
Elementy kinetyki i
termodynamiki
termodynamiki

Kinetyka
•
Kinetyka – badanie szybkości reakcji chemicznych, ustalanie ich mechanizmu oraz możliwości wpływania
na szybkość i kierunek reakcji.
•
Szybkość reakcji chemicznej:
v=
±
dc/dt
v=
±
dc/dt
A→C
v=-d[A]/dt=k*[A];
k-stała szybkość reakcji – zależy od rodzaju reakcji i temperatury (nie zależy od stężenia)
Lub
v=d[C]/dt=k*[C]

Czynniki wpływające na szybkość reakcji
•
Stężenie
•
Temperatura
•
Katalizator
•
Środowisko

układ
• otwarty
• zamkni
ę
ty
• izolowany
otoczenie
• izolowany
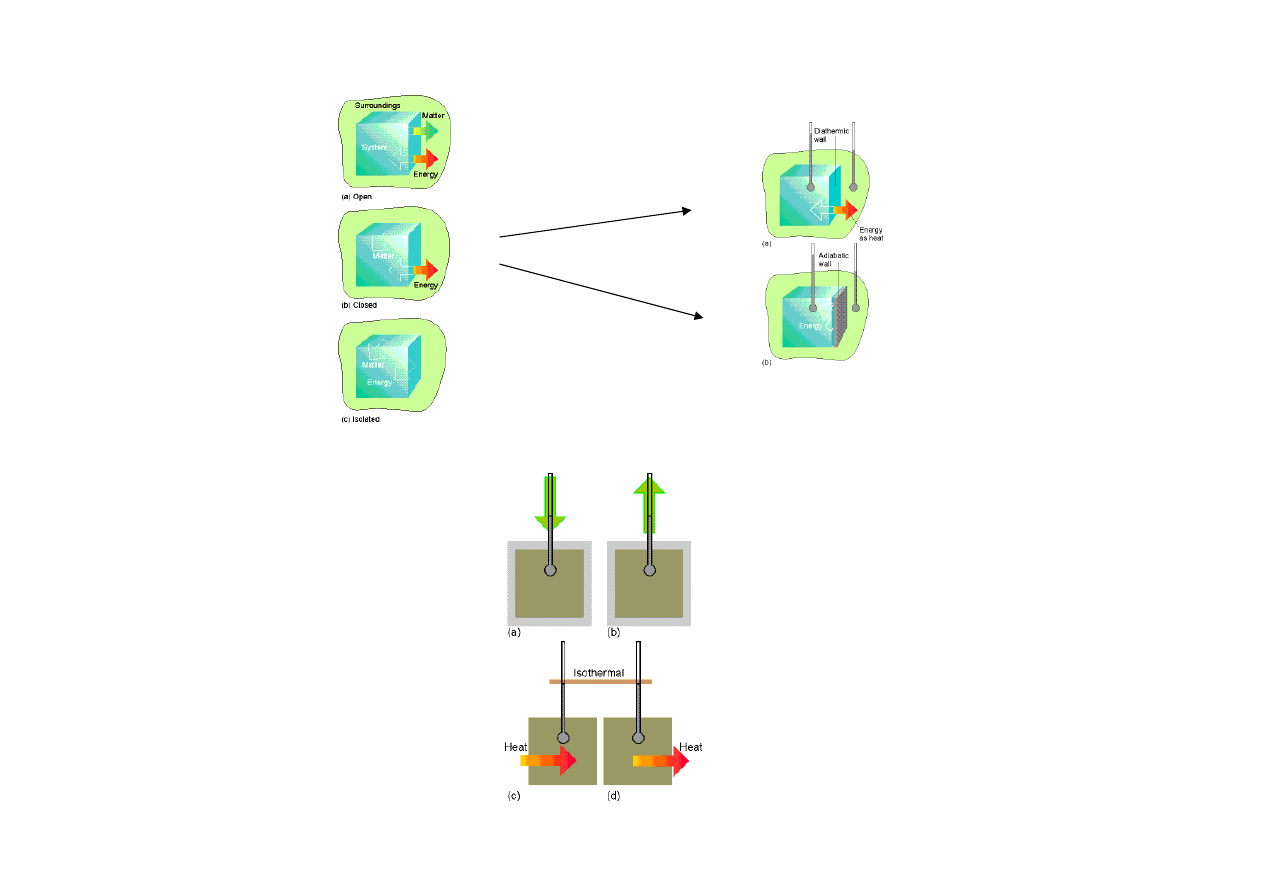
Układy
Przemiany
Przemiany
Endotermiczne-
ciepło jest
pochłaniane
q<0
(parowanie)
Egzotermiczne- ciepło
jest wytwarzane
q>0
(spalanie)

