
IMMUNOGENETYKA
CZĘŚĆ II
Lek. Przemysław Łodej
Zakład Genetyki Klinicznej
Uniwersytetu Medycznego w Lublinie

ANTYGENY GRUPOWE
ERYTROCYTÓW

Antygeny grupowe erytrocytów
Niektóre antygeny grupowe występują
wyłącznie na komórkach krwi (np. Rh, Kell).
Inne wykrywa się niemal na wszystkich
komórkach organizmu np. ABO, Lewis, MNS,
O.
Przeciwciała przeciwko antygenom grupowym
dzieli się na:
Alloprzeciwciała
Autoprzeciwciała

Antygeny grupowe erytrocytów
Alloprzeciwciała:
rozpoznają obce antygeny z tego samego układu grupowego, nie
występujące na własnych krwinkach.
Wyróżnia się alloprzeciwciała odpornościowe i naturalne.
Przeciwciała
odpornościowe
należą zazwyczaj do klasy IgG i mogą
powstawać w następstwie kontaktu z krwinkami z obcej grupy w wyniku
przetoczenia krwi lub w trakcie ciąży.
W niektórych układach grupowych (ABO, MNS, Lewis, P) występują w
osoczu przeciwciała, pomimo braku wcześniejszej immunizacji
odpowiednimi antygenami. Noszą one nazwę przeciwciał
naturalnych
,
są zwykle klasy IgM i powstają wskutek immunizacji antygenami
powszechnie występującymi w otoczeniu. W pewnych układach
gupowych przeciwciała naturalne wykrywa się prawie zawsze u osób o
odpowiednim fenotypie (ABO), w innych znajduje się je tylko u niektórych
ludzi (Lewis).
Autoprzeciwciała:
reagują z antygenami obecnymi na własnych komórkach badanego oraz
z identycznymi antygenami znajdującymi się na krwinkach innych osób.

Antygeny grupowe erytrocytów
Układ grupowy ABO
Antygeny tego układu znajdują się na
krwinkach czerwonych oraz na pozostałych
komórkach organizmu, z wyjątkiem neuronów.
Synteza antygenów ABO pozostaje pod
kontrolą co najmniej 3 niezależnych loci
zawierających jeden z alleli:
H lub h – H koduje enzym, h jest genem niemym
A
1
, A
2
, B i O
Se i se.
Swoistość
antygenu
warunkuje
cukier
zajmujący ostatnią pozycję łańcucha.

Antygeny grupowe erytrocytów
Istnieją
dwie
potencjalne
cząsteczki
prekursorowi antygenów ABO:
typ I
i
typ II
–
zawierają identyczne reszty cukrowe.
Różni je wiązanie między końcowymi resztami
cukrowymi łańcucha:
prekursor typu I ma galaktozę dołączoną do N-
acetylkoglukozaminy wiązaniem α (1-3)-glikozydowym
prekursor typu II – te same cukry wiążą się wiązaniem
α(1-4)-glikozydowym
Na krwinkach czerwonych występuje wyłącznie
typ II łańcucha. Pozostałe komórki organizmu
mają oba rodzaje prekursorów.

Antygeny grupowe erytrocytów
Dołączenie cząsteczki fukozy do końcowej
galaktozy łańcucha typu I lub II powoduje
powstanie substancji (antygenu H),
bezpośredniego prekursora antygenów A i B.
Antygen
grupowy
A
powstaje
po
przyłączeniu do substancji H reszty
N-
acetylogalaktozaminy
.
Antygen
grupowy
B
powstaje
po
przyłączeniu cząsteczki
galaktozy
.
Antygeny układu ABO pojawiają się w 6
tygodniu życia płodowego. Do ich pełnej
ekspresji dochodzi w 6-18 miesięcy po
urodzeniu.

Antygeny grupowe erytrocytów
Poszczególne antygeny układu ABO mają wiele odmian, z których
największe znaczenie ma grupa A
2
, występująca u 20% osób z grupą
krwi A. Gen A
2
różni się od A
1
zamianą nukleotydu w pozycji 467,
przez co przesunięciu ulega ramka odczytu i w cząsteczce enzymu
pojawia się dodatkowa domena zawierająca 21 aminokwasów.
Transferaza A
2
ma kilkakrotnie mniejszą aktywność, wydaje się, że
przenosi reszty N-acetylogalaktozaminy tylko na pewne warianty
typu II łańcucha H.
W rezultacie osobnicy określani fenotypowo jako grupa A
2
mają
czterokrotnie mniej determinant antygenowych na powierzchni
krwinki czerwonej w porównaniu do odmiany A
1
.
Antygen A
1
jest rozpoznawany przez osoby z grupą A
2
jako odrębna
swoistość, co może prowadzić do wstępowania w ich surowicy
„naturalnych” przeciwciał anty-A
1
. Ponadto mniejsza wydajność
transferazy sprawia, że na powierzchni krwinki pozostaje wolny
antygen H.
Pozostałe odmiany antygenów A i B mają niewielkie znaczenie.

