1
Mechanizmy chorób dziedzicznych
Podział chorób dziedzicznych
• Choroby jednogenowe
– Autosomalne
• Dominujące – achondroplazja, neurofibromatoza
• Recesywne – fenyloketonuria, mukowiscydioza
– Sprzężone z płcią
• Dominujące – krzywica oporna na wit. D
• Recesywne – hemofilia, dystrofia mięśniowa Duchenne’a
• Choroby wieloczynnikowe
– Wady rozwojowe
– Schizofrenia
– Miażdzyca
– I inne
Choroby jednogenowe (dominujące)
• Zaburzenia w budowie białek strukturalnych
• Zmniejszenie ilości białek regulatorowych
• Zmniejszenie ilości białek receptorowych
• Delecja genów supresorowych
• Produkcja toksycznych białek
• Hemoglobinopatie dotyczące łąńcucha alfa hemoglobiny
Choroby jednogenowe (recesywne)
• Brak białek enzymatycznych
• Brak białek receptorowych
• Brak białek regulacyjnych
• Hemoglobinopatie dotyczące łańcucha beta
hemoglobiny
2
Bloki metaboliczne
• W organizmie funkcjonują szlaki metaboliczne
będące ciągiem przemian biochemicznych – jedna
substancja (A) przekształcana jest w drugą (Z)
poprzez szereg stadiów pośrednich (B, C, D, itd.).
• Każdy etap przekształcenia katalizowany jest przez
określony enzym.
• Brak enzymu katalizującego określoną reakcję szlaku
metabolicznego wywołuje tzw. blok metaboliczny.
• Blok metaboliczny powoduje nagromadzenie
