
Ć
WICZENIA
Z
O
GÓLNEJ
T
ECHNOLOGII
ś
YWNO
Ś
CI
K
ATEDRA
B
IOTECHNOLOGII
,
ś
YWIENIA
C
ZŁOWIEKA
I
T
OWAROZNAWSTWA
ś
YWNO
Ś
CI
U
NIWERSYTET
P
RZYRODNICZY W
L
UBLINIE
12
LUTY
2010

S
PIS TREŚCI
12 luty 2010
2
PRZEPISY BHP ................................................................................................................................. 4
REGULAMIN OBOWIĄZUJĄCY NA ĆWICZENIACH Z OGÓLNEJ TECHNOLOGII
śYWNOŚCI ................................................................................................................................. 6
1.
REAKCJE MAILLARDA .......................................................................................................... 6
1.1 WPROWADZENIE…………………………………………………………………..........6
1.2.
W
YKONANIE OZNACZENIA
................................................................................................. 132
1.3.
P
RZEDSTAWIENIE WYNIKÓW
.............................................................................................. 143
1.4.
S
PRZĘT I ODCZYNNIKI
........................................................................................................ 143
2.
EKSTRAKCJA W PRZEMYŚLE SPOśYWCZYM ........................................................... 154
2.1.
W
PROWADZENIE
................................................................................................................ 154
2.2.
W
YKONANIE ĆWICZENIA
...................................................................................................... 18
2.3.
P
RZEDSTAWIENIE WYNIKÓW
................................................................................................ 18
2.4.
S
PRZĘT I ODCZYNNIKI
.......................................................................................................... 19
3.
WARUNKI POWSTAWANIA śELU PEKTYNOWEGO ................................................... 20
3.1.
W
PROWADZENIE
.................................................................................................................. 20
3.2.
W
YKONANIE ĆWICZENIA
...................................................................................................... 24
3.3.
P
RZEDSTAWIENIE WYNIKÓW
.............................................................................................. 254
4.
ZAGĘSZCZANIE ROZTWORÓW W TECHNOLOGII śYWNOŚCI ............................ 265
4.1.
W
PROWADZENIE
................................................................................................................ 265
4.2.
W
YKONANIE ĆWICZENIA
.................................................................................................... 343
4.3.
P
RZEDSTAWIENIE WYNIKÓW
.............................................................................................. 354
4.4.
S
PRZĘT I ODCZYNNIKI
........................................................................................................ 354
5.
ROZMRAśANIE śYWNOŚCI ................................................................................................ 35
5.1.
W
PROWADZENIE
.................................................................................................................. 35
5.2.
W
YKONANIE ĆWICZENIA
...................................................................................................... 40
5.3.
P
RZEDSTAWIENIE WYNIKÓW
................................................................................................ 40
5.4.
S
PRZĘT I ODCZYNNIKI
.......................................................................................................... 41
6.
SUSZENIE PRODUKTÓW SPOśYWCZYCH ..................................................................... 42
6.1.
W
PROWADZENIE
.................................................................................................................. 42
6.2.
W
YKONANIE ĆWICZENIA
...................................................................................................... 50
6.3.
P
RZEDSTAWIENIE WYNIKÓW
................................................................................................ 52
6.4.
S
PRZĘT I ODCZYNNIKI
.......................................................................................................... 52
7.
MIKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII śYWNOŚCI ..................... 53
7.1.
W
PROWADZENIE
.................................................................................................................. 53
7.2.
W
YKONANIE ĆWICZENIA
...................................................................................................... 62
7.3.
P
RZEDSTAWIENIE WYNIKÓW
................................................................................................ 63
8.
WODA W PRZEMYŚLE SPOśYWCZYM ........................................................................... 64
8.1.
W
PROWADZENIE
.................................................................................................................. 64
8.2.
W
YKONANIE ĆWICZENIA
...................................................................................................... 78
8.3.
P
RZEDSTAWIENIE WYNIKÓW
................................................................................................ 80
8.4.
S
PRZĘT I ODCZYNNIKI
.......................................................................................................... 80
9.
UTRWALANIE śYWNOŚCI PRZEZ ZAKWASZENIE ..................................................... 81
9.1.
W
PROWADZENIE
.................................................................................................................. 81
9.2.
W
YKONANIE ĆWICZENIA
...................................................................................................... 89
9.3.
P
RZEDSTAWIENIE WYNIKÓW
................................................................................................ 94
9.4.
S
PRZĘT I ODCZYNNIKI
.......................................................................................................... 94
10.
EMULSJE ................................................................................................................................... 95
10.1.
WPROWADZENIE
................................................................................................................... 95

10.2.
W
YKONANIE ĆWICZENIA
.................................................................................................... 105
11.
TERMICZNE UTRWALANIE śYWNOŚCI ....................................................................... 108
03
11.2.
W
YKONANIE ĆWICZENIA
.................................................................................................... 119
11.3.
P
RZEDSTAWIENIE WYNIKÓW
.............................................................................................. 119
11.4.
S
PRZĘT I ODCZYNNIKI
........................................................................................................ 120
ZAŁĄCZNIKI ................................................................................................................................ 121
Z
AŁĄCZNIK
1.
W
ZÓR SPRAWOZDANIA Z ĆWICZEŃ
................................................................... 121
LITERATURA ............................................................................................................................... 122
SPIS AUTORÓW ........................................................................................................................... 123

PRZEPISY BHP
12 luty 2010
4
PRZEPISY BHP
1. Do laboratorium przychodzimy bez płaszczy (kurtek), dużych plecaków, toreb itp., które
zostawiamy w szatni.
2. Osoby przebywające na sali ćwiczeń obowiązuje bezwzględny nakaz stałego używania
fartuchów ochronnych (zapiętych!)
3. Zabrania się spożywania posiłków i picia napojów w laboratorium.
4. Podczas ćwiczeń zachowujemy ostrożność. Chaotyczne i bezmyślne wykonywanie
poszczególnych operacji oraz niedostateczna ich znajomość prowadzi najczęściej do
nieszczęśliwych wypadków.
5. Wszelkie urządzenia włączamy/wyłączamy jedynie za zgodą prowadzącego.
6. Przyjmujemy zasadę, że żadnych substancji w laboratorium nie badamy na smak.
7. Powonieniem badamy tylko substancje wskazane przez prowadzącego. Nie nachylamy się
nigdy bezpośrednio nad naczyniem i nie wdychamy głęboko par substancji, lecz tylko pary
danej substancji kierujemy dłonią w stronę twarzy.
8. Przy ogrzewaniu i przelewaniu cieczy nie nachylamy się nad nimi, ponieważ mogą wyprysnąć i
trafić do oka lub poparzyć twarz.
9. Przy pracy z substancjami, które mogą ulec rozpryskiwaniu lub wybuchowi stosujemy zawsze
okulary ochronne.
10. Ogrzewanie cieczy w probówkach wykonujemy w ten sposób, że wylot probówki skierowujemy
tak, aby ciecz przy ewentualnym wypryśnięciu nie oblała nikogo znajdującego się w
laboratorium.
11. Przy przenoszeniu naczyń i przedmiotów gorących bierzemy je w rękę poprzez ściereczkę lub
za pomocą szczypiec.
12. Podczas przelewania cieczy żrących i przesypywania substancji żrących nakładamy ochronne
rękawice i okulary. Odczynników umieszczonych pod digestorium nie wolno wynosić poza jego
obręb bez wyraźnej zgody prowadzącego ćwiczenia.
13. Prace z substancjami o nieprzyjemnym zapachu oraz wydzielającymi szkodliwe dla zdrowia
pary wykonujemy zawsze pod włączonym wyciągiem (pod digestorium).
14. Gdy mimo zachowania ostrożności dojdzie do kontaktu z substancją niebezpieczną (kontakt z
oczami, skórą, przy spożyciu lub wdychaniu) neutralizujemy jej działanie zgodnie z
wytycznymi
umieszczonymi
w
karcie
charakterystyki
substancji
chemicznej.
O zaistniałym zdarzeniu zawsze w pierwszej kolejności informujemy prowadzącego ćwiczenia.
15. Substancji niebezpiecznych (np. rozpuszczalników organicznych) nie wylewamy do kanalizacji
bez zgody prowadzącego.
16. W przypadku skaleczenia się szkłem lub innym ostrym narzędziem ranę przemywamy 3% wodą
utlenioną i owijamy sterylnym bandażem. Przy poważniejszych skaleczeniach przystępujemy
przede wszystkim do zatamowania krwotoku. W tym celu wykonujemy ucisk powyżej
skaleczenia przy krwotoku tętniczym, a poniżej przy żylnym. Po założeniu ucisku należy udać
się jak najszybciej do lekarza.
17. W przypadku powstania pożaru w laboratorium gasimy ogień odpowiednim środkiem
gaśniczym.
18. Po zakończeniu ćwiczeń student zobowiązany jest uporządkować stanowisko pracy i
doprowadzić je do stanu uniemożliwiającego wystąpienie zagrożeń.
19. W przypadku wystąpienia wątpliwości lub zauważonych nieprawidłowości (np. w działaniu
urządzeń) - należy natychmiast zwrócić się z zapytaniem do osoby prowadzącej ćwiczenia.

REAKCJE MAILLARDA
REGULAMIN OBOWIĄZUJĄCY NA ĆWICZENIACH
Z OGÓLNEJ TECHNOLOGII śYWNOŚCI
Na ćwiczenia:
- przychodzimy punktualnie
- obowiązuje fartuch (zapięty!)
- warunkiem dopuszczenia do wykonywania ćwiczeń jest teoretyczna znajomość materiału
ćwiczeniowego
- obowiązuje posiadanie własnej kopii wykonywanego ćwiczenia
- ćwiczenia wykonywane są w stałych zespołach 2-3 osobowych
- stłuczone kolbki, pipety, zlewki, cylindry, biurety i inny zepsuty, uszkodzony sprzęt zgłaszamy
prowadzącemu ćwiczenia, a następnie zapisujemy w odpowiednim zeszycie
- po każdym z wykonanych ćwiczeń należy oddać sprawozdanie z przebiegu ćwiczenia (wzór
sprawozdania zamieszczono w załączniku 1 na stronie 128)
- w przypadku nie oddania sprawozdania w dniu wykonania ćwiczenia, należy otrzymane wyniki
przedstawić na piśmie prowadzącemu w celu uzyskania parafki.
- sprawozdanie z wykonywanego ćwiczenia przynosimy najpóźniej na następne ćwiczenia (z
dołączonymi parafowanymi w dniu wykonania ćwiczenia wynikami)
Kolokwia:
- kolokwium będzie składać się z pytania/pytań teoretycznych związanych z wykonywanymi
ćwiczeniami, zadań rachunkowych (formę kolokwium ustala prowadzący!)
- pierwsze kolokwium odbędzie się po przeprowadzeniu połowy ćwiczeń, drugie kolokwium
zaplanowane jest po ukończeniu pozostałych ćwiczeń.
- za odpowiedzi na pytania pisane nieczytelnie jak i nie na temat będzie przyznawane 0 punktów
- nieusprawiedliwiona nieobecność na zajęciach pociąga za sobą wystawienie oceny 2,0 z kolokwium
- niezaliczoną teorię należy zaliczyć „ustnie” na następnych zajęciach lub w terminie ustalonym
wspólnie z prowadzącym
Ocena końcowa:
- posiadanie nieusprawiedliwionych lub więcej niż 2 usprawiedliwione nieobecności jest podstawą do
niezaliczenia ćwiczeń
- niezaliczenie więcej niż 2 ćwiczeń (niezaliczone sprawozdania) jest podstawą do niezaliczenia
ćwiczeń (nie ma możliwości odrabiania ćwiczeń praktycznych!)
- ocena końcowa będzie średnią ze wszystkich ocen uzyskanych z teorii, zadań rachunkowych w
przebiegu ćwiczeń
- jeżeli wspomniana średnia będzie mniejsza niż 3,0 wówczas możliwe będzie poprawienie tej oceny
pod koniec semestru
- na ocenę końcową wpływać będzie również postawa studenta na ćwiczeniach (m.in. ilość spóźnień,
terminowe oddawanie sprawozdań, odpowiednia postawa i zaangażowanie w prace)
R
EAKCJE
M
AILLARDA
12 luty 2010
6
1.
REAKCJE MAILLARDA
1.1.
W
PROWADZANIE
Reakcje Maillarda (lub reakcja Maillarda) zwane są również ciemnieniem nieenzymatycznym
albo reakcjami melanoidynowymi. Jest to szereg złożonych reakcji chemicznych, jakim podlegają
różne produkty pochodzenia roślinnego i zwierzęcego, prowadzących do wytworzenia się ciemnego
zabarwienia produktu (grec. melanos - czarny). Po raz pierwszy reakcje te były badane przez
Maillarda (1912 r.) przy użyciu mieszanin cukru redukującego i aminokwasu, i stąd noszą jego
imię. Charakter reagującego cukru prostego, jego budowa, struktura przestrzenna, a przede
wszystkim czynniki sprzyjające przesunięciu się równowagi w kierunku formy acyklicznej z wolną
grupą karbonylową (podwyższone pH roztworu, podwyższona temperatura, znaczne stężenie
substratów) sprzyjają reakcji Maillarda, a także decydują o jej przebiegu i produktach końcowych
(melanoidynach). Zgodnie z sugestią Kostyczewa i Brillianta (1920 r.) reakcje te mogą zachodzić
czasami w żywej komórce.
Różne aldehydy i ketony, w tym cukry redukujące, kondensują łatwo z aminokwasami,
aminami, peptydami, czy też z białkami, tworząc tzw. zasady Schiffa, a następnie W-podstawione
glikozyloaminy.
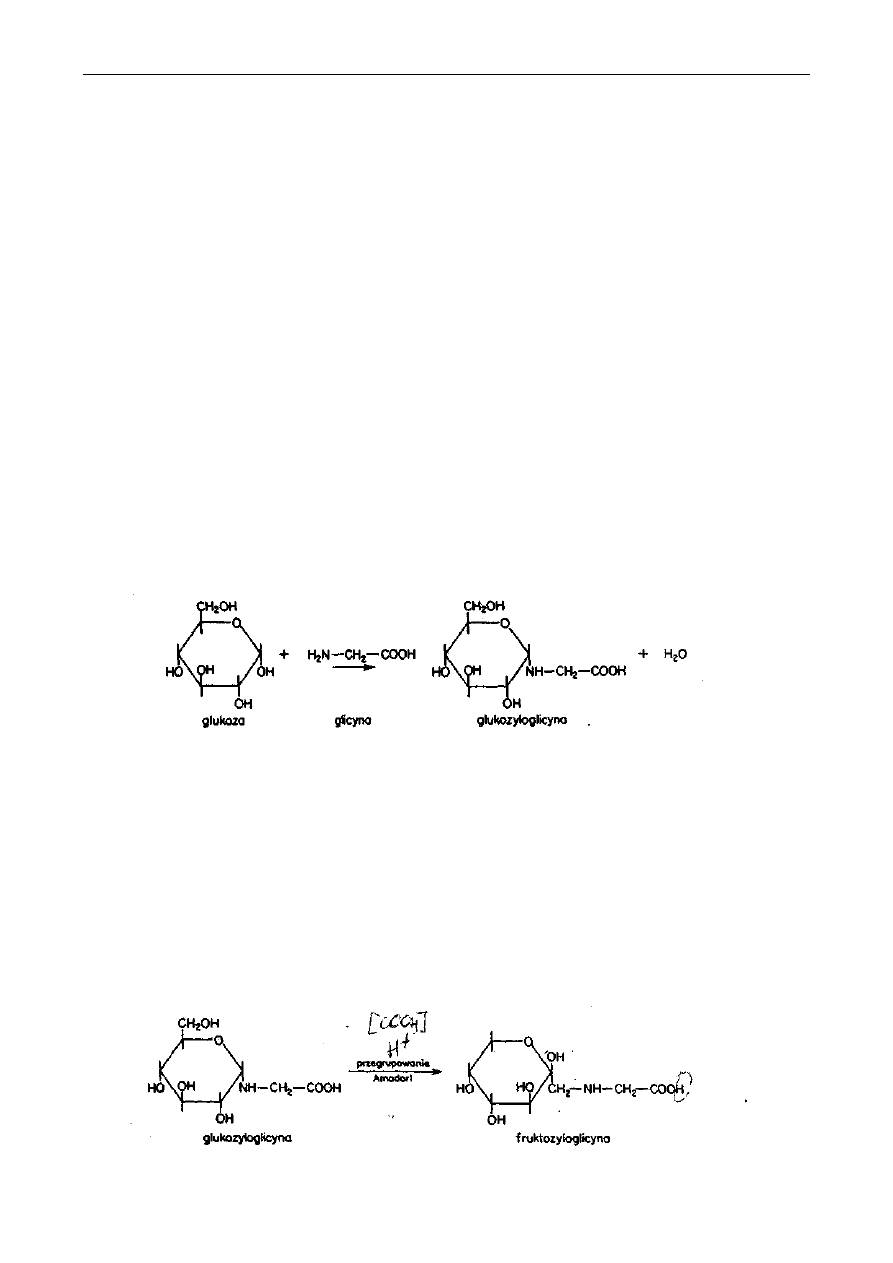
W przypadku prostej mieszaniny glukozy i glicyny można odtworzyć początkowe stadia reakcji
Maillarda.
Glukoza reaguje za pomocą swojej grupy aldehydowej z grupą aminową glicyny, tworząc
glukozyloglicynę:
W czasie tej kondensacji wydziela się cząsteczka wody, dlatego reakcja zachodzi lepiej w
środowisku o małej zawartości wody. Produkt reakcji jest bezbarwny. Pentozy reagują szybciej niż
heksozy, a monosacharydy są aktywniejsze niż redukujące disacharydy. Brak wolnej grupy
hydroksylowej pólacetalu jest przyczyną, że cukry nieredukujące nie reagują, stąd np. sacharoza tak
długo nie wchodzi w ciąg reakcji ciemnienia nieenzymatycznego, jak długo nie zostanie
zhydrolizowana do glukozy i fruktozy. Kwasy uronowe i estry fosforanowe cukrowców reagują
łatwiej niż odpowiadające im cukry wolne. Im dalej od grupy karboksylowej położona jest grupa
aminowa, tym szybciej reaguje aminokwas, przy czym aminokwasy zasadowe są bardziej
reaktywne niż kwaśne. Duża reaktywność lizyny jest przyczyną poważnych strat tego egzogennego
aminokwasu w żywności.
Drugą reakcją jest przegrupowanie Amadori, które prowadzi do wytworzenia się
fruktozoglicyny, będącej chemicznie 1-amino-l-deoksyfruktozą:
REAKCJE MAILLARDA
Przegrupowanie Amadori wymaga jako katalizatora obecności jonów wodorowych. Funkcję tę
może spełniać grupa karboksylowa aminokwasu. Powstały związek jest również bezbarwny.
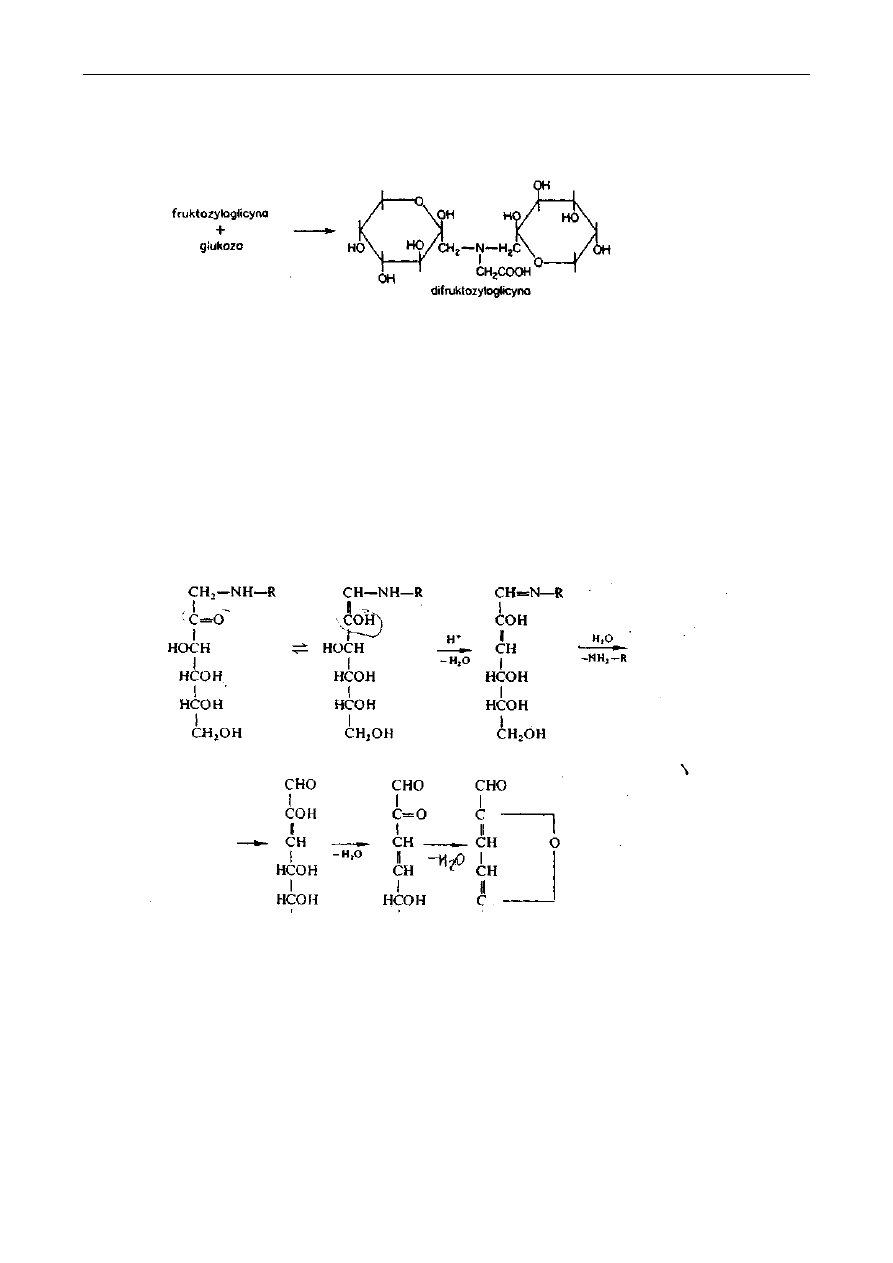
Fruktozyloglicyna, a także inne ketozoaminy mogą reagować z drugą cząsteczką glukozy i po
następnym przegrupowaniu Amadori powstaje difruktozyloglicyna:
Zarówno monoketozoaminokwasy jak i diketozoaminokwasy zostały wyodrębnione z
przecierów owocowych, wykazujących zbrunatnienie nieenzymatyczne. Diketozoaminy są mniej
trwałe od monoketozoamin. Podczas ogrzewania roztworów wodnych powstają ponownie
monoketozoaminy i szereg produktów rozkładu z silnie zaznaczonymi właściwościami związków
nienasyconych z grupami karbonylowymi. Monoketozoaminy również podlegają rozkładowi
podobnego typu.
Fruktozyloglicyna może podlegać albo 1,2-enolizacji, albo też 2,3-enolizacji. Pierwsza
enolizacja zachodzi w głównej mierze w warunkach łagodnych, jakie przeważają w produktach
żywnościowych. Po odszczepieniu grupy hydroksylowej w pozycji 3 (reakcja odwodnienia) tworzy
się zasada Schiffa, której hydroliza prowadzi do 3-deoksyozulozy. Następne odszczepienie grupy
hydroksylowej przy C-4 (odwodnienie) daje nienasyconą ozulozę, która przez ponowne usunięcie
wody przekształca się w hydroksymetylofurfural:
Aminokwas został uwolniony w formie niezmienionej. Jednakże nie musi nastąpić uwolnienie
aminokwasu i wówczas powstaje zasada Schiffa hydroksymetylofurfuralu. 2,3-enolizacja prowadzi
do utworzenia się 2,4-diulozy poprzez nietrwałą 2,3-diulozę:
R
EAKCJE
M
AILLARDA
12 luty 2010
8
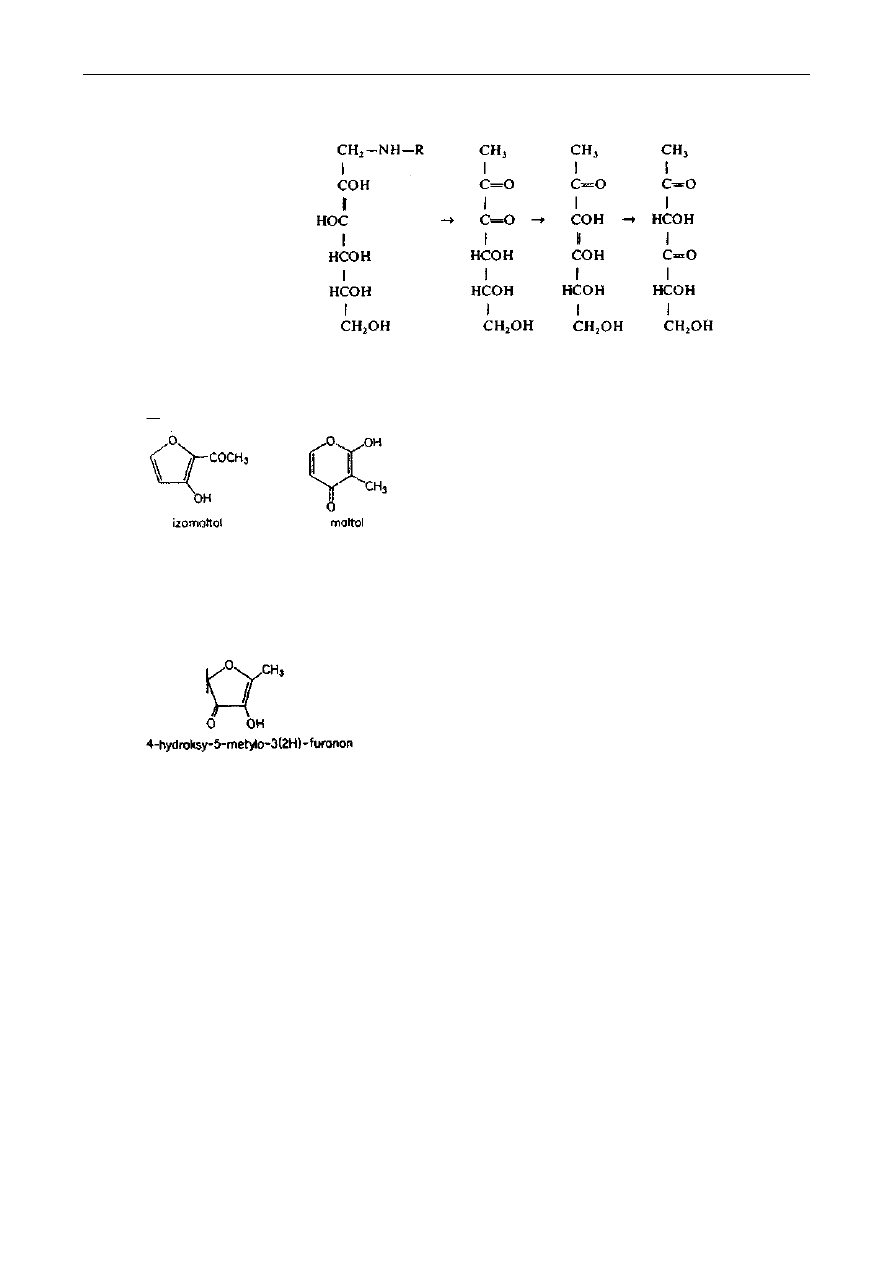
Reakcje powyższe mają mniejsze znaczenie w tworzeniu się barwnych połączeń, lecz
uczestniczą w produkcji lotnych substancji zapachowych. 2,3-Enolizacja może prowadzić do takich
lotnych substancji jak izomaltol i jego pironowy izomer - maltol:
Związki te stanowią składniki zapachu pieczywa.
Natomiast rozkład D-ksylozy lub D-mannozy w obecności związków aminowych prowadzi do
powstania 4-hydroksy-5-rnetylo-3(2H)-furanonu, który to rozkład również poprzedza 2,3-
enolizacja:
Związek ten jest składnikiem zapachu gotowanej wołowiny.
Czynniki wpływające na reakcje Maillarda
Temperatura ma jednoznaczny wpływ na nieenzymatyczne ciemnienie produktów. Wzrost
temperatury o 10°C przyspiesza reakcje ciemnienia 2-3-krotnie, a nawet czasami 3-4-krotnie.
Wynika stąd wniosek, że przechowywanie żywności w podwyższonych temperaturach sprzyja
ciemnieniu nieenzymatycznemu, prowadząc często do dyskwalifikacji produktu.
Wzrost pH również sprzyja reakcji ciemnienia. Reakcja jest powolna poniżej pH 5-6 i szybko
wzrasta wraz z podnoszeniem się pH. Zmiany pH wpływają nie tylko na zmianę szybkości reakcji,
lecz i na jej przebieg jakościowy-w środowisku zasadowym brunatnienie przebiega przez azotowe
związki pośrednie. Podczas reakcji ciemnienia ma miejsce zmiana kwasowości środowiska, reakcja
ma zatem przebieg inny w środowisku zbuforowanym niż niezbuforowanym. Oczywiście sam bufor
może odgrywać rolę katalizatora reakcji, np. kwasy organiczne, fosforany.
W ciemnieniu produktów żywnościowych dużą rolę odgrywa ilość zawartej wody.
Rozcieńczenie roztworów powoduje spadek szybkości ciemnienia nieenzymatycznego. Soki
owocowe ciemnieją wolniej niż ich koncentraty. Natomiast produkty odwodnione, z których
usunięto wodę wolną, nie podlegają ciemnieniu. W tym drugim przypadku czynnikiem
ograniczającym szybkość reakcji jest dyfuzja substratów. W miarę zwiększania się ilości wody
swobodna dyfuzja ułatwia coraz bardziej zajście reakcji, natomiast przy dużych rozcieńczeniach
REAKCJE MAILLARDA
maleje stężenie substratów, hamując szybkość ciemnienia. Efekt zmniejszającej się szybkości
ciemnienia przy małych zawartościach wody trudno wytłumaczyć wyłącznie zmniejszoną dyfuzją
substratów. W pewnych stadiach reakcji Maillarda ma bowiem miejsce wydzielanie się wody,
zatem jej usuwanie powinno sprzyjać reakcji. Z rozkładu jednego mola cukru podczas reakcji
dehydratyzacji tworzy się ponad 3,5 mola wody w różnych fazach ciemnienia nieenzymatycznego,
działając jako inhibitor reakcji. Obecność tlenu nie wpływa zasadniczo na szybkość procesu
ciemnienia nieenzymatycznego. Jednakże w niektórych przypadkach można wykazać zwiększoną
szybkość ciemnienia w obecności tlenu, podczas gdy w innych przypadkach działa on jak inhibitor.
Niewątpliwie tlen sprzyja reakcji ciemnienia spowodowanej rozkładem kwasu askorbinowego.
Fosforany, kwasy karboksylowe i ich sole zwiększają zarówno szybkość ciemnienia, jak też
intensywność końcowej barwy roztworu. Miedź przyspiesza ciemnienie jedynie w przypadku
obecności kwasu askorbinowego. Cyna wydaje się opóźniać ciemnienie soków owocowych
prawdopodobnie z uwagi na wytwarzanie środowiska redukującego. Jednakże najlepszym
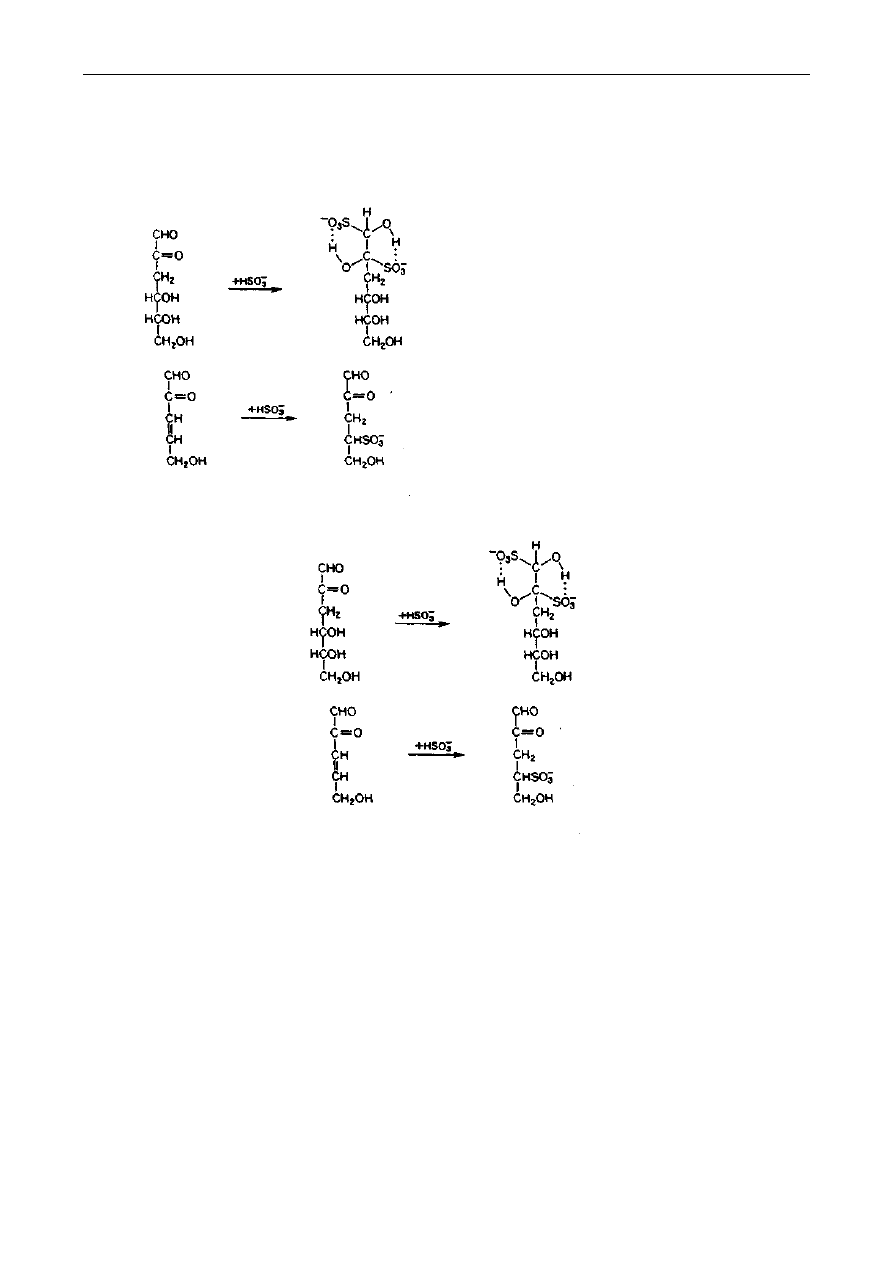
inhibitorem, w praktyce przemysłowej, jest dwutlenek siarki lub siarczyny. Inhibicja procesu
zachodzi w stadium tworzenia się 3-deoksyozuloz, a także nienasyconych ozuloz.
W pierwszym przypadku siarczyn łączy się z deoksyozulozą w sposób odwracalny, opóźniając
reakcje ciemnienia. W drugim przypadku powstaje nieodwracalny produkt addycji trwały tak długo,
jak długo siarczyn nie zostanie utleniony. Ponadto SO
2
wykazuje właściwości redukujące, co jest
szczególnie ważne w przypadku ciemnienia wywołanego rozkładem kwasu askorbinowego:
Reakcje Maillarda w procesach przetwarzania i składowania żywności
Reakcje Maillarda towarzyszą wielu procesom technologicznym w przemyśle żywnościowym.
Często mają one dodatni wpływ na produkt wskutek wytwarzania tzw. aromatów wtórnych oraz
nadawania produktom odpowiedniej barwy. Reakcjom tym przypisuje się polepszenie cech
sensorycznych podczas wypieku chleba, czy przyrządzania tostów, prażenia ziarna kawowego i
orzechów ziemnych, wyrobu płatków śniadaniowych kukurydzianych lub innych, biszkoptów, przy
obróbce termicznej ziarna kakaowego i herbaty, przygotowywaniu słodów w browarnictwie,
wyrobie whisky i wielu innych. Z niepożądanych skutków ciemnienia nieenzymatycznego należy
wymienić spadek wartości odżywczej takich przetworów, jak soki owocowe (straty witaminy C),
wiązanie deficytowej lizyny w produktach zbożowych, podczas smażenia lub pieczenia mięsa itp.
W takich przypadkach zapobieganie ciemnieniu wysuwa się na pierwszy plan.
Najlepszym sposobem zapobiegania ciemnieniu wydaje się schładzanie żywności czy też jej
zamrażanie. Gdy w grę wchodzi kondensacja cukrów z białkami lub aminokwasami, obniżenie pH
sprzyja spowolnieniu procesu ciemnienia. W przypadku soków owocowych, w których ciemnieniu
uczestniczy kwas askorbinowy, obniżenie pH nie jest celowe, gdyż stymuluje ono ciemnienie. Tak,

R
EAKCJE
M
AILLARDA
12 luty 2010
10
więc soki pomarańczowe (pH = 3,4) ciemnieją znacznie wolniej niż soki grapefruitowe (pH = 2,9)
czy cytrynowe (pH = 2,15). Z tego też powodu soki grapefruitowe i cytrynowe zatęża się w
stosunku 4:1, podczas gdy soki pomarańczowe mogą być zatężane w stosunku 6:1.
Jeżeli cukier redukujący stanowi nieistotny składnik artykułu (np. jaja, mięso), to utlenienie
grupy aldehydowej zapobiega skutecznie reakcji Maillarda. Bardzo efektywnie działa dodatek
oksydazy glukozowej i katalazy, dzięki którym powstają produkty nieszkodliwe dla zdrowia (kwas
glukonowy, woda i tlen), a środowisko zostaje pozbawione grup aldehydowych.
Zastąpienie glukozy nieredukującą sacharozą, czy mniej aktywną fruktozą, jest również
efektywnym sposobem zahamowania czy opóźnienia reakcji Maillarda. W tym przypadku jednak
dalsza obróbka technologiczna nie powinna prowadzić do hydrolizy sacharozy.
Ciemnienie soków cytrusowych łączy się ściśle z obecnością powietrza (tlenu). W
doświadczenia Joslyna i in. obserwowano prostą zależność między ciemnieniem a ubytkiem kwasu
askorbinowego. Natomiast w warunkach beztlenowych soki nie ciemniały nawet po upływie
jednego roku. Z kolei soki malinowe, truskawkowe i porzeczkowe ciemniały bardzo szybko mimo
dokładnej deaeracji. Wynika z tego, że ten problem jest bardzo złożony i nie może być wyjaśniony
jednoznacznie.
Ciemnienie suszonych owoców, koncentratów owocowych zwykle łączy się z wydzielaniem
dwutlenku węgla. Można obserwować pojawianie się wybrzuszeń na całkowicie sterylnych
konserwach koncentratów pomarańczowych podczas długotrwałego przechowywania. Istnieje
bezpośredni związek wydzielającego się CO
2
z ciemnieniem koncentratów. Równocześnie ma
miejsce rozkład kwasu askorbinowego. Natomiast dwutlenek siarki powstrzymuje wydzielanie się
CO
2
i w takim samym stopniu hamuje ciemnienie.
Znacznie trudniej jest wykazać wpływ cukrów redukujących na szybkość ciemnienia soków
owocowych. Przyczyna tkwi w zbyt dużym stężeniu cukrów, maskujących ewentualne straty, jakie
można by wykazać analitycznie.
Trzeba, bowiem pamiętać, że wystąpienie reakcji Maillarda, nawet w małym stopniu, pociąga
za sobą znaczne ściemnienie produktu z uwagi na intensywną barwę tworzących się melanoidyn.
Tym można wytłumaczyć niepowodzenie doświadczenia Joslyna i Marsna, którzy badali wpływ
usunięcia cukrów przez fermentacje na szybkość ciemnienia soków pomarańczowych. Zarówno
sfermentowane, jak i niesfermentowane soki ciemniały w tym samym stopniu. Podobne
doświadczenie przeprowadzili Stadtman i in. na syropach morelowych wykazując, że usunięcie
cukrów przez fermentację spowodowało zmniejszenie się intensywności ciemnienia o około połowę
w porównaniu z próbkami nieodfermentowanymi. Dodatek fruktozy i glukozy przywrócił
ciemnienie sfermentowanych syropów do wartości próbek nieodfermentowanych. Doświadczenie
to wykazało, że część reakcji ciemnienia była spowodowana innymi czynnikami niż cukry
redukujące.
Znaczne trudności analityczne napotyka się podczas analizy frakcji azotowych soków
owocowych, suszonych owoców itp. w celu wykazania ich udziału w ciemnieniu
nieenzymatycznym. Trudne są również do interpretacji wyniki badań ź dodanymi ilościami różnych
związków azotowych do soków. Do ciekawych wniosków prowadzi analiza tabeli:
Czas o
Czas ogrzewania
[h]
Barwa
roztworu
jednostki
umowne
Składniki soku pomidorowego
cukry
ogółem
%
azot
całkowity
%
azot
aminowy
%
koloidy
%
0
2
3,33
0,095
0,050
0,295
22
45
3,01
0,088
0,032
-
41
92,1
2,88
0,080
0,025
0,612
49
65,7
2,83
0,070
0,025
0,425
wytraca się
osad

REAKCJE MAILLARDA
Zmiany zawartości niektórych składników puszkowanego soku pomidorowego podczas
ogrzewania zamkniętych puszek z sokiem pomidorowym obserwuje się w czasie od 0 do 49 h
wzrost zabarwienia i w końcu wypadanie osadów (niższy stopień zabarwienia po 49 h w stosunku
do wartości po 41 h łączy się z wytrąceniem osadu). Równocześnie maleje zawartość cukrów i
azotu ogólnego i aminowego. Analiza elementarna wytworzonych melanoidyn (osad) wykazała
54,2% C, 5,4% H, 7,35% N i 33,05% O. Podczas ogrzewania roztworu modelowego mieszaniny
glukozy i glicyny uzyskuje się bardzo podobny skład elementarny osadu: 56,17% C, 5,6% H,
7,87% N i 30,36% O. Reakcje Maillarda powodują zmniejszanie się wartości odżywczej produktów
żywnościowych. Szczególnie podatne na reakcje Maillarda są artykuły mleczarskie z uwagi na
zawartość laktozy (cukier redukujący) oraz względnie niestabilnych białek. Artykuły zbożowe są
również wrażliwe na reakcje Maillarda z uwagi na częściową hydrolizę enzymatyczną i termiczną
skrobi do cukrów redukujących. Mączki rybne natomiast zawierają kwasy nukleinowe, których
hydroliza do wolnej rybozy prowadzi do bardzo aktywnych reakcji tworzenia się melanoidyn.
Natomiast nasiona roślin strączkowych i produkty drożdżowe są stosunkowo trwałe, z uwagi na
małą zawartość cukrów redukujących. Mięso jest również trwałym produktem, że względu na brak
cukrów redukujących i odpowiednią kwasowość.
Podczas wypieku chleba zostaje związane 10-15% lizyny, podczas czerstwienia ulega stracie
dalsze 5%, a przygotowywanie tostów powoduje znów rozkład 5-10%. Melanoidyny mogą również
zmniejszać przyswajalność białek wskutek ich kompleksowania. W doświadczeniach ze szczurami
dieta kazeinowa (mieszanina 0,2% glukozy z glicyną) powodowała spadek przyswajalnego azotu z
49 do 31%, co było przyczyną 40%-owego zmniejszenia ich wzrostu.
R
EAKCJE
M
AILLARDA
12 luty 2010
12
1.2.
W
YKONANIE OZNACZENIA
Celem ćwiczenia jest określenie wpływu pH i rodzaju cukrów na nieenzymatyczne ciemnienie
produktów i ich enolizację.
Wykonanie ćwiczenia.
1. Należy przygotować 3 kolbki stożkowe na 100 mL, a następnie do kolbek odważyć
odpowiednio: do pierwszej 5 g fruktozy, do drugiej 5 g sacharozy i do trzeciej 5 g glicyny.
Kolbki należy jednoznacznie i wyraźnie podpisać. Naważki w każdej kolbce należy rozpuścić w
objętości 50 mL wody destylowanej aż do uzyskania roztworów rzeczywistych.
2. Należy przygotować 25 probówek w statywach. Probówki należy podpisać według planu
doświadczenia tak samo jak opisano próbki w poniższych tabelach. W przypadku pojedyńczych
składników (fruktozy, sacharozy i glicyny, Tab. 1) w każdej probówce będzie znajdowało się 8
mL odpowiedniego buforu i 2 mL odpowiedniego roztworu (pierwsza tabelka), zaś w
przypadku roztworu cukru i aminokwasu (Tab. 2) w każdej probówce będzie znajdowało się 6
mL odpowiedniego buforu, 2 mL roztworu cukru i 2 mL roztworu glicyny (Tab.2).
Tab. 1 Roztwory 1-składnikowe.
Dodaj 2mL
roztworu:
Do probówki dodaj 8 mL buforu o pH:
2
4
6
8
10
fruktozy
B2 F
B4 F
B6F
B8 F
B10 F
sacharozy
B2 S
B4 S
B6 S
B8S
B10 S
glicyny
B2 G
B4 G
B6 G
B8 G
B10 G
Tab. 2. Roztwory zawierające cukier i aminokwas.
Dodaj:
Do probówki dodaj 6 mL buforu o pH:
2
4
6
8
10
2mL r-ru fruktozy
2mL r-ru glicyny
B2 F G
B4 F G
B6 F G
B8 F G
B10 F G
2mL r-ru sacharozy
2mL r-ru glicyny
B2 S G
B4 S G
B6 S G
B8 S G
B10 S G
3. Statywy z probówkami (25 sztuk) należy zakorkować korkami aluminiowymi lub z waty,
umieścić w autoklawie nagrzanym do temp 135°C (0,75 at) i autoklawować przez 30 minut.
Uwaga! Autoklaw obsługuje wyłącznie osoba prowadząca ćwiczenie.
4. Po tym czasie zredukować ciśnienie w autoklawie, wyjąć próbki i ochłodzić wstawiając je do
naczynia z wodą.
5. Należy ocenić aromat poszczególnych próbek- ocena organoleptyczna (wykonać w formie
opisowej- tabelka).
6. Należy zmierzyć ekstynkcję każdej z próbek wobec wody destylowanej. (sporządzić tabelę z
ekstynkcjami).
7. Proszę zanotować wyniki i wykreślić zależność pomiędzy ekstynkcją a wartościami pH (wykres-
ekstynkcja odłożona na osi y, wartość pH użytych buforów- na osi x).

