
Choroby autosomalne
dominujące
- kryteria dziedziczenia oraz
przykłady

Kryteria dziedziczenia
autosomalnego dominującego
• Cecha uwydatnia się u heterozygot, stan homozygotyczności genu
choroby jest najczęściej letalny;
• Cecha uwidacznia się jednakowo często u obu płci;
• Stopień nasilenia lub brak cech klinicznych zależy od stopnia
penetracji patologicznego genu, lub od zmiennej ekspresji genu (w
obrębie rodziny występuje różne nasilenie objawów), może
wystąpić dziedziczenie z przeskokiem pokoleniowym (chorują
dziadkowie i wnukowie, rodzice są zdrowi);
• Nasilenie objawów choroby może zależeć od płci rodzica
przekazującego zmutowany gen;
• Występowanie choroby może być wynikiem mutacji genu de novo,
której główną przyczyną jest wiek ojca
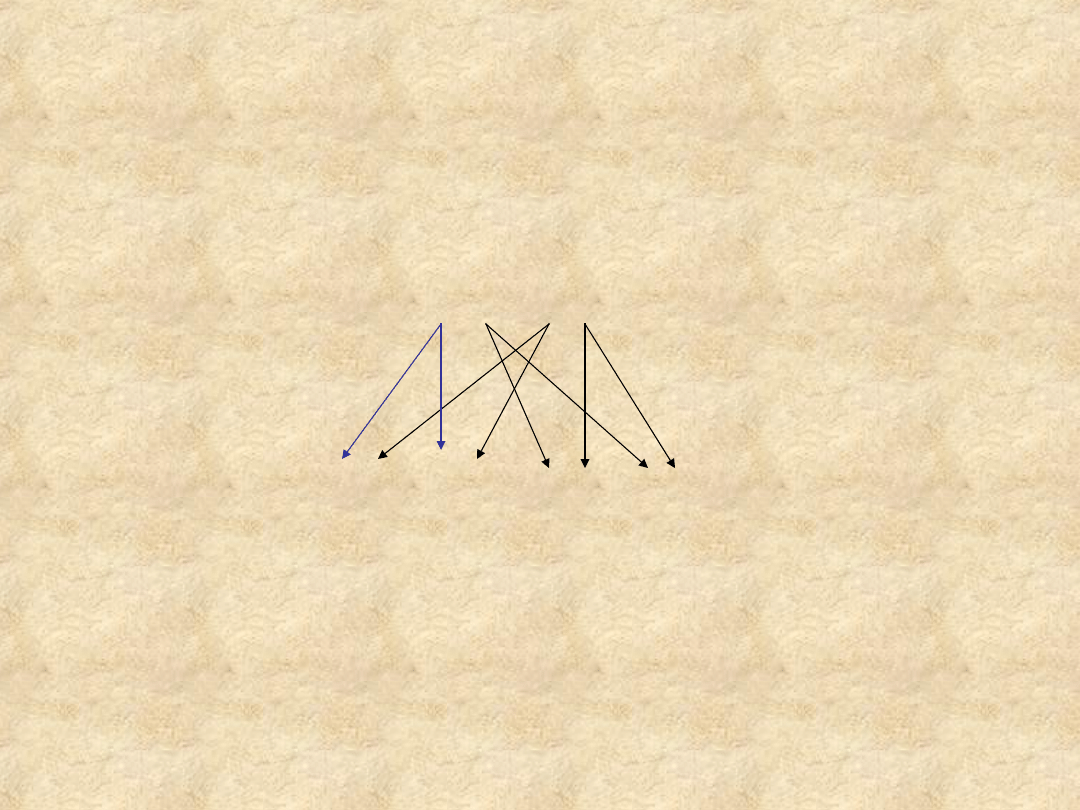
Schematy dziedziczenia
autosomalnego dominującego
(osoba chora)
A
a + aa (osoba zdrowa)
A
a
A
a aa aa
50% potomstwa choruje 50%potomstwa jest zdrowe
I
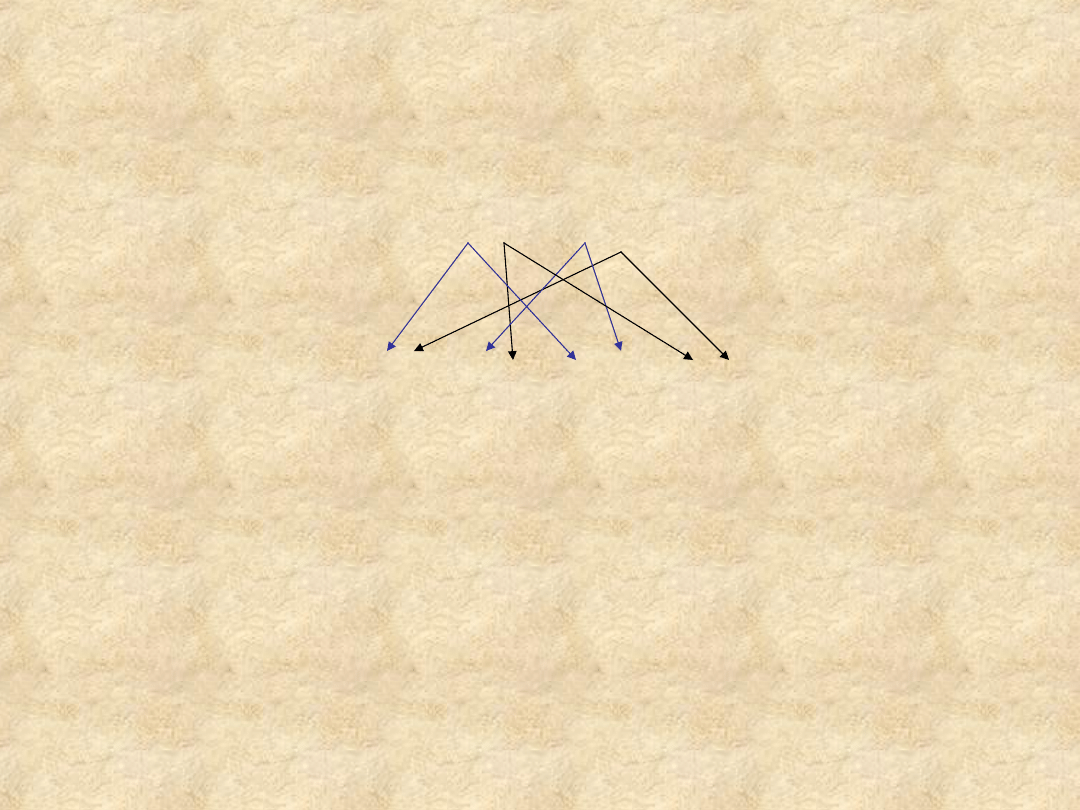
II
(osoba chora)
A
a +
A
a (osoba chora)
A
a
A
a
AA
aa
75% choruje (2 heterozygoty + homozygota dominująca) 25% jest
zdrowe

