P. Gałecki i wsp.
Jony wapnia, kwas glutaminowy, oś podwzgórze-
-przysadka-nadnercza, ATP-aza zależna od wapnia jako przyczyny uszkodzeń oksydacyjnych u chorych na depresję (część II)
PIOTR GAŁECKI1, ANTONI FLORKOWSKI1, MAŁGORZATA MROWICKA2, TADEUSZ PIETRAS3, ELŻBIETA GAŁECKA2
Uniwersytet Medyczny w Łodzi: 1Klinika Psychiatrii Dorosłych, kierownik: prof. dr hab. med. A. Florkowski, 2Katedra i Zakład Chemii i Biochemii Klinicznej, kierownik: prof. dr hab. med. J. Kędziora, 3Klinika Pneumonologii i Alergologii, kierownik: prof. dr hab. med. Paweł Górski Jony wapnia, kwas glutaminowy, oś podwzgórze-Calcium ions, glutaminate acid, hypothalamic-pituitary-
-przysadka-nadnercza, ATP-aza zależna od wapnia
-adrenal axis, calcium dependent ATP-ase as causes jako przyczyny uszkodzeń oksydacyjnych u chorych of oxidative damage in depression patients (part II) na depresję (część II)
Gałecki P.1, Florkowski A.1, Mrowicka M.2, Pietras T.3, Gałecka E.2
Gałecki P.1, Florkowski A.1, Mrowicka M.2, Pietras T.3, Gałecka E.2
Medical University of Łódź, Poland: 1Department of Adults Psychia-Uniwersytet Medyczny w Łodzi: 1Klinika Psychiatrii Dorosłych, try, e-mail: klpsych@o2.pl; 2Chair and Department of Chemistry and e-mail: klpsych@o2.pl; 2Katedra i Zakład Chemii i Biochemii Klinicz-Clinical Biochemistry, 3Department of Pneumonology and Allergo-nej, 3Klinika Pneumonologii i Alergologii
logy
Zaburzenia depresyjne stanowią poważny problem we współcze-Depressive disorder is still a rising and important problem in the snym świecie. Dotykają one 15% populacji. Obecne formy terapii są modern world, it affects about 15% of the population. Present forms skuteczne u około 70% pacjentów, wymagają wielomiesięcznego of treatment are effective in about 70% and require monthly therapy leczenia, które czasem wiąże się z objawami niepożądanymi. Bada-which sometimes causes side effects. Last decade studies paid at-nia ostatniej dekady zwróciły uwagę na inne niż monoaminergiczna tention to theories different to monoaminergic and to neurodegene-teoria depresji oraz na zmiany neurodegeneracyjne głównie w ob-rative changes mainly in the limbic system of hippocampus. In this szarze układu limbicznego. Autorzy przedstawiają wzajemne po-article authors show a relationship between calcium ions, glutami-wiązanie gospodarki komórkowej jonem wapnia, nasilonego prze-nergic transduction and disfunction of hypothalamic-pituitary-adre-kaźnictwa glutaminergicznego, zaburzonej regulacji osi podwzgó-
nal (HPA) axis. They also take into account the activity of calcium rze-przysadka-nadnercza (PPN) oraz aktywności pompy ATP-azo-dependent ATPase and its influence on overproduction of reactive wej zależnej od wapnia – w aspekcie ich wpływu na nadmierne wy-oxygen species in the central neuron system (CNS).
twarzanie reaktywnych form tlenu.
In the second part authors conclude that deregulation of calcium W części drugiej autorzy wnioskują, że zaburzona regulacja stęże-ions concentration in and out of cells and decreased activity of cal-nia jonów wapnia w kompartmentach komórkowych i pozakomórko-cium dependent ATPase stimulate tricarboxylic acid cycle, oxidative wych oraz nieprawidłowa aktywność zależnej od wapnia ATP-azy phosphorilation. Mitochondria work faster and consume more oxy-stymulują cykl kwasów trikarboksylowych, fosforylację oksydacyjną.
gen. It correlates well with overproduction of reactive oxygen spe-Przyśpiesza też pracę mitochondriów i powoduje zwiększone zużycies (RFT). Above process results in neurons apoptosis and necro-cie tlenu, a następnie wytwarzanie zwiększonej ilości reaktywnych sis.
form tlenu. Opisane procesy prowadzą do apoptozy i nekrozy ko-mórek w ośrodkowym układzie nerwowym.
