w łańcuchu cząsteczki kwasu nukleinowego.
zasad azotowych w łańcuchu cząsteczki kwasu nukleinowego.
Białka, podobnie jak kwasy nukleinowe są
Białka, podobnie jak kwasy nukleinowe są
wielkocząsteczkowymi polimerami, złożonymi z liniowo
wielkocząsteczkowymi polimerami, złożonymi z liniowo
połączonych cząsteczek aminokwasów.
połączonych cząsteczek aminokwasów.
Liczba kombinacji 20 rodzajów aminokwasów (występujących
Liczba kombinacji 20 rodzajów aminokwasów (występujących
w przyrodzie), dla przeciętnego białka jest praktycznie
w przyrodzie), dla przeciętnego białka jest praktycznie
nieskończona. Do utworzenia i utrzymania przy życiu
nieskończona. Do utworzenia i utrzymania przy życiu
organizmu jest potrzebne wiele dziesiątków tysięcy różnych
organizmu jest potrzebne wiele dziesiątków tysięcy różnych
białek.
białek.

WYSTĘPOWANIE
WYSTĘPOWANIE
Białka są nieodzownymi składnikami wszelkiej materii żywej.
Białka są nieodzownymi składnikami wszelkiej materii żywej.
Do białek należą m.in. enzymy, niektóre hormony oraz wiele
Do białek należą m.in. enzymy, niektóre hormony oraz wiele
ważnych składników strukturalnych komórki. Typowym
ważnych składników strukturalnych komórki. Typowym
białkiem jest czerwony barwnik krwi – hemoglobina.
białkiem jest czerwony barwnik krwi – hemoglobina.
Obecność białek stwierdzono we wszystkich komórkach
Obecność białek stwierdzono we wszystkich komórkach
żywych, a także u wirusów jako istotny składnik ich
żywych, a także u wirusów jako istotny składnik ich
"organizmu".
"organizmu".
Białka są głównym elementem budulcowym skóry, mięśni,
Białka są głównym elementem budulcowym skóry, mięśni,
ścięgien, nerwów, krwi, mleka, chrząstek, sierści, paznokci,
ścięgien, nerwów, krwi, mleka, chrząstek, sierści, paznokci,
piór, kopyt, a ponadto niezliczonej ilości enzymów,
piór, kopyt, a ponadto niezliczonej ilości enzymów,
receptorów, przeciwciał, antybiotyków, toksyn bakteryjnych,
receptorów, przeciwciał, antybiotyków, toksyn bakteryjnych,
jadu węży i wielu hormonów.
jadu węży i wielu hormonów.

STRUKTURA BIAŁEK
STRUKTURA BIAŁEK
S
S
truktura drugorzędowa białka powstaje gdy
truktura drugorzędowa białka powstaje gdy
aminokwasy sąsiadujące w łańcuchu
aminokwasy sąsiadujące w łańcuchu
polipeptydowym (struktura pierwszorzędowa)
polipeptydowym (struktura pierwszorzędowa)
tworzą wiązania wodorowe
tworzą wiązania wodorowe
.
.
T
T
rójwymiarowa, trzeciorzędowa struktura białka
rójwymiarowa, trzeciorzędowa struktura białka
powstaje na skutek oddziaływań pomiędzy
powstaje na skutek oddziaływań pomiędzy
aminokwasami znajdującymi się w różnych
aminokwasami znajdującymi się w różnych
miejscach skręconej, drugorzędowej struktury
miejscach skręconej, drugorzędowej struktury
białka
białka
.
.
C
C
zwartorzędowa struktura białka powstaje ze
zwartorzędowa struktura białka powstaje ze
zwinięcia dwóch lub więcej łańcuchów
zwinięcia dwóch lub więcej łańcuchów
polipeptydowych.
polipeptydowych.

PODZIAŁ I CHARAKTERYSTYKA
PODZIAŁ I CHARAKTERYSTYKA
Białka proste
Białka proste
Białka globularne
Białka globularne
Białka fibrylarne
Białka fibrylarne
Białka złożone
Białka złożone

BIAŁKA PROSTE
BIAŁKA PROSTE
Są zbudowane tylko z aminokwasów.
Są zbudowane tylko z aminokwasów.
Wyróżniamy białka rozpuszczalne czyli
Wyróżniamy białka rozpuszczalne czyli
globularne i białka włókniste czyli fibrylarne
globularne i białka włókniste czyli fibrylarne
(skleroproteiny).
(skleroproteiny).

BIAŁKA GLOBULARNE
BIAŁKA GLOBULARNE
I
I
ch cząsteczki mają postać kłębków (globul),
ch cząsteczki mają postać kłębków (globul),
mają budowę bardziej złożoną niż białka
mają budowę bardziej złożoną niż białka
fibrylarne, cząsteczki ich charakteryzują się
fibrylarne, cząsteczki ich charakteryzują się
specyficznością nie tylko struktury
specyficznością nie tylko struktury
pierwotnej, lecz także struktur drugo- i
pierwotnej, lecz także struktur drugo- i
trzeciorzędowej w roztworze i wysoką
trzeciorzędowej w roztworze i wysoką
czułością struktury na zmiany własności
czułością struktury na zmiany własności
fizykochemicznych środowiska.
fizykochemicznych środowiska.

BIAŁKA GLOBULARNE
BIAŁKA GLOBULARNE
Wśród nich wyróżniamy:
Wśród nich wyróżniamy:
1) albuminy (roślinne, białko jaja kurzego, mleka, albuminy
1) albuminy (roślinne, białko jaja kurzego, mleka, albuminy
osocza krwi),
osocza krwi),
2) globuliny ((roślinne, globuliny osocza krwi),
2) globuliny ((roślinne, globuliny osocza krwi),
3) gluteiny (ziaren zbóż, gluten),
3) gluteiny (ziaren zbóż, gluten),
4) prolaminy (ziaren zbóż),
4) prolaminy (ziaren zbóż),
5) histony (składowe chromatyny) oraz
5) histony (składowe chromatyny) oraz
6) protaminy (wchodzą w skład jąder komórkowych,
6) protaminy (wchodzą w skład jąder komórkowych,
krwinek krwi czerwonych i białych).
krwinek krwi czerwonych i białych).

ALBUMINY, GLOBULINY, GLUTEINY,
ALBUMINY, GLOBULINY, GLUTEINY,
HISTYNY, PROTAMINY
HISTYNY, PROTAMINY
Albuminy dobrze rozpuszczają się w wodzie,
Albuminy dobrze rozpuszczają się w wodzie,
źle w roztworach kwasów i zasad.
źle w roztworach kwasów i zasad.
Globuliny trudno rozpuszczają się w wodzie,
Globuliny trudno rozpuszczają się w wodzie,
dobrze w rozcieńczonych roztworach
dobrze w rozcieńczonych roztworach
kwasów, zasad i soli.
kwasów, zasad i soli.
Gluteiny dobrze rozpuszczają się tylko w
Gluteiny dobrze rozpuszczają się tylko w
kwasach i zasadach, natomiast prolaminy w
kwasach i zasadach, natomiast prolaminy w
alkoholach.
alkoholach.
Histony rozpuszczają się w wodzie i
Histony rozpuszczają się w wodzie i
rozcieńczonych roztworach kwasów.
rozcieńczonych roztworach kwasów.
Protaminy rozpuszczają się w wodzie.
Protaminy rozpuszczają się w wodzie.

BIAŁKA FIBRYLARNE
BIAŁKA FIBRYLARNE
Z reguły odznaczają się dość
Z reguły odznaczają się dość
dużą sprężystością i trwałością,
dużą sprężystością i trwałością,
w związku z czym organizm
w związku z czym organizm
wykorzystuje je do tworzenia
wykorzystuje je do tworzenia
struktur sprężystych.
struktur sprężystych.

PODZIAŁ BIAŁEK
PODZIAŁ BIAŁEK
FIBRYLARNYCH
FIBRYLARNYCH
Do nich należą:
Do nich należą:
1) Fibroina (buduje jedwab)
1) Fibroina (buduje jedwab)
2) kolagen (składowa tkanki łącznej
2) kolagen (składowa tkanki łącznej
właściwej np. ścięgien)
właściwej np. ścięgien)
3) keratyna (budulec piór, włosów, kopyt,
3) keratyna (budulec piór, włosów, kopyt,
paznokci, rogów).
paznokci, rogów).
Z uwagi na swoją nie rozpuszczalność są
Z uwagi na swoją nie rozpuszczalność są
składową tkanki łącznej (chrzęstnej, kostnej,
składową tkanki łącznej (chrzęstnej, kostnej,
właściwej) oraz włosów, kopyt, rogów itd..
właściwej) oraz włosów, kopyt, rogów itd..

BIAŁKA ZŁOŻONE
BIAŁKA ZŁOŻONE
Są to kompleksy białek ze związkami
Są to kompleksy białek ze związkami
niebiałkowymi (zwanymi grupą
niebiałkowymi (zwanymi grupą
prostetyczną).
prostetyczną).
Proteidy występują znacznie częściej
Proteidy występują znacznie częściej
w przyrodzie niż proteiny.
w przyrodzie niż proteiny.
Oprócz aminokwasów zawierają jedną
Oprócz aminokwasów zawierają jedną
lub więcej grup nie aminokwasowe,
lub więcej grup nie aminokwasowe,
czyli prostetyczne, takich jak np.
czyli prostetyczne, takich jak np.
cukry, tłuszcze, kwasy nukleinowe,
cukry, tłuszcze, kwasy nukleinowe,
barwniki, czy kwas fosforowy.
barwniki, czy kwas fosforowy.

PODZIAŁ BIAŁEK
PODZIAŁ BIAŁEK
ZŁOŻONYCH
ZŁOŻONYCH
W zależności od rodzaju grupy prostetycznej wyróżniamy:
W zależności od rodzaju grupy prostetycznej wyróżniamy:
1) nukleoproteidy przy połączeniu z kwasami
1) nukleoproteidy przy połączeniu z kwasami
nukleinowymi (występują we wszystkich komórkach),
nukleinowymi (występują we wszystkich komórkach),
2) fosfoproteidy przy połączeniu z kwasem fosforowym
2) fosfoproteidy przy połączeniu z kwasem fosforowym
(np. kazeina),
(np. kazeina),
3) glikoproteidy przy połączeniu z węglowodanami (np.
3) glikoproteidy przy połączeniu z węglowodanami (np.
białko jaja kurzego),
białko jaja kurzego),
4) lipoproteidy przy połączeniu z lipidami,
4) lipoproteidy przy połączeniu z lipidami,
5) chromoproteidy przy połączeniu z barwnikami (np.:
5) chromoproteidy przy połączeniu z barwnikami (np.:
hemoglobina, chlorofil),
hemoglobina, chlorofil),
6) metaloproteidy przy połączeniu z jonami metali (np.
6) metaloproteidy przy połączeniu z jonami metali (np.
ceruloplazmina).
ceruloplazmina).

REAKCJE CHARAKTEYSTYCZNE DLA
REAKCJE CHARAKTEYSTYCZNE DLA
WYKRYWANIA BIAŁEK
WYKRYWANIA BIAŁEK
Reakcja biutetowa
Reakcja biutetowa
Reakcja ksantoproteinowa
Reakcja ksantoproteinowa

REAKCJA BIUTETOWA
REAKCJA BIUTETOWA
Jest to reakcja polegająca na powstaniu intensywnego,
Jest to reakcja polegająca na powstaniu intensywnego,
fioletowego zabarwienia po dodaniu do dwumocznika
fioletowego zabarwienia po dodaniu do dwumocznika
(H2NCONHCONH2 - zwanego biuretenem), soli miedzi w
(H2NCONHCONH2 - zwanego biuretenem), soli miedzi w
środowisku alkalicznym.
środowisku alkalicznym.
Barwa jest wywołana tworzeniem się kompleksu z
Barwa jest wywołana tworzeniem się kompleksu z
miedzią.
miedzią.
Reakcja ta jest charakterystyczna dla białek ( ze
Reakcja ta jest charakterystyczna dla białek ( ze
wzrostem liczby wiązań peptydowych w cząsteczce,
wzrostem liczby wiązań peptydowych w cząsteczce,
barwa powstałego kompleksu jest intensywniejsza), jak
barwa powstałego kompleksu jest intensywniejsza), jak
również dla wszystkich związków, które zawierają więcej
również dla wszystkich związków, które zawierają więcej
niż jedno wiązanie amidowe – CONH – lub podobne
niż jedno wiązanie amidowe – CONH – lub podobne
grupy w cząsteczce.
grupy w cząsteczce.
Reakcje biuretowe dają np. aminoalkohole zawierające
Reakcje biuretowe dają np. aminoalkohole zawierające
ugrupowanie. aminohydroksyetylenowe – CH(NH2)CHOH
ugrupowanie. aminohydroksyetylenowe – CH(NH2)CHOH

Węglowodany
Węglowodany
podstawowe wiadomości
podstawowe wiadomości

WĘGLOWODANY
WĘGLOWODANY
(CUKRY,SACHARYDY
(CUKRY,SACHARYDY)
)
P o d z ia ł c u k r ó w z e w z g lę d u n a b u d o w ę :
C U K R Y P R O S T E
( m o n o s a c h a r y d y )
C U K R Y Z Ł O Ż O N E N I Ż S Z E
( o lig o s a c h a r y d y )
C U K R Y Z Ł O Ż O N E W Y Ż S Z E
( p o lis a c h a r y d y )
C U K R Y
( s a c h a r y d y )

Węglowodany-charakterystyka ogólna
Węglowodany-charakterystyka ogólna
Obok białek i tłuszczów są podstawową grupą związków
Obok białek i tłuszczów są podstawową grupą związków
naturalnych
naturalnych
Stanowią ½ masy związków organicznych na Ziemi
Stanowią ½ masy związków organicznych na Ziemi
Powstają w procesie fotosyntezy
Powstają w procesie fotosyntezy
Ich zawartość w organizmach roślinnych dochodzi do 80%
Ich zawartość w organizmach roślinnych dochodzi do 80%
ich suchej masy
ich suchej masy
Spełniają rolę materiału zapasowego, budulcowego i
Spełniają rolę materiału zapasowego, budulcowego i
usztywniającego
usztywniającego
Zbudowane są z węgla, wodoru i tlenu.
Zbudowane są z węgla, wodoru i tlenu.

