GENETYKA
- termin wprowadzony przez Batesona w 1906 r.
Mendel :
- jednostki dziedziczności, zawiązki cech - „merkmallen”
- prawo czystości gamet:
„substancja dziedziczna ma charakter jednostkowy i nie miesza się w zygocie z substancją pochodzącą od drugiego z rodziców”
Pojęcia `gen'. `genotyp', `fenotyp' zaproponował Johansen
Gen - podstawowa jednostka dziedziczenia zajmująca w chromosomie określone miejsce, czyli locus, jest określonym segmentem DNA, który koduje odpowiednią sekwencję aminokwasów polipeptydu
- pierwszy raz zaproponował taką nazwę Johannes (1909 r.)
Genotyp - zespół cech danego osobnika, całość informacji genetycznej organizmu
Każdy człowiek(oprócz bliźniąt monozygotycznych) ma swój charakterystyczny genotyp
Genotyp nie zmienia się od poczęcia
Cały `plan budowy' jest w pierwszej komórce po poczęcia, jest to plan nukleotydowy
Różnorodność jest koncepcją biologiczną
Przenoszenie informacji genetycznej:
DNA -(transkrypcja) RNA -(translacja) białko
Kod genetyczny:
Trójkowy - 3 nukleotydy kodują 1 aminokwas
Kod genet. jest uniwersalny dla wszystkich organizmów
Fenotyp - zespół cech zewnętrznych powstałych na skutek ekspansji genotypu oraz działania środowiska, całość cech fizycznych organizmu
Cytogenetyka - dział genetyki zajmujący się:
- chromosomami
liczbą
klasyfikacją
morfologią
aberracjami
- determinacją płci
chromosomowej
chromatynowej
- relacjami płci - stosunek liczbowy/procentowy płci męskiej i żeńskiej w czasie rozwoju człowieka
- chromosomopatiami = chorobami chromos. spowodowanymi aberracjami chromosomowymi:
1. chromosomopatie autosomalne = Chromosomopatie autosomów:
aneuploidie :
- trisomie - np. zespół Downa
2. chromosomopatie płci = Chromosomopatie heterochromosomów:
aneuploidie :
- monosomie - np. zespół Turnera
- polisomie - np. zespół Klinefertera
Chromosomy:
- odkryte w 1875 r. przez Edwarda Strasburgera
- nazwę `chromosomy' wprowadził Waldayer w 1888 roku - chroma = barwa, soma = ciało
- wielkość chromosomów nie jest związana z ich liczbą
- definicje:
kontener z pulą informacji genetycznej (magazynuje informację genetyczną) służącym do przemieszczania tej informacji w czasie mitozy i mejozy
chromosom jest jednostką segregacji grup sprzężonych genów, które przemieszczają się razem
chromosom to struktura sprzężona, złożona z sekwencji nukleotydowych, spełniająca tę samą funkcją mimo różnic strukturalnych u eukariontów i prokariontów - przenoszenie informacji genetycznej z komórki na komórkę, z pokolenia na pokolenie, kontrolująca funkcję i rozwój komórki
Rodzaje chromatyny:
heterochromatyna - rodzaj chromatyny silnie barwiącej się barwnikami zasadowymi, silnie skondensowana - to chromatyna nieaktywna, niefunkcjonalna
zawsze nieaktywna
konstytutywna (długie ramiona chromosomu Y)
fakultatywna (może przejść w euchromatynę)
euchromatyna - słabo się barwi, jest rozluźniona:
- procaryota - genofor (DNA) - zastępuje właściwy chromosom (nie jest to jednak struktura zaliczająca się do definicji pojęcia chromosomu)
- eucaryota - chromosomy (DNA, białka histonowe)
Rozmiary chromosomów:
- wielkość nie jest związana z ich liczbą komórce ani ze stopniem rozwoju organizmu,
- im organizm na większym stopniu rozwoju tym więcej materiału genetycznego
- u człowieka są małe chromosomy, u cebuli długie, krótkie u roślin z rodziny kapustowatych
Chromosomy olbrzymie - politeniczne:
- taenia (łac.) - wstążka - powstają w wyniku wielokrotnej endomitotycznej replikacji chromosomów bez mitoz, składają się z wielkiej liczby ułożonych równolegle chromosomów o bardzo słabej spiralizacji,
chromosomy ślimakowe - w jądrach nie dzielących się komórek gruczołów ślinowych larw Dwuskrzydłych
chromosomy szczoteczkowe - w oocytach kręgowców
chromosomy tojadu - Aconitum - trujące
Przewężenia chromosomów:
pierwotne - zawierające centromer
- chromatyna słabo się barwi
- rozróżniamy rodzaje chromosomów ze względu na położenie centromeru
Metacentryczne
Submetacentryczne
Akrocentryczne
Telocentryczne (u konia i myszy)
- ramiona chromosomów: p (ramię krótkie), q(ramię długie) xp , xq
- region (kompleks): centromer (DNA) - kinetochor (białko)
- kinetochor - odpowiada za połączenie mikrotubul wrzeciona podziałowego do chromosomu i za właściwy ruch w trakcie podziałów komórki,
- Rodzaje kinetochorów:
a) zlokalizowany = chromosom monocentryczny - to najczęstszy rodzaj kinetochorów
b) niezlokalizowany (= dyfuzyjny = rozproszony) - w chromosomach holokinetycznych - brak przewężenia pierwotnego - mikrotubule przyczepiają się w wielu miejscach
wtórne - organizator jąderkowy Nor - Nuclear Organization Region
- występuje w chromosomach akrocentrycznych z satelitą = trabantem (to oddzielony przez przewężenie wtórne koniec chromosomu)
- chromosomy z przewężeniem wtórnym nazywamy chromosomami jąderkotwórczymi - w telofazie w przewężeniu wtórnym powstają jąderka, przewężenie wtórne zawiera RNA (materiał tworzący jąderko), jest widoczne jako przewężenie, ponieważ RNA barwi się inaczej niż DNA budujące cały chromosom
Liczba chromosomów:
- jest charakterystyczna dla gatunku, ale może być taka sama u różnych gatunków:
- liczba haploidalna - n (w gametach) - u glonów, mszaków, grzybów z chromosomów rozwijają się samodzielne organizmy - organizmy haploidalne
- liczba diploidalna - 2n (w komórkach somatycznych)
- liczba chromosomów z przewężeniem wtórnym jest stała dla danego genomu i odpowiada liczbie jąderek w interfazie, o ile nie nastąpiła fuzja jąderek, np.:
Człowiek 2n=46
Pszenica 2n=46
Cebula 2n=16
Czosnek 2n=16
Drosophila melanogaster 2n=8
n = x n = x = 23
2n = 2x 2n = 2x = 46
Podstawowa liczba chromosomów to x, ale nie zawsze n = x !
n = 2x => 2n = 4x (tetraploidalny)
- na każdą cechę przypadają 2 geny
Tetraploid 2n = 4x !
Aberracje liczbowe chromosomów :
Aneuploidia - aberracje kilku chromosomów (2-4)
Poliploidia - zmiana całego garnituru chromosomowego
Jak powstaje triploid:
2n + n - komórka jajowa zawiera 2n chromosomów, a plemnik n lub odwrotnie
Dispermia - kiedy komórka jajowa jest zapłodniona dwoma plemnikami
Endomitoza - podziały chromosomów bez podziałów komórki
Ad.2 - Poliploidia:
poliploidalność występuje częściej u roślin - 50% roślin to poliploidy
pewne tkanki mogą mieć różną liczbę chromosomów np. merystemy(2x), miękisz asymilacyjny, miękisz spichrzowy - jest zbudowany z komórek poliploidalnych
poliploidalność może być indukowana (przez człowieka)
kolchicyna - alkaloid (Colchicum autumnale - zimowit jesienny)
oryzalina - herbicyd - środek chwastobójczy
poliploidalność jest letalna dla człowieka
rośliny poliploidalne mają lepsze cechy - odporność, większy pyłek
- lecz są niepłodne lub mają obniżoną płodność
Allium porum - por - jest tetraploidem, z kolei hodowlane chryzantemy są dekaploidami
Poliploidalność u człowieka:
naturalna - niektóre tkanki - pojedyncze komórki łożyska, ale nigdy cały osobnik !!!
patologiczna - tkanki nowotworowe
organizmy poliploidalne, to najczęściej triploidy, rzadziej tetraploidy
najczęstsze kariotypy, które są poronieniami XXX, XXY, XYY
10% poronień to triploidy
Kariotyp - zespół chromosomów charakterystyczny dla komórek somatycznych danego osobnika
- kariotyp ≠ genotyp !!!
Zapis kariotypu :
- K(kobieta): 46, XX
- M(mężczyzna): 46,XY
Zespół Klinefertera: 47, XXY
Zespół Downa: dodatkowy 21 chromosom
Mozaikowatość 46 XX , 47 XXX
- część komórek ma zestaw prawidłowy, a część nie
Kariogram = ideogram - zestaw sfotografowanych lub narysowanych chromosomów jednej komórki opracowany wg zasad ustalonych na konferencji w Paryżu 1971 roku
Konferencja Denver (1960 r.):
- zasady ustalono podziału:
- A-G (grupy) - wielkość i położenie centromerów
- 1-22 - pary chromosomów
- chromosom X - grupa C, para 6
- chromosom Y - grupa G
Determinacja płci:
- haploidalna - diploidalna (odrębne) - np. pszczoły: n - męskie (w wyniku partenogenezy), 2n - żeńskie (z jaj zapłodnionych)
Rodzaje płci człowieka:
Rodzaj |
Kobieta |
Mężczyzna |
Genetyczna |
------ |
SDR (odcinek chromosomu Y) |
Chromosomowa |
X |
Y |
Chromatynowa |
chromatyna płciowa, ciałko Barra, pałeczka dobosza |
------ |
Gonadalna |
jajniki |
jądra |
Hormonalna |
estrogeny |
androgeny |
Fenotypowa(somatyczna) |
Noworodek - córka |
Noworodek - syn |
Metrykalna |
Imię żeńskie |
Imię męskie |
Psychiczna = psychoseksualna |
Model zachowania kobiety |
Model zachowania mężczyzny |
Płeć chromosomowa (XY):
Szeroko rozpowszechniona u zwierząt (XY)
Nie ma u roślin (wyjątek u mszaków!)