Antygeny grupowe erytrocytów
Przeciwciała układu ABO
Przeciwciała „naturalne” w przeciwieństwie do „odpornościowych”
powstają bez uprzedniego kontaktu z antygenem. Wydaje się, że
przeciwciała „naturalne” przeciwko antygenom grupowym
powstają dzięki stymulacji powszechnie występującymi w
przyrodzie substancjami podobnymi do antygenów grupowych.
Przeciwciała „naturalne” występują regularnie u osób nie
mających odpowiedniego antygenu na przykład: anty-A u osoby z
grupą B, anty-B w grupie krwi A, anty-A i anty-B w grupie O.
Całkowity brak przeciwciał przeciwko substancjom grupowym
stwierdza się bardzo rzadko (z wyjątkiem osób z grupą AB)
Istnienie naturalnych przeciwciał grupowych wykorzystuje się do
określenia grupy krwi w układzie ABO. W tym celu przeprowadza
się reakcję aglutynacji jego krwinek z surowicami wzorcowymi
(anty-A i anty-B) jak i między jego surowicą a wzorcowymi
krwinkami grupy A i B.

Antygeny grupowe erytrocytów
Przeciwciała „naturalne” należą najczęściej do klasy IgM i nie
przechodzą przez łożysko.
Wytwarzanie przeciwciał „naturalnych” rozpoczyna się
bezpośrednio po urodzeniu, jednak do 3-6 miesiąca życia ich
miano jest zbyt małe, aby mogły zostać wykryte. Największe
stężenie obserwuje się między 5 a 10 rokiem życia, następnie
miano przeciwciał przeciwko antygenom grupowym stopniowo
maleje.
W obrębie populacji stwierdza się znaczne różnice miana
odpowiednich przeciwciał wśród osobników tej samej grupy
krwi. Osoby z grupą O mają przeciwciała reagujące z
krwinkami grupy A lub B nawet po intensywnej absorpcji ich
surowicy erytrocytami tych grup.
Przeciwciała te noszą nazwę anty-A, B są głównie klasy IgG i
często reagują z odmianami antygenu A nie rozpoznawanymi
przez przeciwciała anty-A od osób z grupą B.

Antygeny grupowe erytrocytów
U ludzi mających „słabe” odmiany antygenu A(A
2
, A
3
, A
x
)
mogą pojawić się w surowicy przeciwciała anty-A, które
reagują z A
1
lecz nie wiążą antygenu A
2
. Przeciwciała te różnią
się swoistością od przeciwciał anty-A występujących u osób z
grupą B.
Aglutyniniy anty-H stwierdza się u wszystkich ludzi, którzy nie
mają łańcucha H na powierzchni erytrocytu. Należą do nich
nosiciele antygenu A
1
i/lub B ( antygen H jest wówczas zajęty
przez NAcGal lub Gal) oraz posiadacze niezwykle rzadkiej
grupy krwi Bombay (O
h
), którzy nie mają aktywnej
fukozylotransferazy na skutek mutacji genu H.
U osób z genotypem hh pomimo obecności genów kodujących
grupę krwi A lub B nie występują antygeny na erytrocytach,
choć w ich surowicy są odpowiednie naturalne przeciwciała
dla danej grupy AB0. Jest to tzw. grupa krwi "Bombay"

Antygeny grupowe erytrocytów
Fenotypową
grupę
krwi
kodują
odpowiednio:
Grupę A
1
mają osoby o genotypie: A
1
0, A
1
A
1
,
A
1
A
2
Grupę A
2
mają osoby o genotypie: A
2
A
2
lub
A
2
0
Grupę B mają osoby o genotypie: BB lub B0
Grupę A
1
B mają osoby o genotypie: A
1
B
Grupę A
2
B mają osoby o genotypie: A
2
B
Grupę 0 mają osoby o genotypie: 00

Antygeny grupowe erytrocytów
Poza antygenami każda grupa krwi
charakteryzuje
się
odpowiednim
zestawem naturalnych przeciwciał w
surowicy, należących do klasy IgM:
Grupa A
1
: anty-B
Grupa A
2
: anty-B i niekiedy anty-A
1
Grupa B: anty-A
Grupa 0: anty-A i anty-B
Grupa A
1
B: brak naturalnych przeciwciał
Grupa A
2
B: mogą wystąpić anty-A
1

Antygeny grupowe erytrocytów
Rodzic
Rodzic
0
A
B
AB
0
0
0 lub A
0 lub B
A lub B
A
0 lub A
0 lub A
0, A, B lub AB
A, B lub AB
B
0 lub B
0, A, B lub AB
0 lub B
A, B lub AB
AB
A lub B
A, B lub AB
A, B lub AB
A, B lub AB