Słowa kluczowe: jony wapnia, kwas glutaminowy, oś podwzgórze-Key words: calcium ions, glutaminergic acid, hypothalamic-pituita-
-przysadka-nadnercza, ATP-aza zależna od wapnia, stres oksyda-ry-adrenal axis, calcium dependent APTase, oxidative stress, de-cyjny, depresja
pression
Pol. Merk. Lek., 2008, XXIV, 139, 72
Pol. Merk. Lek., 2008, XXIV, 139, 72
Badania potwierdzające rolę N-metylo-D-asparginianu też nadtlenek wodoru, kwas podchlorawy, jon nadtlenoazo-
(NMDA) w stymulowaniu mitochondriów do wytwarzania anio-tynowy. Wszystkie te rodniki przyczyniają się do uszkodzeń norodnika ponadtlenkowego (O –) przeprowadzili Sengpiel i składników komórkowych: kwasów nukleinowych, białek, li-2
wsp. Do kultur komórek hipokampu dodawali inhibitora pidów. Zaburzają zachodzące w komórce procesy, przyczy-NMDA, jakim jest cyjanek sodu. Związek ten powoli ograni-niają się również do nekrozy i/lub apoptozy komórek [2, 20].
czał indukowane przez NMDA wytwarzanie O – [5, 45].
Przemiany prowadzące do powstawania wolnych rodni-2
Przedłużająca się i nadmierna aktywacja receptora NMDA ków, będące skutkiem zwiększania stężenia jonów wapnia w prowadzi do masowego napływu jonów wapnia do wnętrza komórce, można umownie podzielić na te, które związane są komórki. Dochodzi do przeładowania jonów wapnia w komór-z łańcuchem oddechowym mitochondriów oraz na mechani-ce. Może to powodować zwiększoną generację wolnych rod-zmy wytwarzania wolnych rodników, które związane są z in-ników [25, 44, 48]. Do wolnych rodników należą m.in. anio-nymi przemianami zachodzącymi w komórce (ryc. 1).
norodnik ponadtlenkowy, rodnik hydroksylowy, rodnik perok-Mitochondria to główne miejsce wytwarzania wolnych rod-sylowy, rodnik alkoksylowy, nadtlenek azotu, ditlenek azotu, ników w komórkach [14, 43]. Akumulują one duże ilości jo-a także wiele innych związków, mogących łatwo ulegać przenów wapnia dzięki obecności uniportu transportującego je mianom do wolnych rodników. Do substancji tych zaliczamy zgodnie z gradientem stężeń. Całkowita pojemność uniportu
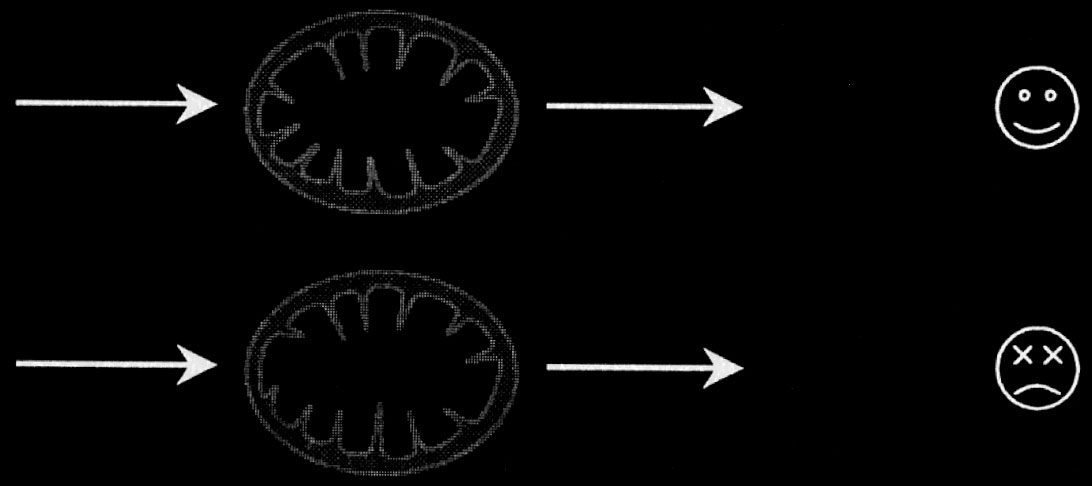
Jony wapnia, kwas glutaminowy, oś podwzgórze–przesadka–nadnercza, ATP-aza zależna od wapnia 73
Ca2+
[Ca2+]m
ATP
Ca2+ plus
patologiczna
RFT
[Ca2+]m
stymulacja
śmierć komórki
Ryc. 1. Jony wapnia jako pośrednik w procesach fizjologicznych i patofizjologicznych zachodzących w mitochondrium, za Brookes i wsp. [6]
Fig. 1. Calcium ions as a mediator in physiological and pathological processes taking place in mitochondrion, Brookes and al. [6]
może być tak duża, że przekracza zdolność mitochondriów (ang. permeability transition pores). Rezultatem tego jest blo-do usunięcia jonów wapnia. Zdolność mitochondriów do wy-kada kompleksu III, mogąca powodować zwiększenie wytwa-chwytu Ca2+ jest ograniczona i może prowadzić do uszko-rzania wolnych rodników [17, 35]. Zwiększenie stężenia jo-dzeń procesów mitochondrialnych i patofizjologicznego wy-nów wapnia w komórce przyczynia się także do hamowania twarzania wolnych rodników tlenowych. Zwykle jest ona skut-kompleksu IV łańcucha oddechowego (oksydoreduktaza zre-kiem rozprzężenia i depolaryzacji mitochondriów [45, 48].
