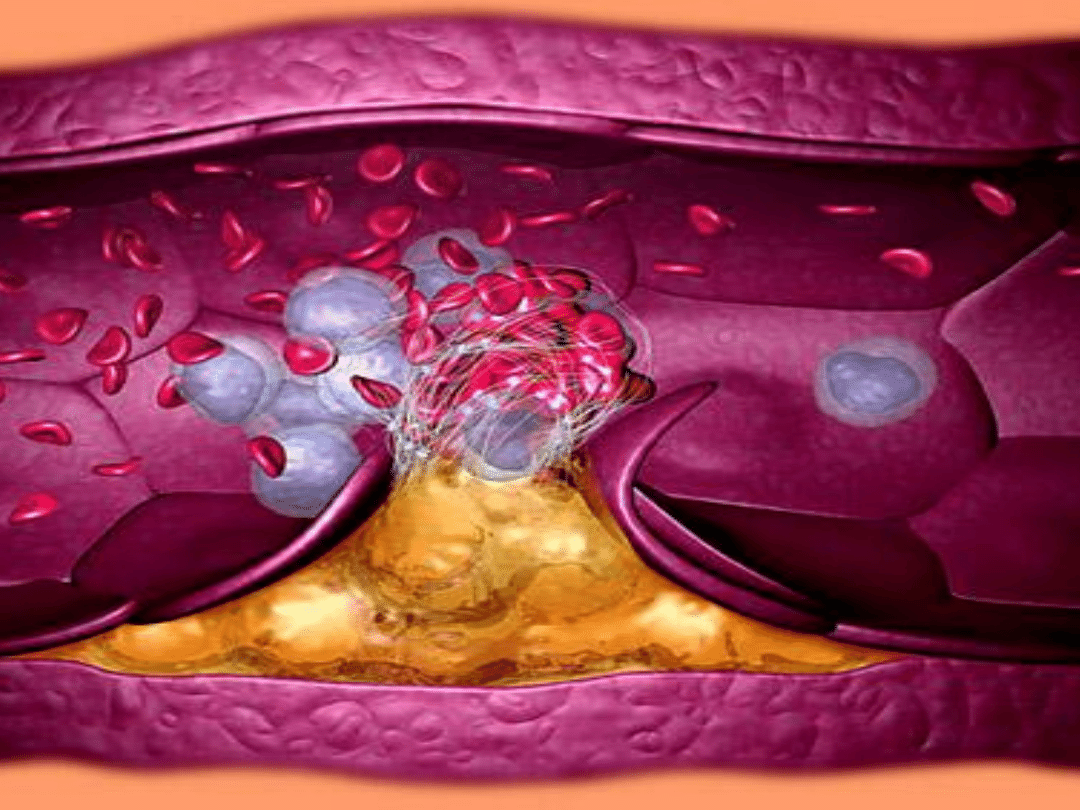
PATOGENEZA MIAŻDŻYCY
Zakład Nadciśnienia Tętniczego
Katedry Nefrologii i Nadciśnienia Tętniczego
Uniwersytetu Medycznego w Łodzi

• Miażdżyca tętnic (łac. atheromatosis, atherosclerosis) (potoczna
nazwa to "arterioskleroza") – przewlekła choroba, polegająca na
zmianach zwyrodnieniowo-wytwórczych w błonie wewnętrznej i
środkowej tętnic, głównie w aorcie, tętnicach wieocowych i
mózgowych, rzadziej w tętnicach kooczyn
• Przyczyną miażdżycy jest odkładanie się złogów cholesterolu w
ścianach naczyo krwionośnych, prowadzące do zmniejszenia lub
całkowitego blokowania światła naczynia.
• Utrudnienie przepływu krwi pociąga za sobą zaburzenia w
funkcjonowaniu wielu tkanek spowodowane ich
niedotlenieniem.
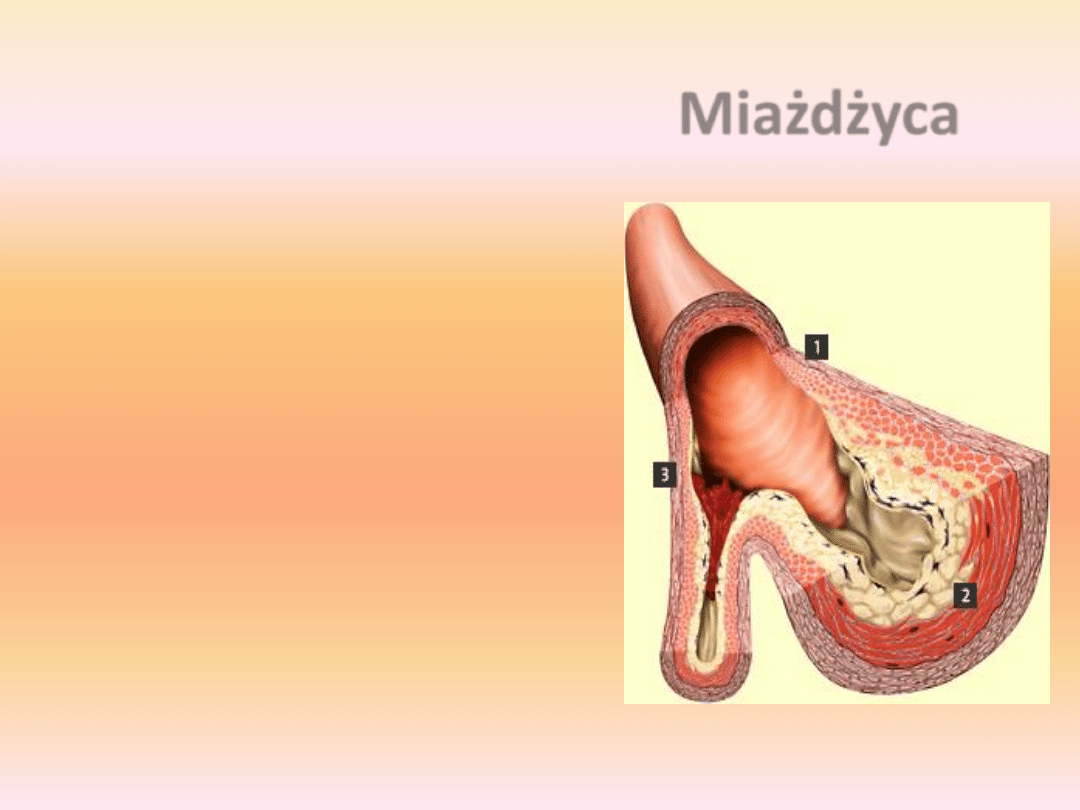
Miażdżyca
Ściana tętnicy składa się z 3 warstw:
•
tkanki łącznej (1)
•
warstwy składającej się z mięśni
gładkich i elastycznej tkanki
łącznej (2)
•
warstwy komórek śródbłonka
naczyniowego, bezpośrednio
kontaktującego się z krwią (3)
Powierzchnia śródbłonka
naczyniowego musi byd
nienaruszona. Bowiem tylko
wtedy możliwy jest przepływ
krwi bez turbulencji
zwiększających ryzyko powstania
zakrzepu krwi.
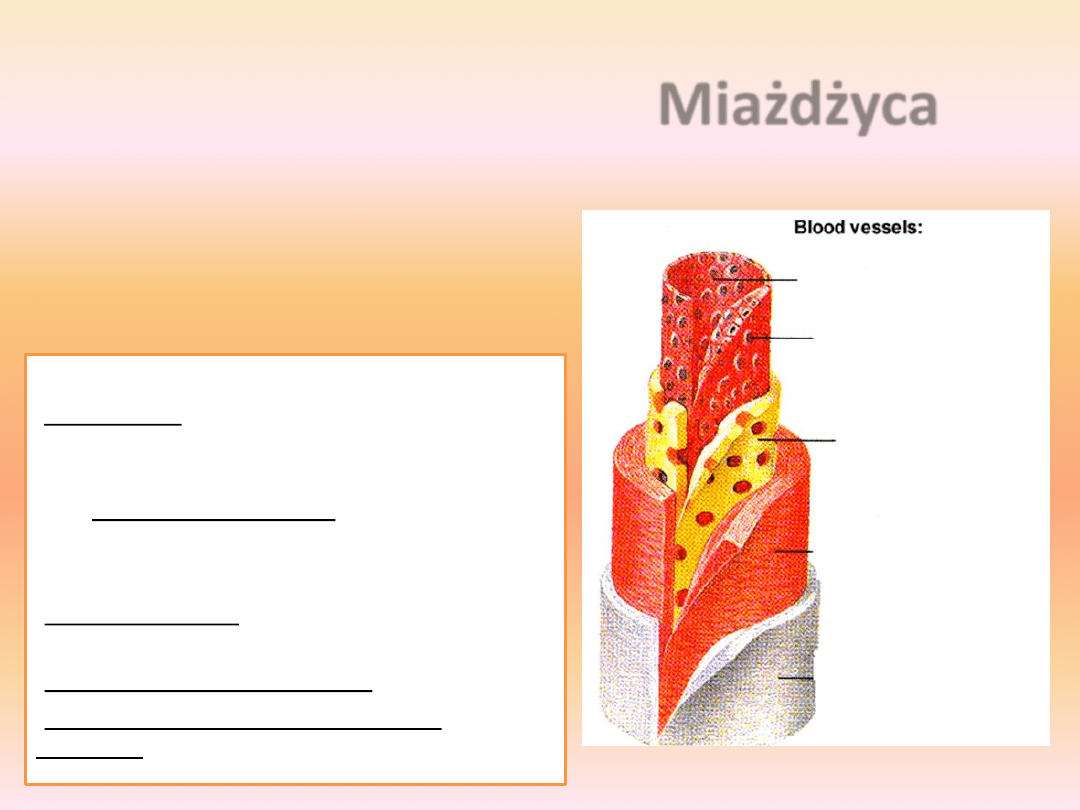
Miażdżyca
światło naczynia
komórki śródbłonka
tunica intima
warstwa komórek
mięśni gładkich
tunica adventitia
Podstawowym pierwotnym
procesem patologicznym leżącym u
podstaw rozwoju blaszek
miażdżycowych jest wywoływana
przez różne czynniki ryzyka
dysfunkcja
śródbłonka.
Należą do nich:
•siły nacisku prądu krwi na ścianę naczyniową
(shear stress
), działające w miejscach
rozdwojenia lub zwiększonej krzywizny tętnic
• towarzyszące hipercholesterolemii, krążące we
krwi lipoproteiny i monocyty
•produkty zaawansowanej glikacji białek
(cukrzyca)
•związki drażniące znajdujące się w dymie
tytoniowym
•noradrenalina i angiotensyna II
•kompleksy immunologiczne i czynniki
infekcyjne
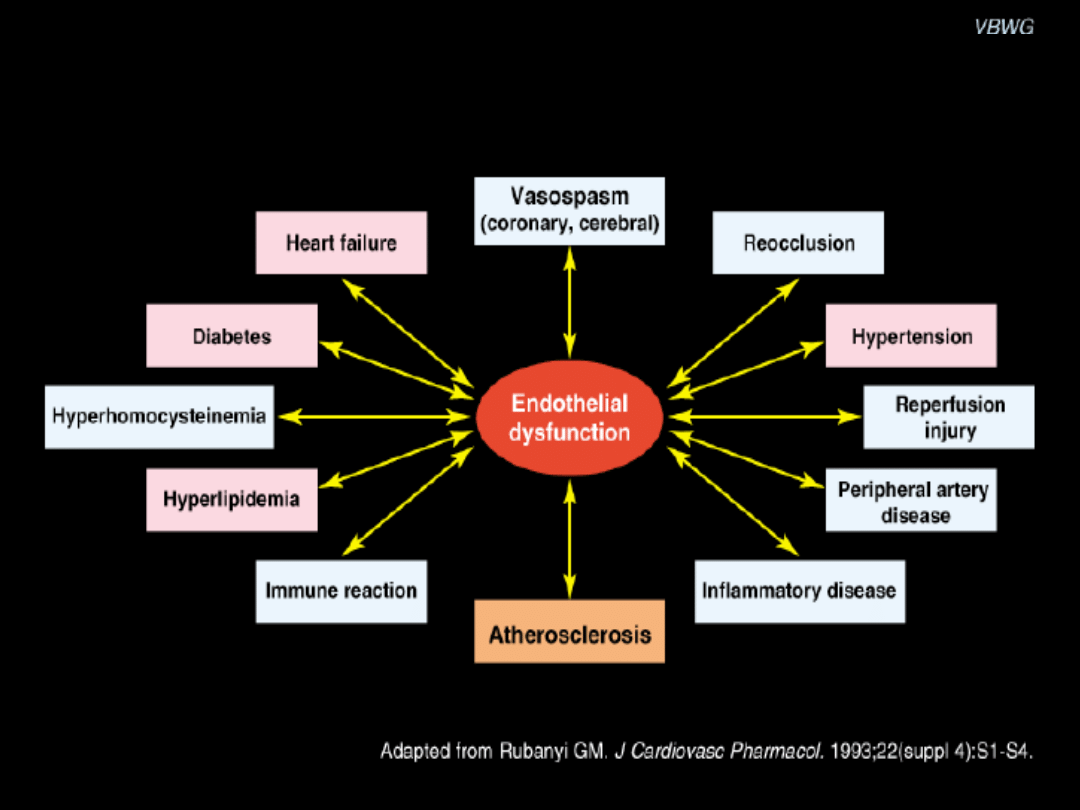
Przyczyny i konsekwencje dysfunkcji śródbłonka
naczyniowego
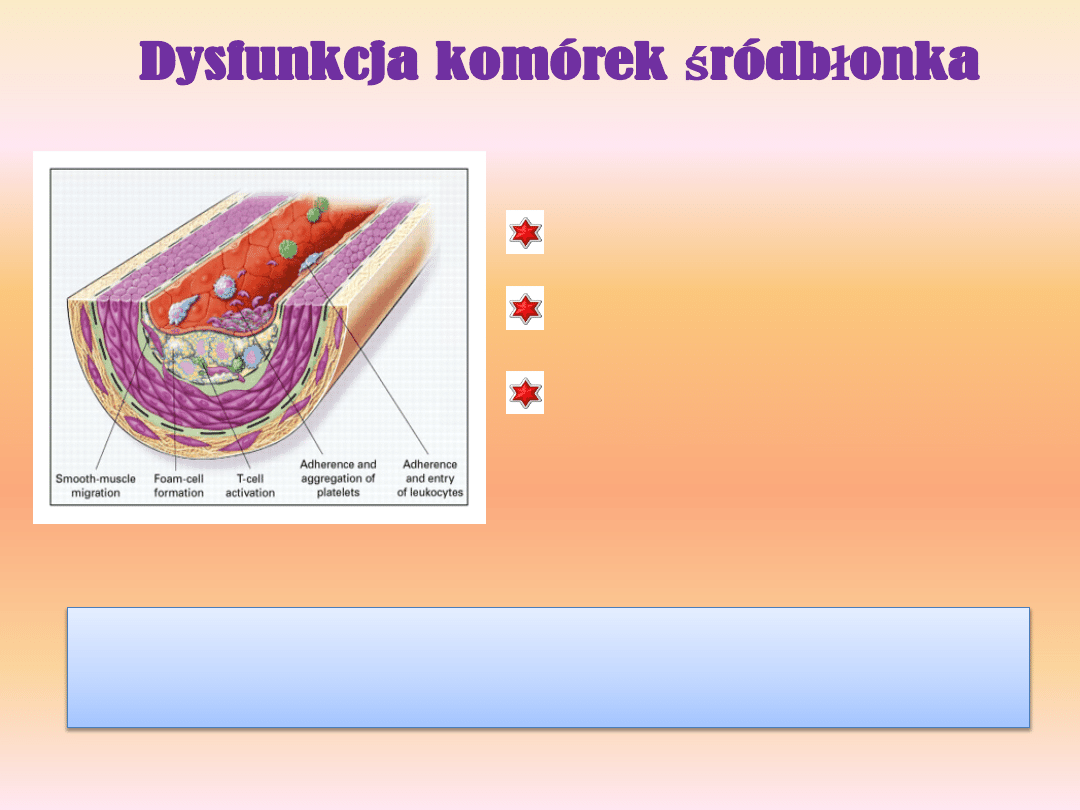
Dysfunkcja śródbłonka charakteryzuje
się
• zwiększoną przepuszczalnością błony
plazmatycznej,
• zmniejszoną syntezą i uwalnianiem
tlenku azotu (NO)
• wzmożoną ekspresją na powierzchni
śródbłonka chemoatraktantów
(substancji, które przyciągają komórki
zapalne) i cząsteczek adhezyjnych
(selektyna P i E, ICAM-1, VCAM-1).
Zmiany te ułatwiają rekrutowanie i późniejszą kumulację komórek zapalnych
w obrębie intimalnej warstwy ściany naczyniowej oraz proliferację komórek
naczyniowych mięśni gładkich.
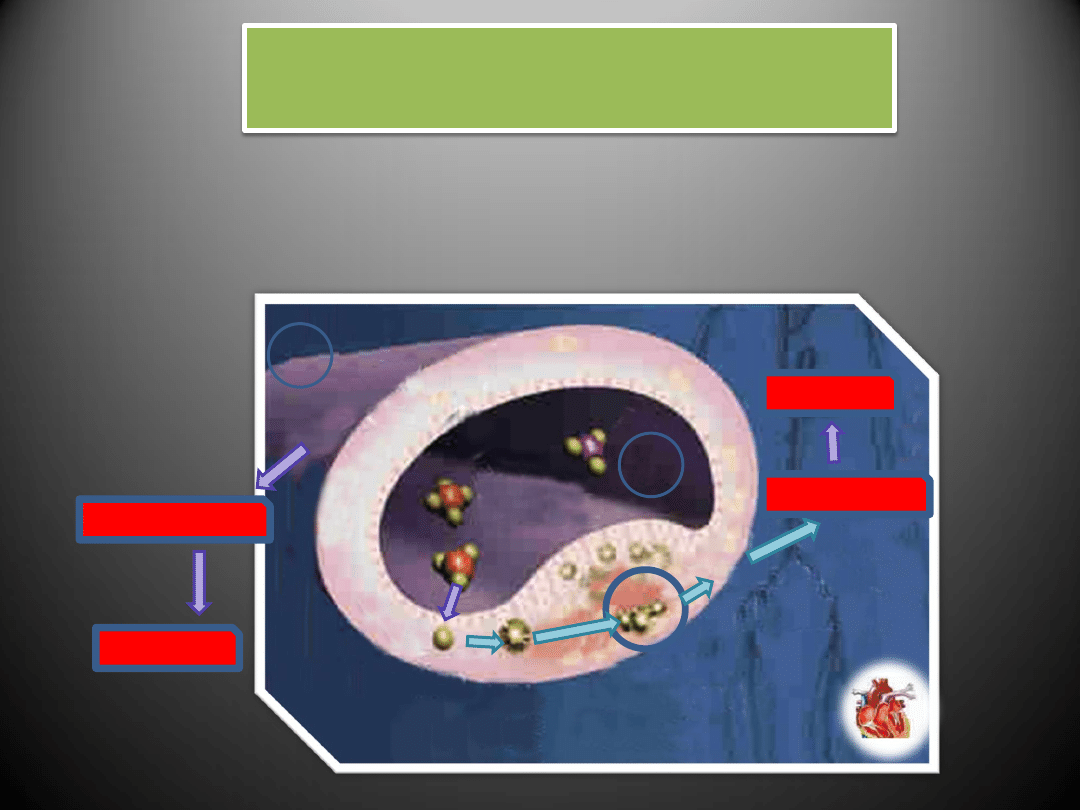
Etapy rozwoju blaszki miażdżycowej
Oksydowane LDL (oxLDL)
nasilają adhezję
przepływających z
prądem krwi
monocytów do komórek
śródbłonka, zwiększając
tym samym skłonnośd
do rozwoju procesów
zakrzepowych.
Ścisły kontakt monocytów z
warstwą śródbłonka hamuje
sekrecję cząsteczek o
własnościach
fibrynolitycznych (np.
tkankowego aktywatora
plazminogenu [tPA]), a z
drugiej strony stymuluje
sekrecję inhibitora aktywatora
plazminogenu (PAI-1)].
Ponadto oxLDL wywierają
bezpośredni toksyczny
wpływ na komórki
śródbłonka – uszkadzając
go oraz zwiększając
przepuszczalnośd dla
komórek zapalnych i
kolejnych cząsteczek LDL.
Pierwsza faza rozwoju miażdżycy
obejmuje deponowanie, kumulację i
następnie utlenianie cząsteczek lipoprotein o niskiej gęstości (LDL cholesterolu)
w komórkach intimy.
Ogniskowo nagromadzone cząsteczki LDL tworzą
naciek tłuszczowy
, który
stanowi wstępną zmianę miażdżycową.
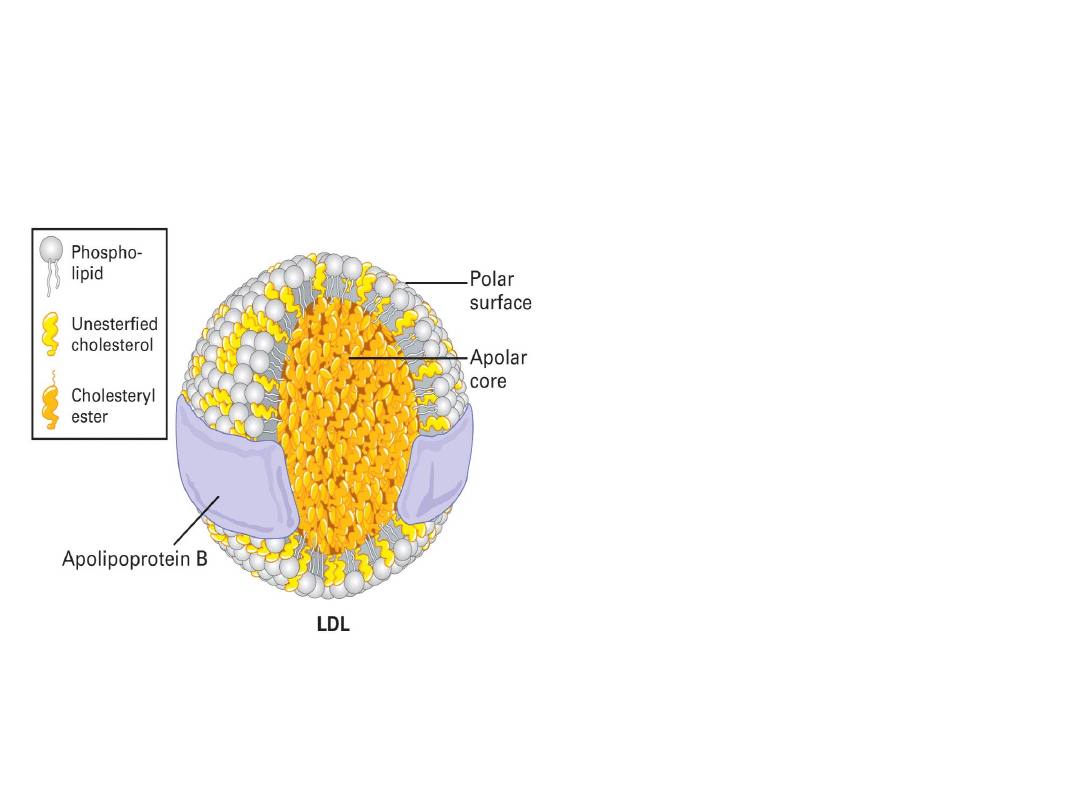
Lipoproteiny osocza są sferycznymi
cząstkami zbudowanymi z
hydrofobowego rdzenia, bogatego w
cholesterol i triglicerydy oraz z płaszcza
utworzonego z apolipoprotein (białka),
fosfolipidów i wolnego
(niezestryfikowanego) cholesterolu.
Gęstośd względna oraz średnica cząstki
zależą od składu chemicznego.
Im większa zawartośd apolipoprotein, tym
mniejsza średnica cząstki (białko
wykazuje duży stopieo upakowania)
oraz wyższa gęstośd względna (białko
posiada dużą masę cząsteczkową w
stosunku do lipidów). Najwyższą
zawartością apolipoprotein cechują się
lipoproteiny o dużej gęstości (HDL), a
najniższą chylomikrony.
LIPOPROTEINY
Głównym składnikiem lipidowym LDL jest
cholesterol w formie zestryfikowanej i
wolnej, który za ich pośrednictwem
przedostaje się do tkanek obwodowych,
służąc tam m.in. do syntezy błon,
prowitaminy D oraz hormonów
steroidowych.
Apolipoproteina B-100 pokonuje drogę od
wątroby, gdzie zostaje wbudowana do
VLDL, poprzez IDL i LDL do tkanek
obwodowych, w których jej interakcja z
receptorem dla apoB/E (receptor
wysokiego powinowactwa) warunkuje
prawidłowy klirens LDL.
Cholesterol LDL (low-density lipoprotein)
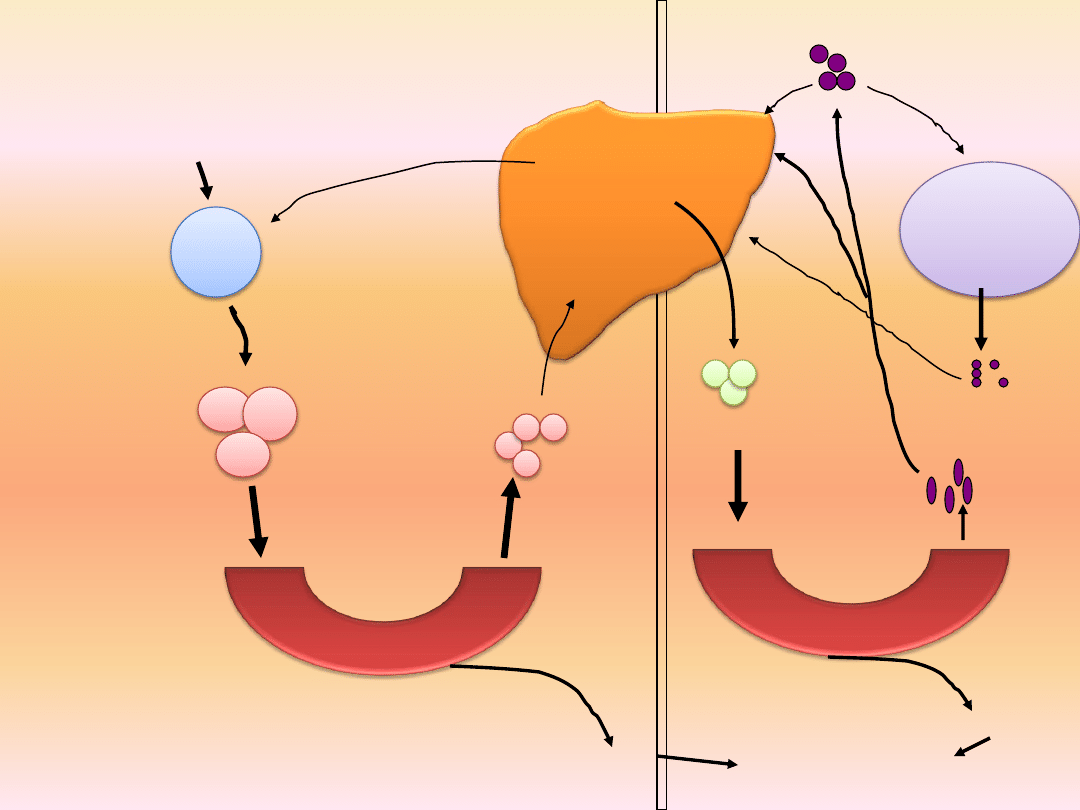
Tłuszcz
pokarmowy
Jelito
cienkie
Naczynia
Lipaza
Lipoproteinowa
WKT
Adipocyty,
mięśnie
chylomikrony
chylomikrony
resztkowe
VLDL
LPL
WKT
Tkanki
poza-
wątrobowe
IDL
HDL
LDL
Endogenny
cholesterol
Egzogenny
cholesterol
Kwasy
żółciowe
Wątroba
Y
Y
Y
Y
LDLR
LDLR
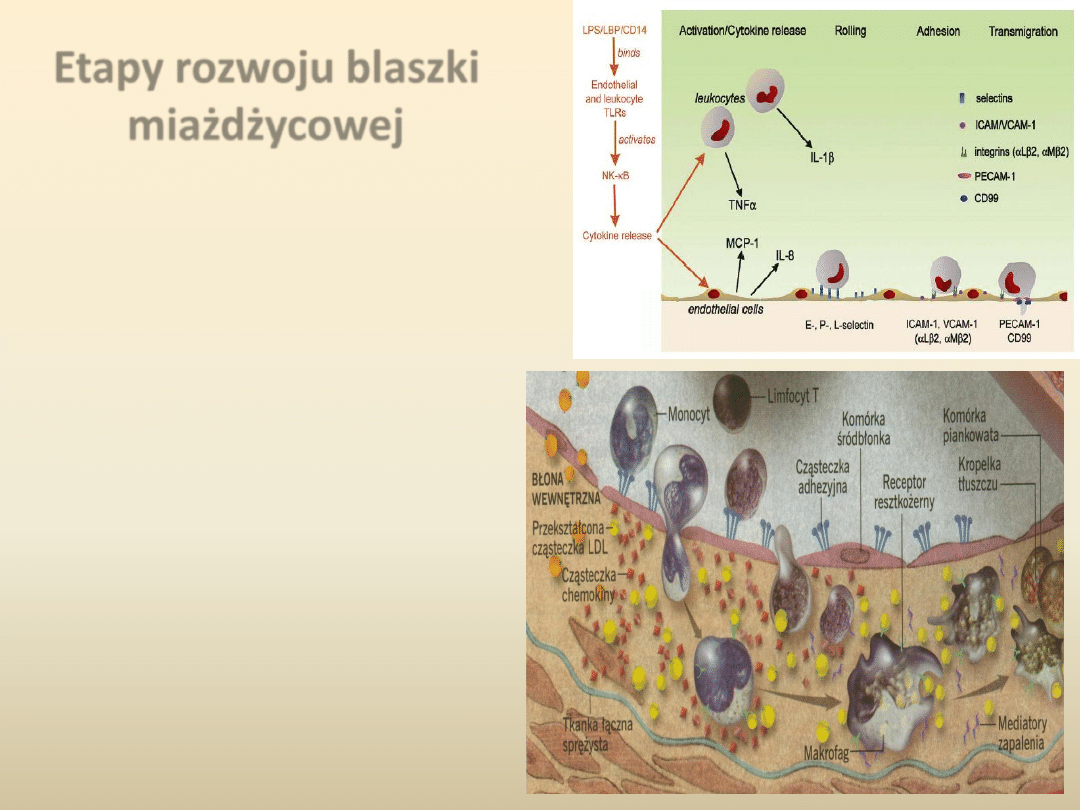
Etapy rozwoju blaszki
miażdżycowej
Druga fazę rozwoju miażdżycy stanowi
rekrutacja komórek zapalnych,
monocytów i limfocytów T, które
przyciągane są do śródbłonka przez
2 rodzaje cząsteczek adhezyjnych
VCAM-1 i ICAM-1. Struktury te
wywołują adhezję monocytów oraz
limfocytów T do warstwy naczynia.
Komórki te migrują następnie z
powierzchni śródbłonka do ściany
tętniczej w towarzystwie innego
zestawu białek, znanych jako
chemotaktyczne cytokiny lub
chemokiny, z których najlepiej
poznano białko chemotaktyczne dla
monocytów- 1 (MCP-1).
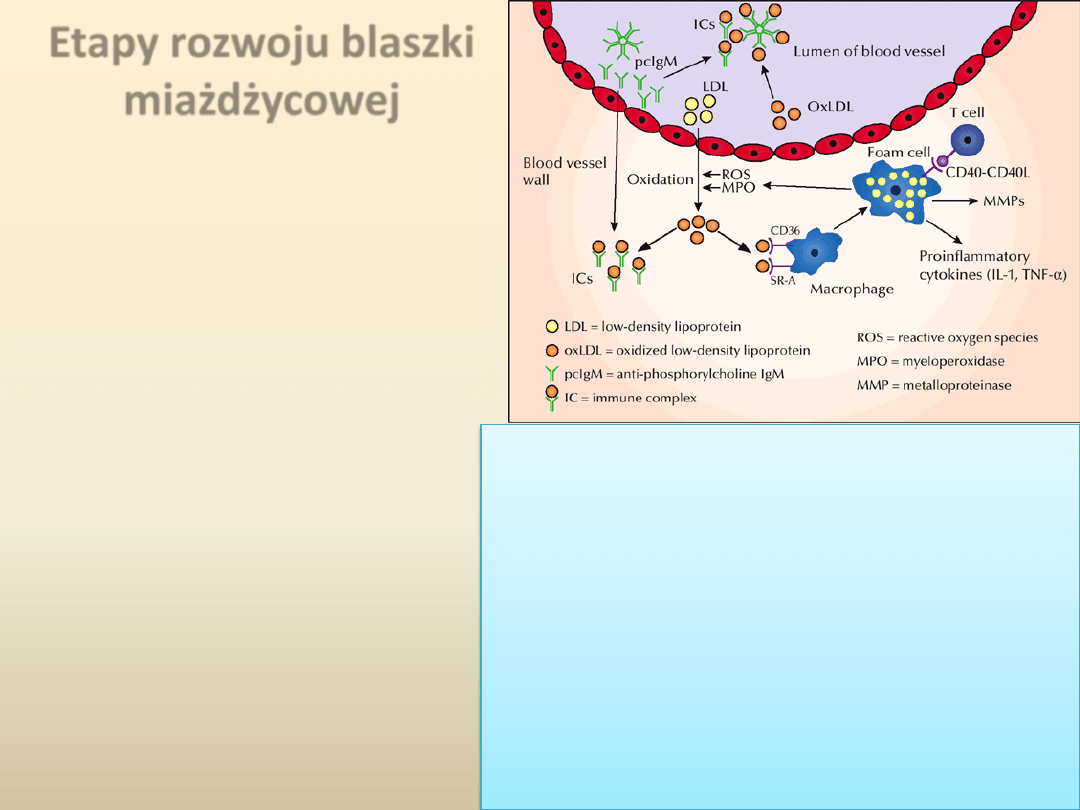
Etapy rozwoju blaszki
miażdżycowej
Trzecią fazę w rozwoju blaszki
miażdżycowej stanowi masywna
migracja monocytów do ściany tętnicy.
Komórki te wykazują ekspresję
receptora o charakterze
scavenger
(zmiatacz),
który wiąże oxLDL.
W następstwie komórki te
przekształcają się w makrofagi i
ulegają podziałom pod wpływem
makrofagowego CSF.
Związane z powierzchnią komórki
oxLDL
pochłaniane są przez
makrofagi, które pod mikroskopem
ujawniają się jako obładowane lipidami
komórki piankowate
, a ich skupienia
widoczne pod warstwą śródbłonka
określa się jako
nacieczenie
tłuszczowe
.
Zmodyfikowane LDL zwiększają następnie aktywność
powstałych komórek piankowatych, a także mogą
powodować rozszerzenie się procesu zapalnego poprzez:
•
stymulację replikacji makrofagów i migrację kolejnych
monocytów do blaszki miażdżycowej
•
wzrost ekspresji metaloproteinaz i hamowanie ekspresji
enzymu syntazy tlenku azotu, odpowiedzialnego za syntezę
tlenku azotu
Uwolnione przez makrofagi mediatory zapalne –
interleukina 1b (IL-1b), czynnik martwicy nowotworow –
TNF-α (
tumor necrosis factor α
) i
M-CSF(czynnik
aktywacji kolonii makrofagów
) nasilają wiązanie LDL do
śródbłonka i mięśniówki naczynia.
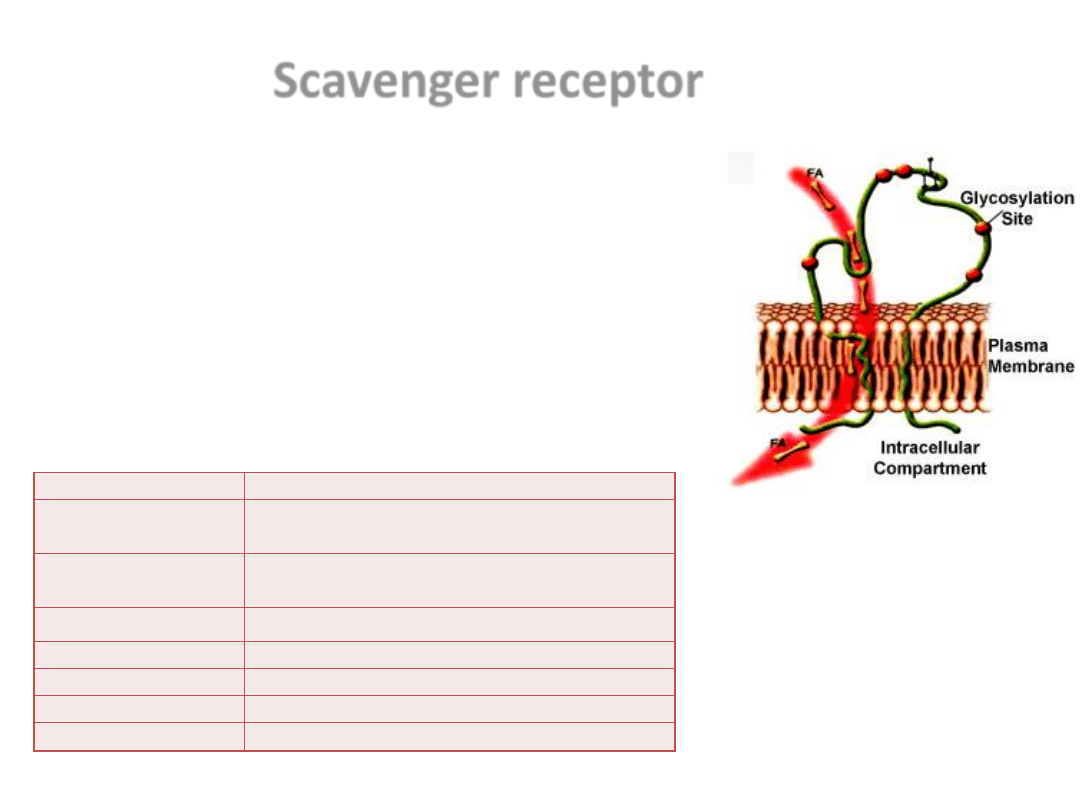
Scavenger receptor
Przyłączanie oxLDL przez makrofagi, poprzedzające ich
fagocytozę odbywa się z udziałem różnorodnych
receptorów znajdujących się na jednojądrowych
komórkach fagocytujących, w tym receptorów typu
scavenger.
Receptory te (SR – scavenger receptor) są grupą białek
wiążących chemicznie lub oksydacyjnie
zmodyfikowane lipoproteiny, polianiony i komórki
ulegające apoptozie.
Receptor CD36
Rodzaj receptora SR
Typ ligandu
SR-AI
SR-AII
acetylowane i utlenione
LDL, polianiony i martwe komórki
CD36
receptor fagocytujący wiąże m.in. acetylowane
i utlenione LDL, fosfatydyloserynę i komórki apoptotyczne
SR-BI
lipoproteiny o dużej gęstości HDL (high density lipoproteins)
SR-CI
acetylowane LDL i polianiony
CD68
(makrosialina) oxLDL
SR LOX-1
oxLDL i polianiony
SR-F
oxLDL, acetylowane LDL i polianiony
Tab.1. Rodzaje receptorów SR zaangażowanych w tworzenie „komórek piankowatych”
Etapy rozwoju blaszki miażdżycowej
Dalszy proces miażdżycowy przebiega z udziałem komórek linii monocytarno-makrofagowej.
Obecne w błonie wewnętrznej oprócz makrofagów liczne limfocyty T aktywują komórki
piankowate, które z kolei wytwarzają wolne rodniki tlenowe oraz czynniki wzrostowe;
•
czynnik wzrostu fibroblastów (FGF, fibroblast growth factor)
•
insulinopodobny czynnik wzrostu (IGF, insulin growth factor)
•
transformujący czynnik wzrostu (TGF, transforming growth factor)
•
interleukinę 1 (IL-1)
•
czynnik martwicy guza (TNF-α, tumor necrosis factor)
Mediatory te stymulują komórki mięśni gładkich do migracji z warstwy mięśniowej do ogniska
miażdżycowego.
Komórki ulęgają tam proliferacji i częściowemu przekształceniu w histiomiocyty produkujące
elementy włókniste tkanki łącznej (elastyna, kolagen).
Komórki m. gładkich naczyo i elementy włókniste tkanki łącznej stopniowo wrastają oraz
otaczają rdzeo lipidowy blaszki, tworząc utwardzony zrąb łącznotkankowy.

Blaszka miażdżycowa
Ukształtowana blaszka miażdżycowa jest zbudowana z włóknistej okrywy, która
zamyka centralnie położony rdzeo. Pokrywa zapewnia strukturalną
integralnośd zmiany miażdżycowej, natomiast rdzeo złożony z
pozakomórkowych lipidów jest miękki i silnie trombogenny.
Destabilizacja blaszki miażdżycowej
Aktywacja komórek zapalnych może w przeważającym stopniu destabilizowad ogniska
miażdżycowe, czego wyrazem są rozpoczynające się destrukcyjne procesy z udziałem
tych komórek, szczególnie w obszarze brzeżnym zmiany miażdżycowej, mogące
osłabiad pokrywę blaszki.
Destabilizacja blaszki
miażdżycowej
limfocyty T
INF-γ
kolagen
proliferacja
INF-γ, TNF-α, IL-1, MCSF, oxLDL,
makrofagi,
komórki mięśni
gładkich
MMP
aktywacja
uwalnianie
degradacja kolagenu, elastyny
Zwiększona aktywnośd
komórek zapalnych →
apoptoza komórek m.
gładkich → zmniejszona
mechaniczna odpornośd
pokrywy blaszki
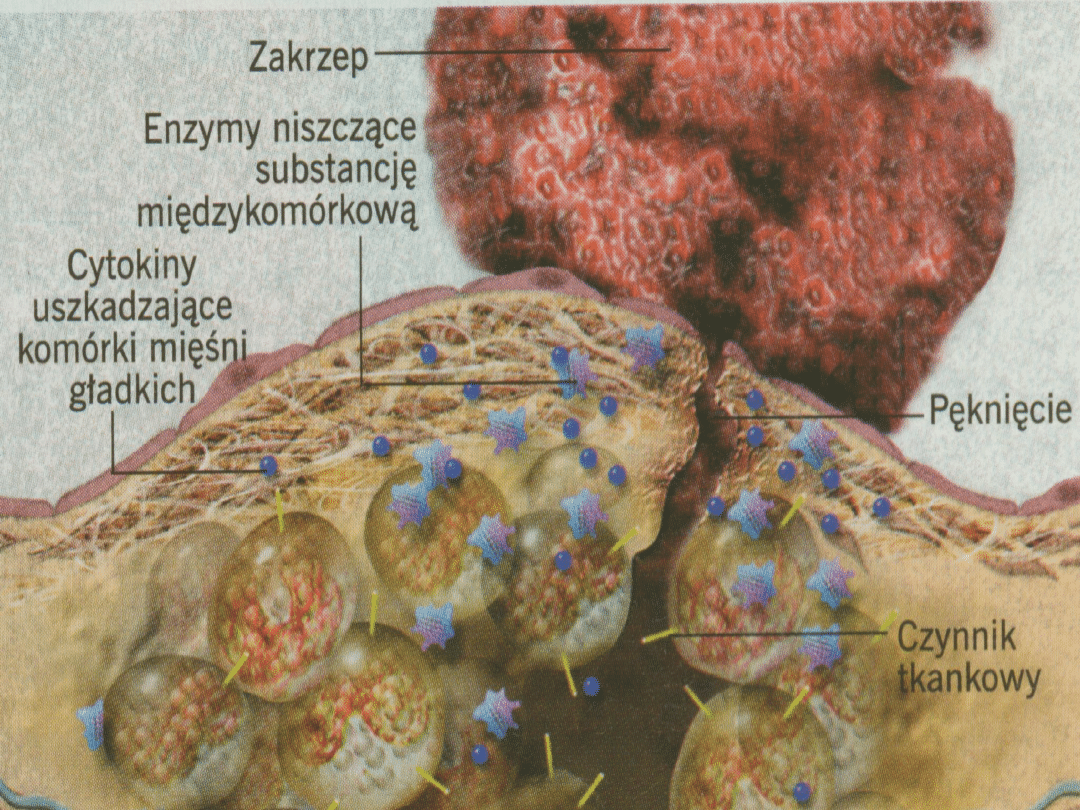
Destabilizacja blaszki miażdżycowej
Pękanie blaszki miażdżycowej
Aktywny
proces działania
enzymów proteolitycznych
uwalnianych z makrofagów na
pokrywę włóknistą
Bierne
pękanie blaszki związane z
naprężeniami okrężnymi i podłużnymi,
działającymi na najcieosze miejsce otoczki
włóknistej, w punkcie przylegania do
„prawidłowej” ściany naczynia
Progresja blaszki miażdżycowej, związana z ewolucją od stabilnej blaszki do ostrego incydentu
wieocowego, zależy od lokalnych i systemowych czynników trombogennych i równowagi między układem
krzepnięcia i fibrynolizy.
Zasadniczymi trombogennymi składnikami blaszki miażdżycowej są kolagen i bogatolipidowy rdzeo z dużą
zawartością czynnika tkankowego. Kiedy włóknista pokrywa blaszki ulega rozerwaniu, ekspozycja
pochodzących z krążącej krwi czynników krzepnięcia (zwłaszcza VIIa i Xa) na czynnik tkankowy, zlokalizowany
w blaszce miażdżycowej, powoduje rozszczepienie protrombiny do trombiny.
Proces ten inicjuje krzepnięcie, deponowanie fibryny i aktywację płytek, prowadząc ostatecznie do
wytworzenia zakrzepu. Adhezja płytek i ich aktywacja powoduje uwalnianie tromboksanu A2, serotoniny,
ADP, PAF, trombiny i czynnika tkankowego, stymulujących dalsze gromadzenie się płytek, narastanie zakrzepu i
w koocu zamknięcie zwężonej tętnicy.
Procesy reparacyjne w stabilizacji blaszek
miażdżycowych
Komórki uszkodzonego śródbłonka, płytki krwi i makrofagi uwalniają
płytkopochodny czynnik wzrostu
(PDGF, platelet - derived growth factor )
wywołujący silne właściwości proliferacyjne. Wszystkie czynniki wzrostowe
rozpoczynają procesy naprawcze, które przeważają w tym okresie rozwoju
blaszki.
Zgodnie z obecnymi poglądami o stabilności blaszek miażdżycowych decyduje
przewaga procesów reparacyjnych nad zapalnymi
. W wyniku procesów
reparacyjnych blaszka zwiększa swoją całkowitą objętośd, ale dzięki
uzyskanej sztywności i stłumieniu odczynu zapalnego może wykazywad
mniejsza skłonnośd do pękania.
Wtórne zmiany blaszek miażdżycowych
Wraz z rozwojem zmian miażdżycowych w obrębie blaszki powstają liczne
siatki drobnych naczyo krwionośnych połączonych
z vasa vasorum tętnic.
Przez te naczynia leukocyty penetrują i opuszczają ogniska miażdżycowe.
Naczynia te są również źródłem krwotoków do wnętrza blaszki.
Utworzone blaszki miażdżycowe często gromadzą
wapo
. Za jego kumulacje
odpowiedzialne są wyspecjalizowane białka wiążące wapo np.:
osteokalcyna, osteopontyna i morfogenetyczne białka, które, podobnie jak
w kości, lokalizują się w zmianie miażdżycowej.
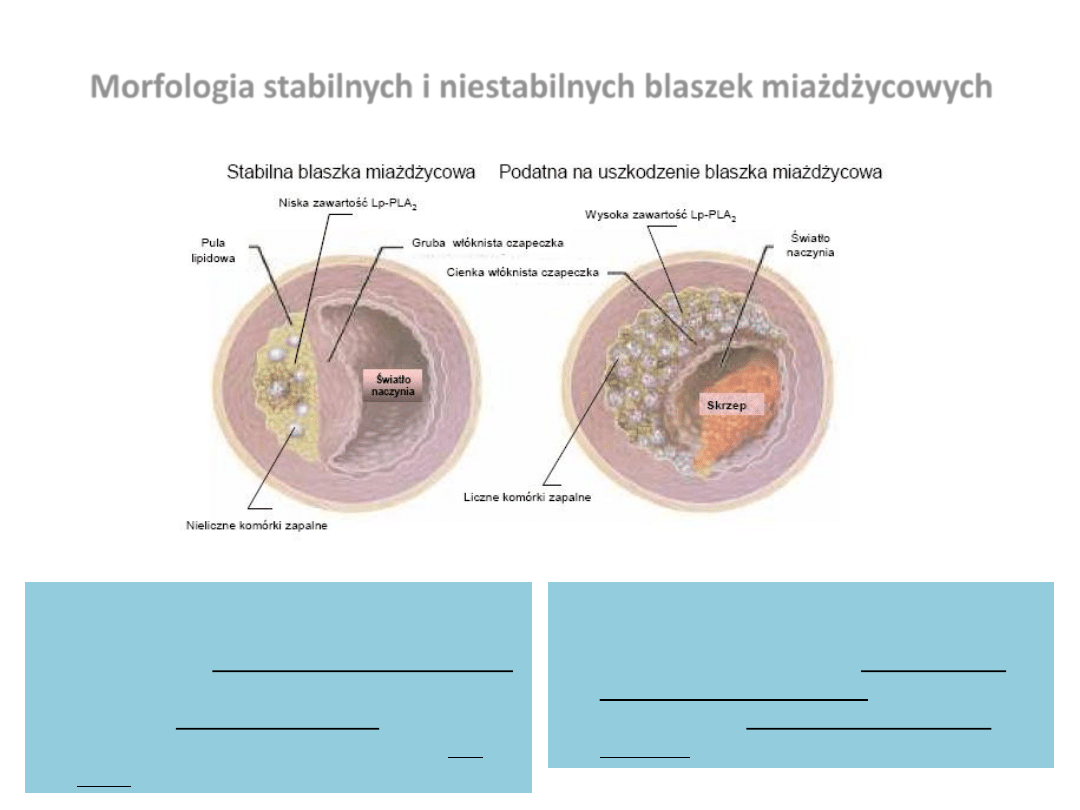
Morfologia stabilnych i niestabilnych blaszek miażdżycowych
Blaszki miażdżycowe u pacjentów ze stabilną
dławicą piersiową są zazwyczaj nieaktywne
biologicznie z przewagą komórek m. gładkich
nad makrofagami, limfocytami T i komórkami
tucznymi. Ryzyko ich pęknięcia bądź
owrzodzenia i wytworzenia skrzepliny jest
małe.
Blaszki miażdżycowe w ostrych zespołach
wieocowych cechują się zwiększoną
aktywnością biologiczną, z gromadzeniem
licznych komórek zapalnych i dużą
gotowością do gwałtownego zamknięcia
naczynia.
koncentryczne zwężanie naczynia
ekscentryczne zwężanie naczynia
Podłoże zapalne miażdżycy
W ostatnich latach wyniki wielu badao coraz wyraźniej wskazują, że miażdżyca
jest procesem o
charakterze zapalnym
.
Koncepcję roli przewlekłego procesu zapalnego w miażdżycy potwierdza
szereg obserwacji:
miażdżycy towarzyszą podwyższone poziomy parametrów zapalnych,
zwłaszcza białka C-reaktywnego (CRP)
miażdżycowe tętnice produkują szereg enzymów hydrolitycznych,
molekuły adhezyjne, cytokiny i czynniki wzrostu podobnie jak obserwuje
się to w innych przewlekłych stanach zapalnych
komórki stwierdzane we wczesnych zmianach miażdżycowych (monocyty,
makrofagi, limfocyty T) są komórkami typowo zapalnymi
Istnieją przekonujące kliniczne i eksperymentalne dowody, że podczas tego
przewlekłego procesu zapalnego nasila się produkcja reaktywnych
pochodnych tlenu (ROS), wynikająca z braku równowagi pomiędzy syntezą
ROS a neutralizującymi antyoksydatywnymi mechanizmami obronnymi.
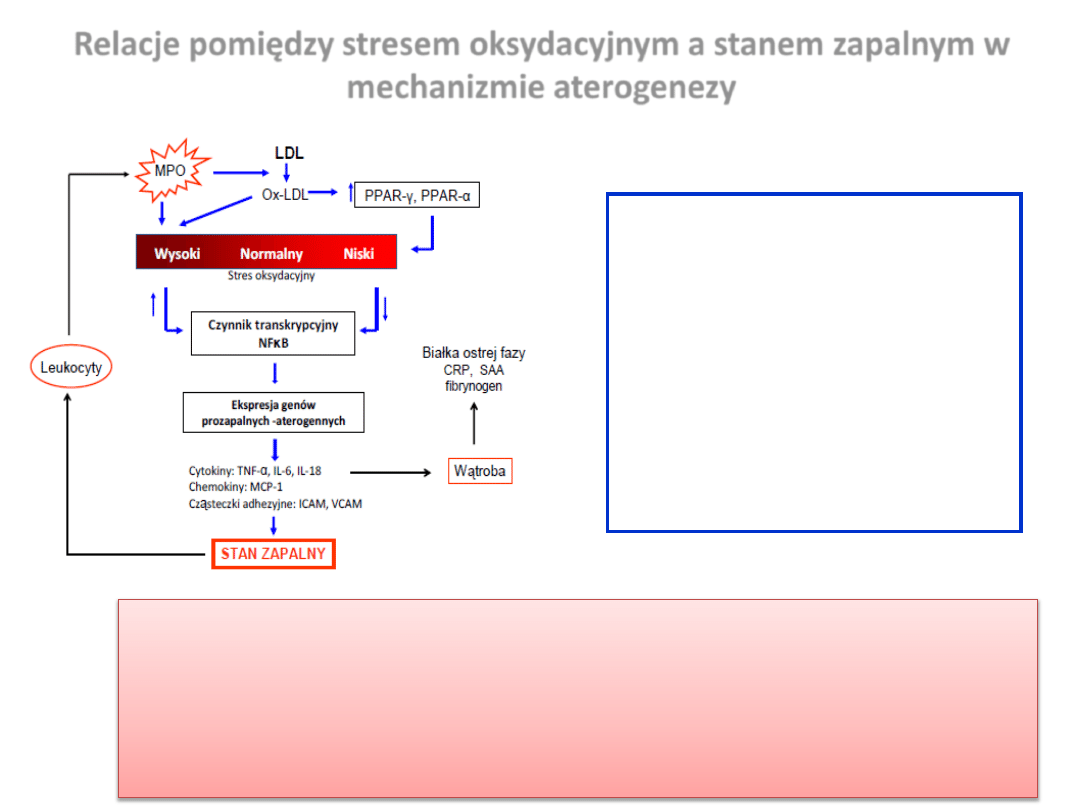
Relacje pomiędzy stresem oksydacyjnym a stanem zapalnym w
mechanizmie aterogenezy
Stres oksydacyjny i reaktywne formy tlenu
(RFT) stanowią drugorzędowy przekaźnik
sygnałów zaangażowanych w regulację
ekspresji prozapalnych genów i ich
produktów takich jak: cytokiny i
molekuły adhezyjne.
Zwiększona ekspresja tych genów promuje
infiltrację monocytów do ściany
naczynia.
Proces ten prowadzi do nasilenia lokalnego
stanu zapalnego i w konsekwencji do
dysfunkcji śródbłonka naczyniowego.
Kluczowym elementem tego mechanizmu wydaje się byd jądrowy czynnik transkrypcyjny NFκB , należący
do rodziny czynników transkrypcyjnych wrażliwych na stres oksydacyjny. Jest on aktywowany przez wiele
różnych czynników, wśród których najważniejsze wydają się koocowe produkty peroksydacji lipidów oraz
prozapalne cytokiny.
Z drugiej strony stymulacja receptorów aktywowanych przez proliferator peroksysomów (PPAR), hamuje
ekspresję prozapalnych genów kodujących cytokiny, metaloproteinazy macierzy (MMP) oraz białka ostrej
fazy poprzez regulację aktywności NFκB
Zakażenie w etiologii miażdżycy
Badania serologiczne, a także niektóre obserwacje epidemiologiczne sugerują, że w aterogenezie pewną rolę
odgrywad mogą rolę bakterie lub wirusy:
• Chlamydia pneumoniae
• Helicobacter pylori
• cytomegalowirusy
• wirusy Herpes simplex i wirusy Hepatitis A
W ludzkich blaszkach miażdżycowych wykrywano obecnośd tych mikroorganizmów, a w surowicy pacjentów z
chorobą wieocową stwierdzano skierowane przeciwko nim przeciwciała.
Możliwe mechanizmy aterogenne związane z zakażeniem obejmują, m.in.:
•
bezpośrednie patogenne inwazje komórek tętnic
•
przewlekłe zakażenia innych obszarów ustroju, prowadzące do produkcji mediatorów
aterogenezy
•
mechanizmy o charakterze autoimmunologicznym, wywołane przez wcześniejszą ekspozycję
patogenów na ustrojowe systemy odpornościowe
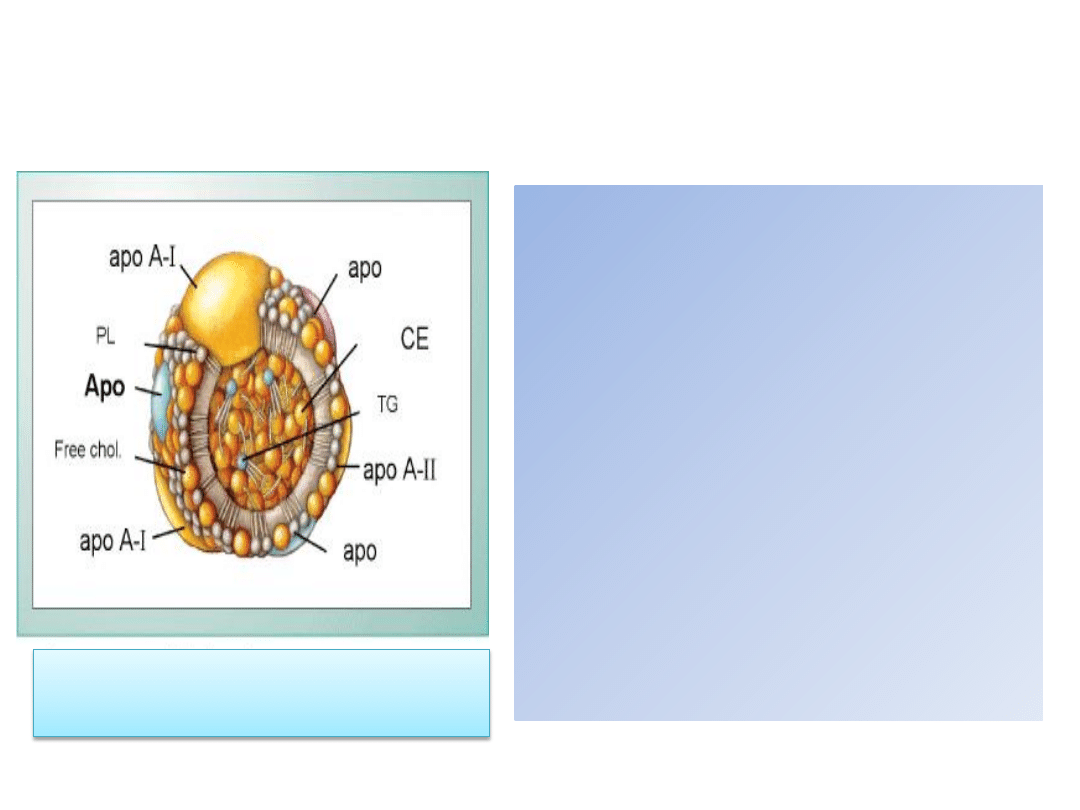
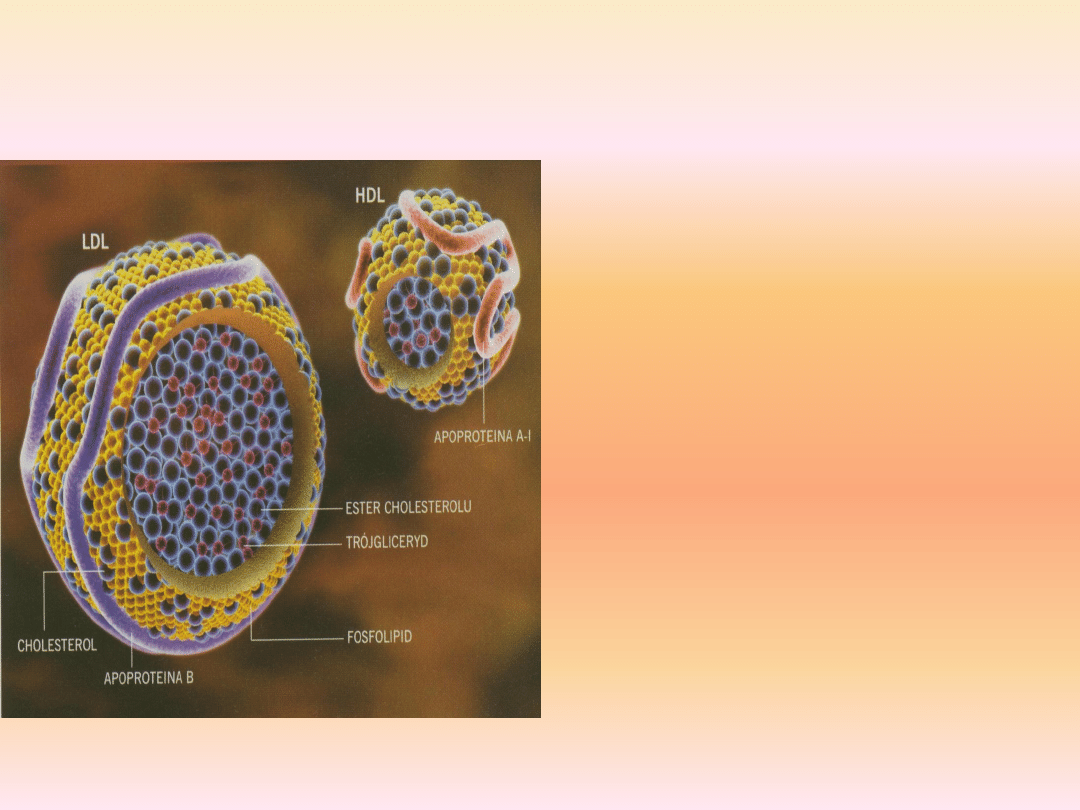
Cholesterol HDL (high-density lipoprotein)
Lipoproteiny o wysokiej gęstości stanowią
frakcję cholesterolu uzyskiwaną w wyniku
ultrawirowania lipoprotein surowicy.
Wysoka gęstośd HDL wiąże się ze znaczną
zawartością apolipoprotein (stanowiącą aż
55% całej cząsteczki), w skład których
wchodzą: apo A-I, apo A-II, apo C-III, apo
C-I, apo D.
Pozostała częśd cząsteczki HDL to lipidy (45%),
w tym: 25% to fosfolipidy, 20% —
cholesterol, a 5% — triglicerydy.
Obecnośd wolnego cholesterolu w rdzeniu
cząsteczek HDL zwiększa ich zdolnośd do
wiązania cholesterolu z innych lipoprotein.
Apo = apolipoproteina; PL =fosfolipidy; chol =
cholesterol; CE= cholesterol zestryfikowany; TG =
triglicerydy
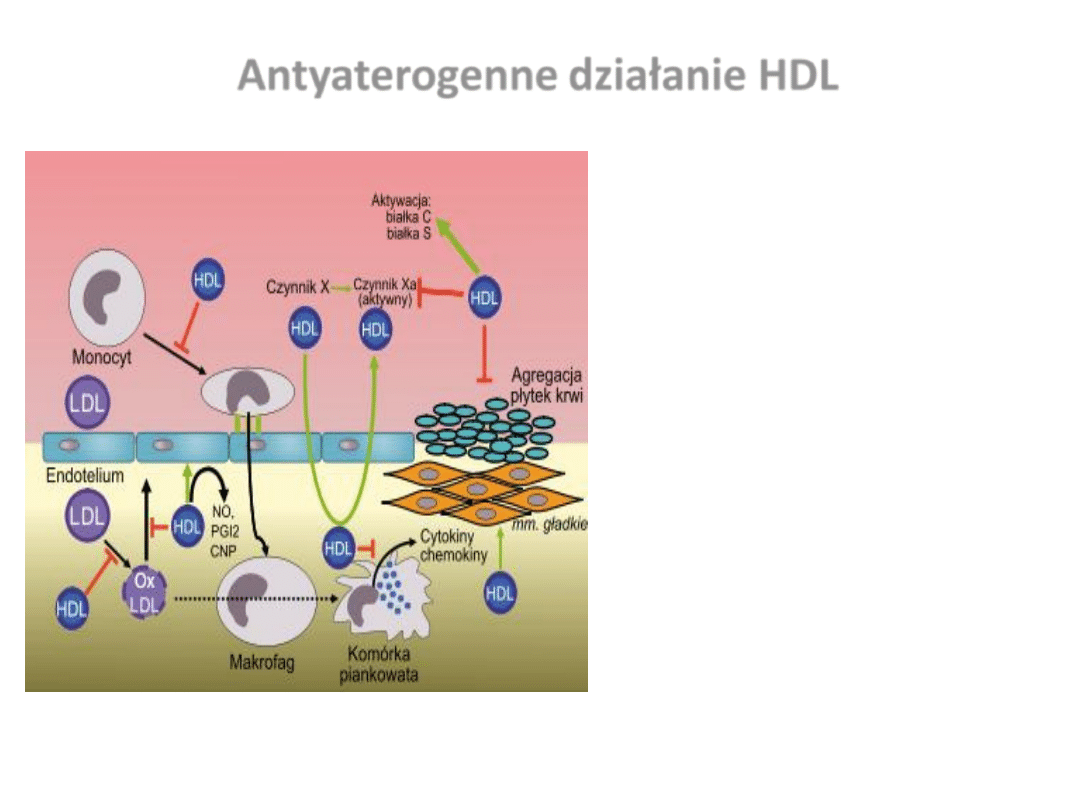
Antyaterogenne działanie HDL
•
Zwrotny transport cholesterolu z
tkanek obwodowych do wątroby
•
Działanie przeciwzapalne - inhibicja
adhezji i aktywacji monocytów (↓
ICAM-1,VCAM-1, selektyna E)
•
Działanie antyoksydacyjne (↓oxLDL)
•
Działanie ochronne i modulujące w
stosunku do śródbłonka (↑NO
2
, PGI
2
,
CNP, hamowanie apoptozy, układu
dopełniacza)
•
Działanie przeciwpłytkowe
(hamowanie wiązania Fg, hamowanie
uwalniania ziarnistości płytkowych,
↑NO
2
,)
•
Działanie fibrynolityczne i
przeciwzakrzepowe (↓ powstawania
kompleksu tenazy i protrombinazy,
↓czynnika X, ↑ białko C i S