substratu dla brakującego enzymu, przy
jednoczesnym braku produktu reakcji przez niego
katalizowanej.
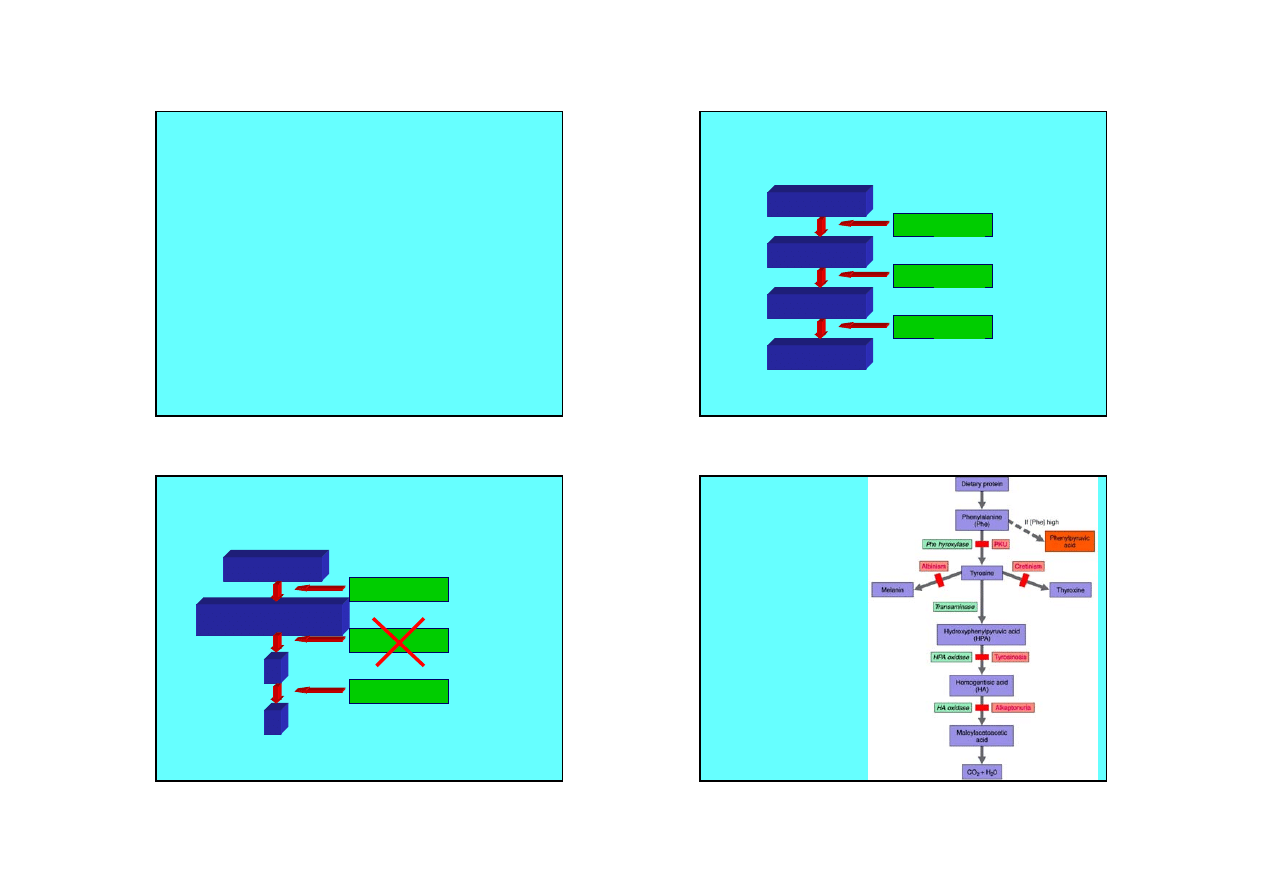
Szlak metaboliczny
A
B
C
D
1
2
3
Bloki metaboliczne
A
B
C
D
1
2
3
Szlak przemian
fenyloalaniny

3
Fenyloketonuria
Albinizm
Alkaptonuria
Choroby spichrzeniowe
• Choroby spichrzeniowe wywołane są niedoborem enzymów
odpowiedzialnych za rozkład polimerów obecnych w
komórkach.
• Pierwszy typ chorób spichrzeniowych związany jest z
niezdolnością do rozkładu glikogenu. Niezdolność do rozkładu
tego związku prowadzi do szybkiego pojawienia się objawów
chorobowych związanych z niezdolnością (wiekszą lub
mniejszą) do uzyskiwania glukozy z glikogenu. W następnej
kolejności pojawiają się objawy wywoływane przez uszkodzenie
komórek przez zmagazynowany nadmiar glikogenu.
• Drugi typ chorób spichrzeniowych związany jest z niezdolnością
do rozkładania i usuwania z komórek związków „wycofanych z
obiegu”. W chorobach tych objawy ujawniają się później i
narastają w miarę gromadzenia się szkodliwych substancji w
komórkach.
4
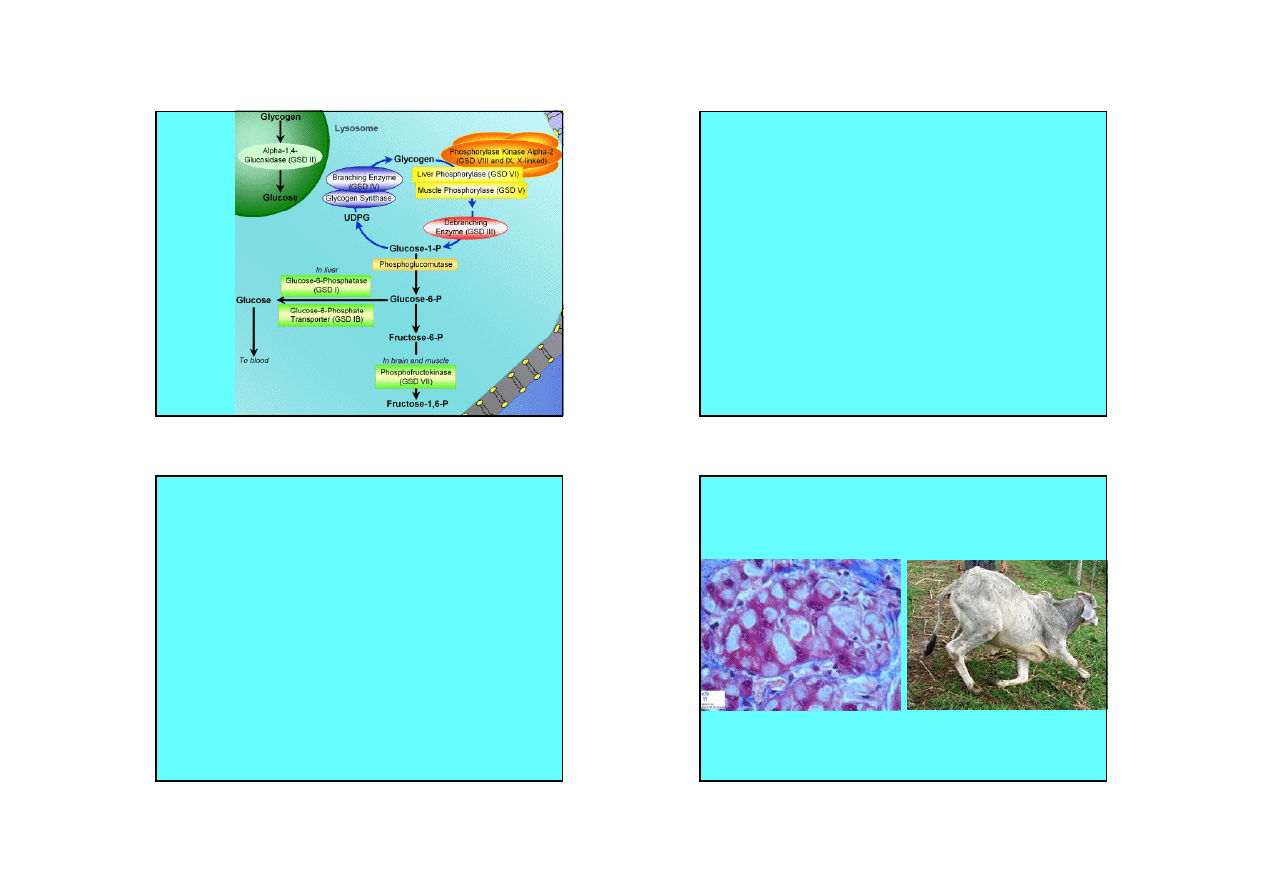
Przemiany
glikogenu
Glikogenozy
• Synteza i rozkład glikogenu wymaga aktywności
wielu enzymów. Niedobór jednego z enzymów
prowadzi do zaburzeń w depolimeryzacji glikogenu, a
