
Principles of Neurosurgery,
edited by Robert G. Grossman. Rosenberg © 1991.
Published by Raven Press, Ltd., New York.
CHAPTER 19
Neural Tube Defects
Michael Pollay
Incidence, 399
Etiology and Embryology, 399
Pathology, 402
Natural History, 404
Clinical Presentation, 405
Myelomeningocele, 405
Occult Spinal Dysraphism, 406
Diagnostic Tests, 406
Prenatal, 406
Radiographic, 407
Urologic Evaluation, 409
Miscellaneous, 410
Indications for Surgery, 410
Operative Management, 410
Position, 410
Anesthesia, 411
Operative Observations, 411
Complications and Postoperative Care, 412
Results, 412
Outcome, 413
References, 413
This chapter on spinal cord disease is limited to those
neural tube defects that are compatible with life and ame-
nable to medical and surgical therapy.
INCIDENCE
In the United States, the incidence of all forms of spi-
nal dysraphism is in the range of 0.7 to 1.0 per 1,000 live
births, although the incidence appears higher in the mid-
eastern United States (1,2). This low incidence contrasts
with those of Celtic lineage in Ireland (approximately
4/1,000) and Wales (approximately 12/1,000). The open
variety of dysraphism is at least seven times greater than
the occult variety. The risk factor in mothers with
previously affected children is 40 to 50 per 1,000 and
rises to 100 per 1,000 with two such children. From the
standpoint of the individual mother, the percentage re-
currence rate usually quoted after the birth of one af-
fected sibling is 5 percent and includes cases of spina
bifida and anencephaly. The chance of producing a
dysraphic child after the birth of two afflicted children
increases twofold, to 10 percent (3). Probably half of the
children included in this group will be stillborn (4). Sig-
nificantly increased risks have been noted for mothers
M. Pollay: Neurosurgical Section, University of Oklahoma
Health Sciences Center, Oklahoma City, Oklahoma 73190.
over the age of 35 and for siblings of mothers who have
given birth to dysraphic children. In the male survivors
who do not have serious disabilities from their dysraphic
condition, about 35 percent produce children. In the
case of female survivors, up to 40 percent have children.
In either group, the incidence of neural tube defects in
their offspring is about 3 percent (4). These high recur-
rent risks suggest the importance of genetic counseling
and the survey of maternal serum and amniotic fluid for
a-fetoprotein and acetylcholinesterase (4-6). There is re-
cent evidence that the occurrence of neural tube defects
is declining in certain parts of the world, including the
United States. This may in part be due to prenatal diag-
nosis, genetic counseling, and nutritional supplementa-
tion (5).
Spinal dysraphism, as a group of conditions, occurs
more frequently in the white population, and females are
affected almost twice as often as males (7). The lumbosa-
cral region is the site of the dysraphic lesion 80 to 90
percent of the time.
ETIOLOGY AND EMBRYOLOGY
The cause of myelodysplasia is unknown, although
there is evidence that both environmental and genetic
influences may affect neural tube development. For the
purposes of this discussion, it is important to understand
399
400 / CHAPTER 19
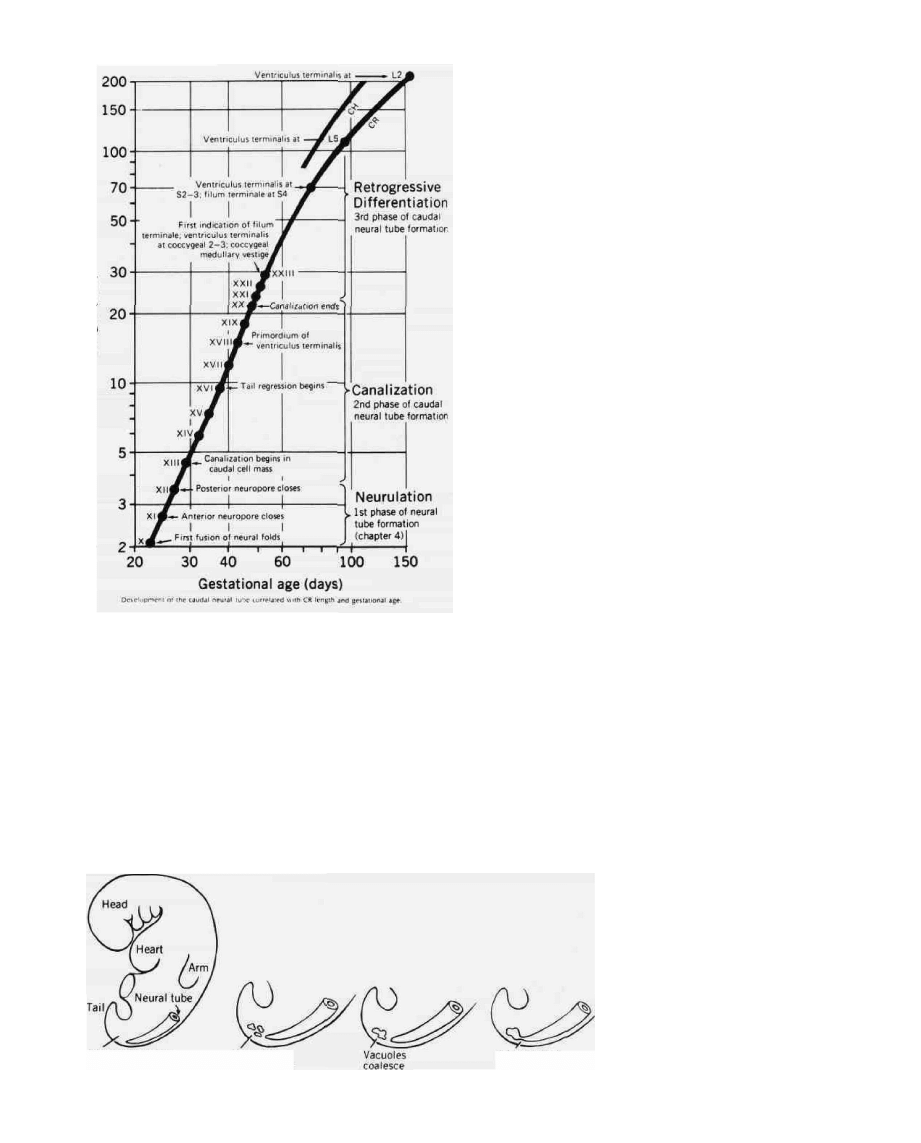
FIG. 1. Relationship of gestational age and the phases of
spinal cord development. (From reference 9.)
the alterations in normal development during various
embryologic phases that result in the clinical expression
of the dysraphic condition (8,9). The development of the
neural tube is presented graphically in chronological
order in Figure 1. The first phase is called neurulation; it
begins with the development of the neural plate and ter-
minates with the closure of the posterior neuropore at a
gestational age of somewhat less than 30 days. The ce-
phalic end of the tube closes a few days earlier, and the
cavity thus formed represents the primitive ventricles,
which are in continuity with the central canal of the spi-
nal cord. One would not expect the spinal malforma-
tions that occur during this period to be covered by in-
tact skin. Furthermore, since the location of the closure
of the neural tube (posterior neuropore) is at the first or
second lumbar segment, one would expect to find mal-
formations occurring during this embryologic period ei-
ther at this level or above.
One of the more popular theories proposed to explain
the origin of open defects of the neural tube is Gardner's
(10). His hydromyelia theory suggests that the neural
tube closes normally in most of the open dysraphic prob-
lems but that reopening or rupture occurs because of the
distension of the central canal of the spinal cord, second-
ary to failure in the development of the outlets from the
fourth ventricle, resulting in the prolongation of the
transient hydrocephalus that occurs as a normal phase in
development. In severe cases, an embryonic syringo-
myelocele will develop prior to rupture of the neural
tube. The same forces, according to Gardner's theory,
could cause impaction of the hindbrain into the upper
cervical region, while in less severe cases hydrocephalus
and/or cystic dilatation of the fourth ventricle could re-
sult. Patten proposed that the development of a myelo-
cele is secondary to overgrowth of the neural tube that
prevents complete closure of the tube (11). The hypothe-
sis that open neural tube defects are caused by nonclo-
sure of the neural tube has gained support based on ob-
servations in human embryos, although Lemire has
suggested that a small number of cases result from re-
opening of the closed neural tube (8). At the present
time, based on the wide range and frequency of central
nervous system anomalies associated with spina bifida
aperta, it is reasonable to conclude that neither the over-
growth, traction, nor hydrodynamic theory fully ex-
plains the full spectrum of pathological features seen
with this condition (12).
The last two phases of neural tube development that
occur after closure of the posterior neuropore result in
elongation of the neural tube by the processes of canali-
Caudal cell mass
(site of canalization)
Vacuoles
differentiate
Connect with
neural tube
FIG. 2. The processes involved in the
second phase of caudal neural tube
development. (From reference 9.)
NEURAL TUBE DEFECTS / 401
30-mtn CR
(Stage XXIII)
67-mm CR
Central
canal
Ventriculus
terminals
Dura
Filum
terminale
Ventriculus
terminalis
Coccygeal medullary vestige
FIG. 3. The relationship of the structures that develop during
the last phase (retrogressive differentiation) of caudal neural
tube development. (From reference 9.)
zation (Fig. 2) and retrogressive differentiation (Fig. 3).
