GENETYKA (prelekcja 1)
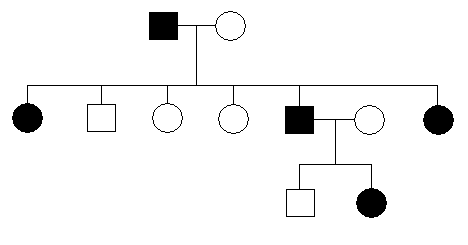
rodowód - graficzne przedstawienie pokoleń danej rodziny oraz danych klinicznych dotyczących występowania cechy;
rodowód cechy dominującej
umożliwia określenie sposobu dziedziczenia i ryzyka genetycznego wystąpienia czy powtórzenia się choroby w rodzinie;
analizujemy od pokolenia najmłodszego;
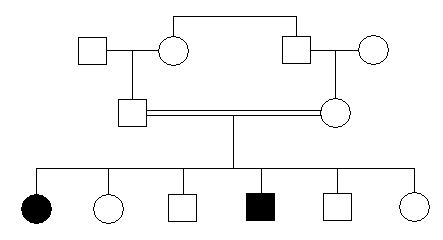
rodowód cechy recesywnej
utrudnienia w analizie rodowodów:
heterogenność genetyczna;
fenokopie - naśladownictwo fenotypowe bez zmiany genotypu;
rzadkie przypadki w mało licznych rodzinach;
niepewność ojcostwa;
osoby spokrewnione - osoby mające wspólnego przodka;
SFEROCYTOZA DZIEDZICZNA (MIKROSFEROCYTOZA)
wrodzona niedokrwistość (żółtaczka) hemolityczna
cecha autosomalna dominująca;
locus 8p;
częstotliwość występowania 1:5000 urodzeń;
wynika z mutacji powodującej niedobór ilości lub nieprawidłową budowę spektryny - białka szkieletowego krwinki czerwonej;
we krwi obwodowej prócz normocytów występują sferocyty;
sferocyty w porównaniu z erytrocytami:
mniejsza średnica - 2/3 średnicy normocytów;
kulisty kształt;
mniejszy stosunek powierzchni do objętości;
krótszy czas życia;
większa zawartość hemoglobiny;
sferocyty są niszczone w śledzionie, co powoduje splenomegalię, niedokrwistość hemolityczną, zwiększone stężenie bilirubiny w surowicy;
są bardziej podatne na rozpad w warunkach obniżonego ciśnienia osmotycznego;
stwierdzono kilka rodzajów nieprawidłowości w budowie białek błon krwinek czerwonych, wynikające z różnorodnych mutacji; najczęstszy - niedobór białka spektryny;
NIEDOKRWISTOŚĆ SIERPOWATOKRWINKOWA (hemoglobinopatia HbS)
cecha autosomalna monogenowa;
locus 11p;
występuje głównie w krajach klimatu równikowego;
częstotliwość występowania ok. 1% urodzeń;
wynika z mutacji punktowej zmiany sensu w obu genach białka β-globiny, wchodzącego w skład hemoglobiny (Glu zamienione na Val w pozycji 6);
hemoglobina prawidłowa HbA zawiera łańcuchy α2 β2, hemoglobina sierpowata HbS zawiera łańcuchy α2 β2S, które przy niskim stężeniu parcjalnym tlenu mają skłonność do polimeryzacji (powstają włókna deformujące erytrocyt, który przyjmuje kształt sierpowaty);
krwinki sierpowate tracą elastyczność, nie mogą przepływać przez naczynia włosowate zaczopowują je i powodują niedokrwienie;
anemia sierpowata HbS/HbS;
sierpowatość krwinek HbA/HbS - nosicielstwo genu HbS;
wykrywanie heterozygot HbA/HbS:
test Emmela;
elektroforeza lub chromatografia hemoglobin erytrocytów;
hybrydyzacja DNA z odpowiednią sondą molekularną;
FENYLOKETONURIA KLASYCZNA (PKU)
cecha autosomalna recesywna;
niedobór lub brak enzymu hydroksylazy fenyloalaninowej;
locus 12q;
częstotliwość występowania 1:7000 - 12000 (Europa);
nieleczeni pacjenci z PKU wykazują podwyższone stężenie we krwi fenyloalaniny i produktów jej przemiany, np. kwasu fenylooctowego i kwasu fenylopirogronowego (tzw. fenyloketonów);
powoduje niedorozwój umysłowy wynikający z zaburzeń wzrostu mózgu i tworzenia osłonek mielinowych nerwów;
