Kinetyka
Zajmuje się badaniem przebiegu reakcji w czasie i pod wpływem różnych czynników wpływających na szybkość reakcji.
![]()
Szybkość reakcji
Reakcje mogą zachodzić w ciągu sekundy, kilku minut, godzin, kilka dni a nawet tys. Lat.
Reakcje I-rzędu - szybkość wprost proporcjonalna do stężenia roztworu
V= K*[A] N2O5->N2O3+O2
Reakcje II-rzędu - szybkość wprost proporcjonalna do kwadratu stężenia substratu A lub do stężenia substratu A i B.
V= K*[A]2 lub V= K*[A][B] 2HI->H2+I2
Reakcje III-rzędu - szybkość wprost proporcjonalna do skł A w 3 potędze, lub do kwadratu substratu A i stężenia B, lub stężenia A, B, C.
V= K[A]3= k*[A]2*[B]= K*[A][B][C] 2Na+Cl2->2NaCl
Reakcje n-tego rzędu nazywamy reakcję, której szybkość jest proporcjonalna do n-tej potęgi stężenia 1go z reagentów lub iloczynu stężeń n-reagentów
Okazuje się, że często rząd reakcji jest liczbą niecałkowitą, co oznacza, że reakcja biegnie przez etapy pośrednie, a w rezultacie o całym biegu procesu, decyduje 1 lub 2 etapy najważniejsze.
2N2O5->N2O4+O2
Reakcja zachodzi wieloetapowo:
N2O5->N2O3+O2 r. powolna
N2O3->NO+NO2 r. szybka
NO+N2O5->3NO2 r. szybka
4NO2->2N2O4 r. szybka
2N2O5->N2O4+O2
Matematyczne wyznaczanie - Reakcje I rzędu:
![]()
![]()
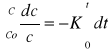
![]()
K to tgα na wykresie
![]()
![]()
![]()
Okres połowicznego rozpadu
Okres półtrwania, stężenie substancji zmniejsza się o połowę
![]()
![]()
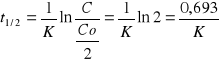
Matematyczne wyznaczanie - Reakcje II rzędu:
![]()
![]()
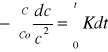
![]()
![]()
![]()
![]()
Czynniki wpływające na szybkość reakcji:
Rodzaj i stężenie reagentów
Temperatura -wzrost przyspiesza przebieg
Ciśnienie- o ile temp. We wszystkich r. tak ciśnienie tylko ze zmianą objętości
Promieniowanie elektromagnetyczne- czynnik podnoszący Ek cząsteczki a także przyspiesza
pH roztworu- czynnik selektywny dotyczy tylko niektórych reakcji, czasąmi przyspiesza
Stopień rozdrobnienia- reakcje pomiędzy ciałami stałymi (większe powierzchnie)
Mieszanie - powoduje wzrost pow., na której zachodzi reakcja
Katalizator- wchodzi w reakcje z substratami a po zakończeniu reakcji odtwarza się
Reakcja z katalizatorem
Dodanie przyspiesza reakcje chemiczną
Inhibitory opóźniają szybkość reakcji
A+B->AB Reakcja chem. Bez katalizatora zachodzi powoli
A+K->AK Reakcja chem. Z katalizatorem zachodzi szybko
AK+B->AB+K Reakcja chem. Z katalizatorem zachodzi szybko
Reakcja syntezy chlorku nitrozynu bez katalizatora:
2NO+Cl2->2NOCl
Reakcja syntezy chlorku nitrozynu z katalizatorem
2NO+Br2->2NOBR
2NOBr+Cl2->2NOCl+Br2
Kataliza homogeniczna
Katalizator występuje w tej sąmej fazie, co reagenty
Utlenianie SO2 do SO3, CO do CO2
Kataliza heterogeniczna
Katalizator występuje w innej fazie niż reagenty (najczęściej w fazie stałej)
Polimeryzacja acetylenu do benzenu wobec platyny, katalizatory spalin w sąmochodzie
Autokataliza
Katalizatorem reakcji jest 1 z produktów reakcji
Utlenianie kwasu szczawiowego
Holoenzym= Apoenzym+koenzym
Aktywność enzymów zależy od:
Temperatury, pH, stężenia enzymu, obecności inhibitorów lub aktywatorów.
Adsorpcja
Zjawisko zmian stężenia substancji na powierzchni międzyfazowej w układzie heterofazowym, najczęściej w układzie faza stała/gaz, faza stała/ciecz.
Adsorbent
Substancja adsorbująca, na powierzchni, której zachodzi zjawisko adsorpcji.
Adsorbat
Substancja adsorbowana
Adsorpcja fizyczna
Adsorbat gromadzi się na powierzchni adsorbenta wskutek oddziaływania sił Van der Wasąla lub wiązań wodorowych (odwracalna, może być wielowarstwowa, małe ciepło adsorpcji).