These processes result in the development of the lower
lumbar and sacrococcygeal segments of the completed
neural tube (13). The canalization phase proceeds from
day 13 to 16 of gestation. The anlagen of the lower cord
segments consist of undifferentiated cells arranged
around vacuoles (Fig. 2). The vacuoles enlarge, coalesce,
and ultimately make contact with the long portion of the
tube previously formed during neurulation. The last
phase is retrogressive differentiation and consists pri-
marily of necrobiosis. The caudal cell mass regresses into
the ventriculus terminalis (the only grossly identifiable
remnant of the caudal central canal), filum terminale,
and coccygeal medullary vestige (13). The filum be-
comes fibrous, although it may contain ependymal or
ganglion cells. It also ultimately pursues an extradural as
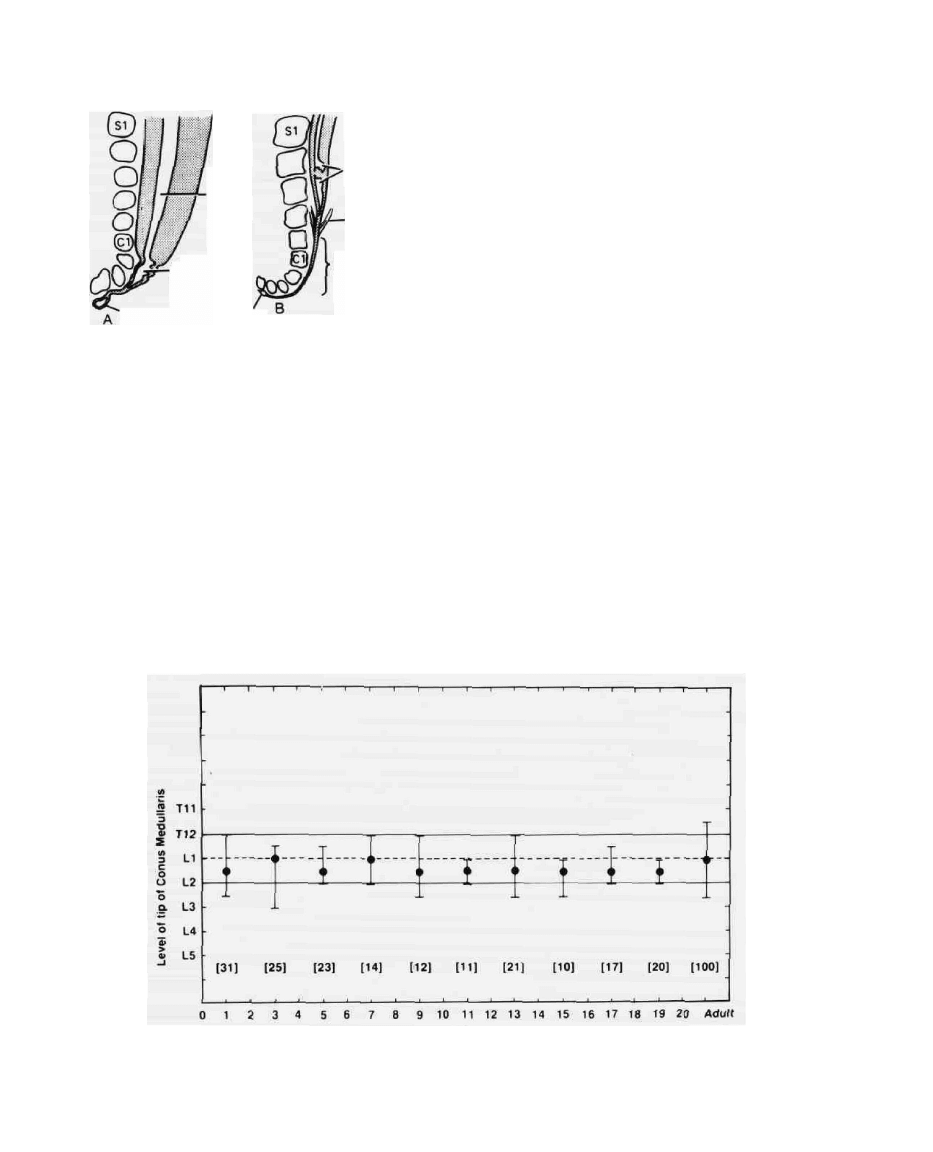
well as intradural course. At birth, the level of the conus
is about L3, but because of the more rapid growth of the
vertebral column it lies at approximately the adult level
of LI-2 by two months after birth (Fig. 4) (14,15).
The spinal malformations occurring during these last
two phases of neural tube development are covered with
intact skin and display a wide variety of conditions
usually included in the general classification of occult
spinal dysraphism (Figs. 5 and 6). The mechanisms in-
volved in the development of most of these spinal mal-
formations are poorly understood, although the persis-
tence of a split notochord may reasonably explain a wide
variety of visceral and central nervous system defects
(e.g., pre- and postvertebral enteric cysts, butterfly verte-
brae, and splitting or duplication of the spinal cord). The
split notochord syndrome was first described by Bentley
and Smith (16). They theorized that the notochord de-
velops in duplicate, allowing a transient or permanent
connection (neurenteric canal) to exist between the dor-
sal surface of the embryo and the embryonic gut. A wide
variety of endodermal, mesodermal, and ectodermal
rests might be expected to reside along the course of this
defect, which would explain not only the enteric cysts
mentioned above but also the splitting of the cord by a
median septum (diastematomyelia) and the intraspinal
presence of certain congenital tumors [Figs. 5(B) and
6 (4)].
Age in Years
FIG. 4. The vertebral level of the conus medullaris at various ages as measured from magnetic reso-
nance images of the spine. (From reference 15.)
402 / CHAPTER 19
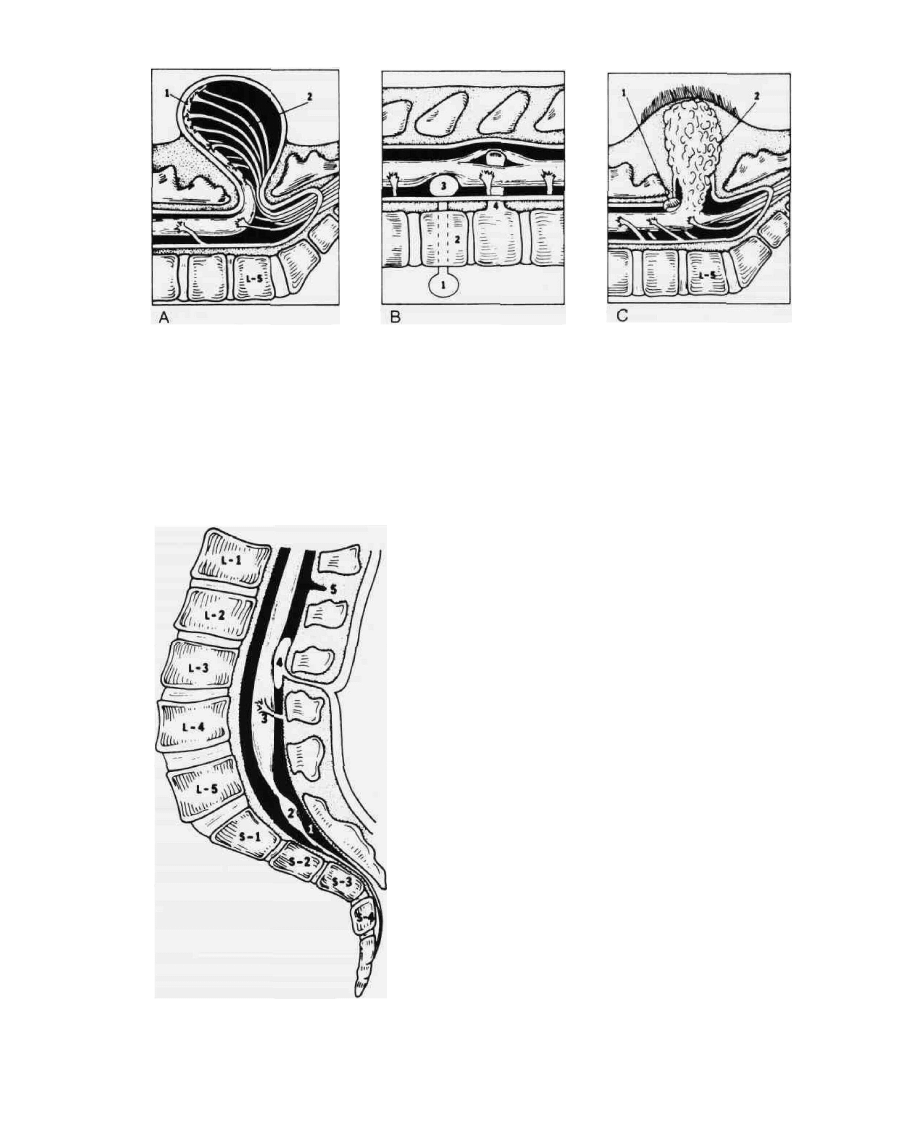
FIG. 5. Variety of common dysraphic conditions. (A) Typical myelomeningocele with neural plaque (1)
and nerve roots (2) attached to the external membrane sac; (B) pre- and postvertebral enteric cysts (1
and 3) communicating by means of residual enteric canal (2) through body of vertebra. Bony diastometa-
myelic septum (4) at lower end of split spinal cord; (C) lipomyelomeningocele (2) beginning beneath area
of hypertrichosis and extending into caudal end of spinal cord by way of a dural defect. The transverse
dural band (1) is shown to illustrate the potential compressive effect of this type of lesion.
FIG. 6. Tethering of spinal cord by thickened filum terminale
(1), with lipomatous inclusion (2), aberrant nerve roots (3), and
external dural band (5). Example of intradural tumor (dermoid,
epidermoid) in continuity with dermal sinus and dimple (4).
PATHOLOGY
The most common dysraphic defect is the absence of a
portion of the posterior elements of the spinal vertebrae.
In most cases, spina bifida occulta is of no clinical im-
portance and is observed as an incidental finding on
plain x-rays of the spine. It should be appreciated, how-
ever, that this usually hidden defect is very often asso-
ciated with other defects of the intraspinal contents.
The most common clinically significant neural tube
defect is the myelomeningocele (meningomyelocele,
spina bifida cystica, or aperta), which has been defined as
a defect in the dorsal elements of the vertebrae, accompa-
nied by herniation of the spinal cord and nerve roots into
a cystic swelling on the surface of the back [Fig. 5A]. This
lesion arises during neurulation, between the 17th and
30th gestational days (8). It is possible to classify this
defect into nine subcategories, depending on the actual
contents of the herniated sac (17). For most purposes,
however, the more general definition is sufficient to un-
derstand the clinical implications of the condition and
the therapeutic possibilities. This group of nervous sys-
tem defects is often associated with malformations of the
brainstem and the early potential for developing hydro-
cephalus. Other abnormalities associated with myelome-
ningoceles include, along with hydrocephalus and the
Arnold-Chiari malformation, dysplasia of the cranial
nerves, agenesis of the corpus callosum, and cortical and
cerebellar dysplasia (12).
