
Uniwersytet Mikołaja Kopernika w Toruniu,
Collegium Medicum im. L. Rydygiera w Bydgoszczy
Wydział Lekarski
Katedra Biofizyki, Zakład Fizyki Medycznej
Karolina Jakołcewicz
Nr albumu: 217507
BADANIE WŁA
Ś
CIWO
Ś
CI ANTYOKSYDACYJNYCH
WYBRANYCH FLAWONOIDÓW METOD
Ą
SPEKTROSKOPII OPTYCZNEJ
PRACA MAGISTERSKA
NA KIERUNKU BIOTECHNOLOGIA
wykonana pod kierunkiem
dr hab. Stefana Kruszewskiego, prof. UMK
Katedra Biofizyki, Zakład Fizyki Medycznej
TORU
Ń
2012
Pracę przyjmuję i akceptuję Potwierdzam złożenie pracy dyplomowej
............................................ .............................................................
data i podpis opiekuna pracy data i podpis pracownika dziekanatu

2
Panu mgr Tomaszowi Wybranowskiemu
składam serdeczne podziękowania
za pomoc przy opracowaniu wyników,
opiekę oraz poświęcony mi czas.

3
Gorące podziękowania pragnę przekazać
Profesorowi Stefanowi Kruszewskiemu
za wyrozumiałość oraz udzielenie
mi wielu życzliwych rad.

4
Wykaz skrótów
AAPH
-
2,2’-diazobis (2-amidinopropan) dichlorowodorku
ATP
-
adenozynotrójfosforan
DMSO
-
dimetylosulfotlenek
FITC
-
izotiocyjanian fluoresceiny
HSA
-
albumina ludzka
LDL
-
lipoproteiny o małej gęstości
NOS
-
syntaza tlenku azotu
ORAC-FL
-
(ang. Oxygen Radical Absorbance Capacity) zdolność
pochłaniania reaktywnych form tlenu przez przeciwutleniacze
OUN
-
ośrodkowy układ nerwowy
PBS
-
zbuforowany roztwór soli fizjologicznej
RFT
-
reaktywne formy tlenu
RNS
-
reaktywne formy azotu
rys.
-
rysunek
tab.
-
tabela

5
Spis treści
Wstęp …………………………………………………………………………...............………7
1.
Wolne rodniki....................................................................................................................... 9
1.1.
Powstawanie reaktywnych form tlenu.............................................................................9
1.2.
Reaktywne formy tlenu w warunkach fizjologicznych.................................................10
1.3.
Szkodliwy wpływ wolnych rodników...........................................................................12
2.
Wpływ reaktywnych form tlenu na białka.......................................................................14
2.1.
Utlenianie łańcucha polipeptydowego..........................................................................14
2.2.
Utlenienie reszt aminokwasowych w białkach..............................................................16
3.
Flawonoidy ………………………………….....………………......……..........................18
3.1.Budowa, klasyfikacja i nazewnictwo.......…..………..............…………………….......18
3.2.
Aktywność biologiczna ...............................................................................................21
3.2.1.
Działanie antyoksydacyjne.................................................................................21
3.2.2.
Działanie prooksydacyjne...................................................................................23
3.2.3.
Działanie przeciwzapalne i przeciwalergiczne...................................................24
3.2.4.
Działanie estrogenne ..........................................................................................25
3.3.
Flawonoidy w profilaktyce i terapii chorób...................................................................26
3.3.1.
Aktywność przeciwnowotworowa......................................................................26
3.3.2.
Flawonoidy w chorobach sercowo-naczyniowych.............................................28
3.3.3.
Flawonoidy a cukrzyca.......................................................................................30
3.3.4.
Flawonoidy w terapii AIDS................................................................................30
3.3.5.
Wpływ na ośrodkowy i obwodowy układ nerwowy...........................................31
3.4.
Kwercetyna....................................................................................................................31
3.4.1.
Budowa i właściwości.........................................................................................31
3.4.1.1.Właściwości antyoksydacyjne......................................................................32
3.4.1.2.Działanie przeciwnowotworowe..................................................................33
3.4.1.3.Działanie przeciwzapalne.............................................................................34
4.
Albumina surowicy krwi....................................................................................................35
4.1.
Struktura białka.............................................................................................................35
4.2.
Synteza..........................................................................................................................36
4.3.
Funkcje..........................................................................................................................36
4.4.
Utlenianie albuminy......................................................................................................37
5.
Metody fizyczne badania cząsteczek.................................................................................38
5.1.
Absorpcja...................................................................................................................... 38
5.2.
Fluorescencja.................................................................................................................39
5.3.
Anizotropia....................................................................................................................42
6.
Cel pracy..............................................................................................................................44
7.
Aparatura pomiarowa i metody badawcze.......................................................................45

6
7.1.
ORAC-FL......................................................................................................................45
7.2.
Metoda pomiaru produktów utleniania białek (AOPP).................................................46
8.
Wyniki i wnioski..................................................................................................................47
8.1.
Przygotowanie roztworów podstawowych....................................................................47
8.2.
Widma absorpcji kwercetyny w obecności rodników generowanych przez AAPH.....47
8.3.
Widma emisji fluorescencji kwercetyny o różnych stężeniach.....................................49
8.4.
Rejestracja widma fluorescencji kwercetyny i HSA metodą ORAC(FL)
....................51
8.5.
Pomiar natężenia fluorescencji kwercetyny w różnych stężeniach HSA......................52
8.6.
Rejestracja widma fluorescencji kwercetyny z HSA.....................................................55
8.7.
Pomiar absorpcji utlenionego HSA...............................................................................56
9.
Podsumowanie....................................................................................................................58
10.
Streszczenie.........................................................................................................................60
11.
Bibliografia.........................................................................................................................61

7
Wstęp
Wolne rodniki to niestabilne cząsteczki zawierające niesparowany elektron.
Określenie „wolny” oznacza, że atom lub cząsteczka są zdolne do samodzielnego istnienia.
Według współczesnego mianownictwa termin wolne rodniki, jest równoznaczny
z określeniem reaktywne formy tlenu (RFT). W ilościach fizjologicznych, czyli w warunkach
równowagi pomiędzy ich wytwarzaniem, a neutralizacją,
pełnią one rolę mediatorów
i regulatorów wielu procesów zachodzących wewnątrz komórki. W przypadku zaburzeń
związanych ze zwiększeniem ilości ich wytwarzania dochodzi do tzw. stresu oksydacyjnego.
Stres oksydacyjny następuje w momencie zaburzenia równowagi pomiędzy wytwarzaniem
RFT, a zdolnością do ich neutralizacji. Ze stresem oksydacyjnym wiążą się takie choroby,
jak: nowotwory, choroby sercowo-naczyniowe, cukrzyca i inne choroby cywilizacyjne [2].
Jedną z form obrony wolnych rodników jest nieenzymatyczna obrona związana z
dostarczaniem organizmowi przeciwutleniaczy. Takimi przeciwutleniaczami są między
innymi flawonoidy.
Flawonoidy, inaczej zwane związkami flawonowymi lub bioflawonoidami, to jeden
z typów substancji fitochemicznych, które znajdują się w roślinach. Pełnią one rolę
barwników, przeciwutleniaczy oraz naturalnych insektycydów i fungicydów (chronią przed
atakami insektów i grzybów). Wszystkie flawonoidy są oparte na szkielecie
2-fenylochromanu. Większość typów flawonoidów (poza katechinami i antycyjanidynami)
zawiera szkielet flawonu, z grupą ketonową w pozycji 4. Flawonoidy różnią się między sobą
liczbą i rodzajem podstawników. Różnice między związkami w poszczególnych klasach
wynikają zazwyczaj z odmiennej budowy tylko jednego skrajnego pierścienia[19]. Większość
flawonoidów zawiera grupy hydroksylowe, z których jedna lub więcej jest zwykle połączona
z cząsteczką cukru i tworzy glikozydy. Prawie wszystkie flawonoidy wykazują różnorodną
aktywność biologiczną zarówno w organizmach roślinnych i w zwierzęcych. Stanowią one
uniwersalne zmiatacze wolnych rodników dzięki właściwościom antyoksydacyjnym [20].
Dotychczasowe badania nad biologiczną aktywnością flawonoidów pokazały, że wzmagają
one w sposób istotny odporność organizmu i łagodzą stany zapalne. Takie działanie tych
związków może wynikać z właściwości antyoksydajnych, aktywności estrogennej oraz
szerokiego działania przeciwzapalnego [19].
Jednym z przedstawicieli flawonoidów jest kwercetyna. Wywiera różnorodne
pozytywne działanie na poziomie komórek i całego organizmu. Posiada właściwości

8
regulujące niektóre mechanizmy prowadzące do powstania nowotworu, skutecznie hamuje
kluczowe etapy w kaskadzie mechanizmów zapalnych. Jako przeciwutleniacz chroni
komórki, błony komórkowe oraz DNA przed uszkodzeniami spowodowanymi przez wolne
rodniki.
Rozwój analitycznych metod fizycznych opartych na spektroskopii optycznej
umożliwił zastosowanie ich u wielu dziedzinach nauki, a w szczególności w biologii
i w medycynie. Zastosowanie tych metod pozwala przewidzieć szereg biofizycznych
właściwości związków, które mogą znaleźć zastosowanie terapeutyczne.
W pracy przeprowadzono badania nad właściwościami antyoksydacyjnymi
flawonoidów. Badania zostały przeprowadzone na przykładzie kwercetyny, flawonoidu
najbardziej rozpowszechnionego w przyrodzie. Jedną z zastosowanych metod badawczych
była metoda ORAC-FL. Dzięki tej metodzie można było przedstawić jaką zdolność
antyutleniającą posiada badany przeze mnie flawonoid. Poza wykazaniem działania
antyoksydacyjnego kwercetyny, badane były również jej interakcje z białkiem surowicy krwi
ludzkiej - albuminą. Albumina stanowi ponad połowę białek znajdujących się w surowicy
i odpowiedzialna jest za transport leków, hormonów i kwasów tłuszczowych. Badania te były
wykonane w związku z wysokim powinowactwem jakie wykazuje kwercetyna w stosunku do
albuminy. Z racji tego, że jest to białko, które odpowiada między innymi za transport leków
w naszym organizmie, ważnym było, aby wykazać jaki wpływ na siebie mają obie te
cząsteczki. Badano właściwości antyoksydacyjne kwercetyny w stanie wolnym i związanej
z albuminą. Zbadano także wpływ anionorodnika ponadtlenkowego na utlenianie albuminy,
metodą opracowaną przez Witko-Sarsat i wsp. Dokładne zdefiniowanie interakcji, które mają
miejsce pomiędzy kwercetyną i albuminą dałoby nadzieję na wykorzystanie flawonoidów
jako środków terapeutycznych.

9
1.
Wolne rodniki
Wolne rodniki, mogą być atomami, jonami lub cząsteczkami. Mają one niesparowany
elektron, czyli charakteryzują się spinem elektronowym różnym od zera. Rodniki mają
zazwyczaj charakter obojętny, ale mogą również przyjmować ładunek dodatni lub ujemny.
Wówczas rodniki obdarowane ładunkiem nazywamy odpowiednio: aniorodnikami (ładunek
ujemny) lub katiorodnikami (ładunek dodatni) [1].
In vivo rodniki powstają w wielu reakcjach i odgrywają ważną rolę zarówno w
fizjologii jak i patofizjologii. W organizmach ssaków uwalniane są w łańcuchu oddechowym,
podczas
utleniania
hemoglobiny
oraz
podczas
wybuchów
tlenowych
komórek
fagocytujących. U roślin mogą powstawać w procesie fotosyntezy. Rodniki mogą pełnić rolę
przekaźników sygnału w komórkach, negatywną stroną ich działania jest udział w
patogenezie i rozwoju ponad dwustu chorób. Wśród tych chorób znajdują się tzw. choroby
cywilizacyjne takie jak cukrzyca, choroby układu krążenia i nowotwory [1,2].
1.1.
Powstawanie reaktywnych form tlenu
Toksyczność tlenu związana jest z powstawaniem w komórkach i ich otoczeniu RFT.
Pod kontrolą mechanizmów enzymatycznych przekształcane jest 98-99% tlenu. 1-2%
zatrzymuje się na etapie produktów pośrednich, czyli tworzy reaktywne formy tlenu.
Reaktywne formy tlenu mogą powstawać w organizmie na skutek działania zewnętrznych
czynników fizycznych takich jak promieniowanie jonizujące, nadfioletowe oraz ultradźwięki.
Wymienione powyżej czynniki fizyczne są źródłem RFT o niewielkim znaczeniu
biologicznym.
Bardziej
istotne
są
wewnątrzkomórkowe
źródła
RFT,
głównie
jednoelektronowe utlenianie zredukowanych form wielu związków przez tlen cząsteczkowy
oraz szereg reakcji enzymatycznych. Pierwotnie w większości istotnych biologicznie sytuacji
tworzony jest anionorodnik ponadtlenkowy. W wyniku jego rozpadu, czy też niektórych jego
reakcji może powstać nadtlenek wodoru. Obecność nadtlenku wodoru i anionorodnika
ponadtlenkowego daje możliwość tworzenia rodnika hydroksylowego jak również innych
wysoce reaktywnych form tlenu.
Związki określane jako fotosensybilizatory (światłouczulacze) po zaabsorbowaniu
światła mogą przejść w stan wzbudzony (to znaczy ze stanu singletowego w trypletowy)

10
i reagować z tlenem trypletowym. W reakcji tej powstaje anionorodnik ponadtlenkowy (np.
fotoredukcja ryboflawiny).
Obecność zredukowanych form niskocząsteczkowych składników komórek (np.
zredukowana ryboflawina, cukry redukujące, związki tiolowe) podobnie jak fotoredukcja
może prowadzić do powstania anionorodnika ponadtlenkowego.
•
+
•
+
+
→
+
2
2
2
O
H
RH
O
RH
(1.7)
Substancje takie jak herbicydy, insektycydy, fungicydy ulegają w komórkach
cyklicznej redukcji. Cykle te napędzone są poprzez metabolizm komórki, a nie światło.
Mechanizm toksyczności tych związków polega właśnie na wytwarzaniu RFT.
W organizmach żywych utlenieniu ulegają również hemoglobina i mioglobina, czyli
białka oddechowe. Obie jako grupę prostetyczną zawierają hem. W skład grupy hemowej
wchodzą jony żelaza Fe
2+
, które pod wpływem tlenu łatwo mogą się utleniać do jonów żelaza
Fe
3+
. W przypadku obu tych białek jony Fe
2+
są silnie związane z ich hemem, dlatego też są
mniej podatne na utlenianie, nie mniej jednak procesowi temu mogą ulec.
•
+
+
+
−
→
+
−
2
3
2
2
O
Fe
hem
O
Fe
hem
(1.8)
90% tlenu w komórce zużywane jest w mitochondriach. W warunkach prawidłowych
cząsteczka tlenu ulega w nich czteroelektronowej redukcji, a energia wytwarzana podczas
tego procesu jest wykorzystana do syntezy ATP. Jednak część cząsteczek w procesie
oddechowym ulega redukcji jednoelektrodowej, w wyniku czego powstaje anionorodnik
ponadtlenkowy. Jest to proces, który stanowi najważniejsze źródło anionorodników
ponadtlenkowych w większości komórek aerobowych [1,2].
1.2.
Reaktywne formy tlenu w warunkach fizjologicznych
W warunkach homeostazy reaktywne rodniki tlenowe są uwalniane w ilości
bezpiecznej dla komórki. Odgrywają rolę mediatorów i regulatorów wielu procesów
komórkowych [3,5]. Reaktywne formy tlenu (RFT) wpływają na syntezę, uwalnianie oraz
inaktywację tlenku azotu, indukują różnicowanie się komórek oraz ich apoptozę a także
pobudzają transport glukozy do komórek. Warunkują prawidłowy przebieg reakcji zapalnej

11
poprzez zwiększenie przepuszczalności ścian naczyń włosowatych. Jedną z najistotniejszych
ról jaką odgrywają RFT jest regulacja procesów przekazywania sygnałów z komórki do
komórki oraz w jej obrębie [3,4]. Idealnymi kandydatami na przekaźniki są jon
ponadtlenkowy i nadtlenek wodoru ze względu na małą reaktywność, wybiórczość i stałą
dostępność w komórce [3,4,5]. Wykazują podstawowe działanie w hamowaniu funkcji
receptorów, głównie tych zawierających grupy –SH. Większość białek posiadających grupy
tiolowe jest inaktywowana przez RFT, ale są też takie których aktywność wzrasta. Zaliczane
są do nich m.in. cyklaza guanylanowa (enzym wytwarzający cGMP) oraz wybrane białka
transportowe np. 5-lipooksygenaza, która jest źródłem wolnych rodników generowanych
przez pobudzone limfocyty. Metabolity powstające w wyniku utleniania wielonienasyconych
kwasów tłuszczowych utrzymują wewnątrz komórki równowagę oksydacyjną aktywując
szlaki przekazywania sygnału i ekspresję genów [3,6,7]. Wolne rodniki tlenowe wykorzystane
są przez komórki fagocytujące do eliminacji patogenów. Proces ten nazywany jest
„wybuchem tlenowym” i wiąże się z kilkudziesięciokrotnym wzrostem zużycia tlenu. Nazwa
wybuch tlenowy wiąże się z wykorzystaniem tlenu do wytworzenia i uwolnienia
anionorodnika ponadlenkowego, który jest prekursorem jonu hydroksylowego. Dodatkowo
RFT uczestniczą w eliminacji pasożytów oraz procesie eliminacji czynników potencjalnie
chorobotwórczych znajdujących się w jamie ustnej, stwierdza się to przez wykazanie
obecności peroksydazy i mieloperoksydazy w ślinie [3,8]. Jednym z najbardziej istotnych
zadań wolnych rodników jest udział w procesie starzenia. RFT nie tylko wpływają na
starzenie się, ale również decydują o śmierci lub przeżyciu komórki. Niskie stężenia RFT
powodują aktywację czynników transkrypcyjnych przez co umożliwiają zróżnicowanie
komórek i przystosowanie się do zmienionych warunków, natomiast wyższe stężenia wolnych
rodników powodują kierowanie komórki na drogę apoptozy, dzięki czemu komórki, które
uległy dużym uszkodzeniom i mogłyby stanowić zagrożenie dla organizmu ulegają
eliminacji [3,5,10].
Reaktywne formy tlenu uczestniczą również w regulacji odporności. Wykazano, że
nasilają aktywację limfocytów T i indukują adhezję komórek limfocytarnych do śródbłonka
co umożliwia ich przenikanie z układu krwionośnego do miejsca reakcji zapalnej [3,4,5,9].

12
1.3.
Szkodliwy wpływ wolnych rodników.
Wpływ wolnych rodników na komórkę zależy od czasu działania i ich stężenia. Niskie
stężenie spełnia warunki fizjologiczne, natomiast wysokie prowadzi do toksycznego
uszkadzania komórek i prowadzi do ich destrukcji [3,5].
Szkodliwe działanie wolnych rodników przejawia się między innymi w zdolności do
utleniania białek [3,5]. Nadtlenki białek powstają w wyniku kontaktu białek z wolnymi
rodnikami powstającymi w reakcjach z udziałem ksantyny i oksydazy ksantynowej,
mitochondrialnego łańcucha oddechowego, czy też pobudzonych komórek fagocytujących.
Nieodwracalnie zmienione białka są selektywnie usuwane przez proteazy, jednak w wyniku
starzenia, gdy aktywność proteolityczna ulega obniżeniu, mogą być gromadzone
w komórce [11,12,13]. Utlenianie białek może prowadzić do pojawienia się zmienionych
reszt aminokwasowych, rozerwania łańcucha polipeptydowego czy też do tworzenia się
dimerów lub agregatów białkowych. Zmiany te w konsekwencji prowadzą do utraty
aktywności
funkcjonalnej
enzymów,
białek
regulatorowych
czy
transporterów
błonowych [11,14,15]. Oddziaływanie RFT z białkami może powodować nie tylko ich
utlenianie, ale również tworzenie się w białkach grup redukujących zdolnych m.in. do
redukcji cytochromu c i jonów metali [5,16].
Kwasy nukleinowe wykazują większą stabilność niż białka czy lipidy. Reakcja
rodnika hydroksylowego i tlenu singletowego z kwasami może prowadzić do uszkodzenia
zasad purynowych i pirymidynowych reszt cukrowych lub do rozerwania wiązań
fosfodiestowych łączących nukleotydy. Prowadzi to do pęknięć nici kwasów
nukleinowych [3,17,18].
Kolejne niebezpieczeństwo ze strony wolnych rodników tlenowych wynika z procesu
peroksydacji lipidów, czyli wolnorodnikowego utleniania lipidów [3]. W przebiegu procesu
peroksydacji lipidów można wyróżnić trzy fazy inicjacji, propagacji i terminacji.
1.
Inicjacja polega na oderwaniu cząsteczki wodoru od cząsteczki nienasyconego kwasu
tłuszczowego wchodzącego w skład fosfolipidów – głównych składników budujących
błonę komórkową. Inicjacja może być zapoczątkowana przez rodniki takie jak:
hydroksylowy (OH‘), nadtlenkowy (LOO‘) i alkilowy (LO‘) oraz tlenek i dwutlenek
azotu.
2.
W reakacji propagacji wolne rodniki alkilowe reagują z tlenem dając wolne rodniki
nadtlenkowe, a w końcu nadtlenek kwasu tłuszczowego. Taki cykl reakcji może się

13
powtarzać wielokrotnie, aż do terminacji i może doprowadzić do przekształcenia w
nadtlenek nawet kilkuset cząsteczek kwasów tłuszczowych.
3.
Reakcja
terminacji
może
zachodzić
między
dwoma
rodnikami
alkilowymi,
nadtlenkowymi lub dwoma różnymi rodnikami występującymi w układzie. Produktami tej
reakcji są uszkodzone cząsteczki lipidów.
Dalsze przemiany prowadzą do rozpadu reszt wielonienasyceniowych kwasów
tłuszczowych i powstania kilku lub kilkunastuwęglowych fragmentów takich jak
dwualdehyd malonowy czy 4-hydroksynonenal [1]. Wiele białek wykazuje zdolność
wiązania się z 4-hydroksynonenalem, które to wiązanie nie jest korzystne dla komórki z
racji tego, że białka występujące w tym kompleksie zmieniają się konformacyjnie i
funkcjonalnie. Produkty peroksydacji lipidów zmieniają właściwości błon komórkowych,
może to prowadzić do zahamowania aktywności enzymów błonowych i białek
transportujących, jak również indukować ekspresję cyklooksygenazy typu 2 (COX-2) w
makrofagach i aktywować potencjał zapalny tych komórek [3].

