Plan ćwiczeń
Ochrona Środowiska Rolniczego, Biochemia, rok akademicki 2011/2012
1) 6,7.10.2011 Ćwiczenia organizacyjne, zasady BHP, nauka posługiwania się pipetami automatycznymi.
2) 13,14. 10.2011 CUKRY CZ. 1 (instrukcja: ćwiczenie 1)
1. Elementy analizy jakościowej cukrów
a. Reakcja Seliwanowa na ketozy (odróżnienie aldoz od ketoz)
b. Reakcja Biala na pentozy (odróżnianie pentoz od heksoz)
c. Reakcja Benedicta (odróżnienie cukrów redukujących od nieredukujących)
3) 20,21.10.2011 CUKRY CZ. 2 (instrukcja: ćwiczenie 2)
1. Hydroliza polisacharydów
a. Otrzymywanie skrobi z ziemniaka
b. Hydroliza skrobi i próba jodowa
2. Oznaczanie zawartości cukrów rozpuszczalnych w tkankach roślinnych metodą antronową
a) Wykonanie krzywej kalibracyjnej
b) Oznaczenia w materiale roślinnym
4) 27,28.10.2011 LIPIDY cz.1 (instrukcja: ćwiczenie 3)
1. Reakcje charakterystyczne tłuszczów prostych
a. Wykazanie obecności kwasów nienasyconych w tłuszczach
b. Wykrywanie glicerolu
2. Ekstrakcja i frakcjonowanie lipidów z żółtka jaja kurzego
a. Wytrącanie pochodnych kwasu fosfatydowego
b. Wykrywanie choliny
c. Wykrywanie jonów ortofosforanowych
d. Wykrywanie steroli (reakcja Libermana i Burcharda)
3. Oznaczanie zawartości tłuszczu w mleku
5) 3,4.11.2011 LIPIDY cz.2 (instrukcja: ćwiczenie 4)
1. Chromatografia cienkowarstwowa (TLC) fosfolipidów (jedno oznaczenie na grupę)
2. Przybliżone oznaczenia niektórych parametrów charakteryzujących tłuszcze
a) Wyznaczanie liczby kwasowej
b) Wyznaczanie liczby zmydlania
3) Zmydlanie tłuszczów, wytwarzanie mydła, wytrącanie mydła
4) Rozdział barwników fotosyntetycznych metodą chromatografii bibułowej
6) 17,18.11.2011 kol 1. cukry, tłuszcze
7) 24,25.11.2011 AMINOKWASY cz.1 (instrukcja: ćwiczenie 5)
1. Analiza jakościowa aminokwasów
a. Odróżnianie aminokwasów od innych związków
b. Wykrywanie wolnych grup tiolowych
c. Wykrywanie obecności pierścieni aromatycznych
d. Wykrywanie obecności hydroksylu fenolowego
2. Miareczkowanie aminokwasu
8) 1,2.12.2011 AMINOKWASY cz. 2, BIAŁKA cz.1 (instrukcja: ćwiczenie 6)
1. Chromatografia bibułowa aminokwasów
2. Oznaczanie zawartości białek rozpuszczalnych w materiale roślinnym metodą Bradford
3. Elektroforeza SDS-PAGE
9) 8,9.12.2011 BIAŁKA cz.2 (instrukcja: ćwiczenie 7)
1. Miareczkowanie białka
2. Wysalanie białek
3. Wyznaczanie wartości punktu izoelektrycznego białka
10) 15,16.12.2011 kol 2. aminokwasy, białka
11) 5,6.01.2012 ENZYMY CZ.1 (instrukcja: ćwiczenie 8)
1. Oznaczanie aktywności kwaśnej fosfatazy (fosfomonoesterazy)
a) Otrzymywanie ekstraktów z ziemniaka
b) Dobranie czasu inkubacji:
c) Wyznaczenie zależności aktywności kwaśnej fosfatazy od stężenia substratu
d) Określanie zależności aktywności kwaśnej fosfatazy od pH
12) 12,13.01.2012 ENZYMY CZ.2 (instrukcja: ćwiczenie 9)
1. Wykazanie aktywności oksydaz w ziemniaku
2. Wykazanie obecności katalazy w ziemniaku
3. Spektrofotometryczne oznaczanie aktywności enzymów
a) Oznaczanie aktywności puli peroksydaz w ekstrakcie z roślin
b) Oznaczanie aktywności katalazy w ekstrakcie z roślin (CAT) (EC: 1.11.1.6.)
13) 19,20.01.2012 KWASY NUKLEINOWE (instrukcja: ćwiczenie 10)
1. Hydroliza, wykrywanie składników kwasów nukleinowych (ćwiczenie znajduje się również w skrypcie)
2. Izolacja DNA z warzyw, owoców metodą uproszczoną
3. Spektrofotometryczne oznaczanie zawartości kwasów nukleinowych i badanie czystości
4. Jakościowa analiza kwasów nukleinowych - reakcja PCR
14) 26,27.01.2012 kol 3 kwasy nukleinowe, enzymy
15) ZALICZENIE
Ćwiczenie nr 1 (13,14. 10.2011)
CUKRY CZ. 1
1. Elementy analizy jakościowej cukrów (Poniższe ćwiczenia znajdują się również w skrypcie)
a. Reakcja Seliwanowa na ketozy (odróżnienie aldoz od ketoz)
Zasada: Pierścieniowe formy cukrów ulegają odwodnieniu pod wpływem mocnych kwasów, tworząc furfural (pentozy) lub 5-hydroksymetylofurfural (heksozy). Hydroksymetylofurfural tworzy następnie barwne połączenie z rezorcyną. Jednakże pod wpływem rozcieńczonego kwasu solnego odwadnianie ketoz następuje znacznie szybciej aniżeli aldoz, co jest podstawą ich odróżniania.
Wykonanie: Do probówek zawierających oddzielnie po 1 cm3 roztworów A, B, C, które zawierają cukier dodać po 1 cm3 odczynnika Seliwanowa (skład odczynnika: 100 cm3 stężonego kwasu solnego, 300 cm3 wody, 150 mg rezorcyny) i ogrzewać na wrzącej łaźni wodnej. W przypadku ketozy po ogrzewaniu przez ok. 0,5-1 min roztwór zabarwia się na kolor czerwony. Aldoza może również tworzyć podobnie zabarwione połączenie z rezorcyną, jednakże po znacznie dłuższym ogrzewaniu.
b. Reakcja Biala na pentozy (odróżnianie pentoz od heksoz)
Zasada: Pierścieniowe formy cukrów ulegają odwodnieniu pod wpływem mocnych kwasów, tworząc furfural (pentozy) lub 5-hydroksymetylofurfural (heksozy). Furfural daje z orcyną w obecności jonów żelazowych zielone lub zielono-niebieskie zabarwienie, podczas gdy 5-hydroksymetylofurfural w tych warunkach daje roztwór zabarwiony na żółto-brązowy kolor.
Wykonanie: Do 3 probówek nalać po 2 cm3 odczynnika Biala (skład odczynnika: 500 cm3 stężonego HCl, 500 cm3 H2O, 1 g orcyny, 20 kropli 10% wodnego roztworu FeCl3) i ogrzewać na łaźni wodnej przez ok. 3 min, a następnie dodać do każdej probówki oddzielnie po 1 cm3 analizowanych roztworów A,B, C. Zamieszać i wstawić do łaźni wodnej. Po kilku minutach zanotować powstające zabarwienie.
Na podstawie wyników reakcji 2a i b zidentyfikować cukry używane w doświadczeniu; rozróżnić cukry A, B, C spośród wymienionych: glukoza, fruktoza, ryboza.
c. Reakcja Benedicta (odróżnienie cukrów redukujących od nieredukujących)
Zasada: Cukry o właściwościach redukujących mają zdolność redukowania jonów miedzi Cu++ do jonów Cu+ o charakterystycznej, czerwonopomarańczowej barwie tlenku Cu2O. Właściwości redukcyjne charakteryzują tylko te cukry, które posiadają wolny węgiel z grupą aldehydową lub ketonową, a więc wszystkie monosacharydy i te spośród oligosacharydów, które mają wolny przynajmniej jeden węgiel półacetalowy (czyli węgiel karbonylowy, uczestniczący w tworzeniu formy pierścieniowej cząsteczki).
Wykonanie: Do dwóch probówek odmierzyć po 1 cm3 cukrów D i E, a następnie dodać po 2,5 cm3 odczynnika Benedicta (skład odczynnika: 173 g cytrynianu sodowego, 100g bezwodnego węglanu sodowego, 800 cm3 wody + roztwór 17,3 g CuSO4 w 100 cm3 wody - po czym uzupełnić wodą do 1 dm3), i ogrzewać na łaźni wodnej przez kilka minut. W obecności cukru redukującego wytwarza się ceglastoczerwony osad. Przy mniejszej zawartości cukru może powstać jedynie zielone zabarwienie albo osad o barwie od żółtej do pomarańczowej.
Na podstawie wyników reakcji 2c zidentyfikować cukry używane w doświadczeniu; rozróżnić cukry D i E spośród wymienionych: sacharoza, maltoza
Sprawozdanie:
Podać jakie związki były oznaczone literami A,B,C,D,E i na jakiej podstawie poszczególne cukry zostały rozróżnione.