dukowany cytochrom c: tlen). Do inhibicji dochodzi w wyniku Łańcuch oddechowy mitochondriów jest głównym miej-zwiększonej jonami wapnia aktywności syntazy tlanku azotu scem syntezy adezynotrifosforanu – ATP (ang. adesine trii wzmożonego wytwarzania rodnika NO, który hamuje łań-
phosphorate). W procesie tym elektrony uwalniane ze zre-cuch oddechowy na tym etapie. Inhibicja oksydazy cytochro-dukowanego substratu są przenoszone na tlen przez łańcuch mowej przez tlenek azotu polega na konkurencyjnym wiąza-pomp protonowych. Dochodzi do czteroelektronowej reduk-niu się NO w miejscu wiązania O [10, 32]. Tlenek azotu może 2
cji tlenu, produktem której są dwie cząsteczki wody. Układ być uznany za substancję sygnałową w powstawaniu RFT.
ten nie jest jednak procesem szczelnym, pewna ilość elek-Blokowanie łańcucha oddechowego przez produkt reakcji NO
tronów wydostaje się z łańcucha oddechowego i następuje i O –, czyli jon ONOO– następuje także w obrębie kompleksu 2
jednoelektronowa redukcja tlenu. Produktem tej reakcji jest V, czyli syntazy ATP [7].Tak więc jeśli chodzi o łańcuch odde-anionorodnik ponadtlenkowy. Głównym źródłem tego rodni-chowy nadmierna ilość jonów wapnia w mitochondriach jest ka jest rodnik ubisemichinonowy. Takiej jednoelektronowej przyczyną stymulacji cyklu kwasów trikarboksylowych i zwięk-redukcji ulega około 1 - 2% tlenu cząsteczkowego [2, 6, 22, szenia ruchu elektronów w proksymalnej części łańcucha 50]. Powstały anionorodnik ponadtlenkowy spontanicznie lub oddechowego. Indukując jednak uwalnianie cytochromu c przy udziale SOD ulega przemianie do H O , która to w powoduje inhibicję części dystalnej łańcucha oddechowego.
2
2
obecności jonów metali takich jak Fe2+ ulega przemianie do Niewykluczony jest także wpływ jonów wapnia na status anionorodnika hydroksylowego. Powstające RFT w ilościach oksydacyjny mitochondriów. Sugerowane jest, że glutation wyższych niż te niezbędne w procesach przekazywania sy-mitochondriów uwalniany jest podobnie jak wiele innych bia-gnału są przyczyną uszkodzenia składników komórkowych łek bardzo wcześnie w wyniku indukowanego właśnie jona-białek, lipidów, kwasów nukleinowych.
mi wapnia otwarcia porów przepuszczalności przejściowej Powstawanie nadmiernej ilości wolnych rodników wynika
[38]. Ponadto powstający w reakcji NO + O – toksyczny i wy-2
również ze zwiększonej jonami wapnia stymulacji cyklu kwa-soce reaktywny związek ONOO– ma zdolność modyfikowa-sów trikarboksylowych i fosforylacji oksydacyjnej. Powoduje nia białek mitochondrialnych. Należą do nich m.in. glutation, to zwiększenie szybkości pracy mitochondriów oraz zużycie manganowa dysmutaza ponadtlenkowa, czyli substancje większej ilość tlenu. Zwiększa się jednocześnie namnażanie będące antyoksydantami [4]. Przez oksydację lub nitrowanie wolnych rodników [6]. Nadmierne wytwarzanie wolnych rod-tych związków dochodzi do uszkodzenia systemu usuwania ników może następować na różnych etapach łańcucha od-wolnych rodników.
dechowego. Inhibicja kompleksu I łańcucha oddechowego W powstawaniu wolnych rodników na skutek ekscytotok-
(oksydoreduktaza NADH: ubichinon) przez działające jedno-sycznego działania kwasu glutaminowego istotne znaczenie cześnie: tlenek azotu i jony wapnia może być przyczyną ma także udział jądrowej poli-ADP-rybozy (PARP1). Enzym zwiększonej generacji RFT [23].
ten katalizuje hydrolizę utlenionego dinukleotydu nikotynami-Choć nie ma jednoznacznych dowodów na to, że inhibi-doadeninowego (NAD+). Reakcja ta przyczynia się więc jed-cja przez te właśnie związki sprzyja powstawaniu RFT, to nocześnie do niedoboru zredukowanego fosforanu dinukle-warto brać pod uwagę taką możliwość, opierając się na ba-otydu nikotynamidoadeninowego – NADH niezbędnego do daniach przeprowadzonych z użyciem inhibitora kompleksu redukcji utlenionej postaci glutationu (GSSG). Dochodzi do I, jakim jest rotenon na mitochondriach z mózgu szczurów.