A more uncommon condition presenting at the body
surface is the meningocele. In this instance, the cystic
NEURAL TUBE DEFECTS / 403
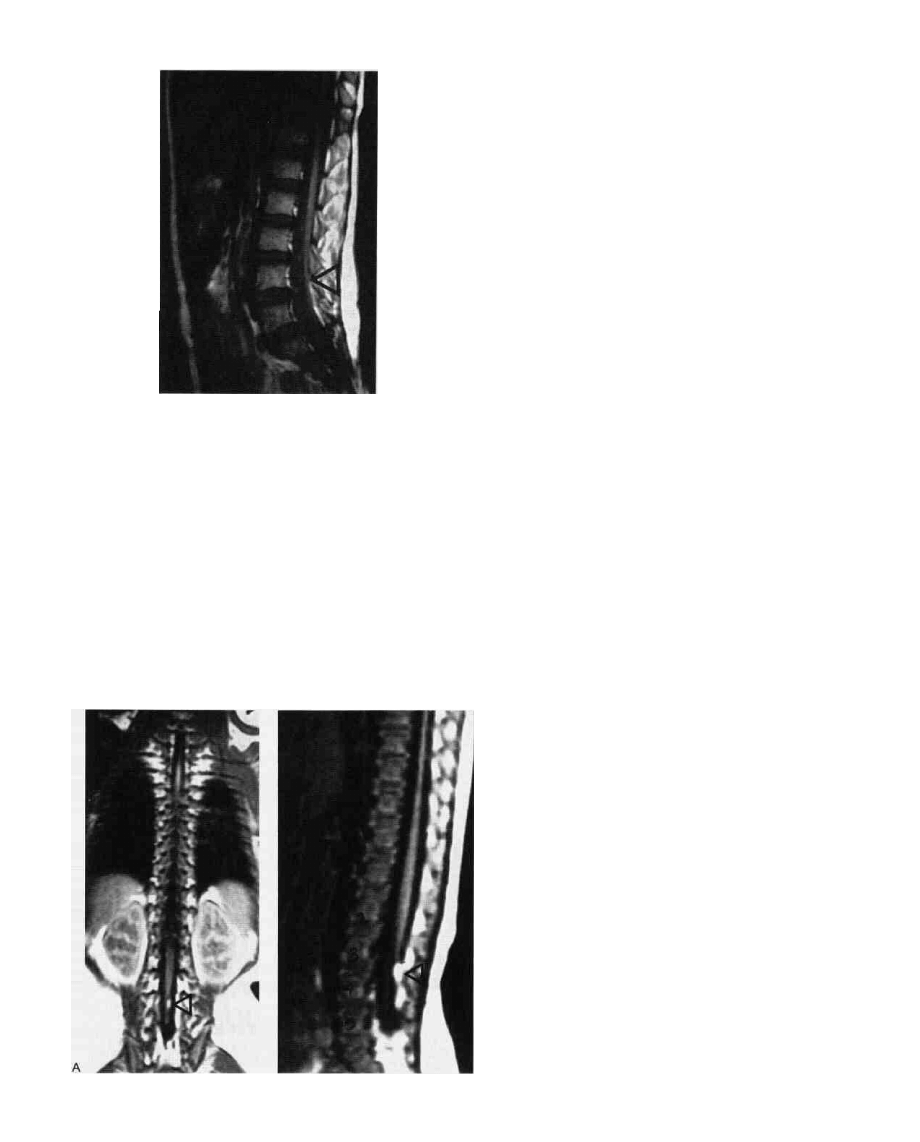
FIG. 7. MRI of spinal cord tethering due to a thickened filum
terminale. Arrow at site of dorsal atachament to dura.
swelling is formed by dura and arachnoid, while the ner-
vous tissue remains within the spinal cord. A simple
meningocele will often be connected to the intraspinal
space by a narrow stalk emerging from a single spine.
The bony defects are usually small and not palpable, and
there occasionally may be virtually complete covering
of the lesion with skin. The term meningocele manque
has been proposed to describe those meningocele lesions
that are small and present little evidence of their exis-
tence externally but have a loop of recurrent nerve roots
or spinal cord tracts adherent to the internal surface of
the dura mater (11). The wide variety of abnormalities in
the central nervous system and other organ systems ob-
served in myelomeningocele patients is rarely encoun-
tered with the usual meningocele defect.
The occult spinal dysraphic conditions are less com-
mon but just as important as the previously described
open defects. They present the greatest diagnostic chal-
lenge because they are often hidden by the intact overly-
ing skin. This group exerts its effect on spinal function by
traction and/or compression on the spinal cord. The
most common pathologic entities in the occult group are
those malformations that interfere with the normal ceph-
alad migration of the spinal cord during the dispropor-
tionate growth of the vertebral column. The fixation of
the spinal cord can be due to an abnormally thickened
filum terminale that is tethered to the inner surface of
the dorsal dura mater [Figs. 6 (1), and 7]. The filum
under these conditions may contain ganglion cells and a
generous vascular bed. There also may be considerable
lipomatous infiltration of the thickened filum [Figs. 6 (2)
and 8]. Extradural extensions of these bands do occur
and may be in continuity with a dermal stalk to the skin
surface [Fig. 6 (4) and (5)].
Another malformation that produces fixation of the
spinal cord is diastematomyelia [Fig. 5 (B4)]. By defini-
tion, this is a congenital anomaly in which the spinal
cord or the filum terminale or both are split dorsoven-
trally into two parts, usually separated by a septum. The
septum may be fibrous, cartilaginous, or bony and gener-
ally extends from the dorsal surface of the vertebral body
(Fig. 9). Such septa are most commonly seen in the tho-
racic region and may be associated with intraspinal neur-
enteric cysts as well as other bony anomalies (e.g., but-
terfly vertebrae, interlaminar fusion, or spinal bifida).
Unlike the duplicated spinal cord (diplomyelia), the cord
FIG. 8. A-P (A) and lateral (B) MRI of spinal cord tether-
ing due to thickened filum terminale with an intraspinal
B lipoma (arrow).
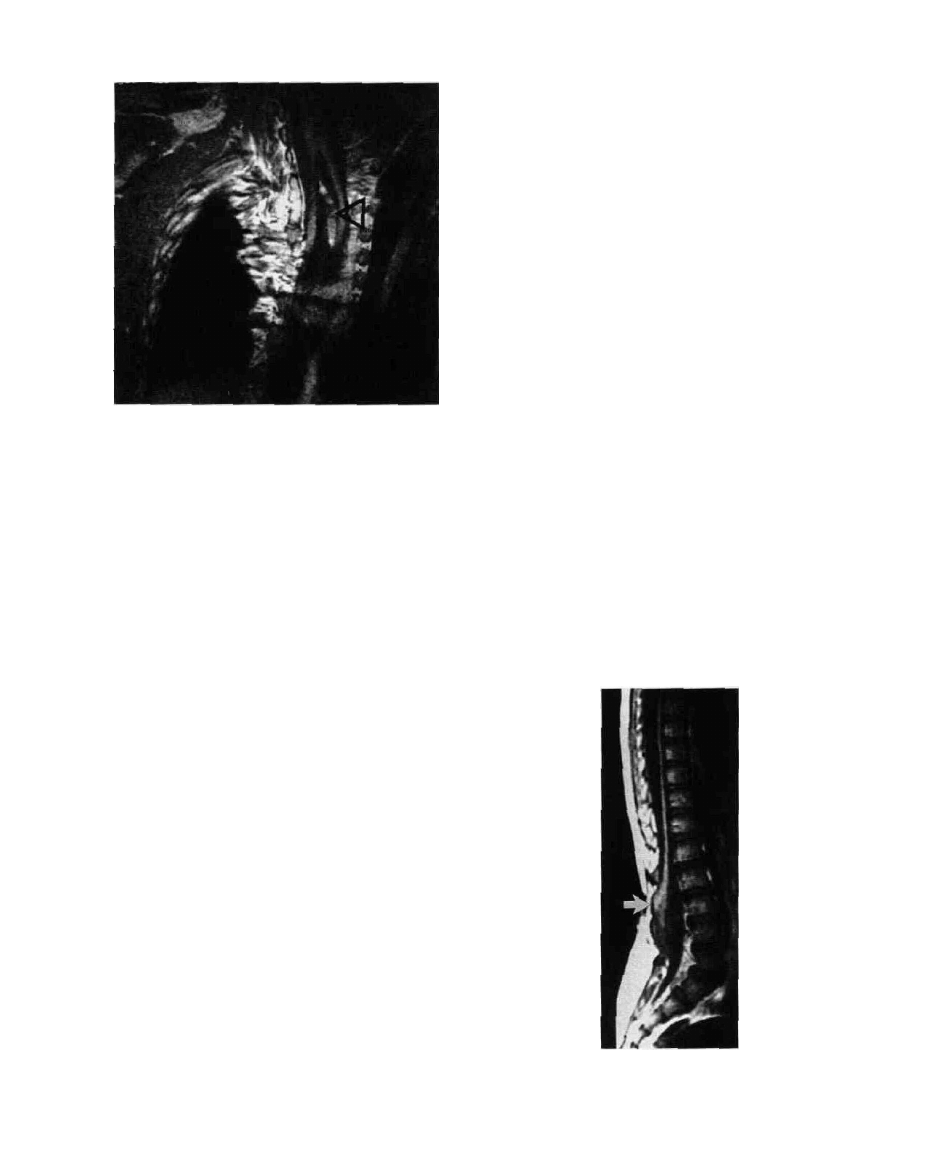
404 / CHAPTER 19
FIG. 9. MRI of diastematomyelia with septum (arrow).
"halves" in diastematomyelia are generally unequal in
size and cellular content (18). In approximately one-half
the cases, the dura is duplicated. In most instances, the
cord above and below the split is normal. Generally the
dividing septum is at the lower limits of the split cord. In
a number of cases, the septum has been found at a consid-
erable distance from the lower end of the divided spinal
cord (19). In those instances where no septum is present,
the tethering effect on the spinal cord results from bands
between spinal cord and dura, either at the split end or
more distally at the conus.
In the fully developed split notochord syndrome, in-
traspinal and mediastinal foregut cysts may also be pres-
ent. The intraspinal cysts may occur without the diaste-
matomyelic defect and produce a neurologic deficit by
means of direct spinal cord compression. Compression
syndromes are also seen with transverse dural bands
[Fig. 5 (Cl)] and intraspinal tumors (epidermoid, der-
moid, and teratomas) [Fig. 6 (4)]. These masses may
have extradural extensions by means of a dermal or fi-
brous tract and may occasionally produce distortion at
the skin surface [Fig. 6 (4)]. A somewhat more frequent
malformation in the mass effect group is the lumbosa-
cral lipoma [Fig. 5 (C2)]. It is thought that these lesions
produce their effect by traction as well as compression.
