Kryptografia molekularna
Krzysztof Maćkowiak
Doradztwo Gospodarcze DGA S.A., Politechnika Poznań ska
www.kryptografia.com
Celem referatu jest przybliżenie innej, alternatywnej metody ochrony danych.
Niekonwencjonalne podejście, wykorzystujące cząsteczki DNA, umożliwia
wykonywanie najważniejszych operacji wykorzystywanych w ochronie danych
takich jak: szyfrowanie, ukrywanie danych (steganografia), tworzenie skrótu,
kryptoanaliza oraz identyfikacja osób. W referacie przedstawione będą
podstawowe pojęcia związane z biologią (budowa cząsteczki DNA, łańcuchowa
reakcja polimerazy, sekwencjonowanie) oraz bioinformatyką (komputer DNA i
jego porównanie z komputerami tradycyjnymi, wykorzystywanie DNA i RNA do
rozwiązywania problemów obliczeniowych). Omówione będą w skrócie
sposoby wykorzystywania cząsteczek DNA w ochronie danych oraz dokładnie
przedstawione zostaną przykładowe metody kryptografii i steganografii z
wykorzystaniem cząsteczek DNA.
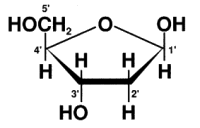
1. Budowa cząsteczek DNA
Wszystkie organizmy żywe mają podobną molekularną budowę biochemiczną.
Składają się z tych samych molekuł: białek i kwasów nukleinowych. Kwasy
nukleinowe kodują informację genetyczną potrzebną do wytwarzania białek i
przekazania tych reguł następnym pokoleniom.
Wyróżniamy dwa rodzaje kwasów nukleinowych:
DNA – deoksyrybonukleinowy (deoxyribonucleic acid),
RNA – rybonukleinowy (ribonucleic acid).
Zasadę budowy DNA odkryli w 1953 roku:
J.D. Watson i F.H. Crick – model helisy – Nagroda Nobla,
M. Wilkins – Nagroda Nobla,
R.E. Franklin.
DNA jest dwuniciową helisą składającą się z prostszych molekuł (nukleotydów).
Pojedynczy nukleotyd zbudowany jest z:
cząsteczki cukru – deoksyrybozy (2’-deoxyribose), zawierającej pięć atomów
węgla, oznaczonych od 1’ do 5’.
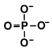
jednej grupy fosforanowej,
zasady azotowej.
Molekuły DNA różnią się zasadami azotowymi. To właśnie zdefiniowana kolejność
zasad zawartych w cząsteczkach DNA stanowi nośnik informacji genetycznych.
Wyróżnia się cztery zasady azotowe: adenina A (adenine), guanina G (guanine),
cytozyna C (cytosine) oraz tymina T (tymine). Adenina i guanina to pochodne
puryny, natomiast cytozyna i tymina to pochodne pirymidyny.
Nukleotydy w zależności od występujących w nich zasad nazywamy:
deoksyadenylanem (z adeniną), deoksyguanylanem (z guaniną),
deoksycytydylanem (z cytozyną) oraz deoksytymidylanem (z tyminą). Łańcuch DNA
jest oligonukleotydem.
Podwójną helisę DNA otrzymujemy dzięki wiązaniom zasad komplementarnych
(wiązania wodorowe, H-bond). Ze względu na budowę helisy, możliwe są purynowo-
pirymidowe pary zasad. Jednym ze składników musi być puryna, drugim zaś
piramidyna. Powstawanie par zasad (hybrydyzacja, parowanie) jest dodatkowo
ograniczone warunkami tworzenia się wiązań wodorowych. Atomy wodoru w
zasadach zajmują dokładnie zdefiniowane położenie. Adenina nie może tworzyć
pary z cytozyną, gdyż w jednej z pozycji wiążących mogłyby znajdować się dwa
wodory, natomiast w drugim brakowałoby wodoru (komplementarność zasad).
Podobnie guanina nie może tworzyć pary z tyminą. Możliwe jest utworzenie
następujących par: A-T (adenina-tymina) oraz G-C (guanina-cytozyna).
Pomiędzy zasadami występują wiązania wodorowe:
podwójne pomiędzy A i T,
potrójne pomiędzy G i C.
Rdzeń DNA jest stały dla całej cząsteczki i składa się z reszt deoksyrybozy
połączonych resztami fosforanowymi. Zasady purynowe i pirymidowe znajdują się
wewnątrz łańcucha, a fosforany i reszty deoksyrybozy – na zewnątrz helisy. Zasady
są skręcone względem siebie pod kątem 36˚. Zatem na całkowity skręt helisy
przypada 10 nukleotydów w każdym łańcuchu.
