
Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and
high physiological fidelity to the human condition
Michael Charlton, Anuradha Krishnan, [...], and Gregory Gores
Abstract
Although there are small animal platforms that recapitulate some of the histological features of nonalcoholic fatty liver disease,
there are no small animal models of nonalcoholic steatohepatitis (NASH) with consistent hepatocellular ballooning and
progressive fibrosis that also exhibit fidelity to the human condition physiologically. We examined the metabolic and histological
effects of a diet on the basis of the composition of “fast food” (high saturated fats, cholesterol, and fructose). Mice (n = 8 in
each group) were assigned to diets as follows: 1) standard chow (SC), i.e., 13% energy as fat [1% saturated fatty acids (SFA)], 2)
high fat (HF), i.e., 60% energy as fat (1% SFA), and 3) fast food (FF), i.e., 40% energy as fat (12% SFA, 2% cholesterol). All
three diets were supplemented with high fructose. All diets produced obesity. T he HF and FF diets produced insulin resistance.
Liver histology was normal in animals fed the SC diet. Steatohepatitis with pronounced ballooning and progressive fibrosis (stage
2) was observed in mice fed the FF diet. Although the HF diet produced obesity, insulin resistance, and some steatosis;
inflammation was minimal, and there was no increase in fibrosis. T he FF diet produced a gene expression signature of increased
fibrosis, inflammation, and endoplasmic reticulum stress and lipoapoptosis. A diet based on high cholesterol, high saturated fat,
and high fructose recapitulates features of the metabolic syndrome and NASH with progressive fibrosis. T his represents a novel
small animal model of fibrosing NASH with high fidelity to the human condition. T hese results highlight the contribution of
dietary composition to the development of nonalcoholic fatty liver disease and NASH.
Keywords:
nonalcoholic steatohepatitis, hepatocellular ballooning, nonalcoholic fatty liver disease
ON
THE
BASIS
OF
CURRENT
PREVALENCES
of obesity and type 2 diabetes mellitus, nonalcoholic fatty liver disease (NAFLD) can
conservatively be estimated to affect >30,000,000 people in the United States, of which >600,000 are likely to have cirrhosis
(
,
,
). It is estimated that 3% of patients with NAFLD will develop liver-related complications (e.g.,
hepatocellular carcinoma) within 10 yr (
). Liver disease secondary to nonalcoholic steatohepatitis (NASH) is already a common
indication for liver transplantation (
). T he scale of the public health burden of NAFLD is likely to increase in parallel with
increases in the prevalence and severity of obesity in the United States and globally. T here are no approved pharmacotherapies
for NAFLD and NASH.
NAFLD, for the great majority of affected individuals, is one of many consequences of chronic overnutrition and obesity.
Hepatic histological findings in chronically overnourished individuals range from entirely normal, to simple steatosis, to
steatohepatitis with progressive fibrosis (
). T he various histological features of NAFLD are thus not inevitable
consequences of overnutrition, obesity, or insulin resistance but are based on the balance between biological mechanisms for
hepatic susceptibility and the physiological consequences of overnutrition. Human studies of the pathophysiology of NAFLD and
NASH have been limited, in part, by difficulties in distinguishing primary cause(s) from secondary effects and epiphenomena
related to obesity and liver disease. A fuller understanding of the physiology of NAFLD and NASH has been impeded by the
absence of an animal model that closely recapitulates the human condition. Although there are an increasing number of animal
models of NAFLD, none of these models fully produces the metabolic profile in concert with the histological patterns seen in
humans (
,
). T he widely employed methionine-choline-deficient mouse (
) produces steatohepatitis and
fibrosis, but in a metabolic context that is distinct from that of humans with NASH. A recent murine model incorporating
prolonged administration of a “Western diet,” with high saturated fat and cholesterol content, was able to reproduce NASH with
some increase in fibrosis markers, but not ballooning (
). T he lack of a substantial content of fructose in the Western diet may
have been important physiologically, as the addition of high fructose content to a diet high in saturated fat and cholesterol has
been seen to reproduce all the features of NASH, including ballooning in large animals (
), and is a typical feature of the diet of
humans with NASH (
). Recreation of NAFLD through genetic manipulation, such as leptin receptor deficiency, can
result in the loss of an important component of fibrosis signaling (
,
). More recent overnutrition-based models have
demonstrated substantial metabolic similarity to humans with NAFLD and NASH but incompletely reproduce the histological
features of NASH (
). Most importantly, none of the small animal models consistently produces NASH with
hepatocellular ballooning and progressive hepatic fibrosis in a context of high fidelity to the physiological profile seen in humans
with fibrosing NASH.
We sought to develop a new rodent model of fibrosing NASH by reproducing the physiological milieu seen in humans with NASH,
i.e., physical inactivity and chronic overnutrition with a high caloric intake rich in saturated fats and fructose. Because fibrosis in
patients with NASH typically evolves over prolonged periods of time, we planned to continue overnutrition and physical
inactivity for longer than dietary interventions in previous studies in overnutrition rodent models of NASH. T he availability of a
more “accurate” mouse model of NASH would greatly facilitate studies of the pathobiology of NAFLD and NASH and also the
screening of potential therapies.
MATERIALS AND METHODS
Animals.
Genetically unaltered C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) were maintained in individual cages (1
mouse per cage to promote sedentary movement patterns) and had free access to standard rodent chow and water for 1 wk until
the start of the experiment. T he animals were then randomly assigned to three groups receiving different diets (
) for 25
wk: 1) fast food (FF) diet (relatively rich in saturated fats and cholesterol and fructose), providing 40% of energy as fat (milk fat,
12% saturated) with 2% cholesterol (AIN-76 Western Diet, T est Diet), 2) high-fat (HF) diet (rich in nonsaturated fats and
fructose), providing 60% of energy as fat (milk fat, 0.8% saturated; DIO Basal Purified Diet, Crisco, T est Diet), and 3) standard
chow (SC) diet, providing 13% of energy as fat (milk fat, 0.9% saturated; PicoLab Rodent Diet 20, Lab Diet).
Diet composition
High-fructose corn syrup (HFCS, 42 g/l final concentration) was also administered in the drinking water of all mice. Detailed diet
compositions are shown in
. At 6 mo (174 days), mice were weighed and euthanized by carbon dioxide inhalation, blood
was drawn by cardiac puncture, and the liver was excised, weighed, and apportioned for RNA/DNA extraction or tissue
histochemistry as flash-frozen tissue or preserved in 10% buffered formalin. All animal procedures were performed in accordance
with the guidelines of the Institutional Animal Care and Use Committee of the Mayo Clinic, which reviewed and approved all
protocols. T he gender makeup of each group is as follows: 5 males and 3 females in the SC diet group, 4 males and 3 females in
the FF diet group, and 4 males and 4 females in the HF diet group. Caloric intake was not recorded.
