Kinetyka chemiczna i kataliza
Spełnienie warunku termodynamicznego samorzutności reakcji
(ΔG0 < 0) wcale nie oznacza, że dana reakcja zachodzi z szybkością mierzalną. Wiele reakcji, choć termodynamicznie możliwych przebiega w temperaturze pokojowej tak powoli, iż praktycznie nie można stwierdzić ich zachodzenia, a inne reakcje przebiegają gwałtownie.
Pojęcie szybkości zachodzenia reakcji wiąże się ze zmianami stężeń poszczególnych reagentów podczas reakcji; kinetyka chemiczna zajmuje się badaniem szybkości reakcji i ich kierunkiem.
W czasie przebiegu reakcji chemicznej maleją stężenia substratów i w równoważnych ilościach rosną stężenia produktów.

Szybkość rzeczywista reakcji:
♦ zmienia się w miarę jej przebiegania (zwykle największa jest na początku reakcji);
♦ zależy od rodzaju reagentów i od warunków prowadzenia reakcji
Wiele reakcji pomiędzy związkami nieorganicznymi przebiega z dużą szybkością, podczas gdy związki organiczne reagują wolniej.
Jeżeli reakcję przedstawimy równaniem:
to zależność szybkości reakcji od stężenia będzie miała postać:
Wykładniki potęgowe α, β, γ ... noszą nazwę rzędów reakcji względem reagentów A, B, ... , ich suma to jest rząd danej reakcji.
Współczynnik proporcjonalności k zależy od temperatury i nazywa się stałą szybkości reakcji.
Reakcje chemiczne są najczęściej pierwszego, drugiego lub trzeciego rzędu. Znane są też reakcje rzędu zerowego, których szybkość nie zależy od stężenia reagentów (np. niektóre reakcje katalitycznego utleniania czy uwodnienia).
Równania kinetyczne o bardziej skomplikowanej postaci wskazują na złożony przebieg reakcji.
Wiele reakcji chemicznych przebiega wieloetapowo poprzez następujące po sobie proste etapy zwane procesami elementarnymi. W etapach tych mogą reagować różne liczby cząsteczek.
Liczbę cząsteczek, atomów, jonów lub rodników biorących udział w procesie elementarnym prowadzącym do reakcji, nazywa się cząsteczkowością reakcji.
Procesy elementarne odbywają się zwykle z udziałem małej liczby cząsteczek (dwóch najczęściej, rzadziej jednej lub trzech).
Pomiędzy rzędowością a cząsteczkowością reakcji jest następująca różnica:
rzędowość jest pojęciem stosowanym tylko w przypadku doświadczalnie wyznaczonych sumarycznych równań kinetycznych;
cząsteczkowość dotyczy procesu elementarnego danej reakcji i wskazuje jaka liczba cząsteczek bierze w nim udział.
REAKCJE CHEMICZNE RÓŻNYCH RZĘDÓW:
Reakcje pierwszego rzędu:
Równanie kinetyczne dla takich reakcji ma postać:
k1 - stała szybkości reakcji pierwszego rzędu
cA - stężenie molowe substratu A
Stała szybkości tych reakcji wyraża się wzorem:
C0A - stężenie początkowe substratu A
CA - stężenie substratu A po czasie t
Po przekształceniu:
Jeżeli reakcja przebiega przez wiele etapów, to o jej szybkości decyduje (tzn. ją limituje) etap najwolniejszy.
Ważną wielkością charakteryzującą kinetykę reakcji chemicznej jest tzw. okres połowicznej przemiany lub okres półtrwania τ½ (t½), który określa czas, po którym stężenie substratu zmniejsza się w wyniku reakcji o połowę w stosunku do stężenia początkowego.
Dla reakcji pierwszego rzędu t½ wynosi:
nie zależy od stężenia !!
Do reakcji pierwszego rzędu należą: hydroliza estrów, inwersja sacharozy, rozkład N2O5.
