Dostępność farmaceutyczna- Ilość substancji leczniczej uwalniającej się z postaci farmaceutycznej (postaci leku) i rozpuszczającej się w otaczającym płynie oraz szybkość z jaką ten proces zachodzi warunki IN VITRO
Dostępność biologiczna
Szybkość i stopień wchłaniania substancji leczniczej lub czynnego farmaceutycznie ugrupowania z postaci farmaceutycznej (postaci leku) do krążenia ogólnego
Parametry
AUC- pole powierzchni pod krzywą zależności c= f(t)- ułamek dawki wchłoniętej do organizmu
Cmax- maksymalne stężenie leku
tmax- czas po jakim osiągnięte jest cmax warunki IN VIVO
Dostępność biologiczna bezwzględna- stosunek dostępności biologicznej po pozanaczyniowym podaniu leku do dostępności biologicznej po podaniu dożylnym- procentowy ułamek wartości AUC.
Dostępność biologiczna względna- stosunek dostępności biologicznej dwóch leków podawanych inną drogą niż dożylna
Cele badania dostępności farmaceutycznej
Badanie jakościowe leku (Serie produkcyjne, Przechowywanie)
Badania biorównoważności (Podobieństwa profili uwalniania)
Narzędzie w pracach rozwojowych nad postacią leku (Symulacja warunków In vivo i korelacja IV/IVC (In vitro/ In vivo correlation)
Dostępność farmaceutyczna
Uwalnianie substancji aktywnej (API)
Kinetyka 0 rzędu (ZOK)
Stała szybkości uwalniania z postaci leku, która nie ulega rozpadowi
![]()
W0- początkowe stężenie API w preparacie
Wt- stężenie API po czasie t
K- stała szybkości
Kinetyka I rzędu
Równanie Noyes- Whitney
![]()
![]()
dc/dt- szybkość rozpuszczania API
C- ilość API rozpuszczona w czasie t
CS- rozpuszczalność API w stanie równowagi w temperaturze badania
K i K1- stałe szybkości
S- powierzchnia API dostępna dla substancji aktywnej
Równanie Noyes- Whitney- model kinetyki uwalniania substancji (dr) aktywnej z postaci leku
![]()
Powierzchnia rozpuszczania
D- współczynnik dyfuzji
h- grubość warstwy dyfuzyjnej (droga dyfuzji)
CS- stężenie roztworu nasyconego (w warunkach testu)
V- objętość rozpuszczalnika
Xd- ilość rozpuszczonego leku
Ct- stężenie w roztworze po czasie t Xd/V
Czynniki wpływające na szybkość rozpuszczania substancji leczniczej
Parametr |
Czynniki fizykochemiczne |
Czynniki fizjologiczne |
A |
Wielkość cząstek Zwilżalność |
Obecność surfaktantów w przewodzie pokarmowym |
D |
|
Lepkość płynów w przewodzie pokarmowym |
H |
Lepkość płynu Wielkość cząstek |
|
CS |
Rozpuszczalność Struktura krystaliczna Stała jonizacji |
|
Xd |
|
Przenikalność |
V |
|
Stan fizjologiczny przewodu pokarmowego |
Czynniki wpływające na zwiększenie dostępności farmaceutycznej:
Modyfikacje chemiczne i przeprowadzenie związku do łatwiej rozpuszczalnych pochodnych
Dobór odpowiedniej odmiany krystalograficznej
Maksymalne rozdrobnienie substancji trudno rozpuszczalnych
Substancje zmniejszające napięcie powierzchniowe
Buforowanie roztworu i „cofanie” dysocjacji leku
Zmniejszenie lepkości środowiska
Zwiększenie powierzchni leku na syntetycznych nośnikach