Biochemistry and lipids.
Serum glucose was measured using a blood glucose monitor (Assure 4, Arkray). Serum aspartate
aminotransferase (AST ) levels were measured using standardized and automated procedures of the diagnostic laboratory of the
Mayo Clinic. Commercial ELISA kits were used to measure levels of insulin, growth hormone, adiponectin (EZRMI-13K,
EZRMGH-45K, and EZMADP-60K, Millipore), cholesterol, and insulin-like growth factor I (MG100, R & D Systems) following
the manufacturers' instructions.
Histology, immunohistochemistry, and digital image analysis.
Formalin-preserved liver tissue samples were embedded in paraffin and
sectioned (4–6 μm thick). Deparaffinized, hydrated serial liver tissue sections were stained with hematoxylin-eosin, Masson's
trichrome, and picrosirius red using standardized protocols of the Pathology Department of the Mayo Clinic. T issue sections
stained with hematoxylin-eosin and Masson's trichrome were analyzed for symptoms of NASH by a pathologist (S. Sanderson)
who was blinded to the study. T he classification of Brunt et al. (
) was used to assign numerical scores to steatosis, inflammation,
and fibrosis. Quantitative analysis of collagen deposition was based on digitally acquired images of picrosirius red-stained slides.
Digital images were acquired on a Leica DMR microscope mounted with an Olympus DP70 camera. A series of images covering
>90% of the total tissue section were generated at uniform settings of magnification, light, and exposure time. Quantitative
analysis was performed using KS400 software (Carl Zeiss). Data are presented as percentage of total tissue area. Additional serial
sections were stained with antibody against lumican (AF2846, R & D Systems) and anti-smooth muscle actin (ASMA; NB110-
55432, Novus Biologicals) at dilutions that were optimized for each stain. Bound antibodies were detected using
diaminobenzidine, and sections were counterstained with hematoxylin.
Apoptotic cells were visualized by T dT dUT P nick end labeling of DNA strand breaks with fluorescein using a commercially
available kit following the manufacturer's instructions (11684795910, Roche). T issue sections were counterstained with 4′,6-
diamidino-2-phenylindole for visualization of nuclei. Four to six images were generated per section. Nuclei were quantified using
KS400 software. Fluorescing apoptotic nuclei were individually verified for colocalization with nuclear staining, and the count
was normalized to total nuclei count.
8-Hydroxydeoxyguanosine.
Genomic DNA was extracted from ~150 mg of flash-frozen liver tissue (Wako Chemicals) and diluted
to a uniform concentration of 1.5 mg/ml. DNA was hydrolyzed/oxidized, and 8-hydroxydeoxyguanosine was measured using a
competitive ELISA according to the manufacturer's instructions (Wako Chemicals).
RNA isolation and quantitative RT-PCR.
T otal RNA was isolated from frozen liver tissue using the RNeasy Plus kit (Qiagen)
according to the manufacturer's instructions. Equal quantities of total RNA were reverse-transcribed into cDNA using random
hexamers (T ranscriptor High Fidelity cDNA synthesis kit, Roche). Real-time PCR for collagen, ASMA, transforming growth
factor-β1 (T GFβ1), T NFα, fatty acid-binding protein (FABP), SOD1, and specificity protein 1 (Sp1) was performed on an
iCycler (Bio-Rad) in a total volume of 20 μl using LightCycler 480 SYBR Green 1 Master Mix (Roche, Indianapolis, IN).
Eukaryotic translation initiation factor 2α kinase 3 (PERK), CCAAT -enhancer-binding protein homologous protein (CHOP),
Bcl-2 binding component 3 (PUMA), activating transcription factor 6 (AT F6), and GRP78, a key regulator of the unfolded
protein response, were analyzed on a Roche LightCycler 480. Primers were generated through software programs available from
the Universal Probe Library, Roche, or the National Center for Biotechnology Information or were commercially available (see
Supplemental T able S1 in Supplemental Material for this article, available online at the Journal website, for primer sequence and
source). 18S gene expression was stable across the three experimental groups, and the 18S gene was used as the reference gene to
normalize target genes. All data are expressed as fold changes over expression in mice reared on the SC diet.
Statistical analysis.
Values are means ± SE representing replications within an experiment. Statistical significance was determined
by Student's t-test using two-tailed analyses. P < 0.05 was considered significant.
RESULTS
Animals reared on the FF diet recapitulate the clinical phenotype of NASH: physical characteristics and serum profile.
Animals reared on
the FF and HF diets were significantly more obese, averaging 42 and 44 g, respectively, than those raised on the SC diet (29 g;
). T he initial rate of increment in weight was greatest for the FF animals, although by 6 mo the weights of the FF and HF
groups were statistically and numerically similar. Weight of the SC animals did not increase significantly during the experiments (
). FF animals developed hepatomegaly, with significantly higher mean liver weights as a proportion of total body weight
(8.5%) than animals fed the HF (4.2%) or SC (4.4%) diet (P = 0.0003). Serum cholesterol was significantly higher in FF than HF
or SC animals. Serum AST was significantly more elevated in FF (488.3 ± 26.8 IU/l) than HF (121.1 ± 9.3 IU/l, P = 0.0003) and
SC (176.9 ± 10.3 IU/l, P = 0.001) animals. AST levels were within the normal range for mice (54–298 IU/l) in the SC and HF
groups. Although numerically higher in the HF group, the difference was not statistically significant. Serum glucose and insulin
levels were similarly and significantly higher in FF and HF than SC animals (
). Consequently, the homeostasis model
assessment of insulin resistance was significantly higher for FF and HF than SC animals. Growth hormone levels were, similarly,
lower in FF and HF than SC animals. Adiponectin levels were significantly lower in FF than HF or SC animals (
).
Phenotype and serum biochemical profile

Weight change over time in animals fed standard chow (SC), fast food (FF), and high-fat (HF) diets. Although initial rate
of increment in weight was greatest for FF animals, by 6 mo, weights of FF and HF animals were statistically and
numerically similar.
...