Achondroplazja
• Najczęściej spotykana postać karłowatości ludzkiej;
• Cecha o pełnej penetracji i małej zmienności ekspresji;
• Najczęściej jest wynikiem mutacji de novo lub dziedziczenia
patologicznego genu od rodziców
• Częstość występowania: 1/15000 – 1/77000;
• Osobniki z genotypem AA mają tak liczne wady, że
najczęściej umierają w 1 roku życia;
• Śmiertelność heterozygot wzrasta od urodzenia do 4 roku
życia oraz w 4-5 dekadzie życia;
• Gen wywołujący znajduje się na ramieniu krótkim
chromosomu 4 (4p16.3);
• W nukleotydzie tego genu może wystąpić tranzycja G w A,
lub transwersja G w C. Konsekwencją jest zamiana glicyny
na argininę w łańcuchu białka G308R, występującego w
błonie komórkowej. Zatem efektem jest uszkodzenie
receptora dla czynnika wzrostu fibroblastów, prowadzące do
achondroplazji.

Objawy:
• Skrócenie kończyn;
• Mikromelia (małe dłonie);
• Szpotawe kolana;
• Ograniczenie prostowania stawu łokciowego;
• Nadmierna lordoza lędźwiowa;
• Małe talerze biodrowe;
• Duża głowa, wypukłe czoło, zapadnięta nasada nosa;
• Wzrost: u kobiet – ok. 123 cm, u mężczyzn – ok. 132 cm;
Rozwój umysłowy jest prawidłowy.