objawy: wzmożone napięcie mięśni, napady padaczkowe, zachowanie przypominające autyzm, nadmierne pocenie się, słaba pigmentacja skóry, hipoplazja szkliwa, `mysi' zapach moczu;
leczenie: dieta z ograniczoną zawartością fenyloalaniny;
test moczowy (pieluszkowy):
dodatnie zakwaszenie moczu osoby z fenyloketonurią; po dodaniu kilku kropli 10% roztworu FeCl3 mocz zmienia barwę na niebiesko-zieloną;
test umożliwia rozpoznanie wady w 4-5 tygodniu życia;
test Guthriego:
rozpoznanie wady już od 4 doby życia;
mikrobiologiczny, półilościowy test Guthriego do niedawna był obowiązującym testem przesiewowym (skriningowym) stosowanym w celu wykrycia fenyloketonurii u noworodków. Obecnie w Polsce obowiązkowym testem przesiewowym jest test enzymatyczny określający stężenie fenyloalaniny w suchej kropli krwi;
wykonanie testu:
na podłoże agarowe niezawierające fenyloalaniny posiewa się mutanty bakterii Bacillus subtilis, wymagające do wzrostu fenyloalaninę;
z bibuły testowej (numerowanej kodem paskowym i opatrzoną danymi dziecka), z próbkami krwi noworodków wycina się krążki, które następnie umieszcza się na podłożu (szereg próbek krwi rutynowo badanych noworodków);
równolegle sporządza się na płytce agarowej szereg kontrolny - wzorzec stref wzrostu B. subtilis dla odpowiednich stężeń fenyloalaniny we krwi (od 2 mg% do 20 mg%);
płytki inkubuje się w 37ºC przez 24 h; po upływie doby odczytuje się wyniki testu, mierząc strefy wzrostu bakterii wokół krążków (strefy zmętnienia); szerokość strefy zmętnienia wzrostu bakterii wokół krążków jest miarą stężenia Fen we krwi badanego noworodka;
interpretacja wyników testu:
u noworodków z klasyczną fenyloketonurią stężenie Fen = 20 mg% i więcej, podczas gdy za normę przyjmuje się do 4 mg%;
jeżeli strefa zmętnienia wokół krążka bibuły z krwią badanego noworodka przewyższa strefę wzorca Fen dla 4 mg% należy powtórzyć badanie; potwierdzenie zwiększonego stężenia Fen we krwi w kolejnym badaniu obliguje do skierowania dziecka na dalsze badania diagnostyczne potwierdzające rozpoznanie fenyloketonurii;
obecnie: badania przesiewowe oznaczania stężenia w suchej kropli krwi noworodka z wykorzystaniem biochemicznych reakcji kolorymetrycznej; krew pobiera się po 24 h życia;
stężenie fenyloalaniny |
kwalifikacja |
postępowanie |
poniżej 2,8 mg/dl |
norma |
brak |
2,8 - 8,0 mg/dl |
małe prawdopodobieństwo wady |
powtórzenie testu; jeśli jeden z wynikó testu jest powyżej 4,0 mg/dl wysyłana jest druga bibuła do rodziców dziecka (jeżeli i ona > 4,0 mg/dl dziecko zostaje skierowane na badania) |
powyżej 8,0 mg/dl |
duże prawdopodobieństwo wady |
bezzwłoczne wezwanie do poradni wad metabolicznych |
fenyloketonuria matczyna:
matki chore na PKU nie stosujące diety rodzą dzieci ze znacznym upośledzeniem umysłowym, mimo że dzieci te nie są chore;
wykrywanie heterozygot w stosunku do genu fenyloketonurii (testy czynnościowe służą do sprawdzenia regulacji Fen-Tyr):