Podział cukrów prostych
Podział cukrów prostych
A L D O Z Y
( - C H O )
c u k r y p r o s te z d o d a tk o w ą
g r u p ą a ld e h y d o w ą
K E T O Z Y
( = C O )
c u k r y p r o s te z d o d a tk o w ą
g r u p ą k e to n o w ą
M O N O Z Y
( m o n o s a c h a r y d y )
-
zawierają 3-7 atomów węgla w cząsteczce,
-uważamy je za alkohole wielowodorotlenowe z dodatkową
grupą funkcyjną: aldehydową (formylową) lub ketonową
(karbonylową).

C
C
n
n
H
H
2n
2n
O
O
n
n
-monozy
-monozy
C
C
3
3
H
H
6
6
O
O
3
3
aldotriozy
aldotriozy
ketotriozy
ketotriozy
C
C
4
4
H
H
8
8
O
O
4
4
aldotetrozy
aldotetrozy
ketotetrozy
ketotetrozy
C
C
5
5
H
H
10
10
O
O
5
5
aldopentozy
aldopentozy
ketopentozy
ketopentozy
C
C
6
6
H
H
12
12
O
O
6
6
aldoheksozy
aldoheksozy
ketoheksozy
ketoheksozy
C
C
7
7
H
H
14
14
O
O
7
7
aldoheptozy
aldoheptozy
ketoheptozy
ketoheptozy

Przykłady cukrów prostych:
Przykłady cukrów prostych:
TRIOZY
TRIOZY
Dihydroksyaceton
Dihydroksyaceton
(ketotrioza)
(ketotrioza)
D-aldehyd glicerynowy
D-aldehyd glicerynowy
(aldotrioza)
(aldotrioza)
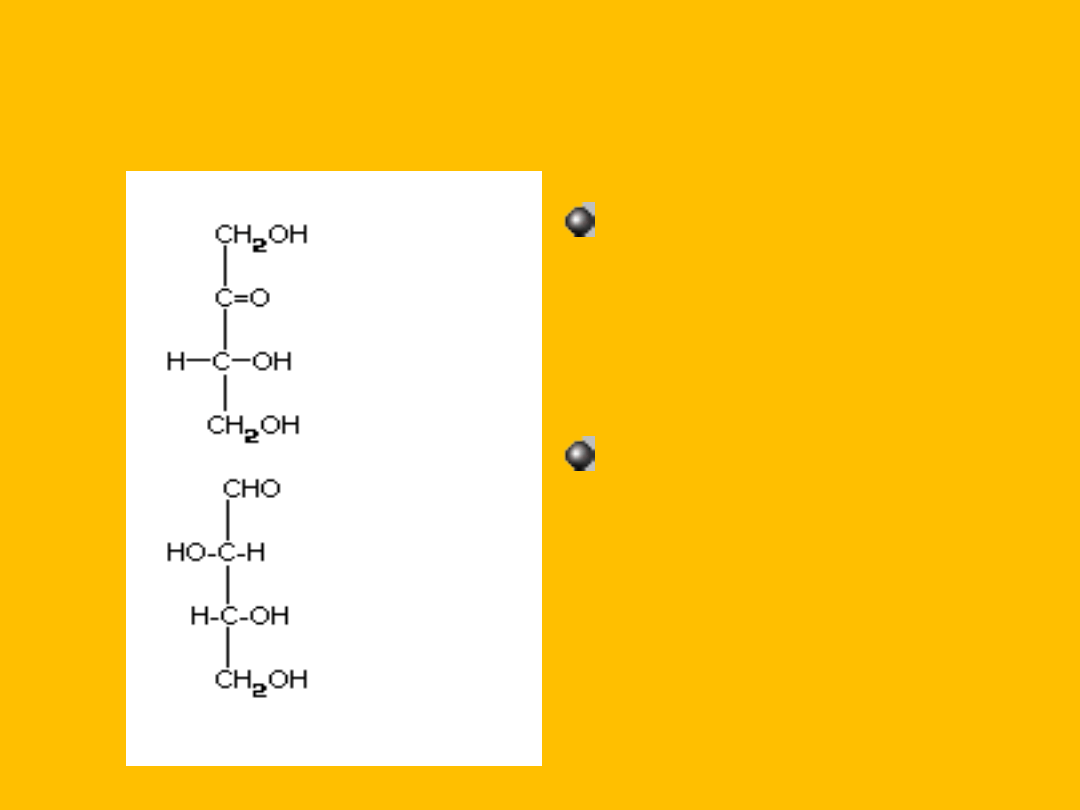
TETROZY
TETROZY
D-erytruloza
D-erytruloza
(ketotetroza)
(ketotetroza)
D-treoza
D-treoza
(aldotetroza)
(aldotetroza)
PENTOZY
PENTOZY
D-ryboza
D-ryboza
(aldopentoza)
(aldopentoza)
D-rybuloza
D-rybuloza
(ketopentoza)
(ketopentoza)

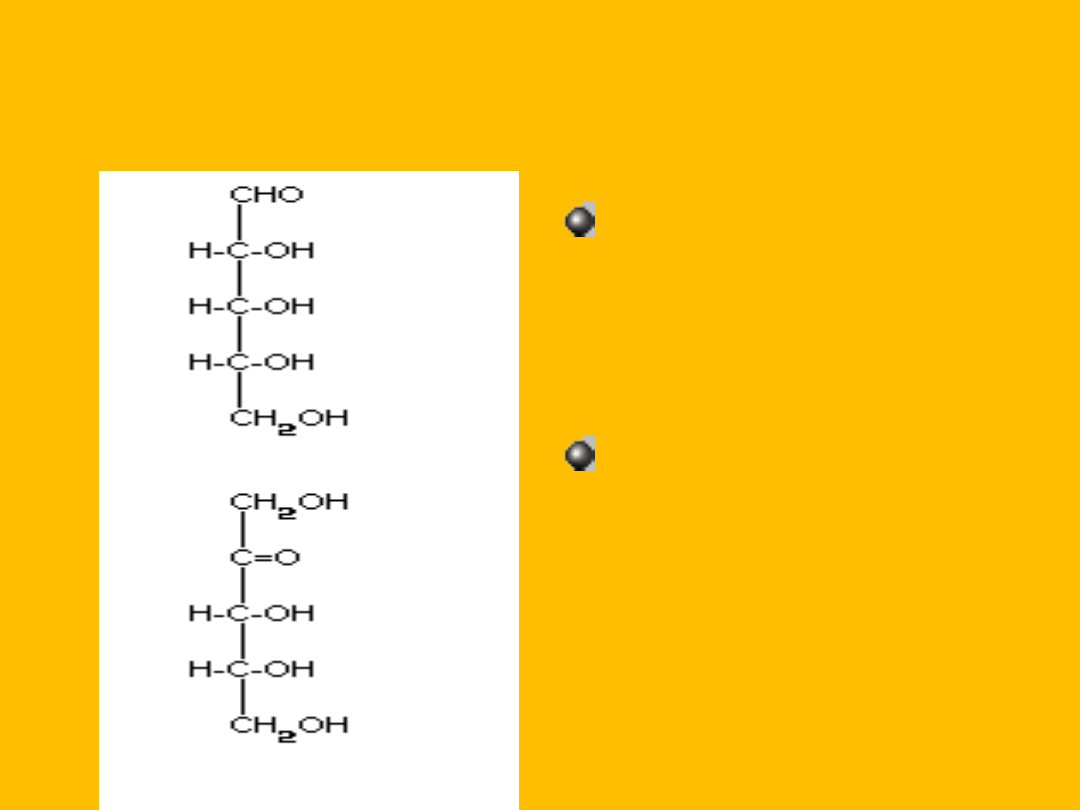
HEKSOZY
HEKSOZY
D-glukoza
D-glukoza
(aldoheksoza)
(aldoheksoza)
D-alloza
D-alloza
(aldoheksoza)
(aldoheksoza)

D-mannoza
D-mannoza
(aldoheksoza)
(aldoheksoza)
D-galaktoza
D-galaktoza
(aldoheksoza)
(aldoheksoza)