obniżenia stężenia glutationu i tym samym zwiększania ge-Blokowanie przez rotenon łańcucha oddechowego na tym neracji wolnych rodników [12].
etapie doprowadziło do zwiększenia RFT [15, 24, 47]. Nie Kolejnym elementem udziału nadmiernej zawartości jo-ma jednak dowodów na blokowanie tego elementu przez nów wapnia jest udział w syntezie tlenku azotu [5]. Nadpro-działający osobno wapń i NO. Powstający w wyniku reakcji dukcja tego rodnika może pośredniczyć w dużej mierze w NO z O – jon ONOO– ma również zdolność inhibicji komplek-uszkodzeniach tkanki nerwowej [49]. Proces zwiększenia 2
su I łańcucha oddechowego w mechanizmie nitrowania tyro-syntezy NO przedstawia się następująco. Na skutek stymu-zyny [46].
lacji receptora NMDA kwasem glutaminowym, wnikające do Kolejny element łańcucha oddechowego to kompleks III komórki jony wapnia wiążą się z kalmoduliną. Skutkiem tego (oksydoreduktaza ubichinol: utleniony cytochrom c). Jony jest aktywacja syntazy tlenku azotu (NOS) i nadmierne wy-wapnia przyczyniają się do zwiększenia dyslokacji cytochro-twarzanie NO. Reakcja katalizowana przez ten enzym jest mu c z wewnętrznej błony mitochondriów. Prawdopodobny procesem wytwarzania tlenku azotu i cytruliny z argininy i mechanizm uwalniania cytochromu c polega na współzawod-tlenu. Pojawiający się w nadmiernej ilości tlenek azotu może nictwie jonów wapnia o miejsce wiązania kardilopiny lub wcze-wejść w reakcję z O –·, wytwarzając ONOO– [44]. Jon ten 2
snym otwarciu porów przepuszczalności przejściowej – PT
powoduje uszkodzenie struktury DNA, aktywację PARP, w
P. Gałecki i wsp.
wodnym środowisku z kolei reaguje spontanicznie wytwarza-MR. Obserwacje te sugerują, że ograniczenie PMCA1 jest jąc rodnik hydroksylowy [41].
jednym z molekularnych mechanizmów, za pomocą których W komórce następują także inne procesy mające udział
steroidy regulują homeostazę jonów Ca2+ w komórce [3].
w powstawaniu wolnych rodników. Zaliczmy do nich, m.in.
reakcje związane z oksydazą ksantynową uznaną za jedno z istotnych źródeł anionorodnika ponadtlenkowego i przemia-GLIKOKORTYKOSTEROIDY I ANTYOKSYDANTY
ny kwasu arachidonowego.
Zwiększenie stężenia jonów wapnia w komórce prowadzi Glikokortykosteroidy zwiększają stężenie reaktywnych form do przemiany dehydrogenazy ksantynowej w oksydazę ksan-tlenu dwojako. Nasilają cykl reakcji wytwarzających wolne tynową. Reakcja ta następuje pod wpływem proteazy zależ-
rodniki i jednocześnie wpływają na generację wolnych rodni-nej od wapnia. Proteazą tą jest kalpaina I [8]. Dalsze przeków, obniżając stężenie antyoksydantów. W celu neutralizo-miany hipoksantyny i ksantyny w kwas moczowy przez oksy-wania stresu oksydacyjnego komórki mają kompleks enzy-dazę ksantynową powoduje generowanie O – i H O [21]. Mc mów antyoksydacyjnych. Należą do nich cynkowo-miedzio-2
2
2
Nally i wsp. wykazali, że H O prowadzi do nieodwracalnej wa dysmutaza ponadtlenkowa – CuZnSOD, dysmutaza man-2
2
przemiany XO do XOD [30]. Zwiększenie stężenia jonów Ca2+
ganowa – MnSOD, katalaza – CAT, peroksydaza glutationowe wnętrzu komórki nerwowej sprzyja aktywacji fosfolipazy wa – GSH-Px [20, 29, 34, 36]. Stres prowadzi do nadmierne-A , która działając na fosfolipidy powoduje uwolnienie kwasu go wytwarzania RFT, takich jak: O –, ·OH, H O , które z kolei 2
2
2
2
arachidonowego. Dalsze przemiany tego związku przy udziale powodują uszkodzenie składników komórek m.in. poprzez lipooksygenaz i cyklooksygenaz do eikozanoidów prowadzą peroksydację lipidów [27]. Uszkodzenia oksydacyjne nie są do wytwarzania anionorodnika ponadtlenkowego [9, 33, 48].
