
BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
W latach 70-ych DNA uważano za najtrudniejszą do analizy makrocząsteczkę i mogło być analizowane tylko
metodami pośrednimi poprzez analizę białek, sekwencjonowanie RNA itp. Obecnie DNA jest najłatwiejszym
materiałem do badań.
Można np. przeciętnie w ciągu 1 dnia wyizolować specyficzny fragment genomu, wyprodukować w laboratorium
dowolną ilość jego identycznych kopii, a także określić jego sekwencję dosłownie przez 1 noc. W oparciu o
podobne techniki taki wyizolowany gen można zmienić (np. wprowadzając mutacje) i wprowadzić ponownie do
dowolnych komórek w taki sposób, by funkcjononował w zgodzie z pozostałymi mechanizmami komórkowymi i
umożliwiał analizę wprowadzonych zmian na poziomie komórkowym, tkankowym, organu bądź całego
organizmu.
Techniki rekombinacji DNA są obecnie podstawowymi technikami BIOLOGII KOMÓRKI i umożliwiają poznanie
tak komórek jak i budujących je makrocząsteczek na tysiące wręcz sposobów. Te same techniki umożliwiają
produkcję na szeroką skalę np. białek, hormonów i szczepionek. Ponadto, ponieważ jest możliwa analiza
regulatorowych regionów genów (promotorów), pozwala to biologom analizować szereg nieraz bardzo
skomplikowanych mechanizmów regulacji genów u wyższych organizmów.
Podstawowe techniki analizy i inżynierii DNA/RNA obejmują:
1. Cięcie DNA w specyficznych miejscach przez ENZYMY RESTRYKCYJNE
2. Klonowanie DNA z użyciem odpowiednich wektorów oraz PCR, w wyniku czego pojedynczy fragment
cząsteczki DNA może być użyty do otrzymania bilionów identycznych cząsteczek
3.
Hybrydyzację kwasów nukleinowych, pozwalającą znaleźć odpowiednią sekwencję DNA lub RNA z
dużą dokładnością, w oparciu o wiązanie komplementarnych sekwencji DNA lub RNA
4.
Sekwencjonowanie wszystkich nukleotydów w oczyszczonej próbce DNA, co pozwala zidentyfikować
geny, a także wydedukować sekwencję aminokwasów dla białka, które kodują
5.
Monitorowanie ekspresji wielu genów w komórce z użyciem tzw. macierzy DNA (z ang. „microarrays”)
itp. narzędzi, które pozwalaja dokonać tysięcy hybrydyzacji równocześnie
ENZYMY RESTRYKCYJNE: Mogą być wyizolowane i oczyszczone np. z
bakterii, różne szczepy bakterii produkują różne enzymy restrykcyjne. Enzymy
te chronią bakterie przed wirusami degradując wirusowe DNA i mają wysoką
specyficzność, tj różne enzymy tną podwójną helisę DNA w różnych
miejscach. Rozpoznają one specyficzną sekwencję 4-8 nukletydów obcego
DNA. Własne DNA bakterii jest chronione przed działaniem ich własnych
enzymów restrykcyjnych poprzez metylację zasad A i C we własnym DNA.
Pochodzenie enzymów na rusunku obok: HpaI: Hemophilus parainfluenzae,
EcoRI: Escherichia coli, HindIII: Hemophilus influenzae, PstI: Providencia
stuartii.
Enzymy restrykcyjne tną nić DNA albo w danym miejscu, albo w pobliżu
określonej sekwencji i zwykle tną DNA tak, że pozostawiają tzw. „lepkie
końce”, czyli wystające jednoniciowe fragmenty DNA po obu stronach
rozpoznawanej sekwencji. W niektórych przypadkach tną jednak nić tak, że
pozostawiają tzw. „tępe końce”, jak np. HpaI

BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
Fragmenty DNA posiadające identyncze lepkie końce mogą być łatwo
dopasowane, jak pokazuje to rysunek poniżej, tępe końce można
również połaczyć z odpowiednim DNA, choć wymaga to więcej pracy.
Połączone ze sobą dwa lub więcej fragmenty DNA powszechnie
nazywane jest rekombinowanym DNA
ROZMIAR (długość) fragmentu DNA służy zwykle do rozpoznania
interesującego nas fragmentu w mieszaninie pociętych fragmentów DNA.