Sposób przekazu energii pomiędzy układem i otoczeniem
Przekaz w formie ciepła – ruch
nieuporządkowany
Przekaz w formie pracy – ruch
uporządkowany
Parametry układu
ekstensywne – zależą od wielkości układu (ilości substancji) – np.” masa,
objętość,– addytywne
intensywne – nie zależą od wielkości układu – temperatura, ciśnienie
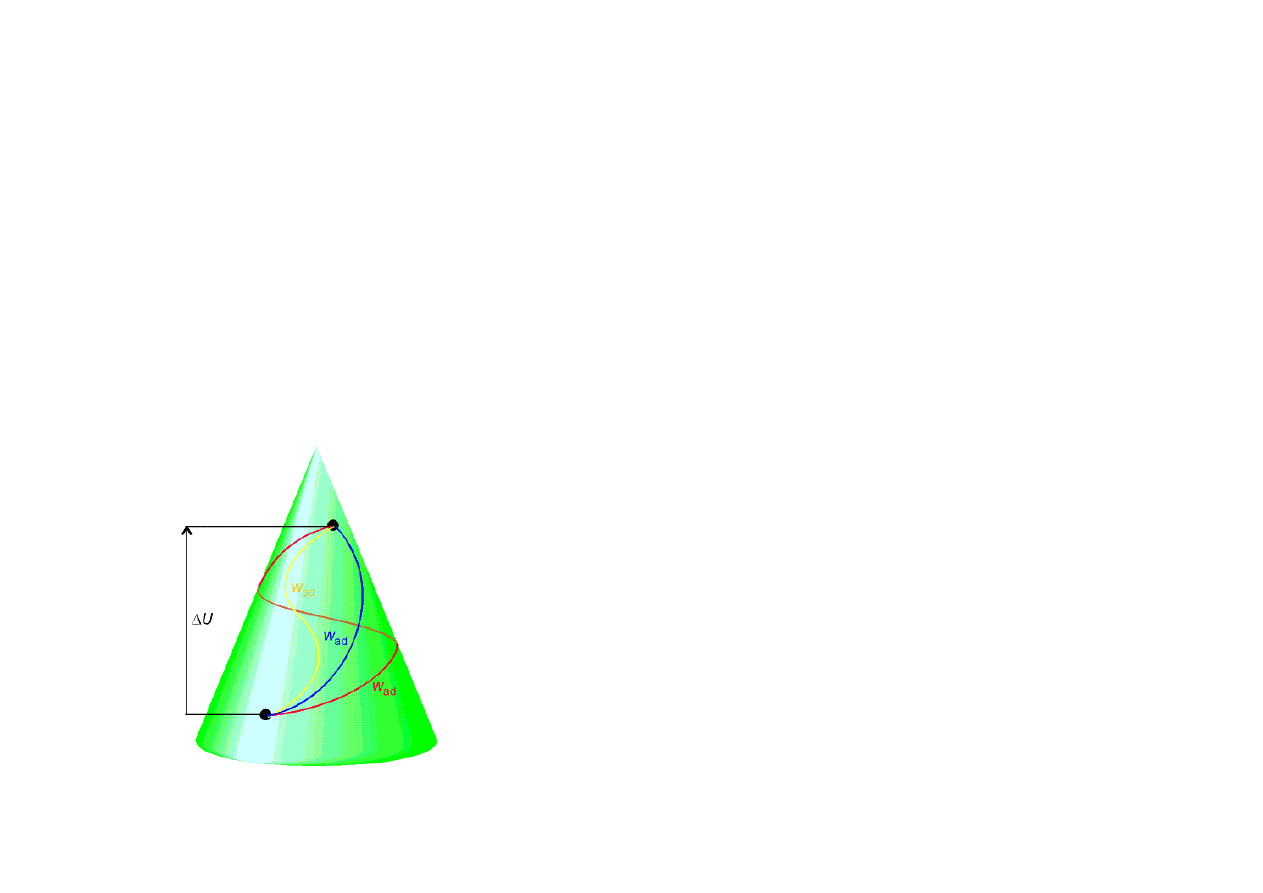
I ZASADA TERMODYNAMIKI
∑U = const – w układzie izolowanym całkowita ilość energii jest wielkością stałą.
Energia wewnętrzna – funkcja stanu – zależy tylko od stanu układu, a nie zależy od drogi po jakiej stan
ten został osiągnięty. Pozwala ona określić czy dana przemiana jest dozwolona, ponieważ tylko takie
przemiany mogą zachodzić dla których energia układu izolowanego pozostaje stała.
Energia wewnętrzna – uśredniona suma energii kinetycznej i potencjalnej wszystkich
cząsteczek układu
∆
U = U
k
- U
p
U
k
U
p
Dla układu otwartego lub
półotwartego:
∆∆∆∆
U = q + w
W > 0 – energia układu
ro
ś
nie – praca wykonana
nad układem
W < 0 – energia układu
maleje – układ wykonuje
prac
ę