• dieta
• palenie tytoniu
• aktywnośd fizyczna
Elementy stylu życia
• ciśnienie tętnicze
• stężenie cholesterolu całkowitego
• stężenie cholesterolu LDL
• stężenie cholesterolu HDL
• stężenie trójglicerydów
• stężenie glukozy/cukrzyca
• nadwaga/otyłośd
• czynniki prozakrzepowe
• markery przewlekłego procesu zapalnego
Czynniki biochemiczne i
fizjologiczne
• wiek
• płed
• wywiad rodzinny (przedwczesne występowanie chorób sercowo-
naczyniowych)
• choroba sercowo-naczyniowa w wywiadzie
• markery genetyczne
Czynniki indywidualne
Czynniki ryzyka miażdżycy
Mechanizmy niewydolności krążenia-
rola układu neurohormonalnego serca
Zakład Nadciśnienia Tętniczego
Katedry Nefrologii i Nadciśnienia Tętniczego
Uniwersytetu Medycznego w Łodzi

Niewydolnośd serca
Zespół kliniczny spowodowany nieprawidłowością serca o
charakterystycznym okresie hemodynamicznym, któremu
towarzyszy upośledzenie funkcji nerek oraz odpowiedź układu
nerwowego i humoralnego.
wg Poole-Wilsona 1985
Niewydolnośd serca jest stanem, w którym serce jako pompa nie
zabezpiecza odpowiedniego do potrzeb ustroju dopływu krwi do tkanek i
narządów, mimo prawidłowego wypełnienia łożyska naczyniowego.
wg Steada D. 1948
Niewydolnośd
serca
objętośd
wyrzutowa
pojemnośd
minutowa
Objętośd wyrzutowa
kurczliwośd mięśnia sercowego
obciążenie wstępne (wielkośd ciśnienia
późnorozkurczowego)
obciążenie następcze (wielkośd ciśnienia
skurczowego w komorach i aorcie lub tętnicy
płucnej)
częstośd i miarowośd rytmu serca
Skurczowa (spadek frakcji
wyrzutowej)
przewlekła
Rozkurczowa (zaburzenie
relaksacji komory)
ostra
Prawokomorowa
Lewokomorowa
Niewydolnośd serca
Niewydolnośd prawokomorowa
•
powikłanie niewydolności lewokomorowej
•
pierwotne przeciążenie prawej komory (nadciśnienie płucne, izolowana niedomykalnośd
zastawki trójdzielnej powodująca napływ dodatkowej krwi do prawej komory podczas
rozkurczu, zaciskające zapalenie osierdzia.
•
zawał prawej komory
Niewydolnośd lewokomorowa
•
stan po zawale mięśnia sercowego
•
nadciśnienie tętnicze,
•
wada mitralna, aortalna
•
choroba niedokrwienna serca
Ostra niewydolnośd serca
•
może byd wywołana nagle pojawiającym się upośledzeniem kurczliwości serca (np. zawał
serca), masywny zator tętnicy płucnej, jego nasilenia (np. nagły wzrost ciśnienia tętniczego)
bądź nałożenia się dodatkowych czynników (np. częstoskurcz) na już istniejące przeciążenie
hemodynamiczne serca.
•
objawami ciężkiej niewydolności serca są obrzęk płuc i wstrząs kardiogenny
Etiologia niewydolności serca
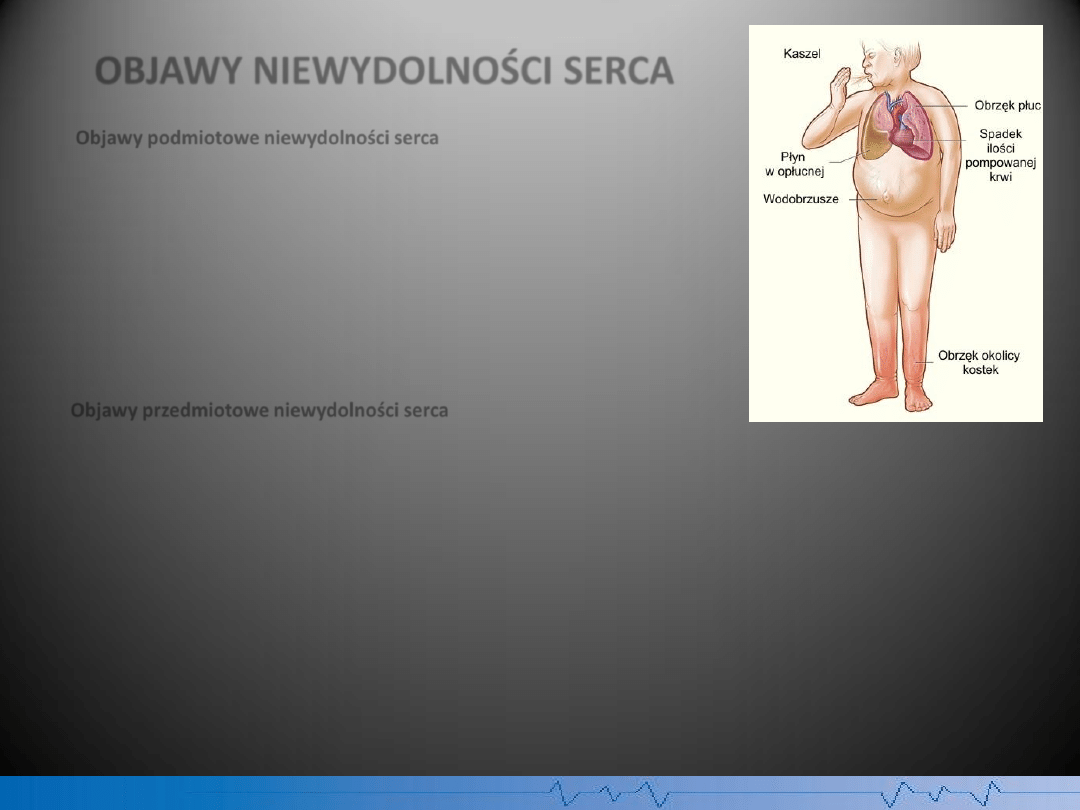
OBJAWY NIEWYDOLNOŚCI SERCA
Objawy podmiotowe niewydolności serca
* pogorszenie tolerancji wysiłku
* dusznośd, dychawica sercowa
* kaszel
* nocne oddawanie moczu, skąpomocz
* obrzęki
* ból brzucha
Objawy przedmiotowe niewydolności serca
* bladośd skóry, ochłodzenie skóry, sinica
* nadmierne wypełnienie żył szyjnych (związane z podwyższonym ciśnieniem żylnym), objaw wątrobowo-
szyjny (często nieobecne w ciężkiej niewydolności serca)
* powiększenie wątroby
* tachykardia (mało swoisty objaw)
* trzeci ton serca (nieswoisty objaw ciężkiej niewydolności serca)
* stwierdzane osłuchowo trzeszczenia nad polami płucnymi (objaw zastoju w płucach): objaw nieswoisty, o
małej wartości prognostycznej
* płyn w jamie opłucnej i jamie otrzewnej
* oddech Cheyne'a-Stokesa (oddech periodyczny)
Rys. Objawy niewydolności prawokomorowej serca.

Niewydolnośd serca
Niewydolnośd spowodowana uszkodzeniem funkcji mięśnia serca charakteryzuje się
zazwyczaj
zredukowanym przepływem krwi w tkankach.
Istotą niewydolności serca nie są
doraźne zaburzenia hemodynamiczne, lecz postępujące pogarszanie się sprawności serca.
Przeciążenie ciśnieniowe lewej komory prowadzi do
przerostu
i następowego
osłabienia
,
bądź ostrego wypadnięcia
czynności skurczowej
znacznego obszaru komory, co w
konsekwencji prowadzi do nadmiernego wydzielania katecholamin, podwyższonego
poziomu angiotensyny II i wazopresyny oraz wzrostu wydzielania mineralokortykoidów.
W wyniku tych procesów następuje uszkodzenie mięśnia sercowego, a zmiany w głównej mierze
dotyczą:
•
degeneracji kardiomiocytów
•
przebudowy zrębu łącznotkankowego (włóknienie)
•
zmniejszenia łożyska naczyniowego
•
wyczerpania mechanizmów adaptacyjnych mięśnia serca
•
spadku zasobów noradrenaliny w włóknach mięśniowych
•
spadku gęstości beta-adrenoreceptorów
•
obniżenia wrażliwości na działanie katecholamin
•
obnizeniu odpowiedzi inotropowej włókien mięśniowych
Efektem tego jest niezdolnośd do poprawy wydolności serca.
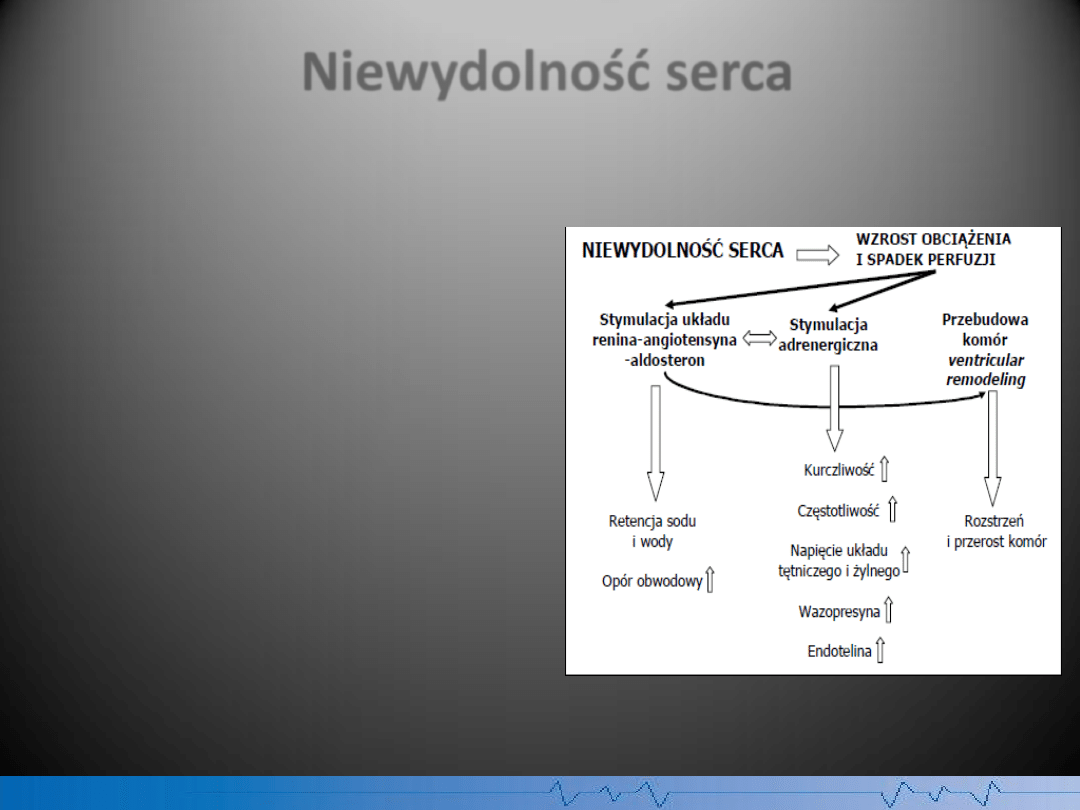
Niewydolnośd serca
Układy regulacyjne, których aktywacja
odgrywa istotną rolę w patogenezie
niewydolności serca, to
układ współczulny
i
układ renina-angiotensyna-aldosteron
(RAA). Efektem ich oddziaływania jest
szereg niekorzystnych zmian
hemodynamicznych w przebiegu
niewydolności serca (wzrost oporu
obwodowego, zwiększenie wolemii,
tachykardia, wzrost naprężenia ściany) oraz
liczne zaburzenia humoralne i
metaboliczne.
Trzecim bardzo istotnym narządem, którego
dysfunkcja odgrywa rolę w patogenezie
niewydolności serca jest śródbłonek.
Dysfunkcja śródbłonka
objawia się między
innymi przesunięciem równowagi pomiędzy
jego właściwościami wazorelaksacyjnymi,
zależnymi od tlenku azotu, a
wazokonstrykcyjnymi, zależnymi od
endoteliny.
Hipoteza zapalna niewydolności serca
•
Badania ostatnich lat wskazują, że przewlekłą niewydolnośd krążenia traktowad
można jako przewlekły proces zapalny, w którym istotną rolę odgrywają cytokiny
prozapalne.
•
Spośród wielu cytokin (interleukiny 1,2,3,4,6, TNF-α, interferon γ), wskazuje się na
kluczową rolę czynnika martwicy nowotworów (TNF-α) oraz interleukiny 1 i 6 (IL-1,
IL-6) w patogenezie niewydolności serca.
•
W badaniu SOLVD (Studies of Left Ventricular Dysfunction) stężenie TNF-α
korelowało z nasileniem objawów wg klasyfikacji NYHA (New York Heart
Association), a wysokie jego stężenie stanowiło niezależny czynnik prognozujący
zgon.
•
TNF-α wywiera biologiczne działania, które mogą odpowiadad za powstawanie
objawów niewydolności serca.
•
Do potencjalnych mechanizmów uszkodzenia i powstawania NS indukowanej przez
czynnik martwicy nowotworów należy m.in.: upośledzenie funkcji skurczowej lewej
komory, obrzęk płuc, indukowany procesu przebudowy, rozprężenie beta-receptora
z cyklazą adenylową, zaburzenie procesów energetycznych w mitochondriach,
apoptoza kardiomiocytów oraz komórek endotelium.
Hipoteza cytokinowa
•
Hipoteza cytokinowa niewydolności serca zakłada, że nadmierna ekspresja cytokin
zarówno w obrębie tkanek mięśnia sercowego, jak również w krążeniu ogólnym i
tkankach obwodowych powoduje powstawanie lub nasilenie już istniejących
zaburzeo i objawów niewydolności serca.
•
Istnieje kilka potencjalnych źródeł, mogących generowad cytokiny w przebiegu
niewydolności serca.
•
Najbardziej oczywista jest hipoteza sugerująca produkcję cytokin przez układ
immunologiczny w odpowiedzi na uszkodzenie tkanek serca.
•
Inna sugeruje możliwośd syntezy cytokin (np. IL-6) w niedokrwionych tkankach
obwodowych.
•
Kolejna koncepcja opiera się na założeniu aktywacji syntezy cytokin przez układ
odpornościowy w wyniku zwiększonego przenikania do krążenia endotoksyn z
przewodu pokarmowego.
•
Wreszcie ostatnia hipoteza wiąże zwiększone wydzielanie cytokin z aktywacją układu
adrenergicznego, która ma miejsce w niewydolności serca. Wiąże się to z obserwacją,
że podwyższone stężenia cAMP (cyklicznego adenozynomonofosforanu) powodują
zwiększoną ekspresję niektórych mediatorów zapalnych.
Dysfunkcja śródbłonka w przebiegu
niewydolności serca
•
Kluczowym zaburzeniem w patofizjologii niewydolności serca wydaje się ogrywad dysfunkcja
śródbłonka.
•
Do mechanizmów, za pomocą których cytokiny powodują dysfunkcję śródbłonka, należy: zaburzona
synteza tlenku azotu (NO), nasilenie stresu oksydacyjnego oraz uruchomienie procesu apoptozy.
•
Podstawowym objawem zaburzonej czynności śródbłonka jest upośledzenie jego właściwości
wazorelaksacyjnych, wyrażające się m.in. zaburzoną reakcją wazodilatacyjną na acetylocholinę, przy
zachowanej reakcji na podanie egzogennego donora NO.
•
Stres oksydacyjny u chorych z niewydolnością serca pojawia się jako wynik nadmiernej syntezy
wolnych rodników tlenowych lub w wyniku upośledzenia komórkowych mechanizmów
antyoksydacyjnych. Jest jednym z mechanizmów remodelingu serca w przebiegu niewydolności,
prowadzi także do apoptozy komórek miokardialnych oraz komórek endotelium, przyczyniając się do
postępu choroby.
•
Mięsieo sercowy podlega wzmożonemu stresowi oksydacyjnemu zarówno w wyniku niedokrwienia,
jak i w momencie reperfuzji.
•
Jedna z hipotez zakłada, że zaburzenie równowagi pomiędzy produkcją wolnych rodników a
mechanizmami antyoksydacyjnymi odpowiada za przejście od fazy przerostu do fazy dekompensacji
serca w procesie powstawania niewydolności serca.
•
Zarówno angiotensyna II, jak i niektóre cytokiny nasilają produkcję wolnych rodników tlenowych,
zwiększając tym samym stres oksydacyjny. Aktywacja układu RAA, będąca skutkiem upośledzenia
wydolności serca, prowadzi do zwiększonej produkcji wolnych rodników tlenowych.
•
W wyniku powstania stresu oksydacyjnego dochodzi do aktywacji genów zależnych od potencjału
oksydo-redukcyjnego. Częśd z tych genów odpowiada za produkcję prozapalnych cytokin (IL-6), co
prowadzi do nasilenia stresu oksydacyjnego tworząc w mechanizmie dodatniego sprzężenia
zwrotnego klasyczne "błędne koło" postępującej dysfunkcji narządu.

Układ renina-angiotensyna-aldosteron (RAAS)
Układ renina-angiotensyna-aldosteron bierze udział w patogenezie wielu chorób układu sercowo-
naczyniowego włączając:
•
zawał mięśnia sercowego
•
hipertrofie
•
kardiomiopatie
•
migotanie przedsionków
•
zastoinową niewydolnośd serca
•
miażdżyca
System RAAS działa jak układ wewnątrzwydzielniczy.
Renina jest syntetyzowana w postaci proreniny, przechowywana w ziarnistościach i uwalniana do krążenia
w nerkach, gdy pojawią się następujące bodźce:
•
obniżenie ciśnienia transmuralnego w tętniczkach doprowadzających
•
zmniejszenie zawartości sodu w plamce gęstej nefronu
•
pobudzenie receptorów beta-adrenergicznych aparatu przykłębuszkowego
•
obecnośd prostaglandyny E2, I2 oraz kininy
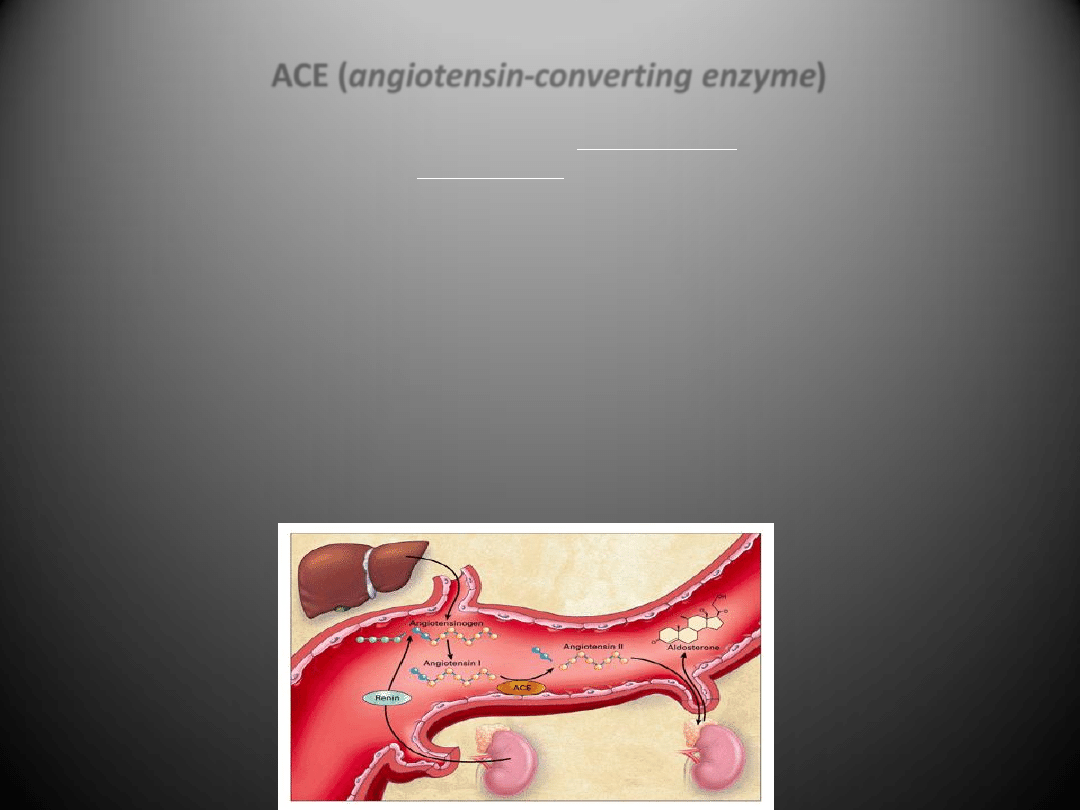
Krążąca renina katalizuje konwersję
angiotensynogenu
do
angiotensyny I
, przekształcanej dalej do
angiotensyny II
.
Układ renina-angiotensyna-aldosteron (RAAS) odgrywa istotną rolę w regulacji
systemu fibrynolitycznego
,
gdyż jak zostało udowodnione zarówno w badaniach in vitro jak i in vivo, angiotensyna II zwiększa
poziom PAI-1, który jest głównym fizjologicznym inhibitorem fibrynolizy
.
Produkcja zarówno ACE jak i
angiotensyny jest znacznie zwiększona miejscu, w którym naczynie jest uszkodzone. Składniki systemu
RAA występują lokalnie w wielu tkankach, gdzie pełnią
rolę prozapalną
i
sprzyjają zwłóknieniu
.
ACE (angiotensin-converting enzyme)
•
Enzym ACE jest odpowiedzialny za powstawanie z angiotensyny I aktywnego
wazokonstrykcyjnego peptydu - angiotensyny II. Odcina on od angiotensyny I C-koocowy
dipeptyd His-Leu
•
ACE może także działad na bradykininę odcinając z jej C-kooca dipeptyd Phe-Arg, co prowadzi do
jej inaktywacji
•
ACE jest głównie białkiem powierzchniowym, jego produkcja w zdrowym sercu jest niewielka i
ogranicza się głównie do śródbłonka naczyo, wzrasta natomiast znacznie w przeroście i
niewydolności serca
•
Może on występowad w tkankach w formie związanej z błoną komórkową lub w formie
rozpuszczalnej w płynach ustrojowych
•
ACE odgrywa znaczącą rolę w regulacji napięcia naczyo, utrzymywaniu homeostazy, zwiększa
aktywację i agregację płytek krwi, oraz wpływa na remodeling naczyniowy

Znaczenie ACE w chorobach układu krążenia
•
Niekorzystny wpływ tego enzymu na układ krążenia jest dwojaki. Poza generowaniem
angiotensyny II
, ACE
hydrolizuje bradykininę
do nieaktywnych związków znosząc jej korzystny
wpływ na śródbłonek naczyo
(bradykinina stymulująca produkcję i wydzielanie prostacykliny,
tlenku azotu i tkankowego aktywatora plazminogenu t-PA, ma działanie wazorelaksacyjne,
przeciwpłytkowe i fibrynolityczne)
•
Zwiększone stężenie ACE może prowadzid do zwiększonego wytwarzania angiotensyny II i w
konsekwencji do rozwoju miażdżycy. Zwiększoną ekspresję ACE odnotowano w makrofagach
kumulujących się wokół osłabionej pokrywy włóknistej blaszki. Przypuszcza się, że wyższa
aktywnośd tkankowa ACE może przyczyniad się do zmniejszenia stabilności blaszki miażdżycowej.
•
Zwiększoną ekspresję i aktywnośd ACE zaobserwowano także w obrębie „przerośniętej” komory
serca oraz w komórkach mięśni gładkich naczyo szczurów z uszkodzonym śródbłonkiem.
•
Udział ACE w patogenezie niewydolności serca potwierdzają liczne badania. Stosowanie
inhibitorów ACE (ACE-I)
przyczynia się do zmniejszenia częstości występowania incydentów
niedokrwiennych
oraz do poprawy funkcjonowania śródbłonka. Inhibitory ACE wpływają
pozytywnie na funkcjonowanie śródbłonka, prawdopodobnie przez to, że zmniejszają poziom
angiotensyny II, a przez to i endoteliny, obniżają stężenie reaktywnych form tlenu oraz zwiększają
stężenie rozszerzającej naczynia i stymulującej produkcję i uwalnianie NO bradykininy i
prostacykliny.
•
Inhibicja ACE przyczynia się do niezależnego od obciążenia zmniejszenia hipertrofii serca,
zapobiega także rozszerzeniu i remodelingowi komory po przebytym zawale mięśnia sercowego.

Budowa i powstawanie białka angiotensyny II
•
Angiotensyna jest hormonem peptydowym o budowie
(H-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-OH).
•
Aktywna angiotensyna II powstaje głównie na drodze odcięcia dipeptydu od angiotensyny I
przez enzym ACE. Peptyd ten może także powstawad w tkankach w sposób niezależny od ACE,
poprzez trawienie przez niespecyficzne karboksypeptydazy lub białka podobne do
chymotrypsyny np. chymazę.
•
Chymazowy szlak generowania angiotensyny II odnotowano w sercu, komórkach śródbłonka i
komórkach tucznych
Angiotensyna może byd generowana przez dwa systemy:
krążący we krwi układ RAA, który odpowiada za regulację krótkotrwałą (renina uwalniana do
krążenia z aparatu przykłębuszkowego pod wpływem obniżonej perfuzji kłębuszkowej
katalizuje transformację angiotensynogenu do angiotensyny I; system ten pełni funkcje
endokrynną)
tkankowy RAA biorący udział w długotrwałych zmianach (bierze udział w narządowym -
serce, naczynia krwionośne, nerki i tkankowym działaniu para-i autokrynnym)
•
W warunkach patologicznych (niewydolnośd serca, przerost lewej komory) działające lokalnie
(np. w sercu) czynniki systemu RAA wydają się mied większe znaczenie niż te krążące w osoczu.
•
Angiotensyna II może ulegad dalszemu przekształceniu w angiotensynę III (powstającą poprzez
odcięcie kwasu asparaginowego od oktapeptydu), a nawet w angiotensynę IV. Jednakże oba te
peptydy mają znacznie słabsze zdolności kurczenia naczyo niż AngII. Angiotensyna III stymuluje
syntezę aldosteronu i procesy zapalne (poprzez receptor AT2, natomiast angiotensyna IV
działając poprzez AT1 wywołuje skurcz naczyo, a poprzez AT4 wzmaga produkcję PAI-1.
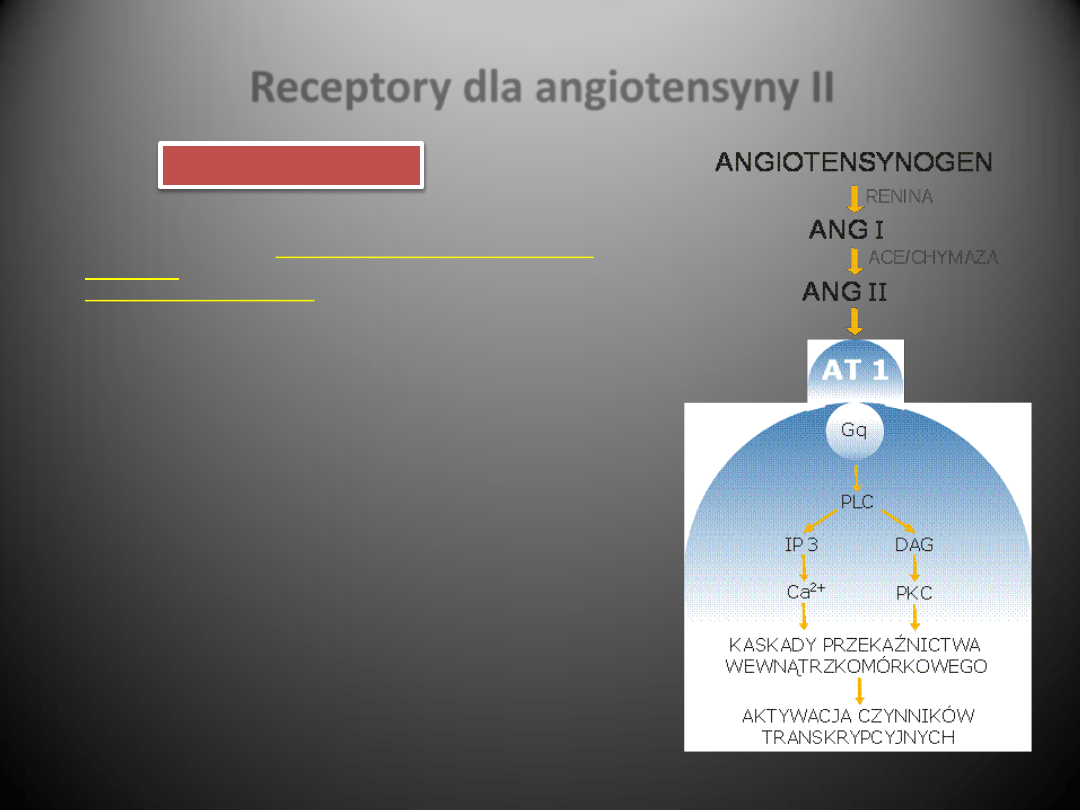
Receptory dla angiotensyny II
•
Receptor
AT1
ulega ekspresji w bardzo wielu tkankach i
narządach włączając
serce, nerki, wątrobę, płuca, mózg,
nadnercza
. Receptory te znajdują się także na powierzchni
monocytów/makrofagów
.
•
Wpływ na ekspresję receptora AT1 poza angiotensyną II,
której podwyższony poziom obniża jego aktywację, mają
czynniki wzrostu (PDGF, FGF, EGF – powodują obniżenie
ekspresji) oraz inne czynniki takie jak: glukokortykoidy,
aldosteron, forskolina, TNF-
, cytokiny, NO, insulina, LDL,
estrogeny, progesteron, sód, wolne rodniki tlenowe, IGF-1 i
izoprenalina.
•
Istnieją co najmniej cztery mechanizmy regulacji związane z
receptorem AT1.
•
Receptor AT1 sprzężony jest z białkiem Gq, jego aktywacja
prowadzi do stymulacji fosfolipazy C (PLC), a w dalszej
kolejności do powstania IP3 i IDG, mobilizacji Ca
2+
i aktywacji
białkowej kinazy C (PKC). IDG prawdopodobnie pośredniczy
w naczynioskurczowym działaniu AngII, a także w aktywacji
kinaz tyrozynowych/serynowych, które z kolei przyczyniają
się do stymulacji wzrostu indukowanego angiotensyną II
RENINA
ACE/CHYMAZA
Receptor AT1
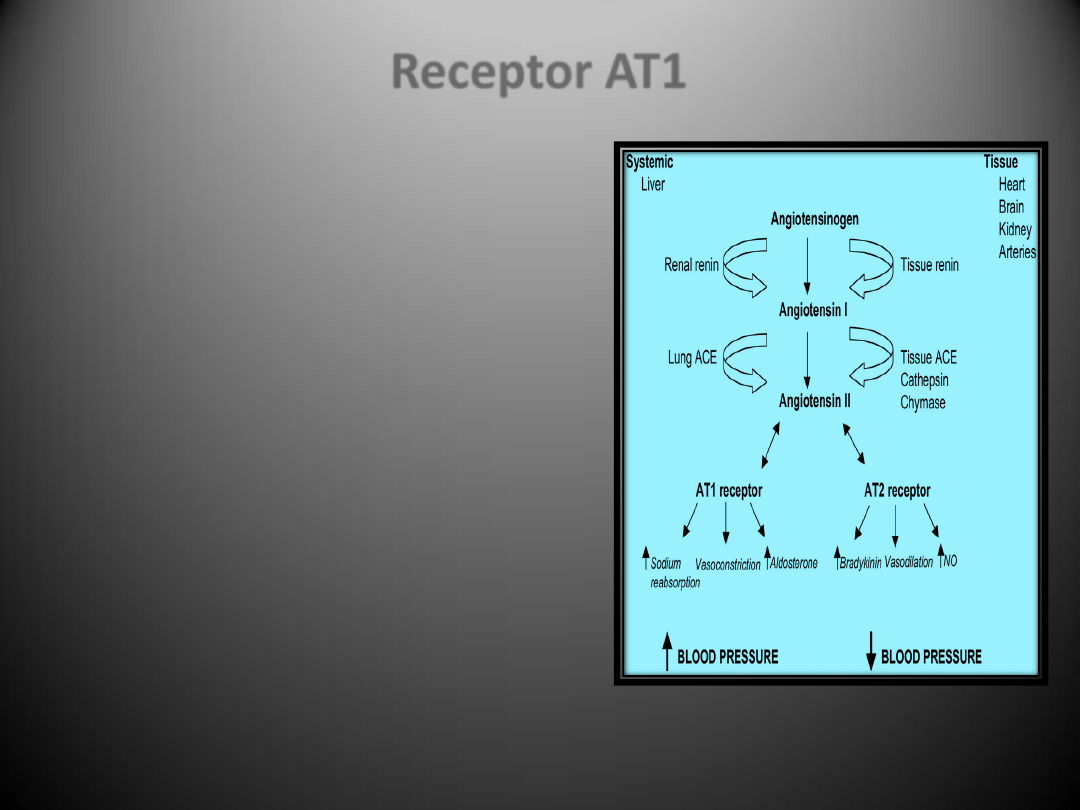
Receptor AT1
Do efektów działania angiotensyny II, w której
pośredniczy AT1 zalicza się:
promowanie wzrostu komórkowego,
regulacja ekspresji substancji bioaktywnych
takich jak: hormony wazokonstrykcyjne, czynniki
wzrostu, cytokiny, aldosteron oraz składniki
macierzy zewnątrzkomórkowej
pośredniczenie w skurczu naczyo,
uwalnianianie katecholamin z zakooczeo
współczulnych włókien nerwowych
inicjowanie reakcji sprzężenia zwrotnego w
systemie RAS np. stymulowanie ekspresji
angiotensynogenu poprzez oddziaływanie na
fragment promotora zwany elementem
odpowiedzi ostrej fazy
hamowanie sekrecję reniny
Zwiększona produkcja angiotensyny II wraz z
podwyższonym stężeniem receptorów AT1 (dzięki
obecności w procesie miażdżycowym czynników, które
przyczyniają się do jego regulacji w górę) prowadzą do
większej wydajności systemu RAA, a przez to do
nasilonego niekorzystnego działania jego wszystkich
składników.
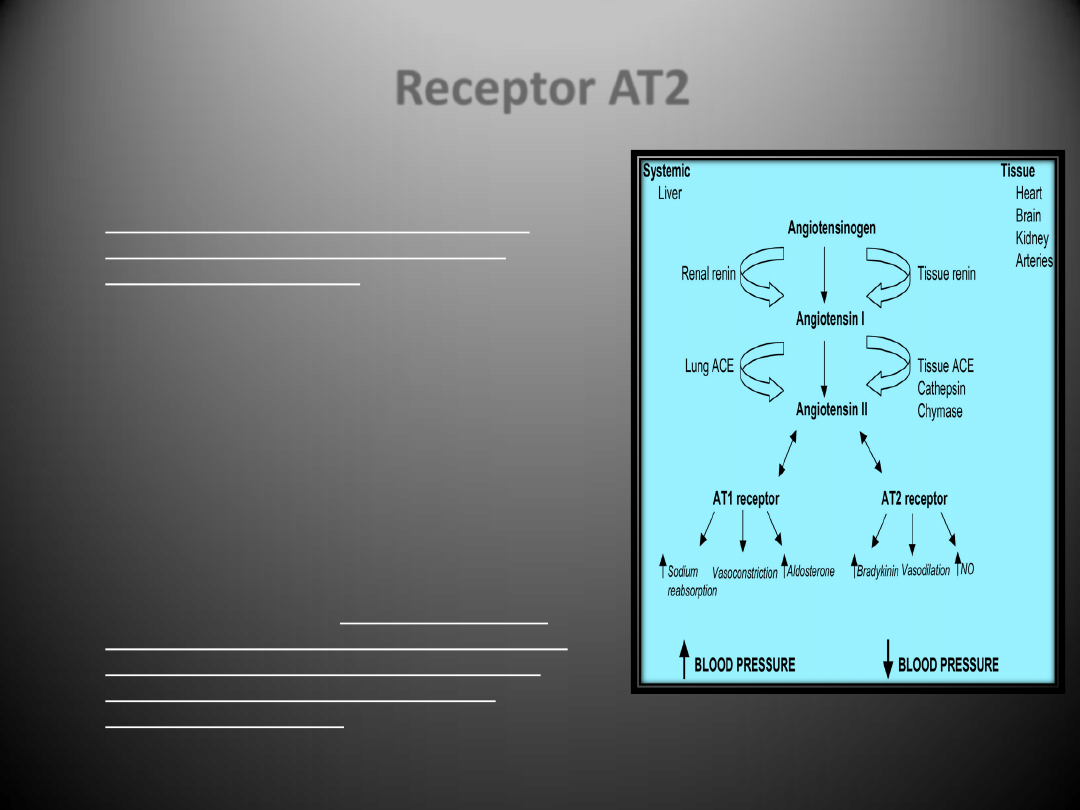
Receptor AT2
Receptor AT2 jest sprzężony z
fosfatazą
fosfotyrozynową
i występuje głównie w
przedsionkach serca, nabłonku naczyo,
macicy, jajnikach, rdzeniu nadnerczy,
trzustce oraz komorach.
Lokalizacja AT2 w śródmiąższowych
fibroblastach w sercu może sugerowad, że
może on także brad udział w rozwoju stanu
zapalnego i zwłóknienia.
Pomimo, iż zazwyczaj receptorowi AT2
przypisuje się pośredniczenie w działaniu
antyproliferacyjnym
i w procesie apoptozy,
może on również odgrywad rolę w stymulacji
wzrostu komórkowego.
Ekspresja receptorów dla angiotensyny II w
sercu jest odmienna: receptory AT2 są w
przewadze w zdrowym sercu, natomiast w
przypadku niewydolności receptory AT1
ulegają regulacji w dół, podczas gdy
ekspresja AT2 wzrasta

Funkcje angiotensyny II
Działanie:
–
syntezy i wydzielania aldosteronu
– skurcz naczyo
–
ADH (wazopresyna)
–
ACTH (kortykotropina)
–
resorbcji Na w kanalikach nerkowych
–
pragnienie
Aktywacja receptora AT1 zlokalizowanego na powierzchni komórek śródbłonka, mięśni gładkich
naczyo oraz monocytów wywołuje nie tylko szybkie uruchomienie szlaku fosfolipazy C i wapnia
wewnątrzkomórkowego, ale i pośredniczy w długofalowym efekcie AngII poprzez indukcje
zmian w transkrypcji genów.
Lokalnie syntetyzowana AngII wywołuje bezpośredni efekt inotropowy i poprawia funkcję
skurczową serca poprzez ułatwianie uwalniania noradrenaliny z zakooczeo nerwów
współczulnych, i o ile takie krótkotrwale działanie jest korzystne w stanie pozawałowym, to
przedłużona aktywacja może prowadzid do postępującego uszkodzenia mięśnia sercowego.
Angiotensyna II może stymulowad syntezę kolagenu typu I i III w ludzkich fibroblastach, a także
hamowad aktywnośd metaloproteinazy I

Rola angiotensyny II w dysfunkcji śródbłonka
AngII bierze udział w rozwoju nadciśnienia tętniczego
(ważnego czynnika ryzyka choroby wieocowej) poprzez
wywołanie skurczu naczyo (bezpośrednia stymulacja komórek
mięśni gładkich) i zwiększenie objętości
wewnątrznaczyniowej wody w organizmie na drodze
stymulacji kory nadnerczy do wydzielania aldosteronu.
Angiotensyna II zwiększa także wydzielanie endoteliny silnie
kurczącej naczynia krwionośne i wazopresyny (z przysadki
mózgowej) oraz zmniejsza biodostępnośd tlenku azotu
(NO), co w znaczący sposób wpływa na zależną od
śródbłonka wazodylatację naczyo.
Angiotensyna II stymulując oksydazę NADPH wytwarzającą
wolne rodniki tlenowe w komórkach mięśni gładkich naczyo i
śródbłonka przyczynia się do powstania stresu oksydacyjnego
oraz zmniejszenia biodostępności i aktywności NO. Efekt
przeciwny jest wywierany poprzez receptor AT2, który zwiększa
wytwarzanie bradykininy i stymuluje aktywnośd syntazy tlenku
azotu.

Rola Ang II w incydentach zakrzepowych
System RAA moduluje
równowagę fibrynolityczną
poprzez hamowanie układu
fibrynolitycznego i aktywowanie
procesów zakrzepowych na
drodze wpływania na kaskadę
koagulacji i aktywnośd płytek.
Zwiększa ekspresję czynnika
tkankowego (TF), który odgrywa
istotną rolę w inicjacji kaskady
krzepnięcia (jest ważnym
kofaktorem czynnika VII w kaskadzie
koagulacyjnej).
Hamuje szlak fibrynolizy
poprzez stymulowanie
ekspresji PAI-1, inhibitora
aktywatora tkankowego
plazminogenu (t-PA)
osłabiając proces
rozpuszczania skrzepu.
RAA przyczynia się także do aktywacji i
agregacji płytek krwi, które posiadają na
swojej powierzchni receptory dla
angiotensyny II, co prowadzi do ich
uwrażliwienia na działanie płytkowych
agonistów i stymuluje uwalnianie
płytkowych czynników naczyniokurczących
(tromboksan A2) i proliferacyjnych (PDG
F).

Rola Ang II w generacji ROS i oksydacji LDL
Angiotensyna II poprzez zwiększanie aktywności oksydazy NADPH wytwarzającej
reaktywne formy tlenu ma duży udział w powstawaniu i rozwoju miażdżycy
(powstają oxLDL)
•
RFT stymulują m.in. aktywowane-mitogenami kinazy – Akt, szlak JAK/STAT, indukują
czynniki transkrypcyjne (białko aktywatora-1), wpływają na funkcjonowanie kanałów
jonowych, a indukcja powyższych szlaków sygnałowych prowadzi do wzrostu i
migracji komórek mięśni gładkich naczyo, regulacji funkcji śródbłonka, ekspresji
czynników prozapalnych i modyfikacji macierzy zewnątrzkomórkowej
Angiotensyna II zwiększa oksydację cząsteczek LDL nie tylko poprzez stymulację oksydazy
NADPH w makrofagach, ale i lipoksygenazy.
•
oxLDL osłabiają tworzenie NO, zwiększają wytwarzanie reaktywnych form tlenowych
i indukują syntezę endotelialnych cząstek adhezyjnych (E-selektyna, ICAM-1, VCAM-
1), chemokin i czynników wzrostu mięśni gładkich
•
angiotensyna przyczynia się do regulacji „w górę” receptora LOX-1 (lectin-like
oxidized LDL receptor) zlokalizowanego na powierzchni komórek śródbłonka i
makrofagów oraz receptora „scavenger” (CD36) przez co odgrywa istotną rolę w
przyswajaniu cząstek ox-LDL przez makrofagi
Rola angiotensyny II w stymulacji procesu zapalnego
Poprzez receptor AT1 aktywuje ona w komórkach mięśni gładkich naczyo, w
monocytach i komórkach endotelium
prozapalny czynnik transkrypcyjny NF-
B,
który z kolei przyczynia się do zwiększenia syntezy komórkowych czynników
adhezyjnych (ICAM-1, VCAM-1), cytokin prozapalnych (MCP-1, IL-6)
Białko MCP-1 działając lokalnie przyciąga monocyty i limfocyty T zawierające
receptor CCR2 do miejsca uszkodzenia naczynia, natomiast interleukina-6 (IL-6)
uwalniana przez aktywowane monocyty i komórki mięśni gładkich naczyo,
stymuluje proliferację tych ostatnich

Rola angiotensyny II w stymulacji wzrostu komórkowego i
niestabilności blaszki miażdżycowej
Angiotensyna II aktywuje wiele szlaków sygnałowych
kinaz białkowych
(np. kinaza
Janusa/STAT, MAPK),
stymuluje wytwarzanie proto-onkogenów
(c-fos, c-jun, c-
myc) oraz wielu
autokrynnych czynników wzrostu
(TGF-
1, PDGF).
•
Angiotensyna II stymuluje proces proliferacji fibroblastów i syntezę kolagenu
poprzez zwiększanie sekrecji TGF-ß. Działając poprzez receptor AT1 angiotensyna II
powoduje hipertrofię komórek mięśni gładkich naczyo, powstawanie lokalnych
ognisk zapalnych prowadzące do rozwoju miażdżycy i pękania blaszki
miażdżycowej.
•
AngII indukuje ekspresję cytokin prozapalnych (IL-6) w hodowlach komórek mięśni
gładkich naczyo (VSCM) i makrofagów, które indukują proliferację VSMC na drodze
aktywacji PDGF i stymulacji degradacji macierzy przez metaloproteinazy.
•
Także stymulowane przez AngII generowanie reaktywnych form tlenu, które
regulują aktywnośd metaloproteinaz MMP-9 i MMP-2 degradujących kolagen,
przyczynia się do zmniejszenia stabilności blaszki miażdżycowej.
•
Angiotensyna II wywołuje
kompensacyjny przerost miocytów
oraz
komórek
mięśni naczyo krwionośnych
, co poprzez zmniejszenie przepływu krwi może
prowadzid do niedokrwienia mięśnia sercowego. Działając poprzez receptor AT2,
który uaktywnia fosfatazy, a nie kinazy tak jak AT1, angiotensyna II może hamowad
przerost mięśni gładkich naczyo i kardiomiocytów.
Aldosteron
• Aldosteron należy do mineralokortykoidowych hormonów kory nadnerczy.
Wydzielany przez warstwę kłębkowatą
kory nadnerczy
, aldosteron pełni
ważną funkcję w
utrzymywaniu prawidłowej równowagi wodno-
elektrolitowej
poprzez regulację transportu jonów w cewkach dystalnych
(retencja Na
+
i Cl
-
oraz wydalanie H
+
i K
+
) oraz ciśnienia tętniczego krwi.
Zwiększenie syntezy aldosteronu zachodzi pod wpływem takich czynników jak:
angiotensyna II
hormon adrenokortykotropowy
endotelina 1
spadek objętości krążącej krwi
obniżenie ciśnienia tętniczego
zmniejszenie stężenia sodu (Na) w organizmie
wzrost aktywności układu adrenergicznego
Swoje działanie hormon może wywierad bezpośrednio poprzez
receptory
mineralokortykoidowe
(MR) bądź pośrednio na drodze zwiększania ekspresji
angiotensyny II, endoteliny, aktywację COX-2, lub zwiększenie stresu
oksydacyjnego
K
+
kora nadnerczy
(warstwa kłębkowata)
angiotensyna II
ACTH
aldosteron
resorbcji Na
+
Na
+
wydalanie K
+
wydalanie H
+
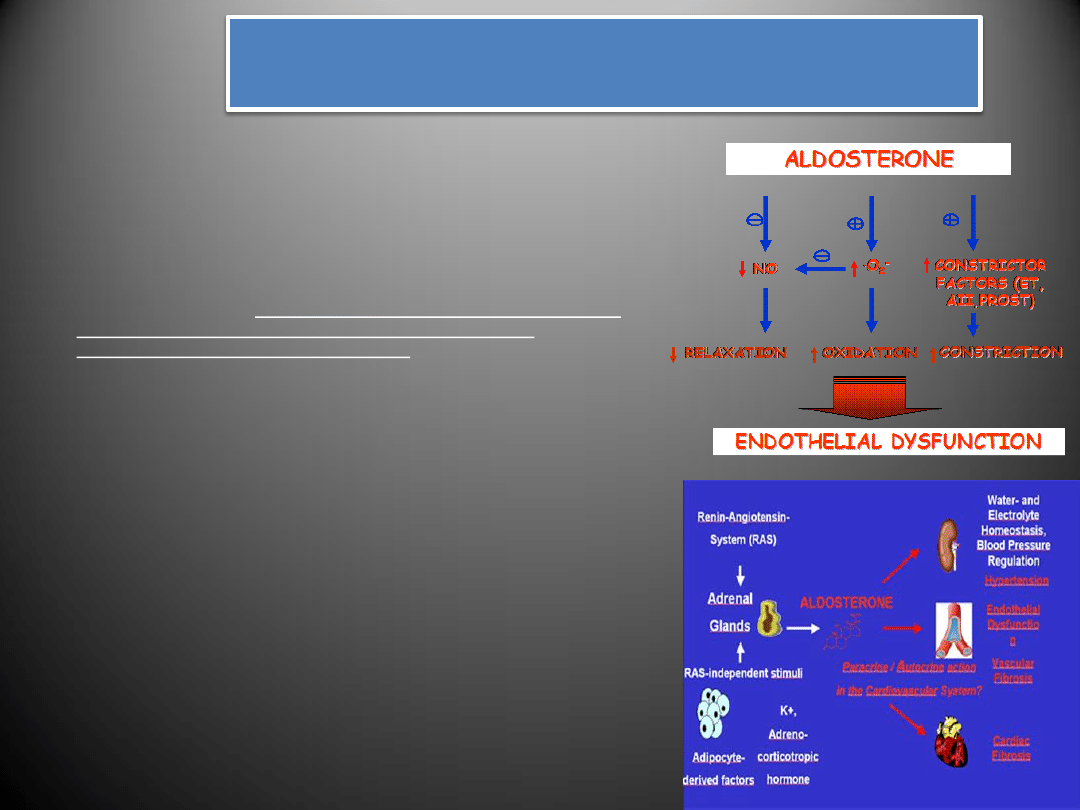
Rola aldosteronu w patogenezie chorób układu
sercowo-naczyniowego
•
Nadmierna synteza aldosteronu, działającego poprzez receptory
mineralokortykoidowe zlokalizowane w sercu, naczyniach
krwionośnych i mózgu przyczyniad się może do
włóknienia serca
i naczyo krwionośnych
poprzez działanie na fibroblasty
naczyniowe, uszkodzeo śródbłonka naczyniowego
oraz ciężkich
uszkodzeo tętnic wieocowych.
•
Uszkodzenia ścian naczyo wywołane przez nadmiar aldosteronu
mogą byd wywołane zwiększonym napływem sodu do miocytów,
zwiększoną aktywacją receptorów dla angiotensyny II oraz
hamowaniem wychwytu katecholamin.
•
Aldosteron nasila także
syntezę czynników wzrostu
, wykazuje
działanie
prozapalne
,
mitogenne
i
prozakrzepowe.
•
Podwyższone stężenie aldosteronu może stymulowad
powstawanie w sercu nacieków zapalnych z komórek
makrofagów, limfocytów T, monocytów oraz białek układu
dopełniacza prowadzących do lokalnych ognisk martwicy lub
niedokrwienia, co sugeruje, że hormon ten może uczestniczyd w
aktywowaniu komórek immunokompetentnych
•
Zwiększenie aktywności oksydazy NADPH przez aldosteron,
wpływa na
zwiększenie stresu oksydacyjnego
, co może
wywierad działanie prozapalne i nasilad syntezę niektórych
czynników wzrostu
•
Poza nasilaniem dysfunkcji śródbłonka i upośledzeniem
zależnego od endotelium rozszerzania naczyo na drodze
zmniejszania wydzielania tlenku azotu
oraz promowaniem
powstawania wtórnych ognisk martwicy, aldosteron zaburza
funkcje baroreceptorów oraz wychwyt norepinefryny przez
miokardium, co prowadzid może do zaburzenia rytmu serca

Endotelina-1
•
Endoteliny są obecne prawie we wszystkich tkankach i narządach, gdzie biorą udział
w regulacji układu krążenia, ośrodkowego i obwodowego układu nerwowego, układu
wewnątrzwydzielniczego i wielu innych
•
Endoteliny pełnią niezwykle ważną rolę w regulacji funkcji układu krążenia,
ośrodkowego i obwodowego układu nerwowego, układu wewnątrzwydzielniczego,
układu oddechowego i pokarmowego, regulacji równowagi kwasowo-zasadowej
ustroju oraz wielu innych narządów i układów
•
Przypisuje im się także udział w patogenezie wielu stanów chorobowych
(nadciśnienie płucne, nadciśnienie tętnicze, samoistne zwłóknienie płuc, astma i
alergie pokarmowe, kardiomiopatia rozstrzeniowa, skurcz naczyo mózgowych)
ET-1
ET-2
ET-3
komórki śródbłonka, mięśni
gładkich naczyo, neurony,
hepatocyty, astrocyty,
komórki mezangialne
kłębuszków nerkowych,
neutrofile, monocyty,
makrofagi, fibroblasty
nerki, jelito, niewielkie
ilości w sercu i komórkach
łożyska
ośrodkowy układ nerwowy,
komórki śródbłonka,
komórki nabłonka
kanalików nerkowych ,
komórki epitelialne jelita
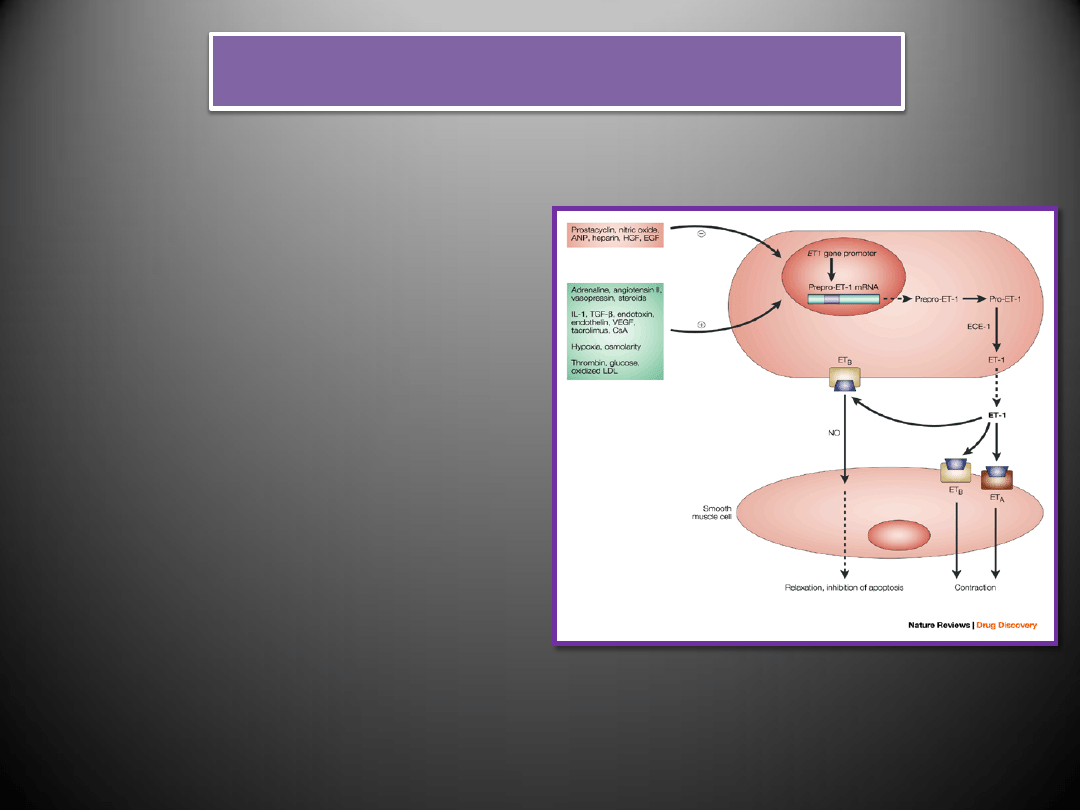
Ekspresja i wydzielanie endoteliny-1
•
Endotelina-1 nie jest magazynowana w komórkach, ale
ulega syntezie de novo pod wpływem takich czynników jak:
rozciąganie komórek mięśniowych, hipoksja,
niedokrwienie, kwasica metaboliczna, angiotensyna II,
trombina, wazopresyna argininowa, cząsteczki HDL i LDL,
cytokiny (IL-1, IL-2, IL-6, TNF-α, THF-β), oraz czynniki
wzrostu (EGF, FGF, IGF-1)
•
Wydzielanie endoteliny zapoczątkowuje aktywacja
fosfolipazy C
prowadząca do zwiększenia stężenia
zewnątrzkomórkowego wapnia
i
stymulacji kinazy
białkowe
j.
•
Indukcja wewnątrzkomórkowej syntezy cGMP wywołana
działaniem przedsionkowego peptydu natriuretycznego
(ANP), tlenku azotu (NO), prostacyklin (PGI2), endoteliny-3,
heparyny, glikokortykosteroidów powoduje obniżenie
produkcji endoteliny-1.
•
Stężenie endoteliny-1 w osoczu jest zazwyczaj niewielkie,
gdyż jest ona głównie wydzielana (przez błonę komórkową
stykającą się bezpośrednio ze ścianą naczynia), a jej reszta
jest szybko wychwytywana przez swoiste receptory
(ETA i
ETB).
•
Działanie endoteliny w warunkach fizjologicznych jest
niewielkie i obejmuje głównie rozszerzanie naczyo działając
przez śródbłonkowe receptory ETB. Jednakże w sytuacjach
patologicznych takich jak: stymulacja mięśnia sercowego
przez neurohormony i cytokiny lub wzrost obciążenia
wstępnego obserwuje się zwiększenie stężenia peptydu,
pobudzenie ich receptorów na komórkach mięśni gładkich
oraz skurcz tych ostatnich.