ELEKTROFOREZA DNA: Opiera się na podobnych zasadach jak
elektroforeza białek. W przypadku DNA jest ona jeszcze prostsza,
ponieważ DNA
jest
naładowane
ujemnie
i nie trzeba nadawać mu ładunku
ujemnego jak w przypadku białek z użyciem SDS.
Na załączonym rysunku użyto dla A: żelu
poliakrylamidowego
o małych
porach
do
frakcjonowania
fragmentów
(10-500
nukleotydów)
jednoniciowego DNA , dla B: żelu
agarozowego
o średniej wielkości porów
dla frakcjonowania średniej wielkości fragmentów dwuniciowego DNA
(300–10 000 nukleotydów), a dla C: zastosowano tzw. „pulsed-field
agarose
gel
electrophoresis” do rozdzielenia fragmentów DNA 220 000 –
2.5 mln nukleotydów.
Ostatnia z wymienionych metod polega na rozdziale dużych cząsteczek
DNA w polu elektrycznym, którego kierunek zmieniany jest okresowo. W
stałym polu elektrycznym takie duże cząsteczki nie mogłyby być
rozdzielone.
W przypadku B i C detekcję DNA w żelu przeprowadzono barwiąc żel
bromkiem
etydyny,
który wbudowuje się w DNA, podświetlając żel
następnie światłem UV. W przypadku A zastosowano odpowiednie
znakowanie izotopowe (tj
32
P; patrz: sekwencjonowanie metodą Sangera).
BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
NORTHERN i SOUTHERN BLOT to sposób transferu RNA i DNA na odpowiednie filtry, które ułatwiają detekcję