Adsorpcja Chemiczna - Chemisorpcja
Adsorbat gromadzi się na powierzchni adsorbenta wskutek tworzenia się wiązań chemicznych z adsorbentem (trudno odwracalna lub nieodwracalna, jednowarstwowa, duże ciepło adsorpcji rzędu ciepła reakcji chemicznej.
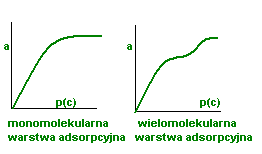
a- Adsorpcja - ilość Adsorbat zaadsorbowana przez jednostkę masy
p(c)- ciśnienie lub stężenie adsorbatu
Etapy adsorpcji fizycznej:
Dyfuzja cząsteczek adsorbatu do powierzchni adsorbenta
Adsorpcja w warstwie monomolekularnej
Ustalenie stanu równowagi w warstwie monomolekularnej
Adsorpcja w warstwie bimolekularnej
Ustalenie stanu równowagi w warstwie bimolekularnej
![]()
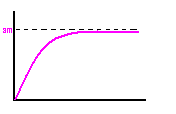
Izoterma adsorpcji Langmuira
a-adsorpcja
am- adsorpcja przy całkowitym pokryciu powierzchni warstwą monomolekularną
p- ciśnienie gazu lub par adsorbentu
![]()
k*p<<1 to am*kp
k*p>>1 to a->am
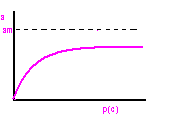
Izoterma adsorpcji Freundlicha
Adsorpcja
p- ciśnienie gazu lub par adsorbentu
c- stężenie wł. W roztworze
am- przy całkowitym pokryciu pow. warstwą monomolekularną
k- wyznaczane doswiadczalnie
![]()
Zlinearyzowana postać izotermy Freundlicha:
Log a= log k+ 1/n log p
Rodzaje adsorbentów:
- nieporowate (czarne sądze, białe sądze, sądze grafitowane) mają zastosowanie głównie jako wypełnianie polimerów, smarów, laków.
- porowate (węgle aktywne, żele krzemionkowe i glinokrzemionkowe, tlenek glinu, sita molekularne) mają zastosowanie jako do pochłaniania gazów i par, rozdzielania składników mieszanin lub jako aktywne katalizatory względnie nośniki katalizatorów.
Powierzchnia właściwa
suma powierzchni geometrycznej oraz wszystkich niejednorodnych powierzchni adsorbenta. Wyrażamy w m2/g. Różnica 20 średnic atomowych to tzw. Powierzchnia idealna, dla powierzchni pozornie gładkich (szlifowany marmur) odchylenie wynosi 10.000 średnic atomowych.
Szkło sodowe ok. 2m2/g
Żel krzemionkowy 300-500 m2/g
Węgiel aktywny 500-1000 m2/g
Typowe zastosowanie węgla aktywnego:
- ścieki - usuwanie koloru, zapachu
- oczyszczanie wody pitnej i procesowej
- oczyszczanie surowców
- oczyszczanie powietrza i gazów procesowych
- odzywanie rozpuszczalników
- filtry papierowe
- maski przeciwgazowe
- jako katalizator
- urządzenia klimatyzacyjne
- przechowywanie owoców
- usuwanie halogenów, rozp. organicznyc
Chromatografia
Zajmuje się metodami rozdzielenia różnych substancji o charakterze zarówno jonowym jak i niejonowym, różniących się miedzy sobą współczynnikiem podziału między formą nieruchoma i ruchomą układu. M. Wiet. W 1906 rozdzielił ekstrakt z zielonych cz. roślin na szereg barwnych pasm podczas przesuwania go przez kolumnę z CaCO3. Podstawą metod chromatograficznych jest podział substancji miedzy fazą nieruchoma (stacjonarną) a fazą ruchomą. Fazą nieruchomą może być ciało stałe lub ciecz naniesiona na odpowiedni nośnik. Fazę ruchomą tworzą ciecze lub gazy.
Wychwytywanie składników ruchomej fazy ciekłej lub gazowej na nieruchomej fazie stałej może być warunkiem adsorpcji, Chemisorpcja lub ekstrakcji.