- u roślin nasiennych - rzadko - są rozdzielnopłciowe - np. szczaw, bniec biały
Determinacja płci u człowieka - moment zapłodnienia(fenotypowo):
Krew obwodowa
Izolacja limfocytów (hodowle in vitro) - 72 godziny
Fitohemaglutynina - stymuluje limfocyty do podziału
Kolchicyna - zatrzymanie mitozy w metafazie poprzez uszkodzenie mikrotubul wrzeciona kariokinetycznego, zamiast niej wykorzystuje się kolcemid - działa jak kolchicyna
Płyny hipotoniczne - chromosomy się rozsypują (szok hipotoniczny)
Utrwalenie
Barwienie metodą Feulgena (jednolite barwienie)
badanie prenatalne - w czasie ciąży
badanie postnatalne - po porodzie
Barr w interfazowych jądrach komórkowych samic kota znalazł nieznany dotąd element wyróżniający się od innych grudek chromatyny jądrowej wielkością i skondensowaniem - ten element to ciałko Barra (zwany tak z powodu nazwiska odkrywcy)
Ciałko Barra - w jądrach interfazowych niemal wszystkich somatycznych komórek samic ssaków. Jest to zasadochłonna (barwiąca się barwnikami zasadowymi) grudka chromatyny - przylega do błony jądrowej bezpośrednio
Wymiary 1,0-1,4 x 0,1-0,7 μm
Hipoteza wyjaśniająca istotę ciałka Barra
Według tej teorii ciałko Barra jest to jeden nieaktywny genetycznie chromosom X (ulega inaktywacji)
Regularna(94% występowania):
Mozaicyzm:
Zrównoważona - nie ma zmiany fenotypu
Niezrównoważona - jest zmiana fenotypu
Chromosomopatie = choroby chromosomowe - 4% ogółu chorób
Genopatie = choroby związane z genami - 96% , można je podzielić na:
monogenowe - uwarunkowane jedną parą alleli - 6% przypadków
poligenowe - uwarunkowane wieloma parami alleli - 90% przypadków
NIEDZIEDZICZNE- powstają poprzez uszkodzenie płodu w łonie matki na skutek różnych czynników np.:
stosowanie niektórych leków - czynniki teratogenne (teratos gr. - potwór) - np. działanie talidomidu
zarażenie toksoplazmozą
zespół alkoholizowanego płodu
DZIEDZICZNE
Autosomalne
Sprzężone z płcią
Przykurcz stawów międzypaliczkowych w jednym lub większej liczbie palców
Różny stopień ekspresji tej cechy, najczęściej dotyczy to palca piątego
Brak niektórych palców i palcozrosty u dłoni
Etiologia genetyczna i ekologiczna (czynniki teratogenne), uważa się także, że ektrodaktylia symetryczna jest dziedziczna, zaś niesymetryczna jest niedziedziczna
Występuje różny stopień ekspresji tej wady
Utrata słuchu w wieku 20-25 lat
Można operacyjnie usunąć skostniałą część
Występują włókniaki i nerwowłókniaki
Możliwa lekka złośliwość
Gen NF-1 w 17q (ramię dłuższe chromosomu), 1/ 3 300 urodzeń
Polipy dolnego odcinka jelita grubego
Ryzyko uzłośliwienia 70-100%
Leczenie objawowe - usunięcie całego jelita grubego
Izoantygeny = alloantygeny = izoaglutynogeny A, B
Substancja podstawowa, prekursorowi H - na błonie erytrocytów i innych komórek (oprócz układu nerwowego)
Są już u 6-tygodniowego płodu
Są glikoproteidami, ale ich aktywność i swoistość antygenowa związana jest z determinantami, którymi są reszty cukrowe, a nie z całą cząsteczką
Przy białaczce - osłabienie antygenów A i B
Sprzężenie grupy 0 z zespołem paznokciowo-rzepkowym:
Brak rzepki (mała kość w stawie kolanowym)
Małe paznokcie u rąk
Cecha autosomalne dominująca
Lokus genu w chromosomie 9, tak jak lokus genu AB0
Choroby:
Rak żołądka - grupa 0:
2x częściej niż u osób z grupy A i B
Bo podobieństwo antygenowe z Helicobacter pylori
Dżuma
Pałeczka dżumy - antygen-H, jak w grupie 0, osoby z grupą krwi 0 nie wytwarzają przeciwciał anty-A
A i AB - ospa
ciąży - pod wpływem krwinek płodu
po transfuzji krwi
pod wpływem pewnych drobnoustrojów (zakażenie), które zamierają antygeny podobne do antygenów A i B
matka grupa krwi 0 ojciec A lub B
u noworodka pojawia się żółtaczka, spowodowana wzrostem stężenia bilirubiny z rozpadu erytrocytów
uczuleni
krwinki płodu przez łożysko do matki tzw. przeciek ciąży
stymulacja przez krwinki płodu do wytwarzania antygenu anty Rh+
pamięć immunologiczna - szybsza i zwiększona odpowiedź immunologiczna
transfer przeciwciał przez łożysko do płodu
połączenie przeciwciał matki z antygenami dziecka
zmiany u dziecka:
mężczyzna |
kobieta |
46 XY |
45 X |
47 XXY |
46 XX |
48 XXXY |
47 XXX |
49 XXXXY |
48 XXXX |
Chromosomy metafazowe (płytka metafazowa):
ad.1
- pobieranie płynu owodniowego - amniopunkcja (amnioceteza)
Amnio - błona, centesis - przebicie
- badanie leukocytów z płynu owodniowego
- limfocyty płodu z krwioobiegu matki
- fibroblasty - komórki twórcze tkanki łącznej
- biopsja trofoblastu - pobiera się kosmki z kosmówki (narząd pośredniczący pomiędzy organizmem matki a organizmem dziecka)
Chromosom Y - 1/3 chromosomu X
- jest nieaktywny (prawie) - jest genetycznie prawie pusty, różni się kształtem, fluorescencją (części dystalne [końcowe] - inaczej świecą)
Techniki prążkowe - nowoczesne
- 1971 r. - schemat metody prążkowej GTG
- metody prążkowe umożliwiają jednoznaczną identyfikację i klasyfikację chromosomów oraz wyszukiwanie chromosomów płci, umożliwiają też wyszukiwanie aberracji chromosomowych
- chromosom barwi się w prążki - różne w zależności od metody
- analiza komputerowa - cytoscan - z zastosowaniem metody prążkowej
- prążki G uzyskuje się poprzez barwienie chromosomów barwnikiem Giemsy po uprzedniej, częściowej ich hydrolizie, GTG - G bo barwią się prążki G, T bo wytrawia się trypsyną, G bo barwi się barwnikiem Giemsy
- prążki Q uzyskuje się metodą fluorescencyjną - stosuje się fluorochormony
- prążki R - są odwrotnością prążków G, uzyskuje się je po wstępnej denaturacji i barwieniu barwnikiem Giemsy, pozwala to na ujawnienie drobnych aberracji niedostrzegalnych po wybarwieniu prążków G czy Q
Ciałko Y
- rozpoznanie chromosomu Y = ustalenie płci chromosomowej
- jest to dystalna część długich ramion chromosomu Y
Wykrywanie ciałka Y:
- metoda fluorescencyjna - fluorescencja po zabarwieniu fluorochromowymi barwnikami zarówno w jądrach interfazowych (nie dzielących się) jak i w czasie podziałów
- są trudności diagnostyczne - pod wpływem barwników barwią się też niektóre odcinki autosomów
- ciałko Y nie jest odpowiednikiem chromatyny X ani odpowiednikiem SDR !!!
- genotyp 46 XX oznacza brak ciałka Y (ciałko Y zależy od ilości chromosomów Y)
Płeć chromatynowa:
Hipoteza Marii Lyon:
Inaktywacja = heterochromatyzacja = lyonizacja:
- euchromatyna (aktywna genetycznie) heterochromatyna (nieaktywna genetycznie)
- długie ramiona chromosomu X centra inaktywacji
- cel - ilościowe zrównoważenie informacji genetycznej u kobiet i mężczyzn
- inaktywacja jest losowa - kwestia przypadku
- może być nieaktywny albo od matki albo od ojca(mozaika)
- przeważnie chromosomy o gorszej strukturze są inaktywowane
- liczba ciałek= X-1 gdzie X to liczba chromosomów, np. u mężczyzn jest 0
- inaktywacji ulegają wszystkie chromosomy dodatkowe
- w komórkach nowotworowych u kobiet niski poziom ciałek Barra
46, XX 1 ciałko Barra
47, XXX 2 ciałka Barra
Wykrywanie ciałek Barra:
- nabłonek jamy ustnej, cebulki włosowe, komórki płynu owodniowego
- bada się w 100 zakwalifikowanych do oceny (tzn. dobrze wybarwionych i nieuszkodzonych) jądrach komórkowych:
- 15-30% - norma dla kobiet
- 0-4% - norma dla mężczyzn
- zmiany samoistne w odsetku jąder z ciałkiem Barra w czasie życia osobnika oraz pod wpływem czynników zewnętrznych, chorób, leków
Pałeczka dobosza:
- chromatyna płciowa w granulocytach obojętnochłonnych w postaci pałeczki dobosza - wypustka jądra komórkowego
- liczba pałeczek dobosza to X-1
Hipoteza Lyon dziś:
- jeden z chromosomów ulega aktywacji, a nie odwrotnie
- jeden z dwóch nieaktywnych chromosomów X uaktywnia się (heterochromatyna euchromatyna)
Płeć molekularna:
- SDR - Sex Determing Region
- mały odcinek chromosomu Y wykrywalny tylko metodami molekularnymi
- delecja odcinka SDR
46, XY - SDR kobieta, ale bezpłodna, jajniki nierozwinięte prawidłowo, infantylne narządy płciowe, niedorozwój piersi, wzrost prawidłowy
- translokacja odcinka SDR:
46, XX + SDR mężczyzna, ale niepłodny, z ginekomastią, atrofią jąder, upośledzeniem spermatogenezy
- antygen H-Y to białko związane z błoną komórkową odpowiedzialne za rozwój pierwotnej gonady w kierunku męskim, brak go u normalnych kobiet
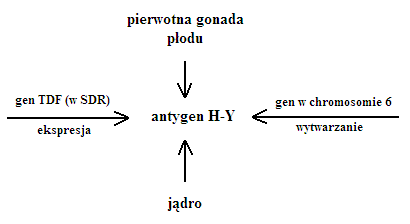
Pierwotna gonada
- za jej rozwój odpowiada antygen H-Y (kierunek męski)
- brak antygenu u kobiet
- za ekspresję SDR jest odpowiedzialny gen TDF
Relacje płci:
- większa przeżywalność kobiet w ciągu całej ontogenezy
- geny letalne sprzężone z płcią
- 106 chłopców/ 100 dziewczynek w momencie urodzenia
- 103 chłopców / 100 dziewczynek w momencie zapłodnienia
Przykłady chromosomopatii:
Zespół Downa
- John Langdon Down - „O etnicznej klasyfikacji upośledzenia umysłowego”
Złe określenia zespołu Downa:
- mongolizm, mongołowatość, idiotyzm mongoidalny - NIE MA TO NIC ZWIĄZANEGO Z RASĄ MONGOLSKĄ !!!
Zespół Downa trisomiczny i translokacyjny(rzadki):
- trisomia 21 pary:
47, XY, + 21
47, XX, + 21
46, XX / 47, XX, + 21
46, XY/ 47, XY, + 21
Przyczyny trisomii:
- zygota trisomiczna powstaje na skutek niewłaściwego zapłodnienia
- rzadko występuje nondysjunkcja w czasie pierwszych podziałów mitotycznych zygoty
- etiologia nondysjunkcji jest niewyjaśniona
- nondysjunkcja w czasie gametogenezy, głównie w 1 podziale mejotycznym
- występowanie:
Oogeneza - 95%
Spermatogeneza - 5%
Uwaga ! :
- część linii komórkowej może ulec zanikowi
- znaczna część ginie w embriogenezie
- według badań najnowszych to 22 para a nie 21, ale zamieniano je kolejnościami wbrew regule układania się chromosomów
Przykłady Trisomii
- trisomie chromosomów 1-7 są letalne
- trisomie letalne u zwierząt to trisomie chromosomów 12 i 19
- u roślin trisomie nie są letalne
Translokacyjny zespół Downa:
- przyczyna 3% przypadków zespołu Downa
Translokacja - to przemieszczenie fragmentu chromosomu na inny chromosom lub ten sam, ale w inne miejsce lub też przemieszczenie całego chromosomu na inny chromosom, wyróżniamy:
- translokację wzajemną - wzajemna wymiana odcinków między chromosomami niehomologicznymi
- translokacja robertsonowska - to fuzja:
- w translokacji zespołu Downa chromosom 21 ma któryś z chromosomów grupy D: 13, 14, 15 lub z grupy G: 21, 22
- kariotyp zespołu translokacji:
46, XX, der(14q, 21q), +21
46, XY, der(14q, 21q), + 21
46, XX, der(15, 21), +21
- osobnik z przemieszczonym chromosomem jest przenosicielem zespołu Downa, nie ma objawów zespołu Downa
- translokacja zrównoważona:
45, XX (14:21)
45, XX (13:21)
- nie ma nadmiaru materiału genetycznego, osobnik nie ma objawów zespołu Downa, osobnik jest nosicielem
- monosomia 21 (45, XX - 21) - „antymongolizm” - nazwa niewłaściwa - monosomia jest letalna
- nullisomia - brak pary 21 - jest letalna
- mozaicyzm - 44, XX, - 21, - 21 - kiedy w jednych komórkach jest jeden zestaw chromosomów prawidłowych i jeden zmienionych
(45, XX, - 21), (47, XX, + 21)
- nondysjunkcja - podziały gamet
A) nondysjunkcja mitotyczna
B) nondysjunkcja mejotyczna
Fenotyp zespołu Downa:
- zespół somatycznych i psychicznych
- krótkie palce i kończyny
- niski wzrost (K- 137, M-153)
- nieprawidłowe proporcje ciała (75% - krótka szyja, 80% - otwarte usta)
- duży język - macroglossia
- zmarszczka nakątna (jest to wąski fałd skóry nad kącikiem wewnętrznym oka) - u 97% chorych
- skośne ustawienie oczu
- wąskie szpary powiekowe
- krótkie kończyny, krótkie palce
- odstający palec i kciuk
- bruzda dłoniowa - bruzda w poprzek dłoni
- zmniejszona liczba listewek skórnych
- nieprawidłowe dermatoglify - odciski linii papilarnych
- częściej niż w zdrowej populacji plamki Brunshfielda (świecące plamki) w tęczówce
- strategia uczenia się bez błędów - metoda pracy z chorymi
IQ poniżej normy (w 100% przypadków zespołu D.) - 35-55 (w normalnej populacji 75-100)
- głębokie upośledzenie gdy IQ~ 25-40
- niezdolność do abstrakcyjnego myślenia
- upór, zdolność do naśladowania
- usposobienie pogodne
- chętnie grają na instrumentach i malują - ale nie mają uzdolnień w tym kierunku (!) - mają tylko możliwość wypowiadania się przez to
- zespół Downa dziedziczy się tylko w wypadku translokacji !!