Choroba Alzheimera
• W 50% przypadków przyczyną otępienia starczego jest
zespół Alzheimera;
• Choroba występuje w dwóch postaciach: wczesnej
(ujawniającej się przed 65 rokiem życia) i późnoobjawowej;
• Postać wczesna dziedziczona jest autosomalnie dominująco z
pełną penetracją genu;
• Choroba polega na nieprawidłowej obróbce prekursora białka
beta-APP (21q21.3), uwalniany jest beta-amyloid,
odkładający się pozakomórkowo w postaci
nierozpuszczalnych złogów;
• Postać późnoobjawowa jest związana z apolipoproteiną E,
wiążącą się z amyloidowym białkiem beta, odkładającym się
w mózgu chorych. Gen kodujący apolipoproteinę E znajduje
się na ramieniu długim chromosomu 19 (19q13.2);
• W wyniku powyższych mutacji w mózgu odkładają się blaszki
amyloidowe, płytki starcze, neurony degenerują się,
nabywają cechy zwyrodnienia nerwowo-włókienniczego.

Objawy:
• Agnozja (nieumiejętność rozpoznawania przedmiotów);
• Afazja (zaburzenia i spowolnienie mowy);
• Apraksja (zaburzenia czynności ruchowych);
• Depresja;
• Zaburzenia pamięci i orientacji;
• Wypełnianie luk w pamięci wymyślonymi treściami
(konfabulacja)

Choroba Huntingtona
• Częstość występowania: 4-7/100000; w Polsce 1/15000
• Gen HD, odpowiedzialny za chorobę, zlokalizowany jest na
ramieniu krótkim chromosomu 4 (4p16.3);
• Mutacja: nieprawidłowa liczba powtórzeń trójki CAG na końcu
5‘ genu kodującego białko huntingtinę, u osób zdrowych to 10-
29 powtórzeń, stan premutacyjny to 30-35 powtórzeń, 36 i
więcej powtórzeń oznacza chorobę;
• Zmiany neuropatologiczne: zanik małych neuronów w jądrze
ogoniastym (istota szara), skorupie (mózgowie), oraz dużych
neuronów w gałce bladej (kresomózgowie);
• W kolejnych pokoleniach choroba występuje w coraz młodszym
wieku i ma coraz cięższy przebieg. Antycypacja ta jest mocniej
wyrażana, gdy zmutowany gen pochodzi od ojca;
• Początek choroby występuje w czwartej dekadzie życia;
• Śmierć następuje w ciągu 10-15 lat od wystąpienia pierwszych
objawów choroby.

Objawy:
• Gwałtowne, niekontrolowane ruchy;
• Zaburzenia mowy;
• Otępienie umysłowe;
• Zaburzenia pamięci;
• Drżenie rąk i nóg;
• Zaburzenia połykania – ich wynikiem jest zachłystowe
zapalenie płuc (przedostawanie się treści żołądka do
drzewa oskrzelowego) – pierwsza przyczyna śmierci
pacjentów z chorobą Huntingtona (85% zgonów)