Antygeny grupowe erytrocytów
Układ grupowy Rh
Antygeny Rh kodowane są przez dwa wysoce homologiczne , lezące
blisko siebie loci nazwane RHD (kodowany jest antygen D oraz G) i
RHCE (antygeny: C, c, E, e).
W błonie erytrocytu produkty wymienionych loci związane są z
glikoproteiną RhAG, która jest niezbędna do prawidłowej ekspresji
antygenów Rh.
Za najważniejszy antygen tego układu uważa się peptyd D ze względu
na jego silną immunogenność.
Antygeny Rh pojawiają się w 6 tygodniu życia płodowego i występują
wyłącznie na krwinkach czerwonych. Od okresu płodowego wykazują
dużą immunogenność, przy czym najsilniej syntezę swoistych
przeciwciał pobudza antygen D. Z tego powodu osoby mające na
powierzchni erytrocytów antygen D mianem Rh-dodatnich, bez względu
na swoistość pozostałych antygenów układu Rh.
Około 20% osób nie ma antygenu D. Określa się ich mianem Rh-
ujemnych, brak antygenu D zaznacza się jako fenotyp d. U osób tych w
genomie nie znajduje się locus RHD.

Antygeny grupowe erytrocytów
Przeciwciała anty-Rh powstają w wyniku uczulenia w
czasie ciąży lub po przetoczeniu krwi niezgodnej w
układzie Rh.
Bezpośrednio po stymulacji antygenem pojawiają
się w krążeniu swoiste IgM, po czym, po 2-3
tygodniach izotop wytwarzanych przeciwciał ulega
przyłączeniu do IgG.
Przeciwciała anty-Rh należą zwykle do klasy IgG1.
Często przeciwciała te wykazują powinowactwo do
innych antygenów układu Rh.
Przeciwciała klasy IgG skierowane przeciwko
antygenom układu Rh często wykrywa się u chorych
z niedokrwistością autoimmunohemolityczną.

Antygeny grupowe erytrocytów
Nazwa Rh wzięła się od małp rezusów Rhesus, u
których po raz pierwszy wykryto ten układ. Obejmuje
ponad 47 antygenów, lecz 5 z nich, które kodowane
są przez 3 geny, ma znaczenie praktyczne:
antygen C: genotypy CC lub Cc
antygen c: genotyp cc
antygen D: genotypy DD lub Dd. Allel d jest genem
niemym i genotyp dd nie koduje żadnego antygenu
antygen E: genotypy EE lub Ee
antygen e: genotyp ee.
Wśród ludzi rasy białej około 85% posiada czynnik
Rh.

Antygeny grupowe erytrocytów
Rodzic
Rodzic
DD (Rh+)
Dd (Rh+)
dd (Rh-)
DD (Rh+)
DD (Rh+)
DD lub Dd (Rh+)
Dd (Rh+)
Dd (Rh+)
DD lub Dd (Rh+)
DD lub Dd (Rh+)
lub dd (Rh-)
Dd (Rh+) lub dd
(Rh-)
dd (Rh-)
Dd (Rh+)
Dd (Rh+) lub dd
(Rh-)
dd (Rh-)

Choroba hemolityczna
noworodków
Choroba hemolityczna noworodka
Choroba, u której podłoża leży reakcja immunologiczna
pomiędzy przeciwciałami klasy IgG, wytwarzanymi przez
matkę a antygenami krwinek płodu.
Występuje ona w przypadku, gdy matka z układem
antygenów krwi Rh(-), uczulona na antygen D, rodzi
dziecko, które posiada grupę krwi Rh(+) – jest to tzw.
konflikt serologiczny.
Konflikt serologiczny pojawia się w momencie, gdy po raz
pierwszy niewielka ilość krwi dziecka dostaje się do
krwiobiegu matki (dojdzie do przecieku płodowo-
matczynego). Zazwyczaj ma to miejsce dopiero w
momencie porodu (bariera łożyskowa). Po przedostaniu się
krwinek Rh(+) do krwiobiegu matki jej organizm zaczyna
wytwarzać przeciwciała (typu IgM i IgG), przeciw
antygenowi D obecnemu na erytrocytach.

Choroba hemolityczna
noworodków
Przeciwciała IgG mają zdolność przenikania bariery
łożyskowej, w następnych ciążach. W przypadku
płodu Rh(+) przeciwciała IgG matki niszczą jego
erytrocyty, powodując głęboką niedokrwistość.
Powoduje to zahamowanie rozwoju płodu. Może
doprowadzić do jego obumarcia a następnie
poronienia.
Choroba hemolityczna noworodka może pojawić
się niekiedy w trakcie trwania pierwszej ciąży (np.
jako powikłanie zabiegów wewnątrzmacicznych).
Ze względu na szeroko stosowaną profilaktykę są
to przypadki sporadyczne, a większość ciąż
"konfliktowych" kończy się urodzeniem zdrowego
dziecka.