Cechy chorobowe zespołu Downa:
- niedrożność nosa
- skłonność do nieżytu dróg oddechowych
- wady wzroku, m.in. zaćma
- wrodzone wady serca - np. `dziura w sercu'
- skłonność do białaczek i Alzheimera (około 30-35 roku życia)
- niedoczynność tarczycy
- niski poziom płodności u kobiet, bezpłodność u mężczyzn
- obniżone napięcie mięśniowe - u blisko 100% przypadków
- w przypadku mozaicyzmu cechy mogą być słabiej zaznaczone
Przeżywalność w zespole Downa:
- śmierć najczęściej z powodu wrodzonych chorób
* choroby układu oddechowego (zapalenie płuc)
* choroby serca
- wzrost przeżywalności związany m.in. ze stosowaniem antybiotyków oraz prowadzeniem operacji serca.
- średnia długość życia osób z zespołem Downa to obecnie 35-40 lat (w Anglii w 1929 r. było 9 lat)
Częstość zespołu Downa:
- 1/500-600 urodzeń
- w Polsce rodzi się rocznie 800 dzieci z zespołem Downa
- zależność między wiekiem matki:
* 30-34 lata - 1/800
* >40 - 1/50
Leczenie:
- tylko objawowo - nie istnieje możliwość leczenia przyczynowego !!!
- witaminy i hormony - zapobiegawczo
- tzw. terapia komórkowa - komórki z płodu owcy
- antybiotykoterapia i kardiochirurgia
- operacje plastyczne - dla poprawienia wyglądu (raczej dla poprawienia samopoczucia rodziców)
Inne trisomie autosomalne
Zespół Edwardsa:
- trisomia 18 pary chromosomów,
- trisomia regularna:
47, XX, + 18 47, XY, + 18
- trisomia mozaikowa:
46, XX / 47, XX, + 18 46, XY / 47, XY, + 18
- 95% przypadków to poronienia samoistne
- im większe chromosomy tym bardziej nasilają się objawy zespołu Edwardsa
Częstość występowania:
- 1/3 000 - 7 500 żywych urodzeń
- z urodzonych 30% umiera w 1 roku życia, 1 rok osiąga tylko 10% dzieci
Cechy fenotypowe:
- małogłowie
- małe, nisko osadzone oczy
- niedorozwoje nerek, oczu
- zniekształcenie stopy
- zachodzące na siebie palce rąk
Zespół Patau:
- trisomia 13 pary chromosomów
- trisomia regularna (75% przypadków) :
47, XX, + 13 47, XY, + 13
- trisomia mozaikowata (5% przypadków) :
46, XX / 47, XX, + 13 46, XY / 47, XY, + 13
- noworodki z zespołem Patau najczęściej są martwe lub umierają w pierwszych miesiącach po urodzeniu
Cechy fenotypowe:
- występuje głęboki niedorozwój umysłowy
- wilcza i zajęcza warga
- zdeformowane, nisko osadzone oczy
- zrosłopalczastość - SYNDAKTYLIA
- wady oczu - cyklopizm
- wady serca
- torbiele nerek
Trisomia 8:
- trisomia regularna:
47, XX, + 8 47, XY, + 8
- trisomia mozaikowata:
46, XX / 47, XX, + 8 46, XY / 47, XY, + 8
Cechy fenotypowe:
- duże małżowiny uszne, nisko osadzone
- duży nos
- niski wzrost
- skrzywienie kręgosłupa
- nadliczbowe kręgi i żebra
- rozszczepienie kręgów
- upośledzenie umysłowe
Inne wiadomości:
- letalne są trisomie regularne (niemozaikowe) największych chromosomów
- u roślin np. u bielunia dziędzierzawy (Datura stramonium) trisomia występuje i nie jest letalna
Aneuploidie heterochromosomalne
- dotyczą chromosomów płci - heterochromosomów (małych chromosomów)
Zespół Klinefeltera:
- 1/1 000 noworodków męskich
- trisomia heterochromosomalna (82%):
47, XXY
48, XXXY
48, XXYY
49, XXXXY - występują wady serca, układu kostnego, niedorozwój umysłowy
- mozaicyzm:
46, XY / 47, XXY
48, XXY/ 49, XXXY
- przyczyny - nondysjunkcja w czasie oo/spermatogenezy lub w czasie pierwszych podziałów mitotycznych
Cechy fenotypowe:
- normalny wygląd do momentu pokwitania
- wysoki wzrost (>180cm)
- długie kończyny
- niepłodność
- ginekomastia - powiększone gruczoły piersiowe
- azoospermia - narządy płciowe słabo rozwinięte
Wykrywanie - badanie chromatyny płciowej
Leczenie - tylko objawowo poprzez podawanie hormonów męskich - androgenów
Zespół Turnera:
- 1938r. - Henry Turner opisał zespół objawów
- monosomia regularna(60% przypadków) - zespół Turnera chromatynoujemny:
45, X lub 45, X0
- mozaicyzm - zespół Turnera chromatynododatni:
45, X / 46, XX
45, X / 47, XXX
- przyczyna - nondysjunkcja w czasie gametogenezy - nieznana przyczyna tej nondysjunkcji
- w 80% przypadków chromosom X w zespole Turnera pochodzi od matki
Cechy fenotypowe:
- u noworodków:
- nadmiar skóry na szyi
- opuchlizna limfatyczna stóp
- u dorosłych:
- niska linia włosów na karku
- opadające powieki
- dysmorfia twarzy - tzw. twarz sfinksa
- nisko osadzone małżowiny
- koślawość łokci i kolan
- puklerzowata klatka piersiowa
- masywne dłonie i stopy
- skrócona, płetwiasta szyja - tzw. „skrzydlik”
- niski wzrost
Cechy chorobowe:
- zmiany barwnikowe na skórze
- nerki uszkodzone, podkowiaste, wędrujące
- dysgenezja gonad - niedorozwój gonad, słabo rozwinięte gruczoły płciowe
- brak miesiączki
- długość życia normalna
- choroby nosa i gardła
- wady wrodzone serca
- skłonność do niedorozwojów tarczycy
- zaburzenia widzenia barwnego
- zaburzenia rozwoju umysłowego rzadkie - tzw. uciekanie w chorobę
Wykrywanie - poprzez badanie chromatyny płciowej X w postaci ciałka Barra
- wynik bliski 0 oznacza zespół Turnera
Częstość występowania zespołu Turnera:
- 1/ 3 000 żywych urodzeń
- w Polsce około 100-200 urodzeń rocznie
- przyjmuje się, że około 8 000 kobiet w Polsce choruje na zespół Turnera
- występuje tu zjawisko teratanazji, czyli samoistnego usuwania przez organizm kobiety nieprawidłowych zarodków i płodów
Leczenie - tylko objawowe
- leczenie otyłości poprzez stosowanie odpowiedniej diety i aktywności fizycznej
- leczenie terapeutyczne hormonami w podanej kolejności (nigdy odwrotnie!) :
1. terapia wzrostowa - przed 10 rokiem życia (w wieku 14-15 lat jedynie `terapia ostatniej chwili')
2. terapia hormonalna - estrogeny dla rozwinięcia cech kobiecych
- operacje kosmetyczno-plastyczne
- wsparcie psychologiczno-pedagogiczne - zwłaszcza w okresie dojrzewania, ponieważ mogą pojawiać się kompleksy oraz depresje
Inne, `ciekawe' kariotypy
- „męski zespół Turnera” - 45, X / 46, XY - bardzo rzadko występuje - wynikiem jest mężczyzna z niedorozwojem narządów płciowych
- 45, Y (= 45, Y0) - letalny (męski odpowiednik zespołu Turnera o kariotypie 45, X0)
- trisomia 47, XXX lub 46, XX / 47, XXX
- kariotyp dawniej zwany `nadkobietą' - termin obecnie niewłaściwy
- kobiety normalne psychicznie i fizycznie, choć może wystąpić obniżona inteligencja
- większość kobiet jest płodna
- częstość występowania: 1/1 000 żywych urodzeń
- 48, XXXX - zaburzenia w narządach płciowych, także w cechach płciowych
- zespół Patrycji Jacobs - 47, XYY:
- tzw. `nadmężczyzna' - supermężczyzna
- wykryty przypadkowo u zdrowego ojca dziecka z zespołem Downa podczas badań diagnostycznych genezy choroby dziecka
- kiedyś uważano, że to kariotyp `wrodzonej przestępczości', obecnie uważa się, że 96% mężczyzn o tym kariotypie jest normalnych - nie są przestępcami !!!
- symptomami zespołu P.J. mogą być: trądzik, wysoki wzrost i problemy z mówieniem
- przyczyną zespołu P.J. jest nondysjunkcja - dotyczy spermatogenezy
Choroby genetyczne
ad. A) - Genopatie monogenowe
A.a) autosomalne dominujące (AA, Aa)
A.b) autosomalne recesywne (aa)
A.c) sprzężone z chromosomem X - dominujące
A.d) sprzężone z chromosomem X - recesywne
Cechy autosomalne - to cechy kodowane przez geny występujące w chromosomach autosomalnych, dziedziczą się według praw Mendla
Dziedziczenie autosomalne - zapis alleli
A - allel dominujący
a - allel recesywny
AA - homozygota dominująca
aa - homozygota recesywna
Aa - heterozygota
Cechy prawidłowego fenotypu - cechy monogenowe:
- np. oczy czarne (CC i Cc) i niebieskie (cc), grupa krwi A (LA LA , LA L0) i grupa 0 (L0 L0)
Cechy monogenowe patologiczne:
- WW i Ww - ujawnia się choroba/wada
- ww - brak objawów wady, osoba zdrowa
P (parentes) : WW x ww
F1(pokolenie pierwsze) : Ww, Ww, Ww, Ww
100% z wadą lub chorobą
- rodzice w sensie genetycznym (krzyżówka Mendla) są homozygotami - jedno jest homozygotą dominującą, a drugie homozygotą recesywną
- rodzice w sensie biologicznym (krzyżówka biologiczna) są heterozygotami - nie oznacza się wtedy literami `P' i `Fx' pokolenia rodzicielskiego i potomnego
Przykłady krzyżówek biologicznych:
WW x Ww
Ww, Ww, WW, WW
Statystycznie 100% dzieci z wadą lub chorobą
Ww x Ww
WW, Ww, Ww, ww
Statystycznie 75% dzieci z wadą lub chorobą
Ww x ww
Ww, Ww, ww, ww
Statystycznie 50% dzieci z wadą lub chorobą
Wady wrodzone
Amelia - brak kończyn
Fokomelia - brak dłoni / stóp
a) monogenowe
b) poligenowe
Przykłady wad autosomalnych monogenowych (wady dłoni i stóp) :
- polidaktylia = wielopalczastość - dodatkowe palce, leczy się operacyjnie, częstość to 1/25 000
- syndaktylia = palcozrost - dotyczy części miękkich lub miękkich i kostnych, powstaje na skutek nie rozdzielenia się kończyn na palce podczas życia płodowego
- kamptodaktylia - skrzywienie palca/palców
- brachydaktylia = krótkopalczastość - skrócenie paliczków pojedynczych (np. środkowych) lub wszystkich palców, cecha dominująca (!), częstość w Europie to 1/1 500 żywych urodzeń
- ektrodaktylia = `szczypce homara'(więcej o tym na ćwiczeniach) :
Przykłady chorób monogenowych dominujących:
- torbielowatość nerek dorosłych - torbiele w nerkach, wątrobie i śledzionie, (nie mylić z torbielowatością nerek wieku dziecięcego - to dziedziczy się monogenowo recesywnie !!!)