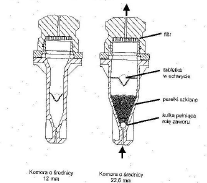
Aparat łopatkowy (No 2)
Badana postać leku- tabletki
Szybkość mieszania: 50-100 obr/ min (nie więcej niż 150 obr/ min)
Pojemność naczynia: 500- 1000 ml
Temperatura: 37 +/- 0,50C
Aparat koszyczkowy (No 1)
Badana postać leku- tabletki flotacyjne i wolno rozpadające, kapsułki
Szybkość mieszania: 100 obr/ min
Pojemność naczynia: 500- 1000 ml
Zalety aparatu przepływowego
Łatwość zmiany pH płynu do uwalnianiaUzyskanie efektu szybszego i lepszego zwilżania postaci leku bez względu na jej zdolność do flotacji
Możliwość badania uwalniania substancji ulegających rozkładowi
Możliwość uzyskania do analizy roztworów bardziej stężonych
Większa powtarzalność wyników
Wady aparatu przepływowego
Problemy techniczne (duża liczba elementów)
Duża ilość płynu stosowanego do uwalniania
Preparaty zawierające substancje trudno rozpuszczalne w wodzie
Zastosowanie jako płynu do uwalniania rozpuszczalników organicznych (etanol) lub substancji powierzchniowo czynnych (30% propanol dla kortyzonu, 0,1% Laurylosiarczan .Na dla spironolaktonu)
Zastosowanie komory przepływowej i porównanie wyników z metodą łopatkową
Zastosowanie układu dwóch nie mieszających się cieczy (bufor i rozpuszczalnik organiczny) w aparacie łopatkowym
Płyny do uwalniania w badaniach uwalniania z doustnych postaci leku
Podstawowe: woda, 0,1 i 0,01 mol/ l HCl, bufor fosforanowy (pH 6,8)
Rzadziej stosowane: bufor fosforanowy (pH 7,6), kwas octowy, kwas winowy, etanol, płyny z LSS lub enzymami
Płyny do uwalniania w badaniach uwalniania z doustnych postaci leku (kapsułki)- płyn żołądkowy pH 1,2; płyn jelitowy pH 7,5, płyn żołądkowy i jelitowy „po posiłku”
Różnica między badaniem rozpuszczania a badaniem uwalniania wg USP
Badanie rozpuszczania- dla substancji szybko uwalniających
Badanie uwalniania- dla substancji o modyfikowanym uwalnianiu
Q- wymagana ilość uwolnionej substancji czynnej wyrażona jako % zawartości deklarowanej (5%, 15%, 25%); wartości procentowe odniesione do wartości deklarowanej. Q- 75% w czasie 45 minut
Modyfikowana szybkość uwalniania
Podział ze względu na kinetykę
Zerowego rzędu
Pierwszego rzędu
Modyfikowane uwalnianie
Kapsułki
Peletki/ granulki
Tabletki/ peletki flotacyjne
Zerowy rząd
Tabletki, kapsułki:
Dojelitowe o przedłużonym uwalnianiu
Dojelitowe (enteric coated; delayed release)
O szybkim i przedłużonym uwalnianiu
O kontrolowanym uwalnianiu (controlled release)
Bardzo szybko uwalniające
O przedłużonym uwalnianiu (prolonged release)
Inne: np. uwalniające w okrężnicy lub uwalniające w sposób przedłużony w żołądku
Plastry
Szybko uwalniające- substancja uwalniana jest i rozpuszczana w płynie ustrojowym, a następnie wchłaniania do krążenia ogólnego. We krwi występuje w postaci wolnej lub związanej z białkami. API ulega rozmieszczeniu w poszczególnych tkankach a następnie metabolizmowi i eliminowana jest z moczem, kałem, potem i wydychanym powietrzem.