• Procesy adiabatyczne:
q=0
∆
U = w
Procesy izochoryczne:
Jeżeli reakcja przebiega w stałej temperaturze, to:
w=p
∆
v, przy czym jeśli v=const, wówczas w=0, natomiast
w=p
∆
v, przy czym jeśli v=const, wówczas w=0, natomiast
zmiana energii wewnętrznej jest równa:
•
∆
U = q
v,

•
Procesy izobaryczne:
∆
U = q
p,
+ p
∆
v
q
p
=
∆
U + p
∆
v
q
p
= (U
2
+pv
2
) – (U
1
+ pv
1
)
∆
H =
∆
U + p
∆
v - entalpia
q
p
=
∆
H
•
Dla reakcji przebiegających w roztworach i fazie stałej (pod stałym ciśnieniem i
przy niewielkiej zmianie objętości:
∆
H =
∆
U = q;
H = U = q;
•
Gazy – jeśli (
∆
(pv)≈0), czyli dla gazów spełniających prawo Boyle’a -Mariotte’a.;
∆
H =
∆
U = q;
•
Jeśli podczas reakcji liczba moli substancji gazowych ulega zmianie wówczas;
∆
H =
∆
U +
∆
(pv) =
∆
U +RT
∆
n

•
Entropia – miara molekularnego nieuporządkowania układu.
•
Procesy odwracalne;
•
∆
S = Q
odwr
./T [J/mol*K]
•
W procesie termodynamicznie odwracalnym zachodzącym w układzie
izolowanym zmiana entropii jest równa zeru.
•
Procesy nieodwracalne;
•
W układzie izolowanym przemianie samorzutnej (nieodwracalnej),
towarzyszy wzrost entropii.
towarzyszy wzrost entropii.
•
Podsumowanie:
–
∆
S ≥0
– W układzie izolowanym przemianie samorzutnej towarzyszy wzrost
entropii, (entropia dąży do maksimum aż ustali się równowaga
termodynamiczna między wszystkimi składnikami układu). W stanie
równowagi każda przemiana ma charakter odwracalny a zmiana
entropii układu w tym stanie jest równa zeru.

•
Teoremat Nernsta – entropia czystych ciał krystalicznych w temperaturze zera
absolutnego równa się zeru.
•
Podsumowanie:
•
Pierwsza zasada termodynamiki posługuje się energia wewnętrzną do
przewidywania, czy dana przemiana jest dozwolona, natomiast druga zasada
wykorzystuje entropię do ustalenia która z dozwolonych przemian jest samorzutna.
•
Entalpia swobodna
∆
G =
∆
H - T
∆
S – entalpia swobodna (potencjał termodynamiczny) – zmiana
potencjału termodynamicznego okre
ś
la prac
ę
u
ż
yteczn
ą
w warunkach
potencjału termodynamicznego okre
ś
la prac
ę
u
ż
yteczn
ą
w warunkach
izotermiczno-izobarycznych i jest kryterium równowagi oraz miar
ą
samorzutno
ś
ci procesu

Czy istnieje funkcja stanu, której przyrost odpowiadałby ciepłu pobranemu lub
wydzielonemu pod stałym ciśnieniem ?
pdV
Dq
dU
−
=
gdy p = const dU<Dq
)
pV
(
d
Dq
Vdp
pdV
Dq
dU
−
=
−
−
=
)
const
p
(
,
Dq
)
pV
U
(
d
=
=
+
pV
U
H
+
=
Entalpia - f. stanu
Vdp
Dq
dH
+
=
Ogólnie:
Dw
Vdp
Dq
dH
+
+
=
Ogólnie:
nobj
Dw
Vdp
Dq
dH
+
+
=
Pomiar ciepła pobranego lub wydzielonego w procesie izobarycznym (p=const)
⇔
pomiar zmiany entalpii
Energia wewnętrzna - f. stanu dla procesów izochorycznych
Entalpia
- f.stanu dla procesów izobarycznych