co za tym idzie, zaburzeń w uruchamianiu
magazynów glukozy.
• Glikogenoza typu I (von Gierkego) – komórki nerek i
wątroby sa przeładowane glikogenem. Adrenalina i
glukagon nie powodują uwalniania glukozy. U
chorych występuje ketoza i hyperlipidemia. U chorych
brak aktywności glukozo-6-fosfatazy.
Glikogenozy
• Glikogenoza typu II (Pompego) – brak alfa-1-4-
glukozydazy degradującej glikogen gromadzący się
w lizosomach. Choroba kończy się śmiercią.
• Glikogenoza typu IV (Andersena) – brak enzymu
rozgałęziającego. Gromadzi się polisacharyd mający
niewiele punktów rozgałęzienia. Choroba kończy się
śmiercią.
• Glikogenoza typu V (McArdle’a) – brak fosforylazy
mięśniowej – mała tolerancja na wysiłek.
Nienormalnie wysoka zawartość glikogenu w
mięśniach szkieletowych.
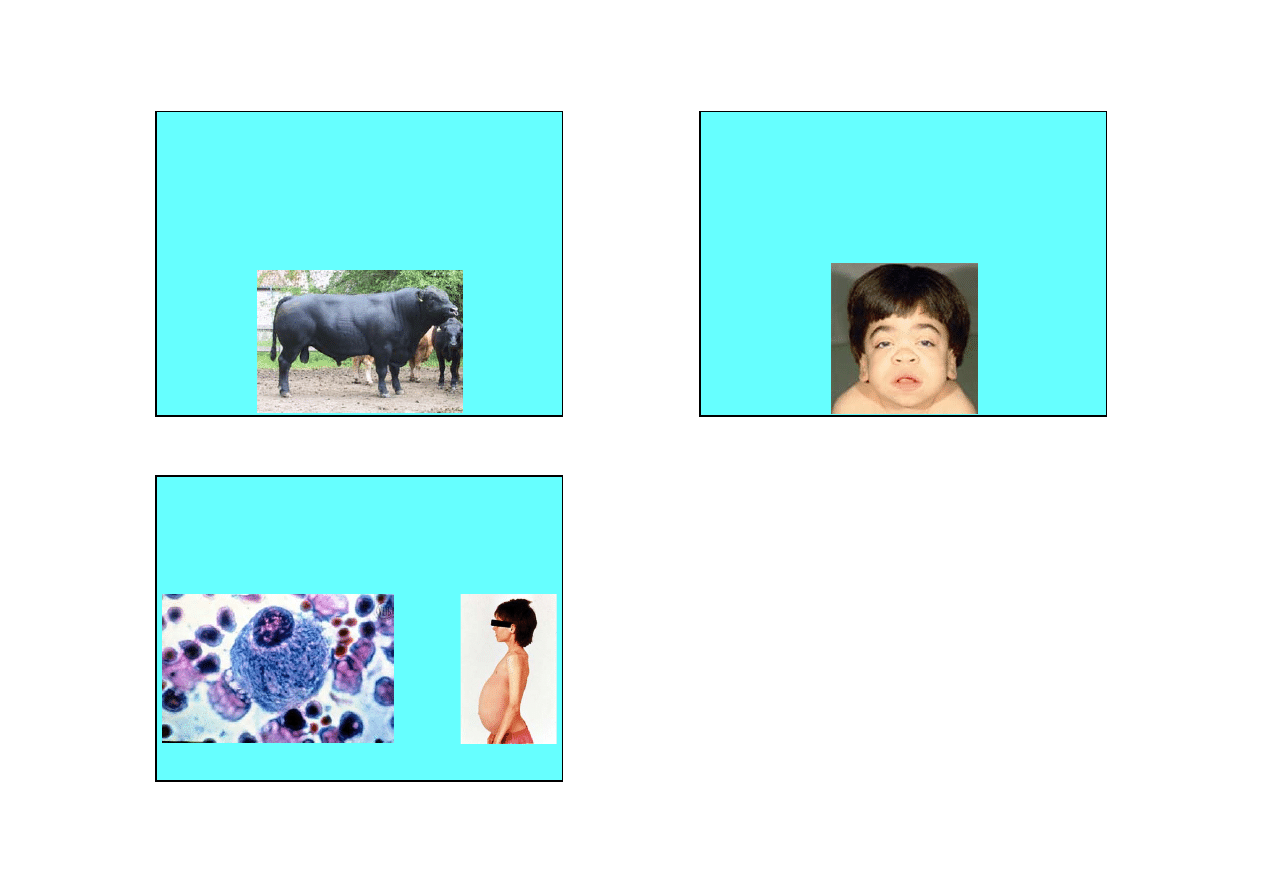
Glikogenozy
Glikogen w komórkach nerki
Glikogenoza typu V u cielęcia

5
Glikolipidy
Gangliozyd GM1
Patogeneza chorób spichrzeniowych
Patogeneza chorób spichrzeniowych
Patogeneza chorób spichrzeniowych
6
Lipidozy i polisacharydozy
• Mannozydoza – występuje w dwu formach – jako
alfa-mannozydoza i beta-mannozydoza. Zaburzenia
rozkładu oligosacharydów zawierających mannozę i
acetylo-glukozaminę. Występuje u człowieka. Alfa-
mannozydoza wystepuje u bydła Aberdeen-Angus.