Ćwiczenie nr 2 (20,21. 10.2011)
CUKRY CZ. 2
1. Hydroliza polisacharydów (ćwiczenie znajduje się również w skrypcie)
a. Otrzymywanie skrobi z ziemniaka
Zasada: W metodzie tej wykorzystuje się znaczny ciężar właściwy ziaren skrobiowych, które uwolnione z komórek łatwo sedymentują.
Wykonanie: Kilka ziemniaków umyć, obrać, zetrzeć na miazgę i rozcieńczyć równą ilością wody. Strzępy tkankowe odsączyć przez kilka warstw gazy, a roztwór pozostawić do dekantacji albo odwirować przy ok. 1000 obr/min przez 5 min. Zlać ciecz znad osadu, a skrobię wypłukać etanolem i wysuszyć na bibule.
b. Hydroliza skrobi i próba jodowa
Zasada: Cząsteczki glukozy tworzą w cząsteczce skrobi rozgałęzione helisy. Do wnętrza tych helis może wnikać jod, rozmieszczając się regularnie po 1 atomie na 1 skręt helisy. Powoduje to powstanie zabarwionego kompleksu jodowo-skrobiowego o barwie od fioletowo-czerwonej (amylopektyna) do niebieskiej (amyloza). Po ogrzaniu zabarwienie znika z powodu rozkręcania się helis i rozpadu kompleksu. Procesowi postępującej hydrolizy skrobi towarzyszy stopniowa zmiana barwy z niebieskiej w fioletową, czerwoną i ostatecznie jej zanik.
Wykonanie: Przygotować 10 probówek zawierających po 1 cm3 0,002% roztworu Lugola (jod w jodku potasu). Do ok. 10 cm3 kleiku skrobiowego otrzymanego ze skrobii wyizolowanej w części a) dodać 1 cm3 stężonego kwasu solnego i ogrzewać na łaźni wodnej. Co ok. 1 min pobierać kilka kropel kleiku i wlewać do kolejnej probówki. Obserwować postępującą hydrolizę skrobi i wywołaną tym zmianę zabarwienia w kolejnych probówkach.
2. Oznaczanie zawartości cukrów rozpuszczalnych w tkankach roślinnych metodą antronową [Ashwell 1975].
a) Wykonanie krzywej kalibracyjnej
W probówkach przygotować po 3 cm3 wzorcowych roztworów glukozy (0,2; 0,1; 0,05; 0,025; 0,0125 mg glukozy na 1 cm3). Odpowiednie rozcieńczenia należy uzyskać poprzez kolejne rozcieńczanie wzorcowego roztworu glukozy o stężeniu 1 mg/cm3.
Pierwsze rozcieńczenie: 2 cm3 wzorca + 8 cm3 wody, kolejne: 3 cm3 poprzedniego rozcieńczenia + 3 cm3 wody).
Następnie pobrać do kolejnych probówek po 1 cm3 każdego z rozcieńczeń (5 probówek). Do każdej z nich dodać 2 cm3 0,2% antronu (Sigma) w stężonym H2SO4 (POCh), wstrząsnąć probówkę na wstrząsarce laboratoryjnej i ogrzewać przez 3 minuty na łaźni wodnej o temperaturze +90°C. Po ostygnięciu próbek do temperatury pokojowej zmierzyć absorbancję przy λ = 620 nm względem ślepej próbki (zamiast roztworu cukru z roztworem antronu zmieszać 1 cm3 wody).
b) Oznaczenia w materiale roślinnym
Ekstrakcja cukrów rozpuszczalnych
Pobrać dwukrotnie po 0,5 g świeżej masy liścia i każdą z prób utrzeć oddzielnie w moździerzu w ciekłym azocie. Po odparowaniu azotu, podczas ucierania, dodawać stopniowo 5 cm3 wody. Homogenat przenieść do szklanej probówki, a moździerz przepłukać jeszcze 5 cm3 wody. Próbki gotować przez 15 minut na wrzącej łaźni wodnej, a następnie zawartość przenieść dokładnie do probówek wirówkowych. Strzępy tkankowe odwirować przez 10 minut przy 15 000 obr/min. Nadsącz zlać i uzupełnić wodą do 10 cm3.
Oznaczanie zawartości cukrów rozpuszczalnych
Następnie pobrać 0.5 cm3 roztworu i rozcieńczyć go przez dodanie 9.5 cm3 wody. Stopień rozcieńczenia należy dobrać tak, by odczytywane wartości absorbancji mieściły się w zakresie krzywej kalibracyjnej. Rozcieńczony ekstrakt wymieszać na wstrząsarce laboratoryjnej i pobrać z niego dwukrotnie po 1 cm3. Do każdej z probówek dodać po 2 cm3 0,2% antronu (Sigma) w stężonym H2SO4 (POCh), wstrząsnąć probówkę na wstrząsarce i ogrzewać przez 3 minuty na łaźni wodnej o temperaturze +90°C. Po ostygnięciu próbki do temperatury pokojowej zmierzyć absorbancję przy λ = 620 nm względem ślepej próbki (zamiast roztworu cukru z roztworem antronu zmieszać 1 cm3 wody).
3. Rozpuszczanie celulozy w odczynniku Schweitzera
Zasada: Celuloza różni się od skrobi przede wszystkim tym, iż cząsteczki glukozy połączone są wiązaniami -glikozydowymi zamiast . Konsekwencją tego faktu jest bardzo duża odporność celulozy na działanie czynników hydrolizujących, rozpuszczających oraz na rozkład enzymatyczny. Dopiero stężone kwasy oraz takie specyficzne rozpuszczalniki jak odczynnik Schweitzera rozpuszczają celulozę.
Wykonanie: Kawałek bibuły, strzępek waty lub skrawek płótna zalać pod wyciągiem 50 cm3 odczynnika Schweitzera (do 50 cm3 0,3 M roztworu CuSO4 dolać mieszając 50 cm3 0,6 M, NaOH, wytrącony osad rozpuścić w 75 cm3 28% roztworu amoniaku) po czym mieszać do rozpuszczenia. Po wlaniu tego roztworu do rozcieńczonego kwasu solnego celuloza ponownie się wytrąca. Jeśli roztwór celulozy wytłaczać przez bardzo wąskie dysze, wówczas może w ten sposób powstać sztuczne włókno celulozowe.
UWAGA! Stężony roztwór amoniaku drażni drogi oddechowe, stąd eksperyment należy wykonywać koniecznie pod sprawnym wyciągiem.
Sprawozdanie
a. Opisać zabarwienie w próbówkach na poszczególnych etapach hydrolizy
b) Za pomocą arkusza kalkulacyjnego MS Excel wyliczyć średnią absorbancję dla każdego stopnia rozcieńczenia (było ich pięć), a następnie wykreślić krzywą kalibracyjną i wyznaczyć równanie regresji liniowej zależności absorbancji od stężenia glukozy (wzorca) w próbce.
b. Obliczyć stężenie cukru z pomocą równania regresji liniowej otrzymanego w podpunkcie a). Następnie stężenie cukru w próbce przeliczyć na jego zawartość w świeżej masie liścia.
Ashwell G., 1975. Colorimetric analysis of sugars. Methods Enzymol., 3: 467-471
Ćwiczenie nr 3 (27,28.10.2011)
LIPIDY cz.1
1. Reakcje charakterystyczne tłuszczów prostych
a. Wykazanie obecności kwasów nienasyconych w tłuszczach
Do probówki zawierającej dwie krople oliwy dodać 5 cm3 Na2CO3 o stężeniu 1 mol·dm-3. Zawartość probówki lekko ogrzać i wstrząsając, dodawać po kropli KMnO4 o stężeniu 0,01 mol·dm-3 aż do wystąpienia trwającego ok. 2 minuty różowego zabarwienia.
Pod wpływem KMnO4 następuje utlenienie nienasyconych kwasów tłuszczowych. W miejscu podwójnego wiązania zostaje przyłączony tlen i kwas tłuszczowy rozpada się na dwa fragmenty, analiza których pozwala ustalić miejsce położenia nienasyconego wiązania w łańcuchu kwasu tłuszczowego.
b. Wykrywanie glicerolu
Do wykrycia glicerolu można wykorzystać następujące reakcje:
b.1. Umieścić w probówce kroplę glicerolu, dodać 3 cm3 5% etanolowego roztworu NaOH i 0.5 cm3 nasyconego roztworu CuSO4. W obecności glicerolu powstaje zielononiebieski osad.
b.2. Do dwóch kropli glicerolu w suchej probówce dodać około 100 mg KHSO4. Zawartość probówki ogrzewać ostrożnie (!) nad płomieniem palnika aż do wystąpienia charakterystycznego ostrego zapachu.
Jedną z reakcji służących do wykrywania glicerolu jest zdolność tworzenia w środowisku zasadowym z jonami miedziowymi kompleksu o zielononiebieskiej barwie (reakcja Wagenaara). Kompleks chelatowy tworzą prawdopodobnie dwie cząsteczki glicerolu i jeden atom miedzi (ćwicz. 1.2.a.).Inny sposób wykrywania glicerolu polega na odwodnieniu glicerolu. Pod wpływem ogrzewania z KHSO4 przekształca się on w akroleinę - nienasycony aldehyd o charakterystycznym drażniącym zapachu (ćwicz. 1.2.b.).
