Układ odpornościowy
Układ odpornościowy stanowi 1/10 masy ciała. W jego skład wchodzi śledziona, grasica, węzły chłonne, szpik, migdałki, kępki Payera, wątroba.
Bariery immunologiczne
fizykochemiczna - skóra, śluzówki, pot, wydzieliny gruczołów, pH płynów ustrojowych
komórkowa
fagocyty - komórki obdarzone właściwościami żernymi. Można podzielić je na dwie grupy:
fagocyty krążące - granulocyty (obojętno, kwaso i zasadochłonne), monocyty
fagocyty osiadłe w tkankach - makrofagi, komórki Kupffera w wątrobie, komórki mikrogleju
limfocyty B, T
humoralna - immunoglobuliny, opsoniny, układ dopełniacza (grupa termolabilnych białek osocza biorąca udział w chemotaksji, fagocytozie i hemolizie).
Wstrząs septyczny - patofizjologia i leczenie
Definicja wstrząsu septycznego
Infekcja - zjawisko mikrobiologiczne charakteryzujące się odpowiedzią zapalną na obecność mikroorganizmów w normalnie sterylnym organizmie gospodarza.
Bakteriemia - jest to stan w którym stwierdza się obecność bakterii we krwi, przy czym nie muszą być obecne kliniczne wykładniki zakażenia (~ wiremia, fungemia).
SIRS (Systemic Inflammatory Response Syndrome) - zespół uogólnionej odpowiedzi zapalnej do której dochodzi w przebiegu różnych stanów klinicznych, takich jak infekcja zapalenie trzustki, niedotlenienie, uraz wielonarządowy, uszkodzenie tkanek, wstrząs krwotoczny i inne. SIRS przejawia się dwoma lub więcej z wymienionych:
temperatura > 38OC lub < 36OC
częstość akcji serca > 90/min
częstość oddechów > 20/min
ciśnienie parcjalne CO2 we krwi tętniczej poniżej 32 mmHg
leukocytoza > 12 000 lub < 4 000
Sepsa (posocznica) - ogólnoustrojowa odpowiedź zapalna na zakażenie. Rozpoznanie sepsy wymaga stwierdzenia co najmniej dwóch kryteriów SIRS oraz zakażenia.
Ciężka sepsa in. zespół septyczny (sepsis syndrome) - posocznica do której dołączyły się objawy kliniczne wywołane niedostateczną perfuzją narządową, np. kwasica mleczanowa, oliguria, zaburzenia psychiczne
Wstrząs septyczny - zespół septyczny oraz zaburzenia krążenia (ciśnienie skurczowe poniżej 90 mmHg lub spadek większy niż 40 mmHg)
Etiologia
Za wystąpienie wstrząsu septycznego odpowiedzialne mogą być różne patogeny, w tym bakterie G(+) i G(-), wirusy, riketsje i grzyby. Do najczęściej wywołujących wstrząs należały dotychczas bakterie G(-), a w szczególności Escherichia coli, Klebsiella i Pseudomonas aeruginosa. Ogniska zakażenia znajdowano zwykle w układzie moczowo - płciowym, przewodzie pokarmowym i płucach. W ostatnich latach wzrasta jednak znaczenie bakterii G(+) w etiologii wstrząsu septycznego, przy czym podkreśla się wyższą śmiertelność, niż w przypadku bakterii G(-). Do G(+) bakterii najczęściej spotykanych we wstrząsie należą Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecium, a pierwotnym źródłem zakażenia okazują się rany skóry, zakażone tkanki miękkie oraz cewniki donaczyniowe.
Do przyczyn wzrastającej częstości zakażeń bakteriami G(+) należy zaliczyć:
szpitalne reżimy antybakteryjne kierowane z zasady przeciwko bakteriom G(-) pozwoliły na wyselekcjonowanie opornych na antybiotyki bakterii G(+),
długotrwałe utrzymywanie cewników naczyniowych,
szerokie stosowanie różnorodnych implantów chirurgicznych (sztuczne stawy, zastawki, protezy naczyniowe),
gwałtowne szerzenie się antybiotykooporności wśród bakterii G(+).
Do czynników ułatwiających wystąpienie wstrząsu septycznego należą:
wiek (wstrząs pojawia się częściej u noworodków i niemowląt oraz u chorych w wieku podeszłym),
współistnienie takich chorób jak: nowotwory, cukrzyca, niewydolność nerek lub wątroby, AIDS,
stosowanie niektórych leków: kortykoidy, leki podwyższające pH treści żołądkowej,
intensywne zabiegi diagnostyczne i lecznicze: cewnikowanie pęcherza, długotrwałe utrzymywanie linii tętniczych i żylnych, intubacja dotchawicza, wentylacja mechaniczna, całkowite żywienie parenteralne
Wieloletnie badania potwierdzają decydującą rolę mikroorganizmów w rozwoju wstrząsu septycznego. Coraz częściej jednak podkreśla się znaczącą rolę zaburzeń immunologicznych (patologiczna reakcja na zakażenie), co w obserwacjach klinicznych wyraża się słabą korelacją pomiędzy rozległością zakażenia, a prawdopodobieństwem wystąpienia wstrząsu.
Mediatory wstrząsu septycznego
Dzieli się je na egzogenne (tj. produkowane przez mikroorganizmy) oraz endogenne (wytwarzane przez zaatakowany ustrój). Z mediatorów egzogennych najlepiej poznano endotoksynę (lipopolisacharyd związany ze ścianą komórki bakterii gram (-) i uwalniany z chwilą jej rozpadu) oraz uwalniane przez bakterie gram (+) egzotoksyny (np. gronkowcowa enterotoksyna B). Do mediatorów endogennych należą m.in. cytokiny, eikosanoidy, kininy.
Gdy drobnoustroje zaczynają się namnażać w ognisku infekcji, zarówno bakteryjne produkty (metabolizmu i rozpadu) jak i mediatory zapalne (leukotrieny, czynniki dopełniacza, cytokiny, kompleksy antygen-przeciwciało) powodują migrację granulocytów obojętnochłonnych i monocytów do tego obszaru. Gromadzenie się leukocytów w obszarze zapalenia jest ułatwione dzięki wydzielaniu przez komórki śródbłonka naczyniowego selektyn, czyli substancji adhezyjnych. Po pojawieniu się w ognisku zapalnym, leukocyty rozpoznają i fagocytują zopsonizowane bakterie i grzyby. Zabijanie bakterii odbywa się dzięki wytwarzaniu wysoce toksycznych i niestabilnych pochodnych tlenu oraz transportowi różnych substancji bakteriobójczych z ziarnistości lizosomalnych do wodniczek fagocytujących. Procesowi temu towarzyszy uwalnianie rozmaitych cytokin z makrofagów (monocytów tkankowych) w obszarze ogniska zapalnego.
Inicjacja odpowiedzi zapalnej układa się następująco: liczne stymulatory egzogenne (endotoksyna, egzotoksyna) jak i endogenne (np. aktywowany dopełniacz, produkty uszkodzenia komórki ... i inne) są w stanie pobudzać monocyty i makrofagi do syntezy kachektyny (TNF-α) i interleukiny-1β, które to cytokiny odgrywają kluczową rolę w rozwoju SIRS.
Funkcja tych cytokin polega na ograniczaniu uszkodzeń, zwalczaniu organizmów patogennych, eliminacji obcych patogenów i wspomaganiu procesów naprawczych.
Z tymi procesami koegzystuje aktywność przeciwzapalna, wyrażająca się obecnością w osoczu ciał przeciwzapalnych, które albo bezpośrednio blokują wiązanie prozapalnych mediatorów do powierzchniowych receptorów (receptorowy antagonista IL-1, rozpuszczalny receptor TNF, receptorowy antagonista leukotrienu B4) albo wykazują własne działanie przeciwzapalne (IL-4, IL-10, TGF-β).
Za uszkodzenie komórek i tkanek w procesie SIRS odpowiedzialne są aktywowane immunocyty, włączając granulocyty, makrofagi i limfocyty. Komórki te wywołują dysfunkcję narządową poprzez migrację z krwią do miejsc odległych od ogniska pierwotnego. Stąd ich funkcja immunologiczna jest zarówno protekcyjna, poprzez niszczenie czynnika zakaźnego, jak i uszkadzająca poprzez uwalnianie do światła włośniczek toksycznych enzymów i wolnych rodników.