REAKCJE MAILLARDA
1.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń umieścić w sprawozdaniu w formie oddzielnych punktów z tabelkami,
wykresem i opisową charakterystyką próbek) i przedłożyć osobie prowadzącej ćwiczenia w celu
sprawdzenia (wzór, załącznik 1 na stronie 121). Należy także zaznaczyć wszelkie odstępstwa od
metod podanych w opracowaniu ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski
w formie zwięzłych punktów.
1.4.
S
PRZĘT I ODCZYNNIKI
Aparatura i odczynniki:
•
autoklaw
•
pH metr laboratoryjny
•
spektrofotometr „Spekol"
•
waga analityczna
•
kolby stożkowe o poj. 100 cm
3
5 sztuk
•
pipeta l cm
3
•
probówki
20 sztuk
•
metalowy statyw do probówek
•
łyżeczka wagowa
1 sztuka
•
cylinder miarowy o poj. 100 cm
3
2 sztuki
•
glukoza
•
fruktoza
•
sacharoza
•
galaktoza
•
glicyna (kwas aminooctowy)-0,1 n roztwór HCL
•
-0,1 n roztwór NaOH
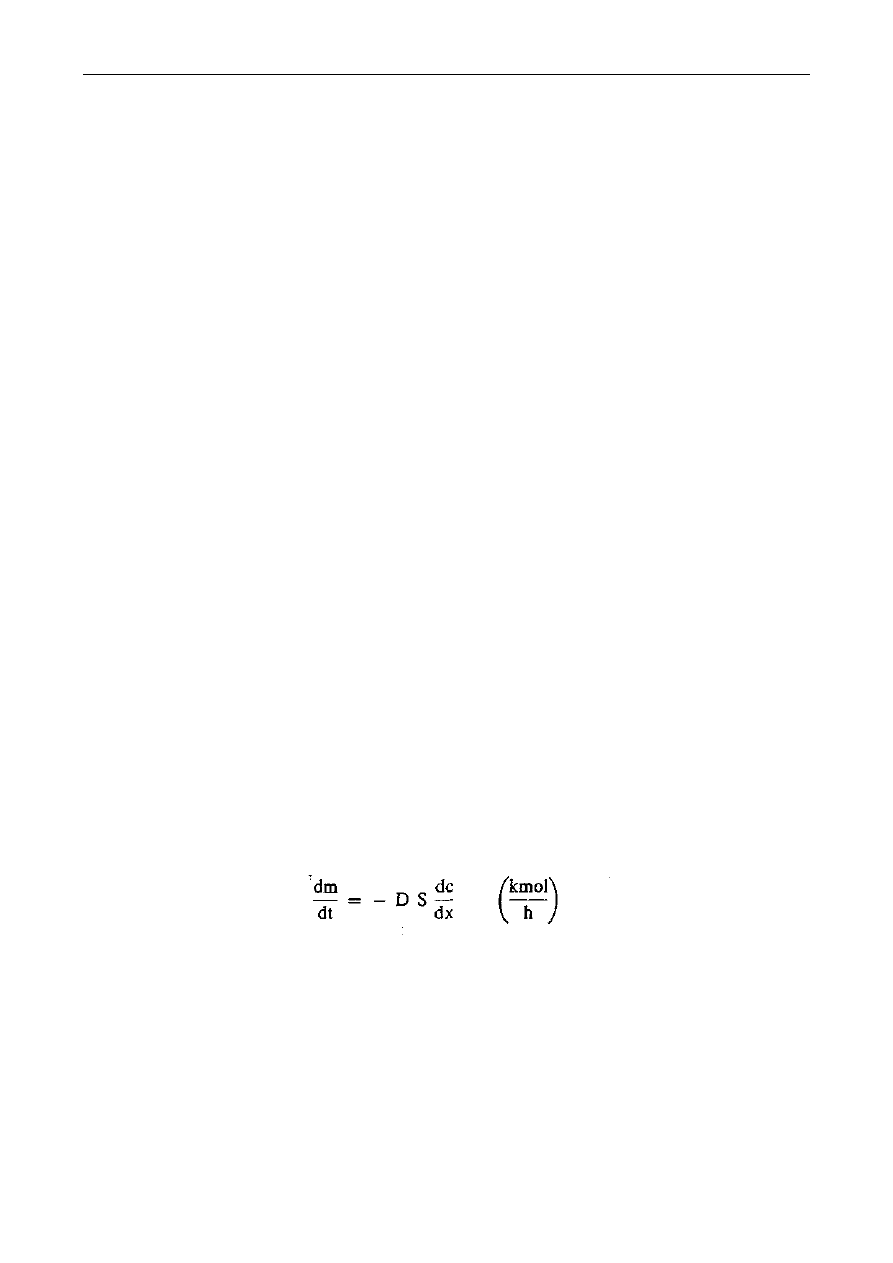
E
KSTRAKCJA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
14
2.
EKSTRAKCJA W PRZEMYŚLE SPOśYWCZYM
2.1.
W
PROWADZENIE
Ekstrakcja polega na częściowym lub całkowitym rozdzieleniu mieszaniny stałej lub ciekłej
przy użyciu rozpuszczalnika, w którym składniki mieszaniny mają niejednakową rozpuszczalność.
W wyniku ekstrakcji otrzymuje się ekstrakt, czyli roztwór składnika w rozpuszczalniku, oraz
rafinat, czyli pozostałość pozbawioną znacznej ilości składnika rozpuszczonego. Ekstrakt i rafinat
tworzą w rozpuszczalniku dwie odrębne fazy, w związku z czym mogą być oddzielone pod
działaniem sił ciężkości lub bezwładności. Fazę rozpuszczalnika poddaje się następnie destylacji
lub rektyfikacji, otrzymując prawie czysty ekstrakt składników i odzyskując znaczną część
rozpuszczalnika użytego do ekstrakcji. Rafinat po oddestylowaniu z niego rozpuszczalnika jest
poddawany suszeniu i wykorzystywany jako produkt uboczny.
W zależności od stanu skupienia rozróżnia się ekstrakcję w układzie ciecz-ciecz i ekstrakcję w
układzie ciało stałe-ciecz. W przemyśle spożywczym ciałem stałym jest materiał biologiczny
charakteryzujący się budową tkankową, w związku z czym w procesie ekstrakcji zjawisku dyfuzji
towarzyszą również zjawiska osmozy i dializy.
W technologii żywności podstawowe znaczenie ma ekstrakcja w układzie ciało stałe-ciecz.
Przykładem tego rodzaju ekstrakcji jest wydzielanie cukru z buraków cukrowych, tłuszczu z nasion
roślin oleistych oraz białek z surowców zwierzęcych i roślinnych. W procesach przetwarzania
ekstrakcja typu ciało stałe-ciecz odgrywa ważną rolę w produkcji koncentratów witaminowych i
mięsnych, napojów alkoholowych, np. wina, piwa, wódek gatunkowych, oraz używek, np. ekstrakty
kawy i herbaty.
Ekstrakcja w układzie ciecz-ciecz jest stosowana w produkcji alkoholu, wina, olejów i do
wydzielania produktów fermentacji. W procesie ekstrakcji w układzie ciecz-ciecz wykorzystuje się
selektywność rozpuszczalnika i zjawisko niecałkowitego rozpuszczania się w sobie obydwu cieczy,
które tworzą wówczas dwie fazy ciekłe.
Selektywność rozpuszczalnika oznacza zdolność niejednakowo intensywnego rozpuszczania
składników mieszaniny ciekłej lub stałej. Selektywność zależy od budowy chemicznej substancji
tworzących dany układ ekstrakcyjny i od temperatury. Selektywność maleje z reguły ze wzrostem
temperatury. Rozpuszczalniki o budowie polarnej stosuje się do ekstrakcji składników polarnych, a
rozpuszczalniki niepolarne do ekstrakcji składników niepolarnych.
Dyfuzja jest zjawiskiem w wyniku którego ustala się równowaga rozkładu stężenia elementów
układu wskutek ich bezładnej wędrówki i wynika z ruchu cieplnego elementów układu (atomów,
cząsteczek, cząsteczek makroskopowych), zawieszonych w gazie lub w cieczy.
Prędkość dyfuzji określa pierwsze prawo Ficka, wyrażone wzorem:
gdzie:
dm/dt - prędkość przenoszenia substancji (kmol/h),
D - współczynnik dyfuzji (m
2
/h),
S - powierzchnia prostopadła do kierunku x (m
2
),
dc/dx - gradient stężenia c (kmol/m
3
), w kierunku x (m).
Znak minus wskazuje, że przenoszenie elementów układu zachodzi w kierunku od większego
do mniejszego stężenia.
W technologii żywności dyfuzja jest wykorzystywana do rozdziału składników żywności
metodami ekstrakcji, destylacji, sorpcji, odwadniania i membranowymi.
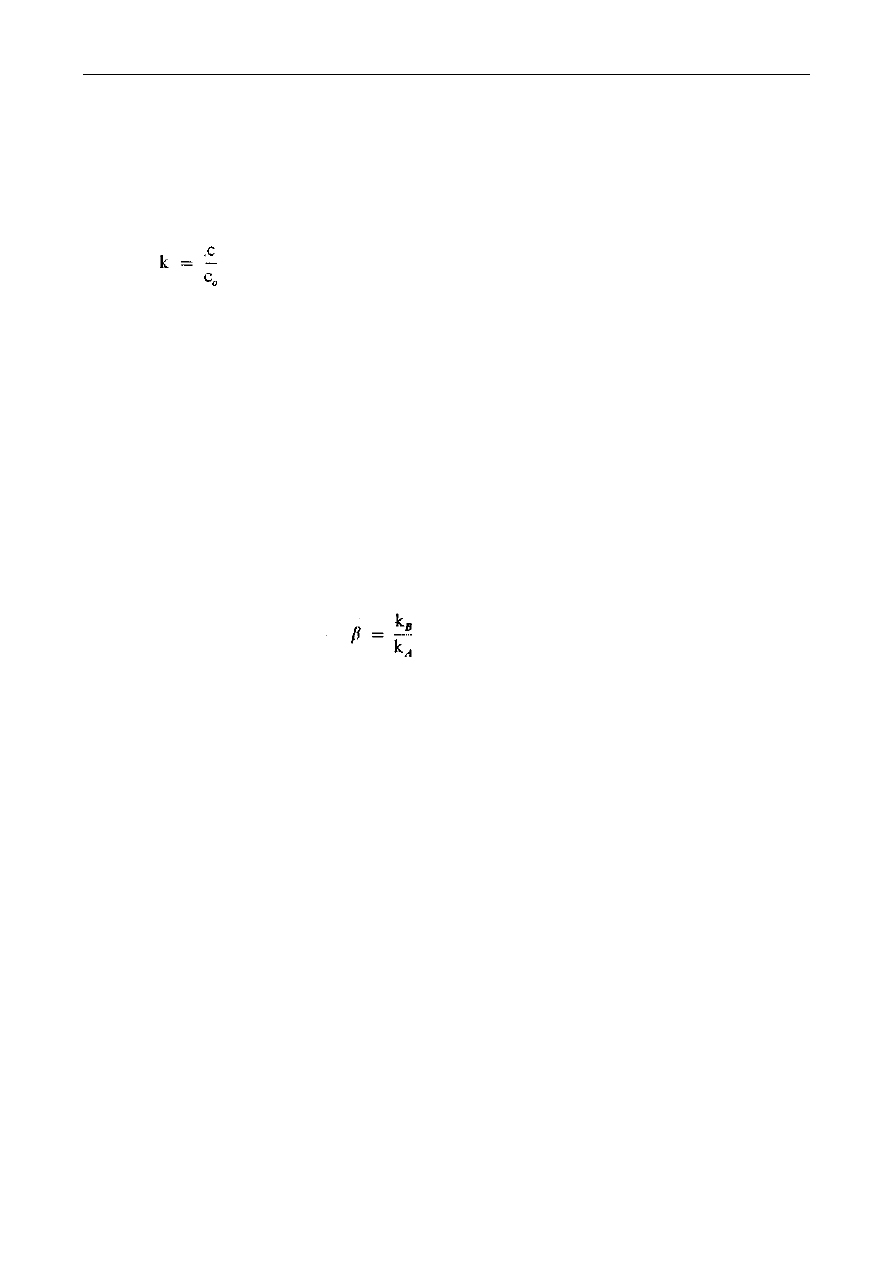
E
KSTRAKCJA W PRZEMY
Ś
LE SPO
ś
YWCZYM
Ruch składnika rozpuszczonego zachodzi z jednej fazy do drugiej wskutek różnicy stężeń do
momentu ustalenia równowagi. Stan równowagi w układach fazowych określa prawo podziału
Nernsta, które brzmi: substancja rozpuszczalna w dwóch cieczach nie mieszających się ze sobą, w
stanie równowagi dzieli się miedzy nimi w stałym stosunku, określanym współczynnikiem
podziału.
Wartość współczynnika podziału wyraża się wzorem:
gdzie:
c - stężenie składnika w ekstrakcie (kmol/m
3
),
c
0
- stężenie składnika w rafinacie (kmol/m
3
).
Teoretycznie wartość współczynnika podziału w danym układzie rozpuszczalników jest
wielkością stałą w stałej temperaturze i nie zależy od stężenia tego składnika jak również od
obecności innych substancji. W rzeczywistości stwierdza się odstępstwa od tej zasady. Przyczyną
zmiany
wartości
współczynnika
podziału
jest
częściowa
wzajemna
rozpuszczalność
rozpuszczalników polarnych i niepolarnych oraz zmiany prędkości rozpuszczania, zachodzące
wskutek solwatacji, asocjacji, dysocjacji, reakcji kompleksowania i reakcji z rozpuszczalnikiem
składnika rozpuszczonego. W roztworach elektrolitów współczynnik podziału zależy również od
stałej dysocjacji i pH. Współczynnik podziału jest, podobnie jak selektywność rozpuszczalnika,
kryterium skuteczności rozdziału składników. Pomiędzy selektywnością a współczynnikiem
podziału istnieje zależność wyrażona wzorem:
gdzie:
β - współczynnik selektywności.
KB - współczynnik podziału składnika 3 mieszaniny w danym rozpuszczalniku,
kA - współczynnik podziału składnika A mieszaniny w danym rozpuszczalniku.
Współczynnik selektywności informuje o stopniu trudności w przebiegu ekstrakcji. Ekstrakcja
jest niemożliwa do przeprowadzenia, ze względu na brak selektywności rozpuszczalnika, jeżeli
β=1. Ekstrakcja rozpuszczalnikiem o współczynniku selektywności większym od jedności zachodzi
łatwo. Gdy wartość współczynnika selektywności jest zbliżona do jedności, proces ekstrakcji jest
utrudniony i wymaga zastosowania ekstrakcji frakcjonowanej, tj. przy użyciu kilku
rozpuszczalników w kolejności zwiększającej się selektywności.
Rozpuszczalniki stosowane do ekstrakcji powinny charakteryzować się inną niż surowiec
gęstością, małą rozpuszczalnością w surowcu i rafinacie oraz dużą selektywnością.
Prędkość ekstrakcji jest wprost proporcjonalna do różnicy stężeń składnika ekstrahowanego w
ekstrakcie i rafinacie oraz odwrotnie proporcjonalna do oporu dyfuzyjnego.
Różnica stężenia składnika ekstrahowanego w ekstrakcie i rafinacie zależy od kierunku
wzajemnego ruchu faz (współprądowego, przeciwprądowego) oraz stosunku ilościowego faz (ilości
surowca i rozpuszczalnika). Przeciwprądowy kierunek przepływu rozpuszczalnika i zwiększenie
ilości rozpuszczalnika wpływają na utrzymywanie względnie wysokiej różnicy stężenia składnika
między substancją ekstrahowaną a ekstraktem, a tym samym intensyfikują proces.
Opór dyfuzyjny jest określany przez współczynnik dyfuzji w przypadku ekstrakcji w układzie
ciecz-ciecz. Podczas ekstrakcji w układzie ciało stałe-ciecz, opór dyfuzyjny zwiększa się wskutek
ruchu masy w substancji ciała stałego. Zmniejszenie oporu dyfuzyjnego uzyskuje się przez
E
KSTRAKCJA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
16
wymuszenie ruchu burzliwego rozpuszczalnika, zwiększenie powierzchni faz oraz eliminowanie
procesów dializy i osmozy.
Wzrost temperatury zwiększa prędkość ekstrakcji wskutek zwiększania prędkości rozpuszczania
składnika, obniżania lepkości roztworu oraz niszczenia błon komórkowych w przypadku ekstrakcji
w układzie ciało stałe-ciecz. Ze wzrostem temperatury zmniejsza się jednak selektywność
rozpuszczalnika, co powoduje ekstrakcję również innych składników surowca.
Czas ekstrakcji zależy od zawartości składnika ekstrahowanego, założonego stopnia
wyekstrahowania, jakości surowca, sposobu przygotowania surowca do ekstrakcji oraz metody
ekstrakcji.
Najprostszym przemysłowym sposobem ekstrakcji w układzie ciało stałe-ciecz jest maceracja.
Maceracja (wymoczenie lub zmiękczenie) polega na zalaniu rozdrobnionego surowca
rozpuszczalnikiem o temp. 15-20°C na określony czas, a następnie oddzieleniu ekstraktu przez
sączenie, wyciskanie lub odwirowanie. Wagowa proporcja rozpuszczalnika do surowca wynosi 5:1
lub 10:1. Rozpuszczalnikami są woda lub wodne roztwory soli i alkoholu. Macerację przeprowadza
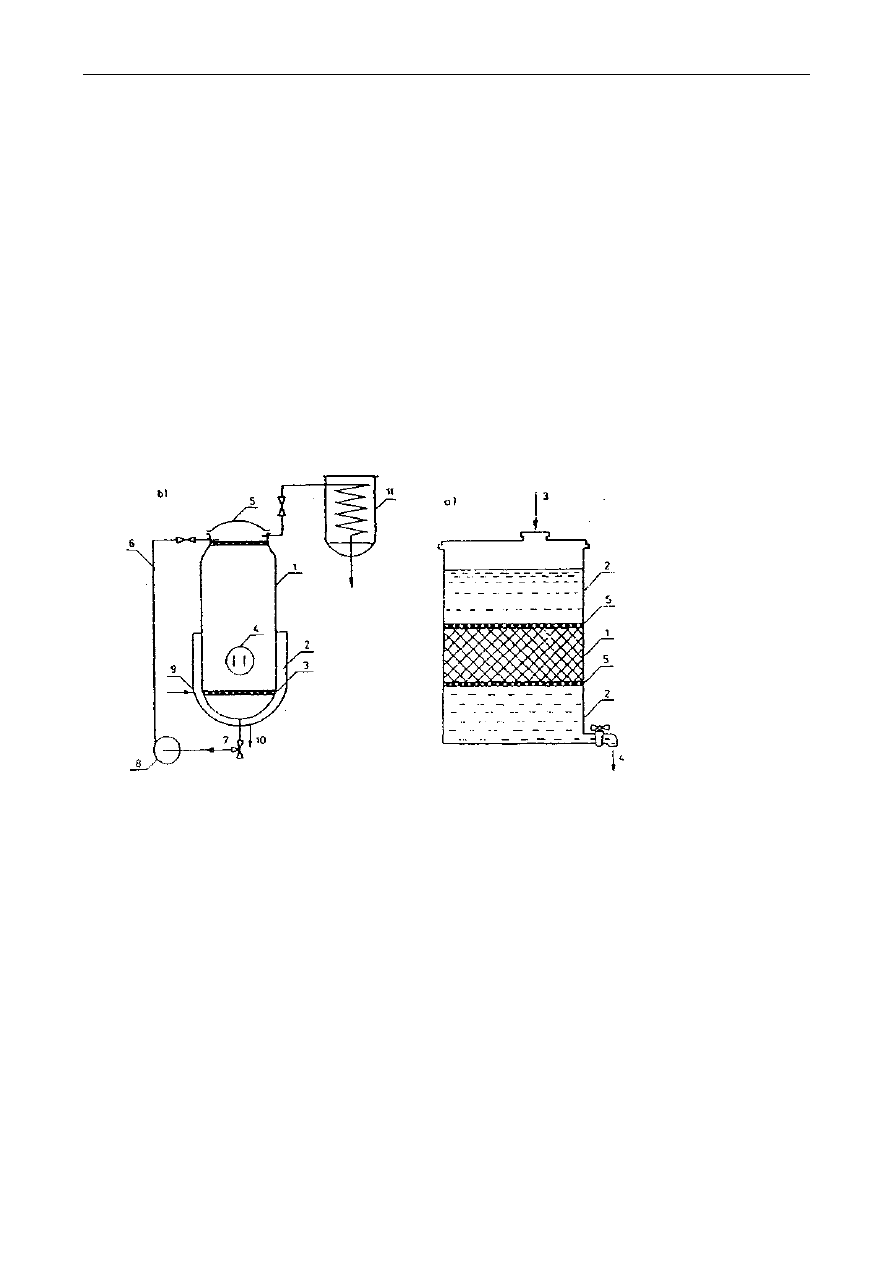
się w maceratorze z perforowaną przegrodą lub maceratorze obiegowym (rys. 1). Formami
maceracji są digestia, infuzja i dekokcja, stosowane głównie w procesach przetwarzania, np.
podczas zacierania słodu na brzeczkę piwną i gotowania w produkcji koncentratów obiadowych.
Rys. 1. Schematy maceratorów:
a - macerator z perforowaną przegrodą; 1-surowiec do ekstrakcji, 2-rozpuszczalnik, 3-dopływ
rozpuszczalnika, 4- odpływ ekstraktu. 5– wkłady perforowane,
b - macerator obiegowy: 1- korpus maceratora, 2- płaszcz parowy grzejny, 3- sita, 4- właz, 5-
pokrywa, 6- doprowadzenie ekstraktu, 7- odprowadzenie ekstraktu, 8- pompa, 9-
doprowadzenie pary grzejnej, 10- odprowadzenie kondensatu, 11– chłodnica
Digestia (wytrawienie) polega na maceracji w podwyższonej temp. 30-40°C. Infuzja
(naparzanie) polega na zalaniu surowca wrzącą wodą i ewentualnie krótkotrwałym ogrzewaniu.
Dekokcja (wygotowywanie) różni się od infuzji dłuższym czasem ekstrakcji. Temperatura podczas
infuzji i dekokcji wynosi powyżej 60°C.
Do bardziej wydajnych metod ekstrakcji należy perkolacja (wypieranie). Podczas perkolacji
rozpuszczalnik spływa pod wpływem siły grawitacji przez masę surowca.
Miernikiem wydajności ekstrakcji jest współczynnik wyekstrahowania lub stopień czystości
rafinatu, który określa się stosunkiem wyekstrahowanej ilości składników do ilości ogółem zawartej
w surowcu. W praktyce przemysłowej stopień czystości rafinatu ustala się na poziomie poniżej 1%.
Czas ekstrakcji dla takiego stopnia wyekstrahowania nie przekracza na ogół 100 min.

E
KSTRAKCJA W PRZEMY
Ś
LE SPO
ś
YWCZYM
Zużycie rozpuszczalnika i energii na jednostkę wagową surowca ma duże znaczenie dla
ekonomiki procesu. Ilość rozpuszczalnika do ekstrakcji nie powinna powodować nadmiernego
rozcieńczenia ekstraktu, ponieważ prowadziłoby to do wzrostu kosztów wydzielania czystego
składnika z ekstraktu. Technicznym rozwiązaniem uwzględniającym ekonomiczną ilość
rozpuszczalnika jest ekstrakcja wielostopniowa ciągła i z zastosowaniem powrotu części ekstraktu.
Ilość rozpuszczalnika (odbierana w warunkach przemysłowych jako ekstrakt wynosi ok. 100%
(± 20%) masy surowca, przy stężeniu ekstraktu na poziomie odpowiadającym zawartości składnika
ekstrahowanego w surowcu. Praktycznie zużycie rozpuszczalnika jest wyższe i zależy od
technicznych rozwiązań ekstrakcji i możliwości regeneracji rozpuszczalnika.
Wielkość strat rozpuszczalników organicznych, określająca zużycie rozpuszczalnika na
jednostkę wagowe surowca, jest wyznaczana przez ich lotność w ekstrakcie i rafinacie. Natomiast
wielkość strat rozpuszczalników nieorganicznych zależy od zasobów wody naturalnej. Straty
rozpuszczalnika zmniejsza się przez jego odzyskiwanie lub oczyszczanie i uzdatnianie, np. wody.
Rozpuszczalniki węglowodorowe odzyskuje się z ekstraktu przez destylację z parą wodną i pod
obniżonym ciśnieniem.
E
KSTRAKCJA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
18
2.2.
W
YKONANIE ĆWICZENIA
Celem ćwiczenia jest określenie zawartości tłuszczu surowego w nasionach roślin oleistych
takich jak: rzepak, słonecznik, len, rzepik, gorczyca, rzodkiew oleista, mak, rącznik itp.
Metodyka.
Oznaczenie zawartości tłuszczu w nasionach roślin oleistych można przeprowadzać metodą
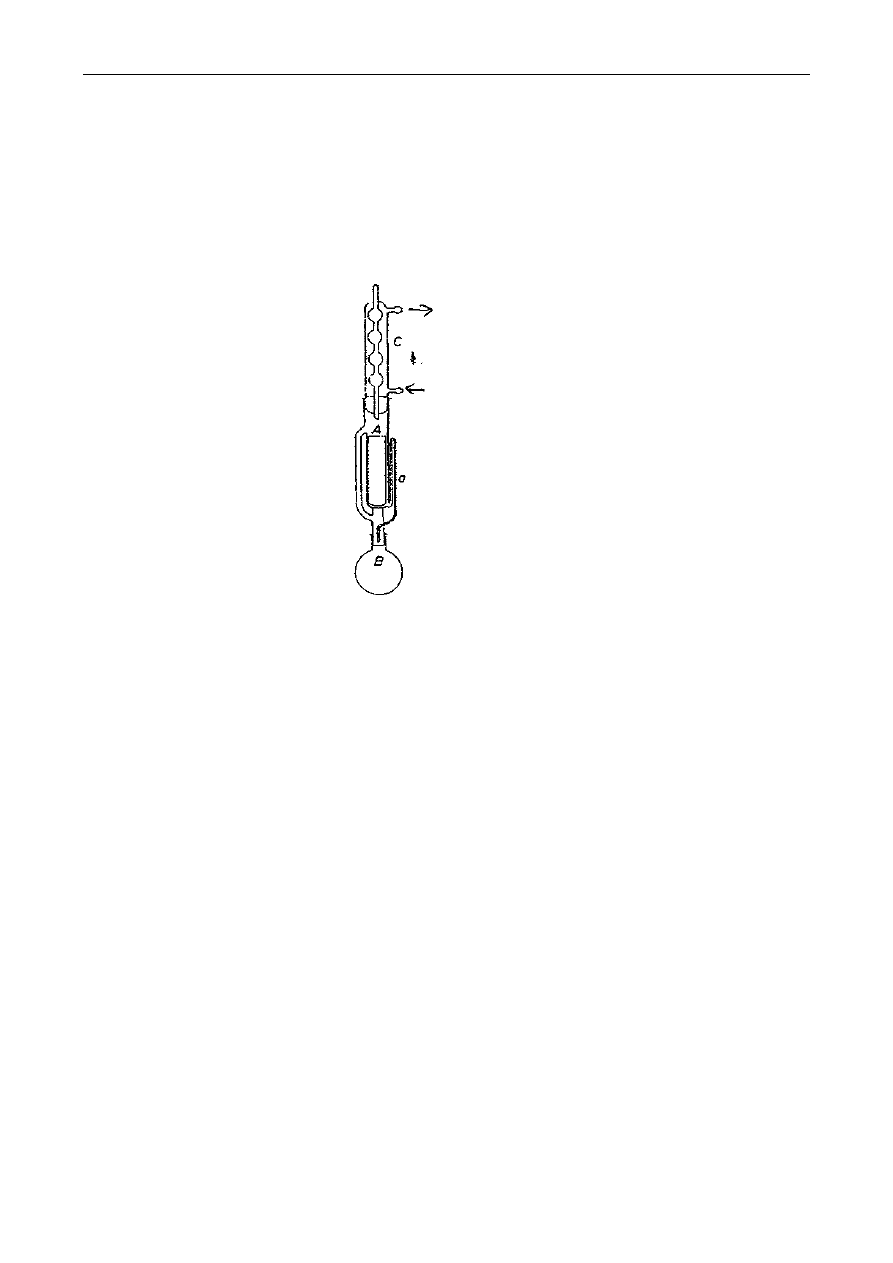
Soxhleta według poniższego opisu:
Rys. 2. Aparat Soxhleta: A-ekstraktor, B-kolba, C-chłodnica, a–syfon
Kolbę ekstrakcyjną (B- patrz rys.) wysuszoną do stałej masy należy zważyć a wynik zanotować.
10g dokładnie rozdrobnionych nasion należy przenieść ilościowo do gilzy ekstrakcyjnej i przykryć
odtłuszczoną watą, żeby zapobiec wypłynięciu części stałych podczas ekstrakcji. Gilzę umieścić w
ekstraktorze (A) aparatu Soxhleta (rys. 2). Kolbę (B) z próbką w gilzie należy podłączyć do zesta-
wu ekstrakcyjnego. Następnie do ekstraktora należy wlewać rozpuszczalnik tak długo, aż rurką
syfonu (a) przeleje się on do kolby, następnie dodać jeszcze połowę tej objętości. Aparat połączyć z
chłodnicą (C) i kolbę podgrzewać w maszynce elektrycznej czaszowej prowadząc ekstrakcję przez
1,5 godziny z prędkością 8-10 przelewów rozpuszczalnika przez rurkę syfonu a (patrz rysunek) na
godzinę.
Po zakończeniu ekstrakcji:
Należy zdjąć chłodnicę z kolby bez zdejmowania kolby z maszynki elektrycznej, umożliwiając
w ten sposób odparowanie rozpuszczalnika, pozostałość w kolbie zaś wysuszyć początkowo pod
dygestorium, a potem w suszarce w temp. 100°C do stałej masy i zważyć. Z różnicy mas kolby po
ekstrakcji i pustej należy obliczyć procentową zawartość tłuszczu surowego w badanej próbce
nasion.
2.3.
P
RZEDSTAWIENIE WYNIKÓW
Wynik oznaczenia należy umieścić w sprawozdaniu wraz z przeliczeniami i przedłożyć osobie
prowadzącej ćwiczenia w celu sprawdzenia (wzór, załącznik 1 na stronie 121). Należy także
zaznaczyć wszelkie odstępstwa od metody podanej w opracowaniu ćwiczenia. Na końcu
sprawozdania z ćwiczeń należy podać wniosek.

E
KSTRAKCJA W PRZEMY
Ś
LE SPO
ś
YWCZYM
2.4.
S
PRZĘT I ODCZYNNIKI
Aparatura i odczynniki:
•
aparat Soxhleta – 1 sztuka
•
statyw laboratoryjny z kompletem uchwytów
•
czasza grzejna – 1 sztuka
•
łaźnia wodna – 1sztuka
•
rurki silikonowe do zasilania chłodnicy
•
gilzy ekstrakcyjne
•
wata celulozowa odtłuszczona
•
waga analityczna
•
zestaw do destylacji prostej
•
rozpuszczalnik organiczny np. chloroform
•
śruta rzepakowa suszona

W
ARUNKI POWSTAWANIA
ś
ELU PEKTYNOWEGO
12 luty 2010
20
3.
WARUNKI POWSTAWANIA śELU PEKTYNOWEGO
3.1.
W
PROWADZENIE
Substancje pektynowe są szeroko rozpowszechnione w świecie roślin. Tworzą one lepiszcze
ścian komórek roślinnych i występują w postaci tzw. protopektyny. Protopekryna jest
nierozpuszczalna w wodzie. Przechodzi w formę rozpuszczania zwaną pektyną pod wpływem
działania słabych kwasów, meta i pirofosforanów oraz enzymów. Pektyna składa się głównie z reszt
kwasu galakturonowego połączonych wiązaniami 1-4-glikozydowymi, w których większość grup
karboksylowych jest zestryfikowana alkoholem metylowym i częściowo zneutralizowana zasadami.
Dalsza degradacja związków pektynowych w wyniku reakcji biochemicznych (np. nasilających się
bardzo w owocach w czasie dojrzewania), jak i reakcji chemicznych prowadzi do powstania
kwasów pektynowych (kwasy poligalaturonowe) i kwasu galakturonowego.
W procesach żelifikacji, w technologii wykorzystuje się związki pektynowe zwane pektyną. W
krajowym przemyśle preparaty produkowane są z wytłoków jabłecznych. Ponadto wykorzystuje się
do tego celu albedo owoców cytrusowych, łuski i kwiatostany słonecznika. Proces otrzymywania
preparatów polega na uwolnieniu pektyn z protopektyny poprzez łagodną hydrolizę, a następnie
rozpuszczone w wodzie pektyny są ekstrahowane, oczyszczane, zagęszczane i suszone.
Pektyna jest estrem metylowym kwasu poligalakturonowego. Preparaty pektynowe są zaliczane
do liofilnych koloidów, które łatwo pęcznieją i rozpuszczają się w zimnej i gorącej wodzie. Wodne
roztwory pektyn odznaczają się wysoką trwałością, którą zawdzięczają zdolności wiązania przez
micele pektynowe wielkiej liczby cząsteczek wody na swej powierzchni, częściowo zaś również
jednolitemu ładunkowi elektrycznemu.
Na temat mechanizmu tworzenia się żelu, a ścisłe powstawania wiązań pektynowych, istnieją
różne hipotezy, jednakże żadna z nich nie wyjaśnia w pełni tego zjawiska. Twierdzi się, że w
przypadku pektyn o wysokim stopniu zestryfikowania w reakcji biorą udział grupy -COOH i -OH i
tworzą się tzw. wiązania poboczne. Przypuszcza się także, że w przypadku pektyn o wyższym
stopniu zestryfikowania w tworzeniu wiązań biorą również udział jony wapniowe i jony innych
metali. W przypadku pektyn o niskim stopniu zestryfikowania w wiązaniu cząsteczek uczestniczą
tylko grupy karboksylowe łączone jonami wapniowymi lub innymi metalami dwu- i mniej
wartościowymi; są to tzw. wiązania główne. Przyjmuje się, że siatka powstała w przestrzeni w
wyniku połączenia łańcuchów pektynowych obudowuje ciecz, którą najczęściej jest roztwór cukru i
innych składników.
Pektyna rozpuszczona w wodzie lub roztworze cukru tworzy układ koloidalny, w którym
cząsteczki kwasów pektynowych są zdysocjowane i przybierają ujemny ładunek elektryczny, a to
powoduje, że są one ciałami odpychającymi się wzajemnie. Dodatkowym zjawiskiem jest, że grupy
karboksylowe i wodorotlenowe są zhydratowane, czyli otoczone osłonkami wodnymi. Cząsteczki
kwasów pektynowych wykonują w roztworze ruchy drgające i ruchy Browna, a energia kinetyczna
tych cząsteczek wzrasta w miarę wzrostu temperatury (podgrzewania roztworu). Wszystkie
wymienione i opisane zjawiska nie sprzyjają łączeniu się łańcuchów pektynowych i wytworzeniu
żelu o prawidłowej strukturze. Chcąc zatem doprowadzić do wzajemnego powiązania
poszczególnych cząsteczek pektyny, należy stworzyć takie warunki technologiczne, podczas
których cząsteczki mogłyby się do siebie zbliżyć i trwale połączyć.
Czynnikami warunkującymi powstanie tych połączeń są:
•
dodatek kwasu w celu osiągnięcia optymalnego pH,
•
odpowiedni dodatek cukru,
•
obniżenie temperatury.