*stężenie Fen w surowicy krwi osób zdrowych (homozygot dominujących) wynosi ok. 1,5 mg/100 cm3;
badanie stężenia fenyloalaniny w teście obciążeniowym po doustnym podaniu Fen w ilości 0,1 g/kg masy ciała; stężenie u homozygot dominujących po godzinie wzrasta do ok. 8 mg/100 cm3, w ciągu następnej godziny spada do 6 mg/100 cm3, zaś po upływie 24 h wraca do normy; w analogicznym teście obciążeniowym stężenie Fen u heterozygot po 2 h wzrasta do ok. 9 mg/100 cm3, a następnie wolniej niż u homozygot dominujących wraca do normy;
badanie stężenia tyrozyny w teście obciążeniowym - doustne podanie Fen w ilości 0,1 mg/kg masy ciała powoduje u homozygot dominujących trzykrotny wzrost stężenia Tyr w ciągu pierwszej godziny, następnie stopniowy spadek i powrót do normy w ciągu 24 h; w analogicznym teście obciążeniowym stężenie Tyr u heterozygot nieznacznie wzrasta, a w ciągu 24 h powraca do wartości wyjściowej;
TYROZYNEMIA NOWORODKÓW
cecha autosomalna recesywna;
częstotliwość występowania 1:100000 - 120000 urodzeń;
niedobór hydroksylazy parahydroksyfenylopirogronianowej utleniającej kwas parahydroksyfenylopirogronianowy do kwasu homogentyzynowego;
wzrost stężenia Tyr i Fen w surowicy krwi, w moczu Tyr i jej metabolitów;
postacie łagodne i ostre choroby (zgon w drugim półroczu życia wskutek uszkodzenia nerek, zaburzeń wodno-elektrolitowych);
wykrywanie: ilościowe oznaczanie Tyr metodami biochemicznymi lub półilościowym mikrobiologicznym testem Guthriego;
metabolity nieprawidłowej przemiany Tyr w moczu dają reakcję barwną z roztworem FeCl3;
ALKAPTONURIA (OCHRONOZA)
cecha autosomalna recesywna;
częstotliwość występowania 1:200000 urodzeń;
brak lub niedobór enzymu oksydazy homogentyzynianowej (dioksygenoza) katalizującej przemianę kwasy homogentyzynowego do kwasu fumaryloacetooctowego;
kwas homogentyzynowy jest wydalany z moczem; w obecności powietrza utlenia się i zmienia barwę na ciemnobrązową;
w 2-3 dekadzie życia pojawiają się objawy ochronozy;
ALBINIZM (BIELACTWO)
hipomelanoza z dziedzicznych wad komórek barwnikowych (melanocytów oczu i skóry);
albinizm oczno-skórny: cecha autosomalna recesywna;
albinizm tyrozynoizoujemny: brak enzymu tyrozynazy, przekształcającej Tyr w jeden z prekursorów melaniny; skóra różowoczerwona, białe włosy, jasnoniebieskie tęczówki lub różowe, zmniejszona ostrość wzroku;
albinizm oczny: cecha recesywna autosomalna lub sprzężona z chromosomem X;
Wyszukiwarka
Podobne podstrony:
prelekcje genetyka medyczna
prelekcja 6 (genetyka)(1)
Prelekcja 10 - cz 2 - Mutacje chromosomowe człowieka, Genetyka
Biologia medyczna prelekcja 1 genetyka stomatologia'13
prelekcja 2 (genetyka)
prelekcja1, Genetyka
prelekcja 4 (genetyka)
prelekcje genetyka dodatek
Prelekcja 10 (Genetyka II)
prelekcja 2 (genetyka)
prelekcja 6 (genetyka)
Genetyka prelekcja prof F
prelekcja4, MEDYCYNA - ŚUM Katowice, III ROK, GENETYKA, genetyka prelekcje
prelekcja 5 (genetyka)
prelekcja 3 (genetyka)
Prelekcja 8 Modele badań genetycznych, biologia
prelekcja1b, Genetyka
więcej podobnych podstron