2. Ekstrakcja i frakcjonowanie lipidów z żółtka jaja kurzego
1 łyżeczkę żółtka jaja kurzego zalać 18 cm3 mieszaniny chloroform:metanol 1:2 i mieszać starannie przez kilka minut. Następnie mieszaninę wlać do plastikowych próbówek wirówkowych i odwirować (5 min., 15.000 obr/min). Odessać (pipetą) ciecz znad osadu, dodać do niej 5 cm3 wody, dokładnie wymieszać, a następnie odessać górną warstwę wodną. Chloroformowy ekstrakt lipidowy wlać do cylindra miarowego i uzupełnić chloroformem do 10 cm3 i pozostawić do dalszych doświadczeń (punkt a i d)
a. Wytrącanie pochodnych kwasu fosfatydowego
7,5 cm3 roztworu otrzymanego powyżej przelać powoli do cylindra z 15 cm3 acetonu. Wytrąca się osad glicerofosfolipidów, głównie lecytyn. Przelać zawartość do 2 plastikowych próbówek wirówkowych (po równo) i odwirować (jak wyżej), a następnie zawiesić w 3 cm3 wody. Wykryć obecność choliny według procedury opisanej w podpunkcie b i jonów ortofosforanowych według procedury opisanej w podpunkcie c.
b. Wykrywanie choliny
Odmierzyć do probówki 1 cm3 roztworu otrzymanego w podpunkcie a , dodać 1 cm3 10% roztworu KOH i ostrożnie (!) ogrzewać nad małym płomieniem palnika aż do pojawienia się charakterystycznego zapachu.
KOH
CHOLINA → TLENEK ETYLENU + TRÓJMETYLOAMINA
c. Wykrywanie jonów ortofosforanowych
Odmierzyć do probówki 1 cm3 roztworu otrzymanego w punkcie a i dodać 0,5 cm3 20% roztworu NaOH. Zawartość probówki dobrze wymieszać, wrzucić "porcelankę" i gotować nad palnikiem przez 2 minuty. Następnie dodać 2 cm3 odczynnika molibdenianiowego (2% (NH4)2MoO4). Roztwór barwi się na kolor żółty, co świadczy o obecności fosforanów.
UWAGA: poza żółknięciem roztworu można obserwować także wydzielanie się warstwy kwasów tłuszczowych na powierzchni cieczy
Obecność fosforanu można wykryć stosując kwaśny roztwór molibdenianu amonowego. Tworzy się w tych warunkach kompleks soli amonowej heteropolikwasu czterotrójmolibdenianofosforowego o żółtej barwie.
(NH4)2MoO4
PO43- → (NH4)3[P(Mo3O10)4]
d. Wykrywanie steroli (reakcja Libermana i Burcharda)
Odmierzyć do suchej (!) probówki 1 cm3 chloroformowego ekstraktu lipidów z żółtka jaja kurzego a następnie ostrożnie nad palnikiem odparować chloroform. Rozpuścić osad w 10 kroplach lodowatego kwasu octowego, wymieszać zawartość probówki i dodać dwie krople stężonego H2SO4. Pojawia się czerwone zabarwienie przechodzące w niebieskozielone.
W reakcji tej, której mechanizm nie jest znany, pod wpływem stężonego kwasu siarkowego i bezwodnika kwasu octowego tworzą się produkty o barwie różowej przechodzącej przez purpurową i niebieskozieloną w zieloną. Reakcję tą dają wszystkie sterole zawierające przynajmniej jedno podwójne wiązanie. Jest ona podstawą ilościowej metody oznaczania cholesterolu i ergosterolu.
3. Oznaczanie zawartości tłuszczu w mleku
3,5 cm3 mleka umieścić w plastikowej probówce wirówkowej a następnie bardzo powoli, wstrząsając, dodać 3 cm3 stężonego kwasu siarkowego. Unikać zagrzania probówki ! Dodać 0,3 cm3 alkoholu amylowego, zmieszać i umieścić probówkę w wirówce. Wirować przez 5 minut przy 10.000 obrotach/min. Po odwirowaniu zaobserwować wysokość słupa tłuszczu w górnej części probówki.
Ćwiczenie nr 4 (3,4.11.2011)
LIPIDY cz.2
1. Chromatografia cienkowarstwowa (TLC) fosfolipidów (jedno oznaczenie na grupę)
Skład fosfolipidowy próbki określić metodą chromatografii cienkowarstwowej na płytkach Kieselgel 60 (0.25 mm) firmy Merck. Przed nałożeniem próbek płytki aktywować w 110°C przez 30 minut (zostało już wykonane!!!!!!).
Komorę chromatograficzną napełnić 60 cm3 mieszaniny rozwijającej (chloroform - metanol - kwas octowy; 65:25:8; v/v/v) i pozostawić zamkniętą przez 30 minut dla zrównoważenia (osiągnięcia stanu równowagi pomiędzy parami a mieszaniną).
Za pomocą mikropipety automatycznej nanieść na płytkę po 5 μl chloroformowego ekstraktu lipidów oraz standardy lipidowe firmy Sigma. Poszczególne roztwory winny być nakładane w formie 5 mm paska (odstęp pomiędzy próbkami 1,5 cm; odstęp od dolnej krawędzi 1,5 cm; odstęp od krawędzi bocznych 1,7 cm), przed zmianą nakładanego roztworu należy bezwzględnie zmieniać końcówki pipety. Płytkę umieścić w komorze rozwijającej. Po rozwinięciu chromatogramu (40-50 minut; czoło mieszaniny rozwijającej ok. 2 cm od górnej krawędzi płytki) płytki wysuszyć na powietrzu, wysuszone płytki umieścić w komorze wypełnionej parami jodu i po uwidocznieniu się plamek obrysować je ołówkiem. Alternatywnie można płytki spryskać 0,6% K2Cr2O7 w 55% H2SO4 i ogrzewać w 180°C przez 30 minut dla uwidocznienia się wszystkich plamek pochodzących od fosfolipidów. (Wykonujemy według metody podkreślonej).
Komentarz: Metoda ta może być również wykorzystana do oznaczeń ilościowych, w tym celu wykonuje się analizę densytometryczną płytek w oparciu o komputerowy program analizy obrazu lub wycina obrysowane miejsca z płytki i ekstrahuje z nośnika do roztworu chloroformu.
2. Przybliżone oznaczenia niektórych parametrów charakteryzujących tłuszcze (Poniższe ćwiczenia znajdują się również w skrypcie)
a) Wyznaczanie liczby kwasowej
Zasada: Liczba kwasowa charakteryzuje ilość wolnych (niezobojętnionych) reszt kwasów organicznych zawartych w tłuszczu. Określa się ją liczbą mg KOH potrzebnych do zobojętnienia wolnych reszt kwasowych zawartych w 1 g tłuszczu. Podwyższona wartość liczby kwasowej może świadczyć o procesach hydrolizy (jełczenia) tłuszczu, np. przy zbyt długim przechowywaniu.
Wykonanie: Do kolbek odważyć po 5 g świeżego i starego tłuszczu. Każdą z próbek rozpuścić dokładnie w 50 cm3 rozpuszczalnika benzynowego (ewentualnie OSTROŻNIE!!! podgrzewając). Po ostudzeniu zmiareczkować 0,1 M etanolowym roztworem KOH w obecności fenoloftaleiny do uzyskania trwałej, różowej barwy. Powtórzyć miareczkowanie w czystym rozpuszczalniku.
b) Wyznaczanie liczby zmydlania
Zasada: Liczba zmydlania charakteryzuje całkowitą ilość zestryfikowanych oraz wolnych (niezobojętnionych) reszt kwasów organicznych zawartych w tłuszczu. Określa się ją liczbą mg KOH potrzebnych do zobojętnienia wszystkich reszt kwasowych zawartych w 1 g tłuszczu. Liczba ta pozwala wyznaczyć średnią masę cząsteczkową kwasów tłuszczowych zawartych w tłuszczu (długość łańcucha węglowodorowego).
Wykonanie: Do kolbek odważyć po 1 g tłuszczu stałego i tłuszczu płynnego, a następnie dodać 10 cm3 0,5 M roztworu KOH i 50 cm3 etanolu, po czym ogrzewać przez 20 min na wrzącej łaźni wodnej i pod chłodnicą zwrotną, albo nakrywając wylot kolby (lub lepiej zlewki) okrągłodenną kolbą, napełnioną w połowie zimną wodą w celu skraplania oparów etanolu. Następuje wówczas hydroliza tłuszczu. Po ostudzeniu zmiareczkować nadmiar niezobojętnionego KOH 0,5 M roztworem HCl wobec fenoloftaleiny do zaniknięcia różowej barwy. Równolegle wykonać próbę kontrolną bez tłuszczu (powtórzyć pomiar w czystym rozpuszczalniku).
3) Zmydlanie tłuszczów, wytwarzanie mydła, wytrącanie mydła
Zasada: tłuszcze jako estry kwasów tłuszczowych i glicerolu są podatne na hydrolizę. Jeśli hydrolizę prowadzi się za pomocą wodorotlenku sodowego wówczas wydziela się wolny glicerol i sodowa sól kwasu tłuszczowego, czyli mydło. Mydła sodowe i potasowe są rozpuszczalne, natomiast mydła magnezowe, wapniowe i innych metali są nierozpuszczalne w wodzie.
Z roztworu wodnego można wydzielić mydło przez wysalanie w nasyconym roztworze NaCl albo przez dodanie roztworu dowolnej soli, która wytwarza nierozpuszczalne mydło.