Łańcuch DNA wykazuje polarność. Jeden z jego końców ma grupę 5’-OH, drugi
3’-OH. Przyjmuje się, że niezwiązana grupa 5’-OH jest ulokowana w nukleotydzie
znajdującym się po lewej stronie zapisu, natomiast grupa 3’-OH po prawej stronie.
Zasady zapisujemy, więc w kierunku 5’->3’.
Obie nici DNA mają przeciwną orientację - są antyrównoległe. Znając jeden łańcuch,
można odtworzyć drugi poprzez operację odwrotnego dopełnienia (reverse
complementation).
3’ … ATCTGAATCCG … 5’
Jest to podstawa mechanizmu replikacji DNA w komórkach. W procesie replikacji,
biorą udział enzymy, zwane polimerazami DNA, które w procesie tym czerpią
instrukcje od matrycowych łańcuchów DNA.
Dwuniciowa helisa stabilizowana jest przez wiązania wodorowe między
komplementarnymi zasadami. Po ich zerwaniu dwa łańcuchy DNA z łatwością się
rozdzielają. Efekt ten można uzyskać, przez ogrzewanie roztworu DNA, jego
zakwaszaniu albo alkalizację, powodujące jonizację zasad tzw. denaturację. Proces
rozplatania dwuniciowej helisy nazywamy topnieniem. Temperatura topnienia zależy
od ilości odpowiednich par zasad. Musi być ona wyższa, gdy mamy więcej par G-C,
które są stabilniejsze od par A-T.
1.1. Reakcja łańcuchowa polimerazy (PCR – polymerase chain reaction)
Reakcja PCR opracowana została przez K.B. Mullisa i M. Smitha, którzy otrzymali za
to odkrycie Nagrodę Nobla w dziedzinie chemii w 1993 roku. Jest to reakcja
powielania (amplifikacji) określonej sekwencji DNA. Reakcja PCR wykorzystuje
enzym (polimerazę DNA), który jest katalizatorem syntezy pojedynczego łańcucha
DNA. Polega ona na rozpleceniu podwójnego łańcucha (etap denaturacji). Etap
anilingu: hybrydyzacja primera komplementarnego do końca 5’ pojedynczego
łańcucha DNA. Kolejny etap: polimeryzacja – do końca 3’ primera zostanie
dobudowana komplementarna nić DNA. W efekcie tych trzech etapów powstanie
dwuniciowa helisa. Poszczególne etapy związane są ze zmianą temperatury,
dlatego w reakcji PCR wykorzystywana jest polimeraza odporna na wysoką
temperaturę.
Jednokrotna reakcja podwaja liczbę kopii. Wielokrotne zastosowanie reakcji PCR
powoduje ekspotencjalny przyrost liczby kopii. Błędy w procesie powielania
występują bardzo rzadko, mniej więcej raz na 10 miliardów zreplikowanych zasad.
1.2 Sekwencjonowanie
Metoda ta, wynaleziona przez naukowców amerykańskich i brytyjskich w roku 1977,
polega na podziale cząsteczki DNA na fragmenty a następnie na odczytaniu
sekwencji nukleotydów, z których składa się ta cząsteczka.
Istnieje wiele metod sekwencjonowania. Przykładowe metody:
1. Elektroforeza DNA w żelu agarozowym – fragmenty łańcuchów DNA
umieszczamy w 4 kieszonkach i poddajemy działaniu silnego pola
elektrycznego, na skutek czego fragmenty DNA migrują w żelu w kierunku
elektrody dodatniej (DNA ma ładunek ujemny) z szybkością zależną od ich
wielkości i kształtu. Następnie odczytujemy sekwencję na podstawie
kolejności prążków. Metoda nie jest pozbawiona błędów.
2. Sekwencjonowanie przez hybrydyzację – wykorzystujemy możliwość
tworzenia helisy z pojedynczej nici. Chcemy odczytać sekwencję nukleotydów
pojedynczej nici o długości n. Wprowadzamy ją do roztworu wraz z pełną
biblioteką oligonukleotydów o długości k (k<<n). Oligonukleotydy, które
wykrywamy na podstawie fluorescencji, przyłączają się do łańcucha. Po
przyłączeniu możemy na podstawie barwy przyłączonych oligonukleotydów
odczytać szukaną sekwencję łańcucha. Bibliotekę nukleotydów można
stworzyć na specjalnym chipie.