Podział metod chromatograficznych:
- ze względu na stan skupienia fazy ruchomej
ch. Cieczowa
ch. Gazowa
- ze względu na sposób nagromadzenia fazy stałej
warstwowa (bibułowa, cienkowarstwowa)
kolumnowa
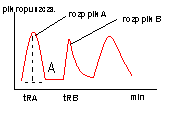
Interpretacja jakościowa chromatografu (na podstawie czasów retencji)
P=1/2a*h
Elektrochemia- zajmuje się współzależnością zjawisk elektrycznych i chemicznych, a zwłaszcza przemianą energii reakcji chemicznych w energię elektryczną i przemianę odwrotną, zajmuje się procesąmi przebiegającymi podczas pracy ogniw galwanicznych lub podczas elektrolizy
Przewodniki- ciała przewodzące prąd elektryczny
Izolatory- ciała nieprzewodzące pradu elektrycznego
Wzór Nernsta
Określa potencjał elektrody odpowiadający stanowi równowagi
![]()
E- potencjał elektrody R- stała gazowa (8,314 J/K*mol)
F- stała Farradaya (96500C) n-liczba wymienianych elektronów
T- temp. W K
Cme- stężenie molowe jonów w metal
Eo- potencjał normalnej elektrody niesiony względem elektrody wodorowej omywanej wodorem pod ciśnieniem 1013hPa w temp 293K przy jednorodnych stęż. Reagentów
Siła elektromotoryczna ogniwa
Jest miara zdolności reakcji ogniwa do spowodowania przepływu elektronów przez obwód. SEM ogniwa definiuje się jako różnice potencjałów elektrody dodatniej (o wyższej wartości potencjału-katoda) i ujemnej (anoda) dla ogniwa otwartego, takiego, w którym obwód elektryczny nie jest zamknięty, a opór między biegunami ogniwa jest nieskończenie wielki
SEM= Katoda- E anoda= E
W zależności od tego, z jakich półogniw tworzymy ogniwo, ta sąma elektroda może być anoda lub katodą.
Mg|Mg2+ Eo=-2,38V
Zn|Zn2+ Eo=-0,76V
Cu|Cu2+ Eo=+0,34V
W układzie
Mg|Mg2+ || Zn2+|Zn
Anoda Katoda- bo ma większe Eo SEM= -0,76-(-2,38)=1,62V
Zn|Zn2+ || Cu2+|Cu
Anoda Katoda SEM=0,34-(-0,76)=1,1V
Szereg napięciowy metali:
K, Na, Mg, Al., Zn, Fe, Ni, Sn, Pb, H, Cu, Hg, Ag, Au
Metale leżące z prawej strony od wodoru wypierają wszystkie metale z r. leżące przed nim
Metale leżące z lewej strony wodoru wypierają go z kwasów.
Reduktor:
Nie mogą być nim substancje na najwyższym stopniu utlenienia (Mn7+,N5+, C6+)
Utleniacz:
Nie mogą być nim subst. Na najniższym stopniu utlenienia.
Elektroda jonoselektywna
Elektrody, na których powstaje potencjał pod wpływem zetknięcia materiału elektrody z roztworem zawierającym oznaczone jony
Zalety: Wady:
- reagują na 1 rodzaj jonów -nie zawsze są jonoselekt. (Zakłócenia 103-105)
- szeroki zakres pomiarowy (do 10-6) - wskazania są proporc. do stęż. (niepew. 30%)
- szybki pomiar
-łatwość powtórzeń
Elektroda prętowa do pomiaru pH
- nakładka z tworzywa sztucznego
-dla różnego rodzaju mediów
- zakres pracy od 0…12 (14pH)
-wytrzymałość 10bar
Elektroliza
Proces wymuszony, polegający na przepływie ładunku elektrycznego przez elektrolit. Nośnikami ładunku elektrycznego mogą być jony dodatnie i ujemne. W czasie elektrolizy zawsze następuje rozkład zw. Chemicznego! Może zachodzić w wodnych roztworach elektrolitów lub w elektrolitach stopionych.
Katoda Płyta połączona z ujemnym biegunem źródła prądu (zachodzą na niej procesy redukcji)
Anoda Płytka połączona z dodatnim biegunem źródła prądu (zachodzą na niej procesy utleniania)
Napięcie rozkładowe
Różnica potencjałów, przyłożona z zew. Źródła przekraczająca określoną wartość. Wtedy wydzielane SĄ produkty elektrolizy
Elektroliza stopionego NaCl daje Na i Cl2
Katoda 2Na+ +2e = 2NA
Anoda 2ClCl2+2e
Elektroliza stężonego NaCl daje H2 i Cl2
Katoda 2Na++2e-> 2Na
Ale 2Na + 2H2O-> 2NaOH + H2
Anoda- 2Cl Cl2+2e
Elektroliza rozcieńczonego NaCl daje O2 i H
Katoda 2H20 +2e->H2+2Oh
2H++2e->H2
Anoda 4OH- - 4e-> 2H20+O2
2H2O->O2+4H++4e
Jeżeli roztwór poddawany jest elektrolizie zawiera kilka kationów i klika anionów, wówczas rozładowaniu ulegają:
Na katodzie- kation o najwyższym potencjale wydzielania
Na anodzie- anion o najniższym potencjale wydzielania
Produkty elektrolizy
Wodnych roztworów kwasów tlenowych oraz wodorotlenków jest O2 i H2. Dlatego, zę potencjały wydzielania większości metali z roztworów wodnych na elektrodach metalicznych są niższe od potencjału wydzielania wodoru, a potencjały wydzielania produktów rozładowywania anionów kwasów tlenowych SĄ najczęściej wyższe od potencjału rozładowania tlenu.