W
ARUNKI POWSTAWANIA
ś
ELU PEKTYNOWEGO
Dodatek kwasu, np. cytrynowego, powoduje cofnięcie dysocjacji cząsteczek pektynowych, a
tym samym ich neutralizację. Jednocześnie dodatek kwasu rozszerza i wzmacnia włókna struktury
żelu, ułatwiając przez to zatrzymanie syropu w tak powstałej siatce. Dodatek cukru likwiduje
osłony wodnej czyli dehydratyzuje grupy -OH i -COOH kwasów pektynowych, które w ten sposób
mogą łatwiej wytworzyć silne wiązania. Obniżające temperaturę, uzyskuje się zmniejszenie
wartości energii kinetycznej cząsteczek kwasu pektynowego a przez to wiązania tworzące siatkę
pektynową są trwalsze.
Warunki tworzenia żelu przez pektyny
Z technologicznego punktu widzenia, najważniejszą właściwością pektyn jest zdolność
tworzenia w określonych warunkach ścisłego i zwartego żelu. Zdolność tworzenia żelu nazywa się
żelowaniem. Biorąc pod uwagę budowę chemiczną pektyny, siła żelowania zależy, przede
wszystkim, od stopnia jej zestryfikowania. Jak już podawano wcześniej, podstawowymi warunkami
powstawania żelu są:
•
właściwe pH roztworu,
•
dodatek cukru, który odbiera wodę z drobin pektyny, umożliwiając tym samym
odpowiednie zbliżanie się cząsteczek obdarzonych ładunkami i powstanie wiązań
chemicznych. Wymagane stężenie ekstraktu roztworu jest w granicach 60-75%.
Trwałość i wytrzymałość żel jest zależna od:
•
długości łańcucha, z którego jest zbudowana drobina pektyn,
•
stopnia estryfikacji grup karboksylowych znajdujących się w cząsteczce pektyny,
•
substancji balastowych,
•
zawartości kwasu.
Pektyny dobrej jakości mają długi łańcuch połączonych glikozydowo reszt kwasu
galakturonowego, który wykazuje w 60-80% zestryfikowanie z alkoholem metylowym. Ciężar
cząsteczkowy takich pektyn powinien być wyższy od 150 000. Wysoki ciężar cząsteczkowy i
odpowiedni stopień estryfikacji wykazują przede wszystkim pektyny otrzymane z owoców
wyrośniętych, lecz niedojrzałych.
Pomimo że pektyny są estrami alkoholu metylowego i kwasu galakturonowego, to w większości
pektyn stwierdza się po hydrolizie również grupy acetylowe (CH
3
CO).
W zależności od stopnia estryfikacji rozróżniamy następujące typy pektyn (tab.1.):
•
szybko żelujące — zestryfikowane w 75-85% (powyżej 2 min);
•
średnio żelujące — zestryfikowane w ok. 70% (powyżej 12 min);
•
wolno żelujące - zestryfikowane w ok. 65% (powyżej 20 min).
Oprócz pektyn wysokometylowanych o zawartości 10-12% grup metoksylowych
(zestryfikowanie 60-75% grup karboksylowych), są produkowane pektyny niskometylowane o
zawartości grup metoksylowych 3-7%, czyli poniżej 44% zestryfikowania.
Bardzo często preparaty pektynowe są zanieczyszczone tzw. substancjami balastowymi.
Najczęściej są to węglowodany, jak np. araban i galaktany, które nie będąc związane chemicznie z
kwasem poligalakturonowym nie mają wpływu na zdolności żelowania pektyny. Ponadto mogą
również występować grupy związków organicznych, które są połączone z cząsteczkami kwasu
galakturonowego trwałymi wiązaniami chemicznymi. Wówczas taka pektyna ma wprawdzie duży
ciężar cząsteczkowy, ale nie tworzy żelu o trwałej strukturze.
W
ARUNKI POWSTAWANIA
ś
ELU PEKTYNOWEGO
12 luty 2010
22
Tabela 1. Podział pektyn w zależności od stopnia zestryfikowania i szybkości żelowania w
określonych warunkach
Stopień
zestryfikowania kwasu
pektynowego
Typ
preparatu
Czas żelowania
[min]
Temperatura
żelowania fC]
pH
Ekstrakt
dżemu
65
wolno
żelujący
37
32
70
70
3,0
2,8
65
65
70
średnio
żelujący
12
17
70
70
3,0
2,8
65
65
75
szybko
żelujący
5-6
3
70
70
3,0
2,8
65
65
Zmniejszanie lub utrata siły żelowania są równoznaczne z obniżeniem ciężaru drobinowego,
czyli rozkładem łańcuchów, z których są zbudowane pektyny. Degradacja łańcucha pektyny może
być spowodowana przez: kwasy, zasady, enzymy i temperaturę. Odporność pektyny na działanie
kwasów zależy od pH roztworu i temperatury środowiska.
Pektyna jest najbardziej wytrzymała w środowisku o pH 3, lecz w niższym pH następuje rozpad
łańcucha na krótkie drobiny. Stabilność pektyny wobec kwasów maleje wraz ze wzrostem
temperatury. Środowisko zasadowe również wpływa niekorzystnie na wielkość cząstek pektyny.
Okazało się jednak, że stabilność pektyny względem zasad jest mniejsza niż względem kwasów.
Innym czynnikiem mogącym powodować skracanie łańcucha pektynowego jest obecność enzymów
pektynolitycznych w tkance owocowej. Zapobieganie szkodliwemu działaniu enzymów polega na
termicznej obróbce wstępnej surowca (80-85°C w ciągu kilku minut) oraz niszczeniu pleśni, które
wydzielają znaczne ilości enzymów hydrolizujących. Wysoka temperatura również wpływa
destrukcyjnie na cząsteczki pektyny. Niebezpieczną granicą jest w tym przypadku temp. 70°C,
powyżej której następuje szybki rozpad drobin. Z tego powodu zaleca się ograniczenie do minimum
czasu gotowania dżemu i galaret oraz schłodzenie częściowo przed rozlewem i dalsze po rozlewie.
Znaczenie pH w tworzeniu żelu pektynowego
Decydujący wpływ na powstawanie żelu pektynowego ma pH środowiska. Optymalne warunki
pH wynoszą:
•
przy zawartości ekstraktu 68-72% - 3,1-3,3
•
przy zawartości ekstraktu 65-68%- 3,0-3,2
•
przy zawartości ekstraktu 60-65%- 2,8-3,0
Poniżej pH 2.8 następuje hydroliza pektyny i nie zachodzi żelowanie dżemu lub galarety.
Regulować pH można dodając kwas, którego ilość musi być ściśle określona, gdyż
przedawkowanie może spowodować synerezę. Do dokwaszania dżemów można stosować kwasy,
takie jak: cytrynowy, winowy i mlekowy. Należy jednak pamiętać, że wymienione kwasy mają
różny wpływ na redukcję pH, a jednocześnie mogą zmieniać smak gotowego wyrobu.
Kwas powinno się dodawać jak najpóźniej, tzn. po zakończeniu gotowania i bezpośrednio przed
rozlewem. Zalecenie takie jest uzasadnione niebezpieczeństwem redukcji siły żelowania oraz
wzrostem inwersji podczas zakwaszenia środowiska.
Cukry odgrywają bardzo ważną rolę w tworzeniu żelu. Do dosładzania wyrobów owocowo-
warzywnych i cukierniczych stosuje się przede wszystkim sacharozę, ale i inne; cukry wywołują
żelowanie roztworów pektynowych. Do produkcji przetworów wysokosłodzonych najchętniej jest
wykorzystywana sacharoza ze względu na jej wysoką rozpuszczalność i słodycz. Obowiązuje
zasada, że przy stałej ilości pektyny, a jednocześnie niskim pH środowiska jest wymagany wyższy

W
ARUNKI POWSTAWANIA
ś
ELU PEKTYNOWEGO
dodatek cukru. Ogólnie przyjmuje się: że optymalne warunki galaretowacenia to: 0,5-0,7%
pektyny, 65-68% cukru i pH środowiska ok. 3.
Wpływ jakości i ilości pektyny na tworzenie żelu
Jakość pektyny określają dwie cechy: zdolność (siła) żelowania oraz szybkość żelowania. Siłę
żelowania pektyn można oznaczać metodą amerykańską Cox-Higby’ego (USA-SAG) lub metodą
Tarr-Bakera (°TB).
W metodzie amerykańskiej mierzy się procent opadu (osiadania) galaretki w określonym czasie
(2 min.), w stosunku do wysokości pierwotnej żelu. Za opad normalny, charakteryzujący
standardową galaretkę, przyjmuje się 23,5% (23,5 jednostek). Metoda ta daje bardziej powtarzalne
wyniki niż metoda Tarr-Bakera i uznaje się ją za metodę odwoławczą w stosunku do innych metod.
W metodzie duńskiej z użyciem żelometru Tarr-Bakera mierzy się nacisk słupa wody, który
powoduje zagłębienie tłoczka żelometru w galarecie. Konsystencja galaretki jest uważana za
naturalną, jeżeli zagłębienie tłoczka żelometru w galarecie nastąpi pod naciskiem 575 mm słupa
wody. Z wyników pomiarów uzyskanych za pomocą specjalnych tablic oblicza się stopnie żelo-
wania Tarr-Bakera (°TB). Siła żelowania w °TB podaje, z jaką ilością cukru (w gramach) 1g
preparatu pektynowego utworzy galaretkę o ekstrakcie 65% i pH 3,0 (galaretkę standardową).
Drugim pojęciem stosowanym podczas charakterystyki preparatów pektynowych są jednostki
żelowania. Jednostka żelowania to iloczyn gramów preparatu i zdolności żelowania w stopniach
(TB).
Dżem prawidłowo wyprodukowany powinien mieć 67,5 jednostki żelowania.
j.ż. = g
preparatu pektyny
•
°TB
Ogólnie można stwierdzić, że wartość preparatu pektynowego nie tylko zależy od zawartości kwasu
pektynowego, ale od jego jakości, która w głównej mierze decyduje o sile żelowania.

W
ARUNKI POWSTAWANIA
ś
ELU PEKTYNOWEGO
12 luty 2010
24
3.2.
W
YKONANIE ĆWICZENIA
Celem ćwiczenia jest porównanie żelowania preparatu pektynowego w różnych warunkach.
Metodyka
Należy wytarować zlewkę wraz z bagietką. Odważyć 0,5-1 g preparatu pektynowego (dokładną
ilość jak i rodzaj pektyny wyznaczy prowadzący ćwiczenia) z dokładnością do 1mg, 30 g cukru i
0,4g kwasu winowego. Cukier należy rozetrzeć w moździerzu. Do zlewki na 100 cm
3
należy
przenieść część cukru (ok. 4g) i naważkę pektyny, dobrze wymieszać, dodać około 18 cm
3
gorącej
wody destylowanej zwilżając najpierw mieszaninę niewielką jej ilością, następnie mieszając
bagietką doprowadzić do całkowitego rozpuszczenia pektyny i cukru. Dodać pozostałą część cukru,
rozpuścić go mieszając i podgrzewając w łaźni wodnej (około 90°C) następnie dodać kwas winowy
i rozpuścić go. Zawartość zlewki uzupełnić do 50 g gorącą wodą destylowaną. Próbę wymieszać i
pozostawić w spokoju przez godzinę w chłodnym miejscu, po czym opisać jej konsystencję.
UWAGA: W zależności od rodzaju otrzymanego preparatu pektynowego należy powyższy
przepis zmodyfikować, bazując na teoretycznej znajomości właściwości pektyn.
Następnie należy powyższe operacje powtórzyć wraz z następującymi modyfikacjami:
•
nie dodając kwasu winowego,
•
dodając połowę cukru (15 g),
•
stosując zimną wodę do rozpuszczenia i uzupełnienia.
Porównać uzyskane wyniki, należy wyjaśnić przyczyny obserwowanych różnic. Konsystencja
prób może być płynna o różnej gęstości lub stała, tj. w postaci żelu. śel ocenia się obserwując jego
kształt po wyjęciu z naczynia i wykonaniu przekroju - słaby żel rozlewa się, a mocniejszy osiada
tylko pod wpływem własnego ciężaru.
3.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń umieścić w sprawozdaniu (wzór, załącznik 1 na stronie 121)w formie
oddzielnych punktów i przedłożyć osobie prowadzącej ćwiczenie w celu sprawdzenia. Należy także
zaznaczyć wszelkie odstępstwa od metod podanych w opracowaniu ćwiczeń. Na końcu
sprawozdania z ćwiczeń należy podać wnioski w formie zwięzłych punktów.

Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
4.
ZAGĘSZCZANIE ROZTWORÓW W TECHNOLOGII śYWNOŚCI
4.1.
W
PROWADZENIE
Większość znanych metod zagęszczania polega na usuwaniu wody, a tylko nieliczne-na
zwiększeniu stężenia składników suchej substancji przez dodatek składników, np. sacharozy w
technologii dżemów lub galaretek owocowych. Często zagęszczanie cieczy jest etapem pośrednim
przed suszeniem i decyduje o jakości gotowego produktu (koncentratu) oraz o zmniejszeniu
kosztów usuwania wody.
Proces zagęszczania charakteryzuje się stopniem koncentracji, który określa się stosunkiem
masy roztworu przed zagęszczaniem do masy roztworu zagęszczonego lub stosunkiem procentowej
zawartości suchej substancji w koncentracie i w roztworze przed zagęszczeniem. Procentową
zawartość suchej substancji oznacza się metodą suszenia w temp. 105°C, refraktometrycznie lub
areometrycznie.
Ilość wody usuwanej podczas zagęszczania oblicza się ze wzorów:
(kg)
)
B
b
-
(1
S
W
lub
B
b
S
S
W
lub
s
S
W
⋅
=
⋅
−
=
−
=
S - ilość jednostek wagowych roztworu (kg),
s - ilość jednostek wagowych koncentratu (kg),
b - zawartość suchej substancji w roztworze (%),
B - zawartość suchej substancji w koncentracie (%).
W technologii żywności roztwory zagęszcza się najczęściej do zawartości 30-45% suchej
substancji w koncentracie, co odpowiada 2-3-krotnemu zagęszczeniu masy roztworu.
Metody zagęszczania
W praktyce stosuje się wiele metod zagęszczania. Do najpowszechniejszych zalicza się
zagęszczanie przez odparowanie lub wymrażanie wody oraz zagęszczanie metodami
membranowymi.
Odparowanie wody z cieczy jest związane z przejściem wody ze stanu ciekłego w postać pary, a
wiec wymaga pokonania sił wiążących cząsteczki w cieczy, które są większe w sianie ciekłym niż
w gazowym. Proces odparowania wody z roztworu wymaga doprowadzenia określonej ilości
energii, która najczęściej jest przekazywana od czynnika grzejnego do roztworu przez konwekcję i
przewodzenie. Niezbędna do tego celu energia odpowiada utajonemu ciepłu parowania wody. które
w temp. 100°C wynosi 2300 kJ/kg. Zakładając, że z S kg cieczy (roztworu o zawartości b % s.s.)
otrzymuje się s kg koncentratu o B % s.s. oraz że proces odbywa się bez strat, można zapisać
następującą zależność:
100
100
B
s
b
S
⋅
=
⋅
Odparowanie wody w cieczy można prowadzić w naczyniach otwartych pod normalnym
ciśnieniem w temp. 100°C co niekorzystnie wpływa na jakość koncentratu i na koszty dehydratacji.
Aktualnie do zagęszczania roztworów stosuje się aparaty wyparne, w których proces prowadzi
się pod obniżonym ciśnieniem, a ciepło niezbędne do odparowania wody dostarcza się za
pośrednictwem pary.
Wyparki, stosowane w przemyśle spożywczym cechują się:
•
wysoką intensywnością wymiany ciepła przy ustalonej różnicy temperatur,
•
minimalnymi stratami ciepła,
•
dużą zawartością konstrukcji,

Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
26
•
ciągłym i skutecznym odprowadzeniem skroplin z komory grzejnej,
•
łatwością czyszczenia powierzchni grzejnej bez zatrzymywania ciągłości pracy.
W zależności od sposobu działania rozróżnia się wyparki o działaniu okresowym i ciągłym. W
wyparkach o działaniu okresowym proces odparowania przebiega w sposób nierównomierny, gdyż
temperatura roztworu, jego gęstość, lepkość oraz ilość odprowadzonego rozpuszczalnika zmieniają
się cyklicznie podczas procesu. W wyparkach o działaniu ciągłym doprowadzenie roztworu przed
zagęszczeniem i odprowadzeniem koncentratu odbywa się w sposób nieprzerwany. Wyparki te ze
względu na mniejsze straty energii związane zużyciem ciepła na okresowe rozgrzewanie aparatu, są
bardziej ekonomiczne od wyparek o działaniu okresowym.
W zależności od ciśnienia wewnątrz aparatu, wyparki można podzielić na pracujące pod ciśnieniem
podwyższonym, atmosferycznym i obniżonym.
Ciśnienie podwyższone stosuje się tylko wtedy, gdy opary mogą być wykorzystane jako czynnik
grzejny w innych aparatach. Obniżone ciśnienie stosuje się wówczas, gdy:
•
w roztworze zagęszczonym mogą wystąpić: denaturacja białek, tworzenie się połączeń
białkowo-cukrowych, rozkład substancji zawartych w roztworze, zmiany barwy lub
zapachu;
•
dostępny jest czynnik grzejny o wymaganych parametrach; konieczne jest zwiększenie
użytecznej różnicy temperatur między czynnikiem grzejnym i ogrzewanym;
•
należy zwiększyć intensywność odparowania wody.
Stosując obniżone ciśnienie, a tym samym niższe od 100°C temperatury wrzenia roztworów, nie
uzyskuje się dużej oszczędności energii niezbędnej przy odparowaniu wody, ponieważ do zamiany
1 kg wody w parę potrzeba więcej wody i ciepła niż pary o temp. 100°C. Podczas zagęszczania w
temp. 100° C zużywa się 2260 kJ/kg. Gdy wrzenie zachodzi w temp. 80°C, zużywa się 2309 kJ/kg,
w temp. 60°C-2356 kJ/kg. W zagęszczaniu przy obniżonym ciśnieniu zużywa się jednak mniej
ciepła na podgrzanie roztworu do temperatury wrzenia, i dlatego - w miarę obniżania ciśnienia i
temperatury wrzenia roztworu-całkowite zużycie ciepła jest coraz mniejsze. Przyspieszenie tempa
odparowania wody przy zastosowaniu obniżonego ciśnienia następuje wskutek znacznie większej
różnicy temperatur czynnika grzejnego i środowiska ogrzewanego, ponieważ wrzenie zachodzi w
temperaturze niższej niż 100°C. Ilość ciepła doprowadzonego zależy od różnicy tych temperatur, i
dlatego ogrzewając roztwór parą (w płaszczu grzejnym) - np. o temp. 120°C, gdy wrzenie zachodzi
przy 100°C-różnica temperatury wynosi 120-100°C=20°C, przy wrzeniu w temp. 60°C różnica
wynosi 60°C i jest 3 razy większa, co w przybliżeniu pozwala doprowadzić w tym samym czasie 3
razy więcej ciepła i odprowadzić 3 razy większą ilość wody.
Obniżenie temperatury wrzenia roztworu zmniejsza zapotrzebowanie na ciepło niezbędne do
doprowadzenia go do wrzenia oraz powoduje mniejsze straty na promieniowanie z powodu
mniejszej różnicy temperatur pomiędzy ośrodkiem wrzącym a otoczeniem. Wadą odparowania przy
obniżonym ciśnieniu są dodatkowe nakłady na urządzenia skraplające i utrzymujące próżnię oraz
wyższe koszty eksploatacyjne, wyrażające się dodatkowym zużyciem wody w skraplaczu oraz
energii na poruszenie pompy próżniowej.
Wyboru typu wyparki dokonuje się przede wszystkim na podstawie właściwości
fizykochemicznych roztworu takich, jak: lepkość, gęstość, napięcie powierzchniowe. Decyzja
wyboru odpowiedniego rozwiązania konstrukcyjnego musi być podjęta po przeprowadzeniu pełnej
analizy warunków techniczno-ekonomicznych.
Oceniając wyparki rozpatruje się trzy podstawowe problemy: wymianę ciepła pomiędzy
czynnikiem grzejnym a roztworem zagęszczanym, oddzielenie roztworu od pary oraz
wykorzystanie energii cieplnej oparów. Energię cieplną oparów można wykorzystać do
zagęszczania tego samego roztworu albo przez obniżenie temperatury wrzenia roztworu, albo przez
podwyższenie temperatury oparów. Pierwszy sposób realizuje się w wielodziałowych instalacjach
wyparnych, drugi-w wyparkach ze sprężaniem. W wyparkach ze sprężaniem również wykorzystuje
się utajone ciepło oparów. Opary spręża się w celu podwyższenia ich temperatury do wymaganego

Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
poziomu. Sprężanie oparów stosuje się zarówno w wyparkach jednodziałowych, jak również w
wyparkach wielodziałowych w celu zmniejszenia zużycia pary.
W obydwu rozwiązaniach opary spręża się za pomocą termo- lub turbosprężarki, wykorzystując
energie pary o wysokich parametrach lub energię mechaniczną, lub elektryczną. Zastosowanie
sprężania oparów w wododziałowej instalacji wyparnej pozwala obniżyć (od 30 do 45%)
zapotrzebowanie pary świeżej. Najkorzystniejsze efekty otrzymuje się stosując termosprężanie.
Wyparki pracujące w polskim przemyśle spożywczym wykazują bardzo dużą energochłonność,
niekiedy sięgająca 11-18 MJ/kg odparowanej wody. Dążąc do racjonalnego gospodarowania
energią, należy zmniejszyć nieuzasadnione wysokie zużycie wody chłodzącej skraplacz i
zagospodarować ciepłą wódę barometryczną.
Postęp w konstrukcji wyparek zmierza do oszczędności energii, jak również do przyspieszenia
tempa zagęszczania np. czas przetrzymywania cieczy w wyparkach tradycyjnych jodnodziałowych
wynosi od 1 do kilku godzin, a w nowoczesnych wyparkach kilkanaście sekund.
Zagęszczanie roztworów zawierających lotne z parą wodna substancje aromatyczne prowadzi
do ich strat, w przybliżeniu proporcjonalnych do ilości usuwanej wody. Straty substancji
aromatycznych podczas zagęszczania w wyparkach powodują pogorszenie jakości koncentratów w
porównaniu z surowcem. Właściwości koncentratów można poprawić przez odzysk substancji
aromatycznych i dodanie ich do gotowego produktu.
Procesy odzysku aromatu można podzielić następująco:
•
procesy prowadzone pod ciśnieniem atmosferycznym,
•
procesy częściowo lub całkowicie prowadzone pod obniżonym ciśnieniem w niskiej
temperaturze,
•
procesy oparte na adsorpcji na węglu drzewnym substancji aromatycznych oraz ich ekstrakcji,
•
procesy oparte na ekstrakcji substancji aromatycznych gazem obojętnym,
•
destylacja frakcjonowana, która jest najbardziej przydatną metodą odzysku składników
aromatycznych, wydzielających się podczas odparowania wody w wyparce.
W celu wyeliminowania strat substancji aromatycznych podczas zagęszczania roztworów, np.
soków owocowych, zaproponowano inne metody, np. kriokoncentrację, czyli zagęszczanie przez
wymrażanie wody. Zagęszczanie przez wymrażanie jest procesem zmierzającym do otrzymania
wysokowartościowych koncentratów przy zmniejszonym zużyciu energii w porównaniu z
odparowaniem w wyparkach. Stwierdzono, że w nowoczesnych rozwiązaniach kriokoncentracji
soków owocowych zużycie energii jest mniejsze niż w trzydziałowej wyparce wyposażonej w
urządzenie do odzysku aromatu, a zawartość suchej substancji w koncentracie wynosi 45%.
Zagęszczanie przez wymrażanie oparte jest na równowadze składników suchej substancji i
cieczy. Roztwór zagęszczany, zawierający dużo składników rozpuszczonych, rozpatruje się jako
pseudodwuskładnikowy, w którym wszystkie substancje rozpuszczone w wodzie uważa się jako
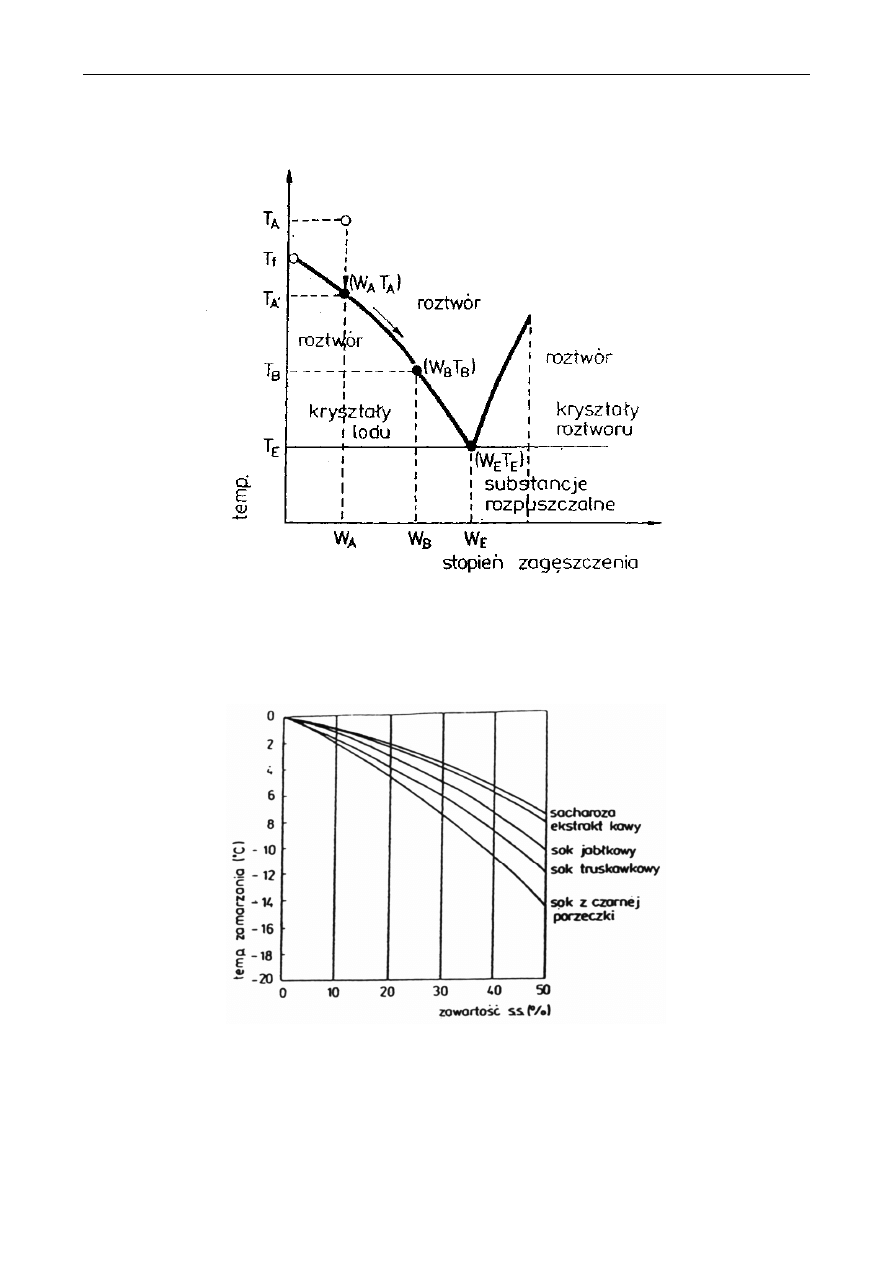
komponent. Uproszczony schemat zamrażania mieszaniny dwuskładnikowej przedstawiono na
rysunku 1. Jeżeli taka mieszaninę ochładzamy w warunkach zbliżonych do równowagi, to „czyste"
kryształy lodu (wody) są wydzielane w punkcie, który odpowiada składowi WA i temperaturze
zamrażania tego roztworu TA. Dalsze obniżanie temperatury powoduje wydzielanie większej ilości
kryształów, a skład cieczy jest zgodny z obszarem wyznaczonym przez linie (WA, TA), (WB, TB).
Natomiast w punkcie WE kryształy mają ten sam skład jak roztwór.
Na rysunku 2 przedstawiono doświadczalne krzywe zamrażania niektórych soków.
Stwierdzono, że temperatura zamrażania soku z czarnych porzeczek o zawartości suchej substancji
10% wynosiła -2°C. Po obniżeniu temperatury do -14°C zawartość suchej substancji w fazie cieczy
(przy stanie równowagi) osiąga 50%, a 90% wody obecnej w soku znajduje się w postaci
wydzielonych kryształów lodu. Zamrażanie soków prowadzi się powyżej punktu eutektycznego.
Przeważnie ze względu na problemy związane z wydzielaniem lodu z roztworów o dużej
lepkości, proces można prowadzić tylko nieco poniżej tego punktu. Niekiedy konieczna jest
depektynizacja soku przed wymrażaniem, co zapobiega jego gęstnieniu podczas zagęszczania i po
Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
28
zagęszczeniu. Przyjmuje się, że zagęszczanie można prowadzić do momentu, kiedy koncentrat ma
konsystencje umożliwiającą jego transport pompą.
Rys. 1. Schemat zamrażania mieszaniny dwuskładnikowej: W
e
- stężenie w punkcie eutektycznym
T
E
- temperatura w punkcie eutektycznym, T
A
- początkowa temperatura roztworu, W
A
-
początkowe stężenie roztworu, T
f
- punkt zamarzania czystego rozpuszczalnika, T
A
- punkt
zamarzania rozpuszczalnika, W
B
- końcowe stężenie roztworu, T
B
- końcowa temperatura
roztworu
Rys. 2. Krzywe zamrażania niektórych soków
Przy obecnym wyposażeniu technicznym kriokoncentrację można prowadzić do zawartości 45-
50% suchej substancji. Ilość wody, którą należy usunąć w celu osiągnięcia wymaganego stopnia
koncentracji składników suchej substancji, oblicza się ze wzoru:

Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
(%)
C
C
C
C
W
i
p
f
p
−
−
=
gdzie:
W - ilość wody usuwanej jako lód (%),
C
p
- stężenie suchej substancji w koncentracie (%),
C
f
- początkowa zawartość suchej substancji w cieczy (%),
C
i
- zawartość suchej substancji w usuwanej wodzie (%).
Zakładając, że ilość strat suchej substancji w usuwanej wodzie jest niewielka, powyższe równanie
można przedstawić następująco:
(%)
C
C
1
W
p
f
−
=
Typowy zestaw urządzenia do zagęszczania metodą wymrażania składa się z trzech
podstawowych części:
1.
krystalizatora, w którym otrzymuje się kryształy lodu,
2.
wirówki do oddzielania kryształów lodu,
3.
wymiennika ciepła do ochładzania cieczy, w celu usuwania ciepła krystalizacji oraz ciepła
powstającego podczas tarcia i oporów transportowanej cieczy.
W praktyce stosuje się krystalizatory z bezpośrednim, tj. kontaktowym, lub pośrednim
odbiorem ciepła. W krystalizatorach bezpośrednich zagęszczoną ciecz wprowadza się do czynnika
zamrażającego, np. do butanu. Krystalizatory tego typu nie są odpowiednie do zagęszczania soków,
ponieważ istnieje możliwość zniszczenia produktu przez kontakt z czynnikiem zamrażającym. W
krystalizatorach pośrednich ciecz zagęszczana kontaktuje się z czynnikiem zamrażającym, którym
jest najczęściej freon, przez metalową ścianę. Typowy krystalizator jest zbudowany jako rurowy
wymiennik ciepła. Wewnątrz każdej rury jest umieszczony obracający się skrobak, który systemem
ciągłym usuwa lód z wewnętrznej powierzchni ścian.
W celu otrzymania regularnych, możliwie dużych kryształów lodu prowadzi się kontrolę
krystalizacji polegająca na regulacji temperatury, strumienia zasilania i turbulencji cieczy.
Kryształy lodu zagęszczanego roztworu oddziela się za pomocą pras, wirówek lub kolumn
przemywających. Stosuje się prasy filtracyjne, wyposażone w pompy tłokowe lub śrubowe. Pras
używa się do oddzielania kryształów lodu z wymrożonych soków owocowych oraz napojów
alkoholowych. Straty składników rozpuszczalnych oznacza się jako ilość zagęszczonego roztworu,
która została zatrzymana w zaprasowanym „płacie" lodu. W zależności od stopnia zagęszczenia i
rozmieszczenia kryształów lodu i ilości zatrzymanych składników roztworu wynosi od 0,03 do 0,1
kg na 1 kg zaprasowanego lodu. Straty substancji aromatycznych są znikome, ponieważ prasy są
szczelne.
Wirowanie jest powszechnym sposobem wydzielania kryształów z zagęszczonego roztworu.
Stosuje się w tym celu wirówki pracujące okresowo lub systemem ciągłym. Konstrukcja wirówek
zapewnia zastosowanie siły odśrodkowej w granicach 1000g, a tym samym skuteczne wydzielanie
kryształów lodu. Niekiedy składniki roztworu zagęszczanego mogą pod wpływem sił powierzch-
niowych pozostawać na powierzchni kryształów. Składniki te można odzyskać stosując
przemywanie kryształów lodu przed ich rozmrożeniem w kondensatorze. Po przemywaniu wodą
kieruje się ponownie do krystalizatora. Do wad przedstawionej metody oddzielania kryształów lodu
można zaliczyć straty aromatu i dlatego wskazane jest wirowanie w układzie zamkniętym
(hermetycznym).
Kolumny przemywające stosuje się do oddzielania lodu podczas zagęszczania cieczy o niskiej
lepkości, np. piwa czy wina. Suspensję z kryształami oraz roztwór zagęszczany wprowadza się z
jednego końca kolumny. Na drugim końcu kolumny zbiera się lód, który jest zaprasowany na siatce
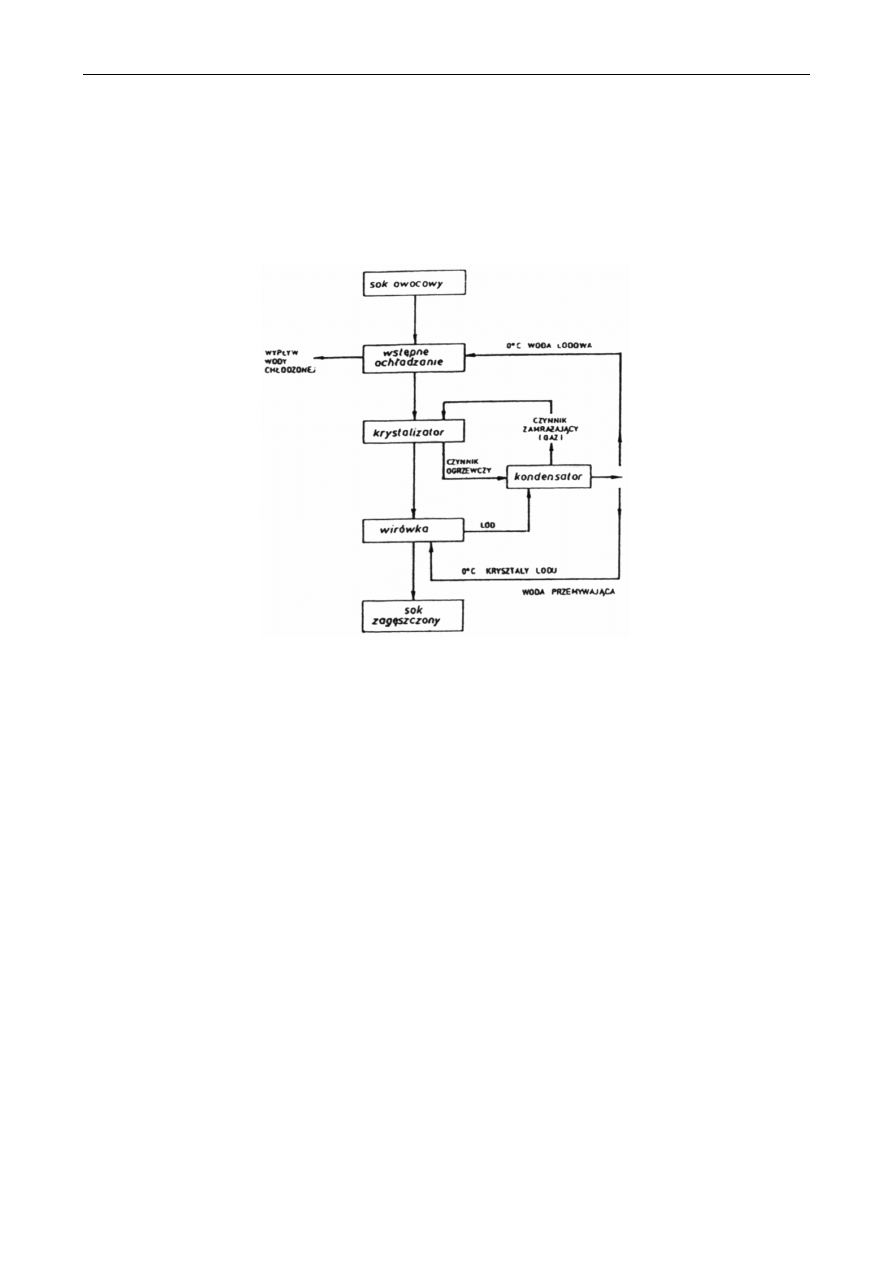
Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
30
umieszczonej wewnątrz specjalnych rur. Około 95-98% lodu topi się, a woda jako czynnik
przemywający ponownie jest zawracana do złoża lodu. Straty składników rozpuszczalny wynoszą
mniej niż 0,01%.
Schematyczny diagram kriokoncentracji soków owocowych przedstawiono na rysunku 3.
Wstępnie ochłodzony sok wprowadza się do krystalizatora, kryształy lodu oddziela się od roztworu
zagęszczonego przez wirowanie. Następnie lód topi się w specjalnym kondensatorze, ogrzanym
czynnikiem zamrażającym. Część wody lodowej wykorzystuje się do przemywania kryształów lodu
w wirówce, z której woda przepływa do krystalizatora.
Rys. 3. Schemat kriokoncentracji soków
Przedstawiony proces ma następujące zalety: minimalne straty produktu (mniej niż 1%),
wysoką ekonomikę procesu-związaną z niewielkim zużyciem energii oraz niskie koszty ogólne.
Do ciekawych rozwiązań należy zaliczyć jednostki ruchome do kriokoncentracji. Niezbędny
sprzęt umieszcza się na platformie i transportuje do miejsca, gdzie będzie prowadzony proces
zagęszczania, pod warunkiem, że jest tam możliwość podłączenia wody i energii elektrycznej.
Charakterystykę soków owocowych zagęszczanych różnymi metodami przedstawiono w
tabelach 1 i 2.
Soki owocowe zagęszczone metodą kriokoncentracji wykazują lepsze właściwości w
porównaniu z odpowiednimi produktami zagęszczanymi w wyparkach próżniowych lub metodą
odwróconej osmozy. Wyższość kriokoncentracji w porównaniu z innymi metodami zagęszczania
wyraża się przede wszystkim tym, że otrzymuje się koncentraty bez znacznych zmian smaku,
zapachu, koloru oraz wartości biologicznej. Potwierdzeniem tego są wyniki dotyczące analizy strat
kwasu askorbinowego, składników aromatycznych które przedstawiono w tabeli 1.
Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
Tabela 1.
Porównanie właściwości soków owocowych zagęszczonych metodą kriokoncentracji i przez
odparowanie w wyparce próżniowej
Wyszczególnienie
Cukry
ogółem, %
Zawartość
kwasów
Zawartość
kwasu
askorbinowego
mg/ 100 g
Substncje
aromatyczne
mg/ 100 g
pH
Sok przed zagęszczaniem
9,24
3,72
169,8
13,20
3,10
Sok po kriokoncentracji
31,23
13,00
596,2
21,36
2,82
Sok po zagęszczeniu
w wyparce
38,70
10,64
493,8
2,08
3,06
Tabela 2.
Straty substancji lotnych z soków zagęszczonych różnymi metodami
Sok
Straty w %
wymrażanie
dyfuzja
membranowa
osmoza
odwrócona
osmoza
Jabłkowy
39
92
88
84
Wiśniowy
31
42
56
74
Gruszkowy
-
44
65
73
Do wad kriokoncentracji można zaliczyć trudności w zagęszczaniu soków zawierających
większe ilości składników nierozpuszczalnych, np. włókna w nektarach. Niedogodności tego
rodzaju można złagodzić przez oddzielanie kryształów lodu w wirówce, a po rozmrożeniu lodu
przez dodanie nierozpuszczalnych składników do koncentratu. Niekiedy zaleca się filtrację soku
przed zagęszczaniem i dodanie wydzielonych składników do koncentratu.
Jakość zagęszczanych soków zależy od tempa zamrażania w krystalizatorze. Powolne
zamrażanie powoduje zwiększoną retencję składników. Większość autorów tłumaczy to
powstawaniem tzw. mikroobszarów wywoływanych asocjacją cząsteczek rozpuszczalnych, np.
węglowodanów, w zależności od wielkości kryształów lodu, która zależy od tempa zamrażania.
Szybkie zamrażanie powoduje powstawanie małych kryształów lodu, czyli dużej liczby
mikroregionów.
Koszty zagęszczania zależą od wielu zmiennych parametrów, np. zawartości suchej substancji
w surowcu, stopnia zagęszczania produktu, dopuszczalnej tolerancji stosowanych temperatur,
właściwości, jakości i przeznaczenia koncentratu. Kriokoncentracja zalicza się do procesów
energochłonnych, a więc w celu zmniejszenia kosztów opracowano metody odzysku utajonego
ciepła topnienia. Stwierdzono, że 7% energii pracy sprężarki można odzyskać wówczas, gdy
zagęszczany sok będzie ochłodzony, np. z 8°C do 0°C, przez wydzielone kryształy lodu.
Oszczędności te będą jeszcze większe, gdy temperatura zagęszczonego soku będzie wynosiła np.
20°C. Autorzy proponowanych rozwiązań twierdzą, że kriokoncentracja może być procesem
bardziej ekonomicznym od zagęszczania w wyparkach próżniowych. Aktualnie koszty inwestycji
typowego urządzenia do kriokoncentracji soków owocowych są dwukrotnie wyższe od kosztów
wyparki rurowej, łącznie z wieżą rektyfikacyjną do odzysku substancji aromatycznych. Natomiast
koszty zagęszczania soków porównywalnymi metodami są zbliżone. Wysokie koszty inwestycyjne
kriokoncentracji mogą być częściowo zrekompensowane przez oszczędności energetyczne.
Korzystne efekty ekonomiczne można uzyskać stosując kriokoncentrację do wstępnego
zagęszczania cieczy przed suszeniem sublimacyjnym, np. soków, kawy i herbaty.
W ostatnich latach szczególną uwagę przywiązuje się do zagęszczania roztworów metodami
membranowymi takimi, jak ultrafiltracja, diafiltracja, osmoza, odwrócona osmoza.

Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
32
Do nietradycyjnych metod zagęszczania zalicza się działanie ultradźwięków. W praktyce
wykorzystano efekty cieplne powstające przy oddziaływaniu ultradźwięków z roztworami.
Podwyższanie temperatury roztworu zależy od jego lepkości.
a)
Tabela 3. Podwyższenie temperatury pod działaniem ultradźwięków
Obiekt działania
Podwyższenie
temperatury w
o
C
Obiekt działania
Podwyższenie
temperatury w °C
śelatyna (roztwór)
żel
1
1
olej parafinowy
kwas steearynowy
10
36
Woda
Alkohol
2
3,5
gliceryna
parafina
10
44
* pochłanianie energii akustycznej wyraźnie wzrasta wraz ze zwiększeniem lepkości ciał,
przyczyniając się do podwyższenia temperatury. Wyjątek sianowi roztwór żelatyny oraz żel
W tabeli 3 przedstawiono dane dotyczące podwyższania temperatury roztworów (2 cm
3
)
poddanych działaniu ultradźwięków w czasie 10s, przy częstotliwości drgań 250 kHz. Drgania -
ultradźwięki są wytwarzane przez specjalne generatory, skąd są przekazywane do roztworu
zagęszczanego. Stosując tę metodę można kilkakrotnie zagęszczać roztwory bez żadnych strat
smaku i zapachu. Zastosowanie ultradźwięków zapewnia pasteryzację koncentratów. Efekty cieplne
wywołane działaniem ultradźwięków umożliwiają wyjałowienie środowiska.

Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
4.2.
W
YKONANIE ĆWICZENIA
Celem ćwiczenia jest określenie wpływu wybranych metod zagęszczania płynnych produków
spożywczych na zawarte w nich składniki takie jak np. witamina C, cukry i białka.
Wykonanie ćwiczenia.
Należy sporządzić 0,1 dm
3
10% roztworu z odpowiedniego koncentratu (rodzaj koncentratu i
jego rozcieńczenie wskazuje prowadzący ćwiczenia). Następnie z tego roztworu należy pobrać
próbkę i oznaczyć w niej zawartość witaminy C (metoda poniżej).
Zagęszczanie w wyparce próżniowej:
0,2 dm
3
sporządzonego 10% roztworu koncentratu przelać do kolby rotowapora (laboratoryjny
aparat wyparny, OBSŁUGIWANY JEST PRZEZ OSOBĘ PROWADZĄCĄ ĆWICZENIE).
Instrukcja uruchamiania wyparki próżniowej:
1.
W łaźni wodnej podgrzewającej kolbę z zagęszczanym płynem ustawić temperaturę na
45°C.
2.
Opróżnić kolbę odbieralnika.
3.
Smarem silikonowym przesmarować uchwyt kolby z próbką.
4.
Nałożyć kolbę z próbką, wlączyć pompkę wodną i przytrzymać chwilę do zassania przez
podciśnienie.
5.
Powoli odkręcić wodę chłodzącą.
6.
Opuścić kolbę do łaźni wodnej.
7.
Ustawić obroty kolbki w łaźni wodnej na 3.
Po zagęszczeniu:
8.
Zredukować ciśnienie w aparacie wyparnym poprzez obrót zaworu przy manometrze,
9.
Zamknąć wodę dochodzącą do pompki wodnej.
10.
Wyłączyć wodę chłodzącą.
11.
Zdjąć kolbkę z próbką z uchwytu.
12.
Wyłączyć łaźnię wodną.
Zagęszczony roztwór należy przelać do cylindra na 200 cm
3
i uzupełnić do kreski
wodą
destylowaną. Próbka ta jest przeznaczona do oznaczania zawartości witaminy C.
Zagęszczanie w otwartym naczyniu pod ciśnieniem atmosferycznym:
Do otwartego naczynia o poj. 1 dm
3
(może być garnek) przelać 0,2 dm
3
sporządzonego 10%
roztworu koncentratu i zagęścić go ok. 5 krotnie gotując na kuchence elektrycznej. Po zagęszczeniu
należy zagęszczony roztwór przelać do cylindra na 200 cm
3
i uzupełnić do kreski
wodą
destylowaną. Jest to trzecia próbka przeznaczona do oznaczania zawartości witaminy C.
Dodatkowo należy określić także barwę i zapach próbek zagęszczanych pod obniżonym
ciśnieniem i w warunkach ciśnienia atmosferycznego.
Oznaczanie zawartości witaminy C.
Do kolby stożkowej odmierzyć 10 cm
3
badanego, klarownego roztworu. Dodać 40 cm
3
2%
roztworu kwasu szczawiowego i szybko miareczkować roztworem 2-6-dwuchlorofenoloindofenolu
(0,250 g barwnika w 1 dm
3
wody w obecności 0,210 g NaHCO
3
) aż do wystąpienia lekko różowego

Z
AG
Ę
SZCZANIE ROZTWORÓW W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
34
zabarwienia, nie znikającego w ciągu 30 sekund. Miareczkowanie powtórzyć 3 krotnie.
Jednocześnie należy wykonać w ten sam sposób próbę zerową, używając do miareczkowania 50cm
3
2% kwasu szczawiowego.
W przypadku gdy na miareczkowanie próby właściwej zużyto objętość barwnika równą objętości
pipety, miareczkowanie należy powtórzyć odpowiednio rozcieńczając miareczkowaną próbkę.
Miano roztworu barwnika należy nastawić używając roztworu kwasu L-askorbinowego w kwasie
szczawiowym (naważka 0,1g kwasu L-askorbinowego/50cm
3
kwasu szczawiowego).
Należy obliczyć zawartość kwasu L-askorbinowego [mg/100cm
3
niezagęszczonego] w próbie bez
zagęszczenia oraz w próbkach zagęszczanych.
4.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń i analiz należy umieścić w sprawozdaniu [wzór, rozdział Załącznik 1.
Załącznik 1. ] w formie oddzielnych punktów wraz z przeliczeniami i przedłożyć osobie
prowadzącej ćwiczenia w celu sprawdzenia. Należy także zaznaczyć wszelkie odstępstwa od metod
podanych w opracowaniu ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski w
formie zwięzłych punktów.
4.4.
S
PRZĘT I ODCZYNNIKI
•
wyparka próżniowa
•
pompka wodna do wytworzenia podciśnienia
•
łaźnia wodna
•
biureta do miareczkowania
•
statyw z uchwytem do biurety
•
zlewka 100 cm
3
•
zlewka 1 dm
3
•
termometr o zakresie pomiarowym 100 °C
•
lejek szklany
•
sączki z bibuły filtracyjnej
•
kuchenka elektryczna
•
cylinder miarowy 1 dm
3
kolba miarowa o pój. 50 cm
3
•
woda destylowana
•
roztwór 0,0015 n 2-6-dwuchlorofenoloindofenolu.

