założenia statystyki Maxwella-Boltzmana:
rozmiary cząstek są zaniedbywanie małe w porównaniu z odległościami między nimi
między cząsteczkami nie występują siły przyciągania
cząsteczki poruszają się po torach prostoliniowych, z wyjątkiem momentu zderzenia. Zderzenia cząstek są doskonale sprężyste, tj energia kinetyczna cząstek zostaje zachowana podczas zderzenia, może jednak być przekazywana od jednej cząstki do drugiej.
Prawo rozkładu Maxwella- Boltzmana:
Rozkład Boltzmana opisuje statystyczne rozłożenie składników (cząstki, atomy) między dostępne stany tego układu.
a) ![]()
b) ![]()
c) ![]()
Σi = Σi +/- Σdi
1,2…Ni…N
N(N-1)(N-2)…=N!
18. Równanie rozkładu prędkości cząstek
Średnia energia cząstki jest stała w stałej temperaturze. Rozkład prędkości:
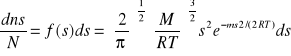
f(s)ds - prawdopodobieństwo, że prędkość cząstki jest zawarta w zakresie (s)-(s+ds.)
N- liczba; M-masa molowa
równanie określające prędkości: najbardziej prawdopodobną, średnią, średnią kwadratową:
najbardziej prawdopodobna: prędkość odpowiadająca maksimum krzywej rozkładu
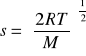
b) średnia kwadratowa: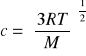
gdzie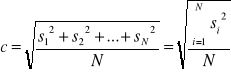
c) średnia: ![]()
współczynnik ściśliwości i rozszerzalności cieplnej:
współczynnik ściśliwości:
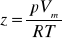
współczynnik rozszerzalności:
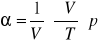
lub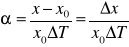
ΔT- przyrost temp; x0 -długość początkowa; x - długość przedmiotu po zmianie temp
Równanie stanu Van der Waalsa:
![]()
p -ciśnienie, a/V2 -poprawka na ciśnienie; V -objętość; b -poprawka na objętość,
T- temperatura; R - stała gazowa
zredukowana postać równania van der Waalsa:
![]()
definicja temperatury krytycznej:
Tkr - Temperatura krytyczna - temperatura, powyżej której gazu nie można skroplić przez sprężarkę; charakterystyczna dla każdego gazu; temp, powyżej której nie istnieje obszar dwufazowy;
równanie 1 zasady termodynamiki:
postuluje istnienie termodynamicznej funkcji stanu (tzw energia wewnętrzna) „U”; cechą charakterystyczną „U” jako funkcji stanu jest to, że w dowolnej przemianie rozpatrywanego układu zmiana jego energii wewnętrznej jest równa sumie ilości energii wymienionej na sposób ciepła i ilości energii wymienionej na sposób pracy: dU=dQ+dW; dla przemiany skończonej ΔU=Q+W
definicje Cv i Cp:
Cp - ciepło molowe w stałym ciśnieniu. Pochodna entalpii układu po temperaturze:
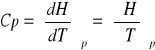
Cv - ciepło molowe w stałej objętości. Pochodna energii wewnętrznej po temperaturze w stałej objętości:
![]()
różnice między Cv a Cp:
Pojemność cieplna pod stałym ciśnieniem Cp różni się od pojemności cieplnej w stałej objętości Cv o wartość pracy potrzebnej do zmiany objętości układu pod stałym ciśnieniem
Równanie określające różnicę Cp-Cv:
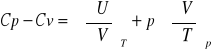
Wykazać, że dla gazu doskonałego 1-atomowego Cp-Cv=R:
![]()
![]()
![]()
entalpia jako termodynamiczna funkcja stanu:
H = U+pV ; dla zmian skończonych: ΔH = ΔU+pΔV
Zmiana entalpii w procesie, równa się ciepłu wymienionemu w układzie pod stałym ciśnieniem. Jest funkcją stanu, której wartości bezwzględnej nie można poznać.