System OROS
Różnice w kinetyce 0 i I rzędu
Warunki uwalniania API z podłoży czopkowych
Stopieniu podłoża czopkowego (podłoża lipofilowe)
Rozpuszczeniu podłoża czopkowego w płynie odbytniczym (czopki hydrofilowe)
API z czopków omija efekt pierwszego przejścia
Czynniki, od których zależy uwalnianie i wchłanianie API z czopków
Właściwości samej substancji leczniczej
Współczynnik podziału o/w
Stopień rozdrobnienia
Rodzaju podłoża czopkowego
Lipofilowe (olej kakaowy, półsyntetyczne glicerydy kwasów tluszczowych)
Hydrofilowe (mieszaniny makrogoli o różnej masie cząsteczkowej, masy glicerynowo- żelatynowe)
Rodzaju substancji pomocniczych (związki powierzchniowo czynne, modyfikujące lepkość i temperaturę topnienia, środki konserwujące, substancje poślizgowe, wypełniające i adsorpcyjne, barwniki)
Stanu fizjologicznego miejsca podania
Metody badania uwalniania API z czopków
Czynniki sprzyjające uwalnianiu API z maści
Czynniki wpływające na wchłanianie API z maści
Wchłanianie przez skórę
Wytyczne do prowadzenia badań dostępności farm. Maści
Komora dyfuzyjna (aparat łopatkowy z komorą ekstrakcyjną, komora dyfuzyjna Franza, komora Mutimera, komora Vankela)
Odpowiednia błona syntetyczna
Nie może wpływać na szybkość uwalniania
Powinna utrzymywać preparat w pierwotnym miejscu
Nie może wykazywać absorpcji lub innej interakcji
Zabezpiecza przed uwalnianiem z podłoża do płynu akceptorowego substancji pomocniczych (polimerów)
Zapobiega zmianom powierzchni uwalniania
Odpowiednia faza receptora
Woda, bufor o pH 5-8; izotoniczny roztwór NaCl
Zachowane warunki SINK
Temperatura 370C lub 320C
Substancje trudno rozpuszczalne- dodatek rozpuszczalników lub substancji powierzchniowo czynnych
Co najmniej 5 punktów czasowych w ciągu co najmniej 6 h- określenie kinetyki uwalniania (pobieranie płynu akceptorowego w odpowiednim czasie, szybkość uwalniania wyrażona w μg/ cm2, zależność powinna być prostoliniowa, miarą szybkości uwalniania- nachylenie prostej)
Aparaty i komory dyfuzyjne do badania uwalniania maści
Aparat łopatkowy z komorą ekstrakcyjną
Błony dyfuzyjne
Komora dyfuzyjna Franza
Objętość płynu: 3-3,5 ml
Możliwość badania w warunkach okluzyjnych lub w modelu bez okluzji (odparowanie rozpuszczalnika)
Badania z zastosowaniem dawki nieskończonej i skończonej
Możliwość automatycznego systemu pobierania próbek
Komora Mutimera- duża powierzchnia ok. 100 cm2, konieczna duża ilość maści, nie zawiera elementu mieszającego płyn
Enhancer cell (komora Vankela)
Objętość płynu do uwalniania: 100- 200 ml
Powierzchnia uwalniania: 0,5- 4 cm2
Wady
Duża powierzchnia (ok. 100 cm2)
Konieczna duża ilość maści
Nie zawiera elementu mieszającego płyn akceptorowy
Na czym polega graficzna metoda wyznaczania rzędowości reakcji?
Kryteria systemu klasyfikacji BCS
Rozpuszczalność
Dobrze rozpuszczalne- największa dawka API zawarta w preparacie o natychmiastowym uwalnianiu rozpuszczalna w ≤ 250 ml wody w zakresie pH 1-7,5 (8,0) w temperaturze 37+/- 10C.