D-fruktoza
D-fruktoza
(ketoheksoza)
(ketoheksoza)
D-sorboza
D-sorboza
(ketoheksoza)
(ketoheksoza)
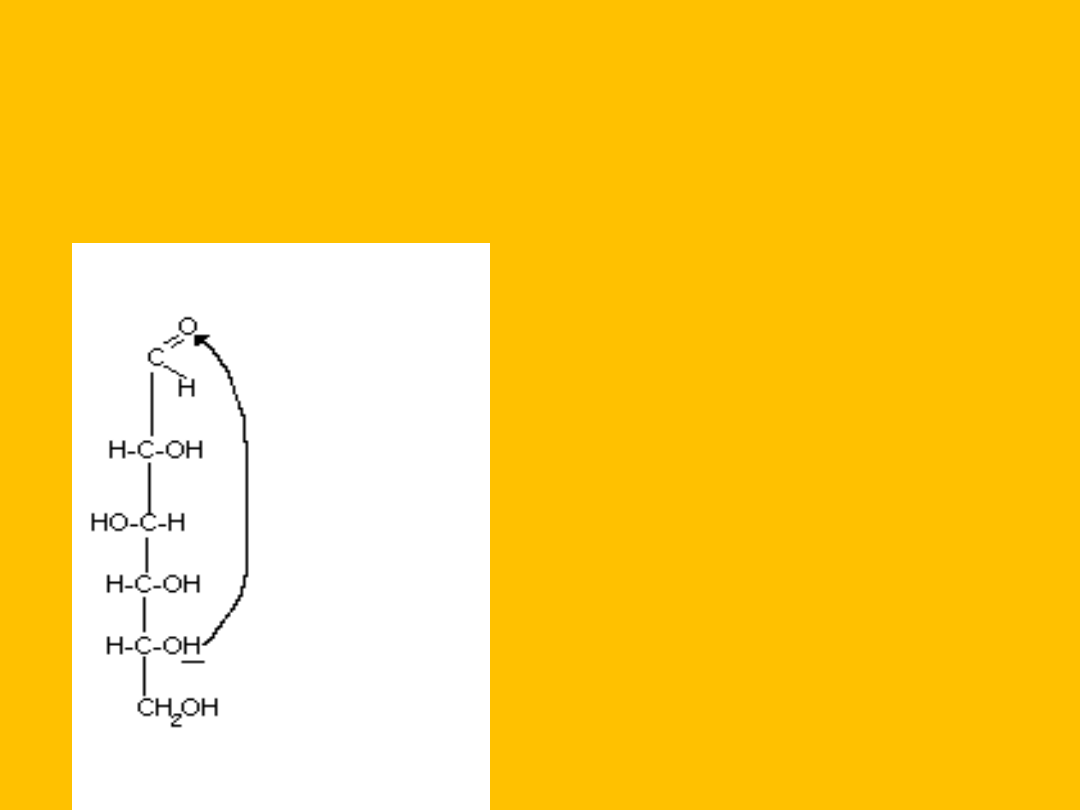
FORMA PIERŚCIENIOWA
FORMA PIERŚCIENIOWA
GLUKOZY
GLUKOZY
W roztworach wodnych cukry proste występują w formach pierścieniowych
W roztworach wodnych cukry proste występują w formach pierścieniowych
-
-
C-O-C-
C-O-C-
wiązanie hemiacetalowe
wiązanie hemiacetalowe
(półacetalowe)
(półacetalowe)
wzór odmiany
wzór odmiany
łańcuchowej D-glukozy
łańcuchowej D-glukozy
Występowanie obok siebie grup –OH i –CHO lub =CO jest
Występowanie obok siebie grup –OH i –CHO lub =CO jest
przyczyną tworzenia się wewnątrz cząsteczkowych wiązań
przyczyną tworzenia się wewnątrz cząsteczkowych wiązań
zwanych półacetalowymi (hemiacetalowymi) lub
zwanych półacetalowymi (hemiacetalowymi) lub
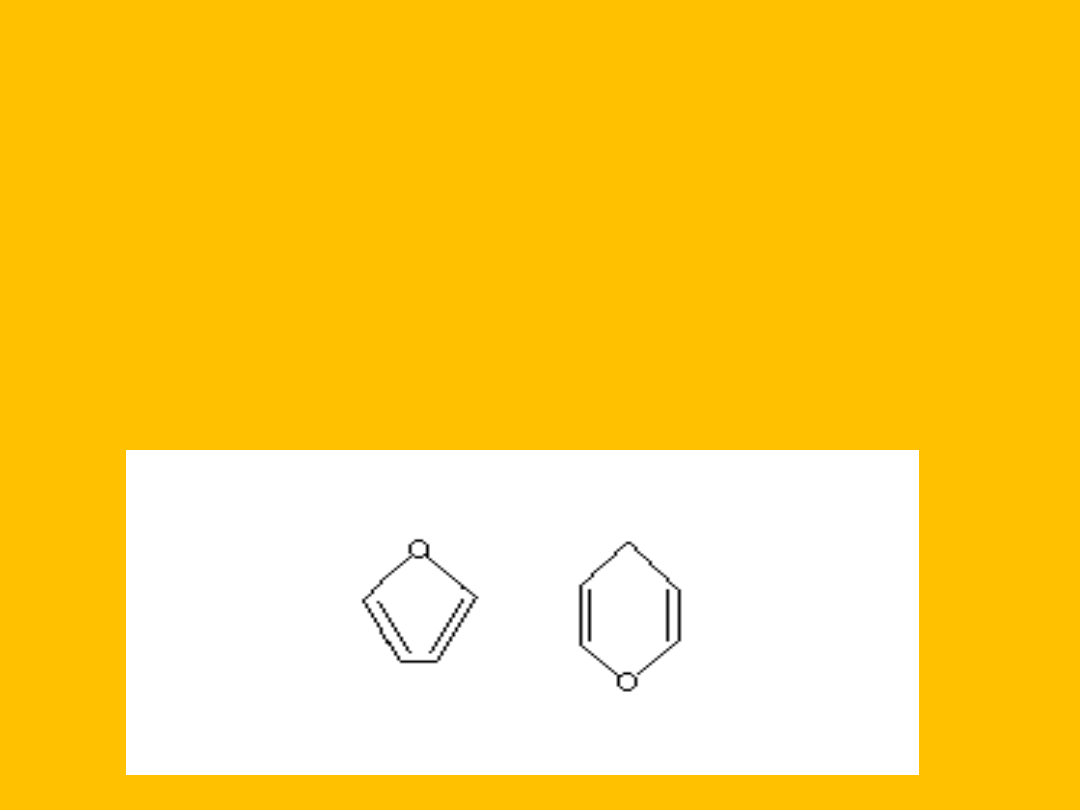
półketalowymi (hemiketalowymi) i powstają heterocykliczne
półketalowymi (hemiketalowymi) i powstają heterocykliczne
układy pięcio- lub sześcioczłonowe będące pochodnymi
układy pięcio- lub sześcioczłonowe będące pochodnymi
furanu lub piranu:
furanu lub piranu:
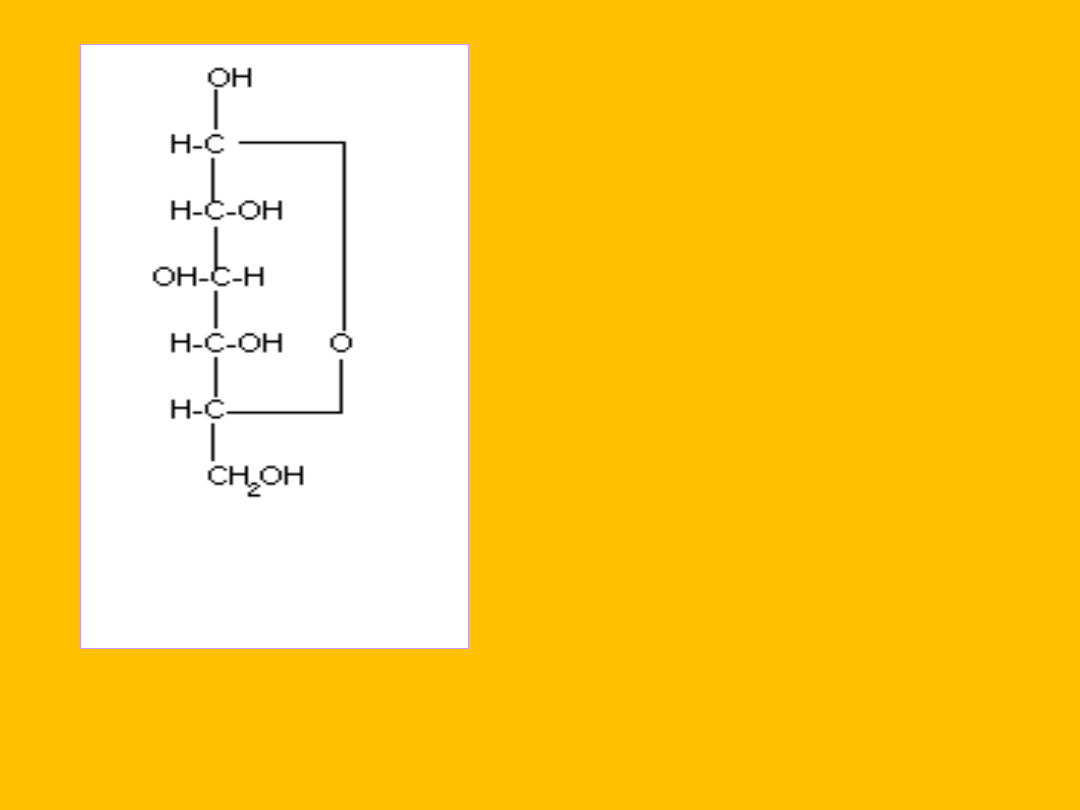
Wzór rzutowy
Wzór rzutowy
Fischera odmiany
Fischera odmiany
pierścieniowej
pierścieniowej
(półacetalowej)
(półacetalowej)
W roztworach wodnych forma łańcuchowa współistnieje z formą
pierścieniową
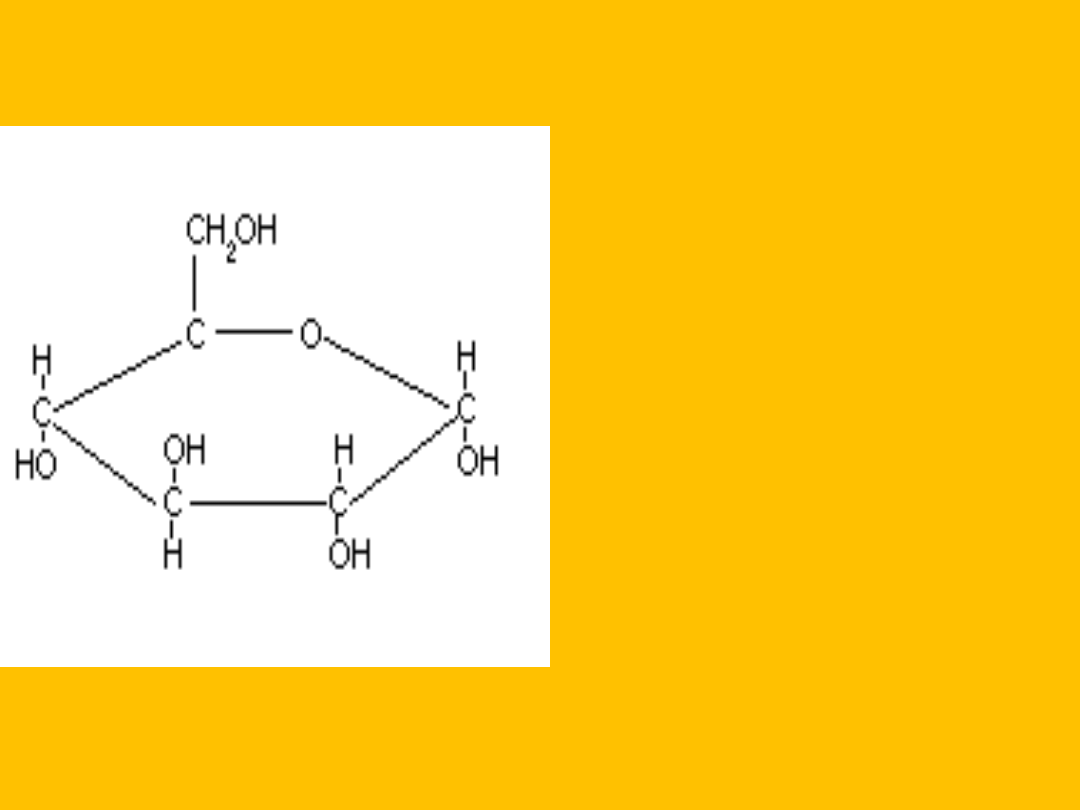
-
-
D-glukopiranoza
D-glukopiranoza
wzór Hawortha
wzór Hawortha
(taflowy)
(taflowy)
Tautomeria-współistnienie w równowadze dwóch
izomerów przechodzących w siebie nawzajem
(łańcuch- pierścień)
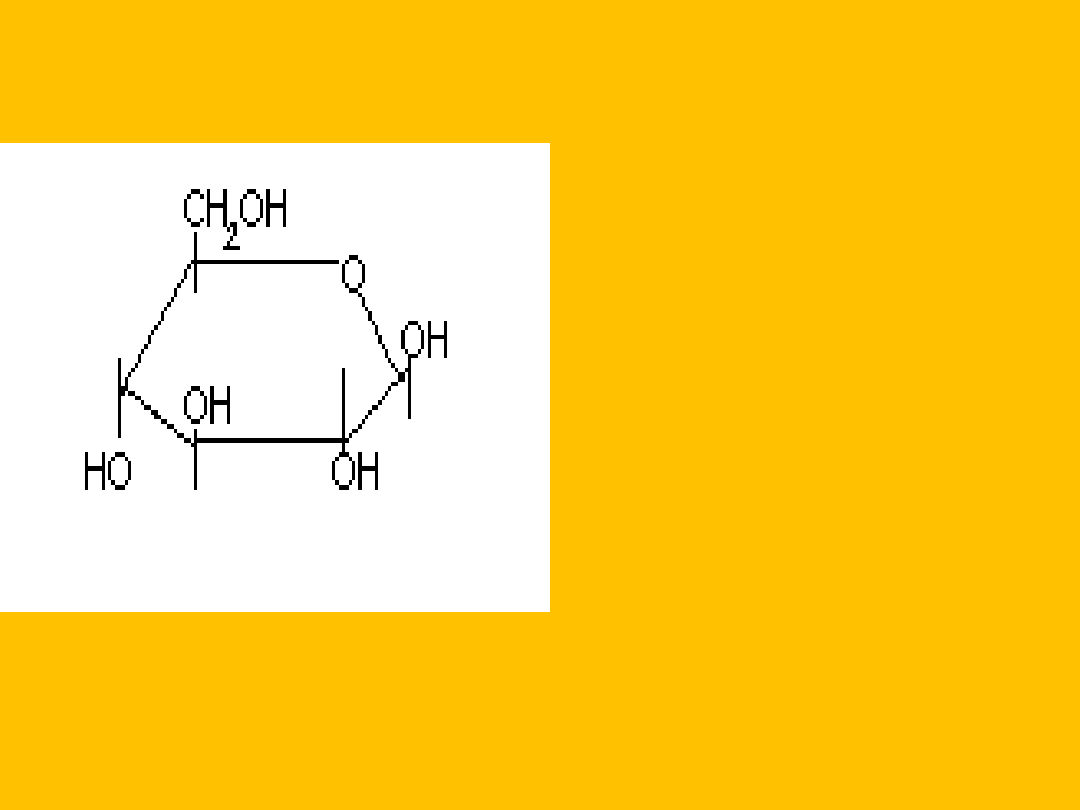
- D-glukopiranoza
- D-glukopiranoza
Stereoizomery różniące się konfiguracją przy
pierwszym węglu to ANOMERY.

Właściwości cukrów
Właściwości cukrów
prostych:
prostych:
1.Właściwości glukozy:
1.Właściwości glukozy:
Słodka, krystaliczna, bardzo dobrze rozpuszczalna
Słodka, krystaliczna, bardzo dobrze rozpuszczalna
w wodzie,
w wodzie,
Ulega fermentacji alkoholowej:
Ulega fermentacji alkoholowej:
C
C
6
6
H
H
12
12
O
O
6 --fermentacja--------
6 --fermentacja--------
2C
2C
2
2
H
H
5
5
OH + 2CO
OH + 2CO
2
2
Daje pozytywne wyniki w próbie Trommera i w
Daje pozytywne wyniki w próbie Trommera i w
próbie Tollensa (wykazuje własności redukujące
próbie Tollensa (wykazuje własności redukujące
wynikające z obecności grupy aldehydowej –CHO)
wynikające z obecności grupy aldehydowej –CHO)