interesujących nas fragmentów RNA lub DNA za pomocą odpowiednich SOND i metod hybrydyzacji DNA/DNA,
RNA/RNA oraz RNA/DNA
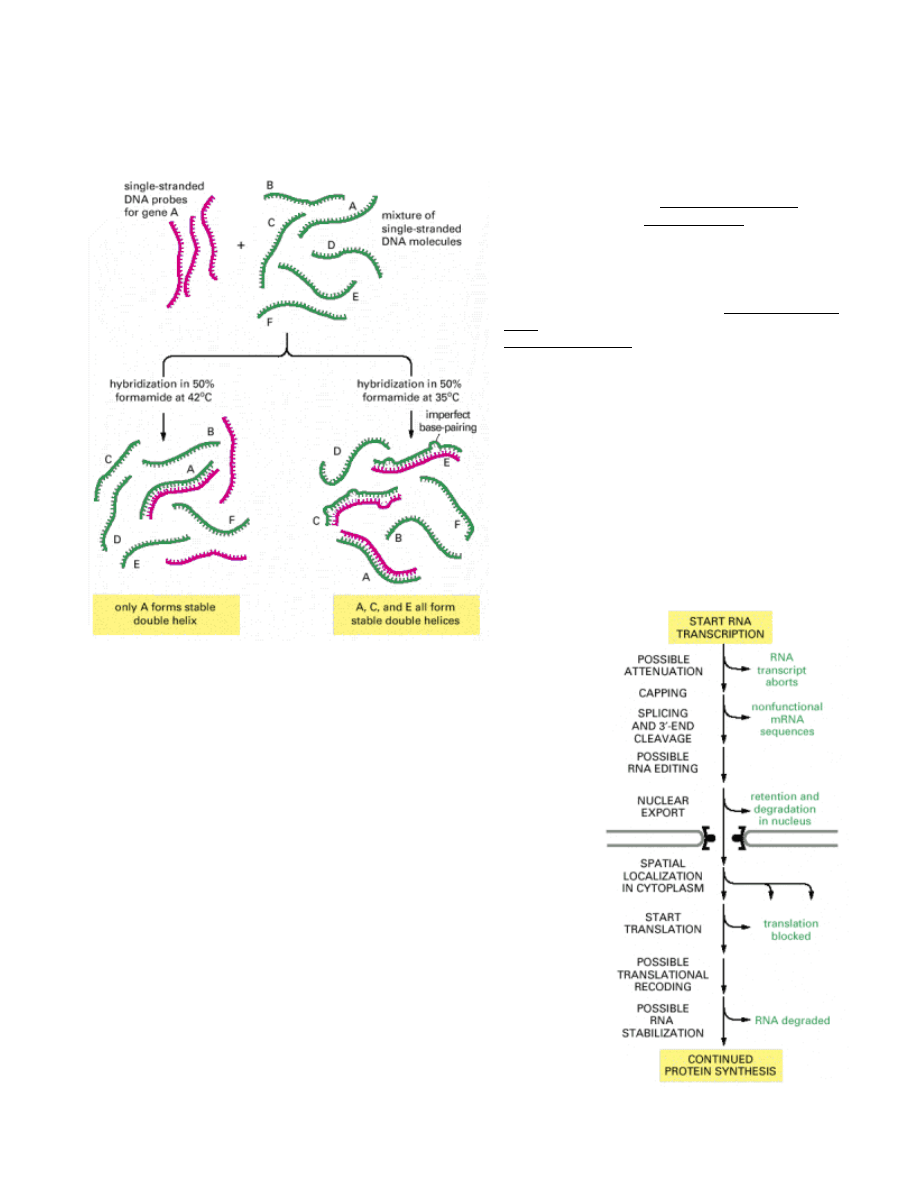
HYBRYDYZACJA: Kiedy roztwór wodny DNA
jest podgrzany do temperatury
100°C
lub
poddany działaniu wysokiego
pH
(pH
≥
13)
komplementarne
zasady
DNA
dysocjują,
pozwalając na rozdzielenie dwuniciowego DNA
na pojedyncze nici. Proces ten nazywany jest
DENATURACJĄ DNA. Jeśli takie zdenaturowane
DNA podgrzeje się ponownie w
temperaturze
65°C następuje renaturacja DNA lub inaczej
hybrydyzacja
DNA
do formy podwójnej helisy.
Hybrydyzacja
może
zachodzić
pomiędzy
komplementarnymi fragmentami DNA, RNA i
DNA, oraz RNA i RNA.
Jednoniciowe
fragmenty
DNA
lub
RNA
komplementarnego do odpowiednij sekwencji
stanowiącej matrycę nazywa się SONDAMI.
Umożliwiają one detekcję wybranego fragmentu
kwasu nukleinowego poprzez znakowanie ich
izotopowo lub chemicznie. Hybrydyzacja jest tak
selektywna, czuła i specyficzna, że pozwala
wykryć nawet jedną cząsteczkę określonego
fragmentu w komórce.
W oparciu o hybrydyzację RNA i DNA można badać nie tylko, czy zachodzi
ekspresja danego genu w komórkach, ale również jakiego rodzaju
modyfikacjom podlega ten fragment w ramach mechanizmów kontroli w