Wpływ endotelin na układ krążenia
Endoteliny w zdrowym sercu wpływają na kurczliwośd miocytów, inotropię, chronotropię
mięśnia sercowego oraz powstawanie arytmii. Mogą one także działad mitogennie na
fibroblasty i wywoływad przerost kardiomiocytów.
ET-1 w komórkach śródbłonka i komórkach mięśni gładkich naczyo bierze udział w
neowaskularyzacji i proliferacji mięśni gładkich
Endotelina może brad udział w patogenezie nadciśnienia tętniczego, nie tylko dlatego, że
zwiększa całkowity opór obwodowy, ale i poprzez upośledzenie prawidłowego
funkcjonowania nerek
Oddziałuje ona także na układ renina-angiotensyna-aldosteron, wazopresynę i peptydy
natriuretyczne, przez co wpływa na homeostazę wodno-elektrolitową.
Zwiększa ona także wydzielanie aldosteronu w wyniku pobudzenia receptorów
zlokalizowanych na powierzchni warstwy kłębuszkowej kory nadnerczy.
Endotelina wpływa także na uwalnianie innych czynników kurczących naczynia takich jak
wazopresyna, angiotensyna II, adrenalina, noradrenalina, tromboksan A2.
Sprzyja ona powstawaniu stanów zapalnych i zwiększa infiltrację makrofagów i leukocytów
poprzez nasilanie produkcji cytokin m.in. czynnika martwicy guza, IL1, IL6, IL8, czynnika
stymulującego kolonie granulocytów i makrofagów.
Pobudza proliferację komórek mięśni gładkich poprzez aktywację receptorów ET-A oraz
adhezję granulocytów obojętnochłonnych i agregację płytek.
Ekspresja ET-1 może przyczyniad się także do pękania blaszki miażdżycowej i powstania
ostrych incydentów niedokrwiennych. Stężenie endoteliny jest podwyższone u pacjentów z
ostrymi zespołami wieocowymi, zawałem mięśnia sercowego i samoistnym nadciśnieniem
.
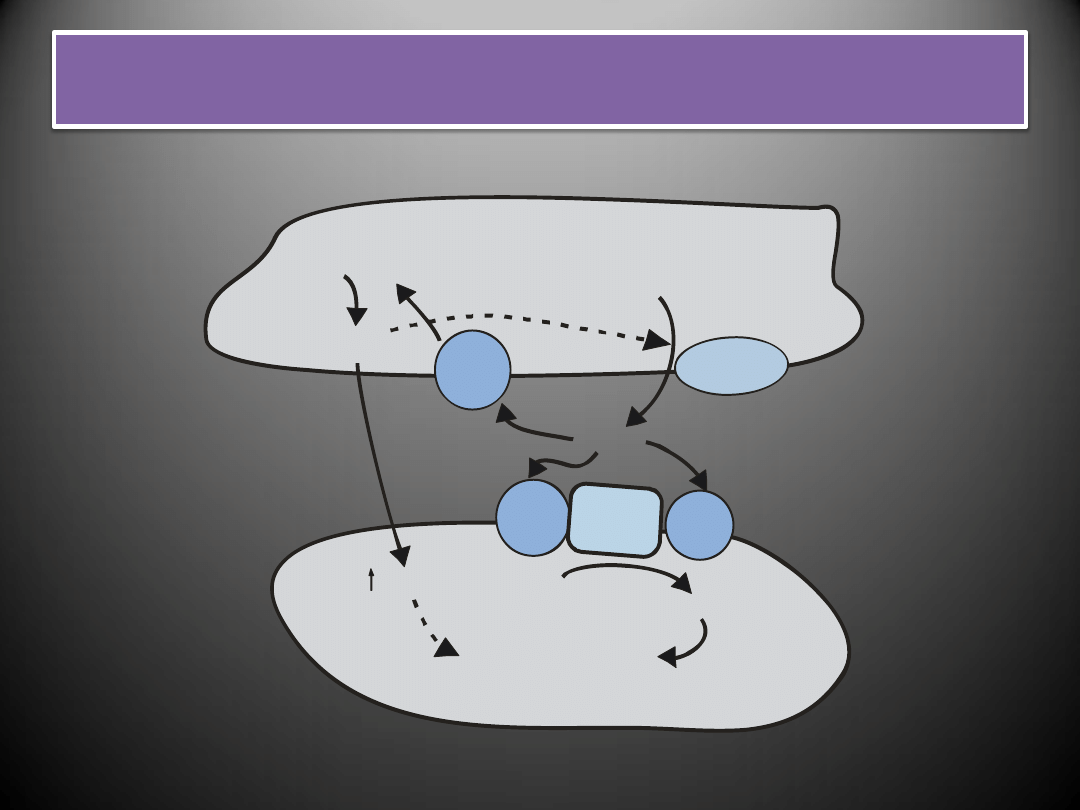
Działanie tlenku azotu i endoteliny na komórki mięśni
gładkich naczyo
Mięśni gładkich naczyń
SKURCZ
Śródbłonek
NOS
ECE
ET
B
ET
B
ET
Duża-ET-1
ET-1
PIP
IP
NO
C
GMP
+
+
+
-
-
L-arginina
G
A
3
q
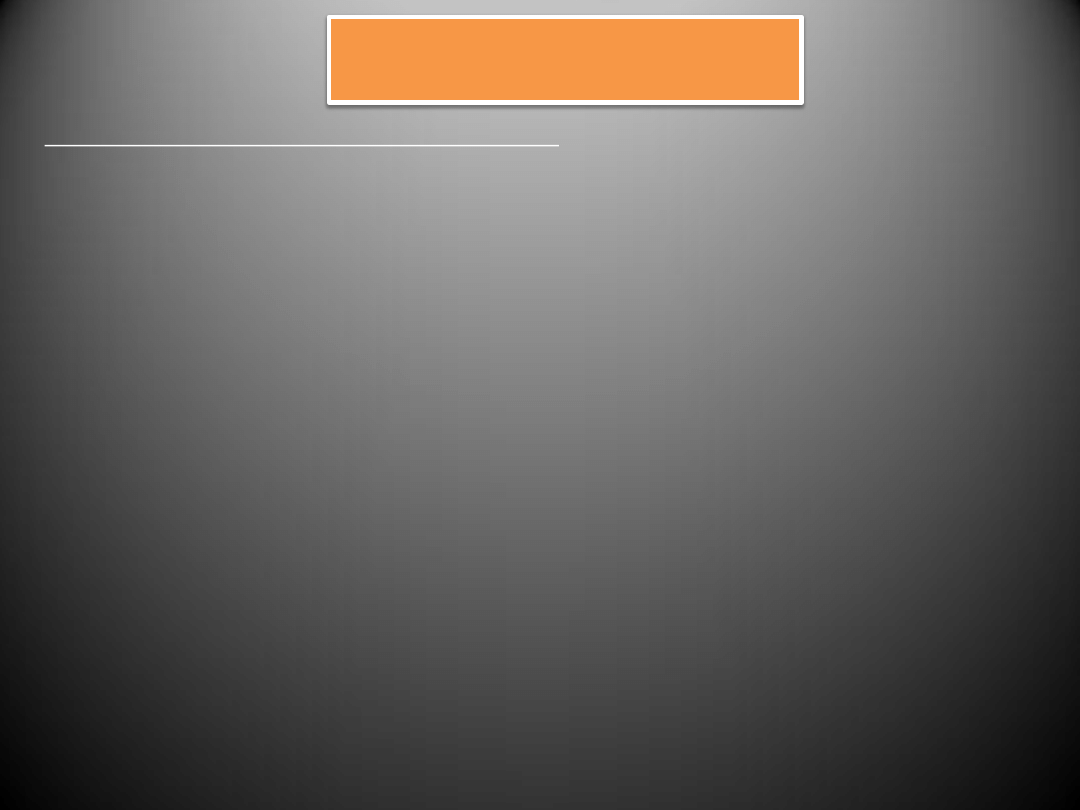
Peptydy natriuretyczne
W skład tej grupy wchodzą następujące hormony:
przedsionkowy peptyd natriuretyczny ANP (atrial natriuretic peptide)
mózgowy peptyd natriuretyczny BNP (brain natriuretic peptide)
peptyd natriuretyczny C CNP (C-natriuretic peptide)
urodilatina i DNP
Do biologicznych działao peptydów natriuretycznych należy
natriureza, rozszerzanie naczyo,
supresja systemu RAA
.
Peptydy natriuretyczne są syntetyzowane i magazynowane w postaci prohormonów
•
ANP
jest wytwarzany głównie w
przedsionkach serca
•
BNP
syntetyzowany jest głównie w
komorach
pomimo, iż po raz pierwszy został odkryty w
mózgu
(stąd nazwa mózgowy peptyd natriuretyczny).
Peptydy ANP i BNP, a także receptory dla nich występują także w rdzeniu kręgowym,
przysadce mózgowej, nerkach, nadnerczu
•
CNP
wytwarzany jest głównie w
komórkach śródbłonka
w
naczyniach
, w mniejszej ilości w
sercu
•
Urodilatyna
jest produkowana głównie w
nerkach
, natomiast obecnośd
DNP
stwierdzono
w ludzkim
osoczu
i
przedsionkach serca
.

Funkcje peptydów natriuretycznych
Peptydy natriuretyczne wpływają na prawidłowe funkcjonowanie śródbłonka poprzez
rozkurcz mięśniówki gładkiej naczyo, działanie antagonistyczne w stosunku do układu
renina-angiotensyna-aldosteron
oraz hamowanie aktywności presyjnej układu
noradrenergicznego.
ANP poprzez rozszerzenie naczyo, zwiększenie filtracji kłębuszkowej i wydzielania
sodu oraz poprzez inhibicję systemu RAA odgrywa istotną rolę w regulacji wodno-
elektrolitowej oraz w kontroli ciśnienia.
ANP zwiększa także przepuszczalnośd naczyo, co umożliwia przepływ płynów z
przestrzeni wewnątrznaczyniowej do tkanki śródmiąższowej.
CNP w ścianach naczyo i fibroblastach wpływa na napięcie i proliferację przyległych
komórek mięśni gładkich sąsiadujących fibroblastów. Nie wykazuje on działania
natriuretycznego, posiada jednak właściwości wazodylatacyjne.
Peptyd DNP obecny w ludzkim osoczu i przedsionkach serca
wykazuje natomiast
działanie natriuretyczne i rozszerza on naczynia krwionośne.
Rola peptydów natriuretycznych w
chorobach układu sercowo-naczyniowego
Peptydy natriuretyczne osłabiają aktywnośd systemu RAA (ograniczenie przebudowy mięśnia
sercowego), hamują wydzielanie aldosteronu, ograniczają syntezę kolagenu, hamują stymulowaną
przez AngII, ET-1, FGF (czynnik wzrostu fibroblastów) i IGF (insulinowy czynnik wzrostu) syntezę DNA.
Hamowanie wydzielania reniny, aldosteronu i endoteliny, obniżanie aktywności ACE oraz rozkurcz
mięśni gładkich naczyo przez peptydy natriuretyczne powoduje zmniejszenie obciążenia następczego i
wstępnego serca oraz obniżenie ciśnienia tętniczego krwi.
Peptydy natriuretyczne wykazują działanie antymitogenne w stosunku do komórek mięśni gładkich
naczyo, komórek śródbłonka i miocytów sercowych.
Peptydy natriuretyczne chronią mięsieo sercowy modulując uszkodzenia niedokrwienno-reperfuzyjne,
włóknienie i przerost.
Wzrost wewnątrzkomórkowego cGMP i aktywacja kinazy białkowej C spowodowane działaniem
peptydów hamują wewnątrzkomórkowe sygnały wzrostowe, przez co ograniczają martwicę w mięśniu
sercowym. Inhibicja przerostu i włóknienia, hamowanie wytwarzania angiotensyny II i endoteliny-1
przez peptydy natriuretyczne są zjawiskami korzystnymi dla niewydolnego serca, jednakże procesy
pro-apoptotyczne stymulowane także przez te peptydy mogą prowadzid do utraty komórek
kardiomiocytów i dalszego uszkadzania serca
Peptydy natriuretyczne są także zaangażowane w regulację systemu koagulacyjnego i fibrynolitycznego
m.in. poprzez hamowanie ekspresji PAI-1 indukowanej angiotensyną II i TF
Peptydy natiuretyczne
Poziom osoczowych peptydów natriuretycznych jest podwyższony u
osób zastoinową
niewydolnością serca
i we
wczesnej fazie zawału mięśnia sercowego
.
Zwiększona sekrecja ANP i BNP u pacjentów z niewydolnością krążenia korelowała z
„ciężkością” choroby, a także z przerostem serca. Utrzymujący się przez dłuższy czas
podwyższony poziom BNP koreluje z
powiększeniem lewej komory
i z jej
zmniejszoną
kurczliwością
, u pacjentów z zastoinową niewydolnością serca i zawałem. Z badao klinicznych
wynika, że BNP może służyd jako specyficzny marker dysfunkcji lewej komory
Stężenie osoczowe
NTpro-peptydów BNP i CNP
może byd wykorzystywane jako marker
diagnostyczny.
Poziom
NTpro-BNP
w osoczu u pacjentów z chorobą mózgowo-naczyniową jest dobrym
markerem kolejnego incydentu niewydolności serca, a poprawnośd prognozy jeszcze wzmacnia
jednoczesne oznaczenie białka C reaktywnego.
Jednakże przy wyciąganiu wniosków dotyczących funkcji lewej komory i rokowania pacjenta na
podstawie pomiaru NTpro-BNP lub BNP należy uwzględnid takie parametry jak m.in.
funkcjonowanie nerek, nadwaga itp.
Oznaczenie poziomu NTpro-CNP może wnieśd dodatkowe informacje dotyczące patofizjologii
niewydolności serca i odpowiedzi krążenia obwodowego na ten stan.

Receptory adrenergiczne
Podstawowymi regulatorami rzutu minutowego serca, zwłaszcza w czasie wysiłku fizycznego i/lub
stresu psychicznego (tzw. sytuacje „walki i ucieczki”, ang. fight-or-flight), są współczulny układ
nerwowy i mechanizm Franka Starlinga. Stymulacja układu sympatycznego powoduje uwolnienie
noradrenaliny ze współczulnych zakooczeo nerwowych w sercu oraz adrenaliny z rdzenia
nadnerczy. Mediatory te, działając poprzez błonowe receptory beta-adrenergiczne (ang. beta-
adrenergic receptor, β-AR) zlokalizowane na komórkach sercowych, powodują przyspieszenie
akcji serca oraz wzrost kurczliwości kardiomiocytów. Oba te mechanizmy są odpowiedzialne za
wzrost rzutu minutowego serca pod wpływem stymulacji współczulnej.
W skład rodziny receptorów adrenergicznych wchodzi dziewięd typów receptorów:
•
receptory
1A
-,
1B
-,
1D
- adrenergiczne oddziałujące głównie na białko Gq (stymulacja
fosfolipazy C)
•
receptory
2A
-,
2B
-,
2C
- adrenergiczne oddziałujące głównie na białko Gi (inhibicja cyklazy
adenylowej)
•
receptory
1
-,
2
- i
3
-adrenergiczne wiążące głównie białko Gs (stymulacja cyklazy adenylowej).
Receptory
adrenergiczne
1
2
1
2
Lokalizacja Komórki presynaptyczne serce mięśnie
docelowe zakończenia
gładkie
nerwowe
Efekt skurcz rozkurcz siły skurczu relaksacja
naczyń naczyń
częstości akcji wydzielania NP
kontrola serca
uwalniania NA przewodzenia
agregacja przez węzeł PK
płytek
rozkurcz naczyń
wydzielania ADH
i reniny

Receptory adrenergiczne
W strukturze receptorów adrenergicznych sprzężonych z białkami G występuje
charakterystycznych
siedem hydrofobowych
obszarów o długości 20-25
aminokwasów przedzielonych ośmioma hydrofilowymi regionami o zmiennej
długości.
Każda z hydrofobowych domen tworzy
transmembranową helisę
, a łączące je
hydrofilowe pętle wystają po stronie zewnątrz- i wewnątrzkomórkowej.
Region N-koocowy receptora znajduje się po stronie zewnątrzkomórkowej błony,
natomiast fragment C-koocowy po stronie cytoplazmatycznej. Sekwencje regionów
hydrofobowych tworzących helisę transmembranową wykazują duże podobieostwo
Receptory sprzężone z
białkami G
są glikoproteinami, wszystkie posiadają
konserwatywne sekwencje (od 1 do 3) miejsc N-glikozylacji w pobliżu N-kooca
receptora.
Aktywacja receptorów
-adrenergicznych
Receptory adrenergiczne występują na powierzchni komórek, gdzie aktywowane wiązaniem
agonistów wywołują uruchomienie szlaków sygnałowych. Transdukcja sygnału rozpoczyna się
od przyłączenia agonisty (aminy katecholowej) do receptora w błonie komórkowej, co
stymuluje interakcje receptorów z białkami G
Wszystkie białka G składają się z trzech podjednostek:
G
, G
i G
i są zlokalizowane po
stronie cytozolowej błony komórkowej.
Kiedy białko
G jest połączone z cząsteczką GDP
znajduje się ono w stanie
nieaktywnym.
W wyniku oddziaływao białko-receptor następuje uwolnienie GDP z miejsca wiążącego GTP
w białku G, i przyłączenie do tego miejsca GTP.
Aktywowane białko G
następnie
oddysocjowuje
od receptora i stymuluje enzym efektorowy. Hydroliza GTP wprowadza białko
G z powrotem w stan nieaktywny. Przyłączenie liganda do receptora wywołuje poprzez
stymulację białka G aktywację lub inhibicję enzymu generującego przekaźnik II typu.
Białka G mogą stymulowad
cyklazę adenylową
i
guanylową
,
fosfolipazy C i A2
,
fosfodiesterazy
oraz
kanały wapniowe i potasowe
.
Rodzaj aktywowanego przekaźnika i uruchomionego szlaku zależy od rodzaju białka, np.
białko Gs stymuluje cyklazę adenylową i przyczynia się do zwiększenia stężenia cAMP, białko
Gq/11 uruchamia szlak fosfolipazy C (PLC) i stymuluje metabolizm fosfoinozytydów, a białko
Gi/o hamuje aktywnośd cyklazy adenylowej i obniża stężenie cyklicznego AMP.
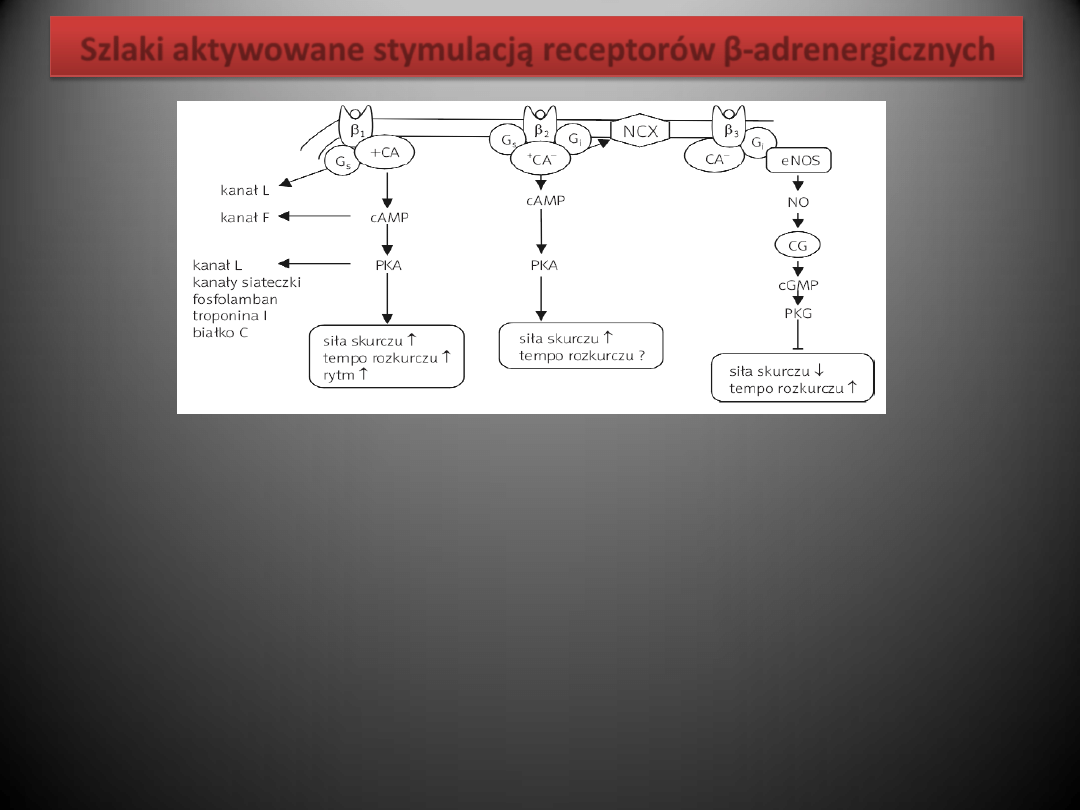
Szlaki aktywowane stymulacją receptorów β-adrenergicznych
Aktywacja β1-AR powoduje aktywację cyklazy adenylowej, wzrost komórkowej produkcji cAMP i stymulację
przez cAMP kinazy białkowej typu A. Enzym ten, poprzez przenoszenie reszt kwasu fosforowego z
cząsteczki ATP na różne białka (fosforylacja), powoduje zmianę właściwości tych białek. Efektem
czynnościowym fosforylacji białek przez kinazę białkową A (ang. protein kinase A, PKA) w sercu jest
przyspieszenie akcji serca i przewodzenia p-k, wzrost siły skurczu i przyspieszenie rozkurczu mięśnia
sercowego oraz szereg efektów metabolicznych.
W sercu ssaków i w sercu ludzkim β2-AR są sprzężone zarówno z białkiem Gs, jak i z białkiem Gi. Jednakże w
zdrowym sercu w sumie przewagę mają efekty związane z aktywacją cyklazy adenylowej i zwiększoną
produkcją cAMP.
NO aktywuje cyklazę guanylową, co prowadzi do wzrostu komórkowego stężenia cyklicznego GMP (cGMP). Z
kolei cGMP aktywuje kinazę białkową G (PKG) oraz fosfodiesterazę typu drugiego (PDEII). Kinaza białkowa
G fosforyluje: a) kanał wapniowy typu L, co przeciwnie do fosforylacji przez PKA zmniejsza prąd
wapniowy i siłę skurczu, oraz b) troponinę I, co – podobnie jak fosforylacja przez PKA – przyspiesza
rozkurcz. PDEII jest enzymem rozkładającym cAMP i tym samym prowadzącym do zmniejszenia
aktywności PKA i stopnia fosforylacji jej substratów.

Znaczenie receptorów
-adrenergicznych w chorobach układu
sercowo-naczyniowego
•
Dysfunkcja lewej komory prowadzi do aktywacji neurohormonalnej (systemu RAA, układu
współczulnego, uwalniania cytokin), co jest korzystne w ostrej fazie, jednakże przedłużająca
się stymulacja prowadzi do stopniowego pogarszania funkcjonowania serca. Zwiększona
stymulacja β-adrenergiczna jest główną przyczyną progresji niewydolności serca. Uważa się,
że stopieo aktywacji współczulnej odwrotnie koreluje z przeżywalnością pacjentów HF.
Główne zmiany obserwowane w układzie receptorów β-AR w niewydolności serca to
:
1) wzrost ekspresji i aktywności kinazy β-ARK;
2) spadek wrażliwości receptorów β1 i β2 na katecholaminy;
3) spadek gęstości β1 (zmniejszenie ilości mRNA o ok. 50%);
4) wzrost gęstości receptorów β3;
5) wzrost ekspresji białka Gi;
6) wzrost powinowactwa receptora β2 do białka Gi.
•
Stymulacja receptorów
1-adrenergicznych w sercu prowadzi do dodatniego efektu
inotropowego, luzitropowego i chronotropowego, w którym pośredniczy białko Gs i wzrost
stężenia cAMP, natomiast aktywacja receptorów β2-adrenergicznych wywołuje relaksację
mięśni gładkich naczyo, a przez to przyczynia się do zmniejszenia całkowitego oporu
obwodowego i obniżenia ciśnienia krwi. W zdrowym sercu ekspresja receptorów
2-
adrenergicznych jest niewielka, natomiast w niewydolnym sercu receptory te mogą stanowid
nawet 40% całkowitej puli receptorów ze względu na drastyczny spadek ilości receptorów
1.

Znaczenie receptorów
-adrenergicznych w chorobach układu
sercowo-naczyniowego
Ciągła aktywacja receptorów adrenergicznych stymuluje remodelling lewej komory,
obumieranie komórek na drodze nekrozy lub apoptozy oraz retencję wody i soli.
Pobudzenie układu adrenergicznego stymuluje uwalnianie reniny, a przez to i
wytwarzanie angiotensyny II, co jeszcze przyspiesza progresje niewydolności serca.
Aktywacja receptorów a1 i
-adrenergicznych przez noradrenalinę może, poprzez
stymulowanie syntezy kolagenu, prowadzid do zwłóknienia oraz przerostu mięśnia
sercowego.
Stymulacja receptorów
1 powoduje przyspieszenie akcji serca i zwiększoną jego
kurczliwośd oraz pobudzenie tworzenia reniny, co z kolei może przyczyniad się do
wystąpienia zaburzeo przepływu krwi i wzrostu napięcia ścian naczyo, prowadząc
do uszkodzenia śródbłonka i pękania blaszki miażdżycowej. W przeciwieostwie do
receptorów
1, receptory
2 mogą mied kardioprotekcyjne działanie, ich
pobudzenie w badaniach na modelach zwierzęcych hamowało apoptozę w
kardiomiocytach przypuszczalnie za pośrednictwem białka inhibitorowego Gi
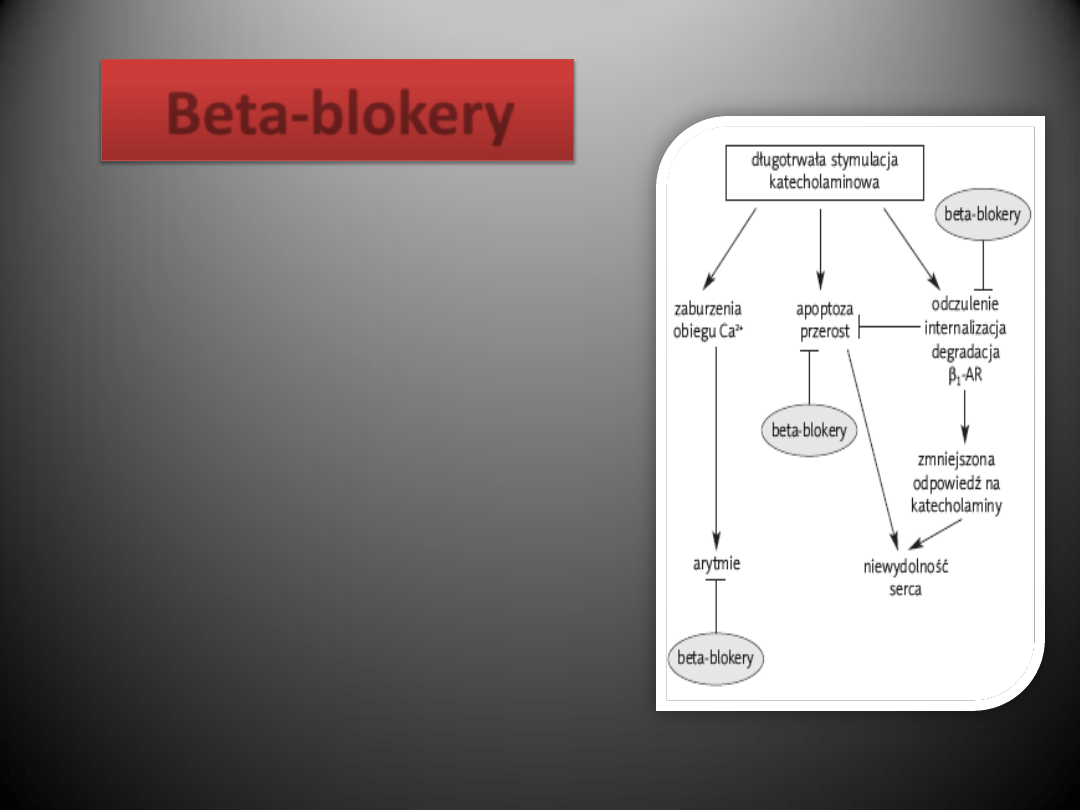
Beta-blokery
Przewlekła zwiększona stymulacja sercowego
układu
-adrenergicznego jak wykazano
jest toksyczna dla serca i może leżed u
podstaw patogenezy zastoinowej
niewydolności serca.
Stosowanie leków blokujących te receptory w
leczeniu osób po zawale zmniejsza ich
śmiertelnośd i ryzyko wystąpienia
powtórnego zawału.
Wykazano, że blokada receptora
1-
adrenergicznego w znacznym stopniu
poprawia stan pacjenta z niewydolnością
serca.

PODZIAŁ, MECHANIZMY POWSTAWANIA,
POWIKŁANIA.
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ.
NADCIŚNIENIE TĘTNICZE

PRZESZŁOŚD A DZIŚ
- U przodków Homo Sapiens poważnym problemem było ryzyko spadku ciśnienia, co
wiązało się z ograniczonym dostępem do wody i soli. Ryzyko to tworzyło presję ewolucyjną,
selekcjonującą osobniki wyposażone w geny, sprzyjające mechanizmom przeciwdziałającym
zmniejszaniu, (a nie zwiększaniu!) ciśnienia tętniczego.
-
Czynniki ryzyka nadciśnienia tętniczego pojawiły się dopiero w warunkach cywilizacji.
-
Nadciśnienie tętnicze znacznie częściej występuje u rasy czarnej, prawdopodobnie jako
skutek genetycznie zaprogramowanej wzmożonej aktywności enzymów syntezy amin
katecholowych, niezbędnych w łaocuchu syntez melaniny.

Ciśnienie tętnicze
– Ciśnienie wywierane przez krew na ściany tętnic.
Ciśnienie skurczowe
(maksymalne, górne) - Ciśnienie krwi
w tętnicach w czasie skurczu serca – gdy mięsień sercowy wpompowuje
krew do naczyń.
Ciśnienie rozkurczowe
(dolne) – Najniższa wartość ciśnienia
w chwili, gdy mięsień sercowy jest rozkurczony, a zastawki
półksiężycowate są zamknięte.
Nadciśnienie tętnicze
– Przewlekła choroba układu krążenia, podlegającą
długotrwałemu leczeniu, w której występuje (utrwalone – wykazane w kilku
pomiarach) zwiększone (zgodnie z zaleceniami WHO powyżej 139 mm HG
dla ciśnienia skurczowego i 89 mm Hg dla ciśnienia rozkurczowego)
ciśnienie krwi w naczyniach tętniczych.
DEFINICJE
CZYNNIKI RYZYKA
CHOROBY NADCIŚNIENIOWEJ
ŚRODOWISKOWE
GENETYCZNE
INNE
Nadwaga i otyłośd
Wiek i płed
Alkohol i papierosy
Napięcie psychiczne
(stres)
Mała aktywnośd fizyczna
Nadmierne spożycie soli
Zaburzenia metaboliczne
Cukrzyca typu 2.
Zaburzenia lipidowe
Insulinoopornośd
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADWAGA I OTYŁOŚĆ
- Otyłość jest bardzo ważnym czynnikiem, predysponującym
do wystąpienia nadciśnienia tętniczego, dlatego poszukiwane
są coraz bardziej skuteczne sposoby jej zapobiegania.
Waga ciała obecnie jest najczęściej mierzona i klasyfikowana wg tzw.
Wskaźnika Masy Ciała BMI
(ang. Body Mass Index), którego wartość oblicza
się z następującego wzoru:
BMI = masa ciała (kg) / wzrost
2
(m
2
)
Według WHO interpretacja wyniku jest następująca:
BMI <18,5 kg/m
2
- niedowaga
BMI 18,5–24,9 kg/m
2
- masa prawidłowa
BMI 25,0–29,9 kg/m
2
- nadwaga
BMI >30,0 kg/m
2
- otyłość
Podstawowe ogniwa łączące otyłość i nadwagę z patogenezą nadciśnienia
tętniczego:
- Zaburzenia hemodynamiczne
- w porównaniu z nadciśnieniem
tętniczym u osób szczupłych, w otyłości nadciśnienie tętnicze
charakteryzuje się zwiększoną objętością krwi i zwiększonym
rzutem serca.
-
Insulinooporność
- komórki tłuszczowe wydzielają substancję
o nazwie TNF-
powodującą insulinooporność.
-
Zwiększenie aktywności układu renina-angiotensyna-aldosteron (RAA)
-
Wzrost aktywności układu współczulnego
- skurcz naczyń obwodowych i
związany z tym wzrost oporu naczyniowego
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADWAGA I OTYŁOŚD
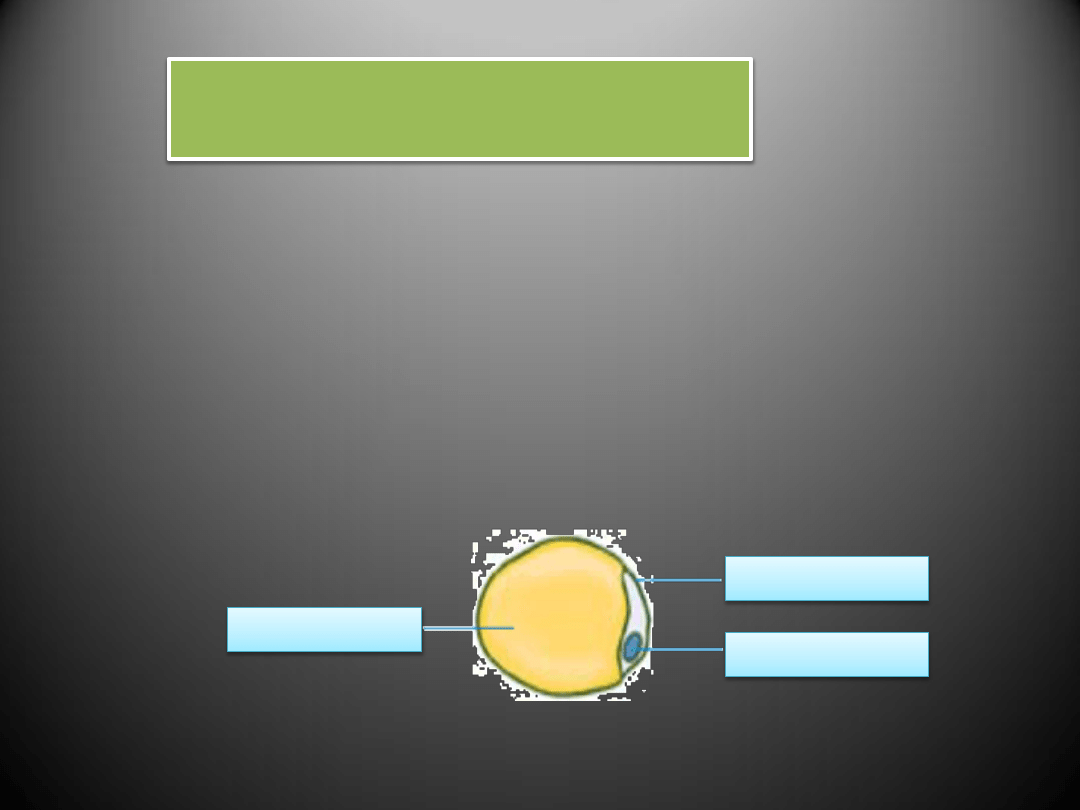
Tkanka tłuszczowa składa się z komórek tłuszczowych (adipocytów).
- Wśród substancji produkowanych przez adipocyty szczególną rolę
w patogenezie chorób układu sercowo-naczyniowego przypisuje się
leptynie.
- Efekt hipertensyjny leptyny dokonuje się przede wszystkim poprzez
pobudzenie układu współczulnego oraz pobudzenie mięśni gładkich
naczyń do rozrostu.
Komórka tłuszczowa – adiopocyt
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADWAGA I OTYŁOŚD
Błona komórki
Trójglicerydy
Jadro komórki
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
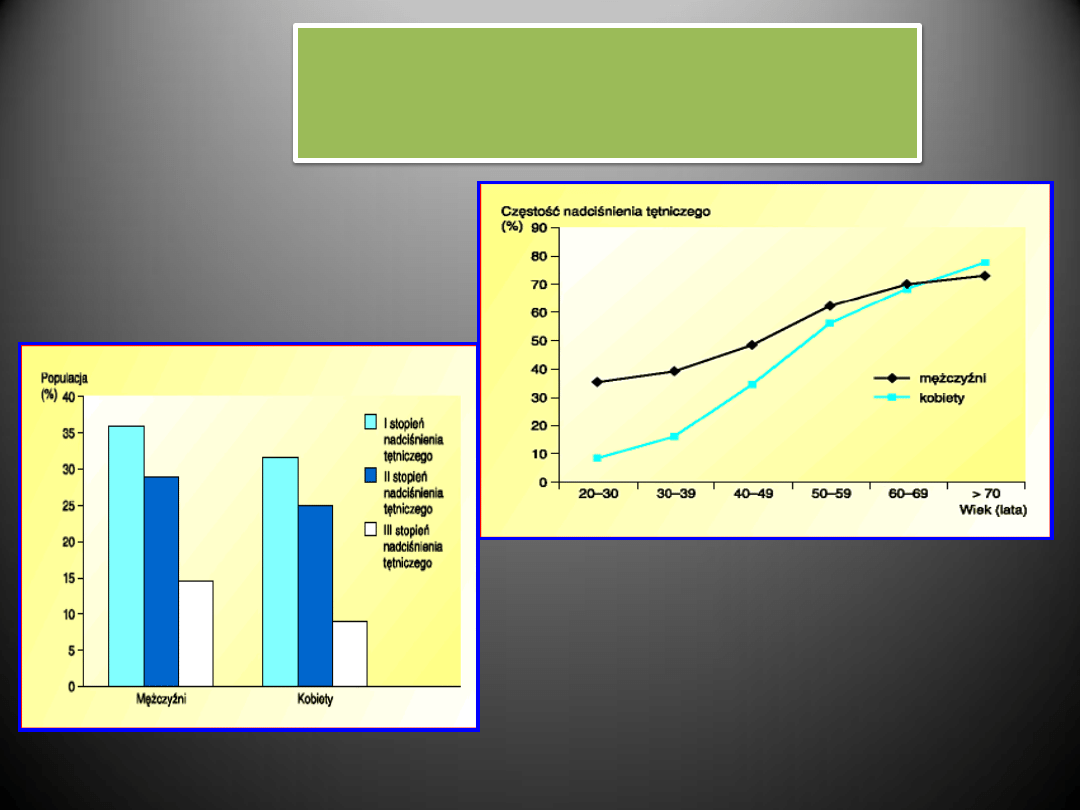
WIEK I PŁEĆ
Przybliżona liczba osób w Polsce chorujących
na nadciśnienie tętnicze
Częstośd występowania nadciśnienia tętniczego w Polsce
w zależności od wieku i płci
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
PALENIE TYTONIU
Nikotyna i jej metabolit powstający w organizmie palacza – kotynina – przyczyniają się do
wzrostu stężenia substancji odpowiedzialnych za zwężanie naczyo – angiotensyny II i
endoteliny I. Skutkiem tego jest upośledzone rozszerzania naczyo krwionośnych i
predyspozycja do występowania stanów zakrzepowych w naczyniach.
Nikotyna
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
PALENIE TYTONIU
Substancje utleniające, pochodzące z dymu tytoniowego ułatwiają proces utleniania
cholesterolu LDL. Powstający w ten sposób związek jest czynnikiem silnie uszkadzającym
komórki śródbłonka.
1
Obniżone
HDL:LDL
Złogi
LDL
Utleniony
LDL
Miejsce
zapalne
Proliferacja
komórek
mięśniowych
Miażdżyca
2
Zmniejszenie
elastyczności
ściany
naczynia
Nadciśnienie
Udar
Zawał
Działanie utlenionego LDL

CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
NADMIERNE SPOŻYCIE SOLI
Na podstawie badao epidemiologicznych przeprowadzonych na licznej populacji udowodniono, że
nadmiar spożywanej soli powoduje wzrost ciśnienia tętniczego. Zjawisko to wiąże się z pojęciem
sodowrażliwości.
Ze względu na nadmierne spożycie soli w przeważającej części populacji ludzkiej, celowe jest
ograniczenie jej spożycia.
Wg zaleceo dieta powinna zawierad < 6g soli dziennie (co odpowiada < niż 100 mmol Na)
Zmniejszenie spożycia soli o 50 mmol/dobę powoduje zmniejszenia:
-o 50% liczby osób wymagających leczenia farmakologicznego
-o 22% liczby zgonów związanych z udarem mózgu
-o 16% liczby zgonów spowodowanych chorobą wieocową
- Szacuje się, że w 50%, na wartość ciśnienia tętniczego wpływają czynniki
genetyczne.
- Okazało się, że u chorych z nadciśnieniem pierwotnym częściej występują
odmiany genów, które u nosiciela zwiększają wchłanianie sodu w nerkach.
Dochodzi wówczas do nadmiernego gromadzenia sodu w organizmie, co
sprzyja podwyższeniu ciśnienia. Przypuszcza się, że taki mechanizm
powoduje wzrost ciśnienia tętniczego u osób spożywających duże ilości soli
kuchennej (chlorku sodu).
- Współczesny stan badań nad pierwotnym nadciśnieniem tętniczym
dowodzi, że niezbędnym warunkiem do jego powstania jest poligenowe
uwarunkowanie genetyczne. Osoby z pierwotnym nadciśnieniem tętniczym
wykazują w 1 chromosomie polimorfizm alleli genu kodującego
angiotensynogen (AGT). Występuje także polimorfizm insercyjno-delecyjny
genu kodującego konwertazę angiotensyny w 17 chromosomie.
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
CZYNNIKI GENETYCZNE
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
ZABURZENIA METABOLICZNE
Cukrzyca
Przyczynami rozwoju nadciśnienia tętniczego towarzyszącego cukrzycy są:
- Zwiększenie oporu obwodowego (stymulacja układu współczulnego),
- Zwiększenie objętości krwi i wzrost resorpcji sodu w nerkach,
- Zaburzenie czynności układu renina-angiotensyna-aldosteron (RAA),
- Uszkodzenie śródbłonka naczyniowego,
- Rozwój miażdżycy (wzrost oporu obwodowego) i angiopatii cukrzycowej,
- Produkty tkanki tłuszczowej wywołujące insulinooporność i zwiększone
wydzielanie insuliny,
- Glomerulopatia cukrzycowa (nadciśnienie nerkowe) jako powikłanie
cukrzycy.

Insulinooporność
- Osoby z opornością na insulinę obciążone są większym ryzykiem
rozwoju nadciśnienia niż pozostała część populacji.
- Insulinooporność może wpływać na rozwój nadciśnienia, między
innymi poprzez zmiany gospodarki jonami wapnia
w komórkach mięśni gładkich naczyń.
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
ZABURZENIA METABOLICZNE
CZYNNIKI RYZYKA CHOROBY NADCIŚNIENIOWEJ
ZABURZENIA METABOLICZNE
Zaburzenia lipidowe
Patogeneza nadciśnienia tętniczego w przypadku zaburzeń lipidowych wiąże się z następującymi
zjawiskami:
- Cząsteczki lipidów o niskiej gęstości (LDL) u chorych z nadciśnieniem tętniczym łatwiej wnikają w
ścianę naczynia, uszkadzając ją.
- Rozrost mięśni gładkich naczyń utrwala nadciśnienie tętnicze.
- Hipercholesterolemia i nadciśnienie tętnicze są przyczyną dysfunkcji śródbłonka naczyń, co
niekorzystnie zmienia wydzielanie substancji naczynioskurczających i naczyniorozszerzających.
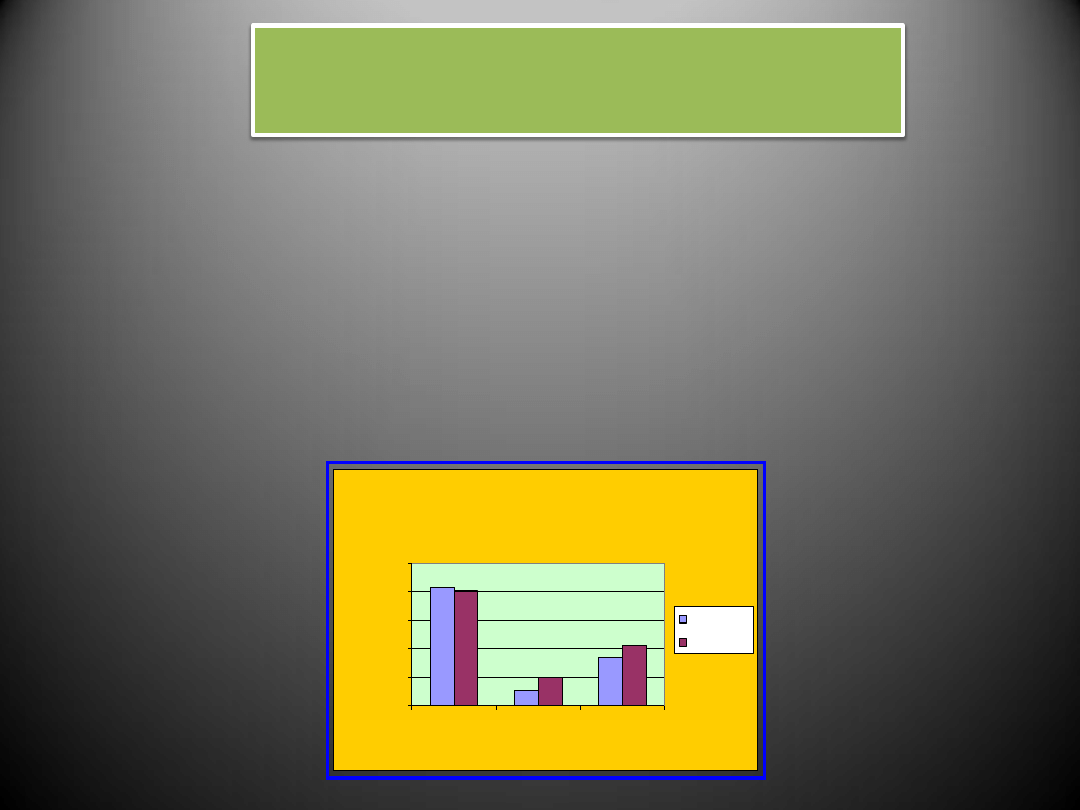
Zaburzenia lipidowe u osób z nadciśnieniem
tętniczym
0%
20%
40%
60%
80%
100%
cholesterol
>200 mg/d
HDL < 40
mg/d
TG > 150
mg/d
%
Kobiety
Mężczyźni

Objawy
NT
Krwawienie
z nosa
Szum
w uszach
Nerwowośd
i większa
potliwośd
Ból i zawroty
głowy
Najczęściej
bezobjawowo!
Zaczerwienienie
skóry
OBJAWY
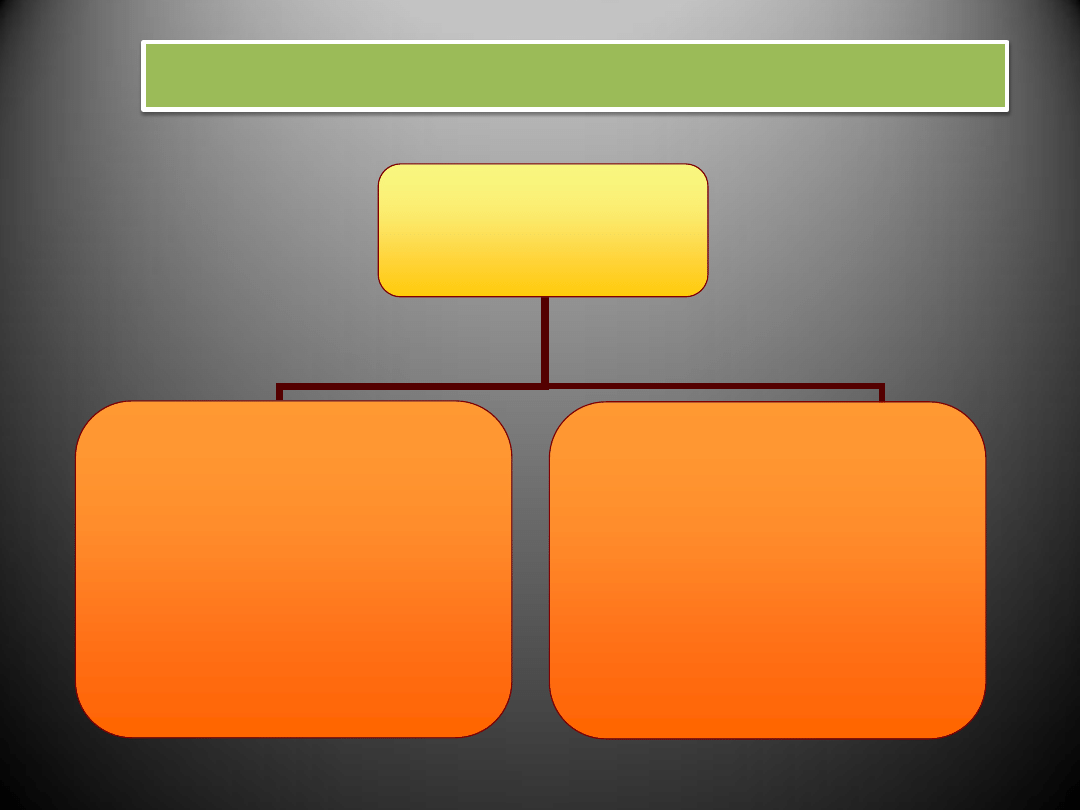
RODZAJE NADCIŚNIENIA TĘTNICZEGO
N A D C I Ś N I E N I E
T Ę T N I C Z E
P I E R W O T N E (idiopatyczne)
- brak bezpośredniej przyczyny
(podłoże genetyczne, menopauza,
zła dieta, stres)
- do 95% przypadków nadciśnienia
- po 40. roku życia
W T Ó R N E
- towarzyszy innym chorobom
(gł. przewlekłym chorobom nerek,
zaburzeniom hormonalnym,
miażdżycy)
- do 30. i po 50. roku życia
- z reguły usunięcie lub wyleczenie
przyczyny nadciśnienia prowadzi
do jego wyeliminowania
MECHANIZM NADCIŚNIENIA TĘTNICZEGO
- Utrwalone nadciśnienie tętnicze charakteryzuje się takim
zwiększeniem całkowitego obwodowego oporu naczyniowego (TPR),
że spoczynkowy przepływ krwi w narządach organizmu pozostaje
prawidłowy.
- Bezpośrednie przyczyny nadciśnienia polegają więc na zmienionej
czynności i strukturze obwodowych tętnic dużego krążenia.
- Występuje nadmierna ekspresja czynników sprzyjających zwężeniu
i przebudowie ściany tętnic lub niedostateczna ekspresja czynników
rozszerzających naczynia obwodowe.
- Istotną rolę w mechanizmie nadciśnienia odgrywa czynnik
neurogenny: spoczynkowa aktywność współczulna skierowana do naczyń
krwionośnych i do serca jest podwyższona, większe jest także uwalnianie
noradrenaliny zwężającej tętnice i neuropeptydu Y (NPY, związek
wzmagający naczyniozwężające działanie noradrenaliny),
przebudowującego jej ściany.
- Tlenek azotu hamuje aktywację genu egr-1 (odpowiedzialny
za proliferacja miocytów i komórek śródbłonka) wywierając tym samym
korzystny wpływ antyproliferacyjny na miocyty, zmniejszając
przebudowę ściany tętnic. Dzięki tym mechanizmom tlenek azotu
przeciwdziała rozwojowi nadciśnienia tętniczego.
- Jednym z mechanizmów nadciśnienia tętniczego jest przestawienie
na wyższy poziom i zwiększona reaktywność odruchu
z chemoreceptorów tętniczych. Wzmaga on aktywność współczulną
adresowaną do naczyń krwionośnych.
MECHANIZM NADCIŚNIENIA TĘTNICZEGO

MECHANIZM NADCIŚNIENIA TĘTNICZEGO
TEORIE MACHANIZMU
POWSTAWANIA
NADCIŚNIENIA TETNICZEGO
T E O R I A F O L K O W ’A
T E O R I A G U Y T O N ’A
MECHANIZM NADCIŚNIENIA TĘTNICZEGO
TEORIA FOLKOW’A
-
Podstawowym mechanizmem nadciśnienia są zgrubiałe ściany tętnic.
-
Podwyższenie stosunku grubości ściany naczyniowej
do wewnętrznego promienia naczynia zmienia ich geometrię tak,
że zwiększa się ich odpowiedź zwężająca na wszystkie bodźce.
-
Powtarzające się stresowo – emocjonalne wzrosty ciśnienia
tętniczego, wywołane aktywnością współczulną i osłabieniem odruchu
z baroreceptorów, hamującym w prawidłowych warunkach nadmierną
aktywność współczulną, powodują po pewnym czasie adaptacyjną
przebudowę i zgrubienie warstwy mięśni gładkich w ścianach naczyń
tętniczych oporowych.
MECHANIZM NADCIŚNIENIA TĘTNICZEGO
TEORIA GUYTON’A
Podwyższenie progu
wydalania Na
Zwiększenie stężenia Na
w płynach ustrojowych
Rozciągnięcia zbiornika
tętniczego
Zwiększenie objętości krwi
(woda podąża za Na)
Autoregulacja – ogólnoustrojowe
zwężenie naczyo
Wzrost ciśnienia tętniczego
Zwiększenie przepływu
i filtracji kłębuszkowej
Zwiększenie ilości Na w moczu
pierwotnym (zw. ze wzrostem
objętości moczu pierwotnego)
Nadmiar Na napływający do
kanalików ułatwia jego
prawidłowe wydalanie
Zachowanie homeostazy objętości krwi kosztem
zwiększenia ciśnienia tętniczego
-
Nadciśnienie jest reakcją fizjologiczną utrzymującą homeostazę
płynów ustrojowych, adaptującą organizm do zmniejszonej zdolności
usuwania sodu przez nerki.
CO WPŁYWA NA WARTOŚĆ CIŚNIENIA
TĘTNICZEGO
- Ciśnienie w układzie tętnic zależy od siły i częstości skurczów serca,
oporu, jaki stawiają ściany naczyń napływającej fali krwi oraz od lepkości
krwi. Wykazuje ono charakterystyczną zmienność, związaną z pracą serca.
- Wartości ciśnienia tętniczego nie są stale jednakowe.
- W nocy ciśnienie tętnicze jest najczęściej nieco niższe niż w ciągu dnia
(spada o co najmniej 10%), co wiąże się ze spadkiem aktywności
i zmniejszeniem pobudzenia układu nerwowego.
- Praca fizyczna lub stres psychiczny mogą zwiększyć wartości ciśnienia
tętniczego. Również wysoka temperatura otoczenia, chłód i ból mogą mieć
wpływ na jego wartość.
- U człowieka zdrowego ciśnienie tętnicze zwiększa się jednak zawsze tylko
na krótki czas, np. w trakcie wysiłku fizycznego, po czym normalizuje się
bardzo szybko.
- „Zespołu białego fartucha" - chwilowego wzrostu ciśnienia podczas
badania przez lekarza.