związane jedynie ze zwiększeniem ilości RFT, ale również z Reakcja powstawania wolnych rodników jest znacząca, gdy obniżeniem aktywności enzymów antyoksydacyjnych [13, 29].
agoniści receptora NMDA powodują zmniejszenie ATP i Przeprowadzono wiele badań wiążących stres, zwiększone zwiększenie AMP. Dochodzi wówczas do podwyższenia gli-stężenie glikokortykosteroidów i aktywność enzymów anty-kolizy i następuje znaczące zwiększenie ilości kwasu mleko-oksydacyjnych.
wego [16]. Zmniejszenie zatem pH środowiska powoduje Badania przeprowadzone przez Rodaka i wsp. dowodzą, uwalnianie komórkowych zapasów jonów Fe2+. Jony te są że mobilizacja układu limbicznego indukuje oksydacyjne niezbędnym substratem reakcji Fentona, w wyniku której z uszkodzenia w strukturach hipokampu [42]. Dalsze badania H O powstaje jedna z najbardziej niebezpiecznych postaci dowodzą zwiększenia stresu oksydacyjnego na skutek obni-2
2
RFT rodnik hydroksylowy ·OH [11].
żenia aktywności SOD, CAT, S-transferazy glutationowej oraz obniżenia stężenia glutationu w komórkach [52]. Wiadomo, że glikokortykosteroidy zwiększają akumulację glutaminianu GLIKOKORTYKOSTEROIDY A ATP-za ZALEŻNA
w szczelinach synaptycznych. Antyoksydanty z kolei pełnią OD WAPNIA
funkcję neuroprotekcyjną w obecności tego neuroprzekaźni-ka.
Wypływ jonów wapnia z komórek odbywa się przy udziale Spekulować można więc powiązanie między zwiększo-dwóch podstawowych systemów. Należą do nich charakte-nym stężeniem kortyzolu, jakie występuje w zaburzeniach ryzujący się niskim powinowactwem do Ca2+ wymieniacz so-depresyjnych, a uszkodzeniami w neuronach. Glikokortyko-dowo-wapniowy (NA+/Ca2+) i zależna od wapnia pompa Ca2+-
steroidy zwiększają toksyczność RTF podnosząc ich podsta-ATP-aza (PMCA) o wysokim powinowactwie do jonów wap-wowe stężenie, a obniżają jednocześnie pojemność antyok-nia [1, 18, 31]. Zależna od wapnia pompa ATP-azowa należy sydacyjną [40, 51]. Przypuszczenia te potwierdzają badania do podtypu P rodziny ATP-az, które zużywając energię ATP
wykonane przez Sapolskiego i wsp. Mówią one, że ciągła transportują jony wbrew ich elektrochemicznemu gradiento-ekspozycja glikokortykosteroidów powoduje obniżenie pod-wi po obydwu stronach membrany [53]. Wśród najlepiej scha-stawowej aktywności CuZnSOD we wszystkich regionach rakteryzowanych odmian tego enzymu postać PMCA1 wy-mózgu. Obniżona była również aktywność GSH-Px w komór-stępuje we wszystkich tkankach, natomiast postacie PMCA2
kach hipokampu i kory.
i PMCA3 znajdują się w mózgu i sercu. Ponadto istotny po-Istotnym elementem potwierdzającym wpływ glikokorty-zostaje fakt, że komórki mózgu zawierają wielokrotnie wię-
kosteroidów na obniżenie aktywności wymienionych enzy-cej tego enzymu niż pozostałe komórki [19]. Aktywność Ca2+
mów jest fakt, że obniżenie ich aktywności nastąpiło mimo ATP-azy regulowana jest przez wiele czynników. Jednym z niestwierdzenia dodatkowych uszkodzeń neurologicznych nich są hormony steroidowe. Mają one zdolność obniżania,
[29]. Zaobserwowano również, że kortykosteron obniża cał-
stymulowanej przez kalmodulinę (CaM) aktywności Ca2+
kowite stężenie glutationu. Prawdopodobnie to właśnie obni-ATP-azy. Dzieje się tak prawdopodobnie na skutek wiązania żenie stężenia zredukowanego glutationu, będącego substra-się steroidów z domeną wiążącą CaM przez Ca2+-ATP-azę tem dla peroksydazy glutationowej, sprzyja obniżeniu jej ak-lub hamowania transkrypcji dla tego białka [53].