The passage of these subcutaneous, benign fatty tumors
through the dura into the spinal cord and/or roots inter-
feres with neural conduction by causing traction during
childhood growth (11). In addition, the intra- and extra-
dural lipoma may cause a mass effect on the involved
neural tissue (20). These intradural extensions of fatty
tissue are properly called lipomyelomeningocele when
there is an associated cystic cavity within the mass. All of
these lesions may on occasion be, connected to the over-
lying skin by a tract.
NATURAL HISTORY
Meningoceles and myelomeningoceles are usually ap-
parent at birth. The neurologic deficit, if present, is
usually well developed. Even when the myelomeningo-
cele is repaired, the patient may later deteriorate neuro-
logically owing to a tethered cord (Fig. 10) (21). The of-
ten associated hydrocephalus may not be apparent at
birth, but usually appears before the third year. When
untreated, this leads to progressive enlargement of the
skull, with associated neurologic dysfunction. The clini-
cal course is often dictated by the associated anomalies.
According to a study by Laurence and Tew, 25 percent
of the myelomeningocele infants are stillborn, and 13
percent die within the first week (13). Untreated, another
47 percent die from complications of this condition;
only 9 percent of those untreated were still alive during
the sixth year of their study. In the United States, Shurt-
leff reported that 23 percent of his untreated group lived
until 6 to 10 years of age (3). Early death generally results
from increased intracranial pressure secondary to hydro-
cephalus or from central nervous system infections, al-
though other severe associated anomalies within and
outside of the nervous system may be responsible as well.
The late mortality is generally caused by urinary tract
dysfunction and infections, as well as the complications
of hydrocephalus. The later cause of death has often
been sudden and is presumably a result of the acute ob-
struction of ventricular shunts or of pulmonary embo-
lism in the case of ventriculovascular shunts (22).
The natural history of concealed dysraphic conditions
is substantially different. These lesions are often not ap-
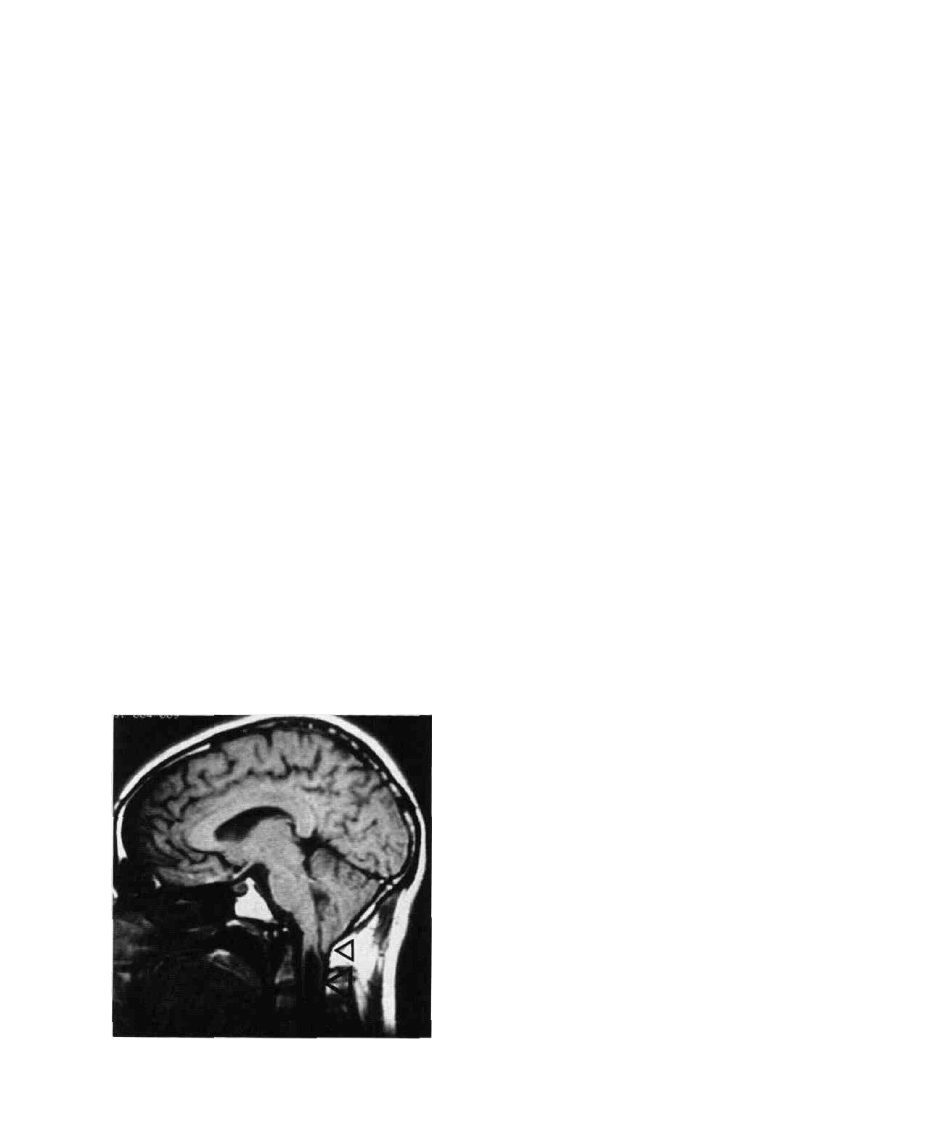
FIG. 10. Lateral view MRI of tethering following a myelome-
ningocele repair (arrow).
NEURAL TUBE DEFECTS / 405
predated at birth because of the intact skin. The develop-
ment of clinical dysfunction is usually delayed and insidi-
ous in onset. In some series, almost 90 percent of the
patients develop significant defects in function in the
lower extremity or of the bowel and bladder (23). In
many instances, once developed, these defects are not
reversible by any therapy (24). There is little question
that, in terms of the natural history of the open and
closed dysraphic lesions, the former is less a challenge in
diagnosis than in treatment, whereas the reverse is true
of the latter.
CLINICAL PRESENTATION
Myelomeningocele
The diagnosis at birth is based on the appearance of
the external cystic lesion, which contains the externally
placed plaque of neural tissue with varying degrees of
intact skin [Fig. 11(A)]. Most of these lesions are in the
lumbar region, with the remaining cases found primarily
in the lower thoracic and upper sacral areas. These le-
sions may, however, occur anywhere along the neuraxis.
Deformity of the spine is common and includes widened
pedicles in the region of the spina bifida, as well as ky-
phosis and scoliosis. Severe deformities are found in 5 to
10 percent of patients and are often caused by muscle
imbalance not associated with vertebral body abnormali-
ties. In more than half the cases there is obvious limb
deformity at birth. As expected, the degree of neurologic
deficit is generally related to the site of the lesion. The
lower the lesion is, the more restricted the neurologic
deficit. Unfortunately, only 10 percent or fewer of the
lesions are found in the sacral region. The clinical mani-
festations, in a general sense, reflect the level of the defect
as follows. Lesions at or above the thoracolumbar junc-
tion result in a flaccid paraplegia without hip dislocation
or limb deformity because there are no deforming forces
present. Involvement of the lower lumbar segments re-
sults in preservation of hip flexors and abductors, but
weak or absent extensors, and abductors with no useful
function at the knee or ankle. As a result of unopposed
muscular activity at the hip, dislocation at this joint is
common. With low sacral myelomeningocele, the deficit
may involve only the ankle or movements of the distal
foot (25). In the newborn, complete evaluation of the
motor system is difficult, but, on close observation, re-
flex movement sometimes can be separated from volun-
tary or purposeful activity. Observing the response to
pulling or extension of the extremities and to superficial
sensory stimulation will often lead to a useful evaluation
of the motor level of the lesion. These lesions may be of
the upper or lower motor neuron variety. Unfortunately,
the motor levels do not always correlate well with the
sensory loss or the radiographic definition of abnormal
vertebra (3).
Altered function of the bowel and bladder is difficult
to evaluate at birth and represents the greatest threat to
early and late survival (1,26). The presence of an anal
wink has little significance in terms of indicating a func-
tional anal sphincter. The tone of a grossly patulous anus
usually will increase with time and often is associated
with rectal prolapse. The spastically tight anal sphincter
can be determined at birth, but, along with the lax
sphincter, functional evaluation of control at birth re-
mains difficult. Evaluation of bladder function is also
impossible, but a large palpable bladder indicates an in-
creased urinary sphincter tone with an associated high
resistance to emptying by abdominal compression. The
opposite might be seen in the case of a sphincter with
little tone.
Most infants with myelomeningocele are afflicted
with the Arnold-Chiari malformation, and it is therefore
not surprising that this potential compromise in the out-
flow of cerebrospinal fluid from the ventricular system
and/or the basal subarachnoid space results in a high
incidence of hydrocephalus. In addition, this brainstem
malformation may result in a failure to thrive owing to
breathing and swallowing difficulties, progressive spastic-
ity, and upper extremity weakness (27). The incidence
and severity of the hydrocephalus is related to the site of
the lesion. In most series, 15 to 20 percent of infants with
a myelomingocele present with hydrocephalus at birth.