5.
ROZMRAśANIE śYWNOŚCI
5.1.
W
PROWADZENIE
Wartość odżywcza i organoleptyczna potraw przygotowanych z mrożonek zależy m.in. od
metody rozmrażania, czy też od sposobu restytucji mrożonej żywności. Proces rozmrażania jest
również ważną operacją technologiczną w przemyśle spożywczym, rybnym, owocowo-
warzywnym, mięsnym, opartym na półproduktach uzyskanych przez zamrożenie. Technologia
rozmrażania może przyczyniać się do pogorszenia jakości i obniżenia wydajności produktu, dlatego
też zasługuje na szczególną uwagę. Celem rozmrażania jest podwyższenie temperatury produktu
zamrożonego powyżej krioskopowej, przy możliwie maksymalnym zachowaniu korzystnych cech
jakościowych surowca wyjściowego,
W procesie rozmrażania produkty ulegają zmianom fizycznym, mikrobiologicznym, a także
biochemicznym. Najważniejszą zmianą fizyczną, zachodzącą podczas podwyższania temperatury
mrożonki jest przemiana fazowa lodu w wodę. Ponieważ całkowita odwracalność całokształtu
procesów zachodzących w poszczególnych fazach obróbki zamrażalniczej nie jest możliwa, dlatego
pewna ilość wody wydzielonej w przemianie fazowej nie jest powtórnie wiązana przez składniki
żywności i wypływa na zewnątrz produktu, jako główny składnik wycieku rozmrażalniczego.
Podczas procesu rozmrażania następuje na ogół zmniejszenie się wymiarów produktów, wynikające
ze wzrostu gęstości wody przy przemianie fazowej. Zjawiska występujące równolegle, takie jak
utrata turgoru, zmniejszenie wymiarów i zmiana kształtu produktów, określane są niekiedy jako
"skurczenie" rozmrażalnicze, które również przyczynia się do wzrostu wielkości wycieku. Wyciek
traktowany jest jako zewnętrzny objaw denaturacji białka i zmian w obrębie innych
wielkocząsteczkowych substancji, będących składnikami żywności. Pośrednio wyciek związany
jest również z częściowym zniszczeniem struktury produktu, już w trakcie procesu rozmrażania.
Przyczyny tego faktu upatruje się we wzroście dużych kryształów kosztem małych w temperaturach
bliskich krioskopowej. Najprawdopodobniej wynika to z większej prężności pary wodnej nad
powierzchnią małych kryształów. W skład wycieku wchodzą składniki rozpuszczalne w wodzie,
takie jak: witaminy, barwniki, sole mineralne, cukry, aminokwasy, niskocząsteczkowe peptydy i
białka oraz inne substancje. Do frakcji wycieku może przechodzić w procesie rozmrażania kilka do
kilkunastu procent całkowitej zawartości tych substancji. Innym procesem mogącym zachodzić
podczas rozmrażania jest odparowywanie wody z powierzchniowych warstw produktu, co jest
równoznaczne
ze
zmniejszeniem
wydajności
i
na
ogół
niekorzystnymi
zmianami
organoleptycznymi. Ważną zmianą o charakterze fizycznym zachodzącą w rozmrażanej żywności
jest zmiana przewodnictwa cieplnego. Produkty rozmrożone w stosunku do zamrożonych
charakteryzują się mniejszym współczynnikiem przewodzenia ciepła λ, co utrudnia wymianę ciepła
i przy zbyt intensywnym dostarczaniu energii może prowadzić do miejscowego przegrzewania
rozmrożonych fragmentów produktu. Wraz z podwyższeniem temperatury produktu wzrasta
szybkość reakcji chemicznych, mogących prowadzić do niekorzystnych zmian jakościowych.
Tempo reakcji enzymatycznych w żywności rozmrożonej jest wypadkową następujących
czynników: stopnia denaturacji białek enzymów, wielkości zniszczenia struktury produktu i
dezagregacji układów koloidalnych, dostępnością substratów, obecnością inhibitorów reakcji itp.
Produkty po rozmrożeniu są narażone na rozwój drobnoustrojów i związane z tym
konsekwencje. Wielu trudności związanych z rozmrażaniem można uniknąć bezpośrednio poddając
mrożonki właściwej obróbce termicznej (gotowanie, smażenie, pieczenie). Odnosi się to jednak
głównie do gotowych potraw lub ich głównych składników (np. mieszanki warzywne). Do
produktów, które muszą być rozmrożone przed dalszym przerobem zalicza się ryby i mięso.
Bezpośrednio przed spożyciem rozmraża się natomiast: owoce, soki owocowe i warzywne, wyroby
piekarskie i cukiernicze, desery i inne.
Istnieją dwie zasadnicze grupy metod rozmrażania: pierwsza, w której ciepło wprowadzane jest
do produktu przez jego powierzchnię oraz druga, w której ciepło wytwarzane jest wewnątrz pro-
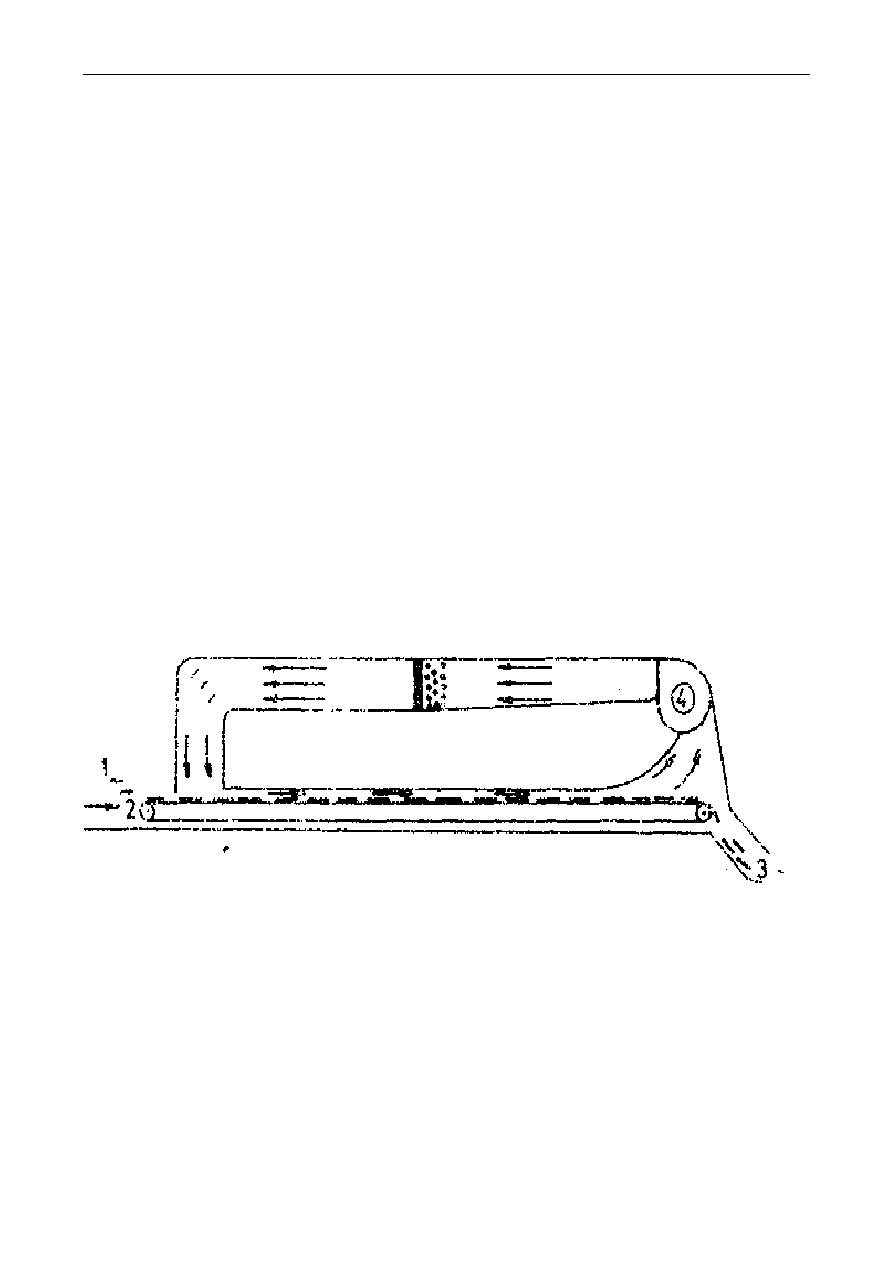
12 luty 2010
36
duktu. W pierwszej grupie stosuje się zabiegi polegające na poddaniu produktu takim działaniom,
jak napromieniowanie od gorących powierzchni, ciepłe powietrze, ciepła woda, ogrzewane płyty
lub działanie parą wodną w podciśnieniu. W drugiej grupie ciepło wytwarzane jest w produkcie
przez zastosowanie zjawiska oporu elektrycznego, ogrzewania dielektrycznego lub mikrofalowego.
W praktyce metody powierzchniowego ogrzewania są stosowane częściej od metod wewnętrznego
ogrzewania.
Metody ogrzewania zewnętrznego
W metodach tych ciepło przenoszone jest za pomocą konwekcji, front topnienia zaś przesuwa
się od warstw zewnętrznych do wnętrza produktu. Stosowane czynniki rozmrażalnicze to:
powietrze, woda lub roztwory wodne soli kuchennej oraz para wodna.
Rozmrażanie powietrzne. Rozmrażanie w powietrzu o ruchu grawitacyjnym w temperaturze
pokojowej jest mało skuteczne (niski współczynnik wnikania ciepła α). Zewnętrzne warstwy
produktu długo pozostają w temperaturze otoczenia, podczas gdy środkowe partie są jeszcze
zamrożone. Sprzyja to rozwojowi mikroflory na powierzchni produktów. Ponadto produkty
przetrzymywane przez kilka lub kilkanaście godzin w powietrzu o średniej wilgotności (50-70%)
ulegają znacznej ususzce. Jedynie do celów domowych można polecić rozmrażanie w spokojnym
powietrzu o średniej wilgotności (w komorze chłodziarki o temperaturze bliskiej 0°C).
W warunkach przemysłowych stosuje się powietrze ogrzewane, nawilżone, o wymuszonej
cyrkulacji. Rozmrażanie odbywa się w tunelach wyposażonych najczęściej w nagrzewnice wodne,
rozpylacze do nawilżania powietrza i wentylatory (rys. 1). Na przykład blok mrożonych ryb o
grubości 10 cm w powietrzu o ruchu grawitacyjnym i temperaturze 20°C rozmraża się ok 20 godz.,
w powietrzu zaś o takiej samej temperaturze, lecz wilgotności bliskiej 100% i dużej liniowej
prędkości przepływu (ok. 5 m/s) - tylko 4 godz., co spowodowane jest polepszeniem warunków
wymiany ciepła (wzrost współczynnika α).
Rys. 1. Schemat tunelu do rozmrażania ryb w przepływie ciepłego wilgotnego powietrza: 1-
doprowadzenie
mrożonych
ryb,
2-przenośnik
taśmowy,
3-odprowadzenie
ryb
rozmrożonych, 4- wentylator, 5-nagrzewnica i nawilżacz powietrza
Podczas rozmrażania powietrznego tusz i półtusz mięsnych w warunkach kontrolowanych
programowane są różnice temperatur pomiędzy powierzchnią produktu a środowiskiem. Zwykle
wraz ze wzrostem temperatury powierzchni produktu obniża się temperaturę powietrza. Aby
uniknąć nadmiernego rozwoju mikroflory w niektórycn tunelach instalowane są promienniki
ultrafioletu.
Rozmnażanie w warunkach kontrolowanych realizowane jest zwykle w dwóch fazach.
W fazie I - różnica temperatur pomiędzy powietrzem a powierzchnią rozmrożonego produktu jest
duża (20-25°C), wilgotność względna powietrza zaś możliwie niska. Wpływa to na
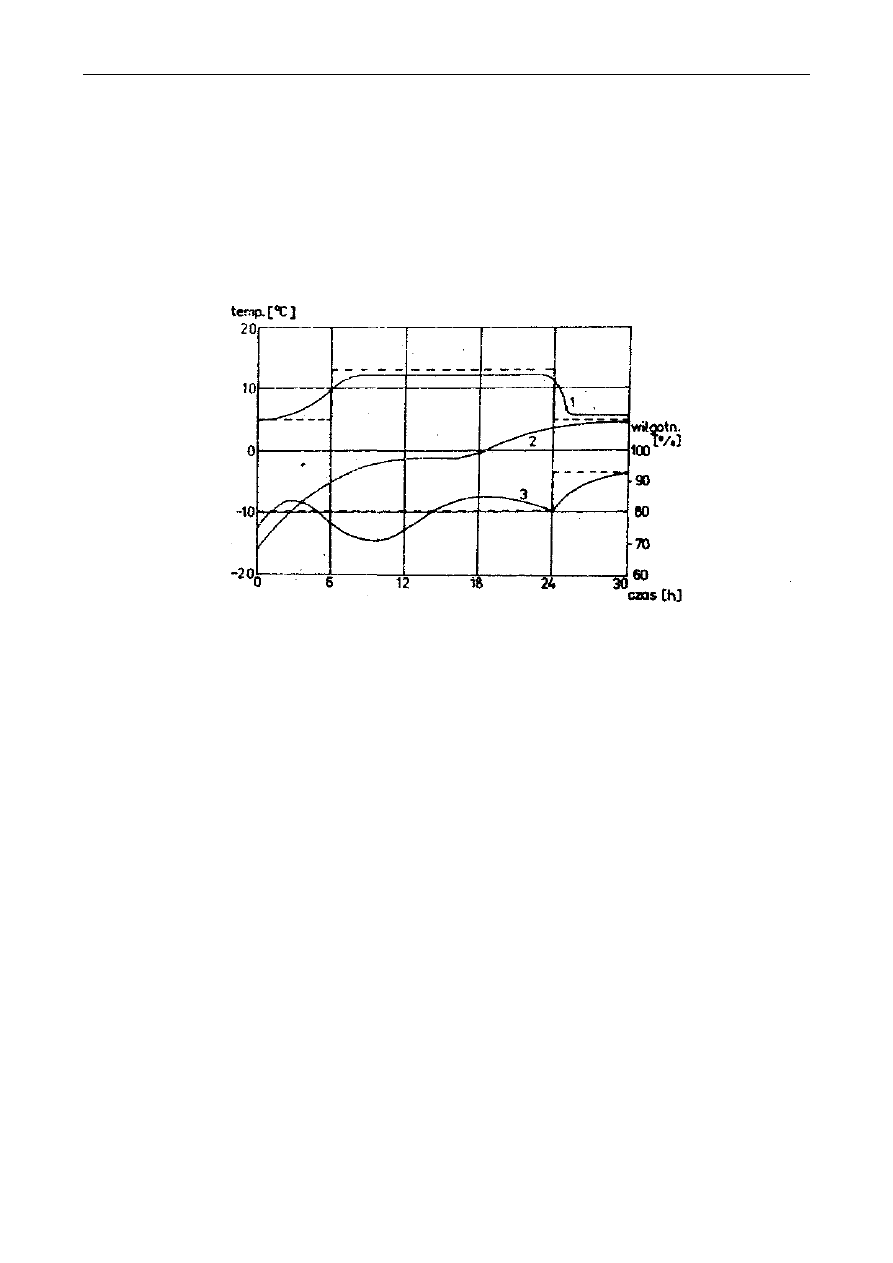
zmniejszenie wykraplania się wilgoci na powierzchni produktu. Woda skraplająca się jest
stosunkowo szybko usuwana, gdyż prędkość przepływu powietrza jest wysoka (5 m/s),
W fazie II - następuje wzrost temperatury produktu i jednoczesne obniżenie temperatury powietrza
oraz wzrost jego wilgotności względnej. Pod koniec tej fazy wyrównują się temperatury w całej
objętości rozmrażanych tusz. W niektórych typach tuneli stosowanych głównie do rozmnażania
mięsa stosuje się wdmuchiwanie cieplejszego powietrza na najgrubsze partie tusz, co wpływa
na wyrównanie czasu rozmrażania. Tunele użytkowane są okresowo. Typowa pojemność tunelu
wynosi od 20 do 30 t mięsa, czas zaś rozmrażania ok. jednej doby. Parametry procesu
kontrolowanego rozmrażania zostały przedstawione na rysunku 2.
Rys 2. Zaprogramowany i faktyczny przebieg kontrolowanego rozmrażania mięsa: 1-temperatura
powietrza, 2-temperatura wewnątrz, tusz, 5-wilgotność powietrza. Uwaga: linią
przerywaną oznaczono zaprogramowany, zaś ciągłą faktyczny przebieg procesu
rozmrażania.
Rozmrażanie wodne. Ten rodzaj rozmrażania stosowany jest zazwyczaj do mięsa, ryb i masy
jajecznej opakowanej w puszki i realizowany jest przez immersję lub dyspersje. Rozmrażanie w ką-
pieli przeprowadza się w wannach w ciągłym powolnym przepływie wody (prędkość wody 0,1-0,3
m/s). W przypadku produktów nie opakowanych rozmrażanie wodne wywiera bardziej
niekorzystny wpływ niż powietrzne, gdyż rozmrażane produkty mogą absorbować wodę i
jednocześnie tracić substancje odżywcze w procesie wymywania i dyfuzji. Przykładem metody
rozmrażania wykorzystującej natrysk wodą jest rozmrażanie bloków ryb. Metoda ta polega na
zraszaniu bloków wodą rozpylaną w dyszach (kropelki o średnicy ok. 0,1 mm). W wyniku
dostarczonego ciepła ryby ulegają rozmrożeniu i dzięki specjalnej konstrukcji pojemników
swobodnie spadają na taśmę przenośnika odprowadzającego je do dalszej obróbki. Po rozmrożeniu
i rozpadnięciu się jednego bloku do pojemnika usuwany jest blok następny. Całkowite rozmrożenie
10 kg bloku o temperaturze -25°C do 0°C trwa ok. 1,5 godz.
W niektórych urządzeniach zamiast wody stosuje się 5% roztwór soli kuchennej. Proces ten
przeprowadzany jest w wannach kwasoodpornych, przy czym stosuje się w nim specjalną obróbkę
cieczy obiegowej mającą na celu zachowanie czystości mikrobiologicznej procesu.
Do metod rozmrażania wodnego można zaliczyć również prosty sposób rozmrażania we
wrzącej wodzie, który stosowany jest do mrożonych potraw, niektórych warzyw i owoców
poddawanych przed spożyciem gotowaniu. Proces rozmrażania przebiega bardzo szybko i
połączony jest z obróbką termiczną. Sposób ten stosowany jest głównie w gospodarstwach
domowych i żywieniu zbiorowym.
Rozmrażanie parowe. Polega na bezpośrednim kontakcie zamrożonego produktu z parą wodną,
która kondensuje się na powierzchni produktów oddając duże ilości ciepła. W przypadku
12 luty 2010
38
stosowania pary o temperaturze 100
o
C lub wyższej, proces powoduje miejscowe, znaczne
przegrzania produktu, prowadzące do obniżenia jego jakości. Ten niekorzystny wpływ
zastosowania pary można wyeliminować stosując rozmrażanie próżniowe z parą wodną, które
polega na dostarczeniu ciepła z kondensującej pod próżnią pary wodnej do zamrożonego produktu.
W ten sposób wykorzystuje się utajone ciepło skraplania przy temperaturach nie powodujących
zmian na powierzchni produktu. Podstawowymi zaletami tej metody jest bardzo wysoka wartość
współczynnika wymiany ciepła, krótki czas procesu (kilkanaście lub kilkadziesiąt minut),
gwarancja nie przekroczenia danej temperatury (temperatura pary jest ściśle skorelowana z
ciśnieniem dającym się łatwo regulować), oraz równomierność procesu i brak ususzki produktu.
Urządzenia do rozmrażania próżniowego są wykorzystywane głównie w przemyśle rybnym. Ich
wydajność jest duża i może wynosić do 2 t/godz.
Rozmrażanie kontaktowe. W tym typie rozmrażania stosowane są urządzenia podobne do
wielopłytowych zamrażarek kontaktowych. Temperatura cieczy krążących w płytach nie powinna
przekraczać +20
o
C. Metoda kontaktowa dotyczy prawie wyłącznie bloków ryb mrożonych w
zamrażarkach płytowych. Na przykład blok ryb o grubości 10 cm rozmraża się tą metodą ok. 5
godz.
Do metod kontaktowych można również zaliczyć przeponowe (zwykle parowe) ogrzewanie
mrożonki w otwartym zbiorniku. Przykładem takiego rozwiązania, stanowiącego połączenie
procesu rozmrażania z dalszym procesem technologicznym, może być produkcja dżemów i
mrożonych owoców.
Metody ogrzewania wewnętrznego
W metodach tych, które są bardziej skomplikowane od rozmrażania powierzchniowego, ciepło
wytwarza się w całej masie produktu. Metody te można podzielić aa następujące grupy:
rozmrażanie dielektryczne, mikrofalowe i opornościowe.
Rozmrażanie dielektryczne. Metoda dielektrycznego rozmrażania polega na poddaniu produktu
działaniu pola wysokiej częstotliwości. Produkty przesuwane są między dwiema płytami - elek-
trodami, do których przyłożone jest napięcie o wysokiej częstotliwości od 10 do 100 MHz.
Rozmrażane produkty są podgrzewane przez zmienne pole elektryczne i zachowują się w nim, jak
dielektryk w kondensatorze. System ten może być stosowany do jednorodnego surowca o
wyrównanym kształcie bloków (rys. 3).
Rys. 3. Schemat urządzenia do dielektrycznego rozmrażania bloków mięsa lub ryb: 1 - przenośnik
taśmowy, 2 - bloki rozmrażanego surowca, 3 - elektrody, 4 - mycie taśmy
System dielektrycznego rozmrażania jest stosowany najczęściej do bloków ryb. Układane są
one na tacach ze specjalnego szkła i po napełnieniu ich wodą przekazywane są na przenośnik
przesuwający je przez tunel. W urządzeniu zainstalowane są elektrody wytwarzające pole
elektryczne oraz automatyczny system spłukiwania i mycia. Metoda ta jest bardzo szybka, wydajna
i umożliwia rozmrażanie produktów wewnątrz opakowań, jest jednak bardzo energochłonna.
Rozmrażanie mikrofalami. Zakres widma elektromagnetycznego zawarty w granicach od 1 mm
do 1m nazywany jest mikrofalami. Aby wyeliminować możliwości zaburzeń w telekomunikacji do
zastosowania przemysłowego dopuszczono promieniowanie o określonej długości fali (λ= 33 cm,
12,5 cm i 1,35 cm). Mikrofale przechodzą przez warstwy powietrza, masy plastyczne, porcelanę i

szkło, odbijają się od powierzchni metalowych i są absorbowane przez substancje dielektryczne,
m.in. składniki żywności.
Mikrofale oddziaływują głównie na dipole wody, powodując szybkie obracanie się ich i
drgania, co wywołuje duże siły tarcia.
Energia wytwarzana w całej masie produktu powoduje bardzo szybki wzrost jego temperatury.
W procesie rozmnażania mikrofalowego występuje niekorzystny czynnik nierównomiernej
absorpcji fal przez produkt. Wynika to z różnych stałych dielektrycznych wody (ε=88) i lodu
(ε=35). Powodować to może miejscowe przegrzania produktu: uwzględniając jednak bardzo krótki
czas rozmrażania, zjawisko to nie mają większego znaczenia. W najnowszych rozwiązaniach
technicznych tej metody powierzchnię produktu chłodzi się przez niezbyt intensywny natrysk
skroplonych gazów, np. ciekłego freonu lub azotu.
Metoda rozmrażania mikrofalowego ma wiele zalet m.in.:
•
bardzo krótki czas procesu,
•
dość dużą równomierność nagrzewania całej masy produktu
•
możliwość rozmrażania produktów opakowanych
•
możliwość zachowania odpowiedniej higieny procesu.
Istnieje wiele rozwiązań technicznych rozmrażania mikrofalowego zarówno o działaniu
okresowym, jak i ciągłym. W typowych urządzeniach energia mikrofal wytwarzana jest w
magnetronie i przesyłana falowodem do osłoniętego ekranem prostopadłościennego pieca, gdzie
uzyskuje się pole wielokrotnych odbić mikrofal. W urządzeniach ciągłych rozmrażany produkt
przesuwany jest na transporterze przez piec. Mała przenikalność mikrofal sprawia, że na
przenośniku lub tacy pieca można umieszczać produkty w warstwie nie przekraczającej kilku
centymetrów.
Metoda ze względu na konieczność wysokich nakładów inwestycyjnych oraz duże zużycie
energii, w warunkach przemysłowych nie jest stosowana na szeroką skalę. Metody mikrofalowego
rozmrażania żywności znalazły zastosowanie w żywieniu zbiorowym, a także w gospodarstwach
domowych.
Rozmnażanie opornościowe. Rozmrażanie przez wykorzystanie oporności elektrycznej polega
na przepuszczaniu prądu zmiennego przez blok zamrożonego produktu. Wykorzystuje się tu
wysoką oporność właściwą produktów zamrożonych w stosunku do rozmrożonych. Na przykład
zamrożona tkanka mięśniowa ryb w temperaturze -30oC wykazuje przy częstotliwości prądu 50 Hz
oporność 25x106 ohm/cm, w temperaturze natomiast 0°C tylko 800 ohm/cm. Metodę rozmrażania
opornościowego wykorzystuje się głównie do bloków ryb. Blok zanurzany jest w wodzie o
niewielkim przepływie, co podnosi jego temperaturę, a zarazem zmniejsza oporność. Jest to
wstępne podgrzanie ryb. Następnie z obu stron bloku, w bezpośrednim z nim kontakcie, umieszcza
się elektrody. Powierzchnia bloku częściowo rozmrożona zapewnia dobry przepływ prądu. Przez
blok przepuszcza się prąd zmienny o napięciu 10-40 V i natężeniu 10-20 A. Metoda ta ma
zastosowanie dc rozmrażania cienkich bloków ryb oraz pojedynczych ryb. Proces rozmrażania trwa
w niej ok. kilkudziesięciu minut.
Rozwój i udoskonalanie metod rozmrażania związane jest głównie z produkcją przemysłu
rybnego. Ryby dostarczane są do przetwórni znajdujących się na lądzie w postaci zamrożonej,
gdzie są przetwarzane i opuszczają przetwórnie, jako mrożone dania, półprodukty, potrawy itp.
Istnieje zatem potrzeba szybkiego i wydajnego rozmrożenia nie obniżającego jakości produktu. W
tabeli 3 została przedstawiona charakterystyka różnych metod rozmrażania żywności.
W przyszłości należy się spodziewać dalszych udoskonaleń technicznych pozwalających
skutecznie, szybko i tanio rozmrażać żywność. Do metod takich będą należały z pewnością
skojarzone sposoby rozmrażania, np. ogrzewania wewnętrznego na początku procesu, a następnie
rozmrażanie w powietrzu kontrolowanym.

12 luty 2010
40
5.2.
W
YKONANIE ĆWICZENIA
Celem ćwiczenia jest zapoznanie się oraz porównanie różnych metod rozmrażania żywności.
Porównanie zostanie przeprowadzone dla różnych rodzajów mrożonek.
Wykonanie ćwiczenia
Wymyć i osuszyć warzywa lub owoce. Warzywa np. marchew, pokroić w kostkę o wymiarze
boku ok. 1 cm. Krojonkę zamrozić w temp.-21°C na tydzień przed zajęciami (ta część ćwiczenia
jest już wykonana i prowadzący ćwiczenie dostarczy zamrożony produkt).
Zadanie 1. Rozmrażanie bezprzeponowe w powietrzu i w wodzie.
Odważyć w sitkach plastikowych uprzednio zamrożone truskawki i kostki marchwi (oba
produkty ok. 100 g z dokładnością do 0,01g). Następnie jedną z próbek należy umieścić w wodzie o
temp. 20°C, a drugą umieścić na szalce Periego i pozostawić na powietrzu do rozmrożenia. Po
rozmrożeniu ponownie zważyć i określić procent odcieku, a także czas rozmrażania.
Zadanie 2. Rozmrażanie mikrofalowe.
Do rozmrażania tą metodą użyć kuchenki mikrofalowej. Na szalkach Petriego należy odważyć
uprzednio zamrożone próbki owoców truskawek i kostek marchwi (oba produkty ok. 100 g z
dokładnością do 0,01g). Szalki umieścić w komorze kuchenki ustawionej na rozmrażanie i
kuchenkę uruchomić na kilka minut. Po rozmrożeniu przenieść próbki na sitca i po odsączeniu
określić procent odcieku (zważyć próbki) oraz czas rozmrażania.
Zadanie 3. Rozmrażanie przeponowe na podgrzewanej powierzchni metalowej.
Odważyć próbki uprzednio zamrożonych owoców truskawek i kostek marchwi do metalowych
naczynek(oba produkty ok. 100 g z dokładnością do 0,01g). Naczynka postawić na płycie
metalowej termostatu nagrzanego do temp. 30
o
C do rozmrożenia. Należy określić czas rozmrażania
i procentowy odciek.
Z przeprowadzonych doświadczeń wyciągnąć wnioski i ocenić przydatność poszczególnych
metod rozmrażania do praktyki przemysłowej.
5.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń należy umieścić w sprawozdaniu [wzór, rozdział Załącznik 1. Załącznik 1. ] w
formie oddzielnych punktów wraz z przeliczeniami i przedłożyć osobie prowadzącej ćwiczenie w
celu sprawdzenia. Należy także zaznaczyć wszelkie odstępstwa od metod podanych w opracowaniu
ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski w formie zwięzłych punktów.

5.4.
S
PRZĘT I ODCZYNNIKI
•
warzywa i owoce (marchew, burak ćwikłowy, jabłka, truskawki itp.)
•
komplet sitek-4 sztuki
•
zlewka 1 dm
3
•
termostat mikrobiologiczny
•
kuchenka mikrofalowa
•
waga techniczna
•
odważniki
•
łyżeczka wagowa
•
szalki Petriego (duże) 5 sztuk
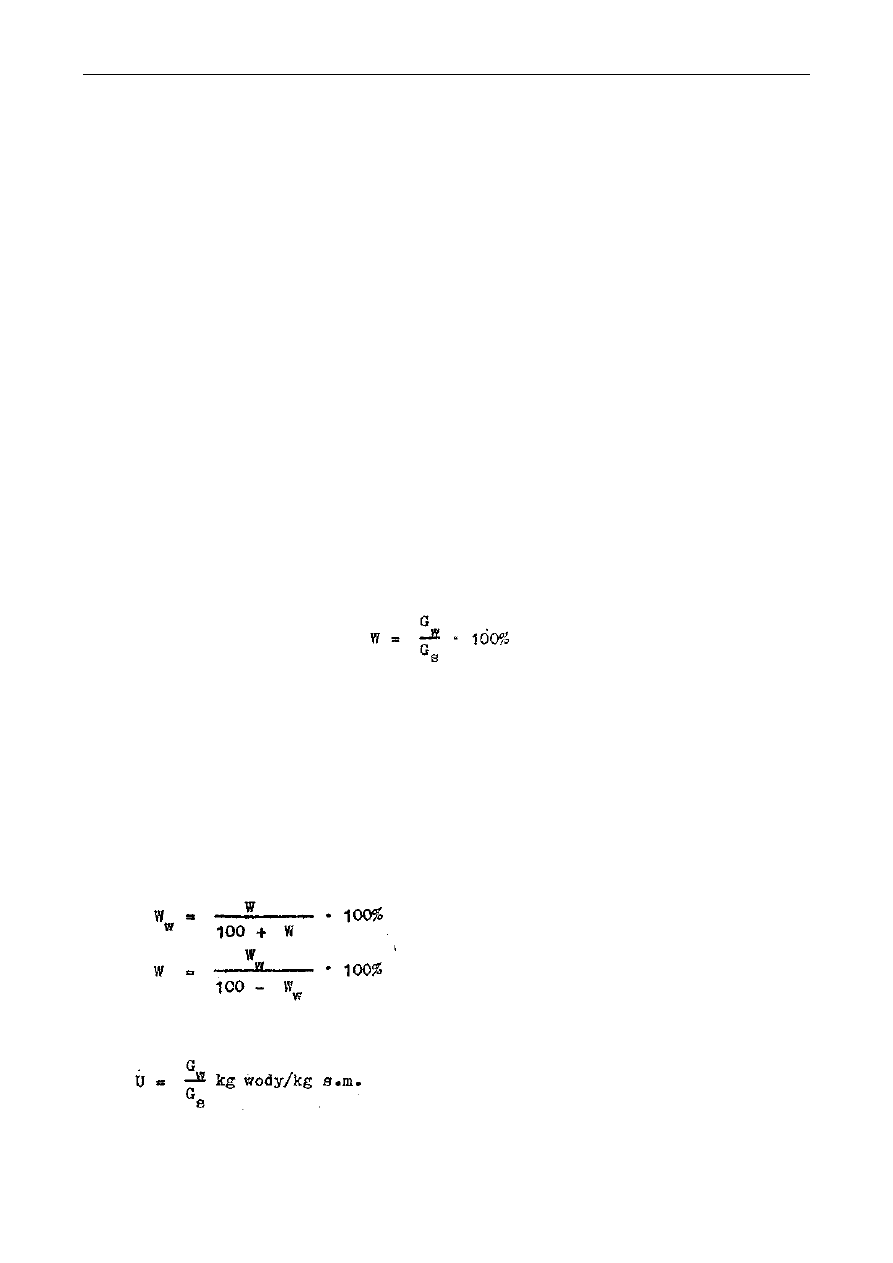
S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
12 luty 2010
42
6.
SUSZENIE PRODUKTÓW SPOśYWCZYCH
6.1.
W
PROWADZENIE
Suszenie jest procesem technologicznym zmierzającym do obniżenia zawartości wody w
produktach do kilku lub kilkunastu procent w celu wyeliminowania lub zwolnienia procesów
mikrobiologicznych, fizykochemicznych i biochemicznych. Głównym celem suszenia produktów
spożywczych (zarówno cieczy jak i ciał stałych) jest dążenie do przedłużenia ich trwałości.
Produkty spożywcze stanowią złożone układy strukturalne, w których woda jest związana z
materiałem:
a/ chemicznie;
b/ fizykochemicznie - woda utrzymywana osmotycznie i woda strukturalna;
c/ mechanicznie - woda utrzymywana przez makrokapilary materiału (średni promień powyżej
10
-5
cm)i mikrokapilary (średni promień poniżej 10
-5
cm).
Różne rodzaje wilgoci warunkują mechanizm jej usuwania podczas suszenia, np. w celu
usunięcia wilgoci związanej adsorpcyjnie wewnątrz materiału trzeba ją przekształcić w parę, która
przemieszcza się wewnątrz materiału, głównie w postaci ciekłej.
Tak zwana woda wolna przemieszcza się wewnątrz materiału głównie w postaci ciekłej bez
uprzedniej zamiany w parę. Woda „kapilarna" przemieszcza się, w zależności od warunków
suszenia, zarówno w postaci ciekłej (kosztem sił kapilarnych), jak również w postaci pary (kosztem
różnicy ciśnień).
Wilgotność produktu W w technice suszarnictwa przyjęto obliczać w odniesieniu do masy
absolutnie suchej substancji, która podczas suszenia nie powinna podlegać zmianie:
gdzie:
G
w
- masa wilgoci w produkcie (kg),
G
s
- masa absolutna suchej substancji w produkcie.
Suchą masę materiału (G
s
) oznacza się zazwyczaj przez suszenie w 105°C aż do momentu
uzyskania nie zmieniającej się wartości. Często w technologii suszarnictwa stosuje się pojęcie
wilgotności względnej. Wilgotność względną W
w
oblicza się w odniesieniu do początkowej masy
wilgotnego materiału. Jest ona powiązana z wilgotnością bezwzględną W następującymi
zależnościami:
Bezwzględna zawartość wody w produkcie U jest to stosunek zawartej w nim wilgoci do suchej
masy produktu:
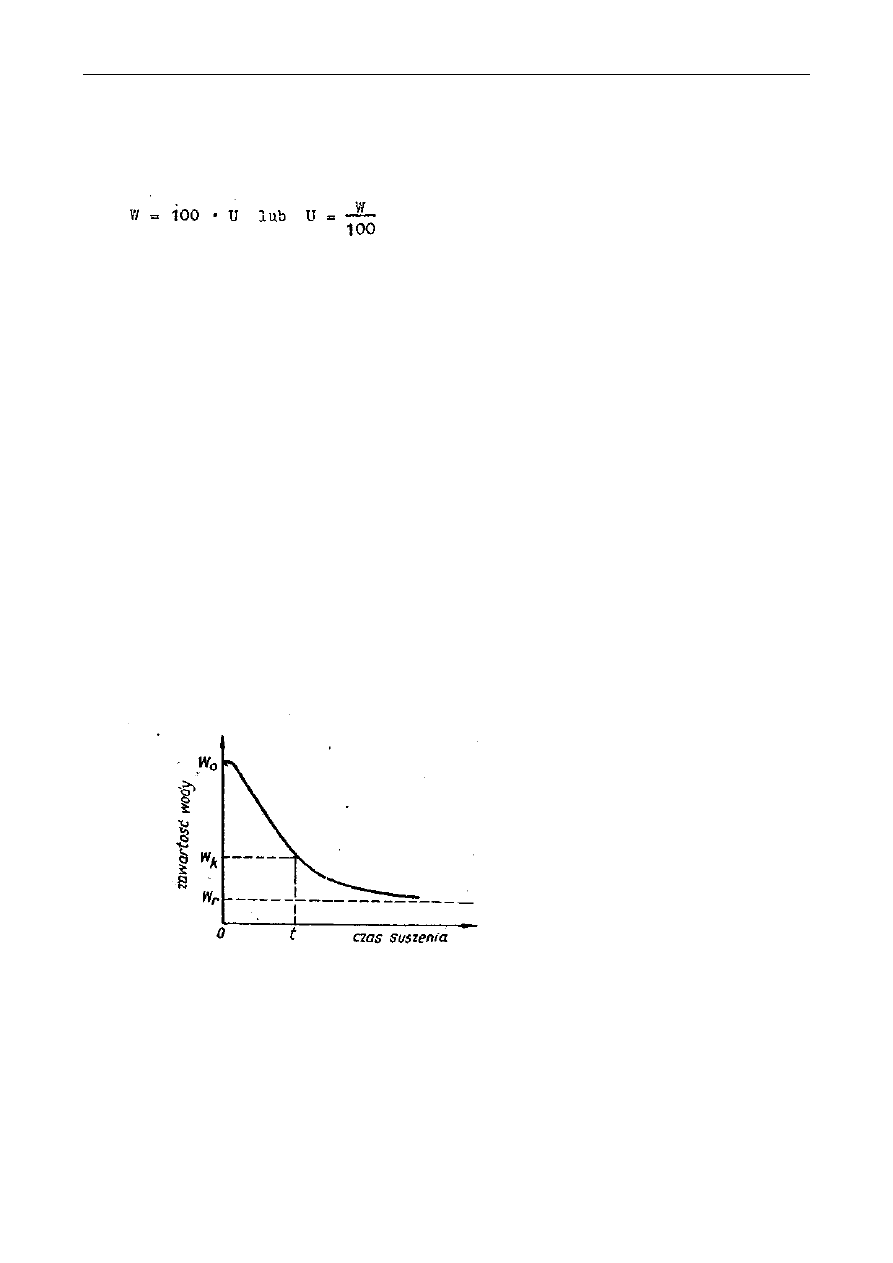
S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
Zawartość wilgoci może charakteryzować nie tylko cały produkt, lecz również dowolną
warstwę lub część jego objętości. Przy równomiernym rozłożeniu wilgoci W
w
całej objętości
produktu możemy przyjąć:
Szybkość suszenia
Szybkością suszenia nazywamy wyrażenie określające wielkość zmiany średniej zawartości
wody w suszonym produkcie w jednostce czasu. Szybkość suszenia jest pochodną średniej
zawartości wody do czasu suszenia i może być przedstawiona w postaci dW/dt.
Jedna z metod oznaczania szybkości suszenia polega na graficznym różniczkowaniu krzywej
suszenia przedstawionej na rysunku 1.
Krzywa suszenia przedstawia zależności miedzy średnią zawartością wody w suszonym
produkcie a czasem suszenia. Przebieg krzywej suszenia najczęściej składa się z dwu odcinków,
tzn. odcinka prostoliniowego od W
o
do W
k
oraz odcinka krzywoliniowego od W
k
do W
r
.
Zawartość wody W
k
nazywamy krytyczną zawartością wody. Krytyczna zawartość wody
oddziela dwa etapy suszenia różniące się intensywnością wysychania produktu.
Zawartość wody W
r
nazywa się równoważną zawartością wody w produkcie i w czynniku
suszącym.
Wykonując graficzne różniczkowanie krzywej suszenia przedstawionej na rysunku 1
przedstawiając wynik w układzie współrzędnych prostokątnych: szybkość suszenia-zawartość
wody, uzyskujemy wykres szybkości suszenia przedstawiony na rysunku 1. Wykres składa się z
trzech części i linii wzrostu szybkości suszenia od 0 do pewnej stałej wartości; linii między W
o
i W
k
(I okres suszenia) oraz linii malejącej szybkości suszenia między W
k
i W
r
(II okres suszenia).
Należy zaznaczyć, że przedstawiony przykład graficznego różniczkowania krzywej suszenia jest
adekwatny dla większości produktów spożywczych suszonych konwencyjnie.
Rys 1. Krzywa suszenia
Proces suszenia powinien przebiegać w warunkach uniemożliwiających powstawanie w
produktach spożywczych nieodwracalnych zmian, które mogłyby doprowadzić do pogorszenia ich
jakości. Zmiany powyższe związane są przede wszystkim z denaturacją białka i ze skleikowaniem
skrobi, co jest następstwem zbyt intensywnego nagrzewania materiału podczas suszenia.