POMIAR CIŚNIENIA TĘTNICZEGO
- Najczęstszą metodą pomiaru ciśnienia
tętniczego jest użycie sfigmomanometru
(metoda Riva-Rocci - stąd skrót RR
określający ciśnienie tętnicze)
z zastosowaniem metody osłuchowej
(Korotkowa).
- Holter (Ambulatoryjne całodobowe
monitorowanie ciśnienia krwi - ABPM) -
automatyczne, całodobowe mierzenie
ciśnienia tętniczego krwi przez specjalny
aparat składający się z mankietu
do zakładania na ramię oraz sprzężonego
z nim rejestratora.
Ciśnienie
skurczowe
Ciśnienie
rozkurczowe
Rozpoznanie
Komentarz
< 120
< 80
Ciśnienie optymalne
- Najlepsze dla zdrowia
120 - 129
80 - 84
Ciśnienie prawidłowe
- Nie powoduje szkód
130 - 139
85 - 89
Ciśnienie wysokie prawidłowe
- Zmiana stylu życia
- U osób z czynnikami ryzyka
leczenie farmakologiczne
140 - 159
90 - 99
Nadciśnienie 1. stopnia - łagodne
- Zmiana stylu życia
- Konsultacja lekarska
(wdrożenie leczenia)
160 - 179
100 - 109
Nadciśnienie 2. stopnia - umiarkowane
180
110
Nadciśnienie 3. stopnia - ciężkie
KLASYFIKACJA NADCIŚNIENIA TĘTNICZEGO
120
/
80
Ciśnienie skurczowe
Ciśnienie rozkurczowe
LECZENIE
FARMAKOLOGICZNE
NIEFARMAKOLOGICZNE
POLITERAPIA
(częściej)
MONOTERAPIA
Diuretyki tiazydowe
i tiazydopochodne
-blokery
Blokery kanału
wapniowego
Inhibitory ACE
Antagoniści receptora
angiotensynowego
ZMIANA STYLU ŻYCIA
beta
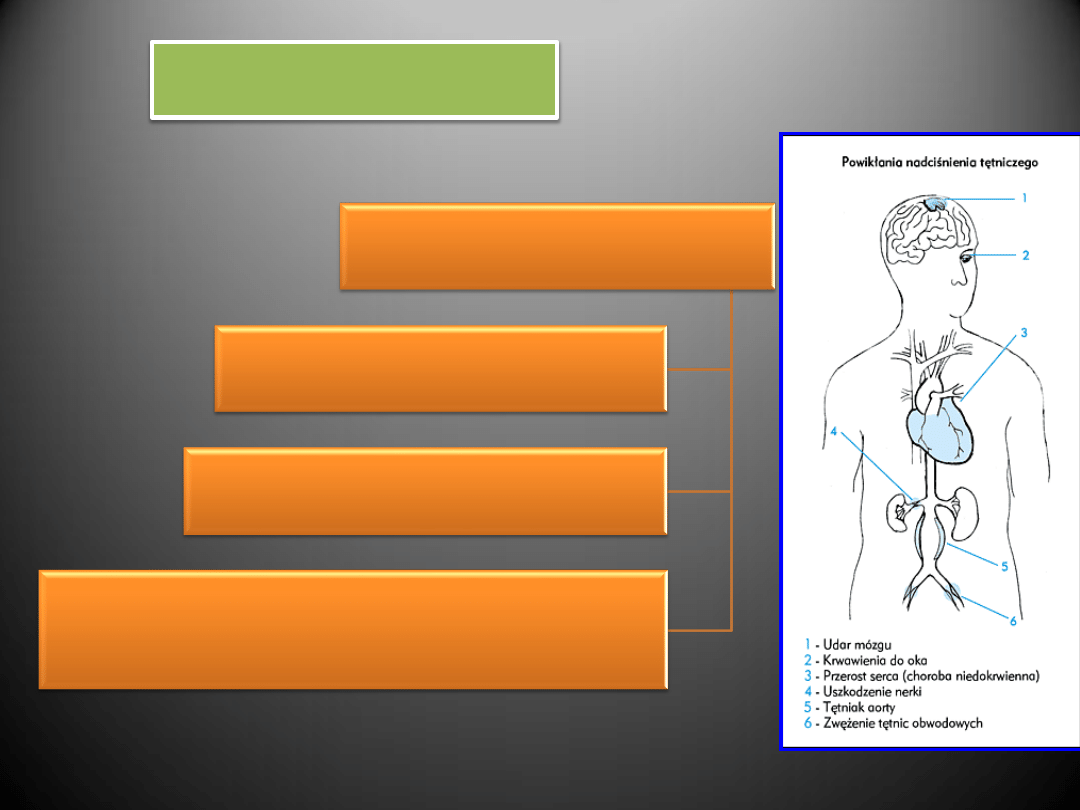
POWIKŁANIA
STADIA NADCIŚNIENIA TĘTNICZEGO
WG WHO
I stadium - brak zmian w narządach
II stadium – przerost lewej komory serca,
retinopatia nadciśnieniowa, białkomocz
III stadium – nadciśnieniowe uszkodzenia: serca
(niewydolność lewokomorowa), nerek (niewydolność nerek),
mózgu i oka (retinopatia)
- UDAR MÓZGU (APOPLEKSJA)
– Uszkodzenie mózgu na skutek zbyt
wysokiego ciśnienia krwi. Objawia się nagłym pogorszeniem lub utratą
możliwości poruszania kończynami (niedowład, paraliż), zwykle po jednej
stronie ciała. Mogą towarzyszyć zaburzenia mowy i czucia. Niekiedy objawy
nie są nasilone, a czasami ustępują same. Udar może być wynikiem
zamknięcia tętnicy (dopływ krwi, a tym samym tlenu, jest odcięty - udar
niedokrwienny) lub przerwania jej ściany i krwawienia do mózgu (udar
krwotoczny). Prawidłowe leczenie nadciśnienia tętniczego zmniejsza ryzyko
wystąpienia udaru w obydwu tych mechanizmach.
- KRWAWIENIA DO OKA
- Do struktur oka krew dociera przez bardzo małe
i delikatne tętniczki. Podwyższone ciśnienie bardzo łatwo uszkadza ich
ściany. Niewielkie krwotoki, które gdzie indziej nie miałyby znaczenia,
w siatkówce oka mogą nawet prowadzić do utraty wzroku. Prawidłowe
leczenie nadciśnienia może więc zapobiec powikłaniom w strukturach oka.
POWIKŁANIA
-
CHOROBA NIEDOKRWIENNA SERCA
- zespół objawów chorobowych będących następstwem
przewlekłego stanu niedostatecznego zaopatrzenia komórek mięśnia sercowego w tlen i
substancje odżywcze. Zaburzenie równowagi pomiędzy zapotrzebowaniem a możliwością ich
dostarczenia, doprowadza do niedotlenienia - niewydolnośd wieocowa, a nawet do zawału
mięśnia sercowego.
-
NEFROPATIA
– ok. 1/5 objętości krwi pompowanej podczas jednego skurczu serca przepływa
przez nerki. Jeśli dzieje się to pod zbyt dużym ciśnieniem, dochodzi do uszkodzenia także i tego
narządu. Nerki coraz słabiej oczyszczają więc organizm ze szkodliwych substancji. Uczestniczą w
regulowaniu poziomu ciśnienia krwi, toteż ich uszkodzenie może prowadzid do dalszego wzrostu
ciśnienia. Podwyższone ciśnienie prowadzi do niewydolności nerek.
- TĘTNIAK AORTY
– poszerzenie aorty o ponad 50% w stosunku
do jej prawidłowej szerokości.
POWIKŁANIA


Źródłem kwasów nukleinowych mogą być komórki
Źródłem kwasów nukleinowych mogą być komórki
prawie wszystkich tkanek:
prawie wszystkich tkanek:
Krew obwodowa:
zabieg pobrania jest mało inwazyjny,
z leukocytów uzyskuje się dużo DNA/RNA
Wymaz z jamy ustnej:
do prostych testów genetycznych
Wycinek skóry i założenie hodowli fibroblastów:
dla szczegółowych i długotrwałych badań
Złuszczone komórki płodu pobierane podczas amniocentezy:
do badań prenatalnych
Komórki guza, szpik kostny:
do badań onkologicznych
Płyn ustrojowy, surowica, płyn mózgowo-rdzeniowy, plwocina,
popłuczyny oskrzelowe, bioptaty tkankowe:
do wykrywania czynników zakaźnych
Nasienie:
kryminalistyka, ustalanie ojcostwa
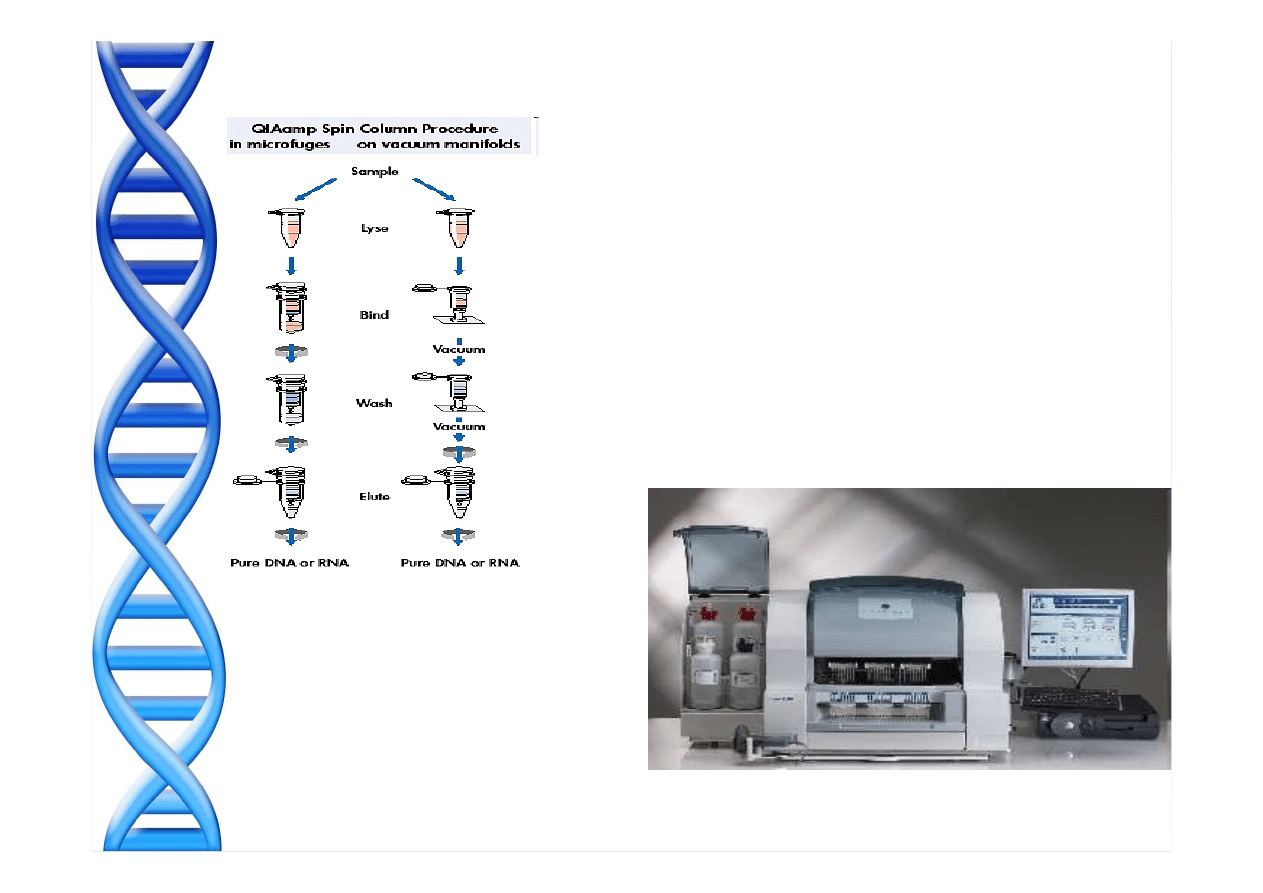
Izolacja DNA
Izolacja DNA
przygotowanie materiału biologicznego
dezintegracja i liza komórek
inaktywacja nukleaz komórkowych oraz uwolnienie
nici DNA z kompleksów białkowych, za pomocą np.
proteinazy K
oddzielenie kwasu nukleinowego od pozostałych
komponentów komórkowych
zagęszczenie preparatu DNA
usunięcie zanieczyszczeń małocząsteczkowych
strącanie wyizolowanego DNA w obecności kationów
jednowartościowych za pomocą etanolu
jednowartościowych za pomocą etanolu
Rys. EasyMAG aparat do automatycznej izolacji kwasów
nukleinowych
Rys. Schemat izolacji DNA
Obecnie stosuje się wiele metod
izolacji, w tym także
izolacje automatyczne
Izolacja DNA
Izolacja DNA
rozdzielenie komórek, rozbicie/strawienie ścian komórkowych
homogenizacja mechaniczna
homogenizacja w ciekłym azocie
trawienie chitynazą
trawienie proteinazą K
liza komórek
detergent w buforze zasadowym
detergent w buforze zasadowym
oczyszczenie DNA – usunięcie białek, lipidów, cukrów
ekstrakcja organiczna (fenol/chloroform) lub wysalanie białek,
następnie strącanie DNA alkoholem
wiązanie DNA na kolumnach wypełnionych krzemionką
przechowywanie DNA
DNA zawiesza się w lekko zasadowym buforze z dodatkiem
EDTA (wychwytuje jony Mg2+) i przechowuje w +4 lub -20°C
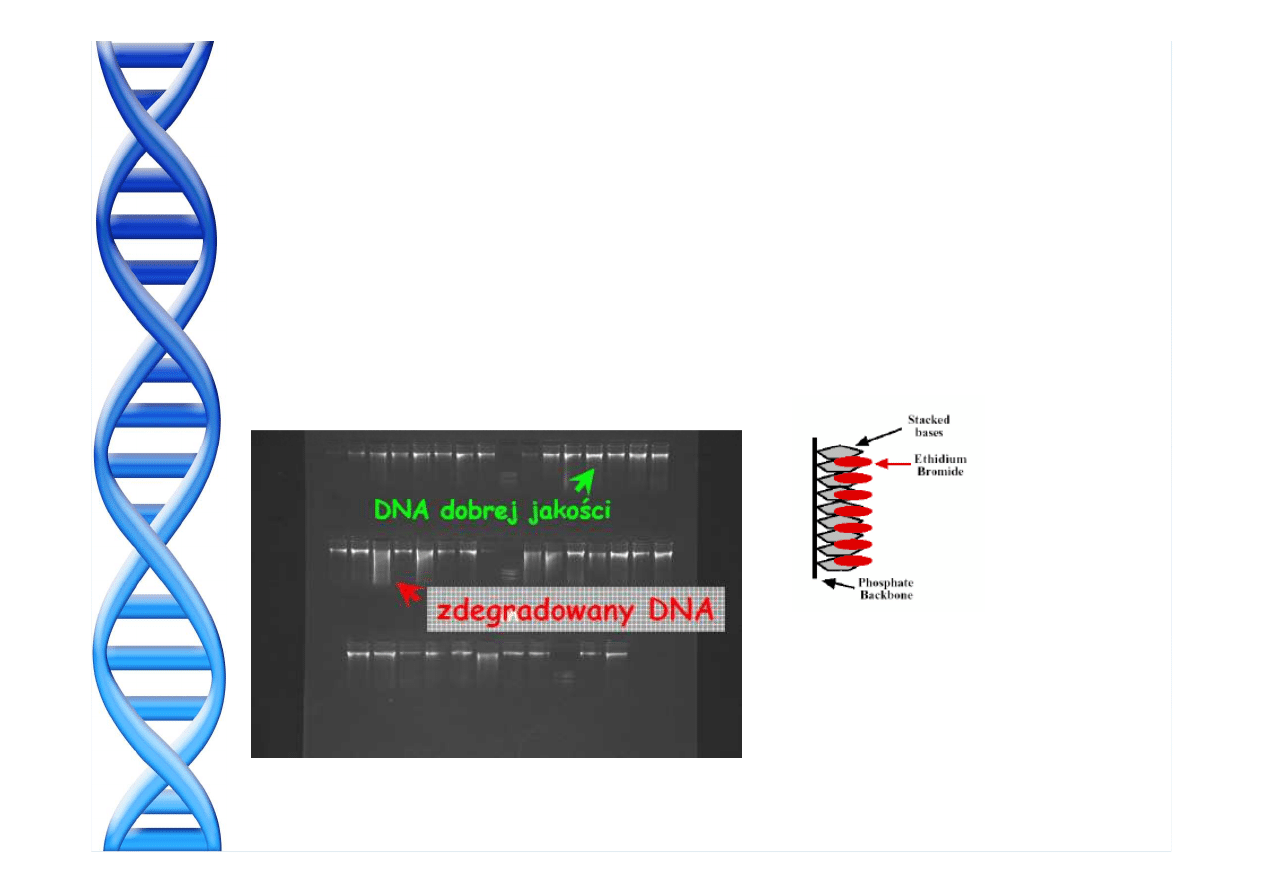
Ocena ilości i jakości wyizolowanego DNA
Ocena ilości i jakości wyizolowanego DNA
Elektroforeza w żelu agarozowym o niskim stężeniu (0.5 – 1 %)
DNA barwi się bromkiem etydyny lub innymi, mniej toksycznymi
barwnikami fluorescencyjnymi SYBR Gold SYBR Safe, Gel Red
itd., które świecą w świetle UV


Metody biologii molekularnej
Metody biologii molekularnej
Reakcja łańcuchowej polimerazy- PCR
Polimorfizm długości fragmentów
restrykcyjnych (RFLP)
Elektroforeza w żelu
Elektroforeza w żelu
Hybrydyzacja kwasów nukleinowych
Mikromacierze (technologie „chip” DNA)
Sekwencjonowanie
Klonowanie DNA
Łańcuchowa reakcja polimerazy
Łańcuchowa reakcja polimerazy --PCR
PCR
Najważniejsza technika biologii molekularnej
Metoda opracowana w latach 80-tych XXw. W 1993 r. Kary Mullis otrzymał za opracowanie jej
założeń Nagrodę Nobla w dziedzinie chemii.
Pozwala uzyskać ze złożonej mieszaniny fragmentów DNA, np. całego genomu człowieka, miliardy
kopii ściśle określonego fragmentu DNA
Metoda PCR polega na przeprowadzeniu wielu cykli syntezy fragmentu DNA, ograniczonego
dwoma oligonukleotydowymi starterami. Dlatego niezbędnym warunkiem przeprowadzenia takiej
syntezy jest znajomość znajomość sekwencji krótkich odcinków DNA po każdej stronie sekwencji
przeznaczonej do powielenia → startery.
O tym, który fragment będzie namnożony decydują startery, krótkie (15-30 bp) jednoniciowe
fragmenty DNA o znanej sekwencji, które przyczepiają się do matrycy DNA definiując fragment
ulegający amplifikacji; startery syntetyzuje się chemicznie.
ulegający amplifikacji; startery syntetyzuje się chemicznie.
Amplifikacja może odbywać się z minimalnej ilości wyjściowego DNA, nawet z 1 cząsteczki
Reakcja zachodzi w jednej probówce, która poza matrycowym genomowym DNA,
komplementarnymi starterami i termostabilną polimerazą DNA (polimeraza Taq) zawiera substraty
do syntezy DNA w formie trifosforanów nukleotydów, MgCl
2
oraz bufor .
Etapy cyklu reakcji PCR: denaturacja, hybrydyzacja i elongacja
Liczba cykli jest uzależniona od wymaganej krotności powielenia wyjściowego DNA
bufor
dwuniciowa matryca DNA
sekwencja docelowa
starter1
starter2
dNTP
polimeraza Taq
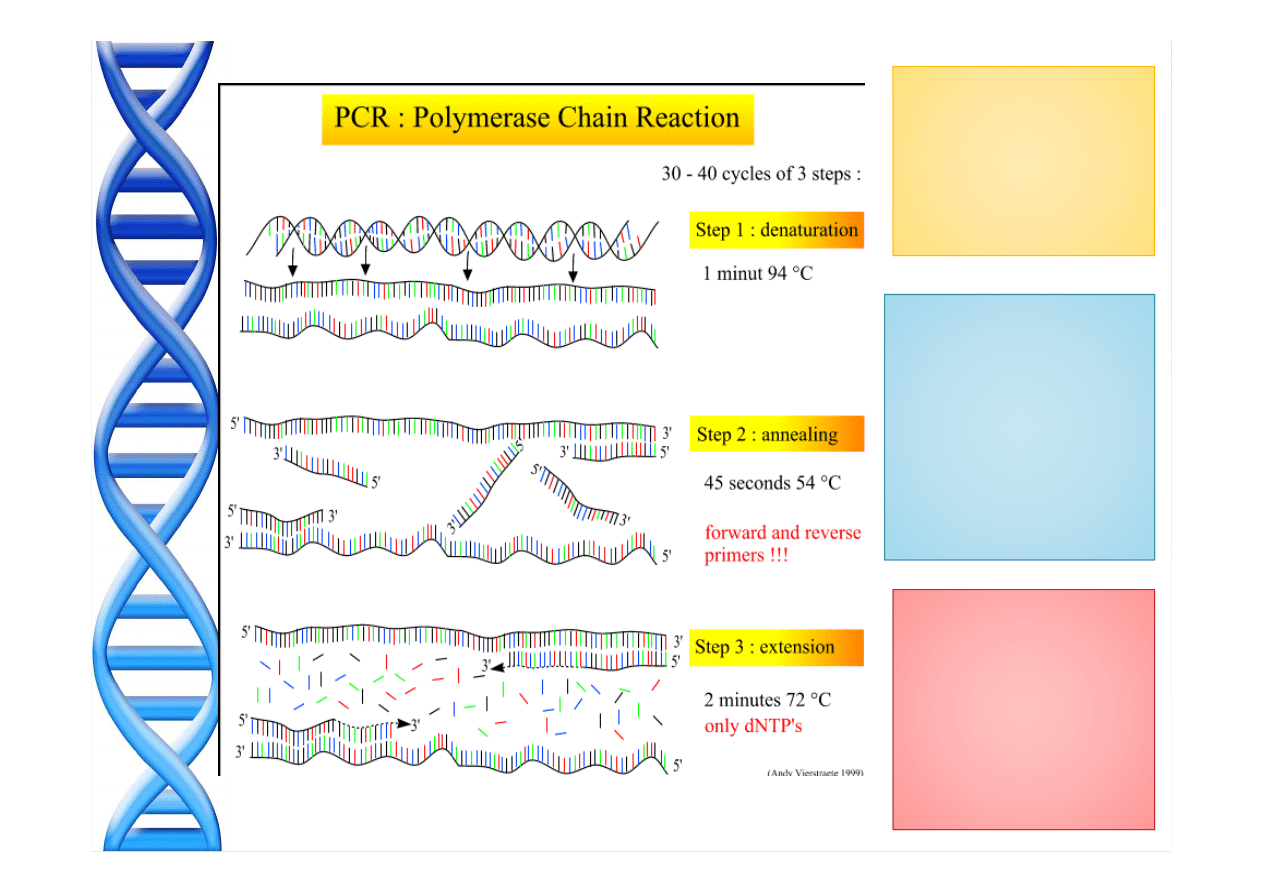
1.Denaturacja termiczna
chromosomalnego DNA- w
temperaturze 90-94
o
C –
dochodzi do rozdzielenia
podwójnej helisy DNA, co
umożliwia przyłączenie
sterterów
2. Przyłączanie
starterów – w
temperaturze ok. 55
o
C,
dochodzi do specyficznego
przyłączenia starterów do
komplementarnych
komplementarnych
odcinków, położonych po
obu stronach powielanego
fragmentu DNA
3. Elongacja –
podniesienie temp. do 72
o
C,
wartości optymalnej dla
polimerazy Taq prowadzi do
syntezy fragmentów DNA
na podstawie sekwencji
matrycy ograniczonej
straterami
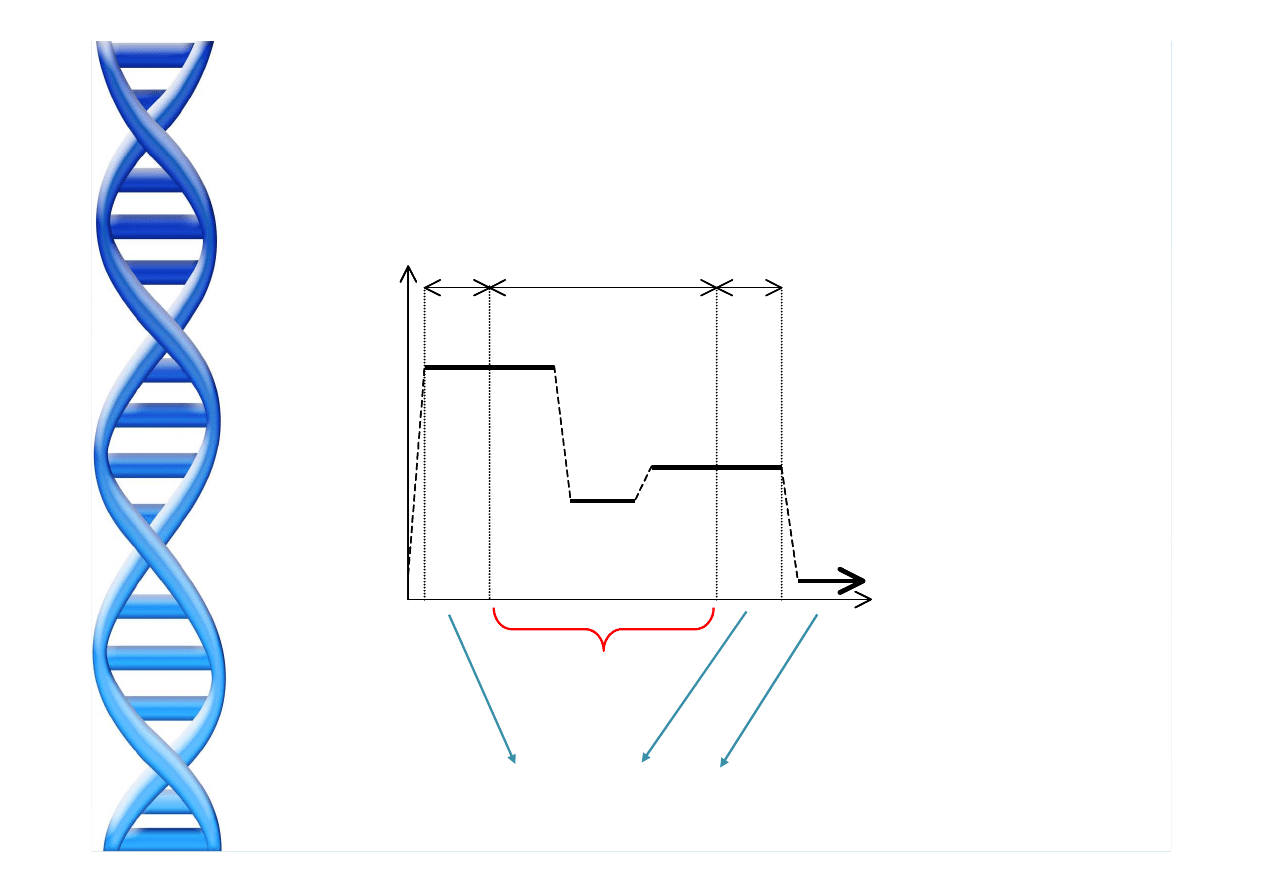
94°C
15s
94°C
3 min
te
m
p
er
at
u
ra
15x
1x
1x
94°C
15s
94°C
3 min
te
m
p
er
at
u
ra
15x
1x
1x
Profil termiczny reakcji PCR
Profil termiczny reakcji PCR
65°C
15s
72°C
30s
czas
72°C
30s
4°C
±∞
65°C
15s
72°C
30s
czas
72°C
30s
4°C
±∞
cykl
podstawowy
cykle dodatkowe

Łańcuchowa reakcja polimerazy
Łańcuchowa reakcja polimerazy --PCR
PCR
Kolejny cykl rozpoczyna się wzrostem temperatury w celu przerwania
reakcji amplifikacji i ponownej denaturacji podwójnej helisy DNA. Wraz z
kolejnymi powtórzeniami cyklu liczba syntetyzowanych fragmentów narasta
wykładniczo →
2
n
(n-liczba cykli)
Po 30-40 cyklach uzyskuje się amplifikację wybranego fragmentu DNA
nawet klika milionów razy.
Uzyskany materiał może być w dalszym ciągu poddawany analizie
hybrydyzacyjnej, restrykcyjnej lub może być sekwencjonowany.
Metodę PCR wykorzystuje się obecnie powszechnie:
•
do wykrywania infekcji wirusowych, również obecności wirusa HIV we krwi
pacjentów chorych na AIDS (lub podejrzewanych o chorobę);
•
do badania samoczynnie powstających nowotworów ludzkich, aby określić
ewentualne zmiany w sekwencji genów kontrolujących wzrost i podział
komórek;
•
przed przeszczepami, do określania typu genów, od których zależy układ
zgodności tkankowej;
•
PCR okazała się również potężnym narzędziem medycyny sądowej, dzięki
temu, że próbka niezbędna do wykonania takich badań może być znacznie
mniejsza niż wymagają tego analizy bezpośrednie.
Najważniejsze odmiany techniki PCR
Najważniejsze odmiany techniki PCR
Asymetryczny PCR
- celem asymetrycznego PCR jest amplifikacja
jednoniciowego DNA o określonej długości.
Amplifikacja allelo-specyficzna
(ASA - allelo-specific amplification)
Wewnętrzny PCR
(nested PCR)
Multipleksowy PCR
(multiplex PCR)
Różnicowy PCR
(differential PCR)
Amplifikacja nieznanych sekwencji
Amplifikacja nieznanych sekwencji

Amplifikacja
Amplifikacja allelo
allelo--specyficzna
specyficzna (ASA
(ASA-- allelo
allelo--specific
specific
amplification)
amplification)
Podstawą tej odmiany techniki PCR jest założenie, że niekomplementarność
nukleotydów przy końcu 3' jednego lub obu stosowanych primerów zapobiega
elongacji końca 3' primera przez polimerazę Taq. Oczywistym jest więc, że
można tutaj stosować tylko polimerazy DNA pozbawione aktywności 3'-5'
egzonukleazy.
Wydajna inicjacja syntezy DNA w takim PCR jest determinowana przez
sekwencję ostatniego lub dwóch ostatnich nukleotydów na końcu 3'
primerów.
primerów.
Jeśli są one komplementarne do matrycy, to badane allele są wydajnie
amplifikowane, a przy braku komplementarności dla innych alleli, amplifikacja
ich jest zahamowana.
Zaleca się stosowanie krótkich primerów (np. 14 nt). Każdy allel musi być
testowany w oddzielnej probówce reakcyjnej. Stąd, ASA wymaga bezwzględnie
koamplifikacji kontrolnego DNA dla sprawdzenia możliwego zahamowania
reakcji. Stosowanie takiej kontroli jest kluczowe w tej metodzie, ponieważ
brak produktu w reakcji ma takie same znaczenie diagnostyczne jak jego
obecność.

wyjaśniania mechanizmu badanej choroby,
wyjaśniania ewolucyjnych powiązań między gatunkami,
badania mechanizmu działania substancji mutagennych,
ASA jest metodą wykrywania polimorfizmu alleli,
ASA jest metodą wykrywania polimorfizmu alleli,
istnienia punktowych mutacji i jest wykorzystywana do:
istnienia punktowych mutacji i jest wykorzystywana do:
wykrywania sprzężonych z chorobą genów w diagnostyce pewnych
chorób,
badania zasad powstawania oporności przeciw chemioterapeutykom u
mikroorganizmów,
badania powiązań genetycznych.
Amplifikacja
Amplifikacja allelo
allelo--specyficzna
specyficzna
5'
3'
5'
3'
allel 1
G
C
primer 1
primer 3
5'
3'
primer 1
produkt PCR
A
5'
3'
5'
3'
primer 3
brak produktu PCR
allel 2
primer 1
C
C
5'
3'
5'
3'
C
primer 1
primer 3
5'
3'
5'
3'
allel 1
G
primer 3
B
primer 2
G
brak produktu PCR
5'
3'
5'
3'
G
C
primer 3
5'
3'
5'
3'
primer 3
produkt PCR
primer 2
allel 2
brak produktu PCR
primer 2
G
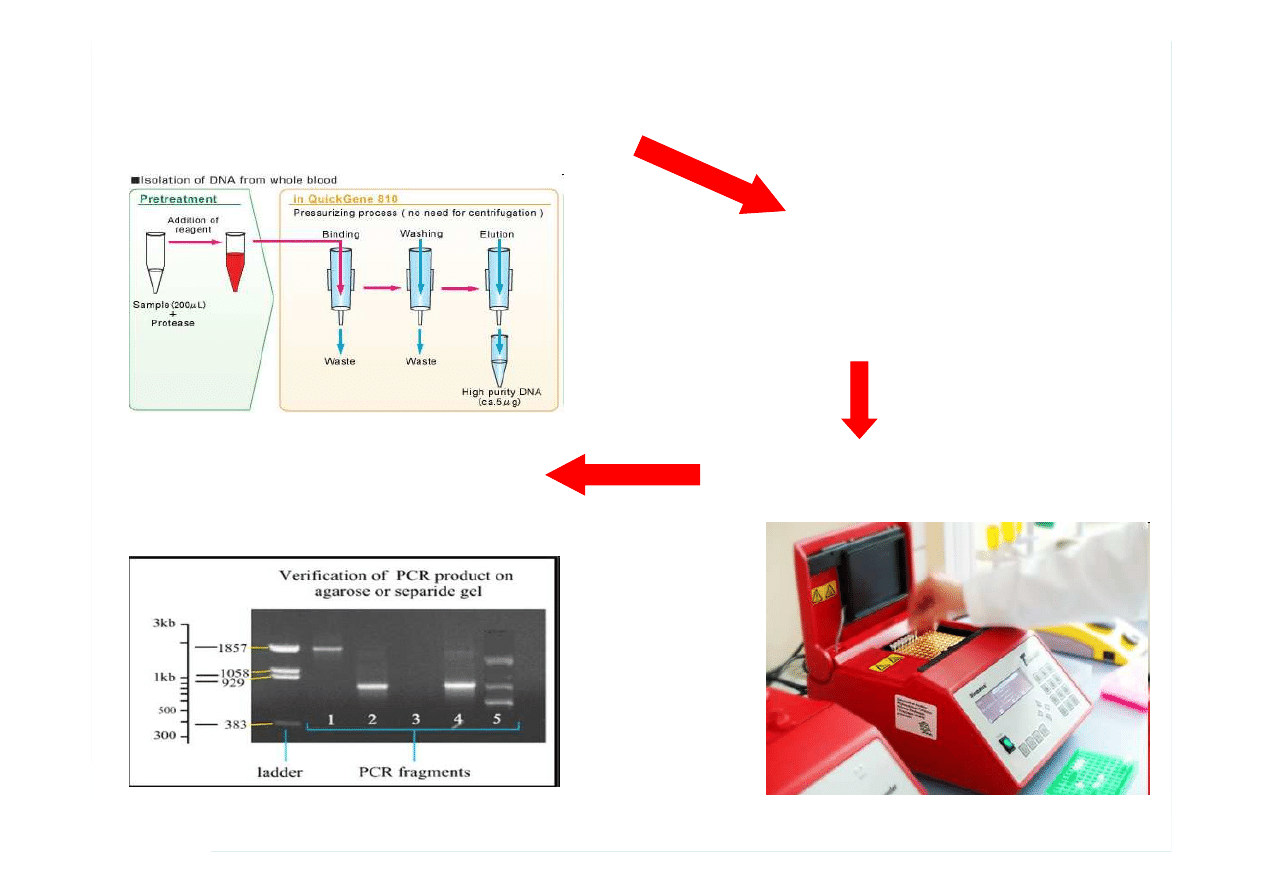
Analiza za pomocą reakcji PCR
Analiza za pomocą reakcji PCR
1. Izolacja materiału, np. DNA z krwi
2. Sporządzenie mieszaniny reakcyjnej, np.
MasterMix+ startery specyficzne dla
fragmentu szukanego genu +DNA badane
Rys. Schemat izolacji DNA z krwi
3. Reakcja PCR w termocyklerze z
określoną ilością cykli
4. Rozdział produktu reakcji PCR
na żelu agarozowym i wizualizacja
w świetle UV
Rys. Zdjęcie termocyklera
Rys. Wynik rozdziału elektroforetycznego
produktu PCR
Analiza restrykcyjna
Analiza restrykcyjna
Enzymy restrykcyjne występują w bakteriach i stanowią element systemu
obronnego, chroniącego te organizmy przed wirusami. Maja one zdolność
specyficznego rozpoznawania sekwencji DNA, składających się z 4-8
nukleotydów i przecinania nici DNA w określonym miejscu tej sekwencji lub w
ściśle określonej odległości od niej.
5’ GGTACCAATTCAAAGCTTATG 3’
3’ CCATGGTTAAGTTTCGAATAC 5’
Alu I
Kpn I
GGTAC
↓↓↓↓
C
C
↑↑↑↑
CATGG
5’
GGTAC
3’
3’
C
5’
5’
C
AATTCAAAGCTTATG 3’
3’
CATGG
TTAAGTTTCGAATAC 5’
Hind III
A
↓↓↓↓
AGCTT
TTCGA
↑↑↑↑
A
5’ GGTACCAATTCA
A
3’
3’ CCATGGTTAAGT
TTCGA
5’
5’
AGCTT
ATG 3’
3’
A
TAC 5’
Alu I
AG
↓↓↓↓
CT
TC
↑↑↑↑
GA
5’
CT
TATG 3’
3’
GA
ATAC 5’
5’ GGTACCAATTCAA
AG
3’
3’ CCATGGTTAAGTT
TC
5’
Polimorfizm długości fragmentów
Polimorfizm długości fragmentów
restrykcyjnych
restrykcyjnych –
– RFLP
RFLP
Allele polimorficzne lub zmiany rodzaju mutacji, najczęściej typu punktowego, często prowadzą
do zanikania lub powstawania nowych miejsc w cząsteczce DNA, rozpoznawanych i
przecinanych przez dany enzym restrykcyjny.
Zjawisko to wykorzystuje się do detekcji tych zmian za pomocą analizy restrykcyjnej.
Najczęściej analiza ta jest poprzedzona namnożeniem przy użyciu metody PCR danego regionu
DNA (PCR-RFLP) np. określonego egzonu badanego genu, gdzie występują różnice w sekwencji
nukleotydów, determinujące zmienność polimorficzną.
Uzyskany produkt amplifikacji jest następnie poddawany działaniu określonego enzymu
restrykcyjnemu. Wielkość i liczba fragmentów DNA, uzyskiwanych w wyniku trawienia danej
restrykcyjnemu. Wielkość i liczba fragmentów DNA, uzyskiwanych w wyniku trawienia danej
cząsteczki DNA, jest zależna od liczby miejsc o sekwencji rozpoznawalnej przez dany enzym
restrykcyjny.
Fragmenty cząsteczek DNA można łatwo rozdzielić, stosując metodę elektroforezy w żelach
agarozowych lub poliakrylamidowych, w których fragmenty DNA poddane działaniu pola
elektrycznego wędrują z prędkością zależną od ich masy.
W ostatecznym efekcie uzyskuje się charakterystyczny dla danego allelu obraz prążków na żelu
Polimorficzne miejsce restrykcyjne
DNA
(allel I)
DNA
(allel II)
4 fragmenty
3 fragmenty
Dodanie nukleazy
restrykcyjnej
PCR
PCR--RFLP
RFLP analiza żelu
analiza żelu
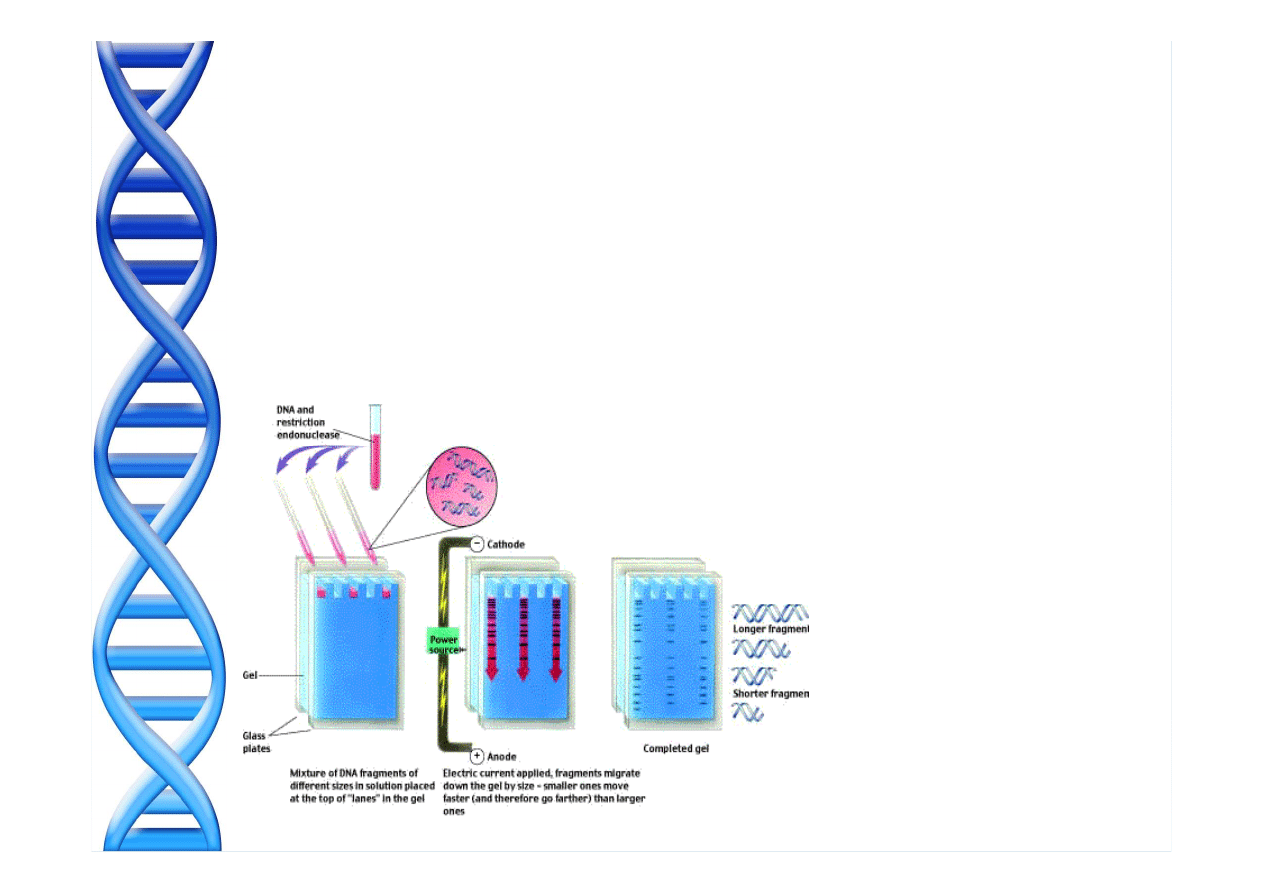
Elektroforeza w żelu
Elektroforeza w żelu
Elektroforeza w żelu to standardowa metoda rozdziału cząsteczek DNA różnej długości. Ma
wiele zastosowań w analizie RFLP, oczyszczaniu pojedynczych fragmentów do klonowania czy
sekwencjonowania, badaniu produktów PCR. Może także służyć do rozdzielania cząsteczek
RNA.
Elektroforeza to przemieszczanie się naładowanych cząsteczek w polu elektrycznym. Ujemnie
naładowane cząsteczki wędrują w kierunku elektrody (+), a dodatnio naładowane - w kierunku
elektrody (-).
Elektroforezę przeprowadza się w żelu, będącym siecią porów przez które cząsteczki DNA
musza się przeciskać, aby dotrzeć do anody. Krótsze cząsteczki są słabiej zatrzymywane przez
pory niż cząsteczki dłuższe, a więc wędrują przez żel szybciej.
W biologii molekularnej używa się
W biologii molekularnej używa się
dwóch rodzajów żeli: agarozowych i
poliakrylamidowych.
Agaroza to polisacharyd
tworzący żele o porach
osiągających 100-300 nm średnicy,
wielkość zależy od stężenia agarozy
w żelu. Stężenie żelu determinuje
rozmiar fragmentów DNA, które
można rozdzielić, np. żelu 0,3%
używa się do cząsteczek między 5-
50 kb, a żelu 5% do cząsteczek 100-
500 kb.
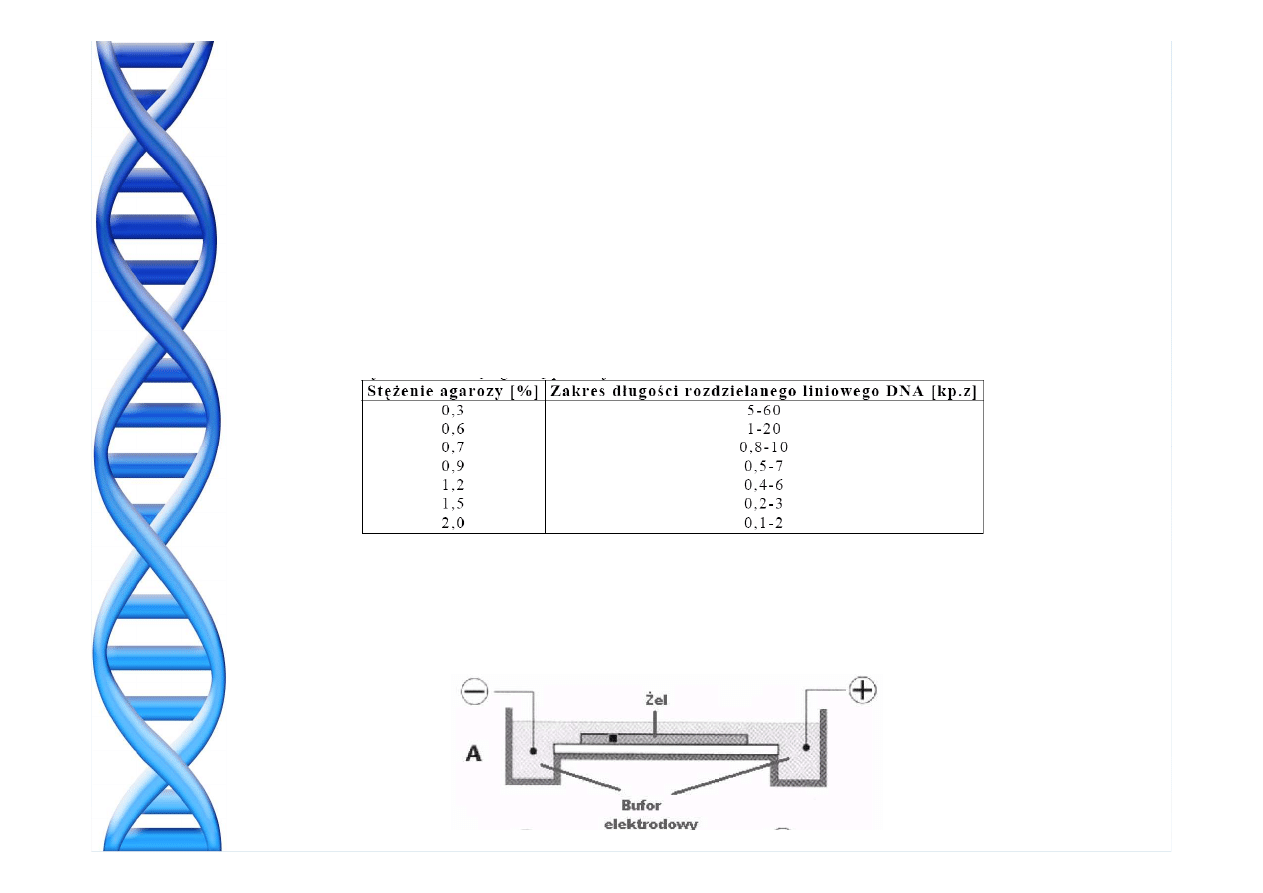
ELEKTROFOREZA W śELU AGAROZOWYM
ELEKTROFOREZA W śELU AGAROZOWYM
Zaletą żeli agarozowych jest łatwość i szybkość ich przygotowania oraz możliwość separacji dużych
makromolekuł np. DNA i RNA.
śel agarozowy stosuję się do rozdzielania DNA o szerokim zakresie mas cząsteczkowych
Wadą zaś jest słaba wytrzymałość mechaniczna żeli oraz trudność ich utrwalania po rozdziale.
Wyschnięte żele rozsypują się pod wpływem bardzo niewielkich sił.
Wadą elektroforezy w żelu agarozowym jest również słaba rozdzielczość frakcji DNA różniącego się
poniżej 5% wielkości.
Elektroforeza w żelu agarozowym prowadzona jest w aparatach ustawionych poziomo, a rozdział
elektroforetyczny prowadzony jest w buforze TBE×1 (90 mM Tris-base, 90 mM kwas borowy, 2 mM
EDTA, pH 8) lub TAE×1(40 mM Tris-base, 40 mM lodowy kwas octowy, 1 mM EDTA, pH 8).

ELEKTROFOREZA W śELU AGAROZOWYM
ELEKTROFOREZA W śELU AGAROZOWYM
Cząsteczka DNA jest naładowana ujemnie w środowisku obojętnym i alkalicznym, a
więc umieszczona w polu elektrycznym, przemieszcza się w kierunku anody.
DNA o tych samych masach cząsteczkowych, ale różnych konformacjach
charakteryzuje różna ruchliwość elektroforetyczna. Im dłuższa jest cząsteczka DNA
lub RNA tym dłużej odnajduje ona drogę poprzez pory żelu. Jeżeli umieścimy DNA w
żelu agarozowym i przyłożymy niskie napięcie to prędkość migracji DNA o różnych
ciężarach cząsteczkowych jest proporcjonalna do napięcia. Aby otrzymać optymalny
rozdział DNA o wielkości większej niż 2 kp.z, elektroforeza prowadzona jest w polu
elektrycznym o natężeniu nie większym niż 5V/cm. Powyżej tego napięcia fragmenty
elektrycznym o natężeniu nie większym niż 5V/cm. Powyżej tego napięcia fragmenty
DNA przemieszczają się z prędkością odwrotnie proporcjonalną do logarytmu ich
ciężaru cząsteczkowego.
Do uwidocznienia DNA po lub w trakcie elektroforezy stosowany jest rutynowo
bromek etydyny (EtBr), który interkaluje pomiędzy sąsiednie pary dwuniciowego DNA
(powinowactwo EtBr do jednoniciowego DNA jest znacznie słabsze). Należy pamiętać,
że obecność związku interaklującego do DNA w żelu agarozowym zmniejsza
ruchliwość elektroforetyczną DNA o około 15% (EtBr migruje podczas elektroforezy
w kierunku elektrody (-), co powoduje jego wypłukiwanie z żelu do buforu podczas
elektroforezy) . Innym barwnikiem DNA jest np. SYBR Green, którego czułość
barwienia dwuniciowego DNA jest około 25 razy większa niż EtBr.
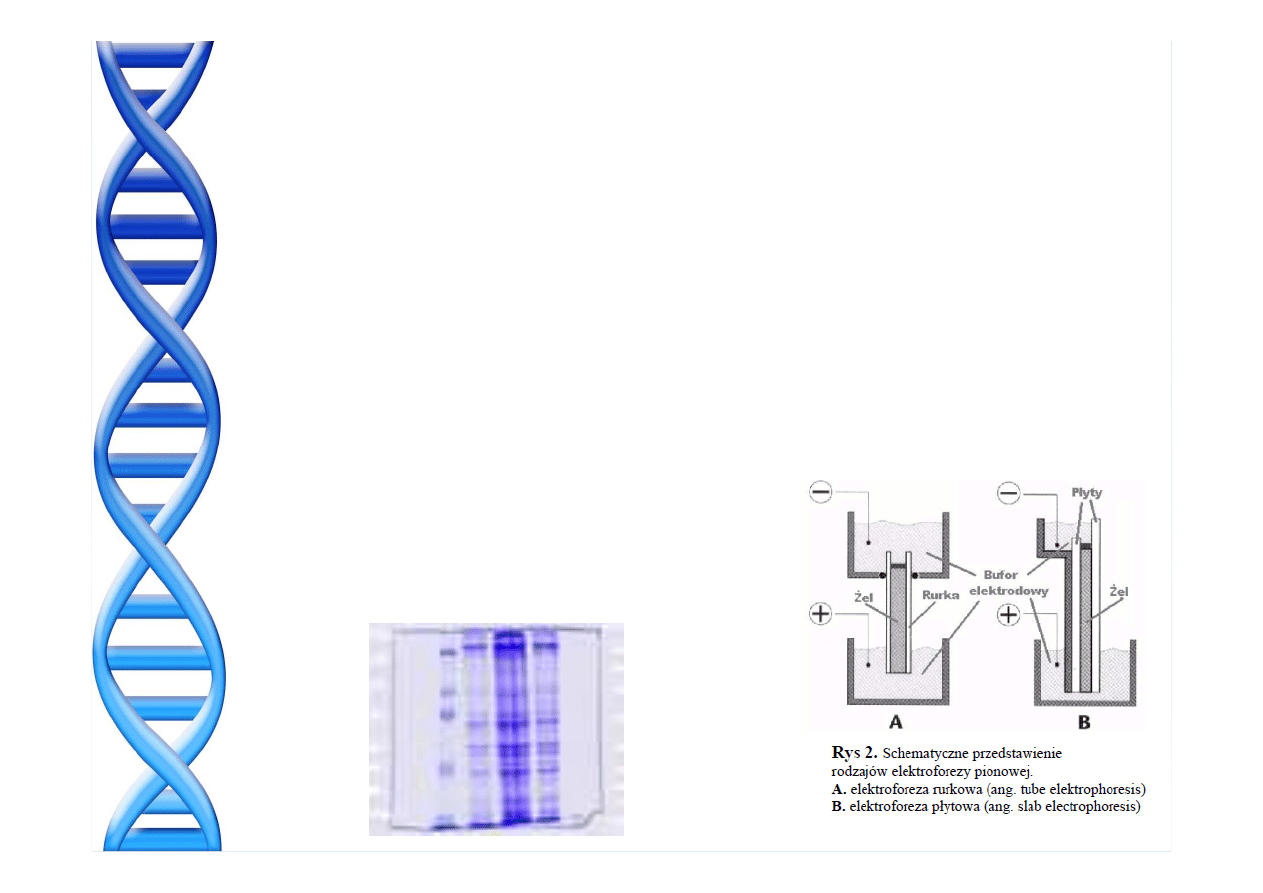
ELEKTROFOREZA W śELU POLIAKRYLOAMIDOWYM
ELEKTROFOREZA W śELU POLIAKRYLOAMIDOWYM
Elektroforezy w żelu poliakryloamidowym używa się do rozdziału cząsteczek DNA otrzymanych
podczas sekwencjonowania metodą terminacji łańcucha. Nie można tutaj zastosować
elektroforezy w żelu agarozowym, gdyż metoda ta nie ma wystarczającej rozdzielczości, to znaczy
zdolności odseparowania od siebie jednoniciowych cząsteczek DNA różniących się długością o
jeden nukleotyd. śele poliakryloamidowe mają mniejsze pory niż żele agarozowe, co umożliwia
dokładniejszy rozdział cząsteczek DNA w zakresie wielkości. 10-1500 bp.
śel poliakrylamidowy zbudowany jest z łańcuchów monomerów akrylamidu poprzecznie
połączonych z cząsteczkami N, N’-metylenobisakryloamidu (bis-akrylamid).
Reakcję polimeryzacji, będącą w istocie wolnorodnikową reakcją polimeryzacji, można zainicjować
chemicznie lub fotochemicznie. Przy chemicznej inicjacji procesu najczęściej stosuje się
nadsiarczan amonu w obecności katalizatora N,N,N’,N’-tetrametyletylenodiaminy (TEMED).
śele poliakryloamidowe dodatkowo zawierają mocznik, którego właściwości denaturujące
zapobiegają tworzeniu się dwuniciowych fragmentów DNA.
zapobiegają tworzeniu się dwuniciowych fragmentów DNA.
śele poliakryloamidowe przygotowuje się pomiędzy dwiema
szklanymi płytkami oddzielonymi od siebie przekładkami. Służy to
m.in. otrzymaniu bardzo cienkich żeli czego efektem jest
zwiększenie ostrości prążków oraz umożliwia równomierną
polimeryzacje żelu (hamowaną przez tlen).
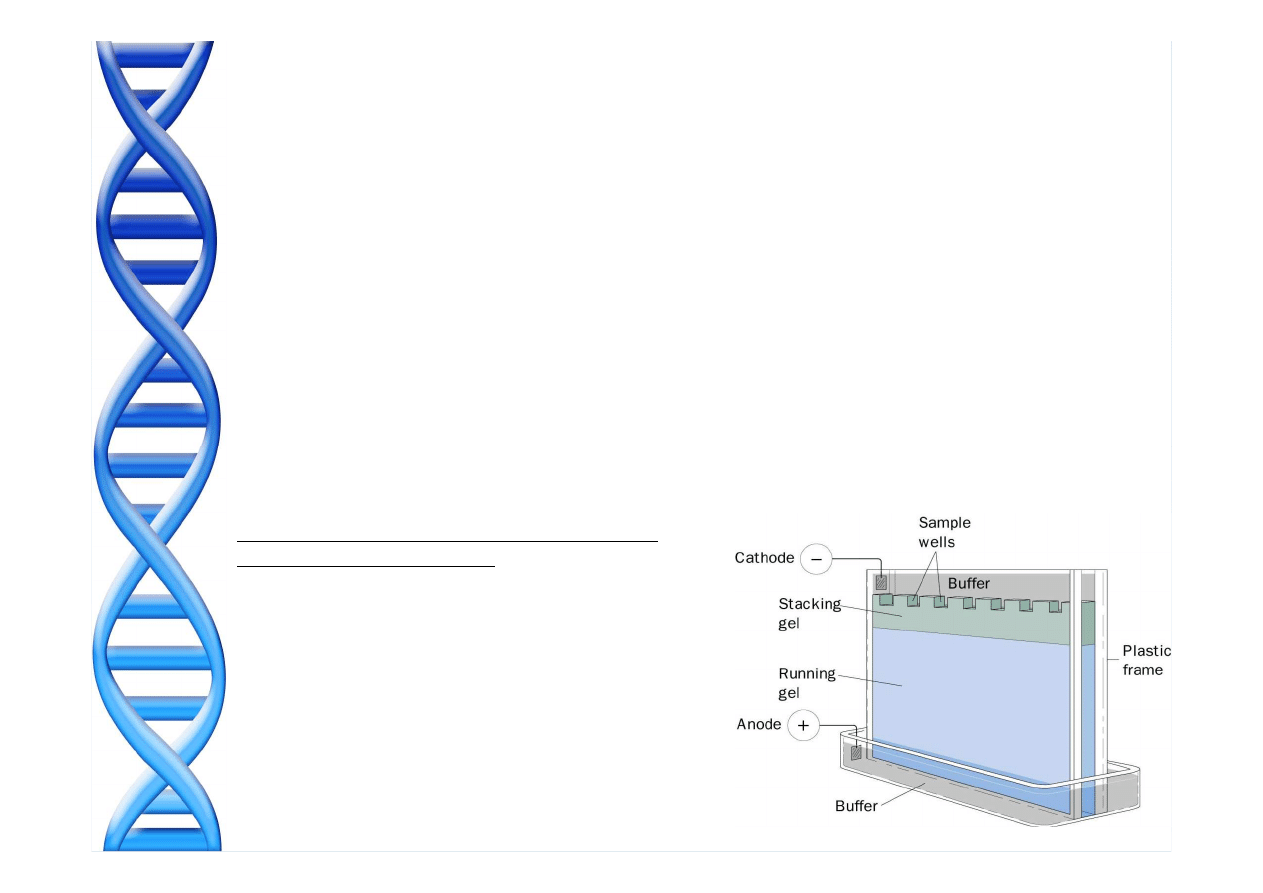
Elektroforeza poliakrylamidowa DNA
Elektroforeza poliakrylamidowa DNA
Najlepszy rozdział uzyskuje się dla DNA o długości mniejszej niż 1000 par zasad.
W żelach tych można efektywnie rozdzielać także jednoniciowe fragmenty
DNA i RNA.
Elektroforezę w żelach poliakrylamidowych prowadzona jest w aparaturze
ustawionej pionowo, a więc migracja cząsteczek DNA jest zgodna z kierunkiem
siły ciążenia, gdzie jednoniciowe DNA charakteryzują się spowolnioną, w
stosunku do dwuniciowego DNA, migracją w żelu.
Rozdział elektroforetyczny prowadzony jest w buforze TBE×1.
DNA w żelu poliakrylamidowym można wybarwiać za pomocą np. EtBr lub
DNA w żelu poliakrylamidowym można wybarwiać za pomocą np. EtBr lub
Sybr Gold.
śele poliakrylamidowe mają przewagę nad żelami
agarozowymi z kilku przyczyn:
zdolność rozdzielcza żeli poliakrylamidowych jest tak
duża, że można rozdzielać cząsteczki DNA różniące się
o 1 p.z.
studzienkę żelu poliakrylamidowego można
załadować znacznie większą ilością DNA niż w żelu
agarozowym
DNA odzyskany z żelu poliakrylamidowego jest
czysty (nie wymaga dodatkowego czyszczenia przy
stosowaniu w biologii molekularnej)
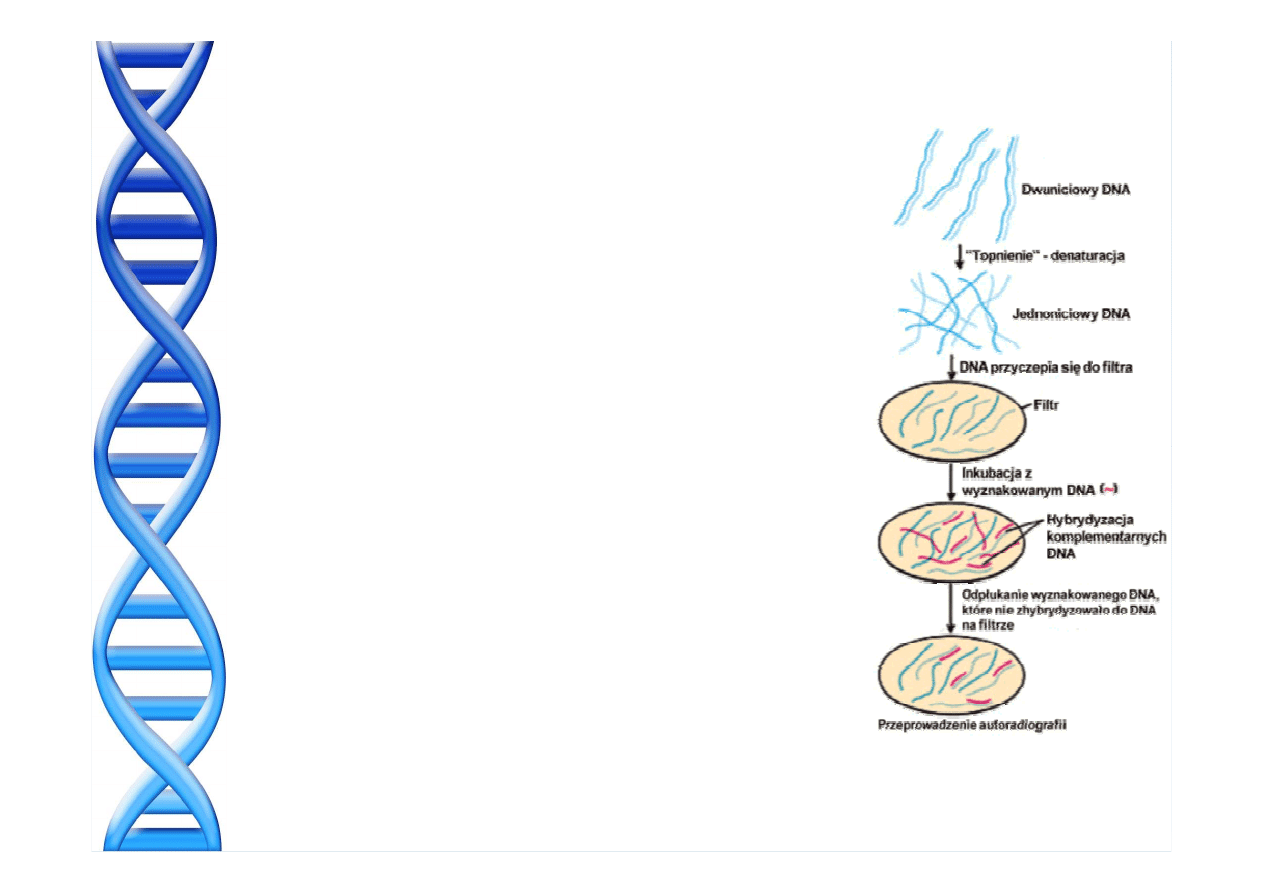
Metody hybrydyzacji
Metody hybrydyzacji
Wykorzystują zdolność łączenia się
jednoniciowych cząsteczek kwasów
nukleinowych o całkowicie lub częściowo
komplementarnej sekwencji w struktury
dwuniciowe
Jedna z nici reprezentuje badany kwas
nukleinowy, a druga pełni funkcje
znakowanej sondy
znakowanej sondy
Hybrydyzacja Southerna
Wykrywanie swoistego fragmentu DNA przez
sondę DNA znakowaną izotopem
promieniotwórczym lub barwnikiem
fluorescencencyjnym
Hybrydyzację Notherna
Rozdział cząsteczek mRNA i badanie ekspresji
genów
Rys. Schemat metody Southern blott
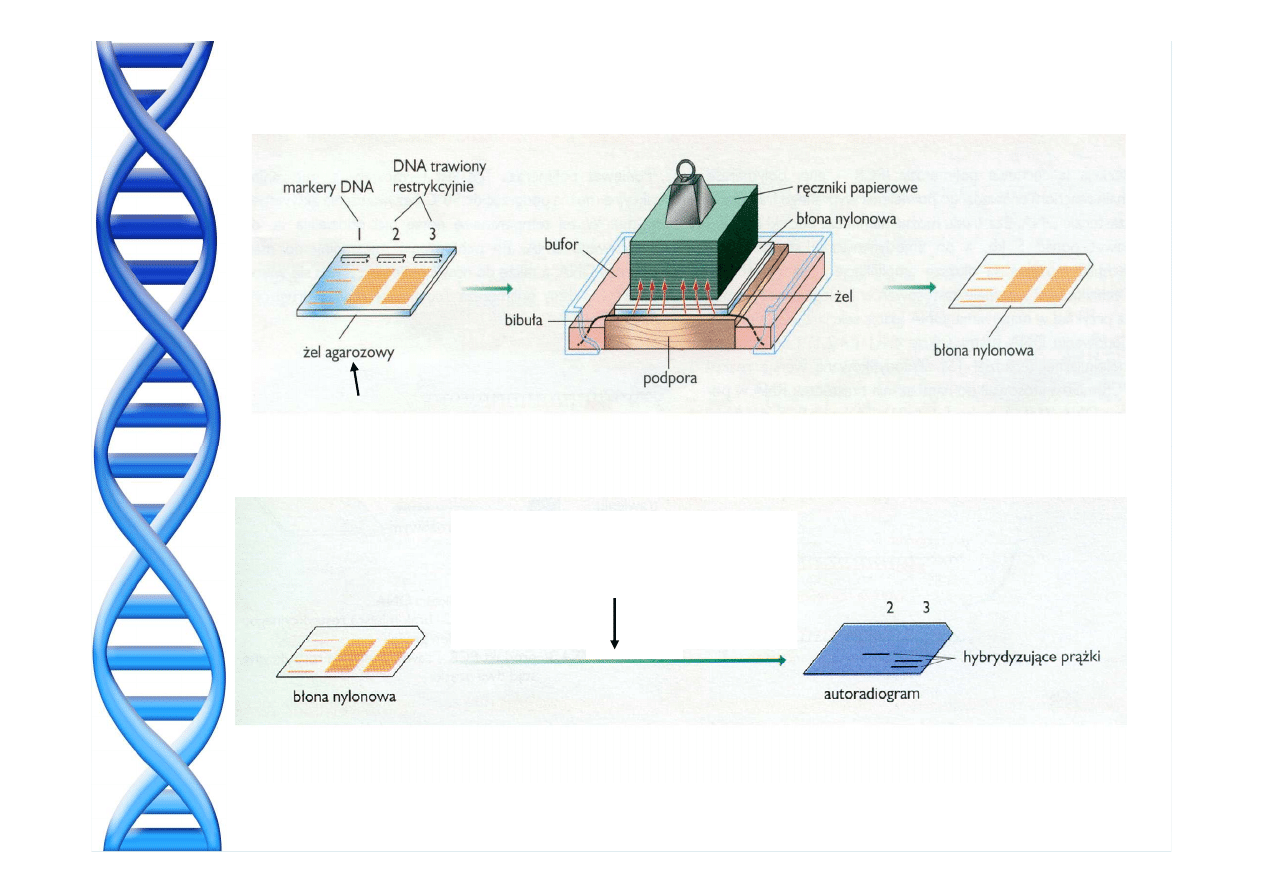
Hybrydyzacja metodą
Hybrydyzacja metodą Southerna
Southerna
denaturacja DNA w celu
uzyskania jednoniciowych cząsteczek
inkubacja błony z jednoniciową
wyznakowaną radioaktywnie sondą DNA
uzyskania jednoniciowych cząsteczek
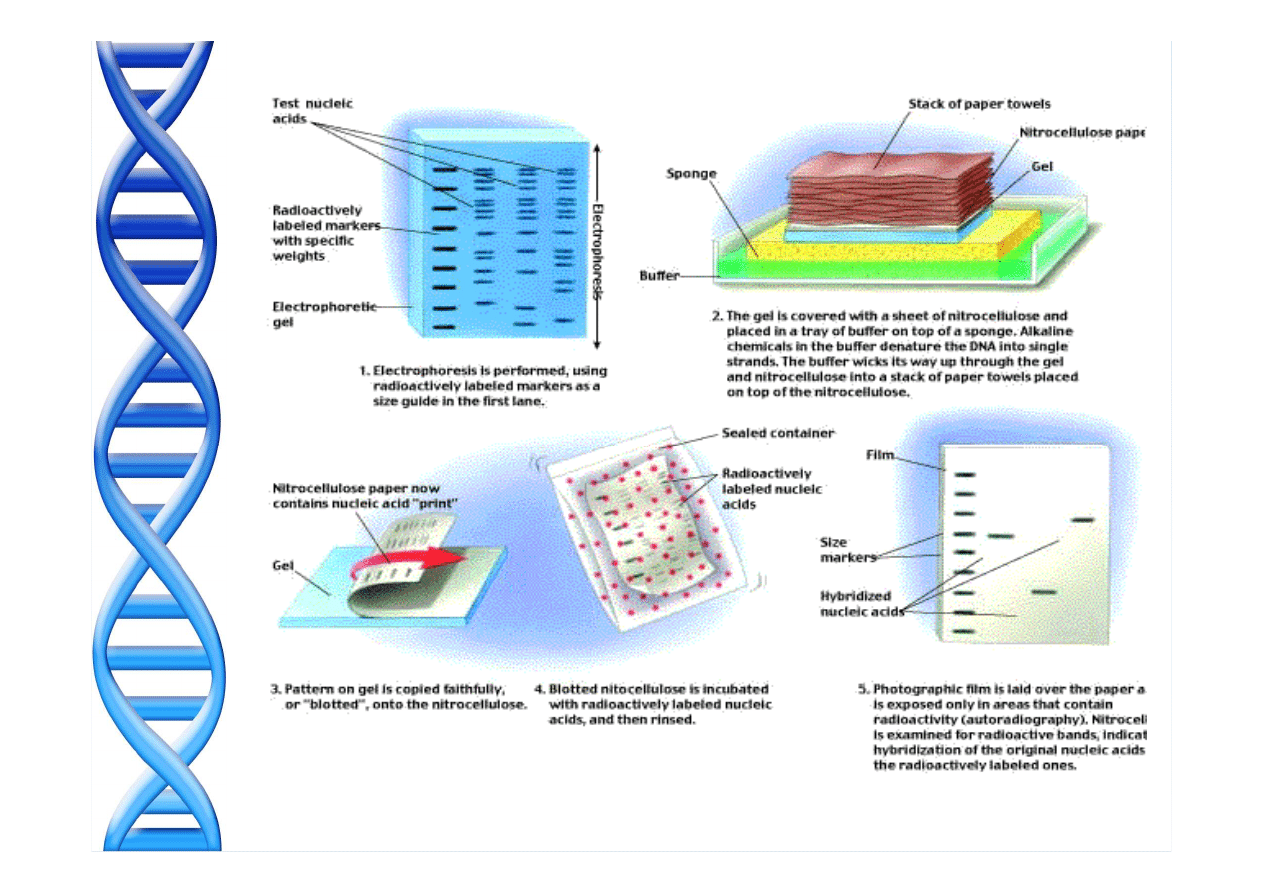
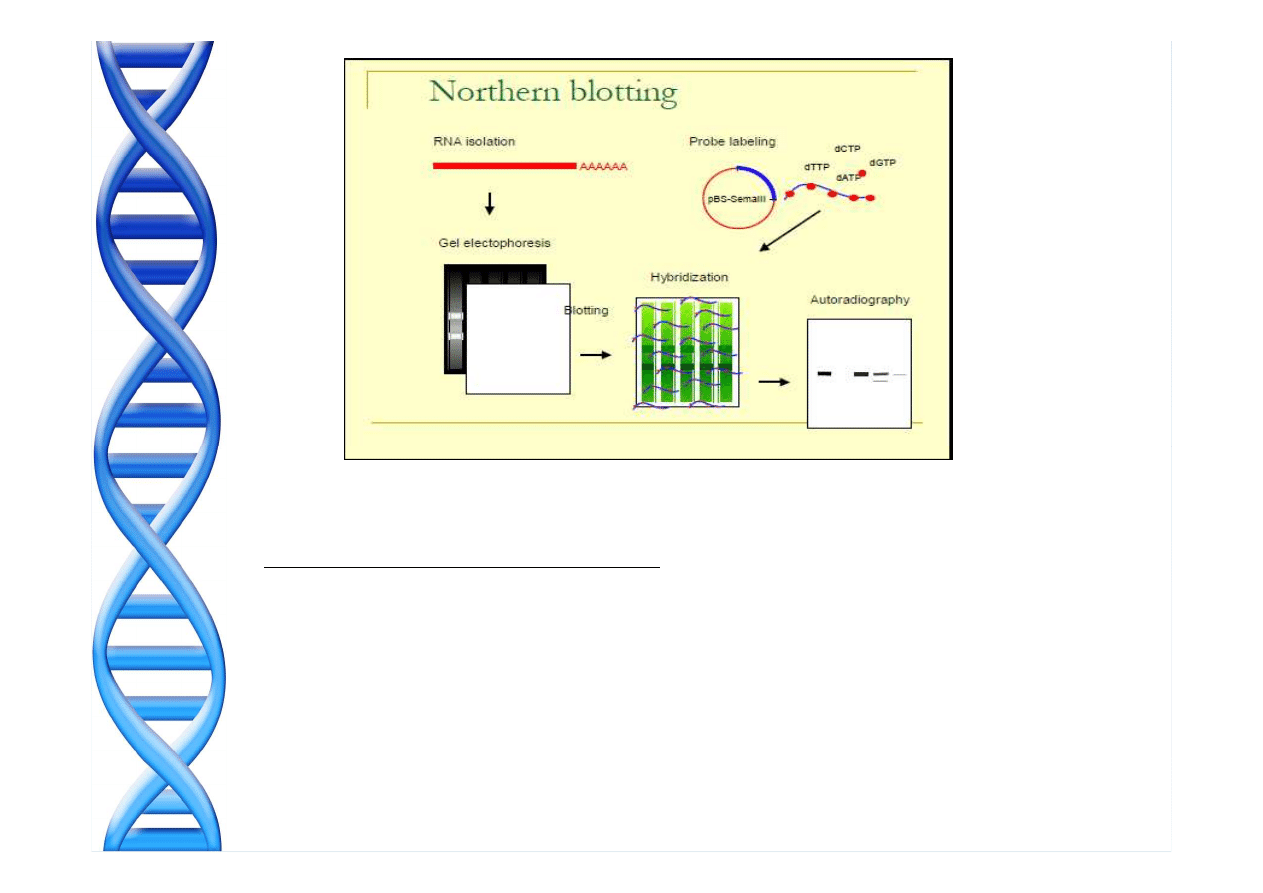
Hybrydyzacja northern (ang. northern blotting) to metoda stosowana w biologii molekularnej,
służąca detekcji kwasów rybonukleinowych (RNA). Najczęściej stosuje się ją do wykrywania mRNA
genów ulegających transkrypcji w komórce.
Metodyka jest zbliżona do hybrydyzacji Southerna:
* RNA poddaje się rozdziałowi elektroforetycznemu (elektroforezie) na żelu
* po rozdziale RNA przenosi się na odpowiednią membranę oraz utrwala na niej
* membranę z RNA inkubuje się z sondą, tzn. cząsteczką DNA lub RNA, która po pierwsze jest
komplementarna do poszukiwanej sekwencji, a po drugie daje się łatwo wykryć
* po inkubacji i odpłukaniu nadmiaru sondy przeprowadza się detekcję, czyli uwidacznianie sondy, która
związała się do poszukiwanej sekwencji RNA na membranie.
Jeżeli nie interesuje nas długość wykrywanego RNA, krok pierwszy (elektroforezę) można pominąć.
Wówczas RNA nanosi się bezpośrednio na membranę.
Sonda daje na ogół sygnał proporcjonalny do ilości RNA obecnego na membranie, stąd po intensywności
sygnału po detekcji można ilościowo oszacować poziom wykrytego RNA.
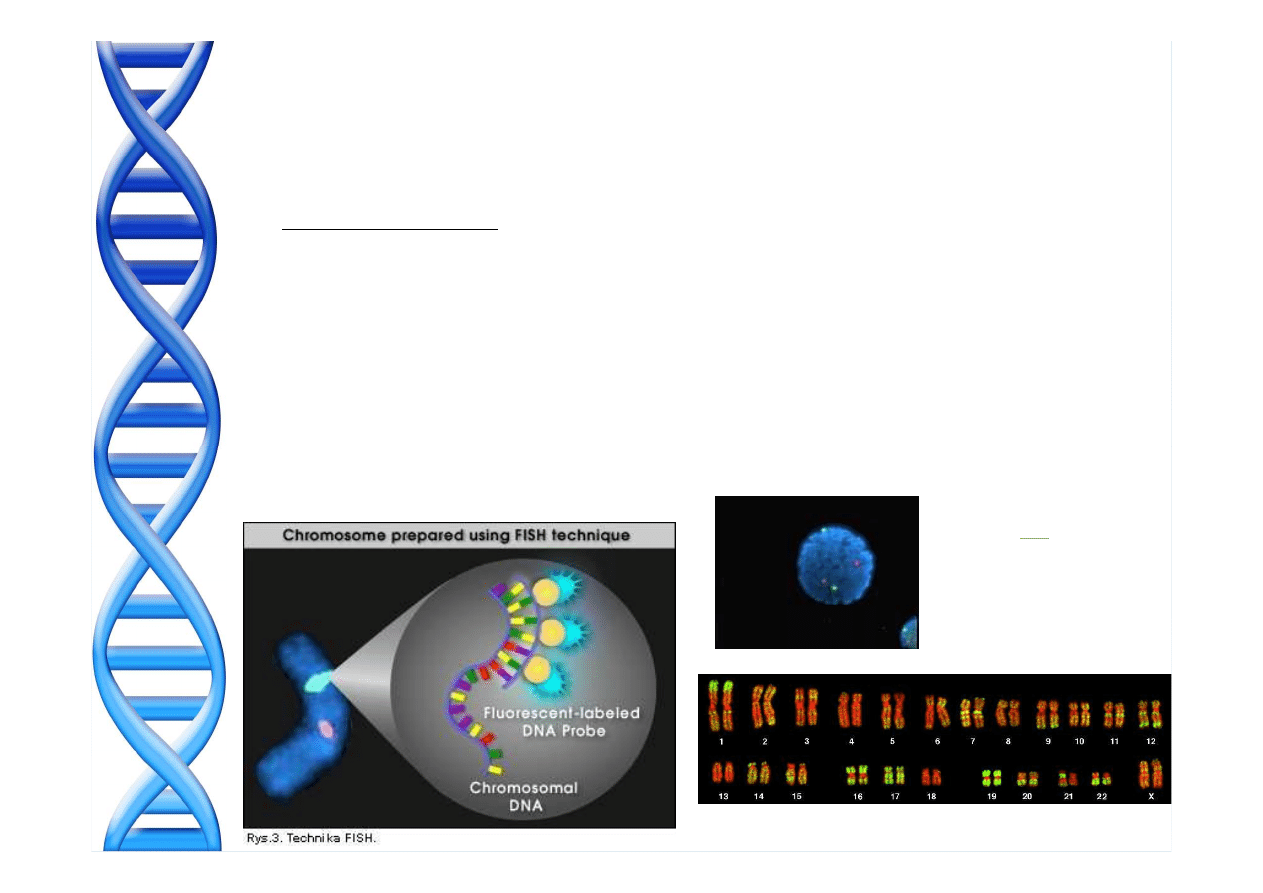
FISH
FISH –
– fluorescencyjna hybrydyzacja
fluorescencyjna hybrydyzacja in
in situ
situ
Technika umożliwia wykrywanie specyficznych sekwencji DNA w chromosomach, pojedynczych
komórkach i tkankach.
Składa się z kilku etapów:
- Przygotowanie wysokiej jakości preparatów histologicznych (polega na usunięciu cytoplazmy i
utrwaleniu jąder komórkowych na szkiełku podstawowym)
- Denaturacja preparatów 70% formamidem o temp. 70ºC
- Dodanie sond molekularnych znakowanych flouroscencyjnie
- Płukanie nadmiaru sond
- Detekcja za pomocą trzech przeciwciał związanych z fluorochromami (popularnymi znacznikami
fluorescencyjnymi, używanymi do znakowania sond są rodamina i fluoresceina, a także
fluorescencyjnymi, używanymi do znakowania sond są rodamina i fluoresceina, a także
kumaryna).
Interfazowe jądro limfocytu
wybarwione
DAPI
z sygnałami
fluorescencyjnymi z chromosomu
13 (zielony, sonda znakowana
fluoresceiną) i 21 (czerwony,
rodamina jako znacznik
fluorescencyjny)
Chromosomy metafazowe człowieka (kariotyp żeński) poddane badaniu FISH