OH,I,Br,Cl,No5,So4,PO4
Wymieniona kolejność rozładowywania jonów może być zmieniona w zależności od stężenia jonów materiału elektrod.
I prawo Farradaya
Masą substancji (m) wydzielonej na elektrodzie podczas elektrolizy jest proporcjonalna do wielkości ładunku (Q), który przepłynął przez elektrolit.
M=K*Q=K*I*t
K- równoważnik elektrochemiczny t- czas trwania procesu
Q- ładunek elektryczny
I- natężenie prądu
m- masą subst. Wydzielona na elektrodzie
K=M/n*F
M- masą atomowa substancji
n- stopień utlenienia
F- stała Farradaya (965000C) - ład. Wydziela 1 równoważnik dowolnego pierwiastka
II Prawo Farradaya
Jednakowe ładunki elektryczne wydzielają z różnych elektrolitów masy substancji proporcjonalne do równoważników elektrochemicznego.
M2/m2=k2/k1
Przemysłowe zastosowanie elektrolizy:
- Otrzymywanie pierwiastków:
Na, Li, Mg - elektr. Stopionych chlorków
Al.- elektroliza stopionego Al2O3
Cl, F, I- elektroliza wodnych roztworów chlorków, litowców, berylowców
Cu, Cr, Mn, Ag, Au - elektroliza wodnych roztworów soli
- Elektrochemiczna rafinacja metali
Miedź, Ni, Co, Pb, Au, Ag
- Otrzymywanie powłok galwanicznych, ochronnych i ozdobnych (srebrzenie, złocenie)
KOROZJA
Proces stopniowego niszczenia zachodzący na powierzchni metali i innych stopów oraz tworzyw niemetalicznych (betonu, drewna, tworzyw sztucznych) wskutek chemicznego lub elektrochemicznego oddziaływania środowiska.
Czynniki przyspieszające korozję metali:
- obecność wody w otoczeniu
- obecność zanieczyszczeń w wodzie, w powietrzu otaczającym metal
- obecność spalin
- obecność grzybów lub bakterii glebie
Korozja chemiczna metalu
Zachodzącej reakcji nie towarzyszy przepływ ładunku elektrycznego przez metal lub elektrolit (np. reakcje metali z gazami zachodzące w podwyższonej temp. W kominach)
Korozja elektrochemiczna metalu
zachodzi w wyniku powstania i działania ogniw galwanicznych. Korozji elektrochemicznej towarzyszy przepływ elektronów przez granicę metal elektrolit.
W procesąch korozji wyróżniamy następujące rodzaje ogniw:
- stykowe - na styku 2 metali katody Cu i anody Fe elektrolit
- ogniwo stężeniowe
- ogniwo temperaturowe- powstaje, gdy części wykonywane SĄ z tego sąmego materiału , pracują na różnych temperaturach (np. wymienniki ciepła)
Ochrona przed korozją:
- dobór odpowiednich materiałów (stale nierdzewne, kwasoodporne)
- stosowanie powłok ochronnych (or., ceramicznych, metalicznych, szkliwa, emalie) ( powłoki z metalem mniej szlachetnym od żelaza, zaczyna się rozwarstwiac i ulega rozpuszczeniu, z bardziej szlachetnego np. z srebra- szybciej korodują)
- stosowanie inhibitorów korozji
- ochrona elektrochemiczna (katodowa i anodowa)
Ochrona Katodowa
Elementy konstrukcji narażone na korozję łączy się z ujemnym biegunem źródła stałego o napięciu 1-2V. Dodatni biegun łączy się grafitową płytą przylegająca do konstrukcji. Doprowadzone ze źródła elektrony zobojętniają powstające jony i nie zachodzi proces Fe-2e=fe2+
Ochrona anodowa
Tworzymy sztuczne ogniwa, w których metal chroniony jest katodą (np. Fe) Rozpuszczeniu ulega anoda (Zn, Mg)
CHEMIA FIZYCZNA- KOLOIDY
Koloidy
Układy, najczęściej 2składnikowe, w których wielkość cząstki mieści się w granicach 1mm do 500 mm. Są stanem pośrednim między roztworami rzeczywistymi, a zawiesinami i mieszaninami niejednorodnymi. Sprawiają wrażenie układów fizycznie jednorodnych, chociaż w rzeczywistości oba składniki nie SĄ zmieszane cząsteczkowo.