S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
12 luty 2010
44
Rys. 2. Wykres szybkości suszenia (I i II okres suszenia).
Maksymalna temperatura suszenia produktów spożywczych powinna być niższa od temperatury
denaturacji białek. Ważne znaczenie ma także szybkość nagrzewania produktu oraz czas suszenia w
dopuszczalnej temperaturze.
Dla produktów zawierających aktywne enzymy, od których zależy ich aktywność biologiczna,
istnieje minimalna granica obniżenia wilgoci do około 5%, przy niższej zawartości wody mogą
występować w produkcie suszonym nieodwracalne zmiany. Podczas suszenia większości
produktów spożywczych niedopuszczalne są zmiany ich zabarwienia.
W doborze odpowiedniej metody suszenia produktów spożywczych zwraca się uwagę warunki
procesu: temperaturę, wilgotność, prędkość przepływu powietrza, jak również na właściwości
suszonych produktów i zdolności dyfuzji wody oraz przenikania masy. Przy wyborze warunków
suszenia uwzględnia się nie tylko odporność termiczną produktu, jego cechy biologiczne, lecz także
właściwości strukturalno-mechaniczne, od których zależy zachowanie kształtu i wytrzymałości
produktu.
Podsumowując należy stwierdzić, że wybór odpowiedniej metody suszenia oraz konstrukcja
suszarki powinny być uzależnione od właściwości suszonego produktu.
Ogólnie suszenie żywności można podzielić na naturalne (słoneczno-powietrzne i wietrzno-
powietrzne) oraz suszenie sztuczne. Do konwecjonalnych sposobów suszenia przyjęto zaliczać
suszenie kondukcyjne i konwekcyjne.
Suszenie kondukcyjne polega na odprowadzeniu wody z produktu poprzez przekazanie kontaktowe
ciepła z ogrzanego wewnętrznie materiału.
Suszenie konwekcyjne odbywa się za pomocą owiewu suszonego produktu gorącym powietrzem
(lub innym gazem). W procesie suszenia konwekcyjnego ważną rolę odgrywa przenoszenie ciepła i
masy między czynnikiem suszącymi a suszonym produktem oraz przenoszenie ciepła i wilgoci
wewnątrz produktu.
Przebieg suszenia konwekcyjnego przedstawia się następująco:
•
przejmowanie ciepła od czynnika suszącego przez suszony produkt,
•
zamiany wody znajdującej się w produkcie na parę dzięki ciepłu przejętemu od czynnika
suszącego,
•
przejmowanie wody (w postaci pary) od ciała stałego przez czynnik suszący,
•
przemieszczanie się wody wewnątrz suszonego produktu od jego wnętrza w kierunku
powierzchni.
Proces suszenia jest więc procesem wymiany ciepła i masy oraz odbywającej się przemiany
fazowej (parowanie wody). Suszenie konwekcyjne jest procesem niestacjonarnym, to znaczy, że
zarówno temperatura produktu suszonego, jak i zawartość wody w nim ulegają ciągłej zmianie w
czasie trwania procesu.
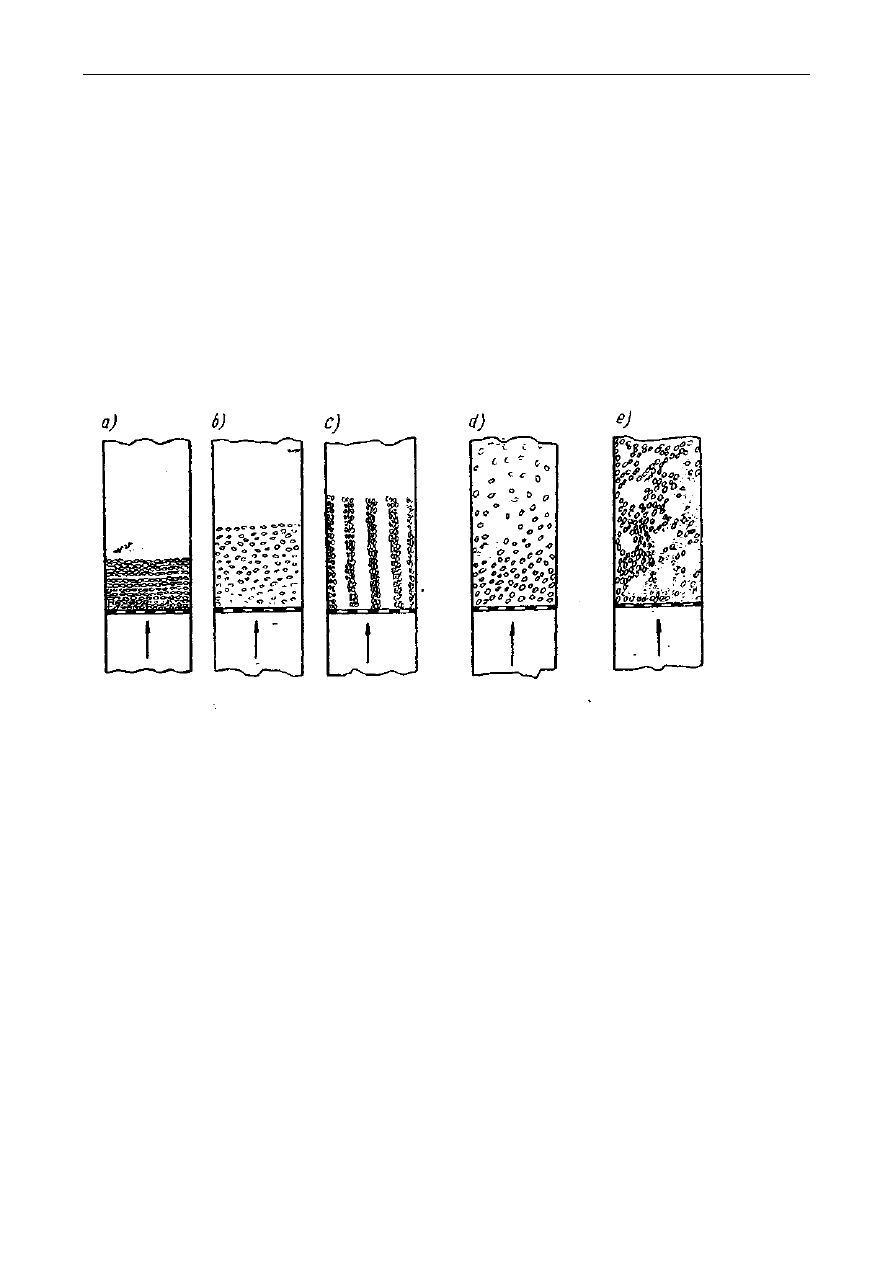
S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
Urządzenia do realizacji suszenia produktów nazwano suszarkami. Są to suszarnie otwarte,
komorowe, tunelowe, karuzelowe, taśmowe, bębnowe, walcowe, rozpyłowe, fluidyzacyjne,
próżniowe i sublimacyjne.
Suszenie fluidyzacyjne
Przy suszeniu rozdrobnionych materiałów w nieruchomej warstwie intensywność procesu jest
określana, przede wszystkim, przez zewnętrzne przenoszenie ciepła między materiałem i
czynnikiem suszącym, unoszącym parę wodną z powierzchni między ziarnami.
W ostatnich latach szerokie zastosowanie znalazło suszenie rozdrobnionych materiałów w stanie
fluidalnym. Zasada suszenia fluidyzacyjnego polega na oddolnym przepuszczeniu przez sypki,
ziarnisty materiał strumienia powietrza ogrzanego (suchego) o takiej prędkości, że cała masa
ziarnista zostaje uniesiona; tworzy stan „półzawieszony" czyli fluidalny, w którym suszony materiał
zachowuje stałą swobodę ruchów. W celu lepszego zrozumienia istoty procesu fluidyzacji na ry-
sunku 3 zobrazowano zmiany zachodzące w strukturze ładunku ziarna w zależności od prędkości
strumienia powietrza.
Rys. 3. Charakter zmian struktury ładunku ziarna w zależności od prędkości strumienia powietrza
V: a-ładunek nieruchomy, b-ładunek spulchniony (ekspandowany), c-początek fluidyzacji
(przepływ powietrza kanałami), d-pierwsze stadium fluidyzacji, e-stadium intensywnej
fluidyzacji („burzliwego wrzenia")
Jeżeli przez warstwę składającą się z ciała stałego w postaci ziarnistej, umieszczoną w cylindrze
z dnem dziurkowanym, przepuszcza się z określoną prędkością powietrze, wówczas sypki ładunek
ulega najpierw spulchnieniu, a następnie przy zwiększaniu prędkości przepływu powietrza,
przechodzi w etan przypominający wrzącą ciecz. Zjawisko upłynniania materiału suszonego
nazywamy fluidyzacją. W stanie fluidalnym materiał suszony podlega intensywnemu mieszaniu, w
następstwie czego wszystkie cząstki materiału są owiewane czynnikiem suszącym. Intensywne
mieszanie się oraz kontakt poszczególnych cząstek z czynnikiem suszącym warunkują
wyrównywanie temperatury w całej masie suszonego materiału, co jest szczególnie ważne podczas
suszenia produktów spożywczych. Istotną zaletą suszenia produktów spożywczych metodą
fluidyzacyjną jest fakt, że załadunek materiału może osiągać 100-120 kg/m
2
. Odpowiednio
załadunek surowca przy suszeniu w nieruchomej warstwie z zastosowaniem suszarek taśmowych,
tunelowych, szafkowych wynosi od 5 do 17 kg/m
2
.
Przenoszenie ciepła w suszarce fluidyzacyjnej jest uwarunkowane nie tylko intensywnym
ruchem cząstek i mieszaniem ładunku, lecz również wysoką zdolnością akumulowania ciepła przez
fazę stałą w porównaniu z tą zdolnością fazy gazowej. Pakt ten umożliwia łatwą regulację
temperatury suszonego materiału. Przy suszeniu produktów spożywczych metodą fluidyzacji
powstaje również wiele trudności spowodowanych z jednej strony szybkością procesu, z drugiej

S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
12 luty 2010
46
strony specyficznymi właściwościami produktów spożywczych. Trudność termicznej obróbki
produktów spożywczych w fazie fluidalnej polega głównie na tym, że intensyfikacja procesu jest
ograniczona zbyt szybkim nagrzewaniem materiału do temperatury granicznie dopuszczalnej.
Ze względu na specyficzne właściwości produktów spożywczych, prowadzone są badania w
celu określenia dopuszczalnych temperatur nagrzewania materiału, sposobu doprowadzenia ciepła,
parametrów hydrodynamicznych oraz konstrukcji suszarki.
Suszarki fluidyzacyjne możemy podzielić następująco:
a/ suszarki o działaniu periodycznym, w których załadunek materiału prowadzony jest okresowo, a
po zakończeniu każdego cyklu pracy suszarka jest całkowicie wyładowywana;
b/ suszarki o działaniu półciągłym - załadunek i wyładunek materiału jest prowadzony w sposób
ciągły,
c/ suszarki o działaniu ciągłym - załadunek i wyładunek materiału przebiegają w sposób ciągły; w
każdym miejscu aparatu wilgotność materiału oraz parametry czynnika suszącego pozostają stałe
podczas całego procesu.
Duże zastosowanie w suszeniu produktów spożywczych znajdują suszarki o działaniu ciągłym przy
zachowaniu oscylujących warunków suszenia. Oscylujące warunki pracy suszarki polegają na
podawaniu na przemian nagrzanego i chłodnego powietrza do odpowiednich stref suszarki. Czas
trwania poszczególnych cykli nagrzewania i chłodzenia materiału określa się czasem jego
przebywania w każdej strefie.
W ostatnich latach do suszenia produktów spożywczych stosuje się tzw. wibrofluidyzację.
Wibrofluidyzacja różni się od aerodynamicznej zasady zwykłej fluidyzacji tym, że potrzebne
spulchnienie i intensywne mieszanie ziarnistego materiału osiąga się zarówno kosztem
przedmuchiwania powietrza przez ładunek, jak również poprzez wibracyjne oddziaływanie na
materiał.
Suszenie promiennikowe w podczerwieni
Metoda ogrzewania za pomocą promieniowania podczerwonego została wykorzystana do
suszenia produktów spożywczych. Suszony materiał umieszcza się w suszarni (najczęściej
suszarnię stanowi tunel z ruchomą taśmą), w ścianach której umieszcza się promienniki pod-
czerwieni. Powierzchnia suszonego materiału szybko nagrzewa się przez napromieniowanie, w
wyniku czego w pobliżu powierzchni odbywa się szybkie suszenie materiału. Jednocześnie przez
suszarnie przepływa powietrze, do którego odparowuje woda z rozgrzanej powierzchni.
Ze względu na fakt, że absorpcja promieniowania cieplnego jest najbardziej intensywna blisko
powierzchni, metoda ta nadaje się głównie do suszenia cienkich warstw materiałów. Omawianą
metodę można również stosować do suszenia grubszych warstw takich materiałów, w których ruch
wilgoci w fazie stałej ułatwiony jest np. działaniem sił kapilarnych. Do suszenia w podczerwieni
natomiast nie nadają się materiały w postaci grubych warstw, głównie ze względu na szybkie
schnięcie miejsc w pobliżu powierzchni. Wówczas intensywne odparowanie wody z powierzchni
utrudnia dopływ wilgoci z wnętrza warstwy suszonego materiału. Duże gradienty stężeń wilgoci
powstające w czasie napromieniowania mogą powodować pękanie wrażliwych na skurcz
materiałów, np. ziarna jęczmienia, rzepaku itp.
W razie stwierdzenia pękania materiału należy zastosować przerywane naświetlanie materiału. W
czasie przerwy w naświetlaniu, ciepło przenika od powierzchni w głąb materiału i ułatwia dyfuzję
wody z wnętrza do powierzchni.
Omawiana metoda znalazła bardzo duże zastosowanie do dosuszania materiału suszonego innymi
metodami.
Ujemną stroną suszenia w podczerwieni jest znaczne zużycie energii-około 1 kWh/1kg
odparowanej wody. Zmniejszenie zużycia energii można osiągnąć przez przerywane naświetlanie
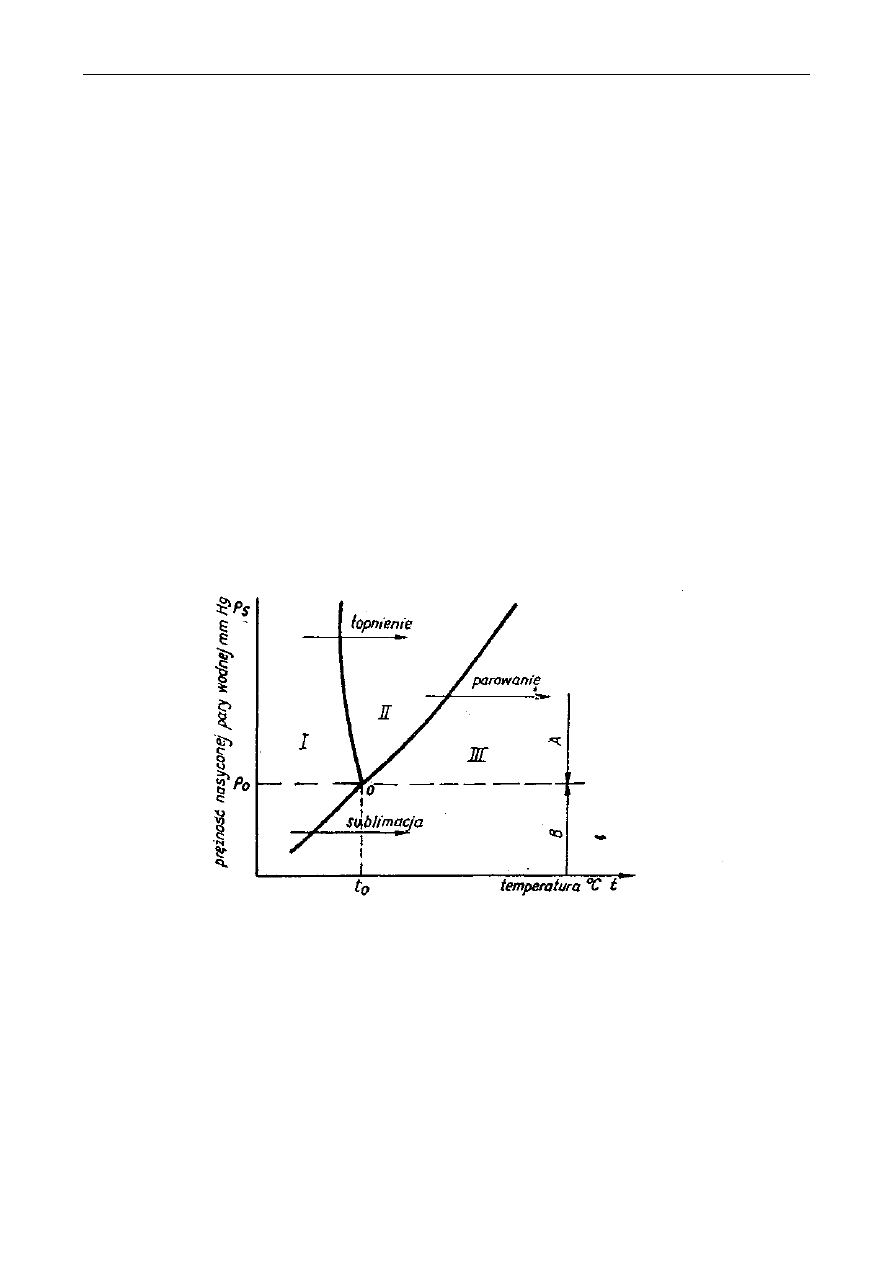
S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
produktu lub zastąpienie promienników zasilanych energią elektryczną na promienniki ogrzewane
gazem świetlnym.
Azeotropowe suszenie w rozpuszczalnikach
Zasada metody suszenia w rozpuszczalnikach polega na wykorzystaniu rozpuszczalników
organicznych, tworzących z wodą mieszaninę azeotropową. W suszeniu produktów spożywczych
jako rozpuszczalnik można stosować octan etylu. Mieszanina azeotropową octan etylu + woda
może być odparowana w wysokiej próżni w temperaturze pokojowej.
Proces suszenia przebiega w trzech etapach:
a/ mieszanina azeotropowa wrze w temp. 24°C i ciśnieniu 100 mm Hg;
b/ ciśnienie obniża się do 3 mm Hg i dalej prowadzi się odwadnianie;
c/ ciśnienie obniża się do 0,1 mm Hg w celu usunięcia śladowych ilości mieszaniny azeotropowej.
Omawiana metoda znalazła zastosowanie do suszenia warzyw i owoców. Koszt suszenia tą metodą
jest niższy w porównaniu z kosztem suszenia metodami konwencjonalnymi. Ostatnio wprowadza
się modyfikację suszenia azeotropowego między innymi w połączeniu z suszeniem sublimacyjnym.
Suszenie sublimacyjne
Przebieg procesu podobny jest do konwencjonalnego suszenia w próżni z tą jednak różnicą, że
woda z produktów usuwana jest na drodze przemiany fazy stałej w parę z pominięciem fazy ciekłej.
W celu zrozumienia istoty suszenia sublimacyjnego przedstawiono wykres równowagi faz dla wody
w układzie współrzędnych ciśnienia–temperatura (rysunek 4).
Rys. 4. Wykres równowagi faz dla wody: I-obszar stanu stałego (lód), II-obszar cieczy, III-obszar
pary
Krzywe odgraniczające trzy fazy skupienia wody: stałą, ciekłą i gazową nazywamy krzywymi
granicznymi. Miejsce przecięcia się krzywych granicznych nazywamy punktem potrójnym. W
punkcie potrójnym istnieje możliwość (przy odpowiednich wartościach ciśnienia i temperatury)
równoczesnego występowania wody w trzech stanach skupienia (stałym, ciekłym i gazowym).
Poniżej punktu potrójnego (pod ciśnieniem pary niższym od 4,60 mm Hg woda występuje tylko w
fazie stałej (lód) lub gazowej (para). Dla prawidłowego przebiegu procesu sublimacji konieczne jest
więc wytworzenie próżni, w komorze suszarniczej, w której umieszczony jest suszony produkt.
Podstawowy schemat urządzeń do suszenia sublimacyjnego przedstawiono na rysunku 5,
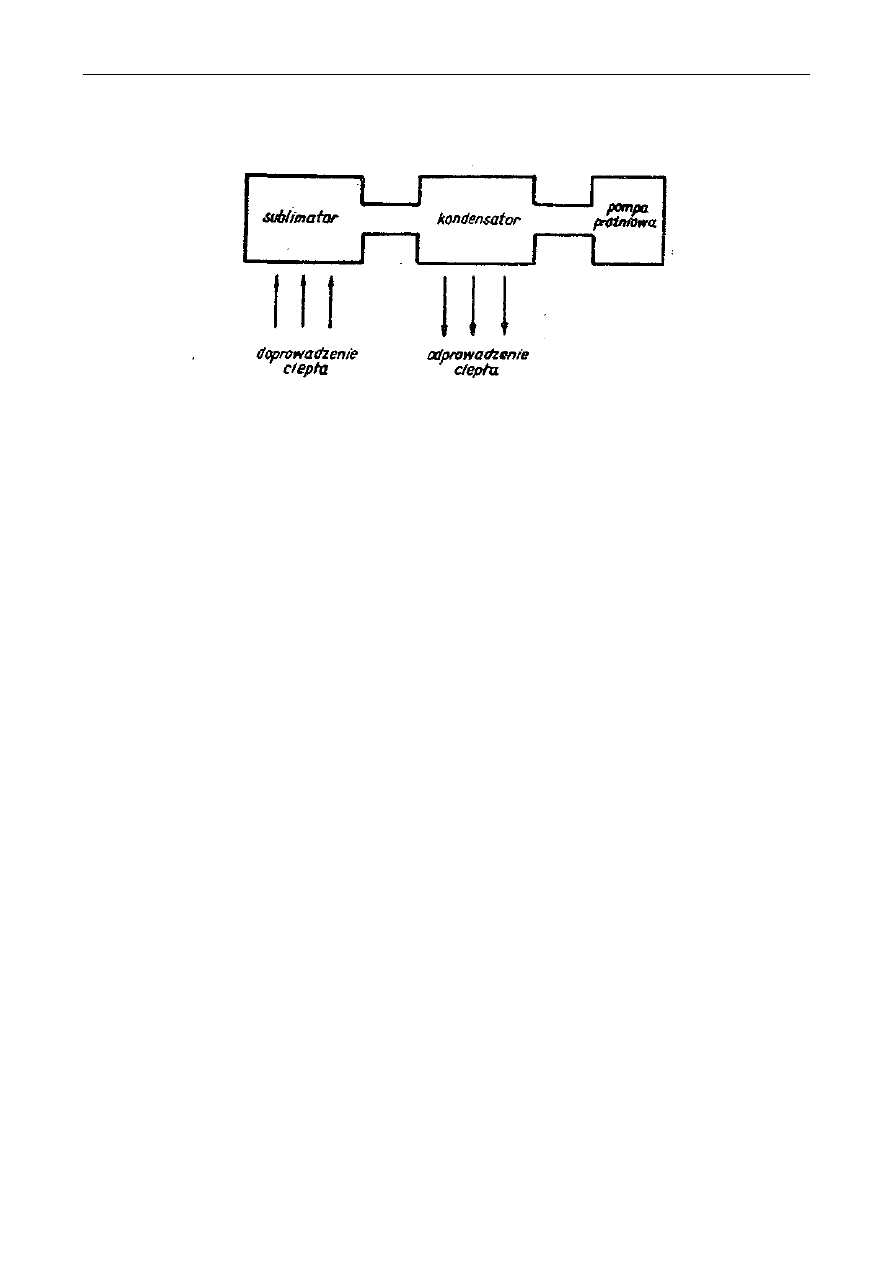
S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
12 luty 2010
48
Urządzenie składa się z: sublimatora, kondensatora i pompy próżniowej, połączonych w zamknięty
układ próżniowy.
Rys. 5. Podstawowy schemat urządzenia do suszenia sublimacyjnego
Wstępnym etapem suszenia sublimacyjnego jest zamrażanie produktów suszonych. Zamrożenie
można prowadzić w dwojaki sposób:
a/ stosując zamrażanie produktów kosztem intensywnego parowania części wody w następstwie
ciągłego zwiększenia próżni w sublimatorze; w zależności od postaci suszonego produktu
(produkt wstępnie zamrożony lub o dodatniej temperaturze) stosuje się odmienną technologię
suszenia;
b/ stosując wstępne zamrażanie produktów przed liofilizacją w specjalnych komorach pod
ciśnieniem atmosferycznym.
W suszeniu materiałów nie zamrożonych stosuje się bardzo niskie ciśnienie, już w pierwszym
etapie suszenia, w celu intensywnego odprowadzenia wilgoci z powierzchni materiału, a tym
samym do ochłodzenia produktu, a następnie zamrożenia oraz ochłodzenia produktu do ujemnej
temperatury. Obniżanie ciśnienia nad powierzchnią parowania powoduje zwiększenie
intensywności parowania. Intensywne parowanie prowadzi do spadku temperatury aż wreszcie
dochodzi do zamrożenia wody. Opisane zjawisko nazywamy samozamrożeniem.
Czas procesu zamrażania zależy od geometrycznych wymiarów próbki. Im mniejsza jest grubość
produktu i większy stosunek jego powierzchni do objętości, tym szybciej przebiega proces
zamrażania.
Wykazano, że samozamrażanie w sublimatorze jest niewskazane w suszeniu sublimacyjnym takich,
produktów spożywczych, jak: surowe mięso, ryby, soki owocowe, niektóre jagody i owoce.
Samozamrażanie prowadzi bowiem w tych wypadkach do znacznych zmian właściwości produktu.
W suszeniu produktów wstępnie zamrożonych proces odwadniania odbywa się z pominięciem
etapu samozamrażania. Podczas suszenia sublimacyjnego następuje intensywne odbieranie ciepła z
produktu, kosztem którego zachodzi proces odwadniania. Na początku procesu suszenia intensywna
sublimacja zachodzi w strefie bezpośrednio przylegającej do wolnej powierzchni produktu. W
miarę przebiegu procesu strefa sublimacji przesuwa się w głąb produktu, a tworząca się para przy
przenikaniu do wolnej powierzchni ponownie napotyka na opór już wysuszonej warstwy. Opory
przepływu są przede wszystkim zależne od takich czynników, jak: struktura suszonego produktu,
rodzaj i budowa kapilar, przez które musi przepłynąć para wodna wewnątrz materiału.
Ważnym zagadnieniem w procesie dehydratacji poprzez sublimację jest dostarczanie ciepła do
suszonego produktu w celu wyrównania jego strat w procesie parowania. Ciepło może być
doprowadzone do produktu przez przewodnictwo lub promieniowanie. Przy stosowaniu ogrzewania
istotne jest znalezienie optymalnej temperatury procesu. Proces ogrzewania winien być tak
prowadzony, aby maksimum ciepła doprowadzić we wczesnych stadiach, tzn. wtedy, gdy materiał
zawiera znaczne ilości wody. Podczas suszenia należy zwracać szczególną uwagę na równomierne
nagrzewanie całej powierzchni parowania. Osobnym zagadnieniem w procesie suszenia

S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
sublimacyjnego jest usuwanie pary wodnej z sublimatora. Usuwanie pary wodnej może odbywać
się:
a/ przez kondensację w chłodnicy w niskiej temperaturze, niższej od temperatury produktu
suszonego;
b/ przez adsorpcję wilgoci materiałem pochłaniającym, np. siarczanem wapniowym, żelem
krzemionkowym itp.;
c/ przez zastosowanie pomp inżektorowych do szybkiego usuwania dużych objętości pary pod
niskim ciśnieniem.
Należy podkreślić, że szybkość dehydratacji produktów spożywczych poprzez sublimację zależy od
wielkości ciśnienia mieszaniny pary wodnej i gazów w sublimatorze, temperatury produktu,
powierzchni produktu.
S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
12 luty 2010
50
6.2.
W
YKONANIE ĆWICZENIA
Celem ćwiczenia jest wyznaczenie wilgotności wybranych surowców rolniczych oraz szybkości
ich suszenia.
Wykonanie oznaczeń.
Zakres
1. Oznaczanie względnej wilgotności początkowej surowca.
2. Wyznaczanie szybkości suszenia.
3. Oznaczanie względnej wilgotności końcowej suszonego produktu.
Szczegółowy plan ćwiczenia, prowadzonego z wykorzystaniem wagosuszarki przedstawi
prowadzący ćwiczenie.
UWAGA!
Maksymalne obciążenie szalki wagosuszarki wynosi 50g
– przekroczenie tej wartości może doprowadzić do uszkodzenia urządzenia
Elementy wagosuszarki:
S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
Klawiatura wagosuszarki
Przycisk Start/Stop – rozpocz
ę
cie / zako
ń
czenie procesu suszenia wg wybranego programu.
Przycisk Esc, słu
ż
acy do rezygnacji z wprowadzanych zmian / wyj
ś
cie o poziom wy
ż
ej w menu wagi.
Grupa przycisków nawigacyjnych - zmiana warto
ś
ci parametrów; poruszanie si
ę
po menu
wagosuszarki.
Przycisk Print/Enter – przesyłanie stanu wy
ś
wietlacza do urz
ą
dzenia zewn
ę
trznego (Print) lub zatwierdzenie
wybranej warto
ś
ci parametru lub funkcji (Enter).
Przycisk TARA - zerowanie wskaza
ń
wagi.
Przycisk On/Off, słu
ż
acy do zał
ą
czenia / wył
ą
czenia wy
ś
wietlacza wagosuszarki. Po wył
ą
czeniu wy
ś
wietlacza
inne podzespoły s
ą
zasilane, a wagosuszarka pozostaje w stanie gotowo
ś
ci.
Przycisk Display - zmienia rodzaj danych eksponowanych w trakcie i po procesie suszenia.
Przycisk Setup - przycisk słu
żą
cy do wej
ś
cia w menu główne.
Przycisk Test menu - przycisk słu
żą
cy do uruchomienia wyboru programów suszenia.
Proces suszenia:
1. Włączyć wagosuszarkę
2. Ustawić odpowiedni program suszenia (wskazuje prowadzący ćwiczenia)

S
USZENIE PRODUKTÓW SPO
ś
YWCZYCH
12 luty 2010
52
3. Nacisnąć przycisk START
4. Umieścić czystą szalkę na uchwycie szalki, zamknąć komorę suszenia
5. Nacisnąć przycisk TARA
6. Umieścić badaną próbkę na szalce, zamknąć komorę suszenia
7. Proces suszenia rozpocznie się i zakończy automatycznie
Wagosusarka zakończy prace jeśli w czasie 240s zmian masy suszonej próbki będzie mniejsza
niż 1 mg.
8 W czasie suszenia, naciskając przycisk Display można dokonywać zmiany rodzaju wyświetlanych
danych
6.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń należy umieścić w sprawozdaniu [wzór, załącznik 1 na stronie 121] w formie
oddzielnych punktów wraz z przeliczeniami i przedłożyć osobie prowadzącej ćwiczenie w celu
sprawdzenia. Należy także zaznaczyć wszelkie odstępstwa od metod podanych w opracowaniu
ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski w formie zwięzłych punktów.
6.4.
S
PRZĘT I ODCZYNNIKI
1.
ziarno zbożowe wstępnie nawilgocone 1 kg
2.
suszarka do suszenia w przepływie powietrza
3.
pojemniki z perforowanym dnem (sitka) 4 sztuki
4.
waga analityczna
5.
naczynka wagowe duże 4 sztuki

M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
7.
MIKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
śYWNOŚCI
7.1.
W
PROWADZENIE
W ostatnich latach obserwuje się tendencję do poszukiwania nowych metod przetwarzania
żywności. Współczesny człowiek poświęcający coraz więcej czasu pracy zawodowej pragnie do
minimum skrócić czas potrzebny do sporządzania posiłków. Nauka i technika starają się
zadośćuczynić temu wyzwaniu. Jedną z propozycji usprawnienia obróbki kulinarnej w warunkach
domowych stały się powszechnie już dzisiaj stosowane kuchenki mikrofalowe. Znajdują one
wszechstronne zastosowanie do gotowania, podgrzewania, pieczenia, suszenia i rozmrażania. Na
skalę przemysłową energię mikrofalową zaczęto stosować niedawno, gdyż wymagało to
wprowadzenia niezbędnych do generowania mikrofal magnetronów o dużej mocy-urządzeń
całkiem nowego typu. Nie znano również dobrze właściwości dielektrycznych żywności i obawiano
się wysokich kosztów wytwarzania mikrofal. Dopiero wzrost cen paliw tradycyjnych pomógł
dostrzec zalety tej metody ogrzewania. Wykazano, że stosowanie mikrofal jest bardzo ekonomiczne
w przypadku temperyzacji mrożonej żywności. Jest to operacja, która polega na doprowadzeniu
głęboko zamrożonego produktu do temperatury ok. -3°C, kiedy daje się on już bez trudu kroić,
odkostniać i rozdrabniać. Inne etapy obróbki technologicznej, w których w większym lub
mniejszym zakresie wykorzystuje się ogrzewanie mikrofalowe, to wstępne podgotowywanie,
gotowanie, pieczenie, suszenie, pasteryzacja, sterylizacja, blanszowanie i rozmrażanie. Czasami
stosuje się układy kombinowane wraz z ogrzewaniem konwencjonalnym, co ma na celu osiągnięcie
pożądanych cech organoleptycznych i poprawę jakości mikrobiologicznej.
Pomimo wielu zalet ogrzewanie mikrofalowe nie zawsze spełnia oczekiwania technologów,
dlatego ciągle prowadzi się badania nad optymalizacją wykorzystania tej formy energii w
przemyśle spożywczym.
Stosowanie promieniowania podczerwonego do przetwarzania żywności ma znacznie dłuższą
historię. Od wieków ludzie wykorzystywali energię słoneczna do suszenia ryb, mięsa i owoców, a
wiadomo, że ok. 48% tej energii przypada na zakres podczerwieni. Obecnie podczerwień stosuje się
na skalę przemysłową w technologii żywności do suszenia produktów o małej zawartości wilgoci,
takich jak: ziarno, mąka, słód, makarony i herbata, do pieczenia oraz ogrzewania opakowaniowej
folii termokurczliwej. Ogrzewanie podczerwienią stosuje się też w kombinacji z mikrofalowym, co
w pewnych przypadkach daje bardzo dobre rezultaty.
Właściwości mikrofal i podczerwieni
Mikrofale i podczerwień są formami energii elektromagnetycznej (rys. 1).
Rys. 1. Umiejscowienie
mikrofal
i
podczerwieni
(IR)
w
spektrum
promieniowania
elektromagnetycznego

M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
54
Rozchodzą się one w postaci fal, które w zetknięciu z żywnością wykazują zdolność jej ogrzewania.
Główne różnice między mikrofalami i podczerwienią są następujące:
•
dla celów przemysłu spożywczego generuje się mikrofale tylko o określonych
częstotliwościach (najczęściej 2450 i 915 MHz), aby wyeliminować możliwość zakłócania
innych urządzeń pracujących w tym zakresie widma elektromagnetycznego (np. urządzenia
radiolokacyjne, diatermia mikrofalowa). Promieniowanie podczerwone natomiast wytwarza
się bez żadnych ograniczeń dotyczących częstotliwości.
•
głębokość penetrowania żywności przez mikrofale ściśle wiąże się z częstotliwością - im
niższa częstotliwość, tym głębsza penetracja. Podczerwień natomiast jest pochłaniana na
powierzchni produktu.
•
mikrofale wywołują rotacje cząsteczek wody i tarcie molekularne między nimi, na skutek
czego wydziela się ciepło, natomiast podczerwień jest bez pośrednio absorbowana i
przetwarzana na ciepło.
•
efekt ogrzewania mikrofalowego jest uzależniony od zawartości wilgoci, a w przypadku
ogrzewania przez promienie podczerwone zależy od charakteru powierzchni i jej koloru.
•
powierzchniowe pochłanianie promieniowania podczerwonego sprawia,
•
że przy tej metodzie ogrzewania termiczne przewodnictwo żywności jest czynnikiem dużo
bardziej istotnym niż przy ogrzewaniu mikrofalowym. Podczerwień nagrzewa tylko
powierzchnię produktu, a głębiej ciepło przekazywane jest przez przewodnictwo i/lub
konwekcję, podczas gdy mikrofale wnikają znacznie głębiej i tam ogrzewają produkt.
•
mikrofale stosuje się m.in. do przedłużania trwałości żywności (np. suszenie, blanszowanie,
pasteryzacja), a podczerwień zwykle do zmiany właściwości organoleptycznych, takich jak
kolor powierzchni, smak, aromat. Mikrofale wytwarza się w urządzeniach zwanych
magnetronami, które przemieniają energię elektryczną o niskich częstotliwościach (50, 60 Hz)
w pole elektromagnetyczne o częstotliwości rzędu miliardów Hz. Jest to cylindryczna dioda,
w której katodą jest gorący, metalowy walec wytwarzający wolne elektrony, umieszczony w
pierścieniowej anodzie stanowiącej rezonator wnękowy. Po przyłożeniu wysokiego napięcia
elektrony tracą energię i generują szybko oscylujące pole mikrofalowe, które dalej jest
kierowane przez elektromagnesy do kanału wprowadzającego je do komory grzewczej. W
celu wyeliminowania nierównomierności w natężeniu mikrofal w komorze stosuje się tzw.
mieszadła mikrofal (wirujące anteny) i/lub produkt wprowadza się w ruch na taśmach lub
specjalnych obrotowych talerzach. O zachowaniu się produktu w polu elektromagnetycznym
decyduje jego przenikalność elektryczna, zwana też stałą dielektryczną, i ściśle z nią
związany współczynnik strat dielektrycznych, które zależne są od składu. Po wniknięciu
mikrofal do produktu oddziaływają one z dipolami wody, co powoduje ich reorientacje,
rozrywanie wiązań wodorowych między sąsiednimi cząsteczkami i generowanie ciepła
poprzez tarcie molekularne. Jony zawarte w żywności (np. Na
+
, Cl
-
) również migrują w polu
mikrofalowym i przez to dodatkowo przyczyniają się do wytwarzania ciepła. Także i niektóre
inne niewodne składniki żywności o budowie polarnej mogą absorbować mikrofale, ale
znacznie słabiej od wody i dlatego ich efekt ogrzewający w produktach o dużej zawartości
wody jest pomijany.
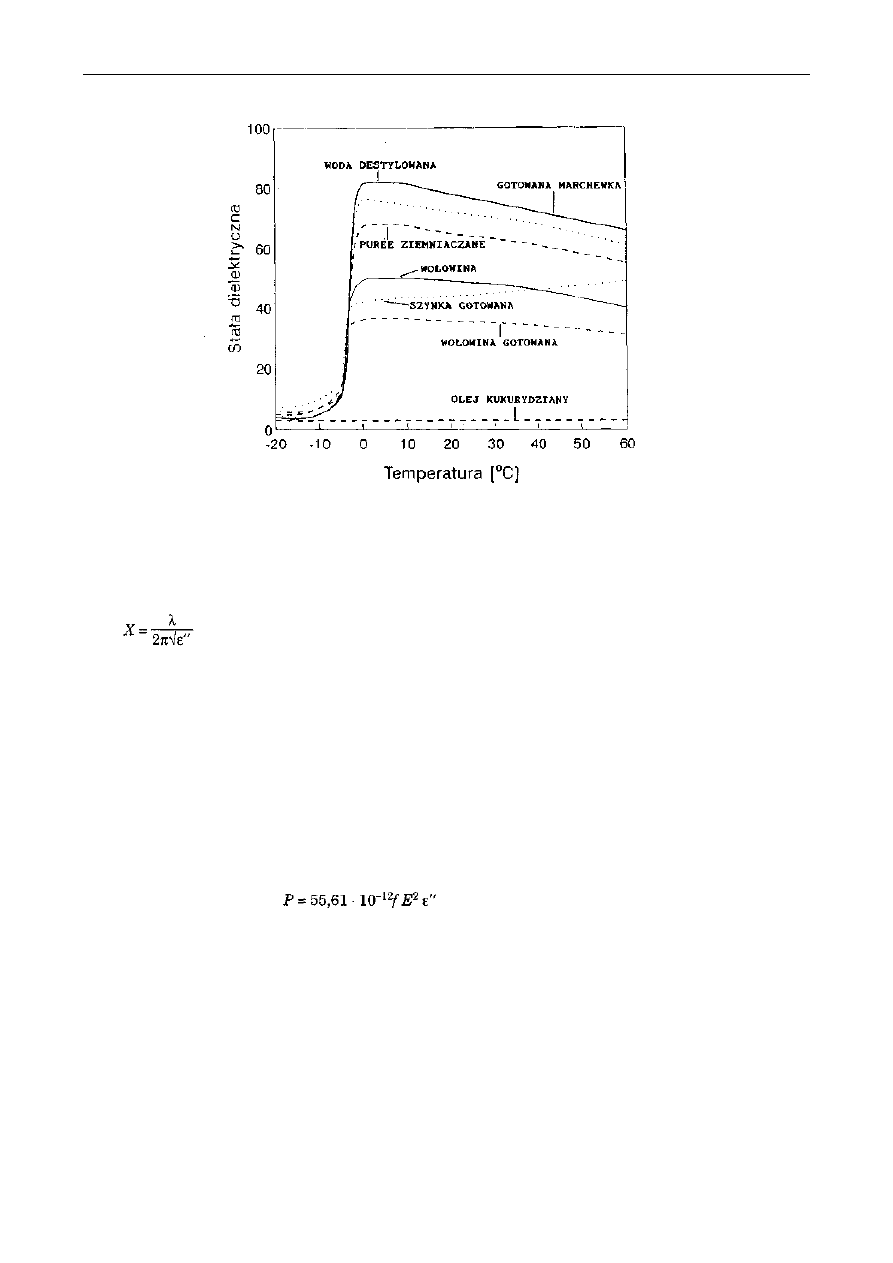
M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
Rys. 2. Zmiany stałej dielektrycznej niektórych produktów spożywczych w zależności od
temperatury.
Stan skupienia w sposób istotny wpływa na właściwości dielektryczne produktu. Na przykład
procesowi zamrażania towarzyszy bardzo wyraźne zmniejszanie się stałej dielektrycznej (rys. 2).
Umowna głębokość wnikania mikrofal do produktu wyraża się wzorem
gdzie: X - umowna głębokość wnikania mikrofal [m], A. — długość fali [m], ε" — współczynnik
strat dielektrycznych.
Tak więc lód, dla którego ε" jest mniejszy niż dla wody, jest bardziej „przezroczysty" dla
mikrofal i żywność mrożona przepuszcza je głębiej od niemrożonej. Znaczna część promieniowania
mikrofalowego jest jednak absorbowana i zamieniana na ciepło. Dlatego natężenie mikrofal maleje
w miarę penetracji przez nie produktu i dla warstw położonych głębiej niż umowna głębokość X
jest praktycznie zaniedbywanie.
Ilość zaabsorbowanej energii także zależy od współczynnika strat dielektrycznych i wyraża się
wzorem:
gdzie: P - moc absorbowana przez jednostkę objętości [W/m
3
], f-częstotliwość mikrofal [Hz], E-
natężenie pola elektrycznego [/m
3
], ε" - współczynnik strat dielektrycznych.
Produkty żywnościowe o dużej zawartości wody mają duży współczynnik ε"-absorbują one
łatwo mikrofale i w miejscu ich pochłaniania ogrzewają się szybko. Natomiast szkło, porcelana i
większość innych tworzyw opakowaniowych charakteryzują się niewielką wartością tego
współczynnika (są przezroczyste dla mikrofal) i dlatego nie ogrzewają się. Metale odbijają
mikrofale.
Głębsze wnikanie mikrofal do produktów, a więc i bardziej równomierne ich ogrzewanie, ma
miejsce w przypadku użycia mikrofal o większej długości fali (mniejsza częstotliwość) oraz gdy
produkty są małych rozmiarów i mają mniejszy współczynnik strat dielektrycznych. Tak więc
mikrofale o częstotliwości 915 MHz wnikają kilkakrotnie głębiej niż o f= 2450 MHz i przyjmuje
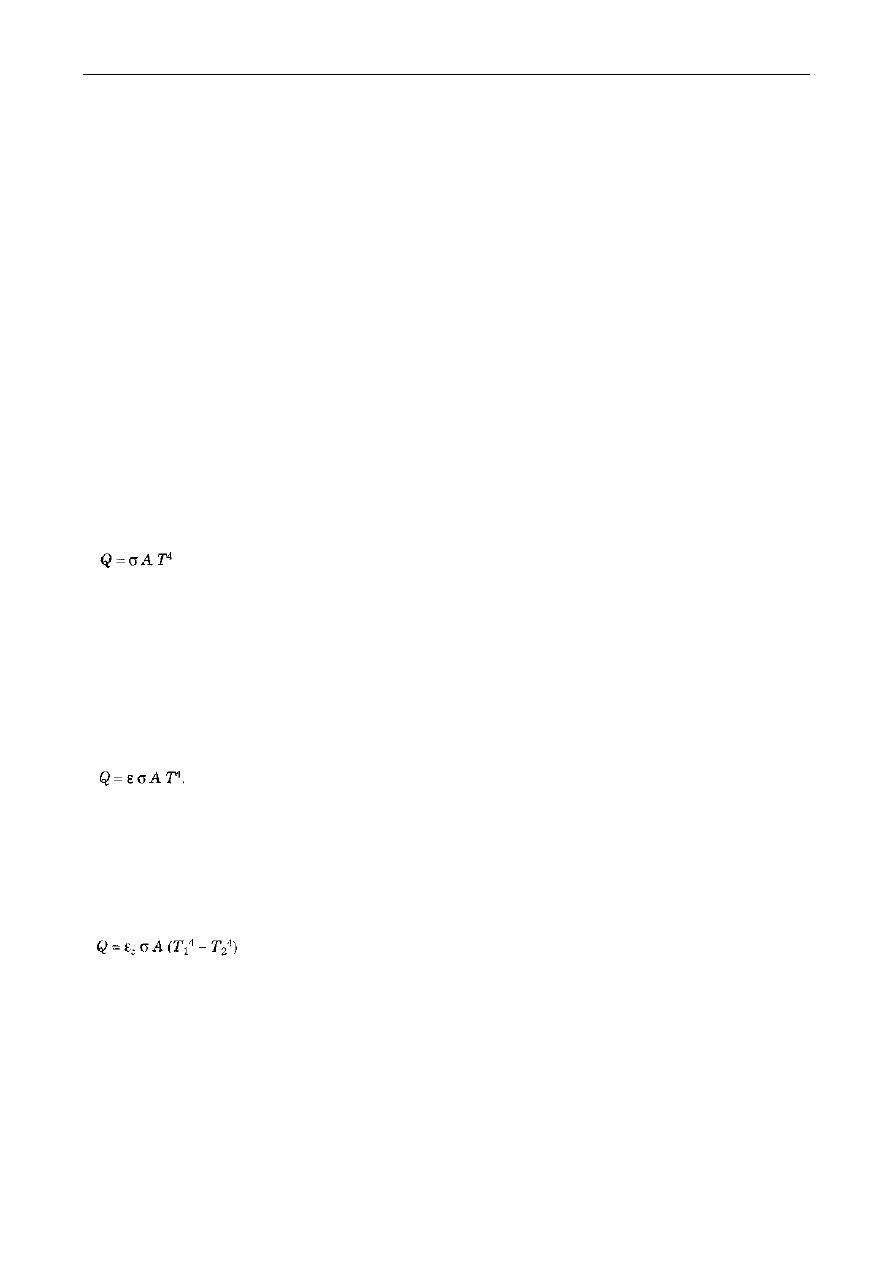
M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
56
się, że mogą dochodzić do głębokości od 10 do 30 cm. Grubość produktu należy dobrać
odpowiednio do możliwości penetrowania go przez mikrofale. Gdy jest on zbyt gruby, to na skutek
absorpcji przez warstwy zewnętrzne mikrofale praktycznie nie osiągają jego środka.
Wytworzone w wyniku działania mikrofal ciepło rozchodzi się dalej poprzez przewodnictwo.
Odbywa się to tym szybciej, im większa jest dyfuzyjność cieplna, czyli im większe jest
przewodnictwo, a mniejsza pojemność cieplna i gęstość produktu. śywność o małej zawartości
wilgoci charakteryzuje się właśnie takimi parametrami i dlatego ogrzewa się ona bardziej
równomiernie od żywności bogatej w wodę. Ponadto w tym ostatnim przypadku na skutek bardziej
intensywnego parowania wody następują ubytki ciepła na powierzchni. Stałe składniki żywności
prawie nie absorbują energii mikrofalowej w produktach o dużej i średniej zawartości wilgoci,
jednakże w żywności suchej jest inaczej-energia mikrofalowa może nawet doprowadzić do
zapalenia się jej. Sposób transformacji energii mikrofalowej w cieplną nie jest jeszcze w tych
przypadkach dobrze poznany.
Reasumując, szybkość i równomierność ogrzewania produktu zależy od jego składu,
temperatury, kształtu, struktury, rozmiarów oraz mocy i częstotliwości padających mikrofal.
Promieniowanie podczerwone jest formą energii elektromagnetycznej emitowanej przez
obiekty gorące. Podczas absorpcji na powierzchni materiału ogrzewanego traci ono swoją energię.
Szybkość ogrzewania tą metodą zależy od temperatury ciała emitującego i absorbującego
podczerwień oraz od kształtu i właściwości powierzchni obu tych ciał.
Równanie Stefana-Boltzmanna określa ilość ciepła emitowanego przez ciało doskonale czarne:
gdzie: Q — ilość energii wyemitowana w jednostce czasu [J/s], σ = 5,710
-8
[J/s·m
2
·K
4
]-stała
Stefana-Boltzmanna, A - czynna powierzchnia emitera [m
2
], T-temperatura absolutna [K].
Powyższe równanie pozwala także wyliczyć ilość ciepła absorbowanego przez ciało doskonałe
czarne. Jednakże ani używane w praktyce źródła podczerwieni nie są idealnymi emiterami, ani też
żywność nie jest idealnym absorberem, w związku z tym tylko część energii określonej powyższym
równaniem jest przenoszona. W celu uwzględnienia tego odstępstwa od stanu idealnego wprowadza
się tzw. współczynnik emisji ε i równy mu liczbowo współczynnik absorpcji, które przyjmują
wartości od 0 do 1. Tak zmodyfikowane równanie Stefana-Boltzmanna przyjmuje postać:
Promieniowanie, które nie zostaje zaabsorbowane, ulega odbiciu. Ilość energii zaabsorbowanej,
a więc i stopień ogrzania, zależy od składu żywności, bowiem różne jej składniki absorbują
podczerwień w różnym stopniu, oraz od długości fali padającego promieniowania. Długość fali z
kolei zależna jest od temperatury źródła-im jest ona wyższa, tym maksimum emitowanej energii
przesuwa się w kierunku fal krótszych. Ilość ciepła netto przekazana do żywności równa jest
różnicy energii zaabsorbowanej i wyemitowanej przez nią i wyraża się wzorem:
gdzie: ε
z
— emisyjność zastępcza, zależna od emisyjności obu ciał i ich geometrii, T
1
- temperatura
emitera [K], T
2
— temperatura żywności absorbującej podczerwień [K].
Źródła promieniowania podczerwonego stosowane w przemyśle ta płaskie i rurowe grzejniki z
metalowym elementem grzejnym, grzejniki ceramiczne oraz urządzenia halogenowe wyposażone w
kwarcowe rury wypełnione żarnikami elektrycznymi. W gastronomii często stosuje się
zminiaturyzowane promienniki podczerwieni o mocy kilkuset W.