B
D
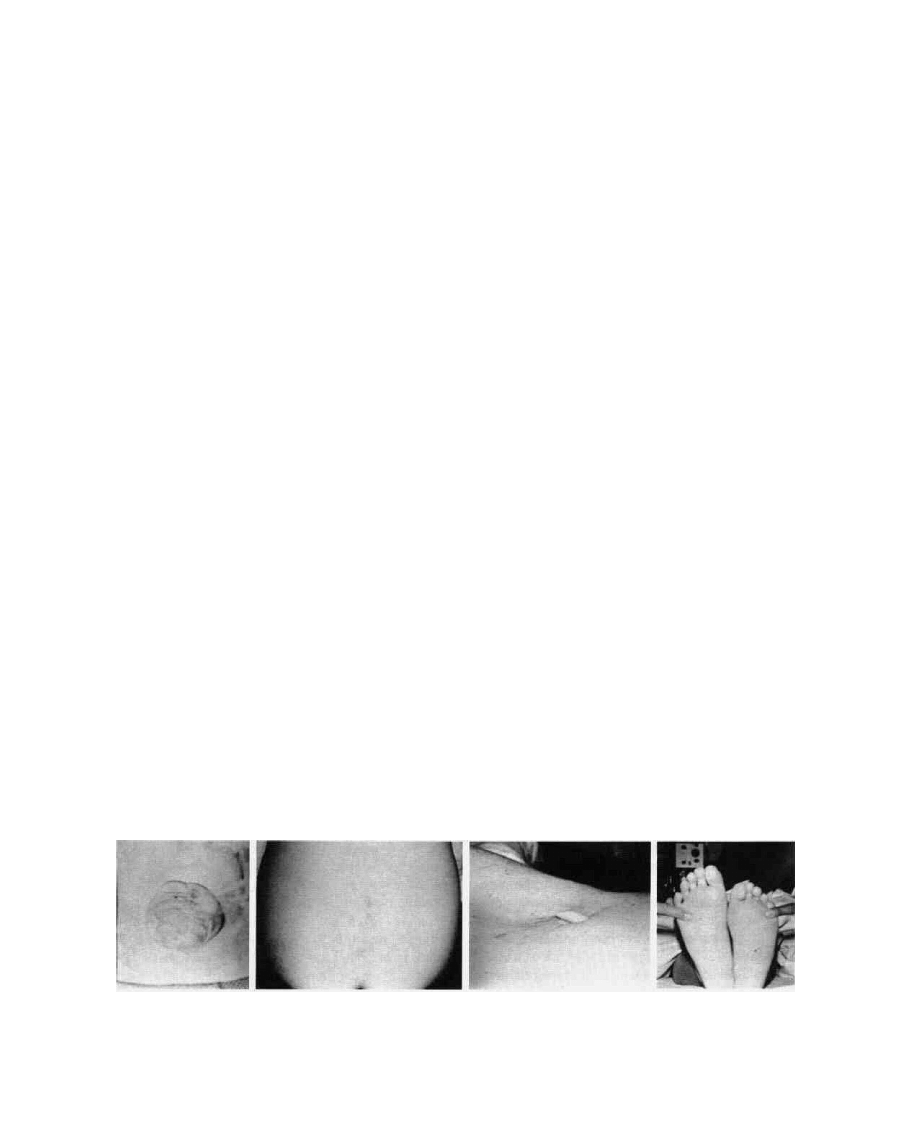
FIG. 11. (A) Large upper lumbar myelomeningocele; (B) dermal sinus, low lumbar, with two skin dimples
above this level; (C) tail-like structure, hypertrichosis, and subcutaneous lipoma; (D) small foot in occult
spinal dysraphic patient.
c
A

406 / CHAPTER 19
Another 40 to 60 percent develop this complication
within three years, although in most it is apparent within
the first month of life (28). A large number of hydroce-
phalic infants have aqueductal stenosis, which may be
secondary to the compression of the tectum by the di-
lated lateral ventricles. Since a relatively small number
of hydrocephalic infants are observed at birth, one nor-
mal head measurement has no predictive value. Serial
measurements must be made to evaluate properly the
early development of hydrocephalus. In this regard, pre-
mature infants may have significantly large changes in
ventricular size with little or no change in the measured
head circumference (28). It is important to examine
these infants for malformations in other organ systems:
polydactyly, limb bone hypoplasia, fused ribs, defects in
the heart, and overt or hidden anomalies of the genitouri-
nary system (3,4).|
Occult Spinal Dysraphism
As mentioned earlier, the diagnosis of these conditions
is often missed until a definite and often irreversible or-
thopedic or urologic syndrome develops, since these
postneurulation lesions are covered with skin (9). In
about half of the cases, however, there are cutaneous
manifestations that act as sentinels for malformations of
bone and nervous tissue deep beneath the skin surface.
Some of these are shown in Figure 11 and include, in
order of decreasing frequency, the following: lipoma, hy-
pertrichosis (hairy patch), dermal dimple or sinus, pig-
mented macule, vascular nevus, and a tail-like cutane-
ous appendage. In a significant number of cases there is a
combination of these cutaneous stigmata. In most in-
stances, recognition of these spinal malformations prior
to the development of clinical syndromes will depend on
recognition of the skin lesions by an astute observer. It
should be appreciated that a sacrococcygeal dermal sinus
(pilonidal sinus) usually terminates in an extradural lo-
cation, although some dermal sinuses with intraspinal
communications have been reported (29). Those occur-
ring cephalad to the lumbosacral region should be
viewed with a high index of suspicion, and the patient
should be evaluated to exclude intraspinal pathology
(30,31). Awareness of the association between an imper-
forate anus and sacral bony and neurologic abnormali-
ties is also important since the associated neurologic le-
sions have been shown to be progressive as a result of
tethering of the lower cord and/or bony and dural steno-
sis (32).
In general, the onset of the spinal dysraphic syn-
dromes occurs in childhood, but they are often accen-
tuated during adolescence. In some instances, however,
the clinical manifestations arise initially in the adult.
Two major clinical syndromes are associated with these
spinal malformations, although very often a combina-
tion is seen. In the orthopedic syndrome there is usually
present an asymmetry of the legs, because of a predomi-
nant unilaterally diminished muscle bulk, and a short
foot [Fig. 11(D)]. The foot is often inverted, with a high
arch and clawed toes. The skin of the plantar surface of
the foot may show trophic ulcers. The deep tendon re-
flexes in the involved extremity may be absent in the
presence of an extensor-plantar response. The sensory
loss may be quite variable, but generally involves the low
lumbar and sacral segments asymmetrically. The exter-
nal examination of the spine (excluding cutaneous man-
ifestations) may be quite normal, although scoliosis and
kyphosis are sometimes seen. The level and complete-
ness of the neurologic abnormalities are generally related
to the location as well as the extent of the malformation.
The urologic syndrome may present itself early by the
complaints of bed-wetting or frequent urinary tract in-
fections (24). Often these complaints are preceded by a
history of a normal voiding pattern.
The varied manifestations of deficits produced by
these occult spinal malformations emphasize the impor-
tance of the roles played by the pediatrician, urologist,
and orthopedist in the early diagnosis of these syn-
dromes (33).
DIAGNOSTIC TESTS
Prenatal
Prenatal tests are increasingly used in high-risk par-
ents to rule out significant dysraphic defects in the fetus
(4,34). A number of methods of diagnosis are used in-
cluding ultrasonography, fetography, fetoscopy, and bio-
chemical analysis of the maternal serum and amniotic
fluid. The latter analysis is based on the observation that,
in some children with neural tube defects, there is an
increase in a-fetoprotein (AFP) in the amniotic fluid.
The main source of this chemical marker is the fetal ur-
ine; it is synthesized in normal embryonal liver cells, the
yolk sac, and the intestinal tract. The concentration of
this marker is maximally different in the dysraphic fetus,
as compared with the normal fetus, between 14 and 16
weeks of gestational age. The normal value of 25 Mg/ml
may be elevated up to tenfold in the dysraphic fetus.
Failure to detect dysraphic conditions with this test can
occur, owing to skin-covered lesions or amniotic fluid
samples taken late in pregnancy. Elevated levels of this
marker resulting from dysraphic conditions can also be
seen in maternal serum, although similar elevations have
been reported in benign and malignant liver disease. The
presence of an open dysraphic lesion should be con-
firmed by ultrasonography when this marker is elevated.
A complementary test to AFP assay is the measurement
of acetylcholinesterase (AChE) in the amniotic fluid.
The presence of elevated levels of both AChE and AFP
NEURAL TUBE DEFECTS / 407
will reduce the already small percent of false results with
either test alone (5).
Radiographic
Among the earliest studies to be obtained in dysraphic
conditions are anteroposterior (AP) and lateral radio-
graphs of the spine. The most common anomaly to be
found is spina bifida, which may be related to the site of
the neural lesion. The most obvious and gross changes in
the posterior elements of the vertebrae are seen in the
open dysraphic conditions (myelomeningocele), as are
the most severe kyphotic and scoliotic deformities.
In patients with a myelomeningocele, scoliosis may be
observed extending over a great number of vertebrae,
with associated fused ribs and kyphosis. In addition, in-
formation derived from routine and contrast radio-
graphic studies is used to evaluate cardiopulmonary,
musculoskeletal, and urologic abnormalities. These le-
sions may be in the form of cardiac septal defects, hip
dislocation, urinary reflux, and hydronephrosis. Routine
contrast myelography, computed tomography (CT), or
magnetic resonance imaging (MRI) probably has little or
no role in the initial evaluation of open neural tube de-
fects, although these diagnostic modalities can be impor-
tant in evaluating delayed complications owing to Ar-
nold-Chiari malformation, syringohydromyelia, and
postmyelomeningocele tethering (Figs. 10 and 12)
(35,36).
Routine radiographs of the spine may be normal in
some cases of occult dysraphism. Usually, however,
there will be evidence of skeletal anomalies, such as de-
fects in the posterior elements of the vertebrae (spina
FIG. 12. Lateral view MRI of Arnold-Chian malformation
(small arrow) and cervical syringomyelia (large arrow).
bifida), malformed or fused laminae, a widened spinal
canal [Fig. 13(A)], or bony median vertebral septa [Fig.
14(A and B)]. These bony abnormalities are more pre-
dictive of the site of the intraspinal pathology than are
cutaneous signs (19). Usually, the midline bony spurs
seen in diastematomyelia are difficult to visualize in rou-
tine films and are appreciated only by linear tomography
of the suspected area of involvement. The combination
of intersegmental laminar fusion and spina bifida often
suggest the level of the diastematomyelia (19). The most
definitive radiographic studies utilize a contrast sub-
stance or computed imaging (CT or MRI). In recent
years the contrast agents generally have been of the
water-soluble variety so that linear tomography can be
easily combined with computed tomography during the
same visit to the radiology department. Perfectly accept-
able films, however, can be obtained with air as the con-
trast substance. The latter can also be combined with a
CT study. Approximately 50 to 60 ml of air is used, but
infused in increments of 10 ml with the patient in a 20-
degree Trendelenburg position. Both air and water-solu-
ble contrast substances generally require the use of linear
tomography for the best definition. Water-contrast me-
dia offer the best overall advantages including denser
images than obtained with air, easy combination with
linear and computed tomography, and lack of need to
remove (as compared to oil) at the end of the study. In
most cases, myelography is performed under local anes-
thesia supplemented with intravenous diazepam. The
total volume of the water-soluble agent (metrizamide)
used is in the range of 6 to 14 ml, at a concentration of
190 mg of iodine per ml. Complications, which may in-
clude headache, vomiting, and/or seizures, are infre-
quent if the patient is properly hydrated and drugs that
lower seizure threshold (phenothiazine derivatives) are
avoided. Recently, newer non-ionic water-soluble con-
trast agents (e.g., iohexol) have been introduced which
produce fewer side effects and which do not require pre-
medication.