Choroba hemolityczna
noworodków
Konflikt grup głównych
Ten rodzaj konfliktu może pojawić się, gdy matka
ma grupę krwi np. 0 a dziecko dziedziczy grupę A
lub B. Krwinki grupy krwi dziecka powodują
powstawanie przeciwciał typu odpornościowego u
matki, a skutki tego procesu są podobne do
konfliktu w zakresie czynnika Rh.
W przeciwieństwie do niezgodności w zakresie Rh,
w konfliktach grup głównych krwi pierwsze dziecko
choruje równie często jak następne.
W następstwie tego konfliktu dochodzi do
niedokrwistości,
nadmiaru
bilirubiny,
czego
skutkiem może być przedwczesna i nasilona
żółtaczka u noworodka.

Choroba hemolityczna
noworodków
Profilaktyka
Konfliktowi serologicznemu zapobiega się
, podając zaraz po porodzie, poronieniu,
zabiegach, w których może dojść do
transfuzji
krwi
płód-matka,
immunoglobulinę anty-D, która niszczy
erytrocyty
Rh(+),
zanim
układ
immunologiczny
matki
zdąży
zareagować.

Rodzaje przeszczepów
Autologiczny (autogeniczny)
: kiedy dawcą i
biorcą jest ten sam osobnik.
Izogeniczny
(syngeniczny)
:
między
identycznymi
osobnikami
tego
samego
gatunku (bliźnięta monozygotyczne, szczepy
wsobne u zwierząt).
Allogeniczny
: między różnymi genetycznie
osobnikami tego samego gatunku.
Ksenogeniczny
:
między
osobnikami
odmiennych gatunków (przeszczepy zgodne –
są wykonywane między zbliżonymi gatunkami;
przeszczepy niezgodne – dawca i biorca
pochodzą z gatunków odległych genetycznie)

Odpowiedź na przeszczep
Różnice genetyczne między dawcą a biorcą sprawiają, że
układ
odpornościowy
biorcy
rozpoznaje
antygeny
przeszczepu jako obce i uruchamia reakcję (odrzucenie)
dążącą do jego zniszczenia.
Odpowiedź na antygeny przeszczepu wymaga rozpoznania
ich i aktywacji odpowiednich, swoistych wobec tych
antygenów klonów limfocytów T.
W jej przebiegu zostają pobudzone limfocyty T pomocnicze,
które stymulują limfocyty B do wytwarzania swoistych
przeciwciał o wysokim mianie i powinowactwie, jak również
zwiększają
aktywność
limfocytów
cytotoksycznych,
makrofagów, komórek NK wobec komórek przeszczepu.
W trakcie reakcji odrzucania dochodzi do bezpośredniej
indukcji
i
proliferacji
swoistych
limfocytów
T
cytotoksycznych.

Odpowiedź na przeszczep
W
przebiegu
odpowiedzi
na
antygeny
przeszczepu można wyróżnić:
Fazę indukcji odpowiedzi (aferentną) w
czasie której dochodzi do prezentacji i
rozpoznania obcych antygenów
Fazę efektorową (eferentną) w której
zostają uruchomione swoiste i nieswoiste
mechanizmy odpowiedzi na przeszczep.

Pierwotne niedobory odporności
Zespół DiGeorge’a
Zespół ten jest spowodowany zaburzeniami organogenezy w
rozwoju embrionalnym, których przyczyną w 90% przypadków są
delecje (a częściej mikrodelecje) fragmentu chromosomu 22.
Takie delecje zaburzają rozwój struktur wywodzących się z 3 i 4
kieszonki gardłowej.
Dzieci urodzone z tym zespołem mają deformacje części
twarzowej czaszki, wady serca i dużych naczyń, brak lub
niedorozwój grasicy i przytarczyc.
Zespół ma charakter wielogenowy.
Gen
Tbx1
kodujący czynnik transkrypcyjny (odpowiedzialny za
wady w układzie sercowo-naczyniowym).
Zakażenia nie są w tym zespole dominujące. Tylko u 20% chorych
obserwuje się zmniejszoną liczbę i/lub aktywność limfocytów T. Z
czasem u chorych pojawiają się limfocyty T i dochodzi do kolekcji
niedoboru (wskutek ektopowego rozwoju grasicy).

Pierwotne niedobory odporności
Zespół Wiskotta-Aldricha
Zespół ten dziedziczy się z płcią.
Jego najbardziej charakterystyczną cechą jest skaza
krwotoczna.
Pierwszymi objawami są zazwyczaj krwawe biegunki
i wybroczyny skórne.
W pierwszym roku życia dołączają się zmiany skórne
o charakterze atopowego zapalenia skóry oraz
infekcje
bakteryjne
(najcześciej
paciorkowce),
zakażenia wirusowe (przeważnie z grupy Herpes)
oraz oportunistyczne (Pneumocytis carinii).
W późniejszych latach obserwuje się choroby
autoimmunizacyjne oraz nowotwory.