Dobór pH zależy od właściwości API, np. jeżeli pKa API 1-3, to badania rozpuszczalności
W roztworze o pH= pKa i w roztworze o pH= pKa +/- 1
W roztworze o pH max i min
Uwalnianie- Szybko uwalniające- ≥85% dawki leku uwolnione w czasie 30 minut w 3 mediach: pH 2,0 (HCl lub SGF- simulated gastrin fluid), pH 4,5 i pH 6,8. Aparat I przy 100 rpm lub aparat II przy 50 rpm. Objętość roztworu buforującego ≤900 ml
Przenikalność- Dobrze przenikalny- zdolność przenikania API przez błonę jelita co najmniej 90% najwyższej dawki API przy braku udokumentowanych danych na temat jej niestabilności w przewodzie pokarmowym
Jak zdefiniować klasę przenikalności BCS
Badania absorpcji (badanie substancji modelowych)
Farmakokinetyka badania biorównoważności
Badania absorpcji dostępności biologicznej
Metoda perfuzji jelitowej
Metody oznaczania przenikalności na poziomie przewodu pokarmowego
In vivo lub In situ ocena jelitowej perfuzji u zwierząt doświadczalnych
In vitro- badanie przenikalności z użyciem ludzkich lub zwierzęcych tkanek jelitowych oraz z użyciem jednowarstwowych kultur komórek nabłonka
Z badań biorównoważności mogą być zwolnione
Leki I klasy- Mogą być zwolnione z badań biorównoważności (In vitro) w przypadku wykazania natychmiastowego uwalniania- 85% dawki leku jest uwalnianie w ciągu 30 minut w 3 standardowych mediach
Leki II klasy- Mogą być zwolnione w przypadku wykazania podobieństwa profili uwalniania pomiędzy produktem badanym a lekiem referencyjnym
Badanie biorównoważności polega na
Ocenie właściwości leku generycznego i oryginalnego- referencyjnego
Stwierdzeniu biorównoważności dwóch produktów farmaceutycznych (wykazanie równoważności farmaceutycznej, wykazanie podobnego zakresu dostępności biologicznej dla tej samej wartości dawki leku)
Parametry używane do korelacji wyników badań ivivc
Korelacja parametrów ivivc polega na
Poziomy korelacji parametrów ivivc
Poziom A- profil uwalniania API i zmiany poziomu leku we krwi są porównywalne we wszystkich punktach czasowych
Bezpośrednie porównanie profilu uwalniania i krzywej stężenia API we krwi po podaniu preparatów o różnych szybkościach uwalniania
Dekonwulacja wyników z badań In vivo w oparciu o model matematyczny (estymacja wchłaniania lub uwalniania API po podaniu In vivo)
Poziom B- analiza momentów statystycznych, np. porównanie średniego czasu uwalniania w warunkach In vitro ze średnim okresem trwania API w organizmie. Brak możliwości uzasadnienia wniosku o odstępstwie od badań równoważności biologicznej ze względu na niemożność odtworzenia zmian stężenia API we krwi.
Poziom C- badanie pojedynczych parametrów In vitro (t50%, t90%) i parametrów farmakokinetycznych (AUC, cmax, tmax); wstępna faza prac rozwojowych
Poziom C poszerzony- powiązanie jednego lub kilku parametrów farmakokinetycznych z parametrami dostępności farmaceutycznej dla kilku punktów czasowych; możliwość uzasadnienia wniosku o odstępstwie badań równoważności biologicznej podobnie jak dla poziomu A.
Znaczenie BCS w przewidywaniu korelacji parametrów i w badaniach biorównoważności
Zalety i ograniczenia korelacji wyników
Biowaiver- co to jest i jakie info w monografii
Właściwości fizykochemiczne: rozpuszczalność w 370C w pH 1,2- 6,8, pKa, logP, polimorfizm, solwaty i sole, dodatkowo badania rozpuszczalności i uwalniania z czystym API
Oznaczenie przenikalności np. wydalanie z moczem, Caco- 2 badania
Przegląd piśmiennictwa na temat biorównoważności istniejących produktów
Interakcje z żywnością i substancjami pomocniczymi
Dane z piśmiennictwa i laboratoryjne porównujące uwalnianie istniejących produktów
Wskazania terapeutyczne, indeks terapeutyczny, typ i ważkość obserwowanych efektów toksycznych
Metody służące do porównywania profili uwalniania
Metody statystyczne- Analiza wariancji, test t- Studenta- w odniesieniu do jednopunktowego badania uwalniania substancji leczniczej oraz wielopunktowych profili uwalniania
Metody statystyczne niezależne od modelu- Metody analizy wielowymiarowej (opartej na odległości Mahalanobisa); Metoda współczynników podobieństwa i różnicy
Metody statystyczne zależne od modelu, np. Metoda funkcji Weibulla
poziom |
Liczba badanych jednostek |
Kryterium akceptacji |
S1 |
6 |
Każda jednostka nie mniej niż Q+ 5% |
S2 |
6 |
12 jednostek (S1+S2)- średnia wartość nie mniej niż Q, wszystkie nie mniej niż Q- 15% |
S3 |
12 |
24 jednostki- średnia wartość nie mniej niż Q, najwyżej 2 jednostki mniej niż Q-15%, wszystkie nie mniej niż Q-25% |
Postać leku |
Liczba punktów czasowych |
charakterystyka |
Przedłużone |
3 lub więcej |
1 pkt- uwolnienie 20-30% 2pkt- 50% 3 pkt- więcej niż 80% |
opóźnione |
indywidualnie |
Badanie przy zwiększających się wartościach pH Postacie dojelitowe- co najmniej 2 pkt czasowe (w badaniu czasowym- po 1 lub 2 h pH kwaśne, po czasie zalecanym pH 6,8) Q>=75% jeśli nie określono inaczej |
Uwalnianie zgodnie z kinetyką zerowego rzędu zachodzi gdy substancja lecznicza uwalnia się z postaci leku ze stałą szybkością. Uwalnianie zachodzi z postaci leku, która nie ulega rozpadowi, a podczas uwalniania nie zmienia się pole powierzchni preparatu.