In those patients in whom a mass lesion is suspected in
the lumbar region, it might be prudent to inject the con-
trast agent into the cisterna magna or into the cervical
subarachnoid space by means of a lateral cervical punc-
ture. Following the instillation of the contrast medium,
the spinal needle is removed and the patient placed in
the supine position for the myelographic study. The CT
scan then follows completion of the myelogram. To
diagnose a tethered conus medullaris, it is necessary to
visualize clearly the lowest point of the conus in order to
relate it to the vertebral level (Fig. 7). The vertebral level
at which the normal conus is found at various times fol-
lowing birth, based on MR imaging of the spine, is pre-
sented in Figure 4. MRI appears to be the radiographic
procedure of choice not only for localization of the
conus but for most intraspinal abnormalities (35). The
size and the site of attachment of the filum or other
408 / CHAPTER 19
B
D
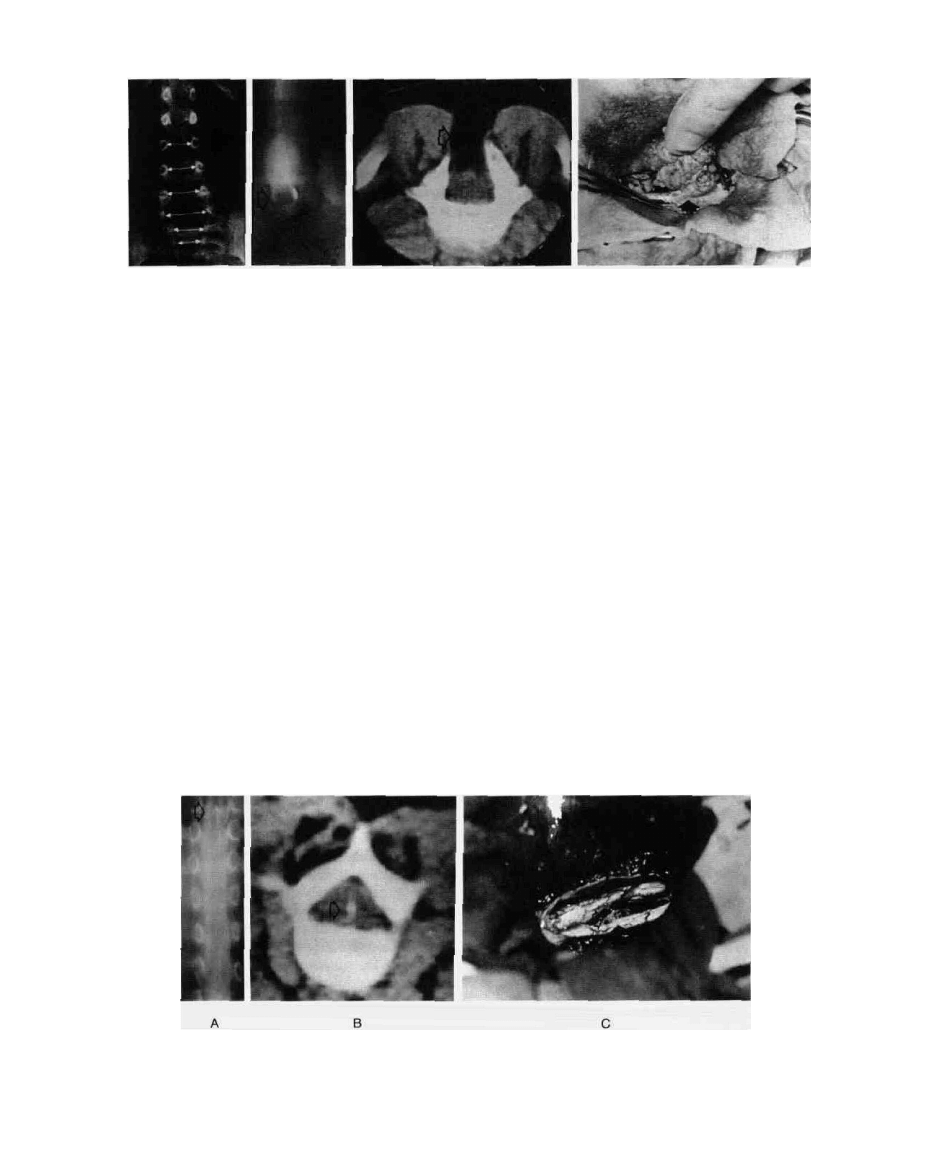
FIG. 13. (A) Wide lumbar canal with defects in lamina at low lumbar level; (B) myelographic evidence of
cystic intraspinal lipoma (lipomyelomeningocele); (C) CT cross-section through lipoma (arrow) passing
into spinal cord by way of laminar and dural defect; density number of lesion consistent with fat;
(D) operative finding of lipoma infiltrating spinal cord through defect in lumbar dura mater (arrow).
bands transversing the space between the spinal cord and
the dura must be determined (10). Based on the ana-
tomic studies of Barson (14) and Gryspeerdt (37), Fitz
and Harwood-Nash (38) suggested the following criteria
for diagnosing a tethered cord (Fig. 15): (1) a low posi-
tion of the conus medullaris (a conus tip below the L2-3
interspace in a child older than five years should be con-
sidered abnormal); (2) a filum terminale greater than 2
mm wide; and/or (3) a dorsal position of the filum ter-
minale. In addition, a widened sac and laterally emerg-
ing roots are often seen with the above radiographic find-
ings.
Intraspinal masses are easy to diagnose with myelogra-
phy, as they usually demonstrate the features of an intra-
dural filling defect. They can be associated directly with
the conus or a malformed filum. The pathologic anat-
omy of these lesions may frequently be determined by
the CT density numbers [Fig. 13(C)] or by the MR image
(Fig. 8). The intradural passage of a lipomyelomeningo-
cele also produces tethering and a rather characteristic
myelogram [Fig. 13(B)], although a lipoma may present
without a cystic component in association with a thick-
ened filum terminale (Fig. 8). The CT or MR scan often
provides evidence of the involvement of the lipoma with
the spinal cord and roots, as well as of the presence of a
cystic cavity within the lipoma. The CT density numbers
are characteristically those of fat.
The most difficult and varied lesion to diagnose is
diastematomyelia, either with or without a median sep-
tum. As mentioned earlier, the vertebral anomalies often
indicate the approximate location of the bony, cartilagi-
nous, or fibrous septum, which produces a definite split
in the contrast shadow [Fig. 14(A)]. The cord halves are
rarely symmetrical, and the conus is found at an abnor-
mally low level (18). There can also be a fibrous or ab-
normal spinal root tethering the cord at or remote to the
site of the split cord (39). In some cases, there is no sep-
tum and these bands, not a spur, provide the posterior
fixation of the cord (39). The myelographic features of
diastematomyelia are well known (19). Generally the le-
sions are located in the thoracic region. If a median sep-
tum is demonstrated, it is often found at the lower mar-
FIG. 14. (A,B) Median septum (arrow) and split halves of spinal cord in diastematomyelia seen in air
myelogram and CT scan; (C) operative finding of split cord (lower arrow) and remnant of bony median
septum (upper arrow).
c
A

NEURAL TUBE DEFECTS / 409
A B
FIG. 15. (A) Myelographic findings in spinal cord tethered by
thickened filum terminale and lipoma (lower arrow); conus
(upper arrow) located approximately at lower end of L3;
(B) operative finding of thick (>3 mm) filum terminale.
gin of the split halves of the cord (Fig. 14). A widened
spinal canal and vertebral anomalies, such as defects in
the posterior arch and interlaminar fusion, often mark
the site of the bony, cartilaginous or fibrous septum. A
CT scan at the site of the septum may demonstrate the
tissue characteristics of the septum, not only by the CT
appearance but also by the density numbers recorded
[Fig. 14(B)]. MRI is especially useful for preoperatively
evaluating the extent of the cord separation, as well as
the location and type of the diastomatomyelic peg (Fig.
9). Demonstration of this is important in planning the
proper operative strategy.
A frequently unrecognized form of spinal cord tether-
ing is that associated with a repaired myelomeningocele
(21). Myelography with polytomography will demon-
strate, in these cases, a thinned or absent dorsal (to the
cord) subarachnoid space cephalad to the tethered cord,
and an obtuse course of exiting nerve roots. Addition-
ally, the vertebral anomalies commonly associated with
myelomeningoceles are usually present in these cases.
MRI also demonstrates the tethering of the spinal cord at
the site of surgical repair and appears to be the procedure
of choice for evaluating delayed neurological deteriora-
tion in this condition (Fig. 10) (40).
Urologic Evaluation
In most cases, the early treatment of open dysraphic
defects is not preceded by an extensive urologic work-up,
since a more precise diagnosis is possible only after the
age of two or three years. It is, however, often appro-
priate to perform intravenous pyelography to rule out
malformations of the genitourinary tract. It is important
to appreciate that over half of the children with myelo-
dysplasia have neurological deterioration of urological
function, although some will have evidence of re- or
neoinnervation of the external sphincter and the blad-
der. Most of these changes occur within the first three
years of life (41). In the occult spinal dysraphic condi-
tions, the evaluation of the genitourinary system is most
important in terms of diagnosis and of intra- and postop-
erative evaluation of surgical therapy. In addition, an
understanding of the pathophysiology allows the ratio-
nal approach to therapy during the postoperative phase
of treatment. At a minimum, this evaluation should in-
clude a urinalysis, blood urea nitrogen, and excretory
urogram. Even in asymptomatic cases, a full urodyna-
mic diagnostic study is necessary when there is evidence
of significant cutaneous or skeletal abnormalities. The
measurement of bladder function (cystometry) is often
combined with external urinary and anal sphincter elec-
tromyography (EMG). When indicated, cystourethro-
scopy and voiding cystourethrography complete the uro-
logic work-up.
Cystometry is performed via an indwelling (transur-
ethral) catheter by infusing either a liquid or carbon
dioxide gas. The gas is more convenient to use and al-
lows rapid filling of the bladder. With either method the
patient is in a supine position, and the bladder is filled at
a constant rate (i.e., 100 ml/min) while the bladder pres-
sure is continually monitored and recorded. A determi-
nation is made of the volume at which the patient per-
ceives bladder fullness, urge to void, and imminent
urination. It should be appreciated that the measured
bladder pressure includes change in intra-abdominal
pressure (IAP) as well, and therefore a pressure trans-
ducer attached to the anal EMG plug will measure IAP
changes so that the intravesicular pressure may be
corrected. The typical pressure-infusion curve is nonlin-
ear, with the early part of the curve characterized by a
relatively greater rise in vesicular pressure per unit vol-
ume of infusion than during the remaining portion of
the infusion phase. Usually a definite volume marks the
first sensation of filling (around 120 ml, but variable)
and the urge to void that precedes voluntary contraction.