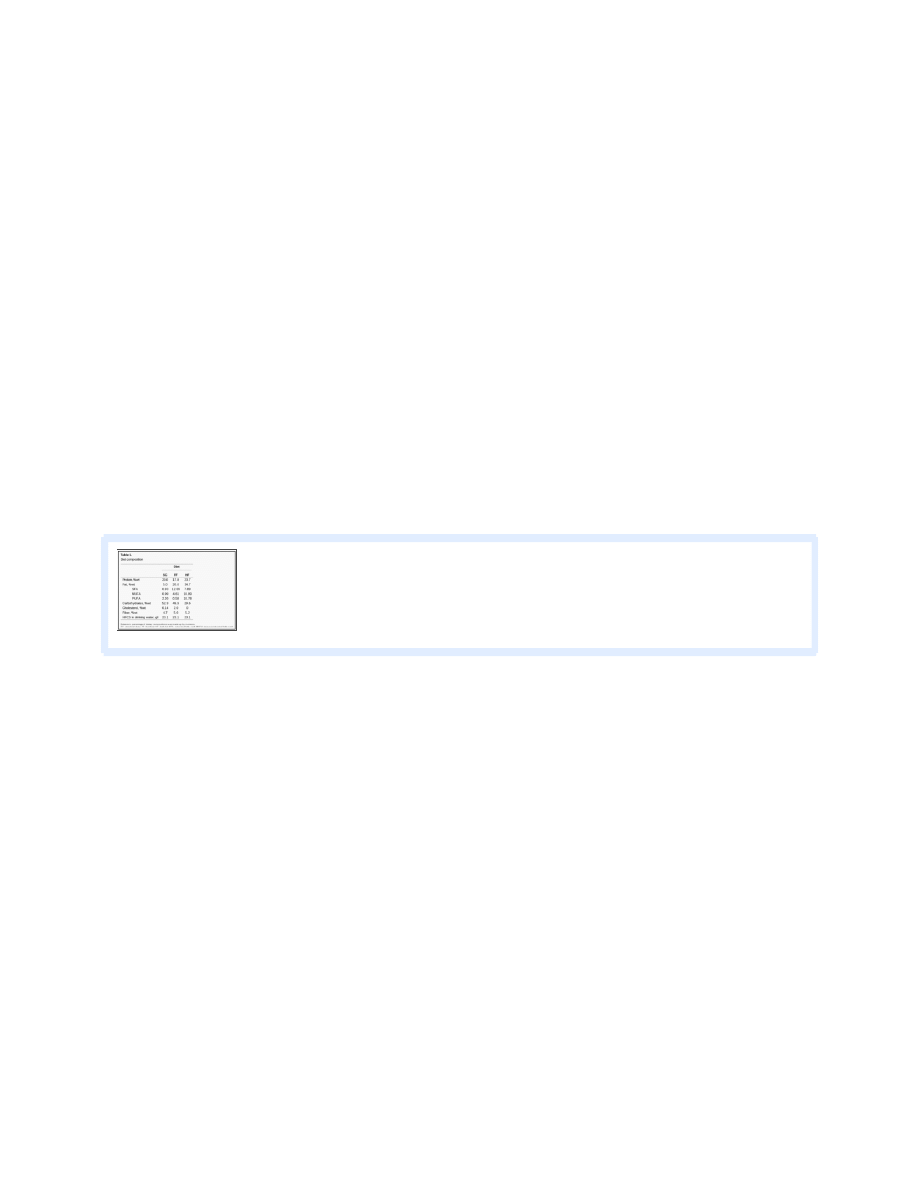
FF diet induces steatosis, pronounced hepatocellular ballooning, and steatofibrosis of the liver.
Hematoxylin-eosin-stained sections of
liver tissue of all animals were scored for symptoms of NASH by a pathologist (S. Sanderson) who was blinded to the study. Scores
for steatosis, inflammation, and fibrosis were assigned according to the classification of Brunt et al. (
). T here was no evidence of
steatosis in SC mice (
). In contrast, all FF animals developed cellular ballooning, paracinar steatosis, and intra-acinar
inflammation commonly associated with severe NASH (
). T he average score for steatosis for FF mice was 2.71 (
).
Although HF animals also showed evidence of steatosis, with an average score of 2.13, steatosis was largely microvesicular, and
there was little or no evidence of inflammation (
). Hepatic triglyceride levels were 3.48 ± 0.40, 3.53 ± 0.33, and
3.99 ± 0.18 nmol in SC, FF, and HF animals, respectively. Although hepatic triglycerides were numerically higher in HF than FF
and SC animals, the difference was not significantly different.
Hematoxylin-eosin (H&E, left)- and Masson's trichrome (right)-stained sections of liver tissue from SC, FF, and HF animals.
There was no steatosis in mice reared on the SC diet. All FF animals developed cellular ballooning, paracinar steatosis,
...
Hematoxylin-eosin scores of liver from SC, FF, and HF mice
Hepatocellular ballooning, which has been difficult to recreate in animal models of NAFLD/NASH, was a pronounced histological
feature in FF animals (
). Furthermore, there was no evidence of fibrosis in SC or HF animals. By comparison, in six of the
seven FF animals, there was evidence of perisinusoidal and pericellular fibrosis (stage 2 of 4;
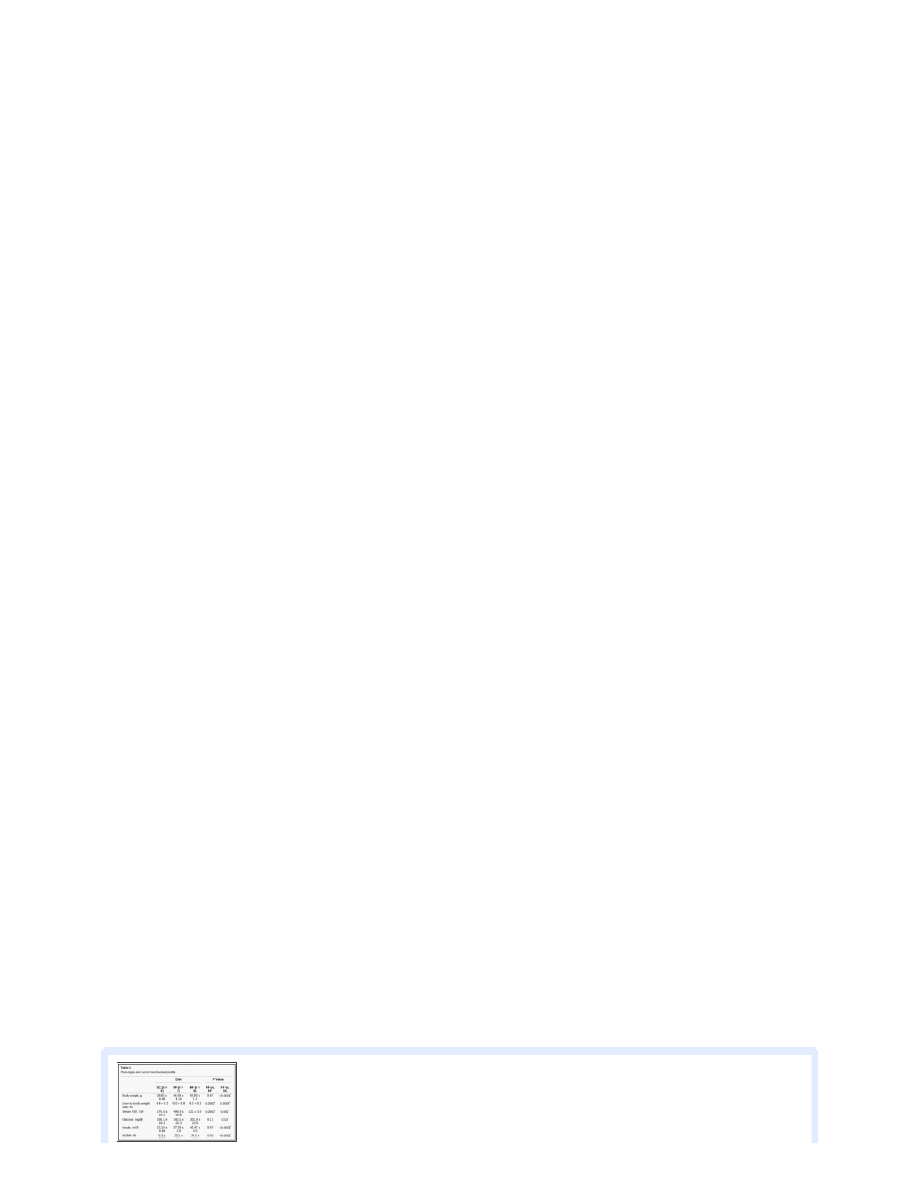
). Picrosirius red-stained tissue
sections were analyzed for collagen distribution. T he increase in collagen-stained area was significantly greater (~2-fold, P <
0.05) in tissue sections from FF than HF or SC mice. Gene expression studies indicated the same directional differences between
study groups, i.e., 17-fold higher in FF than SC or HF animals. Since the major sources of collagen in the liver are hepatic stellate
cells, tissue sections were immunostained for ASMA, a known indicator of their activation. Morphometric analysis of digital
images indicated that ASMA was significantly overexpressed in HF mice (P < 0.05;
). In addition to collagen, lumican, an
extracellular matrix protein involved in collagen fibrogenesis, has been previously shown to be upregulated in NASH (
).