Definicje termodynamicznej funkcji stanu:
Termodynamiczna funkcja stanu - wartość określonej wielkości układu zależy jedynie od stanu tego układu w tym momencie
F- energia swobodna F=U-TS
G- entalpia swobodna G=H-TS
H- entalpia H=U+pV
S- entropia ΔS=nRTln(Vk/Vp)
Q- ciepło Q=ΔU-W
Równanie na różniczkę zupełna entalpii swobodnej: dG=dH-TdS-SdT
określić pochodne cząstkowe (ds./dp)T; (ds./dT)p:
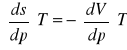
![]()
określić pochodne cząstkowe (ds./dv)T; (ds./dT)V:
![]()
![]()
termodynamiczne równanie stanu na ciśnienie:
![]()
termodynamiczne równanie na objętość:
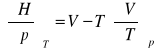
wyprowadzenie równania na entropię mieszania gazów:
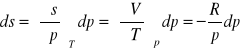
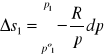
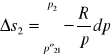
![]()
![]()
![]()
![]()
po1,po2 - przed zmieszaniem; n1,n2 - liczba moli; p1,p2 - ciśnienia cząstkowe, p- ciśnienie całkowite
wyprowadzić równanie określające współczynnik Joule'a - Thompsona:
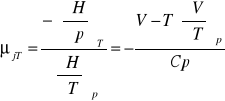
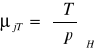
C,T,P H=H(T,P)
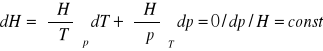
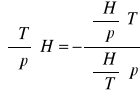
![]()
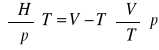
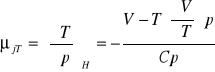
![]()
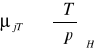
równanie szybkości dla reakcji 1 i 2 rzędu:
1 rz:
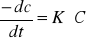
2 rz:
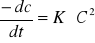
równanie kinetyczne dla reakcji 1 i 2 rzędu:
1 rz:
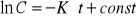
2 rz:
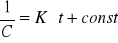
zależność stałej K od temperatury wyrażona równaniem Arheniusa:
![]()
lub ![]()
T- temp; Ea - energia aktywacji; R -stała fazowa; A -czynnik przedwykładniczy
Stała szybkości reakcji zmienia się wraz z temp. Szybkość reakcji wzrasta z temp, gdy Ea jest dodatnia. Im większa Ea, tym większa wrażliwość reakcji na zmiany temp.
podstawowe teorie kinetyczne:
gaz stanowi olbrzymia liczba bardzo małych cząstek, poruszających się szybkim ruchem prostoliniowym i zderzających się ze sobą i ze ściankami naczynia. Ciśnienie wywierane przez gaz na ścianki naczynia jest wynikiem uderzeń cząstek.
Cząstki gazu są doskonale sprężyste, kiedy cząstki się zderzają wymieniają między sobą energie, jednak ich całkowita energia kinetyczna po zderzeniu jest taka sama jak przed zderzeniem;
Cząstki nie oddziałują ze sobą oraz ze ściankami naczynie, jedynie w momencie zderzenia występują siły odpychania między cząstkami;
Objętość zajmowana przez cząstki gazu nie jest istotna w porównaniu z całkowitą objętością. Można przyjąć, że cząstki gazu zajmują nie więcej niż 0,05-0,06% objętości zajmowanej przez fazę gazową;
Średnia energia kinetyczna cząstek jest proporcjonalna do temperatury bezwzględnej gazu. Niezależnie od rodzaju substancji współczynnik proporcjonalności jest taki sam, także średnia energia kinetyczna różnych gazów jest jednakowa w różnej temperaturze: Eśr.kin.=K*T
równanie adiabaty (V,T):
postać różniczkowa: CvdlnT=-RdlnV
postać całkowa:
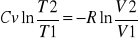
zmiana entalpii swobodnej, gdy przepuszcza się gaz od p1 do p2:
W = W1+W1 = p1V1 - p2V2
ΔU = U2-U1 = W1 +0(dąży do zera) = W= p1V1 - p2V2
U2-U1 = p1V1 - p2V2
U2+p2V2 = U1+p1V1
H1=H2=H=const
równanie określające zależność entalpii reakcji od temperatury:
proces, w którym wzrasta entalpia układu (delta H jest dodatnie) to proces endotermiczny,
proces, w którym entalpia układu maleje (delta H jest ujemne) to proces egzotermiczny
Zmiana entalpii wynikająca ze zmiany temperatury pod stałym ciśnieniem:
![]()
(Cp się nie zmienia)
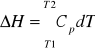
(Cp się zmienia)
definicja termodynamicznej stałej równowagi:
stała równowagi wyrażona za pomocą aktywności (lotności) reagentów nazywa się termodynamiczną stałą równowagi (jest bezwymiarowa):