This portion of the curve becomes more vertical.
An electromyogram (EMG) of sphincter activity is
valuable, since it relates sphincter activity to bladder fill-
ing and records the response to attempts to stimulate or
inhibit voluntary efforts to void. In addition, the electri-
cal pattern (such as polyphasic potentials) can often indi-
cate lower motor neuron disease. Recording of poten-
tials from an anal plug can also be useful, as there can be
considerable disparity between anal and urinary
sphincter involvement in neurologic disease. In general,

410 / CHAPTER 19
the results of the urodynamic studies depend on the cord
segment or root level involved. In the occult dysraphic
patient, a complete lower motor neuron lesion results in
an areflexic bladder and a denervated urinary or anal
sphincter. When the lesion is located above the sacral
area, an upper motor neuron lesion is usually found and
is characterized by a hyperreflexic bladder with or with-
out external sphincter dyssynergy. Cystoscopy and radio-
graphic studies will sometimes demonstrate in these
cases bladder trabeculations and, in severe obstructive
cases, ureteral reflux.
Miscellaneous
In some instances, full evaluation of cardiopulmonary
function is indicated, especially with the multisystem in-
volvement often seen in the myelomeningocele patient.
In cases of suspected hydrocephalus, CT scanning of the
head without enhancement or MRI is required to estab-
lish this diagnosis and other abnormalities of the nervous
system. It has been suggested that, in those patients with
significant anomalies in other organ systems, palm and
fingerprints as well as chromosomal studies be done to
establish the diagnosis of chromosomal errors or field
defects (4).
Recently it has been shown that spinal- and scalp-re-
corded somatosensory evoked potentials (SEPs) are use-
ful in evaluating young adults and children with sus-
pected tethered spinal cord syndromes. Posterior tibial
SEPs are highly predictive of level and laterality of the
lesion. There is also a high correlation between the sever-
ity and extent of the lesion and extent of the abnormality
of the SEP (42).
INDICATIONS FOR SURGERY
There have been wide differences of opinion as to the
use of early surgical intervention in infants born with
open neural tube defects (26,43,44). There are many
moral, social, and medical issues that have been used to
restrict the early repair of myelomeningoceles and other
conditions associated with these neural defects, although
some programs have delivered maximum therapy with-
out selection (43). This selection process, however, has
changed during the past two decades largely because of
the improved results associated with progress in medical
and surgical therapy. The selection of care became a
moot issue in April 1985 under provisions of the Child
Abuse and Neglect and Prevention and Treatment pro-
grams. These rules require that all infants be treated with
all medical and surgical modalities unless (1) the infant is
chronically and irreversibly comatose, (2) treatment will
only prolong dying, and (3) treatment would not alter
the survival of the infant after utilizing an inhumane
treatment (44).
The approach to treatment of the myelomeningocele
has especially changed over the past 25 years, because
prior to adequate control of hydrocephalus the treat-
ment of an open spinal defect was primarily academic
(45). The closure of these defects within 48 hours after
birth was proposed in 1963, in order to prevent infection
and diminish the degree of lower limb paralysis. A re-
duced incidence of central nervous system infection re-
sulted from this policy, although the proposed beneficial
effect on limb function has not occurred (45,46). In gen-
eral, the goals and timing of treatment are based on medi-
cal factors, although social factors are also considered in
the individual case (1,3). The question of the advantages
of immediate (<24 hours), early (two to seven days), or
late surgical repair is still unsettled, but most evidence
supports the view that the incidence of infection and
retained neurological function are not significantly dif-
ferent between the immediate and early repair groups
(46,47). It should be appreciated that treatment of the
myelodysplastic child is invariably a long-term commit-
ment by the medical team and family.
There is little controversy concerning the indications
for operative intervention in the occult group of spinal
malformations, although the timing of such procedures
has not been clearly established. The notion that prophy-
lactic surgery prior to a period of rapid truncal growth
seems reasonable based on our understanding of the role
of traction in the development of neurological dysfunc-
tion. There is little question, however, that the onset of
orthopedic and urologic syndromes with defined myelo-
graphic abnormalities requires timely surgical interven-
tion (48). Most experienced clinicians recommend surgi-
cal relief from traction or compressive lesions that are
capable of adversely affecting spinal cord function. The
final decision to operate, as well as the selection of the
appropriate operative procedure, is based on the radio-
graphic localization and description of the spinal lesion.
OPERATIVE MANAGEMENT
Position
The myelomeningocele patient is generally placed in a
prone position unless a ventricular shunt is to be inserted
at the same time. In that case, the hips are slightly rotated
to expose the flank for the insertion of the abdominal
catheter. In all cases, pressure on the abdomen is mini-
mized by using chest rolls or, in the older patient, an
orthopedic frame. Ideally the abdomen should be freely
suspended. However, contact with the heating pad used
with infants is often lost. It is, therefore, usually neces-
sary to heat the operating room to between 80 and 85°F
in order to control the child's core temperature. External
heating lamps are used primarily during induction, and
temperature loss prevented by the use of a plastic wrap

NEURAL TUBE DEFECTS
411
around the exposed skin outside of the operative field.
The legs of older patients are wrapped with elastic ban-
dages all the way to the groin. In recent years we have
been routinely monitoring SEPs and the electromyo-
graphic activity of the anal sphincter during the opera-
tive procedure. The electrodes are placed following the
final positioning of the patient.
Anesthesia
In newborn infants, the repair of a small myelomenin-
gocele may be carried out under local anesthesia. In most
other cases we generally use endotracheal anesthesia
with a nondepolarizing muscle relaxant (e.g., atracurium
besylate). Recovery from neuromuscular blockade can
be easily and rapidly accomplished with an anticholines-
terase reversing agent prior to stimulating roots for anal
sphincter activity.
Operative Observations
In general, all of these cases are done using magnifica-
tion. Operating telescopes with a power of between 2.5
and 3.5 provide adequate magnification. The fiber-optic
light source has also proven to be a necessity in most of
these cases. The skin incision in the myelomeningocele
patient circumscribes the external lesion, saving as much
normal skin as possible. For large defects, consultation
with plastic surgery allows proper planning for the shape
of the incision, preparation of donor sites, and position-
ing of the patient. The closure of the defect should be
carried out in four layers, as shown in Figure 16 (49).
Great care is taken to preserve functional neural ele-
ments by using stimulation prior to dissection of these
structures, generally accomplished by observing the
movement in the extremities, as well as sphincter and
bladder contraction as visualized on the dual channel
recorder. After separating the neural plaque [Fig. 16(A)]
from the adjacent epithelial strip, the plaque is covered
with the pia-arachnoid (layer one). The remnant of the
dura and/or paraspinal fascia provides the next level of
closure (layer two). It has been suggested that the circula-
tion of cerebrospinal fluid around the neural placode can
be enhanced by utilizing a dural graft. This enlargement
of the dural sac may prevent tethering of the repaired
myelomeningocele (50). Finally the skin is approxi-
mated with subcutaneous sutures (layer three) and fine
skin sutures (layer four). Intravenous fluorescein may be
used to demonstrate adequate circulation of the skin su-
ture line. If it is inadequate, relaxation incisions, skin
grafting, and rotational flaps may be necessary to ensure
that a tight closure is achieved without tension at the
suture line. In some cases, the extent of the spina bifida is
great, and there is difficulty in producing a strong cover-
ing layer for the reconstructed cord and meninges. This
problem can be solved by using a large latissimus dorsi
muscle flap or by the medial rotation of the spread pedi-
cles (with their muscle mass) by surgical fracture (51,52).
The site of the skin incision in the occult dysraphic
patient is dictated by the myelogram. In the case of a
dermal dimple or sinus, a midline elliptical incision is
used in order to follow and remove these structures; the
shape of the incision for a lumbosacral lipoma is dictated
by the geography of the lesion itself. In the case of simple
tethering by a thickened filum terminale, fibrous intra-
dural bands, or aberrant roots, the sectioning of these
structures should occur only after stimulation studies.
Lipomas that enter the dural defect and infiltrate the
spinal cord or cauda equina must be dissected carefully.
In most instances, it is safe to perform only a subtotal
removal. A careful, watertight closure of the dura with or
without a fascial graft should follow the subtotal resec-
tion. A multilayer closure, after removal of most of a
subcutaneous lipoma, will lead to a satisfactory cosmetic
result. The strategy for dealing with diastematomyelia
depends on the demonstration of a median septum. The
median septum, regardless of its composition, should be
removed. The dural sleeve that is present in one-half of
the cases should also be removed. In addition, tethering
bands at the site of the septum or at the conus or filum
should be sectioned. Great care must be taken while re-
moving the septum in order not to damage the adjacent
cord. A large collection of vessels that may cause nui-
sance bleeding is often encountered at the base of the
septum. If anticipated, this bleeding is usually easy to
control under magnification.
B
FIG. 16. (A) The neural plaque has been sepa-
rated from the skin margin. The straight arrow
indicates the direction the neural plaque will
go after its inversion into the open spinal canal
following the closure of the lateral margins
(curved arrows) of the pia-arachnoid mem-
brane. (B) Following closure of the pia-arach-
noid membrane, the lateral margins of the re-
maining dura and/or fascia are closed over the
neural tissue, thus producing a complete
membranous tube. (Modified from reference
49.)

412 / CHAPTER 19
The intraspinal compressive lesions are removed in a
manner similar to that used for intradural tumors. Great
care is taken to preserve the adjacent or involved neural
tissue. In addition, intradural tethering bands sometimes
seen with these lesions must be found and severed. The
extradural fixation bands are technically easy to remove
prior to a multilayered closure.
COMPLICATIONS AND POSTOPERATIVE CARE
The major complications following myelomeningo-
cele repair are poor skin healing, infection, and spinal
fluid leak. Secondary wound repair generally solves these
problems, although the presence of severe hydrocepha-
lus may require ventricular shunting prior to secondary
closure. The problem of spinal fluid leak and stress on
the site of myelomeningocele repair in children with hy-
drocephalus at birth might be alleviated by inserting a
ventricular shunt at the time of the back repair (53). Al-
ternatively, placement of an infant ventricular reservoir
and postoperative tapping can accomplish the same goal
in those infants with grossly contaminated wounds. Post-
operatively, the anal opening is shielded from the repair
site by a plastic drape attached to the skin of the but-
tocks. An abdominal sling or gauze donut can be used to
hold the infant in a prone position. Bladder emptying is
carried out at intervals by a crede maneuver. Oral feed-
ings are started as early as possible, generally within 12
hours of surgical repair. Daily head measurements are
most important, and any change in the rate of growth
and fullness of the open anterior fontanelle indicates the
development of hydrocephalus and the need for a confir-
matory CT study.