tywności. Istnieje także inna możliwość, chodzi tutaj miano-Wiele przeprowadzonych ostatnio badań potwierdza udział
wicie o obniżanie przez glikokortykosteroidy stężenia NADPH, RFT w modyfikacji biologicznych membran. Pewna ilość bia-który jest niezbędnym elementem reakcji redukcji utlenionej łek jest szczególnie wrażliwa na zmiany spowodowane oksy-postaci glutationu do postaci zredukowanej. Zmniejszone dacją. Jednym z tych białek jest PMCA. Pompa ta na skutek bowiem stężenie NADPH przez glikokortykosteroidy może utleniania przez askorbinian i jon żelazowy zmniejsza swoją być wynikiem zmniejszania przez te hormony transportu gli-aktywność. Podobna sytuacja pojawia się po inkubacji z uży-kozy w neuronach hipokampu, a to właśnie dostępność gli-ciem Fe2+ i H O [28, 39]. Nie bez wpływu na prawidłowe funk-kozy reguluje neuronalne stężenie NADPH i stosunek GSH/
2
2
cjonowanie PMCA pozostaje też jon ONOO–. Przyczynia się GSSG. Innym czynnikiem powodującym obniżenie stężenia on do agregacji, nitrowania protein i inaktywacji Ca2+ ATP-azy.
glutationu jest zmiana w wychwycie glutationu przez barierę Inne badania dowodzą zmniejszenia aktywności tego białka krew-mózg spowodowana glikokortykosteroidami oraz zmiany na skutek peroksydacji lipidów błonowych przez ONOO–.
w syntezie tego peptydu [37].
PMCA spełnia istotną rolę w regulacji wewnątrzkomórkowej puli wapnia. Glikokortykosteroidy wpływają na ekspresję ge-nów dla tej ATP-azy.
PODSUMOWANIE
Przypuszczenia te potwierdzają badania przeprowadzone przez Bhargava i wsp. na komórkach hipokampu szczu-Pojawiająca się w zaburzeniach depresyjnych zwiększona rów. Wskazują one, że ekspresja genu dla PMCA1 ograni-aktywność osi podwzgórze-przysadka-nadnercza ma istot-czona jest przez kortykosteron przy udziale receptorów GR i ne znaczenie w wielu zmianach patologicznych, związanych
Jony wapnia, kwas glutaminowy, oś podwzgórze–przesadka–nadnercza, ATP-aza zależna od wapnia 75
z uszkodzeniem neuronów obserwowanych w depresji. Po-24. Kudin A.P., Bimpong-Buta N.Y., Vielhaber S. i wsp.: Charakterization of większone stężenie kortyzolu jest prawdopodobną przyczy-superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem., 2004, 279, 4127-4135.
ną zwiększenia przewodnictwa glutaminergicznego, które z 25. Kushnareva Y.E., Wiley S., Ward M.A. i wsp.: Excitotoxic injury to mito-kolei powoduje liczne uszkodzenia procesów komórkowych.
chondria isolated from cultured neurons. J. Biol. Chem., 2005, 12, 28894-Jednym z efektów nadmiernego przekaźnictwa glutaminer-28902.
gicznego jest zwiększenie stężenia komórkowej puli wapnia 26. Lee Al., Ogle W., Sapolsky.: Stress and depression; possible links to neuron death in the hippocampus. Bipolar Disoredrs, 2002, 4, 117-128.
i uruchomienie całej kaskady procesów przebiegających z 27. Liu J., Wang X., Shigenaga M.K., Yeo H.C. i wsp.: Immobilization stress wytwarzaniem wolnych rodników.
causes oxidative damage to lipid, protein and DNA in brain of rats, FA-Istotne znaczenie ma także negatywny wpływ glikokort-SEB J., 1996, 10, 1532-1538.
kosteroidów na status antyoksydacyjny komórek nerwowych.
28. Marla S.S., Lee J., Groves J.T.: Peroxynitrrite rapidly permeates phospho-lipids membranes. Proc. Natl. Acad. Sci. USA, 1997: 94, 14234-14248.
Zarówno zwiększenie generacji RFT, jak i obniżenie stęże-29. McIntosh L., Cortopassi K., Sapolsky R.: Glucocorticoids my alter antio-nia związków o właściwościach antyoksydacyjnych jest naj-xidant enzyme capacity in the brain: baseline studies. Brain Res., 1998, prawdopodobniej jedną z przyczyn zachodzącego w zabu-791, 209-214.
rzeniach depresyjnych procesu neurodegeneracyjnego.
30. McNally J.S., Saxena A.. Dikalov S.i wsp.: Regulation of xanthine oxidoreductase protein expression by hydrogen peroxide and calcium. Arterio-scler. Thromb. Vasc. Biol., 2005, 25, 8, 623-628.
31. Miller R.J.: The control of neuronal Ca2+ homeostasis. Prog. Neurobiol., PIŚMIENNICTWO
1991, 37, 255-285.
32. Moncada S., Erusalimsky J.D.: Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol., 2002, 3, 214-220.
1. Baimbridge K.B., Celio M.R., Rogers J.H.: Calcium binding proteins in 33. Muralikrishma A.R., Hatcher J.F.: Phospholipase A2, reactive oxygen the nervous system. Trends Neurosci., 1992, 15, 303-308.
species, and lipid peroxydation in cerebral ischemia. Free Radical Biol.