Pierwotne niedobory odporności
Zespół Wiskotta-Aldricha
Średni czas życia chłopców z tym zespołem nie przekracza
15 lat.
Najczęstszą przyczyną zgonów są krwawienia, zakażenia,
nowotwory, głównie chłoniaki (zwłaszcza chłoniaki Burkitta).
Obok trombocytopenii we krwi obwodowej obserwuje się
zmniejszoną liczbę limfocytów T, ale prawidłowe stężenie
przeciwciał.
Zaburzenia dotyczą zakresu antygenów rozpoznawanych
przez przeciwciała.
Molekularną przyczyną jest mutacja białka nazwanego WASP
(Wiskott-Aldricha syndrome protein), którego wybiórczą
ekspresję obserwuje się w limfocytach i megakariocytach.

Pierwotne niedobory odporności
Ciężkie złożone niedobory odporności:
Wszystkie postacie SCID charakteryzują się albo głębokim
upośledzeniem albo całkowitym brakiem odpowiedzi humoralnej i
komórkowej.
Występują dość rzadko (2 na 75 tys. do 100 tys. urodzeń).
U dzieci dotkniętych SCID w ciągu kilku-kilkunastu tygodni od
urodzenia zaczynają pojawiać się kliniczne oznaki niedoboru
odporności: biegunki, nawracające zakażenia, do których później
dołącza się zatrzymanie wzrostu. Najczęściej spotykane są zakażenia
drożdżakami (Candida), adenowirusami, wirusami typu Herpes ( w
tym cytomegalowirusem oraz wirusem Epsteina-Barr), wirusem
paragrypy typu 3, a także patogenami oportunistycznymi
(Pneumocystis carinii, Aspergillus sp.).
Nawet szczepionki zawierające atentowane żywe mikroorganizmy,
np. BCG, stanowią dla chorego dziecka śmiertelne zagrożenie. Dzieci
z SCID ze względu na brak funkcjonującego układu odpornościowego,
nie potrafią odrzucać przeszczepów allogenicznych.

Pierwotne niedobory odporności
Ciężkie złożone niedobory odporności ze zmniejszoną
liczbą limfocytów T i prawidłowym poziomem limfocytów
B: T(-) B(+)
Najczęściej spotykaną postacią SCID (50-60% wszystkich
przypadków) jest zespół dziedziczony z płcią, spowodowany
przez mutację genu dla łańcucha γ (common γ chain)
podjednostki receptora dla interleukin: 2, 4, 7, 9, 15, 21.
Niedobór ten charakteryzuje się brakiem dojrzałych limfocytów
T i komórek NK, zwiększoną liczną limfocytów B, ale
zmniejszonym stężeniem przeciwciał (wyjątkiem mogą być IgM
u niektórych chorych).
W grasicy brak jest podziału na korę i rdzeń, obwodowe narządy
limfatyczne są hipoplastyczne.
Przyczyną tylko śladowych ilości przeciwciał IgG oraz IgA jest
brak kostymulacji ze strony limfocytów T pomocniczych
niezbędnych do przełączenia klas immunoglobulin.

Pierwotne niedobory odporności
Opisano kilka przypadków SCID z mutacją genu
dla łańcucha α receptora dla IL-7. Obraz kliniczny
różni się tym od mutacji γc, że chorzy mają
prawidłową liczbę komórek NK. Dojrzewanie
limfocytów T jest jednak zablokowane, co
wskazuje
na
niezastąpioną
rolę
IL-7
w
różnicowaniu limfocytów T.
Mutacja genu kodującego CD45 jest przyczyną
SCID. Cząsteczka ta jest markerem komórek
hematopoetycznych.
Ta
postać
SCID
charakteryzuje
się
głęboką
limfopenią
z
nieznacznym spadkiem liczby komórek NK i dużą
liczbą limfocytów B.

Pierwotne niedobory odporności
Ciężkie złożone niedobory odporności ze zmniejszoną liczbą
limfocytów T i B oraz komórek NK: T(-) B(-) NK(-).
Około 20% wszystkich przypadków SCID związanych jest z
niedoborem deaminazy adenozynowej (ADA).
Najczęstsza
postać
SCID
spośród
postaci
dziedziczonych
autosomalnie.
Niedobór spowodowany jest mutacją genu dla ADA, leżącym na 20
chromosomie.
ADA jest enzymem uczestniczącym w metabolizmie puryn. ADA
przekształca adenozynę oraz 2’deoksyadenozynę odpowiednio do
inozyny oraz 2’deoksyinozyny. 2’deoksyinozyna może swobodnie
dyfundować i ulegać fosforylacji do deoksyATP (dATP). Niedojrzałe
limfocyty nie potrafią przekształcić z powrotem dATP w
2’deoksyinozynę i są wyjątkowo wrażliwe na jego toksyczne
działanie. dATP hamuje reduktazę rybonukleotydową, enzym
niezbędny do syntezy deoksynukleotydów, przy ich braku synteza
DNA jest niemożliwa.