14
2.
Wpływ reaktywnych form tlenu na białka
Do utlenienia łańcucha polipeptydowego i reszt aminokwasowych w białkach
dochodzi pod wpływem utleniaczy. Prowadzić to może do rozerwania łańcucha
polipeptydowego, utworzenia wiązań krzyżowych w obrębie tego samego lub kilku
łańcuchów
polipeptydowych
i
spowodować
pojawienie
się
zmienionych
reszt
aminokwasowych [59]. Białka w których doszło do takich zmian mogą utracić lub
wykazywać zwiększoną aktywność biologiczną jak również mieć tendencję do tworzenia
agregatów [60]. Agregaty powstałe w wyniku modyfikacji oksydacyjnych posiadają zdolność
do hamowania systemów odpowiedzialnych za ich degradację, co sprzyja ich nagromadzeniu.
Wykazano, że pod wpływem wolnych rodników dochodzi do utraty aktywności takich
enzymów jak np. dehydrogenazy gliceroaldehydofosforanowej czy dehydrogenazy glukozo-
6-fosforanowej [11].
2.1. Utlenianie łańcucha polipeptydowego
Rodnik hydroksylowy, który powstaje w reakcji Fentona, tj. reakcji nadtlenku wodoru
np. z jonami Fe
2+
lub w reakcji Habera-Weissa, czyli reakcji nadtlenku wodoru
z anionorodnikiem ponadtlenkowym, daje początek utlenianiu łańcucha polipeptydowego
poprzez oderwanie atomu wodoru przy węglu α aminokwasu. Powstały w tej reakcji rodnik
alkilowy wchodzi gwałtownie w reakcję z tlenem tworząc poprzez rodnik alkilonadtlenkowy,
alkilowodorondtlenek. Ten przekształca się w rodnik alkoksylowy, który następnie może
przejść w hydroksylowaną przy węglu α resztę aminokwasową lub może doprowadzić do
fragmentacji łańcucha polipeptydowego [62]. Rodniki alkilowy, alkilonadtlenkowy,
alkoksylowy reagują z innymi resztami aminokwasowymi tego samego lub innego łańcucha
polipeptydowego białka, umożliwiając tym samym powstawanie kolejnych rodników.
Tworzenie rodników alkilonadtlenkowych przy niedoborze tlenu jest utrudnione, dlatego
rodniki alkilowe w obrębie tego samego lub różnych białek, reagując ze sobą mogą prowadzić
do powstania wiązań krzyżowych pomiędzy łańcuchami polipeptydowymi [11]. Reakcjom
prowadzącym do rozerwania łańcucha polipeptydowego sprzyja obecność rodników
alkoksylowych. Powstać mogą dwa typy produktów rozpadu i jest to zależne od miejsca
pęknięcia (rys.1) [62].
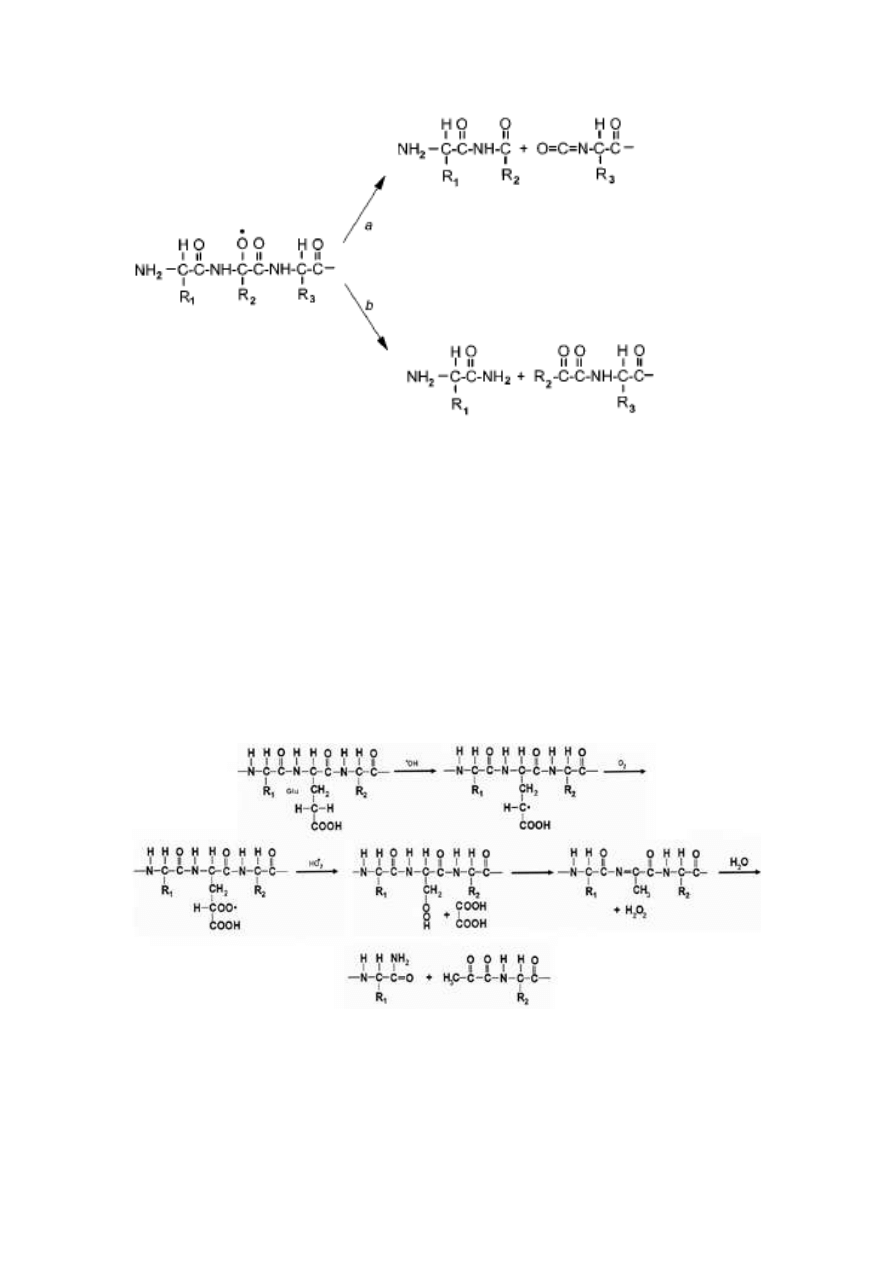
15
Rys.1 Fragmentacja łańcucha polipeptydowego poprzez rodnik alkoksylowy [62].
Do przerwania łańcucha polipeptydowego doprowadzić również może atak
reaktywnych form tlenu na reszty kwasu glutaminowego, kwasu asparaginowego i proliny.
Atom wodoru oderwany od atomu węgla γ reszty kwasu glutaminowego lub kwasu
asparaginowego przez rodnik hydroksylowy może doprowadzić do fragmentacji łańcucha
(rys.2) [11].
Rys.2 Fragmentacja łańcucha polipeptydowego poprzez utlenianie reszty kwasu
glutaminowego [11].
16
Utlenianie reszt proliny prowadzi do przerwania łańcucha polipeptydowego i równocześnie
do powstania 2-pyrolidylonu (rys. 3) [11].
Rys.3 Utlenianie reszt proliny [11].
2.2.Utlenianie reszt aminokwasowych w białkach.
Wszystkie reszty aminokwasowe białek podatne są na utlenianie. Największą
wrażliwość na działanie wolnych rodników wykazują: cysteina, metionina, tyrozyna
i tryptofan. Jedyną modyfikacją w białkach in vivo, która może zostać naprawiona
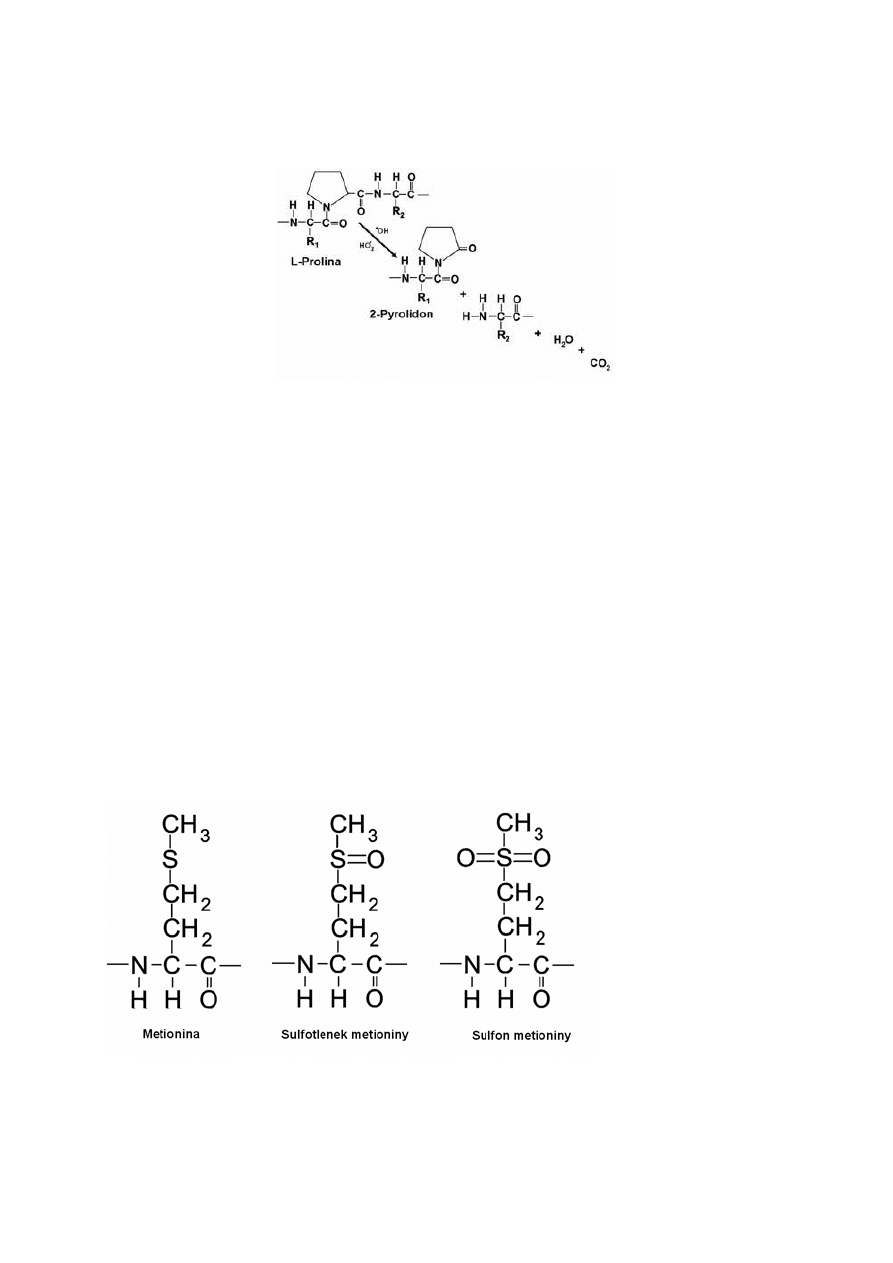
w obecności specyficznych reduktaz jest utlenienie metioniny i cysteiny. Reszty metioninowe
utleniają się do sulfotlenku metioniny (rys. 4), a cysteinowe do reszt dwusiarczkowych. Jako
jeden z mechanizmów zmiatania wolnych rodników może mieć znaczenie utlenianie
i redukcja reszt metioniny [62].
Rys. 4 Utlenianie reszt metioniny [11].
17
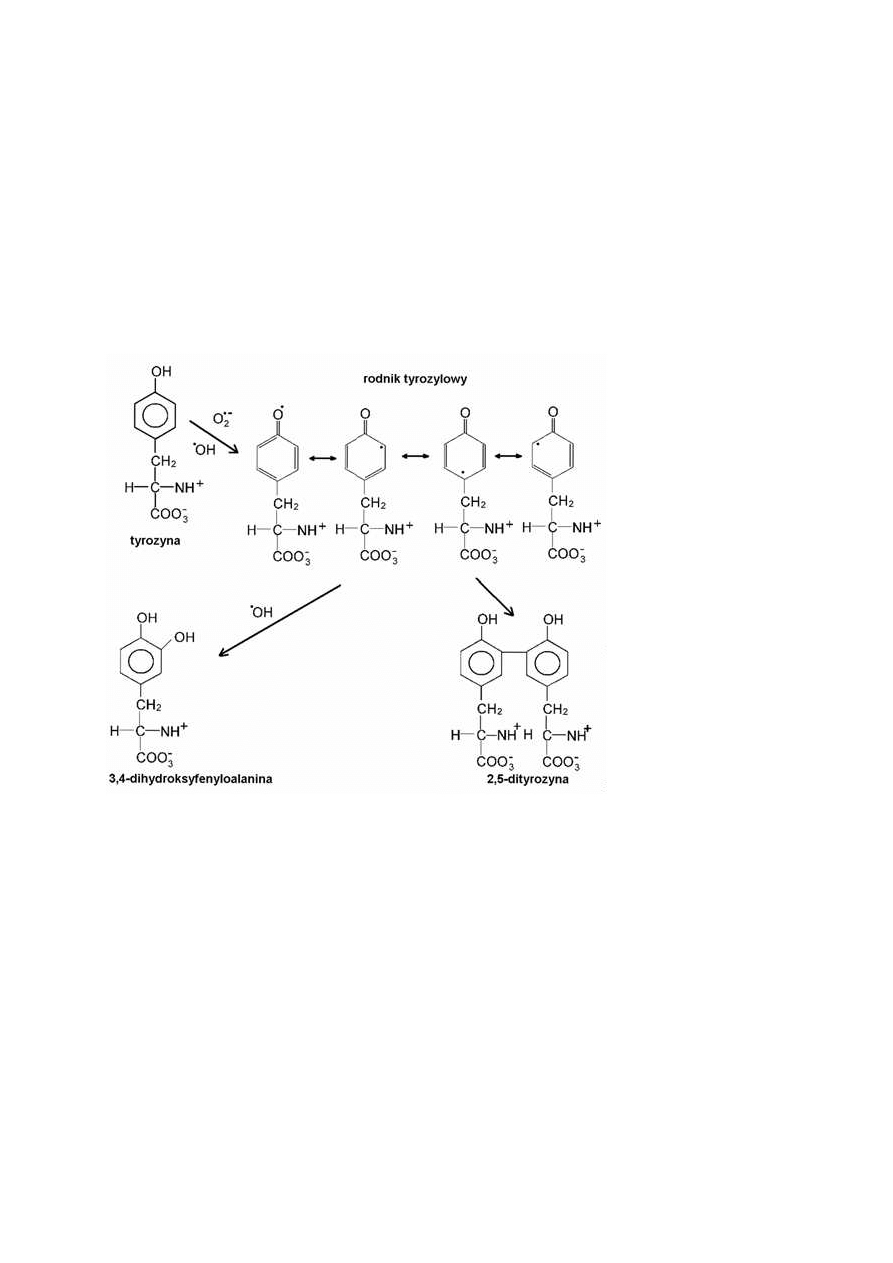
Wzmożoną wrażliwość na atak wolnych rodników wykazują reszty aminokwasów
aromatycznych.
Utlenianie
reszt
tyrozynowych
prowadzi
do
powstania
3,4-
dihydroksyfenyloalaniny (DOPA), która powstaje przez włączenie dodatkowej grupy
hydroksylowej do pierścienia aromatycznego. Dojść może również do wytworzenia wiązań
krzyżowych pomiędzy pierścieniami aromatycznymi dwóch cząsteczek tego samego
aminokwasu dzięki powstaniu 2,5-dityrozyny (rys. 5) [11].
Rys. 5 Utlenianie reszt tyrozyny [11].
Utlenienie reszt tryptofanowych prowadzi do powstania formylokinureniny
i kinureniny. Histydyna natomiast utlenia się do 2-oksohistydyny, asparaginy i kwasu
asparaginowego. Powstawanie pochodnych karbonylowych jest wynikiem utleniania reszt
aminokwasowych z wolną grupą aminową, amidową lub hydroksylową. Pochodne takie mogą
reagować z innymi wolnymi grupami aminowymi reszt lizyny w tej samej lub innej
cząsteczce białka, tworząc wiązania krzyżowe. Reakcja ta jest kolejnym mechanizmem,
w którym tworzone są wiązania krzyżowe [11].

18
3.
Flawonoidy
Badania nad flawonoidami roślinnymi zapoczątkował Albert Szent-Györgyi
biochemik węgierski, laureat Nagrody Nobla w dziedzinie medycyny (1937). Dokonał on
identyfikacji bioflawonoidów w owocach cytrusowych w latach 30. ubiegłego stulecia.
Zaobserwował on także pozytywny wpływ tych związków na naczynia krwionośne
człowieka, który polegał na zmniejszeniu ich przepuszczalności. Zidentyfikowane związki
nazwał witaminą P, od angielskiego słowa permeability, oznaczającego przepuszczalność.
Związki flawonowe, nazywane również flawonoidami lub bioflawonoidami, należą do
szerokiej klasy substancji fitochemicznych, czyli takich, których źródłem są rośliny.
Flawonoidy to składniki wielu substancji pokarmowych niezbędnych w diecie człowieka.
Znajdziemy je w owocach (w szczególności cytrusowych, a także jabłkach, jagodach,
winogronach), warzywach (pomidory, brokuły, papryka, sałata, nasionach roślin
strączkowych, młodym pieprzu) oraz w produktach o pochodzeniu roślinnym (herbata,
czerwone wino czy kasza gryczana). Do tej pory zidentyfikowano ok. 7000 tych związków, z
czego ok.800 zostało dokładnie opisanych. Istnienie tych związków w diecie, a zwłaszcza
zachowanie właściwego poziomu ich spożycia ma istotne znaczenie w profilaktyce
powstawania wielu chorób, między innymi miażdżycy. Dotychczasowe badania prowadzone
na hodowlach komórkowych z wykorzystaniem flawonoidów dają nadzieję na wykorzystanie
ich w profilaktyce i leczeniu nowotworów. Jako antyoksydanty mogą one wpływać w bardzo
zróżnicowany sposób na przykład poprzez: bezpośrednia reakcję z wolnymi rodnikami,
zmiatanie
wolnych
rodników,
hamowanie
lub
wzmacnianie
działania
wielu
enzymów [19,20,21,22,23,24].
3.1.
Budowa, nazewnictwo i klasyfikacja
Flawonoidy są pochodną 2-fenylo-benzo-
γ
-pironu. Podstawową strukturę cząsteczki
związków flawonowych stanowią dwa pierścienie benzenowe (A i B), pomiędzy którymi
znajduje się heterocykliczny pierścień piranu lub piranu (C) [21].
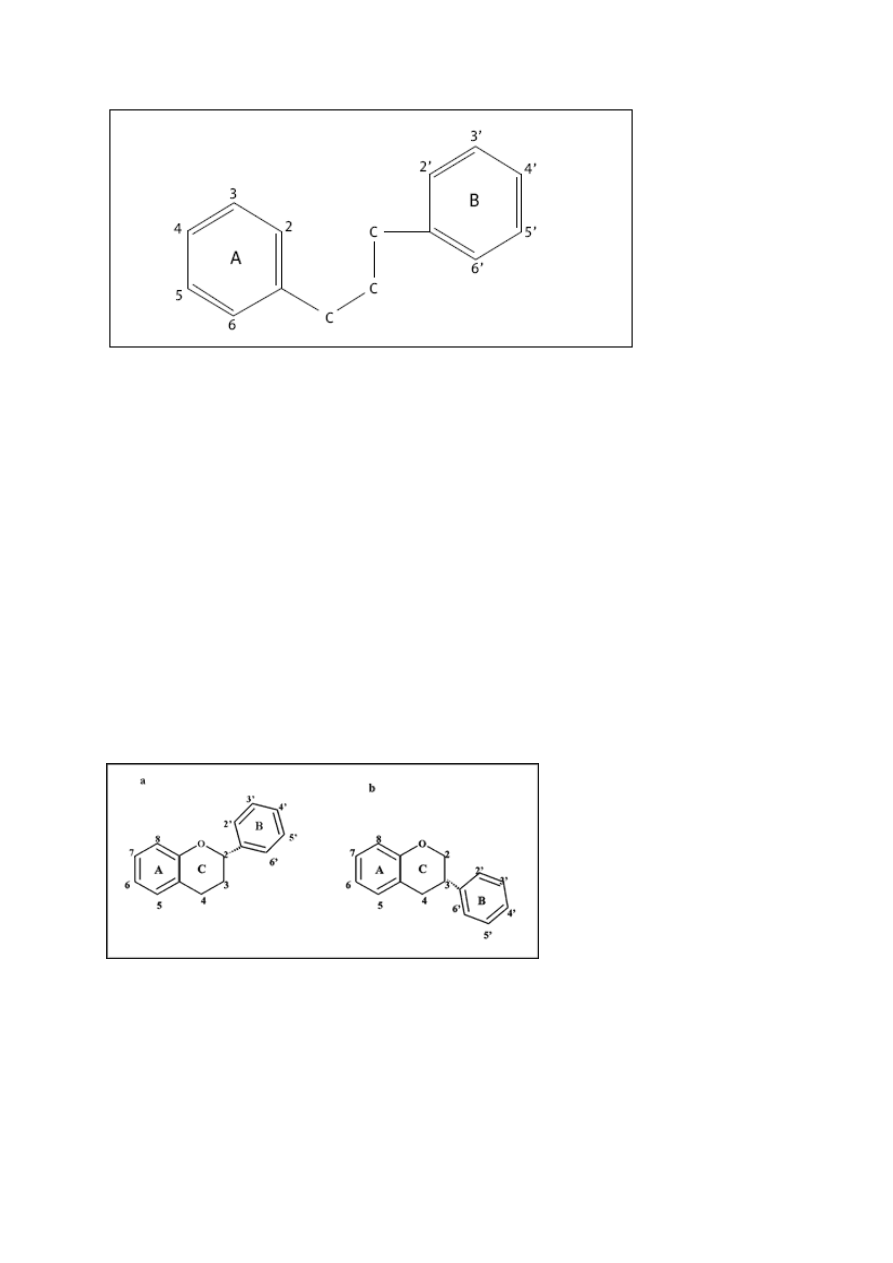
19
Rys.6 Struktura 2-fenylo-benzo-
γ
-pironu [24].
Biosynteza pierścieni A i B zachodzi w dwóch szlakach. Pierścień A powstaje na drodze
szlaku octanowego z 3 cząsteczek malonylo-CoA powstających z przemian glukozy.
Pierścień B powstaje na drodze szlaku szikimowego z 4 kumaroilo-CoA, które powstają
z fenyloalaniny w szlaku szikimowym. Kondensacja pierścieni A i B prowadzi do powstania
chalkonu, który ulega cyklizacji z udziałem izomerazy w wyniku czego powstaje flawanon -
wyjściowy związek do syntezy pozostałych grup flawonoidów [24]. W cząsteczkach
większości naturalnych flawonoidów pierścień A zawiera dwie grupy hydroksylowe
w pozycji 5 i 7, a pierścień B jedną w pozycji 3, która zwana jest grupą katecholową.
W związku z tym flawonoidy należą do polifenoli. Flawonoidy mogą występować w dwóch
postaciach izomerycznych – flawonoidowej i izoflawonoidowej [21].
Rys.7 Podstawowe struktury cząsteczek flawonoidów: a – postać flawonoidowa, b – postać
izoflawonoidowa [21].
W cząsteczce flawonoidów przy atomie węgla w pozycji 2 pierścienia heterocyklicznego,
znajduje się zwykle grupa hydroksylowa lub podstawnik fenylowy, w izoflawonoidach są one
usytuowane przy atomie węgla w pozycji 3. W zależności od występowania lub braku grupy
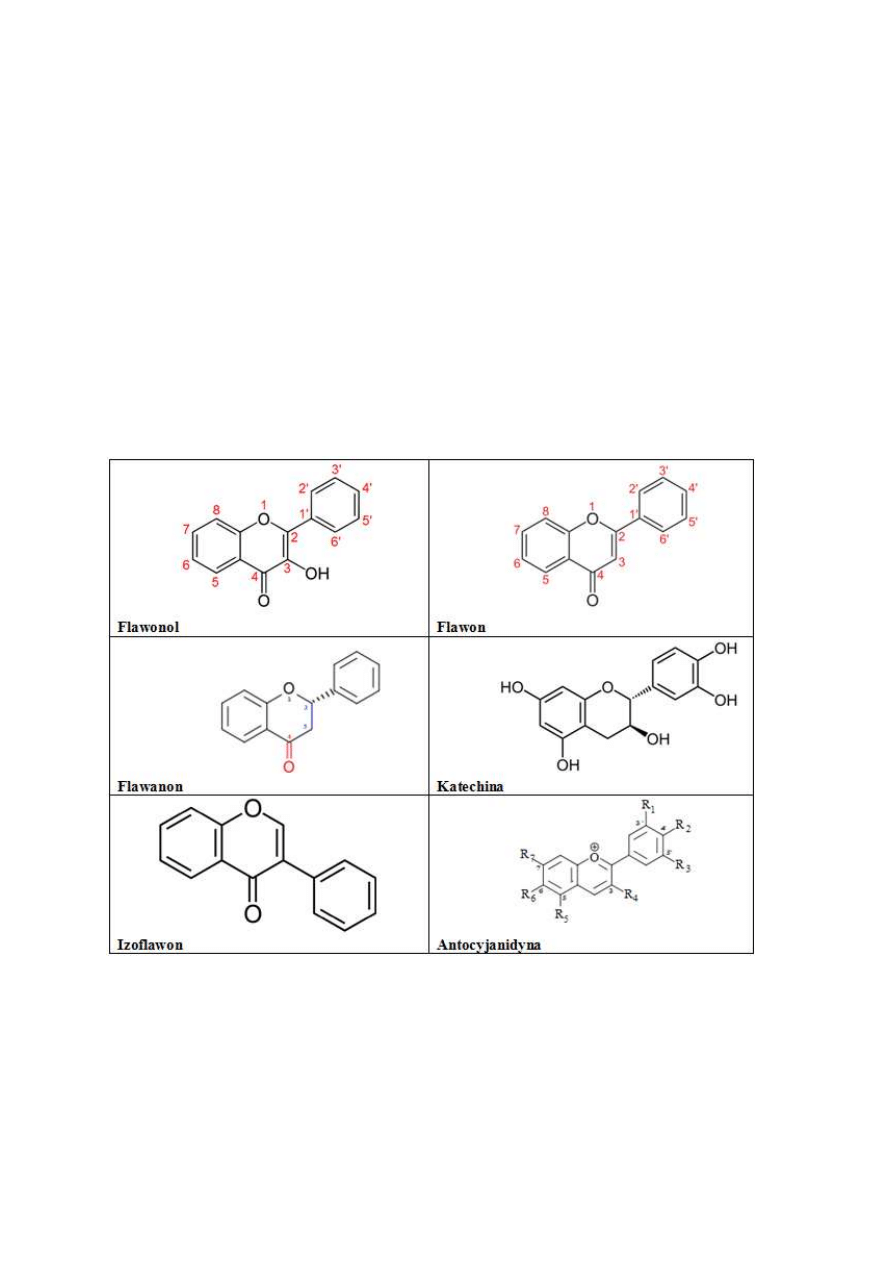
20
karbonylowej w pozycji 4 pierścienia C, występowania lub braku wiązania podwójnego
pomiędzy atomami węgla w pozycji 2 i 3 pierścienia C oraz liczby grup hydroksylowych,
wśród flawonoidów można wyróżnić:
−
flawanony np. naryngenina, naryngina, hesperetyna, hesperydyna
−
flawanole/ katechiny np. epikatechina, epigallokatechina, katechina
−
flawony np. apigenina, diosmetyna, luteolina
−
chalkony np. floretyna, aflorydzyna
−
flawonole np. kwercetyna, kemferol, mirecytyna, fisteina, morina
−
antocyjanidyny np. cyjanidyna, pelargonidyna, malwidin
−
izoflawony np. daizeina, genisteina i glicyteina [19,21,24].
Rys.8 Struktura wybranych flawonoidów
W roślinach flawonoidy występują w dwóch formach: wolnej - aglikonów lub w postaci
połączenia aglikonu z częścią cukrową
β
-glikozydów. Część cukrową stanowi zazwyczaj 1-5
cząsteczek cukrów prostych takich jak np. β-D-glukoza, β-L-ramnoza, β-D-galaktoza. Cukry
przyłączane są zazwyczaj w pozycji C-3, rzadziej C-4’, C-3’, C-5 czy C-7. W formie