trakcie i po transkrypcji DNA do mRNA
BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
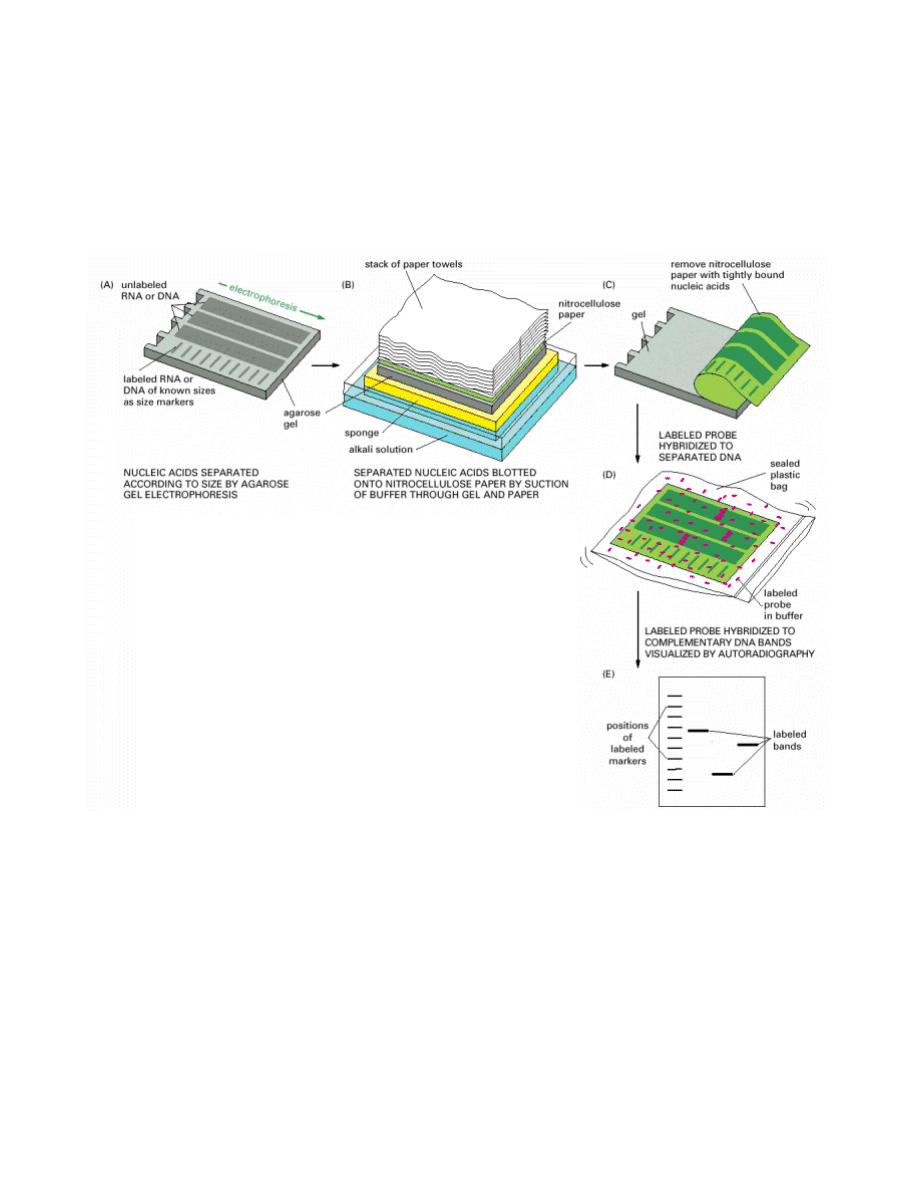
Klasycznego transferu DNA i RNA na odpowiednie filtry nie trzeba dokonywać w aparacie jak w przypadku
białek, a jedynie użyć metody kapilarnej przedstawionej poniżej
BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
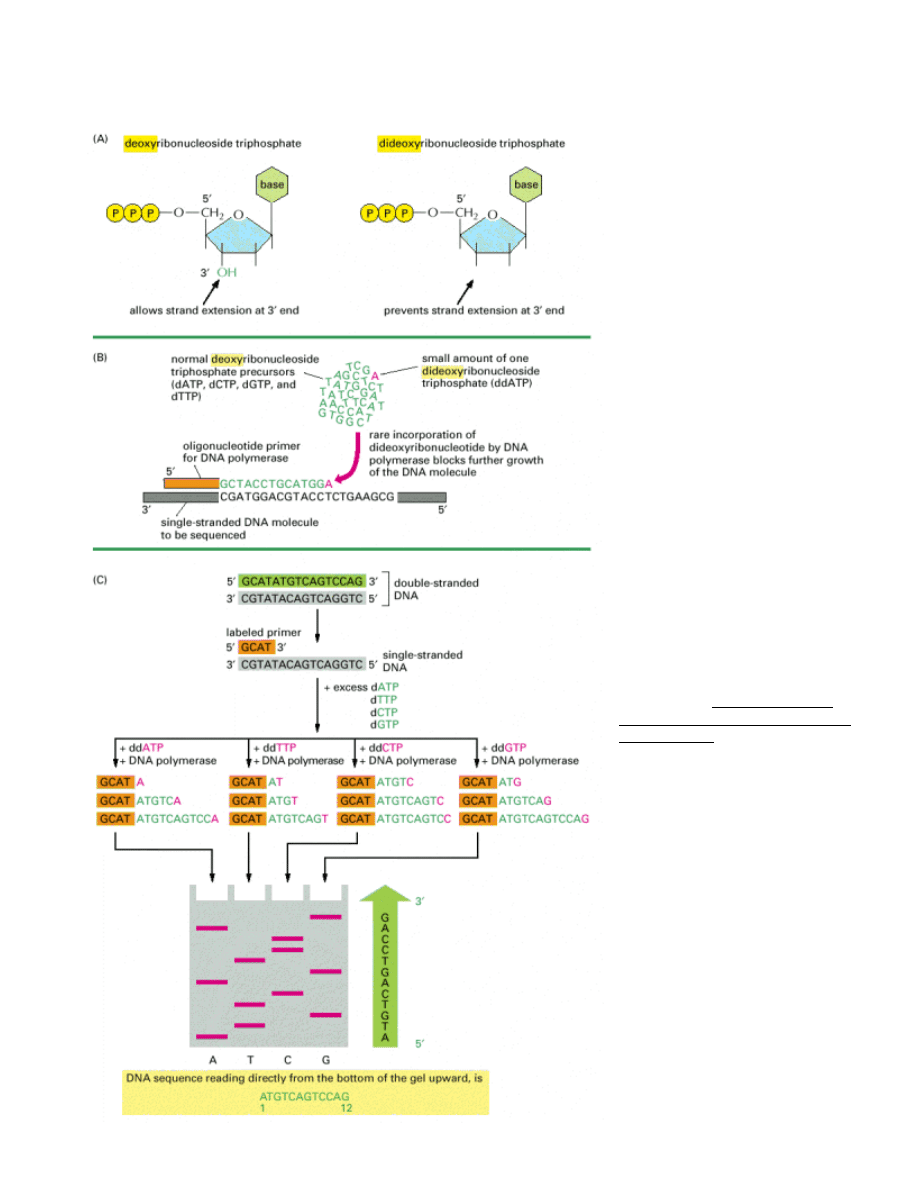
SEKWENCJONOWANIE DNA:
W późnych latach 70-ych
opracowano
technikę
sekwencjonowania
DNA
z
wykorzystaniem tzw. didioxy
pochodnych nukleotydów.
Jak się znajduje region DNA
faktycznie kodujący jakieś
białko?
Dowolny region DNA może
kodować jakieś białko i zwykle
musimy rozważyć 6 różnych
kombinacji sekwencji, ponieważ
mamy do dyspozycji 2 nici DNA
i na każdej 3 ramki odczytu
(gdyż kod genetyczny jest 3-
literowy). Ustalono regułę, że
sekwencję nukleotydów czyta
się zawsze od 5’ do 3’ i
wówczas odpowiadająca im
sekwencja
aminokwasów
czytana będzie od N-końca do
C-końca białka. Dla każdego
odczytu
oszacowuje
się
częstotliwość
występowania
kodonu STOP, jeśli dana
sekwencja koduje jakieś białko,
sygnał STOP nie
będzie
zdarzał
się
częściej
niż
co
21
aminokwasów (czyli raz na 63
nukleotydy).

BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