Reakcje drugiego rzędu:
Dla takich reakcji wzór na ich szybkość może mieć różną postać matematyczną, w zależności od typu reakcji.
Gdy w reakcji występuje tylko jeden substrat A, to szybkość jest proporcjonalna do kwadratu jego stężenia:
oraz:
a także:
Przykładem takiej reakcji może być rozkład jodowodoru.
Gdy w reakcji biorą udział dwa substraty A + B, to:
i w tym przypadku:
gdzie y: ubytek stężenia substratu A równy ubytkowi stężenia substratu B.
Przykładem takich reakcji są np. termiczny rozkład NO2, reakcje dimeryzacji, hydroliza estrów w środowisku zasadowym.
Reakcje trzeciego rzędu:
na przykład:
A + B + C produkty
k3 ma prostą postać w przypadku A = B = C
C0 - stężenie początkowe każdego z trzech substratów
C - stężenie po czasie t.
Przykładem jest: 2NO + Cl2 2NOCl
2NO + H2 N2O + H2O
Dla reakcji: 2A + B produkty
lub: 3A produkty
We wszystkich dokładnych rozważaniach kinetycznych w miejsce stężeń w powyższych równaniach należy wprowadzić aktywności.
Reakcje zerowego rzędu:
W takich reakcjach szybkość nie zależy od stężenia substratu (substratów), lecz od innych czynników, jak np. dyfuzji substratów i produktów w niektórych reakcjach katalitycznych czy od natężenia promieniowania w reakcjach fotochemicznych.
oraz
a także
Przykładem są tu reakcje enzymatyczne i katalityczne oraz fotochemiczne.
REAKCJE ZŁOŻONE:
Reakcje odwracalne:
Każdej reakcji tego typu A B
towarzyszy reakcja biegnąca w kierunku przeciwnym: B A.
Szybkość reakcji z lewa na prawo maleje z czasem, a reakcji odwrotnej - wzrasta. Po pewnym czasie ustala się stan równowagi dynamicznej, w którym:
dla reakcji A → B
dla reakcji B → A
Stąd - jeżeli są to reakcje pierwszego rzędu:
Jeżeli, w danym czasie reakcja zachodząca w jednym kierunku ma inną szybkość niż reakcja odwrotna, to sumaryczna szybkość v wynosi:
W stanie równowagi: υ = 0 → kA ⋅ cA - kB ⋅ cB
Stąd:
Reakcje szeregowe (następcze):
Ten typ reakcji spotykamy, gdy substrat A przereaguje na produkt końcowy nie od razu, a poprzez kolejne etapy pośrednie:
A B C D E
Sumaryczna szybkość takiej reakcji będzie limitowana najwolniejszym etapem (C D).
Do takich reakcji należą np. reakcja hydrolizy estrów kwasów dwukarboksylowych, dwuestrów glikoli, niektóre procesy rozpadu promieniotwórczego.
Reakcje równoległe:
Schematycznie ten typ reakcji można przedstawić jako:
B
A C Z O szybkości takich reakcji decyduje etap
najszybszy.
Jeśli otrzymuje się więcej produktów, to mówimy o reakcji rozgałęzionej, np.:
B X
A C Z
Y
Przykładem takiej reakcji jest rozpad chloranu (V) potasu:
k1 4 KCl + 6 O2
4 KClO3
k2 3 KClO4 + KCl
Niektóre pierwiastki ulegają rozpadowi promieniotwórczemu wg tego mechanizmu.
Reakcje łańcuchowe:
Należą do nich reakcje spalania, rozkładu, polimeryzacji węglowodorów, niektóre reakcje fotochemiczne, np. fotosynteza HCl z chloru i wodoru zachodząca pod wpływem naświetlania.
Cl2 + hν → Cl2*
Cl2* → Cl⋅ + Cl⋅ zapoczątkowanie łańcucha
Cl⋅ + H2 → HCl + H⋅
H⋅ + Cl2 → HCl + Cl⋅ rozwijanie łańcucha
Cl⋅ + H2 → HCl + H⋅ itd.