- otoskleroza - patologiczne skostnienie błędnika, (oto - ucho, skleros - twardy)
- nerkowłóknowatość - choroba Recklinghausena
- polipowatość rodzinna gruczolakowata jelita grubego
- albinizm oczny - brak barwnika w tęczówce, normalny wygląd ogólny, albinizm może być zaliczony do wad
Dziedziczenie cech monogenowych - fizjologicznych
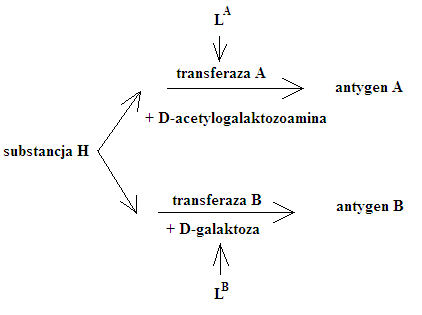
- układ grupowy krwinek AB0
Antygen :
- cząstka wiążąca się selektywnie z przeciwciałami
- izoprzeciwciała
- alloprzeciwciała
anty-A = α
anty-B = β
to są przeciwciała naturalne, są stałymi składnikami osocza krwi, nigdy nie są skierowane przeciw własnym
- od 3 miesiąca życia u człowieka są przeciwciała, wcześniej są matczyne
- przeciwciała anty-A i anty-B powstają w odpowiedzi na materiał A i B w komórkach bakterii
- z wiekiem spada ich stężenie
Reguła Landsteina:
- w surowicy / osoczu - nie występują aglutyniny dla cech grupowych zawartych we własnych krwinkach
- stwierdza się obecność aglutynin dla cech grupowych nieobecnych we własnych krwinkach![]()
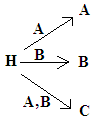
Allelizm wielokrotny, allele wielokrotne, odmiany allelomorficzne genu strukturalnego
- alternatywne formy genu
- 3 różne allele układu AB0 kodujące antygeny A i B, czyli allele strukturalne
- kontrolujące syntezę tych antygenów locus (miejsce genowe 9q 34
różny zapis:
IA = LA = A
IB = LB = B
I0 = L0 = i = 0
Allel amorficzny, nie koduje syntezy jakiegokolwiek antygenu
- antygeny regulatorowe - kontrolują czynności genów strukturalnych
Allelizm wielokrotny, allele wielokrotne
- odczyn zlepny = aglutynacja - zlepianie się antygenów w środowisku zawierającym swoiste przeciwciała
- izoaglutynacja - hemaglutynacja
- LA > L0 dominacja, gr. krwi A
- LB > L0 dominacja, gr. krwi B
- LA = LB kodominacja = współdziałanie w parze alleli - współdominowanie, gr krwi AB
- prawo Dungerna i Hirszfelda - 1910 r. - cechy grupowe krwi dziedziczą się według praw Mendla
- Dungern i Hirszfeld wyodrębnili 2 odmiany antygenu A
- `mocną' A1 u ok. 80% grup krwi A
- `słabszą' A2 u ok. 20% grup krwi A
- podgrupy te wyodrębniają się ok. 6 miesiąca życia, przy czym A1 > A2
- antygeny słabsze od A i antygeny podgrup B są rzadkie
Wydzielanie antygenów grupowych AB0
- antygeny A i B, jak i substancja podstawowa H mogą być wydzielane do wydzielin i wydalin ustrojowych, takich jak: ślina, mocz, mleko, osocze, sok żołądkowy
- substancje grupowe w ślinie reagują swoiście z przeciwciałami, jednak nie posiadają zdolności pobudzania do syntezy przeciwciał
Wydzielacze:
- antygeny w płynach ustrojowych - 80% populacji, w Polsce 84% - uwarunkowania genetyczne
- Se = W - Allel wydzielania
- Se = w - Allel niewydzielania
Genotypy:
- Se Se (W W)
- Se se (W w)
- se se (w w)
Fenotyp:
- Wydzielacie
- Wydzielacie
- Niewydzielacze
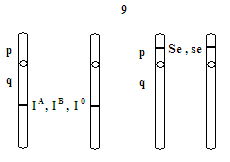
- locus genu kodującego wytwarzanie antygenów A i B w chromosomie 9q, a genu kodującego wydzielanie tych antygenów w chromosomie 19p, a więc kodowanie antygenów układu AB0 jest niezależne od kodowania wydzielania
- w danym locus może być jeden z 3 alleli (allelizm wielokrotny)
- cechy te dziedziczą się niezależnie od siebie, według II prawa Mendla - współdziałanie
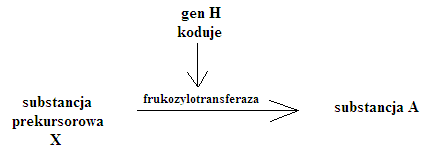
Epistaza - hamowanie w układzie AB0 = grupa krwi Bombay = fenomen bombajski
Matka - grupa krwi B IB IB , IB I0
Ociec - grupa krwi 0 I0 I0
Dziecko może mieć IB I0 lub I0 I0
- badania krwi dziecka: brak aglutynacji z surowicami anty-A i anty-B oraz anty-H
- surowica anty-H aglutynuje krwinki 0, które posiadają substancję H
- zapis genotypu dziecka: IB I0 hh lub IB I0 xx
(nie sprzężone)
h(x) - allel hamujący - epistatyczny
IB - allel hamowany - hipostatyczny
- substancja H jest wytwarzana jeśli są genotypy HH i Hh
- jeśli jest hh to nie ma substancji H i nie jest wytwarzany antygen B, bo w procesie powstawania grupy jest zablokowana produkcja transferazy (frukozylotransferazy)
- rodzice zatem byli heterozygotami:
Matka IB I0 Hh ojciec I0 I0 Hh
|
|
Grupa 0 |
Grupa Bombay |
Genotyp |
|
L0 L0 HH lub L0 L0 Hh |
LB LB hh lub LB L0 hh |
Obecność antygenów |
A |
- |
- |
|
B |
- |
- |
|
H |
+ |
- |
Obecność przeciwciał |
anty-A |
+ |
+ |
|
anty-B |
+ |
+ |
|
anty-H |
- |
+ |
Aglutynacja z surowcami |
anty-A |
- |
- |
|
anty-B |
- |
- |
|
anty-H |
+ |
- |
Transferaza B |
|
- |
+ |
Prawa Mendla:
I) każda gameta wytwarzana przez organizm posiada tylko jeden allel z danej pary alleli genu. Wynika z tego, że każda komórka płciowa musi zawierać po jednym genie z każdej pary alleli.
II) geny dziedziczą się niezależnie od siebie. geny należące do jednej pary alleli są dziedziczone niezależnie od genów należących do drugiej pary alleli.
Grupy układu AB0 a choroby:
Konflikt serologiczny w układzie AB0:
- niezgodność to nie to samo co konflikt (!)
- w układzie AB0 występują zarówno regularne jak i nieregularne przeciwciała
- immunizacja - uodpornianie - może zachodzić w:
- niezgodność matka-dziecko:
dziecko A lub B
Grupa krwi a transfuzjologia:
- próba krzyżowa - test in vitro zgodności pomiędzy krwią dawcy a biorcy
Występowanie grup krwi:
- grupa 0:
- najwięcej w Ameryce Pn.(70-80%) i Pd.(90-100%), bo nie było tam dżumy(atakuje grupę 0)
- rzadziej w Indiach, Mongolii, na terenach ognisk dżumy
Fitochemaglutyniny:
- substancje białkowe wiążące cukry
- występują w roślinach (wyciągi z nasion i liści) i grzybach (wyciągi)
- charakteryzują się działaniem fitogenicznym
- stosuje się je do oddzielania białych od czerwonych krwinek
fasola |
bób |
Kolcosit europejski |
zlepia krwinki A |
zlepia krwinki A |
zlepia krwinki A |
- z grzybów
Lakówka pospolita |
Huba pospolita |
zlepia krwinki A i 0 |
zlepia krwinki B |
Układ MNSs
- składa się z układów MN i Ss
- antygeny grupowe krwi M i N, dziedziczą się niezależnie od układu AB0
- mają małą aktywność
- znaczenie mają przeciwciała anty-M i anty-N
- przeciwciała produkuje się z krwi królików
MN:
- fenotypowo - M (36%) , MN (48%) - najczęstszy , N (16%)
- allele - LM = M LN = N = MN
- występuje tu zjawisko kodominacji (= współdziałania)
- genotypy:
LM LM = MM
LN LN = NN = MN MN
LM LN = MN = M MN
- układ MN nie ma wyraźnego znaczenia klinicznego
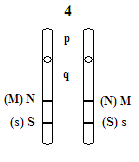
Ss:
- tu też występuje zjawisko kodominacji
- allele - S i s S = s
- dziedziczą się wbrew prawom Mendla
- może wystąpić konflikt
- przeciwciała anty-S mają charakter odpornościowy
- lokus 4q 31, rzadko crossing-over
Genotypy |
Fenotypy |
LMS LMS |
MS |
LNS LNS |
NS |
LMS LMS |
MNS |
LMS LMs |
MSs |
LNS LNs |
NSs |
LMS LNs |
MNSs |
Układ Rh
- Landsteiner i Wiener w 1940 r. uodparniając króliki i świnki morskie orkryli nowe przeciwciała aglutynujące krwinki małp, nazwano go genem Rh
- allele Rh i rh:
rh rh - Rh -
Rh Rh
- Rh +
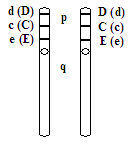
Rh rh
- genotypy - zapis rozwinięty
- allele C, D, E, c, d, e
- dominacja C > c , D > d , E > e
- przykłady genotypów (jest możliwych 35):
CDE / CDE , CDe / Cde , cDe / CDE itp. - Rh +
cde / cde - Rh -
- Allel D - jest najsilniejszym, o znaczeniu leczniczym
- Allel d - amorficzny, nie koduje żadnego antygenu
- antygeny są na błonie erytrocytów
- genotypy - 2-gi zapis uproszczony:
* DD i Dd - Rh+ - w Polsce odpowiednio - 35% i 49%
* dd - Rh- - w Polsce - 17%
- jeżeli oboje rodzice mają Rh+ to dziecko mimo tego może mieć zarówno Rh- jak i Rh+
- wykrywanie:
* dawniej za pomocą surowic zwierzęcych
* obecnie z surowic ludzkich:
a) w przypadku konfliktu serologicznego
b) od osób poddanych immunizacji
Konflikt serologiczny grupy Rh:
- to choroba płodu, a później noworodka wywołana przez przeciwciała matki
- konflikt występuje, gdy u kobiety jest immunizacja
- dopiero w 1924 r. Hirszfeld i Zborowski przedstawili hipotezę na temat Rh
Immunizacja kobiety:
- ciąża
- poronienie samoistne
- poronienie sztuczne (aborcja)
- cesarskie cięcie
- heterohemoterapia - lecznicze domięśniowe wstrzykiwanie krwi
- amniopunkcja
- biopsja trofoblastu
- przetaczanie krwi
- narkomania
Etapy konfliktu serologicznego:
- konflikt serologiczny = choroba hemolityczna noworodków - objawy:
* niedokrwistość
* żółtaczka
* obrzęk uogólniony - 100% śmiertelności, nie leczona choroba hemolityczna - 75% śmiertelności
uwolniony ze zniszczonych krwinek hem przekształca się w bilirubinę
uwolniony CO2 łączy się z hemoglobiną w innych erytrocytach i obniża ich zdolność transportową
obrzęk płodu w 18-19 tygodniu ciąży
skutkiem choroby hemolitycznej jest przewlekła niedokrwistość
może dojść do obumarcia płodu
po urodzeniu objawy konfliktu są mocniejsze
część bilirubiny jest odprowadzana do organizmu matki
Wykrywanie:
identyfikacja krwinek płodu z krwi matki
badanie krwi płodu
określenie genotypu
odczyn antyhemoglobinowy Coombsa
Leczenie:
- pierwotnie stosowano przedwczesny poród (ze względu na wzrastającą przepuszczalność łożyska)
- perinatologia:
a) transfuzja fetoskopowa
b) transfuzja dootrzewnowa
c) transfuzja wewnątrzotrzewnowa
- transfuzja krwi u noworodków
- plazmoforeza
- immunoforeza - osocze dawców immunizowanych dla uzyskania większego miana krwinek
Profilaktyka:
- zapobieganie immunizacji kobiet
- osłabienie odpowiedzi immunologicznej - leki immunosupresyjne
- podawanie surowicy anty-D zaraz po urodzeniu dziecka
Albinizm (=bielactwo) uogólniony tyrozynoujemny:
- cecha monogenowe recesywna
- defekt przemiany tyrozyny, brak enzymu - tyrozynazy - przemieniającego tyrozynę
- zahamowanie syntezy melaniny w melanocytach (brak w skórze, włosach, tęczówce):
* melanosomy - pęcherzyki gromadzące melaninę