Bioinformatyka
(bioinformatics, biocomputing) zajmuje się symulacjami
komputerowymi w biochemii i biologii molekularnej, tworzeniem i zarządzaniem
bazami danych, poszukiwaniem, wyciąganiem, analizą i interpretacją informacji z
biologicznych baz danych, tworzeniem nowych algorytmów i metod statystycznych
do analizy danych biologicznych oraz innymi technikami informatycznymi
związanymi z naukami biologicznymi.
Informatyka DNA (DNA computing) określana również jako informatyka
molekularna, jest nową alternatywą dla równoległych systemów komputerowych. Jej
początek sięga 1994 roku, kiedy to Leonard M. Adleman (współtwórca znanego
algorytmu szyfrowania asymetrycznego RSA) po raz pierwszy zademonstrował
możliwość wykorzystania cząsteczek molekularnych do rozwiązywania problemów
matematycznych. Z użyciem cząsteczek DNA rozwiązał on siedmiowierzchołkowy
(14 dróg) problem poszukiwania ścieżki Hamiltona.
Ścieżka Hamiltona jest ścieżką wychodzącą z dowolnego, ustalonego wierzchołka
grafu i przechodzącą przez wszystkie wierzchołki dokładnie jeden raz (przez
pojedynczą krawędź może przejść wielokrotnie). Ścieżka kończy się w ustalonym
wierzchołku docelowym (w przypadku cyklu Hamiltona jest to ten sam wierzchołek,
w którym rozpoczęto poszukiwanie).
Algorytm Leonarda Adlemana dla grafu o n wierzchołkach:
I)
Stwórz duży zbiór losowych ścieżek, przechodzących przez graf.
II)
Dla każdej ścieżki sprawdź, czy:
a) zaczyna się w wierzchołku początkowym i kończy w docelowym, jeżeli
nie to usuń ją ze zbioru,
b) przechodzi dokładnie przez n wierzchołków, jeżeli nie to usuń ją ze
zbioru,
c) przechodzi dokładnie przez każdy wierzchołek, jeżeli nie to usuń ją ze
zbioru.
I)
Jeżeli powstały zbiór zawiera elementy to istnieje szukana ścieżka
Hamiltona, w przeciwnym razie (zbiór jest pusty) ścieżka nie istnieje.
Problem ten zaliczamy do grupy problemów NP-zupełnych, których nie można
rozwiązać w czasie wielomianowym. Adleman rozwiązał ten problem generując
wszystkie możliwe kombinacje jako odrębne łańcuchy DNA. Dla siedmiu
wierzchołków rozwiązanie jest trywialne i można je szybko otrzymać, stosując
normalne komputery lub obliczając ręcznie. Przykład ten ilustruje jednak potencjalne
możliwości komputerów i informatyki DNA.
Inne doświadczenie przeprowadzone w Mount Sinai School of Medicine w Nowym
Yorku pokazuje możliwość wykonywania operacji dodawania liczb binarnych
reprezentowanych przez łańcuchy DNA. Powstał również komputer DNA, z którym
można zagrać w grę „kółko i krzyżyk”.
W 2000 w Princeton przedstawiono możliwość zastosowania cząsteczek RNA do
rozwiązywania problemów (problem skoczków szachowych na szachownicy o
wielkości 3x3) oraz budowy komputerów molekularnych.
2. Komputer DNA
Komputer DNA (molekularny) jest to zbiór specjalnie wyselekcjonowanych
łańcuchów DNA, których kombinacja spowoduje rozwiązanie postawionego
problemu. Nadzieją pokładaną w komputerach DNA jest ich wysoki stopień
równoległości, co potencjalnie powinno umożliwić rozwiązanie problemów
wymagających wielu obliczeń poprzez obliczenia równoległe.
W 1973 roku Charles Benett zaproponował model programowalnego komputera
molekularnego zdolnego do realizacji dowolnego algorytmu. W praktyce pierwsze
komputery powstały w 2001 roku. Autorami jednego z nich są naukowcy z Instytutu
Weizmanna w Rehovot, którzy wykorzystali w swoich doświadczeniach cząsteczki
DNA, które pełnią zarówno rolę „oprogramowania”, sygnału wejścia-wyjścia, jak
również dostarczają potrzebnej energii. W roku 2003 komputer ten został
udoskonalony i osiągał prędkość reakcji molekularnych 330 TFLOPS w objętości 5
mililitrów (mała łyżeczka płynu). W komputerze tym rolę sprzętu pełnią enzymy
restrykcyjne, które rozpoznają ściśle określone sekwencje DNA i w ich obrębie
przecinają cząsteczkę.