Cl⋅ + Cl⋅ → Cl2
zakończenie reakcji łańcuchowej
H⋅ + Cl⋅ → HCl
Cl2* - cząsteczka chloru w stanie wzbudzenia
Cl⋅ , H⋅ - rodniki chloru i wodoru
Reakcja łańcuchowa przebiega ze wzrastającą szybkością, aż do wyczerpania substratu, niekiedy przebieg jej jest gwałtowny lub wybuchowy.
Wpływ temperatury na szybkość reakcji.
Energia aktywacji.
Wg empirycznej reguły van't Hoffa wzrost temperatury o 10°C zwiększa szybkość reakcji 2 do 4 razy (prawdziwe dla temperatur bliskich temperatury pokojowej).
Dokładniejsza zależność podana przez Arrheniusa:
R - stała gazowa T1, T2 - temperatura reakcji
k - stała szybkości reakcji
Ea - wielkość charakterystyczna dla danej reakcji zwana energią aktywacji.
Im energia aktywacji większa tym stała szybkości k w danej temperaturze jest mniejsza, a więc i szybkość reakcji będzie mniejsza.
Energia aktywacji jest energią, jaką muszą pobrać reagujące cząsteczki, aby zderzenia między nimi były efektywne,
tzn. doprowadzały do produktów reakcji (energię tę odnosi się do
1 mola substancji).
Teoria kompleksu aktywnego
(inaczej zwana teorią stanów przejściowych)
Wg tej teorii zakłada się, że cząsteczki reagujące ze sobą tworzą w trakcie reakcji aktywny kompleks, który może ulec rozpadowi wytwarzając produkty lub odtwarzając substraty:
A B A - B * A B
+ +
C D C - D C D
substraty kompleks aktywny produkty
Kompleks aktywny ma dużą energię i jest w stanie równowagi z substratami. Zmiany energii zachodzące w danym układzie reaktywnym podczas tworzenia się kompleksu aktywnego można przedstawić rysunkiem następującym:
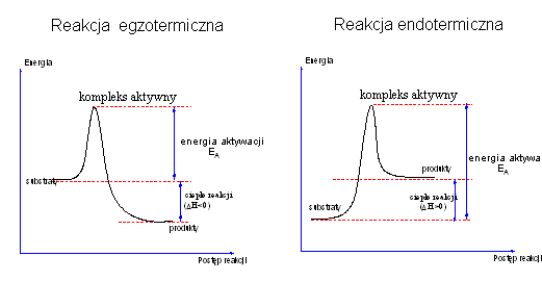
Aby przejść od stanu początkowego A do stanu produktu końcowego B cząsteczki muszą pokonać barierę energetyczną E1, która jest równa energii aktywacji Ea. Tylko cząsteczki mające odpowiednio dużą energię, równą co najmniej Ea, mogą pokonać tę barierę i przejść na poziom produktów. Różnica poziomów energetycznych produktów i substratów jest równa efektowi energetycznemu reakcji:
Q = E2 - E1
Im E1 jest większe, tym reakcja przebiega trudniej.
Dla rozpadu produktów B z odtworzeniem substratów A potrzeba jeszcze większej energii
E2 = E1 + Q
Reakcje katalityczne:
Działanie katalizatora można przedstawić na rysunku:
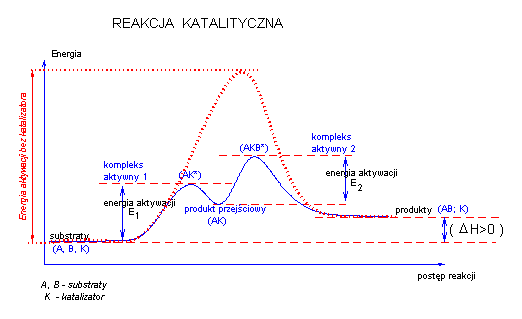
Mechanizm przebiegu reakcji chemicznej z udziałem katalizatora jest inny niż bez niego.