- u różnych raz są różne ilości melanosomów oraz melaniny:
* u rasy nigroidalnej brak enzymów rozkładających tyrozynę

Fenotyp:
- skóra różowa
- włosy bezbarwne
- tęczówki różowe lub niebieskie
- zmniejszenie ostrości wzroku
Mukowiscydoza = zwłóknienie, zwyrodnienie torbielowate trzustki = Cystic Fibrosis = CF
- najczęstsza choroba autosomalne rasy kaukaskiej
- częstość 1/2 000 - 1/3 000, w Polsce 1/2 300, Azjaci 1/90 000, Murzyni 1/17 000
- gen CFTR - 25 tys. nukleotydów, gen znajduje się w chromosomie 7q 23,2
* gen również koduje białko o nazwie CFTR lub białko kanału chlorkowego(reguluje procesy wydzielania wewnętrznego
- jest więcej jak 500 mutacji tego genu:
* mutacja najczęstsza - trójnukleotydowa delecja
* brak kodonu CTT brak w białku CFTR w pozycji 508 aminokwasu fenyloalaniny
* ta jedna delecja powoduje silne zaburzenia w regulacji procesów wydzielniczych organizmu (zaburzenia w transporcie jonów chlorkowych przez błony)
- do krzyżówki:
* mm - chory
* MM - zdrowy
* Mm - nosiciel
Fenotyp:
- jest zmienny, w różnych chorych jest różny
- wielonarządowa manifestacja choroby:
* zasadniczo dotyczy układu oddechowego i pokarmowego
* nadmierne wydzielanie śluzu, zwłaszcza w płucach
- duża śmiertelność - średnia wieku to około 30 lat
- występuje również niewydolność trzustki
- w ślinie i pocie wzrasta stężenie jonów chlorkowych
- heterozygoty Mn mają zwiększoną odporność na zakażenia bakteryjne dróg oddechowych i jelit
Wykrywanie:
- ocena zmian płucnych
- testy trzustki
- poziom jonów Cl- w ślinie, ogólne badanie Na+
- badanie DNA w płynach prenatalnych,
- prowadzi się badania przesiewowe noworodków
- do tej pory jedynie 1 opisany nowy przypadek mutacji
Leczenie:
objawowe:
- leczenie dietą wysokokaloryczną
- stosuje się enzymatyczne preparaty trzustkowe
- antybiotykoterapia
- przeszczepy płuc, płucoserca
- inhalacja płuc, odsysanie śluzu
przyczynowe:
- „poprawianie genu” - genowa terapia somatyczna - wprowadza się `dziki', nie zmutowany gen
- transfekcja - podawanie genu do komórek nabłonka oddechowego
- retro lub adeno wirusy mogą być stosowanie w leczeniu
Fenyloketonuria
- blok metaboliczny:
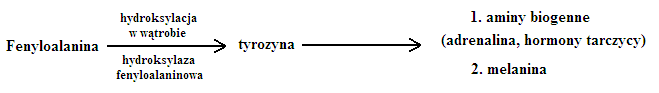
- zdiagnozowana - 1934 r. Fölling
- genotypy:
* FF - zdrowy
* ff - chory
* Ff - nosiciel
- lokus 12q 24
- częstość to 1/2 000 - 1/20 000 żywych urodzeń
- u chorych:
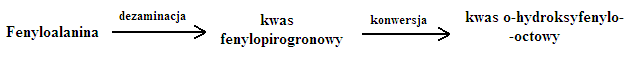
Fenotyp:
- objawy ujawniają się dopiero około 3 miesiąca życia
- obniżone IQ, jasne włosy, jasna skóra
- wymioty, napięcie mięśniowe
Wykrywanie:
- test moczu - oliwkowe zabarwienie moczu
- test Guthriego - zastosowanie Bacillus subtillis żywiących się fenyloalaniną
- DNA - badania prenatalne
Leczenie:
- dietetyczne - bez fenyloalaninowa dieta (bez mleka):
* jeśli stosuje się dietę odpowiednio wcześnie, to można nawet wyleczyć
- fenokopia
Galaktozemia

- zaburzenia w przemianie galaktozy:
- lokus 9p
- występowanie 1/12 000 - 1/30 000 żywych urodzeń
- nietolerancja laktozy i mleka krowiego
Wykrywanie:
- oznaczanie aktywności urydynotransferazy
Leczenie:
- dietetyczne
- plejotropia
Niedokrwistość sierpowatokrwinkowa =
= hemoglobinopatia S
- występowanie 1/250 urodzeń, Afryka - murzyni
- hemoglobina A (normalnie we krwi) = hem + globina A(2α, 2β) - pozycja 6 w łańcuchu β - kwas glutaminowy
- hemoglobina S = hem + globina S (2α, 2β) - zmieniony - pozycja 6 w łańcuchu β-waliny
Genotyp |
Fenotyp |
HbS / HbS |
- niedokrwistość sierpowatokrwinkowa - 50% krwinek sierpowatych - zamiast hemoglobiny A jest hemoglobina S - erytrocyty uszkodzone (zawierają stronty, łatwo hemolizują, zmienny ładunek elektryczny) |
HbA / HbS |
- zdrowa osoba - krwinek sierpowatych 1% - posiada obie hemoglobiny (A i S) - występuje wrażliwość na obniżanie ciśnienia tlenu |
HbA / HbA |
- zdrowa osoba - posiada hemoglobinę A |
- kodominacja - wytwarzanie hemoglobiny HbA = HbS, podobnie było z grupami krwi A i B, układ…
Objawy (fenotyp):
- bóle głowy, żółtaczka
- niewydolność nerek, owrzodzenie goleni
- powiększenie wątroby i nerek
- zagrożeniem jest:
*duży wysiłek fizyczny
* znaczna wysokość
* wysiłek, podróż samolotu
Wykrywanie:
- badanie krwinek (inkubacja w 37*C:
* 0,5-1 godzina - HbS / HbS
* 4-6 godzin - HbA / HbS
- elektroforeza - badanie krwinek za pomocą prądu
- diagnostyka molekularna - badanie płynu owodniowego - bada się DNA za pomocą restryktaz, które dzielą DNA na fragmenty
Uprzywilejowanie heterozygot:
- preferowane na terenach malarycznych
- zarodziec malarii nie może się prawidłowo rozwijać
- 10% Murzynów HbA / HbS
Inne hemoglobinopatie - talasemia
- występowanie - rejon morza Śródziemnego:
- talasemia α - niedostateczna synteza łańcucha α
- talasemia β - niedostateczna synteza łańcucha β
Fenotyp:
- erytrocyty tarczowate
- niedokrwistość, powiększenie śledziony
Cechy sprzężone z płcią:
- to cechy zależne od genów znajdujących się w chromosomach płciowych:
* hemizygotyczność - pojedynczo allele w genotypie diploidalnych
* hemizygota - osobnik hemizygotyczny (hemi-połowa)
- są to cechy zależne od genów autosomalnych, ale możliwość ich rozwoju zależy od płci
Hemofilia = krwawiączka = choroba królewska
- genetyczny defekt układu krzepnięcia - skaza krwotoczna
- hemofilia A - brak czynnika VIII (globulina antyhemolityczna = globulina przeciwkrwawiączkowa)
* synteza w wątrobie
- w Polsce około 1/12 000 urodzeń
- królowa Wiktoria była nosicielką i jej córki także
- hemofilia A - lokus genu 10p 28, gen strukturalny, ……………….
- hemofilia B - lokus 10q 27.1-27.2 , w Polsce 1/30 000 urodzeń, brak czynnika X
- w lokusie hemofilii A jest też umiejscowiona ślepota na barwy
- hemofilia możliwa jest również u zwierząt
Objawy:
- objawy hemofilii ujawniają się w 1-2 roku życia
- ogólnonarządowe, pourazowe krwawienia
- krwawienie z błon śluzowych
|
Genotyp |
Fenotyp |
homozygoty |
XH XH |
Kobieta zdrowa |
|
XH Y |
Mężczyzna zdrowy |
|
Xh Xh |
Kobieta chora |
|
Xh Y |
Mężczyzna chory |
|
XH Xh |
Kobieta zdrowa, ale nosicielka, obniżona zawartość czynnika VII |
Częstość występowania homozygot:
Xh Xh = 1/10 000 x 1/10 000 = 1/100 000 000
Hipoteza Lyon a hemofilia:
- jeden chromosom jest aktywny, drugi nieaktywny
Xmat Xpat
1) |
XH Xh act |
50% |
|
Kobieta zdrowa |
|
XH act Xh |
50% |
czynnik VIII + |
|
2) |
XH act Xh |
100% |
czynnik VIII + |
Wszystkie……………………………….(?) Aktywny chromosom z allelem zmutowanym |
3) |
XH Xh act |
100% |
czynnik VIII - (brak czynnika) |
Jawna heterozygota - hemofilia |
Wykrywanie:
- diagnostyka DNA
Leczenie:
objawowe:
- leczenie substytucyjne - podajemy czynnik VII dożylnie, podawanie okresowe
- czynniki VIII jako produkt inżynierii genetycznej
przyczynowa:
- terapia genowa (gen typu dzikiego - transfekcja do szpiku)
Daltonizm
- ślepota na barwy
- według Younga-Hernholza - widzenie 3 barw - 3 fotoreceptory
- achromatyzm - całkowita ślepota na barwy, wada autosomalna recesywna
- monochromatyzm -
- dichromatyzm - daltonizm - widzenie dwóch barw:
a) tritanopia - niewidzenie barwy niebieskiej, dziedziczenie autosomalne monogenowe
b) protanopia - niewidzenie czerwnieni
c) deuteranopia - niewidzenie zieleni
- malie - niedowidzenie barw:
- tritanomalia
- protanomalia
- deuteranomalia
- wykrywanie daltonizmu za pomocą metody pigmentowej
P: Xd Y x XD XD Xh Y IA IB (hemofilik) x XH XH I0 I0 (zdrowa)
F1: XD Xd , XD Y XH Xh IA I0, XH Xh IB I0, XH Y IA I0, XH Y IB I0
Dystrofia mięśniowa postępująca typu Duchenne'a
- najcięższa i najczęstsza odmiana dystrofii
- częstość - 1/3 000 chłopców
- gen DMD - jeśli jest niezmutowany to koduje białko zwane dystrofiną
- Allel typu zmutowanego (delecja) Xdst
- Allel typu dzikiego XDst
- genotyp z dystrofią: Xdst Xdst , Xdst Y
- wzrost poziomu enzymów: kinazy kreatynowej i dehydrogenazy mleczanowej
Objawy:
- ujawnia się w 2-3, a nawet 5-6 roku życia
- osobie trudno wstać z siadu
- dziwny, kaczkowaty chód
- charakterystyczny rozrost łydek (tkanka łączna a nie mięśniowa!)
- dawniej śmierć około 10 roku życia, obecnie najczęściej około 20, a czasem 30 roku życia
- śmierć z powodu niewydolności oddechowej oraz niewydolności krążenia
Wykrywanie:
- badania DNA:
a) prenatalne
b) postnatalne
- elektromiogram
Leczenie:
- podaje się steroidy anabolizujące, kwas glutaminowy
Głuchota - cecha sprzężona z płcią !
Dominujące choroby sprzężone z płcią:
- istnieją cechy sprzężone z chromosomem Y, ale są one niezwykle rzadkie
- niektóre cechy mylnie są przypisywane do chromosomu Y
Krzywica odporna na witaminę D = hipofosatemia
- wydzielana duża ilość fosforanów z moczem
Objawy:
- takie jak w zwykłej krzywicy, ale nie leczy się jej witaminą D, bo jest odporność
Istniał spór między zwolennikami Galtona i Mendla
Genetyka mendlowska Mendelizm
- dziedziczenie jakościowe
- krzyżówki mendlowskie
- prawa Mendla
- sukcesy:
1. czyste linie grochu
2. analiza matematyczna wyników badań
Genetyka Galtona dziedziczenie ilościowe
- lekarz, twórca eugeniki, ojciec daktyloskopii
- zajmował się cechami t.j wzrost, inteligencja, uzdolnienia, linie papilarne
Fischer:
- kuzyn Darwina, matematyk, eugenik
- zajmował się dziedziczeniem poligonowym, genetyka populacji
- „inbreeding” - chów wsobny
Dziedziczenie ilościowe spowodowane współdziałaniem dużej liczby genów
Współdziałanie w dziedziczeniu - rodzaje:
- w parze alleli - kodominacja
Genów niesprzężonych:
współdziałanie 2 genów:
epistaza np. grupy krwi Bombaj
komplementarność
poligenia:
jakościowa = dziedziczenie
ilościowa = dziedziczenie addytywne = ciągłe
głównie chirurgiczno-plastyczne - plastyka warg
foniatryczne (?)