Energia
Węglowodany ~ 4
kcal/g
Lipidy ~ 9.5
kcal/g
Białka ~ 5 kcal/g

Glukoza
5 - 6.5 mM (90-120
mg/dL)
Glikogen i tłuszcze
(zapasy energii)
Glikoliza
cykl kwasów
trójkarboksylo
wych
(energia)
Cykl
pentozofosforano
wy
(energia,
nukleotydy)
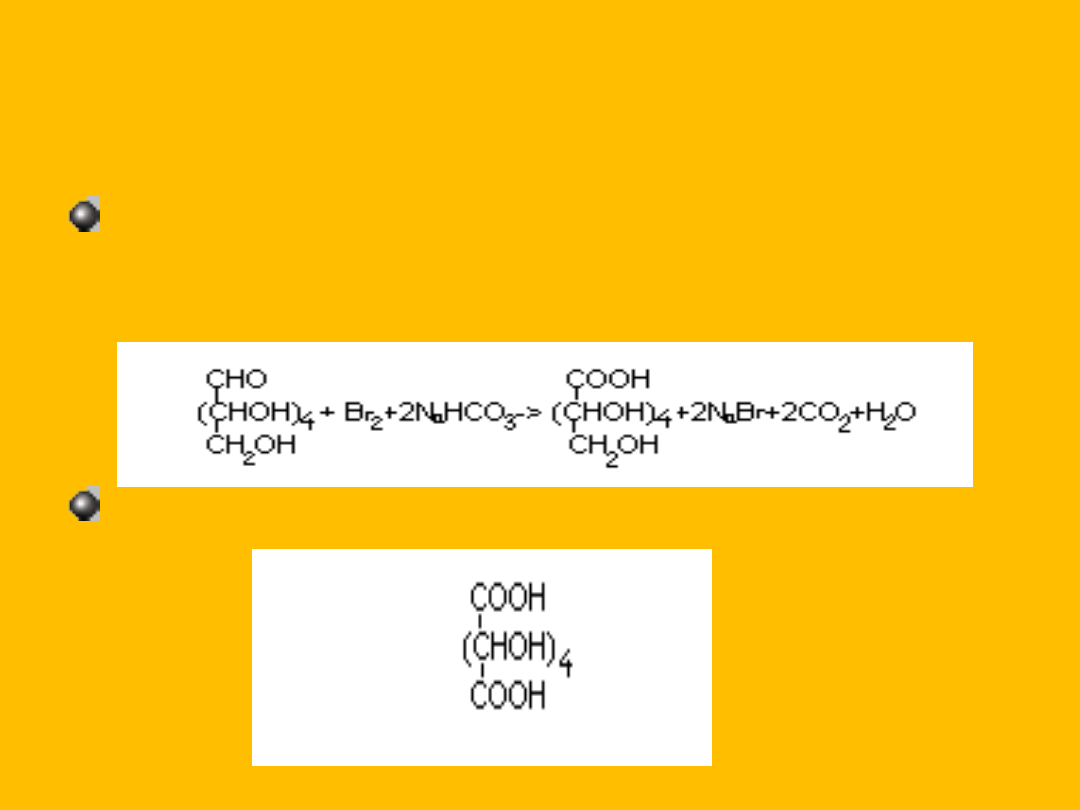
Inne łagodne utleniacze utleniają glukozę analogicznie
Inne łagodne utleniacze utleniają glukozę analogicznie
np. woda bromowa (łagodne utlenianie utlenia tylko
np. woda bromowa (łagodne utlenianie utlenia tylko
grupę aldehydową do grupy karboksylowej)
grupę aldehydową do grupy karboksylowej)
Silny utleniacz np..HNO
Silny utleniacz np..HNO
3
3
utlenia glukozę do kwasu
utlenia glukozę do kwasu
glukarowego
glukarowego
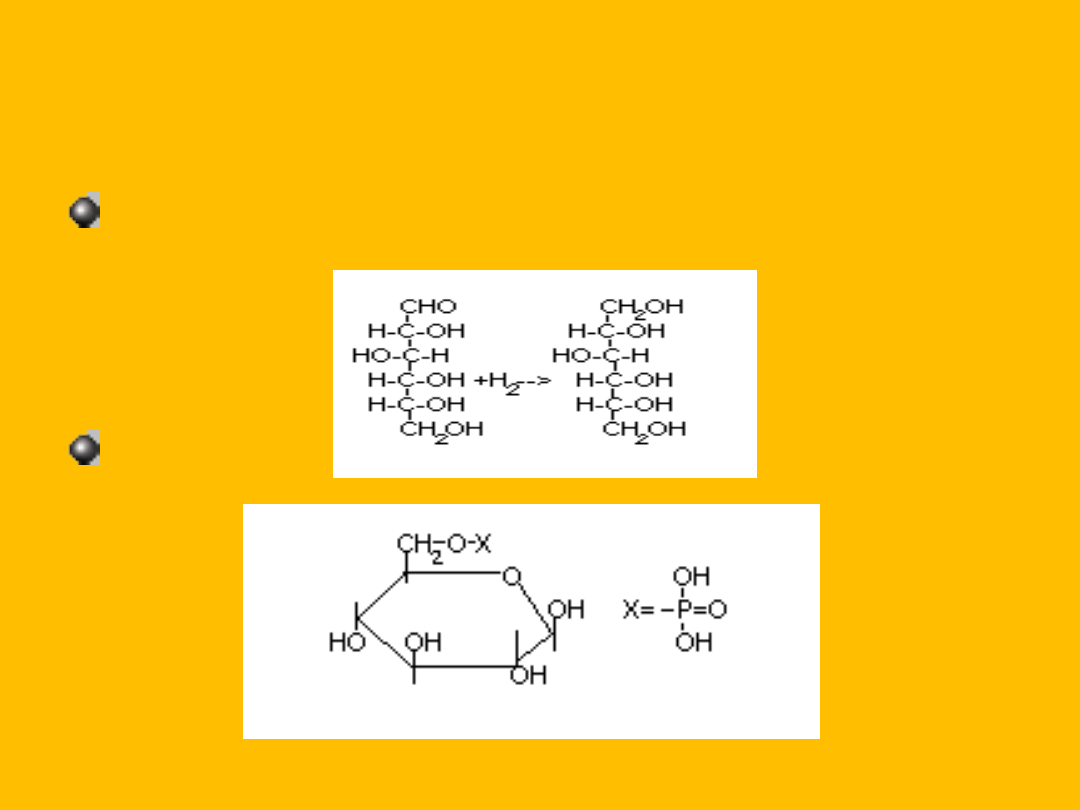
Ulega redukcji wodorem w obecności katalizatora palladowego
Ulega redukcji wodorem w obecności katalizatora palladowego
Tworzy estry z H
Tworzy estry z H
3
3
PO
PO
4
4
D-glukopiranozylo – 6-fosforan (V)
D-glukopiranozylo – 6-fosforan (V)
Tworzy glikozydy; grupa –OH przy pierwszym
Tworzy glikozydy; grupa –OH przy pierwszym
węglu to tzw. glikozydowa grupa wodorotlenowa
węglu to tzw. glikozydowa grupa wodorotlenowa
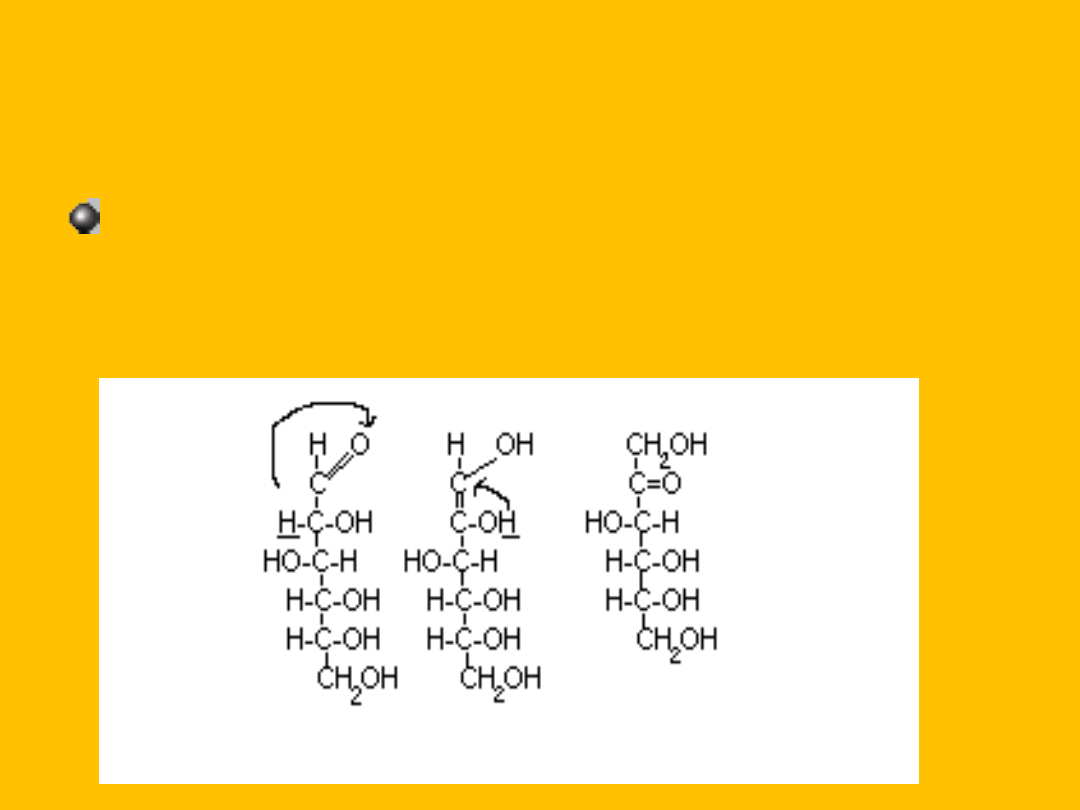
Ulega enolizacji w środowisku zasadowym (aldoza
Ulega enolizacji w środowisku zasadowym (aldoza
przechodzi w ketozę za pośrednictwem
przechodzi w ketozę za pośrednictwem
przejściowej formy endiolowej)
przejściowej formy endiolowej)

2.Właściwości fruktozy:
2.Właściwości fruktozy:
słodka, krystaliczna, dobrze
słodka, krystaliczna, dobrze
rozpuszczalna w wodzie,
rozpuszczalna w wodzie,
ulega fermentacji mlekowej,
ulega fermentacji mlekowej,
Daje pozytywne
Daje pozytywne
wyniki w próbie
wyniki w próbie
Trommera i
Trommera i
Tollensa(jest to
Tollensa(jest to
wynikiem specyficznej
wynikiem specyficznej
budowy fruktozy- jest
budowy fruktozy- jest
ona
ona
–
–
hydroksykwasem) ale
hydroksykwasem) ale
jej utlenianie
jej utlenianie
przebiega z
przebiega z
rozerwaniem łańcucha
rozerwaniem łańcucha
węglowego
węglowego

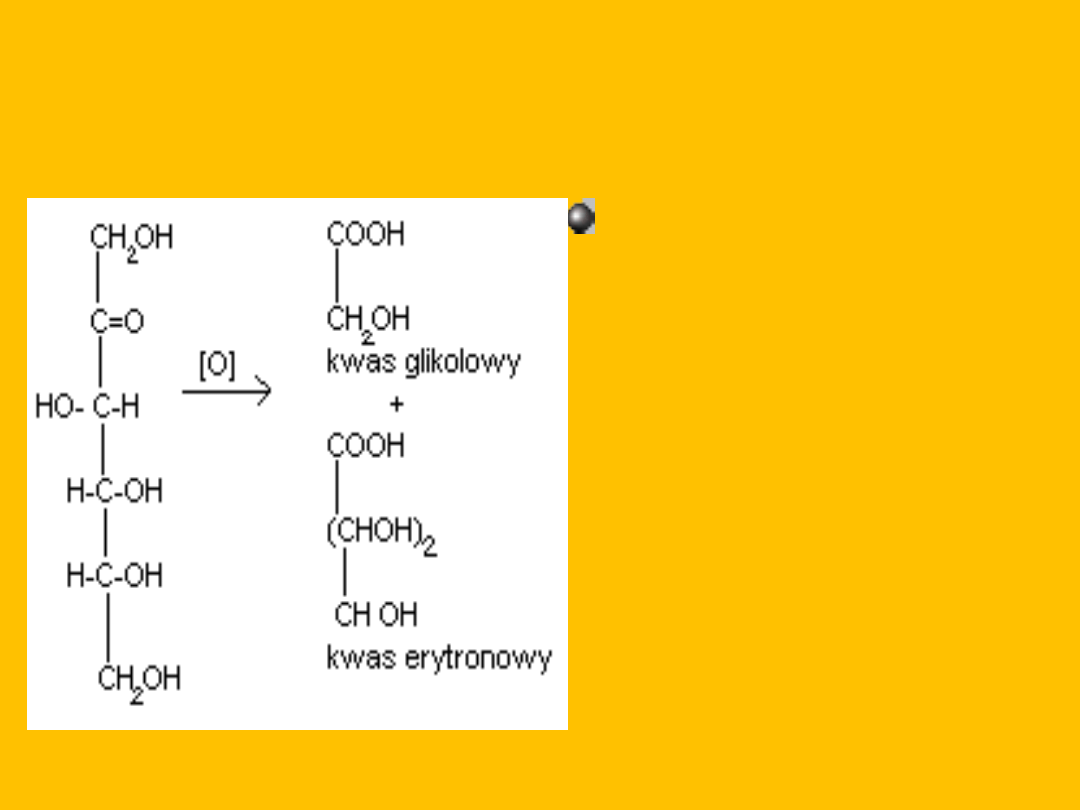
I dalej pod
I dalej pod
wpływem silnego
wpływem silnego
utleniacza HNO
utleniacza HNO
3
3
powstają :
powstają :