M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
Wpływ mikrofal na drobnoustroje
Uważa się powszechnie, że energia mikrofal niszczy drobnoustroje jedynie na skutek
wywoływanego przez nią wzrostu temperatury. Zostało to stwierdzone na podstawie badań
przeżywalności komórek wegetatywnych i spor poddawanych działaniu mikrofal i ogrzewaniu
konwencjonalnemu w tym samym zakresie temperatur. Najnowsze badania wskazują jednak na
możliwość istnienia pewnych efektów atermicznych. Przedmiotem obserwacji były wegetatywne
komórki Staphylococcus aureus, Escherichia coli, spory Bacillus stearothermophilus, a także
kolonie Aspergillus niger. Odnotowano większe uszkodzenia komórek poddawanych działaniu
mikrofal, wyższy poziom białek w uwalnianych płynach wewnątrzkomórkowych, stwierdzono
także wpływ mikrofal na aktywność enzymów. Zmian tych nie udało się wyjaśnić jedynie efektem
działania temperatury. Z drugiej strony istnieją publikacje dokumentujące większą przeżywalność
drobnoustrojów na powierzchni produktów ogrzewanych mikrofalowo w porównaniu z
ogrzewaniem konwencjonalnym. Na nieopakowanym mięsie wieprzowym ogrzewanym
mikrofalami do osiągnięcia temperatury 77°C w środku termicznym stwierdzono przeżywanie
włośni (Trichinelta spiralis), podczas gdy już po dwuminutowym tradycyjnym ogrzewaniu do 60°C
były one niszczone. Przypuszcza się, że zjawisko to spowodowane było obniżeniem temperatury
powierzchni na skutek pobierania z niej ciepła parowania. Natomiast przy ogrzewaniu
konwencjonalnym, gdzie ciepło w głąb produktu transportowane jest przez powierzchnię, jej
temperatura przez cały czas pozostaje wysoka. Operacja ogrzewania mikrofalowego trwa zwykle
krócej niż przy wykorzystaniu metod tradycyjnych, a domowe kuchenki mikrofalowe często
wykorzystuje się do odgrzewania potraw. Aby wyeliminować możliwość niedogrzania powierzchni
na skutek pobierania z niej ciepła parowania, wskazane jest nieco dłuższe ogrzewanie produktów w
opakowaniach lub równoczesne stosowanie ogrzewania konwencjonalnymi elementami grzejnymi,
w które coraz częściej wyposaża się nowoczesne kuchenki mikrofalowe. W przypadku
przygotowywania do spożycia mrożonych lub chłodzonych dań gotowych (tzw. żywność wygodna)
ze względów bezpieczeństwa mikrobiologicznego wymagane jest utrzymywanie temperatury
powyżej 70°C przez co najmniej 2 minuty w każdym miejscu produktu. Użytkownik kuchenki
mikrofalowej powinien to osiągnąć poprzez ustawienie mocy i czasu ogrzewania zgodnie z
instrukcją umieszczoną na opakowaniu zakupionego przez siebie wyrobu.
Bezpieczeństwo mikrofal dla człowieka
Pomimo spotykanych w literaturze przedmiotu doniesień biologów informujących o tym, że
mikrofale, podobnie jak promieniowanie jonizujące, wywoływać mogą efekty atermiczne, takie jak
interakcje z udziałem DNA i białek, powszechnie uważa się, że oddziaływają one na organizmy
wyższe głównie poprzez efekt temperaturowy. Energia kwantowa mikrofal jest bowiem znacznie
mniejsza od energii wymaganej do rozrywania kowalencyjnych wiązań chemicznych. Dlatego
możliwość zachodzenia wywoływanych przez mikrofale reakcji, które mogłyby prowadzić do
tworzenia się w żywności produktów toksycznych, jest bardzo mało prawdopodobna.
Bezpośrednie działanie na człowieka mikrofal o dużej mocy objawia się hipertermią. Ponieważ
absorpcja energii mikrofalowej w środowisku zależy od stałej dielektrycznej i współczynnika strat
dielektrycznych, to te tkanki i organy, które zawierają dużo wilgoci i soli, ogrzewają się najsilniej i
pierwsze ulegają zniszczeniu. Szczególnie jest to widoczne w organach, w których ze względu na
ich budowę cyrkulacja płynów ustrojowych jest ograniczona (oczy, uszy, jądra). W badaniach na
zwierzętach wykazano, że na skutek działania mikrofal najwcześniej następuje denaturacja białek
soczewek ocznych, powstawanie katarakty, uszkodzenie słuchu i sterylizacja spermy.
W medycynie, w diatermii mikrofalowej, bez żadnych ubocznych efektów szkodliwych
rutynowo wykorzystuje się mikrofale o mocy do l W/cm
2
. Ekspozycja całego ciała człowieka w
polu mikrofalowym o mocy 10 mW/cm
2
jest bezpieczna przez nieograniczony czas. Jednak
wprowadzono dodatkowy margines bezpieczeństwa, ograniczając w normach „wyciek" mikrofal w
czasie eksploatacji urządzeń domowych i przemysłowych do 5 mW/cm
2
w odległości 5cm od ich

M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
58
powierzchni. Konstrukcja aparatury mikrofalowej zawiera podwójne zabezpieczenia i w razie
niewłaściwej obsługi magnetron wyłącza się.
Zastosowanie mikrofal w technologii żywności
W ostatnich latach stosowanie urządzeń mikrofalowych w przemyśle staje się bardziej
opłacalne, gdyż koszty kapitałowe ulegają zmniejszeniu dzięki rosnącej produkcji magnetronów o
budowie modułowej i wysokiej niezawodności. Postępy w konstruowaniu wyposażenia
mikrofalowego, trendy w kształtowaniu się cen energii elektrycznej w stosunku do innych jej form
oraz coraz lepsza znajomość właściwości dielektrycznych żywności pozwalają na takie
modelowanie procesów mikrofalowych, aby mogły być one zastosowane w przemyśle spożywczym
w coraz większym zakresie. Większość urządzeń mikrofalowych przypada na temperyzację mięsa i
ryb, suszenie produktów o niskiej zawartości wilgoci oraz podgotowywanie i gotowanie mięsa. Inne
operacje z udziałem mikrofal- suszenie próżniowe, liofilizacja, pasteryzacja, sterylizacja, pieczenie,
blanszowanie, wytapianie tłuszczu są coraz częściej wdrażane. Wiele z powyższych procesów łączy
ogrzewanie mikrofalowe z tradycyjnym. W takich przypadkach konwencjonalne źródło ciepła służy
do wytwarzania pożądanego czasem zbrązowienia oraz chrupkości powierzchni, a także do
szybszego zniszczenia obecnych na niej drobnoustrojów.
Instalacje mikrofalowe z reguły wykorzystują częstotliwość 2450 lub 915 MHz i mają moc od
30 do 120 kW. Przy niższej z tych częstotliwości penetracja mikrofal sięga do głębokości ok. 10-30
cm, podczas gdy przy wyższej tylko do ok. 3 cm. W zależności od charakteru surowca
poddawanego obróbce i jego ilości stosuje się odpowiednią częstotliwość i moc. Ogrzewanie
omawianą tu metodą jest szybkie i nie powoduje przegrzewania się powierzchni, co mogłoby
prowadzić do jej uszkodzeń. Instalacje są małe, zwarte i łatwe do montażu, a produkt przez cały
czas przebywa w warunkach higienicznych.
Poniżej omówiono krótko najważniejsze aspekty zastosowania mikrofal w niektórych procesach
technologicznych.
Temperyzacja
Temperyzacja mrożonej żywności stosowana jest często w warunkach przemysłowych zamiast
całkowitego rozmrażania. W urządzeniu mikrofalowym przystosowanym do tego celu może być
wykonana w czasie od kilku do kilkudziesięciu minut w porównaniu z wieloma godzinami, jakie
wymagane są do rozmrażania dużych elementów w tradycyjnej rozmrażalni. Z tego względu oraz z
uwagi na to, że temperyzację mikrofalową prowadzić można bez usuwania opakowań, znacznie
mniejsze są możliwości powierzchniowego rozwoju mikroorganizmów. Do zalet należy zaliczyć
także ograniczenie strat wagowych, dużą retencję soków komórkowych, utrzymywanie pH mięsa
na właściwym poziomie i większą elastyczność procesu produkcyjnego, co oznacza dzie-
sięciokrotne zmniejszenie potrzebnej powierzchni produkcyjnej oraz umożliwienie dalszego
przerobu głęboko zamrożonych produktów w ciągu bardzo krótkiego czasu. Wadą wykorzystania
mikrofal do temperyzacji jest to, że w temperaturach bliskich 0°C warstwa zewnętrzna absorbuje
znaczną ilość energii i produkt na powierzchni może ulec przegrzaniu. Aby ograniczyć to zjawisko,
mikrofalową temperyzację przeprowadza się czasami stosując dodatkowo owiew zimnego
powietrza. Najczęściej stosuje się ją do mięsa, jego przetworów, masła oraz innych tłuszczów
jadalnych. Typowe urządzenia o mocy od 30 do 120 kW umożliwiają przetworzenie w ciągu
godziny od l do 4 ton mięsa lub od 1,5 do 6 ton masła.
Suszenie
Najlepsze efekty osiąga się przy suszeniu produktów o zawartości wilgoci mniejszej niż 20%.
Mechanizm suszenia z użyciem mikrofal istotnie różni się od konwencjonalnego, ponieważ z
łatwością przechodzą one przez warstwy wysuszone, docierając do nieodparowanej wilgoci i tam
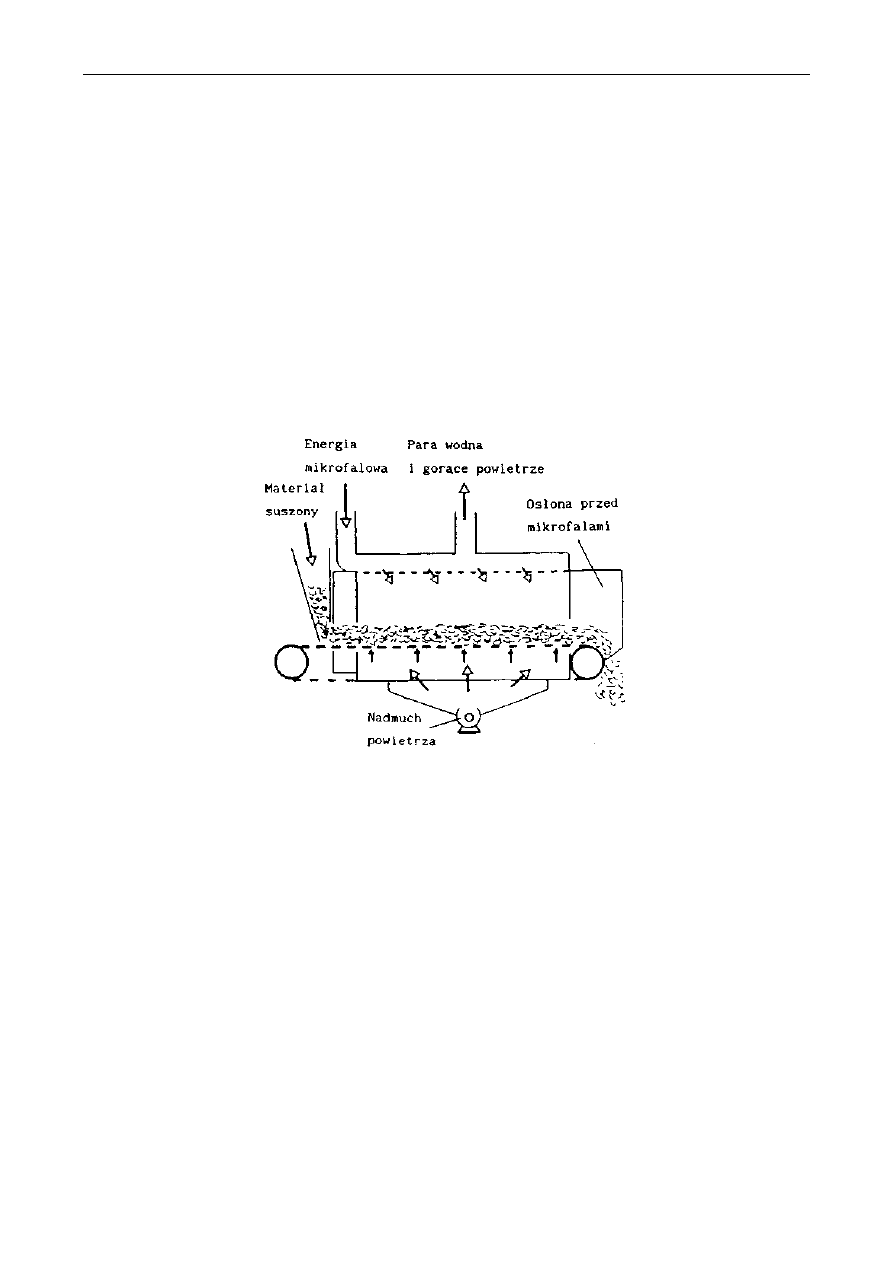
M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
generują ciepło. Ponadto obserwuje się kilkakrotne zmniejszenie czasu trwania procesu i ok. 30-
procentowe zmniejszenie zużycia energii, m.in. dlatego że ogrzewaniu ulegają jedynie mokro części
produktu, natomiast części suche, powietrze w suszarni i jej wnętrze nie są ogrzewane. Wysuszony
produkt charakteryzuje się mniej twardą powierzchnią, gdyż nie styka się ona z otaczającym
gorącym środowiskiem, jak ma to miejsce przy metodach tradycyjnych. Najczęściej suszone
mikrofalowo asortymenty to makaron, przyprawy, koncentrat pomidorowy, ryż, bekon i żywność
przekąskowa (snack foods). W niektórych przypadkach stosuje się razem z mikrofalami
konwencjonalne źródła ciepła, aby osiągnąć zamierzony cel technologiczny.
Na rycinie 3 przedstawiono schemat urządzenia do ciągłego mikrofalowego dosuszania
żywności. Przemieszcza się ona na perforowanej taśmie w strumieniu powietrza wytwarzanym
przez wentylator. Mikrofale padają na produkt od góry, generując w nim ciepło i zwiększając
prężność pary wodnej, która opuszcza urządzenie specjalnym otworem. Produkt wysuszony
odbierany jest z taśmy perforowanej na końcu urządzenia. Szczególnie istotne jest zapewnienie
szczelności instalacji, aby mikrofale nie wydostawały się na zewnątrz i nie stwarzały zagrożenia dla
zdrowia obsługi.
Rys. 3. Schemat urządzenia do ciągłego mikrofalowego dosuszania żywności.
Gotowanie
Mikrofale stosuje się z dobrym skutkiem do wstępnego gotowania bekonu, mięsa i części drobiu
z przeznaczeniem na rynek detaliczny i dla żywienia zbiorowego. Zaletami tej operacji jest duża
wydajność, krótki okres przygotowywania, mała pracochłonność oraz wysoka jakość produktów.
Moc i czas gotowania mikrofalowego należy dobierać stosownie do asortymentu. Większe i
grubsze elementy wymagają więcej energii niż małe i cienkie. Podobnie jak w przypadku
mikrofalowego suszenia, czasem dodatkowo stosuje się jednocześnie ogrzewanie konwencjonalne.
Wypiek pieczywa
Wypiek pieczywa przeprowadza się najczęściej tradycyjnymi metodami, niemniej jednak
istnieją także linie technologiczne wykorzystujące do tego celu ogrzewanie mikrofalowe. Już sam
etap rośnięcia ciasta można przyspieszyć dzięki zastosowaniu mikrofal. W trakcie wypieku
natomiast mikrofale mogą działać równocześnie z ogrzewaniem konwencjonalnym lub poprzedzać
go. Pozwala to na skrócenie czasu wypieku nawet o 60% i istotne zaoszczędzenie energii. Pieczywo
takie jest bardziej wyrośnięte, a jego wartość odżywcza jest większa niż produkowanego
tradycyjnie, ze względu na ograniczenie zachodzenia reakcji Mailarda i strat lizyny. Dobre efekty

M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
60
osiąga się przy ciągłej produkcji pieczywa cukierniczego typu biskwitów. Mikrofale, które
wykorzystuje się tu dopiero w ostatniej fazie wypieku, powodują dopieczenie i usunięcie nadmiaru
wilgoci bez dalszych zmian koloru powierzchni.
Blanszowanie mikrofalowe znalazło zastosowanie w ograniczonym stopniu, gdyż nie uzyskuje
się tą metodą lepszych jakościowo produktów niż przy blanszowaniu tradycyjnym. Spodziewany
jest natomiast rozwój liofilizatorów z ogrzewaniem mikrofalowym, konstrukcja taka bowiem
pozwala na szybszy i bardziej ekonomiczny przebieg suszenia, co w dobie coraz większego zapo-
trzebowania na produkty liofilizowane stwarza duże możliwości rozwoju. Obserwacja kilku
instalacji mikrofalowych do pasteryzacji pieczywa i soków owocowych, które pracują w przemyśle,
pozwala na prognozowanie szerszego ich zastosowania w przyszłości. Wysokiej jakości smalec i łój
wytapia się w prototypowym urządzeniu zainstalowanym w jednym z zakładów mięsnych w USA.
Na skalę półtechniczną praży się kawę i kakao uzyskując po 5-10 minutach produkt z większą
wydajnością i wyższej jakości niż przy prażeniu tradycyjnym. Na podstawie pozytywnie
zakończonych prób technicznych przewiduje się w ciągu najbliższych kilku lat wzrost
wykorzystania mikrofal do sterylizacji mleka i różnych półstałych produktów bezpośrednio w
opakowaniach z tworzywa sztucznego. W przemyśle mięsnym z powodzeniem stosuje się
prototypowe mikrofalowe urządzenia do produkcji parówek bezosłonkowych, uzyskując znaczne
skrócenie czasu i oszczędność energii.
Domowe kuchenki mikrofalowe
Duże zainteresowanie kuchenkami mikrofalowymi na świecie sprawiło, że przeróżne ich
modele produkuje się obecnie w ogromnych ilościach. Ich pojemność waha się najczęściej od 10 do
40l, a moc od 400 do 1000W. Badania wykazały, że stosowanie kuchenek mikrofalowych pozwala
zaoszczędzić średnio 63% energii w porównaniu z ogrzewaniem tradycyjnymi metodami, niemniej
jednak są potrawy, których przyrządzanie metodami tradycyjnymi jest mniej energochłonne.
Odpowiadając na wzrost zainteresowania kuchenkami mikrofalowymi, technolodzy
ukierunkowali swoją produkcję na wytwarzanie żywności nadającej się do wygodnego i szybkiego
przyrządzania z ich wykorzystaniem (micro-waveable foods). Obecnie już na szeroką skalę
produkuje się chłodzone, mrożone i suszone dania obiadowe, zupy, zakąski i desery pakowane
oddzielnie, na specjalnych tackach.
Tworzywa opakowaniowe muszą spełniać wymagania norm dotyczących ewentualnej migracji
materiału opakowaniowego do żywności. Często stosuje się wprowadzanie do nich ferrytu lub
proszku aluminiowego w miejscach, gdzie wymagane jest osłonięcie przed mikrofalami lub
zogniskowanie ich w celu bardziej jednorodnego ogrzewania produktu o niejednorodnej zawartości
wilgoci (np. pizza). Z uwagi na to, że wiele gospodarstw domowych w Europie Zachodniej i
Ameryce wyposażonych jest już w skomputeryzowane kuchenki mikrofalowe, producenci często
podają na opakowaniach swoich wyrobów parametry, jakie należy wprowadzić do wejścia
mikroprocesora, aby uzyskać produkt o najwyższej jakości i gotowy do spożycia.
Użytkownik kuchenki mikrofalowej wie, że istotną różnicą między ogrzewaniem tradycyjnym a
mikrofalowym jest to, że mikrofalówka zwykle nie daje zbrązowienia i kruchości produktu.
Różnica ta wynika z faktu, że powietrze wewnątrz kuchenki mikrofalowej nie jest ogrzewane przez
mikrofale, podczas gdy w metodach tradycyjnych jest gorące, ponadto na skutek odparowywania
wilgoci z powierzchni dodatkowo obniża się jej temperatura.
Czasami przeprowadzane w warunkach domowych rozmrażanie, ogrzewanie lub gotowanie
przebiega niewłaściwie, co jest związane z jednym z poniższych efektów.
a)
efekt powierzchniowy - wiąże się z tym, że większość mikrofal jest absorbowana i
przekształcana w ciepło przy powierzchni (zwykle do głębokości 3 cm). Zatem w niektórych
przypadkach, szczególnie przy stosowaniu częstotliwości 2450 MHz, obserwuje się
mocniejsze ogrzewanie powierzchni niż warstw głębszych.
b)
efekt ostrego rogu - polega na nadmiernym ogrzewaniu części wystających (np. skrzydełko
kurczaka), które wyeksponowane są na działanie mikrofal ze wszystkich stron, podczas gdy

M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
na resztę produktu padają one tylko z jednego kierunku. Efekt ten można osłabić przez
ekranowanie (np. folią aluminiową) fragmentów narażonych na nadmiar mikrofal.
c)
efekt różnicowy - obserwuje się go w żywności posiadającej obszary o różnej aktywności
dielektrycznej. Jeden region absorbuje mniej lub więcej energii niż inny i w konsekwencji
ogrzewają się one z różną szybkością. Przykładem może być mikrofalowe ogrzewanie pizzy.
Aby ograniczyć wpływ tego efektu, stosuje się dodatki do materiału opakowaniowego, które
poprzez ekranowanie wybranych fragmentów przed mikrofalami lub ich zogniskowanie do
prowadzają do bardziej równomiernego ogrzewania. Można także przesłaniać składniki
łatwiej ogrzewające się składnikami słabiej absorbującymi mikrofale (tzw. cieniowanie).
Mrożona żywność poddawana ogrzewaniu mikrofalowemu może wykazywać specyficzne dla
niej zachowanie wynikające z różnicy w penetracji i absorpcji mikrofal przez wodę i lód oraz
roztwory wodne. Mikrofale penetrują głębiej lód niż wodę, a ich absorpcja przez wodę jest znacznie
większa. Dlatego obszary, które zaczęły się już rozmrażać, absorbują więcej energii niż nie
rozmrożone, a zatem ulegają przegrzewaniu, podczas gdy fragmenty lodu pozostają niedogrzane.
Ponadto jeśli w czasie składowania zamrażalniczego występowały wahania temperatury, to na
skutek rekrystalizacji lodu powstać mogły obszary o zwiększonym stężeniu soli (tzw. kieszenie
solne). Obszary takie podczas rozmrażania mikrofalowego topnieją pierwsze i w ich okolicach
występuje nadmierne przegrzewanie produktu. Aby ograniczyć występowanie powyższych zjawisk,
rozmrażanie należy przeprowadzać powoli przy niewielkiej mocy mikrofal lub stosować przerwy w
celu umożliwienia wyrównywania się temperatury przez przewodzenie. Kuchenki mikrofalowe
mają najczęściej podany zakres mocy, przy jakiej należy rozmrażać żywność, lub są tak zapro-
gramowane, że proces ten przebiega z przerwami.
Zachowanie wartości odżywczej przez żywność ogrzewaną mikrofalowe było tematem wielu
prac badawczych. Eksperci z Instytutu Technologów śywności (IFT) z USA wydali opinię, że
żywność taka zachowuje więcej witamin i termicznie labilnych składników, ponieważ ogrzewanie
mikrofalowe z reguły trwa krócej niż tradycyjne i nie wywołuje zbrązowienia powierzchni. Istnieją
jednak i inne opracowania, które sugerują, że ten korzystny efekt żywieniowy jest niewielki.
Jakkolwiek szybkość jest najbardziej atrakcyjną cechą ogrzewania mikrofalowego, nie należy
bezkrytycznie jej wykorzystywać. Rozmrażanie, gotowanie, pieczenie i inne czynności kuchenne są
złożonymi procesami fizykochemicznymi wymagającymi często zajścia właściwych przemian i
reakcji. Powinny one następować w odpowiedniej kolejności i we właściwych relacjach czasowo-
temperaturowych. Nadmiernie szybkie ogrzewanie może zaburzać tok przemian, co przejawiać się
może pęknięciami produktu, przegrzewaniem oraz występowaniem niekorzystnych cech tekstury.
M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
12 luty 2010
62
7.2.
W
YKONANIE ĆWICZENIA
1. Określanie równomierności nagrzewania mikrofalami:
a) przy użyciu miazgi ziemniaczanej.
Utarte i odciśnięte ziemniaki ułożyć równomierną warstwą o grubości ok. 0,5 cm na całej
powierzchni płytki Petriego i umieścić w komorze roboczej. Prowadzić nagrzewanie mikrofalami
przez kolejne okresy 2-minutowe przy drugim poziomie nagrzewania, aż do chwili wystąpienia
pierwszych wyraźnych zmian w wyglądzie powierzchni ziemniaków. Po każdym cyklu
nagrzewania notować zmiany w wyglądzie warstwy ziemniaków nanosząc odpowiednio oznaczenia
na arkuszu odpowiadającym powierzchni komory roboczej kuchni.
W
DOŚWIADCZENIACH NR
1A,
2
I
3
NALEśY UśYWAĆ WODY DESTYLOWANEJ
.
b) przy podgrzewaniu wody.
Należy umieścić w komorze roboczej (zgodnie z rysunkiem) pięciu identycznych zlewek
szklanych, zawierających po 150 cm
3
wody. Prowadzić nagrzewanie przez kolejne okresy 2-
minutowe przy drugim poziomie nagrzewania i mierzyć temperaturę w zlewkach po zakończeniu
każdego cyklu ogrzewania. Termometry można wprowadzać do komory tylko podczas przerw w
działaniu magnetronu.
Rys. 1. Schemat rozmieszczenia zlewek z wodą w komorze grzejnej.
Pomiar zakończyć z chwilą uzyskania w jednej ze zlewek temp. 100°C. Badanie przeprowadzić
dwukrotnie. Należy sporządzić wykres wzrostu temperatury w funkcji czasu w poszczególnych
punktach pomiarowych. Na podstawie przeprowadzonych pomiarów należy wykreślić mapę
równomierności nagrzewania powierzchni komory.
2. Określanie wpływu masy produktu na czas nagrzewania
W środku komory roboczej należy umieszczać kolejno zlewki zawierające: 100, 200 i 300 cm
3
wody. Prowadzić nagrzewanie przez kolejne okresy 2-minutowe przy drugim poziomie
nagrzewania aż do chwili uzyskania temp. 100°C. Badania przeprowadzić dwukrotnie. Należy
sporządzić wykres wzrostu temperatury w funkcji czasu w poszczególnych porcjach nagrzewanej
wody.
3. Określanie wpływu kształtu naczynia na czas nagrzewania
Przygotować 5 naczyń szklanych o zróżnicowanym kształcie, ale posiadających ścianki
jednakowej grubości, np. zlewka, erlenmajerka, kolba płaskodenna itp. Każde naczynie napełniać
wodą w ilości 200 cm
3
o określonej temperaturze. Następnie należy umieszczać naczynia kolejno w
geometrycznym środku komory roboczej i nagrzewać przez 2 minuty przy drugim poziomie
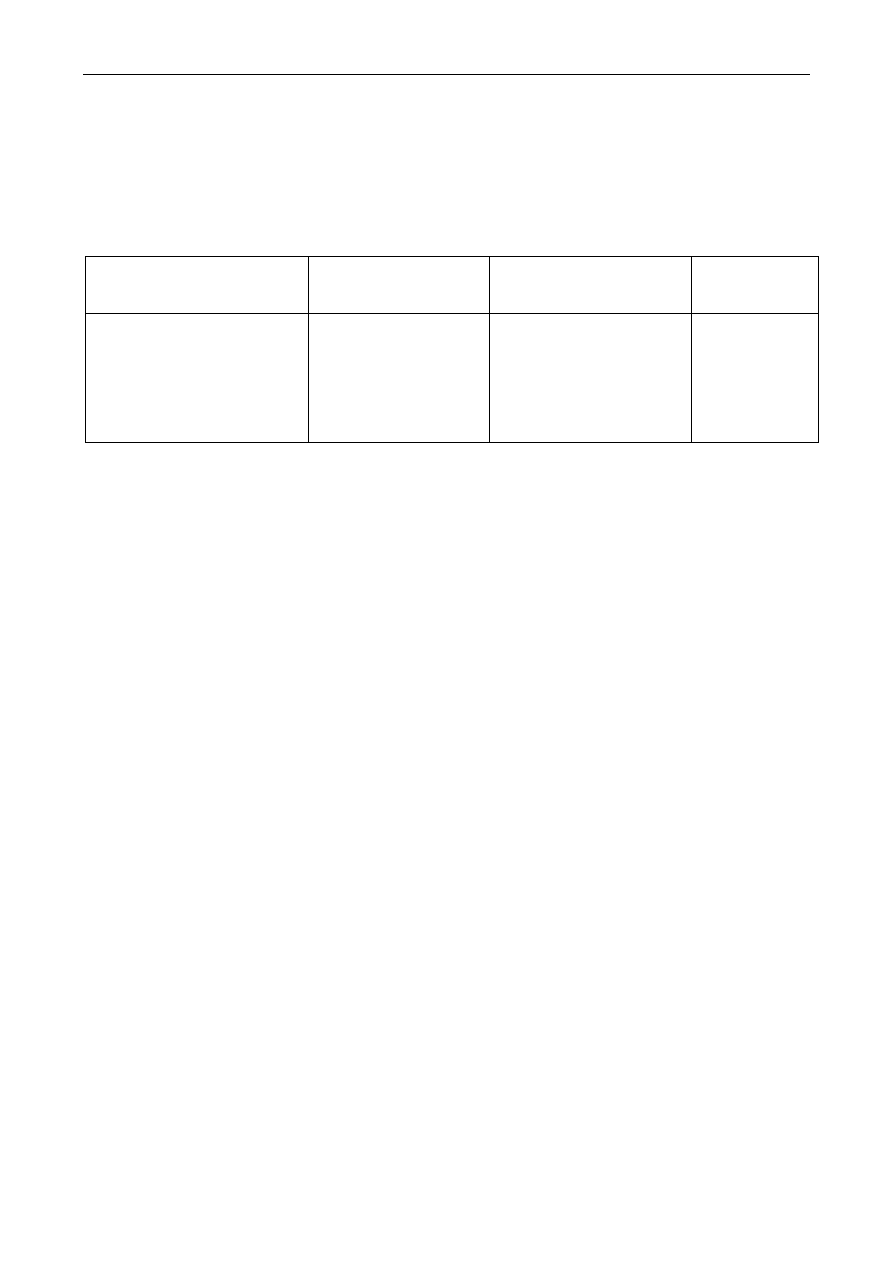
M
IKROFALE I ICH ZASTOSOWANIE W TECHNOLOGII
ś
YWNO
Ś
CI
nagrzewania i mierzyć temperaturę wody w każdym naczyniu po zakończeniu procesu
nagrzewania. Badanie wykonać w dwóch powtórzeniach.
W identyczny sposób proszę wykonać ogrzewanie kolby miarowej (200cm
3
) i dokonać pomiaru
temperatury wody w jej szyjce, środku kolby i przy dnie.
Wyniki pomiarów należy zebrać w tabeli według załączonego wzoru:
Wzór tabeli:
Rodzaj naczynia
Temperatura
początkowa
(T
p
) [°C]
Temperatura
po nagrzewaniu- końcowa
(T
k
) [°C]
Temperatura
średnia
(T
śr
) [°C]
1. …………………………
2. …………………………
3. …………………………
4. …………………………
7.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń należy umieścić w sprawozdaniu w formie oddzielnych i przedłożyć osobie
prowadzącej ćwiczenia w celu sprawdzenia. Należy także zaznaczyć wszelkie odstępstwa od metod
podanych w opracowaniu ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski w
formie zwięzłych punktów.