Lipidozy i polisacharydozy
• Zespół Hurlera – brak alfa-iduronidazy – zaburzenia
rozpadu glikozaminoglikanów. Dochodzi do
gromadzenia związków macierzy pozakomórkowej.
Występuje u człowieka, ale była również stwierdzana
u psa i kota.
Lipidozy i polisacharydozy
• Choroba Gauchera – brak glukocerebrozydazy –
gromadzenie glukocerebrozydów w komórkach
wątroby i śledziony. Występuje również u psów.
Leukocyt ze złogami glukocerebrozydu
Hepatosplenomegalia
Wyszukiwarka
Podobne podstrony:
Mechanizmy chorób dziedzicznych 1
Mechanizmy chorob atopowych, Medycyna, Med3, Alergologia (pajro)
Choroby dziedziczne, Szkoła, przydatne w szkole
Choroby dziedziczne
Wykłady z genetyki (Choroby, dziedziczność) by Kusy
MECHANIZMY STABILNEGO DZIEDZICZENIA PLAZMIDÓW(1)
2006 hemochromatoza dziedziczna najczestsza choroba dziedz c
8. Mechanizm chorób zakaźnych, szkoła medyczna
X-meni albo choroba dziedziczna, Naja Snake
Mechanizmy chorˇb dziedzicznych
CYSTYNURIA choroba dziedziczna
Zwyrodnienia siatkówki Choroby dziedziczne, Okulistyka-Optometria, Choroby siatkówki (umkc)
Mechanizmy chorob atopowych, Medycyna, Med3, Alergologia (pajro)
Choroby dziedziczne, Szkoła, przydatne w szkole
Podstawy mechanizmów zmienności i dziedziczenia
więcej podobnych podstron