21
glikozydów najczęściej występują flawonole oraz flawony i w takiej formie są spożywane
przez człowieka. Pośród form glikozydowych wyróżnia się formy O-glikozydowe np. rutyna
– 3-O-(6’’-ramnozylo)-glukozyd kwercetyny oraz rzadziej występujące formy C-glikozydowe
np. witeksyna – 8-C-glukozyd apigeniny. Spotykane są również glikozydoestry
flawonoidowe, flawonolignany np. sylibina, prenyloflawonoidy, a także pochodne
biflawonoidowe np. ginkgetyna. Przyłączenie do formy aglikonowej cukru zwiększa
polarność związków flawonoidowych. Flawonoidy mogą również łączyć się między sobą,
tworząc cząsteczki biflawonoidów, a także oligomery np. procyjanidyny, bądź duże,
nieulegające hydrolizie polimeryczne cząsteczki, w których jednostką podstawową są
flawanole połączone wiązaniami C-C np. taniny [19,21,24].
3.2.
Aktywność biologiczna
Flawonoidy to interesująca grupa związków naturalnych, ze względu na szeroki zakres
działania jaki posiadają. Obecność w ich strukturze chemicznej różnych grup i ugrupowań
sprawia, że wykazują one szeroki zakres aktywności biologicznej. Dzięki takiej budowie
mogą w różny sposób oddziaływać na metabolizm komórkowy. Głównym powodem dla
którego stwierdzono pozytywny wpływ flawonoidów na organizm człowieka jest ich
aktywność
antyoksydacyjna,
czyli
zdolność
do
neutralizacji
wolnych
rodników [19,20,21,22,23,24].
3.2.1.
Działanie antyoksydacyjne
Aktywność antyoksydacyjna flawonoidów wynika z ich budowy pierścieniowej oraz
obecności w ich strukturze grup hydroksylowych, szczególnie w pozycji C-3, C-5, C-7, C-3’,
wiązania podwójnego w pozycji C-2 i C-3 oraz grupy karbonylowej w pozycji C-4.
Aktywność antyoksydacyjna poszczególnych flawonoidów wynika z ilości oraz położenia
grup hydroksylowych. Większa ilość grup hydroksylowych, to silniejsze działanie
antyoksydacyjne, również położenie tych grup w pozycji orto i para wzmaga to działanie.
Badania in vitro wykazały, że ze względu na zablokowanie przez resztę cukrową grup
hydroksylowych, glikozydy wykazują słabsze właściwości antyoksydacyjne w porównaniu do
odpowiednich im aglikonów. Ponadto różnice we właściwościach antyoksydacyjnych zależą
również od rodzaju reszty sacharydowej obecnej w cząsteczce. Glikozydacja grupy
hydroksylowej przy węglu w pozycji 3 cząsteczką monosacharydu nie obniża zdolności

22
antyoksydacyjnych flawonoidów, podczas gdy aktywność antyoksydacyjna jest istotnie niższa
w przypadku glikozydacji resztą disacharydową. Jednak badania przeprowadzone
w warunkach in vivo nie potwierdzają zależności pomiędzy obecnością grupy metoksylowej
lub
reszty
węglowodanowej
w
cząsteczkach
flawonoidów,
a
ich
działaniem
antyoksydacyjnym w organizmie zwierząt. Może to być spowodowane tym, że w jelitach
obecne są bakterie zawierające glikozydazy, enzymy katalizujące reakcje hydrolizy reszt
cukrowych z glikozydów flawonoidów. Powstałe w wyniku działania glikozydaz aglikony
ulegają następnie metabolizmowi w sposób właściwy dla flawonoidów [21,22,23,24].
Ugrupowanie aktywne
Działanie antyoksydacyjne
Grupa
(o-dihydroksylowa)
katecholowa w pierścieniu B
wykazuje dużą zdolność wychwytywania rodników
tlenowych, jednak nie przyczynia się przez swe reakcje do
ochrony przed peroksydacją lipidów w mitochondriach
mózgu szczura
Ugrupowanie
pirogalolowe
(trihydroksylowe) w pierścieniu
B
nadaje cząsteczce wyższą aktywność antyoksydacyjną.
Podwójne wiązanie pomiędzy węglem C2 i C3
w pierścieniu C przyczynia się do wzrostu zdolności
wychwytywania
rodników,
ponieważ
po
reakcji
z rodnikiem powstaje stabilny rodnik fenoksylowy
Ugrupowanie
4-okso
(grupa
ketonowa, podwójne wiązanie
pomiędzy
węglem
C4
pierścienia C i atomem tlenu)
szczególnie w obecności podwójnego wiązania pomiędzy
C2 i C3, wzrasta zdolność wychwytywania rodników
dzięki zdelokalizowanym elektronom pierścienia B
Grupa hydroksylowa przy węglu
C3 pierścienia C
wykazuje szczególnie silne zdolności wychwytywania
rodników spotęgowane obecnością podwójnego wiązania
pomiędzy węglem C2 i C3 oraz grupowania 4-okso (jest to
najbardziej korzystne połączenie dla nadania cząsteczce
zdolności wychwytywania rodników)
Grupy hydroksylowe przy węglu
C5 i C7
mogą także zwiększać zdolności do wychwytywania
wolnych rodników w wielu reakcjach wolnorodnikowych.
Tabela.1 Ugrupowania obecne we flawonoidach, które nadają im największą aktywność
antyoksydacyjną [21].

23
Flawonoidy mogą działać z wolnymi rodnikami poprzez reakcje bezpośrednie i pośrednie.
Bezpośrednie działanie antyoksydacyjne flawonoidów polega na zmniejszeniu reaktywności
tlenu do bardziej stabilnych, niereaktywnych form. Mechanizmy bezpośrednie polegają na:
−
Wychwytywaniu/wymiataniu wolnych rodników i ich reaktywnych form (RFT)
−
Ograniczeniu wytwarzania wolnych rodników poprzez hamowanie aktywności
enzymów biorących udział w powstawaniu RFT (oksydazy ksantynowej, błonowej
oksydazy NAD(P)H, mieloperoksydazy)
Działanie pośrednie prowadzi do zahamowania powstawania reaktywnych form tlenu, polega
ono na:
−
Chelatowaniu jonów metali przejściowych (miedzi i żelaza), co zapobiega
powstawaniu w komórkach reaktywnego rodnika hydroksylowego
−
przerywaniu kaskady reakcji wolnorodnikowych w enzymatycznej i nieenzymatycznej
peroksydacji lipidów;
−
ochronie niskocząsteczkowych antyoksydantów (np. askorbinianu w cytosolu,
α-tokoferolu w błonach biologicznych) przed utlenianiem
Zdolność do wychwytywania reaktywnych form tlenu i chelatowania jonów metali
przejściowych może mieć istotne znacznie w stanach patologicznych takich jak miażdżyca,
cukrzyca, choroby neurodegeneracyjne, nowotwory, którym towarzyszy stres oksydacyjny.
Flawonoidy takie jak kwercetyna czy rutyna działają ochronnie na witaminę C i E.
Chelatowanie jonów metali przejściowych powoduje hamowanie utleniania askorbinianu,
stabilizuje jego cząsteczkę, a także zwiększa jego wchłanianie z przewodu pokarmowego.
Lokalizacja związków flawonoidowych w pobliżu błon komórkowych powoduje zwiększenie
ich stabilności poprzez zwiększenie ich odporności na czynniki utleniające. Flawonoidy
przyczyniają się również do obniżenia aktywności enzymów takich jak fosfolipaza A2,
cyklooksygenaza, lipooksygenaza, które biorą udział w enzymatycznej peroksydacji
błonowych fosfolipidów [21,24].
3.2.2.
Działanie prooksydacyjne
Niektóre związki flawonoidowe w zależności od warunków tlenowych panujących
w komórce mogą wykazywać działanie prooksydacyjne. Dotyczy to flawonoidów, które mają
ugrupowanie pirogalolowe – trzy grupy OH w pierścieniu B lub katecholowe – grupa OH
w pozycji 3’ w pierścieniu B, gdyż one w obecności tlenu i jonów miedzi Cu
2+
ulegają
autooksydacji. W wyniku tego powstaje utleniona forma flawonoidu w formie rodnika
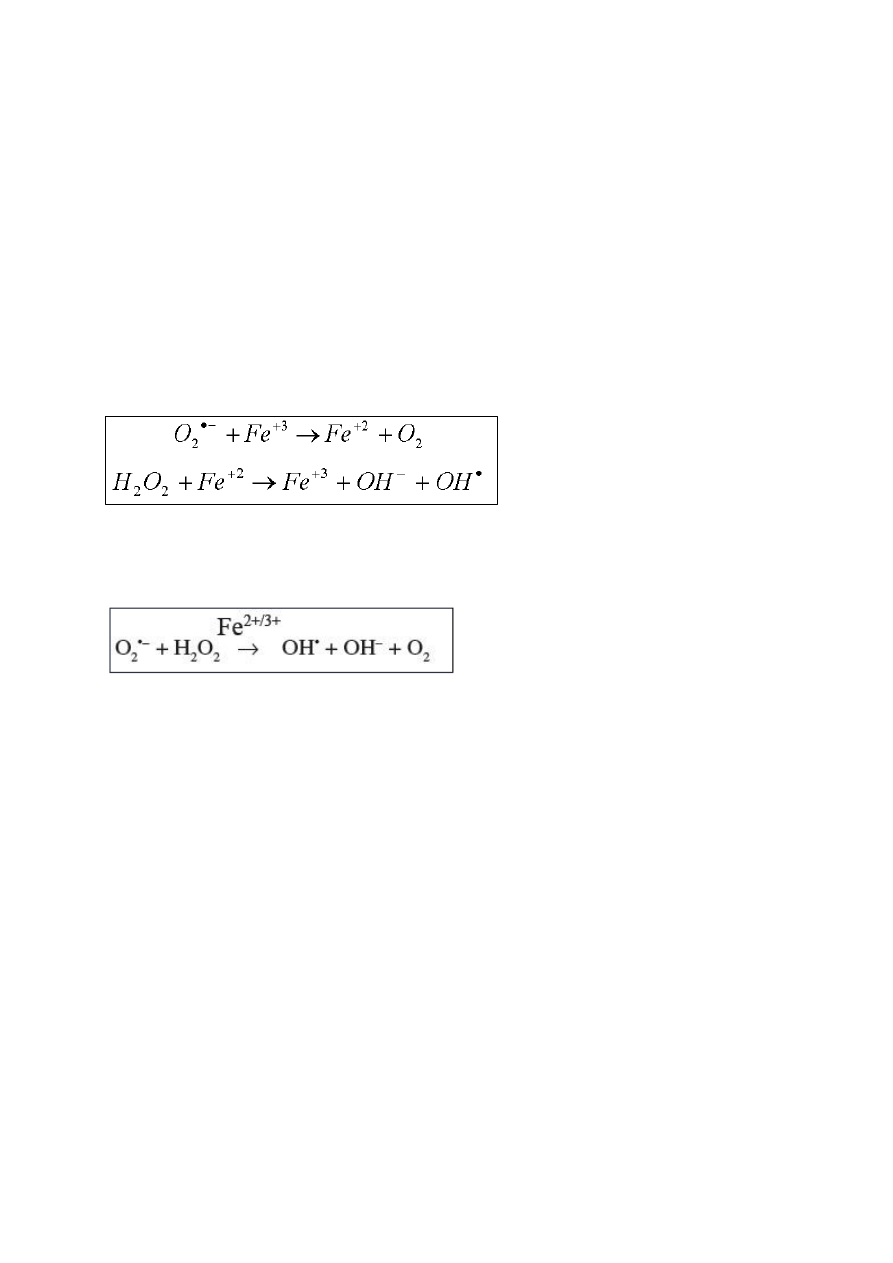
24
semichinonowego i jony miedzi Cu
+
. Przy udziale NADH rodnik semichinonowy jest
redukowany co prowadzi do powstania cyklu reakcji redoks i generowania RFT. Rodniki
semichinonowe powstające podczas utleniania flawonoidów pomimo, że charakteryzują się
stabilnością, mogą wykazywać działanie cytotoksyczne. Jony miedzi (I), powstałe na skutek
autooksydacji flawonoidów, reagują z tlenem wytwarzając rodnik ponadtlenkowy, z którego
może powstać nadtlenek wodoru a dalej w reakcji Habera Weissa czy Fentona może
powstawać reaktywny chemicznie rodnik hydroksylowy odpowiedzialny za oksydacyjne
modyfikacje DNA, białek czy lipidów[21,22,24, 25].
Reakcja Fentona
Reakcja Habera Weissa
[26]
3.2.3. Działanie przeciwzapalne i przeciwalergiczne
Mechanizm działania przeciwzapalnego związków flawonoidowych polega
głównie na hamowaniu aktywności 5-lipooksygenazy i cyklooksygenazy. Zahamowanie tych
enzymów prowadzi do inhibicji napływu leukcytów i wyrównania napięcia naczyń
włosowatych i zmniejszenia odczynu zapalnego. Podczas reakcji zapalenej dochodzi do
powstania wielu RFT, które uszkadzają ściany naczyń krwionośnych przez degradacje
kolagenu, dlatego przeciwzapalne i antyoksydacyjne działanie flawonoidów poprawia stan
naczyń krwionośnych [24].
Poza działaniem przeciwzapalnym niektóre flawonoidy wykazują działanie
przeciwalergiczne. Ich przeciwalergiczne działanie polega na oddziaływaniu z komórkami
układu odpornościowego. Polega ono na hamowaniu proliferacji limfocytów, zahamowaniu
syntezy immunoglobulin klasy E, G, M, A oraz na uwalnianiu cytokin. Flawonoidy mogą

25
również hamować aktywność enzymów lizosomalnych, biorących udział w procesach
zapalnych i alergicznych. Przykładami związków flawonoidowych o właściwościach
przeciwalergicznych są kwercetyna i luteolina, które oprócz obniżania syntezy mediatorów
zapalnych, hamują również uwalnianie histaminy z mastocytów pobudzonych wcześniej
immunoglobulin klasy E [23,24,27,28].
3.2.4. Działanie estrogenne
Flawonoidy należą do fitoestrogenów, czyli związków niesteroidowych pochodzenia
roślinnego o budowie podobnej do naturalnych estrogenów. Najbardziej znane i poddawane
różnego typu badaniom są flawonoidy grupy izoflawonów, np. genisteina czy daidzeina.
Podobieństwo ich budowy do estrogenów sprawia, że wykazują powinowactwo do
receptorów estrogenowch α (ER-α) oraz w dużo większym stopniu do receptorów β (ER-β).
ER-α występują głównie w gruczole sutkowym, endometrium czy w zrębie jajników,
natomiast ER-β w mózgu, naczyniach krwionośnych, nerkach, pęcherzu moczowym,
płucach, kościach oraz w jelitach. Dzięki temu łagodzą objawy menopauzy. W badaniach
epidemiologicznych wykazano, że uderzenia gorąca pojawiają się znacznie rzadziej w grupie
kobiet spożywających znaczne ilości soi zawierającej fitoestrogeny. Doświadczenia
laboratoryjne dowiodły, że podawanie zwierzętom jedynie izoflawonów pobudziło przerost
macicy, co wskazało na ich działanie estrogenne. Natomiast podawanie ich łącznie
z estrogenami wykazało działanie antyestrogenowe, np. poprzez hamowanie wychwytu
estradiolu przez macicę. Przeprowadzone dotychczas badania w większości skupiają się na
korzystnym działaniu izoflawonów w opóźnianiu menopauzy oraz łagodzeniu jej następstw,
np.
obniżaniu
zachorowalności
na
osteoporozę
kobiet
w
okresie
postmenopauzalnym [20,24,28].
3.3.
Flawonoidy w profilaktyce i terapii chorób
Fitozwiązki od bardzo dawna wykorzystywane są jako naturalne leki w terapii różnych
schorzeń. W ostatnich latach wzrasta zainteresowanie profilaktyką i leczeniem za ich pomocą.
Szeroki zakres farmakologicznego działania flawonoidów powoduje, że podejmowane są

26
również próby zastosowania tych związków jako terapii wspomagającej w wielu
chorobach [22,24].
3.3.1.
Aktywność przeciwnowotworowa
Badania prowadzone w latach 70. i 80. XX wieku dowiodły o aktywności
przeciwnowotworowej flawonoidów. Wykazano, że niektóre z nich obniżały aktywność
mutagenną wybranych promutagenów/prokancerogenów in vitro, a także zmniejszały
częstość występowania nowotworów u zwierząt doświadczalnych [29]. Obserwacje
epidemiologiczne wskazują na odwrotną korelacje pomiędzy spożyciem flawonoidów
w diecie a ryzykiem powstawania niektórych typów nowotworów u ludzi. Zaobserwowano,
że dieta bogata w izoflawony powoduje zmniejszenie ryzyka powstania u kobiet nowotworu
piersi, a u mężczyzn nowotworu prostaty. Ostatnie badania wskazują na możliwość
profilaktycznego działania izoflawonów w nowotworach tarczycy, głowy i szyi. Natomiast
ryzyko wystąpienia nowotworu płuc można zmniejszyć poprzez picie zielonej herbaty
zawierającej katechiny [24].
Działanie przeciwnowotworowe flawonoidów jest możliwe dzięki ich właściwościom
antyoksydacyjnym, a także dzięki: oddziaływaniu na aktywność enzymów I i II fazy
biotransformacji
endo- i egzogennych związków, blokowaniu replikacji DNA przez
hamowanie aktywności enzymów biorących udział w tym procesie (np. polimerazy II DNA,
topoizomerazy I i II). Kwercetyna i kempferol są inhibitorami polimerazy II DNA, luteolina
topoizomerazy
I,
natomiast
mirycetyna,
kwercetyna
czy
baikalina
topoizomerazy II [20,21,24].
Flawonoidy poprzez blokowanie cyklu komórkowego w fazie G1/S lub G2/M, mogą
hamować proliferację oraz indukować apoptozę komórek nowotworowych. Jest to możliwe,
gdyż związki te wykazują wpływ na aktywność:
1. białek odpowiedzialnych za regulację cyklu komórkowego (np. cykliny),
2. białek proapoptotycznych i antyapoptotycznych (np. p21, p53, Bcl-2),
3. enzymów odpowiedzialnych za biotransformację mutagenów i kancerogenów.
Flawonoidy
wykazują
zdolność
do
modulowania
aktywności
enzymów
odpowiedzialnych za metabolizm ksenobiotyków, których to aktywność biologiczna zmienia
się pod wpływem ich działania. Flawonoidy mogą zarówno aktywować, jak i hamować
aktywność różnych izoform cytochromu P-450, czyli enzymów I fazy biotransformacji.
Odpowiedzialne są także za pobudzenie aktywności enzymów II fazy. Zaobserwowano,