Wykonanie:
a) Do zlewki odważyć ok. 5 g tłuszczu, a następnie dodać 8 cm3 30% NaOH i 5 cm3 etanolu. Podgrzewać łagodnie przez kilka minut, stale mieszając, aż utworzy się jednolita masa. Wówczas dodać ok. 200 cm3 gorącej destylowanej wody i dalej podgrzewać aż do rozpuszczenia mydła.
b) Do 20 cm3 otrzymanego powyżej roztworu mydła dodać stopniowo stały chlorek sodowy aż do uzyskania stanu wysycenia. Z roztworu wytrąca się wówczas osad mydła sodowego nierozpuszczalnego w tym roztworze, które można wydzielić przez dekantacje, wirowanie albo sączenie.
c) Do 4 probówek wlać po 2 cm3 roztworu mydła otrzymanego w podpunkcie a), dodać po 1 cm3 rozcieńczonych roztworów Ca(NO3)2 lub CaCl2, BaCl2, CuSO4 i Pb(CH3COO)2. W każdym przypadku wytrącają się osady mydeł nierozpuszczalnych pomimo dodawania większej objętości wody.
4. Rozdział barwników fotosyntetycznych metodą chromatografii bibułowej
Wykonanie: W moździerzu z piaskiem kwarcowym i CaCO3 utrzeć w acetonie 2g świeżych liści pszenicy, a następnie przenieść zawartość moździerza na sączek papierowy. UWAGA: homogenizacja ma być wykonywana pod włączonym digestorium. Otrzymany przesącz nanosić kroplami roztwór barwników z liści. Miejsce naniesienia powinno znajdować się około 2 cm od końca bibuły i na środku szerokości paska. Po naniesieniu każdej kropli należy poczekać, aż wyschnie. Czynność tą powtórzyć należy około 8 razy w taki sposób, by wymiary plamy nie uległy zwiększeniu.
Do probówki nalać 2-3 cm3 mieszaniny benzen:eter naftowy:aceton w stosunku objętościowym 10:2,5:2. Pasek bibuły umieścić w probówce tak, by naniesiona plama znajdowała się ponad mieszaniną. Po około godzinie wyjąć pasek bibuły, wysuszyć i zaobserwować rozdział barwników.
Komentarz: Metody chromatograficzne służą do rozdziału związków chemicznych przy wykorzystaniu faktu, iż ich cząsteczki różnią się wielkością lub właściwościami fizycznymi takimi jak: polarność czy ładunek elektryczny. Każdy rozdział chromatograficzny to rozdział substancji między dwie fazy: stałą (podłoże) oraz ruchomą (nośnik - gaz lub ciecz) której przepływ przez podłoże jest wymuszany bądź przez siłę grawitacji czy adhezji (chromatografia bibułowa), bądź przez specjalne pompy. Znajdujące się w - umieszczonej w takim układzie - próbce, substancje będą w zależności od swoich właściwości silniej lub słabiej adsorbowane na fazie stałej. Na skutek tego po pewnym czasie obserwować będziemy przestrzenny rozdział składników próbki. W przypadku stosowanych przez nas metod fazę stałą stanowią odpowiednio: skrobia i bibuła, a ruchomą - mieszaniny mniej od nich polarnych cieczy. Karoten, jako najmniej polarny z rozdzielanych barwników, nie jest adsorbowany na skrobi i najszybciej wypływa z kolumny. W dalszej kolejności układają się pasma ksantofili (w przypadku pszenicy dwa), chlorofilu "a" (niebieskozielone) i najbardziej polarnego (zawierającego w porównaniu z chlorofilem "a" dodatkową grupę aldehydową) chlorofilu "b" (żółtozielone). Przed ksantofilami może pojawić się brunatne pasmo feofityn. W przypadku chromatografii bibułowej odległość wędrówki poszczególnych barwników z nośnikiem jest odwrotnie proporcjonalna do ich polarności.
Sprawozdanie:
a) Wyznaczyć liczbę kwasową. Dla każdego z tłuszczów obliczyć ilość KOH zużytą na zobojętnienie wolnych kwasów tłuszczowych, następnie wyznaczyć liczbę mg KOH potrzebną do zobojętnienia 1 g każdego z tłuszczów.
b) Wyznaczyć liczbę zmydlania. Dla każdego z tłuszczów obliczyć ilość KOH zużytą na zobojętnienie wszystkich cząsteczek kwasów tłuszczowych, następnie wyznaczyć liczbę mg KOH potrzebną do zobojętnienia 1 g tłuszczu.
W obliczeniach przyjąć masę cząsteczkową KOH równą 56,11. Zinterpretować otrzymany wynik !!!
Ćwiczenie nr 5 (24,25.11.2011)
AMINOKWASY cz.1
1. Analiza jakościowa aminokwasów (ćwiczenie znajduje się również w skrypcie)
Na podstawie reakcji a, b, c odróżnić następujące związki: prolinę, metioninę, cysteinę, tyrozynę, kwas bursztynowy
a. Odróżnianie aminokwasów od innych związków
Odmierzyć do 5 probówek po 0,5 cm3 roztworów oznaczonych jako A, B, C, D, E. Do wszystkich probówek dodać po 0,5 cm3 0.1% roztworu ninhydryny w acetonie i ogrzewać przez trzy minuty na łaźni wodnej. Pojawia się intensywne zabarwienie fioletowe w obecności wolnego aminokwasu lub żółte w obecności wolnego iminokwasu. Reakcja ta może być używana do ilościowego oznaczania wolnych aminokwasów, a reakcja z tzw. kwaśną ninhydryną (roztwór ninhydryny w mieszaninie kwasu octowego i fosforowego) do ilościowego oznaczania wolnej proliny.
b. Wykrywanie wolnych grup tiolowych
Odmierzyć do probówek po 0,5 cm3 roztworów niezidentyfikowanych w punkcie a. Do każdej próby dodać po 1 cm3 20% roztworu NaOH oraz po 0,25 cm3 2% roztworu octanu ołowiawego. Ogrzewać przez 3 - 5 minut we wrzącej łaźni wodnej. W obecności grup tiolowych tworzy się brunatnoczarny osad.
c. Wykrywanie obecności pierścieni aromatycznych
Odmierzyć do probówek po 0,5 cm3 roztworów niewyeliminowanych w podpunktach a i b. Do każdej próby dodać po 0,5 cm3 stężonego kwasu azotowego i ogrzewać przez 5 minut we wrzącej łaźni wodnej. Po ostudzeniu dodać ostrożnie (!), małymi porcjami, i ciągle mieszając po 1,75 cm3 20% roztworu NaOH. W obecności pierścieni aromatycznych pojawia się pomarańczowe zabarwienie.
2. Miareczkowanie aminokwasu
Do zlewki o pojemności 50 cm3 nalać 20 cm3 roztworu glicyny o stężeniu 1 mol⋅dm-3. Zlewkę ustawić na mieszadle magnetycznym, wrzucić do środka „element mieszający”, uruchomić mieszadło i umieścić w zlewce elektrodę pH-metru w sposób uniemożliwiający jej rozbicie przez pracujące mieszadło. Zapisać wartość pH. Dodawać stopniowo (po 0,5 cm3) 10 cm3 1 molowego roztworu HCl. Każdorazowo, po ustabilizowaniu się odczytu pH zapisywać jego wartość. Powtórzyć procedurę używając do miareczkowania roztworu NaOH o stężeniu 1 mol⋅dm-3.
Sprawozdanie:
a) Podać jakie związki były oznaczone literami A,B,C,D,E i na jakiej podstawie poszczególne aminokwasy zostały rozróżnione.
b) Na podstawie obydwu wyników narysować krzywą miareczkowania: na osi x wartości pH, na osi y ilość dodanego HCl (wartości dodatnie) i NaOH (wartości ujemne). Odczytać z wykresu wartość punktu izoelektrycznego (odczyt pH przed miareczkowaniem), oraz pKa i pKb (punkty przegięcia krzywej). Narysować na wykresie w jakich formach jonowych występuje glicyna na poszczególnych odcinkach krzywej, oraz wyjaśnić jej charakterystyczny przebieg.