Endotoksyna
Endotoksyna jest lipopolisacharydem (LPS) związanym ze ścianą komórki bakterii G(-) i uwalnianym z chwilą jej rozpadu. Po uwolnieniu ze ściany komórkowej wiąże się najpierw z produkowanym przez wątrobę białkiem ostrej fazy, a następnie kompleks ten aktywuje granulocyty i makrofagi, które uwalniają całą kaskadę mediatorów endogennych (cytokiny, eikosanoidy, proteazy, wolne rodniki, NO). LPS aktywuje ponadto układ krzepnięcia i fibrynolizy.
Egzotoksyna
W ostatnich latach wzrósł do prawie 50% udział bakterii G(+) w etiologii wstrząsu septycznego. Głównym mediatorem, uwalnianym przyżyciowo przez komórkę bakteryjną gram (+), jest egzotoksyna. Aktywuje ona przede wszystkim limfocyty, które następnie uwalniają kaskadę mediatorów endogennych.
Eikosanoidy
Są metabolitami kwasu arachidonowego (niezbędny kwas tłuszczowy pochodzący z rozpadu fosfolipidu błon komórkowych) i zalicza się do nich prostaglandyny i tromboksan (szlak cyklooksygenazy) oraz leukotrieny (szlak lipooksygenazy). Źródłem eikosanoidów są neutrofile, makrofagi, płytki krwi, komórki tuczne i komórki śródbłonka naczyniowego.
W warunkach ograniczonego dostępu tlenu następuje napływ jonów Ca do cytozolu i aktywacja fosfolipazy A2 która z kolei powoduje „wyłuskiwanie” z komórek endothelium kwasu arachidowego, jednego z głównych składników błon komórkowych, który następnie ulega lipooksgenacji i cyklooksygenacji - powstają leukotrieny, prostaglandyny (w endothelium) i tromboksan (w trombocytach).
PGI2 - prostacyklina - produkowana jest przez śródbłonek naczyniowy. Powoduje ona:
rozszerzenie naczyń poprawiając przepływ wieńcowy, trzewny, nerkowy, w kończynach oraz w tkance płucnej
zwiększenie rzutu serca, a przez to wzrost dostarczania tlenu do tkanek
hamowanie agregacji płytek krwi
stabilizowanie błony komórkowej
zmniejsza adhezję leukocytów do śródbłonka naczyń.
TxA2 - syntetyzowany w trombocytach, jest środkiem najsilniej kurczącym naczynia i pobudzającym płytki krwi do agregacji
PGD2 - uwalniana jest głównie przez komórki tkanki płucnej, silnie kurczy naczynia i oskrzela, hamuje agregację trombocytów
PGF2α - silnie kurczy naczynia, głównie mózgowe
PGE2 - stymuluje miejscowy i ogólny odczyn zapalny, najważniejsza prostaglandyna pośrednicząca w odczynie zapalnym.
Leukotrieny LTC4, D4 i E4 - pobudzają do chemotaksji i agregacji granulocyty, zwiększają przepuszczalność endothelium 1000 razy silniej od histaminy, powodują kurcz mięśni gładkich drzewa oskrzelowego, kurcz naczyń krwionośnych płuc, nerek, naczyń wieńcowych i mózgowych.
--> W [Author:SG] warunkach fizjologicznych głównym źródłem prostanoidów produkowanych przez śluzówkę żołądka, komórki nerek, płytki krwi i śródbłonek naczyniowy jest COX-1. COX-2 pojawia się w toku procesów zapalnych, indukowana przez endotoksynę albo cytokiny prozapalne (TNF, IL-1) i to jej aktywność, a nie COX-1, jest odpowiedzialna za produkcję znacznych ilości prostaglandyn wywołujących objawy zapalenia, takie jak ból, wzrost przepuszczalności naczyń, obrzęk, gorączka.
Wolne rodniki i nadtlenki
Układ kinin
kininogen (α2 globulina osoczowa syntetyzowana w wątrobie) - w formę czynną (kininy) przechodzi pod wpływem kalikreiny (uwalnianej z limfocytów lub powstającej pod wpływem aktywnego czynnika Hagemana, układu dopełniacza albo endotoksyny)
Kininy pobudzają do aktywacji fosfolipazę A2, czego następstwem jest wzmożona produkcja prostaglandyn PGI2 i PGE2 (produkcja leukotrienów nie jest pobudzana)
Układ endogennych endorfin
receptory opiatowe w mózgu (μ, δ, κ) zlokalizowane są w układzie limbicznym, podwzgórzu, przysadce, rdzeniu kręgowym, odpowiadają za emocje, ból, reakcje wegetatywne
μ - mają powinowactwo do β-endorfiny i do opiatów. Odpowiadają za analgezję i depresję oddechową
δ - mają powinowactwo do leu-enkefaliny i odpowiadają za spadek ciśnienia spowodowany rozszerzeniem naczyń
κ - dinorfina - odpowiada za sedację
Cytokiny
Jest to grupa białek uwalnianych głównie z limfocytów i makrofagów, które działają modulująco na układ immunologiczny. Głównymi cytokinami uwalnianymi we wstrząsie, przede wszystkim pod wpływem endotoksyny, zakażeń bakteriami G(+), urazu, niedotlenienia, są:
interleukiny - (główną rolę odgrywają 1, 2 i 6) - stymulują wątrobę do produkcji białek ostrej fazy, pobudzają dojrzewanie i uwalnianie do krążenia krwinek białych, pobudzają limfocyty T i B. Efekty biologiczne obejmują: gorączkę, leukocytozę, katabolizm mięśni poprzecznie prążkowanych. Wywierają również ujemny efekt inotropowy na mięsień sercowy.
interleukina 10 - posiada właściwości przeciwzapalne, hamując syntezę TNF-α oraz IL-1 i IL-6.
kachektyna (TNF-α) - w małych stężeniach ma korzystny wpływ poprzez podwyższanie naturalnej zdolności komórek do zabijania bakterii, w dużych stężeniach wywiera wielokierunkowe, niekorzystne działania przejawiające się depresją układu krążenia, wzmożoną adhezją granulocytów, uwalnianiem dużych ilości wolnych rodników, wzmożonym przesiękaniem (szczególnie w płucach), kwasicą mleczanową, hiperglikemią, gorączką, supresją erytropoezy. Wywołuje zmiany martwicze w wątrobie i nerkach.
Białka ostrej fazy
CRP - podnosi zdolności opsonizacyjne osocza, podwyższa zdolności fagocytarne komórek
ceruloplazmina - zawiera duże ilości miedzi, ma właściwości antyoksydacyjne
α1-antytrypsyna i α2-makroglobulina - stanowią przeciwwagę dla uwalnianych we wstrząsie wielu kwaśnych proteaz
fibrynogen
haptoglobina
Czynnik aktywujący płytki (PAF)
syntetyzowany głównie przez makrofagi płucne, pod wpływem niedotlenienia, urazu, endotoksyny. Spełnia funkcję lokalnej hemostazy. Aktywność jego przejawia się w
działaniu kurczącym oskrzela i naczynia płucne,
kardiodepresyjnym i arytmogennym działaniu na serce,
pobudzaniu płytek krwi i granulocytów do agregacji,
uszkadzaniu błon komórkowych,
zwiększaniu przesiękania przez śródbłonek
i wywoływaniu hipotensji.
Pod wpływem endotoksemii PAF jest produkowany miejscowo w jelitach, powoduje zmiany martwicze i uszkodzenie ściany jelit.
Tlenek azotu (NO)
Fizjologicznie NO pełni wiele ról: jest neuroprzekaźnikiem w OUN, kontroluje napięcie naczyń krwionośnych, współdziała w niszczeniu komórek bakteryjnych i nowotworowych. Mediatory zapalne stymulują intensywną produkcję NO przez komórki śródbłonka, kardiocyty, makrofagi i granulocyty obojętnochłonne. Tworzy się mechanizm błędnego koła - NO nasila z kolei uwalnianie cytokin z komórek fagocytarnych.
Udowodniono, że uogólniona wazodilatacja oraz pogorszenie funkcji komór serca we wstrząsie septycznym spowodowane są w dużej części nadmierną produkcją NO. Nadmiar tlenku azotu w sercowych miocytach stymuluje cyklazę guanylową, która hamując wewnątrzkomórkowe przesunięcia jonów wapnia osłabia kurczliwość mięśnia sercowego. Ponadto zalew NO zaburza mitochondrialny łańcuch oddechowy, co prowadzi do zmniejszenia produkcji ATP w mięśniu sercowym.