RTlnK=-ΔrGθ Aa+bB↔cC+dD ![]()
K- stała równowagi; a -aktywność cząstki
cykl Carnota składa się z przemian:
odwracalne rozprężenie izotermiczne od stanu A do stanu B w temperaturze T;
odwracalne rozprężenie adiabatyczne z B do C; ciepło nie opuszcza układu, zmiana entropii jest równa zeru; temperatura spada do temperatury zbiornika;
odwracalne sprężanie izotermiczne w temperaturze zbiornika od C do D, ciepło odpływa;
odwracalne sprężanie adiabatyczne od D do A, ciepło nie wnika do układu, entropia wynosi zero, temperatura rożnie do temperatury układu:
ciepło właściwe i ciepło molowe:
a) ciepło właściwe - energia termiczna potrzebna do podniesienia temperatury jednej jednostki masy ciała o jedną jednostkę temperatury [J/kgK] ![]()
m-masa subst, Q-ciepło dostarczone do układu, T -temp;
b) ciepło mlowe - ilość ciepła potrzebna do ogrzania 1 mola danej substancji o jedną jednostkę temperatury Cm=c*M
c- ciepło właściwe, M-masa molowa substancji
współczynnik Joule'a-Thompsona:
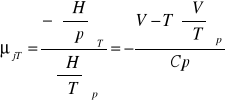
wzór na entropię:
S = kBlnW
najczęściej stosowaną substancja nadkrytyczną jest: woda
przemiany odwracalne:
to proces, który można odwrócić przez infinitezymalną zmianę parametrów układu
dW = -pzewdv = -pdV
potencjał termodynamiczny:
funkcja układu makroskopowego parametrów charakteryzujących stan termodynamiczny układu. Określany jest przez różne zespoły zmiennych: U-energia wewnętrzna, F-energia swobodna Helmholtza, G-entalpia swobodna Gibasa, H-entalpia, μ-potencjał chemiczny
szybkość reakcji
„r,v” - zmiana (powstawanie lub ubytek) ilości reagenta (ΔR) przypadająca na jednostkę czasu (Δt). To szybkość zmiany ilości reagenta: ![]()
nR - współczynnik stechiometryczny dla danego reagenta
okres połowicznej przemiany:
![]()
0 rz: ![]()
1 rz: ![]()
2 rz: ![]()
K -stała szybkości reakcji, C- stężenie substratu po czasie, Co- stężenie początkowe substratu
Standardowa reakcja tworzenia:
Reakcja syntezy 1 mola związku w warunkach standardowych (p=1atm, T=298K) z pierwiastków wziętych w warunkach normalnych. Ilość energii, która zostaje wydzielona na sposób ciepła w trakcie syntezy 1 moja związku z pierwiastkiem
entalpia parowania:
![]()
k- stała określająca straty cieplne w aparacie [J/s]; Q- energia dostarczana do układu [J]; n- liczba moli odparowanej cieczy [mol]; t- czas [s]
entalpia mieszania:
ΔHmiesz = Hroztw - Σni(Hm)i
![]()
![]()
ΔSm - entropia mieszania, n- ułamek molowy składnika, ΔH- entalpia mieszania, G1- entalpia swobodna składnika
entalpia swobodna
entalpia swobodna (energia Gibbsa) G=H-TS (w stałej temp i pod stałym ciśnieniem)
różniczkowa postać równania Clausiussa-Clapeyrona:
![]()
standardowa entalpia tworzenia:
![]()
izobara van't Hoffa:
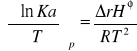
Reakcje następcze:
A( k1) → B (k2) → C (k3) → ….,
dla t=0, cA =a , cB= cC= 0,
dla czasu t: a= CA + CB + CC,
-dcA/dt = k1CA,
-dlncA= k1dt,
lna= const,
czyli: CA= ae-kt,
liczymy dla dcC/dt= k2CB ,
dcB= -dcA/dt,
-dcc/dt=k1CA-k2CB,
cB= (k1a/ k2-k1) e-k1t - e-k2t,
cc= a - CA - CB cc = a - a [ e-k1t + (k1/ k2-k1) e-k1t + e-k2t ].
pojęcia:
chemia fizyczna - dział chemii, której zadaniem jest rozwijanie praw i zależności służących do wyjaśniania zjawisk i obserwacji dotyczących fizycznych i chemicznych właściwości materii. Ważna rola w interpretacji wyników i rozwijaniu nowoczesnych metod badawczych wykorzystywanych do określenia struktury i właściwości substancji, jak nowe materiały syntetyczne czy makrocząsteczki biologiczne
energia - najmniejsza porcja energii wymieniana między układem atmosferycznym a otoczeniem
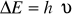
(ΔE - kwant energii, h- stała Plancka, ν- częstość promieniowania)E[kJ/mol]
mol - miara ilości materii, stosowany powszechnie w chemii. 1 mol substancji to taka ilość, która zawiera tyle samo cząstek, co dokładnie 12g izotopu węgla C12.