Lumican gene expression was significantly increased in FF compared with SC and HF animals (P < 0.01;
).
Immunohistochemical analysis also showed that lumican was overexpressed (2-fold, P < 0.05) in FF compared with HF and SC
mice.
Top: higher-magnification view of Masson's trichrome-stained sections from FF mice. Bottom: high-magnification view of
representative hepatocellular ballooning (arrows). Hepatocellular ballooning has been difficult to recreate consistently in
small animal
...
Collagen staining with picrosirius red (left), anti-smooth muscle actin (ASMA, middle), and lumican (right) in SC, FF, and
HF animals. On digital image analysis, collagen-stained area was significantly more abundant (2-fold, P < 0.05) in FF than
...
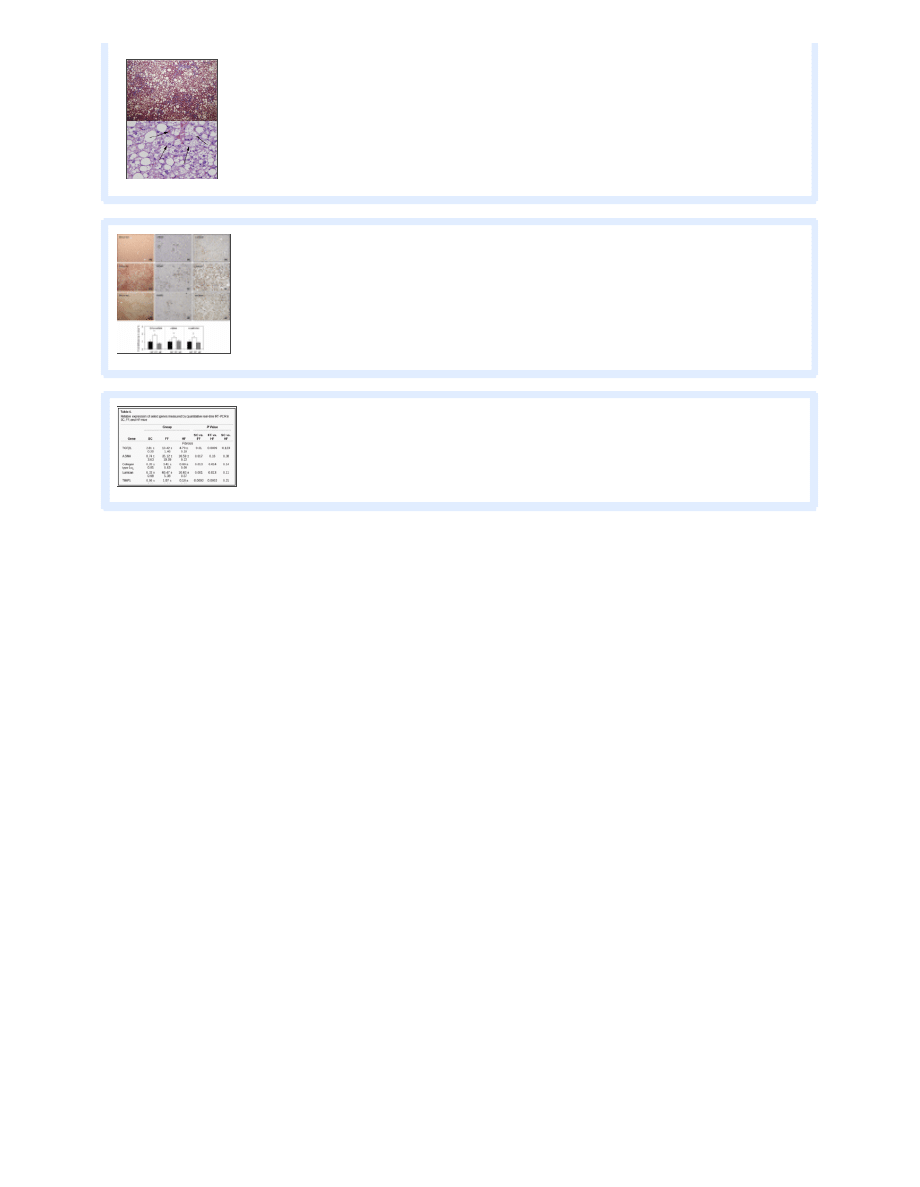
Relative expression of select genes measured by quantitative real-time RT-PCR in SC, FF, and HF mice
Profibrotic and proinflammatory pathways are activated in FF animals.
Expression levels of the profibrotic and proinflammatory genes
were similar between SC and HF animals. Hepatic expression was fivefold higher for T GFβ1 (P = 0.0009) and ninefold higher for
T NFα (P = 0.0001) in FF than HF animals (
). FF animals also demonstrated increased hepatic expression of tissue
inhibitor of metalloproteinase 1 (T IMP1, ~30-fold, P = 0.0002) compared with HF and SC animals. Hepatocyte growth factor
was also upregulated in FF mice compared with HF and SC animals.