Zastosowanie metody FISH
Zastosowanie metody FISH
Technika FISH może być wykorzystana do badania wielu typów komórek i tkanek.
Mogą to być limfocyty (jądra metafazowe oraz interfazowe) uzyskane w wyniku hodowli
komórkowej, jak również w bezpośrednim rozmazie krwi, rozmazy szpiku kostnego, preparaty
moczu, komórki guzów nowotworowych, blastomery, polocyty, czy amniocyty.
Technikę FISH stosuje się chętnie w przypadkach, gdy zawodzi klasyczna analiza cytogenetyczna,
innymi słowy gdy w badaniu kariotypu niemożliwe jest ustalenie dokładnego rodzaju bądź
miejsca powstania mutacji w genomie. Zastosowanie unikalnych sond umożliwia zlokalizowanie
miejsc pęknięć chromosomów czy określenie rodzaju translokacji.
miejsc pęknięć chromosomów czy określenie rodzaju translokacji.
FISH jest również narzędziem w badaniu anomalii genetycznych, będących markerami nowotworów
(cytogenetyka onkologiczna).
Metoda ta, obok techniki PCR, jest także nieocenioną pomocą w diagnostyce preimplantacyjnej,
gdzie badanie pojedynczej komórki polocytu czy blastomeru pozwala uniknąć podania zarodka,
któremu jeden lub oboje rodzice przekazali określone wady genetyczne . W badaniach
prenatalnych z wykorzystaniem amniocytów jako analizowanego materiału, możliwe jest
określenie wady genetycznej płodu.
Hybrydyzacja fluorescencyjna jest także z powodzeniem wykorzystywana do detekcji
mikroorganizmów w materiale biologicznym.
Każda sonda jest specyficzna tylko dla jednego genu
Na płytki nanoszone są sondy (kilka tysięcy)
Na płytki nanoszone jest badane cDNA lub mRNA wyznakowane np.
fluorescencyjnie
Mikromacierze
Mikromacierze DNA (
DNA (microarrays
microarrays, DNA chip)
, DNA chip)
„
Chip” DNA zaprojektowano, by umożliwić przeprowadzenie jednocześnie wielu
eksperymentów hybrydyzacyjnych. Jego głównym zastosowaniem jest
poszukiwanie polimorfizmów, takich jak SNP (polimorfizmy punktowe) i
porównywanie populacji RNA z różnych komórek. Może być także stosowany
w nowych metodach sekwencjonowania DNA.
fluorescencyjnie
Następuje wiązanie badanego cDNA lub mRNA do komplementarnych sond
Analiza wyników za pomocą odpowiedniego czytnika i komputera
Możliwa jest analiza praktycznie całego genomu badanego organizmu
Rys. Płytki do mikromacierzy
Rys. Wynik analizy za pomocą mikromacierzy

Mikromacierze
Mikromacierze DNA (
DNA (microarrays
microarrays, DNA chip)
, DNA chip)
Podstawą działania mikromacierzy jest tak, jak w tradycyjnej hybrydyzacji Southerna,
komplementarność kwasów nukleinowych.
Mikromacierz zawiera sekwencje komplementarne do badanych sekwencji. Próbkę kwasu
nukleinowego wyznakowuje się (jednym lub dwoma znacznikami fluorescencyjnymi) i
hybrydyzuje z mikromacierzą
Cząsteczki wyznakowanego kwasu nukleinowego wiążą się do komplementarnych sekwencji.
Obraz sczytuje się ilościowo (za pomocą lasera lub mikroskopu). Intensywność sygnału dla
poszczególnych sond mikromacierzy jest proporcjonalna do ilości kwasu nukleinowego o
danej sekwencji w próbce.
Z mikromacierzami DNA hybrydyzuje się cDNA zsyntetyzowany na matrycy badanego RNA,
Z mikromacierzami DNA hybrydyzuje się cDNA zsyntetyzowany na matrycy badanego RNA,
cRNA uzyskany na podstawie cDNA lub DNA (dla mikromacierzy genotypujących).
Schemat przebiegu doświadczenia:
* zebranie próbki i izolacja RNA (standardowo kilka mikrogramów, zaawansowane techniki
amplifikacji pozwalają użyć RNA z zaledwie kilkuset komórek)
* synteza cDNA na matrycy wyizolowanego RNA
* synteza wyznakowanego cRNA na podstawie cDNA (lub wyznakowanie cDNA)
* hybrydyzacja wyznakowanego kwasu nukleinowego z mikromacierzą
* płukanie mikromacierzy, w celu usunięcia niesparowanych sekwencji
* skanowanie obrazu mikromacierzy
* przekształcenie obrazu w zbiór danych wartości ekspresji dla każdej sondy

Zastosowanie
Zastosowanie mikromacierzy
mikromacierzy
w medycynie:
w medycynie:
Poznawanie mechanizmów chorobotwórczych, np.
wykorzystując
mikromacierze wykryto m.in. geny związane ze stwardnieniem rozsianym, ciężką
dziecięcą dystrofią siatkówki
Identyfikacja wskaźników pozwalających na ocenę ryzyka
rozwoju chorób
Określenie nowych obiektów, na których powinno
Określenie nowych obiektów, na których powinno
koncentrować się działanie leków
Postawienie dokładniejszej diagnozy i dostosowanie leczenia do
indywidualnego przypadku pacjenta
Diagnostyka chorób zakaźnych, np.
detekcja i identyfikacja rodzaju
wirusa, wykrywanie genów odporności na antybiotyki w szczepach bakteryjnych
Onkologia, np. odróżnienie łagodnej formy raka prostaty od
formy złośliwej
SEKWENCJONOWANIE
SEKWENCJONOWANIE
SEKWENCJONOWANIE
- to ustalenie rodzaju i kolejności nukleotydów, składających
się na zapis informacji w DNA.
Na podstawie sekwencji istnieje możliwość przewidzenia kolejności aminokwasów w
białku, które jest kodowane przez dany gen
Metody sekwencjonowania:
Metoda terminacji łańcucha
, zwana również metodą Sangera lub dideoksy, w
której sekwencje cząsteczki jednoniciowego DNA określa się dzięki enzymatycznej
której sekwencje cząsteczki jednoniciowego DNA określa się dzięki enzymatycznej
syntezie komplementarnych łańcuchów polinukleotydowych, a reakcje są
zatrzymywane losowo w pozycjach określonych nukleotydów.
Metoda chemicznej degradacji DNA
, nazywana metodą Maxama i Gilberta, w
której sekwencję określa się działając na dwuniciowy DNA związkami chemicznymi
rozszczepiającymi cząsteczki w specyficznych pozycjach poszczególnych nukleotydów.
Metoda sekwencjonowania oparta jest na zdolności do rozdziału jednoniciowych
cząsteczek DNA, różniących się między sobą długością tylko o jeden nukleotyd,
poprzez elektroforezę w żelu poliakrylamidowym. Co oznacza, że możliwe jest
rozdzielanie cząsteczek o dł. 10-1500 nukleotydów do postaci prążków widocznych
na żelu.
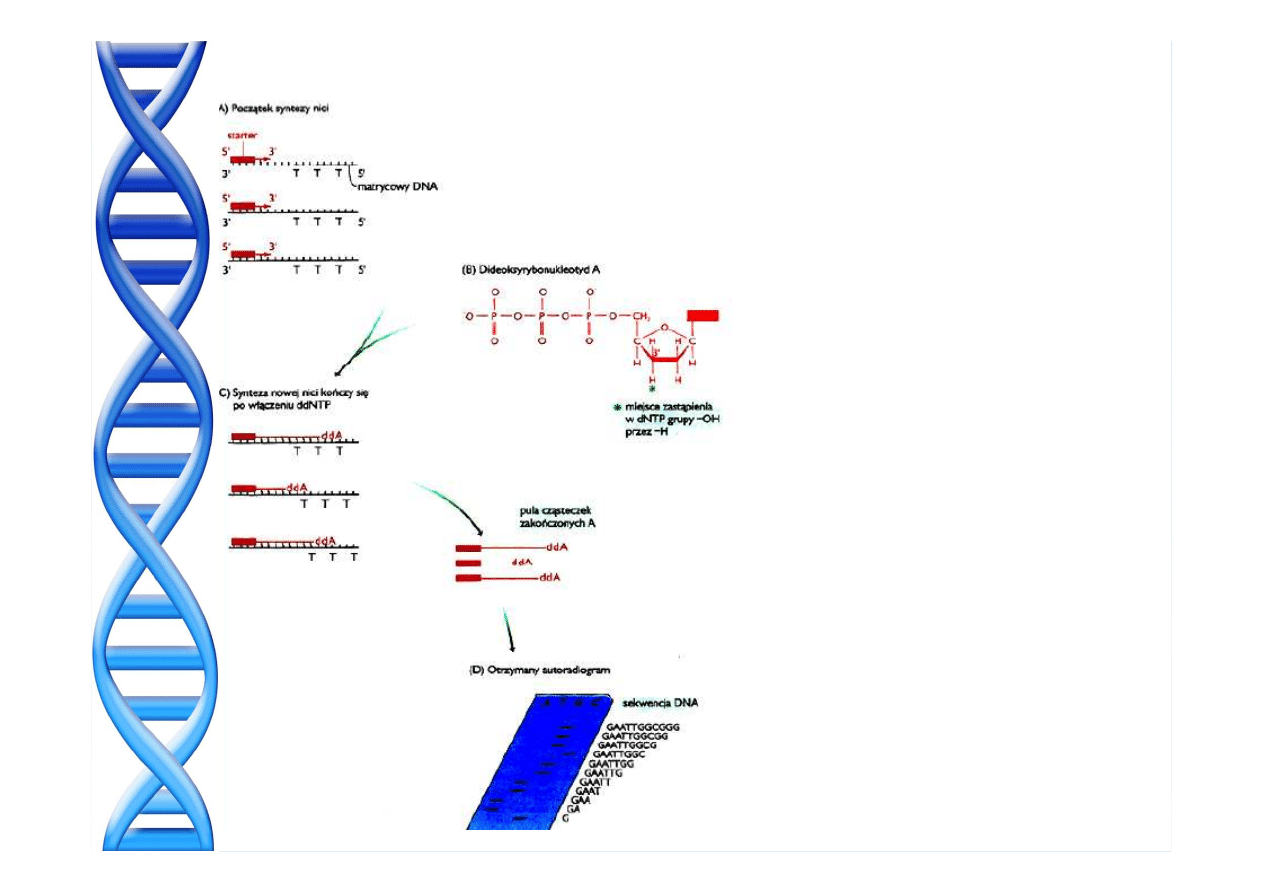
Sekwencjonowanie
Sekwencjonowanie metodą
metodą Sangera
Sangera
Materiałem wyjściowym do sekwencjonowania jest
pula identycznych jednoniciowych cząsteczek DNA.
Pierwszym etapem reakcji jest przyłączenie krótkich
oligonukleotydów w tym samym miejscu każdej
cząsteczki. Oligonukleotydy te wykorzystywane są
jako startery do syntezy nowej nici DNA,
komplementarnej do matrycy
Reakcja syntezy nici katalizowana jest przez
polimerazę DNA i wymaga, jako substratów czterech
trifosforanów deoksyrybonukleotydów (dATP, dCTP,
dGTP, dTTP) oraz małe ilości
dideoksyrybonukleotydów (ddNTP) wyznakowanych
fluorochromem, które hamują elongację ze względu
na występowanie wodoru zamiast grupy
na występowanie wodoru zamiast grupy
hydroksylowej w pozycji 3’.
Efektem dodania do reakcji np. ddATP jest losowe
zakończenie (terminacja) syntezy nowej nici w
jednym z miejsc T matrycowego DNA. Pulę
cząsteczek zakończonych ‘A’ nanosi się an pierwsza
ścieżkę żelu, a obok nanosi się cząsteczki zakończone
T, G, C.
Mieszaninę jednoniciowych produktów syntezy DNA
wyznakowanych barwnikami fluorescencyjnymi
poddaje się elektroforezie w żelu, co uszeregowuje je
rosnąco w zależności od ich długości (uzyskuje się
drabinkę fragmentów DNA różniących się jednym
nukleotydem.) i odczytuje się rodzaj włączonego
ddNTP technikami fluorescencyjnymi.
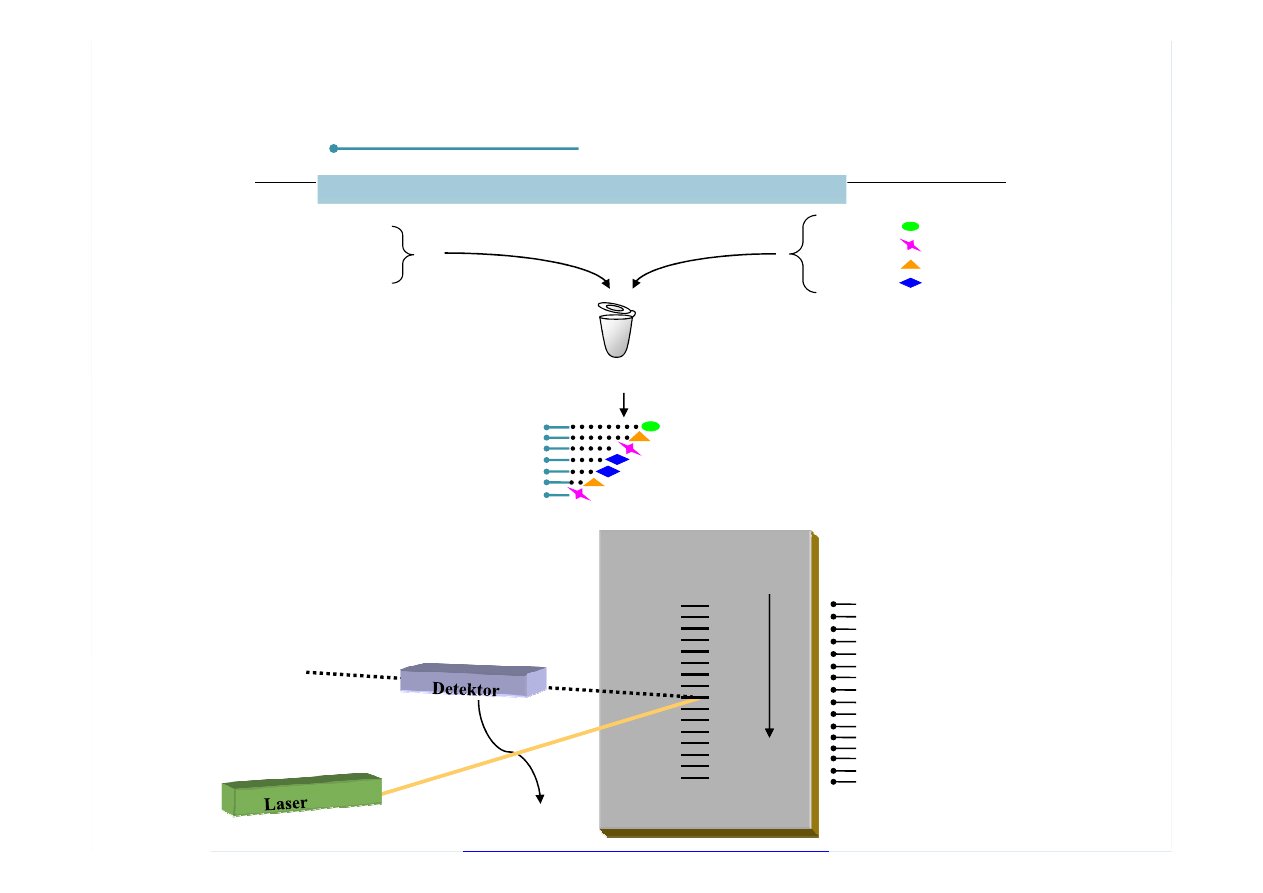
Starter komplementarny do matrycy DNA
3’
5’
5’
3’
GCCTAGGGTTTGCGCTAC
Wydłużanie startera przy użyciu polimerazy DNA
+
CGGATCCCAAACGCGATG
CAGGCATGCAATGACC
Schemat automatycznego
Schemat automatycznego sekwencjonowania
sekwencjonowania DNA
DNA
dATP
dGTP
dTTP
dCTP
ddCTP
ddTTP
ddATP
ddGTP
Terminatory znakowane
fluorescencyjnie
Rozdział produktów reakcji w automatycznym
sekwenatorze
dd
G
GTdd
C
Gdd
T
GTCCdd
G
GTCdd
C
GTCCGdd
T
GTCCGTdd
A
GTCCGTACdd
G
GTCCGTAdd
C
GTCCGTACGdd
T
GTCCGTACGTdd
T
GTCCGTACGTTdd
A
GTCCGTACGTTAdd
C
GTCCGTACGTTACdd
T
GTCCGTACGTTACTdd
G
GTCCGTACGTTACTGdd
G
Elektroforeza na żelu


Znaczenie metody
Znaczenie metody sekwencjonowania
sekwencjonowania DNA
DNA
• rekonstrukcja filogenezy
• genomika
• filogeografia
• ocena bioróżnorodności
• ewolucja eksperymentalna
• medycyna sądowa
• medycyna
Znaczenie sekwencjonowania ciągle rośnie a nowe metody pozwalają na skokowe
zwiększenie wydajności i zmniejszenie kosztów – genomika personalna, ale też
ogromne perspektywy dla badania gatunków niemodelowych.
ogromne perspektywy dla badania gatunków niemodelowych.
Nowe
Nowe sekwenatory
sekwenatory
Rozdział w kapilarach, a nie w żelu poliakryloamidowym
96 kanałów – 96 sekwencji (2 godziny); 1000 sekwencjowań w ciągu doby
Sekwencjonowanie cykliczne (Sears i wsp. 1992)
Wyróżnia się dwiema cechami, których nie ma metoda tradycyjna:
materiałem wyjściowym jest dsDNA (np. produkty PCR)
nie ma konieczności klonowania przed sekwencjonowaniem
wystarcza niewielka ilość DNA
Pirosekwencjonowanie
Klonowanie DNA
Klonowanie DNA
Klonowanie zrewolucjonizowało nauki biologiczne w latach 70. umożliwiając badanie
pojedynczych genów. Szeroki zakres zastosowań klonowania można podzielić na trzy
kategorie:
Izolowanie genów i innych sekwencji DNA do metod in vitro, takich jak
hybrydyzacja z sondą i sekwencjonownie DNA
Tworzenie zmodyfikowanych wersji genów do ponownego wprowadzenia do
organizmu w celu badania ekspresji genu i jego regulacji
Przenoszenie genów do innego organizmu w celu otrzymania dużych ilości białka
kodowanego przez ten gen lub modyfikacji fenotypu biorcy
Klonowaniem DNA nazywa się wyodrębnianie i namnażanie
fragmentów kwasów nukleinowych za pośrednictwem wektorów,
którymi są z reguły cząsteczki plazmidów lub wirusów, liczące kilka
tyś. par nukleotydów. Przed klonowaniem wektory poddaje się
zwykle zabiegom biotechnologicznym , polegającym na dołączeniu
krótkiego fragmentu kwasu nukleinowego (łącznika) zawierającego
wiele miejsc rozpoznawanych przez różne enzymy restrykcyjne
oraz geny oporności na antybiotyki. Obecność łącznika ułatwia
wprowadzenie do wektora fragmentu obcego DNA, a geny
oporności na antybiotyki ułatwiają selekcję odpowiednich klonów.
W procesie klonowania zwielokrotnienie kopii danego fragmentu
DNA otrzymuje się w wyniku samopowielania cząsteczki wektora
wraz z włączonym w jego strukturę obcym fragmentem kwasu
nukleinowego.
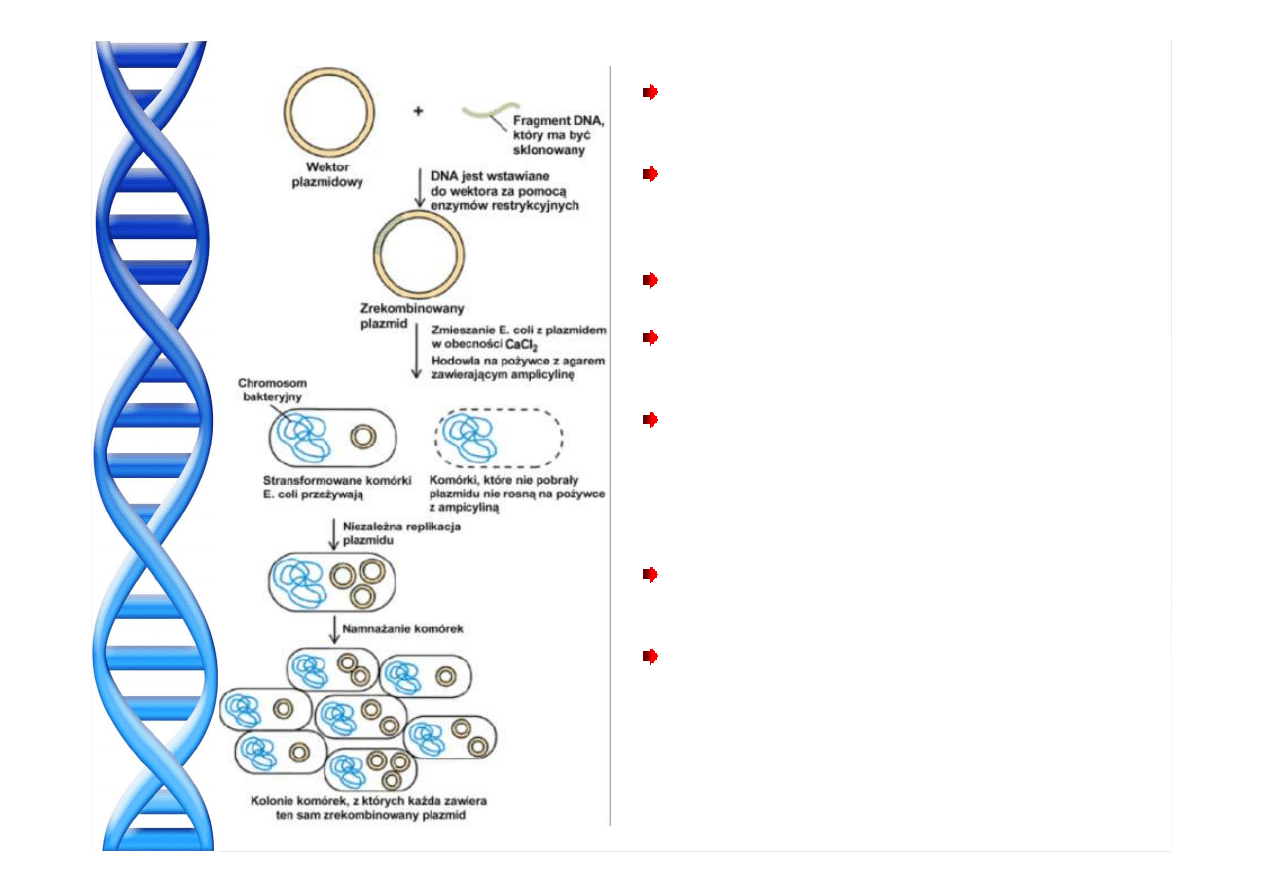
W jednej z klasycznych technik klonowania i
tworzenia banków fragmentów genomu
wykorzystywanym wektorem są plazmidy.
W pierwszym etapie klonowania DNA
chromosomowy jest cięty wybranym enzymem
restrykcyjnym. Tym samym enzymem są cięte
cząsteczki wektora.
Następnie oba preparaty miesza się i dodaje ligazę
– enzym, który łączy fragmenty DNA.
Otrzymuje się w ten sposób populację plazmidów
z włączonymi w ich strukturę różnymi odcinkami
klonowanego DNA.
W celu namnożenia wystarczy niewielką część tak
przygotowanej mieszaniny wprowadzić do
przygotowanej mieszaniny wprowadzić do
komórek biorcy (np. bakterii) w warunkach
umożliwiających plazmidom swobodną replikację i
wyselekcjonować te klony, które zawierają plazmid
z poszukiwanym fragmentem DNA.
Ostatnim etapem jest izolacja, na masową skalę
DNA plazmidowego zawierającego pożądany
odcinek DNA i wycięcie go ze struktur plazmidu.
Wielkość klonowanych w plazmidach fragmentów
DNA nie przekracza z reguły 10 kpz. Większe
fragmenty DNA mogą być klonowane na innych
wektorach: bakteriofagi, sztuczny chromosom
drożdżowy (YAC).
Klonowanie zarodków ssaków
Klonowanie zarodków ssaków
Dzięki temu możliwe jest uzyskanie wielu identycznych osobników. Wzbudza to
ogromne zainteresowanie m.in. hodowców mających nadzieje na stworzenie jak
największej liczby zwierząt o pożądanych cechach.
METODY
1. Metoda izolacji blastomerów.
2. Metoda agregacji blastomerów.
2. Metoda agregacji blastomerów.
3. Metoda podziału zarodków.
4. Metodą transplantacji jąder komórek zarodkowych.
Metoda izolacji blastomerów.
W miarę kolejnych podziałów zygoty powstaje wiele komórek potomnych -
blastomerów. Początkowo każdy z nich jest taki sam i teoretycznie posiada
możliwość utworzenia wszystkich pozostałych komórek. Znaczy to, że w tym
stadium z każdego blastomeru może powstać cały organizm. Doświadczalnie
zostało to potwierdzone dla stadium nie przekraczającego ośmiu blastomerów.
Jest to także uzależnione od gatunku.

Metoda transplantacji jąder komórek zarodkowych
Metoda transplantacji jąder komórek zarodkowych
Jest to jedyna metoda pozwalająca na uzyskanie klonów liczących
większą liczbę osobników. Usuwane są oba przedjądrza zygoty lub
chromosomy z nie zapłodnionych oocytów a następnie
wprowadzane są na ich miejsce jądra z blastomerów zarodków.
Wprowadzenie uzyskuje się rożnymi metodami : mikrochirurgiczną
transplantacją , metodą fuzji komórkowych czego odmianą jest
elektrofuzja itd.
elektrofuzja itd.
Procedura klonowania Dolly :
-pobranie jądra komórkowego z komórki wymienia dorosłej
owcy,
- połączenie tegoż jądra z pozbawioną uprzednio jądra komórką
jajową (oocytem).
- wszczepienie tak spreparowanej komórki jajowej matce
zastępczej.
Dolly urodziła się za 277próbą.
Dolly nie była klonem żadnej z trzech owiec użytych w tym
eksperymencie, bowiem ma dwa genomy od dwóch różnych
owiec A i B (jądrowy i mitochondrialny) oraz „matkę” zastępczą.

Klonowanie terapeutyczne
Klonowanie terapeutyczne
Proces klonowania terapeutycznego polega na wprowadzeniu jądra "dorosłej"
komórki do "pustej" (pozbawionej własnego jądra) komórki jajowej. Taka
hybryda może następnie stworzyć nowy zarodek identyczny genetycznie z
dawcą jądra komórkowego.
Klonowanie terapeutyczne - po co?
Aby z powstałych zarodków pozyskiwać komórki macierzyste.
Komórki te potrafią przekształcić się w dowolną tkankę organizmu, np. we
włókna nerwowe, komórki krwi czy włókna mięśni.
włókna nerwowe, komórki krwi czy włókna mięśni.
Komórki macierzyste mogą stać się niewyczerpanym źródłem tkanek
zastępczych. Początkowo niewielkie grupy komórek wszczepiano by do
organizmu w celu naprawy uszkodzeń, np. po zawale serca.
Docelowo z komórek macierzystych można by hodować całe narządy.
wątroba,
krew (przeszczep szpiku,)
siatkówka (retinopatie)
mózg (choroba Parkinsona , Alzheimera)
serce
skóra
Podstawy genetyczne chorób
układu krążenia
Zakład Nadciśnienia Tętniczego
Katedry Nefrologii i Nadciśnienia Tętniczego
Uniwersytetu Medycznego w Łodzi

Zmiennośd materiału genetycznego
MUTACJE
Mutacja jest to jakościowa lub ilościowa zmiana materiału genetycznego
komórki.
• Mogą one dotyczyd liczby i struktury chromosomów , pojedynczych
genów lub ich fragmentów, lub mogą byd ograniczone do zmiany
pojedynczej zasady w DNA.
• Mogą zaburzad procesy transkrypcji i translacji, prowadząc do zmiany
poziomu ekspresji lub funkcji produktu białkowego danego genu.
• Niektóre mogą prowadzid do śmierci komórki lub całego organizmu.
• Zdarzają się również mutacje nieme czynnościowo.

Typy i mechanizmy mutacji
1.
Aberracje chromosomowe
– zaburzenia liczby lub strukturv chromosomów.
Zaburzenia liczby chromosomów dzielą się na:
•
Poliploidalnośd, czyli zwiększenie się lub zmniejszenie liczby chromosomów o wielkośd całkowitej haploidalnej
liczby chromosomów.
•
Aneuploidalnośd, czyli niewielkie odchylenia od podstawowej liczby chromosomów np.: nullisomia (brak w
komórce jakiejkolwiek kopii danego chromosomu), monosomia (obecnośd jednej kopii chromosomu), disomia,
trisomia itd.
Zaburzenia struktury chromosomów dzielą się na:
•
Translokacje (przegrupowanie fragmentów między dwoma chromosomami)
•
Inwersje (odwrócenie fragmentu chromosomu między dwoma miejscami złamao w chromosomie)
•
Delecje
•
Duplikacje
2.
Duże rearanżacje
genowe:
•
Delecje (brak genu lub jego fragmentu)
•
Duplikacje (powielanie fragmentu DNA, w obrębie genu)
•
Insercje (polegające na wstawieniu do genu sekwencji zwykle pochodzącej spoza jego obszaru)
3.
Mutacje punktowe
, polegające na zmianie pojedynczego nukleotydu:
•
Insercja (wstawienie pojedynczego nukleotydu do sekwencji genu)
•
Delecja (wypadnięcie pojedynczego nukleotydu z sekwencji genu)
•
Substytucja (zastąpienie jednego nukleotydu innym)
Transwersja – zamiana puryny na pirymidynę i odwrotnie
Tranzycja – zamiana puryny na purynę i pirymidyny na pirymidynę

Następstwa mutacji
1.
Mutacja zmiany sensu
zachodzą zwykle, gdy w obrębie kodonu dojdzie do zmiany
pojedynczego nukleotydu, która przekłada się na zmianę pojedynczego aminokwasu w
sekwencji białka. W efekcie funkcja takiego białka może byd nieprawidłowa.
2.
Mutacje nonsensowne
powstają na skutek zmian nukleotydów prowadzących do
przedwczesnego pojawienia się kodonu terminacyjnego translacji.
3.
Mutacje ciche
zachodzą zwykle, gdy dojdzie do zmiany trzeciej zasady kodonu i z powodu
degeneracji kodu genetycznego nie następuje zmiana aminokwasu w nukleotydzie.
4.
Mutacje zmiany ramki odczytu
powstają na skutek insercji lub delecji w regionie
kodującego genu, prowadzą do zmiany kroku odczytu mRNA i błędnego odczytania szeregu
kodonów. Taka nieprawidłowa translacja prowadzi do syntezy polipeptydu często o
całkowicie zmienionej sekwencji aminokwasów.
5.
Mutacje transkrypcyjne
dotyczą obszarów promotorowych i regulatorowych genów. Ich
konsekwencją może byd obniżony poziom transkrypcji danego genu i w konsekwencji
niedostateczna ekspresja produktu białkowego lub zwiększona aktywnośd transkrypcyjna i
nadmierna ekspresja.
6.
Mutacje dotyczące procesów dojrzewania mRNA
prowadza do zaburzeo procesu cięcia i
składania RNA, które są krytyczne dla powstania prawidłowego dojrzałego mRNA. Mutacje
tego typu w efekcie koocowym mogą prowadzid do zmiany ilości syntezowanego białka lub
do powstania białka o nieprawidłowej funkcji.

Polimorfizm genetyczny
• Polimorfizm genetyczny oznacza występowanie w populacji dwóch lub
więcej form danego genu – alleli z częstością większą niż oczekiwana,
wynikającą z ogólnej częstości mutacji w danej populacji.
• Ponieważ częstośd mutacji w danej populacji jest trudna do
sprecyzowania, przyjęto ze o polimorfizmie możemy mówid, gdy najrzadszy
wariant alleliczny w danym locus występuje z częstością większą niż 1 %.
• Polimorfizm podobnie jak mutacja jest efektem zmian sekwencji DNA.

Rodzaje polimorfizmów
Polimorfizm powtórzeo
wielokrotnych
Polimorfizm krótkich powtórzeo
tandemowych
(STRP, short tandem
repeat polymorphism) polega na
występowaniu segmentów DNA o
dł. do 100 pz (zawierających różna
liczbę powtórzeo krótkich sekwencji
– najczęściej *CACA+n )
Zmienna liczba powtórzeo
tandemowych
(VNTR, variable
number of tandem repeats) polega
na występowaniu segmentów DNA
o dł. od 1 kpz do 30 kpz,
zawierających różną liczbę
powtórzeo bardziej złożonych i
wielonukleotydowych sekwencji)
Polimorfizm pojedynczych
nukleotydów (SNP)
Insercja, delecja
fragmentów genu
Może polegad na insercji lub delecji
pojedynczych nukleotydów,
zlokalizowanych w części kodującej
lub niekodującej genu. Następstwa
tej zmienności mogą dotyczyd
zmiany budowy aminokwasowej
białka lub poziomu jego ekspresji

Podwyższone stężenie LDL i choroba wieocowa
•
Lipoproteiny o małej gęstości (LDL) należą do głównych lipoprotein transportujących
cholesterol w surowicy i są powszechnie uznane za czynniki bezpośrednio związane z chorobą
wieocową.
•
Z podwyższonym stężeniem LDL związane są 4 monogenowe schorzenia.
•
Wynika ono ze zmiany aktywności receptorów wątrobowych dla LDL, które wychwytują
cząsteczki LDL z surowicy.
Monogenowe choroby związane z wysokim stężeniem cholesterolu LDL
Choroba
Zmutowany gen
Przybliżone stężenie
cholesterolu [mg/dl]
Rodzinna hipercholesterolemia
• osoby homozygotyczne
• osoby heterozygotyczne
Rodzinny defekt Lp B-100
• osoby homozygotyczne
• osoby heterozygotyczne
Autosomalna recesywna
hipercholesterolemia
Sitosterolemia
LDLR
APOB100
ARH
ABCG5 i ABCG8
650
300
325
275
450
150-650
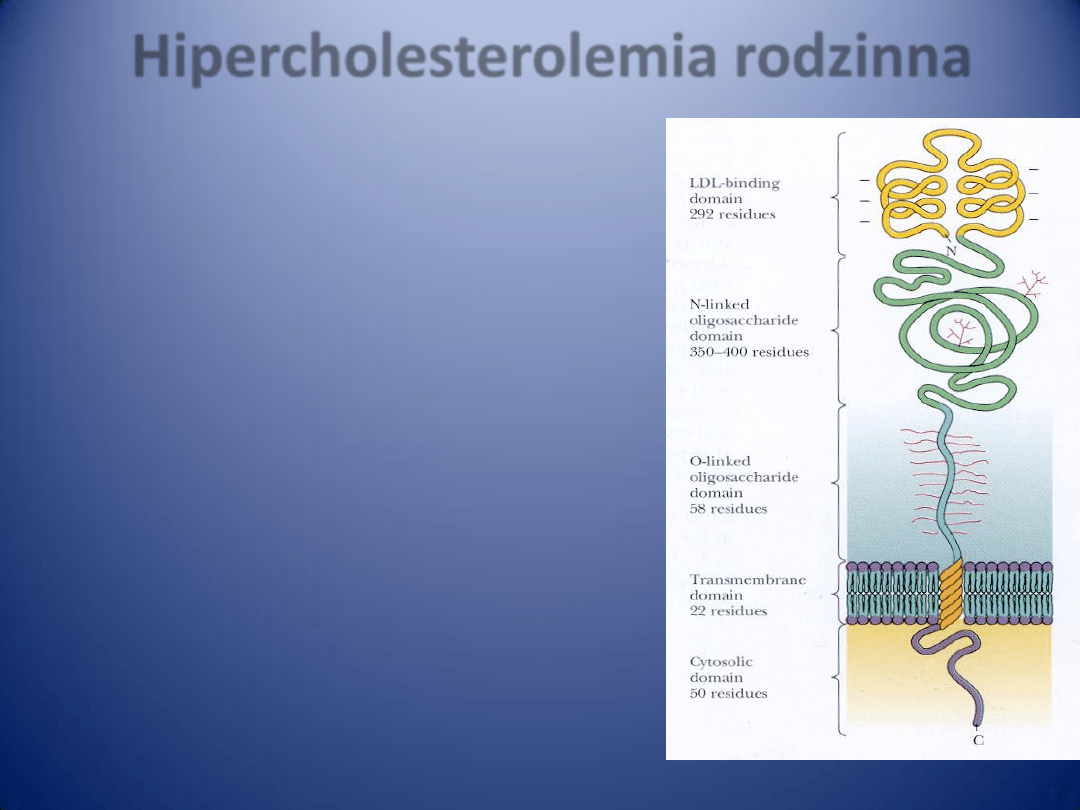
Hipercholesterolemia rodzinna
•
Była pierwszym schorzeniem monogenowym, w
przypadku którego wykazano genetyczne uwarunkowany
wzrost stężenia cholesterolu w surowicy.
•
Pierwotnym defektem hipercholesterolemii rodzinnej jest
deficyt receptorów LDL – dotychczas opisano 600 mutacji
genu receptora LDLR wśród chorych z tym schorzeniem.
•
W przybliżeniu 1/500 ma postad heterozygotyczną
przynajmniej jednej mutacji tego genu, natomiast tylko
jedna osoba na milion jest obciążona homozygotyczną
postacią hipercholesterolemii rodzinnej
•
Osoby z heterozygotyczną postacią mutacji genu LDLR
wytwarzają połowę prawidłowej ilości receptorów LDL,
co prowadzi do 2-3 krotnego wzrostu poziomu LDL w
surowicy.
•
U osób z postacią homozygotyczną stężenia cholesterolu
są 6-10 razy wyższe niż przeciętnie w populacji.
•
Osoby te są obciążone niezwykle wysokim ryzykiem
choroby wieocowej i najczęściej ich zgon następuje w
dzieciostwie z powodu zawału serca.
Rys. Receptor dla LDL

Rodzinny defekt Lp B-100
Niedobór możliwości transportowych lipoprotein prowadzi również do
wzrostu absorpcji cholesterolu i syntezy cząsteczek LDL.
Mutacje w regionie APOB-100, kodującego Apo B-100 ograniczają połączenie
apolipoproteiny B-100 z receptorem LDL, co zwalnia klirens osoczowego
LDL.
Ocenia się, że 1/100 m a postad heterozygotyczną przynajmniej jednej mutacji
tego genu.
Profil lipidowy jest podobny jak w przypadku hiperliporoteinemi rodzinnej
cząsteczka LDL

Sitosterolemia
• Rzadkie schorzenie autosomalne, jest wynikiem mutacji prowadzącej do
utraty funkcji genów kodujących dwa transportery typu ATP-binding casette
(ABC) – ABCG5 i ABCG8 – które współdziałają w wydzielaniu cholesterolu do
światła jelit i powodują zmniejszenie absorpcji cholesterolu.
• Cechuje się nadmierną absorpcją jelitową i zmniejszonym wydzielaniem z
żółcią steroli zawartych w pokarmach, co prowadzi do hypercholesterolemii,
rozwoju żółtaków (żółtawe zmiany grudkowe umiejscowione najczęściej w
okolicach powiek), przedwczesnego rozwoju miażdżycy, i nieprawidłowych
wyników badao hematologicznych i wątrobowych.
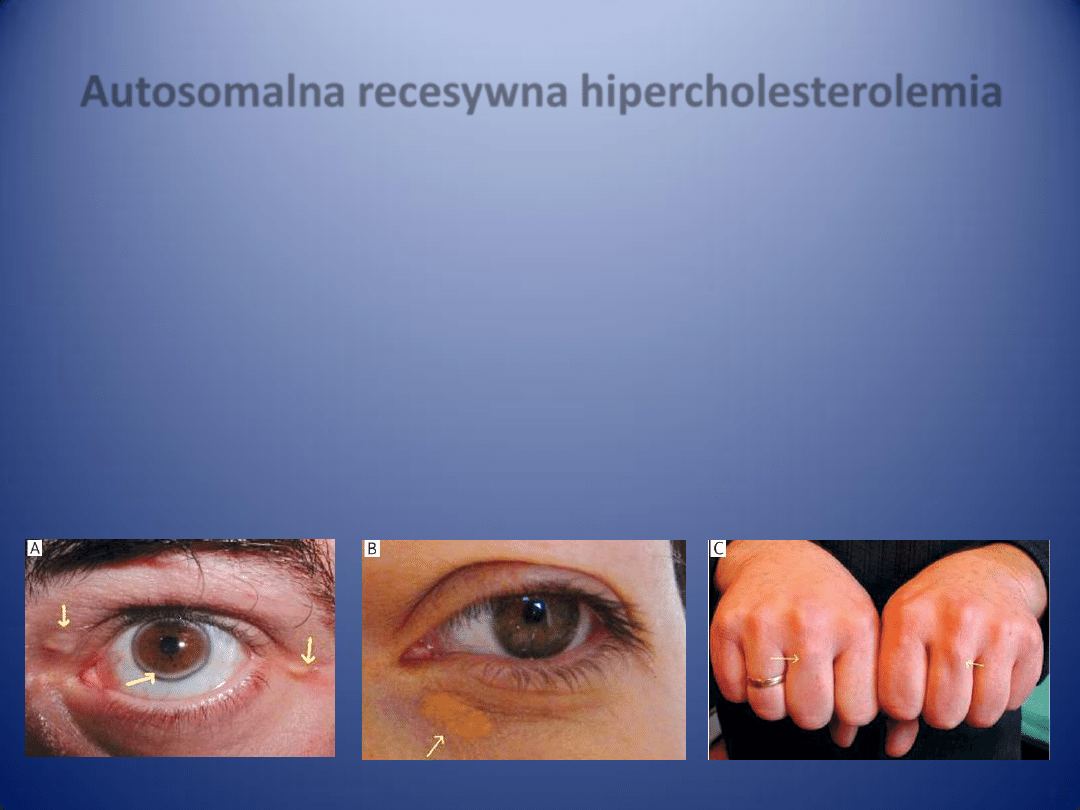
Autosomalna recesywna hipercholesterolemia
• Jest schorzeniem niezwykle rzadkim, występuje rzadziej niż raz na 10 mln
urodzeo.
• Przypuszczalnie molekularnym podłożem tej choroby jest obecnośd
defektu wątrobowego białka adaptującego (hepatic adapter protein), co
uniemożliwia wychwyt cząsteczek LDL z surowicy przez wątrobowe
receptory LDL.
• Mutacja genu kodującego to białko (AHR) związana jest z podwyższeniem
stężenia cholesterolu w surowicy do wartości obserwowanych w
homozygotycznej postaci hipercholesterolemii rodzinnej.
A
– xanthelasma, arcus senilis (rąbek rogówki), B – xanthelasma, C – xanthoma ścięgien
Nadciśnienie tętnicze
• Mutacje, które upośledzają nerkową regulację gospodarki sodowej,
stanowią podstawę zrozumienia patogenezy nadciśnienia tętniczego.
• Badanie rodzin z wysokimi wartościami ciśnienia tętniczego lub
niedociśnieniem umożliwiło wykrycie mutacji genów, mających znaczący
udział w regulacji gospodarki sodowej.
wielotorbielowate zwyrodnienie nerek
zespół MEN 2 (multiple endocrine neoplasia
type 2 - zespół gruczolakowatości
wewnątrzwydzielniczej typu 2)
zespół Liddle’a
zespół Gordona
rodzinny hiperaldosteronizm (reagujący na
leczenie glikokortykosteroidami)
pozorny nadmiar mineralokortykosteroidów
wrodzony przerost tarczycy
mutacja w genie PPAR-γ
Nadciśnienie
tętnicze
Wybrane jednostki chorobowe, w których nadciśnienie tętnicze spowodowane jest mutacjami pojedynczego genu

Zespół Gordona
Zespół Gordona czyli
pseudohipoaldosteronizm typu II
jest schorzeniem
autosomalnym dominującym, charakteryzującym się nadciśnieniem,
hiperkalemią, zwiększoną reabsorpcją nerkową sodu oraz zaburzeniami
wydalania potasu i jonu wodorowego.
Zidentyfikowano 2 geny – oba kodują białka rodziny WNK1 serynowej
kinazy treoninowej.
Mutacje WNK1 maja charakter intronowej delecji na chromosomie 12p lub
są to mutacje zmiany sensu w locus WNK4 na chromosomie 14.
Badania immunofluorescencyjne wykazały, że białka te zlokalizowane są w
dystalnej części nefronu i mogą zwiększyd przepuszczalnośd
pozakomórkową sodu w cewkach zbiorczych, prowadząc do zwiększonej
reabsorpcji sodu, zwiększonej objętości osocza oraz redukcji wydzielania K
+
i H
+
.

Aldosteronizm podatny na leczenie
glikokortykosteroidami
Hiperaldosteronizm steroidozależny
, hiperaldosteronizm poddający się leczeniu
glikokortykosteroidami (GRA - Glucocorticoid-Remediable Aldosteronism) -
dziedziczony jest autosomalnie dominująco.
Związany jest z podatnością do krwotocznych udarów mózgu, wczesnym
wystąpieniem nadciśnienia tętniczego, zahamowaną aktywnościa reninową oraz
prawidłowym lub podwyższonym stężeniem aldosteronu.
Przyczyną tego zespołu jest pojawienie się nowego genu, który jest chimerą
zbudowaną z promotora genu 11 beta-hydroksylazy (CYP11B1) i części kodującej
genu syntazy aldosteronu (CYP11B2). W tej sytuacji ekspresja genu syntazy
aldosteronu podlega regulacji przez ACTH (hormon adenokortykotropowy). W
wyniku pojawienia się genu chimerycznego CYP11B1/B2 dochodzi do ektopowej
syntezy aldosteronu w warstwie pasmowatej kory nadnerczy, a w konsekwencji do
nadciśnienia z powodu zatrzymywania sodu i wody.
Mutacje, które powodują aktywnośd syntazy aldosteronu upośledzają nerkową
reabsorpcję sodu oraz nasilają wydalanie K+ i H+, co skutkuje ciężkim
niedociśnieniem z powodu zmniejszonej objętości wewnątrznaczyniowej.