Pierwotne niedobory odporności
Niedobór ADA charakteryzuje się wcześniejszymi objawami
klinicznymi w porównaniu do pozostałych postaci SCID.
Defekt dotyczy limfocytów T, B oraz komórek NK,
limfopenia jest głębsza w porównaniu do innych postaci
SCID.
Oprócz
klasycznych
objawów
w
postaci
zakażeń,
zatrzymania wzrostu, u połowy chorych dołączają się
zaburzenia w układzie kostno-szkieletowym, a opisywane
są też zaburzenia neurologiczne: ślepota korowa, dystonia.
Zdarzają się przypadki niedoboru ADA o opóźnionych
objawach klinicznych ( występują nawet po kilku latach od
urodzenia). U chorych limfocytopenia rozwija się stopniowo,
nierzadko
obserwuje
się
towarzyszące
choroby
autoimmunizacyjne.

Pierwotne niedobory odporności
Ataksja teleangiektazja
Choroba dziedziczona autosomalnie recesywnie,
ma
złożony
obraz
kliniczny
o
dużej
heterogenności.
Charakterystyczne cechy: objawy neurologiczne
(ataksja), teleangiektazje (rozszerzone obwodowe
naczynia krwionośne), hipogonadyzm, zwiększona
częstość występowania nowotworów.
W surowicy wykrywa się zwiększone stężenie α-
fetoproteiny. Niedobór odporności nie zawsze
pojawia się już w dzieciństwie, ale w końcu rozwija
się u 70% chorych. Ma bardzo zróżnicowany obraz.

Pierwotne niedobory odporności
Częste są nawracające zakażenia dróg oddechowych. Ma
to związek ze zmniejszonym stężeniem przeciwciał.
Selektywny niedobór IgA wykrywa się u ponad połowy
chorych, podobnie duża część chorych ma niedobór
IgG2.
Niedobór odpowiedzi humoralnej związany jest z
defektem dojrzewania limfocytów B, a nie z ich liczbą.
Niedobór
odpowiedzi
komórkowej
łączy
się
z
niedorozwojem grasicy.
Limfopenia jest wynikiem zaburzenia proliferacji pod
wpływem stymulacji mitogenami.
Komórki chorych na ataksję teleangiektazję są zwykle
wrażliwe na promieniowanie, mają liczne anomalie
chromosomowe, o charakterze inwersji czy translokacji.

Pierwotne niedobory odporności
Agammaglobulinemia
Brutona
(agammaglobulinemia
sprzężona
z
chromosomem
X)
zwana
również
hipoimmunoglobulinemią – zespół należący do pierwotnych
niedoborów odporności, charakteryzujący się całkowitym brakiem
przeciwciał i śladową obecnością limfocytów B w krążeniu (poniżej
1%).
Przyczyną choroby są mutację pojedynczego genu znajdującego się
na chromosomie X nazywanego kinazą tyrozynową Brutona (Btk).
Białko kodowane przez ten gen odgrywa rolę w dojrzewaniu
prekursorów limfocytów B i aktywacji komórek tucznych.
W szpiku kostnym wykrywa się zwiększona liczbę niedojrzałych
limfocytów pre-B, nie mających receptorów immunoglobulinowych.
Poziom limfocytów T jest prawidłowy.
Ze względu na sprzężenie z płcią agammaglobulinemia Brutona
występuje przede wszystkim u chłopców, których matki były
zdrowymi nosicielkami defektywnego genu. Częstość występowania
wynosi ok. 1 na 100 tys. narodzin chłopców.

Pierwotne niedobory odporności
Objawy
Kliniczne objawy choroby pojawiają się około 4–6
miesiąca życia, kiedy z krążenia zaczynają znikać
przeciwciała matczyne.
Dominują nawracające zakażenia bakteryjne dróg
oddechowych. Gdy nie podejmuje się leczenia,
prowadzi to do przewlekłego zapalenia zatok i
zmian
rozstrzeniowych
oskrzeli.
Odporność
przeciwwirusowa jest sprawna, z niewiadomych
przyczyn nie dotyczy to enterowirusów.
Najczęstsza przyczyna śmierci w tym zespole jest
właśnie przewlekłe enterowirusowe zapalenia opon
mózgowych i mózgu.