Również w Polsce trwają badania nad stworzeniem komputera molekularnego. W
roku 2002 we Wrocławiu powstała tzw. Grupa Inicjatywna Konstrukcji Prototypu
Opartego na DNA.
2.1. Komputery DNA a komputery tradycyjne
Porównanie komputerów tradycyjnych i komputerów zbudowanych z cząsteczek
DNA jest bardzo trudne i może doprowadzić w wielu sytuacjach do rozbieżnych
wyników. Poniższe dane należy zatem traktować jedynie jako dane przybliżone.
DNA jako nośnik informacji – pojemność pamięci biologicznej jest znacznie większa
niż stosowanych dzisiaj nośników. Małe rozmiary DNA sprawiają, że w objętości 1
mm3 mieści się 10 mld MB informacji – 10 PB (zakładając, że jedna para
nukleotydów stanowi jeden bit informacji – 0 lub 1). Jeden gram DNA zawiera 1021
zasad DNA, co odpowiada 108 TB danych. Kilka gramów DNA może kodować
wszystkie informacje dostępne na ziemi.
DNA jako superkomputer. Zakładając, że przodujące komputery umożliwiają
działania z prędkością 100 MIPS (milion instrukcji na sekundę), łańcuchy DNA
działają z prędkością ponad 10 razy szybszą.
Komputery DNA zapewniają duży stopień równoległości przetwarzania. W jednej
kropli roztworu wodnego może znajdować się ponad bilion molekularnych
procesorów, wykonujących miliard operacji na sekundę.
Komputery DNA nie potrzebują zasilania elektrycznego i są wysoce
energooszczędne.
Na podstawie tych informacji widać, że komputery DNA stanowią ciekawą
alternatywę dla komputerów stacjonarnych.
2.2. Kodowanie DNA a kodowanie binarne
W przypadku kodowania binarnego operujemy dwoma bitami 0 i 1. W przypadku
kodowania DNA mamy możliwość skorzystania z 4 nukleotydów A,T,G,C.
W zależności, ile znaków będziemy chcieli zakodować, taki długi będzie ciąg
nukleotydów przypadający na każdy znak. W przypadku, gdy chcemy zakodować 64
różne znaki potrzebujemy ciągu składającego się z 3 nukleotydów.
Możemy również traktować A i T jako 0 a G i C jako 1. Korzystając z alfabetu ASCII
znak A można zapisać jako 6510=10000012=GTTATAC. Nie musimy tworzyć
specjalnego alfabetu. W tym przypadku nie wykorzystujemy czterech zasad do
kodowania tylko dwie.
Przy budowie alfabetu należy stosować metody charakterystyczne dla szyfrów
homofonicznych (najczęściej występujące litery kodowane są za pomocą kilku
czwórek). Zapobiega to sytuacji, w której charakterystyczny kawałek tekstu, mógłby
być traktowany przez kryptoanalityka jako nowy primer. Dzięki temu mógłby on
otrzymać część szukanego tekstu. Inną możliwością jest zastosowanie kompresji lub
innej metody do zmiany układu liter w tekście jawnym przed jego zamianą na ciąg
nukleotydów. Metoda ta podobnie jak klucz nie może zostać ujawniona.
3. DNA a ochrona danych
Biotechnologia znajduje swoje zastosowanie również w zagadnieniach związanych z
ochroną danych.
DNA może być wykorzystywane w:
kryptografii – stosowany algorytmy jednorazowy (one-time-pad) z
wykorzystaniem operacji podstawienia lub XOR,
steganografii – bezpieczne ukrycie wiadomości a następnie jej odtworzenie
dzięki posiadanej wiedzy o kluczu,
tworzeniu molekularnej sumy kontrolnej z wykorzystaniem obrazów żelowych,
jako odpowiednik skrótu (funkcji haszującej), wykorzystany do znakowania
przedmiotów,
systemach identyfikacji osób,
kryptoanalizie – do łamania konwencjonalnych algorytmów symetrycznych
(np.DES) oraz asymetrycznych.
Algorytm jednorazowy (One-time-pad)
Tekst jawny jest szyfrowany przy użyciu cząsteczek DNA za pomocą algorytmu
jednorazowego, który po spełnieniu trzech podstawowych warunków jest
algorytmem zapewniającym bezwzględne bezpieczeństwo. Trzy podstawowe
warunki, które musi spełniać łańcuch DNA stanowiący klucz:
musi być przynajmniej tak długi jak szyfrowany tekst (przy dużym
upakowaniu danych w cząsteczkach DNA nie stanowi to większego
problemu),
musi być losowy,
może być użyty tylko jeden raz.
