WSTĘP TEORETYCZNY
Kinetyka zajmuje się zmiennością w czasie reagujących ze sobą układów chemicznych. Badania kinetyczne dostarczają informacji o przebiegu reakcji chemicznych oraz stanowią podstawę projektowania reaktorów.
Wyniki pomiarów kinetycznych opisujemy równaniami , które w większości przypadków wyrażają zależność szybkości reakcji r od stężeń reagentów i od temperatury. Jeśli chcemy znać nie tylko postać równań kinetycznych , lecz zrozumieć również ich powiązanie z molekularnym przebiegiem reakcji , musimy poznać mechanizm reakcji.
Szybkość reakcji chemicznej r w układzie reakcyjnym o stałej objętości definiujemy jako zmianę stężenia c reagenta w czasie t :
![]()
(1)
Równanie kinetyczne jest zależnością czysto empiryczną , którą można się bezpiecznie posługiwać tylko w warunkach , dla których została wyznaczona.
Stała szybkości reakcji zależy wykładniczo od temperatury (równanie Arrheniusa) :
![]()
(2)
gdzie R jest stałą gazową , E energią aktywacji , a A - czynnikiem przedwykładniczym.
Równanie Arrheniusa jest równaniem wykładniczym , co wskazuje na znaczny wpływ temperatury na szybkość reakcji chemicznej. Im większa jest energia aktywacji tym większy jest ten wpływ. Logarytmując równanie Arrheniusa otrzymujemy
![]()
(3)
W celu wyznaczenia stałych E i A bada się szybkość reakcji w różnych temperaturach , a znalezione stałe szybkości reakcji k przedstawia się na wykresie w układzie współrzędnych : ln k - odwrotność temperatury bezwzględnej.
Kąt skręcenia α substancji optycznie czynnej w danej temperaturze i dla danej długości światła zależy od długości d drogi światła i od stężenia c danej substancji
α = β dc (4)
gdzie β - skręcalność właściwa.
Oznaczając przez α0 wartość kąta skręcenia w momencie początkowym ( po czasie t1 ) , przez α w danym momencie czasu ti , a przez α∞ - po zakończeniu reakcji ( t = ∞ ) , można równanie
![]()
(5)
przedstawić w postaci
![]()
(6)
Różnica kątów skręcenia (α0 - α∞) jest bowiem proporcjonalna do początkowego stężenia c0 substratu , a (α - α∞) do stężenia aktualnego c.
Równanie [6] można przekształcić do postaci
ln (α0 - α∞) - ln (α - α∞) = k0t
- ln (α - α∞) = - ln (α0 - α∞) + k0t (7)
co daje równanie prostej o współczynniku kierunkowym k0 i rzędnej przecięcia z osią y równej ln (α0 - α∞).
Reakcja inwersji sacharozy jest przykładem reakcji katalizowanej przez kwas.
OPRACOWANIE WYNIKÓW
1. Wyniki zestawiono w tabeli :
Nr pomiaru |
Czas [min] |
Kąt skręc. [0] |
-∞ |
y=-ln(∞) |
t2 |
yt |
1. |
5 |
8,2 |
11,6 |
-2,451 |
25 |
-12,255 |
2. |
10 |
7 |
10,4 |
-2,342 |
100 |
-23,418 |
3. |
15 |
5,5 |
8,9 |
-2,186 |
225 |
-32,791 |
4. |
20 |
4,4 |
7,8 |
-2,054 |
400 |
-41,082 |
5. |
25 |
3,5 |
6,9 |
-1,932 |
625 |
-48,288 |
6. |
30 |
2,6 |
6 |
-1,792 |
900 |
-53,753 |
7. |
40 |
0,9 |
4,3 |
-1,459 |
1600 |
-58,345 |
8. |
50 |
-0,35 |
3,05 |
-1,115 |
2500 |
-55,757 |
9. |
60 |
-1,2 |
2,2 |
-0,788 |
3600 |
-47,307 |
10. |
nieskon |
-3,4 |
|
|
|
|
suma |
255 |
- |
- |
-16,118 |
9975 |
-372,996 |
(suma t)2 |
65025 |
|
|
|
|
|
liczba pomiarów n = 10
2. Wykreślenie zależności - ln (α - α∞) = f(t)
3. Na podstawie równania prostej umieszczonego na wykresie odczytuję stałą szybkości reakcji katalizowanej przez kwas k , wiedząc , że jest ona równa współczynnikowi nachylenia prostej :
k = 0,0304 ![]()
4. Stała szybkości reakcji k obliczona metodą najmniejszych kwadratów wynosi :
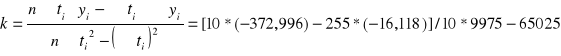
k = 0,011 ![]()
Stała szybkości reakcji k różni się od wyniku uzyskanego w punkcie 3.
5. Skręcalność roztworu α0 w momencie zmieszania roztworów ( t = 0 ) można obliczyć dwoma metodami korzystając z zależności:
![]()
a = - ln(α0 - α∞)
α0 = exp(-a) +α∞
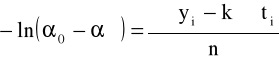
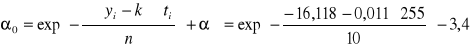
α0 = -5,292
6. Czas po którym przereaguje połowa substratów wynosi :
![]()
minut.
Zakład Chemii Fizycznej
Laboratorium Studenckie
Ćwiczenie nr 20
SACHARYMETRIA. Wyznaczanie stałej szybkości inwersji cukru.
Tomasz Dziadosz
Lukasz Daniel
Inżynieria Chemiczna
rok II grupa I
1
Wyszukiwarka
Podobne podstrony:
Chf30, RADZION DOROTA
ZADANIE A7(5), Radzion Dorota , technologia chemiczna , rok III , grupa III
MOJE17, RADZION DOROTA
ZADANIE A7(3), Radzion Dorota , technologia chemiczna , rok III , grupa III
MOJE17, RADZION DOROTA
ZADANIE A7(3), Radzion Dorota , technologia chemiczna , rok III , grupa III
ZADANIE A7(10), Radzion Dorota , technologia chemiczna , rok III , grupa III
ZADANIE A7(7), Radzion Dorota , technologia chemiczna , rok III , grupa III
ZADANIE A7(6), Radzion Dorota , technologia chemiczna , rok III , grupa III
ZADANIE A7(13), Radzion Dorota , technologia chemiczna , rok III , grupa III
Anka i Halina, RADZION DOROTA
ZADANIE A7(12), Radzion Dorota , technologia chemiczna , rok III , grupa III
ZADANIE A7(2), Radzion Dorota , technologia chemiczna , rok III , grup
więcej podobnych podstron