Ćwiczenie nr 6 (1,2. 12. 2011)
AMINOKWASY, BIAŁKA
1. Chromatografia bibułowa aminokwasów
Do dolnej części szalki Petriego wlać rozpuszczalnik (mieszanina trzeciorzędowego butanolu, 88% kwasu mrówkowego i wody w stosunku objętościowym: 15:15:70). W odległości 2 mm od środka krążka zakreślić ołówkiem na naciętym pasku kółeczko o srednicy 1,5 mm (rysunek b). Na narysowaną kroplę nakładać powoli 1% roztwory: glicyny, fenyloalaniny, seryny i badanego aminokwasu. Każda z grup przygotowuje dwie chromatografie: jeden wzorzec i jeden z roztworów nieznanych aminokwasów: X, Y, Z. Nanoszenie przeprowadzamy stopniowo (tak by mokra plama nie wyszła poza zaznaczone pole) z pomocą pipety o pojemności 0,2 do 10 μl. Po dodaniu każdej kolejnej kropli bibułę należy przesuszyć. Łącznie należy nanieść 20 μl aminokwasu.
a) widok ogólny b) nakładanie aminokwasu c) sposób odczytu Rf
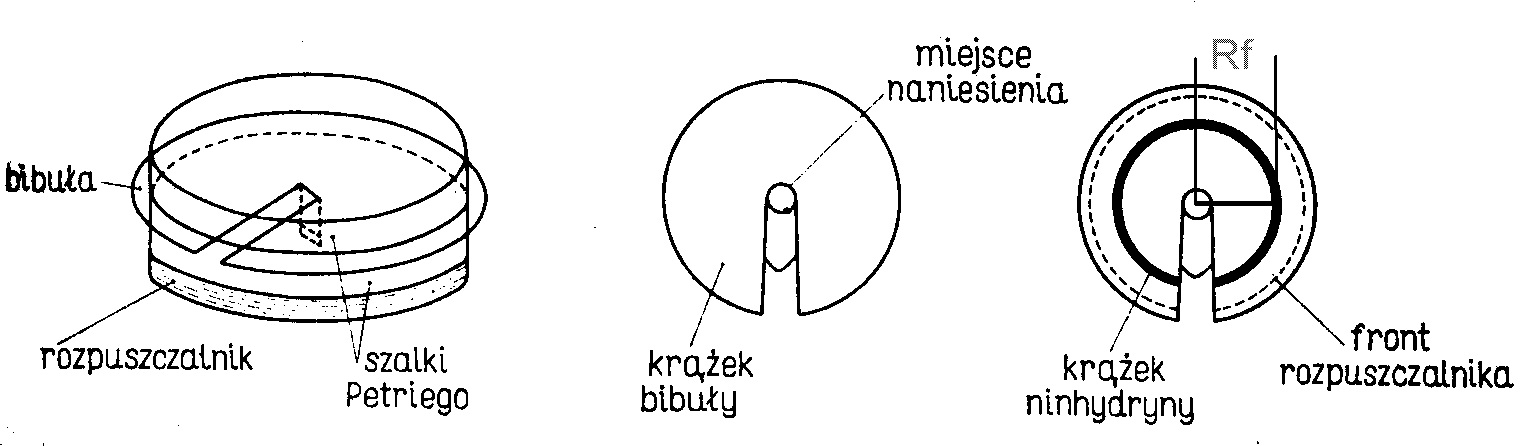
Krążek bibuły umieścić między dwoma częściami szalki, tak, aby koniec zgiętego paska zanurzył się w rozpuszczalniku, a brzegi wystawały poza szalkę (rysunek a).
Po około godzinie, gdy czoło rozpuszczalnika zbliży się do brzegu bibuły, wyjąć chromatogram i zaznaczyć ołówkiem front rozpuszczalnika. Całość wysuszyć w 110°C i spryskać 0,1% roztworem ninhydryny w 95% etanolu. Wysuszyć w 110°C i obrysować ołówkiem barwny krążek. Obliczyć współczynnik migracji Rf dla każdego wzorcowego i badanego aminokwasu według wzoru:
(rysunek c). Zidentyfikować nieznany aminokwas Porównując wartości Rf nieznanego aminokwasu z Rf wzorców.
BIAŁKA
2. Oznaczanie zawartości białek rozpuszczalnych w materiale roślinnym
Spośród kilku powszechnie stosowanych metod oznaczeń ilościowych białka w materiale roślinnym poznają Państwo chyba najbardziej popularną metodę Bradford (1976). Wszystkie metody są w gruncie rzeczy podobne. Do roztworu białka dodaje się jakiegoś odczynnika łączącego się ilościowo z białkiem. Następnie porównuje się absorbancję roztworu badanego z krzywą wzorcową wykreśloną w oparciu o roztwory najczęściej BSA (albumina z osocza krwi bydlęcej). Warto zapamiętać jeszcze istnienie metody Lowrye'go (bardzo czuła, nawet zbyt czuła, co czasami może być kłopotliwe i prowadzić do błędów zważywszy na konieczność stosowania dużych rozcieńczeń) i bardzo podobna do metody Bradford metodą Sedmaka i Grossberga (1977). Różni się ona drobnym szczegółem przy sporządzaniu odczynnika, który jest bardziej specyficzny, tzn. trudniej reaguje ze związkami niebiałkowymi, głównie wielowartościowymi kationami, ale również mniej trwały, co również ogranicza jej stosowanie: ponieważ krzywą kalibracyjną trzeba dla każdego odczynnika sporządzać na nowo, przy wielkoseryjnych oznaczeniach rośnie błąd.
Wykonanie:
Pobrać 1 g świeżej masy liścia rzepaku i utrzeć w moździerzu w ciekłym azocie. Po odparowaniu azotu, podczas ucierania, dodawać stopniowo 3 cm3 buforu ekstrahującego Tris-HCl (0,0625 mol⋅dm-3, pH 6,8). Homogenat przenieść do probówki wirówkowej, a moździerz przepłukać kolejnymi 3 cm3 buforu. Następnie próbki odwirować przez 10 minut przy maksymalnych obrotach wirówki.
Równolegle przygotować krzywą kalibracyjną w oparciu o roztwór BSA (albuminy wołowej) o stężeniu 10 mg⋅cm-3 w buforze ekstrahującym. Przygotować po 4 cm3 roztworów zawierających po 2; 1; 0,5; 0,25; 0,125 mg BSA w 1 cm3.
Do każdej z kuwet spektrofotometrycznych dodać po 0,5 cm3 wody destylowanej, 0,5 cm3 odczynnika Bradford oraz 0,18 cm3 buforu ekstrakcyjnego (Tris-HCl) a nastepnie po 0,02 cm3 z każdego z roztworów wzorcowych oraz z supernatantu. Po zmieszaniu na wstrząsarce laboratoryjnej kuwety odstawić na 5 minut, po czym zmierzyć wartość absorbancji przy λ = 595 nm względem ślepej próby sporządzonej z 200 μl buforu i 1 cm3 odczynnika.
a) Obliczyć Rf dla poszczególnych roztworów wzorców i nieznanych aminokwasów oraz je zidentyfikować.
b) Za pomocą arkusza kalkulacyjnego MS Excel wyliczyć średnią absorbancję dla każdego stopnia rozcieńczenia, wykreślić krzywą kalibracyjną i wyznaczyć równanie regresji liniowej zależności absorbancji od stężenia BSA (wzorca) w próbce oraz obliczyć stężenie białka. Następnie stężenie białka w próbce wyrażone w mg/cm3 przeliczyć na jego zawartość w świeżej masie liścia (mg/g św. masy).
3. Elektroforeza SDS-PAGE
Elektroforeza to przemieszczanie się obdarzonych ładunkiem cząstek (jonów) w polu elektrycznym, np. cząsteczek białek lub kwasów nukleinowych. Większość metod elektroforetycznych wykorzystuje specyficzne nośniki, tj. żele poliakrylamidowe lub agarozowe, bibułę. Przemieszczanie się jonów w polu elektrycznym zależy od ich ładunku, wielkości i kształtu. Im większy jon lub bardziej rozbudowany jego kształt tym wolniej porusza się w polu elektrycznym, a im bardziej naładowany - tym porusza się szybciej. Jony naładowane ujemnie migrują do anody, a naładowane dodatnio - do katody. W przypadku cząstek zawierających w swym składzie ugrupowania naładowane dodatnio i ujemnie (np. białka), o kierunku migracji decyduje ich ładunek wypadkowy.
Białka najczęściej rozdzielamy metodą elektroforezy w żelach poliakryloamidowych w obecności SDS (SDSPAGE, polyacrylamide gel electrophoresis), przy której ich rozdział jest zależny tylko od ich wielkości. SDS (dodecylosiarczan sodu) jest detergentem anionowym denaturującym i opłaszczającym białko, w wyniku czego łańcuch polipeptydowy:
(a) traci struktury wyższego rzędu
(b) staje się anionem.
Dodatkowo, przeprowadza się redukcję wiązań dwusiarczkowych białka i w wyniku tego jego poszczególne łańcuchy polipeptydowe funkcjonują w roztworze niezależnie od siebie. W efekcie, wszystkie białka (łańcuchy polipeptydowe) mają taki sam, liniowy kształt oraz ładunek ujemny, a szybkość ich migracji w żelu zależy wyłącznie od ich wielkości. Białka na ogół są bezbarwne. W związku z tym po przeprowadzonym rozdziale żel trzeba zabarwić, aby uwidocznić obecne w nim białka. W tym celu najczęściej stosuje się barwienie błękitem kumasyny lub związkami srebra.
SDS-PAGE wykorzystuje się także do wyznaczania masy cząsteczkowej analizowanego białka (przez porównanie z wielkością białek wzorcowych) oraz do określania liczby jego podjednostek polipeptydowych.
Ćwiczenie nr 7 (8,9.12.2009)
BIAŁKA
1. Miareczkowanie białka (ćwiczenie znajduje się również w skrypcie)
Do 2 zlewek o pojemności 100 cm3 wlać po 40 cm3 wody, a do dwóch następnych po 40 cm3 1% kazeiny. Zlewki ustawić na mieszadle magnetycznym, wrzucić do środka „myszkę”, uruchomić mieszadło i umieścić w zlewce elektrodę pH-metru w sposób uniemożliwiający jej rozbicie przez pracujące mieszadło. Zapisać wartość pH. Następnie jedną parę (woda, białko) miareczkować 0,5 M HCl (do osiągnięcia pH <4), a drugą parę 0,5 M NaOH (do pH > 10). Kwas solny i wodorotlenek sodu dodawać porcjami (po 1 cm3 za pomocą pipety automatycznej). Każdorazowo, po ustabilizowaniu się odczytu pH zapisywać jego wartość.