Różnica między
∆
U i
∆
H

2
Wniosek:
Różnica pomiędzy
∆
U i
∆
H istotna dla przemian w której uczestniczy faza
gazowa. Dla faz skondensowanych - różnica jest zaniedbywalnie mała jeżeli
przemiana nie odbywa się pod wysokim ciśnieniem.
Podstawy termochemii
Analiza cieplna w przemianach chemicznych
w stałej objętości
w stałym ciśnieniu
energia wewnętrzna
entalpia
V
q
dU
=
p
q
dH
=
0
dU
>
podczas przemiany ciepło jest
dostarczane do układu
p.endotermiczna
0
dH
>
0
dU
<
0
dH
<
podczas przemiany układ wydziela
0
dU
<
0
dH
<
podczas przemiany układ wydziela
ciepło
p.egzotermiczna
1
2
funkcje stanu - funkcje parametrów stanu, określone z dokładnością do stałej ⇒ musimy
określić warunki w jakich będziemy je definiować ⇒ stan standardowy substancji
Substancja w czystej postaci w danej temperaturze pod ciśnieniem
p
∅
∅
∅
∅
= 1 bar
1
2
21
H
H
H
−
=
∆

N.p. w stanie standardowym w temperaturze pokojowej 25 ºC
(298.15 K) objętośc molowa gazu doskonałego:
1
3
m
mol
dm
79
.
24
p
RT
V
−
∅
∅
⋅
=
=
Standardowa entalpia przemiany
zmiana entalpii w procesie przemiany od substratów do produktów w stanach
standardowych
H
Entalpia standardowa przemiany złożonej jest sumą entalpii standardowych reakcji
składowych
∅
∅
∅
∆
+
∆
=
∆
H
H
H
A
B
C
∅
→
∅
→
∅
→
∆
+
∆
=
∆
H
H
H
B
C
C
A
B
A
∅
∅
∅
∆
+
∆
=
∆
H
H
H
par
top
sub
H
Entalpia standardowa procesu odwrotnego
B
A
∅
→
∅
→
∆
−
=
∆
H
H
A
B
B
A