woda bromowa nie utlenia ketoz
woda bromowa nie utlenia ketoz
(próba taka jest dogodnym
(próba taka jest dogodnym
odczynnikiem służącym do
odczynnikiem służącym do
rozróżniania aldoz i ketoz)
rozróżniania aldoz i ketoz)
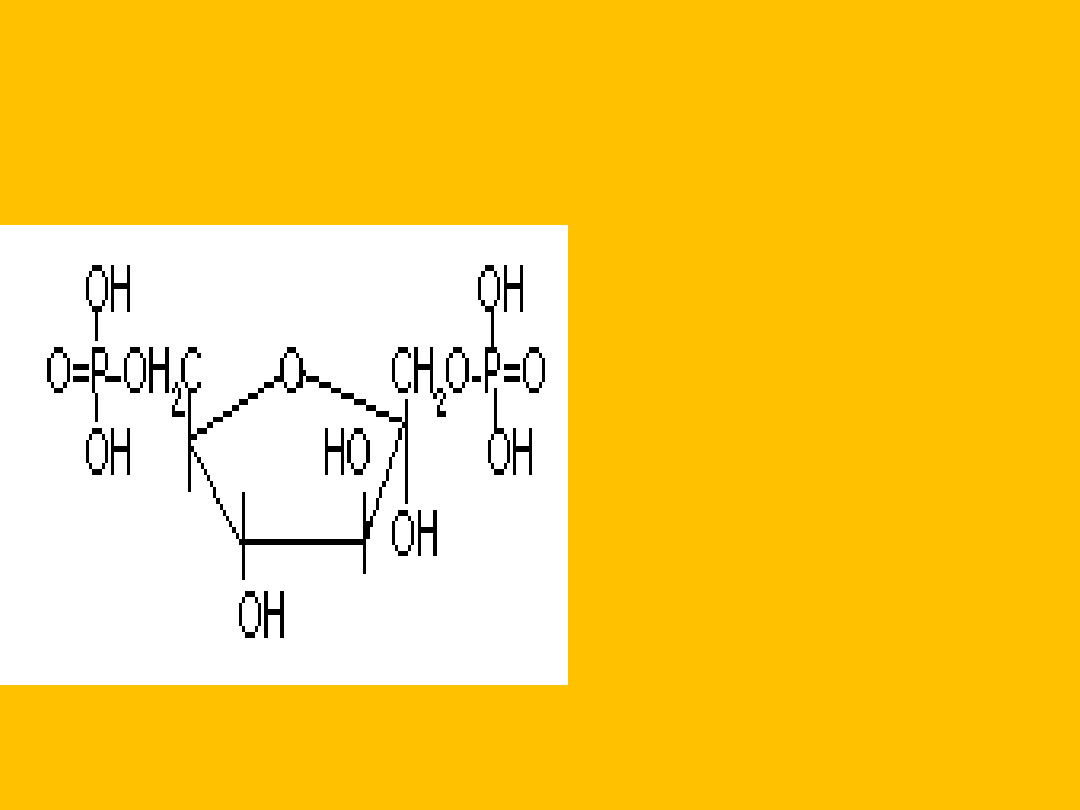
Reakcja estryfikacji z H
Reakcja estryfikacji z H
3
3
PO
PO
4
4
D-
D-
fruktofuranozylo-
fruktofuranozylo-
1,6-difosforan
1,6-difosforan
reakcja fruktozy daje mieszaninę
reakcja fruktozy daje mieszaninę
heksytów: sorbitolu i mannitolu,
heksytów: sorbitolu i mannitolu,
grupa –OH przy drugim węglu to
grupa –OH przy drugim węglu to
glikozydowa grupa wodorotlenowa,
glikozydowa grupa wodorotlenowa,
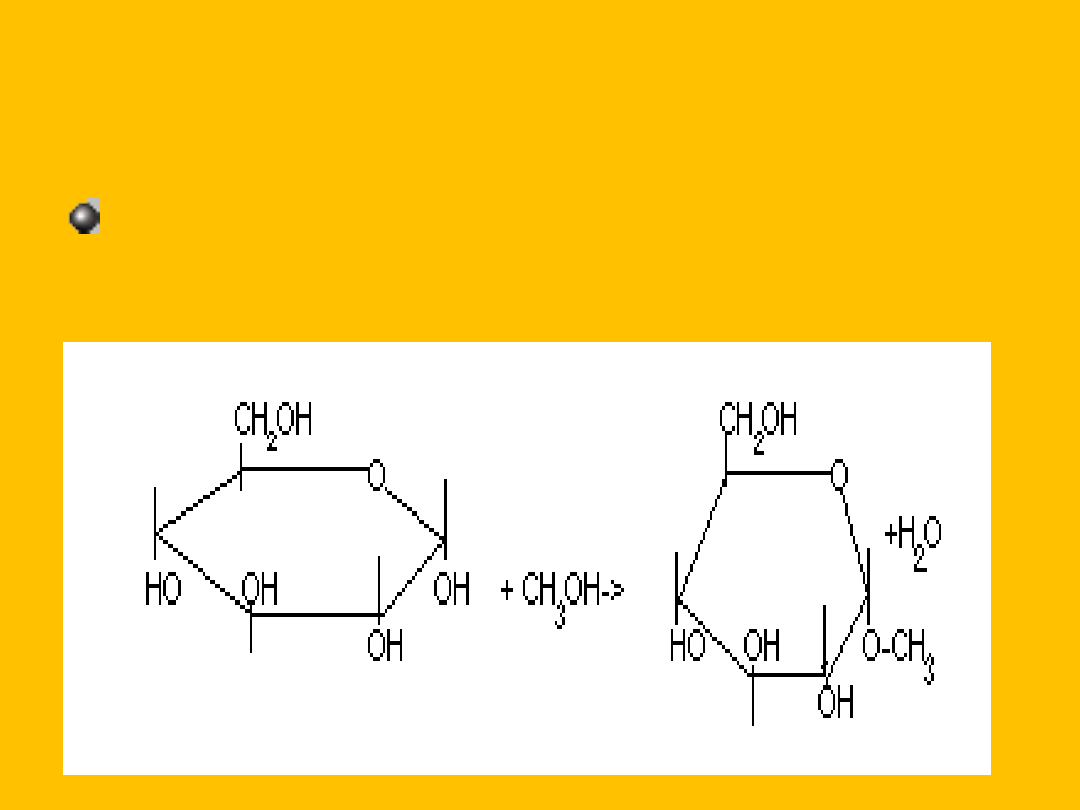
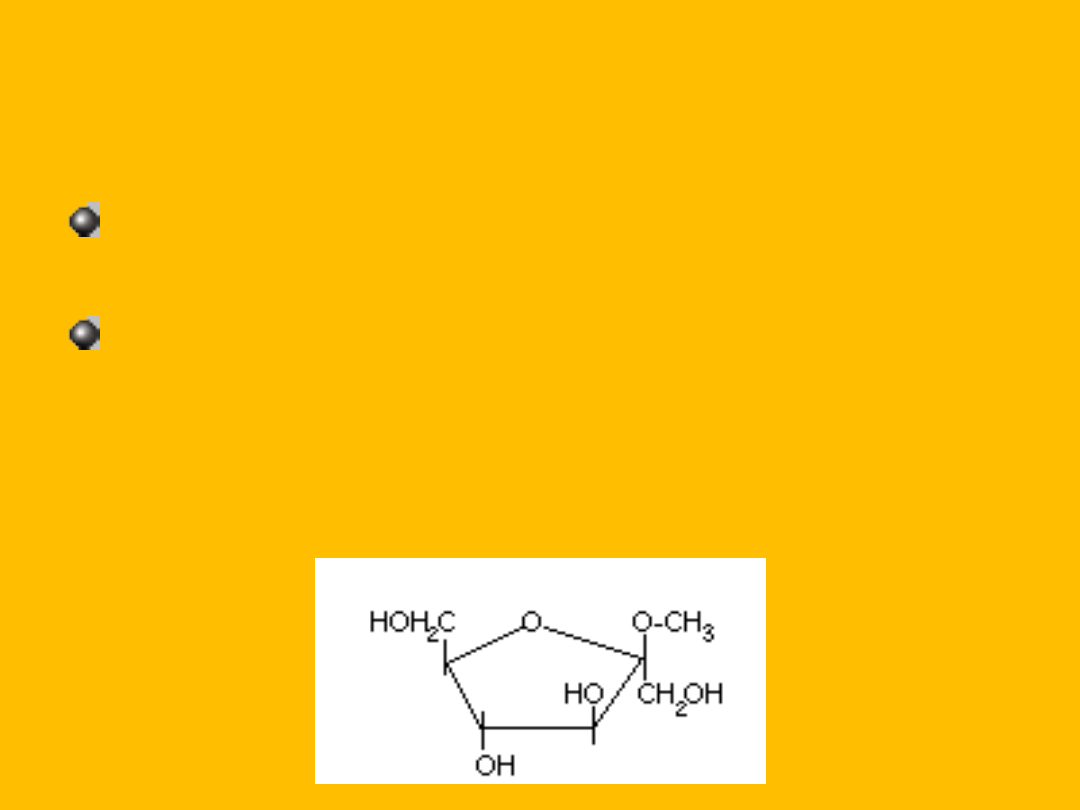
dlatego fruktoza tworzy fruktozydy np. w
dlatego fruktoza tworzy fruktozydy np. w
reakcji z metanolem, w obecności HCl
reakcji z metanolem, w obecności HCl
powstaje
powstaje
:
:

BISACHARYDY
BISACHARYDY
Dwucukry- to produkty
Dwucukry- to produkty
kondensacji dwóch cząsteczek
kondensacji dwóch cząsteczek
cukrów prostych.
cukrów prostych.
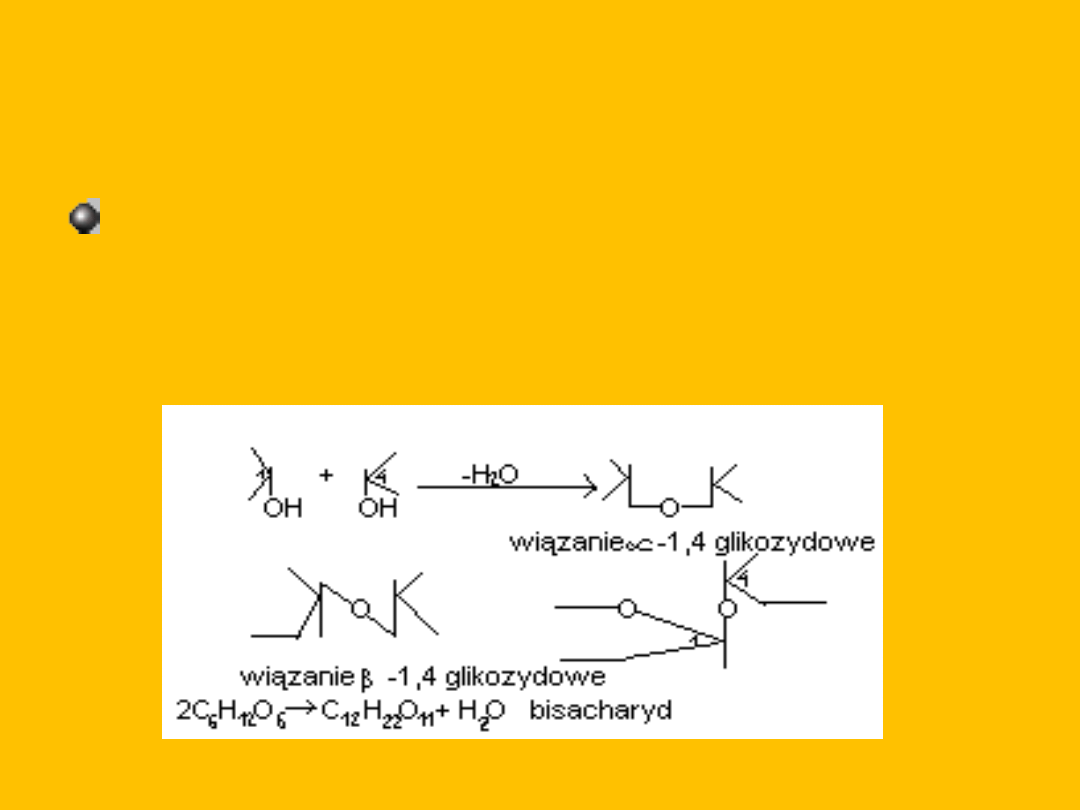
Powstają w wyniku kondensacji dwóch cząsteczek
Powstają w wyniku kondensacji dwóch cząsteczek
monoz- wiązanie wytwarza się z glikozydowej
monoz- wiązanie wytwarza się z glikozydowej
grupy –OH jego monosacharydu i dowolnej grupy
grupy –OH jego monosacharydu i dowolnej grupy
–OH drugiego monosacharydu.
–OH drugiego monosacharydu.
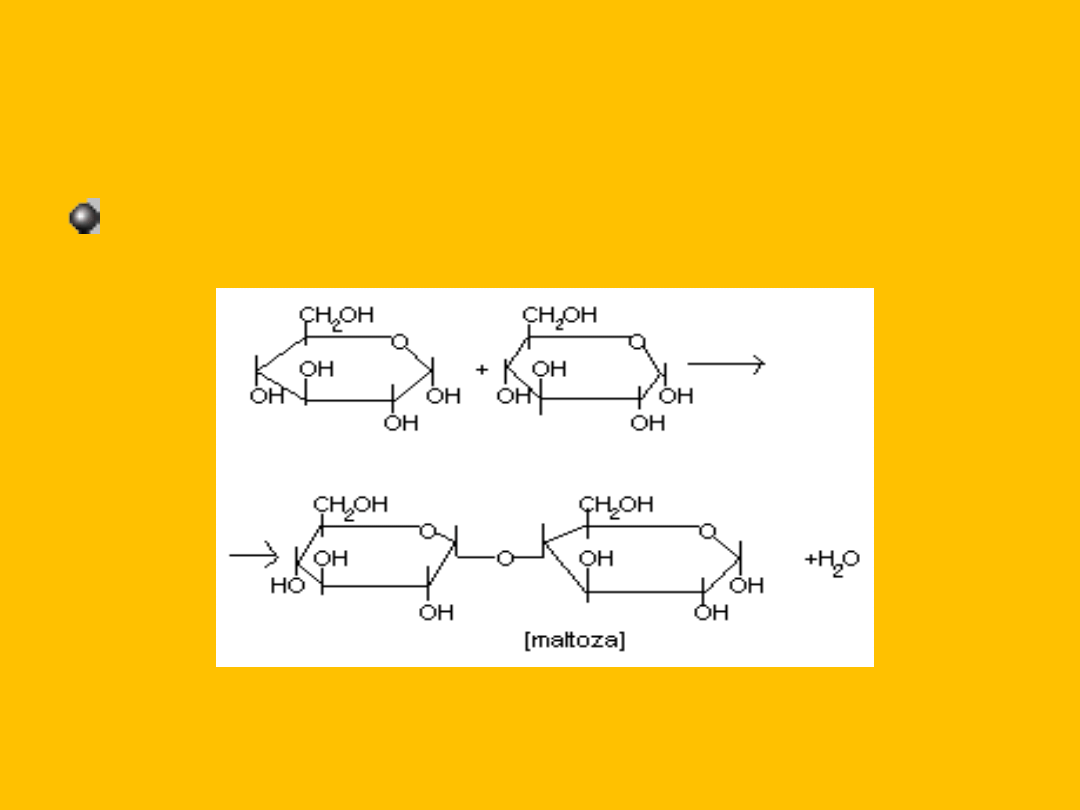
2
2
glukopiranoza
glukopiranoza
maltoza + H
maltoza + H
2
2
O
O
D- glukopiranozylo- 1,4-
D- glukopiranozylo- 1,4-
D- glukopiranoza
D- glukopiranoza
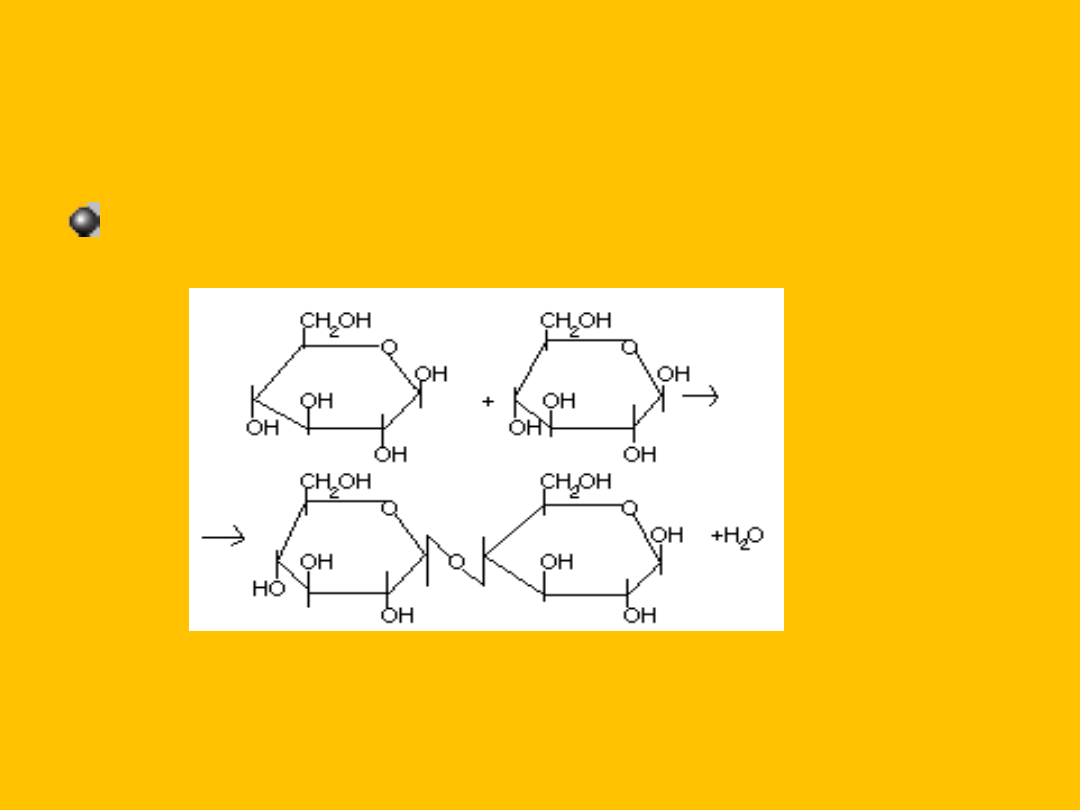
2
2
glukopiranozy
glukopiranozy
celobioza + H
celobioza + H
2
2
O
O
- D-glukopiranozylo-1,4-
- D-glukopiranozylo-1,4-
D-glukopiranoza
D-glukopiranoza
[celobioza]
[celobioza]
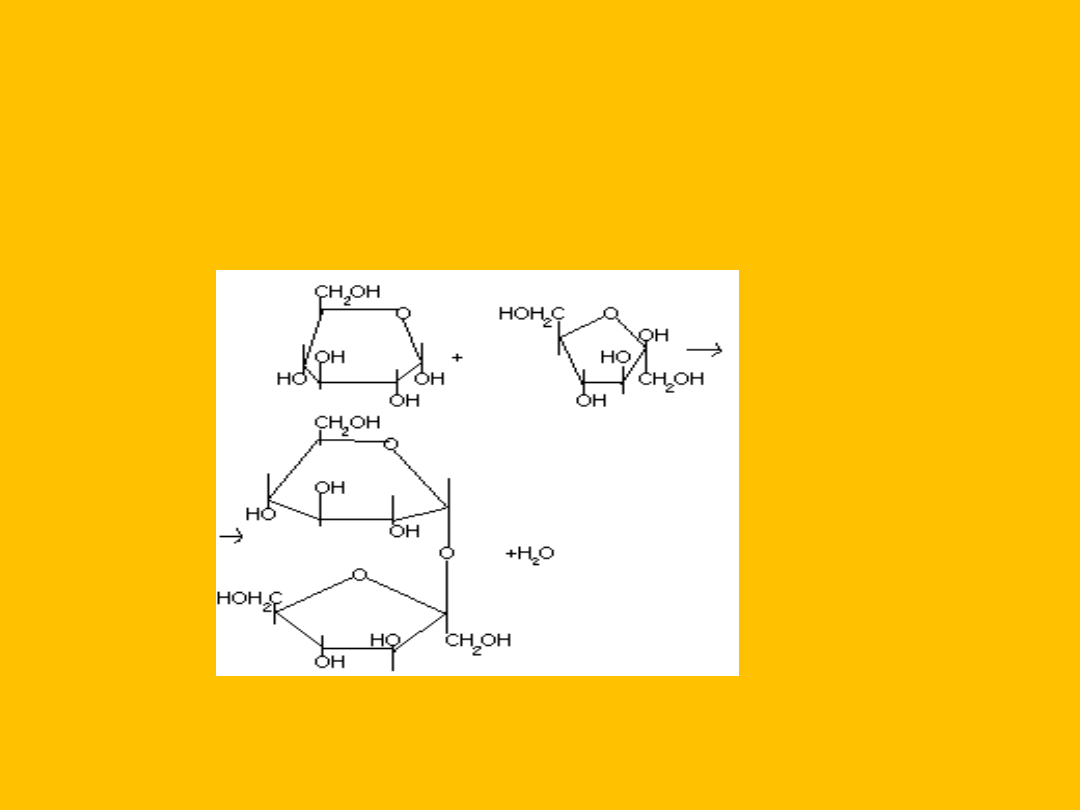
D glukopiranoza+
D glukopiranoza+
–D- fruktofuranoza
–D- fruktofuranoza
sacharoza+
sacharoza+
H
H
2
2
O
O
D-glukopiranozylo-1,2-
D-glukopiranozylo-1,2-
D-fruktofuranoza [sacharoza]
D-fruktofuranoza [sacharoza]
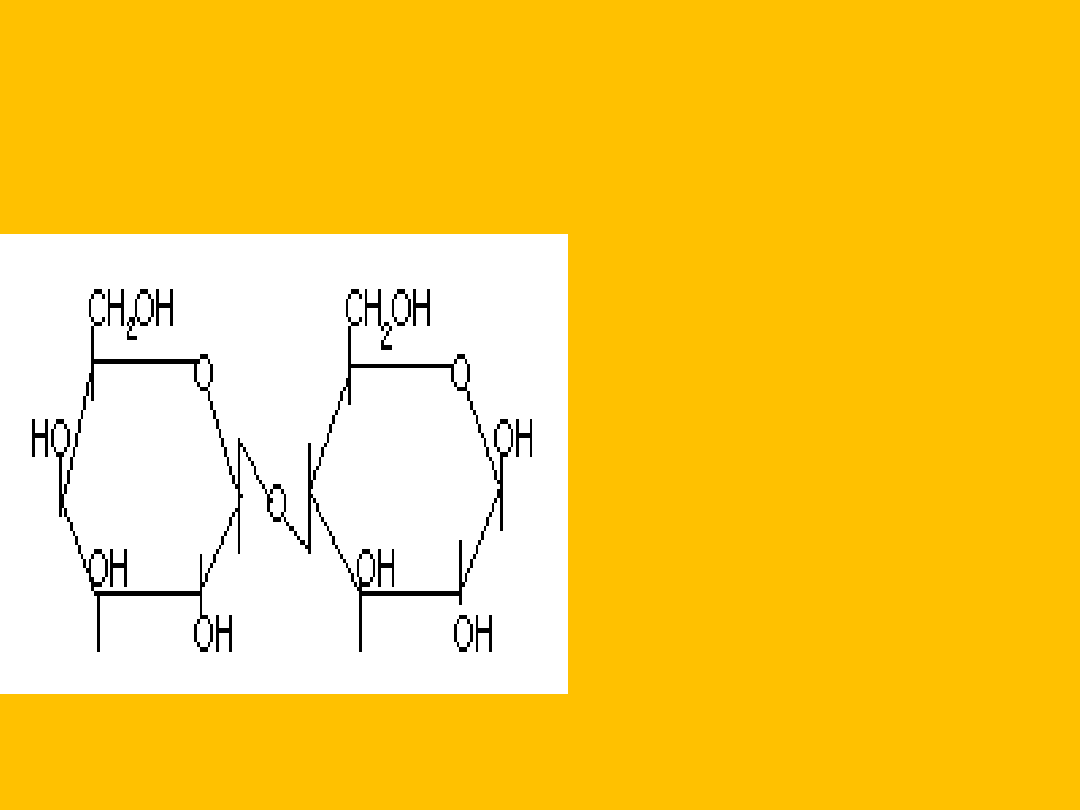
D-galaktopiranozylo-
D-galaktopiranozylo-
1,4-
1,4-
D-glukopiranoza
D-glukopiranoza
(laktoza)
(laktoza)
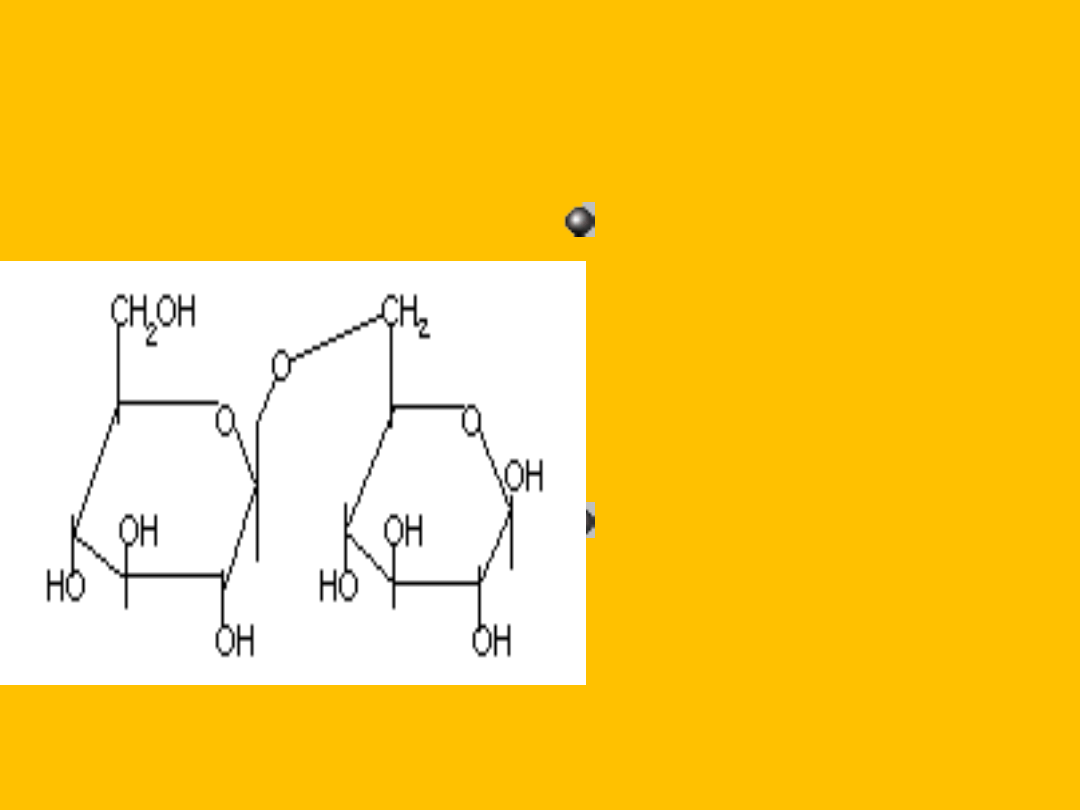
GENCJOBIOZA
GENCJOBIOZA
D- glukopiranozylo-
D- glukopiranozylo-
1,6-
1,6-
D-glukopiranoza
D-glukopiranoza
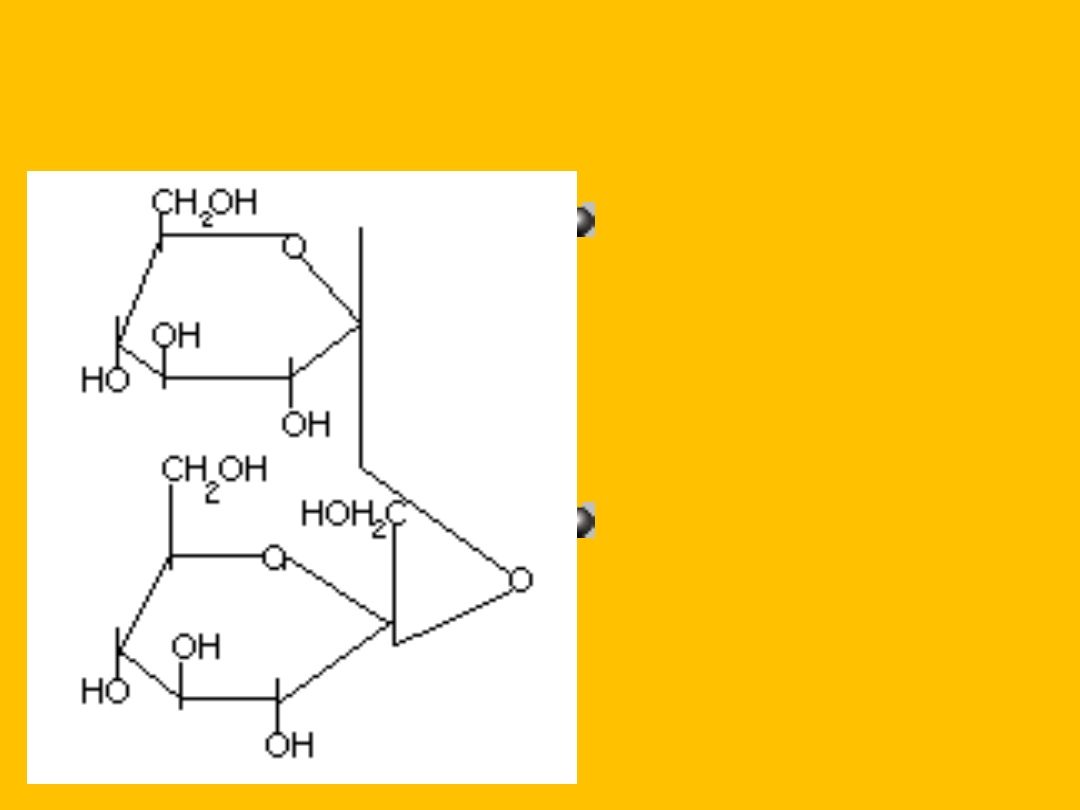
TREHALOZA
TREHALOZA
D- glukopiranozylo-
D- glukopiranozylo-
1,1-
1,1-
D-
D-
glukopiranoza
glukopiranoza