27
że działanie niektórych flawonoidów, np. tangretyny czy chryzyny podwyższa aktywność
transferazy
glutationowej
czy
UDP-glukuronowej,
czyli
enzymów
II
fazy
biotransformacji [24,30].
Poza bezpośrednim działaniem na enzymy biorące udział w nowotworzeniu,
flawonoidy są zdolne do modyfikowanie metabolizmu komórkowego poprzez:
1.
obniżenie aktywności czynników transkrypcyjnych AP-1 i NF-κB, które kontrolują wiele
genów regulujących proliferację, apoptozę czy angiogenezę. Hamujące działanie
flawonoidów na te czynniki wynika z ich właściwości przeciwutleniających, a także
zdolności do:
1.1.
hamowania aktywności kinaz, które aktywują czynnik NF-κB poprzez jego
fosforylację i odłączenie od inhibitora (IκB)
1.2.
hamowania aktywności MAP kinaz, które odpowiedzialne są za aktywację czynnika
AP-1;
2.
obniżenie aktywności kinazy C (PKC), która katalizuje fosforylację seryny, treoniny czy
też kinaz tyrozynowych (PTK) uczestniczących w powstawaniu stanów zapalnych
i zmian nowotworowych;
3.
hamowanie glikoproteiny P, która w zdrowych komórkach usuwa szkodliwe substancje,
natomiast podczas chemioterapii usuwa cytostatyki [29].
Zgromadzone dotychczas dane dotyczące przeciwnowotworowej aktywności
flawonoidów nie są jednoznaczne. Mechanizm ich przeciwnowotworowego działania
poznano przede wszystkim na układach doświadczalnych w warunkach in vitro i tylko dla
kilku flawonoidów takich jak genisteina i daidzeina, w mniejszym stopniu dla kwercetyny,
czy luteoliny. Doświadczenia pokazały, że genisteina i daidzeina dostarczone w diecie są
zdolne do hamowania wzrostu i podziału komórek zależnego od receptorów EGF oraz do
hamowania angiogenezy. Hamowanie kinaz tyrozynowych zaburza przekazywanie sygnału
pomiędzy komórkami, co prowadzi do zaburzenia wzrostu i podziału komórek – jest to
istotne w ograniczeniu namnażania komórek nowotworowych. Izoflawony mogą także
hamować syntezę aromatazy i jednocześnie pobudzać syntezę globuliny wiążącej hormony
płciowe (SHBG), co prowadzi do zahamowania wytwarzania endogennych estrogenów
i androgenów,
a
tym
samym
do
zahamowania
wzrostu
nowotworów
hormonozależnych [21,24].
Ciekawych wyników dostarczyły badania dotyczące zastosowania flawonoidów
w chemioterapii nowotworów. Stwierdzono, że w opornych na działanie chemioterapeutyków

28
liniach komórkowych flawonoidy mogą zwiększać stężenie niektórych z zastosowanych
cytostatyków [24].
3.3.2.
Flawonoidy w chorobach sercowo-naczyniowych
W licznych badaniach epidemiologicznych wykazano odwrotny związek pomiędzy
ilością spożywanych w diecie flawonoidów a zapadalności na choroby układu sercowo-
naczyniowego. Fakt ten potwierdza zjawisko tzw. paradoksu francuskiego. Osoby
mieszkające w rejonie Morza Śródziemnego pomimo diety bogatej w tłuszcze znacznie
rzadziej zapadają na miażdżycę tylko dlatego, że równocześnie dostarczają wraz
z warzywami i owocami związki polifenolowe, w tym flawonoidy, a także spożywają
czerwone wino, które obfituje w rezweratrol i katechiny. Działalność antyoksydacyjna tych
związków zapobiega peroksydacji lipidów błon komórkowych, ochrania lipoproteiny o małej
gęstości (LDL) przed utlenianiem, a także prowadzi do zwiększenia stężenia korzystnego
cholesterolu (HDL) [20,21,22,24].
Flawonoidy mają zdolność do uszczelniania naczyń krwionośnych. Wraz z witaminą
C biorą udział w tworzeniu wiązań poprzecznych pomiędzy łańcuchami polipeptydowymi
włókien kolagenu. W ten sposób uelastyczniają i wzmacniają m.in. naczynia krwionośne.
Uszczelnianie naczyń wynika także ze zdolności do hamowania hialuronidazy (niszczy
połączenia międzykomórkowe) i oksydazy lizosomowej (niszczy wiązania krzyżowe
kolagenu). Hamowanie tych enzymów zmniejsza łamliwość i przepuszczalność naczyń
krwionośnych. Dzięki ich uszczelnieniu LDL nie przechodzą do ścian tętnic i nie tworzy się
blaszka miażdżycowa [20,23,24].
Niektóre z tych związków mają zdolność obniżania ciśnienia tętniczego krwi. Wynika
to z hamowania aktywności fosfodiesterazy cAMP i cGMP oraz aktywności kinazy
proteinowej (np. kwercetyna powoduje relaksacje mięśni gładkich i hamuje napływ jonów do
wnętrza komórek, natomiast wewnątrz komórek zapobiega przenikaniu wapnia do retikulum
sarkoplazmatycznego). Flawonoidy powstrzymują także procesy zapalne, a także proliferację
miocytów w ścianach tętnic co jest uznawane za czynniki rozwoju miażdżycy [23,24].
Rutyna jak i inne flawonoidy wpływa dodatnio na pracę mięśnia sercowego - skurcze
serca stają się wydatniejsze i zwiększa się pojemność minutowa. Flawonoidy dodatkowo
działają korzystnie w przypadku zatrucia substancjami chemicznymi np. alkoholem
metylowym czy też nadmiernym nagromadzeniem kwasu mlekowego [22,23].

29
Miażdżyca jest to choroba wieloczynnikowa o złożonej patogenezie. Jednym
z czynników inicjującym powstawanie miażdżycy jest zaburzenie funkcji śródbłonka naczyń
krwionośnych. Tlenek azotu jest jednym z głównych mediatorów tego procesu. W warunkach
fizjologicznych, w naczyniach krwionośnych, ma on działanie antyoksydacyjne
przeciwzapalne, a także działa rozkurczowo. W warunkach stresu oksydacyjnego, który
towarzyszy stanom zapalnym, jest prekursorem silnych związków prooksydacyjnych.
Flawonoidy mogą zmniejszać odczyn zapalny w procesach miażdżycowych poprzez
wymiatanie reaktywnych form tlenu (RFT) i tlenku azotu (NO) wraz z jego pochodnymi,
a także poprzez hamowanie napływu leukocytów do miejsca zapalenia, które wchodzą
w skład blaszki miażdżycowej [24]. Rozwojowi miażdżycy sprzyja także występowanie we
krwi dużych ilości utlenowanych lipoprotein o niskiej gęstości (LDL), które odkładają się
w ścianach naczyń krwionośnych. Flawonoidy pełnią funkcję ochronną w stosunku do
lipoprotein osocza krwi. Ich właściwości chelatujące zmniejszają występowanie RFT w
osoczu co zapobiega utlenianiu frakcji LDL, a w konsekwencji zapobiega powstawaniu
blaszek miażdżycowych. Mechanizmy przeciwmiażdżycowego działania flawonoidów
polegają również na:
−
hamowaniu reduktazy HMG-CoA (np. hesperytyna), co prowadzi do
obniżenia poziomu cholesterolu we krwi;
−
obniżeniu przez np. kwercetynę czy luteolinę zdolności monocytów do
adhezji do nabłonka naczyń i przenikania przez ściany naczyń;
−
hamowaniu przez np. kwercetynę i baikaleinę proliferacji mięśni gładkich
naczyń;
−
hamowaniu
przez
mirycetynę
czy
kwercetynę
agregacji
trombocytów [24,31,37].
Flawonoidy wykazują korzystny wpływ na czynność płytek krwi. Oddziaływują
z integrynami płytek utrudniając tym samym ich zlepianie. Istnieją dane potwierdzające,
że aktywność antyagregacyjna flawonoidów jest związana z metabolizmem NO. Stymulują
one powstawanie NO w śródbłonku naczyniowym i równocześnie hamują syntezę 12-HETE
(związek upośledzający czynność śródbłonka). Przyczyniają się przy tym do zahamowania
syntezy tromboksanu A i aktywności fosfolipazy C. Działanie antyagregacyjne uzasadnia się
również zdolnością flawonoidów (np. kwercetyny, rutyny, trokserutyny) do hamowania
aktywności takich enzymów, jak fosfodiesteraza i cyklooksygenaza [24,32,37].

30
3.3.3.
Flawonoidy w leczeniu cukrzycy
Cukrzyca jest chorobą, której powstawanie wiąże się z upośledzeniem produkcji
i wydzielania insuliny lub niewrażliwością komórek docelowych na ten hormon, co prowadzi
do zaburzenia poziomu glukozy we krwi. Badania doświadczalne wykazały, że niektóre
flawonoidy wykazują właściwości przeciwcukrzycowe.
Badania przeprowadzone in vivo i in vitro wykazały, ze epikatechina może
stymulować syntezę insuliny i podwyższać poziom cAMP w komórkach
β
trzustki, co
zwiększa wydzielanie tego hormonu. Ponadto przekształcanie proinsuliny w insulinę jest
intensywniejsze i poziom insuliny we krwi się zwiększa. Działanie hipoglikemiczne natomiast
wykazuje 3-galusan epigalokatechiny, który hamuje syntezę glukozy w hepatocytach.
Daidzeina, luteolina i 7-O-glukozyd luteoliny, hamują aktywność enzymów α-amylazy
i α-glukozydazy, a glikozydy kwercetyny osłabiają działanie transporterów glukozy, dzięki
czemu mogą spowolnić wchłanianie glukozy w jelicie, co zapobiega to gwałtownemu
zwiększeniu ilości glukozy we krwi po posiłku. Wykazano również, że flawonoidy (głównie
kwercetyna) chronią cukrzyków przed pojawieniem się zaćmy. Główną przyczyną tego
objawu jest odkładanie się w gałce ocznej sorbitolu, którego synteza jest katalizowana przez
reduktazę aldozolową. Kwercetyna jest inhibitorem tego enzymu przez co może opóźniać
utratę wzroku [24,33].
3.3.4. Flawonoidy w terapii AIDS
Flawonoidy wydają się być potencjalnym czynnikiem terapeutycznym w terapii
chorych na AIDS. Wykorzystanie tych związków jest możliwe dzięki temu, że są one
w stanie ograniczyć namnażanie wirusa, co jest najważniejszym czynnikiem w terapii tej
choroby. Hamowanie proliferacji wirusa HIV jest możliwe dzięki takim właściwościom
flawonoidów:
−
(-)-epikatechina, baikalina, baikaleina, kwercetyna i mirycetyna mogą działać jak
inhibitory odwrotnej transkryptazy – kluczowego enzymu koniecznego do rozwoju
HIV;
−
(-)-epikatechina, EGCG i baikalina mogą hamować wnikanie cząsteczki wirusa do
wnętrza komórki poprzez zaburzenie interakcji białek otoczki wirusa z cząsteczkami
powierzchniowymi atakowanych komórek;

31
−
kwercetyna może hamować aktywność wirusowego białka Vpr, odpowiedzialnego za
zwiększenie wydajności namnażania wirusa w komórkach gospodarza oraz takich
enzymów, jak integrafy oraz proteinazy. [23,24]
3.3.5.
Wpływ na ośrodkowy i obwodowy układ nerwowy
Działanie związków fenolowych na ośrodkowy i obwodowy układ nerwowy może
wynikać z ich powinowactwa do receptorów benzodiazepinowych GABA i aktywowania
tych receptorów.
Ostatnio prowadzone badania wskazują, że galusan epikatechiny w znacznym stopniu
zmniejsza uszkodzenia OUN w przebiegu niedokrwienia dzięki właściwościom
antyoksydacyjnym i przez modulowanie jednego z istotnych dla stanu zapalnego enzymów –
syntetazy tlenku azotu (NOS) [23].
3.4.
Kwercetyna
Kwercetyna jest flawonoidem szeroko rozpowszechnionym w przyrodzie. Nazwa
pochodzi z łac. quercetum – las dębowy i po raz pierwszy została użyta w roku 1857 roku.
Zaliczana jest do grupy flawonoli, najczęściej występuje w postaci glikozydu, a zwłaszcza
rutozydu, jednakże często występuje również w postaci wolnej, czyli aglikonu. Kwercetyna
w materiale roślinnym występuje w ponad 140 różnych strukturalnie pochodnych. Występuje
między innymi w jabłkach, cebuli (zwłaszcza czerwonej), herbacie. Jest to naturalnie
występujący inhibitor polarnego transportu auksyny. Jest ona również budulcem dla innych
flawonoidów [34].
3.4.1.
Budowa i właściwości
Kwercetyna, czyli 3,5,7,3´,4´-pentahydroksyflawon jest jednym z najbardziej
rozpowszechnionych związków flawonoidowych. Jej wzór strukturalny to C
15
H
10
O
7.
Składa
się z trzech pierścieni i pięciu grup hydroksylowych. Otrzymana z surowca roślinnego
w postaci krystalicznej jest praktycznie nierozpuszczalna w wodzie, a ponadto ma dobrą
wchłanialność z przewodu pokarmowego i posiada charakter lipofilny. Dobrze rozpuszcza się
w DMSO, alkaliach i pirydynie, a jej stała dysocjacji dla grupy OH w pozycji C-3 wynosi 5,
dla pozostałych grup OH przy pierścieniach A i B wynosi 10. Jak większość związków z tej
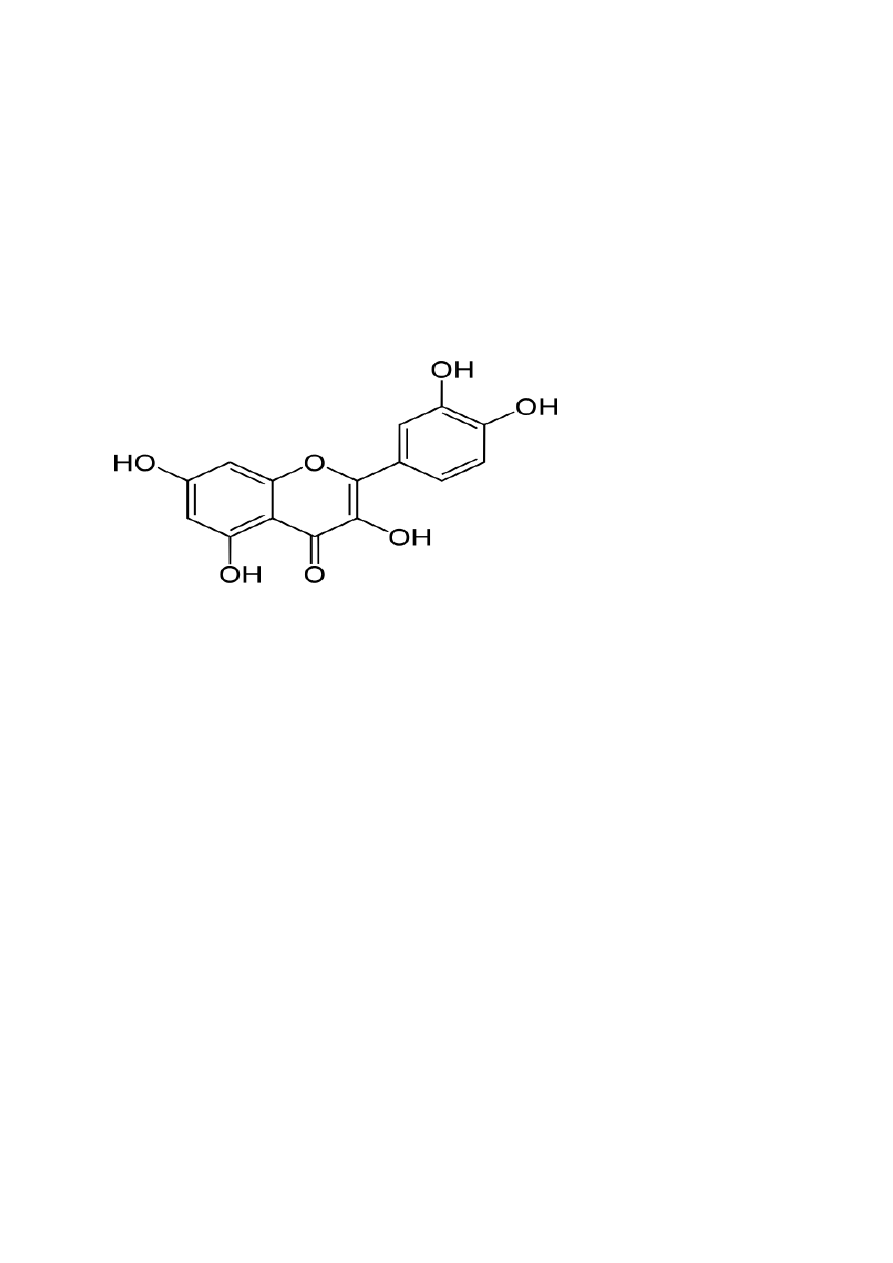
32
grupy posiada ona właściwości antyoksydacyjne, zaliczana jest do grupy tzw. „wymiataczy
wolnych rodników”, w związku z tym ma ona zastosowanie w spowalnianiu procesów
starzenia jak i w samej geriatrii zwłaszcza z witaminą C. Stwierdzono, że poprzez hamowanie
enzymu hialuronidazy zmniejsza ona przepuszczalność naczyń krwionośnych. Ponadto,
dzięki łagodzeniu skutków napromieniowania, może mieć zastosowanie w łagodzeniu
skutków ubocznych radioterapii nowotworów [35].
Rys.9 Struktura chemiczna kwercetyny.
3.4.1.1.
Właściwości antyoksydacyjne
Kwercetyna jako przeciwutleniacz chroni komórki, błony komórkowe oraz DNA
przed uszkodzeniami spowodowanymi przez wolne rodniki. Odgrywa to ważną rolę
w profilaktyce wielu chorób, w tym raka. Przyczynia się również do spowalniania procesów
starzenia się organizmu. Poprzez ochronę lipoprotein transportujących tłuszcze we krwi
przyczynia się do hamowania rozwoju miażdżycy i choroby sercowo-naczyniowej. Chroni
komórki oczu przed szkodliwym promieniowaniem UV i przed innymi czynnikami
oksydacyjnymi.
Swoje właściwości antyoksydacyjne kwercetyna zawdzięcza dzięki obecności
grup hydroksylowych w pierścieniu B. Wykazane zostały one dzięki badaniom
porównawczym nad właściwościami antyoksydacyjnymi pochodnych C(3)-OH i C(4’)-OH
kwercetyny. W reakcji z DPPH kwercetyna jest donorem dwóch atomów wodoru i zostaje
przekształcona w formę pośrednią reakcji, czyli chinon. Obecność grupy hydroksylowej przy
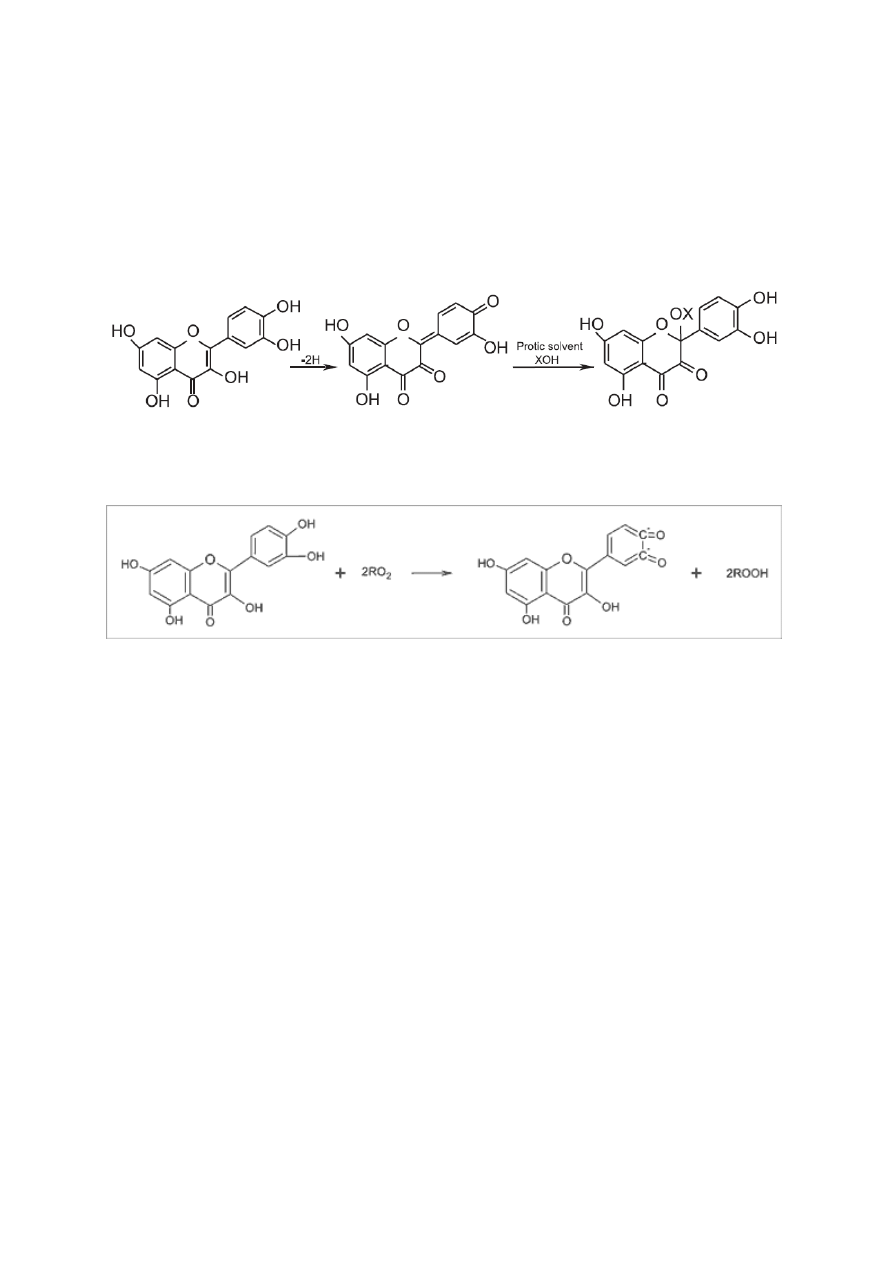
33
węglu C-3 kwercetyny pozwala na jej regenerację poprzez reakcję z protonami zawartymi
w roztworze. W przypadku pochodnych kwercetyny, glikozylacja węgla C(4’)-OH zmniejsza
jej zdolność do bycia donorem atomów wodoru, podczas gdy pochodne C(3)-OH wykazują
obniżenie potencjału w porównaniu z jej formą aglikonu [38].
Rys.10 Zmiany oksydacyjne kwercetyny w reakcji z rodnikami DPPH [38].
Rys.11 Schemat blokowania rodników nadtlenowych przez kwercetynę [39].
3.4.1.2. Działanie przeciwnowotworowe
Kwercetyna posiada właściwości regulujące niektóre mechanizmy prowadzące do
powstania nowotworu. Może ona miedzy innymi hamować niekontrolowane podziały
komórkowe, obniżać ekspresje niektórych onkogenów, czy indukować procesy prowadzące
do śmierci komórek nowotworowych [40].
W badaniach nad zastosowaniem flawonoidów w chemioterapii kwercetyna
zwiększała in vitro w komórkach raka piersi stężenie doksorubicyny, a genisteina cisplatyny.
Natomiast in vivo kwercetyna podwyższała przeciwnowotworowe działanie cisplatyny
i busulfanu, ale nie wpływała na aktywność doksorubicyny i etopozydu [24].
Na podstawie dotychczasowych obserwacji in vitro i in vivo stwierdza się, że stężenie
kwercetyny w przedziale 1-40 mM w komórkach sprzyja procesom antyoksydacyjnym.
Natomiast gdy, jej stężenie przekracza 40 mM działanie zmienia się na prooksydacyjne.
W zakresie niskich stężeń kwercetyna przeciwdziała oksydacyjnym uszkodzeniom materiału