Atrial Natriuretic Factor
Układ dopełniacza
Aktywacja dopełniacza jest brzemienna w skutki, ponieważ prowadzi do wewnątrznaczyniowego pobudzenia neutrofili, które po adhezji do śródbłonka włośniczkowego uwalniają śródnaczyniowo zawartość swoich ziarnistości (proteazy, eikosanoidy, rodniki tlenowe). Prowadzi to do uszkodzeń włośniczek i parenchymy co wyraża się zmianami strukturalnymi narządów, a w konsekwencji zaburzeniami wymiany gazowej w płucach i na poziomie tkankowym. Aktywacja neutrofili jest wyjściową przyczyną uogólnionego odczynu zapalnego (SIRS) prowadząc do zaburzeń wielonarządowych.
Układ krzepnięcia i fibrynolizy
Antytrombina III
Białko C
jest fizjologicznym inhibitorem czynnika krzepnięcia V i VIII
aktywuje go trombina
Fibronektyna
Myocardial Depressant Factor
Pojawia się w niedokrwionym obszarze trzewnym, szczególnie w hipoperfundowanej trzustce. Działa depresyjnie na siłę skurczu mięśnia sercowego.
Hemodynamika wstrząsu septycznego
Faza „ciepła”
Uwalnianie toksyn bakteryjnych do krążenia powoduje początkowo spadek oporu krążenia systemowego i wzrost rzutu minutowego serca. Odpowiedzialnością za spadek oporu obciąża się tlenek azotu, histaminę i bradykininę. Ta hyperdynamiczna („ciepła”) faza wstrząsu septycznego może być słabo wyrażona u chorych z hypowolemią oraz jawną lub ukrytą niewydolnością serca.
Od początku wstrząsu septycznego stwierdza się wyraźny spadek frakcji wyrzutowej komór oraz wzrost objętości końcoworozkurczowej i końcowoskurczowej obu komór, którym to zmianom towarzyszy zwykle zwiększony rzut minutowy. Ten skojarzony obraz dilatacji komór i hiperdynamii krążenia, tak charakterystyczny dla wstrząsu septycznego, jest nazywany „nadkompensowaną” niewydolnością mięśnia sercowego. Nadkompensacja odbywa się kosztem rezerw czynnościowych, stąd stan mięśnia sercowego sprzed wystąpienia wstrząsu jest istotnym czynnikiem warunkującym pojawienie się fazy „ciepłej”.
gorące, suche kończyny chorego
rozszerzone obwodowo naczynia tętnicze
zmniejszony opór obwodowy
zwiększona pojemność minutowa serca
wysokie SAP i niskie DAP → skrajnie np. 160/0 mmHg
bardzo duży przeciek obwodowy
C(a-v)O2 bliskie zeru
Faza „zimna”
W miarę rozwoju wstrząsu septycznego dochodzi do stopniowego spadku rzutu serca, spowodowanego zarówno zmniejszeniem nawrotu żylnego jak i pogorszeniem stanu myokardium. Uwalniane obficie katecholaminy obkurczają zwieracze zawłośniczkowe, a mediatory zwiększają przepuszczalność naczyń, co prowadzi najpierw do sekwestracji płynu w mikrokrążeniu, a później jego przejścia do przestrzeni pozanaczyniowej. Ucieczka płynu pogłębia zaburzenia perfuzji tkanek, nasila beztlenową przemianę materii i zwiększa akumulację mleczanów. Jest to obraz hypodynamicznej („zimnej”) fazy wstrząsu septycznego.
Za pogłębianie depresji mięśnia sercowego odpowiedzialne są również krążące mediatory antyinotropowe, a w szczególności MDF, TNF, IL-1, NO. Niekorzystny wpływ na myokardium wywierają także zmienione warunki metaboliczne, a przede wszystkim kwasica mleczanowa.
W stanach septycznych metabolizm tlenowy komórki ulega zaburzeniom z następujących przyczyn:
Wzrasta zapotrzebowanie na tlen w związku z przyspieszoną przemianą materii w procesie SIRS, czego wyrazem jest zwiększone zużycie O2 (VO2).
Pogarsza się ekstrakcja tlenu (oxygen extraction ratio - O2ER), co łączy się z uogólnionym uszkodzeniem mikrokrążenia przez mediatory endogenne wstrząsu.
Zmniejsza się dostawa tlenu (DO2) do tkanek wskutek depresji septycznej mięśnia sercowego.
Wymienione trzy mechanizmy prowadzą do nierównowagi pomiędzy zapotrzebowaniem na tlen a jego dostawą. W warunkach fizjologicznych, przy spadku DO2 spowodowanym zmniejszeniem rzutu serca nie zmienia się zużycie tlenu, ponieważ pogłębia się jego ekstrakcja. Przy postępującym spadku rzutu serca i dostawy tlenu kompensująca rola ekstrakcji kończy się (jest to tzw. krytyczna wartość DO2) i od tego miejsca zużycie tlenu zaczyna spadać (VO2 staje się zależne od DO2) co implikuje pojawienie się kwasicy mleczanowej. Tę bardzo niekorzystną zależność VO2 od DO2 udało się wykazać we wstrząsie septycznym nawet u chorych ze względnie stabilnym układem krążenia i wysokim wskaźnikiem sercowym (CI powyżej 4 l/min/m2).
Zaburzenia narządowe we wstrząsie septycznym
Zespół zaburzeń wielonarządowych (MODS - multiple organs dysfunction syndrome) jest bezpośrednią przyczyną zgonu we wstrząsie septycznym. Mechanizm powstawania zmian narządowych obejmuje dwie kategorie zjawisk: przerwanie perfuzji tkankowej spowodowane hipowolemią i spadkiem ciśnienia tętniczego w początkowej fazie wstrząsu oraz miejscowe zmiany tkankowe wywołane endotoksyną i innymi mediatorami w fazie późnej. Hipoperfuzja tkankowa uszkadza śródbłonek włośniczkowy, powoduje agregację płytek krwi, gromadzenie się neutrofili, aktywację układu kalikreina - kininy oraz uwalnianie cytokin. Wzrost przepuszczalności uszkodzonego śródbłonka powoduje przejście płynu poza naczynia, co nie tylko pogłębia hipowolemię, ale również jest przyczyną obrzęku śródmiąższowego.
Zmiany zapalne pojawiają się najpierw w płucach, a następnie w wątrobie i jelitach. Kulminacja zapalnych zmian narządowych ma miejsce 48 - 72 godzin od zadziałania toksyn bakteryjnych.
Zaburzenia perfuzji prowadzą do załamania równowagi pomiędzy całkowitą dostawą tlenu do tkanek (DO2) i zużyciem tlenu (VO2). Stan niedotlenienia komórkowego wyrażający się dysfunkcją, a w końcu niewydolnością narządową określany jest terminem „dysoksji”. Metabolicznym odbiciem dysoksji jest obecność kwasicy mleczanowej spowodowanej zablokowaniem glikolizy tlenowej. W warunkach fizjologii VO2 utrzymuje się na stałym poziomie bez względu na wielkość dostawy (o ile DO2 nie spadnie poniżej wartości krytycznej około 8 ml/kg/min). We wstrząsie septycznym wymiana tlenu odbywa się w strefie wartości krytycznych, niezależnie od wielkości oksygenacji tętniczej i rzutu serca (zjawisko permanentnego „głodu tlenowego”).
Układ krążenia
Spadek napięcia naczyń
Spadek napięcia naczyń pojawia się w sepsie jako odpowiedź na liczne mediatory egzo- i endogenne. Zarówno TNF-α jak i IL-1 powodują wazodilatację poprzez efekt bezpośredni oraz hamowanie wrażliwości naczyń na katecholaminy. Obecnie główną rolę w septycznej wazodilatacji przypisuje się tlenkowi azotu. W warunkach fizjologicznych NO produkowany jest w endotelium naczyniowym z L-argininy, następnie dyfunduje do przylegających komórek naczyniowej mięśniówki gładkiej i aktywuje cyklazę adenylową do produkcji cGMP (cyklicznego guanozylomonofosforanu), który jest mediatorem rozkurczu tejże mięśniówki. W septycznym procesie zapalnym dochodzi do silnej aktywacji syntezy NO, który uwalniany w dużej ilości powoduje wazodilatację objawiającą się spadkiem ciśnienia tętniczego. Nadmiar NO jest też najprawdopodobniej przyczyną zmniejszenia wrażliwości naczyń na stymulację adrenergiczną.