ortodontyczne
pediatryczne
insulinozależna = typu I - tzn. młodzieńcza albo wieku młodzieńczego
insulinoniezależna = typu II - tzn. starcza, dorosła,
b)
transgresja dodatnia
transgresja ujemna
test wiadomości
„braki w obrazkach”
powtarzanie cyfr - pamięć krótkotrwała
„porządkowanie obrazków”
definiowanie słów - „słownik”
klocki
arytmetyka
rozumienie
układanki
symbole cyfr
test podobieństwa
wariancja genetyczna
wariancja środowiskowa:
migracja (wieś-miasto)
odżywianie
przeżywalność
wykształcenie
dobór panmiktyczny (panmiksja) - dobór nieprzypadkowy = selektywny = asortatywny
brak mutacji
spontaniczne
indukowane
szkodliwe
obojętne
pozytywne
osobnika tego samego gatunku
osobnika innego gatunku
może być genem syntetycznym - heterologiczny gen - gen obcy - wbudowany do materiału genetycznego i przekazywany zgodnie z prawami genetyki
zmodyfikowane mikroorganizmy:
enzymy:
soja - pierwsza roślina transgeniczna
pomidor
kukurydza (koncern Monsanto )
ziemniak
bawełna
rzepak
burak cukrowy
ryż
batat
zwiększenie tempa wzrostu i jakości mięsa
zwiększenie wydajności mlecznej i jakości mleka
żywe bioreaktory - do produkcji ludzkich białek o działaniu leczniczym
w gruczołach mlecznych
we krwi
w moczu
transgeniczne kury znoszące jajka z białkami do produkcji leków przeciwnowotworowych
transgeniczne zwierzęta do badania ludzkich chorób
transgeniczne zwierzęta do uzyskiwania ksenoprzeszczepów
medyczne:
plany życiowe:
możliwość zmiany trybu życia pod kątem choroby
niekorzystne reakcje na wynik
konflikty rodzinne
naruszenie poufności
utrata zatrudnienia
stygmatyzacja społeczna
izolacja DNA
powielanie = replikacja fragmentu DNA in vitro
badanie w sekwencjach DNA
wynik na ekranie
w medycynie sądowej:
identyfikacja sprawców przestępstw
dochodzenie ojcostwa
ustalenie rodziców zaginionych
badanie szczątków ludzkich - powielanie materiału metodą PCR - badanie m.in.:
mumii egipskich
mózgu człowieka z Florydy
9300-letniej czaszki człowieka z Kennevick
Fryderyk II - średniowiecznego władcy, cesarza Niemiec i króla Sycylii w celach ustalenia pochodzenia
mitochondrialne
chloroplastowe
wyizolowanie DNA
trawienie restryktazami - otrzymanie fragmentów DNA
rozdział fragmentu DNA na żelu agarowym
wprowadzenie fragmentów DNA do komórki bakterii za pomocą wektorów plazmidowych lub fagowych dla prokariontów lub niższych eukariontów a dla wyższych eukariontów chromosomu drożdżowego
glikozylacja
formowanie połączeń dwusiarczkowych (=mostków disiarczkowych)
fałdowanie łańcucha polipeptydowego
oligomeryzacja
metabolicznej aktywności komórki
gatunku organizmu
typu komórki (brak porów w osłonkach plemników)
DNA
Białka histonowe i niehistonowe
euchromatyna:
heterochromatyna:
mitochondria
peroksysomy
glioksysomy
chloroplasty
liczne podobieństwa między współczesnymi mitochondriami
geny bakteryjnych prekursorów mitochondrialnych wbudowane w genom komórki Eukariontów (genom mitochondrialny pozostałością genomu bakteryjnego
alternatywne = nieciągłe
- poligeny wykryte na początku XX wieku, dziedziczenie barwy ziarniaków przenicy zależnie od 3 pary alleli:
ABC - dominujące (aktywne)
Abc - recesywne (nieaktywne)
Np.:
P: ABCDEFGHIJ x abcdefghij .
F1: AaBbCcDdEeFfGgHhIiJj
Poligenia = dziedziczenie poligenowe - uwarunkowanie wieloczynnikowe
- kontrola jednej cechy fenotypowej odbywa się przez wiele współdziałających genów
Wady dziedziczone poligenowo:
Rozszczepy = rozszczepienie (brak ciągłości anatomicznej i niedorozwój tkanek):
a) podniebienia tzw. wilcza warga
b) wargi tzw. zajęcza warga
c) oba jednocześnie
- częstość występowania 0,4-2/1 000
- zaburzenia oddychania, ssania, mowy
- model wieloczynnikowy (genetyczno - środowiskowy)
- w Łodzi jest ośrodek leczenia wad rozwojowych:
- na 6070 pacjentów 3010 miało rozszczep podniebienia
Leczenie:
- plastyka warg około 5-6 miesiąca życia, plastyka podniebienia około 16 - 24 miesiąc życia
- rozwój twarzy kończy się w wieku 18 lat
Choroby poligenowe jakościowe:
1. Choroba wrzodowa żołądka:
- owrzodzenia błony śluzowej penetrujące przez warstwę mięśniową śluzówki (też rodzaje dziedziczone monogenowo)
- czynniki środowiskowe:
* alkohol
* papierosy
* stres
* niektóre leki
* bakterie Helicobacter pylori (70% przypadków),
zwiększone ryzyko raka żołądka
2. Cukrzyca (Diabetes Mellitus)
- choroba nieuleczalna w dzisiejszych czasach
- zaburzenia gospodarki węglowodanowej
- występuje u około 2-4% populacji
Typy cukrzycy:
- monogenowe dominująca
- upośledzenie wytwarzania insuliny przez trzustkę
- rozwija się po 3,5 roku zycia
- Jest uwarunkowana wieloma czynnikami
- DIABETOLOGIA - zajmuje się leczeniem cukrzycy
- typ I - leczy się insuliną
- insulinę można uzyskać drogą inżynierii genetycznej
3. Nadczynność tarczycy:
- choroba autoimmunologiczna tzn. własne antygeny rozpoznawane są jak obce
4. Alergie:
- atopia i astma (dychawica) - astma w około 70% występuje rodzinnie
- czynnikiem środowiskowym są np. pyłki roślin, rozkruszki
5. Schizofrenia
6. Łuszczyca
7. Nowotwory:
- mutacje dotyczące komórek somatycznych
- komórki dzielą się i pasożytują własny organizm
8. Choroba wieńcowa (niedokrwienna mięśnia sercowego)
9. Miażdżyca:
- odkładanie blaszek miażdżycowych
- może być monogenowe dominująca, genotyp wtedy dotknięty ryzykiem, lecz kombinacja wpływu środowiska i genotypu powoduje przejście progu od zdrowia do choroby
Poligenia ilościowa:
- natężenie cechy, geny się kumulują (geny kumulatywne)
- geny dominujące (A, B, C, …..) są aktywne, ale o małym efekcie każdy
1. Dziedziczenie barwy skóry:
- AA ………………………………… HH - Murzyn
- aa …………………………………... hh - Biały
- Aa …………………………………..Hh - Mulat
- cecha zależy co najmniej od 8 par alleli (!):
P: AA …….. EE x aa ……..ee (Murzyn x Biały)
F1: Aa Bb Cc Dd Ee (Mulat)
Gdy P: Mulat x Mulat to w F1 będzie „jeszcze większy Mulat”
Trójkąt Pascala:
1
1 2 1 jedna para alleli
1 3 3 1
1 4 6 4 1 dwie pary alleli
1 5 10 10 5 1
1 6 15 20 15 6 1 trzy pary alleli
1 7 21 35 35 21 7 1
itd.
Zapis rozkładu cech:
- 1 para alleli: 1 : 2 : 1
- 2 para alleli: 1 : 4 : 6 : 4 : 1 ( 6/suma = prawdopodobieństwo heterozygot)
- liczba różnych fenotypów: 2n + 1
- 3n - liczba różnych genotypów
- częstość genotypów skrajnych w F2 to: (1/4)n n = liczba par alleli
Zjawisko transgresji
1. występowanie fenotypów bardziej skrajnych niż rodzice:
Murzyn Mulat Biały
- wzrost i inteligencja też dziedziczy się poligenowo ilościowo
2. wzrost - wpływ czynników środowiskowych ma bardzo duży wpływ na fenotyp cechy
3. suma linii papilarnych
4. ciężar ciała:
- uwarunkowanie wieloczynnikowe: poligenowe + czynniki środowiskowe np. dieta
5. dziedziczenie inteligencji:
- inteligencja = pojętność
- to ogólna zdolność jednostki do celowego działania, racjonalnego myślenia i efektywnego radzenia sobie w otaczającym nas środowisku
- inteligencja ogólna - 45% geny + 35% środowisko + 20% genetyczno-środowiskowa = 100%
Rodzaje inteligencji:
- inteligencja praktyczna:
a) inteligencja płynna - zdolność uczenia się
b) inteligencja skrystalizowana - inteligencja odzwierciedlająca doświadczanie się
- inteligencja akademicka - mierzona testami
Polska adaptacja sklai Werschlera - 11 testów:
IQ w Japonii - czynniki wpływające na wzrost ilorazu inteligencji
odziedziczalność = wpływ czynników ilościowych
H = 0% - 100%
0% - wyłączny wpływ czynników środowiska
100% - wyłączna zależność od dziedziczenia
Szerokość głowy H = 95%
Długość głowy H = 80%
Typ sylwetki H = 70%
- badanie bliźniąt dla określenia odziedziczalności danej cechy:
* częstość bliźniąt w Europie - 1/90
* 33% bliźniąt to monozygotyczne klony - ten sam genotyp
Wzory odziedziczalności:
- badania dzieci adoptowanych - porównanie cech genotypowych dzieci adoptowanych z cechami……
Genetyka populacji
Prawo równowagi genetycznej = prawo Hardy'ego i Weinberga
- `w każdej populacji organizmów diploidalnych utrzymuje się na stałym poziomie określona częstość występowania zarówno poszczególnych alleli jak i genotypów, gdy nie działają czynniki zakłócające tą równowagę i gdy populacja jest dostatecznie duża”
- różnorodność genetyczna populacji utrzymuje się więc na stałym poziomie, o ile nie działają czynniki zakłócające tą równowagę
- warunki spełnienia prawa H. & W. :
I. cechy monogenowe - 1 lokus
II. populacja mendlowska ( populacja idealna) - czyli populacja w stanie równowagi
Kryteria rodzajów mutacji:
I
a) somatyczne
b) genetyczne
II
III
Częstość mutacji = liczba zmutowanych loci / 1 mln gamet:
- wg tego prawa nie może występować dryf genetyczny (w podręczniku Drewy jest błąd, bo mówi się dryf a nie dryft)
- prawdopodobieństwo p + q = 1 lub 100%
- Allel A - jego częstość p
- Allel a - jego częstość q
- osoby-nosiciele mukowiscydozy mają podwyższoną odporność na cholerę i salmonellozę
- uprzywilejowanie heterozygot występuje też w niedokrwistości sierpowatej
- jeżeli występuje rzadka cecha recesywna, to rzadki allel jest recesywny, nie podlega prawom eugeniki ani prawom doboru naturalnego, nie ulega ekspresji
Terapia genowa (genoterapia)
- przyczynowe leczenie chorób genetycznych
* polega na wprowadzeniu do komórki prawidłowej informacji lub usunięciu niepożądanej cechy
* wektor - służy do wprowadzania właściwych informacji - jest on nośnikiem
Rodzaje wektorów:
1. wirusy (pozbawione cech zjadliwości)
a) retrowirusy (pojedynczy t-RNA, uwalnia się i syntezuje na nim DNA) - przykładowy wirus to wirus białaczki mysiej
b) adenowirusy - są problemy, nie konieczna informacja do komórek dzielących się, ale może być reakcja odpornościowa
c) wirusy stowarzyszone z adenowirusami - nie wywołują reakcji odpornościowej, ale mają małą pojemność
d) herpeswirusy - drobne, ale istnieje ryzyko powstania wirusa patogennego
2. wektory niewirusowe:
a) formy DNA: plazmidy (kołowy DNA z komórek bakteryjnych)
i antysensy (zmodyfikowane DNA)
b) liposomy = lipokompleksy - niska skuteczność
c) polipleksy (??)
d) kapsuły biobalistyczne - mikrokulki z polimerów lub metali szlachetnych
- żeby nastąpiła ekspresja genu (żeby gen zadziałał) musi zajść biosynteza białka, innymi słowy gen leczniczy musi być tak wstrzyknięty, by doszło do powielenia
Zastosowanie terapii genowej:
- można leczyć zarówno błędy genetyczne jak i niektóre z chorób nabytych
- terapii genowej podlegają np. choroby takie jak:
* choroby nowotworowe
* mukowiscydozy
* hemofilia
* AIDS
* choroby krążenia
- szczepionki genetycznie zmodyfikowane:
a) w chorobach nowotworowych (wprowadza się cytokiny, interleukiny i inne czynniki stymulujące odpowiedź immunologiczną
b) w AIDS dla podwyższenia odporności
- terapia genetyczna mukowiscydozy - gen CTFR dostarczany in vivo przez liposomy
- w Polsce prowadzone są badania nad „szczepionką” na czerniaka - duże osiągnięcia
Inżynieria genetyczna
- biologia kreatywna - tworzenie nowych, nieznanych genetycznie naturze organizmów
GMO - genetycznie zmodyfikowane organizmy
Transgen - gen z nową informacją, może pochodzić od:
transformacja - technika wprowadzania obcego genu
* powstają organizmy transgeniczne
* może być transformacja protoplastów
* geny dawcy do biorcy trafiają na wektorach
Znaczenie praktyczne GMO:
* wytwarzają hormony i enzymy
* awaryjnie usuwają plamy ropy naftowej
* produkują odczynniki chemiczne
* dostarczają substancji wykorzystywanych w …………………………………………………..