ROZDROBNIENIE I JEGO WPŁYW NA WŁAŚCIWOŚCI UKŁADU
Mechaniczne
>500mm
Układ dwufazowy, cząstki fazy rozproszonej nie przechodzą przez błony półprzepuszczalne, nie dyfundują, nie wykazują ruchów Browna
Koloidalne
1-500 mm
Układ pozornie jednofazowy, cząstki fazy rozproszonej widoczne tylko w mikroskopie, przechodzą przez sączki z bibuły, nie dyfundują, wykazują ruchy Browna
Cząsteczkowe
<1mm 1 fazowy układ, cząstki fazy rozproszonej przechodzą przez sączki z bibuły, dyfundują przez błony półprzepuszczalne, wykazują ruchy Browna
W koloidzie wyróżniamy:
- ośrodek dyspersyjny (rozpraszający)- część układu, tworzy fazę ciągłą
- faza zdyspergowana (rozproszona) - cząstki rozproszone
Podział koloidów ze względu na wielkość cząstek:
- monodyspersyjne - cząstki fazy rozproszonej mają jednakową wielkość
- polidyspersyjne - cz. Fazy rozproszonej maja różna wielkość
Podział ze względu na to, co rozprasza i jest rozpraszane:
FAZA ROZPRASZAJACA FAZA ROZPROSZONA PRZYKŁADY NAZWA
GAZ Gaz - -
Ciecz Mgły, chmury, dezodorant GAZOZOLE
AEROZOLE
Ciało stałe Kurz, dymy, pyły koloidalne GAZOZOLE
CIECZ Gaz Piany PIANY
Ciecz Żelatyna, mleko, białka, emulsje olejów w wodzie EMULSJE
Ciało stałe Zole Tl. I wodorotl. Metali ZOLE
CIAŁO STAŁE Gaz Pumeks, okluzje gaz. w miner. PIANY ST.
Ciecz Kwarce mleczne, woda okludowała w kryształach EMULSJE STAŁE
Ciało stałe Kryształy, ametyst, rubin ZOLE STAŁE
Podział ze względu na budowę cząstki koloidalnej:
Koloidy fazowe- cząstka koloidalna nie jest cząstka chemiczną fazy rozproszonej (AgCl, Agi)
Koloidy cząsteczkowe- cząstka koloidalna jest cząstką chemiczną fazy rozproszonej (cz. białek, polimerów, hemoglobina)
Koloidy asocjacyjne- powstają przez sąmorzutne skupienie się dużej liczby małych cząstek w agregaty o wymiarze koloidalnym po przekroczeniu pewnego stężenia (asocjaty oleinianu sodowego)
O tym czy dany koloid jest 0, +,- decyduje 1 warstwa adsorpcyjna
Koloid fazowy
Podział w zależności od powinowactwa cząstki fazy rozproszonej do rozpuszczalnika:
- KOLOIDY LIOFILOWE - HYDROFILOWE - dla układów, gdzie ośrodkiem rozpraszającym jest woda, mające duże powinowactwo względem rozpuszczalnika BIAŁKA, Żelatyna, Tanina)
-KOLOIDY LIOFOBOWE - HYDROFOBOWE- dla układów, gdzie ośrodkiem rozpraszającym jest woda, mające małe powinowactwo względem rozpuszczalnika (Zole metali, soli, wodorotlenków)
Właściwość LIOFILOWE LIOFOBOWE
Otrzymywanie Rozpuszczanie Metody dyspersji i kondensącji
Struktura cząstek Makrocząsteczki Przeważnie zespoły zwykłych cząstek
Stężenie fazy rozproszonej Może być duże Nieznaczne
Ruchy Browna Bardzo niewyraźne Występują wyraźnie
Efekt Tyndalla Niewyraźny Wyraźny
Barwa układu Bezbarwne Często barwne
Ładunek elektryczny Brak lub nieznaczny Często/zawsze naładowane
Lepkość Duża Nieznaczna
Tworzenie pian Łatwo tworzą piany Nie tworzą pian
Pęcznienie Pęcznieją znacznie zwiększając objętość Nie pęcznieją
Tworzenie galaret Łatwo tworzą galarety Nie tworzą galaret
Wrażliwość na elektrolity
Mała wrażliwość, koagulacja przy dużych stężeniach elektrolitów Koagulacja pod wpływem małych stężeń elektrolitów
Wrażliwość na odwadnianie Przy dużych stężeniach znaczna Nieznaczna
Charakter koagulacji Odwracalna Nieodwracalna
Właściwości kinetyczne ukł koloidalnych:
Ruchy Browna
Dyfuzja
Sedymentacja
Lepkość
Ciśnienie osmotyczne
![]()
Cząstki koloidalne znajdują się w nieustannym ruchu będącym wynikiem zderzeń cząstek w fazie rozpraszającej
Δx- średnie przesunięcie cząsteczki w czasie Na- licz. Avogadra
r- promień pow. O okrągłym kształcie T- temperatura R- stała gazowa 8,31J/mol*K n-lepkość
Dyfuzja
Ruch cząstek substancji stanowiącej fazę rozproszona pod wpływem różnicy stężeń pomiędzy różnymi częściami układu. Miarą szybkości zachodzenia jest współczynnik zachodzenia dyfuzji, który podaje jaka masą substancji będącej fazą rozproszoną przedyfunduje przez pow. 1cm2 w czasie 1 s pod wpływem gradientu stężenia
Czynnik dyfuzji jest odwrotnie proporcjonalny do lepkości.