Reasumując, w septycznych zaburzeniach krążenia wiodącym problemem staje się utrata autoregulacji w mikrokrążeniu większości narządów.
Septyczna wazodilatacja prowokuje wzrost produkcji mediatorów wazokonstrykcyjnych: tromboksanu i endoteliny. Stan napięcia naczyń u chorych we wstrząsie septycznym wynika więc z wypadkowego efektu wazodilatatorów i wazokonstriktorów.
Depresja mięśnia sercowego
Jest u chorego we wstrząsie septycznym mniej oczywista od spadku oporu naczyniowego, ponieważ rzut serca jest z początku wysoki, bądź normalny. Jednak kurczliwość myokardium jest wyraźnie upośledzona od początku wstrząsu. To upośledzenie znajduje kliniczne odbicie w spadku frakcji wyrzutowej lewej komory (LVEF - left ventricular ejection fraction), ocenianym za pomocą badania ECHO. U chorych septycznych stwierdza się również wzrost objętości końcowo-rozkurczowej (rozstrzeń) lewej komory. Jest to prawdopodobnie mechanizm wyrównawczy, pozwalający utrzymać rzut serca przez wykorzystanie rezerwy preload (mechanizm Franka-Starlinga), jego nieobecność prowadzi do dekompensacji układu krążenia.
Podobne zmiany dotyczą również czynności prawej komory.
Septyczne poszerzenie jam serca prowokuje wzrost produkcji atriopeptyny (α-ANP - atrial natriuretic peptide), której poziom służyć może jako wskaźnik nasilenia kardiodepresji, podobnie jak wzrost poziomu endoteliny (reakcja na hipotensję) jest wskaźnikiem wazodilatacji.
Zaproponowano szereg hipotez tłumaczących septyczną depresję mięśnia sercowego:
spadek gęstości receptora β-adrenergicznego
mediatory kardiodepresyjne
MDF (jednak pomimo wieloletnich poszukiwań ten hipotetyczny mediator nie został zidentyfikowany)
TNF-α
IL-1β (stwierdzono bardzo wyraźny synergizm z TNF-α)
a także: PAF (czynnik aktywacji płytek), wolne rodniki tlenowe, interferon-γ i metabolity kwasu arachidonowego
tlenek azotu - jednak obok działania wyraźnie kardiodepresyjnego, chroni mięsień sercowy przed wazokonstrikcją wieńcową
zaburzenia perfuzji wieńcowej
pobudzenie naturalnych inhibitorów układu krzepnięcia, które pozwalają na zlokalizowanie procesu krzepnięcia i utrzymanie homeostazy (np. poziom AT-III podczas sepsy spada bardzo wcześnie)
pobudzenie fibrynolizy (komórki śródbłonka są podstawowym źródłem tkankowego aktywatora plazminogenu)
dreszcze, temperatura > 38OC lub < 36OC,
częstość akcji serca > 90/min,
częstość oddechów > 20/min,
ciśnienie parcjalne CO2 we krwi tętniczej poniżej 32 mmHg,
leukocytoza > 12 000 lub < 4 000, trombocytopenia, hiperbilirubinemia, wzrost poziomu mocznika i kreatyniny,
objawy kliniczne wywołane niedostateczną perfuzją narządową, np. kwasica mleczanowa, oliguria, zaburzenia psychiczne,
zaburzenia krążenia (ciśnienie skurczowe poniżej 90 mmHg lub spadek większy niż 40 mmHg),
różnica temperatury centralnej (przełyk, odbyt) i obwodowej,
tonometria (pHi) - jest względnie nieinwazyjną metodą oceny perfuzji trzewnej. Wykazano ponadto, że żołądkowa funkcja egzokrynna we wstrząsie septycznym jest uzależniona od perfuzji śluzówki żołądkowej, której to perfuzji odbiciem jest właśnie pHi. Stąd pH soku żołądkowego może w pewnym stopniu odbijać adekwatność perfuzji trzewnej, tak jak diureza jest wskaźnikiem przepływu nerkowego.
Leczenie zaburzeń krążenia
koloidy (HES, dekstran, roztwory żelatyny)
krystaloidy
albuminy
hipertoniczny roztwór NaCl (7,5% NaCl przetoczony w ilości 4 ml/kg) - połączenie z koloidem (dekstran albo HAES) pozwala przedłużyć efekt z 30 do 180 minut
osmotyczna mobilizacja płynu pozanaczyniowego oraz płynu z komórek endotelium i erytrocytów
zmniejszenie obrzęku komórek endotelium → poprawa warunków reologicznych,
zmniejszenie obrzęku okołonaczyniowego → poprawa DO2 do tkanek
stymulacja mięśnia sercowego oraz przywrócenie wazomotoryki tętniczej
zmniejszenie ciśnienia śródczaszkowego (istotne zwłaszcza u chorych po urazach czaszkowo-mózgowych lub obrzękiem mózgu)
jest prekursorem adrenaliny,
w dawkach 0,03-5 μg/kg/min wyraźnie podwyższa SVR i MAP (działanie α-adrenergiczne). Dodatni efekt inotropowy, w którym pośredniczą receptory β1 adrenergiczne, jest mniej pewny, ale spodziewać się można poprawy funkcji obu komór serca wskutek wzrostu przepływu wieńcowego. Zwiększając zużycie tlenu przy niezmienionej jego dostawie powoduje cofanie się laktemii, poprawę tonometrii oraz ustępowanie zaburzeń narządowych bez potrzeby forsowania rzutu serca. Następstwem stabilizacji układu krążenia jest ustępowanie hiperlaktemii. Reakcja krążenia trzewnego na noradrenalinę jest szczególnie ważna, z uwagi na rolę jelit i wątroby w patogenezie niewydolności wielonarządowej. Wskazane jest ostrożne jej użycie u chorych z nie wyrównaną hipowolemią,
efekt działania zależny jest od dawki, w niższych dawkach przeważa wpływ na receptory β1, co wywołuje przyspieszenie rytmu serca i wzrost rzutu serca, w większych dawkach dominuje efekt stymulacji receptorów α1, czego wynikiem jest wzrost oporu obwodowego szczególnie w płucach, nerkach i rejonie trzewnym,
wskazania do stosowania noradrenaliny są bardzo ograniczone i dotyczą:
pacjentów ze znacznie nasiloną hipotensją, nie reagującą na przetaczane płyny lub inne środki o działaniu inotropowym,
we wstrząsie kardiogennym dla podniesienie MAP do 70-80 mmHg,
faza hiperdynamiczna wstrząsu septycznego
ma silne właściwości α- i β-adrenergiczne
poprawia perfuzję tkankową zarówno przez zwiększenie MAP jak i CI. Ten korzystny efekt uzyskuje się przy niskich dawkach (0,05-0,15 μg/kg/min), które nie powinny znacząco przyspieszać czynności serca. W większych dawkach adrenalina działa silnie wazokonstrikcyjnie i traci przewagę nad noradrenaliną,
efektem jej działania na serce, poprzez pobudzenie receptorów β1, jest ułatwienie przewodzenia bodźców, w związku z tym skraca bardziej czas skurczu niż rozkurczu oraz usposabia do niemiarowości,
podawana we wlewie dożylnym z szybkością 0,005-0,02 μg/kg/min działa przede wszystkim na receptory β. Efektem takiego działania jest poprawa hemodynamiki mięśnia sercowego. W większych dawkach, używanych podczas resuscytacji, silnie pobudza receptory α, co wyraża się zwiększeniem oporu obwodowego. Podwyższenie ciśnienia rozkurczowego zapewnia lepszą perfuzję naczyń wieńcowych,
wywiera silne działanie na układ oddechowy. Poprzez pobudzenie receptorów β2 rozszerza mięśnie gładkie oskrzeli i hamuje degranulację komórek wytwarzających histaminę,
silnie kurczy naczynia nerkowe. Przy podawaniu z szybkością 0,035 μg/kg/min przepływ nerkowy spada o 10%, jednak diureza może być zwiększona wskutek poprawy krążenia centralnego,
wywiera działanie metaboliczne w postaci hiperglikemii, kwasicy mleczanowej i podwyższenia stężenia kwasów tłuszczowych,
poprzez stymulowanie receptorów β2 obniża poziom potasu
jest katecholaminą o mieszanym efekcie α i β adrenergicznym.