2. Bartosz G.: Druga twarz tlenu. PWN. Warszawa, 2004.
and Med., 2006, 40, 376-387.
3. Bhargava A., Meijer O., Dallman M. i wsp.: Plasma membrane calcium 34. Okado-Matsumoto A., Fridivich J.: Subcellular distribiuton of superoxide pump isoform gene expression is repressed by corticosterone and stress dismutses (SOD) in rat liver, Cu, Zn-SOD in mitochonfria. J. Biol. Chem., I rat hippocampus. J. Neurosci., 2000, 20, 9, 3129-3138.
2001, 276, 38388-38-393.
4. Blomgren K., Hagberg H.: Free radicals, mitochondria and hypoxia-ische-35. Ott M., Robertson J.D., Gogvadze V., Zhivotovsky B. i wsp.: Cytochrome mia in the developing brain. Free Radical Biol. And Med,. 2006, 40, 388-c release from mitochondria proceeds by a two-step process.: Proc. Natl.
397.
Acad. Sci. USA, 2002, 1259-1263.
5. . Bonfoco E., Krainc D., Ankarcrona M. i wsp.: Apoptosis and necrosis: 36. Pajović S.B., Pejić S., Stojiljković V. i wsp.: Alteration in hippocampal antio-Two distinct events induced, respectively by mild and intense insults with xidant enzyme activities and sympathoadrenomodullary system damage N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures.
to lipid, protein and DNA in brain of rats. FABES J., 1996, 10, 1532-1538.
Proc. Natl. Acad. Sci. USA, 1995, 92, 7162-7166.
37. Patel R., McIntosh L., Mc Laughin J. i wsp.: Disruptive effects of gluco-6. Brookes P.S., Yoon Y., Robotham J.L. i wsp.: Calcium, ATP and ROS: a corticoids on gltathione peroxidase biochemistry in hippocampal cultu-mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol., 2004, 287, res. J. Neurochem., 2002, 82, 118-125 .
817-833.
38. Patterson S.D., Spahr C.S., Daugas E. i wsp.: Mass spectrometric iden-7. Brown G.C.: Nitric oxide and mitochondrial apoptosis. Biochim. Biophys.
tification of proteins released from mitochondria undergoing permeability Acta, 1999, 1411, 351-369.
transition. Cell death Differ., 2000, 7, 137-144.
8. Cheng Y., Sun A.Y.: Oxidative mechanism involved in kainite induced 39. Pereira C., Ferreira C., Carvalho C. i wsp.: Contribution of plasma mem-cytotoxicity in cortical neurons. Neurochem. Res., 1994 19, 1557-1564.
brane and endoplasmic reticulum Ca2+-ATPases to the synaptosomal Ca2+
9. Christensen T., Brunth T., Balchen T. i wsp.: Evidence for formation of increase during osidative stress. Brain Res., 1996, 713, 269-277.
hydroxyl radicals during reprfusion after global ischemia in rats using sa-40. Periera B., Rosa L., Safi D. i wsp.: Hormonal regulation of superoxide licylate trapping and microdialysis. Neurobiol. Dis., 1994, 1, 131-138.
dismutase, catalase and glutathione peroxidase activities in rat macro-10. Cleeter M.W., Cooper J.M., Darley-Usmar V.M. i wsp.: Reversible inhibi-phages. Biochem. Pharmacol., 1995, 2093-2098.
tion of cytochrom c oxidase the terminal enzyme of the mitochondrial 41. Pieper A.A., Verma A., Zang J. i wsp.: Poly (ADP-ribose) polymerase, respiratory chain by nitric oxide. Implication for neurodegenerative dise-nitric oxide and cell death. Trends Pharmacol. Sci., 1999, 20, 171-181.
ases. FEBS Lett., 1994, 345, 50-54.
42. Radak Z., Sasvari M., Nyakas C. i wsp.: Single Bout of exercise elimina-11. Coyle J., Puttfarcken P.: Oxidative stress, glutamate and neurodegene-tek the immobilization-induced oxidative stress in rat brain. Neurochem.
rative disorders. Science, 1993, 262, 29, 689-695.
Int., 2001, 39, 33-38.
12. Crompton M.: The mitochondrial permeability transition pore and its role 43. Reynolds I., Hastings T.: Glutamate induces the production of reactive in cell death. Biochem. J., 1999, 341, 233-249.
oxygen species in culture forebrain neurons following NMDA receptor 13. Dringen R.: Metabolism and functions of glutathione in brain. Prog. Neu-application. J. Neurosci., 1995, 15, 3318-3327.
robiol., 2000, 62, 649-671.