![]()
Sedymentacja
W wyniku działania sił ciężkości w układach koloidalnych następuje opadanie cząstek fazy rozproszonej, powodujące w konsekwencji zniszczenie struktury koloidu
![]()
d- gęstość fazy rozproszonej n- lepkość f. rozpraszającej
do- gęstość ośrodka rozpraszającego
Proces opadania można przyspieszyć przez zastosowanie wirowania:
![]()
gdzie: w- prędkość wirówki
x- promień wirówki
g- przyspieszenie ziemskie
Lepkość
W wyniku działania sił kohezji (wzajemnego oddziaływania) między cząsteczkami cieczy występuje przy przemieszczaniu się jednych warstw cieczy względem drugich pewne tarcie wewnętrzne zwany lepkością.
Lepkość koloidu
Jest na ogół większa niż lepkość ośrodka dyspersyjnego, przy czym dla koloidów liofobowych różnice te są nieznaczne, a dla koloidów liofilowych mogą być bardzo duże. Układ, w którym cząstki fazy rozproszonej maja większy ładunek elektryczny, wykazują większą lepkość
Ciśnienie osmotyczne
Ciśnienie, jakie należy przyłożyć od strony roztworu koloidalnego, aby zahamować przechodzenie rozpuszczalnika (fazy rozpraszającej do koloidu). Ciśnienie o. koloidów wykazuje niższe wartości niż dla roztworów rzeczywistych
Właściwości optyczne koloidów:
Rozpraszanie światła
Zmętnienie roztworu
Efekt Tyndalla
to zjawisko fizyczne polegające na rozpraszaniu światła przez koloid z wytworzeniem charakterystycznego stożka świetlnego
Właściwości elektrokinetyczne koloidów:
Elektroosmoza- ruch ośrodka dyspersyjnego pod wpływem pola elektrycznego, wykorzystuje się do odwadniania skór, oczyszczania żelatyny, demineralizacji wody, osuszania budynków (ciecz + przesuwa się do ośrodka -, - do +, kapilara naładowana odwrotnie do ładunku cieczy)
Elektroforeza- ruch naładowanych cząstek koloidalnych pod wpływem pola elektrycznego wzdłuż nieruchomego ośrodka rozpraszającego (wykorzystuje się do rozdzielania białek, witamin, izomerów optycznych, w analityce chemicznej, przy oznaczaniu pestycydów (- do +, + do -, kapilara naładowana zgodnie z ładunkiem cieczy)
Katoforeza- ruch cząstek w kierunku katody
Anoforeza- ruch cząstek w kierunku anody
Potencjał przepływu- potencjał elektryczny uzyskany na elektrodach przy wymuszonym przepływie cieczy (odwrócenie zjawiska elektroosmozy)
METODY OTRZYMYWANIA KOLOIDÓW
Rozdrobnienie mechaniczne koloidalne cząsteczkowe
Dyspersja -> KOLOID <- Kondensącja
Koagulacja <- ROZPAD KOLOIDÓW -> Rozpuszczanie
Metody dyspersyjne
Rozdrobnienie do wielkości rozdrobnienia koloidalnego, osiąga się mechanicznie, elektrycznie, przy pomocy ultradźwięków lub przy użyciu peptyzatorów
Metody kondensącyjne
Maja zapewnić połączenie małych cząstek w zespoły o rozmiarach koloidu. Do osiągnięciu efektu wykorzystuje się reakcje polimeryzacji, redukcji, utleniania, wymiany lub zmniejszenia rozpuszczalności.