Pozorny nadmiar mineralokortykoidów
Pozorny nadmiar mineralokortykoidów (AME - Apparent Mineralocorticoid Excess)
dziedziczony jest autosomalnie recesywnie, prowadząc już we wczesnym okresie życia
do rozwoju nadciśnienia z typowym, bardzo niskim stężeniem aldosteronu w
surowicy.
Przyczyną nadciśnienia jest stymulacja przez kortyzol receptora mineralokortykoidów
w kanaliku dystalnym.
Ponieważ kortyzol i aldosteron mają zbliżone powinowactwo do receptora
mineralokortykoidów, stąd też istnieje fizjologiczny mechanizm ochrony receptora
przed pobudzeniem go przez kortyzol.
Mechanizm ten polega na przemianie kortyzolu do kortyzonu w reakcji katalizowanej
przez dehydrogenazę 11 beta-hydroksysteroidową typu 2 (HSD11B2).
Przemiana kortyzolu zapewnia selektywną stymulację receptora mineralokortykoidów
przez aldosteron.
Przyczyną zespołu AME są mutacje genu HSD11B2, które prowadzą do utraty
aktywności enzymu.
Aktywnośd enzymu HSD11B2 może byd również obniżona w wyniku działania
czynników środowiskowych.
Zespół Liddle'a
W 1963 roku Grant Liddle opisał przypadek chorego rasy kaukaskiej z
nadciśnieniem tętniczym, hipokaliemią, niską aktywnością reninową
osocza oraz niskim osoczowym stężeniem aldosteronu.
Chory nie reagował na leczenie spironolaktonem, podczas gdy
ograniczenie spożycia soli znormalizowało zarówno wartości ciśnienia, jak i
wskaźniki biochemiczne.
W latach 90-tych stwierdzono, że przyczyną zespołu Liddle`a są mutacje C-
koocowych fragmentów genów podjednostek b lub g nabłonkowego
kanału sodowego (ENaC, Epithelial Sodium Channel).
Mutacje te prowadzą do konstytutywnej aktywacji ENaC i wzrostu
reabsorpcji sodu w kanaliku dystalnym.
Jest to schorzenie autosomalne dominujące z charakterystycznym
wczesnym rozwojem nadciśnienia tętniczego.
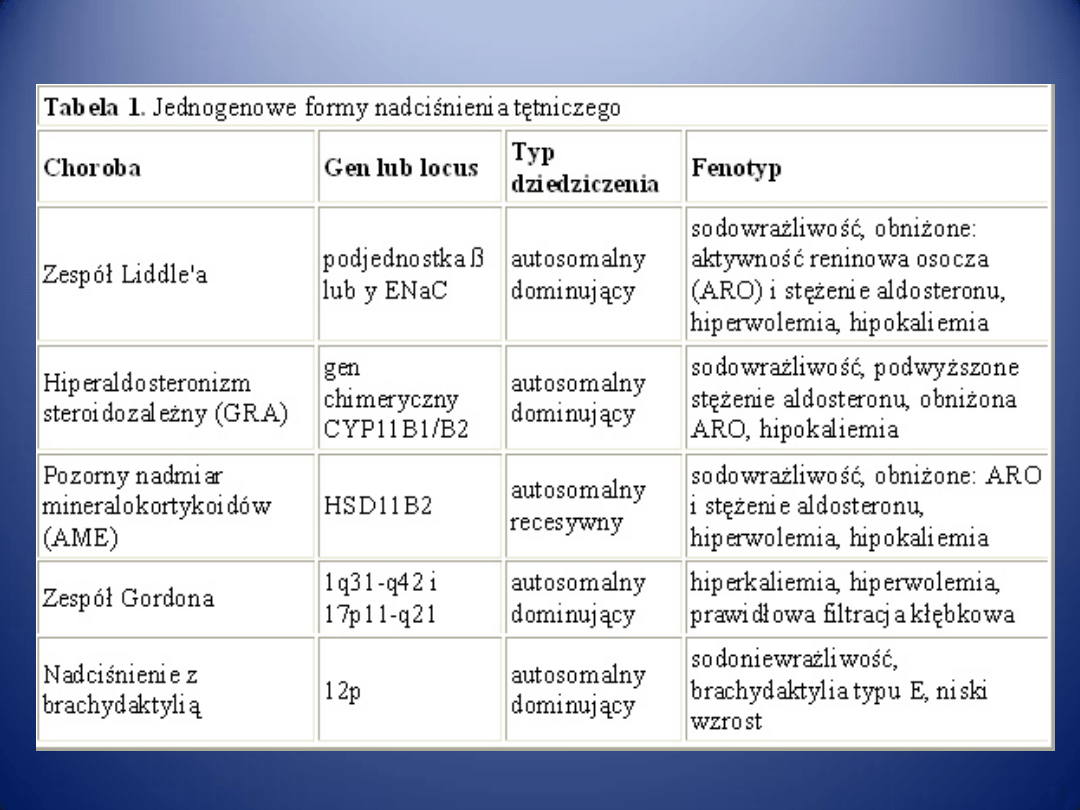
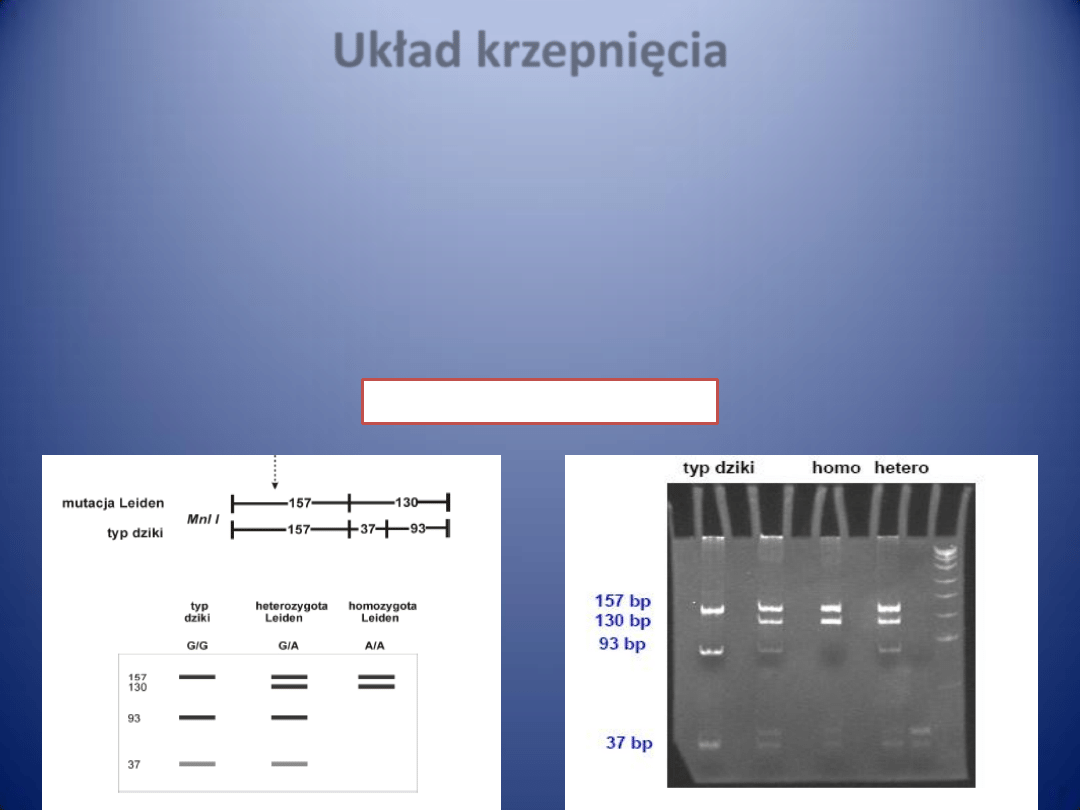
Układ krzepnięcia
Układ krzepnięcia wymaga precyzyjnej kontroli elementów wewnętrznych i zewnętrznych kaskady
krzepnięcia w celu uniknięcia niebezpiecznego krwawienia lub wykrzepiania.
Wariant genu czynnika V, kodujący substytucję glutaminy w miejsce argininy w pozycji 506,
zapobiega degradacji czynnika V i stymuluje powstawanie skrzepliny. Substytucja ta znana jest
także jako czynnik V Leiden i występuje w populacji europejskiej z częstością 2-7%, natomiast
wśród pacjentów z ŻCHZZ z częstością 20-50%.
Zmutowany czynnik V w chwili aktywacji przez trombinę staję się niewrażliwy na proteolityczne
działanie białka C upośledzając w ten sposób hemostazę i powodując zwiększenie ryzyka
powstania zmian zakrzepowo-zatorowych w organizmie.
Mutacja Leiden cz V
Mutacja Leiden cz V
Opornośd na aktywowane białko C powodowana jest punktową mutacją genu
substratu białka C
-
czynnika V
. Mutacja ta dotyczy dinukleotydu CpG (1691→A) i wywołuje zamianę Arg506,
znajdującej się w rozpoznawanym przez aktywne białko C miejscu hydrolizy czynnika V, na
glutaminę (Gln).
Zmutowany aktywny czynnik Va (czynnik V Leiden) wykazuje niezmienioną aktywnośd
prokoagulacyjną, jest natomiast o wiele mniej podatny na inaktywację przez aktywne białko C.
Hydroliza czynnika V przez aktywne białko C dotyczy przynajmniej trzech różnych wiązao
peptydowych. Kolejnośd hydrolizy poszczególnych wiązao jest procesem losowym, zaś miejsce
hydrolizy przy Arg506 nie jest uprzywilejowane.
Jak dowiodły badania molekularne, mutacja 1691→A sprawia, że hydroliza czynnika V przez
aktywne białko C jest o wiele mniej wydajna. Prowadzi to w następstwie do stabilizacji kompleksu
protrombinazy, obserwuje się zwiększone wytwarzanie aktywnej trombiny, oraz dalszą aktywację
czynników V i VIII na drodze dodatniego sprzężenia zwrotnego. Ta zwiększona aktywnośd
prokoagulacyjna, wespół z utratą wydajności działania białka C, sprzyjają powstawaniu stanu
nadkrzepliwości.
W ponad 90% przypadkach genetycznie uwarunkowana opornośd na aktywowane białko C jest
uwarunkowana pojedynczą punktową mutacją (Arg506 →Gln) genu czynnika V. Heterozygotycznośd
pod względem tej mutacji związana jest z 5-10-krotnym wzrostem ryzyka choroby zakrzepowej,
podczas gdy homozygotycznośd podwyższa to ryzyko aż 50-100-krotnie.
Gen czynnika Leiden dziedziczy się jako autosomalna cecha dominująca. Ponieważ niemal wszystkie
wykrywane przypadki oporności na aktywowane białko C wynikają z pojedynczej mutacji Arg506 →
Gln, to homozygoty występują w populacji ze stosunkowo dużą częstością (1-2 na 2000 przypadków
oporności).
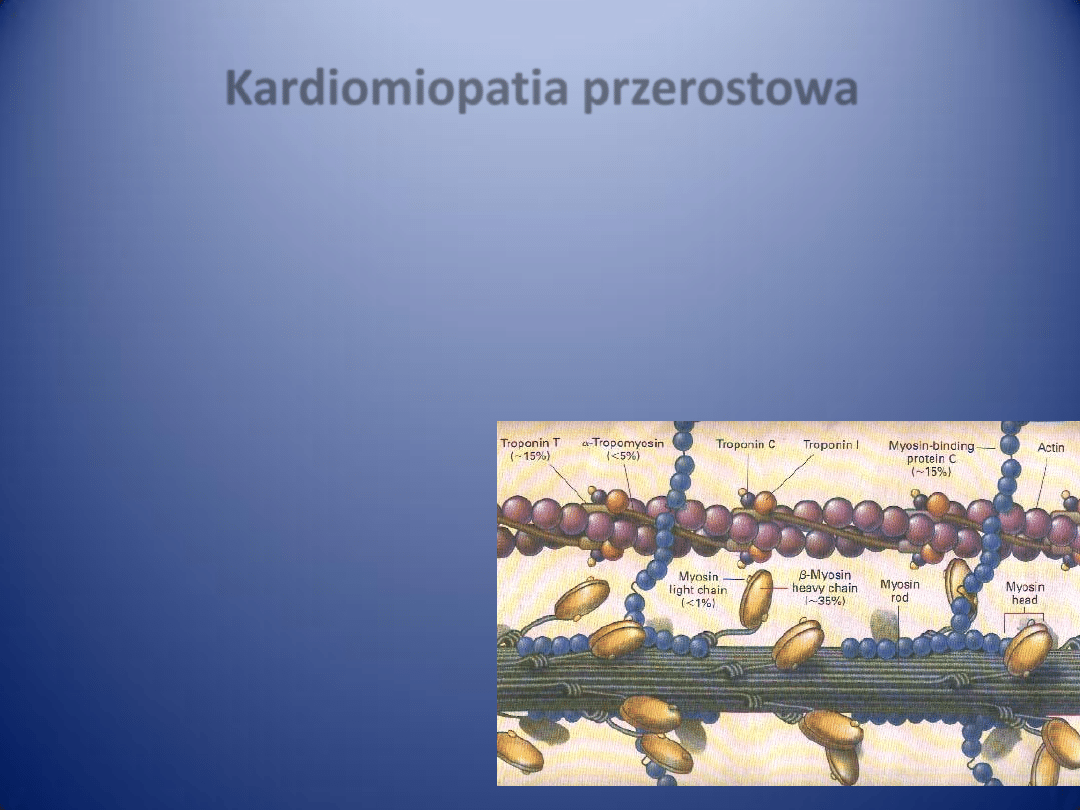
Kardiomiopatia przerostowa
•
Kardiomiopatia przerostowa jest najczęstszym monogenowym schorzeniem kardiologicznym i najczęstszą
przyczyną nagłego zgonu wśród dzieci i młodzieży.
•
Na podstawie badao echokardiograficznych, przeprowadzonych w dużej populacji młodych osób, częstośd
tej patologii ocenia się na ok. 1/500 zachorowao.
•
Dziedziczona jest w sposób autosomalny dominujący.
•
Schorzenie to spowodowane jest przez mutacje genów związanych z białkami aparatu kurczliwego m.
sercowego.
•
Genetycy odkryli liczne mutacje przynajmniej 10 różnych elementów sarkomeru, takich jak:
Łaocuch ciężki sercowej β – miozyny
Sercowe białko wiążące miozynę
Sercowa troponina T
sercowa troponina I
α – tropomiozyna
Sercowe białko C
Niezbędne i regulatorowe łaocuchy lekkie
Sercowa aktyna

Kardiomiopatia przerostowa
Na obraz patologiczny kardiomiopatii przerostowej składa się przede
wszystkim znaczny przerost lewej komory serca, pogrubienie przegrody
międzykomorowej, powiększenie lewego przedsionka oraz zwykle mała
jama lewej komory.
Przerost i dezorganizacja w układzie miocytów oraz włóknienie
śródmiąższowe są obecne w całym m. sercowym.
Obok bezobjawowego lub łagodnego przebiegu choroby, spotyka się
osoby z objawami ciężkiej niewydolności serca, a nagły zgon występuje
zwykle w młodym wieku.
Pierwsza mutacja opisana w rodzinnej kardiomiopatii dotyczyła genu
kodującego łaocuch ciężki miozyny, natomiast dzisiaj znanych jest ponad
100 mutacji odpowiadających za jej rozwój.
Na ostateczny obraz choroby mają wpływ inne geny modyfikujące oraz
czynniki środowiskowe z tego względu nie u wszystkich osób z defektem
genetycznym będą występowad kliniczne objawy choroby.
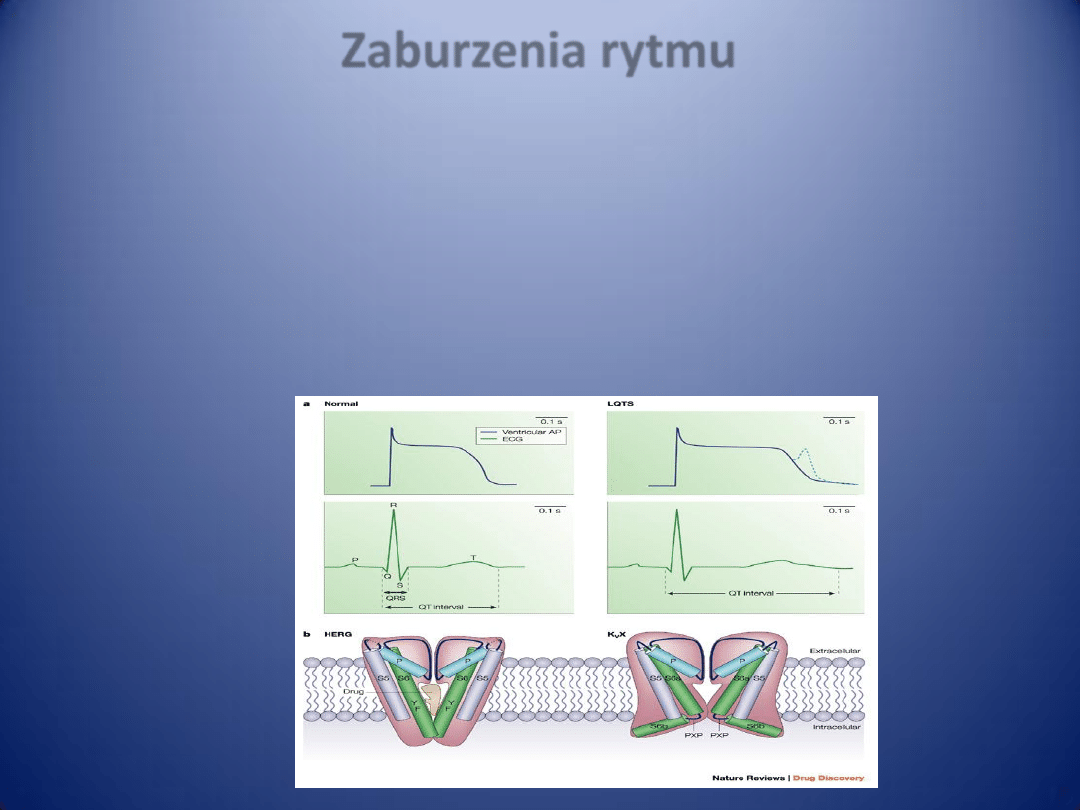
Zaburzenia rytmu
Nagłe zgony związane z groźnymi zaburzeniami rytmu stanowią bardzo istotny problem kliniczny
w kardiologii, przede wszystkim ze względu na ich wielka liczbę.
Czynniki genetyczne mogą modyfikowad ryzyka arytmii związane z typowym podłożem
patologicznym.
Zespół wydłużonego QT (LQTS)
jest genetycznie uwarunkowaną chorobą kanałów jonowych,
która w większości przypadków prowadzi do wydłużenia repolaryzacji co manifestuje się
wydłużeniem skorygowanego odstępu QT (QTc) w zapisie EKG. U większości pacjentów z LQTS
stwierdza się mutację genu kodującego błonowy kanał potasowy lub sodowy.

Genetyka zespołu wydłużonego QT
•
We współczesnej klasyfikacji opartej na kryteriach molekularnych wyodrębnia się aż 12 postaci
wrodzonego zespołu długiego QT.
•
Mutacje będące przyczyną choroby udało się zidentyfikowad u blisko 2/3 chorych, u których
prowadzono diagnostykę genetyczną
Obecnie można stwierdzid, że LQTS jest heterogenną chorobą genetyczną, u której podłoża leżą
mutacje genów kodujących powstanie podjednostki a, która po połączeniu z podjednostką b
tworzy kanał potasowy Kr lub Ks w sercu.
Geny te to:
— KCNH2(HERG)
— KCNJ2
— KCNQ1
I
Na
SCN5A
I
kr
HERG i KCNE2
I
Ks
KCNQ1 + KCNE1
Zespół LQTS można podzielić pod względem rodzaju
mutacji na 3 typy :
LQTS1
— mutacja dotycząca genu KCNH2(HERG):
prowadzi do przyspieszenia szybkiego prądu potasowego
Ikr;
LQTS2
— mutacja dotycząca genu KCNQ1: prowadzi
do przyspieszenia wolnego prądu potasowego Iks
LQTS3
— mutacja dotycząca genu KCNJ2: prowadzi
do przyspieszenia prądu potasowego Ik1; jest to zarazem
wariant LQTS z unikalnym obrazem EKG w postaci
niesymetrycznych załamków T.
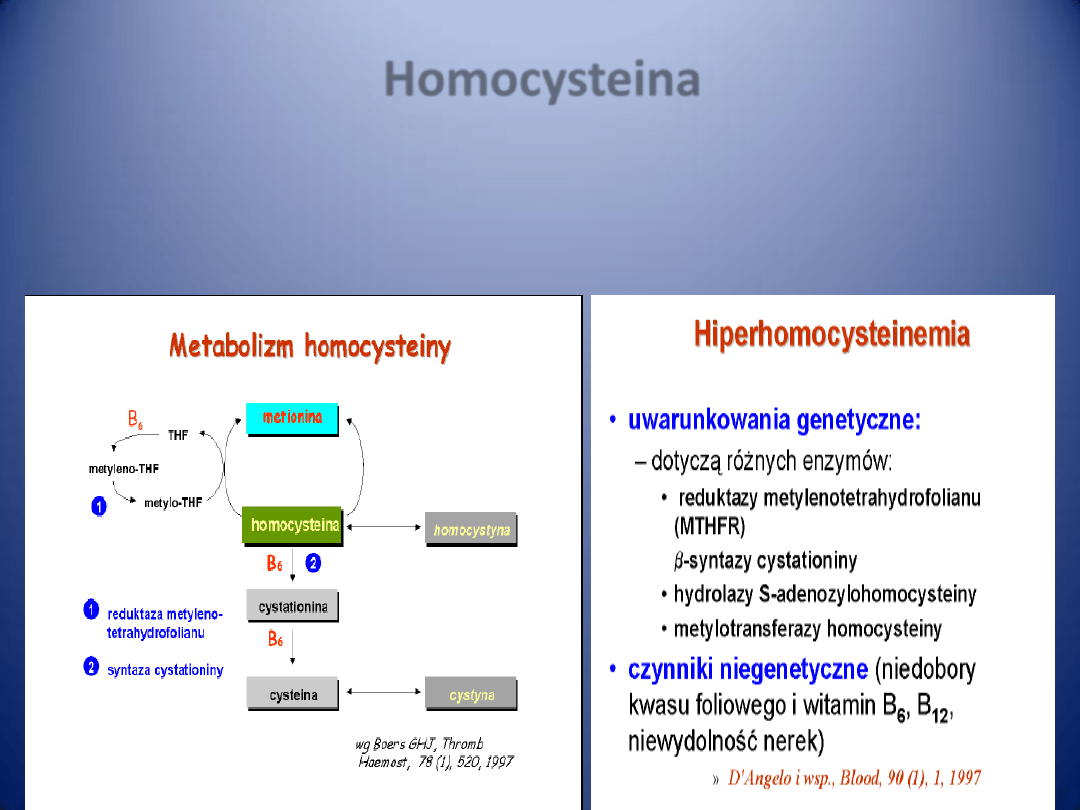
Homocysteina
Homocysteina to aminokwas, który nie wchodzi w skład białek organizmu.
Powstaje w wyniku demetylacji metioniny. W następnej kolejności może
byd przekształcana w cysteinę lub z powrotem w metioninę. Kofaktorami
tych reakcji są witaminy B6 i B12 oraz kwas foliowy.

Znaczenie kliniczne
miażdżyca
zakrzepica naczyo sercowych, mózgowych i obwodowych
zatory naczyo tętniczych i żylnych
zakrzepica żył głębokich
zawał serca, udar niedokrwienny mózgu
ważny czynnik ryzyka u pacjentów po przeszczepie serca
Homocystynuria
o
Homocystynuria – heterogenna etiologicznie, uwarunkowana genetycznie choroba
metaboliczna, polegająca na nieprawidłowym metabolizmie aminokwasu metioniny.
o
Homocystynuria charakteryzuje się podwyższonym poziomem homocysteiny w
surowicy i w moczu.
o
Najczęstszą postacią schorzenia jest homocystynuria spowodowana niedoborem i
niską aktywnością enzymu syntazy β-cystationionowej, który katalizuje reakcję
przekształcenia homocysteiny do cysteiny poprzez cystationinę.
o
Reakcja katalizowana przez syntazę β-cystationionową wymaga udziału pirydoksyny
(witaminy B6), dlatego w części przypadków homocystynurii obserwuje się poprawę
po suplementacji pirydoksyny.
o
Znanych jest dodatkowo przynajmniej siedem innych, znacznie rzadszych chorób
genetycznych powodujących podobny blok metaboliczny; aby odróżnid niedobór
CBS od tych rzadszych przyczyn używa się terminu klasycznej homocystynurii albo
homocystynurii z powodu niedoboru syntazy β-cystationionowej.
o
Dziedziczenie choroby jest autosomalne recesywne, obydwa allele genu CBS muszą
byd zmutowane, aby homocystynuria ujawniła się klinicznie. Heterozygotyczni
nosiciele mutacji w jednym allelu genu CBS nie chorują. Ryzyko urodzenia kolejnego
dziecka z homocystynurią wynosi 25%.
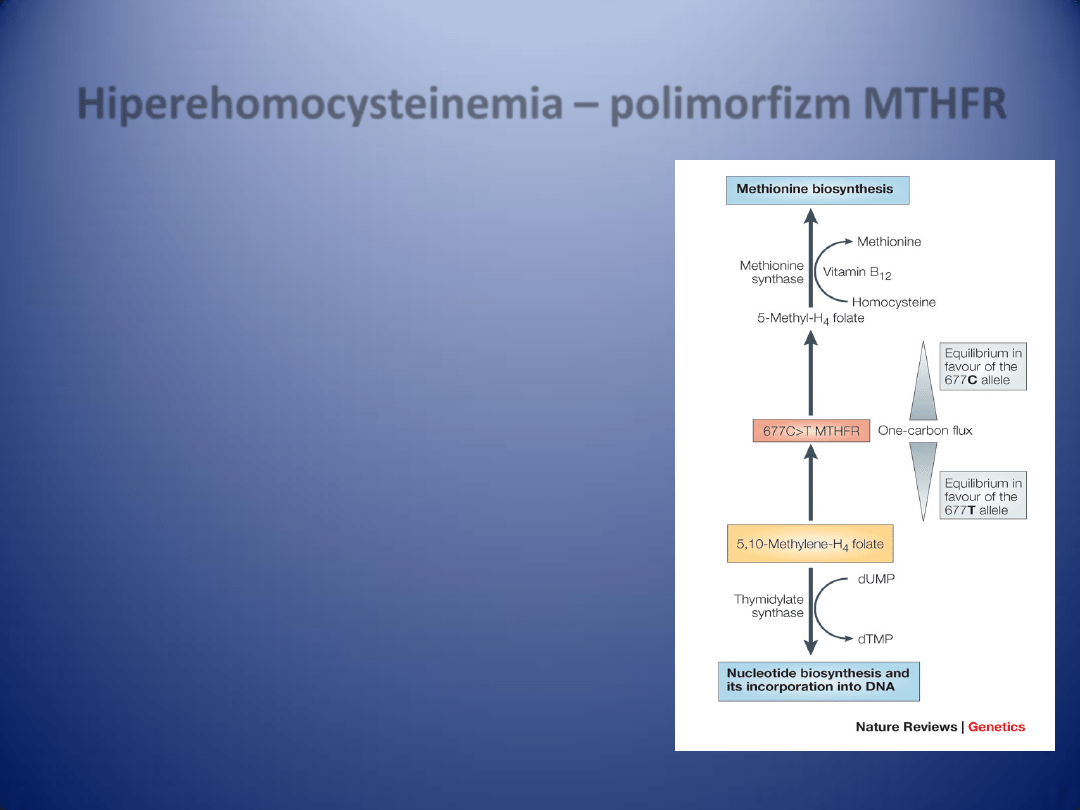
Hiperehomocysteinemia – polimorfizm MTHFR
•
Spośród polimorfizmów genetycznych
reduktazy metylenotetrahydrofolianu
(MTHFR), enzymu, który odpowiada za
konwersję homocysteiny do metioniny w
reakcji remetylacji, najwięcej uwagi w
literaturze poświęcono występowaniu
termolabilnej formy enzymu, w której za
zamianę Ala-Val w cząsteczce białka
odpowiada podstawienie 677C/T w genie
MTHFR.
•
Chociaż wielu autorów uważa ten
polimorfizm (częstośd homozygot w populacji
ogólnej szacuje się na 5-20%) za dziedziczny
czynnik ryzyka choroby zakrzepowo-zatorowej,
jego znaczenie jest ostatnio kwestionowane.
•
Zwraca się uwagę na potencjalne znaczenie
innych polimorfizmów genu MTHFR, np.
1298C/A.

Analiza złożonych cech (fenotypów)
kardiologicznych
•
Analiza złożonych fenotypów kardiologicznych koncentruje się obecnie na poszukiwaniu
wariantów genetycznych, które mogą zwiększad ryzyko wystąpienia najczęstszych schorzeo
sercowo-naczyniowych.
•
Markerami genetycznymi wykorzystywanymi w badaniach poligenowego podłoża choroby
wieocowej, są warianty polimorficzne, których produkty białkowe biorą udział w rozwoju
miażdżycy tętnic wieocowych, procesach trombogenezy, fibrynolizy oraz innych procesach
istotnych dla rozwoju oraz przebiegu miażdżycy i jej powikłao.
•
Geny, których warianty polimorficzne są brane pod uwagę jako potencjalnie związane z
występowaniem danej choroby, określa się mianem genów kandydatów.
Geny
odpowiedzialne
(kandydaci)
Czynniki
środowiskowe
Geny
modyfikujace
Choroba
wieńcowa
Polimorfizmy
•
Analiza genetycznego, wieloczynnikowego podłoża chorób napotyka na wiele trudności.
•
W większości przypadków dla ujawnienia się choroby niezbędna jest kumulacja więcej niż
jednego czynnika genetycznego (poligenowośd).
•
Ponadto ujawnienie się choroby, jej obraz kliniczny i przebieg są w znacznym stopniu
modyfikowane przez czynniki środowiskowe.
•
Badania populacyjne polegają na porównaniu częstości występowania markera lub markerów
genetycznych wśród chorych i w populacji ogólnej lub kontrolnej.
•
Jeśli badany marker występuje znamiennie częściej u chorych niż w grupie kontrolnej, można
podejrzewad istnienie związku między danym markerem a chorobą.
Zaburzenia patofizjologiczne, w których poszukuje się genów kandydatów do
choroby wieocowej
Geny kandydaci, których produkty białkowe zaangażowane są w :
• metabolizm lipidów (apolipoproteiny, enzymy lipolityczne, receptory dla lipoprotein
• układ krzepnięcia i fibrynolizy (fibrynogen, czynniki krzepnięcia, PAI-1, itp.)
• glikoproteiny płytkowe GP IIb/IIIa, GPIa/IIa
• układ RAA (angiotensynogen, ACE, receptor AT1, syntaza aldosteronu)
• czynniki wazoaktywne (ANP, BNP, CNP)
• czynniki adhezyjne i migracyjne dla monocytów i makrofagów
• czynniki zapalne (cytokiny, TNF-α)
• czynniki proliferacji komórek mięśni gładkich

Polimorfizmy wybranych genów
Układ
RAA
Polimorfizm
A1166C genu
AT1R
Polimorfizm
I/D genu ACE
Polimorfizm
M235T genu
ATG
Polimorfizm
T (-344)C genu
CYP11B2
Płytki
krwi
Polimorfizm
Pl
A
lub Zw
GP IIIa
ANP
Scal
ANP

Polimorfizmy wybranych genów
A.
I/D genu ACE
Polimorfizm insercyjno/delecyjny genu kodującego konwertazę angiotensyny (obecnośd w allelu I
lub brak w allelu D 278 par zasad w 16 intronie genu ACE)
Częśd naukowców sugeruje, że w regionie delecji zlokalizowany jest 13-nukleotydowy motyw
„wyciszacza” ekspresji genu tj. miejsce wiążące białko regulatorowe hamujące ekspresję
genu. Brak tego motywu w osób z allelem D ma prowadzid do zwiększonej ekspresji i
aktywności ACE
B.
M235T genu ATG
Substytucja metioniny w pozycji 235 łaocucha polipeptydowego angiotensynogenu przez treoninę
stanowi predyspozycję do nadciśnienia
Polimorfizm ten nie determinuje bezpośrednio stężenia ATG w osoczu, ale pozostaje w sprzężeniu
z innym polimorfizmem tego genu, położonym w promotorze 6 nukleotydów od miejsca
inicjacji transkrypcji - G(-6)A . Obecnośd adeniny w pozycji (-6) ułatwia przyłączenie
czynników transkrypcyjnych i prowadzi do nasilenia transkrypcji genu ATG i w konsekwencji
podwyższenia stężenia tego białka w osoczu.
C.
A1166C genu AT1R
Transwersja adeniny na cytozynę w pozycji 1166 w obrębie regionu nie podlegającego translacji w
koocu 3’ pozostaje w sprzężeniu z innym polimorfizmem modyfikującym wrażliwośd
naczyniozwężające działanie angiotensyny II może mied wpływ na rozwój nadciśnienia
tętniczego, niewydolności serca i zawału serca.
D.
T(-344)C genu CYP11B2
Zastąpienie tyminy przez cytozynę w miejscu (-344) powoduje natężenie aktywności syntazy
aldosteronu.

Polimorfizmy wybranych genów
Polimorfizm Pl
A
lub Zw GP IIIa
• Gromadzące się przyściennie płytki głównie pod wpływem kolagenu,
trombiny, noradrenaliny ulegają aktywacji, zmieniają swój kształt,
uwalniają wiele mediatorów krzepnięcia oraz aktywują receptory
GPIIb/IIIa odpowiedzialne za agregację płytek krwi.
• Polimorfizm Pl
A
wiąże się z częstszym występowaniem zawału serca i
niestabilnej choroby wieocowej, zespołów wieocowych związanych ze
wzmożoną aktywacją i agregacją płytek krwi.
Scal ANP
• Charakteryzuje się występowaniem T lub C w pozycji 2238 genu ANP w
regionie kodonu stop. Zamiana T na C prowadzi do utraty miejsca
restrykcyjnego dla ScaI i utratę właściwości stop kodonu, co potencjalnie
prowadzi do wydłużenia ANP o dwie dodatkowe Arg.
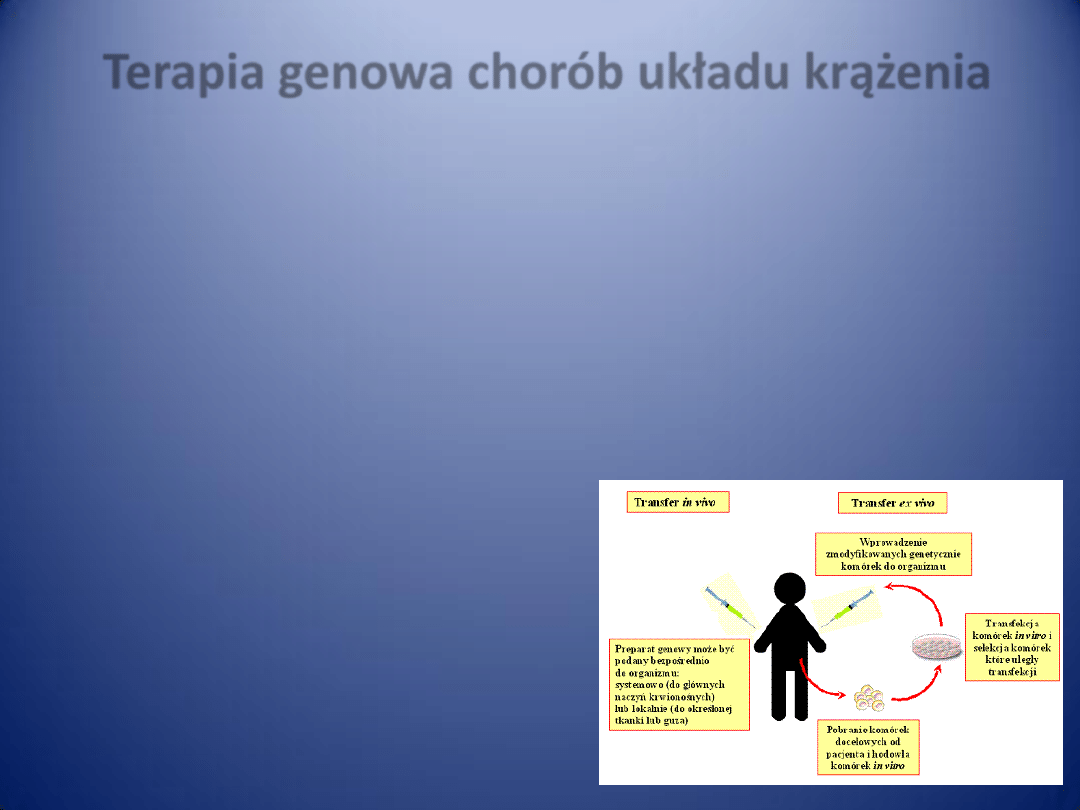
Terapia genowa chorób układu krążenia
Terapia genowa jest działalnością medyczną, polegającą na wprowadzeniu do żywych komórek
egzogennego materiału genetycznego (DNA lub RNA), co powoduje pożądaną zmianę
ekspresji danego genu w celu osiągnięcia efektu terapeutycznego.
Intencyjne zwiększanie lub zmniejszanie ekspresji genu prowadzi do pojawienia się w tkance
większej lub zmniejszonej ilości białka kodowanego przez ten gen. W ten sposób, wpływając
na kluczowe białka zaangażowane w patomechanizm chorób można korygowad defekty
genetyczne lub wywierad efekt terapeutyczny np.:
•
Transfer genu receptora LDL do wątroby w hipercholesterolemii rodzinnej
•
Indukowanie powstawania naczyo krążenia obocznego za pomocą transferu genu
naczyniowo-śródbłonkowego czynnika wzrostu (VEGF) do obszaru niedokrwionego mięśnia
sercowego
Transfer egzogennego materiału genetycznego może
odbywad się :
ex vivo (transfer odbywa się poza organizmem
chorego i dotyczyd może komórek lub tkanek
pobranych, a następnie, po modyfikacji genetycznej,
reimplantowanych do ciała pacjenta)
in vivo (za pomocą wektorów wirusowych i
niewirusowych)
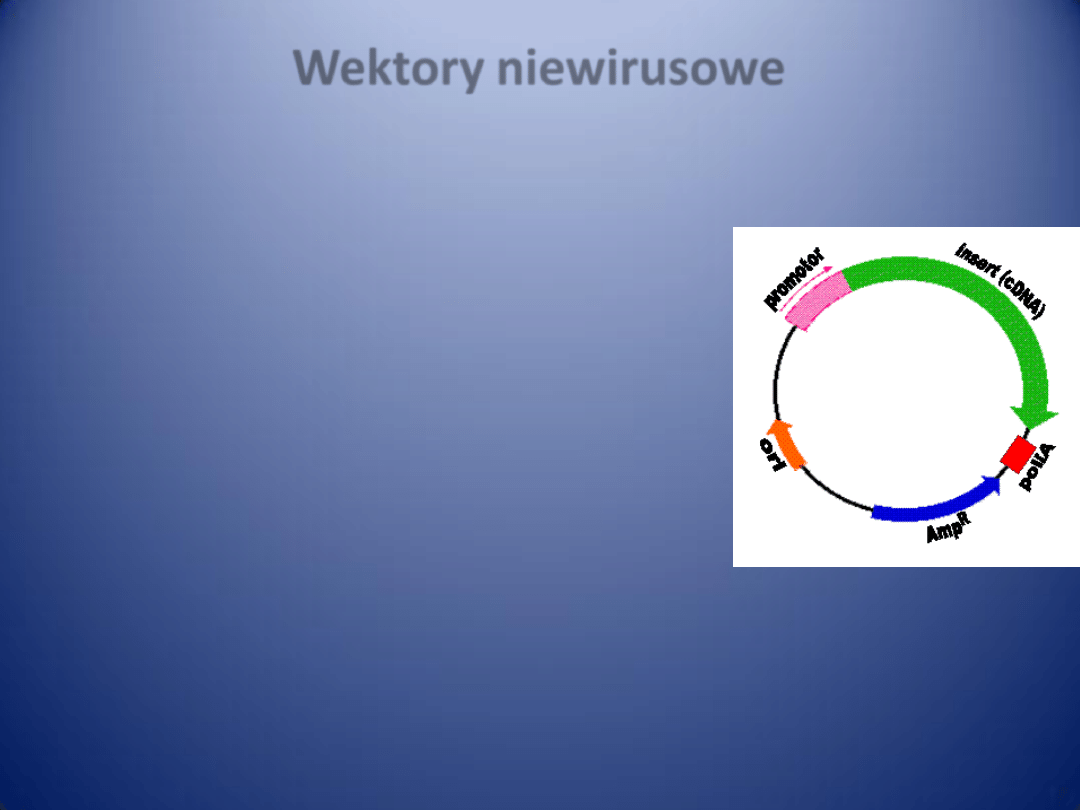
Wektory niewirusowe
Plazmid jest kolistą cząsteczką DNA, która poza sekwencją
terapeutycznego genu określaną jako transgen – zwykle
zawiera odpowiedni promotor, sekwencje regulatorowe i
wybrany gen antybiotykooporności.
Promotor jest rodzajem regulatorowego DNA, odpowiedzialnego
za aktywacje transkrypcji transgenu na mRNA, który ulega
następnie translacji.
Dodatkowe sekwencje regulatorowe najczęściej służą zwiększeniu
ekspresji genu terapeutycznego w określonym typie komórek,
tkance lub specyficznych warunkach metabolicznych.
Geny antybiotykooporności zawarte w plazmidzie służą jego
selekcji i namnażaniu w hodowlach bakteryjnych.
Plazmidy należą do wektorów, które nie ulegają integracji z
materiałem genetycznym komórki docelowej (nie wchodzą w
strukturę chromosomu), lecz pozostają w formie episomalnej
– wektory nieintegrujace. Konsekwencją braku integracji jest
krótkotrwała i przemijająca ekspresja po transferze
mediowanym przez palzmidy.
Plazmidowy wektor ekspresyjny
(promotor
– sekwencja promotora,
insert -
cDNA kodujące
„terapeutyczny gen”, poliA -
sekwencja poliadenylacji, AmpR -
gen oporności na ampicylinę (gen
selekcyjny), ori - miejsce inicjacji
replikacji)
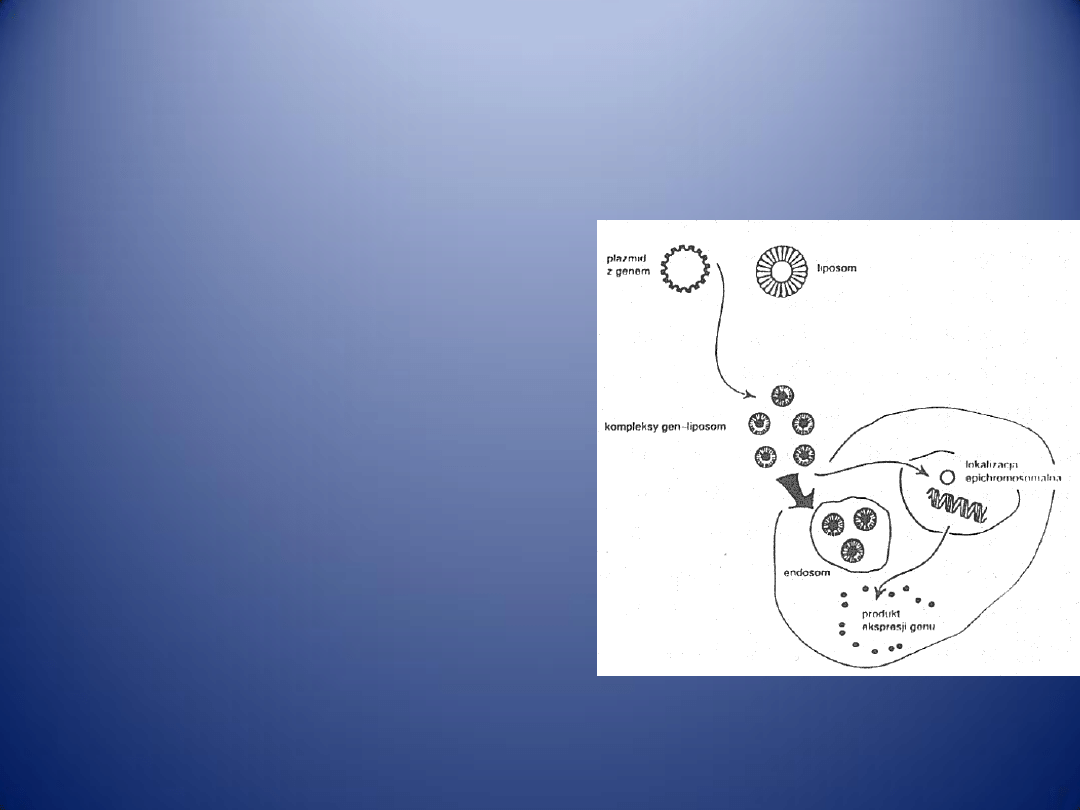
Plazmidy jako wektory
Plazmidy jako wektory dla terapii genowej,
posiadają wiele pozytywnych cech:
•
Są stosunkowo proste do skonstruowania i
łatwe w produkcji w dużych ilościach.
•
Brak infekcyjności i niska toksycznośd
warunkują bardzo korzystny profil
bezpieczeostwa.
•
Przy użyciu plazmidów można dokonad
transferu genów m.in. do kardiomiocytów,
komórek ściany naczyo oraz komórek m.
szkieletowych.
Poważnym ograniczeniem jest natomiast ich
niska efektywnośd w warunkach in vivo
(<1%).
Zastosowanie molekuł nośnikowych, polimerów o
dodatnim ładunku, lipidów i peptydów w celu
ochrony DNA przed degradacją i ułatwienia
integracji z komórka docelową zwiększyło
nieco ich efektywnośd.

Wektory wirusowe
•
Rekombinowane wirusy wykorzystywane w terapii genowej to przede wszystkim
retrowirusy, adenowirusy, wirusy związane z adenowirusami.
•
Wirusy są tak modyfikowane, aby efektywnie wprowadzały geny do docelowych
komórek nie wywołując efektów ubocznych zagrażających życiu pacjenta.
•
Modyfikacje najczęściej polegają na usunięciu genów wirusowych
odpowiedzialnych za wywoływanie choroby (geny wirulencji) oraz za replikację
materiału genetycznego wirusa i jego namnożenie w komórkach gospodarza. W to
miejsce wprowadzany jest transgen kodujący „terapeutyczne białko”.
•
Ponadto naturalne białka otoczki wirusa są zastępowane innymi białkami lub
modyfikowane w celu zwiększenia powinowactwa wektora do określonych typów
tkanek, dzięki czemu infekcji ulegną tylko komórki docelowe.
•
Modyfikacje obejmują także usunięcie lub zmutowanie genów, które wywołują
najsilniejszą reakcję układu odpornościowego.
•
Wykorzystanie naturalnych zdolności wirusów do wprowadzania kwasów
nukleinowych do określonego typu komórek i zdolności do integracji z genomem
gospodarza umożliwia wydajną transfekcję wielu typów komórek oraz zapewnia
wysoki poziom ekspresji transgenu.
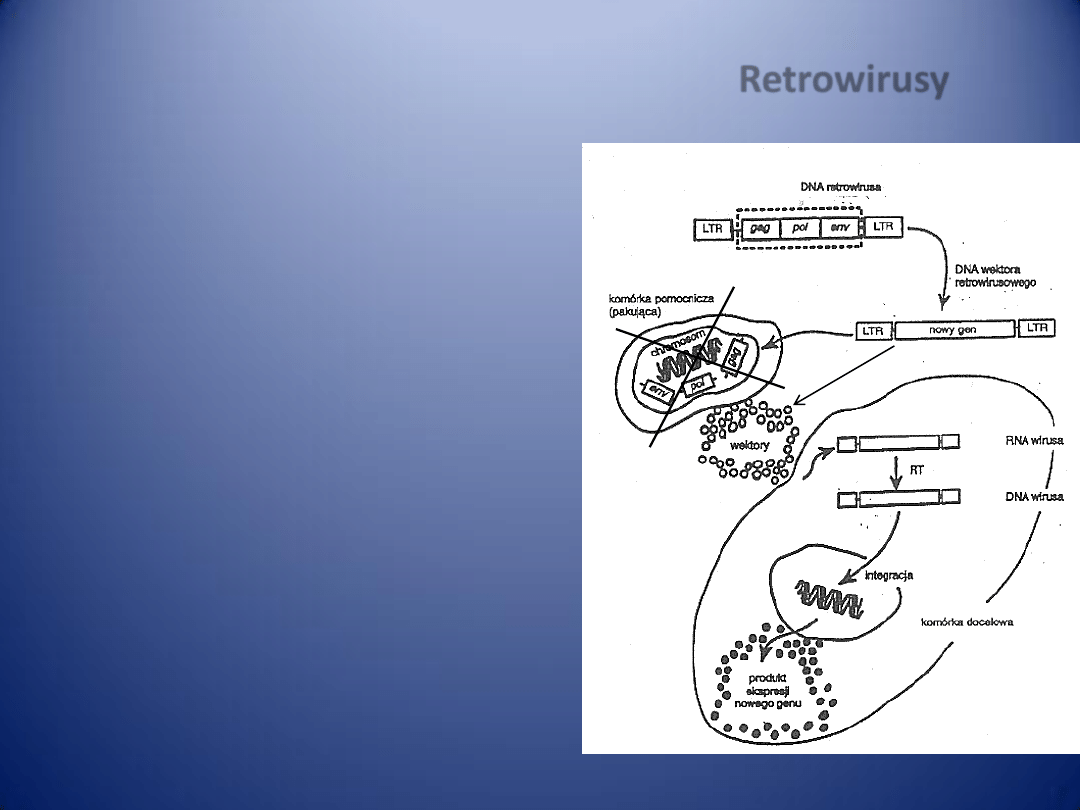
Retrowirusy
Są grupą wirusów powszechnie stosowanych jako
wektory dla transferu genów w badaniach
eksperymentalnych i klinicznych.
Zalety: genom łatwo poddający się manipulacjom
umożliwiającym akomodację dużych
fragmentów egzogennego materiału
genetycznego do 800pz, możliwośd transfekcji
różnorodnych typów komórek, długotrwała
ekspresja, wynikająca z trwałej integracji
transgenu do chromosomalnego DNA
gospodarza.
Wady: zdolne są do transfekcji wyłącznie komórek
aktualnie dzielących się (w ścianie n.
krwionośnych oraz miokardium niewielki
odsetek komórek znajduje się w fazie
podziału), duże ryzyko transformacji
nowotworowej (przypadkowa integracja
transgenu do genomu gospodarza w rejonie
protoonkogenu), niska efektywnośd transferu.
Geny:
-gag (koduje białka kapsydu)
-pol (koduje odwrotna transkryptazę, integrazę , proteazę)
- env (zawiera informację o GP otoczki)
Wprowadzenie
transgenu
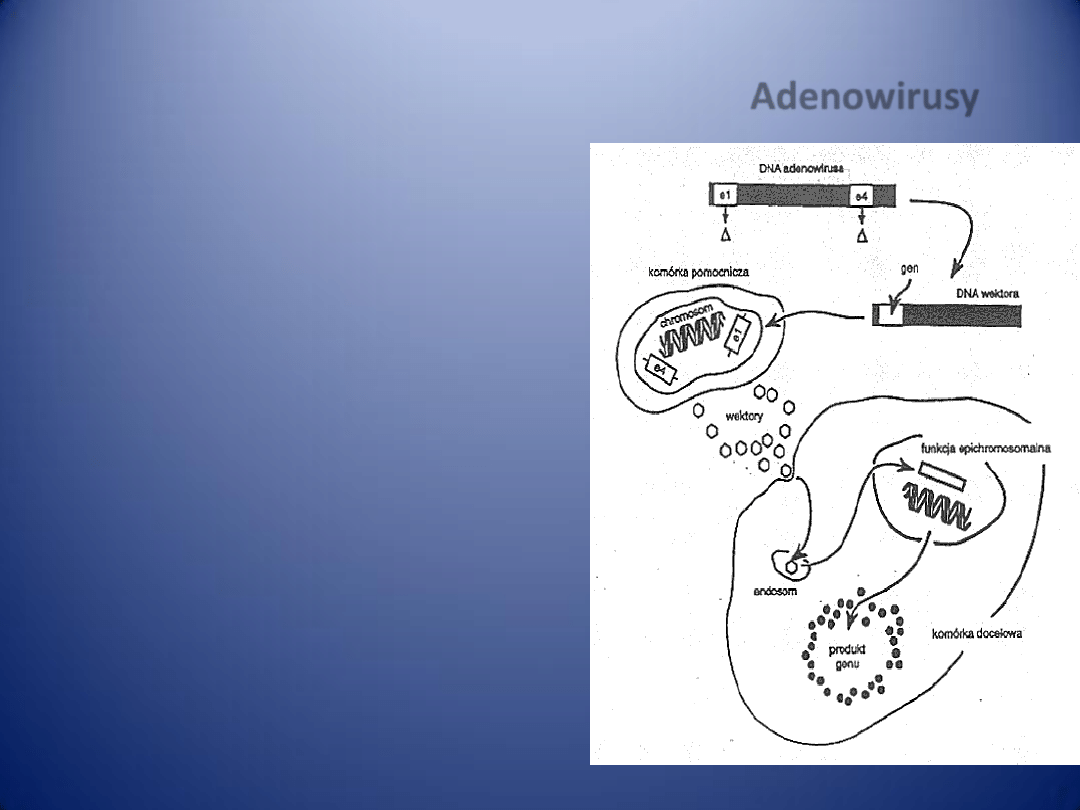
Adenowirusy
Wektory oparte na adenowirusach należą do
najbardziej efektywnych narzędzi transferu
genów.
Ądenowirusy są bezotoczkowymi wirusami DNA
Pomimo, że nabłonek dróg oddechowych jest
pierwotnym celem dla tej grupy wirusów, są
one również zdolne do infekowania wielu
innych typów komórek.
Transfekują z wysoką efektywnością zarówno
komórki dzielące się, jak i komórki już dojrzałe,
niezdolne do proliferacji.
Poważnym ograniczeniem zastosowania tych
wirusów jest krótkotrwałośd mediowanej
ekspresji, która wynika z braku intergracji
transgenu z genomem gospodarza oraz dużym
odczynem immunologicznym skierowanym
przeciwko transfekowanym komórkom.
Materiał genetyczny wirusa tworzą odwrócone
terminalne sekwencje powtórzeniowe (ITR),
sekwencje promotorowe oraz geny wirusowe.

Problemy terapii genowej
Przede wszystkim należy zapewnid bezpieczeostwo procedury poprzez przygotowanie
nietoksycznych, nieimmunogennych preparatów genowych zgodnych z wymogami
farmakopealnymi.
Należy także zaprojektowad sposób podania (łatwy, bezpieczny i efektywny) oraz określid
dawkę preparatu.
Ponadto preparaty genowe powinny byd łatwe i tanie w syntezie i przechowywaniu.
Poważnym problemem jest także brak wydajnego systemu transferu genów do jąder
komórkowych komórek docelowych. Z tym z kolei są związane problemy z uzyskaniem
długotrwałej ekspresji transgenu na odpowiednim poziomie.
Przyczyną niestabilnej ekspresji są także trudności z uzyskaniem ukierunkowanej integracji
wprowadzanego DNA, przez co konieczne staje się wielokrotne podawanie preparatu
genowego, co z kolei przyczynia się do powstania odpowiedzi immunologicznej ze strony
organizmu.
Wydajna ekspresja na stałym poziomie wymaga także dobrania odpowiedniego systemu
ekspresyjnego tzn. odpowiedniego konstruktu wektora, przy czym szczególną uwagę należy
zwrócid na wybór promotora inicjującego transkrypcję.
Biochemiczne markery
kardiologiczne
i sposoby ich detekcji

Markery martwicy kardiomiocytów
Wskaźniki uszkodzenia mięśnia sercowego to średnio i drobnocząsteczkowe białka o
funkcji:
•
budulcowej (troponiny)
•
enzymatycznej (kinaza kreatynowa i jej izoenzymy)
•
transportowo-magazynujacej (białko wiążące kwasy tłuszczowe – H-FABP, mioglobina)
, które wydostają się z uszkodzonych kardiomiocytów.
Do uszkodzenia, najczęściej kooczącego się martwicą, dochodzi w wyniku niedokrwienia,
niedotlenienia, zapalenia, działania toksycznego lub urazu.
W jego wyniku zwiększa się przepuszczalnośd błony komórkowej, a następnie dochodzi
do jej perforacji i wycieku białka z wnętrza kardiomiocyta do krwi. Początkowo
wydostają się białka cytozolowe o małej masie cząsteczkowej np. H-FABP i
miloglobina, następnie izoenzym sercowy kinazy kreatynowej, a na koocu białka
strukturalne – troponiny.
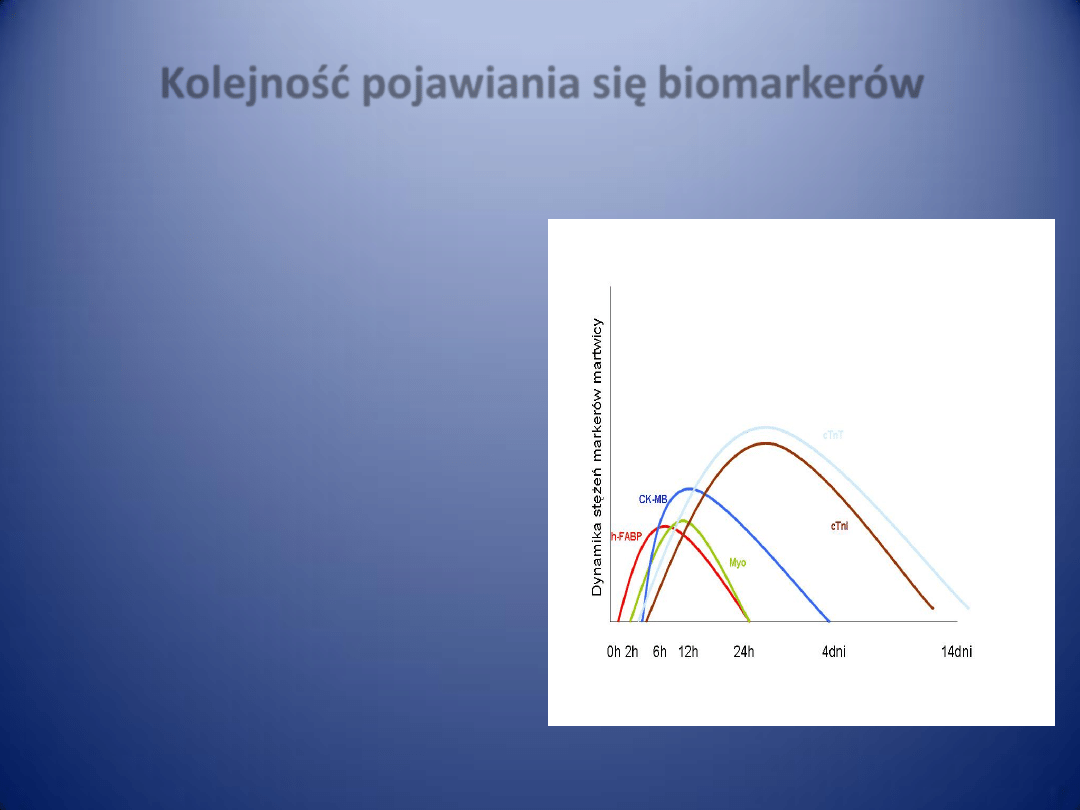
Kolejnośd pojawiania się biomarkerów
Biomarkery wczesne osiągające wykrywalne
stężenia średnio po 1,5 – 3 h od
wystąpienia objawów klinicznych –
białko wiążące kwasy tłuszczowe i
mioglobina
Markerem średniowczesnym pojawiającym
się w krwi po 2-4 h jest izoenzym
sercowy kinazy kreatynowej, oznaczany
jako stężenie białka (a nie aktywnośd
enzymatyczna).
Markerami stosunkowo najpóźniejszymi są
pojawiające się po 4-6 h troponiny.
Do biomarkerów klasycznych, ale o
marginalnym dziś znaczeniu zalicza się:
kinazę kreatynową (aktywnośd
całkowitą), dehydrogenazę mleczanową
(LDH) i aminotransferazę
asparaginianową (AspAT)
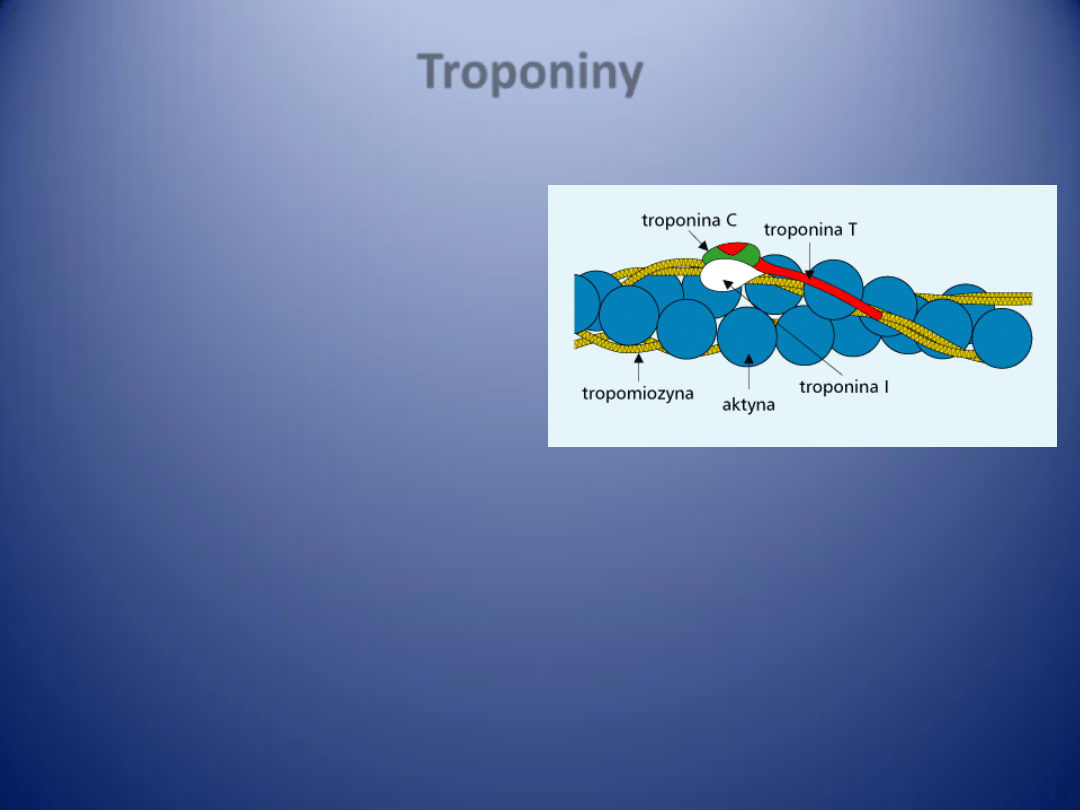
Troponiny
Kompeks troponin wraz z tropomiozyną i aktyną tworzy strukturę filamentu cienkiego
mięśni i jest zaangażowany w zależną od jonów Ca2+ regulację interakcji aktyna-
miozyna.
W czasie skurczu mięśnia, na skutek wiązania jonów Ca2+ do TnC, następują zmiany
konformacyjne kompleksu: wzmocnienie wiązania TnC-TnI i zerwanie wiązania
pomiędzy TnI a kompleksem tropomiozyna-aktyna. W wyniku tego dochodzi do
odsłonięcia miejsc aktywnych w aktynie i przyłączenia główek miozyny. Odwrotny
cykl przemian, który prowadzi do rozkurczu mięśnia, jest zapoczątkowany przez
spadek stężenia jonów Ca2+
Troponina T — (37 kD) wiąże się z tropomiozyną,
jest najbardziej zasadowym składnikiem
kompleksu troponiny.
Troponina I — (23 kD) hamuje interakcję aktyny
z miozyną, jest białkiem zasadowym.
Troponina C — (18,3 kD) wiąże jony wapnia w
czterech miejscach aktywnych, jest kwaśnym
składnikiem kompleksu troponin.