Indexes of cellular stress, apoptosis, and unfolded protein response are differentially expressed in FF animals.
Oxidative stress, free
radicals, and endoplasmic reticulum (ER) disturbances have been associated with the onset and development of NASH in clinical
investigations. Hepatic gene expression of PERK, CHOP, and AT F6, proteins that are upregulated during ER stress, was
significantly (P < 0.05) increased in FF animals compared with SC animals (
). However, expression of PUMA, a protein
associated with ongoing lipoapoptosis in clinical NASH (
), was similar between groups. SOD1, an enzyme associated with
removal of free radicals from the cytoplasmic milieu, has been previously shown to be downregulated in clinical NASH (
). Gene
expression of SOD1 was similar between all experimental groups (
). Abundance of 8-hydroxydeoxyguanosine, an indicator
of oxidative DNA damage induced by oxidative radicals, was similar in all three groups (
). FF animals experienced a
greater degree of hepatic apoptosis activity as measured by the number of T dT dUT P nick end label-positive cells per 1,000 cells
(1.55 ± 0.21, 10.34 ± 1.73, and 4.90 ± 2.14 in SC, FF, and HF, respectively, P = 0.002 for FF vs. SC and P = 0.08 vs. HF).
GRP78, a key regulator of the unfolded protein response, was similarly expressed in all three groups.
The transcription factor Sp1 is upregulated in FF mice.
Sp1 regulates and interacts with a number of other transcription factors,
including Smad, which regulates T GFβ1 signaling (
). Sp1 can also regulate collagen and lumican gene transcription (
),
both of which were overexpressed in FF mice. We therefore looked at gene expression levels of Sp1 in these groups of mice. Sp1
gene expression was significantly (P < 0.05) upregulated in the FF mice compared with SC or HF mice.
DISCUSSION
A small animal model that produces NASH with hepatocellular ballooning and fibrosis in a physiological environment with
fidelity to the condition in humans with NASH has been a longstanding need. While an increasing number of animal models have
been reported to develop features of NAFLD and NASH, none consistently and simultaneously recapitulates the combined

metabolic, physical, and histological features in humans with NASH with progressive fibrosis. We had hypothesized that
recreating the nutritional and physical environment seen in NASH, with progressive fibrosis and a sedentary lifestyle in
conjunction with chronic overnutrition with a diet high in calories and enriched with saturated fats and fructose, might produce
the clinical and histological phenotype of NASH with fibrosis. T he primary result of this study is that the FF mouse exhibits all
the hallmarks of fibrosing NASH most commonly observed in humans: obesity, metabolic syndrome, steatohepatitis,
hepatocellular ballooning, and progressive fibrosis. T he most significant new feature of this model of NASH is the presence of
ballooning with the frequent development of fibrosis. An approximately twofold increase in collagen-stained area was observed in
FF mice compared with HF and SC animals. Evidence of increased hepatic fibrosis in the FF mouse model was present
histologically by Masson's trichrome and picrosirius red staining (with digital analysis) and also by expression levels of the
profibrotic and proinflammatory gene T GFβ1, with associated increased stellate cell activation and increased abundance of
mRNA for procollagen.
It is, of course, important to consider how this model is distinct from other animal models of NASH and what the potential
significance of this model might be. Although the prevalence of NAFLD is indisputably high, ~25–50% among obese individuals
(
,
,
), the prevalence of NASH with progressive fibrosis is proportionally low. T wo recent large, prospective
cross-sectional studies reported the frequency of stage 2 or higher fibrosis among obese individuals to be 2.5–5%, with <1%
developing stage 3 or higher fibrosis. Liver-related clinical consequences of NAFLD and NASH (e.g., related to portal
hypertension and cirrhosis) are unlikely in patients without progressive fibrosis (
). T herapeutic modalities for NASH will need
to prevent or reverse progressive fibrosis to confer clinical benefit in terms of preventing liver-related morbidity and mortality.
Given the inherent limitations and complexities of studying the biology of NAFLD and NASH in humans, a fuller understanding
of the mechanistic basis of steatofibrosis and the identification of methods to prevent/reverse steatofibrosis are likely to be
expedited by the availability of a small animal model that also exhibits steatofibrosis. Existing models of NAFLD/NASH are
characterized by 1) production of steatohepatitis (with or without fibrosis), but without features of the metabolic syndrome, or 2)
reproduction of the metabolic syndrome, but with incomplete histological features of NASH. T he methionine-choline deficiency
model, for example, produces NASH and even fibrosis but is associated with substantial weight loss, low serum leptin levels, and
lack of insulin resistance (
). T he ob/ob mouse develops obesity but is leptin-deficient and lacks the inflammatory and
fibrosis components of NASH (
). Overfeeding models, while fairly efficient in producing obesity, have been highly variable
in resulting hepatic histology (1- to 7-mo range in duration of feeding) (
,
,
). Overfeeding with fat in a mouse strain
susceptible to obesity and insulin resistance has produced encouraging results. A model of a high-fat diet in male C57BL/6J mice
led to the development of features of the metabolic syndrome and steatohepatitis but only mild fibrosis after 50 wk (
). A
model utilizing male C57BL/6 mice that were fed high-fat chow containing trans fats and a HFCS for up to 16 wk produced
obesity, features of the metabolic syndrome, and hepatic steatosis with associated necroinflammatory changes. Although signals
for hepatic fibrosis were increased, hepatic fibrosis was not. An important observation of this study was that HFCS promoted
food consumption (
). In our current study, we also used C57BL/6 mice and a high-fat, HFCS diet. T he basis of development of
NASH and progression to stage 2 fibrosis (
) in animals fed a FF diet merits consideration. T he diet chosen for our study included
a relatively high abundance of saturated fats, 12% of total calories, with 2.2% as cholesterol, and >23 g/l HFCS. In addition, we
housed the mice singly (1 mouse per cage) to promote sedentary behavior. Because FF mice were exposed to greater amounts of
saturated fats/cholesterol in addition to higher carbohydrate intake, it is impossible to know the relative contributions of high
carbohydrate and high saturated fat exposure to the response; it is only possible to know that the combination of the two
produced the observed phenotype. In addition, because HFCS was included in all three diets but caloric intake was not reported,
defining a specific contribution of HFCS would require further study. T he fact that our animals fed the HF diet, in which saturated
fats were only minimally present, developed obesity, insulin resistance, and steatosis, but not NASH or fibrosis, highlights the
importance of dietary fat type in the development of NASH and fibrosis. Dietary cholesterol has been identified as an essential
determinant of progression from NAFLD to NASH in a murine model of NASH (
). T he inflammatory effects of dietary
cholesterol are thought to be mediated by cholesterol oxidation products sensitizing the liver to T NFα and nuclear factor-κB
signaling (
,