* do produkcji soków owocowych
* do dojrzewania serów
* w piekarnictwie
* w przemyśle cukierniczym
* w przetwórstwie mięsnym
Zmodyfikowane genetycznie rośliny - konstrukcje roślin:
- na rynku powszechnie używane są zarówno naturalne jak i transgeniczne organizmy
- transformację przeprowadza się za pomocą (najczęściej) Agrobacterium tumefaciens (plazmidy) [bakterie te normalnie powodują guzowatość w rodzinie różowatych]. Pod wpływem tego DNA następuje replikacja i uwidacznianie
- można też stosować wirusy (wykorzystywany np. wirus mozaiki kalafiora) lub mikroiniekcje
- można także stosować `karabinek genetyczny' (kulki metalu - wolframu z informacją)
Kraje najbardziej zaawansowane w produkcji modyfikowanych roślin:
- USA
- Argentyna
- Kanada
- Chiny
- Brazylia
- w USA (2003 r.) produkcja GMP (% całości upraw) wynosiła:
- 69% bawełny
- 68% soi
- 26% kukurydzy
- w Japonii wyhodowano ponad 50 odmian ryżu zmodyfikowanego
Znaczenie transgenicznych roślin - odporność na:
* herbicydy
* wirusy
* grzyby
* owady (np. ziemniak nie zjadany przez stonkę)
- wprowadza się gen kodujący bakteryjną truciznę pochodzący z bakterii Gran+ Baccillus thuringlensis do genów ziemniaka, kukurydzy, bawełny, ryżu w celu odstraszenia owadów - szkodników danych upraw:
* odporność na mróz (gen ryb zimnych wód w roślinach)
* wzbogacenie roślin w korzystne składniki np.:
- olej rzepaku z korzystnymi kwasami tłuszczowymi
- wzbogacenie pomarańczy wapniem
- ryż z karotenem
- słodki pomidor
- wzbogacenie taumatyną (??) ogórków
* uzyskanie specjalnych, korzystnych cech np. długość przechowywania
* rośliny o odpowiednim składzie farmakologicznym
* uzyskanie roślin ozdobnych o odmiennych cechach
* produkcja roślin produkujących doustne szczepionki
* produkowanie białek zwierzęcych przez rośliny
* olbrzymie zyski:
- z upraw (odporność na szkodniki itp.)
- tańsza produkcja (brak konieczności stosowania herbicydów itp.)
- jedynie w Chinach nie ma kontroli społecznej nad wprowadzaniem upraw GMO
Rośliny transgeniczne w Polsce:
- 1993 r. - pierwsze pozwolenie
- na naszych terenach prowadzono badania polowe
Zagrożenia:
- jest ich sporo
1.
2.
3.
4.
5.
6. wartość nowych produktów opartych na roślinach transgenicznych może budzić zastrzeżenia np.:
7. uniezależnienie krajów uprzemysłowionych od surowców wytwarzanych w krajach rozwijających się np. wytwarzanie …………………….. masła kakaowego z tanich olejów
8. wymiar etyczny inżynierii genetycznej - `czy powinniśmy robić wszystko co potrafimy?'
9. zmniejszenie bioróżnorodności
10. stosowanie dużych dawek herbicydów niszczących, wyjaławiających glebę
Zwierzęta transgeniczne
- zawierają obcy gen - transgen z dodatkową informacją
Transgeneza ssaków:
- obcy gen podaje się na nośnikach wirusowych lub metodą mikroiniekcji, najczęściej wektorami są retrowirusy
- najlepsze wyniki uzyskuje się, gdy obcy DNA wstrzykuje się do przedjądrzy zapłodnionej komórki jajowej
Najważniejsze osiągnięcia w drodze do uzyskania transgenicznych zwierząt gospodarczych:
1980 r. - pierwsze transgeniczne myszy
1982 r. - pierwszy fuzyjny gen wprowadzony do genomu myszy
1985 r. - uzyskanie pierwszych transgenicznych zwierząt gospodarczych
1986 r. - uzyskanie pierwszych transgenicznych ryb
1987 r. - uzyskanie transgenicznych myszy wytwarzających gruczołach mlecznych białko ludzkie o działaniu leczniczym
1988 r. - uzyskanie transgenicznych zwierząt gospodarczych (owiec) wytwarzających w gruczołach mlecznych ludzkie białko o działaniu leczniczym
1991 r. - uzyskanie pierwszego transgenicznego buhaja
1992 r. - pierwsze ryby o znaczeniu gospodarczym wykazują większe tempo wzrostu
Przewidywane znaczenie transgenicznych zwierząt:
- większość zwierząt jest jednak obarczona bezpłodnością oraz zaburzeniami chorobowymi
- małpie podano gen GFP fluoryzujący z meduzy, małpie fluoryzowały włosy i paznokcie
- GFP jest używany jako marker - do identyfikacji komórek transformowanych
Diagnostyka molekularna chorób genetycznych dla celów klinicznych:
- polega na analizie próbki DNA pacjenta w celu wykrycia genotypu związanego z chorobą uwarunkowaną genetycznie
- sondy DNA i badanie polimorfizmu fragmentów uzyskanych po trawieniu restryktazami
- RFLP - polimorfizm długości odcinków pomiędzy miejscem restrykcji
- w 1979 r.. - odkryto bakteryjne enzymy restrykcyjne - endonukleazy restrykcyjne tnące DNA w odpowiedni sposób
Diagnostyka molekularna chorób genetycznych:
- mukowiscydozy
- achondroplazja (choroba ukł. kostnego prowadząca do karłowatości)
- dziedziczna postać raka sutka i jajnika
- zespół nadciśnienia tętniczego
- dystrofia mięśni typu Duchene'a
- fenyloketonuria
- zaburzenia determinacji płci
- cukrzyca insulinozależna
- rodzinna polipowatość jelita
- osteoporoza
- hipercholesterolemia rodzinna
- choroba Huntingtona
- rodzinny defekt apo B
Achondroplazja - rodzaj karłowatości
- mutacja punktowa,
- dziedziczenie autosomalne monogenowe dominujące
- G-A tranzycja lub G-C translacja
- zmienione białko w 380 pozycji łańcucha
- genotyp: AA, Aa, aa (????)
- fenotyp:
* niski wzrost,
* skrócenie kończyn
* duża głowa z wypukłym czołem
* także liczne duże zmiany wewnętrzne w organizmie
* rozwój umysłowy prawidłowy
- często następuje śmierć około 1 roku życia
Choroba Huntingtona:
- nieuleczalna choroba zwyrodnieniowa komórek nerwowych prążkowia
- częstość występowania: 1/10 000
- dziedziczenie monogenowe autosomalne dominujące
- genotyp: HH, Hh, hh
- gen kodujący białko - huntingtynę znajduje się w chromosomie 4 p
- gen zmutowany - zwielokrotnienie kodonu CAG (odpowiada glutaminie)
- zmienione białko - zwiększona ilość glutaminy
- objawy ujawniają się około 30-40 roku życia
- etapy choroby - objawy:
1. zaburzenia ruchu, ruchy mimowolne, upośledzenie pamięci
2. zmienne nastroje
3. postępujące otępienie
4. upośledzenie fizyczne
5. przedwczesna śmierć po 10-20 latach od pojawienia się pierwszych objawów klinicznych
- leczenie - jest tylko objawowe, polega na hamowaniu objawów drgawek oraz depresji związanych z postępem choroby, jest ono mało skuteczne
Korzyści i zagrożenia związane z wykonywaniem przedobjawowych testów molekularnych:
„+”
- zapobieganie groźnym konsekwencjom chorób genetycznych np. rodzinnej polipowatości jelita
- programy profilaktyczne i systematyczne badania w celu wczesnego wykrycia i chirurgicznego usunięcia zmienionych nowotworowo tkanek
- uniknięcie przekazania „złych” genów potomstwu
„-„
Wytyczne dla poradnictwa genetycznego (dotyczy testu molekularnego):
- własna decyzja
- test przedkliniczny tylko dla dorosłych
- test powszechny i bezpłatny
- wynik testu jest poufny
- odbieranie wyników odbywa się tylko w obecności osoby towarzyszącej
- prowadzi się opiekę psychologiczną badanego
- osoba badana może w każdej chwili zrezygnować z odebrania wyników testu
W niektórych wypadkach ubezpieczalnie wymagają informacji na temat testów na choroby genetycznej
Klonowanie organizmów
- klon - zbiór komórek lub organizmów będących swoimi wiernymi kopiami genetycznymi powstałych przez rozród wegetatywny
- klony posiadają ten sam genotyp
Klonowanie roślin
- naturalne klonowanie roślin częste jest w przyrodzie:
* odrosty korzeniowe topoli białej
* osobniki wyrosłe z kłącza konwalii
* poziomka
- klonowanie roślin w hodowli tradycyjnej:
* sępolia
* pelargonia
* begonia
- klonowanie roślin w kulturach in vitro:
* storczyki
* szałwia
* nowe odmiany drzew leśnych
* rosiczka
Klonowanie zwierząt
- klonowanie występuje także w naturze
- klonowanie ssaków:
* do niezapłodnionej komórki jajowej, z której usunięto jądro wprowadza się jądro somatycznej komórki dorosłego osobnika
- klonowanie:
a) klon zwierząt o wyjątkowej użyteczności gospodarczej
b) klonowanie zwierząt transgenicznych
c) klonowanie wymierających lub wymarłych zwierząt
Klonowanie ludzi
- naturalne klonowanie - bliźnięta
- ewentualne cele klonowania ludzi:
* sposób na bezpłodność
* klonowanie przedwcześnie zmarłych dzieci
* klonowanie żołnierzy i niewolników
* klonowanie utalentowanych jednostek
* tworzenie żywych banków narządów
- klonowanie ludzi jest kontrowersyjne
Identyfikacja molekularna ludzi
- zsekwencjonowano w całości genom człowieka
- podstawowe cechy kodu genetycznego:
a) indywidualność
b) niezmienność
Etapy badania:
Znaczenie:
Dziedziczenie pozajądrowe
- małe znaczenie, mało materiału
- mitochondrialne dziedziczenie - głównie od matki, ponieważ plemnik porzuca wić wraz z umiejscowionymi tam mitochondriami
Program ochrony zasobów genowych
- biblioteki genowe - to zbiór klonów bakterii zawierających rekombinowane wektory niosące fragmenty DNA
- biblioteki genów = banki genów = genoteki = bank z DNA, zbiór sekwencji kodujących białka wprowadzanych za pomocą wektorów do bakterii
Etapy konstrukcji biblioteki:
Genomowej:
Genowej:
1. wyizolowanie mRNA
2. przeniesienie informacji z RNA na DNA (z DNA tylko sekwencje kodujące eksony = egozny)
3. wprowadzenie do bakterii za pomocą wektorów
- Międzynarodowa Rada d.s. Roślinnych Zasobów Genowych
- na skutek industrializacji i postępującej degradacji środowiska zagrożone są stare formy i populacje miejscowe roślin
- Bank Genów Roślin Warzywnych w Skierniewicach - zbieranie i zabezpieczanie przed wyginięciem dawnych odmian i populacji miejscowych oraz form dzikich warzyw
- bank spermy noblistów w Stanach Zjednoczonych - 15 uczonych
Genetyczne uwarunkowania procesów starzenia i śmierci:
- długość życia jest uwarunkowana przez:
1. wyposażenie genetyczne
2. płeć (kobiety - 79 lat, mężczyźni - 72 lata)
3. czynniki ekologiczne - czynniki skracające długość życia:
* palenie, używki
* stres, złe odżywianie
* zatrucie środowiska
- zwiększanie wraz z wiekiem ilości chorób takich jak: osteoporoza, reumatyzm, nadciśnienie, cukrzyca II typu
- przedwczesne starzenie z powodu chorób neurodegeneracyjnych:
a) dziedzicznych (np. choroba Alzheimera, ch. Huntingtona)
b) niedziedzicznych (np. …………………………………………………………………….)