Pierwotne niedobory odporności
Leczenie agammaglobulinemii Brutona polega
na okresowym substytucyjnym uzupełnianiu
ludzkich immunoglobulin dożylnie (rzadziej
domięśniowo lub podskórnie).
Leczenie powinno być prowadzone przez całe
życie i prowadzi do przedłużeniu długości i
jakości życia.
Teoretycznie
trwalsze
efekty
mogłaby
przynieść terapia genowa, jednak obecnie nie
jest ona metodą leczniczą.

Główny układ zgodności tkankowej
MHC obejmuje wiele genów odznaczających
się
największym
polimorfizmem
z
dotychczas poznanych.
Maja one podstawowe znaczenie zarówno w
inicjacji jak i w fazie efektorowej odpowiedzi
immunologicznej.
Cząsteczki MHC są glikoproteinami. Istnieją
cząsteczki MHC klasy I i II różniące się pod
względem budowy i funkcji, a także
cząsteczki klasy III i inne.

Główny układ zgodności tkankowej
Cząsteczki klasy I występują na powierzchni wszystkich komórek
jądrzastych, a w niewielkich ilościach również na erytrocytach.
Cząsteczki klasy II występują głównie na limfocytach B,
makrofagach, komórkach dendrytycznych, w tym na komórkach
Langerhansa, a także na komórkach nabłonkowych grasicy.
W wyniku aktywacji lub oddziaływania niektórych cytokin np.
IFN-γ, mogą się pojawić jednak na wielu innych komórkach np.
pobudzonych limfocytach T, komórkach śródbłonka, nabłonka
tarczycy, komórkach nabłonka jelitowego, fibroblastach,
keratynocytach.
U człowieka cząsteczki MHC klasy II występują konstytutywnie
na komórkach śródbłonka naczyń (w niektórych narządach np. w
sercu, nerce). Mogą być syntetyzowane selektywnie np. na 90%
monocytów znajdują się cząsteczki HLA-DR, lecz brak HLA-DQ.

Główny układ zgodności tkankowej
MHC klasy III stanowią różne cząsteczki,
niezwiązane
z
procesem
prezentacji
antygenu.
O ile między klasą I i II widoczne są wybitne
podobieństwa strukturalne, o tyle MHC klasy
III nie są podobne ani do dwóch pozostałych
klas, ani do siebie nawzajem.
Ludzkie MHC określane są mianem HLA
(ang. human leukocyte antigens – ludzkie
antygeny leukocytarne).

Główny układ zgodności tkankowej
U człowieka geny kodujące białka MHC znajdują się na 6
chromosomie.
Klasyczne cząsteczki MHC klasy I są kodowane przez
geny HLA-A, -B i -C, natomiast nieklasyczne – przez
geny HLA-E, -F, -G, MICA i MICB.
Nieklasyczne cząsteczki MHC klasy I mogą być także
kodowane poza regionem MHC (np. CD1).
Klasyczne cząsteczki MHC klasy II są kodowane przez
geny, leżące w regionach HLA-DP, -DQ i -DR, natomiast
nieklasyczne – HLA-DM i HLA-DO.
Pomiędzy regionem MHC klasy I, a regionem MHC klasy
II, znajduje się region kodujący MHC klasy III, choć
niektóre z tych cząsteczek są kodowane przez geny
leżące w locus MHC klasy II.
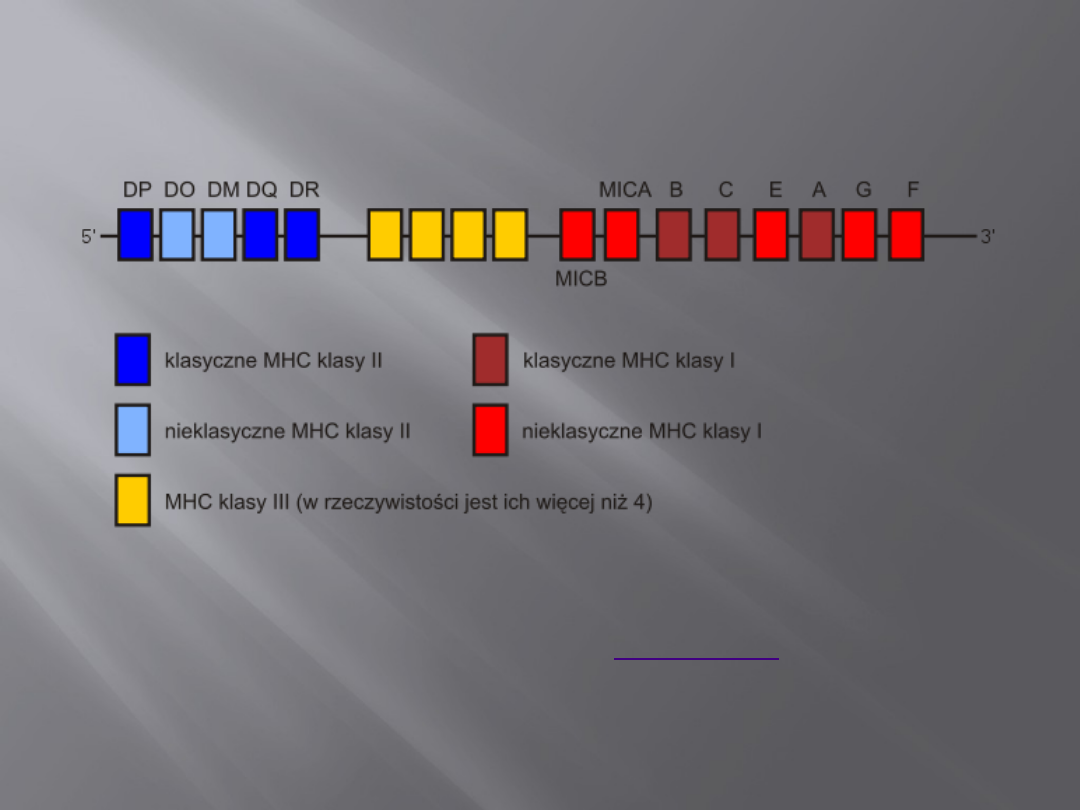
Główny układ zgodności tkankowej
UWAGA: geny MHC klasy II występują w większej ilości loci, ale ta
ilość jest zmienna. Obecne są także
. Przykładowo, w
obrębie HLA-DR występuje 10 genów, przy czym 5 z nich to
pseudogeny. Ponadto, region MHC klasy II zawiera zarówno geny dla
łańcucha α, jak i β.