Zanim zaczniemy szyfrowanie za pomocą tej metody musimy stworzyć długi łańcuch
DNA (klucz) zbudowany z losowo wybranych krótkich sekwencji oligonukleotydów.
Ten łańcuch stanowi podstawę naszej metody. Będzie używany jako tablica (klucz),
za pomocą, której będziemy szyfrować i deszyfrować wiadomości. Musi być ona
znana przez obie komunikujące się strony (łatwość w wymianie stanowi tutaj
mikroskopijna wielkość tego łańcucha) i nie może być ujawniona nikomu innemu.
Metoda podstawieniowa
Tekst wejściowy stanowiący ciąg binarny o długości n dzielony jest na ciągi znaków
o różnych długościach. Tablica podstawieniowa one-time-pad zbudowana jest w taki
sposób, aby wszystkie możliwe ciągi wejściowe zostały zamienione na ciągi znaków
zaszyfrowanych o różnych długościach.
Szyfrowanie za pomocą metody podstawieniowej polega na zamianie każdego ciągu
wejściowego na podstawie tablicy podstawieniowej na tekst wyjściowy
(zaszyfrowany). Deszyfrowanie jest operacją odwrotną.
Zastosowanie cząsteczek DNA w algorytmie.
Tekst wejściowy – probówka zawierająca krótkie odcinki DNA.
Tekst zaszyfrowany – probówka zawierająca inne krótkie odcinki DNA.
Szyfrowanie polega na losowej, lecz odwracalnej zamianie odcinków
reprezentujących tekst wejściowy na odcinki DNA reprezentujące tekst
zaszyfrowany. Oryginalne cząsteczki są usuwane.
Budowa tablicy podstawieniowej:
Tworzymy długi łańcuch DNA składający się z wielu segmentów. Każdy segment
składa się z dwóch części: ciągu znaków reprezentujących tekst jawny oraz ciągu
znaków reprezentujących odpowiadający mu tekst zaszyfrowany.
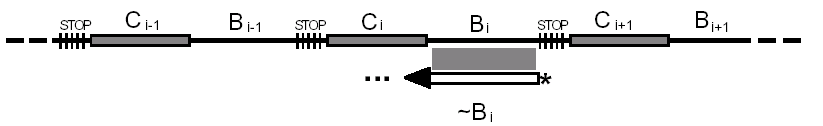
Reprezentacja łańcucha:
Długość łańcucha: n.
Każdy segment to odcinek ograniczony z obu stron stoperem. Łańcuch składa się z
d = n / (L1+L2+L3) powtarzających się segmentów.
Bi – ciąg o długości L1=c1log n, reprezentujący tekst zaszyfrowany.
Ci – ciąg o długości L2=c2 log n, reprezentujący tekst jawny.
Każdy segment unikalnie odwzorowuje ciąg tekstu jawnego na ciąg zaszyfrowany.
STOP – primer - ciąg nukleotydów o długości L3=c3.
Chcąc wygenerować sekwencję nukleotydów, odpowiadających tekstowi
zaszyfrowanemu na podstawie tej tablicy, jako primera używamy ~Bi. Na jego
podstawie określamy ciąg odpowiadający tekstowi jawnemu Ci (reakcja PCR).
Stoper zapobiega dalszemu rozszerzaniu się reakcji ponad interesujący nas ciąg
jawny.
3.2 Steganografia DNA
Ludzie już od czasów starożytnych posiadali tajemnice, które chcieli ukryć przed
innymi. W wielu przypadkach uciekali się do metod, które powodowały, że tekst był
niewidoczny dla innych. Przykładem może być tutaj atrament sympatyczny, używany
przez szpiegów czy też miniaturowe zdjęcia wklejane do dokumentów jako kropki
kończące zdania. Taki sposób ukrywania tekstu nazywamy steganografią.
Prezentowany algorytm wykorzystuje w tym przypadku cząsteczki DNA.
Metoda I
Ukrywanie informacji
1. Tworzymy alfabet reprezentujący znaki za pomocą ciągów nukleotydów o
długości 4.