Progeria - przedwczesne starzenie się
- syndrom Hutchinsona-Gilforda
- częstość to 1/8 000 000 żywych urodzeń
- niezależnie od płci i rasy
- osoby dotknięte tym syndromem dożywają maksymalnie wieku 14-20 lat
Cechy:
- dysmorfia ogólna (niski wzrost, sztywne stawy, dysmorfia paznokci)
- w wieku 10 lat - fenotyp osoby starej
Hipotezy starzenia się:
1. stochastyczne - starzenie na skutek zmian skokowych na skutek nagromadzenia się produktów przemiany materii:
* hipoteza zużycia
* hipoteza wolnych rodników
* hipoteza mutacji somatycznych
Erwin Shroedinger:
- w 1939 r. stwierdził, że nigdy człowiekowi nie uda się wykorzystać energii zawartej w atomie
Cytologia
- Robert Hooker - fizyk, astronom, biolog
* komórka - cellula (ang. cell)
* 1965 r. - „Mikrografia” - komórka korka
Antony Leeuwenhoek:
- przyrodnik holenderski
- konstruktor mikroskopu
- prowadził systematyczne badania i odkrył m.in. bakterie, drożdże, plemniki, erytrocyty
- wraz z Malpighim odkrył naczynia włosowate
Podstawowe składniki komórki:
- ściana komórkowa
- błona komórkowa
- cytoplazma
- jądro komórkowe
- plastydy
- mitochondria
- rybosomy
- retikulum endoplazmatyczne
- centrosomy
- aparat Golgiego
- mikrotubule
- sferosomy
- substancje ergastyczne
Ściana komórkowa:
- produkt endoplazmy
- zasadnicze znaczenie: podstawowy element szkieletowy komórek roślinnych (sztywny pancerz)
- przeciwstawienie dużemu ciśnieniu osmotycznemu we wnętrzu komórki
- u prokariontów - gruba ściana
- ściana może być:
a) pierwotna:
* cienka i elastyczna
* skład: włókna celulozy (20% suchej masy)
b) wtórna:
* celuloza - 60 do 90 %
* może być inkrustowana węglanem wapnia, krzemionką
- micelle - włókna uporządkowane
- ściany mogą ulegać zdrewnieniu (odkładanie ligniny = drzewnika):
* mało wody, ściana twarda, sztywna, odporna mechanicznie
* słabo przepuszczalna dla wody i powietrza
- skorkowaciała - wysycona suberyną:
1. ściana pierwotna
2. suberyna
3. ściana wtórna
- skutynizowana - wysycona kutyną (substancja tłuszczowa, mieszanina wyższych kwasów tłuszczowych)
Blaszka środkowa - scala komórki, jest z pektynianu wapnia
* rozpuszczanie blaszki środkowej: gotowanie, dojrzewanie owoców
Plazmodesmy - przestwory międzykomórkowe, cytoplazmatyczne nici łączące protoplasty komórek, zapewniają integrację komórek w obrębie tkanek
Jamki - ściana pierwotna - blaszka środkowa - ściana pierwotna w pierwotnym polu jamkowym
![]()
- rodzaje jamek:
1. jamki proste - np. komórki kamienne
2. jamki lejkowate
Przestwory międzykomórkowe - powstają w wyniku rozpuszczenia blaszek środkowych lub przez obumieranie komórki
Błona komórkowa = plasmolema:
- właściwości:
* słaba przepuszczalność wody
* selektywna przepuszczalność substancji i jonów
- budowa:
* kompleks białkowo-lipidowy (30-50%, melocyty 80%, komórki bakterii - 25% lipidów)
* dwie warstwy lipidowe o właściwościach hydrofilowo-hydrofobowych (polarno-polarnych)
- zadania:
* bariera,
* selektywna przepuszczalność
* przekazywanie sygnałów chemicznych, energii
* środowiska dla enzymów
Retikulum endoplazmatyczne = siateczka endoplazmatyczne:
- dynamiczny system rurek i cystern
- rodzaje:
* siateczka gładka - synteza lipidów, metabolizm glikogenu, synteza lipidów (trójglicerydy, glikolipidy itd.)
* siateczka ziarnista = szorstka - synteza białek, bo na powierzchni ma rybosomy, inaczej mówiąc są to cysterny z rybosomami
# synteza i modyfikacja białek eksportowych
# białka rezydujące - wewnątrz RER, odpowiedzialne za procesy zachodzące w RER:
- cząsteczki białek utworzone w błonach RER stają się składnikami błon siateczki
Aparat (układ) Golgiego:
- system błon gładkich z cystern, rurek, pęcherzyków
- centralnie znajduje się diktiosom, wkoło są pęcherzyki
- rodzaje:
* forma siateczkowata - u zwierząt kręgowych
* forma diktiosomowa (łuskowata) - u bezkręgowców i u roślin
- zadania:
* potranskrypcyjna modyfikacja białek i fosfolipidów,
* synteza pektyn i hemicelulozy, śluzów,
* tworzenie przegrody pierwotnej w czasie podziału komórki, blaszki środkowej, wodniczek,
* recyklizacja błony komórkowej po endocytozie
Jądro komórkowe:
- z reguły pojedyncza organella komórkowa
- wielkość zależy od gatunku, rodzaju i stanu komórki
- główne procesy zachodzące w jądrze:
* samopowielanie (replikacja) DNA
* przekazywanie informacji genetycznej na RNA
Budowa jądra:
- otoczka jądrowa:
* dwubłonowa u Eucaryota
* brak u Procaryota
* błona zewnętrzna połączona z siateczką endoplazmatyczną szorstką
* wewnętrzna błona zawiera białka receptorowe
* między błonami jest przestrzeń wokół jądrowa = cysterna perynuklearna
* otoczka ma pory z kompleksem białkowym (nukleoporyny):
^ umożliwiają transport między wnętrzem a cytoplazmą
^ około 30% osłonki zajmują pory
^ liczba porów zależy od:
^ kompleksy białkowe rozpadają się podczas mitozy i odtwarzają później
* nukleoplazma = macierz jądrowa:
a) struktura dynamiczna biorąca udział we wszystkich procesach jądrowych
b) skład: roztwór białek, cukrów, nukleotydów, jonów
* chromatyna jądrowa
^ interfazowa forma chromosomów komórki
^ skład:
^ rodzaje:
- postać chromatyny czynna transmisyjnie
- skład - całkowicie rozwinięte odcinki chromosomów
- przejściowy skondensowany stan euchromatyny przypominający skondensowaną heterochromatynę - w ciałku Barra i pałeczce dobosza
- forma stale skondensowana, bez odcinków kodujących, inaczej mówiąc - niekodująca część chromatyny
- odcinki chromosomów nierozwinięte w interfazie
- występowanie:
* w jądrach interfazowych
* w chromosomach przy centromerach i telomerach
- Arabidopsis - rzodkiewnik - roślina wykorzystywana w badaniach molekularnych
Jąderko :
- wielkość zależna od aktywności metabolicznej komórki
- tu zachodzi synteza RNA (tRNA)
- organizatory jąderka w chromosomach jąderkowych
- liczba jąderek = liczba chromosomów jąderkowych: mniej, gdy następuje fuzja jąderek
- u człowieka występuje 5 par chromosomów jąderkowych
-
Rybosomy:
- drobne organella (10-30 nm)
- centra syntezy białek
- synteza rybosomów w jąderkach
- budowa: podjednostka mała (40S) + podjednostka duża (60S)
- są bardzo liczne - nawet do kilku mln
- wyróżniamy rybosomy:
* cytoplazmatyczne
* siateczki śródplazmatycznej ziarnistej (szorstkiej)
* mitochondrialne
* jądrowe
* chloroplastowe
- rybosomy większe u Eukariontów (80S)
Organella metabolizmu energetycznego:
Mitochondria:
- tylko u Eukariontów, transformatory energii
- zmienne kształty
- można obserwować mitochondria żywe
- wybarwiają się hematoksyliną żelazistą lub fuksyną kwaśną
- typy mitochondriów:
a) nitkowate (komórki kanalików nerkowych)
b) ziarniste (oocyty)
c) rozgałęzione (komórki nerwowe człowieka)
- liczba mitochondriów w komórce zależna od zapotrzebowania energetycznego:
* bardzo duże - komórki mięśnia sercowego
* bardzo małe - komórki tkanki tłuszczowej
* brak np. u rzęsistków (zamiast tego są hydroksysomy)
* pojedyncze u świdrowca gambijskiego
- budowa:
A) błona zewnętrzna |
Razem tworzą błony mitochondrialne |
B) błona wewnętrzna |
|
C) między błonami jest przestrzeń peryferyczna = przestrzeń międzybłonowa |
Przestrzeń peryferyczna - są tu kinazy, bardzo dużo enzymów, całe kompleksy |
D) macierz (matrix) mitochondrialna - skład: |
a) kompleksy enzymów |
|
b) dehydrogenaza pirogronowa |
|
c) dehydrogenaza glutaminowa i asparginowa |
- rybosomy mitochondrialne w macierzy mitochondrialnej mają 55S
- genom mitochondrialny - liniowe lub koliste, dwuniciowe DNA:
* liniowa - u grzybów i pierwotniaków
* kolista - w mitochondriach roślin
- uzyskiwanie i magazynowanie energii
- utlenianie substratów takich jak: glukoza, kwasy tłuszczowe, aminokwasy
- fosforylacja oksydacyjna - prowadzi do syntezy ATP - końcowy wynik H2O i CO2
- transport - błona wewnętrzna - stosunkowo szczelna
- pochodzenie mitochondriów (w aspekcie ewolucyjnym):
I. żywe organizmy
II. hipoteza „o symbolicznym pochodzeniu mitochondriów
III. mitochondria powstały z bakterii wchłoniętych na drodze fagocytozy do komórek Eukariontów - premitochondria
hipoteza o fagocytowaniu i o symbiotycznym wchłanianiu:
- pochodzenie mitochondriów (w ontogenezie):
I. hipoteza o powstaniu mitochondriów de novo - z podstawowych związków organicznych
II. hipoteza o powstaniu mitochondriów z innego rodzaju organelli
III.
Peroksysomy:
- dawniej mikrociała
- pęcherzyki o średnicy 0,5-1,5 mikro metra
- ketolaza i oksydaza - zawartość peroksysomów
- budowa:
* lipidowo-białkowa, pojedyncza błona komórkowa, zawiera około 10 rodzajów białek
* macierz ziarnista z rdzeniem - nukleoidami
- nukleoid - budowa rurkowata, rurki I i II rzędu
- zadania:
- rozkładają H2O2 (katalaza) w komórkach wątroby utleniają 10% alkoholu etylowego,
- w komórce mięśnia sercowego utlenianie alkoholu etylowego głównie w peroksysomach,
- syntezują cholesterol
- metabolizują aminokwasy
Peroksysomy roślinne:
- zawartość różnych enzymów zależnie od występowania:
a) peroksysomy liściaste - mają aminotransferazy i współdziałają z chloroplastami oraz mitochondriami w procesie fotooddychania
b) glioksysomy - cykl betaoksydacyjny kwasów tłuszczowych
Wykłady z Parazytologii i Genetyki (rocznik 2007/2008)
wykłady w niniejszej wersji są nieoficjalne i mogą zawierać błędy literowe bądź merytoryczne. Jest to tylko i wyłącznie pomoc w notowaniu zagadnień wykładowych, A NIE ŹRÓDŁO WIEDZY EGZAMINACYJNEJ!!!!
1
Wyszukiwarka
Podobne podstrony:
wykłady z genetyki i parazytologii 2007-2008 część 2 - paraz, farmacja, parazytologia
wykłady z genetyki i parazytologii 2007-2008 część 1 - syste, farmacja, parazytologia
wykłady z genetyki i parazytologii 2007-2008 część 2 - paraz, farmacja, parazytologia
wyklad 11 12 2007 2008
Maści wykład 2007-2008, Farmacja, Technologia postaci leków
wyklady, Psychologia rozwojowa dzieci i młodzieży - wykład 7, PSYCHOLOGIA ROZWOJOWA DZIECI I MŁODZIE
Chemia wyklady 2007 2008(1) id Nieznany
wyklady, Psychologia rozwojowa dzieci i młodzieży - wykład 13 , PSYCHOLOGIA ROZWOJOWA DZIECI I MŁODZ
C Wykład I 2007 2008 M Ch
wyklady, Psychologia rozwojowa dzieci i młodzieży - wykład 8, PSYCHOLOGIA ROZWOJOWA DZIECI I MŁODZIE
wyklady, Psychologia rozwojowa dzieci i młodzieży - wykład 5, PSYCHOLOGIA ROZWOJOWA DZIECI I MŁODZIE
GENETYKA i parazyty KOLOKWIUM 2009, farmacja cm umk
PODSTAWY FINANSÓW 2007 2008 WYKŁADY I ROK
C++ Wykład II 2007 2008 M Ch
Genetyka 2007.2008 zaliczenie I (termin II), genetyka, testy, testy genetyka kolokwium nr 1
więcej podobnych podstron