34
genetycznego, nie dopuszczając tym samym do zainicjowania procesu kancerogenezy,
natomiast w dużych dawkach, stymulując procesy utleniania, prowadzi w komórkach
nowotworowych do oksydacyjnych efektów cytotoksycznych prowadząc ostatecznie do ich
unicestwienia na drodze apoptozy lub nekrozy. Dodatkowo, odkryto mechanizm pozwalający
selektywnie niszczyć komórki nowotworowe, również za sprawą efektów prooksydacyjnych.
W tym przypadku cząsteczka kwercetyny tworzy kompleks trójskładnikowy z DNA i jonem
metalu, miedzią lub żelazem. Kompleks ten, za sprawą cyklu oksydacyjno-redukcyjnego
metalu jest źródłem RFT, w tym rodnika hydroksylowego, co prowadzi ostatecznie do śmierci
komórki nowotworowej [41,42].
3.4.1.3. Działanie przeciwzapalne
Kwercetyna skutecznie hamuje kluczowe etapy w kaskadzie mechanizmów
zapalnych. Znacząco hamuje również aktywność enzymów i pośredników stanów zapalnych,
przez co ma pozytywny wpływ na przebieg wszystkich procesów zapalnych. Inhibicja COX-2
przez kwercetynę, powoduje zmniejszenie syntezy między innymi prostaglandyny PGE
2
,
leukotrienu B
4
i trombosanu A co prowadzi do zahamowania napływu leukcytów
i wyrównania napięcia naczyń włosowatych i zmniejszenia odczynu zapalnego. Wraz
z witaminą E uzupełniają się w modulowaniu procesów zapalnych np. przy artrozie, ponadto
może ona blokować replikację wirusów w komórce [24].
35
4
Albumina surowicy krwi
Albumina surowicy krwi ludzkiej (ang. human serum albumin, HSA) jest białkiem
najobficiej występującym w układzie krążenia i odgrywa ważną rolę w transporcie kwasów
tłuszczowych, metabolitów, i leków [44]. Jako białko transportowe odgrywa ważną rolę
w wiązaniu wielu leków, co ma kluczowe znaczenie w rozprowadzaniu ich w
organizmie [45]. Brak tego białka znaczenie utrudniłby walkę z chorobą, w związku z tym, że
odpowiada ono za transport leków takich jak antybiotyki Albumina pozwala na związanie
substancji leczniczych i rozprowadzenie ich po organizmie. Stanowi ona ok. 60% wszystkich
białek zawartych w osoczu. Można ją również znaleźć w mleku i białku jaja kurzego.
Charakteryzuje się okresem półtrwania ok. 20 dni i dobrą rozpuszczalnością w wodzie [44].
4.1
Struktura białka
Struktura albuminy jest bardzo prosta, ale reprezentuje model struktury
czwartorzędowej. Nie posiada ona żadnych grup prostetycznych, glikanów, czy lipidów. Gen
albuminy ludzkiej ma długość 16,961 nukleotydów od domniemanego miejsca czapeczki do
miejsca przyłączenia ogona poli(A). Masa molowa białka wynosi 67000 Da i jest dosyć duża
jak na fakt, że białko zbudowane jest z 585 aminokwasów [64].
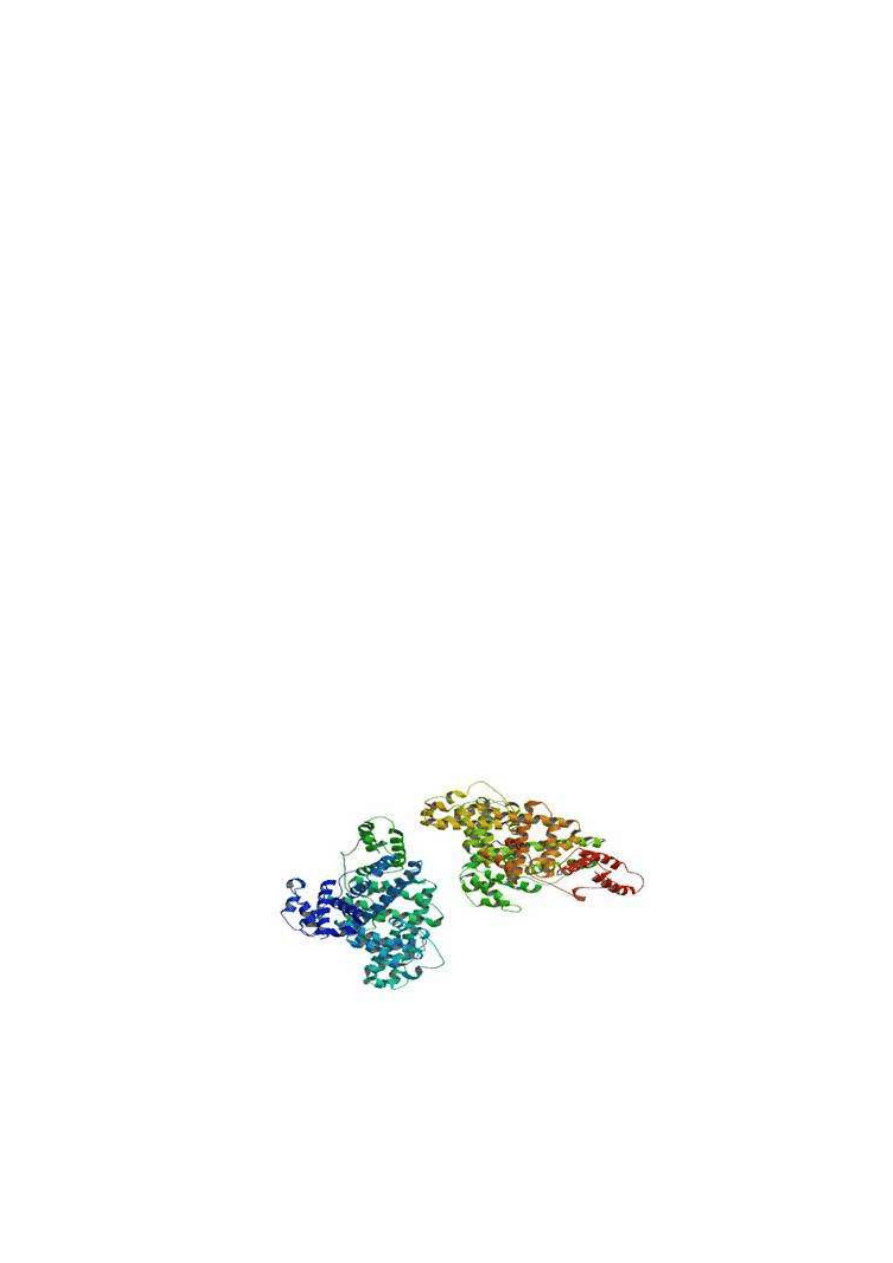
Rys.12 To jest skrystalizowana wersja albuminy ludzkiej surowicy. Reprezentowane są
łańcuchy A i B [Protein Data Bank].

36
Jak widać na powyższym rysunku albumina w około 67% składa się z α-helis. Struktura ta
pozwala na wiązanie cząsteczek o dużej różnorodności. Albumina składa się z kilku głównie
heliakalnych
domen,
które
połączone
są
ze
sobą
siedemnastoma
mostkami
disiarczkowymi [64].
4.2 Synteza
U ludzi synteza albuminy ma miejsce w wątrobie w ilości 12g do 20g dziennie. Po
zsyntetyzowaniu jest przenoszona do wnętrza naczyń jak i poza naczynia. W naczyniach,
w osoczu znajduje się około 40% całkowitej ilości albuminy, podczas gdy w płynach
tkankowych gdzie stężenie albuminy jest niższe, ale pojemność znacznie wyższa znajduje się
jej 60%. W skórze i mięśniach zawiera się około 15% całkowitej albuminy. Nie jest ona
gromadzona w wątrobie, lecz jest wydzielana do krążenia wrotnego tak długo jak długo jest
syntetyzowana. HSA jest produkowana w wątrobie jako pre-proalbumina, czyli tetrameryczne
białko zbudowane z czterech jednakowych łańcuchów. N-końcowy peptyd jest usunięty
zanim powstające białko zostanie uwolnione z szorstkiego retikulum endoplazmatycznego.
Proalbumina w aparacie Golgiego przechodzi proces glikozylacji, w którym do cząsteczki
białka dołączane są węglowodany. Ta modyfikacja prowadzi do powstania albuminy, która
jest wydzielana do krwioobiegu [64].
4.3 Funkcje
Albumina jest białkiem najliczniej występującym w osoczu. Albuminy dzięki
przewadze aminokwasów kwaśnych posiadają silny ładunek ujemny, co nie wykazuje
żadnego związku z wiązaniem molekuł [65]. Jej giętka struktura pozwala substancjom na
tworzenie z nią kompleksów. Cząsteczkami, które ulagają najmocniejszemu wiązaniu są
hydrofobowe aniony organiczne jak i kwasy tłuszczowe. Kwasy tłuszczowe są kwasami
karboksylowymi
z
długimi
nierozgałęzionymi
łańcuchami
alifatycznymi
[64].
Niezestryfikowane kwasy są transportowane przez HSA i dzięki temu łatwiej mogą zostać
usunięte z komórek.
Nie tylko kwasy tłuszczowe są transportowane przez albuminę, ale również
hormony tarczycy [44]. Większość hormonów tarczycy znajdujących się we krwi jest
połączona z białkami, w tym z HSA. Hormony tarczycy występują prawie we wszystkich

37
komórkach i są ważnym czynnikiem w rozwoju ciała ludzkiego. Powodują one wzrost
wskaźnika podstawowej przemiany materii, syntezy białek, regulują wzrost i dojrzewanie
kości. HSA transportuje również hormony rozpuszczalne w tłuszczach [64].
Pełnią one kluczową rolę w utrzymaniu ciśnienia onkotycznego niezbędnego do
utrzymania prawidłowych proporcji miedzy ilością wody zawartą we krwi, a ilością wody
w płynach tkankowych [64].
Albumina ludzkiej surowicy odgrywa kluczową rolę w transporcie leków i mocno
wpływa na ich dystrybucje w osoczu. Łączenie leków z HSA było dokładnie badane przez 30
lat. Naukowcy ustalili, że w HSA istnieją dwa główne miejsca wiążące dla leków.
Miejsce I Sudlow’s i miejsce II [45]. Dystrybucja leków kontrolowana jest przez albuminę ze
względu na to, że większość z nich ulega związaniu z jej domenami wiążącymi [44].
4.4 Utlenianie albuminy
Analiza składu aminokwasowego oksydacyjnie zmodyfikowanej albuminy
(AOPP-HSA) wykazuje znaczące modyfikacje reszt tyrozyny i aminokwasów zasadowych
(lizyna i arginina), tryptofanu i aminokwasów zawierających siarkę. Uważa się,
że modyfikacje te są głównie odpowiedzialne za zmiany konformacyjne obserwowane
w HSA (np. zmniejszenie liczby α-helis). AOPP-HSA najczęściej występuje w formie
wielocząsteczkowych agregatów, które prawdopodobnie są wynikiem wiązań krzyżowych
i/lub mostków disiarczkowych dityrozyny [66].

38
5
Metody fizyczne badania cząsteczek
Spektroskopia optyczna jest sposobem badania właściwości fizycznych, opartym
na pomiarze emisji światła przez badane cząsteczki, a także na pomiarze ich interakcji ze
światłem. Przy użyciu tej techniki możemy mierzyć skład chemiczny, strukturę oraz zmiany
zachodzące w badanych cząstkach. Spektroskopia opiera się na badaniu różnych właściwości
światła. W różnych warunkach różne substancje wytwarzają, odbijają lub pochłaniają światło.
Światło mierzone po odbiciu, przejściu lub emisji przez badany obiekt jest rejestrowane
w postaci linii widmowych. Oprócz długości fali inne cechy światła, takie jak intensywność,
mogą również dostarczyć użytecznych informacji.
5.1Absorpcja
Absorpcją światła nazywamy zjawisko pochłaniania światła w ośrodku
materialnym. Fizyczny mechanizm absorpcji zależy od rodzaju promieniowania oraz
właściwości ośrodka. Absorpcja promieniowania zwiększa energię układu. W procesie
absorpcji światło może być pochłaniane przez cząstkę tylko w porcjach zależnych od jej
struktury energetycznej. Zjawisko to poprawnie opisane jest przez mechanikę kwantową [48].
Kwant energii fali przenoszony jest przez foton, który zderza się z np. elektronem, czy jądrem
atomowym. Cząstka pochłania zawsze całą energię fotonu i tylko wtedy gdy, pozwalają na to
jej dopuszczalne stany kwantowe.
W wyniku absorpcji światła przechodzącego przez substancje (np. gaz) z widma
światła, które pada na daną substancję zostają usunięte pochłaniane częstotliwości. Na tej
podstawie można stwierdzić przez jakie substancje przechodziło światło. Zjawisko to służy do
badania składu chemicznego mieszanin związków chemicznych, gazów otaczających
gwiazdy, obłoków gazowych we wszechświecie.
Wielkość absorpcji światła można obliczyć na podstawie prawa Lamberta-Beera,
które brzmi: jeśli współczynnik absorpcji rozpuszczalnika jest równy zeru, to wiązka
promieniowania monochromatycznego, po przejściu przez jednorodny roztwór substancji
absorbującej o stężeniu c, ulega osłabieniu według równania:
cx
e
I
I
α
−
=
0

39
w którym I
0
- oznacza natężenie wiązki promieniowania monochromatycznego padającego na
jednorodny ośrodek absorbujący, I – natężenie promieniowania po przejściu przez ośrodek
absorbujący, x – grubość warstwy absorbującej, α – współczynnik absorpcji, e – podstawę
logarytmów naturalnych. Stosunek natężenia światła przechodzącego I do padającego I
0
nazywamy transmisją. Współczynnik transmitancji oznaczany jest literą T. Transmitancja
może przyjmować wartości od 0 do 1 lub jeżeli wyrażamy ją w procentach od 0 do 100%.
Wskazuje ona jaka część promieniowania została przepuszczona przez substancję [47,48,49].
Inną ważną wielkością jest absorbancja (A). Absorbancja jest miarą absorpcji
promieniowania i wyraża się wzorem:
T
I
I
A
1
log
log
0
=
=
gdzie: I
0
- oznacza natężenie wiązki promieniowania monochromatycznego padającego na
jednorodny ośrodek absorbujący, I – natężenie promieniowania po przejściu przez ośrodek
absorbujący, T – współczynnik transmitancji.
Cząsteczki różnych związków wykazują odmienne rozkłady poziomów energetycznych, ich
właściwości absorpcyjne różnią się między sobą, co daje duże możliwości identyfikacji tych
cząsteczek, określania stanu elektronów [47].
5.2
Fluorescencja
Na całkowitą energię cząsteczki składają się:
1.
Energia elektronowa, która związana jest z położeniem elektronów, rodzajem
utworzonych wiązań i odległością pomiędzy jądrami atomów wchodzących w skład
cząsteczki.
2.
Energia oscylacyjna, która związana jest z ze
zbliżaniem lub oddalaniem atomów
budujących
daną cząsteczkę.
3.
Energia rotacyjna, która związana jest z ruchami obrotowymi jakie mogą wykonywać
molekuły cieczy lub gazu.
Cząsteczka nie może wykonywać dowolnych ruchów oscylacyjnych i rotacyjnych. Ruchy te
nie mogą również być wykonywane z dowolną częstością. Wszystkie te energie przyjmują
określone wartości.
Zatem cząsteczka może znajdować się jedynie na ściśle określonym,
charakterystycznym dla niej poziomie energetycznym, a przejścia na wyższe lub niższe
poziomy wiążą się odpowiednio z absorpcją i emisją kwantów energii [48]
.

40
Sposobem dezaktywacji wzbudzonych stanów elektronowych, gdy są one
następstwem absorpcji światła, jest fotoluminescencja. W zależności od czasu pomiędzy
pochłonięciem, a wyemitowaniem energii wyróżnia się fluorescencję i fosforescencję.
Fluorescencja to emisja światła powstająca przy przejściach cząsteczek ze wzbudzonych
stanów singletowych do stanu podstawowego. Gdy czas pomiędzy pochłonięciem energii,
a jej emisją jest dłuższy mówimy wówczas o fosforescencji. Jest to zjawisko emisji światła
zachodzące przy przejściu cząsteczki ze stanu trypletowego do stanu podstawowego. Ze
zjawiskiem fosforescencji mamy do czynienia, gdy po relaksacji oscylacyjnej następuje
przejście międzysystemowe ze wzbudzonego stanu singletowego do wzbudzonego stanu
tripletowego T 1 (elektrony nie są sparowane, ich spiny zwrócone są w tym samym kierunku)
i dopiero później do stanu podstawowego. Przejście promieniste między stanami T
1
i S
0
jako
stanami o różnej multipletowości jest zabronione, więc cząsteczka przez długi czas przebywa
w elektronowym stanie wzbudzonym, nim wreszcie, łamiąc multipletową regułę wyboru,
wypromieniuje foton i wróci do podstawowego stanu elektronowego [47,48].
Światło fluorescencji jest zazwyczaj emitowane przez cząsteczki aromatyczne i
heterocykliczne. Cząsteczka absorbuje promieniowanie wówczas, gdy występuje w niej
element struktury zwany chromoforem. W elemencie tym możliwe są przejścia elektronowe
typu:
*
π
π
→
,
*
π
→
n
,
*
σ
→
n
. Są to przejscia pomiędzy orbitalami molekularnymi: π -
wiążącym, π* - antywiążącym, n – niewiążącym. Chromoforem może w takim razie być
związek, który posiada elektrony π i n [50].
Orbital molekularny, podobnie jak atomowy, może być obsadzony przez maksymalnie
dwa elektrony walencyjne o antyrównoległych spinach. Orbital molekularny łączący dwa
atomy jest wynikiem kombinacji dwóch orbitali atomowych. Absorpcja promieniowania
powoduje przejście elektronu z wiążącego orbitalu molekularnego o niższej energii, na
molekularny orbital antywiążący o wyższej energii. Orbital antywiążący oznacza się
gwiazdką. Do najważniejszych chromoforów należą grupy: azowa, nitrowa, nitrozowa,
karbonylowa, tiulowa, etylenowa, układ p-chinoidowy, układ benzenowy. Czynnikiem
decydującym o wielkości absorpcji jest charakter grup chromoforowych jakie występują
w cząsteczkach. Często wystarcza jeden silny chromofor, aby substancja wykazywała
absorpcję. Większa liczba grup chromoforowych powoduje silniejszą absorpcję, a także
przesunięcie maksimum absorpcji w kierunku fal dłuższych. Przesunięcie to nosi nazwę
przesunięcia batochromowego i może być wywołane występowaniem kilku sprzężonych ze
sobą wiązań podwójnych lub obecnością w cząsteczce grup auksochromowych takich jak –
OH lub NH
2.
Grupy te same w sobie nie wywołują barwy, ale dzięki przesunięciom
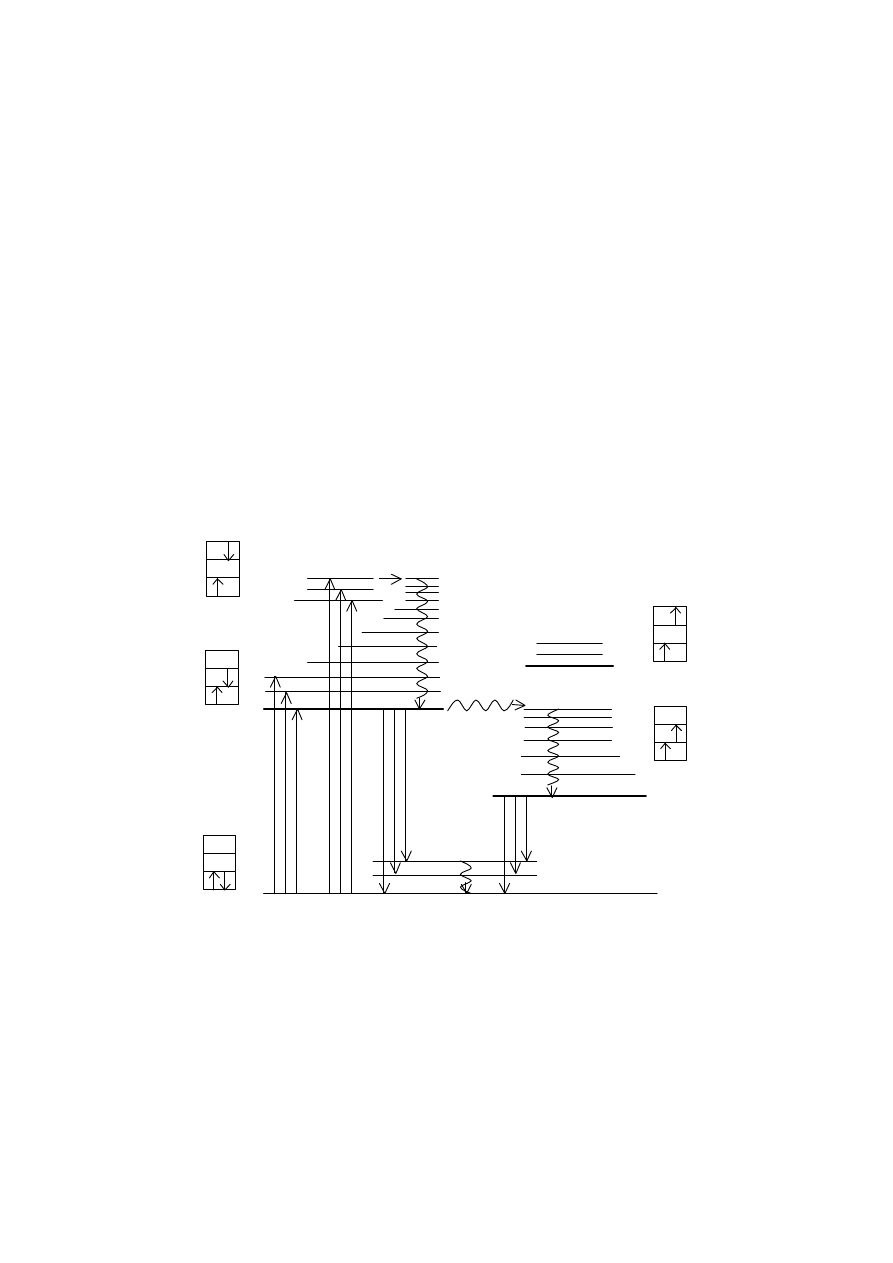
41
batochromowym zwiększają intensywność absorpcji. Przesunięcie absorpcji w kierunku fal
dłuższych rośnie w szeregu: -CH
3
< -OH < -O-CH
3
< -NH
2
< -NH-CH
3
< N(CH
3
)
2
[50]
Właściwości fluorescencyjne związków organicznych zależą od ich struktury.
Przykładowo, widma fluorescencji węglowodorów aromatycznych o pierścieniach
skondensowanych liniowo (np. antracen) leżą w zakresie fal dłuższych niż widma
fluorescencji węglowodorów o tej samej liczbie pierścieni skondensowanych kątowo (np.
fenantren). Podstawienie cząsteczek związków aromatycznych fluorowcami obniża
fluorescencje. Obniżenie fluorescencji przez podstawienie danym atomem fluorowca F, Cl, Br
i I wzmacnia się w podanym porządku. Obecność grupy - NO
2
zwykle powoduje zanik
fluorescencji, natomiast grupy -OH oraz –OR często wzmacniają fluorescencję cząsteczek
aromatycznych, powodując jednocześnie przesuniecie długofalowe widm [63]. Do substancji
fluoryzujących należy między innymi rodamina, eozyna, rywanol, chinina, fluoresceina [48].
fluorescencja
absorpcja
fosforescencja
przejście
międzysystemowe
T
1
T
2
S
0
S
1
S
2
konwersja
wewnętrzna
Rys.13 Diagram Jabłońskiego [49].
Na powyższym rysunku przedstawiony jest diagram Jabłońskiego, który obrazuje
drogi powrotu cząsteczki ze stanów wzbudzonych do stanu podstawowego. Schemat
przedstawia kolejne poziomy energetyczne cząsteczki wieloatomowej. Poziomem
podstawowym jest poziom S
0
. Pozostałe rozmieszczone są według wzrastającej energii.
Wzbudzone stany singletowe oznaczone są symbolami S
1
, S
2
, natomist tripletowe T
1
, T
2
.
Każdy stan elektronowy zawiera poziomy oscylacyjne. Promieniste przejścia pomiędzy

42
stanami elektronowymi na diagramie zaznaczono pionowymi liniami, aby podkreślić ich
natychmiastowy charakter. Przejścia absorpcyjne trwają około 10
-15
s, jest to czas zbyt krótki,
aby jądro zdążyło ulec znaczącemu przesunięciu.
Wydajność kwantowa fluorescencji jest ważnym pojęciem, które pozwala
scharakteryzować daną substancję fluoryzującą. Definiuje się ją jako stosunek liczby
kwantów promieniowania wyemitowanego do liczby kwantów zaabsorbowanych
promieniowania wzbudzającego. Substancje charakteryzujące się wysoką wydajnością
kwantową, bliską jedności, emitują światło o dużym natężeniu [49].
5.3
Anizotropia fluorescencji
W pomiarach anizotropii fluorescencji próbkę wzbudza się światłem ciągłym,
spolaryzowanym. W izotropowym roztworze fluorofory są zorientowane przypadkowo.
Wzbudzeniu z największym prawdopodobieństwem
ulegają te cząsteczki fluoroforu, których
dipol przejścia absorpcyjnego jest równoległy do wektora pola elektrycznego światła
padającego na próbkę.
Zjawisko to nosi nazwę fotoselekcji. Podstawą pomiaru anizotropii jest
fakt, że wzbudzone cząsteczki emitują światło zdepolaryzowane. Intensywność światła
emitowanego mierzy się przy dwóch ustawieniach polaryzatora obserwacyjnego: równolegle
i prostopadle do wektora elektrycznego wzbudzenia.
Wie1kość mierzonej maksymalnej anizotropii fluorescencji w rzeczywistości
uzyskiwana w pomiarach jest niższa od maksymalnej, ze wzg1ędu na częściową
depolaryzację światła emitowanego. Główną tego przyczyną jest dyfuzja rotacyjna cząsteczki.
Jeżeli pomiędzy absorpcją i emisją nie nastąpi rotacja cząsteczki, to I
||
> I
⊥
, a anizotropia
fluorescencji będzie miała wartość maksymalną jaką można uzyskać dla roztworu, równą 0,4.
Ruchy rotacyjne występujące w czasie trwania stanu wzbudzonego zależą od 1epkości
rozpuszczalnika oraz rozmiaru i kształtu rotujących cząsteczek. Im większa szybkość rotacji,
tym wyższy stopień depolaryzacji i niższa, mierzona anizotropia [49].
Korelacyjny czas rotacji małych molekuł jest znacznie krótszy niż czas życia
fluorescencji. Anizotropia fluorescencji ma wartość bliską 0. Z kolei anizotropia światła
fluorescencji emitowanego przez duże, wolno obracające się cząsteczki (lub cząsteczki
przywiązane do dużych układów - membran lub cząsteczek białek) zanika wolno,
a anizotropia fluorescencji przyjmuje w takim wypadku wartość bliską 0,4 [48].