PCR (Polymerase Chain Reaction): W metodzie tej dwa zestawy oligonukleotydów (tzw. starterów, albo z ang.
„primers”) syntetyzuje się z użyciem metod chemicznych tak, by były komplementarne do początku i końca
sekwencji, którą chcemy powielić (zamplifikować). Oligonukleotydy te są następnie wykorzystywane do
cyklicznych reakcji PCR przeprowadzanych przez specjalną, odporną na zmiany temperatury polimerazę DNA.
Reakcje te przeprowadzane są w odpowiednich probówkach laboratoryjnych - obecnie z użyciem aparatów
PCR, które umożliwiają nic innego, a po prostu szybkie zmiany temparatur w zakresie od ok. 40 – 100°C
Zwykle 20-30 cyklicznych zmian temparatur wystarczy, by efektywnie powielić dany fragment, a
najnowocześniejsze aparaty PCR pozwalaja na przeprowadzenie tych reakcji w przeciągu 1-2 godzin.

BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
Różnice pomiędzy klasycznym
PCR, w którym materiałem
wyjściowym jest DNA a RT-
PCR, w którym materiałem
wyjściowym
jest
RNA
pokazane są na rysunku po
lewej.

BIOLOGIA KOMÓRKI; 16-20 STYCZNIA 2012
Podstawy ANALIZY KWASÓW NUKLEINOWYCH
Poniższa tabela podsumowuje najważniejsze wydarzenia dotyczące rozwoju technik rekombinacji DNA i
tworzenia organizmów transgenicznych.