2. Tekst jawny kodujemy przy pomocy stworzonego alfabetu.
3. Tworzymy klucz, który musi pozostać tajny.
4. Klucz kodujemy według tego samego alfabetu, którego użyliśmy do
kodowania tekstu jawnego. Klucz stanowi starter (primer), który umożliwia
znalezienie tekstu wśród innych cząsteczek DNA poprzez zastosowanie
reakcji PCR. Ważne jest, aby po zamianie na ciąg nukleotydów, miał on
długość minimum 20 nukleotydów, aby z dużym prawdopodobieństwem
wśród innych cząsteczek nie było więcej takich ciągów. Kryptoanalityk musi
sprawdzić 420 (240) możliwych primerów, aby odnaleźć wiadomość. Nie jest to
aż tak dużo, więc najlepiej aby ciąg reprezentujący klucz składał się z ponad
35 nukleotydów. Przyjmując 4 nukleotydy na jeden znak, klucz powinien mieć
około 9 znaków.
5. Budujemy nić klucz_komplementarny-tekst-klucz_komplementarny oraz drugą
nić, którą stanowi klucz.
Przykładowe metody tworzenia pojedynczych nici DNA:
•
synteza na podłożu stałym,
•
metoda fotolitograficzna.
5. Wykonujemy reakcję PCR, na skutek czego otrzymujemy dwuniciową
cząsteczkę DNA.
6. Stworzoną cząsteczkę umieszczamy wśród wielu innych cząsteczek o
podobnej budowie.
Druga strona musi znać alfabet, którego użyto do kodowania oraz klucz (primer).
Trudność odnalezienia tekstu polega na przejrzeniu ogromnej ilości cząsteczek
DNA. Odnalezienie właściwej cząsteczki w tym przypadku jest równoznaczne ze
złamaniem tej metody i odnalezieniem szukanego tekstu jawnego.
Odczytywanie ukrytej informacji
1. Chcąc znaleźć właściwą cząsteczkę DNA należy wykonać reakcję PCR,
używając łańcucha nukleotydów reprezentującego klucz jako primera.
Reakcję PCR należy przeprowadzić wielokrotnie, w celu zwielokrotnienia
liczby cząsteczek zawierających ukryty tekst.
2. Po otrzymaniu pojedynczej nici z cząsteczki zawierającej ukryty tekst,
odczytujemy ciąg nukleotydów (sekwencjonowanie).
3. Zamieniamy ciąg nukleotydów na poszukiwany tekst przy pomocy alfabetu,
na podstawie którego kodowaliśmy tekst jawny w fazie ukrywania informacji.
Najlepiej wyjaśnić tą metodę na przykładzie.
Chcemy ukryć tekst jawny IT. Po zamianie tekstu na ciąg nukleotydów, otrzymujemy
następujący ciąg: TATAGTCC.
Tworzymy hasło H2 (ze względu na czytelność przykładu hasło składa się tylko z
dwóch znaków, natomiast w praktyce powinno być dłuższe). Po zamianie na ciąg
nukleotydów ma ono postać: TTACACCA.
Następnie tworzymy następujące nici:
•
AATGTGGT TATAGTCC AATGTGGT,
•
TTACACCA.
Z wykorzystaniem enzymu polimerazy tworzymy podwójną helisę DNA. Cząsteczkę
tę umieszczamy w probówce z określoną substancją.

Odbiorca musi również dokonać zamiany hasła na ciąg nukleotydów. Następnie po
otrzymaniu probówki wielokrotnie wykonuje reakcję PCR, jako primer stosując ciąg
nukleotydów reprezentujących hasło.
Rysunek 1. Działanie reakcji PCR.
W wyniku reakcji PCR liczba cząsteczek DNA zawierających ukryty tekst zostaje
zwielokrotniona. W kolejnym kroku odbiorca wykonuje sekwencjonowanie i
otrzymuje ciąg TATAGTCC. Znając alfabet zamienia go na tekst IT. Bezpieczeństwo
tej metody oparte jest na tajności klucza.
Najtrudniejszą i najdroższą reakcją z punktu widzenia biologii jest tworzenie długich
nici o określonej sekwencji nukleotydów.
Metoda II
Inne podejście podobne jest do kryptografii wizualnej. Tekstu nie zamieniamy teraz
według specjalnego alfabetu, lecz pewne odcinki DNA lub pojedyncze nukleotydy są
w tym przypadku odpowiednikami zer i jedynek. Również w tym przypadku
cząsteczkę, którą chcemy ukryć umieszczamy wśród wielu innych cząsteczek.
Wszystkie cząsteczki mają jednak podobną budowę. Na początku i końcu znajduje
się primer – ten sam dla wszystkich cząsteczek. W poprzedniej metodzie primer
używany był jako klucz potrzebny do deszyfrowania wiadomości. W tym przypadku
nie odgrywa on takiej roli. Jest on potrzebny, aby umożliwić wykonanie w
późniejszym etapie reakcji PCR. To właśnie pola pomiędzy primerami w fikcyjnych
łańcuchach są używane zarówno do szyfrowania jak i deszyfrowania właściwego
tekstu. Wykonując elektroferezę żelu otrzymujemy dla każdej cząsteczki oddzielne
obrazy dla 0 i 1.