43
Obrót fluoryzujących molekuł w czasie życia stanu wzbudzonego prowadzi do
dalszego zmniejszania się anizotropii zgodnie z zależnością
gdzie
Θ
oznacza korelacyjny czas rotacji, t czas. Krzywe zaników anizotropii dostarczają
informacji o tempie rotacji fluoryzujących molekuł.
Θ
−
=
t
e
r
r
0

44
6
Cel pracy
Celem pracy było:
1.
wykazanie właściwości antyoksydacyjnych wybranego flawonoidu – kwercetyny
2.
pokazanie interakcji pomiędzy kwercetyną a białkiem surowicy ludzkiej – albuminą
3.
przebadanie wpływu anionorodnika ponadtlenkowego na utlenianie HSA

45
7
Aparatura pomiarowa i metody badawcze
Widma emisji fluorescencji oraz anizotropię fluorescencji rejestrowano za pomocą
spektrofluorymetru PTI (Photon Technology International Inc. – Birmingham USA). W celu
zmierzenia anizotropii fluorescencji stosowano przystawkę do spektrofluorymetru PTI
zawierającą polaryzator (umieszczony w kanale wzbudzenia) i analizator (umieszczony w
kanale emisji). Widma absorpcji były rejestrowane za pomocą spektrofotometru UV-VIS
JASCO V-550. Stała temperatura badanych roztworów równa 37ºC utrzymywana była za
pomocą ultratermostatu TW2.03 (ELMI).
7.1
ORAC-FL
ORAC (ang. Oxygen Radical Absorbance Capacity), jest to zdolność pochłaniania
reaktywnych form tlenu przez przeciwutleniacze. W metodzie ORAC-FL mierzona jest
pojemność antyutleniająca. W roli sondy fluorescencyjnej wykorzystywana jest fluoresceina.
Metoda pomiaru ORAC-FL jest oparta na technice zastosowanej po raz pierwszy przez
Glazera [52]. Wykorzystuje się a niej reakcję utleniania cząsteczek substancji
fluorescencyjnej po zmieszaniu jej ze związkiem dostarczającym wolnych rodników.
W metodzie mierzony jest zanik świecenia (fluorescencji) fluoresceiny w wyniku
reakcji z rodnikami generowanymi przez azoinicjatora jakim jest AAPH (2,2'-azobis(2-
amidino-propane) dihydrochloride). W obecności antyoksydantu fluoresceina jest chroniona
przed rozpadem oksydacyjnym, w wyniku czego opóźniony zostaje zanik fluorescencji.
Intensywność fluorescencji spada w miarę postępowania procesu utleniania. Zanik
fluorescencji rejestrowany jest w postaci wykresów przedstawiających zależność
intensywności fluorescencji od czasu. Pojemność antyoksydacyjna próbki obliczana jest przez
odjęcie od pola pod wykresem widma fluorescencji próbki z antyoksydantem pola pod
wykresem widma fluorescencji próbki bez antyoksydantu [52,53].
Fluoresceina jest organicznym związkiem chemicznym będącm barwnikiem o masie
molowej 332,21 g/mol. W roztworach zasadowych wykazuje zielonożółtą fluorescencję.
Posiada maksimum absorpcji dla 494 nm i maksimum emisji dla 521 nm.
FITC - izotiocyjanian fluoresceiny – pochodna fluoresceiny posiadająca aktywną
grupę izotiocyjanową podstawioną w miejscu atomu wodoru w dolnym pierścieniu struktury.

46
Modyfikacja ta przeprowadzona jest aby zwiększyć powinowactwo FITC do białek, poprzez
zwiększenie specyficzności w kierunku amin pierwszorzędowych. Widma absorpcji i emisji
wykazują maksimum odpowiednio 495 nm/521 nm. Tak samo jak fluoresceina wykazuje
zielonożółta fluorescencję [54].
AAPH - 2,2’-diazobis (2-amidinopropan) dichlorowodorku – rozkład AAPH generuje
azot molekularny i dwa rodniki węglowe, które mogą reagować z tlenem cząsteczkowym
generując rodniki peroksylowe. Okres połowicznego rozpadu wynosi 175 godzin
w temperaturze 37°C w neutralnym pH, masa molowa wynosi 271,19 g/mol [55].
Kwercetyna - organiczny wielopierścieniowy związek aromatyczny pochodzenia
roślinnego o wzorze C
15
H
10
O
7
z grupy flawonoidów glikozydowych, będący pochodną
flawonu. Stosowany w analizie chemicznej jako odczynnik. Żółte, bezwonne, igiełkowate
kryształy o masie molowej 302,24 g/mol [34].
HSA (human serum albumin) – albumina ludzka; najobficiej występujące białko we
krwi ludzkiej, produkowane przez wątrobę. Stanowi połowę białek krążących w osoczu krwi.
Transportuje szereg ligandów takich jak hormony, kwasy tłuszczowe, miedź i hem. Organizm
ludzki produkuje ok. 3 g na dobę. Masa molowa wynosi 66000 g/mol [56].
7.2 Metoda pomiaru produktów utleniania białek (AOPP)
Stężenie AOPP (advanced oxidation protein products) oznaczono metodą
spektrofotometryczną. Pomiar oparty był na barwnej reakcji końcowych produktów oksydacji
białek z jodkiem potasu w środowisku kwaśnym jakie tworzy kwas octowy – reakcja Witko-
Sarsat.

47
8. Wyniki i wnioski
8.1 Przygotowanie roztworów podstawowych
Kwercetyna i AAPH byłay zakupione w Sigma-Aldrich.
Próbka roztworu podstawowego kwercetyny o stężeniu 25mM została przygotowana
przez rozpuszczenie proszku 99,8% w alkoholu etylowym. Dodatkowo każdorazowo przed
dodaniem do próbki podlegającej badaniu, kwercetyna była 100-krotnie rozcieńczana w PBS
o pH=7,4 w celu eliminacji obecności alkoholu z próbki. Stężenie roztworu podstawowego
fluoresceiny wynosiło 5mM i zostało uzyskane przez rozpuszczenie proszku w wodzie
destylowanej. Stężenie AAPH wynosiło 555mM i zostało uzyskane przez rozpuszczenie
proszku w buforze PBS o pH 7,4. Stężenie roztworu podstawowego HSA wynosiło 100µM
i zostało uzyskane przez rozpuszczenie proszku w wodzie destylowanej. W badaniach użyty
został także 30% roztwór kwasu octowego oraz 1,16 M roztwór jodku potasu.
Widma absorpcji i fluorescencji były rejestrowane dla próbek znajdujących się w
temperaturze 37
o
C, w roztworze PBS o pH=7,4. Pomiary były wykonywane bezpośrednio po
dodaniu odpowiednich objętości roztworów podstawowych do roztworu PBS. Po każdym
dodaniu odpowiedniego roztworu podstawowego próbka była starannie mieszana za pomocą
Vortex’u.
8.2 Widma absorpcji kwercetyny w obecności rodników generowanych przez
APPH.
W badaniu widm fluorescencji wykorzystano próbki roztworów
podstawowych kwercetyny i AAPH.
Do badania użyto 120µl roztworu podstawowego kwercetyny i 450µl roztworu
podstawowego AAPH. Każda z nich była uzupełniana buforem PBS pH 7,4 do objętości 3ml.
Przed
rozpoczęciem
pomiarów
spektrofotometr
kalibrowano
za
pomocą
rozpuszczalnika, którym był PBS.
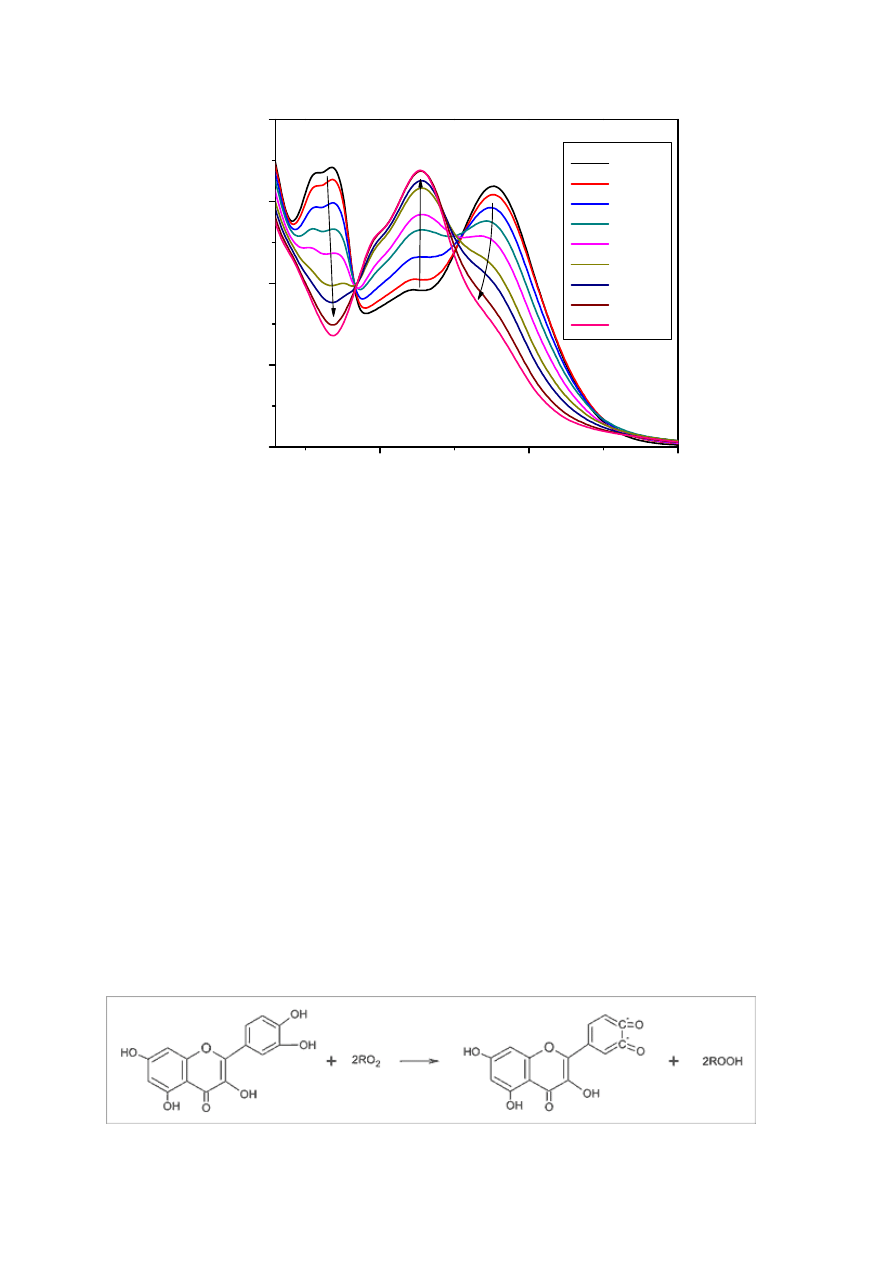
48
300
400
500
0,0
0,1
0,2
0,3
0,4
A
b
s
o
rb
a
n
c
ja
Długo
ść
fali
0 min
5 min
10 min
15 min
20 min
30 min
40 min
50 min
60 min
Rys.14 Absorbancja kwercetyny w różnych przedziałach czasowych. Widma przedstawiają
ewolucję czasową kwercetyny z formy nieutlenionej do formy utlenionej. Utlenianie
kwercetyny było wywołane obecnością wolnych rodników generowanych przez AAPH.
Pomiary były prowadzone dla roztworu kwercetyny o stężeniu 10µM w PBS
o temperaturze 37
o
C i pH=7,4.
Widma utleniania kwercetyny zostały przedstawione na rysunku 14. Rejestracja
pojedynczego widma wykonywana była przez pierwsze 20 min co 5 min, a po upływie
20 min co 10 min. Zmiana odstępu czasu pomiędzy rejestracją widm, po upływie 20 minut od
rozpoczęcia pomiaru, była wywołana spowolnieniem procesu utleniania kwercetyny. Na
powyższym rysunku możemy zaobserwować wzrost natężenia pasma przy długości fali
327nm oraz spadek pików przy długości fal 268 nm i 376 nm. Pik przy długości fali 327 nm
powstał w wyniku oddania przez kwercetynę dwóch atomów wodoru znajdujących się przy
węglu C3 i C4 w pierścieniu B. Skutkiem tego po upływie 60 min nastąpiło całkowite
utlenianie kwercetyny. Zatem możemy potwierdzić działanie przeciwutleniające związku.
Rys.15 Schemat blokowania rodników nadtlenowych przez kwercetynę [39].
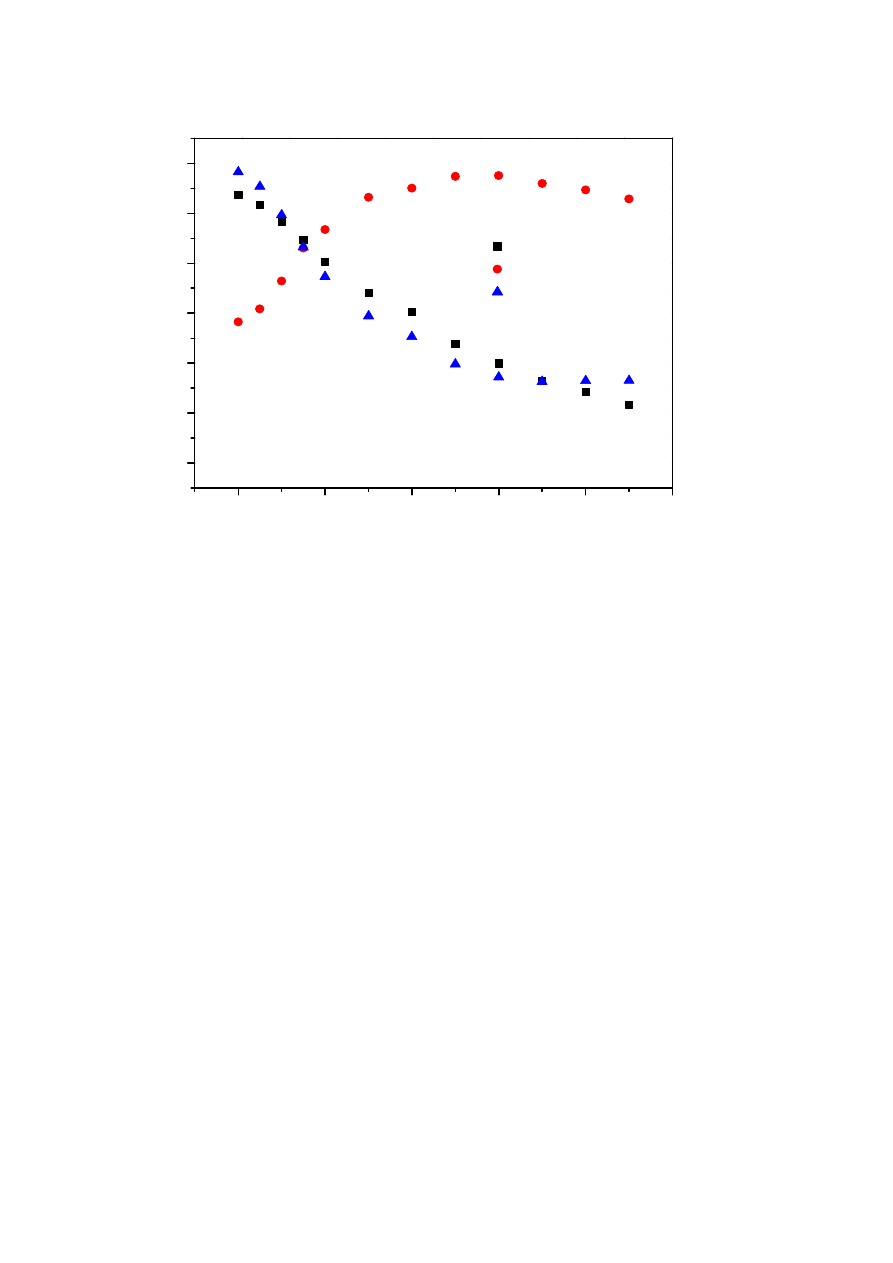
49
0
20
40
60
80
100
0,05
0,10
0,15
0,20
0,25
0,30
0,35
Długo
ść
fali:
376 nm
327 nm
268 nm
A
b
s
o
rb
a
n
c
ja
Czas [min]
Rys.16 Zależność trzech pików absorbancji z rysunku 14 w funkcji czasu. Zmiana
absorbancji kwercetyny w funkcji czasu spowodowana była dodaniem AAPH.
Zaobserwować można wyraźny wzrost pasma przy długości fali 327 nm, który
związany jest z utlenianiem kwercetyny.
8.3 Widma emisji fluorescencji fluoresceiny w obecności kwercetyny o różnych
stężeniach
W badaniu widm fluorescencji wykorzystano próbki roztworów podstawowych
kwercetyny, fluoresceiny i AAPH.
Próbki kwercetyny o stężeniach 3,75 µM; 6,25 µM; 8,75 µM; 9,375 µM były
przygotowane przez dodanie odpowiednio 30 µl, 50 µl, 70 µl, 75 µl roztworu podstawowego
kwercetyny, a także 450 µl roztworu podstawowego AAPH i 10 µl roztworu podstawowego
fluoresceiny. Każda z nich była uzupełniana buforem PBS pH 7,4 do objętości 2 ml. Próbka
stanowiąca krzywą wzorcową nie zawierała kwercetyny.
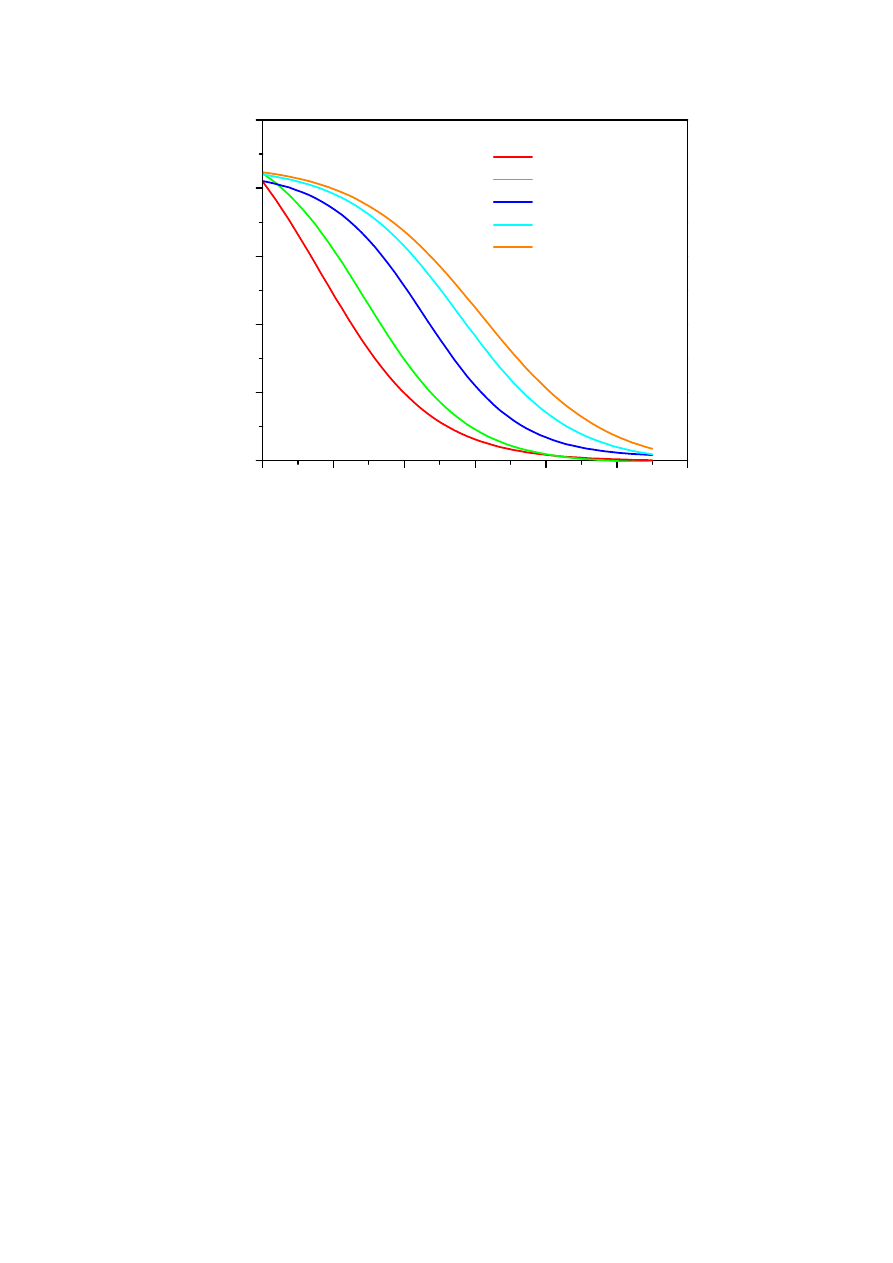
50
0
10
20
30
40
50
60
0,0
0,2
0,4
0,6
0,8
1,0
St
ęż
enie kwercetyny:
krzywa wzorcowa
3,75
µ
M
6,25
µ
M
8,75
µ
M
9,375
µ
M
N
a
t
ę
ż
e
n
ie
f
lu
o
re
s
c
e
n
c
ji
Czas [min]
Rys.17 Natężenie fluorescencji fluoresceiny w funkcji czasu w obecności kwercetyny
o różnych stężeniach. Krzywa wzorcowa (czerwona linia) nie zawierała kwercetyny.
Zaobserwować można wydłużanie czasu fluorescencji fluoresceiny, który jest
powiązany ze wzrostem stężenia kwercetyny. Pomiary były prowadzone w roztworze
PBS o pH=7,4 i temperaturze 37
o
C.
Doświadczenie miało na celu wykazanie właściwości antyoksydacyjnych kwercetyny,
poprzez wykazanie jej ochronnego wpływu na fluoresceinę. Pomiary natężenie fluorescencji
dla każdego stężenia kwercetyny były powtarzane co 5 minut. Kwercetyna miała
spowodować wydłużenie czasu fluorescencji fluoresceiny, przez jej ochronę przed rodnikami
generowanymi przez AAPH. Na rysunku 17 przedstawiono zależność szybkości spadku
fluorescencji fluoresceiny w czasie od stężenia kwercetyny. Im większe stężenie kwercetyny
tym wolniejszy spadek fluorescencji, co potwierdza właściwości antyoksydacyjne związku.