Katalizator tworzy z substratem reakcji odpowiednie kompleksy aktywne oraz związek przejściowy. Sam katalizator praktycznie nie wchodzi do sumarycznego równania reakcji, nie zmienia położenia równowagi reakcji ani nie może spowodować przebiegu reakcji dla której ΔG> 0.
Katalizator zmienia jedynie szybkość dochodzenia do równowagi reakcyjnej poprzez obniżenie bariery energetycznej reakcji.
Jednym z etapów reakcji katalizowanej jest związanie się substratu z katalizatorem. Gdy takie wiązanie jest silne, to bariera energetyczna reakcji
może ulec podwyższeniu w porównaniu z reakcją bez katalizatora.
Jest to przykład katalizy ujemnej, a katalizator opóźniający reakcję nazywa się inhibitorem.
Kataliza jednorodna (homogeniczna) - katalizator występuje w tej samej fazie co reagenty reakcji.
Na przykład:
utlenianie SO2 lub CO w obecności śladowych ilości NO2:
NO2 + SO2 → NO + SO3 i NO + ½ O2 → NO2
NO2 + CO → NO + CO2 i NO + ½ O2 → NO2
Ostateczna ilość NO2 pozostaje bez zmiany.
W układach jednorodnych najczęściej spotyka się katalizę kwasowo-zasadową, np. reakcje hydrolizy estrów.
Działanie katalityczne kwasów i zasad polega na tworzeniu związków pośrednich z jonami H3O+ i OH- , które w dalszym etapie ulegają przegrupowaniu do produktów końcowych.
Kataliza niejednorodna (heterogeniczna) - katalizator występuje w innej fazie niż reagenty reakcji.
W tym przypadku ważną rolę odgrywają procesy, np. adsorpcja, zachodzące na powierzchni stałego katalizatora tworzącego w układzie reagującym odrębną fazę. Taki katalizator nazywa się KONTAKTEM.
Kataliza hetergeniczna ma bardzo duże zastosowanie w przemyśle chemicznym w reakcjach syntezy wielu związków nieorganicznych i organicznych, a także w procesie „dopalania” spalin samochodowych do CO2 i wody w nowoczesnych silnikach spalinowych.
Szybkość reakcji katalitycznej w układzie niejednorodnym jest proporcjonalna do stopnia pokrycia powierzchni katalizatora Θ:
υ = k ⋅Θ
Jeżeli: b ⋅ c
Θ = ---------- gdzie: k - stała szybkości reakcji
1 + bc b - stała charakterystyczna dla
danego układu
c - stężenie molowe adsorbatu
to dla dużego stopnia pokrycia powierzchni b⋅c >> 1
reakcja jest rzędu zerowego
dla małego stopnia okrycia powierzchni, gdy b ⋅ c << 1:
reakcja jest rzędu pierwszego!!
Najczęściej sytuacja jest pośrednia i obserwuje się ułamkowy rząd reakcji katalitycznej:
0 < n < 1
Autokataliza - katalizatorem reakcji jest jej produkt.
Na przykład: zmydlanie octanu metylu (powstający kwas octowy katalizuje reakcję), utlenianie kwasu szczawiowego za pomocą KMnO4.
Szczególnym przykładem reakcji katalitycznych są reakcje enzymatyczne. Tu kataliza ma charakter pośredni pomiędzy katalizą homogeniczną i heterogeniczna, gdyż enzymy stanowią specyficzny koloid białkowy (białko globularne). Enzymy wyróżniają się wybitną selektywnością i efektywnością działania - działają tylko na określone wiązania, a nawet na określoną formę enancjomorficzną tego samego związku, Aktywność enzymów jest niesłychanie wrażliwa na temperaturę i pH reakcji.