The most devastating complication in the surgical re-
pair of an occult spinal malformation is increasing the
neurologic deficit. This occurs in only a small percentage
of cases and is more often associated with lipomyelo-
meningoceles than simple tethering bands. The use of
lower extremity and sphincter monitoring should de-
crease this complication. Infection and wound dehis-
cence are relatively infrequent complications, as most of
these patients are in good health with a proper nutri-
tional status. Spinal fluid leaks are also uncommon and
are more often associated with those cases with a congeni-
tal dural defect (such as a lipomyelomeningocele). The
development of a spinal fluid leak following repair of
these defects is more common when the dural patch is
composed of artificial or dehydrated material. Fascial
autografts may prevent this complication.
Early ambulation and institution of intensive bladder
care and physical therapy are initial goals in most of the
surgically treated occult lesions. The postoperative care
of the bladder depends on the level of the neurologic
lesion. Initially, in the newborn, simple crede of the blad-
der provides adequate emptying. In the older child and
in many of the occult spinal dysraphic patients with
bladder involvement, continence can be achieved by in-
termittent catheterization. In some cases, pharmacologic
agents have been used alone or in combination with a
program of intermittent catheterization. In many of
these cases there is a urinary storage problem owing to
vesical hyperreflexia. Oxybutynin has proven useful in
this situation by producing muscle relaxation. If there is
an associated spasm of the external sphincter, polysyn-
aptic inhibitors, such as baclofen or diazepam, can be
used. The choice of pharmacologic agent depends on an
understanding of the pathophysiology as determined
from the urodynamic studies. The primary goal of a ther-
apeutic program for bowel training is to achieve con-
trolled spontaneous defecation by taking advantage of
the gastrocolic reflex, and by adjusting the diet to include
natural laxatives and high fiber content. Combined with
this approach is the judicious use of suppositories, ene-
mas, and oral purgatives (4). The use of electrical stimula-
tion to achieve bowel or bladder continence is still in the
early phases of development and has no role in the early
postoperative treatment of sphincter disorders of the
bowel or bladder.
RESULTS
It is difficult to quote accurate figures for results in
myelomeningocele patients because of the complexity
and variability of the syndromes associated with these
myelodysplastic conditions. In those patients treated ag-
gressively for both the dysraphic problem and hydroceph-
alus, the rate of survival is now in the range of 90 percent
or better. The quality of survival is very difficult to evalu-
ate. It can be shown that the final outcome is related to
early and aggressive treatment, level of the neural lesion,
state of the hydrocephalus, and presence or absence of
infection. For example, in one study 64 percent of pa-
tients with a midlumbar lesion had normal to high intel-
ligence: this was true of only 38 percent with lesions in
the thoracic area (3). It should be understood that even
with a normal intelligence quotient (IQ), the myelodys-
plastic survivor has moderate to severe visual-motor per-
ceptual defects (1). Ames and Schut have reported that
following surgical therapy of 79 children with myelome-
ningocele and hydrocephalus, 71 percent were ambula-
tory, with most using crutches (54). In this group, 48
percent were considered competitive, with IQs of 80 or
better. This figure was 90 percent in the 36 dysraphic
children who were not hydrocephalic. Similar results
have been presented by ShurtlefTin a much larger group
of patients (3). In this study, almost 94 percent of the
dysraphic children with significant or well-controlled hy-
drocephalus had IQs of 80 to 110 or more. The IQ level
was much lower, however, in the patients who were
shunt dependent or who had a history of infection or

NEURAL TUBE DEFECTS / 413
hemorrhage into the cerebrospinal fluid (3,53). Satisfac-
tory socioeconomic adjustment and educational achieve-
ment were also related to IQ and level of the lesion. Over
70 percent of the patients with a normal IQ achieved a
happy adjustment, with 63 percent becoming indepen-
dent in all activities. In the patients whose lesions were at
the high lumbar and thoracic level, only 50 percent were
happy, whereas 75 percent of the patients with sacral
lesions were described as happy.
The results in the surgical treatment of occult spinal
dysraphism are a little easier to quantify. James and
Lassman have reported that, in a variety of occult dysra-
phic cases followed from 1 to 13 years after surgical treat-
ment, 17 percent remained normal, 43 percent were ei-
ther questionably unchanged or improved, 38 percent
were unchanged, and 2 percent were worse (11). In a
later study, James and Lassman reported that in seven
uncomplicated cases of diastematomyelia, all remained
normal after surgery, whereas in three patients with dia-
stematomyelia and incontinence, only one remained
unimproved (56). In those patients with diastematomye-
lia and a history of progressive neuropathy, 24 percent
were worse following operative removal of the median
septum, while 35 percent remained the same and 41 per-
cent improved. In reviewing the world experience for
this condition, Kennedy reported that a corrective opera-
tive procedure for diastematomyelia resulted in improve-
ment in 75 percent of the cases, while in 15 percent the
patients were not improved (57). In this same review,
there was a worsening of the neurologic status in 6 per-
cent of the cases. In a large series of treated intraspinal
lipomas, Bruce and Schut reported that in 27 of their
cases with normal preoperative examination, some 89
percent remained normal after surgery (23). In the 23
cases with preoperative neurologic deficits, some 35 per-
cent improved, whereas 56 percent were unchanged and
9 percent had an increase in their deficit. The high inci-
dence of urinary complications seen at the time of diag-
nosis in patients with a lipomyelomeningocele is unfortu-
nate, since surgical treatment seems not to reverse these
deficits in many cases (24). IJ should be appreciated that
success is measured not only by improvement in the neu-
rologic deficits but also by prevention of further neuro-
logic deterioration. On this basis, the expected success of
surgical intervention in occult spinal dysraphism is in
excess of 90 percent.
OUTCOME
The progress that will occur in dealing with severe
myelodysplasia will be in the field of genetic counseling
and the antenatal screening for a-fetoprotein. The com-
mitment of our society for the required orthopedic, uro-
logic, and neurosurgical care of the myelomeningocele
patient is great and continues during the lifespan of the
individual. It has been shown that actively treating the
myelodysplastic child can result in great satisfaction for
all those involved in the care of these children, as in-
creasing numbers of the survivors fill important roles in
the family unit and useful places in society (1,43).
The children with closed lesions of the spine are gener-
ally of normal intelligence and easily enter the main-
stream of society. There is, however, a continued role for
orthopedics in reconstructing abnormalities of the limbs
and spine in some of these cases. Urologic evaluation
continues to be most important, although most of these
children can obtain satisfactory results with use of inter-
mittent catheterization, biofeedback therapy, and phar-
macologic treatment of the vesicosphincter abnormali-
ties. There is little question that the comprehensive
diagnostic and therapeutic approach advocated will ulti-
mately result in early operative intervention in order to
preserve normal function rather than attempts, some-
times in vain, to restore lost function (33).
REFERENCES
1. McLaughlin JF, Shurtleff DB. Management of the newborn with
myelodysplasia. Clin Pediatr (Phila) 1979:18:463-476.
2. Vogter DM, Kaufman HH. Spinal dysraphism—a review. W V
MedJ 1985:81:142-145.
3. Shurtleff DB. Myelodysplasia: Management and treatment. Curr
Probl Pediatr 1980; 10:1 -98.
4. Fishman MA. Recent clinical advances in the treatment of dysra-
phic states. Pediatr Clin North Am 1976;23:517-526.
5. Brock DGH, Barren L, Van Heynington V. Prenatal diagnosis of
neural-tube defects with a monoclonal antibody specific for acetyl-
cholinesterase. Lancet 1985; 1:5-8.
6. Goldberg MF, Oakley, GP Jr. Interpreting elevated amniotic fluid
alpha fetoprotein levels in clinical practice: use of the predictive
value positive concept. Am J Obstet Gynecol 1979:133:126-132.
7. Friede RL. Developmental neuropathology. New York: Springer-
Verlag, 1975.
8. Lemire RJ. Neural tube defects. JAMA 1988;259:558-562.
9. Lemire RJ, Loeser JE, Leech RW, et al. Normal and abnormal
development of the human nervous system. Hagerstown, MD:
Harper and Row, 1975.
10. Gardner WJ. Myelocele: Rupture of the neural tube? Clin Neuro-
surg 1967:15:57-79.
11. James CCM, Lassman LP. Spinal dysraphism. New York: Apple-
ton-Century-Crofts, 1972.
12. Gilbert JN, Jones KL, Rorke LB, ChernofTGF, James HE. Central
nervous system anomalies associated with meningomyelocele. hy-
drocephalus, and the Arnold-Chiari malformation—Reappraisal
of theories regarding the pathogenesis of posterior neural tube de-
fects. Neurosurgery 1986;18:559-564.
13. Laurence KM, Tew BJ. Natural history of spina bifida cystica and
cranium bifidum cysticum. Arch Dis Child 1971:46:127-138.
14. Barson AJ. The vertebral level of termination of the spinal cord
during normal and abnormal development. J Anal 1970;106:489-
497.
15. Wilson DA, Prince JR. MR imaging determination of the location
of the normal conus medullaris throughout childhood. AJNR
1989;10:259-262.
16. Bentley FJR, Smith JR. Developmental posterior enteric remnant
and spinal malformations. Arch Dis Child 1960;5:76-86.
17. Talwaker VC, Dastur DK. "Meningoceles" and "myelomeningo-
celes" (ectopic spinal cord). J Neural Neurosurg Psychiatry
1970:33:251-262.
18. Herren RY, Edwards JE. Diplomyelia (duplication of the spinal
cord). Arch Pathol 1940:30:1203-1214.
Wyszukiwarka
Podobne podstrony:
neuralgia trigemen cap 11
19 Mikroinżynieria przestrzenna procesy technologiczne,
Prezentacja1 19
19 183 Samobójstwo Grupa EE1 Pedagogikaid 18250 ppt
19 Teorie porównanie
Sys Inf 03 Manning w 19
19 piątek
19 Emptio venditio ppt
PRCz Wyklady 19 21a
Neural networks in non Euclidean metric spaces
12 19 Life coaching
14 19 (3)
19 Substancje toksyczne
więcej podobnych podstron