51
0,0
0,2
0,4
0,6
0,8
1,0
0
1
2
3
4
5
∆
P
St
ęż
enie kwercytyny
Rys. 18 Zależność wartości pola pod wykresem (∆P) od stężenia kwercetyny. ∆P otrzymano
poprzez obliczenie różnicy miedzy polem pod krzywą z antyoksydantem, a polem pod
krzywą wzorcową. Wartości pola były obliczone dla stężeń kwercetyny użytych w
badaniu widm emisji fluoresceiny, które zostały przedstawione na rysunku 17.
∆P otrzymano poprzez obliczenie różnicy miedzy polem pod krzywą fluorescencji
z antyoksydantem, a polem pod krzywą wzorcową. Obliczeń dokonano przy użyciu narzędzia
Microsoft Excel.
Na rysunku 18 obserwujemy liniowy wzrost właściwości antyoksydacyjnych. Wzrost
ten jest wprost proporcjonalny w stosunku do stężenia kwercetyny. Co oznacza, że dwukrotne
zwiększenie stężenie kwercetyny zwiększa nam dwukrotnie właściwości antyoksydacyjne.
8.4 Pomiar natężenia fluoryzacji kwercetyny w różnych stężeniach HSA
W doświadczeniu użyto roztworów podstawowych HSA w ilościach odpowiednio
0 µl, 60 µl, 90 µl, 150 µl, 200 µl, 450 µl, 600 µl, 900 µl oraz roztworu podstawowego
kwercetyny w ilości 120 µl.
Fluoryzacja kwercetyny jest zależna od tego czy jest ona związana czy też nie.
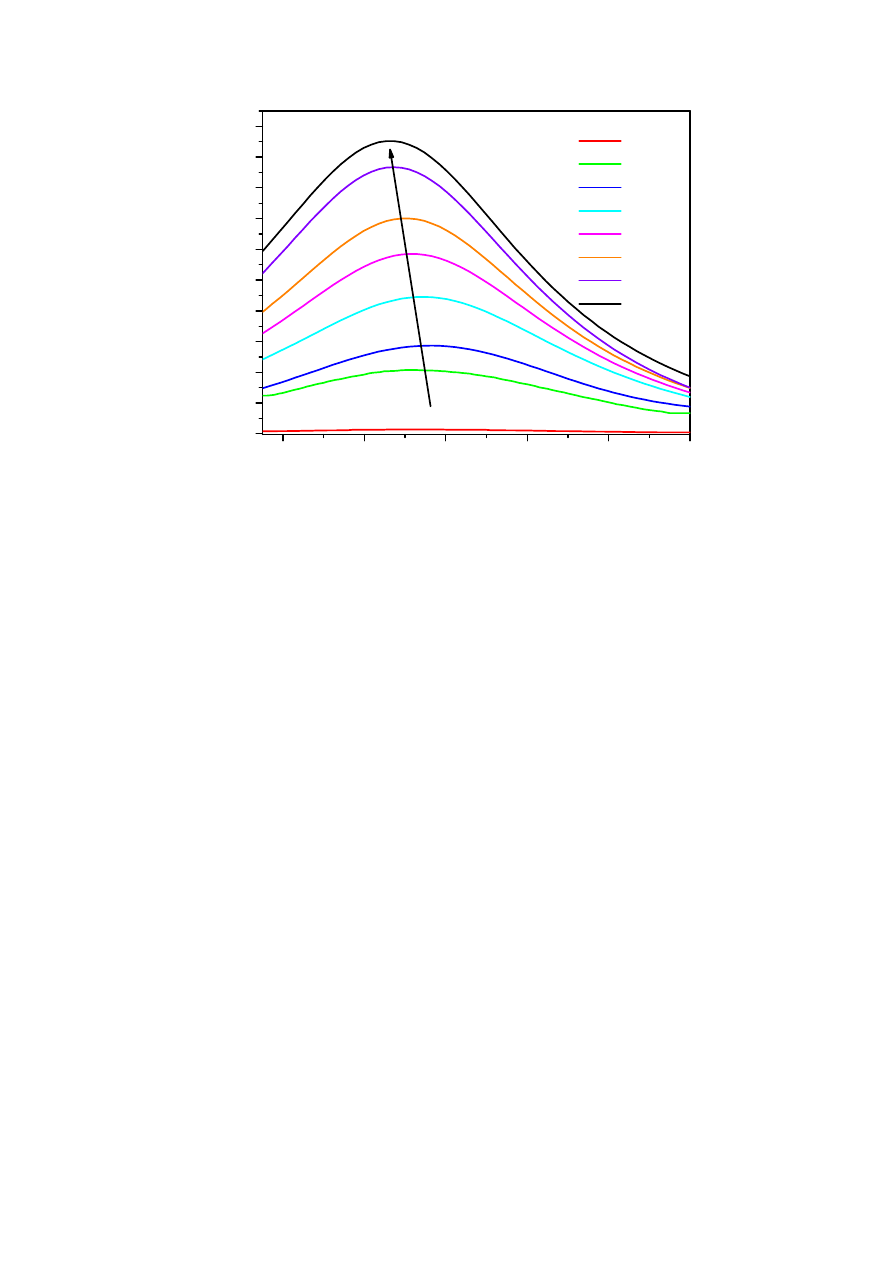
52
500
520
540
560
580
600
0
5
10
15
20
25
30
35
40
45
50
N
a
t
ę
ż
e
n
ie
f
lu
o
re
s
c
e
n
c
ji
Długo
ść
fali
0
µΜ
2
µΜ
3
µΜ
5
µΜ
10
µΜ
15
µΜ
20
µΜ
30
µΜ
St
ęż
enie HSA
Rys. 19 Fragmenty widma zależności natężenia fluoryzacji kwercetyny od stężenia HSA.
Widmo nie zawierające HSA (linia czerwona) wykazuje fluoryzację bliską zeru. Każde
z kolejnych widm zawiera HSA. Wraz ze wzrostem stężenia albuminy możemy
zaobserwować wzrost natężenia fluoryzacji kwercetyny. Pomiary były prowadzone dla
roztworu kwercetyny o stężeniu 10µM w PBS o pH=7,4 i temperaturze 37
o
C.
Fluoryzacja kwercetyny jest zależna od tego czy jest ona związana czy też nie. Jak
widać na rysunku 19 wraz ze wzrostem stężenia HSA zauważalny jest wzrost natężenia
fluoryzacji. Powodem tego wzrostu jest to, że w wyższym stężeniu HSA występuje więcej
molekuł, które przyłączają kwercetynę i dlatego świecenie jest intensywniejsze. Przy stężeniu
białka 30 µM nastąpiło całkowite wysycenie kwercetyny i zaobserwowano maksimum
natężenia fluorescencji. Możemy też zauważyć przesuwanie się widma z długości fali 535 nm
do długości 520 nm.
8.5
Rejestracja widma fluoryzacji kwercetyny i HSA metodą ORAC(FL)
Rejestracja widm emisji fluorescencji była przeprowadzana dla próbek znajdujących
się w temperaturze 37
o
C w roztworze PBS o pH=7,4. Pomiary były wykonywane
bezpośrednio po dodaniu odpowiednich objętości roztworów podstawowych do roztworu
PBS.
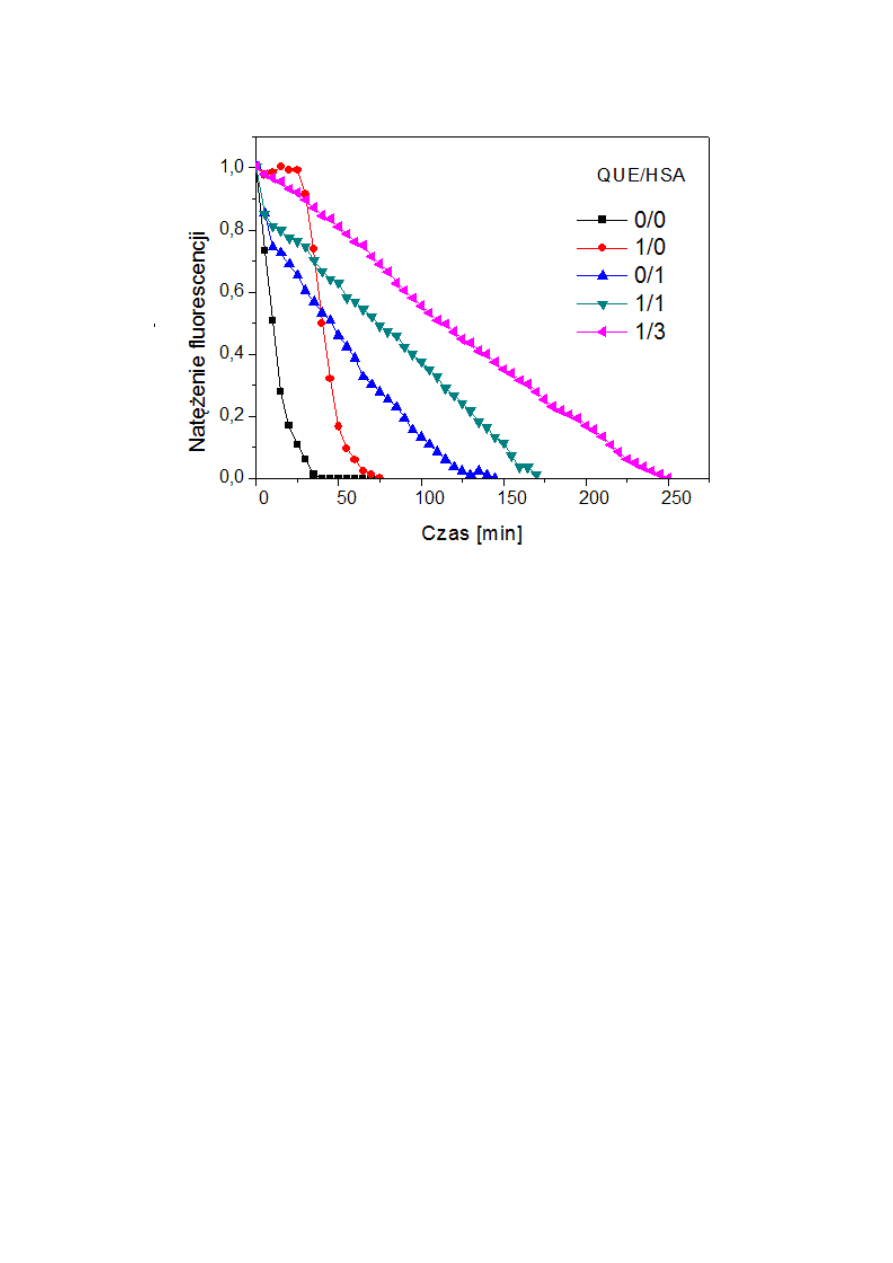
53
Rys.20 Natężenie fluorescencji fluoresceiny w funkcji czasu w zależności od stężenia HSA
i kwercetyny. Przedstawione zostały pomiary natężenia fluorescencji dla roztworów
kwercetyny i HSA oraz dla roztworów kwercetyny i HSA w różnym stosunku
stężeniowym. Linie przedstawiają: krzywa wzorcowa (czarna linia), kwercetyna
(czerwona linia), HSA (niebieska linia), kwercetyna i HSA w stosunku 1:1 (zielona
linia, kwercetyna i HSA w stosunku 1:3 ( różowa linia). Użyto kwercetyny w stężeniu
10µM i HSA w stężeniu 10 µM i 30 µM.
Próbki kwercetyny i HSA o stężeniu 10 µM były przygotowane przez dodanie
odpowiednio 80 µl roztworu podstawowego kwercetyny i 200 µl roztworu podstawowego
HSA, a także 450 µl roztworu podstawowego AAPH i 10 µl roztworu podstawowego
fluoresceiny. Każda z nich była uzupełniana buforem PBS pH 7,4 do objętości 2 ml. Po
każdym dodaniu odpowiedniego roztworu podstawowego próbka była starannie mieszana za
pomocą Vortex’u
Rysunek 20 przedstawia spadek fluorescencji fluoresceiny w czasie dla kwercetyny,
HSA i połączenia kwercetyny z HSA w stosunku 1:1 i 1:3. Zaobserwować można wzrost
właściwości antyoksydacyjnych kwercetyny wraz ze wzrostem stężenia białka. Z rysunku
możemy również wywnioskować, że HSA również posiada właściwości antyoksydacyjne, a
wspólne występowanie obu związków powoduje połączenie ich siły antyoksydacyjnej.
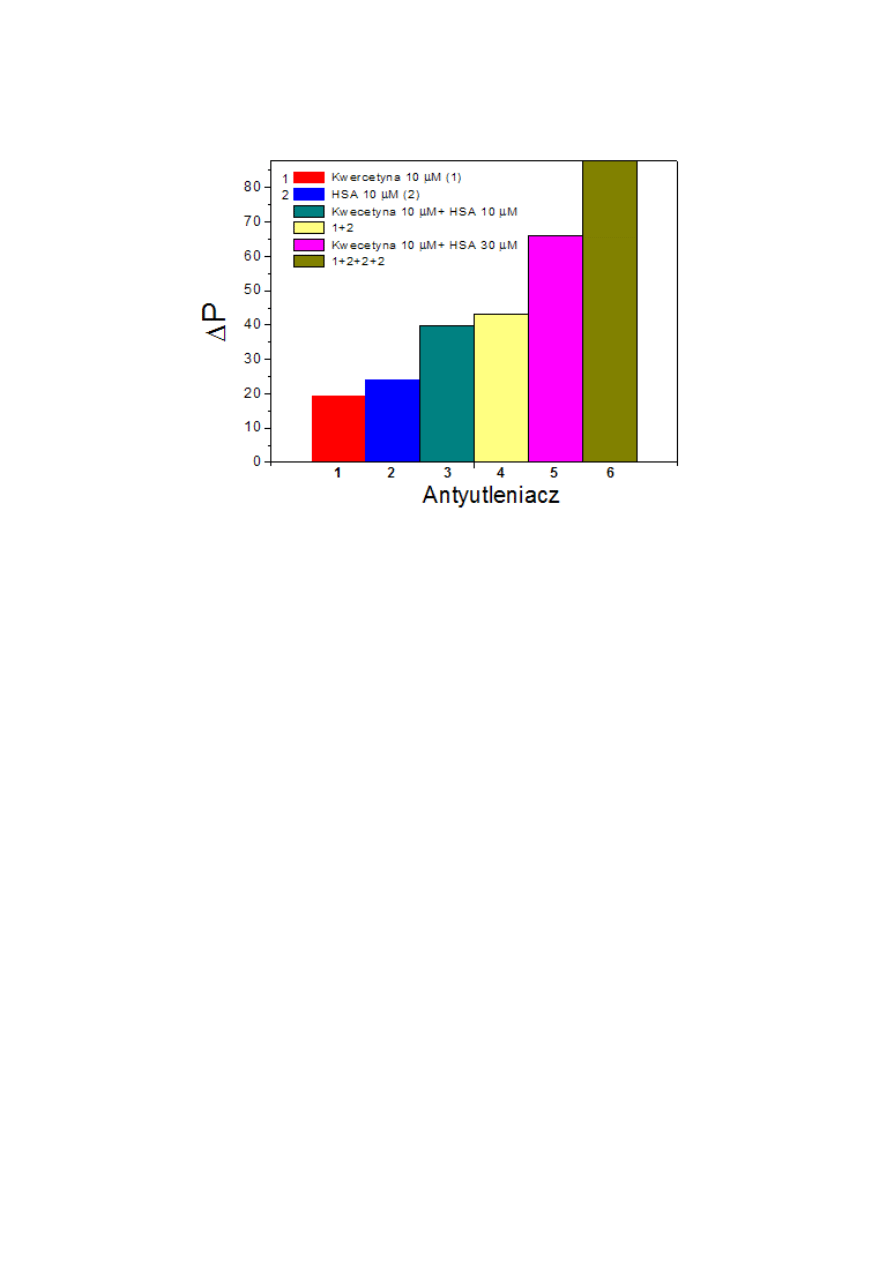
54
Rys.21 Zależność wartości pola pod wykresem od pojemności antyutleniającej kwercetyny
z HSA. ∆P otrzymano poprzez obliczenie różnicy między polem pod krzywą
z antyoksydantem, a polem pod krzywą wzorcową. Obliczenia pola były wykonane dla
stężeń kwercetyny i HSA użytych w badaniu widm emisji fluorescencji, które zostały
przedstawione na rysunku 20. Słupek numer 4 i 6 powstał poprzez odpowiednie
dodanie wartości słupków 1 i 2 i przedstawia wartości teoretyczne.
∆P
otrzymano
poprzez
obliczenie
różnicy
miedzy
polem
pod
krzywą
z antyoksydantem, a polem pod krzywą wzorcową.
Rysunek 21 przedstawia wartości pola pod wykresem badanych próbek. Słupek numer
1 odpowiada wartość pola pod wykresem kwercetyny, numer 2 wartość pola pod wykresem
HSA. Kolejny, czyli numer 3, przedstawia wartość pola pod wykresem kwercetyny i HSA
w stosunku 1:1, natomiast słupek numer 4 powstał przez dodanie wartości pola pod wykresem
kwercetyny (słupek nr 1) do wartości pola pod wykresem HSA (słupek nr 2). Kolejny piąty
słupek odpowiada za pole pod wykresem kwercetyny i HSA w stosunku 1:3. Ostatni słupek,
numer 6, został obliczony analogicznie do słupka czwartego, czyli przez dodanie wartości
pola pod wykresem HSA (słupek nr 2) pomnożonej 3-krotnie, do wartości pola pod wykresem
kwercetyny (słupek nr 1). Po porównaniu słupków 3 i 4 oraz 5 i 6 możemy stwierdzić,
że kwercetyna i HSA w połączeniu wykazują obniżone działanie antyoksydacyjne w stosunku
do działania jakie wykazują osobno.
55
0
10
20
30
40
50
60
0,05
0,10
0,15
0,20
0,25
0,30
FLUORESCEINA-HSA
FITC-HSA
A
n
iz
o
tr
o
p
ia
f
lu
o
re
s
c
e
n
c
ji
Czas [min]
Rys.22 Wartość anizotropii fluorescencji dla fluoresceiny i FITC. Brak połączenia
fluoresceiny z białkiem. Pomiar miał za zadanie wykluczyć możliwość ochronnego
wpływu białka na fluoresceinę.
Rysunek 22 przedstawia pomiar anizotropii fluorescencji dla fluoresceiny z HSA.
Pomiar ten miał za zadanie wykazać, czy fluoresceina łączy się z białkiem, miało to na celu
wykluczyć możliwość ochronnego wpływu białka na fluoresceinę. Ochronny wpływ miałby
polegać na niedostępności, związanych przez białko, molekuł fluoresceiny dla rodników
generowanych przez AAPH. Pomiar anizotropii był bliski zeru, co oznacza, że nie doszło do
związania cząsteczek fluoresceiny z białkiem. Dla porównania umieszczono pomiar
anizotropii fluorescencji dla HSA z FITC, w którym to nastąpiło połączenie pomiędzy
molekułami. FITC jest zmodyfikowaną formą fluoresceiny, która ma zwiększone
powinowactwo do białek.
8.6 Rejestracja widma fluorescencji kwercetyny z HSA
Próbki zawierały stężenie kwercetyny w stosunku do HSA 1:1. Do badania użyto
80 µl roztworu podstawowego kwercetyny i 200 µl roztworu podstawowego HSA , a także
450 µl roztworu podstawowego AAPH. Każda z nich była uzupełniana buforem PBS pH 7,4
do objętości 2 ml.
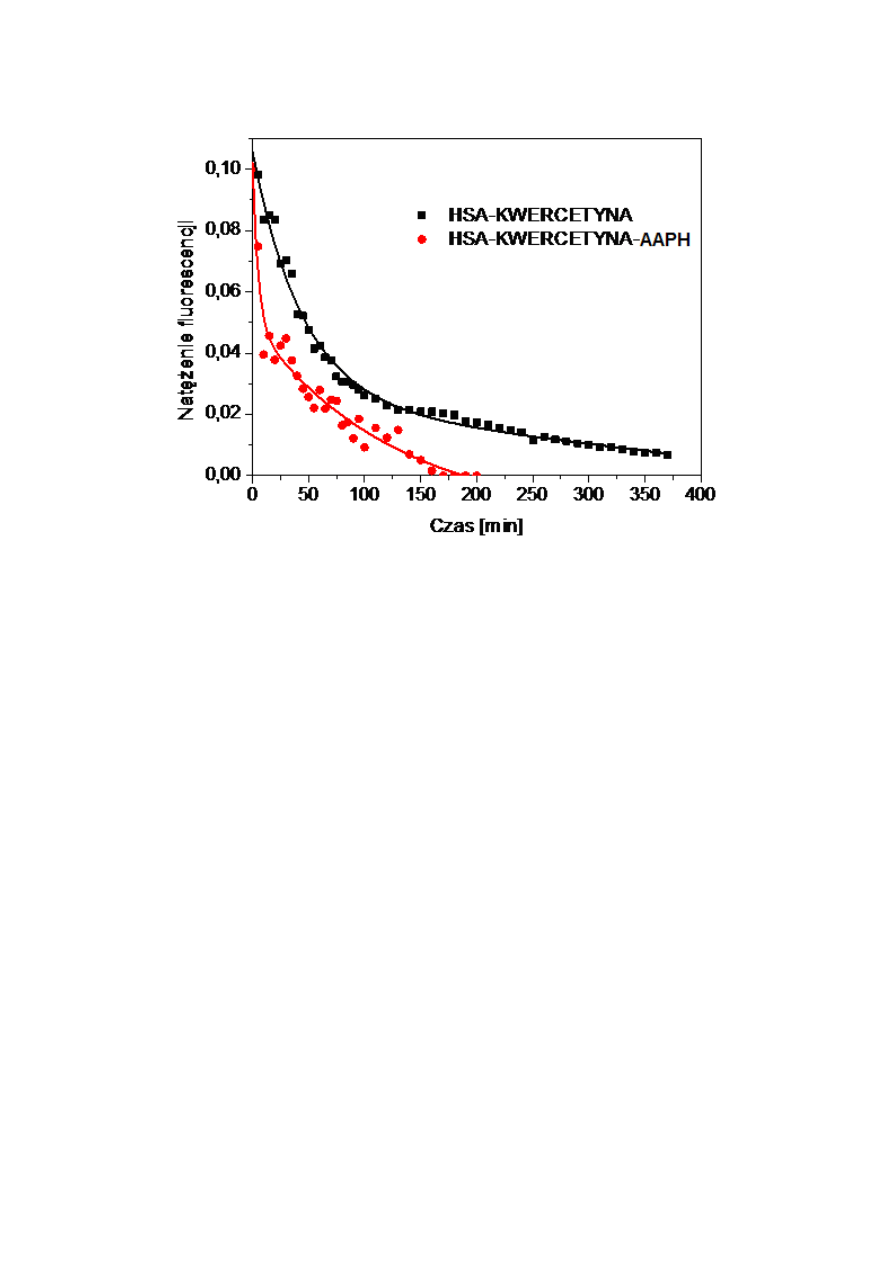
56
Rys.23 Natężenie fluoryzacji kwercetyny z HSA w funkcji czasu w zależności od obecności
AAPH. Zaobserwować można wyraźną różnicę w czasie fluoryzacji kwercetyny w
momencie pojawienia się rodników (czerwona linia) w porównaniu z czasem
fluoryzacji kwercetyny przy braku rodników (czarna linia). Stężenie kwercetyny i HSA
wynosiło 10µM.
Doświadczenie miało na celu wykazać, czy właściwości antyoksydacyjne kwercetyny
są obserwowane gdy jest ona połączona z HSA. Kwercetyna wykazuje fluoryzację tylko w
przypadku połączenia z białkiem, w przypadku utleniania kwercetyny fluorescencja spada. Na
rysunku 23 czas fluoryzacji kwercetyny w obecności rodników jest krótszy, co świadczy o
tym, że pomimo połączenia z HSA wykazuje ona zdolność do blokowania działania rodników
poprzez szybsze utlenianie. Oznacza to, że ma ona zdolność do odłączania się od białka, gdy
w pobliżu pojawiają się wolne rodniki.
8.7 Pomiar absorpcji utlenionego HSA
Zaawansowane produkty utleniania białek (AOPP - advanced oxidation protein
products) zostały zidentyfikowane na podstawie metody opracowanej przez Witko-Sarsat
57
i wsp.. Metoda oparta jest na reakcji tych związków z jodkiem potasu w środowisku kwaśnym
i pomiar absorbancji przy długości fali 340 nm.
325
350
375
400
0,05
0,10
0,15
0,20
0,25
A
b
s
o
rb
a
n
c
ja
Długo
ść
fali [nm]
5 min
10 min
15 min
20 min
25 min
30 min
Rys.24 Absorbancja HSA w różnych przedziałach czasowych w zależności od stopnia
degradacji białka. Fragmenty widma absorbancji przedstawiają ewolucję czasową
HSA wywołaną utlenianiem białka przez rodniki generowane przez AAPH. Rejestracja
poszczególnego widma absorbancji była prowadzona co 5 minut.
Badana próbka uzupełniona była roztworem PBS pH=7,4 do objętości 3 ml i zawierała
200 µl kwasu octowego, 100 µl jodku potasu, 200 µl HSA, 450 µl AAPH oraz 80 µl
kwercetyny.
Stężenie AOPP (advanced oxidation protein products) oznaczono metodą
spektrofotometryczną opartą na reakcji barwnej końcowych produktów oksydacji białek
z jodkiem potasu w środowisku kwaśnym jakie tworzył nam kwas octowy. Na rysunku 24
możemy zaobserwować wzrost absorbancji przy długości fali 360 nm, co jest ściśle
skorelowane z oksydacją białka. Oksydacja albuminy wpływa na modyfikacje jej składu
aminokwasowego. Uważa się, że modyfikacje te są głównie odpowiedzialne za zmiany
konformacyjne obserwowane w HSA (np. zmniejszenie liczby α-helis) [57].