Modyfikowane uwalnianie- uwalnianie jest modyfikowane technologicznie przez spowolnienie uwalniania (lub przyśpieszenie) tak, że zachodzi na całej długości przewodu pokarmowego, w ciągu kilku- kilkunastu godzin. Właściwe stężenie leku we krwi przez dłuższy czas można uzyskać pod warunkiem, że w tabletce zawarta jest odpowiednio duża dawka substancji leczniczej niż w formie szybko uwalniającej.
Dozowanie leku następuje na skutek wytworzenia wysokiego ciśnienia osmotycznego, powodującego wypychanie roztworu substancji leczniczej ze zbiornika w kształcie tabletki powlekanej z otworem w otoczce tabletek
Uwalnianie zgodnie z kinetyką zerowego rzędu zachodzi gdy substancja lecznicza uwalnia się z postaci leku ze stałą szybkością. Uwalnianie zachodzi z postaci leku, która nie ulega rozpadowi, a podczas uwalniania nie zmienia się pole powierzchni preparatu.
Uwalnianie zgodnie z kinetyką I rzędu opisuje równanie N-W, dotyczy leków o niemodyfikowanej szybkości uwalniania.
Działanie miejscowe substancji leczniczej- Dopochwowe, do cewki moczowej, do ucha, do zęba
Działanie ogólne- Doodbytnicze
Rozpuszczenie substancji leczniczej w środowisku odbytnicy po:
Uwolnienie- Rozpuszczenie substancji leczniczej w płynie odbytniczym (pH 7,6- 8,0)
API dostaje się bezpośrednio do krwioobiegu przez nabłonek odbytnicy. Rozchodzi się po całym organizmie, ponieważ naczynia żylne są połączone z krążeniem dużym. Do wątroby trafia ułamek dawki leku wraz z krążeniem wrotnym wątroby. Ponadto substancja lecznicza nie jest rozkładana w wątrobie, przez co dawki leku mogą być mniejsze niż dawki leku doustnego. Dodatkowo działanie jest szybsze ze względu na bogate unaczynienie odbytnicy. Tą drogą można stosować leki o działaniu drażniącym na przewód pokarmowy lub nietrwałe w kwaśnym pH soku żołądkowego.
Aparat łopatkowy z siatką i spiralą
Aparat przepływowy FP
Aparat przepływowy K-H
Temperatura 37/ 32
pH 5-7
rozpuszczalność API w podłożu i płynie akceptorowym
współczynnik podziału /w
lepkość podłoża, hydrofobowość, reologia
hydrofilowe API z podłoży hydrofilowych
wielkość cząstek
Wielkość cząstek API
Właściwości podłoża i leku
Sposób aplikacji
Stan skóry
Grubość warstwy rogowej
Droga transfolikularna- przez przydatki skóry (mieszki włosowe, gruczoły), 1000 razy szybciej niż transepidermalnie, ale mały udział
Droga Transepidermalna- przenikanie przez naskórek z warstwą rogową. Wiele substancji stosowanych na skórę zwiera API wywierające działanie farmakologiczne w naskórku. Leki muszą przejść barierę lipidową, zawierającą przestrzenie międzykomórkowe, dzięki którym możliwy jest transport bierny przez skórę.