). In addition, excess dietary cholesterol has been shown to induce T GFβ1 (
) and T IMP1 expression (
). We
observed relative increases in T GFβ1 and T IMP1 in FF animals. T he effect of cholesterol appears to be relatively specific, as
mitochondrial loading of free cholesterol, but not triglycerides and free fatty acids, decreases mitochondrial glutathione and

sensitizes it to the T NFα-mediated apoptosis of hepatocytes (
). Our model is similar in many respects to that recently
reported by Matsuzawa et al. (
), who fed C57BL/6 mice a diet containing 1.25% cholesterol and 0.5% cholate for 24 wk. T he
model of Matsuzawa et al. also produced NASH with fibrosis. T he fibrosis was, however, not characterized/staged. T he use of
HFCS, a common element of the Western diet and ubiquitous component of fast food products, also distinguishes our model from
that reported by Matsuzawa et al. Because the endocrinologic profile of the mice used by Matsuzawa et al. was also not reported,
the fidelity of the model to the human condition is unknown. HFCS is thought to be important in the pathogenesis of NAFLD
and NASH in mice (
), promoting food consumption, obesity, increased T NFα activation (
), and
impaired insulin sensitivity (
). T his difference may account for the much greater mean body weight at 24 wk in our FF model
(44.9 ± 1.14 g) than in the HFCS-free high cholesterol/atherogenic diet model of Matsuzawa et al. (26.4 ± 1.1 g). Insulin
resistance, as measured by homeostasis model assessment of insulin resistance, was also numerically much greater in our FF model
than in the model described by Matsuzawa et al. Similarly, a recent murine model incorporating prolonged administration of a
Western diet, containing high levels of saturated fat and cholesterol without a high level of fructose, was able to reproduce NASH
with some increase in fibrosis markers but not hepatocellular ballooning (
). Lack of a substantial content of fructose in the
Western diet is also suggested to have been important physiologically, as addition of a high level of fructose to a diet high in
saturated fat and cholesterol has been seen to reproduce all the features of NASH, including ballooning in large animals (
), and
is a typical feature of the diet of humans with NASH (
,
). T hus, although atherogenic, high-fat/high-cholesterol diets
replicate human histopathology, the metabolic status appears to be distinct. Hebbard and George (
) commented that “Further
studies are required to address whether alterations in fat composition or the addition of other dietary factors associated with
metabolic status, such as fructose, can lead to this model attaining the relevant human metabolic parameters.” T he specific
contribution of HFCS to the metabolic and histological features observed in our mice remains to be determined.
Histological features other than fibrosis that define NASH in humans, assessed according to the classification of Brunt et al. (
),
were also seen in FF mice. All FF animals developed cellular ballooning, paracinar steatosis, and intra-acinar inflammation.
Although animals reared on the HF diet also showed evidence of steatosis, this was largely microvesicular, and there was little or
no evidence of inflammation (
). Since hepatic fibrosis is characterized by aberrant collagen deposition, picrosirius
red-stained tissue sections were analyzed for collagen distribution. Gene expression studies indicated the same trends: 17-fold
higher collagen expression in FF than SC or HF animals. Since the major source of collagen in the liver is hepatic stellate cells,
tissue sections were immunostained for ASMA, a known indicator of their activation. Morphometric analysis of digital images
indicated that ASMA was significantly overexpressed in HF mice (
). In addition to collagen, lumican, an extracellular
matrix protein involved in collagen fibrogenesis, has been previously shown to be upregulated in NASH (
). Lumican gene
expression was significantly increased in FF animals compared with SC and HF animals. Immunohistochemical analysis also
showed that lumican was overexpressed (2-fold) in FF mice compared with HF and SC mice.
In addition to a histological picture similar to the human condition, it is desirable that small animal models of NASH would also
have a metabolic profile that has fidelity to that of humans who develop NASH with fibrosis, specifically, insulin resistance (
,
), high systemic leptin levels (
), and low levels of growth hormone (
,
), dehydroepiandrosterone (
), and
adiponectin (
). Circulating cytokines are also abnormal in NASH, with increased circulating levels of T NFα and markers of
apoptosis (
). T he FF diet model recreates each of these features. Although the link(s) between hepatic steatosis,
inflammation, and fibrosis is not fully known, increased oxidative stress is a feature of animal models of steatohepatitis (
) and
humans with NAFLD (
) with free fatty acid sensitization of hepatocytes to T NFα-related apoptosis-inducing ligand (
).
Hepatic gene expression of PERK and CHOP, two proteins that are upregulated during ER stress in NASH (
), was significantly
increased in FF animals. Similarly, AT F6 was relatively overexpressed in FF animals compared with HF animals. Expression of
GRP78/BiP, a key regulator of the unfolded protein response, was greatest in FF animals, but the difference was not statistically
significant. SOD levels, shown to be reduced in humans with NASH with progressive fibrosis (
), were also comparatively low in
FF animals. NASH with progressive fibrosis in humans has recently been reported to be associated with increased intrahepatic
expression of lumican and decreased FABP1 (
). T hese proteins were similarly differentially abundant in the livers of the FF
mice, suggesting that the increased lumican and attenuated FABP1 expression in NASH expression may be mediated by dietary
cholesterol and hypercholesterolemia.

Several technical aspects of the study merit consideration. Reproducibility is a key determinant of the success of any animal
model of disease. T he strain of mice used, C57BL/6, is, of course, commercially available. T he FF diet comprised a standard,
commercially available chow (AIN-76 Western Diet, T est Diet) with added fructose. While many variables may have contributed
to the net effect of the FF diet model, the conditions are not difficult to reproduce. We housed animals singly to encourage
sedentary behavior. Being housed singly may also have provoked a stress response and contributed to the overall observed
changes in the FF group. As animals in all the study groups were housed singly, we do not believe that the stress of being housed
singly could explain the relative differences between the three groups, however.
In conclusion, a diet based on the composition of “fast food” (high cholesterol, high saturated fat, and high fructose)
administered for 6 mo recapitulates features of the metabolic syndrome and NASH with progressive fibrosis in C57BL/6 mice.
T his represents a novel small animal model of fibrosing NASH with high fidelity to the human condition. T hese results highlight
the contribution of dietary composition in the development of NAFLD and NASH.
GRANTS
T his work has been supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants RO1 DK-41876 (to G.
Gomes) and DK-069757-05 (to M. Charlton) and General Clinical Research Center Grant RR-00585.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
Supplementary Material
Supplemental Table:
Article information
Am J Physiol Gastrointest Liver Physiol. Nov 2011; 301(5): G825–G834.