Ukrywanie wiadomości
Nadawca tworzy cząsteczkę DNA z tekstem jawnym oraz primerem na jego
początku i końcu. Następnie preparuje inne cząsteczki, które mają podobną budowę
(długość, primery). Na podstawie obrazu żelowego tych cząsteczek (a) budowana
jest cząsteczka X (b) stanowiąca zaszyfrowany tekst. Cząsteczka X powstaje przez
zmieszanie cząsteczek A,B,C. Następnie nadawca tworzy cząsteczkę Y (b), która
powstaje przez zmieszanie cząsteczek B i C. Stanowi ona klucz potrzebny do
odczytania zaszyfrowanego tekstu. Nadawca musi przekazać odbiorcy tą cząsteczkę
lub jej obraz żelowy.
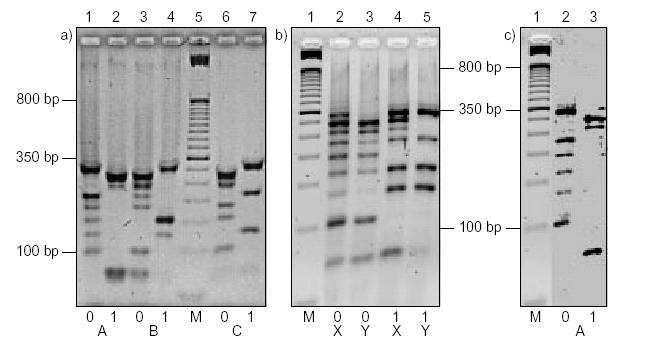
Odkrywanie wiadomości
Za pomocą elektroforezy żelu otrzymujemy obraz żelowy cząsteczek X oraz Y.
Następnie odejmujemy od siebie obrazy X i Y, aby otrzymać wiadomość –
cząsteczkę A (c).
Ta metoda może być również łączona z innymi metodami jak np. wcześniej
omówioną metodą steganografii. Cząsteczki ze wspólnym primerem, potrzebne do
odszyfrowania wiadomości mogą być umieszczane wśród wielu innych cząsteczek o
innej budowie. Primer musi stanowić wtedy tajemnicę.
Przykładowe obrazy żelowe (odczytujemy od dołu do góry):
M - molekularny znacznik wagowy.
Na obrazie żelowym a) mamy przedstawione 3 cząsteczki reprezentujące 9-bitowe
liczby.
1 i 2 = 1000001102 = 26210 - ten ciąg chcemy ukryć
3 i 4 = 0011000012 = 9710
6 i 7 = 1010010012 = 32910
Obraz żelowy b przedstawia cząsteczki X (zmieszane A,B i C) i Y (cząsteczki B i C).
Obraz c) prezentuje cząsteczkę A, którą otrzymaliśmy przez odjęcie Y od X.
Stworzony został również, jak na razie czysto teoretyczny model algorytmu
szyfrowania asymetrycznego z wykorzystaniem DNA.
Wszystkie numery seryjne sprzętu, płyt muzycznych, płyt z oprogramowaniem
można by kodować w postaci cząsteczek DNA a następnie dołączać do przedmiotu,
którego numer ten dotyczy. Wtedy numer seryjny stałby się częścią fizyczną sprzętu
i płyt. Takie oznaczenia zastosowano na szeroką skalę na olimpiadzie w Sydney.
Wszystkie towary związane z olimpiadą: koszulki, czapeczki a nawet kubki do kawy
zostały oznaczone specjalnym atramentem, zawierającym DNA australijskiego
sportowca. Do sprawdzenia autentyczności przedmiotów służył skaner ręczny. W ten
sposób oznaczono ponad 50 milionów przedmiotów. Koszt oznaczenia jednego
przedmiotu wyniósł 5 centów.
3.4 Identyfikacja z użyciem DNA
Cząsteczki DNA wykorzystywane są do identyfikacji ludzi, szczególnie w
kryminalistyce. Fakt, że każdy człowiek ma unikalny kod DNA odkrył w 1985 roku
Alec Jeffreys. Już rok później test DNA pozwolił skazać pierwszych przestępców. W
Polsce identyfikację genetyczną śladów zastosowano na początku lat 90. m.in. w
sprawie zabójstwa taksówkarza w Katowicach w 1994 roku. W przypadku badań
związanych z popełnieniem przestępstwa wykorzystuje się introny, czyli tzw.
niekodujące fragmenty łańcucha DNA. Nie zawierają one informacji o cechach
człowieka a jednocześnie umożliwiają porównanie dwóch fragmentów DNA i
stwierdzenie z dużym prawdopodobieństwem czy pochodzą od tej samej osoby.