Polega na wykreśleniu krzywej zależności stężenia uwolnionej substancji leczniczej z postaci leku (100-AR%) od czasu.
Jeśli krzywa ma przebieg prostoliniowy na skali półlogarytmicznej- I rząd
Gdy krzywa ma przebieg liniowy na skali liniowej to jest to kinetyka 0 rzędu.
AUC, tmax, Cmax, t0,5
To substancje, które powinny charakteryzować się szybkim uwalnianiem, wysoką rozpuszczalnością, wysoką przenikalnością, szerokim indeksem terapeutycznym. Monografia biowaivera powinna uwzględniać następujące informacje
Metoda współczynników podobieństwa i różnicy
Parametry kwalifikacji aparatu do badania uwalniania
Uwzględnienie wymiarów i granic tolerancji przewidzianych dla danego aparatu
Kontrole pracy aparatu- okresowe lub ciągłe
Badanie produktu referencyjnego wrażliwego na zmianę warunków hydrodynamicznych (tabletki a ASA, prednizonem)
Udział z badaniach porównawczych międzylaboratoryjnych.
Cele badania dostępności farm z półstałych postaci leku
Warunki niezbędne do porównania profili uwalniania z maści
Pobieranie płynu akceptorowego w odpowiednim czasie (5 pkt czasowych)
Najwłaściwsza prezentacja wyników: szybkość uwalniania [μg/cm2]=f(
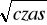
)Zależność powinna być prostoliniowa
Miarą szybkości uwalniania - nachylenie prostej (im większy kąt tym większa szybkość uwalniania)
Warunki niezbędne do porównania profili uwalniania API z tabletek
Preparat porównywany- co najmniej 12 profili uwalniania
Identyczne warunki prowadzenia badań (ten sam czas pobierania próbek)
Tylko w 1 punkcie czasowym ilość uwolnionej API może być większa od 85%
Współczynnik zmienności
≤20% w początkowej fazie uwalniania
≤10% w późniejszych fazach
Zbyt duża zmienność wyników- metoda bootstrappingu w celu obliczenia przedziału ufności
Brak konieczności matematycznego porównywania profili uwalniania- 85% API uwolnionej w czasie < 15 minut
Wyszukiwarka
Podobne podstrony:
OPRACOWANE ZAGADNIENIA NA 2013, Budownictwo, wytrzymałość materiałów, WYTRZYMALOSC POPRAWKA
Zagadnienia Kryminologia - Zagadnienia z opracowaniem, Sudia - Bezpieczeństwo Wewnętrzne, Semestr II
temp krytyczna, TRANSPORT PWR, STUDIA, SEMESTR II, FIZYKA, fizyka-wyklad, zagadnienia opracowane, za
zagadnienia opracowane przeze mnie
gramatyka opisowa zagadnienia opracowane (morfologia, fleksja, składnia)(1)
102147-zagadnienia-egzminacyjne-2013, administracja UMCS, 2 rok
Zagadnieniaa opracowane
zagadnienia opracowane na kolokwium nr3 (marynaty, soki)
zagadnienia opracowane panstwo
Fleksja zagadnienia, opracowania, pomoc 2
I kolokiwum zagadnienia opracowane
NEUROFIZJOLOGIA ćw. 1 - zagadnienia opracowane, Dietetyka CM UMK, Fizjologia
zagadnienia opracowywane, Praca socjalna UMK, andragogika
Tob zagadnienia opracowane, AGH Imir materiały mix, Studia
polityka społ zagadnienia - opracowanie, Dokumenty- PRACA SOCJALNA, Polityka Społeczna
zagadnienia opracowane ZP-1, Zamówienia publiczne UEK
ZAGADNIEnia Opracowane
3 zagadnienia opracowanie Patki
więcej podobnych podstron