Published online Aug 11, 2011. doi:
PMCID: PMC3220319
Divisions of Gastroenterology and Hepatology and
Anatomic Pathology, Mayo Clinic and Foundation, Rochester, Minnesota
Corresponding author.
Address for reprint requests and other correspondence: M. Charlton, Div. of Gastroenterology and Hepatology, Mayo Clinic and Foundation CH-10, 200 First
St. SW, Rochester, MN 55905 (e-mail:
).
Received April 12, 2011; Accepted August 3, 2011.
© 2011 the American Physiological Society
other articles in PMC.
Articles from American Journal of Physiology - Gastrointestinal and Liver Physiology are provided here courtesy of American Physiological Society
REFERENCES
1. Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, Diehl AM. Increased fructose consumption is associated with
1
2

fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 51: 1961–1971, 2010. [
2. Adams LA, Lymp JF, St Sauver, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a
population-based cohort study. Gastroenterology 129: 113–121, 2005. [
]
3. Angulo P. Nonalcoholic fatty liver disease and liver transplantation. Liver Transpl 12: 523–534, 2006. [
4. Angulo P. Nonalcoholic fatty liver disease. J Gastroenterol Hepatol 346: 1221–1231, 2002 [
5. Angulo P, Alba LM, Petrovic LM, Adams LA, Lindor KD, Jensen MD. Leptin, insulin resistance, and liver fibrosis in human nonalcoholic fatty
liver disease. J Hepatol 41: 943–949, 2004. [
6. Arsov T, Larter CZ, Nolan CJ, Petrovsky N, Goodnow CC, Teoh NC, Yeh MM, Farrell GC. Adaptive failure to high-fat diet characterizes
steatohepatitis in Alms1 mutant mice. Biochem Biophys Res Commun 342: 1152–1159, 2006. [
]
7. Batts KP, Ludwig J. Chronic hepatitis: an update on terminology and reporting. Surg Pathol 19: 1409–1417, 1995 [
]
8. Bell LN, Theodorakis JL, Vuppalanchi R, Saxena R, Bemis KG, Wang M, Chalasani N. Serum proteomics and biomarker discovery across the
spectrum of nonalcoholic fatty liver disease. Hepatology 51: 111–120, 2010. [
] [
9. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging
the histological lesions. Am J Gastroenterol 94: 2467–2474, 1999. [
10. Byron D, Minuk GY. Clinical hepatology: profile of an urban, hospital-based practice. Hepatology 24: 813–815, 1996. [
11. Carmiel-Haggai M, Cederbaum AI, Nieto N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J
19: 136–138, 2005. [
]
12. Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, Kahraman A, Garrison SP, Zambetti GP, Charlton MR, Gores GJ.
JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem 284: 26591–26602, 2009. [
13. Charlton M. Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol 2: 1048–1058,
2004. [
14. Charlton M, Angulo P, Chalasani N, Merriman R, Viker K, Charatcharoenwitthaya P, Sanderson S, Gawrieh S, Krishnan A, Lindor K. Low
circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology 47: 484–492, 2008.
15. Charlton M, Viker K, Krishnan A, Sanderson S, Veldt B, Kaalsbeek AJ, Kendrick M, Thompson G, Que F, Swain J, Sarr M. Differential
expression of lumican and fatty acid binding protein-1: new insights into the histologic spectrum of nonalcoholic fatty liver disease. Hepatology 49:
1375–1384, 2009. [
] [
]
16. Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, Karim R, Lin R, Samarasinghe D, Liddle C, Weltman M,
George J. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 35: 373–
379, 2002. [
]
17. Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol 40: S5–S10, 2006. [
18. Colles SM, Irwin KC, Chisolm GM. Roles of multiple oxidized LDL lipids in cellular injury: dominance of 7β-hydroperoxycholesterol. J
Lipid Res 37: 2018–2028, 1996. [
19. DeLeve LD, Wang X, Kanel GC, Atkinson RD, McCuskey RS. Prevention of hepatic fibrosis in a murine model of metabolic syndrome with
nonalcoholic steatohepatitis. Am J Pathol 173: 993–1001, 2008. [
] [
20. Deng QG, She H, Cheng JH, French SW, Koop DR, Xiong S, Tsukamoto H. Steatohepatitis induced by intragastric overfeeding in mice.
Hepatology 42: 905–914, 2005. [
]
21. Ellenrieder V. TGFβ regulated gene expression by Smads and Sp1/KLF-like transcription factors in cancer. Anticancer Res 28: 1531–1539,

2008. [
22. Flegal KM. The obesity epidemic in children and adults: current evidence and research issues. Med Sci Sports 31 Suppl 11: S509–S514, 1999
23. Fraser A, Longnecker MP, Lawlor DA. Prevalence of elevated alanine aminotransferase among US adolescents and associated factors: NHANES
1999–2004. Gastroenterology 133: 1814–1820, 2007. [
] [
24. Ground KE. Liver pathology in aircrew. Aviat Space Environ Med 53: 14–18, 1982. [
]
25. Harris MI, Flegal KM, Cowie CC, Eberhardt MS, Goldstein DE, Little RR, Wiedmeyer HM, Byrd-Holt DD. Prevalence of diabetes, impaired
fasting glucose, and impaired glucose tolerance in US adults: the Third National Health and Nutrition Examination Survey, 1988–1994. Diabetes
Care 21: 518–524, 1998. [
26. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 8: 35–44, 2011. [
27. Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology 40:
46–54, 2004. [
]
28. Hultcrantz R, Glaumann H, Lindberg G, Nilsson LH. Liver investigation in 149 asymptomatic patients with moderately elevated activities of
serum aminotransferases. Scand J Gastroenterol 21: 109–113, 1986. [
29. Ichikawa T, Hamasaki K, Ishikawa H, Ejima E, Eguchi K, Nakao K. Non-alcoholic steatohepatitis and hepatic steatosis in patients with adult
onset growth hormone deficiency. Gut 52: 914, 2003. [
30. Ichikawa T, Nakao K, Hamasaki K, Furukawa R, Tsuruta S, Ueda Y, Taura N, Shibata H, Fujimoto M, Toriyama K, Eguchi K. Role of
growth hormone, insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 in development of non-alcoholic fatty liver disease.
Hepatol Int 1: 287–294, 2007. [
] [
31. Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, Lang T, Fukuda T, Yamashina S, Kitamura T, Sato N. Leptin receptor-
mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 122: 1399–1410, 2002.
32. Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, Hirose H, Ito M, Ishihara A, Iwaasa H, Kanatani A. Longitudinal analysis of
murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res 37: 50–57, 2007. [
]
33. Larter CZ. Not all models of fatty liver are created equal: understanding mechanisms of steatosis development is important. J Gastroenterol
Hepatol 22: 1353–1354, 2007. [
]
34. Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol 23: 1635–1648,
2008. [
35. Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol
37: 206–213, 2002. [
]
36. Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, Sturek M, Chalasani N. Nutritional model of steatohepatitis and
metabolic syndrome in the Ossabaw miniature swine. Hepatology 50: 56–67, 2009. [
] [
37. Li JM, Datto MB, Shen X, Hu PP, Yu Y, Wang XF. Sp1, but not Sp3, functions to mediate promoter activation by TGF-β through canonical
Sp1 binding sites. Nucleic Acids Res 26: 2449–2456, 1998. [
]
38. Li Y, Aoki T, Mori Y, Ahmad M, Miyamori H, Takino T, Sato H. Cleavage of lumican by membrane-type matrix metalloproteinase-1
abrogates this proteoglycan-mediated suppression of tumor cell colony formation in soft agar. Cancer Res 64: 7058–7064, 2004. [
39. Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM. Leptin
regulates proinflammatory immune responses. FASEB J 12: 57–65, 1998. [
]
40. Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 56: 1124–

1131, 2007. [
] [
41. Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free
cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab 4: 185–198, 2006. [
42. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakanuma Y, Miyamoto K, Kaneko S.
Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 46: 1392–1403, 2007. [
43. Neuschwander-Tetri BA, Clark JM, Bass NM, Van Natta ML, Unalp-Arida A, Tonascia J, Zein CO, Brunt EM, Kleiner DE, McCullough AJ,
Sanyal AJ, Diehl AM, Lavine JE, Chalasani N, Kowdley KV. Clinical, laboratory and histological associations in adults with nonalcoholic fatty
liver disease. Hepatology 52: 913–924, 2010. [
] [
]
44. Noel M, Hickner J, Ettenhofer T, Gauthier B. The high prevalence of obesity in Michigan primary care practices. An UPRNet Study. Upper
Peninsula Research Network. J Fam Pract 47: 39–43, 1998. [
45. Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, Johnson RJ, Abdelmalek MF. Fructose consumption as a risk factor for
non-alcoholic fatty liver disease. J Hepatol 48: 993–999, 2008. [
] [
]
46. Popkin BM, Udry JR. Adolescent obesity increases significantly in second and third generation US immigrants: the National Longitudinal
Study of Adolescent Health. J Nutr 128: 701–706, 1998. [
]
47. Rangnekar AS, Lammert F, Igolnikov A, Green RM. Quantitative trait loci analysis of mice administered the methionine-choline deficient
dietary model of experimental steatohepatitis. Liver Int 26: 1000–1005, 2006. [
]
48. Ribeireiro T, Swain J, Sarr M, Kendrick M, Que F, Sanderson S, Krishnan A, Viker K, Watt K, Charlton M. NAFLD and insulin resistance
do not increase the risk of postoperative complications among patients undergoing bariatric surgery—a prospective analysis. Obes Surg 21: 310–
315, 2011. [
]
49. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol 40: 47–
51, 2004. [
]
50. Romestaing C, Piquet MA, Bedu E, Rouleau V, Dautresme M, Hourmand-Ollivier I, Filippi C, Duchamp C, Sibille B. Long term highly
saturated fat diet does not induce NASH in Wistar rats. Nutr Metab (Lond) 4: 4, 2007. [
] [
]
51. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic
steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120: 1183–1192, 2001. [
52. Schwartz Y, Khoshchenko OM, Dushkin MI, Feofanova NA. Effects of cholesterol and nuclear hormone receptor agonists on the production of
transforming growth factor-β in macrophages. Bull Exp Biol Med 148: 406–409, 2009. [
53. Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, Staels B, Maeda N, van Bilsen M, Hofker MH. Early diet-induced
non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol 44: 732–741, 2006. [
54. Silverman JF, O'Brien KF, Long S, Leggett N, Khazanie PG, Pories WJ, Norris HT, Caro JF. Liver pathology in morbidly obese patients
with and without diabetes. Am J Gastroenterol 85: 1349–1355, 1990. [
55. Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced
hepatic steatosis in mice. Hepatology 50: 1094–1104, 2009. [
]
56. Sreekumar R, Rosado B, Rasmussen D, Charlton M. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis.
Hepatology 38: 244–251, 2003. [
]
57. Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A,
Pacetti D, Sigmund S, Benedetti A, Casini A. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-
activated receptor-α and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol 169: 846–860, 2006. [
] [
58. Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed
trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol 295: G987–G995, 2008. [
] [

59. Tous M, Ferre N, Camps J, Riu F, Joven J. Feeding apolipoprotein E-knockout mice with cholesterol and fat enriched diets may be a model of
non-alcoholic steatohepatitis. Mol Cell Biochem 268: 53–58, 2005. [
]
60. Tyagi SC, Kumar S, Katwa L. Differential regulation of extracellular matrix metalloproteinase and tissue inhibitor by heparin and cholesterol in
fibroblast cells. J Mol Cell Cardiol 29: 391–404, 1997. [
]
61. Vos MB, McClain CJ. Fructose takes a toll. Hepatology 50: 1004–1006, 2009. [
]
62. Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology 12: 1106–
1110, 1990. [
]
63. Weltman MD, Farrell GC, Liddle C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation.
Gastroenterology 111: 1645–1653, 1996. [
64. Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel
biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 44: 27–33, 2006. [
]
65. Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease
and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study.
Gastroenterology 140: 124–131, 2011. [
66. Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, van Bilsen M,
Shiri-Sverdlov R, Hofker MH. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of
nonalcoholic steatohepatitis. Hepatology 48: 474–486, 2008. [
67. Yang SQ, Lin HZ, Mandal AK, Huang J, Diehl AM. Disrupted signaling and inhibited regeneration in obese mice with fatty livers:
implications for nonalcoholic fatty liver disease pathophysiology. Hepatology 34: 694–706, 2001. [
68. Zou Y, Li J, Lu C, Wang J, Ge J, Huang Y, Zhang L, Wang Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci
79: 1100–1107, 2006. [
]
Wyszukiwarka
Podobne podstrony:
Differences between the gut microflora of children with autistic spectrum disorders and that of heal
Letter of complain fast food(1)
Rewolucja Na Talerzu s02e01 Fast Food 14 10 2010
fast food
EW Naleśniki a`la fast food
Zizek And The Colonial Model of Religion
Modeling complex systems of systems with Phantom System Models
FAST FOOD
01 Mathematical model of power network
APA practice guideline for the treatment of patients with Borderline Personality Disorder
R 6 2 1 Mathematical model of enterprise, przyklad 1
Discussion of miracles with hume
Fast food
Five?ctor model of personality
TwoWorlds Model of Reality
Fast Food
The algorithm of solving differential equations in continuous model of tall buildings subjected to c
Zoledronic acid improves femoral head sphericity in a rat model of perthes disease
więcej podobnych podstron