Troponiny
W komórkach mięśnia sercowego występują izomeryczne formy troponin.
Troponina C, specyficzna dla serca (cTnC), ma tylko 3 miejsca wiązania dla jonów Ca2+
i ulega ekspresji także w mięśniach szkieletowych. Tak więc spośród białek
regulatorowych kandydatami na specyficzne markery sercowe pozostają jedynie
cTnI i cTnT.
Lokalizacja wewnątrzkomórkowa troponin sercowych oraz ich specyficznośd wskazują
na to, że troponiny (zarówno cTnT, jak i cTnI) spełniają warunki definitywnych
markerów uszkodzenia serca.
Badania różnicowe przeprowadzone na populacji pacjentów z uszkodzeniem tkanki
mięśniowej (trauma, rabdomioliza), chroniczną miopatią i po intensywnym wysiłku
fizycznym dowiodły wysokiej swoistości oznaczenia cTnI .
Podwyższone stężenie cTnI zaobserwowano w takich schorzeniach związanych z
uszkodzeniem serca jak: zapalenie mięśnia sercowego, ostre zespoły wieocowe z
niestabilną dławicą piersiową, zawałem bez załamka Q, a także z zawałem
pełnościennym

Troponiny
Nie ulega zatem wątpliwości, że oznaczenie troponiny sercowej (cTnI i cTnT)
przedstawia dużą wartośd w diagnostyce zawału serca, bez względu na to,
którą z troponin przyjmie się za marker diagnostyczny.
Obserwowany wzrost stężenia troponiny przypada na 3.–6. godzinę od
wystąpienia objawów, osiągając maksimum w 14.–20. godzinie, dlatego
też najwyższą czułośd testu (0,98) uzyskuje się 12 godzin od pierwszych
symptomów zawału. Duża stabilnośd troponin we krwi powoduje, że
wysokie stężenia tych markerów mogą się utrzymywad 5–15 dni po
rozpoznanym zawale, utrudniając tym samym rozpoznanie fazy
dokonanego zawału, tworzenia się blizny pozawałowej, jak też dorzutu
zawału.
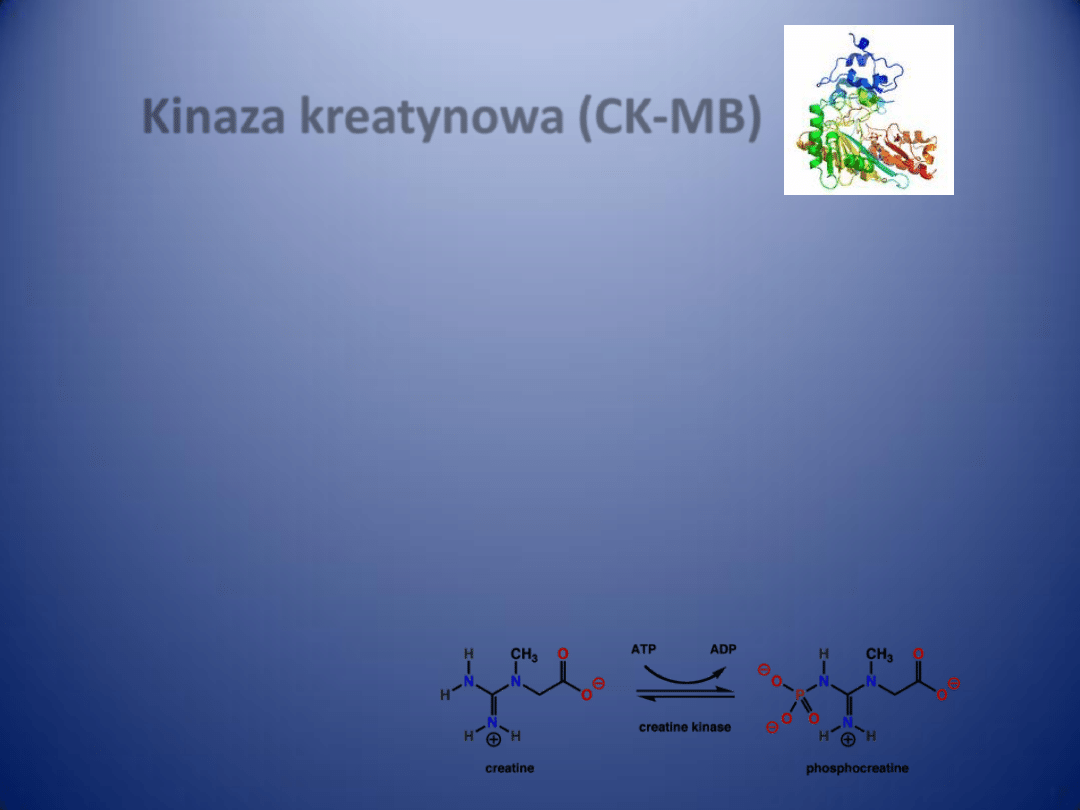
Kinaza kreatynowa (CK-MB)
N-fosfotransferaza kreatyny, inaczej określana kinazą fosfokreatynową (CPK lub CK), jest enzymem
występującym w cytoplazmie komórkowej, który katalizuje odwracalną reakcje fosforylacji
kreatyny przez transfer wysokoenergetyczny wiązania między ATP a kreatyną. Produkt reakcji,
fosfokreatyna, jest głównym „zapasem” energii dla komórek mięśniowych w procesach skurczu,
relaksacji i transportu komórkowego .
Cząsteczka enzymu — kinazy fosfokreatynowej — ma masę 86 kD i jest zbudowana z podjednostek,
które występują w 2 izoformach: CK-M i CK-B.
Z tych podjednostek składają się 3 izoenzymy :
•
CK-MM - mięśnie szkieletowe
•
CK-MB - mięsieo sercowy
•
CK-BB - tkanka mózgowa
W diagnostyce zawału serca najważniejszą rolę odgrywa CK-MB, tak zwana sercowa kinaza
kreatynowa.

Kinaza kreatynowa (CK-MB)
W diagnostyce chorób serca wykonuje się 2 typy oznaczeo: aktywnośd CK-MB (CK-
MBakt) lub pomiar całkowitego stężenia (CK-MBmass).
Pomiar aktywności opiera się zazwyczaj na teście hamowania aktywności CK-M,
natomiast w przypadku oznaczeo całkowitego stężenia enzymu wykorzystuje się
metody immunochemiluminescencyjne.
Słabą stroną oznaczeo kinazy kreatyny jest to, że enzym ten nie jest swoisty wyłącznie
dla serca, dlatego też, aby odróżnid pierwotne uszkodzenie mięśnia sercowego od
innych uszkodzeo tkanki mięśniowej, wprowadzono kryterium dla pomiaru
aktywności CK-MB powyżej 6% całkowitej aktywności CPK. W przypadku pomiaru
całkowitego stężenia sercowej kinazy kreatyny (CK-MBmass) za wartośd odcięcia
dla zawału serca (AMI cut off) przyjmuje się stężenie powyżej 5 ng/ml.

Sercowe białko wiążące
kwasy tłuszczowe (H-FABP)
Białko wiążące wolne kwasy tłuszczowe (h-FABP), które jest małym cytozolowym
białkiem zaangażowanym w transport i metabolizm kwasów tłuszczowych i obficie
występuje w komórkach miokardium, może byd obiecującym bardzo wczesnym
markerem ostrego MI.
Białko h-FABP jest uwalniane z martwiczo zmienionych kardiomiocytów bardzo szybko
(ok. 50% czułośd już w pierwszej godzinie po rozpoczęciu objawów) i może byd bardzo
użyteczne zarówno do wczesnego potwierdzania, jak i wykluczania MI, z tym że wymaga
późniejszego oznaczenia bardziej swoistych markerów – Tn i CK-MB.
Swoistośd tego biomarkera w stosunku do serca sięga 80%.
W warunkach prawidłowych h-FABP jest wykrywane w surowicy w stężeniach do
12μg/L, co jest wynikiem jego uwalniania z m. poprzecznie prążkowanych.
Podwyższone stężenie h-FABP utrzymuje się do 24 h od początku objawów.
Uszkodzenie m. porzecznie prążkowanych i choroba nerek mogą byd przyczyna wyników
fałszywie dodatnich.
Mioglobina
Mioglobina (17,8 kD),
podobnie jak hemoglobina, wiąże tlen, przez co jest jego rezerwuarem w
mięśniach szkieletowych, a także w sercu. Jest ona, tak jak kinaza kreatynowa, białkiem
cytoplazmatycznym, stanowi około 5–10% wszystkich białek cytoplazmy. Stężenie mioglobiny w
surowicy krwi jest odzwierciedleniem masy mięśniowej i dlatego wartości referencyjne dla
mężczyzn są wyższe niż dla kobiet i wynoszą odpowiednio: 19–90 ng/ml i 12–76 ng/ml.
W przypadku zawału serca spośród wszystkich znanych markerów uszkodzenia serca najszybciej
wzrasta stężenie mioglobiny (już w pierwszych 30 min) i trwa około 3 godziny, po 10–12
godzinach następuje powrót do wartości prawidłowych.
Zaletą mioglobiny jako markera niedokrwienia mięśnia sercowego jest jej bardzo szybki wzrost w
surowicy krwi. Wartośd predykcyjna ujemna (NPV, negative predictive value — odsetek
zdrowych osób z ujemnym testem) dla mioglobiny oznaczanej w pierwszych 4 godzinach zawału
jest bardzo wysoka i wynosi 0,89. Porównywalny poziom NPV dla oznaczenia CK-MBmass
uzyskuje się po 7–8 godzinach.
Wadą oznaczeo mioglobiny jest to, że nie jest specyficzna dla serca i powinna byd oznaczana wraz z
innymi wskaźnikami , tak zwanymi definitywnymi wskaźnikami uszkodzenia serca, które
wykazują wyższą swoistośd diagnostyczną (troponiny sercowe, CK-MBmass).
Mankamentem jest też krótki przedział czasu, w którym stwierdza się podwyższone stężenie
mioglobiny, co powoduje często fałszywie negatywne wyniki u pacjentów zgłaszających się
późno do szpitala.
Fałszywie dodatnie wyniki oznaczeo mioglobiny mogą byd skutkiem nie tylko istotnego uszkodzenia
mięśni, ale również iniekcji domięśniowych i wysiłku fizycznego.

Klasyczne wskaźniki uszkodzenia mięśnia
sercowego
Do klasycznych markerów uszkodzenia m. sercowego zalicza się:
całkowitą aktywnośd kinazy kreatynowej (CK)
aktywnośd aminotransferazy asparaginowej (AspAT)
aktywnośd dehydrogenazy mleczanowej (LDH)
Całkowita aktywnośd CK nie ma swoistości w stosunku do mięśnia sercowego.
Aktywnośd tego biomarkera może byd znacznie podwyższona w uszkodzeniu m.
poprzecznie prążkowanych na różnym tle (mechanicznym, toksycznym, w
przebiegu chorób mięśni, drgawek). Fałszywie dodatnie wyniki mogą byd
także efektem występowania tzw. makroform CK.
Oznaczanie aktywności AspAT i LDH w diagnostyce zawału serca ma znaczenie
tylko historyczne. Oba markery nie wykazują pożądanej swoistości w
stosunku do m. sercowego, bowiem ich podniesiony poziom towarzyszy
także wielu innym schorzeniom, np. wątroby.

Wskaźniki przeciążenia mięśnia sercowego
Są to substancje, które wydzielane w zwiększonej ilości w odpowiedzi na przeciążenie
m. sercowego i mogą byd oznaczane we krwi:
•
Pewne biomarkery (peptydy natiuretyczne typu A i B) są uwalniane bezpośrednio z
kardiomiocytów komór i przedsionków.
•
Inne produkowane są poza sercem np. w korze i rdzeniu nadnerczy, we
współczulnym układzie nerwowym, w nerkach, śródbłonku w odpowiedzi na
zmniejszoną perfuzję narządową spowodowana niewydolnością serca. Są to m.in.
aldosteron, noradrenalina i endotelina
Fizjologiczne rola peptydów natiuretycznych polega prawdopodobnie na
przeciwstawianiu się spaczonym, mechanizmom kompensacyjnym uruchamianym
w niewydolności serca za pośrednictwem układy RAA i układu współczulnego,
czego skutkiem jest wzrost stężenia aldosteronu, noradrenaliny i endoteliny. W
odpowiedzi na te zjawiska m. sercowy wytwarza peptydy natiuretyczne, które
zwiększają natiurezę i diurezę, rozszerzają naczynia, hamują układ RAA.
Peptydy natiuretyczne
• NT-proBNP i BNP wykrywają niewydolnośd serca z czułością i swoistością
sięgająca 80%.
• Prawidłowe stężenia peptydów natiuretycznych praktycznie wykluczają
niewydolnośd serca.
Badanie kliniczne, EKG, RTG klatki piersiowej, echokardiografia
BNP <100 pg/ml
NT-proBNP < 400 pg/ml
Przewlekła niewydolnośd
serca bardzo mało
prawdopodobna
BNP 100-400 pg/ml
NT-proBNP 400-2000 pg/ml
Rozpoznanie
niewydolności serca
BNP > 400 pg/ml
NT-proBNP >2000 pg/ml
Przewlekła niewydolnośd
serca bardzo
prawdopodobna
Peptydy natiuretyczne

Biochemiczne wskaźniki zagrożenia miażdżycą
U podstaw zmian miażdżycowych leża przewlekłe procesy zapalne toczące się w śródbłonku w
odpowiedzi na kumulujące się jego uszkodzenia spowodowane czynnikami ryzyka
(lipidowymi, hemodynamicznymi, hemostatycznymi i in.). Ta przewlekła odpowiedź zapalna
ma na ogół niewielkie nasilenie, ale dotyczy właściwie całego śródbłonka.
Białko C – reaktywne (CRP)
Do niedawna CRP wykorzystywano tylko jako białko ostrej fazy w diagnostyce i monitorowaniu
ostrych stanów zapalnych (10-1000 mg/l).
Wiele badao retrospektywnych i prospektywnych wskazuje na związek stężeo CRP (0-10 mg/l) z
częstością występowania choroby wieocowej, zawałów i zgonów z powodów sercowo-
naczyniowych.
Koniecznośd oznaczania bardzo niskich steżeo CRP wymusiła zwiększenie czułości analitycznej i
wprowadzenia wysokoczułego CRP (hs-CRP).
Hs-CRP jest niezależnym markerem ryzyka chorób sercowo –naczyniowych.
•
U pacjentów ze stabilna choroba wieocową lub OZW oznaczenia hs-CRP mogą byd
przydatne jako niezależny wskaźnik prognostyczny takich powikłao jak: zawał serca,
restenoza po angioplastyce wieocowej, nawracające incydenty niedokrwienne.
•
Oznaczenia hs-CRP powinny byd wykonywane dwukrotnie, w odstępie 2 tyg. Wyniki należy
uśrednid i podad w mg/l.
Nie ma jednak jasności jakie jest graniczne stężenie CRP pozwalające przewidzied niekorzystny
przebieg i zgon u osób z OZW.

Biochemiczne wskaźniki zagrożenia miażdżycą
Homocysteina
• Osoby z podwyższonym stężeniem homocysteiny są bardziej zagrożone
miażdżycą, zawałem, udarem i zgonem. Zagrożenie powikłaniami
miażdżycy jest większe u osób wyjściowo zdrowych i pacjentów z już
rozpoznaną miażdżycą. Wydaje się, że hiperhomocysteinemia w większym
stopniu koreluje z powikłaniami zakrzepowymi miażdżycy niż z jej
zaawansowaniem i progresją.
• Mimo łatwości w modyfikowaniu stężeo homocysteiny (wit. grupy B i kwas
foliowy) nie udowodniono, żeby farmakologiczna redukcja jej stężeo
przekłada się na zmniejszenie zagrożenia powikłaniami miażdżycy.
• Oznaczenia homocysteiny można rozważyd u osób z rozpoznaną miażdżycą
lub jej powikłaniami i brakiem ewidentnych klasycznych czynników ryzyka.

Biochemiczne wskaźniki zagrożenia miażdżycą
Lipoproteina (a)
Mechanizm dziania miżdżycorodnego może polegad na nasilonym utlenianiu (z
powodu obecności w osoczu) i wychwytywaniu przez receptory zmiataczowe na
makrofagach, co sprzyja powstawaniu komórek piankowatych i nacieczeo
tłuszczowych. Inna hipoteza zakłada interferencję Lp(a) z aktywacją plazminogenu,
co prowadzi do upośledzenia fibrynolizy.
Stężenie Lp(a)jest bardzo trudno modyfikowalne. Ani dieta ani leki hipolipemizujace, z
wyjątkiem kwasu nikotynowego, nie obniżą stężenia tej lipoproteiny.
Oznaczenie Lp(a) wydaje się celowe u osób z przedwczesna chorobą wieocową, w
wywiadzie rodzinnym. Sugeruje się też oznaczanie jej w rodzinnej
hipercholesterolemii, w której podwyższone stężenia Lp(a) wykazuje synergizm
aterogenny z podwyższonym stężeniem cholesterolu LDL. Względnym wskazaniem
może byd przebycie zawału serca lub udaru mózgu, zwłaszcza u osób z
normolipidemią.
Aby oznaczenie było miarodajne, nie można go wykonywad przed upływem 4 tyg. Od
zawału.

Nowe markery w predykcji OZW
W przeciągu kilku ostatnich lat poznano wiele nowych endogennych substancji,
głównie białek, spośród których nieliczne posiadają właściwości
potencjalnych laboratoryjnych markerów predykcyjnych OZW. Związane są
one z procesem zapalnym toczącym się w obrębie blaszki miażdżycowej.
Markery destabilizacji blaszki
miażdżycowej
Markery pęknięcia blaszki
miażdżycowej
MPO (mieloperoksydaza)
MMP-9 (międzykomórkowa
metaloproteinaza -9 )
PAPP-A (związane z ciążą
osoczowe białko typu A)
sCD40L (rozpuszczalna forma
ligandu CD40)

Metaloproteinaza macierzy -9 (MMP-9)
W obrębie serca MMP-9 uczestniczy w remodelingu naczyo oraz struktury komór po zabiegach
chirurgicznych, a także bierze udział w procesach destabilizacji blaszki miażdżycowej.
MMP-9, znana jako żelatynaza, jest proteinazą zależną od Zn, składająca się z 3 podjednostek. Ma
zdolnośd do łączenia się z żelatyną, kolagenem i lamininą. Aktywacji MMP-9 odbywa się na
drodze proteolitycznej, bądź w wyniku ekspozycji na tlenek cynku.
MMP-9:
•
jest generatorem angiostatyn – po interakcji z plazminogenem zwiększa jego powinowactwo
do kolagenu
•
wiąże się z ICAM-1
•
jej przeciwzapalny charakter wyraża się poprzez aktywację IL-1β hamowanie odpowiedzi IL-2
W tkance naczyniowej MMP-9 jest zlokalizowana w ramieniu blaszki, będącym miejscem
szczególnie narażonym na rozerwanie.
Wzrost stężenia MMP-9 obserwuje się podczas zawałów z uniesieniem odcinka ST (STEMI) oraz w
fazach zaostrzenia choroby wieocowej. Pojawiły się też doniesienia, że wzrost stężenia
MMP-9 koreluje ze stanem zapalnym w blaszce miażdżycowej.
Mieloperoksydaza (MPO)
•
Jest hemoproteina o masie 140 kDa, składającą się z par łaocuchów: lekkiego i
ciężkiego.
•
Magazynowana jest w azurochłonnych ziarnistościach neutrofilów i
monocytów/makrofagów.
•
Katalizuje reakcję łączenia chloru z wodorem w procesie wybuchu tlenowego, z
wytworzeniem kwasu chlorowego oraz modyfikuje oksydacyjnie tyrozynę do
rodnika tyrozynowego.
•
Jest mediatorem uwalnianym przez komórki biorące udział w odpowiedzi zapalnej ,
włączając w to makrofagi zlokalizowane w blaszce miażdżycowej.
•
Chociaż rola neutrofilów w tworzeniu blaszki miażdżycowej nie jest wielka, jednak
ich degranulacja w stanach zaostrzenia choroby wieocowej jest jednoznaczna z
uwolnieniem mieloperoksydazy, która inicjuje wiele procesów prowadzących do
pękania blaszki miażdżycowej:
•
Aktywacja proteaz
•
Oksydacja lipidów w obrębie blaszki
•
Wzmaganie trombogeniczności struktur blaszki

Związane z ciążą białko osoczowe A (PAPP-A)
•
Jest wysokocząsteczkową glikoproteiną syntetyzowaną przez scyntiotrofoblast,
rutynowo oznaczana w diagnostyce prenatalnej zespołu Downa.
•
Jest proteazą białka 4 wiążącego insulinopodobny czynnik wzrostu, a pod
względem enzymatycznym jest metaloproteinazą.
•
Naukowcy wykazali obecnośd PAPP-A w niestabilnych blaszkach miażdżycowych
pacjentów zmarłych nagle w przebiegu OZW i opisali zjawisko podwyższonego
stężenia PAPP-A u pacjentów z niestabilną dusznicą bolesną i ostrym zawałem
serca.
•
Stężenie PAPP-A we krwi osób z OZW zaczyna narastad najwcześniej w ciągu 2,a
najpóźniej po 30 h od pojawienia się objawów, przy czym tylko u 2/3 pacjentów
pozostaje na podwyższonym poziomie.
•
PAPP-A obecne we krwi w związku z OZW różni się strukturalnie od jego formy
wyizolowanej z surowic ciężarnych. W warunkach fizjologicznych PAPP-A krąży we
krwi jako heterotetramer składający się z 2 podjednostek enzymu, związanych
kowalencyjnie z dwiema podjednostkami jego inhibitora – proMBP.
•
PAPP-A pochodzące z niestabilnej blaszki jest homodimerem, co czyni je
potencjalnie trudnym do wykrycia w testach immunochemicznych, któer są
konstruowane, by wykrywad cząstki nieczynne biologicznie (związane z proMBP).

Rozpuszczalny ligand receptora CD40 (sCD40L)
•
Jest trimerycznym białkiem transbłonowym. Podobnym do TNF-α, występującym
na powierzchni płytek krwi.
•
po stymulacji płytki ulega natychmiastowej ekspresji na jej powierzchni, stąd po
odcięciu przedostaje się do osocza w formie rozpuszczalnej.
•
Związanie się CD40L lub sCD40L ze swoim receptorem prowadzi do aktywacji
limfocytów T i makrofag, dając tym samym sygnał zapoczątkowujący
prozakrzepową i prozapalną odpowiedź organizmu.
•
Bilogicznym następstwem utworzenia kompleksu białko-receptor jest sekrecja
prozapalnych cytokin i chemokin, aktywujących reakcje zapalną w komórkach
śródbłonka naczyo.
•
Receptor dla CD40L jest również obecny na limfocytach B, monocytach i
makrofagach, a także na komórkach śródbłonka i m. gładkich.
•
Połączenie którejś z form Cd40L z receptorem na tych komórkach prowadzi do
uwolnienia mieloperoksydaz macierzy pozakomórkowej MMPs, a w konsekwencji
do destabilizacji blaszki. Z kolei, będące następstwem pęknięcia blaszki uwolnienie
TF aktywuje wtórnie płytki, które uwalniają więcej sCD40L, nasilając stan zapalny i
odpowiedź prozakrzepową w krwioobiegu.
Zakład Nadciśnienia Tętniczego
Katedry Nefrologii i Nadciśnienia Tętniczego
Uniwersytetu Medycznego w Łodzi

HEMOSTAZA
Hemostaza jest jednym z wielu znanych systemów zapewniających stałe
warunki wewnętrznego środowiska organizmu człowieka (homeostazy).
Według powszechnie akceptowanej definicji hemostaza to zespół
mechanizmów fizjologicznych, które zapewniają:
sprawne hamowanie krwawienia po przerwaniu ciągłości ściany naczyń
krwionośnych.
szczelność łożyska naczyniowego.
płynność krążącej krwi
Oprócz wymienionych powyżej funkcji, układ hemostazy odgrywa także pewną
rolę w regulacji funkcji łożyska naczyniowego i mechanizmów
odpornościowych.

Funkcjonalny model układu hemostazy
Model ten opiera się na opisie kolejnych etapów działania układu
hemostazy od momentu przerwania ciągłości naczynia krwionośnego do
chwili uruchomienia mechanizmów antykoagulacyjnych (hemostaza
pierwotna, hemostaza wtórna, fibrynoliza).
Uszkodzenie naczynia krwionośnego zapoczątkowuje sekwencję zdarzeń –
uruchamiane są kolejne elementy (mechanizmy) układu hemostazy.
Tworzenie czopu hemostatycznego podzielone zostało na dwa etapy:
hemostazę pierwotną i wtórną
.
W pierwszym etapie ujawnia się prokoakulacyjne działanie naczyń
krwionośnych i płytek krwi, tworzony jest pierwotny czop hemostatyczny
składający się głownie z płytek krwi.

Hemostaza pierwotna
Pierwsza faza aktywacji układu hemostazy: płytki krwi rozpoznają uszkodzenie naczynia i
tworzą czop płytkowy (pierwotny czop hemostatyczny) uwalniając substancje
wspomagające układ krzepnięcia. Czop płytkowy nie jest w stanie przez dłuższy czas
zablokować wypływ krwi z uszkodzonego naczynia, stąd konieczne jest wzmocnienie
jego struktury przez sieć włóknika.
Hemostaza wtórna
Druga faza aktywacji układu hemostazy. Uszkodzona ściana naczynia jest źródłem czynnika
tkankowego (TF, tissue factor), który uaktywnia układ krzepnięcia. W ramach hemostazy
wtórnej uruchamiana jest kaskada czynników krzepnięcia i tworzony jest właściwy czop
hemostatyczny składający się z płytek krwi włóknika, leukocytów i erytrocytów.
Fibrynoliza
Trzecia faza aktywacji układu hemostazy. Jest to ostatni etap działania układu hemostazy w
modelu funkcjonalnym. Jako, że model ten powstał w połowie XX w stąd w schemacie
działania hemostazy jako jedyny mechanizm antykoagulacyjny przedstawiona została
fibrynoliza. Przy obecnym stanie wiedzy ten etap należałoby określić jako faza
ograniczania nadmiernego wzrostu czopu hemostatycznego.

Strukturalny model układu hemostazy
Składniki hemostazy:
ściana naczyń krwionośnych
płytki krwi
układ krzepnięcia
układ fibrynolizy
innych komórki mające znaczenie w
hemostazie (leukocyty – układ fagocytarny)
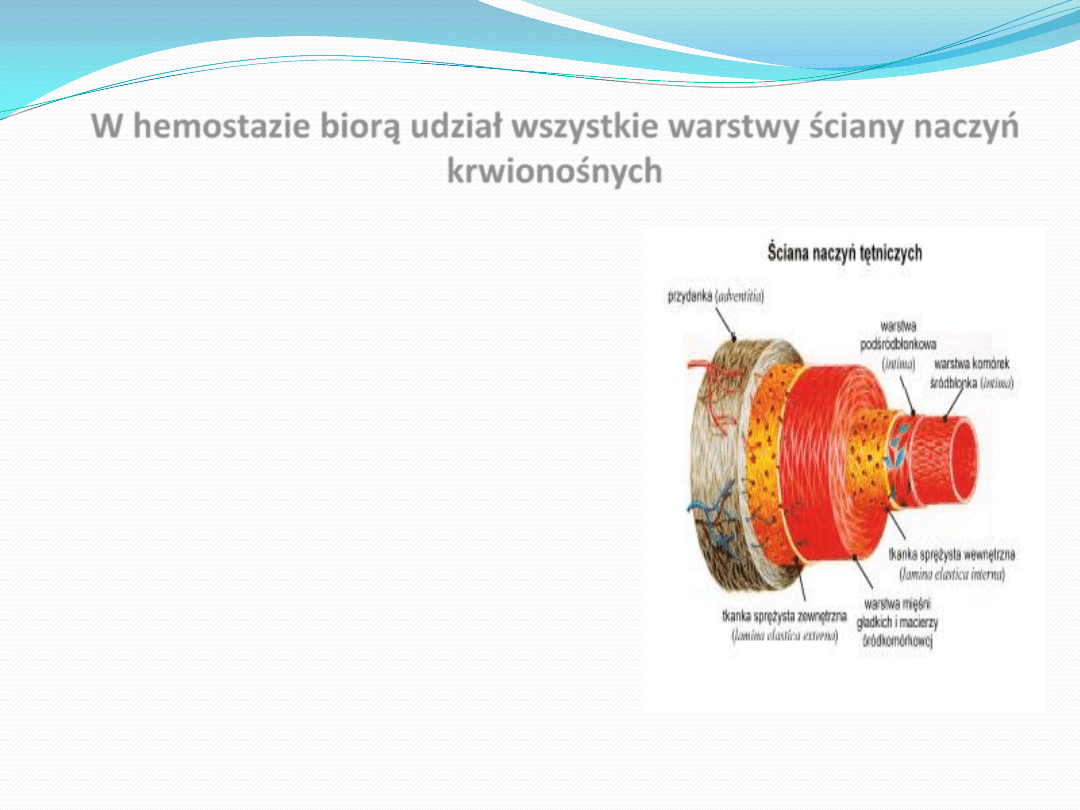
W hemostazie biorą udział wszystkie warstwy ściany naczyo
krwionośnych
Główną rolę odgrywa jednak błona wewnętrzna
ściany naczyniowej (intima) składająca się z
pojedynczej warstwy komórek śródbłonka i
błony podstawnej - podśródbłonkowej tkanki
łącznej.
Śródbłonek pełni głównie rolę
przeciwzakrzepową, a błona podstawna, po jej
odsłonięciu w przebiegu uszkodzenia
śródbłonka - aktywuje płytki krwi.
Warstwa środkowa (media), składająca się
warstw włókien kolagenu i mięśni gładkich o
układzie spiralnym lub okrężnym odgrywa rolę
w regulacji skurczu naczyń krwionośnych po
przerwaniu jego ciągłości.
Warstwa zewnętrzna (adventitia), ze względu na
znaczną ekspresję czynnika tkankowego na
powierzchni fibroblastów, odpowiada za
aktywację układu krzepnięcia.

Śródbłonek naczyniowy (endothelium)
Śródbłonek składa się z pojedynczej warstwy płaskich komórek
wyściełających łożysko naczyń krwionośnych i stanowiących barierę
między krwią a trombogenną warstwą podśródbłonkową.
Zważywszy na całkowitą masę komórek śródbłonka (1-2 kg) oraz na jego
dużą aktywność w zakresie syntezy i wydzielania substancji o wysokiej
aktywności biologicznej, śródbłonek coraz częściej opisywany jest jako
największy gruczoł endokrynny.
Śródbłonek odgrywa kluczową rolę w homeostazie naczyniowej:
reguluje napięcie mięśniówki naczyniowej (
wazodylatacja i
wazokonstrykcja
)
uczestniczy w
angiogenezie
(tworzenie nowych naczyń
krwionośnych)
reguluje działanie
układu hemostazy

Funkcje śródbłonka
Funkcja komórek śródbłonka w fizjologicznej hemostazie sprowadza się do
zapewnienia równowagi między mechanizmami pro i antyzakrzepowymi.
Regulacja ta ma znaczenie w dwóch różnych przypadkach:
po pierwsze, w warunkach nienaruszonej ciągłości ściany naczyniowej
śródbłonek „kontroluje” podprogową aktywację płytek krwi i generację
trombiny,
po drugie, w sytuacji przerwania ciągłości naczynia krwionośnego komórki
śródbłonka zapobiegają niekontrolowanemu rozprzestrzenianiu się aktywacji
hemostazy. Komórki te biorą udział w ograniczaniu obszaru tworzenia czopu
hemostatycznego do miejsca uszkodzenia naczynia.
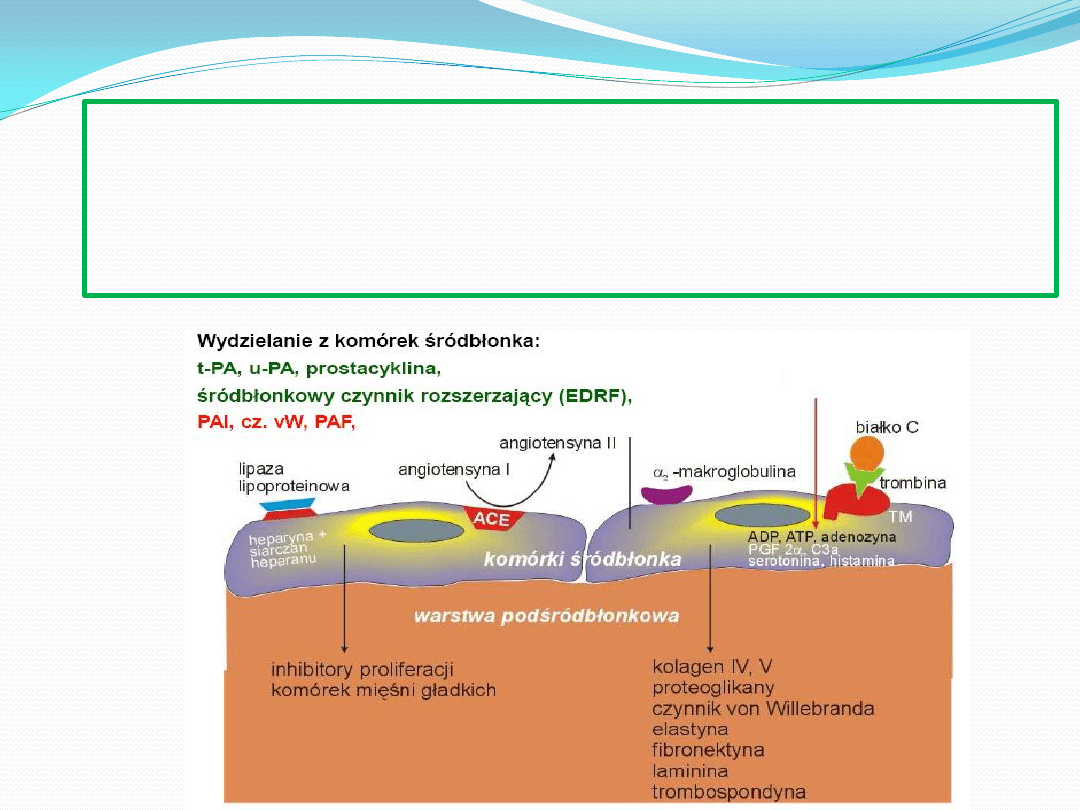
Spełnianie rozlicznych funkcji komórek śródbłonka jest możliwe głównie dzięki aktywności biologicznej
związków w nich syntezowanych.
Należy zwrócić uwagę na substancje:
pokrywające śródbłonek,
ulegające ekspresji na powierzchni błony komórkowej śródbłonka,
wydzielane do światła naczynia krwionośnego

Glikokaliks
Komórki śródbłonka od strony światła naczynia pokryte są
glikokaliksem
(warstwa proteoglikanów), składającym się głownie z
glikozaminoglikanów
(siarczan heparanu, siarczan dermatanu).
Glikokaliks dzięki obecności grup siarczanowych, nadaje wewnętrznej
ścianie naczynia ujemny ładunek elektrostatyczny co skutkuje
„odpychaniem” ujemnie naładowanych powierzchni komorek krwi (np.
płytek krwi).
Główny składnik glikokaliksu -
proteoglikany
są dużymi kompleksami
polisacharydów (95%) i białek (5%). Część sacharydową stanowią
glikozaminoglikany (GAG), które są polimerami jednostek
dwusacharydowych złożonych z heksozoamin (aminocukrow) i kwasów
uronowych.
Biologiczna rola glikokaliksu w regulacji układu jest niesłychanie różnorodna
i zależna od interakcji z elementami macierzy zewnątrzkomórkowej,
takimi jak kolageny, laminina, czy fibronektyna oraz od interakcji z
białkami osocza biorącymi udział hemostazie.
Około 80% glikozoaminoglikanow stanowi siarczan heparanu (SH), związek
posiadający zdolność aktywowania antytrombiny. Inny związek z tej
grupy to siarczan dermatanu (SD), który aktywuje kofaktor II heparyny.
siarczan
chondroityny
siarczan
keratanu
kwas
hialuronowy
białka rdzenia
białka wiążące
Rys. Schemat budowy proteoglikanu

Komórki śródbłonka – związek między budową a funkcją
Na powierzchni komorek śródbłonka znajdują się konstytutywne i indukowane
cząsteczki uczestniczące w oddziaływaniach międzykomórkowych takich jak toczenie się
i adhezja, należą do nich:
białka adhezyjne selektyna E (CD62E) i selektyna P (CD62P),
cząsteczki międzykomórkowej adhezji ICAM-1 i ICAM-2,
cząsteczki komorek śródbłonka i płytek krwi PECAM-1,
cząsteczki adhezji komórkowej naczyń – VCAM-1.
Aktywność biologiczna śródbłonka określana jest także przez enzymy znajdujące się na
jego powierzchni. Najważniejsze z punktu widzenia regulacji hemostazy to:
ADPaza
,
kinaza lipoproteinowa
oraz
konwertaza angiotensyny
.
Pierwszy z wymienionych enzymów (ADP-aza) jest ektonukleazą, która rozkładając
wydzielane przez aktywowane płytki krwi ADP, ogranicza przekazywanie sygnału
aktywacji do płytek znajdujących się w stanie spoczynku.
W komórkach śródbłonka znajdują się liczne pęcherzyki pinocytarne oraz
charakterystyczne dla tych komorek ciałka Weibela-Palade’a zawierające duże ilości
czynnika von Willebranda i selektyn.

Środbłonek - mechanizmy antykoagulacyjne
Prostacyklina (PGI2)
jest najsilniejszym fizjologicznym czynnikiem hamującym aktywacje płytek (głównie
adhezję)
powstaje przy udziale syntazy prostacykliny z nadtlenków lipidowych (PGG2, PGH2)
wytwarzanych w procesach utleniania kwasu arachidonowego
PGI2 hamuje aktywację płytek zwiększając stężenie cAMP , jest także bardzo silnym
czynnikiem rozkurczającym naczynia krwionośne
na powierzchni płytek krwi prostacyklina działa na specyficzny receptor, który
rozpoznaje także inne prostaglandyny o działaniu przeciwzakrzepowym, takie jak PGE1 i
PGE2
Synteza i działanie prostacykliny pozostaje pod wpływem licznych czynników – zarówno
fizjologicznych, jak i pozaustrojowych i farmakologicznych. W warunkach
fizjologicznych prostacyklina jest bardzo szybko (czas biologicznego półtrwania w
organizmie wynosi ok. 3 minut) hydrolizowana do 6-keto-PGF1α. Metabolit ten można
oznaczać jak marker syntezy prostacykliny
Zaburzenia syntezy prostacykliny obserwowane są w niektórych chorobach, podczas ciąży i
pod wpływem palenia tytoniu

Tlenek azotu (NO∙)
Cząsteczka ta znana także jako EDRF (Endothelium-Derived Relaxing Factor) jest
najważniejszym fizjologicznym, wytwarzanym w komórkach śródbłonka, czynnikiem
rozszerzającym naczynia krwionośne.
NO
•
jest wolnym rodnikiem stąd jest cząsteczką krótkotrwałą i bardzo reaktywną
jego przeciwpłytkowe działanie związane ze wzrostem stężenia cGMP dotyczy jedynie
fizjologicznych stężeń
w czasie stanów zapalnych dochodzi do wzmożonej generacji zarówno NO
•
oraz
anionorodnika ponadtlenkowego, oba związki wchodzą w reakcję tworząc nadtlenoazotyn
(ONOO-), niezwykle toksyczna cząsteczka uczestnicząca w reakcjach stresu oksydacyjnego
(np. tworzenia ox-LDL)
Z punktu widzenia regulacji hemostazy najważniejszą właściwością NO• jest działanie w
zakresie jego fizjologicznych stężeń.
Tlenek azotu :
obniża hydrolizę fosfolipidów i uwalnianie kwasu arachidonowego,
hamuje adhezję płytek, hamuje aktywację kompleksu GPIIb/IIIa (receptora dla
fibrynogenu),
hamuje reakcję uwalniania płytek i obniża ekspresję selektyny P.
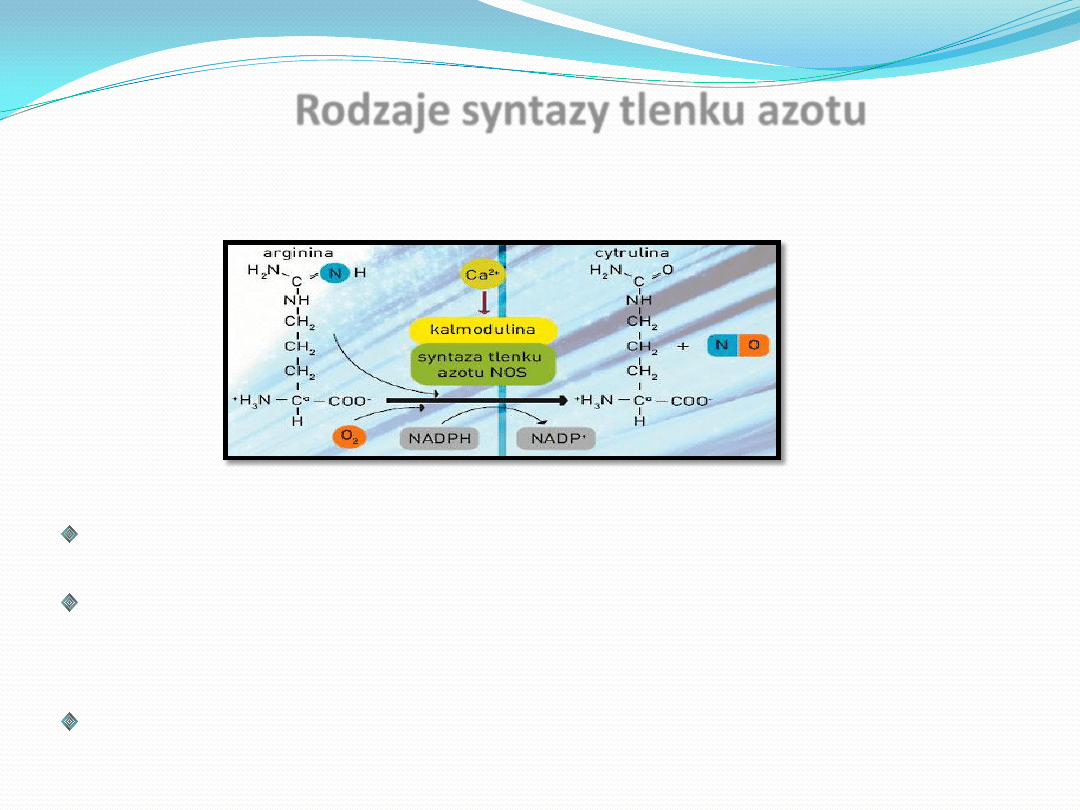
Rodzaje syntazy tlenku azotu
Wytwarzanie tlenku azotu, kontroluje utrzymywanie prawidłowego ciśnienia krwi i jest zależne od
enzymu zwanego syntazą tlenku azotu
.
Wyróżnia się trzy typy syntazy tlenku azotu:
śródbłonkowa (konstytutywna
) forma syntazy
(cNOS lub eNOS)
jest ciągle syntetyzowana w
organizmie; występuje ona w śródbłonku we względnie stałym stężeniu.
indukowalna forma syntazy (iNOS)
-
obecna głównie w komórkach układu odpornościowego.
Zwiększona synteza NO
•
w makrofagach związaną jest z niszczeniem patogenów. Nadmierna synteza
enzymu w śródbłonku (wzrost stężenia NO
•
) uważana jest za kluczowy mechanizm odpowiedzialny
za rozwój zależnego od śródbłonka stresu oksydacyjnego (synteza ONOO-).
neuronalna forma (nNOS)
związana z układem nerwowym. Powstający w komórkach nerwowych
NO
•
pełni funkcje przekaźnika międzykomórkowego.
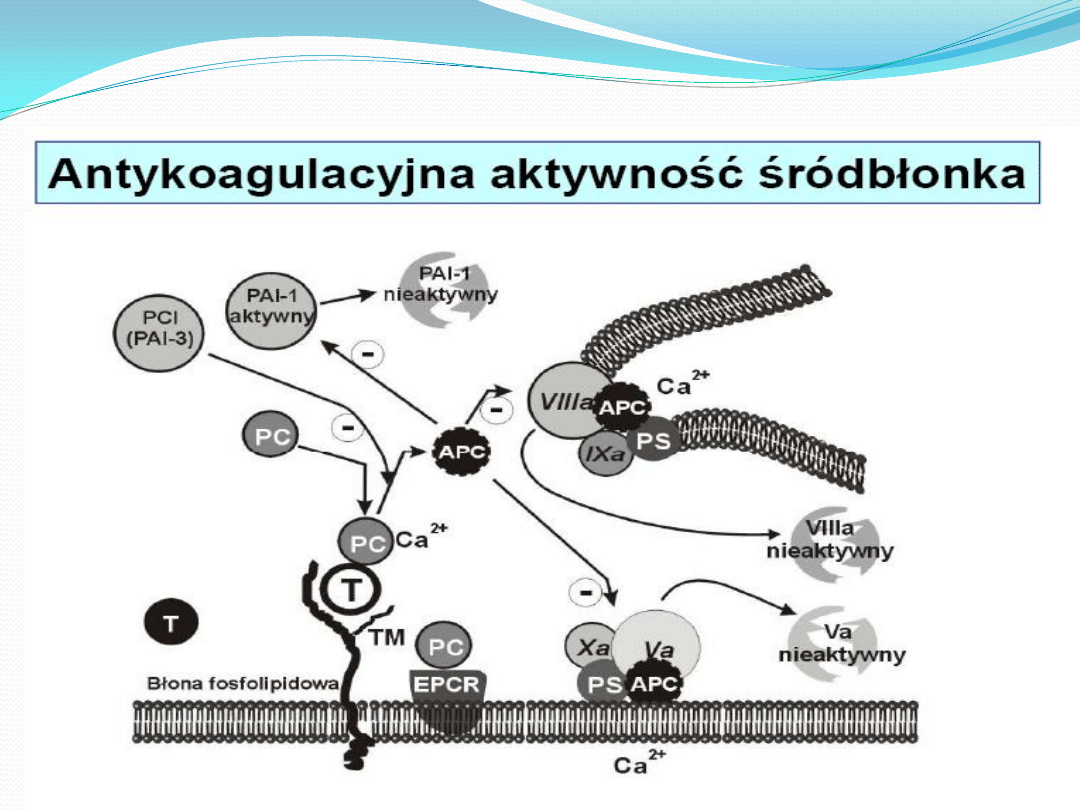
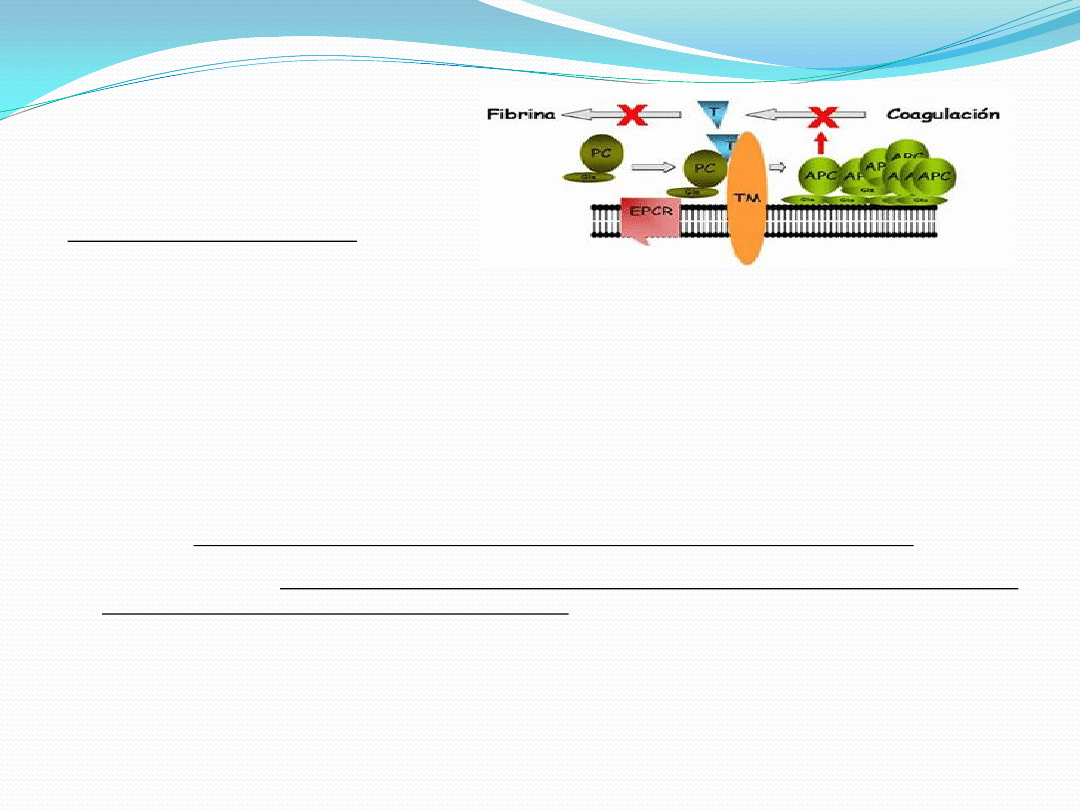
Trombomodulina (TM)
Jest jednołańcuchową glikoproteiną eksponowaną głównie na powierzchni komórek śródbłonka.
TM jest kofaktorowym białkiem, które odpowiada za regulację procesu aktywacji białka C oraz
regulację fibrynolizy.
W organizmie występuje w dwóch formach: tkankowej oraz osoczowej.
Forma osoczowa zachowuje wymaganą aktywność jako kofaktor aktywacji białka C, uważa się ją za
pochodną natywnej formy “tkankowej”.
Trombomodulina poprzez wiązanie się z
trombiną
(w stosunku stechiometrycznym 1:1)
aktywuje
białko C
. Trombina związana z trombomoduliną nie posiada aktywności prozakrzepowej.
Powstały kompleks hamuje fibrynolizę poprzez indukowanie przejścia aktywowanego przez trombinę
inhibitora fibrynolizy (TAFI) w jego formę aktywną.
W warunkach nadkrzepliwości i/lub wzmożonej fibrynolizy osoczowa trombomodulina może pełnić
funkcję osłonową względem śródbłonka naczyniowego, przeciwdziałając nadmiernej generacji
plazminy.
Stężenie rozpuszczalnych fragmentów trombomoduliny w osoczu wynosi 5-20 ng/ml, a jego wzrost może
świadczyć o uszkodzeniu śródbłonka.
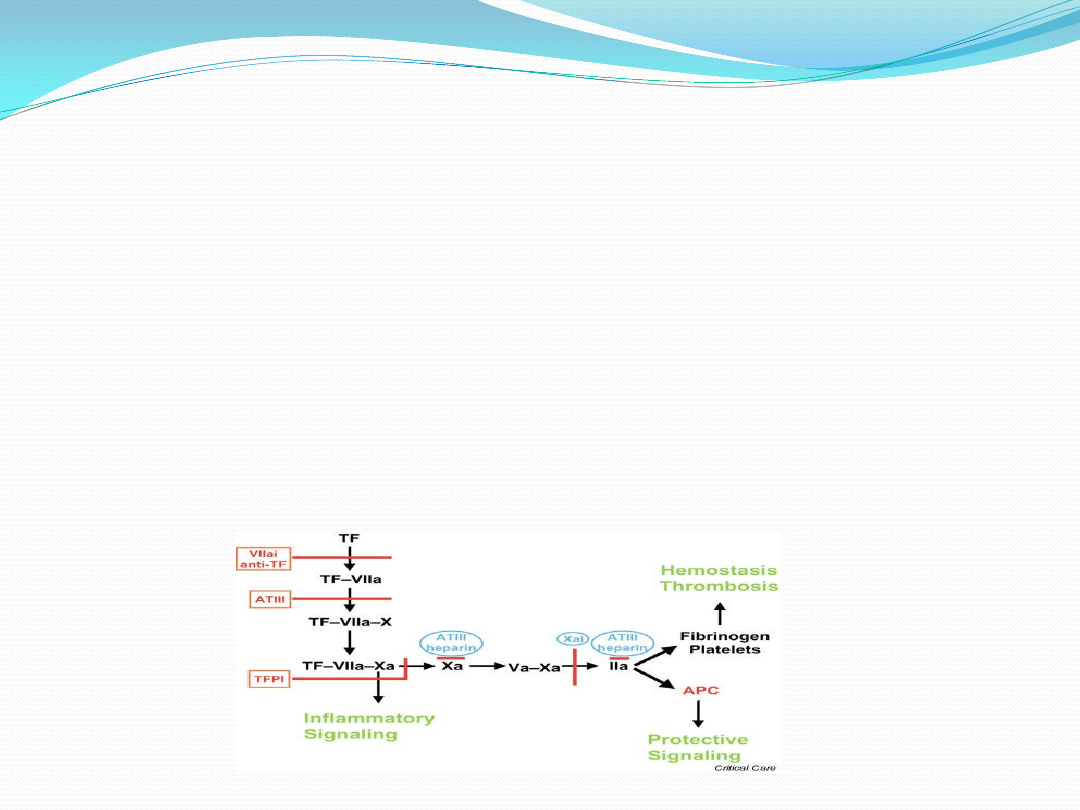
Inhibitor szlaku zależnego od czynnika tkankowego (TFPI)
Jest jednołańcuchową glikoproteiną syntezowaną w wątrobie, śródbłonku i megakariocytach,
posiadającą zdolność hamowania kompleksu TF-VIIa.
TFPI wiąże się nieodwracalnie z czynnikiem Xa, po czym łączy się docelowym kompleksem. Swoją
aktywność wykazuje dopiero po przyłączeniu Xa.
W osoczu krąży w połączeniu z lipoproteinami, związany jest także z powierzchnią śródbłonka.
Jego obecność powoduje zablokowanie szlaku aktywacji zależnej od czynnika tkankowego, od chwili
wygaszenia fazy inicjacji krzepnięcia na powierzchni komórek zasobnych w TF, za generacje trombiny
odpowiedzialne są kompleksy tenazy i protrombinazy zlokalizowane na płytkach krwi.
Jest głównym aktywatorem plazminogenu, odpowiadającym za przebieg naczyniowej fibrynolizy.

Tkankowy aktywator plazminogenu (t-PA)
Tkankowy aktywator plazminogenu (t-PA)
Jest proteazą serynową i podstawowym fizjologicznym aktywatorem plazminogenu.
t-PA syntetyzowany jest w komórkach śródbłonka jako jednołańcuchowa glikoproteina
hydrolizowana w obecności plazminy do dwóch łańcuchów połączonych mostkiem
disiarczkowym.
Tkankowy aktywator plazminogenu pełni kluczową rolę w aktywacji osoczowego
systemu fibrynolitycznego powodując przekształcenie nieaktywnego plazminogenu w
plazminę trawiącą fibrynę.
t-PA aktywuje plazminogen tylko w obecności włóknika, związaniu podlega już jego
forma jednołańcuchowa i to decyduje o wyjątkowej selektywności działania t-PA oraz
ograniczaniu aktywności do miejsca powstania czopu hemostatycznego.
W stanie dynamicznej równowagi wolny aktywny t-PA jest wydzielany przez komórki
śródbłonka, ale jego poziomy w osoczu są niskie, głównie z uwagi na szybkie
kompleksowanie z PAI-1 oraz wychwytywanie przez komórki wątroby
Wzrost stężenia t-PA postrzegany jest jako czynnik ryzyka choroby zakrzepowej.

Śródbłonek - mechanizmy prokoagulacyjne
Prawidłowy śródbłonek pełni role głównie antykoagulacyjną, jednakże na skutek stymulacji
zewnętrznej lub zmian patologicznych bierze udział w mechanizmach prozakrzepowych.
Czynnik von Willebranda (vWF)
Jest białkiem polimerycznym o masie cząsteczkowej od 500 do 20000 kD, a wielkość jego
polimerów kontrolowana jest przez swoiste mataloproteinazy.
vWF jest syntezowany w komórkach śródbłonka i megakariocytach. Przechowywany jest w
ciałkach Weibel-Paladea śródbłonka i ziarnistościach α płytek krwi. W warunkach spoczynku
układu hemostazy w osoczu krwi znajduje się vWF pochodzenia śródbłonkowego, dopiero w
czasie aktywacji dołącza się pula płytkowa.
vWF uczestniczy w procesach adhezji płytek do białek warstwy podśródbłonkowej oraz
agregacji płytek krwi w warunkach dużych sił ścinających.
W osoczu czynnik von Willebranda występuje w kompleksach z czynnikiem VIII,
Czynnik von Willebranda jest uznanym markerem uszkodzenia komórek śródbłonka.

Śródbłonek - mechanizmy prokoagulacyjne
Czynnik aktywujący płytki (PAF)
Jest fosfolipidem (pochodna fosfatydylocholiny) syntezowanym głownie przez leukocyty i komórki
śródbłonka. Związek ten jest aktywatorem leukocytów i regulatorem odczynu zapalnego; aktywacja
płytek, mimo nazwy tego aktywatora, jest drugoplanową funkcją PAF.
Wewnątrzkomórkowe efekty działania PAF nasilają przemiany fosfatydyloinozytolu do diacyloglicerolu i
trifosforanu inozytolu, pobudzają kaskady przemian kwasu arachidonowego, powstanie nadtlenków i
fosforylację białek, głównie miozyny, lipokortyny i proteaz. Prowadzi to do zmian w metabolizmie i
aktywności komórek.
Czynnik VIII
Synteza cz. VIII odbywa się w wątrobie, ale obecnie przyjmuje się, że niektóre grupy komórek
śródbłonka (śródbłonek naczyń płuc) także syntezują czynnik VIII, kluczowy składnik kompleksów
aktywacyjnych układu krzepnięcia. W związku z powyższym zaburzenia funkcji wątroby nie są
związane z niedoborem tego czynnika.
W osoczu czynnik VIII, występuje w kompleksach z czynnikiem vWF niedobór którego (choroba von
Willebranda) przekłada się na obniżone stężenie cz. VIII.
W czasie aktywacji układu krzepnięcia, kompleks tych dwóch czynników rozpada się pod wpływem
trombiny, po czym trombina aktywuje cz. VIII.

Inhibitor aktywatora plazminogenu (PAI-1)
Inhibitor ten może występować w trzech rożnych formach:
jako aktywna cząsteczka białka, potencjalnie zdolna do wiązania cząsteczek wolnego t-PA
w osoczu
w kompleksach z t-PA (t-PA/PAI-1, w stosunku stechiometrycznym 1:1)
w postaci nieaktywnej, która występuje głownie w płytkach krwi, i która może być
uwalniana podczas aktywacji płytek
PAI-1 jest jednołańcuchową glikoproteiną, należącą do rodziny serpin i jest syntetyzowany w
komórkach śródbłonka i hepatocytach, jest także uwalniany przez płytki krwi. Tworzy trwałe
kompleksy z t-PA i urokinazą w proporcjach stechiometrycznych.
Jego stężenie podlega regulacji przez dwie grupy modulatorów:
związki działające poprzez interakcje komórkowe wpływają na szybkość syntezy PAI-1
(trombina, IL-1, niektóre endotoksyny i czynniki wzrostu),
oddziaływujące bezpośrednio z cząsteczką PAI-1 i wpływające na jego aktywność (aktywowane
białko C).
Sądzi się, że wzrost stężenia PAI-1 w krążeniu, obserwowany np. w miażdżycy, zawale mięśnia
sercowego, cukrzycy, wiąże się ze zwiększonym ryzykiem powikłań naczyniowych. Nasilenie
syntezy PAI-1 i jego stężenie w osoczu krwi pozostaje pod wpływem czynników genetycznych.