Szybkość rzeczywista reakcji chemicznej w danej chwili gdy Δt → 0 jest równa stosunkowi nieskończenie małej zmiany stężenia dc substratu lub produktu do nieskończenie małego przedziału czasu, w którym ta zmiana nastąpiła i wyraża się zawsze liczbą dodatnią:
dc
υ = ± -----
dt
a A + b B ↔ c C + d D
υ = k ⋅ cAα ⋅ cBβ ⋅ cCγ ......
W celu określenia szybkości reakcji stosuje się
różne metody. Zazwyczaj w stałej temperaturze i w określonych odstępach czasu z reagującego układu pobiera się próbki i oznacza w nich stężenie substratu lub produktu (np. przez miareczkowanie czy wytrącanie osadów). Można też określać niektóre wielkości fizykochemiczne, jak: zmianę barwy, przewodnictwo elektryczne, współczynnik załamania światła, lepkość, absorpcja promieniowania itp. Na podstawie zmian tych wielkości wyznacza się równanie kinetyczne dla danej reakcji (czyli zmianę stężenia w czasie), a na jego podstawie określa się rzędowość reakcji.
υ = k1 ⋅ cA
2,303 C0A
k1 = ------------ log ---------
t CA
C0A k1 ⋅ t
log ------- = ----------
CA 2,303
0,693
t1/2 = -----------
k1
υ = k2 ⋅ cA2
1 1 1
k2 = ---- --- - ----
t CA C0A
1
t1/2 = -----------
k2⋅ C0A
υ = k2 ⋅ cA ⋅ cB
2,303 C0B (C0A - y)
k2 = ---------------- ⋅ log -------------------
t (C0A - C0B) C0A (C0B - y)
υ = k3 ⋅ cA ⋅ cB ⋅ cC
1 (C02 - C2)
k3 = ---- ⋅ -----------------
t 2 ⋅ C02 ⋅ C2
3
t1/2 = ----------------
2 ⋅k3 ⋅ C02
υ = k3 ⋅ cA2 ⋅ cB
υ = k3 ⋅ cA3
υ = k0
C0 - C
k0 = -----------
t
C0
t1/2 = ----------
2 ⋅k0
υ1 = kA ⋅ cA
υ2 = kB ⋅ cB
kA ⋅ cA = kB ⋅ cB
kA CB
------- = ------- = Kc
kB CA
Czyli, że w reakcji odwracalnej typu A B stała równowagi K jest równa ilorazowi szybkości kA i kB.
Wyszukiwarka
Podobne podstrony:
5 Wiazania chemiczne, Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
6 Zależność właściwości substancji od rodzaju wiązania chemicznego, Budownictwo PK, Chemia, Chemia n
WRZENIE I KRZEPNICIE ROZTWORW, Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
BIOPIERWIASTKI 19.11 (22.11), Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
1 Wprowadzenie – podstawowe pojęcia, Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
ROWNOWAGI, Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
SYSTEMATYKA ZWIAZKOW NIEORGANICZNYCH 5.11 (8.11), Budownictwo PK, Chemia, Chemia nieorganiczna od Ma
UKŁADY KOLOIDALNE (22.01), Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
ELEKTROCHEMIA, Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
ADSORPCJA JONOWYMIENNA (22.01), Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
4 Układ okresowy pierwiastków, Budownictwo PK, Chemia, Chemia nieorganiczna od Marysi
ściaga+chemia, Budownictwo PK, Chemia Budowlana
TEST z 22.01.09 pytania, Budownictwo PK, chemia
Instrukcja kinetyka 2013, Biologia UJ, Chemia nieorganiczna, instrukcje
wzór Sprawozdanie, budownictwo sem3, Chemia, materiały od prowadzącej
zadania rachunkowe rok akad. 2011 2012, budownictwo sem3, Chemia, materiały od prowadzącej
Zaliczenie-11.05 rozwiazania, PK, chemia, nieorganiczna, nieorg zadania lab
zal termin3, PK, chemia, nieorganiczna, nieorg zadania lab
3.kinetyka chemiczna, Politechnika Rzeszowska Budownictwo, IBD, Chemia
więcej podobnych podstron