Koagulacja
Polega na zmniejszeniu stopnia dyspersji układów koloidalnych, a więc na łączeniu się cząstek fazy rozproszonej w większe zespoły. Dzielimy je na odwracalne (piana, żelatyna) i nieodwracalne (białko kurze, zole Tl. I wodorotl. Metali)
Można je wywołać:
Dodaniem elektrolitu- próg koagulacji - minimalna liczba milimoli elektrolitu potrzebna do skoagulowania 1dm3 r. koloid.
Zmiana temp. - płyn do kąpieli - ciepła woda- pieni się, zimna piana ucieka
Czynnikiem mechanicznym (mieszając, wytrącając- otrz. masła niszczenie struktur k) Dodatkiem nieelektrolitu- wlanie roztworu wodnego do alkoholu
Przepływem prądu, działalnością światła.
Przewodniki I rodzaju (metale i ich stopy)
W których przenoszenie prądu odbywa się poprzez przemieszczanie się elektrolitów tworzących gaz elektroosmozy w strukturze kryształu metalicznego wskutek przyłożonej różnicy potencjałów.
Przewodniki II rodzaju (wodne roztwory elektrolitów lub stopione)
W których nośnikami prądu są obecne w roztworze jony, a przepływ prądu zawsze jest związany z zajściem reakcji RED-OX.
Zawsze góruje przechodzenie z metalu roztworzonego na metal w postaci płytki i na odwrót. W przypadku Zn góruje przechodzenie do roztworu, zatem płytka ładuje się ujemnie. Miedź z roztworu osądza się na płytce/ ładuje się dodatnio. Taki sposób to półogniwa.
Ogniwa galwaniczne
są układami, z których można czerpać energię elektryczną. Ogniwo chemiczne, w którym źródłem prądu są reakcje chemiczne zachodzące między elektrodą, a elektrolitem. Dwie elektrody zanurzone w elektrolicie (półogniwa) tworzą ogniwo galwaniczne
Anoda Elektroda, na której zachodzą procesy utleniania
Katoda Elektroda, na której zachodzą procesy redukcji
METODY OTOCZENIA KOLOIDÓW
Dializa (elektrodializa) Zdolność przechodzenia niektórych cząsteczek i jonów przez błonę półprzepuszczalną, rozdzielającą dwa roztwory; metoda badania składu niektórych roztworów zawiesin, oparta na zdolności przechodzenia niektórych ich składników przez błonę półprzepuszczalną.
Filtracja (ultrafiltracja) metoda oddzielania substancji stałych od cieczy i gazów, poprzez mechaniczne zatrzymanie jednej z substancji (zwykle ciała stałego) w przegrodach porowatych (filtrach) przy użyciu odpowiednich aparatów. Ciecz lub gaz otrzymywane po filtracji nazywa się filtratem.
Ultrafiltracja (filtracja molekularna) - to proces filtracji z użyciem sit molekularnych, membran i wszelkich materiałów porowatych, o porach których rozmiary są zbliżone do wielkości pojedynczych cząsteczek. Ultrafiltracja wymaga zwykle stosowania znacznych ciśnień i jest czasochłonna
CHEMIA FIZYCZNA - Spektroskopia
SPEKTROSKOPIA - nauka o powstawaniu i interpretacji widm, nauka badająca oddziaływania promieniowania elektromagnetycznego z materią
![]()
E- zmiana energii h- stała Plancka v- częstotliwość
c- prędkość światła lambda- dł. Fali
Energia dostarczona atomowi
Zgodnie z zasądą zachowania energii dostarczenie atomowi lub cząsteczce kwantu energii powoduje zmianę energii wew. Atomu (lub cz.) Dostarczona energia może zmieniać różne składowe elementy energii wew. Atomu lub cząsteczki
- e. kinetycznych ruchów postępowych
- e. ruchów obrotowych
- e. drgań oscylacji
- e. elektronów znajdujących się na orbitach atom. I cząst.
- e. związaną z ukierunkowaniem spinu jąder (gdy cząst. SĄ umieszczone w polu magnetycznym)
Metody spektroskopowe:
Spektroskopia jądrowa - wł. Energetyczne jąder atomowych
Spektroskopia atomowa- wł. Energetyczne atomów
Spektroskopia molekularna - poziomy energetyczne i struktury cząsteczek (UV, VIS, IR)
Zakres widma
100-390 UV 390-850 VIS 850-3500 IR
Zakres widma widzialnego
430- 549 niebieskie 560-580 zielone 580-620 pomarańczowe 620-850 czerwone
Pochłonięcie (Absorpcja)
Przez atom lub cząsteczkę kwantu energii powoduje osiągnięcie wyższego poziomu energetycznego. Mówimy, ze atom znajduje się w stanie wzbudzonym.