jako pojedynczy lek jest mniej przydatna w zwiększaniu CI i MAP u chorych z niewydolnym sercem, ponieważ 50% jej efektu inotropowego uzyskiwane jest pośrednio: przez biotransformację do noradrenaliny oraz uwalnianie noradrenaliny z zakończeń nerwowych. Zwiększenie dawki powyżej 10 μg/kg/min prowadzi do tachykardii, tachyarytmii, hipertensji płucnej i wazokonstrikcji nerkowej.
w niskiej dawce pobudza receptory dopaminergiczne DA1 i DA2 powodując rozszerzenie naczyń nerkowych, trzewnych i wieńcowych.
w średnich dawkach (5-10 μg/kg/min) dominuje działanie β1-adrenergiczne powodujące zwiększenie rytmu serca i kurczliwości,
w wysokich dawkach powoduje obkurczenie naczyń poprzez pobudzenie receptorów α1
jest syntetyczną katecholaminą o selektywnym działaniu na receptory β1 z niewielkim tylko wpływem na naczynia obwodowe,
efektem nieznacznego pobudzenia receptorów α i β2 jest nieznaczne zmniejszenie oporu obwodowego, w tym również oporu płucnego, co można uznać za zaletę w leczeniu wstrząsu, szczególnie septycznego,
głównym efektem podawania dobutaminy jest dodatnie działanie inotropowe z relatywnie małym przyspieszeniem czynności serca. Skutkiem tego jest obniżenie OCŻ i ciśnienia zaklinowania, będącego wynikiem poprawy funkcji komór,
stosowana w dawce 2-15 μg/kg/min obniża opór płucny i systemowy, redukując tym samym obciążenie następcze (afterload) obu komór serca. Wzrost przepływu nerkowego i diurezy są wtórne do wzrostu rzutu serca. Największą niedogodnością użycia dobutaminy jest spowodowana wazodilacją hipotensja tętnicza, ujawniająca się łatwo w hipowolemii - należy więc zadbać o właściwe wypełnienie łożyska naczyniowego. Korzystnym rozwiązaniem jest równoległa infuzja dobutaminy i noradrenaliny,
stosowanie dobutaminy zalecane jest w przypadkach konieczności podwyższenia kurczliwości mięśnia sercowego bez jednoczesnego wyraźnego wpływu na naczynia obwodowe.
Leczenie zaburzeń oddychania
Kortykosteroidy w leczeniu wstrząsu septycznego
Leczenie zaburzeń krzepnięcia
Niekonwencjonalne metody leczenia wstrząsu septycznego
usunięcie krążących mediatorów (hemofiltracja)
przeciwciała monoklonalne anty-TNF
prostaglandyna E1 - wykorzystanie jej silnego działania przeciwstawnego do tromboksanu
pentoksyfilina - wygasza pobudzenie neutrofili i makrofagów oraz osłabia większość biologicznych efektów kachektyny i interleukiny-1
definicja (wg Programu Zakażeń Szpitalnych w Atlancie z III 2000 roku) - zakażeniem szpitalnym jest każde zakażenie, które nie występowało w formie jawnej, bądź w okresie inkubacji, w czasie przyjęcia chorego do szpitala
stanowią coraz większy problem ze względu na częstość występowania, koszt leczenia i problemy terapeutyczne
są one powiązane ze stosowaniem inwazyjnych metod diagnostycznych i leczniczych, przede wszystkim u pacjentów z obniżoną odpornością
dotyczą najczęściej układu oddechowego, następnie dróg moczowych, jamy brzusznej i ran
u pacjentów w OIT leczonych ponad 2 tygodnie liczba ich wzrasta do prawie 100%
najczęściej wywołują
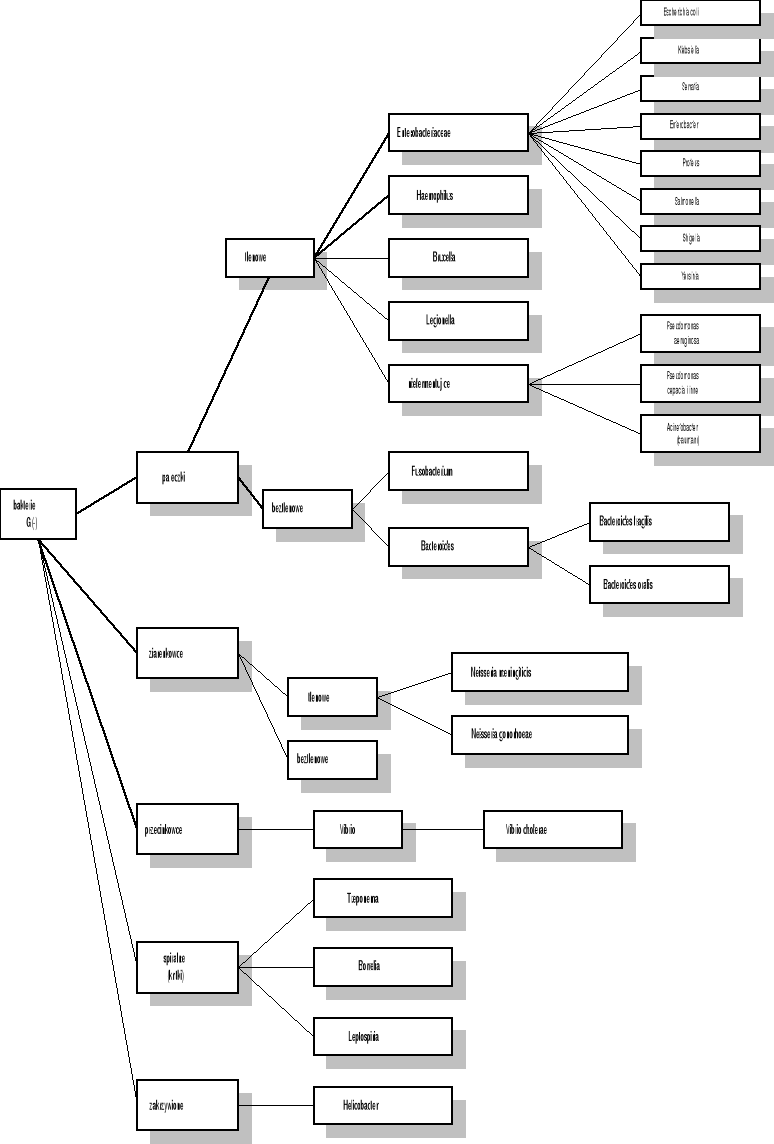
G(-) tlenowe pałeczki
Pseudomonas, Acinetobacter
Enterobacteriaceae - gł. Klebsiella. Przy nagminnym stosowaniu antybiotyków β-laktamowych doszło do wyselekcjonowania szczepów opornych, wśród Enterobacteriaceae, na β-laktamy (tzw. szczepy ESβL+) - w tych sytuacjach jedynie karbapenemy zachowują właściwości bakteriobójcze.