Entalpie reakcji chemicznych
∅
∆
H
r
standardowa entalpia reakcji przebiegającej od substratów do produktów
reakcji w stanach standardowych
Reakcja:
∑
∑
→
produkty
p
p
substraty
s
s
A
A
ν
ν
∑
∑
∅
∅
∅
−
=
∆
substraty
m
,
s
s
produkty
m
,
p
p
r
H
H
H
ν
ν
Alternatywnie:
0
A
j
j
j
∑
→
ν
∑
∅
∅
=
∆
j
m
,
j
j
r
H
H
ν
współczynniki stechiometryczne
ν
j
> 0 dla produktów
ν
j
< 0 dla substratów
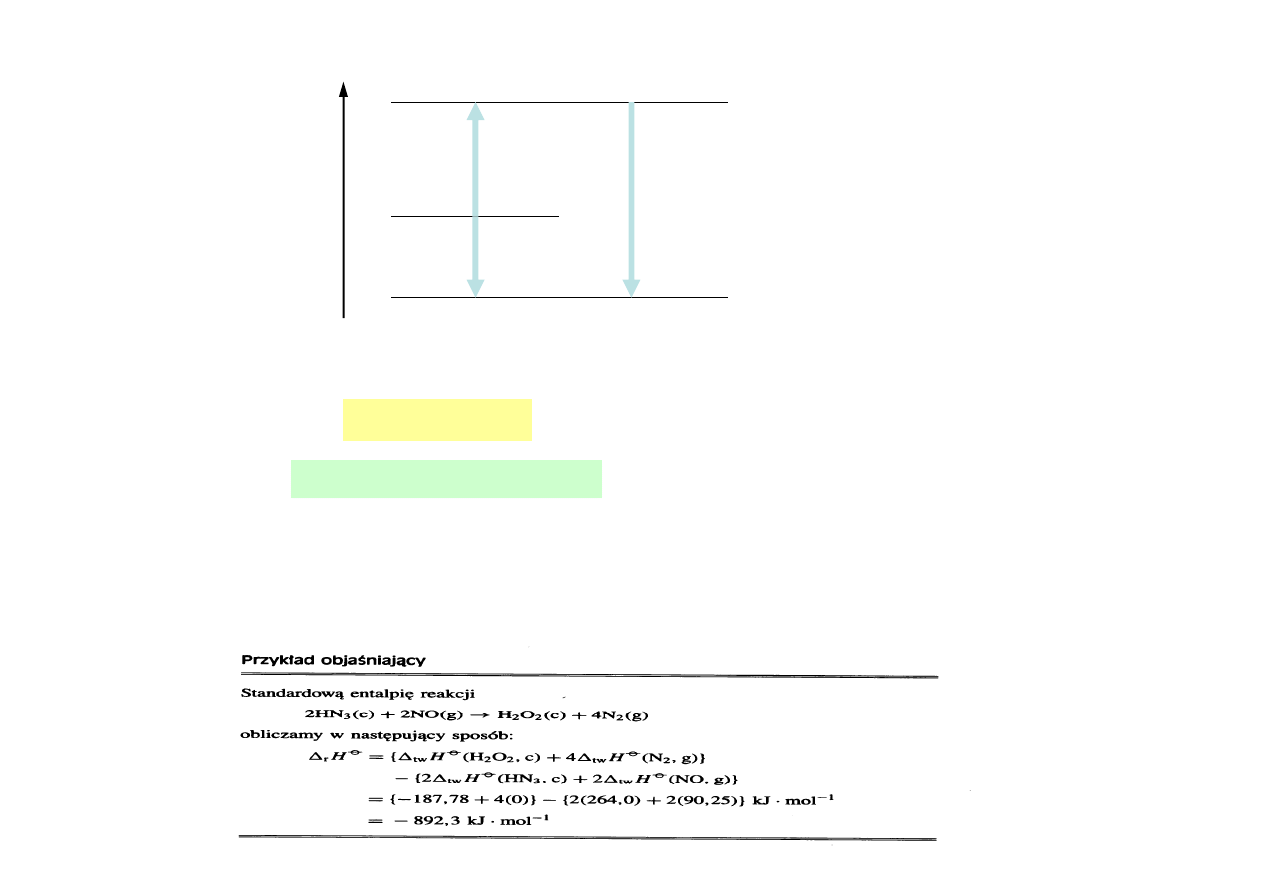
N.p. standardowa entalpia procesu spalania
1
r
2
2
2
6
12
6
mol
kJ
2808
H
)
l
(
O
H
6
)
g
(
CO
6
)
g
(
O
6
)
s
(
O
H
C
−
∅
⋅
−
=
∆
+
→
+
Entalpia jest funkcją stanu
Standardowa entalpia reakcji sumarycznej jest sumą entalpii standardowych reakcji
składowych, na które daną reakcję sumaryczną można rozłożyć - prawo Hessa
Dla danej reakcji:
Możemy policzyć standardowe entalpie potrzebne do wytworzenia produktów i substratów i
Możemy policzyć standardowe entalpie potrzebne do wytworzenia produktów i substratów i
odjąć je od siebie.
Standardowe entalpie tworzenia -
Entalpie tworzenia związków z pierwiastków w ich stanach podstawowych (najbardziej
trwałej formie pierwiastka w danej temperaturze i pod ciśnieniem 1 bar
H
pierwiastki
produkty