Krzywa zmian pH podczas miareczkowania mocnego kwasu mocną zasadą ma charakterystyczny, esowaty kształt z bardzo stromym przebiegiem przy pH około 7. Miareczkowanie aminokwasu daje przebieg z dwoma skokami pH przy zobojętnianiu grup aminowych i karboksylowych. Natomiast w przypadku miareczkowania białka składającego się z różnych aminokwasów krzywa miareczkowania ma przebieg łagodny, pozwalający wyznaczyć ich pojemność buforową (ilość kwasu lub zasady powodującą określoną zmianę pH). Własności buforujące białek w komórkach łagodzą zmiany pH spowodowane np. wymianą jonową, tworzeniem i rozkładem różnych kwasów, spożyciem kwaśnych pokarmów, zmianą równowagi pomiędzy jonami węglanowymi i wodorowęglanowymi itp.
2. Wysalanie białek
Do probówki odmierzyć 2 cm3 roztworu białka jaja i dodać 2 cm3 nasyconego roztworu siarczanu amonowego. Obserwować wytrącanie się kłaczkowatego osadu globulin. Następnie dodać do probówki ok. 10 cm3 wody i wymieszać. Osad ulega rozpuszczeniu.
Do 50 - mililitrowej kolby stożkowej odmierzyć 10 cm3 roztworu białka jaja, dodać 10 cm3 nasyconego siarczanu amonu i wymieszać w celu wytrącenia globulin. Osad odwirować, a przesącz zebrać do suchej kolby stożkowej. Odmierzyć 2 cm3 przesączu do probówki i dodawać małymi porcjami tyle stałego siarczanu amonu aby uzyskać roztwór nasycony. Obserwować wytrącanie się osadu albumin. Po dodaniu ok. 10 cm3 wody destylowanej osad ulega rozpuszczeniu.
Pozostałą ilość przesączu, uzyskaną po odwirowaniu globulin, zadawać powoli stałym siarczanem amonu aż do wysycenia roztworu. Wysycony osad albumin odwirować, a przesącz zebrać do probówki. Do przesączu dodać kroplę kwasu octowego i ogrzać do wrzenia. Brak osadu świadczy o usunięciu białek z roztworu.
Zdolność wytrącania się albumin i globulin przy różnym nasyceniu ich roztworów siarczanem amonu pozwala zarówno odróżnić te białka, jak i rozdzielić ich mieszaninę stosując frakcjonowane wysalanie.
3. Wyznaczanie wartości punktu izoelektrycznego białka
Do 8 probówek odmierzyć wodę destylowaną i kwas octowy o stężeniu w ilościach podanych w tabeli 2, zmierzyć pH w probówkach i zanotować w tabeli. Następnie dodać po 1 cm3 roztworu kazeiny w octanie sodowym. Zawartość probówek wymieszać i po około 5 minutach ocenić stopień zmętnienia płynów w poszczególnych probówkach.
Substancje [cm3] |
Nr probówki |
||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|||||
woda destylowana |
8,9 |
8,8 |
8,5 |
8,0 |
7,0 |
5,0 |
1,0 |
0,0 |
|||||
kwas octowy 0,05 mol dm-3 |
0,1 |
0,2 |
0,5 |
1,0 |
2,0 |
4,0 |
8,0 |
9,0 |
|||||
roztwór kazeiny w octanie sodowym |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
|||||
pH roztworu |
|
|
|
|
|
|
|
|
|||||
stopień zmętnienia |
|
|
|
|
|
|
|
|
|||||
Wiele właściwości białek jest związanych z ich strukturą pierwszorzędową, wynikającą z sekwencji (kolejności) aminokwasów, między którymi powstają wiązania peptydowe. Większość aminokwasów posiada jedną grupę aminową (o charakterze zasadowym) i jedną grupę karboksylową (o charakterze kwasowym). Grupy te łącząc się ze sobą tworzą wiązania peptydowe i w związku z tym tracą dotychczasowe właściwości. W powstających łańcuchach polipeptydowych zbudowanych z reszt aminokwasowych takiego typu występowałaby tylko jedna wolna grupa aminowa na N-końcu łańcucha i jedna grupa karboksylowa na C-końcu. Ponieważ w skład cząsteczek białek wchodzą również reszty aminokwasów jednoamino-dwukarboksylowych (kwas asparaginowy, kwas glutaminowy) oraz dwuamino-jednokarboksylowych (lizyna, arginina, histydyna), w białku pozostaje pewna liczba wolnych grup aminowych i karboksylowych. Gdy więcej jest wolnych grup karboksylowych niż aminowych, wówczas cząsteczki dysocjując w środowisku o odczynie obojętnym wykazują właściwości kwasów. W obrębie cząsteczek występuje w takich warunkach więcej ładunków ujemnych niż dodatnich. Natomiast w cząsteczkach białek zasadowych, znajdujących się w środowisku o odczynie obojętnym, występuje więcej ładunków dodatnich niż ujemnych. Przewaga ładunków jednoimiennych (dodatnich lub ujemnych) sprzyja wiązaniu wody przez cząsteczki białka na zasadzie oddziaływania elektrostatycznego na dipole wody. Prowadzi to do tworzenia się otoczek wodnych wokół cząsteczek białka. Roztwór koloidowy białka jest wówczas trwały, gdyż poszczególne drobiny nie łączą się w agregaty, które mogłyby tworzyć zawiesinę wypadającą z roztworu (koagulacja). Zwiększanie stężenia jonów wodorowych w środowisku powoduje cofanie dysocjacji grup karboksylowych i ułatwianie dysocjacji grup aminowych, a obniżenie - wywołuje efekt odwrotny. Dla każdego białka można dobrać taki odczyn środowiska, przy którym jest ono w jednakowym stopniu zdysocjowane jako kwas i jako zasada. Wartość takiego odczynu (w jednostkach pH) jest określana mianem punktu izoelektrycznego. Dla białek kwaśnych wartości punktu izoelektrycznego są niższe od 7, a dla białek zasadowych wyższe. W punkcie izoelektrycznym liczby ładunków ujemnych i dodatnich w obrębie cząsteczek białka są jednakowe, wskutek czego wypadkowy ładunek jest równy zero. Z tego powodu zanika oddziaływanie elektrostatyczne cząsteczek białka na cząsteczki wody. Cząsteczki białka w takich warunkach z łatwością tracą otoczki wodne, co sprzyja koagulacji. Białka w punkcie izoelektrycznym wykazują zatem zmniejszone powinowactwo do wody i dlatego odczyn środowiska może również wpływać na pęcznienie koloidów białkowych. W układach biologicznych skutki tego oddziaływania zazwyczaj nie są wyraźnie widoczne w związku z dużą różnorodnością białek posiadających różne wartości punktu izoelektrycznego.
Sprawozdanie
Na podstawie wyników z ćw. 1 narysować krzywą miareczkowania: na osi x wartości pH, na osi y ilość dodanego HCl (wartości dodatnie) i NaOH (wartości ujemne). Odczytać z wykresu wartość punktu izoelektrycznego (odczyt pH przed miareczkowaniem), oraz pKa i pKb (punkty przegięcia krzywej) oraz wyjaśnić jej charakterystyczny przebieg.
Ćwiczenie nr 8 (5,6.01.2012)
ENZYMY CZ.1
1. Oznaczanie aktywności kwaśnej fosfatazy (fosfomonoesterazy) (Poniższe ćwiczenia znajdują się również w skrypcie)
a) Otrzymywanie ekstraktów z ziemniaka
Ekstrakt w buforze: Zetrzeć na tarce 10 g ziemniaka, dodać ok. 15 cm3 0,1 M buforu octanowego o pH = 5,5, wycisnąć sok przez warstwę płótna, po czym odwirować przez 15 min przy ok. 10.000 obr/min.
Ekstrakt wodny: przygotować jak wyżej ale zamiast buforu octanowego użyć wody).
b) Dobranie czasu inkubacji:
Do trzech probówek wlać po 0,1 cm3 ekstraktu enzymatycznego w buforze, dodać po 0,9 cm3 0,1 M buforu octanowego o pH = 5,5 i po 1 cm3 0,4% roztworu PNP w tym samym roztworze, zamieszać, odstawić na czas inkubacji reakcji enzymatycznej (odpowiednio: 3, 6 lub 9 min w temperaturze pokojowej), po czym zastopować reakcję 1 cm3 0,3 M NaOH i zmierzyć absorbancję przy = 400 nm. Wynik wyrazić jako liczbę optycznych jednostek aktywności enzymu. Za optymalny uznać należy czas inkubacji, po którym wartość absorbancji osiągną wartość pomiędzy 1 a 1,5. Jeśli żaden z czasów inkubacji okaże się nieodpowiedni, proszę dokonać interpolacji liniowej.
c) Wyznaczenie zależności aktywności kwaśnej fosfatazy od stężenia substratu
Zasada: Wynik aktywności enzymu, tj. masa przetworzonego substratu zależy między innymi od stężenia substratu, ilości enzymu oraz czasu reakcji. Przedłużenie czasu reakcji prowadzi do zużycia substratu, w wyniku czego szybkość reakcji maleje asymptotycznie do zera. Zwiększenie stężenia substratu przy zachowaniu stałości pozostałych parametrów prowadzi początkowo do wzrostu szybkości reakcji aż do uzyskania maksymalnej prędkości, przy której wszystkie cząsteczki enzymu są maksymalnie aktywne i dalsze zwiększanie stężenia substratu nie przyspiesza reakcji. Osiągnięta została tzw. maksymalna aktywność enzymu.