Właściwości dwucukrów:
Właściwości dwucukrów:
Stałe,krystaliczne,słodkie,dobrze
Stałe,krystaliczne,słodkie,dobrze
rozpuszczalne,
rozpuszczalne,
W środowisku kwaśnym lub w
W środowisku kwaśnym lub w
obecności enzymów ulegają
obecności enzymów ulegają
hydrolizie:
hydrolizie:
C
C
12
12
H
H
22
22
O
O
11
11
+ H
+ H
2
2
O
O
2 C
2 C
6
6
H
H
12
12
O
O
6
6
maltoza +woda
maltoza +woda
2
2
D-
D-
glukopiranoza
glukopiranoza

Dwucukry,w których glikozydowa grupa -OH jest
Dwucukry,w których glikozydowa grupa -OH jest
zablokowana (sacharoza,trehaloza) nie wykazują
zablokowana (sacharoza,trehaloza) nie wykazują
własności redukujących, inne dają pozytywne
własności redukujących, inne dają pozytywne
wyniki w próbie Trommera lub w próbie Tollensa
wyniki w próbie Trommera lub w próbie Tollensa
(przykładem takiego cukru jest glukoza)
(przykładem takiego cukru jest glukoza)
Dwucukry redukujące wykazują mutarotację,
Dwucukry redukujące wykazują mutarotację,
tworzą również glikozydy, dwucukry
tworzą również glikozydy, dwucukry
nieredukujące takich cech nie posiadają.
nieredukujące takich cech nie posiadają.

REAKCJA
REAKCJA
KSANTOPROTEINOWA
KSANTOPROTEINOWA
Charakterystyczna reakcja barwna
Charakterystyczna reakcja barwna
na białka. Polega na powstawaniu
na białka. Polega na powstawaniu
żółtego zabarwienia przy działaniu
żółtego zabarwienia przy działaniu
na białko stężonego kwasu
na białko stężonego kwasu
azotowego (V) i następnym
azotowego (V) i następnym
zalkalizowaniu. Żółknięcie skóry
zalkalizowaniu. Żółknięcie skóry
ludzkiej po zetknięciu z kwasem
ludzkiej po zetknięciu z kwasem
jest wynikiem tej reakcji.
jest wynikiem tej reakcji.

Choroba Andersen
Choroba Andersen.
.
Choroba Andersen (glikogenoza typu IV,
Choroba Andersen (glikogenoza typu IV,
amylopektynoza) - rzadka choroba
amylopektynoza) - rzadka choroba
genetyczna, dziedziczona autosomalnie
genetyczna, dziedziczona autosomalnie
recesywnie, polegająca na zaburzeniu
recesywnie, polegająca na zaburzeniu
spichrzania glikogenu.Choroba została
spichrzania glikogenu.Choroba została
nazwana na cześć amerykańskiej lekarki
nazwana na cześć amerykańskiej lekarki
Dorothy Hansine Andersen.
Dorothy Hansine Andersen.

Choroba jest spowodowana defektem
Choroba jest spowodowana defektem
enzymu rozgałęziającego glikogen.
enzymu rozgałęziającego glikogen.
Powstaje glikogen o nienormalnie
Powstaje glikogen o nienormalnie
długich łańcuchach podobnych do
długich łańcuchach podobnych do
tych jakie znajdują się w
tych jakie znajdują się w
amylopektynie.
amylopektynie.

Efektem tego jest nagromadzenie
Efektem tego jest nagromadzenie
nieprawidłowego strukturalnie
nieprawidłowego strukturalnie
glikogenu w narządach, głównie w
glikogenu w narządach, głównie w
wątrobie i mięśniach (mimo na ogół
wątrobie i mięśniach (mimo na ogół
prawidłowej jego zawartości w
prawidłowej jego zawartości w
wątrobie).
wątrobie).

Objawy:
Objawy:
hepatomegalia i splenomegalia
hepatomegalia i splenomegalia
marskość wątroby
marskość wątroby
niewydolność wątroby
niewydolność wątroby
hipotonia mięśniowa
hipotonia mięśniowa
upośledzenie wzrostu
upośledzenie wzrostu
niewydolność serca.
niewydolność serca.

Choroba Coriego
Choroba Coriego.
.
Choroba Coriego (choroba Forbesa;
Choroba Coriego (choroba Forbesa;
glikogenoza typu III; GSD III) - rzadka
glikogenoza typu III; GSD III) - rzadka
choroba genetyczna, dziedziczona w
choroba genetyczna, dziedziczona w
sposób autosomalny recesywny
sposób autosomalny recesywny
spowodowana brakiem enzymu
spowodowana brakiem enzymu
odszczepiającego glikogen (oligo-1,4:1,4-
odszczepiającego glikogen (oligo-1,4:1,4-
glukozotransferaza).
glukozotransferaza).

Niedobór ten prowadzi do
Niedobór ten prowadzi do
nadmiernego odkładania
nadmiernego odkładania
nieprawidłowego glikogenu w
nieprawidłowego glikogenu w
mięśniach, wątrobie a także w sercu.
mięśniach, wątrobie a także w sercu.
Występuje z częstością 1/100 000
Występuje z częstością 1/100 000
żywych urodzeń.
żywych urodzeń.

Typy Choroby Coriego:
Typy Choroby Coriego:
Ze względu na rodzaj zajętego
Ze względu na rodzaj zajętego
organu i demonstrowane objawy
organu i demonstrowane objawy
dzieli się chorobę Coriego na
dzieli się chorobę Coriego na
następujące typy:
następujące typy:
GSD IIIa - zajęcie mięśni i wątroby
GSD IIIa - zajęcie mięśni i wątroby
GSD IIIb - zajęcie tylko wątroby
GSD IIIb - zajęcie tylko wątroby
GSD IIIc i GSD IIId - rzadsze fenotypy.
GSD IIIc i GSD IIId - rzadsze fenotypy.

Objawy Choproby Coriego:
Objawy Choproby Coriego:
Choroba Coriego jest glikogenozą o raczej
Choroba Coriego jest glikogenozą o raczej
łagodnym przebiegu. Występujące objawy
łagodnym przebiegu. Występujące objawy
są raczej słabo nasilone w porównaniu do
są raczej słabo nasilone w porównaniu do
innych chorób tego typu:
innych chorób tego typu:
Hepatomegalia
Hepatomegalia
Hipoglikemia
Hipoglikemia

znaczne podwyższenie transaminaz
znaczne podwyższenie transaminaz
wątrobowych
wątrobowych
Hiperlipoproteinemia
Hiperlipoproteinemia
niewielkie włóknienie wątroby
niewielkie włóknienie wątroby
osłabienie siły mięśniowej
osłabienie siły mięśniowej
kardiomiopatia
kardiomiopatia

Choroba Hersa.
Choroba Hersa.
Choroba Hersa (glikogenoza typu VI; GSD VI) -
Choroba Hersa (glikogenoza typu VI; GSD VI) -
rzadka choroba genetyczna, dziedziczona w
rzadka choroba genetyczna, dziedziczona w
sposób autosomalny recesywny spowodowana
sposób autosomalny recesywny spowodowana
brakiem fosforylazy glikogenowej
brakiem fosforylazy glikogenowej
(wątrobowej). Niedobór ten prowadzi do
(wątrobowej). Niedobór ten prowadzi do
nadmiernego odkładania glikogenu w wątrobie
nadmiernego odkładania glikogenu w wątrobie
(przy prawidłowej strukturze narządu).
(przy prawidłowej strukturze narządu).

Nazwa pochodzi od nazwiska
Nazwa pochodzi od nazwiska
odkrywcy, belgijskiego fizjologa i
odkrywcy, belgijskiego fizjologa i
biochemika Henriego-Géry'ego
biochemika Henriego-Géry'ego
Hersa.
Hersa.

Objawy:
Objawy:
Choroba przebiega stosunkowo
Choroba przebiega stosunkowo
łagodnie.
łagodnie.
Może wystąpić:
Może wystąpić:
Hepatomegalia
Hepatomegalia
Hipoglikemia
Hipoglikemia
Hiperlipidemia.
Hiperlipidemia.

Choroba McArdle'a.
Choroba McArdle'a.
Choroba McArdle'a (glikogenoza typu
Choroba McArdle'a (glikogenoza typu
V; GSD V) - rzadka choroba
V; GSD V) - rzadka choroba
genetyczna, dziedziczona
genetyczna, dziedziczona
autosomalnie recesywnie,
autosomalnie recesywnie,
spowodowana niedoborem enzymu -
spowodowana niedoborem enzymu -
mięśniowej fosforylazy glikogenowej.
mięśniowej fosforylazy glikogenowej.

Polega na nadmiernym gromadzeniu
Polega na nadmiernym gromadzeniu
glikogenu w mięśniach. Została
glikogenu w mięśniach. Została
odkryta w 1951 roku przez Briana
odkryta w 1951 roku przez Briana
McArdle'a z Guy's Hospital w
McArdle'a z Guy's Hospital w
Londynie.
Londynie.

Objawy:
Objawy:
bóle mięśniowe
bóle mięśniowe
Mioglobinuria
Mioglobinuria
podwyższenie poziomu enzymów
podwyższenie poziomu enzymów
mięśniowych we krwi po wysiłku.
mięśniowych we krwi po wysiłku.

Choroba von Gierkego
Choroba von Gierkego
Choroba von Gierkego (glikogenoza typu Ia,
Choroba von Gierkego (glikogenoza typu Ia,
ang. glycogen storage disease Ia, von Gierke
ang. glycogen storage disease Ia, von Gierke
disease, GSD Ia) – najczęstsza z glikogenoz. Jest
disease, GSD Ia) – najczęstsza z glikogenoz. Jest
stosunkowo rzadką chorobą genetyczną,
stosunkowo rzadką chorobą genetyczną,
dziedziczoną w sposób autosomalny recesywny.
dziedziczoną w sposób autosomalny recesywny.
Polega na braku glukozo-6-fosfatazy, enzymu
Polega na braku glukozo-6-fosfatazy, enzymu
niezbędnego w procesie glukoneogenezy.
niezbędnego w procesie glukoneogenezy.

Wśród objawów klinicznych
Wśród objawów klinicznych
dominuje:
dominuje:
hepatomegalia,
hepatomegalia,
hipoglikemia,
hipoglikemia,
kwasica mleczanowa.
kwasica mleczanowa.

Choroby spichrzeniowe
Choroby spichrzeniowe
glikogenu
glikogenu.
.
Choroby spichrzeniowe glikogenu
Choroby spichrzeniowe glikogenu
(glikogenozy) (ang. GSD - glycogen
(glikogenozy) (ang. GSD - glycogen
storage diseases) - genetycznie
storage diseases) - genetycznie
uwarunkowane choroby metaboliczne,
uwarunkowane choroby metaboliczne,
prowadzące do nieprawidłowego
prowadzące do nieprawidłowego
magazynowania glikogenu w wątrobie,
magazynowania glikogenu w wątrobie,
nerkach i mięśniach.
nerkach i mięśniach.