Siła - w układzie Si wielkością podstawową, którą nazywamy siła jest 1N. 1N to siła, która masie 1kg nadaje przyspieszenie 1m/s2. F=m*a m-masa kg, a- przyspieszenie m/s2
Ciśnienie - jeżeli siła 1N działa na powierzchnię 1m2, wywiera ciśnienie 1Pa
P=F/s=1N/1m2 = 1Pa (p- ciśnienie, s-powierzchnia)
Stan standardowy - p=1bar, T=298,15K
Energia - równa jest pracy, która wykona siła 1N na odległości 1m; 1J=1N*1m; 1cal=4,184J
Elektronowolt - energia, którą uzyska elektron przyspieszony przez różnicę potencjałów
1cV=96,485kJ/mol
Hipoteza Avogadro - jednakowe objętości gazów w tej samej temp i pod takim samym ciśnieniem zawierają jednakową ilość cząsteczki
Prawo Daltona ciśnień cząstkowych - ciśnienie wywierane przez mieszaninę gazu zachowujących się jak gaz doskonały, jest sumą ciśnień jaką wywierałby każdy gaz gdyby sam zajmował całą objętość
Gaz - zbiór cząstek, które oddziałują i przyciągają się z siłą odwrotnie proporcjonalną do odległości F=1/r
Dyfuzja - samorzutne rozprzestrzenianie cząstek jednego gazu do drugiego gazu
Efuzja - wypływ gazu przez bardzo mały otwór;
Reguła stanów - jeżeli dwie substancje w stanie gazowym znajdują się w stanie określonym przez takie same wartości ciśnienia i temperatur zredukowanych, to mają takie same wartości objętości zredukowanej
Układ - część przestrzeni fizycznej, dla której przeprowadzamy analizę termodynamiczną
Układ homogeniczny - układ, w którego każdym punkcie właściwości fizyko-chemiczne są takie same lub zmieniają się w sposób ciągły od punktu do punktu
Układ heterogeniczny - układ, w którym występują dwie lub więcej faz
Faza - część układu heterogenicznego, w której właściwości są takie same w każdym punkcie
Stan układu - jest jednoznacznie określony wtedy, gdy określone są parametry stanu (p,V,T)
Ze względu na wymianę masy z otoczeniem układy dzielimy na: a) izolowane - nie ma wymiany energii i masy; b)zamknięte - nie ma wymiany masy, ale może być wymieniona energia; c) otwarte, które z otoczeniem mogą wymieniać masę i energie
0 zasada termodynamiki - postuluje istnienie wielkości fizycznej nazywanej temperaturą termodynamiczna; cechą charakterystyczna tej wielkości jest to, żę różnica temperatur lub gradient temperatur stanowi siłę napędową procesu przenoszenia ciepła dQ=k*ΔT; dQ=k'(υT/υx)
2 zasada termodynamiki - postuluje istnienie termodynamicznej funkcji stanu nazywanej entropię „S”, której cehcą charakterystyczna jest to, że w dowolnej przemianie rozpatrywanego układu zmiana „S” tego układu to dS≥dQ/T; dQ=TdS
ds.=dQ/T - przemiany odwracalne; ds.>dQ/T - przemiany nieodwracalne
3 zasada termodynamiki - postuluje, że entropia substancji czystych tworzących doskonałą fazę krystaliczną dąży do zera, gdy jej temperatura dąży do zera: limS(st kryst dosk)=0
Temperatura bezwzględna - parametry termodynamiczne oparte na tzw bezwzględnej skali temperatury (T). Bezwzględna skala temp oparta jest na zależności między zmiennymi (p,V,T) gazu doskonałego, która określona jest równaniem stanu gazowego: pV=RT
Punkt potrójny - stan w jakim dana substancja może istnieć w trzech stanach skupienia równocześnie w równowadze termodynamicznej
Kryteria samorzutności przemian - a) gdy następuje samorzutna przemiana przy stałej objętości i temperaturze, to energia swobodna układu F maleje i dąży do osiągnięcia minimum; b) jeżeli w układzie następuje samorzutna przemiana przy stałej temperaturze i ciśnieniu, to entalpia swobodna G maleje.