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
64
8.
WODA W PRZEMYŚLE SPOśYWCZYM
8.1.
W
PROWADZENIE
Wymagania dotyczące wody stosowanej w przemyśle spożywczym
Woda jest jednym z najważniejszych surowców w życiu codziennym człowieka. Związana
bezpośrednio z wieloma dziedzinami życia gospodarczego zapewnia jego prawidłowe
funkcjonowanie i możliwości dalszego rozwoju. Woda jest potrzebna w każdej branży przemysłu
spożywczego. Może stanowić mniej lub bardziej istotny składnik otrzymywanego produktu lub
stykać się z nim w określonej fazie produkcji - mówimy wtedy o stosowaniu wody do celów
bezpośrednich, a wodę taką nazywamy wodą technologiczną. Wodę stosowaną do zasilania kotłów
parowych, mycia naczyń i pomieszczeń, tzn. tę wodę, która nie bierze bezpośredniego udziału w
produkcji, nazywa się wodą energetyczną (do kotłów) lub wodą technologiczną do celów
pomocniczych (do mycia).
Wymagania stawiane wodzie zależą od jej przeznaczenia i zmieniają się nie tylko w obrębie
poszczególnych branż przemysłu spożywczego, ale nawet w poszczególnych działach tego samego
zakładu. Fakt, że woda wchodzi w skład organizmu i pożywienia człowieka oraz to, że woda
występująca w przyrodzie zawiera rozpuszczalne związki chemiczne, zobowiązuje do stałej
kontroli jakości wody używanej do produkcji w określonym zakładzie przemysłowym.
Postawą charakterystyki wody są wskaźniki organoleptyczne, fizyczne, mikrobiologiczne i
chemiczne.
•
wskaźniki organoleptyczne – zaliczamy do nich smak i zapach. Woda do picia powinna być bez
zapachu i bez obcego posmaku. W wodzie najczęściej występują następujące zapachy: gnilny -
pochodzący od siarkowodoru, roślinny- spowodowany obecnością wodorostów i mchów,
specyficzny - wywołany związkami chemicznymi.
•
wskaźniki fizyczne –do wskaźników tych charakteryzujących jakość wody zaliczamy:
temperaturę, przezroczystość, mętność oraz barwę. Zmętnienie wody jest spowodowane
występowaniem w niej zawiesiny piasku, drobnych cząsteczek roślinnych. Woda do picia oraz
bezpośredniej produkcji środków żywności nie może wykazywać żadnego zmętnienia. Barwa
wody pochodzi od rozpuszczalnych w niej substancji organicznych i mineralnych oraz od ich
formy. Naturalną barwą wody klarownej jest barwa zielonkawożółtawa z różnymi odcieniami.
•
wskaźniki mikrobiologiczne – oceniają jakość wody na podstawie ogólnej liczby zawartych w
niej drobnoustrojów i miana coli.
•
wskaźniki chemiczne – do tych wskaźników zaliczamy odczyn, twardość, zasadowość,
utlenialność, pozostałość po odparowaniu, zawartość związków azotowych, związków żelaza,
manganu i krzemu oraz soli (chlorków, siarczanów), a także zawartość rozpuszczonych gazów i
innych pierwiastków.
Odczyn wód naturalnych waha się w granicach pH 6,8-7,3. Inne wartości pH wody świadczą o
sztucznym jej zanieczyszczeniu.
Twardość jest to właściwość wody spowodowana zawartymi w niej jonami, głównie
wapniowymi i magnezowymi. Sole tych pierwiastków nie są szkodliwe dla zdrowia, lecz większe
ich stężenie może wykluczyć zastosowanie wody do celów technicznych i technologicznych. Za
jednostkę twardości wody przyjmuje się twardość, jaką nadaje wodzie l miligramorównoważnik
jonów Ca
2+
lub Mg
2+
w l dm
3
wody (tj. 20,04 mg/dm
3
Ca lub 12,16 mg/dm
3
Mg). Dopuszcza się
wyrażanie twardości w stopniach niemieckich (°n), odpowiadających ilości gramów CaO w 100dm
3
wody (1°n=0,357mval/dm
3
lub l mval/dm
3
= 2,804°n).
Rozróżnia się następujące rodzaje twardości wody:
•
twardość wapniową (tw
Ca
) spowodowaną obecnością jonów Ca
2+

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
•
twardość magnezową (tw
Mg
) spowodowaną obecnością jonów Mg
2+
•
twardość węglanową (tw
w
) spowodowaną obecnością kwaśnych węglanów, siarczanów i
krzemianów wapnia i magnezu;
•
twardość przemijającą (tw
przem
) tj. twardość wody ustępującą po rozkładzie wodorowęglanów
wskutek zagotowania:
Ca(HCO
3
)
2
→ CaCO
3
+ CO
2
+ H
2
O
Mg(HCO
3
)
2
→ MgCO
3
+ CO
2
+ H
2
O
•
twardość stałą (tw
st
) tj. twardość pozostającą po zagotowaniu wody
•
twardość ogólną (tw
og
) - wyrażającą ogólną zawartość jonów twardości wody.
Zależność pomiędzy poszczególnymi rodzajami twardości wody można przedstawić
następująco:
tw
og
= tw
Ca
+ tw
Mg
= tw
w
+ tw
nw
= tw
przem
+ tw
st
Przyjmując za kryterium twardość, wody naturalne można podzielić na: miękkie (do 10°n),
średnio twarde (10-15°n), twarde (15-30°n), bardzo twarde (powyżej 30°n).
Zasadowość jest to właściwość wody spowodowana obecnością wodorotlenków, kwaśnych
węglanów, węglanów wapniowych i potasowców. Rozróżnia się zasadowość wobec fenoloftaleiny
(Z
p
), będącą miarą zawartości w wodzie jonów wodorotlenowych i węglanowych, oraz zasadowość
wobec oranżu metylowego (Z
m
), spowodowaną obecnością wodorowęglanów.
Duże znaczenie podczas oceny wody ma obecność związków azotowych, tj. amoniaku,
azotynów i azotanów. Występowanie ich świadczy o zanieczyszczeniu wody produktami rozkładu
azotowych substancji organicznych, które wskutek działalności drobnoustrojów ulegają
mineralizacji.
Chlorki i siarczany ze względu na dużą rozpuszczalność występują we wszystkich wodach
naturalnych, lecz nadmierna ich zawartość wpływa ujemnie na smak wody. Wykazują one działanie
korodujące na konstrukcje betonowe i stalowe urządzeń.
śelazo występuje w wodzie w postaci żelazawych soli nieorganicznych (głównie jako
Fe(HCO
3
)
2
, rzadziej jako FeSO
4
) lub organicznych (w połączeniu z kwasami humusowymi). Wody
naturalne mogą zawierać dziesiątki miligramów żelaza i kilkanaście mg manganu w l dm
3
.
Zależnie od przeznaczenia woda stosowana w przemyśle powinna odpowiadać określonym
wymaganiom pod względem fizycznym, chemicznym i mikrobiologicznym.
Do zasilania kotłów wykorzystuje się skropliny pary technologicznej, uzupełniając ich niedobór
wodą surową. Obecność w wodzie kotłowej zawiesin krzemionki, soli wapnia i magnezu oraz
rozpuszczonych gazów (O
2
, CO
2
, H
2
S) powoduje korozję ścian kotła oraz powstawanie mułu i
kamienia kotłowego. Przy wytwarzaniu pary wodnej należy stosować czystą, miękką wodę, przy
czym w miarę jak wzrasta ciśnienie pary w kotle i obciążenie powierzchni ogrzewalnej, jakościowe
wymagania stawiane wodzie zasilającej kotły są coraz wyższe.
Woda stosowana do chłodzenia nie powinna zawierać zawiesin i drobnoustrojów oraz mieć
właściwości korodujących. Zasadniczym warunkiem jakościowym tej wody jest niska temperatura,
warunkująca
odprowadzanie
ciepła.
Woda
chłodnicza
powinna
charakteryzować
się
termostabilnością, tj. zarówno podczas ogrzewania (40-60°C), jak i chłodzenia (10-0°C) nie
powinna tworzyć osadu węglanu wapniowego. Do wód termostabilnych należą wody, których
twardość węglanowa nie przekracza 2,8 mval/dm
3
, czyli 7,84°n.
Woda używana do celów porządkowych, mycia opakowań i picia powinna odpowiadać
warunkom wody do picia i potrzeb gospodarczych, zawartym w rozporządzeniu Ministra Zdrowia i
Opieki Społecznej:
•
ogólna liczba kolonii bakterii nie może przekraczać 25 po 48 h hodowli w temperaturze 20
o
C
przy wysiewie 1cm
3
nierozcieńczonej wody na podłoże z żelatyną,
•
ogólna liczba kolonii bakterii nie może przekraczać 5 po 24 h hodowli w temperaturze 37
o
C
przy wysiewie 1cm
3
nierozcieńczonej wody na podłoże agarowe,
•
miano coli nie może być mniejsze niż 100,

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
66
•
zapach może należeć tylko do grupy zapachów naturalnych roślinnych,
•
mętność wody w skali krzemionkowej nie może przekraczać 3 mg/dm
3
,
•
barwa wg skali platynowo-kobaltowej nie może przekraczać 20 mg Pt/dm
3
,
•
temperatura wody powinna wahać się od 7 do 12°C,
•
odczyn pH powinien wynosić od 6,5 do 9,0
•
zawartość związków żelaza nie może przekraczać 0,3 mg Fe/dm
3
•
zawartość związków manganu nie może przekraczać 0,1 mg Mn/dm
3
,
•
twardość ogólna nie powinna przekraczać 7,1 mval/dm3 (20°n),
•
woda nie może zawierać agresywnego CO
2
,
•
sucha pozostałość po odparowaniu w temp. 105°C nie powinna przekraczać 500 mg/dm
3
,
•
woda używana w przemyśle spożywczym powinna być uprzednio chlorowana.
Niezależnie od tego, w niektórych branżach przemysłu spożywczego woda używana do ściśle
określonych celów musi odpowiadać dodatkowym wymaganiom związanym ze specyfiką danej
branży.
W przemyśle drożdżowym woda technologiczna jest stosowana do przygotowania brzeczki
drożdżowej oraz płukania drożdży i powinna odpowiadać warunkom wody do picia. Nie może
zawierać azotanów i azotynów, które hamują rozwój drożdży. Obecność żelaza i manganu
powoduje ciemnienie gotowego produktu, co ma szczególnie ujemne znaczenie podczas produkcji
drożdży piekarskich.
Woda technologiczna stosowana w przemyśle spirytusowym do przygotowania zacierów
powinna odpowiadać jakości wody do picia i wymaganiom dla wody technologicznej w przemyśle
drożdżowym. Podczas przygotowania nalewów, wapń, magnez oraz metale ciężkie pochodzące z
wody i używanych naczyń reagują z pektyną, garbnikami oraz kwasami pochodzącymi z owoców.
Powstałe związki wytrącają się w postaci osadów, powodujące wydatne obniżenie właściwości
smakowych nalewów. Węglany i siarczany wapnia i magezu oraz sole żelaza mogą powodować
zmętnienie o charakterze fizycznym wskutek zmniejszenia się ich rozpuszczalności po zmieszaniu
spirytusu i wody.
W przemyśle owocowo-warzywnym woda technologiczna stosowana do produkcji klarownych
pitnych soków owocowych, szczególnie napojów uzyskiwanych z uprzednio zagęszczonego soku
owocowego, wymaga specjalnej korekty. Woda używana do produkcji konserw groszkowych,
blanszowania groszku i zalewy konserw powinna mieć twardość poniżej 20°n. W celu
wyeliminowania zmętnień wywołanych przechodzeniem skrobi z ziaren stosuje się utwardzanie
wody solami wapnia. Powoduje to minimalne utwardzanie skórki ziaren, ale dzięki powstałym w
skórce nierozpuszczalnym pektynianom wapnia czyni ją mniej przepuszczalną dla skrobi.
Wytworzenie pektynianów wapnia powoduje poprawienie konsystencji niektórych owoców
miękkich, np. truskawek.
Podstawowe wymagania stawiane wodzie technologicznej stosowanej w przemyśle
piwowarsko-słodowniczym, oprocz wymagań przewidzianych dla wody do picia, dotyczą głównie
zasadowości i twardości, obecności azotynów, azotanów i krzemianów. Zasadowość ogólna
powinna być niska, mniejsza od twardości ogólnej. Zasadowość alkaliczna powinna być równa
zero, ponieważ w przeciwnym razie następuje alkalizacja zacieru i brzeczki. Niekorzystna jest
obecność wapnia i magnezu w postaci kwaśnych węglanów (działanie alkalizujące) korzystna zaś w
postaci siarczanów i chlorków. Obecność magnezu powoduje zwiększenie intensywności goryczki
piwa. Azotyny i azotany wpływają niekorzystnie na proces fermentacji, utrudniając rozwój drożdży,
a oprócz tego działają ujemnie na smak piwa. Krzem, podobnie jak żelazo, działa niekorzystnie na
przebieg fermentacji i powoduje zmętnienie piwa.
Woda technologiczna stosowana w przemyśle cukrowniczym do ekstrakcji powinna mieć
możliwie niską suchą pozostałość, niską zawartość siarczanu wapnia i chlorku magnezu, azotanów

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
oraz związków organicznych. Siarczany, chlorki, a szczególnie azotany, utrudniania krystalizację
cukru.
Woda technologiczna stosowana w przemyśle ziemniaczanym do produkcji krochmalu powinna
odpowiadać wymaganiom stawianym dla wody do picia, a ponadto powinna mieć możliwie niska
temperaturę, małą twardość i małą zawartość soli mineralnych oraz nie powinna zawierać
związków żelaza i manganu. Duża twardość i duża ilość soli mineralnych zwiększają zawartość
popiołu w krochmalu. Związki żelaza i manganu są przyczyną żółtego zabarwienia krochmalu oraz
ciemnych plam powstałych z żelaza.
W przemyśle mleczarskim ze względu na dużą podatność surowca na zakażenia zaostrza się
wymagania mikrobiologiczne dla wody technologicznej. Największe wymagania stawia się wodzie
służącej do płukania masła. Woda ta nie powinna zawierać żelaza, manganu i miedzi, których
obecność wpływa katalizująco na proces utleniania tłuszczu. Związki żelaza nadają masłu
specyficzny posmak oraz mogą być źródłem rdzawych plam na maśle, serze i twarogu. Również
duża zawartość soli magnezowych nadaje masłu nieprzyjemny, gorzkawy smak, w związku z tym
ogranicza się zawartość magnezu w wodzie do 40 mg MgO/dm
3
. Ogólna twardość wody do
płukania masła nie powinna przekraczać 10°n.
Inne branże przemysłu spożywczego, jak: mięsna, drobiarska, rybna zużywają duże ilości wody
do celów technologicznych. Woda ta, oprócz wymagań dla wody do picia, powinna być czysta pod
względem mikrobiologicznym. Pożądana jest niższa twardość stosowanej wody oraz brak w niej
żelaza, które nawet w minimalnej ilości może wpływać na zmianę smaku i zapachu wyrobów
produkowanych w tych przemysłach.
Uzdatnianie wody na potrzeby przemysłu spożywczego
Dobra woda, nadająca się dla przemysłu spożywczego, nie powinna wywierać ujemnego
wpływu na organizm ludzki, przebieg procesów technologicznych i jakość wytwarzanego produktu.
Ponadto nie może powodować korozji przewodów i urządzeń oraz dawać osadów na ich
powierzchniach.
1. Zmiękczanie wody
Najczęściej stosowanym procesem uzdatniania wody w zakładach przemysłu spożywczego jest
zmiękczanie wody przeznaczonej do zasilania kotłów oraz niektórych celów technologicznych.
Zmiękczanie wody polega na całkowitym lub częściowym usunięciu z wody kationów, głównie
wapnia i manganu, powodujących twardość.
Znane są następujące metody uzdatniania wody:
•
termiczne
•
chemiczne
•
fizykochemiczne.
Metody termiczne stosuje się do zmiękczania wody o dużej twardości przemijającej, a
jednocześnie znikomej twardości stałej. Pod wpływem podwyższonej temperatury, już powyżej
40°C, następuje termiczny rozkład wodorowęglanów wapnia i magnezu i wytrącenie obojętnych
węglanów wapnia i magnezu, zgodnie z reakcjami:
Ca(HCO
3
)
2
→ CaCO
3
+ CO
2
+ H
2
O
2Mg(HCO
3
)
2
→ Mg
2
CO
3
(OH)
2
+ 3CO
2
+H
2
O
Mg
2
CO
3
(OH)
2
+ H
2
O → 2Mg(OH)
2
+ CO
2
Rozkład Mg(HCO
3
)
2
przebiega 1,5 raza wolniej niż Ca(HCO
3
)
2
. Najpierw powstaje zasadowy
węglan magnezu, który po dłuższym gotowaniu ulega hydrolizie i strąca się w postaci trudniej
rozpuszczalnego wodorotlenku magnezowego Mg(OH)
2
.
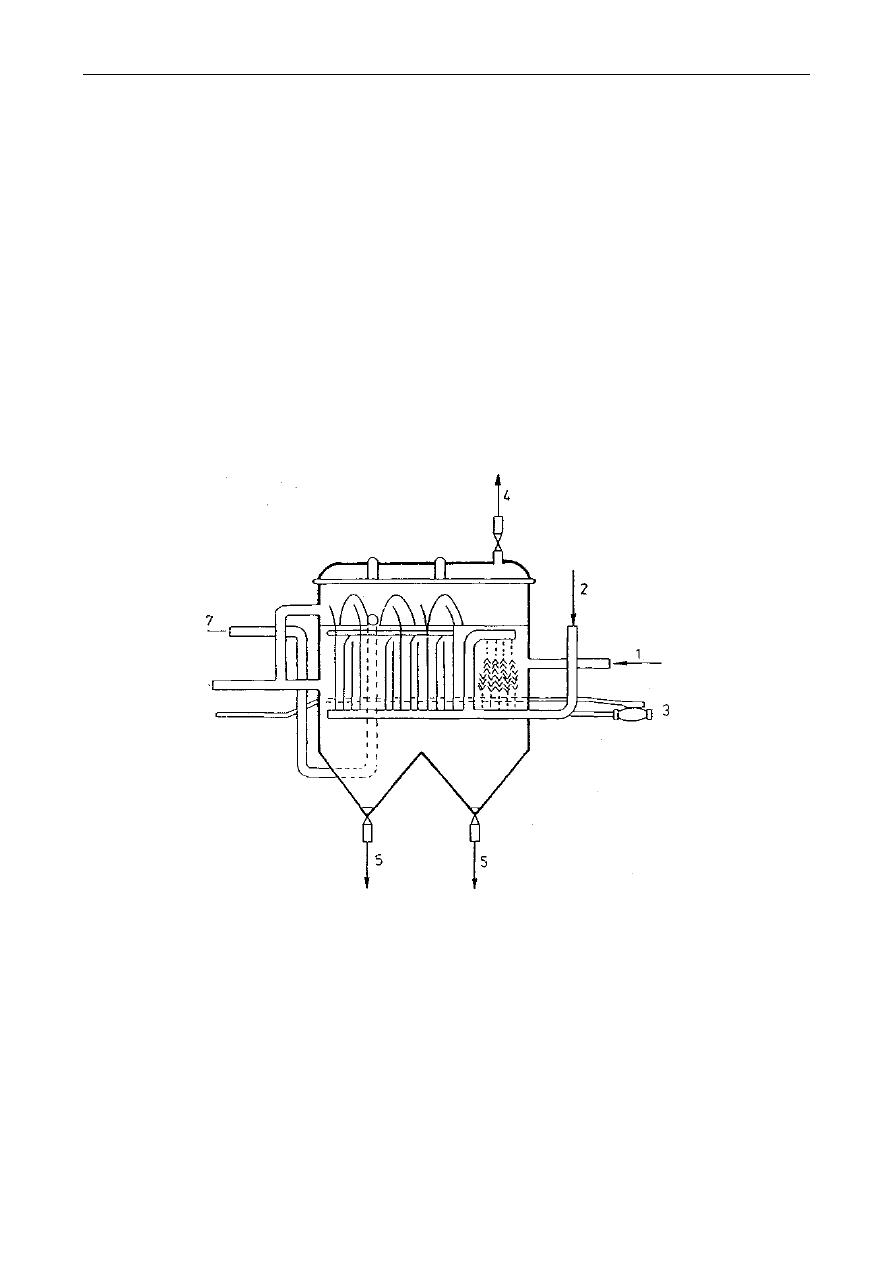
W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
68
Schemat urządzenia do termicznego zmiękczania wody przedstawiono na rys. 1. Woda surowa
o temp. 100°C przepływa między elastycznymi płytami, umocowanymi w ramkach do
wyjmowania. Strącający się osad CaCO
3
opada do lejów osadnikowych, częściowo zaś osiada na
płytach, które są elastycznie umocowane i okresowo wyjmowane w celu oczyszczenia.
Metody chemiczne polegają na strąceniu nierozpuszczalnych osadów lub wiązaniu
kompleksowe związki jonów wapnia i magnezu za pomocą różnych reagentów, jak np.
wodorotlenek wapnia (wapno), węglan sodu (soda), wodorotlenek sodu (soda kaustyczna),
fosforany, sole baru.
Najtańszym i najbardziej rozpowszechnionym chemicznym sposobem zmiękczania wody jest
wytrącanie jonów odpowiedzialnych za twardość wody za pomocą wapna i sody. Wapno dodaje się
zwykle w postaci mleka wapiennego, które strąca kwaśne węglany wapnia i magnezu oraz wiąże
wolny CO
2
, zgodnie z reakcjami:
Ca(HCO
3
)
2
+ Ca(OH)
2
→ 2CaCO
3
+ 2H
2
O
Mg(HCO
3
)
2
+ Ca(OH)
2
→ MgCO
3
+ CaCO
3
+ 2H
2
O
MgCO
3
+ Ca(OH)
2
→ Mg(OH)
2
+ CaCO
3
Ca(OH)
2
+CO
2
→ CaCO
3
+H
2
O
Rys. 1. Schemat aparatury do termicznego zmiękczania wody: l - dopływ wody, 2 - para grzejna, 3
- świeża para, 4 - opary, 5 - odszlamianie, 6 - odpływ wody, 7 - przelew
Proces wytrącania składników twardości nazywa się dekarbonizacacją. Twardość stałą usuwa
się przez dodanie sody, zgodnie z reakcjami:
CaCl
2
+ Na
2
CO
3
→ CaCO
3
+ 2NaCl
MgSO
4
+ Na
2
CO
3
→ MgCO
3
+ Na
2
SO
4
Proces zmiękczania wody tą metodą przebiega w dwóch fazach:
•
powstawanie trudno rozpuszczalnych osadów CaCO
3
i Mg(OH)
2
,
•
krystalizacji CaCO
3
i narastania kryształów, które mogą być zatrzymane w osadniku lub na
powierzchni filtru.
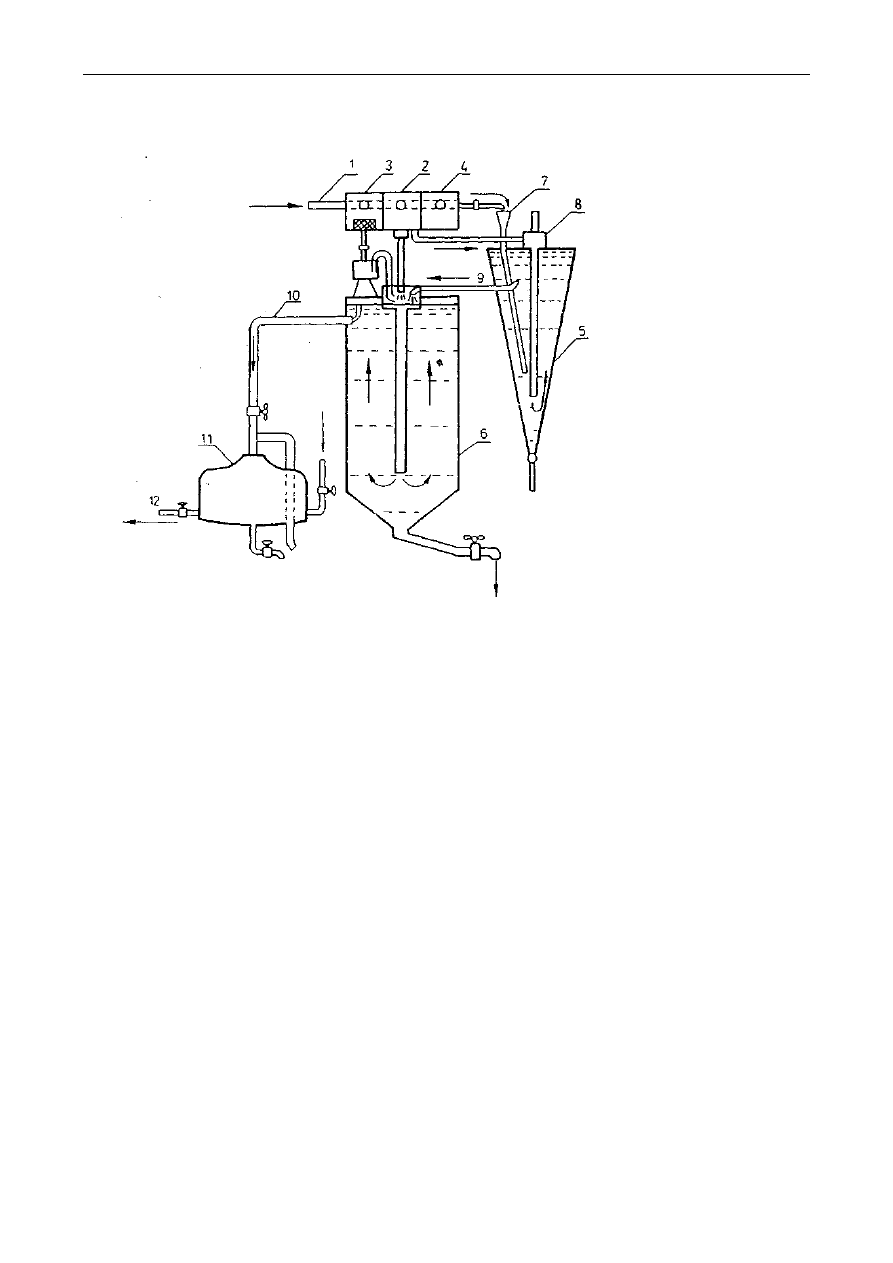
W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
Proces można przyspieszyć przez podgrzanie wody oraz dodanie do wody katalizatora w postaci
zawiesiny węglanu wapniowego.
Schemat instalacji do zmiękczania wody metodą wapienno-sodową przedstawiono na rysunku 2.
Rys. 2. Schemat instalacji do zmiękczania wody metodą wapienno-sodową: l - przewód
doprowadzający wodę surową, 2 - zbiornik rozdzielczy, 3 - zbiornik nasyconego roztworu
sody, 4 - zbiornik na wodę wapienną, 5 - saturator, 6 - reaktor, 7 - przewód, 8 - przewód, 9 -
rura przelewowa, 10-przelew z reaktora na filtr, 11 - filtr żwirowo-piaskowy, 12 - przewód
odpływowy
Woda surowa wprowadzana jest przewodem (1) do zbiornika rozdzielczego (2), skąd rurą
centralną jest kierowana do reaktora (6), a przewodem (8) do saturatora (5). W saturatorze surowa
woda miesza się z mlekiem wapiennym, doprowadzonym do zbiornika (4) przewodem (7). Rurą
przelewową (9) roztwór wodorotlenku wapniowego dopływa do reaktora. Ze zbiornika (3) przez
filtr jest dozowany do reaktora nasycony roztwór sody. Woda w reaktorze jest ogrzewana
bezpośrednio parą, aby proces zmiękczania przebiegał w temp. 90-95°C Wytrącane osady
gromadzą się w stożkowym dnie reaktora i w postaci szlamu są okresowo usuwane do kanału.
Zmiękczona woda odpływa z reaktora przelewem (10) na filtr żwirowo-piaskowy (11). Po
oddzieleniu osadów na filtrze czysta, zmiękczona woda odpływa przewodem (12).
Zmiękczanie wody za pomocą fosforanów polega na strącaniu praktycznie nierozpuszczalnych
fosforanów wapnia i magnezu (ich iloczyny rozpuszczalności są wielokrotnie mniejsze niż CaCO
3
),
dzięki czemu woda charakteryzuje się bardzo małą twardością resztkową.
Do zmiękczania stosuje się fosforan trójsodowy Na
3
PO
4
•10H
2
O, który reaguje z solami Ca i
Mg, zgodnie z równaniami reakcji:
3Ca(HCO
3
)
2
+ 2Na
3
PO
4
→ Ca
3
(PO
4
)
2
+ 6NaHCO
3
3Mg(HCO
3
)
2
+ 2Na
3
PO
4
→ Mg
3
(PO
4
)
2
+ 6NaHCO
3
3CaSO
4
+ 2Na
3
PO
4
→ Ca
3
(PO
4
)
2
+ 3Na
2
SO
4
3MgCl
2
+ 2Na
3
PO
4
→ Mg
3
(PO
4
)
2
+ 6NaCl

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
70
Powstający w reakcjach wodorowęglan sodowy w wyższych temperaturach rozkłada się,
tworząc węglan sodowy obojętny, który może likwidować twardość niewęglanową, zmniejszając w
ten sposób zużycie fosforanu. Rozkład termiczny NaHCO
3
zachodzi wg reakcji:
6NaHCO
3
→ 3Na
2
CO
3
+ 3CO
2
+ H
2
O
Metoda fosforanowa zapewnia 2-3-krotnie większą szybkość procesu zmiękczania niż metoda
wapno-soda, nie wymaga też bardzo ścisłego dozowania fosforanu. Fosforany nie powodują korozji
blachy żelaznej, a w wodzie zapobiegają tworzeniu się kamienia kotłowego.
Metody fizyczno-chemiczne – znalazły tu zastosowanie wymieniacze jonowe, czyli jonity.
Jonity są to ciała stałe nierozpuszczalne w wodzie, które maja zdolność wymiany własnych jonów z
jonami otaczającego je roztworu elektrolitu. Jonit poglądowo można przedstawić jako gąbkę,
zawierającą w porach wodę albo inny rozpuszczalnik. Do ścianek tej gąbki są „przytwierdzone”
wiązaniami atomowymi grupy jonogenne. W warunkach sprzyjających jonizacji grupy te dysocjują
na jony, przy czym tylko jony jednego znaku wykazują ruchliwość (jony znaku przeciwnego
połączone są ze szkieletem jonitu wiązaniami atomowymi). Mogą one być wymienione przez inne
jony tego samego znaku i dostawać się do wnętrza „gąbki” z zewnętrznego roztworu. Jeśli pewna
ilość jonów z zewnętrznego roztworu przechodzi do wnętrza fazy jonitu, to równocześnie
równoważna ilość jonów opuszcza fazę jonitu, przechodząc do roztworu zewnętrznego, podczas
wymiany bowiem musi być zachowana elektryczna obojętność układu. Jony jednakowego znaku z
jonami nieruchliwymi mają bardzo utrudniony „wstęp” do wnętrza fazy jonitu, gdyż są odpychane
siłami kulombowskimi.
Wymieniacze jonowe można podzielić na nieorganiczne i organiczne, a następnie na naturalne,
półsyntetyczne i syntetyczne. Zależnie od znaku ładunku zjonizowanych grup rozróżnia się
kationity i anionity. Kationity jako grupy czynne mają następujące grupy ujemne: sulfonowe -SO
3
,
karboksylowe –COO
–
, aminodwuoctanowe -N(CH
2
COO
–
)
2
, fosforanowe -PO
3
. Anionity mają
grupy czynne, charakteryzujące się ładunkiem dodatnim: czwartorzędowe amoniowe -NR
3
+
,
drugorzędowe aminowe –NR
2
+
, fosfoniowe -PR
3
+
. Kationity mające grupy funkcyjne ujemne będą
wymieniać kationy, anionity zaś dzięki dodatnio naładowanym grupom funkcyjnym wymieniają
aniony.
W zależności od właściwości grup funkcyjnych kationity dzielimy na silnie i słabo kwasowe,
anionity zaś na silnie zasadowe i słabo zasadowe (średnio zasadowe). Kationity silnie kwasowe są
mocnymi kwasami lub solami mocnych kwasów. Anionity silnie zasadowe są mocnymi zasadami
lub solami mocnych zasad. Kationity słabo kwasowe i anionity słabo zasadowe (średnio zasadowe)
to odpowiednio słabe kwasy lub słabe zasady i ich sole.
Oprócz jonitów monofunkcyjnych znane są jonity polifufikcyjne, mające grupy funkcyjne o
różnych właściwościach, np. grupy karboksylowe i sulfonowe lub amoniowe i aminowe.
Jonity amfoteryczne (amfolity) mogą zawierać grupy ujemne i dodatnie, które występują obok
siebie. Przejawiają one wówczas w zależności od pH roztworu, albo charakter kationitu, albo
anionitu.
W zależności od metody otrzymywania polimeru, stanowiącego szkielet jonitu, jonity dzielą się
na polimeryzacyjne i polikondensacyjne. Jeżeli suchy jonit umieści się w roztworze elektrolitu, to
zachodzą następujące zjawiska:
•
pęcznienie jonitu,
•
przenikanie elektrolitu do wnętrza jonitu,
•
wymiana jonów.
Jeżeli jonit RA umieścić w roztworze zawierającym jony B, to zastąpią one częściowo jony A,
którymi początkowo był obsadzony jonit. Nastąpi, więc wymiana jonów, zgodnie z ogólnym
równaniem:
RA + B
w
→ RB + A
w

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
a w konkretnym przypadku wymiana jonów Na
+
na jony Ca
2+
:
2RNa + Ca
2+
→ R
2
Ca + 2Na
+
Osiągnięta równowaga nie zależy od kierunku wymiany. Stan równowagi, czyli podział jonów
pomiędzy jonit i roztwór, zależy od takich czynników, jak:
•
ładunek jonów - w roztworach rozcieńczonych jony o większym ładunku są silniej wiązane
przez jonit, np. Ca
2+
jest silniej wiązany niż Na
+
,
•
wielkość jonów - większe jony organiczne są zwykle wiązane silniej, natomiast
powinowactwo jednowartościowych kationów nieorganicznych maleje ze wzrostem
promienia jonu uwodnionego i rośnie w miarę powiększania się liczby atomowej pierwiastka,
•
hydratacja - im mniejszy jest stopień hydratacji jonu, tym silniej jest on wiązany przez jonit,
•
charakter grup funkcyjnych - kationity słabo kwasowe wykazują duże powinowactwo do
jonów wodorowych, a anionity słabo zasadowe do jonów wodorotlenowych. Jony tworzące
kompleksy z grupami funkcyjnymi są wiązane bardzo silnie (wymieniacze jonowe
chelatujace),
•
stopień usieciowania jonitu - w miarę powiększania się stopnia usieciowania zmniejsza się
zdolność jonitu do wiązania jonów większych. Ogólna zdolność wymienna takiego
wymieniacza wzrasta, ponieważ zwiększa się ilość grup funkcyjnych,
•
stężenie roztworu - podział jonów między jonit i roztwór zależy od stężenia roztworu. Wpływ
stężenia jest szczególnie duży jeśli jony uczestniczące w wymianie mają różną wielkość
ładunku, np. równowaga pomiędzy jonami Ca
2+
i Na
+
jest przesunięta na korzyść wapnia
tylko w roztworach rozcieńczonych.
Powinowactwo kationitów silnie kwasowego R-SO
3
–
do poszczególnych jonów przedstawia się
następująco:
Li
+
> H
+
> Na
+
> K
+
> NH
4
+
> Ag
+
> Ca
2+
Powinowactwo kationitów słabo kwasowych R-COO
–
do poszczególnych jonów przedstawia się
następująco:
Na
+
> Mg
2+
> Ca
2+
> H
+
Kationit słabo kwasowy wykazuje szczególnie duże powinowactwo do jonów wodorowych.
Powinowactwo anionitów słabo zasadowych do jonów OH wzrasta odpowiednio w miarę obniżania
się zasadowości amin.
Wymiana jonowa może zachodzić w warunkach statycznych lub dynamicznych. Wymiana
jonowa w warunkach statycznych polega na wstrząsaniu roztworu z jonitem aż do momentu
ustalenia równowagi stężeń wymienianego jonu w roztworze i w fazie jonitu. Dynamiczna
wymiana jonowa zachodzi podczas przepływu roztworu przez kolumnę wypełnioną jonitem.
Przebieg wymiany jonów w czasie przepływu roztworu chlorku sodowego przez złoże kationitu w
formie wodorowej pokazano na rysunku 3. Jony Na
+
wprowadzone na kolumnę są wymieniane
początkowo w górnej warstwie złoża jonitowego. Po całkowitym obsadzeniu grup funkcyjnych
wymieniacza górne jego warstwy tworzą tzw. strefę powymienną. W dół kolumny spływa roztwór
uboższy w wymieniane jony i kontaktuje się zawsze ze świeżymi warstwami jonitu (strefa wymiany
właściwej). Proces wymiany jonów przebiega więc każdorazowo przy maksymalnej różnicy stężeń,
gdyż w dolnych warstwach złoża (strefa przedwymienna) stężenie wymienianego jonu w fazie
jonitu wynosi zero. W miarę przepuszczania roztworu przez kolumnę następuje przesuwanie się
strefy wymiany ku dołowi, a w poszczególnych warstwach kolumny ustala się od góry stan
równowagi miedzy stężeniem jonów Na
+
w roztworze i w fazie jonitu. Jeżeli nastąpi ustalenie
równowagi przy znacznej przewadze wymienianego jonu w roztworze, pojawi się on w wycieku z
kolumny. Jest to tzw. punkt przebicia, na którego podstawie określana jest robocza zdolność
W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
72
wymienna kolumny jonitowej, wyrażająca w gramorównoważnikach ilość wymienionego jonu do
momentu przebicia.
Rys. 3. Przebieg wymiany jonów w czasie przepływu roztworu chlorku sodowego przez kolumnę
kationową w formie H
+
: a - schematyczny obraz rozmieszczenia jonów Na
+
i H
+
w strefach
kolumny (strzałka wskazuje kierunek przepływu roztworu chlorku sodowego), b- wykres
zmian stężenia jonów Na
+
wzdłuż kolumny: h - wysokość kolumny, C
o
- stężenie jonów Na
+
w roztworze chlorku sodowego, C - stężenie jonów Na
+
w roztworze przepływającym przez
kolumnę.
Robocza zdolność jonowymienna jest zawsze mniejsza od całkowitej zdolności wymiennej
złoża i zależy od warunków procesu wymiany, jak: temperatura, pH roztworu i jego stężenie, rodzaj
innych jonów obecnych w roztworze, szybkość przepływu roztworu, wielkość ziarn jonitu i
geometryczny kształt kolumny.
Zmiękczanie wody za pomocą jonitów jest najprostszym sposobem jej uzdatniania,
polegającym na usunięciu z wody jonów wapnia i magnezu przez wymianę ich na jony zasadowe
kationitu w formie sodowej. Wymianę w cyklu sodowym można opisać następującymi reakcjami:
2KtNa + Ca(HCO
3
)
2
→ Kt
2
Ca + 2NaHCO
3
2KtNa + Mg(HCO
3
)
2
→ Kt
2
Mg + 2NaHCO
3
2KtNa + CaSO
4
→ Kt
2
Ca + Na
2
SO
4
2KtNa + MgSO
4
→ Kt
2
Mg + Na
2
SO
4
2KtNa + CaCl
2
→ Kt
2
Ca + 2NaCl
2KtNa + MgCl
2
→ Kt
2
Mg + 2NaCl
W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
Schemat jonitowego zmiękczania wody przedstawiono na rysunku 4.
Rys. 4. Schemat jonitowego zmiękczania wody
Proces odsalania wody polega na usunięciu z niej kationów i anionów dzięki przepuszczaniu ich
przez złoże kationowe w formie wodorowej, a następnie przez złoże anionitowe w formie
wodorotlenowej, wg reakcji:
KtH + A
+
X
–
= Kt – A + H
+
X
–
AtOH + H
+
X
–
= AtX + H
2
O
Gdzie: A
+
- kation, X
–
- anion.
W zależności od przebiegu procesu rozróżnia się dwie metody odsalania. W metodzie zwanej
demineralizacją są usuwane wszystkie jony z wyjątkiem bezwodnika kwasu krzemowego (SiO
2
) i
kwasu węglowego (CO
2
). W metodzie zwanej dejonizacją są usuwane wszystkie jony.
Układ do demineralizacji składa się z dwóch kolumn połączonych szeregowo i wypełnionych
kolejno silnie kwaśnym kationitem w formie wodorowej i słabo zasadowym anionitem w formie
wodorotlenowej (Rys. 5). Układ jest uzupełniony desorberem dwutlenku węgla oraz kolumną
buforową, wypełnianą zazwyczaj słabo kwaśnym kationitem. Pozwala to na osiągnięcie wody
demineralizowanej o pH ok. 7 przez cały czas trwania procesu.
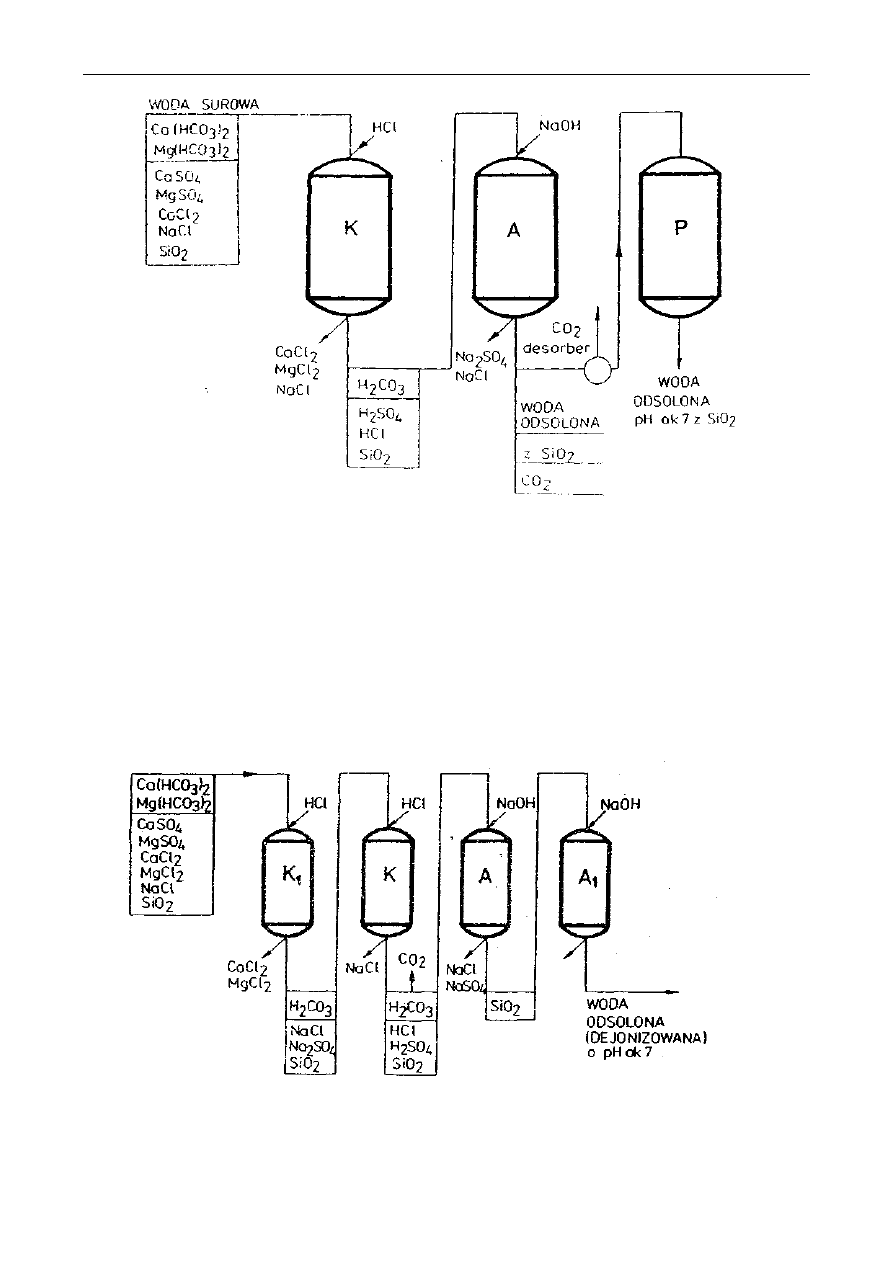
W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
74
Rys. 5. Przebieg demineralizacji wody wraz z desorpcją CO
2
i kolumną buforową: K - złoże silnie
kwaśnego kationitu, A - złoże słabo zasadowego anionitu. P - bufor (słabo kwaśny kationit)
Najprostszy układ kolumn do dejonizacji składa się z dwóch kolumn połączonych szeregowo:
pierwszej (K), wypełnionej silnie kwaśnym kationitem w formie wodorowej i drugiej (A
1
),
wypełnionej silnie zasadowym anionitem w formie wodorotlenowej. W celu odciążenia złoża (Y
1
)
od pochłaniania anionów silnych kwasów, przed te kolumnę włącza się jeszcze jedną kolumnę,
wypełnioną słabo zasadowym anionitem (A), wówczas złoże (A
1
) służy do odkrzemiania. Gdy
włączymy do takiego układu na wejściu dodatkowo kolumny (K
1
) wypełnione słabo kwaśnym
kationitem (Rys. 6), otrzymujemy zestaw pracujący bardzo ekonomicznie, szczególnie przy
znacznym zasoleniu i dużej twardości węglanowej wody.
Rys. 6. Przebieg dejonizacji wody wraz ze wstępnym zmiękczaniem, desorpcją CO
2
oraz
pochłanianiem silnych kwasów: K - złoże silnie kwaśnego kationitu, A
1
- złoże silnie
zasadowego anionitu, K
1
- złoże słabo kwaśnego kationitu, A - złoże słabo zasadowego
anionitu
Woda Surowa

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
2. Odżelazianie wody
Spożywanie wody żelazistej w zasadzie nie jest szkodliwe dla zdrowia, jednakże ze względów
smakowych woda do picia nie powinna zawierać więcej niż ok. 0,3 mg Fe/dm
3
. Wody żelaziste
wywierają ujemny wpływ na jakość produktów spożywczych. Do najważniejszych metod
odżelaziania wody należą:
Napowietrzanie i filtrowanie - żelazo w postaci wytrącalnej jako Fe(HCO
3
)
2
ulega hydrolizie i
utlenianiu w czasie napowietrzania. Kolejne fazy zachodzących reakcji ilustrują równania:
Fe(HCO
3
)
2
+ 2H
2
O → Fe(OH)
2
+ 2H
2
O + 2CO
2
Dzięki temu, że tworzący się w wyniku hydrolizy CO
2
uchodzi do atmosfery, proces hydrolizy
przebiega praktycznie do końca. W czasie napowietrzania tlen rozpuszcza się w wodzie i utlenia
żelazo:
4Fe(OH)
2
+ O
2
+ 2H
2
O → 4Fe(OH)
3
↓
Strącający się wodorotlenek żelazowy odfiltrowuje się.
Odżelazianie przez napowietrzanie i nawapnianie - niekiedy w wodach naturalnych żelazo
może występować w postaci związków bardzo trudnych do wytrącania lub nie dających się wcale
wytrącać przez zwykłe napowietrzanie. Gdy związki żelaza są pochodzenia nieorganicznego, dobre
wyniki daje nawapnianie, polegające na dodawaniu wody wapiennej bezpośrednio po
napowietrzeniu. Zachodzą wówczas następujące reakcje:
2FeSO
4
+ 2Ca(OH)
2
→ 2Fe(OH)
2
+ 2CaSO
4
4Fe(OH)
2
O
2
+ 2H
2
O → 4Fe(OH)
3
↓
Odżelazianie za pomocą koagulacji - sposób ten stosuje się do wód, w których żelazo występuje
w postaci związków koloidowych albo bardzo drobnych zawiesin Fe(OH)
3
, Fe(OH)
2
, FeS albo w
postaci koloidowych związków organicznych. Do odżelaziania metodą koagulacji stosuje się
koagulanty: Al
2
(SO
4
)
3
, Fe
2
(SO
4
)
3
, FeCl
3
, przy czym pH środowiska powinno być utrzymywane w
zakresie 5,7 - 7,5.
Odmanganianie wody - w wodach naturalnych związkom żelaza często towarzyszą związki
manganu, zwykle jako Mn(HCO
3
)
2
, rzadziej jako MnSO
4
. Według większości higienistów,
zawartość związków Mn w większych ilościach jest szkodliwa dla zdrowia.
Odmanganianie wody polega na hydrolizie jonów Mn
2+
do związków Mn
4+
w środowisku
alkalicznym, zgodnie z reakcjami:
2Mn(HCO
3
)
2
+ O
2
+ 4Ca(OH)
2
→ 2Mn(OH)
4
+ 4CaCO
3
+2H
2
O
2MnSO
4
+ O
2
+ 2H
2
O + 2Ca(OH)
2
→ 2Mn(OH)
4
↓ + 2CaSO
4
Wytrącający się wodorotlenek manganowy przechodzi następnie w uwodniony dwutlenek
manganu tzw. brunatniał:
Mn(OH)
4
→ MnO
2
+ 2H
2
O
Najczęściej stosuje się metody odmanganiania: napowietrzania z nawapnianiem i filtrowaniem,
przepuszczanie przez kationit manganowy, koagulacje, filtrowanie przez aktywne złoże.
3.