Główny układ zgodności tkankowej
Znaczenie medyczne MHC:
Znaczenie medyczne MHC wynika przede wszystkim z ich udziału
w procesach odrzucania przeszczepu. Białka te są niezwykle
silnymi, immunogennymi antygenami, w związku z tym komórki,
na których występują, są natychmiast rozpoznawane jako obce.
Stąd też dopasowanie białek MHC ma kluczowe znaczenie w
doborze dawcy i biorcy przeszczepu.
Im większa niezgodność pomiędzy allelami u dawcy i biorcy, tym
większe prawdopodobieństwo odrzucenia przeszczepu i tym
szybciej proces ten następuje
Jednak nawet wtedy, gdy MHC są idealnie dopasowane,
odrzucanie będzie zachodzić, gdyż MHC będą brały udział w
prezentacji słabych antygenów zgodności tkankowej. Jeśli będą się
one różnić (a z wyjątkiem bliźniaków monozygotycznych będą się
różnić praktycznie na pewno), to będą one rozpoznawane jako
obce i w rezultacie komórki będą niszczone przez limfocyty Tc.

Główny układ zgodności tkankowej
Repertuar białek MHC wykazuje także
powiązania z pewnymi chorobami. W tym
przypadku wystąpienie określonych alleli
zwiększa prawdopodobieństwo pojawienia
się
niektórych
chorób
(zwłaszcza
nieinfekcyjnych), np. allel HLA-B13 stwierdza
się częściej u pacjentów z łuszczycą, zaś
HLA-B8 jest częstym allelem u pacjentów z
miastenią. Podłoże tego zjawiska nie jest
znane, co wiąże się z wieloczynnikową
patogenezą tych chorób oraz koniecznością
analizy wielu alleli i ogromnej liczby ich
kombinacji

Główny układ zgodności tkankowej

Document Outline
- Slide 1
- Slide 2
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Antygeny grupowe erytrocytów
- Choroba hemolityczna noworodków
- Choroba hemolityczna noworodków
- Choroba hemolityczna noworodków
- Choroba hemolityczna noworodków
- Rodzaje przeszczepów
- Odpowiedź na przeszczep
- Odpowiedź na przeszczep
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Pierwotne niedobory odporności
- Główny układ zgodności tkankowej
- Główny układ zgodności tkankowej
- Główny układ zgodności tkankowej
- Główny układ zgodności tkankowej
- Główny układ zgodności tkankowej
- Główny układ zgodności tkankowej
- Główny układ zgodności tkankowej
- Główny układ zgodności tkankowej
- Slide 47
Wyszukiwarka
Podobne podstrony:
Błony śluzowe – stan gotowości immunologicznej Część II
Immunogenetyka cześć I
IMMUNOLOGIA I ALERGOLOGIA CZESC I
egzamin IMMUNOLOGIA 15 OPRACOWANE (część)
IMMUNOLOGIA I ALERGOLOGIA czesc II
88 Leki przeciwreumatyczne część 2
SEMINARIUM IMMUNOLOGIA Prezentacja
Testy immunologiczne
Seminarium 6 Immunologia transplantacyjna farmacja 2
guzy część szczegółowa rzadsze
Stomatologia czesc wykl 12
więcej podobnych podstron