44. Salińska E., Danysz W., Nazarewicz J.W.: The role of extotoxicity in neu-14. Dugan L., Sensi S., Canzoniero L., Handran S. i wsp.: Mitochondrial pro-rodegeneration. Folia Neuropathologica, 2005, 43, 4 322-339.
duction of reactive oxygen species in cortical neurons following exposure 45. Sengpiel B., Preis E., Krieglstein J.H. i wsp.: NMDA – induced superoxi-to N-methyl-D-aspartate. J. Neurosc., 1995, 15, 6377-6388.
de production and neurotoxicity in cultured rat hippocampal neurons: role 15. Fiskum G., Starkov A., Polster B.M. i wsp.: Mitochondria mechanism of of mitochondria. Eur. J. Neurosci., 1998, 10, 1903-1910.
neuronal cell Heath and neuroprotective interventions in Parkinson’s di-46. Schöpfer F., Riobó N., Carreras M. i wsp.: Oxidation of ubiquinol by pero-sease. Ann. NY. Acad. Sci., 2004, 1011, 86-100.
xynitrite: implications for protection of mitochondrial against nitrosative 16. Gagliardi R.J.: Neuroprotection, excitotoxicicity and NMDA antagonists.
damage. Biochem. J., 2000, 349, 35-42.
Arq. Neuropsiquiatr., 2000, 58, 2-B, 583-588.
47. Sousa S.C., Maciel E.N., Vercesi A.E. i wsp.: Ca2+ – induced oxidative 17. Grijalba M.T., Vercesi A.E., Schreier S.: Ca2+ induced increased in submi-stress in brain mitochondria treated with the respiratory chain inhibitor tochondrial particles. A possible early step in the mechanism of Ca2+-
rotenone. FEBS Lett., 2003, 543, 179-183.
stimulated generation of reactive oxygen species by the respiratory cha-48. Tibor K., Sesji B.: Calcium in ischemic cell Heath. Stroke, 1998, 29, 705-in. Biochemistry, 1999, 38, 13279-13287.
718.
18. Guerini D.: The Ca2+ pumps and Na+/Ca2+ exchanger. BioMetals, 1998, 49. Toskiahi K., Hiroshi K., Akinori A.: Endogenous factors regulating neuro-11, 319-330.
nal death induced by radical stress. Biol. Pharm., 2004, 27, 7, 964-967.
19. Guerini D.: The significance of the isoform of plasma membrane calcium 50. Turrens J.F.: Mitochondrial formation of reactive oxygen species. J. Phy-ATP-ase. Cell Tissue Res., 1998, 292, 191-197.
siol., 2003, 552, 335-344.
20. Halliwell B, Cross C.E.: Oxygen-derived species: their relation to human 51. Yao X., Rarey K.: Detecion and regulation of Cu/ZnSOD and MnSOD in disease and environmental stress. Environmental Health Perspectives, rat cochlear tissues. Hearing Res., 1996, 96, 199-203.
1994, 102, suppl. 10, 5-12.
52. Zadi S., Banu N.: Antioxidant potential of vitamins A, E and C in modula-21. Harrison R.: Structure and function of xanthine oxidoreductase: where ting oxidative stress in rat brain. Clin. Chim. Acta, 2004, 340, 229-233.
are we now? Free Radicl, Biol. and Med., 2002, 33, 774-797.
53. Żylińska L., Soszyńska M.: Plasma membrane Ca2+-ATP-ase in exitable 22. Inue M., Sato E.F., Nishikawa M., Park A.M. i wsp.: Mitochondrial gene-and nonexcitable cells. Acta Biochim. Pol., 2000, 47, 3, 529-539.
ration of reactive oxygen species and its role in aerobic life. Curr. Med.
Chem., 2003, 10, 2495-2505.
Otrzymano 22 maja 2007 r.
23. Jekabsone A., Ivanoviene L., Brown G.C. i wsp.: Nitric oxide and calcium Adres: Piotr Gałecki, Uniwersytet Medyczny w Łodzi: 1Klinika Psychiatrii together inactivate mitochondrial complex I and induce cytochrome c re-Dorosłych, ul. Aleksandrowska 159, 91-229 Łódź, tel. (0 42) 652 12 89, lease. J. Mol. Cell Cardiol., 2003, 35, 803-809.
faks (0 42) 640 50 58, e-mail: klpsych@o2.pl
Wyszukiwarka
Podobne podstrony:
ca mit
Mit polityczny
020 AC CA
18 Mit mityzacja mitologie wsp Nieznany (2)
zestawy glosnikowe cz1 MiT 10 2007
Legendy Mit o stworzeniu
Mit monogamii Jak poradzic sobie ze zdrada partnera mitmon
mit
Lori Foster Rendezvous mit Risiko
Powerprojekte mit Arduino und C
Ca ka niew a ciwa
Odpowiedzi do tego drugiego ca dałem i jest na 38 pytań, instytucje i źródła prawa w UE
Mit o Syzyfie
Mit o Prometeuszu streszczenie, Ściągi
mit o ucieczce hathor, Egipt
mit o ozyrysie, Egipt
mit. celtycka legendy arturiańskie postacie, Celtowie
więcej podobnych podstron