Haemophilus
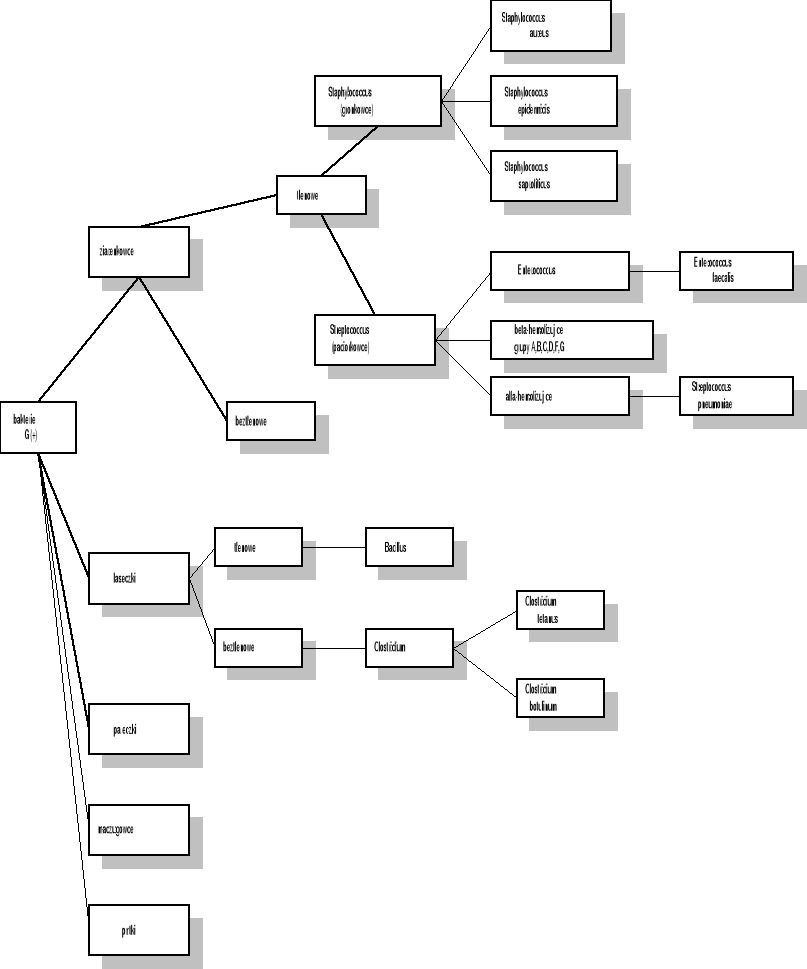
G(+) tlenowe
Staphylococcus aureus - coraz częściej metycylinooporne
gronkowce koagulozoujemne
Streptococcus (paciorkowce) - Enterococcus. Wśród enterokoków pojawiają się szczepy oporne na wankomycynę (tzw. szczepy WRE), zdarza się, że oporność ta przenoszona jest na inne bakterie G(+) np. gronkowce metycylinooporne. Pacjenci z takim zakażeniem wymagają bezwzględnej izolacji (ze względu na łatwe przenoszenie zakażenia drogą kontaktową).
zakażenia grzybicze
Candida albicans
postępowanie aseptyczne w okresie stosowania oddechu zastępczego i zakładania cewników do żył, tętnic lub pęcherza moczowego
prawidłowe zaopatrzenie ran
stosowanie celowanej antybiotykoterapii
przeprowadzenie selektywnej dekontaminacji przewodu pokarmowego
jest przeprowadzana ponieważ własne środowisko chorego jest źródłem zakażenia
w trakcie dekontaminacji usuwa się potencjalnie patogenne bakterie tlenowe, głównie gram(-), oraz grzyby z zachowaniem flory beztlenowej
minusem tej metody jest brak spektrum dla enterokoków i gronkowców oraz koszt
stosuje się ją co 6 godzin przez sondę żołądkową
100 mg polimiksyny E lub colistyny
80 mg tobramycyny
500 mg amfoterycyny B lub nystatyny
antybiotyki β-laktamowe - penicyliny, cefalosporyny, monobaktamy, karbapenemy,
aminoglikozydy,
glikopeptydy (wankomycyna, teikoplanina)
erytromycyna,
linkozamidy (linkomycyna, klindamycyna),
tetracykliny,
chloramfenikol,
sulfonamidy
Antybiotyki β-laktamowe
można je łączyć bezpiecznie z aminoglikozydami, jest to szczególnie istotne w leczeniu zakażeń enterokokami, bowiem same β-laktamy nie wykazują dostatecznego efektu, natomiast same aminoglikozydy są bardzo mało aktywne,
w zakażeniach z udziałem beztlenowców łączy się je z linkozamidami (linkomycyną, klindamycyną) lub metronidazolem,
penicyliny mogą też być kojarzone między sobą oraz z cefalosporynami, pod warunkiem, że użyte antybiotyki nie są induktorami β-laktamaz. Dobrych efektów należy się spodziewać po skojarzeniu np. piperacyliny z cefotaksymem (Claforan), bowiem piperacylina ma znikome właściwości indukcyjne i znakomitą aktywność wobec Pseudomonas.
Penicyliny
Penicyliny o wąskim spektrum G(+) ziarenkowce tlenowe: Streptococcus pneumoniae, viridans, pyogenes i wrażliwe gronkowce (w zakażeniach szpitalnych nie odgrywają znaczącej roli). G(-) bakterie tlenowe Neisseria meningitidis, gonorrhoeae. Bakterie beztlenowe Clostridium, Fusobakterium, Bacteroides z wyłączeniem Bacteroides fragilis, odpowiedzialnego za zakażenie wychodzące z jelit (wyrostek robaczkowy). Treponema pallidum (syfilis).
penicylina benzylowa (krystaliczna, prokainowa)
V-cylina
Penicyliny oporne na działanie penicylinazy - obejmują swoim spektrum działania paciorkowce, gronkowce (oporne, wytwarzające penicylinazę), mniej aktywne w stosunku do bakterii beztlenowych
nafcylina
metycylina
kloksacylina
Penicyliny szeroko wachlarzowe
α-aminopochodne spektrum obejmuje G(+) Streptococcus, enterokoki, G(-) pałeczki tlenowe Enterobacteriaceae - E. Coli, Proteus mirabilis, Salmonella, Shigella
ampicylina
amoksycylina
ampicylina + sulbaktam (Unasyn) - zwiększona skuteczność poprzez zablokowanie działania β-laktamazy. Poszerza to spektrum działania w porównaniu do samej ampicyliny o gronkowce, Acinetobacter i bakterie beztlenowe, w tym Bacteroides fragilis. W stosunku do enterokoków nie wykazuje lepszych właściwości.
karboksypochodne - wykazują silniejsze działanie przeciwko G(-) pałeczkom tlenowym Pseudomonas a także z rodziny Enterobacteriaceae - Proteus, oraz G(-) pałeczkom beztlenowym Bacteroides Fragilis (w leczeniu antybiotyki drugiego rzutu).
karbenicylina
tykarcylina
tykarcylina + kwas klawulanowy (Timentin)
ureidopenicyliny - poszerzone spektrum przeciwko bakteriom G(-). Antybiotyki z tej grupy nie nadają się do monoterapii ciężkich zakażeń i powinny łączone być z aminoglikozydami.
mezlocylina
azlocylina (Securopen)
piperacylina
Inhibitory β-laktamaz
kwas klawulanowy + amoksycylina (augmentin)
kwas klawulanowy + tykarcylina (timentin)
sulbaktam + ampicylina (unasyn)
Cefalosporyny - niezależnie od grupy nie wykazują działania przeciw gronkowcom opornym na metycylinę, paciorkowcom kałowym, słabo działają na bakterie beztlenowe (nie nadają się do leczenia zakażeń wynikających z uszkodzenia jelita)
I generacja - w spektrum przeciwbakteryjnym znajdują się ziarniaki G(+) paciorkowce (jednak brak skuteczności w stosunku do enterokoków), gronkowce oraz niektóre G(-) ziarenkowce tlenowe - Neisseria gonorrhoeae, pałeczki tlenowe - Haemophilus, Enterobacteriaceae (E. coli, Proteus, Klebsiella). Wobec pozostałych rodzajów pałeczek z rodziny Enterobacteriaceae, Pseudomonas i Acinetobacter są nieaktywne. Leki z tej grupy źle przenikają do płynu mózgowo - rdzeniowego, są wrażliwe na β-laktamazy.
cefalotyna
cefradyna (Sefril)
cefazolina (Kefzol)
II generacja - posiadają szersze spektrum w stosunku do G(-) pałeczek tlenowych (Enterobacteriaceae - E. coli, Proteus, Klebsiella; Haemophilus influenzae), wykazują też działanie na ziarniaki G(+), w tym gronkowce oporne na penicylinę, są bardziej oporne na β-laktamazy. Cefoksytyna działa również na beztlenowe bakterie G(-) z rodzaju Bacteroides. Cefuroksym natomiast dobrze penetruje do płynu mózgowo - rdzeniowego. II generacja jest nieaktywna wobec Pseudomonas, Acinetobacter.
cefaklor
cefamandol (Mandol)
cefuroksym (Zinacef)
cefoksytyna (Mefoxin)
III generacja - charakteryzują się szczególną aktywnością wobec tlenowych bakterii G(-) głównie ziarenkowce Neisseria meningitidis i gonorrhoeae, oraz pałeczki Enterobacteriaceae (Enterobacter, Klebsiella, Proteus), Pseudomonas, Acinetobacter, Haemophilus. Posiadają największą wśród cefalosporyn, równą penicylinie, aktywność w stosunku do paciorkowców β-hemolizujących (najczęstsza przyczyna zapalenia wsierdzia), nieaktywne w stosunku do enterokoków. Antybiotyki te penetrują do wszystkich narządów i tkanek, przechodzą także przez barierę krew - opony mózgowe.
cefotaksym (Claforan)
ceftriakson (Rocephin)
ceftazydym (Fortum)
cefoperazon (Cefobid)
IV generacja
cefpirom
cefepim
Monobaktamy
aztreonam - działa bakteriobójczo tylko w stosunku do tlenowych bakterii G(-), zarówno ziarenkowców (Neisseria) jak i pałeczek (Enterobacteriaceae, Pseudomonas, Haemophilus ale nie Acinetobacter). Aktywność ta jest porównywana z aminoglikozydami.