Glikogenozy są chorobami genetycznymi,
Glikogenozy są chorobami genetycznymi,
dziedziczonymi głównie autosomalnie recesywnie
dziedziczonymi głównie autosomalnie recesywnie
(z wyjątkiem glikogenozy typu IX, która jest
(z wyjątkiem glikogenozy typu IX, która jest
dziedziczona w sposób sprzężony z
dziedziczona w sposób sprzężony z
chromosomem X).Nieprawidłowe spichrzanie jest
chromosomem X).Nieprawidłowe spichrzanie jest
spowodowane brakiem jednego z enzymów,
spowodowane brakiem jednego z enzymów,
uczestniczących w metabolizmie glikogenu.
uczestniczących w metabolizmie glikogenu.

Podział.
Podział.
W zależności od głównego miejsca
W zależności od głównego miejsca
spichrzania glikogenu wyróżnia się
spichrzania glikogenu wyróżnia się
dwa rodzaje glikogenoz:
dwa rodzaje glikogenoz:
glikogenozy wątrobowe
glikogenozy wątrobowe
glikogenozy mięśniowe.
glikogenozy mięśniowe.

Objawy:
Objawy:
glikogenozy wątrobowe:
glikogenozy wątrobowe:
hipoglikemia
hipoglikemia
hepatomegalia
hepatomegalia
kwasica mleczanowa
kwasica mleczanowa
hiperlipidemia
hiperlipidemia
Wyjątkiem jest GSD II, w której
Wyjątkiem jest GSD II, w której
metabolizm glukozy nie jest
metabolizm glukozy nie jest
zaburzony.
zaburzony.

glikogenozy mięśniowe:
glikogenozy mięśniowe:
nie występuje hipoglikemia, a objawy ograniczone
nie występuje hipoglikemia, a objawy ograniczone
są do mięśni
są do mięśni
hipotonia mięśniowa
hipotonia mięśniowa
osłabienie siły mięśniowej
osłabienie siły mięśniowej
bóle mięśniowe
bóle mięśniowe
mioglobinuria.
mioglobinuria.

Cukrzyca.
Cukrzyca.
Cukrzyca (łac. diabetes mellitus) to, zgodnie z
Cukrzyca (łac. diabetes mellitus) to, zgodnie z
definicją Światowej Organizacji Zdrowia, grupa
definicją Światowej Organizacji Zdrowia, grupa
chorób metabolicznych charakteryzująca się
chorób metabolicznych charakteryzująca się
hiperglikemią wynikającą z defektu wydzielania lub
hiperglikemią wynikającą z defektu wydzielania lub
działania insuliny. Przewlekła hiperglikemia wiąże
działania insuliny. Przewlekła hiperglikemia wiąże
się z uszkodzeniem, zaburzeniem czynności i
się z uszkodzeniem, zaburzeniem czynności i
niewydolnością różnych narządów, szczególnie
niewydolnością różnych narządów, szczególnie
oczu, nerek, nerwów, serca i naczyń krwionośnych.
oczu, nerek, nerwów, serca i naczyń krwionośnych.

Najczęstsze postacie cukrzycy wynikają ze zmniejszonej
Najczęstsze postacie cukrzycy wynikają ze zmniejszonej
wrażliwości tkanek na insulinę (insulinooporność) oraz
wrażliwości tkanek na insulinę (insulinooporność) oraz
upośledzenia wydzielania insuliny (w cukrzycy typu 2),
upośledzenia wydzielania insuliny (w cukrzycy typu 2),
niedoboru insuliny związanego z niszczeniem komórek β
niedoboru insuliny związanego z niszczeniem komórek β
wysp trzustki (w cukrzycy typu 1) bądź zmian hormonalnych
wysp trzustki (w cukrzycy typu 1) bądź zmian hormonalnych
związanych z okresem ciąży (cukrzyca ciężarnych).
związanych z okresem ciąży (cukrzyca ciężarnych).
Ostatecznie wszystkie postacie cukrzycy wynikają z
Ostatecznie wszystkie postacie cukrzycy wynikają z
niezdolności komórek beta do produkcji wystarczającej ilości
niezdolności komórek beta do produkcji wystarczającej ilości
insuliny, która mogłaby zapobiec hiperglikemii.
insuliny, która mogłaby zapobiec hiperglikemii.

Zasadą współczesnej terapii cukrzycy jest leczenie
Zasadą współczesnej terapii cukrzycy jest leczenie
wszystkich zaburzeń towarzyszących chorobie, a nie tylko
wszystkich zaburzeń towarzyszących chorobie, a nie tylko
kontrola gospodarki węglowodanowej. Dążenie do
kontrola gospodarki węglowodanowej. Dążenie do
normalizacji masy ciała, zwiększenie aktywności fizycznej,
normalizacji masy ciała, zwiększenie aktywności fizycznej,
właściwa dieta, leczenie częstych w cukrzycy zaburzeń
właściwa dieta, leczenie częstych w cukrzycy zaburzeń
lipidowych, nadciśnienia tętniczego i innych chorób układu
lipidowych, nadciśnienia tętniczego i innych chorób układu
krążenia oraz utrzymywanie glikemii w przedziale wartości
krążenia oraz utrzymywanie glikemii w przedziale wartości
możliwie najbardziej zbliżonym do niecukrzycowych
możliwie najbardziej zbliżonym do niecukrzycowych
(normoglikemia) zmniejsza ryzyko rozwoju powikłań
(normoglikemia) zmniejsza ryzyko rozwoju powikłań
choroby.
choroby.

Podstawowym objawem
Podstawowym objawem
cukrzycy jest…
cukrzycy jest…
…
…
podwyższenie stężenia glukozy we krwi. W zależności od
podwyższenie stężenia glukozy we krwi. W zależności od
zaawansowania choroby może ono występować jedynie po
zaawansowania choroby może ono występować jedynie po
spożyciu węglowodanów lub niezależnie od niego.
spożyciu węglowodanów lub niezależnie od niego.
Zawartość cukru we krwi (glikemię) podaje się w
Zawartość cukru we krwi (glikemię) podaje się w
miligramach na 100 ml krwi (mg%) lub w milimolach na litr
miligramach na 100 ml krwi (mg%) lub w milimolach na litr
(mmol/l); związek między nimi określany jest wzorem
(mmol/l); związek między nimi określany jest wzorem
[mmol/l] x 18 = mg%. I tak prawidłowa glikemia na czczo to
[mmol/l] x 18 = mg%. I tak prawidłowa glikemia na czczo to
60-99 mg/dl (3,4-5,5 mmol/l), w 2 godzinie testu doustnego
60-99 mg/dl (3,4-5,5 mmol/l), w 2 godzinie testu doustnego
obciążenia glukozą glikemia poniżej 140 mg/dl (7,8 mmol/l).
obciążenia glukozą glikemia poniżej 140 mg/dl (7,8 mmol/l).

Hipoglikemia – poniżej 2.5 mM (45 mg/dL)
problemy ze skupieniem uwagi,
zawroty, zimny pot,
podwójne widzenie,
drgawki, śpiączka

utrata wody
osłabienie, senność,
nudności, wymioty,
bóle brzucha i głowy,
uczucie pieczenia w jamie
ustnej, utrata apetytu,
chudnięcie,
wzrost glikacji i glikozylacji
białek
Hiperglikemia – powyżej stanu normalnego (>120
mg/dL)
~ 180 mg/dL (10 mM)

Źródła glukozy:
- pokarm
-
glikogen
- glukoneogeneza
(gł. wątroba, nerki):
- synteza z mleczanu,
- aminokwasów glukoneogennych,
- glicerolu

Zespół Fanconiego-Bickela.
Zespół Fanconiego-Bickela.
Zespół Fanconiego-Bickela (glikogenoza typu XI, GSD XI,
Zespół Fanconiego-Bickela (glikogenoza typu XI, GSD XI,
ang. Fanconi-Bickel syndrome, FBS) – rzadka choroba
ang. Fanconi-Bickel syndrome, FBS) – rzadka choroba
genetyczna, dziedziczona w sposób autosomalny
genetyczna, dziedziczona w sposób autosomalny
recesywny, spowodowana mutacją w obrębie genu
recesywny, spowodowana mutacją w obrębie genu
kodującego białko błonowe transportujące glukozę -
kodującego białko błonowe transportujące glukozę -
GLUT2.Charakteryzuje się nadmiernym spichrzaniem
GLUT2.Charakteryzuje się nadmiernym spichrzaniem
glikogenu w wątrobie, zaburzeniami w obrębie
glikogenu w wątrobie, zaburzeniami w obrębie
proksymalnych cewek nerkowych i nieprawidłowym
proksymalnych cewek nerkowych i nieprawidłowym
metabolizmem glukozy i galaktozy.
metabolizmem glukozy i galaktozy.

Objawy:
Objawy:
ciężka tubulopatia
ciężka tubulopatia
krzywica hipofosfatemiczna
krzywica hipofosfatemiczna
kwasica mleczanowa
kwasica mleczanowa
hepatomegalia
hepatomegalia
zahamowanie wzrostu
zahamowanie wzrostu
rozwój psychiczny nie jest zaburzony.
rozwój psychiczny nie jest zaburzony.

Objawy:
Objawy:
Objawami nieleczonej choroby są:
Objawami nieleczonej choroby są:
znacznego stopnia upośledzenie rozwoju umysłowego i
znacznego stopnia upośledzenie rozwoju umysłowego i
motorycznego.
motorycznego.
Niedobór melaniny jest przyczyną występowania jasnej karnacji
Niedobór melaniny jest przyczyną występowania jasnej karnacji
skóry, jasnych włosów i niebieskich tęczówek.
skóry, jasnych włosów i niebieskich tęczówek.
Poza tym mogą występować napady drgawkowe (padaczka),
Poza tym mogą występować napady drgawkowe (padaczka),
hipotonia mięśniowa, zaburzenia chodu, postawy, ruchy
hipotonia mięśniowa, zaburzenia chodu, postawy, ruchy
atetotyczne, zesztywnienie stawów.
atetotyczne, zesztywnienie stawów.
Do obrazu chorobowego dołącza charakterystyczny "mysi" zapach
Do obrazu chorobowego dołącza charakterystyczny "mysi" zapach
oraz częste występowanie wysypek.
oraz częste występowanie wysypek.

Mukopolisacharydoza.
Mukopolisacharydoza.
Mukopolisacharydoza (MPS) - jest bardzo rzadko
Mukopolisacharydoza (MPS) - jest bardzo rzadko
występującą chorobą przemiany materii, dziedziczną
występującą chorobą przemiany materii, dziedziczną
i bardzo trudną do zdiagnozowania. Występuje raz
i bardzo trudną do zdiagnozowania. Występuje raz
na 100 tysięcy urodzeń. Jej przyczyną jest wada
na 100 tysięcy urodzeń. Jej przyczyną jest wada
metabolizmu, polegająca na gromadzeniu się w
metabolizmu, polegająca na gromadzeniu się w
organizmie mukopolisacharydów, które uszkadzają
organizmie mukopolisacharydów, które uszkadzają
komórki i narządy ciała. W efekcie prowadzi to do
komórki i narządy ciała. W efekcie prowadzi to do
wyniszczenia niemal całego organizmu dziecka.
wyniszczenia niemal całego organizmu dziecka.

Objawy:
Objawy:
Wczesnym objawem choroby jest przepuklina pępkowa i
Wczesnym objawem choroby jest przepuklina pępkowa i
pachwinowa, polipy błony śluzowej, problemy ze
pachwinowa, polipy błony śluzowej, problemy ze
słuchem.
słuchem.
W późniejszym życiu problemem jest nadmierna
W późniejszym życiu problemem jest nadmierna
ruchliwość dzieci, usztywnienie stawów, biegunki,
ruchliwość dzieci, usztywnienie stawów, biegunki,
zmętnienie rogówki.
zmętnienie rogówki.
Występują również przypadki drgawek z odpowiednimi
Występują również przypadki drgawek z odpowiednimi
zmianami w wykresie fal mózgowych (EEG).
zmianami w wykresie fal mózgowych (EEG).
Później występuje twarz groteskowa (maszkarowata).
Później występuje twarz groteskowa (maszkarowata).
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
- Slide 99
- Slide 100
- Slide 101
- Slide 102
- Slide 103
- Slide 104
- Slide 105
- Slide 106
- Slide 107
- Slide 108
- Slide 109
- Slide 110
- Slide 111
- Slide 112
- Slide 113
- Slide 114
- Slide 115
- Slide 116
- Slide 117
- Slide 118
- Slide 119
- Slide 120
- Slide 121
- Slide 122
- Slide 123
- Slide 124
- Slide 125
- Slide 126
- Slide 127
- Slide 128
- Slide 129
- Slide 130
- Slide 131
- Slide 132
- Slide 133
- Slide 134
- Slide 135
- Slide 136
- Slide 137
- Slide 138
- Slide 139
- Slide 140
- Slide 141
Wyszukiwarka
Podobne podstrony:
Aminokwasy i białka wykład (1)
Aminokwasy i białka wykład (1)
04) Kod genetyczny i białka (wykład 4)
01 Aminokwasy, peptydy, białka, enzymyid 3054 ppt
SKROT BIALKA WYKLAD IIC DLA STUDENTOW
68 Aminokwasy peptydy i bialka(1)
68 Aminokwasy peptydy i bialka
cd wyklIII i wyklad IV, Immunologia
BCH Aminokwasy, peptdy i bialka Nieznany (2)
Aminokwasy, poptydy i białka
Aminokwasy,peptydy,białka i inne
Aminokwasy, peptydy, białka
Chemia 6 Aminokwasy Peptydy i Białka
AMINOKWASY, PEPTYDY, BIAŁKA
AMINOKWASY BIAŁKOWE I BIAŁKA, MATURA !!!, CHEMIA, Chemia R, Różne zadania i testy
Aminokw,peptydy,białka,enzymy
więcej podobnych podstron