58
9
Podsumowanie
Na podstawie przeprowadzonych badań można wysunąć następujące wnioski:
−
badania widma absorpcji kwercetyny wykazały że posiada ona właściwości
antyoksydacyjne. Wzrost absorbancji przy długości fali 327 nm świadczył o tym, że
kwercetyna uległa utlenieniu. W związku z tym możemy stwierdzić, że oddała ona dwa
atomy wodoru w celu neutralizacji wolnych rodników.
−
na podstawie badań przeprowadzonych metodą ORAC-FL można stwierdzić, że
kwercetyna jest antyoksydantem, gdyż jest w stanie chronić cząsteczki fluoresceiny przed
szkodliwym działaniem wolnych rodników generowanych przez AAPH. Ponadto czas
ochrony fluoresceiny zależał od stężenia kwercetyny jakie zostało zastosowane. Im
wyższe stężenie kwercetyny, tym czas ochrony się wydłużał, co oznacza, że kwercetyna
chroniła cząstki fluoresceiny do momentu, w którym uległa całkowitemu utlenieniu. Po
obliczeniu pola pod wykresem dla poszczególnych stężeń kwercetyny jakie zostały
zastosowane, można powiedzieć, że jej właściwości antyoksydacyjne rosną wraz ze
wzrostem stężenia, a co więcej wzrost ten jest wprost proporcjonalny do wzrostu jej
stężenia.
−
pomiar natężenia fluorescencji kwercetyny z HSA wykazał, że kwercetyna łączy się
z albuminą. Fluorescencja kwercetyny jest zależna od tego czy jest ona związana czy też
nie. Wraz ze wzrostem stężenia HSA zauważalny jest wzrost natężenia fluorescencji.
Powodem tego wzrostu jest to, że w wyższym stężeniu HSA występuje więcej molekuł,
które przyłączają kwercetynę i dlatego fluorescencja jest intensywniejsza. Przy stężeniu
białka 30 µM nastąpiło całkowite wysycenie kwercetyny i zaobserwowano maksimum
natężenia fluorescencji.
−
pomiar natężenia fluorescencji kwercetyny w połączeniu z HSA, w obecności wolnych
rodników i przy ich braku, wykazał, że pomimo połączenia z białkiem kwercetyna nie
traci swoich właściwości antyoksydacyjnych, jednak ulegają one zmniejszeniu. Po
obliczeniu pól pod wykresem można powiedzieć, że w połączeniu kwercetyny z białkiem,
oba związki wykazują obniżone działanie antyoksydacyjne w stosunku do działania jakie
wykazują osobno.
−
Czas fluoryzacji kwercetyny związanej z HSA w obecności rodników jest krótszy, co
świadczy o tym, że pomimo połączenia z HSA wykazuje ona zdolność do blokowania
działania rodników. Stwierdzić też można, że połączenie kwercetyny z HSA nie jest

59
wiązaniem kowalencyjnym, czyli jest wiązaniem odwracalnym. Oznacza to, że wykazuje
ona zdolność do odłączania się od białka, gdy w pobliżu pojawiają się wolne rodniki.
−
Pomiar absorbancji metodą spektrofotometryczną wykazał szkodliwość wolnych
rodników generowanych przez AAPH. Pomiar oparty był na barwnej reakcji końcowych
produktów oksydacji białek z jodkiem potasu w środowisku kwaśnym jakie tworzył nam
kwas octowy – reakcja Witko Sarsat. Na wykresie zaobserwowano wzrost absorbancji
przy długości fali 360 nm, co jest ściśle skorelowane z oksydacją białka.
Badania przeprowadzone w pracy metodami spektroskopii optycznej potwierdziły
aktywność antyoksydacyjną kwercetyny. Także wykazały pozytywne interakcje pomiędzy
HSA i kwercetyną.

60
10
Streszczenie
Zarówno promieniowanie ultrafioletowe, jak i wiele innych czynników środowiska,
takich jak wysoka lub niska temperatura, zanieczyszczenia powietrza, dym papierosowy,
wywołują uszkodzenia wolnorodnikowe, a przede wszystkim procesy biochemiczne
zachodzące w organizmach, zwłaszcza te związane z oddychaniem, prowadzą do
powstawania wolnych rodników tlenowych. Wolne rodniki w organizmie są przyczyną
uszkodzeń DNA, białek, lipidów i są rozważane jako przyczyna starzenia się organizmu.
Reakcjom rodnikowym zapobiegać mogą niektóre związki tzw. zmiatacze wolnych rodników,
czyli substancje reagujące z rodnikami i obniżające ich stężenie w danym układzie. Do
substancji takich zaliczane są flawonoidy.
Aktywność antyoksydacyjna flawonoidów zależy głównie od rodzaju i ilości
podstawników przy pierścieniu B, również od ich umiejscowienia w pozostałej części
cząsteczki. Zdolności do wyłapywania wolnych rodników rosną, kiedy do pierścienia B
przyłączona jest np. grupa katecholowa C
6
H
4
(OH)
2
lub pirogallolowa C
6
H
3
(OH)
3
. Największą
jednak rolę w usuwaniu wolnych rodników odgrywają grupy hydroksylowe, a ich ilość jest
ściśle skorelowana z siłą antyoksydacyjną związku.
Metody spektroskopii optycznych zastosowano w pracy z uwagi na to, że pozwalają
one określić bądź przewidzieć szereg biofizycznych właściwości związków o znaczeniu
terapeutycznym. Przedstawiona praca wskazuje na szerokie możliwości eksperymentalne
jakie dają metody fizyki w badaniach biomedycznych.
Badania przeprowadzone metodą ORAC-FL wykazały zdolności antyoksydacyjne
kwercetyny, a także wykazały, że są one wprost proporcjonalne do wzrostu jej stężenia.
Pomiar natężenia fluorescencji kwercetyny z HSA wykazał zdolność do łączenia się tych
dwóch cząsteczek. Pomiar natężenia fluorescencji kwercetyny w połączeniu z HSA wykazał,
że kwercetyna pomimo połączenia z białkiem wykazuje aktywność atyoksydacyjną, ale jest
ona mniejsza w stosunku do aktywności jaką wykazuje kwercetyna występująca w roztworze
bez białka. Działa to na takiej samej zasadzie w stosunku do białka tzn. białko w roztworze
bez kwercetyny posiada wyższą aktywność antyoksydacyjną. Reakcja prowadzona metodą
opracowaną przez Witko-Sarsat wykazała szkodliwy wpływ wolnych rodników na HSA.
Przeprowadzone badania potwierdziły właściwości antyoksydacyjne kwercetyny.
Wykazały również pozytywne interakcje pomiędzy kwercetyną i HSA. Potwierdziły również
destrukcyjny wpływ anionorodnika ponadtlenkowego na białko surowicy ludzkiej - albuminę.

61
11
Bibliografia
1.
Bartosz G., Druga twarz tlenu. Wyd. Naukowe, PWN, Warszawa 2003.
2.
Puzanowska-Tarasiewicz H., Starczewska B., Kuźmicka L., Reaktywne formy tlenu,
Bromat. Chem. Toksykol. – XLI, 2008, 4, str. 1007–1015.
3.
Zabłocka A., Janusz M., Dwa oblicza wolnych rodników, Postepy Hig Med Dosw.
(online), 2008; 62: 118-124
4.
Drőge W., Free radicals in the physiological control of cell function. Physiol. Rev.,
2002; 82: 47–95
5.
Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J., Free radicals
and antioxidants in normal physiological functions and human disease. Int. J.
Biochem. Cell Biol., 2007; 39: 44–84
6.
Bonizzi G., Piette J., Merville M.P., Bours V., Cell type-specifi c role for reactive
oxygen species in nuclear factor kB activation by IL-1. Biochem. Pharmacol., 2000;
59: 7–11
7.
Los M., Schenk H., Hexel K., Baeuerle P.A., Drőge W., Schulze-Osthoff K., IL-2
gene expression and NF-kB activation through CD28 requires reactive oxygen
production by 5-lipoxygenase. EMBO J., 1995;14: 3731–3740
8.
Hampton M.B., Kettle A.J., Winterbourn C.C., Inside the neutrophil phagosome:
oxidants, myeloperoxidase, and bacterial killing. Blood, 1998; 92: 3007–3017
9.
Los M., Drőge W., Stricker K., Baeuerle P.A., Schulze-Osthoff K., Hydrogen
peroxide as a potent activator of T lymphocyte functions. Eur. J. Immunol., 1995; 25:
159–165
10.
Weinert B.T., Timiras P.S., Theories of ageing. J. Appl. Physiol., 2003; 95: 1706–
1716
11.
Ponczek M.B., Wachowicz B., Oddziaływanie reaktywnych form tlenu i azotu z
białkami. Post. Biochem., 2005; 51: 140–145
12.
Schuessel K., Frey C., Jourdan C., Keil U., Weber C.C., Müller-Spahn F., Müller
W.E., Eckert A., Aging sensitizes towards ROS formation and lipid peroxidation in
PS1M146L transgenic mice. Free Radic. Biol. Med., 2006; 40: 850–862
13.
Sohal R.S., Role of oxidative stress and protein oxidation in the aging process. Free
Radic. Biol. Med., 2002; 33: 37–44

62
14.
Lushchak V.I., Free radical oxidation of proteins and its relationship with functional
state of organisms. Biochemistry (Mosc.), 2007; 72: 809–827
15.
Hensley K., Robinson K.A., Gabbita S., Salsman S., Floyd R.A., Reactive oxygen
species, cell signaling, and cell injury. Free Radic. Biol. Med., 2000; 28: 1456–1462
16.
Alvarez B., Radi R., Peroxynitrite reactivity with amino acids and proteins. Amino
Acids, 2003; 25: 295–311
17.
Higuchi Y., Chromosomal DNA fragmentation in apoptosis and necrosis induced by
oxidative stress. Biochem. Pharmacol., 2003; 66: 1527–1535
18.
Cooke M.S., Evans M.D., Dizdaroglu M., Lunec J., Oxidative DNA damage:
mechanisms, mutation and disease. FASEB J., 2003; 17: 1195–1214
19.
Stołowska M., Kłobus G., Charakterystyka flawonoidów i ich rola w kosmetyce i
terapii. Postępy kosmetologii, 2010; 1(1): 8-12
20.
Miller E., Malinowska K., Gałęcka E., Mrowicka M., Kędziora J., Rola flawonoidów
jako przeciwutleniaczy w organizmie człowieka, Pol. Merk. Lek., 2008, XXIV,
144: 556 - 560
21.
Ostrowska J., Skrzydlewska E., Aktywność biologiczna flawonoidów., Postępy
Fitoterapii 2005; 16(3-4): 71-79
22.
Małolepszy U., Urbanek H., Flawonoidy roślinne jako związki biochemicznie czynne,
Wiadomości botaniczne 2000; 44(3/4): 27-37
23.
Lek. med. Miktus Małgorzata, Barwy natury – roślinni sprzymierzeńcy witaminy C,
Nutrition & Heath 2010; 2(51): 1-12
24.
Majewska M, Czeczot H., Flawonoidy w profilaktyce i terapii, Farm Pol, 2009, 65(5):
369-377
25.
Cao G., Sofic E., Perior R.L., Antioxidant and prooxidant behavior of flavonoids:
Structutre –activity relationships. Free Radic. Biol. Med. 1997, 22: 749-760.
26.
Wolski T., Kalisz O., Gerkowicz M., Morawski
M., Rola i znaczenie antyoksydantów
w medycynie ze szczególnym uwzględnieniem chorób oczu, Postępy Fitoterapii
2007, 2: 82-90
27.
Czeczot H., Biological activities of flavonoids – a review. Pol. J. Ford Nutr. Sci. 2000,
50(4): 3-13.
28.
Olszewska M., Flawonoidy i ich zastosowanie w lecznictwie. Farm. Pol. 2003,
59(9): 391-401.

63
29.
Middleton J.E., Kandaswami C., Theoharides T.C., The effects of plant flavonoids on
mammalian cells: implications for inflammation, hart disease, and cancer. Pharmacol.
Rev. 2000, 52(4): 673–751.
30.
Moon Y.J., Wang X., Morris M.E., Dietary flavonoids: effects on xenobiotic and
carcinogen metabolism. Toxicol. In Vitro. 2006, 20(2): 187-210.
31.
Fuhrman B., Aviram M., Flavonoids protect LDL from oxidation and attenuate
atherosclerosis. Curr. Opin. Lipidol. 2001, 12,(1): 41-48.
32.
Violi F., Pignatelli P., Pulcinelli F.M., Synergism among flavonoids in inhibiting
platelet aggregation and H
2
O
2
production. Circulation.2002, 105(8): 53-54.
33.
Sanderson J., McLauchlan R.W., Williamson G., Quercetin inhibits hydrogen
peroxide-induced oxidation of the rat lens. Free Radic. Biol.Med. 1999, 26(5/6): 639-
645.
34.
http://www.sigmaaldrich.com/catalog/product/sigma/q0125?lang=pl®ion=PL
35.
Polifenole roślinne w terapii schorzeń układu krążenia, Panacea2004, 3(8): 22-26
36.
Pałgan K., Sinkiewicz W.: Ochronna rola flawonoidów w chorobach układu sercowo-
naczyniowego. Czynniki ryzyka, 1998.
37.
Majewska-Wierzbicka M., Czeczot H., Flawonoidy w prewencji i leczeniu chorób
układu sercowo-naczyniowego, Pol. Merk. Lek., 2012, XXXII(187): 50-53
38.
Matreska Małgorzata, Quercetin and its derivatives: chemical structure and
bioactivity – a review, Pol. J. Food Nutr. Sci. 2008, 58(4): 407-413
39.
Kwiatkowska Edyta, Składniki herbat w zapobieganiu chorób układu krążenia,
Postępy Fitoterapii 2007, 2: 91-94
40.
Ketan V. Hirpara, Pawan Aggarwal, Amrita J. Mukherjee, Narendra Joshi, Anand C.
Burman, Quercetin and Its Derivatives: Synthesis, Pharmacological Uses with Special
Emphasis on Anti-Tumor Properties and Prodrug with Enhanced Bio-Availability,
Anticancer Agents Med Chem. 2009, 9 (2): 138-161
41.
Vargas A.J., Burd R.: Hormesis and synergy: pathways and mechanisms of quercetin
in cancer prevention and management. Nutrition Reviews. 2010, 68(7): 418-428.
42.
Hadi S.M. i wsp.: Oxidative breakage of cellular DNA by plant polyphenols: A
putative mechanism for anticancer properties. Seminars in Cancer Biology. 2007,
17: 370-376.
43.
Olaf J. Rolinski, Andrew Martin, and David J. S. Birch, Human serum albumin and
quercetin interactions monitored by time-resolved fluorescence: evidence for
enhanced discrete rotamer conformations, J. Biomed. Opt.2007, 12(3): 1-7

64
44.
Bhattacharya, A. A.; Curry, S.; Franks, N. P. Binding of the General Anesthetics
Propofol and Halothane to Human Serum Albumin. The Journal of Biological
Chemistry 2000, 275(49): 38731–38738
45.
Yang, Feng, et al. Effect of human serum albumin on drug metabolism: Structural
evidence of esterase activity of human serum albumin. Journal of Structural Biology
2006, 157: 348-355
46.
Jóźwiak Z., Bartosz G., Biofizyka. Wybrane zagadnienia wraz z ćwiczeniami, PWN
Warszawa 2005.
47.
Kęcki Zbigniew, Podstawy spektroskopii molekularnej, PWN Warszawa 1992
48.
Lakowicz J.R., Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum
Publishers: New York, 2006.
49.
Haken W., Wolf H.Ch., Fizyka molekularna z elementami chemii kwantowej, PWN
Warszawa 1998.
50.
R. Kocjan, Chemia analityczna. Podręcznik dla studentów. Analiza instrumentalna.
Tom 2, PZWL, Warszawa
51.
Badanie przejść fazowych w błonach biologicznych metodą pomiaru anizotropii
fluorescencji,
http://www.zbf.wbbib.uj.edu.pl/documents/3312903/e4024f4e-dddb-
4a34-ada0-95de2d7521fc
52.
Kohri S, Fujii H, Oowada S, Endoh N, Sueishi Y, Kusakabe M, Shimmei M,
Kotake Y. An oxygen radical absorbance capacity-like assay that directly quantifies
the antioxidant's scavenging capacity against AAPH-derived free radicals. Anal.
Biochem. 2009, 386(2): 167–171.
53.
Badanie pojemności antyoksydacyjnej produktu GanoCafe Classic metodą
fluorymetryczną, test ORAC-FL.,Warszawski Uniwersytet Medyczny w Warszawie,
Zakład chemii fizycznej, Wydział farmaceutyczny
54.
http://www.caymanchem.com/app/template/Product.vm/catalog/82235
55.
http://www.sigmaaldrich.com/catalog/product/fluka/46955?lang=pl®ion=PL
56.
http://www.sigmaaldrich.com/life-science/metabolomics/enzyme-explorer/enzyme-
reagents/human-albumin.html
57.
Piwowar A., Zaawansowane produkty utleniania białek. Część I. Mechanizm
powstawania, struktura i właściwości., Pol. Merk. Lek., 2010, XXVIII, 164: 166-169
58.
Bhattacharya, A. A.; Curry, S.; Franks, N. P. Binding of the General Anesthetics
Propofol and Halothane to Human Serum Albumin. The Journal of Biological
Chemistry 2000, 275(49): 38731-38738

65
59.
Stadtman ER, Levine RL (2003) Free radical-mediated oxidation of free amino acids
and amino acid residues in proteins. Amino Acids 25: 207-218
60.
Grune T, Merker K, Sandig G, Davies KJ (2003) Selective degradationof oxidatively
modified protein substrates by the proteasome. Biochem Biophys Res Commun 305:
709–718
61.
Ryazanov AG, Nefsky BS (2002) Protein turnover plays a key role in aging. Mech
Ageing Dev 123: 207–213
62.
Berlett BS, Stadtman ER (1997) Protein oxidation in aging, disease, and oxidative
stress. J Biol Chem 272: 20313-20316
63.
http://www.chem.univ.gda.pl/kchfiz/assets/Uploads/kchfiz/files/luminofory_organiczn
e/cwiczenie%209.pdf
64.
Vincent, J. L. Revelance of albumin in modern critical care medicine. Baillière's best
practice & research. Clinical anaesthesiology 2009, 23(2):183- 191
65.
Nicholson, J P, M R Wolmarans and G R Park. The role of albumin in critical illness.
British Journal of Anaesthesia 2000, 85: 599-610.
66.
Iwao Y., Anraku M., Hiraike M. i wsp.: The structural and pharmacokinetic
properties of oxidized human serum albumin, advanced oxidation protein products
(AOPP). Drug Metab. Pharmacokinet., 2006, 21: 140-146.
Wyszukiwarka
Podobne podstrony:
Karolina Jakołcewicz praca
multimedialana pomoc dydaktyczna Praca magisterska Karoli
Praca magisterska karolina olech
praca magisterska Akty kończące ogólne postępowanie administracyjne
praca-magisterska-a11406, Dokumenty(2)
praca-magisterska-a11222, Dokumenty(2)
praca-magisterska-6811, Dokumenty(8)
praca-magisterska-a11186, Dokumenty(2)
praca-magisterska-7383, Dokumenty(2)
Metody treningowe, Mikołaj praca magisterska
praca-magisterska-a11473, Dokumenty(2)
praca-magisterska-6699, Dokumenty(8)
praca-magisterska-7444, Dokumenty(2)
praca-magisterska-6435, Dokumenty(8)
praca-magisterska-wa-c-7459, Dokumenty(2)
d druku BIBLIOGRAFI1, cykl VII artererapia, Karolina Sierka (praca dyplomowa; terapia pedagogiczna z
praca-magisterska-wa-c-7525, Dokumenty(2)
więcej podobnych podstron