substraty
∅
∆
H
r
Dla reakcji:
0
A
j
j
j
∑
→
ν
∑
∅
∅
∆
=
∆
j
j
tw
j
r
)
A
(
H
H
ν
pr. Hessa
w praktyce:
Rozkładamy daną reakcję na reakcje cząstkowe znajdujemy entalpie tworzenia
substratów i produktów w reakcjach cząstkowych i stosujemy prawo Hessa
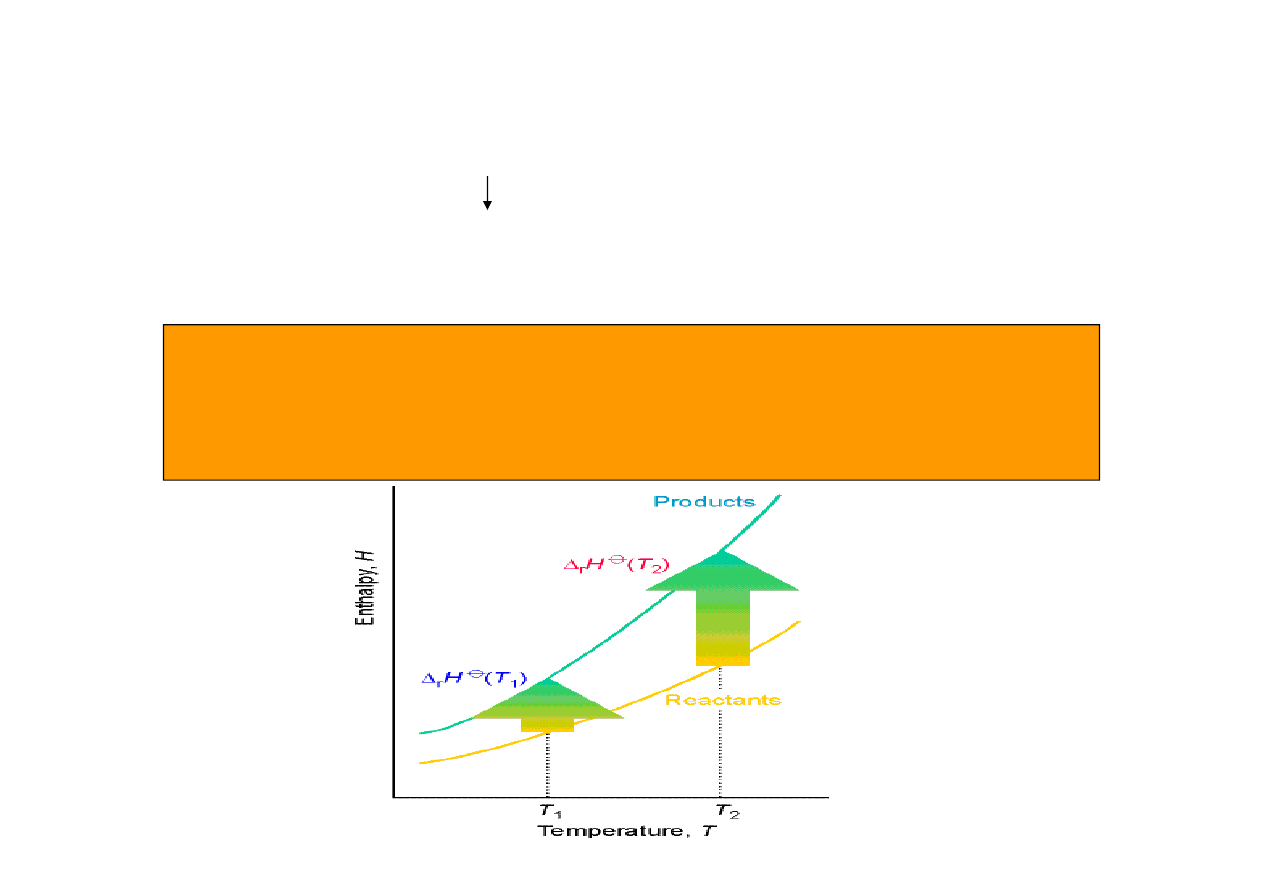
Co zrobić jeżeli znamy entalpię standardową w jednej temperaturze a chcemy określić ją w
innej ?
)
const
p
(
dT
C
dH
p
=
=
( )
∫
+
=
2
1
T
T
p
1
2
dT
C
T
H
)
T
(
H
Ponieważ całkowanie jest operacją liniową, stosując zależność (*) do prawa Hessa
otrzymujemy:
(*)
∫
∅
∅
∅
∆
+
∆
=
∆
2
1
T
T
p
r
1
r
2
r
C
)
T
(
H
)
T
(
H
)
j
(
C
C
m
,
p
j
j
p
r
∅
∅
∑
=
∆
ν
prawo Kirchhoffa

Wyszukiwarka
Podobne podstrony:
kinetyka i termodynamika
Fizyka dynamika, kinetyka, termodynamika, pole magnetyczne
17 kinetyczna teoria gazów i termodynamika II
Kinetyczna teoria gazów i termodynamika I
16 kinetyczna teoria gazów i termodynamika I
Kinetyczna teoria gazów i termodynamika II
Klucz do testu Kinetyka i równowaga chemiczna, termodynamika
Kinetyka i równowaga chemiczna, termodynamika test
Termodynamika i kinetyka chemiczna
17 kinetyczna teoria gazów i termodynamika II
więcej podobnych podstron