Some Major Steps in the Development of Recombinant DNA and Transgenic Technology
1869
Miescher first isolates DNA from white blood cells harvested from pus-soaked bandages obtained from
a nearby hospital.
1944
Avery provides evidence that DNA, rather than protein, carries the genetic information during bacterial
transformation.
1953
Watson and Crick propose the double-helix model for DNA structure based on x-ray results of
Franklin and Wilkins.
1955
Kornberg discovers DNA polymerase, the enzyme now used to produce labeled DNA probes.
1961
Marmur and Doty discover DNA renaturation, establishing the specificity and feasibility of nucleic acid
hydridization reactions.
1962
Arber provides the first evidence for the existence of DNA restriction nucleases, leading to their
purification and use in DNA sequence characterization by Nathans and H. Smith.
1966
Nirenberg, Ochoa, and Khorana elucidate the genetic code.
1967
Gellert discovers DNA ligase, the enzyme used to join DNA fragments together.
1972-
1973
DNA cloning techniques are developed by the laboratories of Boyer, Cohen, Berg, and their
colleagues at Stanford University and the University of California at San Francisco.
1975
Southern develops gel-transfer hybridization for the detection of specific DNA sequences.
1975-
1977
Sanger and Barrell and Maxam and Gilbert develop rapid DNA-sequencing methods.
1981-
1982
Palmiter and Brinster produce transgenic mice; Spradling and Rubin produce transgenic fruit flies.
1982
GenBank, NIH's public genetic sequence database, is established at Los Alamos National Laboratory.
1985
Mullis and co-workers invent the polymerase chain reaction (PCR).
1987
Capecchi and Smithies introduce methods for performing targeted gene replacement in mouse
embryonic stem cells.
1989
Fields and Song develop the yeast two-hybrid system for identifying and studying protein interactions
1989
Olson and colleagues describe sequence-tagged sites, unique stretches of DNA that are used to make
physical maps of human chromosomes.
1990
Lipman and colleagues release BLAST, an algorithm used to search for homology between DNA and
protein sequences.
1990
Simon and colleagues study how to efficiently use bacterial artificial chromosomes, BACs, to carry
large pieces of cloned human DNA for sequencing.
1991
Hood and Hunkapillar introduce new automated DNA sequence technology.
1995
Venter and colleagues sequence the first complete genome, that of the bacterium Haemophilus
influenzae.
1996
Goffeau and an international consortium of researchers announce the completion of the first genome
sequence of a eucaryote, the yeast Saccharomyces cerevisiae.
1996-
1997
Lockhart and colleagues and Brown and DeRisi produce DNA microarrays, which allow the
simultaneous monitoring of thousands of genes.
1998
Sulston and Waterston and colleagues produce the first complete sequence of a multicellular
organism, the nematode worm Caenorhabditis elegans.
2001
Consortia of researchers announce the completion of the draft human genome sequence.
Wyszukiwarka
Podobne podstrony:
Cwiczenia 13 Podstawy analizy bialek
Sprawozdanie ćwiczenie nr 14, Tż, Analiza żywności II, Sprawozdania
Analizy molekularne DNA i RNA w wykrywaniu
Ćwiczenie 14-te, Tż, Analiza żywności II, Sprawozdania
Podstawy analizy 2 Cwiczenia, zadania
Ćwiczenie 1 14 03 2015 Analizy ekonomiczne
cwiczenie 14 id 125164 Nieznany
PODSTAWY REKREACJI CZASU WOLNEGO- ćwiczenia, GWSH, podstawy rekreacji i czasu wolnego
cwiczenia 14 28.03.2008, cwiczenia - dr skladowski
podstawy analizy niepewności pomiarowych
Podstawy analizy fundamentalnej Nieznany
II wirusy wszystkie RNA DNA=HSV, VZV, Adenowirusy
fiz cwiczenia 14
Cwiczenia 14, Ekonometria, Ekonometria, Egzaminy + Testy, Egzaminy, ekonometria 2009, Ekonometria za
Ankieta CWICZENIA, matura podstawowa pisemna
spoleczenstwo mas - Mills, Studia (europeistyka), nauka o polityce, Teoria polityki, ćwiczenia 14
karta podst analiz.stacj, gik, gik, I sem, podstawy analiz sieci pomiarowych
więcej podobnych podstron