Kinetyka chemiczna - jak szybko układ reakcyjny będzie dążył do równowagi
wielkości:
stała Plancka h - 6,626*10-34J/s
ciśnienia: 1atm = 101325N*m-2 = 101325Pa
1atm = 101,325kPa
pө=1*10-5Pa=1bar
1atm=1,01325bar
1Tor=1/760atm=1mmHg
stała Avogadro NA=6,022*1023mol-1
1eV=96,485kJ/mol
Stała gazowa - R=8,31451J*K-1*mol-1
przyspieszenie ziemskie g=9,81m*s-1
cząstkowe prawa gazowe:
Boy'la -Mariotta [n,T] => bv=const
Jeżeli przyjmujemy, że liczba moli I temp są wielkościami stałymi, to iloczyn ciśnienia i objętości jest stały
Gay-Lucassa [n,p]=>V~T
Charlesa [n,V]=>p~T
„~“ proporcjoalne
prawo Daltona:
![]()
równania wirialne - równania stanu gazu:
a)względem ciśnienia: ![]()
b) względem odwrotności objętości ![]()
równania:
a) Berthelota: 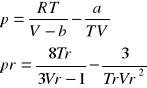
Deterici:
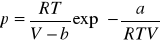
c) Reni Redlicka-Kwouga: ![]()
różniczki zupełne:
dU=TdS-pdV
dH=TdS+Vdp
dG=SdT+Vdp
dF=-Sdt-pdV
różniczki zupełne formalne:
![]()
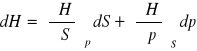
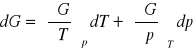
![]()
pochodne magisterskie:
![]()
![]()
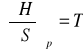
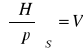
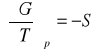
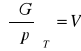
![]()
![]()
równania adiabaty:
(T,V)
Różniczkowa: ![]()
całkowa: ![]()
(T,p)
Różniczkowa: ![]()
Całkowa: ![]()
(p,V)
Całkowa: ![]()
Kappa:
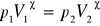
pochodne Maxwella, równania stanu na ciśnienie i objętość:
równanie stanu na objętość:
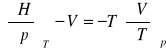
pochodna Maxwella:
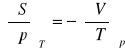
równanie stanu na ciśnienie:
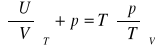
postulat Maxwella:
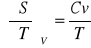
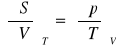
współczynnik Joule'a:
![]()
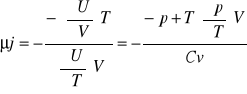
Równanie Clauciussa-Clapeyrona dla równowag:
sublimacji:
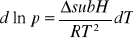
krzywej topnienia:
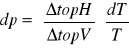
równanie Kirchoffa:
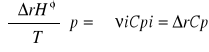
potencjał chemiczny wg Gobisa:
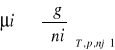
4. izoterma van't Hoffa określona równaniem:
![]()
równanie Clausiussa dla równowagi ciecz-para:
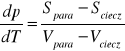

całkowa postać równania Clausiussa-Clapeyrona:
![]()
Wyszukiwarka
Podobne podstrony:
ZAKAAD CHEMI FIZYCZNEJ, Studia, Politechnika
ZAKAAD CHEMI FIZYCZNEJ, Studia, Politechnika
103, Studia Politechnika Poznańska, Semestr II, I pracownia fizyczna, LABORKI WSZYSTKIE, FIZYKA 2, F
303 aga303, Studia Politechnika Poznańska, Semestr II, I pracownia fizyczna, LABORKI WSZYSTKIE
Chemia fizyczn - nr 10, Studia, Politechnika
CHEMIA FIZYCZNa v 2 1SCIAGA, POLITECHNIKA ŚLĄSKA Wydział Mechaniczny-Technologiczny - MiBM POLSL, Se
Półprzewodniki, Studia Politechnika Poznańska, Semestr II, I pracownia fizyczna
105A, Studia Politechnika Poznańska, Semestr II, I pracownia fizyczna, LABORKI WSZYSTKIE, FIZYKA 2,
301 Aga203q, Studia Politechnika Poznańska, Semestr II, I pracownia fizyczna, LABORKI WSZYSTKIE
Chemia fizyczna - nr 22, Studia, Politechnika
302 abulec, Studia Politechnika Poznańska, Semestr II, I pracownia fizyczna, LABORKI WSZYSTKIE, FIZY
Chemia fizyczna - nr 21, Studia, Politechnika
201 sprawozdanie-fizyka, Studia Politechnika Poznańska, Semestr II, I pracownia fizyczna, LABORKI WS
więcej podobnych podstron