Karbapenemy - antybiotyki o bardzo szerokim spektrum, hamujące wzrost bakterii G(+) i G(-), zarówno tlenowych jak i beztlenowych. Szczepy oporne to: Streptococcus faecalis, gronkowce metycylinooporne. Wykazują antagonizm z innymi antybiotykami β-laktamowymi, synergizm z aminoglikozydami.
imipenem (Tienam)
meropenem
Aminoglikozydy - działają przede wszystkim na tlenowe bakterie G(-) z rodzaju Enterobacteriaceae, Pseudomonas, Acinetobacter oraz bakterie G(+) głównie paciorkowce łącznie z enterokokami i częściowo gronkowce. Aminoglikozydy nie działają na bakterie beztlenowe.
Aminoglikozydy naturalne
streptomycyna
neomycyna
gentamycyna
tobramycyna
Aminoglikozydy półsyntetyczne
netylmycyna
amikacyna
Linkozamidy - skierowane są przeciw gronkowcom i paciorkowcom grupy A (jednak ich aktywność wobec ziarenkowców tlenowych jest mniejsza niż penicyliny). Ich największym walorem jest wysoka aktywność wobec bakterii beztlenowych (łącznie z pałeczką G(-) Bacteroides fragilis i G(+) laseczką Clostridium)
linkomycyna
klindamycyna (Dalacin C)
Glikopeptydy - działają synergicznie z aminoglikozydami, jednak należy pamiętać o możliwości wystąpienia spotęgowanego efektu nefro i ototoksycznego. Są jedynymi, chociaż nie w pełni skutecznymi, antybiotykami w leczeniu rzekomobłoniastego zapalenia jelita grubego o etiologii Clostridium difficille.
wankomycyna - spektrum działania ograniczone jest do bakterii G(+) głównie ziarenkowców tj. gronkowców (w tym metycylinoopornych) i paciorkowców (również enterokoków i opornych na penicylinę) oraz G(+) laseczek beztlenowych z rodzaju Clostridium.
teikoplanina - zakres działania obejmuje bakterie G(+) zarówno tlenowe i beztlenowe. Jest bezużyteczna w zakażeniach wywołanych przez bakterie G(-). Dawkowanie - raz dziennie (200 - 400 mg), terapia skojarzona - antybiotyki aminoglikozydowe, chinolony (te ostatnie w oddzielnym wlewie)
Chloramfenikol
Tetracykliny
Antybiotyki makrolidowe
Chemioterapeutyki
Nitroimidazole
Chinolony
cyprofloksacyna
ofloksacyna - Tarivid
norfloksacyna
pefloksacyna
enoksacyna
kwas pipemidynowy
kwas nalidyksowy
Antybiotyki przeciwgrzybicze - ketokonazol, diflucan, amfoterycyna
Chemioterapeutyki przeciwwirusowe
Płuca
Należą do pierwszych uszkodzonych narządów w przebiegu wstrząsu septycznego, głównie za sprawą delikatnej struktury połączeń komórek śródbłonka włośniczkowego. Ponadto płuca filtrując cały rzut minutowy serca, eksponowane są szczególnie na krążące mediatory. Przez uszkodzony śródbłonek płyn przesącza się do przestrzeni śródmiąższowej płuc, a następnie pęcherzyków, tworząc obrzęk śródmiąższowy i ogniska rozsianej niedodmy. Zmniejszenie powietrzności płuc prowadzi do wzrostu pracy oddychania i pogorszenia wymiany gazowej i w konsekwencji do niewydolności oddechowej.
Układ pokarmowy
Jest nie tylko celem dla mediatorów wstrząsu, ale sam staje się źródłem toksycznych metabolitów. Zaburzenia krążenia trzewnego, pojawiające się wcześnie w przebiegu wstrząsu, powodują erozję śluzówki żołądka i jelit, co prowadzić może do krwotoków i perforacji. Niedokrwiona trzustka uwalnia proteazy i wolne rodniki, które uaktywniają dopełniacz oraz układ kalikreina - kininy. Niedokrwiona trzustka jest też źródłem kardiodepresyjnych peptydów (MDF). Niedostateczne krążenie krezkowe prowadzi z kolei do przełamania immunologicznej bariery jelitowej i przedostania się bakterii do węzłów chłonnych krezki i krążenia wrotnego. Proces translokacji bakterii pogłębia septyczne zmiany w wątrobie oraz nasila SIRS przez aktywację komórek Kupffera i hepatocytów.
Zaburzenia czynności wątroby o różnym stopniu nasilenia spotyka się u wszystkich chorych we wstrząsie septycznym. Hiperbilirubinemia jest często nieproporcjonalnie wysoka w porównaniu do wzrostu poziomu enzymów wątrobowych. W rzadkich przypadkach septyczne uszkodzenie wątroby prowadzi do śpiączki.
Nerki
Ostra niewydolność nerek w wyniku wczesnych zaburzeń krążenia jest obecnie rzadko bezpośrednio przyczyną śmierci chorych we wstrząsie septycznym. Zaburzenia wewnątrznerkowego rozdziału krwi oraz uszkodzenie miąższu nerek przez krążące mediatory są przyczyną spadku filtracji nerkowej i oligurii na każdym etapie wstrząsu. Hipoksemia tętnicza oraz stosowane w leczeniu wstrząsu leki (furosemid, antybiotyki aminoglikozydowe) są częstym powodem uszkodzenia cewek nerkowych i osłabienia zdolności zagęszczania moczu.
Zaburzenia układu krzepnięcia i fibrynolizy
Cytokiny aktywują układ krzepnięcia u pacjentów z sepsą. Podczas infekcji dochodzi do zwiększenia ekspresji czynnika tkankowego (TF- tissue factor) na powierzchni krążących monocytów i być może części komórek śródbłonka naczyniowego doprowadzając do aktywacji zewnątrzpochodnego toru krzepnięcia i zakrzepicy w mikrokrążeniu. Aktywacja układu krzepnięcia wydaje się niemal zjawiskiem powszechnym w przebiegu sepsy.
Jednocześnie następuje
Rozpoznanie i monitorowanie
Leczenie wstrząsu septycznego
W leczeniu wstrząsu septycznego priorytetami są przywrócenie perfuzji tkankowej oraz opanowanie zakażenia. Poza tymi głównymi celami, wiele problemów terapeutycznych nastręczają zaburzenia krzepnięcia, niewydolność nerek czy nieprawidłowości w obrębie układu pokarmowego.
Celem resuscytacji krążenia jest zapewnienie dostatecznego ciśnienia perfuzji przez przywrócenie napięcia ścian naczyń, szybką infuzję płynów oraz zwiększenie kurczliwości myokardium.
Terapię wazopresorami należy rozpocząć po wstępnym przetoczeniu płynów, starając się uzyskać skurczowe ciśnienie tętnicze powyżej 90 mmHg (albo średnie powyżej 70 mmHg). Do niedawna w leczeniu wstrząsu septycznego priorytet przysługiwał raczej rzutowi serca niż ciśnieniu tętniczemu - obserwacje wykazały jednak, że utrzymanie ciśnienia tętniczego ma decydujący wpływ na przeżycie we wstrząsie septycznym. Okazało się ponadto, że korekcja hipotensji z użyciem wazopresorów zwiększa ekstrakcję O2 bez potrzeby zwiększania dostawy tlenu.
Za minimum średniego ciśnienia tętniczego (MAP) przyjmuje się zwykle 60 mmHg, ponieważ poniżej tej wartości przestaje funkcjonować autoregulacja krążenia wieńcowego, mózgowego i nerkowego - przepływ w tych narządach staje się proporcjonalny do ciśnienia tętniczego.