Zastosować ekstrakt z ziemniaka w buforze octanowym. Do probówki pobrać 4 cm3 roztworu substratu (PNP) o stężeniu 4% w buforze octanowym. Następnie z tej probówki (1) pobrać do kolejnej (2) 2 cm3; do probówki (2) dodać 2 cm3 buforu octanowego (pH 5,5), zamieszać i pobrać 2 cm3 do probówki (3). Fragment wyróżniony pogrubioną czcionką powtórzyć jeszcze 8-krotnie, uzyskując w efekcie końcowym 10 probówek zawierających po 2 cm3 PNP o stężeniach: 4, 2, 1, 0,5, 0,25, 0,125, 0,064, 0,032, 0,016 i 0,008% (w ostatniej probówce będą 4 cm3 roztworu). Następnie do 10 probówek pobrać po 1 cm3 poszczególnych roztworów PNP oraz po 0,9 cm3 buforu octanowego o PH 5,5. Reakcję enzymatyczną rozpocząć przez dodanie po 0,1 cm3 ekstraktu z ziemniaka w buforze octanowym. Następnie przeprowadzić inkubację w czasie dobranym w podpunkcie b) i w tej samej temperaturze.
d) Określanie zależności aktywności kwaśnej fosfatazy od pH:
Zasada: Bufor (np. octanowy) składa się ze składnika kwaśnego (kwas octowy) i alkalicznego (octan sodu). Zmiana proporcji tych składników pozwala uzyskać różne wartości pH buforu, w którym jest mierzona aktywność enzymu.
Wykonanie: W 11 probówkach przygotować mieszaniny 0,4 M kwasu octowego i 0,4 M octanu sodu w proporcjach podanych w tabeli.
|
Nr probówki |
||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
octan sodu [cm3] |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
kwas octowy [cm3] |
10 |
9 |
8 |
7 |
6 |
5 |
4 |
3 |
2 |
1 |
0 |
pH buforu |
|
|
|
|
|
|
|
|
|
|
|
Zmierzyć wartości pH każdego z buforów.
Do nowych 11 probówek pobrać po 0,9 cm3 poszczególnych buforów oraz po 1 cm3 0,4% wodnego roztworu PNP. Reakcję enzymatyczną rozpocząć przez dodanie po 0,1 cm3 wodnego ekstraktu z ziemniaka. Następnie przeprowadzić inkubację w czasie i temperaturze jak w poprzednim doświadczeniu. Reakcję zastopować przez dodanie po 1 cm3 0,3 M NaOH.
Sprawozdanie:
a) Na podstawie otrzymanych wyników wykreślić funkcję zależności aktywności enzymu od stężenia substratu. Wykres zinterpretować.
b) Na podstawie otrzymanych wyników wykreślić wykres zależności aktywności enzymu od pH. Wykres zinterpretować.
Ćwiczenie nr 9 (12,13.01.2012)
ENZYMY CZ.2
1. Wykazanie aktywności oksydaz w ziemniaku (ćwiczenie znajduje się również w skrypcie)
Zasada: W tkankach występują różne enzymy powodujące utlenianie substratów, a wśród nich oksydazy (utleniające substraty przy udziale tlenu cząsteczkowego) i peroksydazy (wykorzystujące nadtlenek wodoru jako źródło tlenu dla utleniania substratów). Produkty utlenienia mają zwykle ciemne zabarwienie, stąd ciemnienie przekrojonego i poddanego działaniu powietrza jabłka lub ziemniaka. Zwilżenie powierzchni tkanki substratem dla działania enzymów utleniających (p-fenylenodwuamina), jak i rozcieńczonym, ok. 0,1% roztworem nadtlenku wodoru przyspiesza proces ciemnienia tkanki. Oksydazy i peroksydazy są enzymami, w których istotną rolę odgrywają jony aktywujące. Stąd też zwilżenie powierzchni ok. 1% roztworem EDTA (kwas etylenodwuaminoczterooctowy) wiążącego w trwały kompleks różne dwuwartościowe kationy, opóźnia zjawisko ciemnienia powierzchni przekrojonej tkanki.
Wykonanie: Połówkę jabłka lub ziemniaka zwilżyć roztworem p-fenylenodwuaminy, inną zwilżyć roztworem nadtlenku wodoru, jeszcze inną zwilżyć roztworem EDTA i wreszcie ostatnią pozostawić jako kontrolną. Porównać szybkość ciemnienia poszczególnych połówek.
2. Wykazanie obecności katalazy w ziemniaku (ćwiczenie znajduje się również w skrypcie)
Zasada: Katalazy są enzymami rozkładającymi nadtlenek wodoru powstający jako produkt uboczny w niektórych procesach metabolicznych. Ponieważ nadtlenek wodoru jest szkodliwy ze względu na jego utleniające właściwości, przyjmuje się, że katalazy pełnią w organizmie funkcję ochronną przed szkodliwym działaniem nadtlenku wodoru.
Wykonanie: Do miazgi ziemniaczanej dodać kroplami wodę utlenioną. Następuje burzliwe wydzielanie się pęcherzyków tlenu świadczących o aktywności katalazy. Wydzielanie pęcherzyków tlenu następuje również podczas przemywania wodą utlenioną skaleczenia; oznacza, że katalaza występuje nie tylko w tkankach roślinnych.
3. Spektrofotometryczne oznaczanie aktywności enzymów
a) Oznaczanie aktywności peroksydaz w ekstrakcie z roślin
Zasada: Peroksydazy są powszechnie występującymi enzymami utleniającymi różne substraty przy udziale nadtlenku wodoru H2O2. Najczęściej substratem są różne związki fenolowe oraz dwuaminy, których produkty utleniania charakteryzują się wyraźną barwą. Zasada analizy polega na zmieszaniu enzymu z substratem i następnie powtarzaniu pomiaru gęstości optycznej mieszaniny, co pozwala na wykreślenie zależności absorpcji od czasu inkubacji. Liniowy odcinek krzywej służy do wyznaczenia linii regresyjnej, której nachylenie jest miarą aktywności enzymu.
Wykonanie:
Odważyć 350 mg świeżej masy liści. Ucierać w ciekłym azocie stopniowo dodając bufor ekstrakcyjny (3 x 0,5 mL = 1,5 mL 0,05 M buforu fosforanowego pH 7.0 z 0,01 M EDTA). Następnie ekstrakt wirować przy 14000 × g przez 10 min w temp. 4°C.
Aktywność peroksydazy mierzy się spektrofotometrycznie jako wzrost absorbancji przy długości fali 460 nm.
Pomiar aktywności - do kuwety jednorazowej dodajemy:
2 mL buforu ekstrakcyjnego, 12 µL 0,5% roztworu p-fenylenodiaminy w buforze ekstrakcyjnym, 12 µL ekstraktu i następnie zerujemy spektrofotometr. Natychmiast dodajemy 12 µL zbuforowanego H2O2 (50 mL buforu ekstrakcyjnego + 0,15 mL 30% H2O2).
Odczytujemy tzw. slope (kąt nachylenia krzywej) i notujemy jego wartość.
b) Oznaczanie aktywności katalazy w ekstrakcie z roślin (CAT) (EC: 1.11.1.6.)
Zasada: Katalazy są enzymami rozkładającymi nadtlenek wodoru powstający jako produkt uboczny w niektórych procesach metabolicznych. Ponieważ nadtlenek wodoru jest szkodliwy ze względu na jego utleniające właściwości, przyjmuje się, że katalazy pełnią w organizmie funkcję ochronną przed szkodliwym działaniem nadtlenku wodoru.
Wykonanie:
Odważyć 350 mg świeżej masy liści. Ucierać w ciekłym azocie stopniowo dodając bufor ekstrakcyjny (3 x 0,5 mL = 1,5 mL 0,05 M buforu fosforanowego pH 7.5 z 0,01 M EDTA). Następnie ekstrakt wirować przy 14000 × g przez 10 min w temp. 4°C.
Aktywność katalazy mierzy się spektrofotometrycznie jako spadek absorbancji przy długości fali 240 nm, co odpowiada spadkowi zawartości H2O2. Pomiar przeprowadza się w następujący sposób:
Pomiar aktywności -do kuwety spektrofotometrycznej (kwarcowej!) dodajemy kolejno:
2 mL zbuforowanego H2O2 (50 mM buforze fosforanowym o pH 8.0 + 0.3 mL 30% H2O2), a następnie bardzo szybko mieszając 200 µL ekstraktu.
Odczytujemy tzw. slope (kąt nachylenia krzywej) i notujemy jego wartość.
Sprawozdanie
a) Aktywność enzymu peroksydazy wyrazić jako różnicę absorbancji jaka następuje w ciągu 1 minuty w przeliczeniu na 1g świeżej masy liścia.
b) Aktywność enzymu katalazy wyrazić jako spadek H2O2 w ciągu 1 minuty w przeliczeniu na 1g świeżej masy liścia.