Warstwa podśródbłonkowa (subendothelium)
Podstawą prokoagulacyjnego działania subendothelium jest jego trombogenna
powierzchnia. W warunkach fizjologicznych powierzchnia ta jest osłonięta
śródbłonkiem, stąd uszkodzenie tej ochronnej warstwy w naturalny sposób
sprzyja aktywacji układu hemostazy.
Warstwę podśródbłonkową stanowi błona podstawowa (podstawna), błonopodobna
struktura oddzielająca warstwę komórek śródbłonka od wewnętrznej warstwy
elastycznej (lamina elastica interna).
Podstawowe składniki warstwy podśródbłonkowej to:
kolagen
fibronektyna
trombospondyna
laminina
witronektyna
elastyna
Kolagen
Jest najpowszechniej występującym białkiem w
organizmach ssaków.
Kolagen jest polimeryczną glikoproteiną (przeważnie
o niewielkim stopniu glikozylacji) tworzącą
nierozpuszczalne włókna o dużej wytrzymałości.
Włókna kolagenowe są głównym budulcem tkanki
łącznej i macierzy międzykomórkowej.
Rożne typy kolagenu zawierają rożne kombinacje
podjednostek kolagenu. W ścianie naczyń
krwionośnych występuje kilka typów kolagenu, ale
dominujące znaczenie w warstwie podśródbłonkowej
(błona podstawna) ma kolagen IV typu.
Poza kolagenem w macierzy zewnątrzkómorkowej
występują liczne białka (m.in. trombospondyna,
laminina, witronektyna) uczestniczące w
oddziaływaniach płytek ze ścianą naczyń w tym
także czynnik von Willebranda.

Warstwa środkowa (media) i warstwa zewnętrzna (adventitia)
Media (warstwa środkowa/mięśniówkowa ściany naczynia) – obejmuje trzy
warstwy, których budowa jest zróżnicowana w zależności od wielkości i funkcji
naczynia.
Sprężystość nadają naczyniom krwionośnym włókna kolagenu, głównie typu I i
III, a za kurczliwość naczyń odpowiadają komórki mięśni gładkich. Warstwa
mięśni w momencie przerwania ciągłości naczynia reaguje jego obkurczeniem.
Na powierzchni komórek mięśniowych znajduje się czynnik tkankowy
aktywujący krzepnięcie, ponadto płytki krwi aktywowane są przez kolagen.
Aktywacja hemostazy w przypadku warstwy środkowej nie stanowi jej
zasadniczej roli.
Ekspresja TF w tej warstwie jest relatywnie słaba, a główna rolę aktywatora płytek
odgrywa kolagen warstwy podśródbłonkowej. Wysoka ekspresja czynnika
tkankowego związana jest z warstwą zewnętrzną ściany naczyń (przydanka).
Uważa się, że fibroblasty posiadają na swojej powierzchni największą ilość TF
ze wszystkich badanych komórek ściany naczyń, stąd kluczowa rola tej
warstwy (adventiny) w aktywacji układu krzepnięcia.

Ocena funkcji śródbłonka
Aktualnie brak jest powszechnie dostępnego badania testującego sprawność hemostatyczną ściany
naczyń krwionośnych. Badaniem o praktycznie historycznym znaczeniu jest test opaskowy
Rumpela-Leedego, stosowany jest również czas krwawienia, a z badań specjalistycznych
biopsja skóry.
Diagnostyka sprawności śródbłonka obejmuje przede wszystkim czynnościowe i obrazowe
badania hemodynamiczne. Do najważniejszych należą:
I.
FMD (Flow Mediated Dilatation) - badanie zdolności rozkurczowej naczyń krwionośnych
po zwolnieniu kontrolowanego ucisku
II.
Pletyzmografia – rejestracja zmian tętna (ciśnienia) w naczyniach
PAT (Peripheral ArteryTonometry) to uproszczona wersja tej metody wykorzystywana do
badań śródbłonka
III.
Pomiary IMT (Intima Media Thickness) – pomiary grubości kompleksu błona
wewnętrzna- błona środkowa, wykonywane dla tętnicy szyjnej.
Biochemiczne markery uszkodzenia śródbłonka:
w osoczu oznaczane są stężenia białek adhezywnych, uwalnianych z powierzchni
uszkodzonego śródbłonka (ICAM-1, VCAM-1, endoteliny, selektyny).
w czasie uszkodzenia śródbłonka wzrasta we krwi stężenie czynnika von Willebranda i kwasu
moczowego.
Badania opisanych markerów są kosztowne i mało swoiste, stąd nie znalazły szerszego
zastosowania w diagnostyce laboratoryjnej.

Płytki krwi
Płytki krwi (krwinki płytkowe, trombocyty)
opisane zostały w 1881 roku przez Bizzozero.
Nazwa „płytki krwi” dotyczy jedynie ssaków, u których komórki te nie posiadają jądra i są
najmniejszymi (średnica 2-5 Um), o dyskoidalnym kształcie, upostaciowionymi
elementami krwi obwodowej.
Powstawanie płytek z fragmentów megakariocytów szpiku kostnego regulowane jest
przez cytokiny: interleukiny, głównie przez czynnik wzrostowy – trombopoetynę.
Najczęściej podawana liczba płytek krwi u zdrowych ludzi wynosi 130-400 x 109/L.
W warunkach fizjologicznych płytki krwi przeżywają we krwi obwodowej 8-12 dni i są
usuwane z krwioobiegu głownie w śledzionie i wątrobie przez układ fagocytarny
(siateczkowo-śródbłonkowy).

Budowa płytek krwi
Błona zewnętrzna płytek krwi, zbudowana z podwójnej warstwy lipidów (głównie
fosfolipidów) otoczona bezpostaciowym glikokaliksem, który ze względu na obecność reszt
kwasu sjalowego nadaje powierzchni płytek ujemny ładunek elektrostatyczny.
W błonie zewnętrznej zlokalizowane są glikoproteiny o właściwościach receptorów dla
czynników aktywujących i hamujących funkcję płytek oraz enzymy odpowiedzialne za
transport jonów.
Z błoną zewnętrzną łączy się układ kanalików otwartych, poprzez który przemieszczają się
do otaczającego środowiska składniki uwalniane z ziarnistości płytek. Układ ten, łącznie z
układem kurczliwych włókienek (mikrofilamenty), tworzy tzw. cytoszkielet płytkowy, który
przyczynia się do zachowania kształtu płytki, do tworzenia wypustek w fazie adhezji i bierze
udział w retrakcji skrzepu.
Stężenie białek kurczliwych w płytkach jest bardzo duże. Aktyna stanowi 15-30% całkowitej
zawartości białka płytki.
Cechą charakterystyczną budowy płytek krwi, oprócz braku jądra, jest obecność wielu
struktur magazynujących substancje biologicznie czynne. Związki te biorą udział w
aktywacji płytek i regulacji funkcji pozostałych elementów układu hemostazy.

Płytkowe struktury magazynujące substancje biologicznie czynne
W wewnątrzkomórkowym układzie kanalików gęstych (tzw. układzie kanalików zamkniętych)
magazynowane są jony wapniowe, fosfolipidy, fosfolipazy, cyklooksygenaza (COX), lipooksygenaza i
syntaza tromboksanu A2.
W czasie aktywacji płytek dochodzi do przemieszczenia z ziarnistości α na powierzchnię płytek
selektyny P (CD62P) - glikoproteiny o właściwościach receptora wiążącego płytki krwi z komórkami
śródbłonka i leukocytów.
Składniki ziarnistości płytkowych
Ziarnistości gęste
Ziarnistości α
•ADP, ATP, GDP, GTP,
•serotonina,
•fosfoinozytole
• jony wapnia i magnezu
• białka swoiste płytek:
czynnik płytkowy
4 (PF4), β-tromboglobulina,
• białka adhezyjne
: fibronektyna, czynnik
von Willebranda (vWF), trombospondyna,
witronektyna,
• białka biorące udział w hemostazie :
fibrynogen, czynnik V, czynnik VIII,
wielkocząsteczkowy kininogen (HMWK),
białko S, białko C, inhibitor aktywatora
tkankowego plazminogenu 1 (PAI-1),
• mitogeny :
czynnik wzrostu pochodzący
z płytek (PDGF), czynnik wzrostu
komórek śródbłonka pochodzący z płytek
(PDECGF).
• W lizosomach dominują
kwaśne
hydrolazy
, zaś w peroksysomach
katalazy
.
• Ponadto wewnątrz płytek krwi
znajdują się
nieliczne struktury aparatu
Golgiego
,
ziarna glikogenu i
mitochondria.

Receptory płytkowe
Ze względu na występowanie receptorów na powierzchni płytek krwi w cyklu
życiowym komórek, można je także podzielić na:
1.
receptory stale
występujące na powierzchni płytek krwi (także w stanie
podstawowym, w płytkach nieaktywowanych):
receptor dla fibrynogenu
receptory dla kolagenu
receptor dla czynnika von Willebranda
receptor dla ADP
receptor dla trombiny
2.
receptory pojawiające się w następstwie aktywacji płytek krwi
– do tej grupy
należą markery aktywacji płytek krwi:
selektyna P (CD62P, GMP-140)
glikoproteina 53 (CD63)

Receptory płytkowe
Receptor czynnika von Willebranda
Receptor ten pełni dwie funkcje: odpowiada za tworzenie pierwotnych,
przejściowych połączeń płytek krwi z warstwą podśródbłonkową (adhezja
pierwotna) w naczyniach o szybki przepływie krwi oraz funkcjonuje jako
receptor trombiny.
Receptory kolagenu
GPIa/IIa (integryna α2β1) oraz GPVI
przyłączenie kolagenu do jego z receptorów jest jednym z pierwszych etapów
aktywacji
najważniejszą rolę w pobudzeniu komórki po kontakcie z kolagenem, odgrywa
GPVI, natomiast, udział receptora GPIa/IIa (integryna α2β1) jest wspomagający

Receptory płytkowe
Receptor fibrynogenu
kompleks glikoprotein IIb/IIIa, tworzący integrynę αIIbβ3, jest najliczniej reprezentowany
receptorem na powierzchni błony płytkowej (40000-50000 kopii w płytkach w stanie
spoczynkowym)
w sposób typowy dla wszystkich integryn receptor ten złożony jest dwóch podjednostek: αIIb i
β3
receptor ten po aktywacji nabywa zdolności wiązania nie tylko fibrynogenu, ale także innych
białek posiadających sekwencje aminokwasów arginina-glicyna-kwas asparaginowy (RGD) czyli:
vWF
fibronektynę
trombospondyne
witronektynę
Dzięki tej zdolności receptor fibrynogenu uczestniczy zarówno w adhezji jak i agregacji płytek krwi.
W czasie następujących po sobie etapów tworzenia czopu płytkowego nie zmienia się drastycznie
ilość receptorów dla fibrynogenu, lecz głownie ich konformacja i rozmieszczenie na błonie
cytoplazmatycznej. Receptory te przechodzą ze stanu spoczynkowego w pobudzony, nabywając
zdolność trwałego przyłączania ligandów.

Receptory płytkowe
Receptory ADP
Płytki krwi posiadają na swojej powierzchni trzy receptory przyłączające ADP:
P2X1
– kanał jonowy dla Ca2+ otwierany po przyłączaniu ADP, wiążący również ATP
P2Y1
– receptor związany z białkiem Gq odpowiedzialny za wywołanie sygnału
aktywacji (zmiana kształtu, odwracalna agregacja)
P2Y12
– związany z białkiem Gi, po aktywacji odpowiedzialny za inhibicję cyklazy
adenylanowej oraz za utrwalanie agregacji i potęgowanie sekrecji.
W czasie tworzenia pierwotnego czopu hemostatycznego występuje degranulacja, uwalniane jest
ADP, które, obok tromboksanu jest, najważniejszym mediatorem aktywacji, wysłanym przez
agregujące płytki w kierunku komórek znajdujących się w stanie spoczynku.
Przyłączanie agonisty do receptorów ADP uruchamia rożne szlaki aktywacji:
• hamowanie cyklazy adenylanowej za pośrednictwem Gi,
• aktywacja fosfolipazy C poprzez Gs,
• napływ jonów Ca2+ do wnętrza komórki
Inhibitory receptorów P2Y12 (tiklopidyna, klopidogrel) są jednymi z najbardziej skutecznych leków
przeciwpłytkowych.

Receptory pojawiające się w następstwie aktywacji płytek
selektyna P (CD62P), uwalniana z ziarnistości α płytek
ze względu jednak na oznaczenia cytometryczne i wykorzystanie w nich
przeciwciał rozpoznających antygen CD62P, dość często stosuje się
określenie - receptor CD62P
GP53 (CD63), uwalniana z lizosomów
fizjologiczne znaczenie glikoproteiny 53 nie jest ostatecznie ustalone, nie
ma pewności czy przejawia aktywność receptorową, jednak, ponad wszelką
wątpliwość, podobnie jak selektyna P, GP53 jest markerem aktywacji
płytek.
Obie glikoproteiny są powszechnie uważane za markery aktywacji i reakcji uwalniania
płytek krwi, a określanie ekspresji GP53 i selektyny P ma istotne znaczenie w
testach monitorowania degranulacji i aktywacji płytek w krążeniu.
Obecnie oznaczanie glikoprotein pojawiających się w czasie aktywacji płytek krwi jest
jedną z najczęściej stosowanych metod badania aktywacji i reaktywności płytek
krwi

Szlaki przekazywania sygnału aktywacji
Przekazywanie sygnału w czasie aktywacji płytek jest wielostopniowe.
Zgodnie z kolejnymi etapami uruchamiania funkcji płytek, biorące w nich
udział cząsteczki chemiczne można zaszeregować do jednej z czterech
grup:
A.
receptory
B.
enzymy efektorowe (np. cyklazy adenylanowa i guanylanowa,
fosfolipazy, kinaza inozytolo-3-fosforanu)
C.
wtórne przekaźniki sygnału (np. cAMP, cGMP, DAG, IP3, PIP3,
Ca2+)
D.
efektory wewnątrzkomórkowe (enzymy docelowe, takie jak np.
kinazy czy fosfatazy)

Kinazy tyrozynowe
Po połączeniu kolagenu czy fibrynogenu ze swoim receptorem jego
cytoplazmatyczne domeny uruchamiają łańcuch reakcji.
W aktywowanych płytkach dochodzi do gwałtownej fosforylacji reszt
tyrozynowych białek wzbudzających i regulatorowych.
Wywoływanie tych reakcji może zachodzić bezpośrednio, jak ma to miejsce w
przypadku kolagenu i GPVI lub pośrednio – przy udziale kinaz tyrozynowych
związanych z receptorami fibrynogenu (GPIIb/IIIa) oraz większości pozostałych
glikoprotein odpowiedzialnych za adhezję i aktywację płytek krwi.
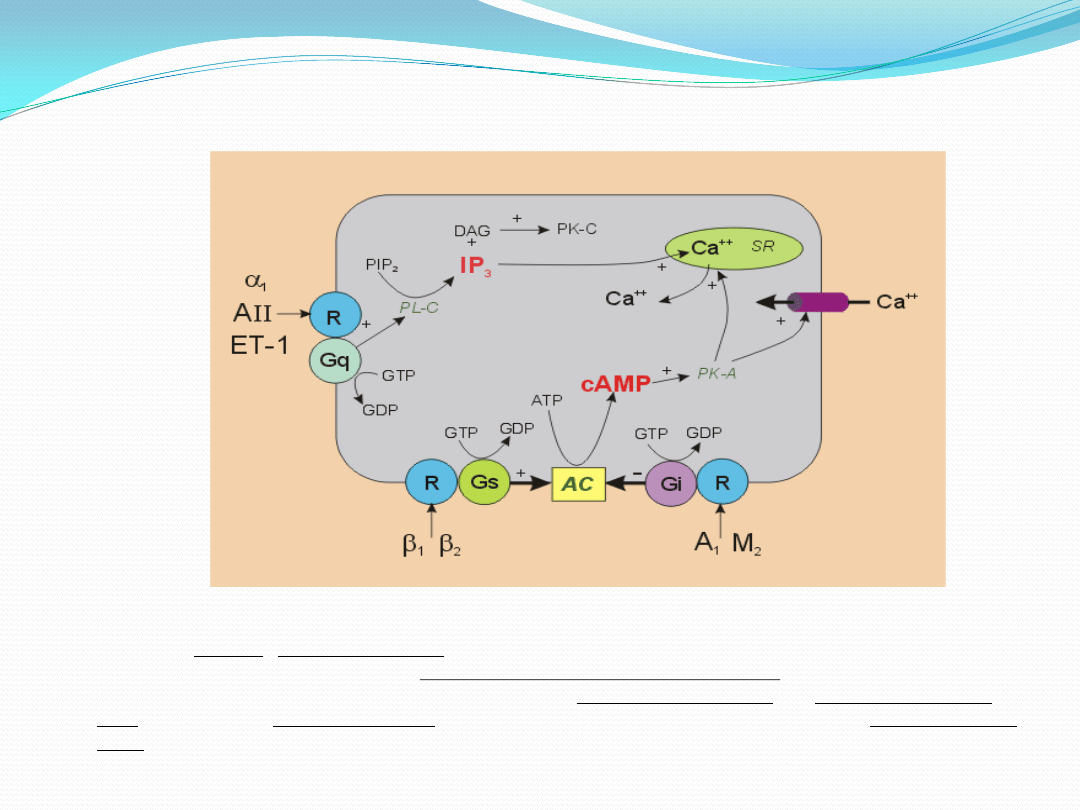
Przemiany inozytoli
Przekazywanie sygnału aktywacji z receptorów do wnętrza komórki odbywa się w tym przypadku za pośrednictwem
błonowych białek G i fosfolipazy C (PLC) odpowiedzialnej za hydrolizę fosfolipidów błonowych. Zlokalizowana
w błonie komórkowej PLC hydrolizuje fosfatydyloinozytolo-4,5-difosforan (PIP2), co prowadzi do powstania
dwóch kluczowych wewnątrzkomórkowych mediatorów: diacyloglicerolu (DAG) oraz inozytolotrifosforanu
(IP3). DAG aktywuje kinazę białkową C, a ta z kolei inicjację degranulację. IP3 odpowiada za uwalnianie jonów
Ca2+ z układu kanalików gęstych oraz aktywację cytoszkieletu i metabolizmu kwasu arachidonowego.
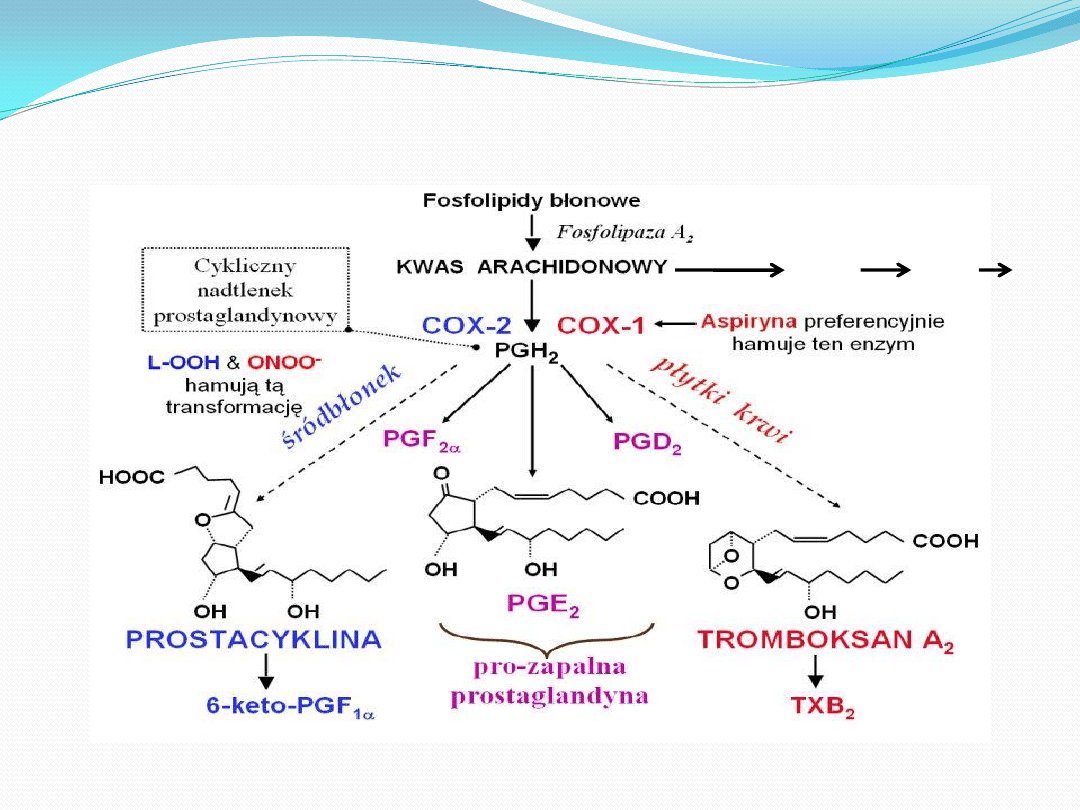
Metabolizm kwasu arachidonowego
HPETE
HETE
12-lipoksygenaza
LT
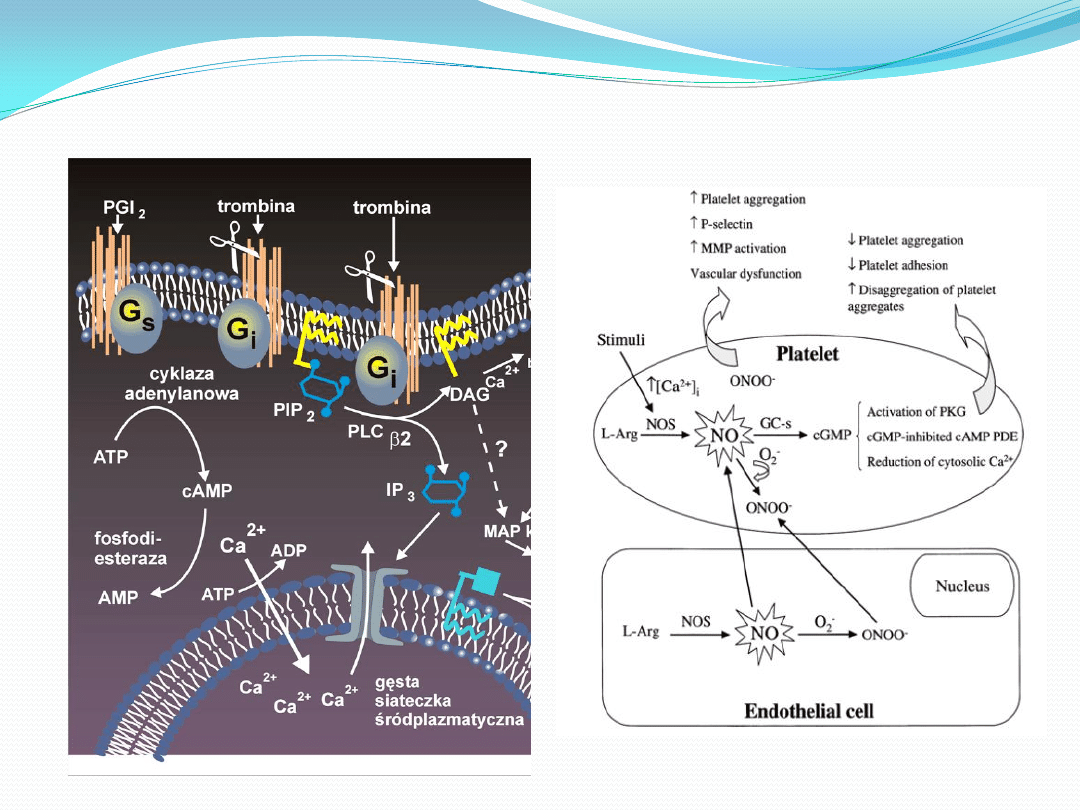
Układ cyklazy adenylanowej i guanylanowej
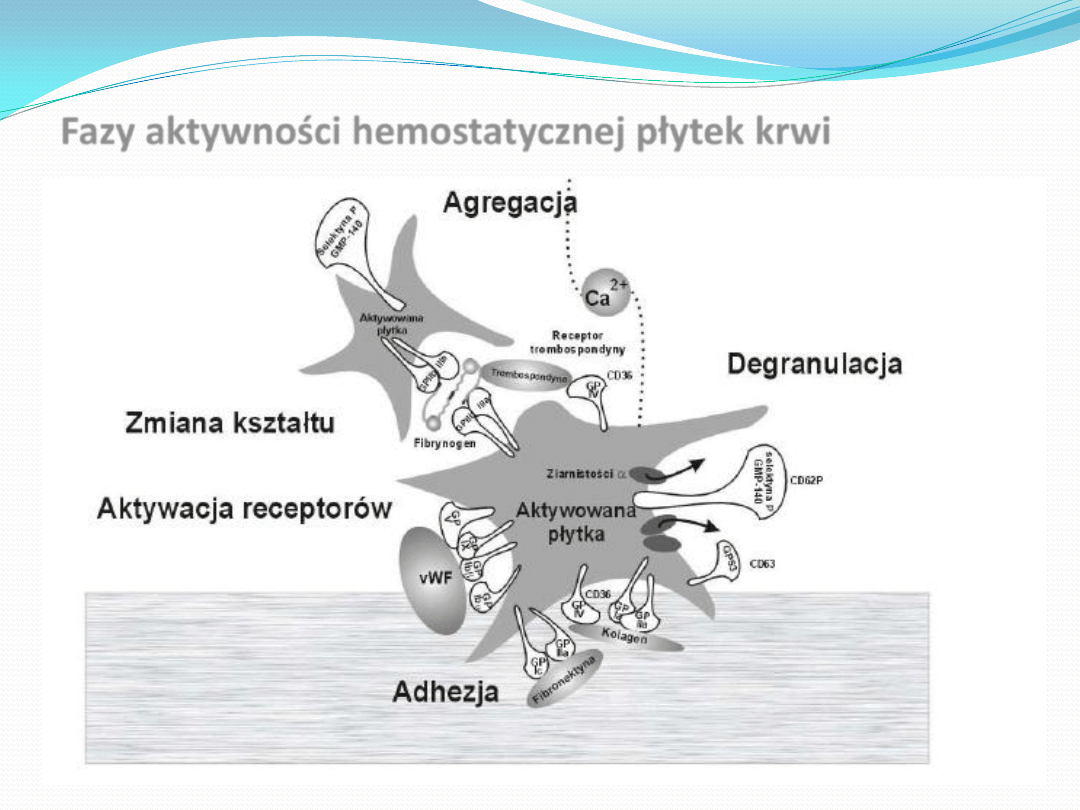
Fazy aktywności hemostatycznej płytek krwi
Tworzenie przez płytki krwi struktury czopu hemostatycznego
Adhezja
In vivo płytki krwi przylegają do elementów
warstwy podśródbłonkowej w miejscach
uszkodzenia komórek śródbłonka naczyń.
Płytki krwi mogą ulegać adhezji do
kolagenu,
fibronektyny, lamininy, witronektyny i
trombospondyny
.
W dużych naczyniach krwionośnych, przy
niskim module ścinania płytki wiążą się
bezpośrednio do białek tkanki łącznej.
Natomiast przy wysokim module ścinania,
wiążą się one z białkami za pośrednictwem
multimerycznych form vWF.
W naczyniach tętniczych adhezja płytek krwi
przebiega w dwóch etapach.
Najpierw za pośrednictwem receptora vWF
wytwarzane są nietrwałe połączenia, następnie
kilka glikoprotein płytkowych (GPІa/ІІa, GPVI,
GPIV, GPІІb/ІІІa) kolagen i inne składniki
podśródbłonkowej tkanki łącznej tworzy trwałe
połączenia i nieodwracalną adhezję płytek krwi.
W pierwszym etapie (adhezja pierwszorzędowa)
uczestniczą tylko czynnik vWF i jego receptor, w
drugim (adhezja drugorzędowa), receptory
wiążące kolagen oraz receptor fibrynogenu
przyłączający się do fibronektyny.
Adhezja płytek do kolagenu i innych białek
warstwy podśródbłonkowej uruchamia
wewnątrzkomórkowe systemy przekazywania
sygnałów aktywujących, w wyniku czego płytki
zmieniają swój kształt z dyskoidalnego w
nieregularny, zaczynają się kolejne fazy aktywacji
płytek krwi.
W przekazywaniu sygnału do wnętrza komórki
pośredniczą białka błonowe G. W zaktywowanych
płytkach dochodzi do zmian metabolicznych (np.:
kaskada kwasu arachidonowego), architektury
cytoszkieletu oraz do translokacji białek i
aktywacji receptorów integrynowych. Zmiany te
warunkują uwolnienie substancji biologicznie
czynnych zgromadzonych w ziarnistościach
wewnątrzpłytkowych, agregację płytek i ich udział
w retrakcji czopu hemostatycznego.

Reorganizacja cytoszkieletu i zmiana kształtu
W stanie spoczynku pęczki mikrofilamentów aktynowych (włókna napięciowe)
sieciowane przez miozynę tworzą podstawę cytoszkieletu płytki. Aktyna, stanowiąca 20-
30% wszystkich białek płytki, występuje także w postaci globularnej, która pod
wpływem pobudzającego bodźca wyzwolonego po przyłączeniu ligandu do receptora
ulega polimeryzacji, a przyłączając się do włókien napięciowych o zreorganizowanej
strukturze wytwarza „superfilamenty aktynowe”.
W czasie aktywacji lekkie łańcuchy miozyny, po ufosforylowaniu przez kinazy zależne
od jonów Ca2+, wytwarzają w interakcji z ciężkimi łańcuchami filamenty miozynowe
sieciujące włókna aktyny.
Płytki krwi zmieniają kształt z dyskoidalnego na ameboidalny. Tworzone wypustki
cytoplazmatyczne zwiększają powierzchnie kontaktu z miejscem uszkodzenia naczynia
krwionośnego.

Agregacja
Do pojedynczej warstwy płytek krwi, które uległy adhezji, przyłączają się
kolejne płytki, zaczyna się agregacja i tworzenie pierwotnego czopu
hemostatycznego.
Płytki łączą się ze sobą poprzez fibrynogenowe mostki międzypłytkowe
tworzone między receptorami błonowymi GPIIb/IIIa sąsiadujących płytek.
W stanie spoczynku receptor ten nie posiada zdolności przyłączania
ligandów, aktywacja receptorów fibrynogenu wywołana jest przez
wewnątrzpłytkowe mediatory powstałe w czasie adhezji.
Także w przypadku agregacji, możemy mówić o dwóch fazach w przebiegu
tego zjawiska. Wyróżniamy: pierwotną – odwracalną agregację oraz wtórną -
nieodwracalną.
Agregacja nieodwracalna i utrwalenie czopu płytkowego pojawia się po
degranulacji, za pośrednictwem mediatorów „wtórnej aktywacji”, czyli ADP i
tromboksanu. W tworzeniu agregatów płytkowo-płytkowych wiodącą role
odgrywają receptory GPIIb/IIIa i fibrynogen, pomocniczą rolę, w naczyniach
o szybszym przepływie krwi, pełni czynnik von Willebranda i jego receptor.
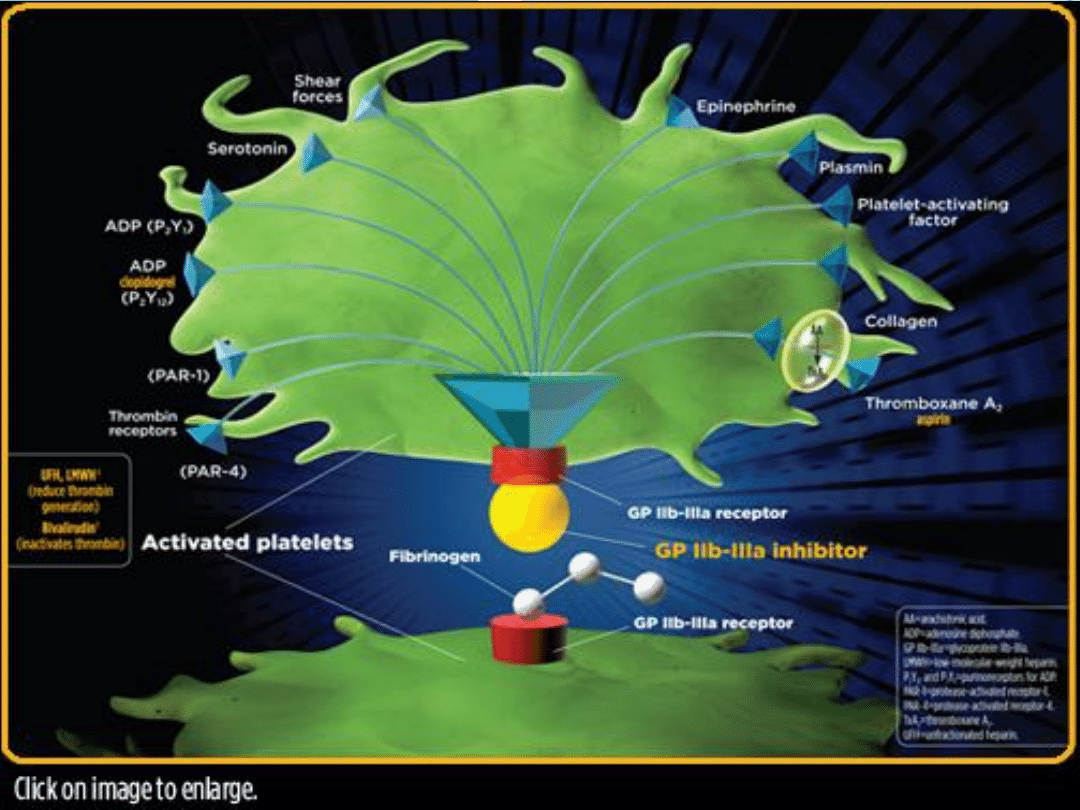

Diagnostyka laboratoryjna
Oznaczenie czasu krwawienia jest najprostszą metodą badania sprawności
hemostatycznej płytek krwi. Metoda ta jest mało czuła i specyficzna,
szczególnie w wersji znanej jako metoda Duke’a. Wersja klinicznie użyteczna to
metoda Ivy’ego. W obecnej chwili nie ma wskazań do rutynowego zlecania
wykonania czasu krwawienia. Badanie to wypierane jest przez instrumentalne
metody badania płytek krwi.
W przypadku podejrzenia występowania skazy krwotocznej istnieją wskazania
do wykonania czasu krwawienia.
W przypadku wydłużonego czasu krwawienia konieczne jest zbadanie funkcji
płytek krwi.
Metody pomiaru agregacji płytek krwi:
I.
agregacja turbidymetryczna
II.
agregacja we krwi pełnej: metoda impedancyjna, określanie ubytku płytek po
aktywacji
III.
pomiar czasu okluzji – PFA-100 (adhezja/agregacja płytek)
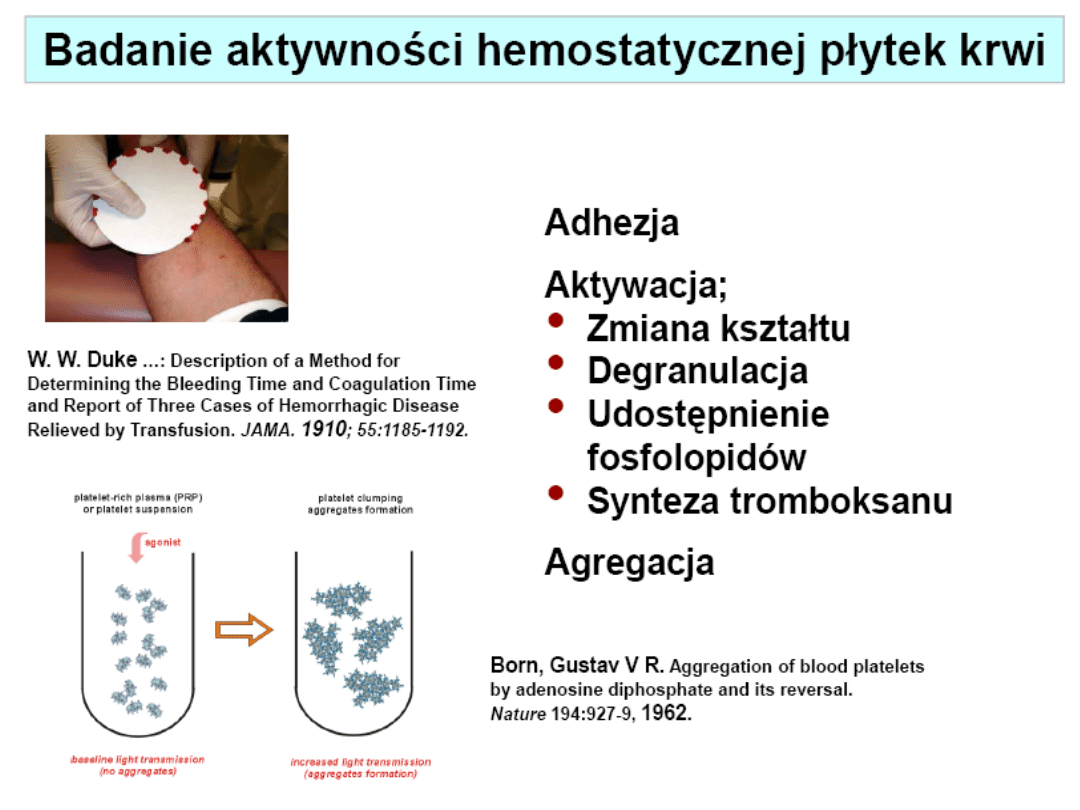
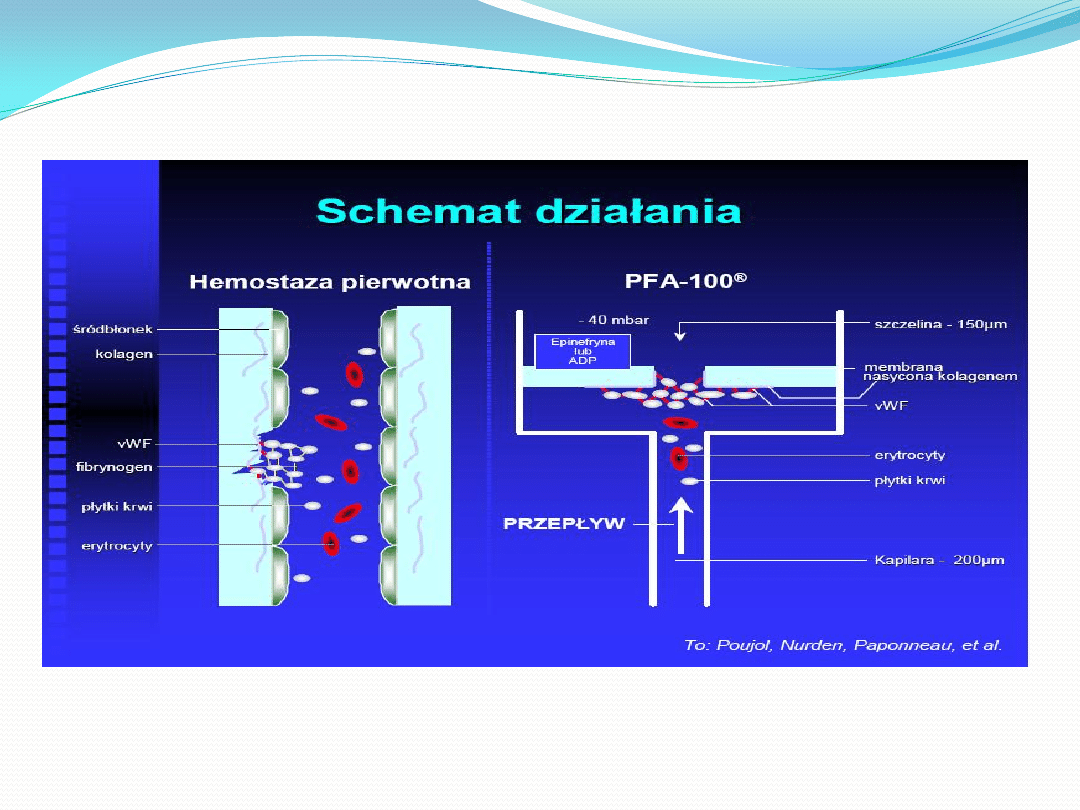
PFA (Platelet Function Analyzer) – pomiar czasu okluzji w układzie symulujacym
uszkodzone naczynie
PFA-100™, pozwalają badać reaktywność płytek krwi w warunkach przepływu zbliżonych do naturalnych. W analizatorze PFA-100™
zastosowano dwa rodzaje kaset pomiarowych (z kolagenem i epinefryną lub z kolagenem i ADP) z układem przepływowym
symulującym uszkodzone naczynie krwionośne. Mierzonym parametrem jest czas przepływu pełnej krwi przez układ
pomiarowy symulujący uszkodzone naczynie krwionośne, czyli czas od zetknięcia się krwi z membraną nasyconą agonistami
do momentu zaczopowania szczeliny pomiarowej w membranie, tzw. czas okluzji - CT
Choroba zakrzepowo-zatorowa
W warunkach prawidłowej hemostazy czop hemostatyczny nie powinien powstawać
wewnątrz szczelnych, nieuszkodzonych naczyń krwionośnych, a jeżeli w nich powstaje,
mamy do czynienia z zakrzepicą.
W zależności od rodzaju naczyń objętych zakrzepicą określamy ja jako zakrzepicę żylną lub
tętniczą.
W niektórych stanach fizjologicznych i chorobowych mamy do czynienia ze zwiększoną
gotowością pokoagulacyjną, stan ten określa się także jako nadkrzepliwość i w pewnych
warunkach może rozwinąć się w zakrzepicę.
Rys.
Rozmieszczenie płytek krwi i
erytrocytów w strumieniu krwi w
naczyniu

Zakrzepica
Zakrzepica to zjawisko powstawania skrzeplin w nieuszkodzonych naczyniach
krwionośnych na skutek zachwiania równowagi układu hemostazy. Może
ona dotyczyć:
• naczyń tętniczych (zakrzepica tętnicza),
• naczyń żylnych (zakrzepica żylna), i/lub
• naczyń włosowatych i przedwłosowatych (rozsiane krzepnięcie
śródnaczyniowe, DIC)
Trzy zasadnicze przyczyny warunkujące rozwój zakrzepicy, zwane potocznie triadą
Virchowa, to:
zmiany w przepływie krwi (zmiany reologiczne),
zmiany dotyczące ściany naczyniowej,
zmiany składu krwi
O różnicach w etiopatogenezie zakrzepicy żylnej i tętniczej decydują odmienności
dwóch składników triady Virchowa:
a) parametrów przepływu krwi (reologii krwi) w naczyniach żylnych i tętniczych, oraz
b) roli ściany naczyniowej, wynikającej z jej odmiennej budowy w naczyniach żylnych i
tętniczych.

Etiopatogeneza zakrzepicy żylnej
W jej przebiegu wyróżnia się następujące etapy:
dochodzi do naruszenia integralności warstwy komórek śródbłonka; niekiedy proces
zakrzepicy żylnej rozpoczyna się bez widocznych wyraźnych przyczyn inicjujących -
mechanizmy tego rodzaju zakrzepicy pozostają niewyjaśnione;
neutrofile przylegają do komórek śródbłonka i wnikają do warstwy podśródbłonkowej;
neutrofilom mogą towarzyszyć płytki krwi i włókna polimeryzującej fibryny;
adhezja płytek krwi do uszkodzonych komórek śródbłonka nasila aktywację płytek oraz
inicjuje proces tworzenia skrzepliny;
aktywacja płytek sprzyja uwalnianiu substancji chemotaktycznych i aktywujących dla
innych płytek, co wzmaga agregację płytek i prowadzi do wytworzenia czopu płytkowego
(zlepu płytkowego);
na powierzchni błony komórkowej płytek zachodzi bardzo wydajnie aktywacja kaskady
krzepnięcia, prowadząc do polimeryzacji fibrynogenu i tworzenia włóknika;
włókna fibryny stabilizują czop płytkowy;
narastająca płytkowo-włóknikowa skrzeplina zwęża, a z czasem zamyka zupełnie, światło
naczynia; w miarę zwężania światła naczynia przez narastającą skrzeplinę przeważa w
niej coraz bardziej udział włóknika; propagacja skrzepliny płytkowo-włóknikowej jest
szybka aż do miejsca gdzie występuje aktywny przepływ krwi żylnej, który uniemożliwia
dalsze szybkie odkładanie włóknika; w miejscach takich skrzeplina narasta wolniej, aż do
ostatecznego zamknięcia światła naczynia, a jej skład jest mieszany.

Zakrzepica żylna
Samoistne udrożnienie światła naczynia zamkniętego przez zakrzep żylny jest utrudnione głównie
ze względu na wolny prąd płynącej w żyłach krwi. Udrożnienie takie może nastąpić dzięki:
a/ rozpuszczeniu skrzepliny przez endogenne czynniki fibrynolityczne - proces ten występuje
stosunkowo rzadko i najczęściej dotyczy jedynie małych skrzeplin żylnych;
b/ oderwaniu narastającej skrzepliny od ściany naczynia przez prąd krwi i przeniesieniu jej w inne
miejsce - powoduje to miejscowe udrożnienie naczynia, ale stwarza ogromne ryzyko powstania
zatoru w innym miejscu przez oderwaną skrzeplinę lub jej fragment; zależnie od wielkości
oderwanej skrzepliny i miejsca jej przemieszczenia w łożysku naczyniowym epizod taki może
być śmiertelny w skutkach (np. w przypadku bardzo niebezpiecznego zatoru tętnicy płucnej).
Skrzepliny, których propagacja ustaje lub spowalnia się mogą zostać wchłonięte lub związanie ze
ścianą naczynia. Występujące w sąsiedztwie skrzepliny komórki śródbłonka nabrzmiewają,
ulegają podziałom i wraz z innymi komórkami warstwy podśródbłonkowej wrastają w
skrzeplinę. Taka skrzeplina, poprzerastana bogato unaczynioną tkanka łączną, ulega
stopniowo zwiotczeniu, a komórki są wypierane przez elementy włókniste substancji
międzykomórkowej. Z czasem naczynia włosowate także zanikają, a poprzerastana naczyniami
i tkanką łączną skrzeplina bliznowacieje i wypełnia światło naczynia (tzw. zwłóknienie
skrzepliny).

Przepływ krwi w naczyniach żylnych
Przepływ krwi w naczyniach żylnych, zwłaszcza naczyniach kończyn dolnych, jest bardzo
powolny, ponieważ kształtowany jest niemal całkowicie przez:
wydolność mięśnia sercowego
skuteczność tłoczenia krwi przez naczynia tętnicze i naczynia włosowate,
napięcie mięśniówki ściany naczyniowej, które w naczyniach żylnych jest niewielkie.
Skuteczny przepływ krwi w naczyniach żylnych zapewniają skurcze mięśni, natomiast
zastawki w żyłach utrudniają cofanie się słupa krwi w czasie relaksacji mięśni.
W przypadkach kiedy regularne skurcze mięśni nie występują, np. w czasie pooperacyjnego
unieruchomienia lub porażeń mięśniowych, mechanizm wymuszonego przepływu krwi
w naczyniach żylnych przestaje funkcjonować. Jest to podstawowy czynnik sprzyjający
rozwojowi zakrzepicy żylnej.
Ryzyko zainicjowania procesu zakrzepowego jest największe w miejscach o niewielkim
przepływie krwi, gdzie turbulencje przepływu krwi są minimalne a zastój krwi duży, np.
w zatokach żylnych naczyń kończyn dolnych lub w miejscach występowania głębokich
kieszonek zastawek naczyń żylnych. Samo osłabienie lub nawet zatrzymanie przepływu
nie wystarcza jednak do powstawania skrzepliny w naczyniu.

Dysfunkcje śródbłonka w zakrzepicy żylnej
Dysfunkcje śródbłonka w zakrzepicy żylnej są o wiele mniej wyraźne niż w zakrzepicy
tętniczej i w zależności od czynnika inicjującego (a/ w warunkach fizjologicznych, bez
ingerencji z zewnątrz lub b/ na skutek uszkodzenia/urazu mechanicznego ściany
naczyniowej) mogą mieć nieco odmienny charakter:
1.
zwolnienie przepływu krwi w naczyniu upośledza dotlenienie komórek śródbłonka i w
następstwie zwalnia tempo ich metabolizmu;
2.
mechaniczne uszkodzenie ściany naczyniowej w wyniku zabiegu operacyjnego lub
długotrwałego rozciągnięcia i napięcia żył może prowadzić do naruszenia ciągłości
warstwy okładzinowej komórek śródbłonka w naczyniach żylnych.
Długotrwałe miejscowe niedotlenienie komórek śródbłonka, występujące np. w obszarach
zastoju żylnego, może:
a/ upośledzać miejscowo procesy syntezy czynników odpowiedzialnych za naturalne
mechanizmy ochronne, takich jak prostacyklina, tlenek azotu (EDRF), t-PA,
b/ zmniejszać ekspresję/aktywność trombomoduliny niezbędnej w aktywacji białka C,
c/ obniżać syntezę i aktywność proteoglikanów, głównie siarczanu dermatanu i siarczanu
heparanu, działających przeciwzakrzepowo.

Zmiany w układzie krzepnięcia
Przyjmuje się, że to nie nadmiar czynników krzepnięcia biorących udział w generacji
trombiny, ale raczej niedobory lub dysfunkcje inhibitorów krzepnięcia oraz
wynikające stąd zachwianie równowagi układu hemostazy odgrywają decydującą
rolę w etiopatogenezie zakrzepicy żylnej. Największe znaczenie przypisuje się:
niedoborom inhibitorów krzepnięcia (białka C i białka S, antytrombiny, oporność
na aktywowane białko C) oraz dysfunkcjom mechanizmów antykoagulacyjnych;
zwiększonej aktywacji i reaktywności płytek krwi i/lub nieprawidłowo dużej liczbie
płytek - może być czynnikiem sprzyjającym zakrzepicy w połączeniu z innym
czynnikiem ryzyka, np. szczególnym zagrożeniem mogą być znaczne
nadpłytkowości występujące po uszkodzeniu ściany naczynia są szczególnie
zagrażające;
hiperfibrynogenemii osocza, która przyczynia się do zwiększonej lepkości krwi i
stwarza szczególne zagrożenie w przypadku znaczącego spowolnienia przepływu
krwi w miejscach zastoju żylnego;
obecności przeciwciał antyfosfolipidowych.

Zmiany aktywności fibrynolitycznej
Zmniejszona aktywność fibrynolityczna w zakrzepicy żylnej na skutek
osłabionego wytwarzania t-PA przez niedotlenione i/lub uszkodzone
komórki śródbłonka sprawia, że powstające złogi włóknika nie zostają
skutecznie rozpuszczone i mogą inicjować narastanie skrzepliny.
Efekt obniżonej syntezy i uwalniania t-PA może być niekiedy spotęgowany
przez towarzyszący wzrost stężenia PAI-1, zwłaszcza po zabiegach
operacyjnych, lub wzrost stężenia lipoproteiny (a).
Uważa się, że zarówno PAI-1, jak i Lp(a) mogą być uznane za genetyczne
czynniki ryzyka, jakkolwiek obserwacje kliniczne pozostają
niejednoznaczne i niespójne.
Udział dysfunkcji układu fibrynolitycznego w rozwoju zakrzepicy żylnej
pozostaje dyskusyjny. Ogólnie, nieprawidłowości w układzie
fibrynolitycznym dotyczą znacznie częściej przypadków zakrzepicy żylnej
pojawiającej się bez jakichkolwiek wyraźnych przyczyn (tzw. idiopatyczna
zakrzepica żylna).

Czynniki ryzyka ŻChZZ
1) wiek >40 lat (ryzyko wzrasta z wiekiem)
2) otyłość (BMI >30 kg/m2)
3) ŻChZZ w wywiadzie rodzinnym
4) urazy (zwłaszcza wielonarządowe lub złamania miednicy, bliższego odcinka kości
udowej i innych kości długich kończyn dolnych)
5) niedowład kończyn dolnych, długotrwałe unieruchomienie
6) nowotwory złośliwe (ryzyko ŻChZZ wzrasta wraz zaawansowaniem nowotworu)
7) przebyta ŻChZZ
8) trombofilia wrodzona lub nabyta
9) sepsa
10) obłożna choroba leczona zachowawczo (np. ciężkie zapalenie płuc)
11) niewydolność serca III i IV klasy NYHA niewydolność oddechowa
12) choroba Leśniowskiego i Crohna, wrzodziejące zapalenie jelita grubego
13) zespół nerczycowy
14) zespoły mieloproliferacyjne
15) nocna napadowa hemoglobinuria
16) ucisk na naczynia żylne (np. guz, krwiak, malformacja tętnicza)
17) ciąża i połóg
18) długotrwały lot samolotem
19) żylaki kończyn dolnych
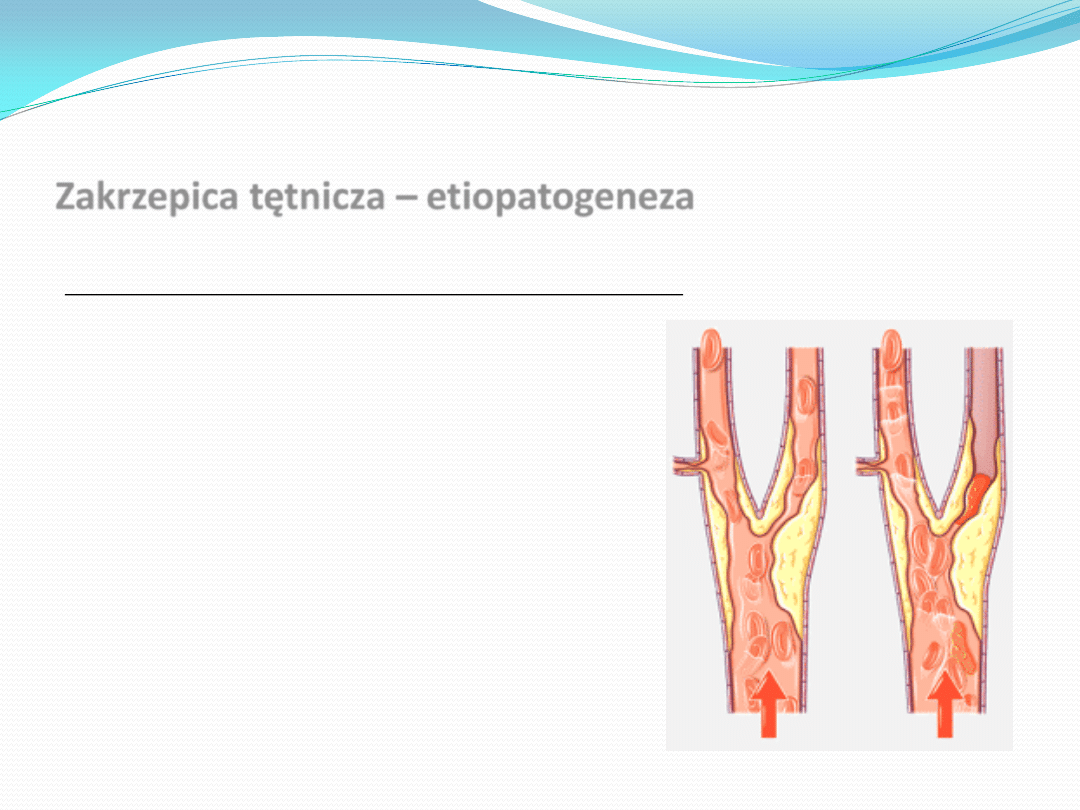
Zakrzepica tętnicza – etiopatogeneza
Do rozwoju zakrzepicy tętniczej przyczyniają się:
a)
Zmiany w ścianie naczyniowej tętnic
b)
Zmiany w przepływie krwi w tętnicach
c)
Zmiany w układzie hemostazy

Zmiany w ścianie naczyniowej tętnic
Ciśnienie krwi, do którego utrzymania przyczyniają się zarówno napięcie ścian naczyniowych
tętnic i drożność naczyń, jak i wydolność lewej komory serca, zapobiega tworzeniu skrzeplin w
naczyniach tak długo, jak długo integralność ściany naczyniowej, a zwłaszcza okładzinowa
warstwa komórek śródbłonka, pozostaje nienaruszona.
W przeciwdziałaniu powstawania skrzepliny we wczesnych etapach istotną rolę odgrywa także
układ fibrynolityczny, ale w etiopatogenezie zakrzepicy naczyń tętniczych jego rola wydaje się
nie być tak kluczowa jak w zakrzepicy żylnej. Podwyższone stężenia najważniejszego
fizjologicznego inhibitora fibrynolizy - PAI-1 - syntetyzowanego w komórkach śródbłonka,
przyczynia się do obniżenia sprawności układu fibrynolizy i mniej wydajnej proteolizy
włóknika w skrzeplinach. Fakt, że skrzepliny tętnicze są zbudowane gównie z płytek krwi - z
niewielkim udziałem włóknika - może tłumaczyć fizjologicznie niższą aktywność
fibrynolityczną tętnic w porównaniu z żyłami.
Do podstawowych czynników zagrażających naruszeniem integralności ściany naczyniowej w
tętnicach i przerwaniem ciągłości okładzinowej warstwy komórek śródbłonka można zaliczyć:
urazy mechaniczne warstwy komórek śródbłonka/ ściany naczyniowej (także te w czasie
zabiegów chirugicznych),
miażdżycę tętnic, która jest znacznie częstszą naturalną przyczyną biologiczną, związaną ze
starzeniem się komórek i tkanek oraz z zaburzeniami metabolizmu lipidów.

Zmiany w przepływie krwi w tętnicach
Zwarta i gruba warstwa mięśniówki w tętnicach sprawia, że ściany naczyń
tętniczych pozostają pod znacznym napięciem pozwalającym na utrzymywanie
podwyższonego ciśnienia krwi w tych naczyniach.
Takie podwyższone ciśnienie decyduje o właściwościach reologicznych przepływu
krwi oraz hemodynamice ruchu poszczególnych elementów morfotycznych
krwi w tętnicach. W warunkach wysokich wartości modułu ścinania w
naczyniach tętniczych płytki krwi są spychane w kierunku ściany naczyniowej,
ulegają łatwiejszej aktywacji, a ryzyko mechanicznego uszkodzenia komórek
śródbłonka, zwłaszcza w miejscach rozgałęzień naczyń, jest większe.
Szybki przepływ krwi warunkuje powstawanie regionów o zwiększonej
turbulencji, zwłaszcza w miejscach nieregularności czy wybrzuszeń ściany
naczyniowej (np. w sąsiedztwie blaszki miażdżycowej).
Duże siły ścinające w tętnicach mają także wpływ na aktywacje krzepnięcia, w
szczególności generację trombiny.

Czynniki ryzyka zakrzepicy tętniczej
Hemostatyczne czynniki ryzyka:
1.
wzrost aktywności prokoagulacyjnej (nasilenie krzepnięcia krwi):
fibrynogen
czynnik VII
czynnik VIII i von Willebranda,
czynnik XIII,
inhibitory krzepnięcia,
dodatkowe czynniki o nieustalonej jednoznacznie roli
2.
obniżenie aktywności układu fibrynolitycznego;
3.
nasilenie aktywacji płytek krwi,
4.
dysfunkcje komórek śródbłonka naczyń.

Czynniki ryzyka zakrzepicy tętniczej
Metaboliczne czynniki ryzyka:
a/
nadciśnienie tętnicze
- jeden z najważniejszych czynników ryzyka chorób
naczyniowych; ciśnienie skurczowe > 150 mm Hg i rozkurczowe powyżej 90-95 mm
Hg zwiększają ryzyko zgonu 2-3-krotnie (dla populacji polskiej, POL-MONICA
1993);
b/
otyłość
- szczególne znaczenie dla etiopatogenezy chorób sercowo-naczyniowych ma
otyłość wisceralna, związana z wieloma zaburzeniami metabolicznymi (cukrzyca
typu 2, hiperinsulinemia, insulinooporność, zaburzona tolerancja glukozy,
hipertriglicerydemia, hipercholesterolemia), które wspólnie określa się mianem
zespołu Reavena lub zespołu polimetabolicznego (X); całokształt tych zaburzeń lub
jego wybrane składniki wpływają na niektóre parametry układu hemostazy (np.
obniżanie sprawności układu fibrynolizy lub zwiększona synteza PAI-1;
c/
hipercholesterolemia
- szczególne znaczenie ma wzrost stężenia cholesterolu frakcji
aterogennej LDL przy obniżonym stężeniu cholesterolu HDL; niskie stężenie
cholesterolu przeciwmiażdżycowej frakcji HDL uważa się niekiedy za lepszy
wskaźnik zagrożenia chorobą wieńcową niż cholesterol LDL;
d/ dodatkowe metaboliczne czynniki ryzyka o nie w pełni ustalonym znaczeniu (np.
lipoproteina (a), ferrytyna
);
e/ znaczenie metabolicznych czynników ryzyka nasilają znacząco czynniki ryzyka
zależne od trybu życia, np.
palenie tytoniu, mało ruchliwy tryb życia
.

Czynniki ryzyka zakrzepicy tętniczej
Czynniki ryzyka zależne od trybu życia:
palenie tytoniu
- wpływa na stężenie w osoczu fibrynogenu, PAI-1, czynnika
von Willebranda, reaktywność i aktywację płytek krwi,
mało ruchliwy tryb życia
,
stosowanie używek
(kofeina) i alkoholu,
niskie stężenia antyoksydantów (witaminy E, A i C)
Czynniki biologiczne nie podlegające modyfikacji:
a/
genetyczne
czynniki ryzyka dotyczące: białek układu hemostazy, stężenia
lipidów osocza (hipercholesterolemia), aktywności konwertazy angiotensyny
(ACE), cech morfologicznych (położenie bruzd płatków uszu i łysienie są
czynnikami ryzyka choroby wieńcowej),
b/
wywiad rodzinny
: ujawnianie się chorób układu krążenia w młodym wieku u
członków najbliższej rodziny,
c/
wiek
- u mężczyzn powyżej 45 roku życia, u kobiet - powyżej 50 r.ż.
Wyszukiwarka
Podobne podstrony:
Bio Mol prezentacje
Bio Mol prezentacje
Bio Mol
artykół bio mol ?ncer as a multistep process
program bio mol
Prezentacja bio
Tekst do prezentacji z Bio, inne
prezentacja z bio longevity
Prezentacja bio
prezentacja finanse ludnosci
prezentacja mikro Kubska 2
Religia Mezopotamii prezentacja
Prezentacja konsument ostateczna
Strategie marketingowe prezentacje wykład
motumbo www prezentacje org
lab5 prezentacja
Prezentacja 18
więcej podobnych podstron