Wystarczy porównać próbkę pobraną z miejsca przestępstwa z tą uzyskaną od
oskarżonego. Przykładowa metoda to analiza VNTR (Variable Number of Tandem
Repeats) polegająca na wyszukaniu w łańcuchu DNA szeregu identycznych
sekwencji (np.CACACA) i zliczaniu ich długości (ilości powtórzeń par). Liczba takich
powtórzeń jest różna i charakterystyczna dla danej osoby. Inne zastosowanie tych
metod to testy w sprawach o ustalenie ojcostwa oraz badania medyczne (schorzenia
genetyczne). Podobne metody mogłyby być również wykorzystywane jako
biometryczne metody uwierzytelniania użytkowników w systemach. Istnieje taka
możliwość, lecz w porównaniu z identyfikacją na podstawie obrazu tęczówki wydaje
się być droższa, trudniejsza w implementacji i zarządzaniu oraz wykazuje większe
prawdopodobieństwo błędu identyfikacji.
3.5 Kryptoanaliza algorytmów z wykorzystaniem cząsteczek DNA
Interesująca jest również możliwość wykorzystania komputerów molekularnych w
kryptoanalizie, dzięki ich wysokiemu stopniu zrównoleglenia. Leonard M. Adleman
pokazał, że komputer DNA o wielkości kilku probówek umożliwia odnalezienie klucza
o długości 256 (metoda przeszukiwania wyczerpującego, atak brutalny) algorytmu
DES. Rozwiązanie to nie jest jednak pozbawione wad. Problem w tym przypadku
stanowi implementacja algorytmu w biochemii oraz dokładność wykonywania
obliczeń z wykorzystaniem cząsteczek DNA. Należy również pamiętać, że
komputery molekularne mogą jedynie przyspieszyć rozwiązywanie problemów,
poprzez wysoki stopień równoległości. Dla dłuższych kluczy również komputery
molekularne nie umożliwiają odnalezienia klucza. Przykładowo Beaver zgodnie z
podejściem Adlemana (atak brutalny) oszacował, że komputer potrzebny do
faktoryzacji 1000-bitowej liczby miałby pojemność 10200000 litrów.
[1] Stryer Lubert, Biochemia. PWN, Warszawa 1999
[2] Stepkiewicz O., Flohr M., Spirala bitów i bajtów CHIP, Grudzień 2000
[3] Adleman Leonard, Computing with DNA, 1998,
http://www.usc.edu/dept/molecular-science/fp-sciam98.pdf
[4] Adleman Leonard, Molecular Computations Of Solutions To Combinatorial
Problems. , http://www.usc.edu/dept/molecular-science/fp-sci94.pdf
[5] Adleman Leonard, Rothemund Paul W. K., Roweis Sam , Winfree Erik, On
Applying Molecular Computation To The Data Encryption Standard., 1999,
http://www.usc.edu/dept/molecular-science/fp-des96.pdf
[6] Unold Olgierd, Wrocławski komputer molekularny,
http://pryzmat.pwr.wroc.pl/Pryzmat_170/170dna.html
[7] Richter Ch., Leier A., Banzhaf W., Rauhe H., Private and Public Key DNA
steganography, h
ttp://ls11 - w
ww.cs.uni - d
ortmund.de/people/banzhaf/PubKey.pdf
[8] Gaurav Gupta, Nipun Mehra & Shumpa Chakraverty, DNA Computing, 2001,
http://www.theindianprogrammer.com/technology/dna_computing.htm
Wyszukiwarka
Podobne podstrony:
Kryptografia molekularna
w4 orbitale molekularne hybrydyzacja
kryptologia w bankowości (power point)
Biologia molekularna
W03b Komórkowe i molekularne podłoże zapaleń
Wprowadzenie do Kryptografii
kryptografia
Biologia molekularna koniugacja
genetyka molekularna
elementy genetyki molekularnej biologia 2
Przenikanie firewalli w tunelach kryptograficznych
Met. izol. oczysz.DNA dla studentów, Biologia molekularna
molekuły, egzamin - stare pytania
gielda-3, B.molekularna
seminaria biol mol onkogeneza, Płyta farmacja Poznań, III rok, Biologia molekularna, 2009, sem 6
więcej podobnych podstron