Ćwiczenie nr 10 (19,20.01.2012)
KWASY NUKLEINOWE
1. Hydroliza, wykrywanie składników kwasów nukleinowych (ćwiczenie znajduje się również w skrypcie)
Po hydrolizie kwasów nukleinowych można wykrywać poszczególne ich składniki jak pentozę, kwas fosforowy i zasady azotowe.
Hydroliza kwasów nukleinowych: Do 5 cm3 0,5 % roztworu RNA z drożdży dodać 5 cm3 HCl o stężeniu 1 mol⋅dm-3 i ogrzewać na wrzącej łaźni wodnej przez 30 minut od momentu zagotowania.
a) Wykrywanie obecności cukru:
Do probówki nalać 2 cm3 odczynnika Biala i ogrzewać na łaźni wodnej przez ok. 3 min, a następnie 1 cm3 hydrolizatu. Zamieszać i wstawić do łaźni wodnej. Po kilku minutach zanotować powstające zabarwienie.
b) Wykrywanie jonów ortofosforanowych:
Do 1 cm3 hydrolizatu dodać 2 cm3 2,5% roztworu molibdenianu amonowego i całość ogrzać do wrzenia. Wytwarza się żółty osad fosforomolibdenianu amonowego. Po dodaniu paru kropel 1% roztworu kwasu askorbinowego (witamina C) następuje redukcja molibdenianu amonowego do mieszaniny tlenków molibdenu (Mo2O5⋅MoO3), tzw. błękitu molibdenowego.
c) Wykrywanie puryn:
2 cm3 hydrolizatu ogrzać do wrzenia, dodać kilka kropel roztworu CuSO4 0,25 M i ostrożnie kroplami nasycony roztwór Na2SO3. W obecności puryn wytrąca się żółtobiały osad nierozpuszczalnych soli miedziowych I (miedziawych) puryn. (Na2SO3 zredukował Cu2+ do Cu+).
2. Izolacja DNA z warzyw, owoców metodą uproszczoną
1 pomidor (zgnieść) lub jabłko (zetrzeć na tarce) a następnie umieścić w zlewce i dodać łyżeczkę płynu do mycia naczyń.
Zlewkę wraz z zhomogenizowanym materiałem wstawić na 15 minut do łaźni wodnej (60°C) - uważać, żeby nie było za ciepło bo się DNA zdenaturuje, nie może też być za chłodno bo nie zdenaturuje RNA co sprawi, że izolat może być nim zanieczyszczony. Przełożyć do łaźni lodowej (szybko schłodzić) i dodać łyżkę stołową soli zmiękczającej do mięsa firmy Kamis (w tym etapie chodzi o wytrącenie białek, które podobnie jak DNA rozpuszczają się w wodzie). Przesączyć otrzymany ekstrakt przez lejek z sączkiem. Wlać przesącz do naczynia (cylindra) zawierającego 50 cm3 denaturatu.
3. Spektrofotometryczne oznaczanie zawartości kwasów nukleinowych i badanie czystości
Aromatyczne pierścienie purynowe i pirymidynowe absorbują światło w zakresie UV. Właściwość tę można zastosować do pomiaru stężenia RNA w roztworze. Wprawdzie poszczególne zasady różnią się nieco pod względem położenia maksimum absorpcji UV, to jednak różnice te są niewielkie i można przyjąć, że = 260 nm jest optymalną długością fali przy pomiarze ilościowym RNA.
Spektrofotometrię w UV wykorzystać można również do ilościowych oznaczeń białek, jednak jedynie w przypadku, jeśli oznacza się zawartość konkretnego białka względem konkretnego wzorca (białko badane i wzorzec zawierać muszą taką samą ilość absorbujących UV aminokwasów aromatycznych, pomiar przy = 280 nm).
Krzywa kalibracyjna: Do 2 probówek wlać 3 cm3 roztworu wzorcowego (w 100 cm3 buforu octanowego o pH = 5,5 rozpuścić na gorąco 10 mg RNA z drożdży) do drugiej dodać 3 cm3 buforu octanowego. Po zmieszaniu powtórzyć rozcieńczanie tą samą metodą pobierając 3 cm3 roztworu z poprzedniej probówki i 3 cm3 buforu octanowego, uzyskując kolejne stężenia RNA : 100, 50, 25, 12.5, 6.2, 3.1, 1.5, 0.8, 0.4 i 0.2 g RNA/cm3. Następnie wykonać pomiar absorpcji UV (w kuwecie kwarcowej) dla = 260 nm każdego roztworu RNA.
Pomiar stężenia RNA w nieznanym roztworze: Wykonać pomiar spektrofotometryczny absorpcji przy = 260 dla roztworu RNA o nieznanym stężeniu (x, y lub z).
Sprawozdanie
a) Za pomocą arkusza kalkulacyjnego MS Excel wykreślić krzywą kalibracyjną i wyznaczyć równanie regresji liniowej zależności absorbancji od stężenia RNA (wzorca).
b) Obliczyć stężenie RNA roztworów x,y,z za pomocą równania regresji liniowej otrzymanego w podpunkcie a).
4. Jakościowa analiza kwasów nukleinowych - reakcja PCR
Technika Real-Time PCR jest powszechnie stosowana w badaniach poziomu ekspresji genów, poprzez pomiar ilościowy kopii RNA w komórkach i tkankach. Różni się ona od „konwencjonalnego PCR”, tym że istnieje możliwość monitorowania przyrostu produktu w czasie rzeczywistym. Umożliwia to specjalnie zaprojektowany składnik mieszaniny reakcyjnej - sonda sprzężona z fluoroforem, która generuje fluorescencję o intensywności proporcjonalnej do ilości powstałego produktu PCR. Instrument dokonuje pomiaru w początkowej fazie przyrostu produktu, kiedy to kinetyka reakcji wchodzi w fazę wykładniczego wzrostu. W celu analiz ekspresji genów konieczne jest wyizolowanie z badanych tkanek całości mRNA zawartego w komórkach z możliwie najmniejszą ilością zanieczyszczeń (białka, genomowe DNA). Próbka tkanki poddana zostaje lizie i homogenizacji w warunkach denaturujących, co dezaktywuje nukleazy będące zagrożeniem dla RNA. Śledzenie przyrostu produktu reakcji umożliwia zastosowanie związków fluorescencyjnych. Natężenie sygnału fluorescencji jest proporcjonalne do ilości powstałego produktu. W związku z tym oznaczenie wymaga specjalnie przystosowanego termocyklera, którego sprzężenie z fluorymetrem umożliwia detekcję sygnału fluorescencji. Według teoretycznych założeń ilość produktu wzrasta dwukrotnie w każdym kolejnym cyklu łańcuchowej reakcji polimerazy. W celu oznaczenia początkowej ilości kopii matrycy wyznacza się wartość CT, którą stanowi numer cyklu PCR odpowiadający poziomowi sygnału nieco powyżej zakresu detekcji analizatora fluorescencji. Im więcej kopii matrycy znajdowało się w mieszaninie reakcyjnej na początku reakcji, tym mniejsza liczba cykli PCR jest wymagana w celu osiągnięcia granicznej wartości fluorescencji CT. Zbieraniem danych i ich analizą poprzez matematyczne kalkulacje zawiaduje specjalne oprogramowanie. Na podstawie kinetyki reakcji automatycznie wyznacza się kluczowe parametry charakteryzujące dynamikę przyrostu produktu- baseline, threshold, a także zdefiniowana zostaje wartość CT. Dokonywane są także dodatkowo normalizacje sygnału (w celu ujednolicenia ilości RNA wyekstrahowanego z różnych próbek, a także wyeliminowania wpływu fluorescencji nie wynikającej bezpośrednio z reakcji PCR). Zastosowanie kontroli endogennej, polegające na porównaniu ekspresji analizowanych genów względem genu, którego ekspresja jest porównywalna we wszystkich próbkach, pozwala na normalizację sygnału pochodzącego ze wszystkich próbek względem ilości cDNA dodawanych do reakcji. Kontrolę endogenną stanowić będzie gen aktyny. Jest to gen kodujący białko cytoszkieletu, ulegający ekspresji w każdej próbce i zgodnie z założeniem liczba kopii transkryptu tego genu jest jednakowa w tkankach liścia.
Wyszukiwarka
Podobne podstrony:
Giełda biochemia# 01 2012
Biochemia egzamin 2012 id 86181
Biochemia WF 2012 Program, Studia, AWF - Wychowanie Fizyczne, Biochemia
biochemia-osr2013-2014 PLAN CW, Studia, UR OŚ, semestr III, biochemia
Giełda biochemia# 01 2012
Biochemia egzamin 2012
pytania biochemia egzamin 2012 I termin
zagadnienia kol I 2012-2013, Studia, UR OŚ, semestr III, biochemia
Lista pytań na zaliczenie uzupełniające 2011 2012, biochemia
2012 I termin, medycyna, II rok, biochemia, giełdy
egzamin biochemia 2012, BIOCHEMIA D, egzamin biochemia
Program cwiczen OAM II rok 2012- 2013, Prywatne, biochemia
Biochemia 22.01.2012 2 cwiczenia, Biochemia
Egzamin 2012 biochamia, Medycyna, Biochemia
Cwiczenia zagadnienia OAM II rok 2012-13, Prywatne, biochemia
Egzamin poprawkowy Biochemia (2012)(1)
więcej podobnych podstron