Zespół Marfana
(Arachnodaktylia)
• Choroba wywołana mutacją genu FBN1 (gen fibrylliny),
zlokalizowanego na ramieniu długim chromosomu 15
( 15q21.1). Gen powodujący powstawanie tego zespołu
charakteryzuje się wysokim stopniem penetracji i zmienną
ekspresją. W 25% przypadków są to nowe mutacje;
• Zespół Marfana jest przykładem działania genów
plejotropowych u człowieka, - choroba uwarunkowana jest
obecnością jednego patologicznego genu dominującego,
którego pierwotnym efektem działania jest synteza
nieprawidłowego kolagenu, wtórnymi efektami są zmiany w
układzie kostno-stawowym, w gałce ocznej i układzie
krążenia;
• Skutkiem mutacji genu FBN1 są zmiany w układzie kostno-
stawowym, w układzie krążenia i gałkach ocznych.
Przyczyną zespołu jest uwarunkowane genetycznie
uszkodzenie włókien sprężystych i zaburzenie w tworzeniu
kolagenu oraz substancji podstawowej tkanki łącznej.
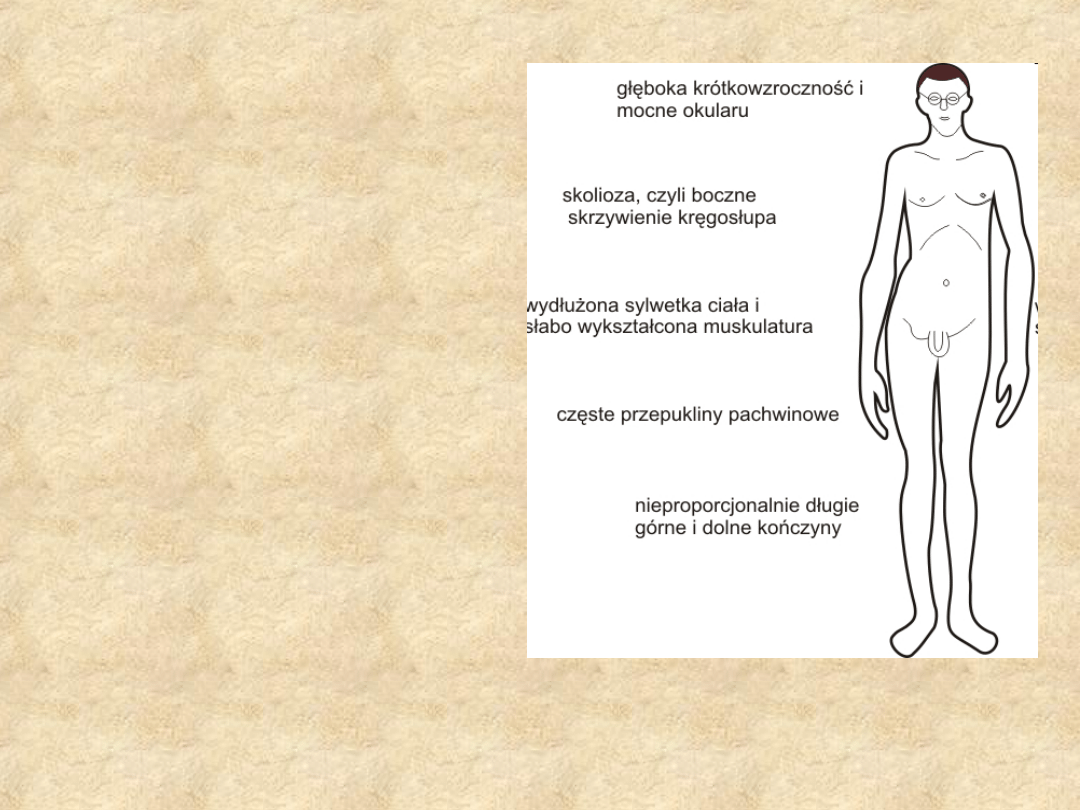
Objawy:
• Nadmierny wzrost kości
długich;
• Zaburzony stosunek długości
tułowia do długości kończyn;
• Charakterystyczna, smukła
sylwetka, wysoki wzrost;
• Nadmiernie długie palce u
rąk i stóp (,,pajęcze palce";
• Kurzy lub lejkowaty kształt
klatki piersiowej;
• Nadmiernie elastyczna
skóra;
• Podwichnięcie soczewki,
krótkowzroczność;
• Wady wrodzone serca,
tętniaki aorty, wypadanie
płatka zastawki mitralnej i
przepukliny.

Zespół Ehlersa-Danlosa
• Podłożem zaburzeń obserwowanych w EDS są
nieprawidłowości w syntezie lub obróbce potranslacyjnej
kolagenu. Defekty są wynikiem mutacji pojedynczych
genów; w większości przypadków dziedziczone są
autosomalnie dominująco;
• Wyróżnia się przynajmniej 10 typów EDS. Różnorodność
zaburzeń leżących u podstawy zespołów Ehlersa-Danlosa
spowodowana jest występowaniem wielu typów
kolagenu oraz wysokim stopniem skomplikowania jego
syntezy i dojrzewania.