Atomy niechętnie przebywają w stanie wzbudzonym, wracają więc do swoich stanów podstawowych pozbywając się energii (EMISJA) w postaci promieniowania o określonej długości fali. W zależności od tego, jaki etap procesu (absorpcja czy emisja) stanowi podstawę danej metody spektroskopowej dzielimy na:
Metody emisyjne - o budowie i składzie próbki wnioskuje się na podstawie analizy promieniowania emitowanego przez substancję badaną ( rzadziej stosowne, ponieważ w norm. Temp. Cz. nie emitują promieniowania, a próby ogrzania substancji badanej do temp. Emisji mogą się zakończyć zniszczeniem badanego związku
Metody absorpcyjne- o budowie i składzie próbki wnioskuje się na podstawie analizy promieniowania pochłoniętego z wiązki promieniowania przepuszczonej, przez subst. Badaną
Barwa biała
to najjaśniejsza z barw. Jest to zrównoważona mieszanina barw prostych, która przez człowieka odbierana jest jako najjaśniejsza w otoczeniu odmiana szarości.
Barwa czarna
To najciemniejsza z barw. W teorii oznacza brak jakiegokolwiek promieniowania z zakresu fal świetlnych. W praktyce miejsce tak ciemne, że poprzez kontrast z resztą otoczenia nie możemy określić jego barwy z powodu niedoboru światła tego kierunku.
Barwa związku
Zależy od obecności w nim pewnych grup, tzw. Chromoforów, czyli grup atomów o małych e. Wzbudzenia, ponieważ wzbudzenie elektronów Pi wymaga mniejszej e. Wzbudzenia niż wzbudzenie elektronów Sigma. Chromofory zawierają zwykle wiązania podwójne.
-C=C- -N=N- =C=O -NO2 -C6H4-
Io=Ir+Ia+Ip
Gdzie:
Io=nat. Prom. Padającego na warstwę częściowo przykrytą
Ir- nat. Prom. Rozproszonego
Ia- nat. Prom. Zaabsorbowanego
Ip- nat. Prom. Przechodzącego
I PRAWO ABSORPCJI ŚWIATŁA- prawo Lamberta
Gdzie l= grubość alfa- WSP. Absorpcji
![]()
Przepuszczalność promieniowania monochromatycznego przechodzącego przez jednorodny ośrodek absorbujący maleje wraz ze wzrostem grubości warstwy
II prawo absorpcji światła- prawo Lamberta- Berga
Absorbuancja promieniowania monochromatycznego przechodzącego przez jednorodny ośrodek absorbujący jest wprost proporcjonalna do grubości warstwy i stężenia substancji
c- stężenie roztworu
l- grubość warstwy roztworu
![]()
k-wsp. absorbancji
![]()
A- Absorbancja
III prawo absorpcji światła
A=A1+A2+A3+A4+….An
Absorbancja roztworu wieloskładnikowego, którego składniki absorbują światło przy tej sąmej dł. Fali jest równa sumie absorbancji poszczególnych składników
KRZYWA WZORCOWA
Odchylenia od prawa Lamberta Berga
Obszar poprawnych pomiarów
Odchylenia aparaturowe
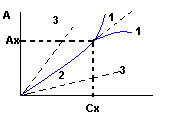
Wyznaczanie zależności wielkości Absorbancji od stężenia substancji
METODY OBLICZENIOWE z prawa Lamberta- Berga
Awz=kwz*Cwz*Lwz i Ax=kx*Cx*Lx
Dla tej sąmej substancji przy jednakowej grubości warstwy mamy:
Kwz=kx Lwz=Lx
![]()
![]()
![]()
15
![]()
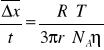
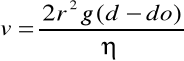
![]()
Wyszukiwarka
Podobne podstrony:
CHEMIA FIZYCZNA- spektrografia sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA-Proces analityczny sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA- koloidy sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA-zanieczyszczenia sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA- Elektrochemia sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA - kinetyka reakcji sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA- srodowisko sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA- spektrografia sc, Ochrona Środowiska pliki uczelniane, Chemia
CHEMIA FIZYCZNA-Proces analityczny sc, Ochrona Środowiska pliki uczelniane, Chemia
alkalodiy, Ochrona Środowiska pliki uczelniane, Chemia
Alkaloidy cz, Ochrona Środowiska pliki uczelniane, Chemia
Alkaloidy c1, Ochrona Środowiska pliki uczelniane, Chemia
genetyka21, Ochrona Środowiska pliki uczelniane, Chemia
Natura 2000 a autostrada A1, Ochrona Środowiska pliki uczelniane, Natura 2000
113MOJA, Ochrona Środowiska pliki uczelniane, Fizyka
Niszczenie drobnoustrojów, Ochrona Środowiska pliki uczelniane, Mikrobiologia
zestawy opracowane eko, Ochrona Środowiska pliki uczelniane, Ekologia lądowa
więcej podobnych podstron