Dotąd uznawano maksymalizację DO2 za główny cel leczenia wstrząsu septycznego. Tzw. „optymalne” albo „supranormalne” parametry hemodynamiczne (CI > 4,5 l/min/m2 i DO2 > 600 ml/min/m2) uznawane były za obowiązujące w leczeniu wstrząsu septycznego. Okazało się jednak, że w grupie chorych leczonych dobutaminą dla uzyskania parametrów „optymalnych” śmiertelność była wyższa niż u chorych leczonych konwencjonalnie.
Jest oczywiste, że stymulacja serca dla uzyskania większej wartości DO2 jest nieefektywnym sposobem korekcji zaburzeń dystrybucji przepływu, ponieważ znaczna część nadmiarowego DO2 kierowana jest raczej do tkanek z już wysoką perfuzją, niż do obszarów hipoksemicznych.
Podstawowym celem terapii wstrząsu septycznego jest zapewnienie perfuzji tkankowej, z adekwatnym dowozem tlenu i substratów. W ocenie perfuzji tkankowej pomocne były dotąd mało specyficzne wykładniki narządowe (np. diureza) i ogólnoustrojowe (np. laktemia).
Krążenie trzewne jest pierwszym, które ulega zakłóceniom zarówno we wstrząsie hypowolemicznym jak i septycznym. Nieinwazyjną metodą oceny perfuzji trzewnej jest tonometria żołądkowa i jelitowa (pHi).
Przetaczanie płynów - umiejętna płynoterapia pozwala szybko uruchomić tkwiącą w preload czynnościową rezerwę mięśnia sercowego. Problemem jest tylko wybór płynu oraz objętość i szybkość jego przetaczania.
Stosowanie katecholamin - z klinicznego punktu widzenia użyteczny jest ich podział na inokonstriktory (noradrenalina, adrenalina, dopamina) i inodilatory (dopeksamina, dobutamina, isoproterenol).
Noradrenalina, adrenalina i dopamina (katecholaminy endogenne) są silnymi α1-agonistami. Przy wyższych dawkach efektowi inotropowemu towarzyszy wazokonstrikcja naczyń obwodowych, nerkowych, trzewnych i płucnych. Ta właściwość może być korzystna u chorych, u których niewydolności mięśnia sercowego towarzyszy spadek SVR (co ma miejsce we wstrząsie septycznym). Jednakowoż nadmierna wazokonstrikcja prowadzi do tkankowej hipoperfuzji, kwasicy metabolicznej i na koniec narządowej ischemii.
Dopeksamina, dobutamina i isoproterenol (katecholaminy syntetyczne) posiadają silne właściwości β2-agonistyczne. Efektowi inotropowemu towarzyszy więc systemowa i płucna wazodilatacja. Korzystnym jej aspektem jest spadek afterload prawej i lewej komory, co samo przez się zwiększa rzut serca. Za efekt niekorzystny uznać trzeba hipotensję tętniczą i odruchową tachykardię. Chociaż perfuzja obwodowa wzrasta po zastosowaniu inodilatorów, to pewna część przepływu jest tracona wskutek otwarcia shuntów tętniczo-żylnych.
W pierwszej fazie wstrząsu, przed ukończeniem resuscytacji objętościowej, bezpieczniej użyć katecholaminy o szerokim spektrum działania (adrenalina, dopamina), niż czystego β-agonistę (dobutamina), który nasilić może wazodilację i tętniczą hipotensję poprzez stymulację receptora β2.
Noradrenalina
Adrenalina
Dopamina
Dobutamina
Rola kortykosteroidów jest ostatnio podkreślana ze względu na koncepcję względnej niewydolności kory nadnerczy we wstrząsie septycznym i ARDS.
W odpowiedzi na infekcję organizm uruchamia liczne mechanizmy obronne: zapalenie, koagulację, immunizację, aktywację osi podwzgórzowo-przysadkowo-nadnerczowej których rolą jest zwalczanie patogenów i reperacja uszkodzonych tkanek. Jeśli aktywacja tych mechanizmów jest nieproporcjonalnie duża do zakażenia, efektem będą raczej zaburzenia narządowe, niż przywrócenie homeostazy.
Glukokortykoidy hamują wymienione mechanizmy obronne na wszystkich poziomach i chronią przed niekorzystną dla organizmu hiperreakcją na zakażenie.
Przedłużona terapia glukokortykoidami prowadzi do znacznego spadku krążących mediatorów zapalnych takich jak: TNF, IL-1, IL-6. Co więcej taka przedłużona terapia powoduje wzrost cytokin przeciwzapalnych: IL-4 i IL-10.
Przedwczesne przerwanie leczenia steroidami we wstrząsie septycznym i ARDS wiązało się z pogorszeniem stanu ogólnego chorych ze wzrostem stężenia cytokin zapalnych, również zbyt krótkotrwała terapia nasila odpowiedź cytokinową na endotoksynę.
Proponowane jest stosowanie jest hydrocortisonu w dawce 100 mg co 8 godzin przez 5-6 dni.
Zaburzenia krzepnięcia są częstym powikłaniem wstrząsu septycznego, których skrajną postacią jest DIC. Głównym inhibitorem kaskady krzepnięcia jest antytrombina III, której poziom jest we wstrząsie septycznym zwykle obniżony. We wstrząsie septycznym należy monitorować: stężenie antytrombiny III, liczbę płytek krwi, czas kaolinowo-kefalinowy oraz stężenie produktów degradacji fibrynogenu.
Zakażenia wewnątrzszpitalne
Podstawową strategią w zapobieganiu zakażeniom szpitalnym powinna być profilaktyka
Selektywna dekontaminacja przewodu pokarmowego
Antybiotykoterapia
Antybiotyki bakteriobójcze
Antybiotyki bakteriostatyczne
Empiryczny wybór antybiotyku zależnie od rodzaju bakterii
Rodzaj bakterii |
Antybiotyk I rzutu |
Antybiotyk II rzutu |
Staphylococcus aureus - metycylinooporny |
glikopeptydy |
Biseptol |
Staphylococcus aureus - wrażliwe na metycylinę, oporne na penicylinę |
oksacylina, nafcylina |
cefalosporyny, glikopeptydy, Augmentin, Unasyn, karbapenemy |
Streptococcus D (Enterococcus) |
penicylina, Ampicylina + aminoglikozydy |
glikopeptydy |
Streptococcus |
penicylina |
cefalosporyny erytromycyna glikopeptydy |
Neisseria |
penicylina |
III generacja cefalosporyn |
Clostridium |
penicylina |
linkozamidy, Metronidazol |
Bacteroides |
linkozamidy, Metronidazol |
ureidopenicyliny karbapenemy |
Pseudomonas |
III generacja cefalosporyn i aminoglikozydy |
monobaktamy, karbapenemy i aminoglikozydy |
Acinetobacter |
karbapenemy |
aminoglikozydy i ureidopenicyliny lub cefalosporyny III generacji |
Proteus |
Cefotaksym Ceftriakson |
aminoglikozydy, monobaktamy, karbapenemy, ureidopenicyliny |
E. coli |
Ampicylina III generacja cefalosporyn ureidopenicyliny |
aminoglikozydy karbapenemy monobaktamy |
16
Medycyna po dyplomie - wydanie specjalne - luty 2000
Wyszukiwarka
Podobne podstrony:
Zakażenia prionowe II, Patologia i choroby
Zakażenia wirusowe skóry, skóra patologiczna
Zakażenia prionowe V, Patologia i choroby
Zakażenia bakteryjne skóry, skóra patologiczna
Zakażenia prionowe I, Patologia i choroby
Najbardziej prawdopodobny przebieg zakażenia u człowieka, Patologia i choroby
Zakażenia prionowe III, Patologia i choroby
analiza złożonych aktów ruchowych w sytuacjach patologicznych
PATOLOGIA GLOWY I SZYI
Zakażenia grzybicze skóry cz2
norma i patologia
zakazenia ukladu moczowego
01 Pomoc i wsparcie rodziny patologicznej polski system pomocy ofiarom przemocy w rodzinieid 2637 p
Cw 3 patologie wybrane aspekty
WykĹ‚ad ochrona pacjenta przed zakażeniem
więcej podobnych podstron