Objawy:
• Dotyczą głównie skóry, więzadeł i stawów, co jest wynikiem
dużej zawartości kolagenu w tych strukturach;
• Skóra nadmiernie elastyczna i podatna na urazy, nadmierne
siniaczenie się;
• Zwiększona ruchomość stawów – obserwowane są ruchy w
płaszczyznach odmiennych niż fizjologiczne;
• Może dojść do uszkodzeń naczyń, pęknięć jelit, defektów
gałki ocznej (np. odklejenia siatkówki).

CHOROBA RECKLINGHAUSENA
(NERWIAKOWŁÓKNIAKOWATOŚĆ)
• Nerwiakowłókniakowatość jest chorobą heterogenną. Poznano
dokładnie dwie postacie tej choroby: NF-1 i NF-2;
• Gen NF-1 (locus 17q11.2), warunkujący tę chorobę, jest
genem autosomalnym dominującym; przejawia pełną
penetrację, ale zmienną ekspresję. W obrębie genu mutacje
typu delecje występują z jednakową częstością u kobiet i u
mężczyzn. Mutacje de novo, najczęściej pochodzenia
ojcowskiego, są przyczyną 50% przypadków nowych
zachorowań. Obniżony poziom produktu tego genu
(neurofibromina) sprzyja rozwojowi nowotworów. Białko
syntezowane w prawidłowej ilości wykazuje właściwości
supresorowe;
• Częstość występowania 1:3500 urodzeń.
• Gen NF-2 znajduje się na chromosomie 22 (22q12.2) i
dziedziczy się autosomalnie dominująco z pełną penetracją.
Produktem genu NF-2 jest merlina, będąca białkiem
cytoszkieletu;
• Częstość występowania choroby wynosi: 1:35000-40000
urodzeń.

Objawy postaci NF-1:
• Wczesne dzieciństwo - zmiany barwnikowe na skórze,
określane jako plamy koloru ,,kawy z mlekiem" (cafe au
lait);
• W okresie dojrzewania rozwój licznych guzków
wywodzących się z nerwów obwodowych (włókniaki i
nerwiakowłokniaki oraz glejaki nerwu wzrokowego). Guzy te
są zwykle łagodne, jednak w 3-13% ulegają zezłośliwieniu;
• Dość często występuje niedorozwój umysłowy i padaczka;
• U 5% chorych występuje skrzywienie kręgosłupa.
Objawy postacji NF-2 :
• Pierwsze objawy pojawiają się w okresie dojrzewania lub
drugiej dekadzie życia;
• Powstawanie nerwiaków osłonkowych nerwu słuchowego i
oponiaków rdzenia;
• Zmętnienie soczewek.

Bibliografia:
• Podstawy genetyki dla studentów i lekarzy, pod
red. Gerarda Drewy i Tomasza Ferenca, wydanie II
poprawione, Wrocław, Wydawnictwo Medyczne
Urban & Partner, rozdział 10, Dziedziczenie
jednogenowe u człowieka, str. 194-200
• Podstawy genetyki dla studentów i lekarzy, pod
red. Gerarda Drewy i Tomasza Ferenca, wydanie II
poprawione, Wrocław, Wydawnictwo Medyczne
Urban & Partner, rozdział 11, Cechy
uwarunkowane wieloczynnikowo, str. 230-232
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
Wyszukiwarka
Podobne podstrony:
Mutacje genowe autosomalne dominujące i recesywne
PREZ metody wykrywania mutacji
Mutacje chromosomowe strukturalne
Materiał genetyczny, mutacje, systemy naprawy DNA, test Amesa
1-Kefir chroni przed mutacjami w DNA, ZDROWIE-Medycyna naturalna, Poczta Zdrowie
ZMIENNOSC I MUTACJE, fizjoterapia, biologia medyczna
Wśród poławianych ryb w morzach i oceanach dominują doc
Podział mutacji
12 uokik problem naduzywania pozycji dominujacej
7 mutacje genomowe id 45020
Mutacje założycielskie artykuł
Aberracje chromosomowe i mutacje
Prelekcja 10 - cz 2 - Mutacje chromosomowe człowieka, Genetyka
Dominus Iesus
Spółka dominująca, rachunkowość pwsz piła
10446-mutacje genomu ludzkiego i czynniki je wywołujące, semestr IV, genetyka, Genetyka
więcej podobnych podstron