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
76
Dezynfekcja wody
Dezynfekcja czyli odkażanie wody ma na celu zniszczenie i usuniecie z wody bakterii
chorobotwórczych. Bakterie obecne w wodzie zostają częściowo usunięte razem z zawiesinami w
procesach koagulacji i filtrowania. Całkowite ich usuniecie wymaga zastosowania specjalnych
metod fizycznych i chemicznych.
Do metod fizycznych odkażania wody należą: ogrzewanie, naświetlanie promieniami
ultrafioletowymi, stosowanie filtrów ceramicznych i oddziaływanie ultradźwiękami na wodę.
Dezynfekcja metodami chemicznymi polega na wprowadzeniu do wody substancji
chemicznych, przeważnie silnych utleniaczy, które przenikają do wnętrza bakterii i utleniają
substancje wchodzące w skład jej protoplazmy. Metody chemiczne umożliwiają także utlenianie
zawartych w wodzie związków organicznych i mineralnych (żelaza, manganu), a także poprawę
smaku wody.
Skuteczność działania środków chemicznych zależy od:
•
rodzaju, stężenia i czasu kontaktu środka odkażającego z wodą,
•
ilości i rodzaju mikroorganizmów w wodzie,
•
rodzaju i stężenia substancji zanieczyszczającach wodę.
Do metod chemicznych odkażania wody zalicza się ozonowanie i chlorowanie. Ozonowanie jest
jednym z najbardziej przyszłościowych sposobów dezynfekcji wody. Zaletą tej metody jest bardzo
duża aktywność utleniająca ozonu. Ponadto ozon nie pozostawia w wodzie szkodliwych produktów
ubocznych i korzystnie wpływa na cechy organoleptyczne wody. Wadą ozonowania jest
krótkotrwale działanie tego zabiegu oraz stosunkowo wysoki koszt.
Chlor i niektóre jego związki po wprowadzeniu do wody działają silnie utleniająco i
bakteriobójczo. Ponadto chlor dodany do wody w odpowiedniej ilości usuwa obce posmaki, ułatwia
usuwanie żelaza i manganu, zapobiega - w pewnym stopniu - korozji, usuwa śluz z urządzeń oraz
ułatwia mycie urządzeń, pomieszczeń i opakowań.
Chlor w wodzie surowej podlega następującym przemianom:
•
ulega hydrolizie zgodnie z równaniem:
Cl
2
+ H
2
O → HClO + HCl
•
przy czym tworzący się kwas podchlorawy ulega dysocjacji:
HClO ↔ H
+
+ ClO
–
•
łączy się ze związkami azotowymi, tworząc chloroaminy lub chloroiminy:
NH
3
+ Cl
2
↔ NH
2
Cl + HCl
NH
3
+ 2Cl
2
↔ NHCl
2
+ 2HCl
Chlor zawarty w chloroaminach jest chlorem użytecznym związanym, który może stopniowo, w
wyniku hydrolizy, przemienić się w działający bakteriobójczo chlor użyteczny wolny;
NH
2
Cl + H
2
O → NH
4
OCl → HClO
3
+ NH
3
Bakteriobójcze i utleniające działanie chloru jest spowodowane rozpadem kwasu podchlorowego, z
wydzielaniem aktywnego tlenu:
HClO → HCl +O
Do chlorowania wody można używać samego chloru lub jego związków, zawierających tzw.
czynny chlor, takich jak: wapno chlorowane, dwutlenek chloru, chloraminy organiczne.
W zależności od pochodzenia wody oraz jej przeznaczenia stosuje się różne metody
chlorowania:
•
chlorowanie stałe w celu dezynfekcji i oczyszczania wody wodociągowej;
•
chlorowanie okresowe do zwalczania odrostów biologicznych oraz dezynfekcji przewodów,
różnych zbiorników i urządzeń;

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
•
chlorowanie wstępne;
•
chlorowanie wielostopniowe wykonuje się przed kolejnymi procesami uzdatniania w celu
utleniania zawartych w wodzie zanieczyszczeń i poprawienia jej właściwości
organoleptycznych (barwy, smaku);
•
chlorowanie końcowe przeprowadza się po uprzednim uzdatnieniu wody pod względem
fizykochemicznym;
•
chlorowanie normalną dawką stosuje się przeważnie przy chlorowaniu końcowym wody
wodociągowej, przeznaczonej do picia. Normalna dawka jest to ilość miligramów chloru,
jaką trzeba dodać do l dm
3
wody, aby po związniu części chloru przez związki znajdujące się
w wodzie, czyli po pokryciu tzw. zapotrzebowania na chlor, pozostało 0,1 mg Cl/dm
3
w
postaci chloru użytecznego. W tych ilościach chlor jest prawie niewyczuwalny w wodzie
surowej, a zanika zupełnie podczas jej ogrzewania;
•
chlorowanie zwiększoną dawką ma na celu skuteczniejsze odkażanie wody, poprawę jej
właściwości i zabezpieczenie urządzeń przed obrostami hydrobiologicznymi. Stosowane
bywa podczas wstępnego chlorowania wody do picia silnie zanieczyszczonej, a ponadto do
wody chłodniczej i przeznaczonej do celów sanitarno-porządkowych.
Nadmiar chloru w wodzie przeznaczonej do picia musi być po zakończeniu uzdatniania
zredukowany do 0,1-0,3 mg/dm
3
aktywnego chloru. Dechlorację wody przeprowadza się przez
napowietrzanie albo za pomocą węgla aktywnego, albo tiosiarczynu sodu oraz siarczynu sodu.
Ilość chloru pozostałego w wodzie stosowanej w przemyśle spożywczym reguluje się zależnie
od przeznaczenia wody. Woda używana w przemyśle owocowo-warzywnym do celów
technologicznych powinna zawierać 2-5 mg/dm
3
użytecznego chloru, natomiast woda do mycia -
10-20 mg/dm
3
.

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
78
8.2.
W
YKONANIE ĆWICZENIA
Zakres ćwiczenia obejmuje:
1. Przygotowanie wody dejonizowanej
2. Oznaczenie twardości ogólnej wody: wodociągowej, destylowanej i dejonizowanej metodą
Wartha-Pfeiffera
3. Oznaczenie zawartości chlorków metodą Mohra w wyżej wymienionych próbkach wody.
1. Przygotowanie wody dejonizowanej.
Aparatura:
- Stanowisko do dejonizacji wody: dwie kolumny w kaskadzie, wypełnione anionitem i kationitem,
- statyw laboratoryjny,
- uchwyty, rurki silikonowe
Pobrać próbkę wody wodociągowej o objętości 0,5 dm
3
. Poddać ją procesowi dejonizacji
wprowadzając na kolumny ze złożem jonowymiennym. Odciek poddać poniższym analizom.
2. Oznaczenie twardości ogólnej wody z użyciem mieszaniny sodowej (metoda
Warha-Pfeiffera)
Zasada metody:
Mieszanina sodowa węglanu sodu z wodorotlenkiem sodu (Na
2
CO
3
+ NaOH) dodana do wody
pozbawionej węglanów i dwutlenku węgla wytrąca z jonami wapnia i magnezu nierozpuszczalne
osady w postaci węglanów i wodorotlenków. Twardość wody oblicza się na podstawie ilości
zużytej mieszaniny.
Oznaczanie przeprowadza się w dwóch etapach: w pierwszym usuwa się węglany i dwutlenek
węgla na drodze miareczkowania 0,1-molowym roztworem kwasu solnego wobec metyloranżu, w
drugim oznacza się twardość ogólną przez dodanie mieszaniny sodowej, która reaguje z jonami
wapnia i magnezu, dając nierozpuszczalny osad. Osad odsącza się, a przesącz miareczkuje 0,l-
molowym roztworem kwasu solnego.
Przygotowanie mieszaniny sodowej.
Zmieszać równe objętości 0,l-molowego roztworu wodorotlenku sodu i 0,05-molowego
roztworu Na
2
CO
3
i sprawdzić miano. W tym celu do kolby stożkowej odmierzyć 100cm
3
wody
destylowanej, dodać pipetą 25cm
3
mieszaniny sodowej, gotować w ciągu 3 min, przenieść do kolby
miarowej o poj. 200 cm
3
i uzupełnić do kreski wodą destylowaną wolną od CO
2
(świeżo
przegotowaną). Do kolby stożkowej o pój. 300 cm
3
odmierzyć 100 cm
3
tego roztworu, dodać 3
krople metyloranżu i miareczkować 0,l-molowym roztworem kwasu solnego.
Współczynnik miana (f) mieszaniny sodowej oblicza się według wzoru:
25
a
2
f
⋅
=
gdzie: a - liczba cm
3
0,l-molowego roztworu kwasu solnego zużytego do miareczkowania.
Jeśli roztwór NaOH jest ściśle 0,1-molowy, a roztwór Na
2
CO
3
jest ściśle 0,05-molowy, to
współczynnik f = 1.
Wykonanie oznaczenia
Do kolby stożkowej o pój. 300 cm
3
odmierzyć 100 cm
3
badanej wody, dodać 3 krople oranżu
metylowego i miareczkować 0,l-molowym roztworem kwasu solnego do wyraźnej zmiany
zabarwienia. Następnie zmiareczkowaną próbkę ogrzewać do wrzenia, gotować 3 min i ostudzić.
Jeśli żółte zabarwienie powróci, to próbkę należy miareczkować ponownie do wyraźnej zmiany
W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
zabarwienia. Odnotować ilość zużytego 0,1 molowego roztworu HCL, na podstawie której można
obliczyć zasadowość ogólną M próbki. Następnie do zmiareczkowanej próbki dodać 25 cm
3
mieszaniny sodowej. Ponownie ogrzewać do wrzenia i gotować w ciągu 3 min. Całość ostudzić,
przenieść ilościowo do kolby miarowej o pój. 200 cm
3
, dopełnić do kreski wodą destylowaną wolną
od CO
2
(świeżo przegotowaną) i odstawić na kilkanaście minut Po opadnięciu osadu sączyć
zawartość kolby do suchego naczynia przez suchy sączek, odrzucając pierwsze 20 cm
3
przesączu.
Następnie do kolby miarowej o poj. 250 cm
3
odmierzyć 100 cm
3
przesączu, dodać 2 krople oranżu
metylowego i miareczkować 0,l-molowym roztworem kwasu solnego.
Obliczanie wyniku.
Twardość ogólną wody (T
o
) w stopniach twardości, oblicza się według wzoru:
V
100
c)
2
a
(f
T
O
⋅
⋅
−
⋅
=
gdzie:
f - współczynnik miana mieszaniny sodowej,
a - ilość mieszaniny sodowej dodana do próbki (w cm
3
),
c - ilość 0,1-molowego roztworu kwasu solnego zużyta do miareczkowania 100 cm
3
przesączu
(w cm
3
),
v - ilość próbki wody zużyta do oznaczania (w cm
3
).
3. Oznaczenie zawartości chlorków metodą Mohra
Zasada metody:
Jony chlorkowe miareczkuje się azotanem (V) srebra wobec chromianu (VI) potasu K
2
CrO
4
jako wskaźnika. W roztworze obojętnym lub słabo zasadowym (pH = 6,5-10) azotan (V) srebra
strąca najpierw biały osad chlorku srebra, a po całkowitym strąceniu chlorków jon srebrowy reaguje
z dwuchromianem (VI) potasu, wytracając czerwonobrunatny osad chromianu (VI) srebra. Zmiana
zabarwienia z żółtozielonego na czerwonobrunatne świadczy o całkowitym strąceniu jonów
chlorkowych.
W oznaczaniu przeszkadzają: siarkowodór, siarczki, barwa powyżej 30mg/dm
3
Pt i mętność
powyżej 10 mg/dm
3
.
Wykonanie oznaczenia
Odczynnik 1. Przygotowanie roztworu mianowanego chlorku sodu:
1,6486 g NaCl wysuszonego do stałej masy rozpuścić w wodzie destylowanej, przenieść do
kolby miarowej o poj. l dm
3
i uzupełnić do kreski wodą destylowaną (l cm
3
roztworu zawiera l mg
Cl
–
).
Odczynnik 2. Roztwór mianowany azotanu (V) srebra
4,791 g AgNO
3
rozpuścić w wodzie destylowanej, przenieść do kolby miarowej o poj. l dm
3
i
uzupełnić do kreski wodą destylowaną (l cm
3
tego roztworu powinien odpowiadać l mg Cl
–
).
Miano roztworu azotanu (V) srebra ustala się za pomocą mianowanego roztworu NaCl (odczynnik
1).
W tym celu odmierzyć dokładnie 10cm
3
mianowanego roztworu NaCl, dodać 1cm
3
roztworu
K
2
CrO
4
(odczynnik 3), dopełnić wodą destylowaną do obj. 100 cm
3
i miareczkować roztworem
AgNO
3
do zmiany zabarwienia na czerwonobrunatne. Miano roztworu (f), w mg na l cm
3
Cl
–
oblicza się według wzoru:
a
10
f
=
gdzie a - ilość roztworu AgNO
3
zużyta do zmiareczkowania 10cm
3
roztworu NaCl (w cm
3
).

W
ODA W PRZEMY
Ś
LE SPO
ś
YWCZYM
12 luty 2010
80
Odczynnik 3. Roztwór 10-procentowy chromianu (VI) potasu.
Do kolby stożkowej o pój. 300 cm
3
odmierzyć 100 cm
3
badanej wody (jeżeli zawartość
chlorków w próbce przekracza 30 mg, należy odmierzyć odpowiednio mniejszą ilość i uzupełnić do
100 cm
3
wodą destylowaną). Następnie dodać do kolby l cm
3
roztworu K
2
CrO
4
i miareczkować
mianowanym roztworem AgNO
3
do zmiany zabarwienia na czerwonobrunatne.
Obliczanie wyniku. Zawartość jonów chlorkowych w wodzie (X), w mg na l dm
3
Cl
–
oblicza się
według wzoru:
V
1000
0,3)
(a
f
X
⋅
−
⋅
=
gdzie: f - miano roztworu AgNO
3
,
a - ilość mianowanego roztworu AgNO
3
zużyta do zmiareczkowania próbki (w cm
3
),
v - ilość próbki wody zużyta do oznaczania (w cm
3
),
0,3 - ilość mianowanego roztworu AgNO
3
potrzebna do wytworzenia chromianu (VI) srebra
AgCrO
4
w 100cm
3
wody destylowanej
8.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń i analiz należy umieścić w sprawozdaniu w formie oddzielnych punktów wraz
z przeliczeniami i przedłożyć osobie prowadzącej ćwiczenia w celu sprawdzenia (wzór, załącznik 1
na stronie 121). Należy także zaznaczyć wszelkie odstępstwa od metod podanych w opracowaniu
ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski w formie zwięzłych punktów.
8.4.
S
PRZĘT I ODCZYNNIKI
1. Oznaczenie twardości ogólnej wody z użyciem mieszaniny sodowej (metoda
Warha-Pfeiffera)
Aparatura i odczynniki:
- 0,1 M roztwór wodorotlenku sodowego
- 0,05M roztwór węglanu sodu
- roztwór metylooranżu
- 0,1M roztwór kwas solnego
- kolba stożkowa 200cm
3
-2 sztuki
- pipeta 25 cm
3
- 2 sztuki
- kolba miarowa 200 cm
3
- 2 sztuki
- biureta do miareczkowania
- palnik gazowy
- trójnóg
- siatka azbestowa
- statyw + uchwyt
2. Oznaczenie zawartości chlorków metodą Mohra
Aparatura i odczynniki
- 1,6486 NaCl w 1,9 dm3 wody destylowanej
- 4,791 g AgNO3 w 1,0 dm3 wody destylowanej
- 10 % roztwór chromianu potasu (IV)
- kolba stożkowa 300cm3 - l sztuka
- pipeta 1,0 cm3 - l sztuka
- biureta do miareczkowania
- statyw + uchwyt

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
9.
UTRWALANIE śYWNOŚCI PRZEZ ZAKWASZENIE
9.1.
W
PROWADZENIE
Zakwaszanie należy do najstarszych (tradycyjnych) metod utrwalania żywności znanych od
wieków. W ostatnich latach było ono rzadziej stosowane ze względu na obecność wielu innych
sposobów utrwalania, ale obecnie zaznacza się tendencja do częstszego wykorzystania tej grupy
metod.
Utrwalające oddziaływanie środowiska kwaśnego
Środowisko kwaśne działa antymikrobiologicznie. Większość drobnoustrojów wykazuje
optimum wzrostu przy pH wynoszącym pomiędzy 6,5-7,5, czyli w warunkach zbliżonych do
środowiska obojętnego. Są oczywiście również drobnoustroje, które rozwijają się w środowisku
kwaśnym oraz zasadowym. Kwasolubne są na przykład bakterie fermentacji mlekowej i octowej, a
także bakterie siarkowe. W środowisku kwaśnym mogą także rozwijać się liczne drożdże i grzyby
strzępkowe których wzrost notuje się w szerokim zakresie pH wynoszącym od 2 do 9. Ogólnie
przyjmuje się, że pH środowiska o wartości 4,2-4,5 jest wystarczające do zahamowania rozwoju
bakterii gnilnych, enteropatogennych i częściowo bakterii fermentacji masłowej.
Oddziaływanie hamujące na rozwój drobnoustrojów jest związane z obecnością w środowisku
cząsteczek niezdysocjowanych kwasów i ich zdolnością do przenikania do komórek
mikroorganizmów. Cząsteczki niezdysocjowane wykazują bowiem cechy związków lipofilnych,
dzięki czemu mogą przenikać przez osłony do wnętrza komórki, gdzie przy wyższym pH
(zbliżonym do obojętnego) ulegają dysocjacji zakwaszając treść komórki. Obecność jonów
wodorowych (H
+
) w komórce hamuje wiele przemian metabolicznych oraz powoduje zakłócenie
proporcji pomiędzy wytwarzaniem a zużyciem ATP w komórce (czyli w przemianach
energetycznych), działa więc toksycznie.
Liczba niezdysocjowanych cząsteczek kwasu jest związana z pH środowiska. Dotyczy to
wszystkich kwasów wykorzystywanych do utrwalania żywności (czyli mlekowego, octowego, czy
konserwantów chemicznych – np. kwasu sorbowego czy benzoesowego). Najwięcej cząsteczek w
formie niezdysocjowanej notuje się przy pH w granicach 4, w związku z czym taki odczyn
środowiska jest już skutecznym czynnikiem utrwalającym.
Obniżone pH jest korzystne również z innych względów – wynikających z oddziaływania
podwyższonej kwasowości środowiska na przemiany enzymatyczne zachodzące w komórkach
surowców. Zmiana pH powoduje zahamowanie procesów oddechowych w tkankach żywności –
zmienia tempo wszystkich procesów enzymatycznych prowadzących do utleniania się (np.
witaminy C) lub brunatnienia powierzchni surowców. Zahamowane zostają zmiany hydrolityczne
(także enzymatyczne), które mogą być przyczyną niekorzystnych przemian sensorycznych w
surowcach, czyli mięknięcia i rozpadu tkanek bądź zmian składników odpowiedzialnych za cechy
smakowo-zapachowe. Kiszonkę czy marynatę dodatkowo stabilizują warunki beztlenowe. Dlatego
na skutek zakwaszania uzyskuje się produkt trwały przez długi okres, o dobrze zachowanych
cechach sensorycznych oraz (w przypadku procesów fermentacyjnych) niewiele zmienionej w
stosunku do surowca wartości odżywczej.
W praktyce przemysłowej oraz przetwórstwie domowym utrwalanie surowców spożywczych przez
zakwaszenie można podzielić na dwie grupy:
•
wykorzystanie procesów fermentacyjnych,
•
stosowanie kwasów organicznych.
Obydwie grupy metod są zupełnie różne i w ich wyniku uzyskuje się produkty o
różnych cechach sensorycznych i różnej wartości odżywczej. Ich cechą wspólną jest natomiast
utrwalenie przez obniżenie pH środowiska.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
12 luty 2010
82
Procesy fermentacyjne
Historia wykorzystania procesów fermentacyjnych do utrwalania żywności bądź w celu nadania
jej pożądanych cech sięga zamierzchłych czasów. Stosowano je zdecydowanie wcześniej niż
opisano sam proces fermentacji, który odkryto stosunkowo niedawno (w XIX w.).
Z zastosowaniem fermentacji utrwala się wiele surowców roślinnych (w Polsce głównie kapustę
i ogórki, na świecie: buraki ćwikłowe, kalafiory, cebulę, selery, pomidory zielone i czerwone,
paprykę i wiele innych), jabłka nasiona roślin strączkowych (fasola, soja i sos sojowy), oliwki,
kawę, herbatę, mięso (fermentowane wędliny, mięso, ryby), mleko (uzyskuje się mleczne napoje
fermentowane), a także sery czy pasze dla zwierząt. Procesy fermentacyjne wykorzystuje się do
produkcji pieczywa oraz alkoholu (wódki, piwa, wina), a w ostatnich latach zaczyna rozwijać się
produkcja fermentowanych soków owocowych (z buraków, marchwi, selerów, pietruszki,
pomidorów i ich mieszanek).
Końcowym produktem fermentacji jest zazwyczaj kwas mlekowy fermentacja mlekowa), ale
także inne związki, np. kwas octowy (fermentacja octowa), alkohol etylowy (fermentacja
alkoholowa) lub ich mieszaniny. Skład związków otrzymanych po fermentacji jest zależny od
charakteru procesu i rodzaju utrwalanych (czy przerabianych) surowców.
Procesom fermentacji mlekowej można poddawać wiele surowców spożywczych, zwłaszcza
roślinnych. Głównym warunkiem ich przydatności do zakwaszania jest odpowiednia zawartość
węglowodanów, które są substratem do przebiegu procesu. Podczas fermentacji wykorzystywane są
cukry proste, disacharydy oraz niektóre tri- i polisacharydy. Rozkład cukrów następuje na skutek
działania enzymów wydzielanych przez drobnoustroje rozkładających heksozy, a końcowym
produktem reakcji jest kwas mlekowy i niekiedy inne związki.
Kwas mlekowy oraz inne substancje będące wynikiem fermentacji, jak wspomniano powyżej,
działają utrwalająco na produkt przez zahamowanie wzrostu drobnoustrojów, a także ograniczając
zmiany zachodzące w tkankach surowców.
Oprócz oddziaływania utrwalającego, istotny jest również wpływ kwasu mlekowego na ustrój
człowieka. Powoduje on zmiany pH w organizmie, aktywując działanie enzymów oddechowych w
mitochondriach komórek, wywołuje zmiany ciśnienia osmotycznego oraz wydzielanie
niemetabolicznego nadmiaru CO2.
Związek ten ma również istotne znaczenie technologiczne. W procesach przemysłowych kwas
mlekowy stosuje się do dokwaszania, konserwowania, regulowania pH, poprawiania smaku,
zapachu i struktury produktów spożywczych. Charakteryzuje się on niskim progiem wyczuwalności
kwaśności. Jest mniej „ostry" w smaku niż kwas octowy, w związku z czym jego zastosowanie do
marynat zdecydowanie polepsza ich smak, wzmagając jednocześnie działanie utrwalające.
W badaniach naukowych stwierdzono ponadto, że przetrzymywanie warzyw w roztworze
kwasu mlekowego o pH 3-4 w ciągu 30-60 minut powoduje usunięcie z nich szkodliwych dla
zdrowia azotanów.
Cechy produktów utrwalonych na skutek fermentacji mlekowej
Produkty utrwalone na drodze fermentacji mlekowej charakteryzują się wieloma zaletami, które
częściowo podano już powyżej. Kiszonki, oprócz tego, że są trwałe, zawierają wiele dodatkowych
substancji powstających w wyniku fermentacji, nadających produktowi końcowemu szczególne i
niepowtarzalne cechy smakowo-zapachowe. Produkty fermentowane charakteryzują się wysoką
wartością odżywczą i składem zbliżonym do surowca. Kiszenie stabilizuje witaminę C i
prowitaminę A, dzięki czemu nie ulegają one rozkładowi. Podczas fermentacji mlekowej powstają
witaminy B2 i PP, czyli produkty fermentowane są wzbogacone w te składniki w stosunku do
surowca wyjściowego. Substancją powstającą podczas fermentacji jest również acetylocholina,
która korzystnie oddziałuje na mechanizm przekazywania bodźców nerwowych, poprawia
perystaltykę jelit i obniża ciśnienie krwi.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
Ponadto, składniki żywności z produktów fermentowanych są lepiej przyswajalne przez
organizm - kiszenie zwiększa strawność i pozwala wyeliminować niektóre substancje z surowców
roślinnych uważane za niepożądane (hemaglutyniny, cyjanki, tioglikozydy, substancje
gazotwórcze). Wraz z żywnością uzyskaną na drodze fermentacji dostarcza się do organizmu
drobnoustroje kwasu mlekowego, które korzystnie wpływają na procesy metaboliczne i na
środowisko w jelitach, regulując skład mikroflory przewodu pokarmowego, hamują rozwój bakterii
patogennych. Kolejną niezwykle korzystną cechą jest niewielka wartość energetyczna takich
produktów (dotyczy to fermentowanych surowców roślinnych i mleka), w przypadku surowców
roślinnych - znaczna zawartość błonnika oraz ich atrakcyjne właściwości sensoryczne (te ostatnie
dotyczą także przetworów mlecznych).
Produkty utrwalone na drodze fermentacji zalicza się do żywności przetworzonej w sposób
naturalny (za pomocą metod biologicznych), co w dobie wyrobów otrzymywanych w złożonych
procesach technologicznych ma ogromne znaczenie. Kiszenie ponadto jest tanie - nie wymaga
kosztownych nakładów energetycznych i finansowych. Wymaga natomiast stałej kontroli przebiegu
procesu i przechowywania.
Wobec powyższych zalet produkcja żywności fermentowanej ma przed sobą przyszłość i
pozwala na zwiększenie asortymentu wyrobów na rynku.
Drobnoustroje fermentacji mlekowej
Mikroorganizmami odpowiedzialnymi za przebieg fermentacji mlekowej a nieprzetrwalnikujące
bakterie kwasu mlekowego. Jest to grupa drobnoustrojów, których wspólną cechą jest zdolność do
przeprowadzania beztlenowej fermentacji mlekowej. Do podstawowej grupy bakterii fermentacji
mlekowej zalicza się ziarniaki i pałeczki należące do rodzajów Lactobacillus, Lactococcus,
Leuconostoc, Pediococcus, Streptococcus oraz niektóre gatunki z rodzajów Enterococcus i
Carnobacterium.
Wśród drobnoustrojów fermentacji mlekowej są bakterie homo- i heterofermentatywne. W
wyniku procesów metabolicznych przeprowadzanych udziałem bakterii homofermentatywnych
powstaje jedynie kwas mlekowy.
Na skutek rozwoju bakterii heterofermentatywnych dodatkowo produktami ubocznymi procesu
mogą być: alkohol etylowy, kwas octowy, mannit, gliceryna i inne związki. Nadają one kiszonkom
korzystne cechy aromatyczne i smakowe.
Fermentację można przeprowadzać, wykorzystując mikroflorę rodzimą produktu, jest to tzw.
fermentacja spontaniczna, a jej przebieg zależy od składu chemicznego środowiska i warunków
otoczenia. Fermentację spontaniczną prowadzi się przy kiszeniu kapusty i ogórków (chociaż nie
zawsze), natomiast do otrzymywania innych produktów (np. mleczne napoje fermentowane, wę-
dliny, soki) wykorzystuje się szczepionki uzyskiwane na skalę przemysłową w procesach
biotechnologicznych. Są to tzw. startery lub zakwasy, które zawierają zagęszczoną liczbę komórek
w formie utrwalonej przez liofilizację lub zamrożenie.
Produkcja kiszonej kapusty
O jakości uzyskanego produktu końcowego decyduje wiele czynników, do których należy
zaliczyć dobrą jakość surowca, dobór odpowiedniej odmiany, sposób uprawy, prawidłowy przebieg
procesu technologicznego oraz właściwe warunki przechowywania surowca po ukwaszeniu.
Proces technologiczny rozpoczyna się od mechanicznego poszatkowania kapusty i dodatku do
surowca soli w ilości 1,5-3%. Dodatek soli jest niezbędny - powoduje wyciek soku komórkowego
zawierającego cukry i sole mineralne. Cukru w kapuście jest zazwyczaj od 3,5 do 6,5%. Jeżeli
kapusta jest uboga w cukry, należy zastosować 1% dodatek sacharozy. Proces przebiega bez do-
datku wody. Poszatkowanie dezintegruje komórki i usprawnia wydzielanie soku. Takie środowisko
stwarza odpowiednie warunki do rozwoju pożądanej mikroflory. Sól ponadto wzmaga rozwój
bakterii mlekowych, a hamuje rozwój drobnoustrojów niepożądanych.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
12 luty 2010
84
W początkowym etapie pH tak przygotowanej mieszaniny wynosi ok. 6,5, co sprawia, że rozwijają
się różne formy drobnoustrojów. Mogą wówczas oprócz bakterii fermentacji mlekowej rozwijać się
drobnoustroje z grupy coli (pałeczki), bakterie gnilne (tlenowe bakterie proteolityczne) oraz
drożdże wytwarzające alkohol i dwutlenek węgla.
W miarę wzrostu kwasowości rozwój bakterii gnilnych i coli jest hamowany. Przeważają wówczas
heterofermentatywne bakterie mlekowe z gatunku Leuconostoc mesenteroides. Jednocześnie
następuje rozwój paciorkowców homofermentatywnych.
Już w pierwszym okresie fermentacji oprócz kwasu mlekowego powstają różnorodne estry nadające
produktowi smak i zapach. Temperatura fermentacji )rzez pierwsze dwa dni procesu powinna
wahać się w granicach 18-20°C.
Intensywny rozwój drobnoustrojów mlekowych powoduje nasilenie fermentacji mlekowej -
zachodzi wówczas fermentacja burzliwa. Zaczynają przeważać bakterie homofermentatywne -
Streptobacterium plantarum. Następuje intensywne wydzielanie gazów i powstawanie piany na
powierzchni. Pianę należy usuwać, gdyż sprzyja ona rozwojowi mikroflory gnilnej. Gazy również
należy sukcesywnie odprowadzać, w przeciwnym razie następuje gorzknienie kapusty.
W tym czasie na skutek produkcji kwasu mlekowego pH obniża się z 6 do około 4. Zwiększające
się stężenie kwasu hamuje intensywną fermentację mlekową. Zaczyna się etap fermentacji cichej.
Rozwijają się pałeczki heterofermentatywne z gatunku Betabacterium brevis, które wytwarzają
kwas mlekowy octowy. Następuje wówczas dofermentowywanie cukrów i wytworzenie związków
aromatycznych nadających produktowi cechy smakowo-zapachowe.
Czas trwania fermentacji jest ściśle skorelowany z temperaturą i może rwać 5-60 dni.
Podczas przemysłowej produkcji kapusty należy kontrolować skład mikroflory, pH (po
tygodniu powinno ono wynosić 3,5 - 4) oraz kwasowość miareczkową, która w przeliczeniu na
kwas mlekowy po upływie 2 tygodni powinna wynosić około 1,5% (jest to końcowa zawartość
kwasu mlekowego).
Po zakończeniu fermentacji należy obniżyć temperaturę w pomieszczeniu, - którym jest
składowana kapusta. Warunki chłodnicze zdecydowanie zwiększają trwałość produktu.
Temperaturę należy kontrolować i regulować tak, żeby nie następował wzrost kwasowości.
Temperatura przechowywania powinna być bliska 0°C.
Podczas przechowywania należy kontrolować również ilość piany i obecności kożucha, który
świadczy o przebiegu procesów gnilnych. Ważna jest pointo ilość i jakość soku. Powierzchnia
niepokryta sokiem, ułatwiająca dostęp powietrza do kapusty, powoduje rozwój drożdży i pleśni
(stąd obecność kożucha. Drożdże i pleśnie rozkładają kwasy, powodując wzrost pH i psucie się
produktu. Podczas przechowywania kiszonek niezwykle istotne jest zachowane czystości w
pomieszczeniach.
Trwałość kiszonej kapusty można zwiększyć przez pasteryzację, którą przeprowadza się w
opakowaniach jednostkowych. Przed pasteryzacją ukwaszoną kapustę należy poddać blanszowaniu.
Zblanszowaną kapustą napełnia się pakowania i zalewa gorącym sokiem. Pasteryzację
przeprowadza się w temperaturze 80-95°C. Pasteryzowana kapusta jest trwalsza, ale charakteryzuje
się gorszymi cechami smakowo-zapachowymi od niepasteryzowanej.
Skład prawidłowo ukwaszonej kapusty jest następujący:
•
kwasy nielotne (w przeliczeniu na kwas mlekowy) 1-1,3%,
•
kwasy lotne (w przeliczeniu na kwas octowy) 0,2 - 0,3%,
•
alkohol 0,5 - 0,6%,
•
cukier 0,0 - 0,2%,
•
związki azotowe 1,5%,
•
sól 2-3%,
•
sucha masa 10-12%,
•
witamina C 20-30 mg/100 g.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
Produkcja kiszonych ogórków
Do kiszenia powinno się wybierać właściwe odmiany ogórków (np. Przybyszewskie,
Monastyrskie), których cechy sensoryczne nie będą ulegały niekorzystnym zmianom podczas
przechowywania produktu końcowego. Ogórki zawierają mniej cukru niż kapusta (1-2%), co
powoduje, że ich pH po ukwaszeniu jest nieco wyższe niż pH kapusty.
Operacją wstępną w produkcji kiszonych ogórków jest ich moczenie. Pozwala ono na częściowe
usunięcie zanieczyszczeń z ich powierzchni i poprawia jędrność tkanek. Moczenie trwa zwykle od
30 minut do 4 godzin. Po wymoczeniu ogórki należy opłukać, przebrać i poddać kalibracji w celu
ujednoliceni.: ich wielkości. Korzystną operacją jest nakłuwanie ogórków, co ułatwia dyfuzję soku
komórkowego z ich wnętrza.
Do kiszenia ogórków wykorzystuje się przyprawy. Wśród nich najważniejszymi są korzeń i
liście chrzanu, koper, czosnek, liście laurowe oraz inne liście (np. dębu, wiśni, porzeczki,
winorośli). Przyprawy muszą być czyste - nie mogą stanowić źródła dodatkowego zakażenia
mikrobiologicznego. Dodatek przypraw przyczynia się do lepszego przebiegu procesu fermentacji,
wpływając na zwiększenie trwałości ogórków oraz nadając im korzystne cechy smakowo -
zapachowe.
Ogórki zalewa się roztworem soli o stężeniu 4-7%. Woda stosowana kwaszenia powinna mieć
odpowiednią twardość - nie mniejszą niż 10° niemieckich (jeden stopień twardości niemiecki - °n -
oznacza ilość jonów wapnia i magnezu równoważną zawartości 10 mg CaO w 1 dm3 wody). W
razie potrzeby twardość wody można zwiększyć, dodając węglan wapnia (CaCO3) lub chlorek
wapnia (CaCl2). Odpowiednia twardość wody zapobiega mięknięciu ogórków - jon wapniowy
przez połączenie się z pektyną i kwasem pektynowym podwyższa twardość skórek i miąższu.
Zwiększenie twardości wody umożliwia przefermentowanie całej ilości cukru znajdującego się w
surowcu.
Sól powoduje dyfuzję cukrów i soli mineralnych z komórek, stwarzając warunki do rozwoju
mikroflory fermentacji mlekowej.
Bakterie przetwarzają cukier zawarty w ogórkach na kwas mlekowy. Etapy fermentacji są
analogiczne jak w przypadku kiszenia kapusty: najpierw rozwijają się bakterie z grupy coli i
peptonizujące (czyli rozkładające białko), później zaczynają przeważać bakterie fermentacji
mlekowej. Najpierw rozwijają się heterofermentatywne Leuconostoc menesteroides, następnie
homofermentatywne Lactobacillus plantarum, na koniec heterofermentatywne Lactobacillus brevis.
Optymalna temperatura procesu powinna wynosić 15-18°C. W temperaturze 20°C rozwijają się
bakterie gnilne i pleśnie, stąd zachowanie odpowiednich warunków temperaturowych jest
gwarancją prawidłowego przebiegu procesu. Korzystne jest nawet obniżenie temperatury do ok.
12°C lub niższej, co wydłuża proces, ale pozwala na wyeliminowanie ewentualnych zmian w pro-
dukcie.
pH ogórków po procesie jest równe 3,4-4, zawartość kwasu mlekowego waha się w granicach
od 0,8 do 1%, ilość kwasu octowego wynosi ok. 0,15-0,25%, natomiast zwartość soli: 1,5-3,5%.
Po fermentacji ogórki magazynuje się w temperaturze 6-8°C, a najlepiej w warunkach
chłodniczych (około 0°C).
Podczas przechowywania ukwaszone ogórki należy kontrolować i w razie potrzeby uzupełniać
zalewę lub ją wymieniać, bądź też spasteryzować, gdyby była śluzowata.
Marynaty
Marynaty są drugą grupą produktów utrwalanych za pomocą zakwaszania. Ich wytwarzanie
polega na dodawaniu roztworów kwasów organicznych do surowców bez przeprowadzania procesu
fermentacji.
Kwasem dodawanym najczęściej jest kwas octowy, rzadziej stosuje się inne kwasy lub ich
mieszaniny. Innymi kwasami mogą być kwasy: cytrynowy, jabłkowy, winowy. Dobre efekty
utrwalające i korzystną jakość sensoryczną marynat można otrzymać, łącząc kwas octowy z

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
12 luty 2010
86
mlekowym. Połączenie takie daje silniejszy efekt antymikrobiologiczny przy mniejszej
wyczuwalnej kwasowości produktu.
Spośród wyżej wymienionych kwasów organicznych kwas octowy najsłabiej dysocjuje w
środowisku wodnym, w związku z czym jego działanie utrwalające jest najskuteczniejsze, co
wyjaśniono opisując mechanizm oddziaływania środowiska kwaśnego na drobnoustroje.
Uważa się, że marynaty, ze względu na zawartość kwasu octowego, są produktem mniej
wartościowym pod względem żywieniowym niż kiszonki lub surowce, z których zostały
sporządzone. Stanowią głównie dodatek do potraw, np. serwuje się je jako produkt pobudzający
apetyt oraz wydzielanie soków trawiennych i uatrakcyjniający posiłek. Niektórzy uważają je za
używki.
Marynaty sporządza się głównie z surowców roślinnych (warzywa i owoce), rzadziej z ryb.
Spośród warzyw wykorzystuje się ogórki, buraki ćwikłowe, dynię, cebulę, paprykę i inne oraz ich
mieszanki. Owoce najczęściej stosowane do produkcji marynat to: śliwki węgierki, gruszki i ich
mieszanki. Popularną marynatą są grzyby leśne i produkowane znacznie częściej pieczarki.
Niekiedy surowcem do sporządzania marynat są kiszonki. Produkty takie (wytworzone na bazie
kiszonek) charakteryzują się specyficznymi, korzystnymi cechami sensorycznymi.
W produkcji marynat odpowiednio przygotowany (umyty i rozdrobniony) surowiec łączy się z
zalewą czyli roztworem kwasu octowego w wodzie z dodatkami smakowymi. Kwas octowy
wykorzystywany do produkcji marynat ma postać 10% wodnego roztworu, natomiast stężenie
kwasu mlekowego, jeżeli się go dodatkowo stosuje, wynosi zazwyczaj 50-80%.
Podstawowym składnikiem zalewy, oprócz octu, jest woda, której jakość wpływa na cechy
uzyskiwanego produktu końcowego. Należy pamiętać, że woda musi spełniać wszelkie wymagania
normatywne wody pitnej.
Składnikami dodatkowymi zalewy są zazwyczaj cukier, sól kuchenna oraz przyprawy -
stosowane w odpowiednich ilościach. Do sporządzania marynat warzywnych spośród przypraw
wykorzystuje się kwiatostany i suche nasiona kopru, liście i korzenie chrzanu, estragon, majeranek,
liście laurowe, czosnek, pieprz, ziele angielskie, gorczycę i inne. Marynaty owocowe natomiast
mają w swoim składzie przyprawy korzenne: goździki, cynamon i imbir.
Zalewa, którą dodaje się do surowca powinna być zawsze gorąca (najodpowiedniejsza
temperatura wynosi 90°C). W ten sposób usuwa się powietrze z opakowań. Przy sporządzaniu
zalewy należy pamiętać, że kwas octowy jest lotny, dlatego można dodawać go dopiero w
końcowej fazie przygotowywania zalewy.
Marynaty, ze względu na zawartość kwasu octowego, dzieli się na następujące grupy:
•
łagodne, zawierające w swoim składzie 0,45-0,8% kwasu octowego, 0,5-2% cukru oraz 0,5-1,5 % soli;
•
średnio kwaśne o składzie: 1-3% kwasu octowego, 0,7-2% cukru oraz sól;
•
ostre: 3% i więcej kwasu octowego, 3% cukru i 2% soli.
Marynaty charakteryzują się ograniczoną trwałością dlatego powinno się przechowywać je w
warunkach chłodniczych. Podczas składowania w temperaturze pokojowej mogą rozwijać się
bakterie octowe, które w środowisku pozbawionym etanolu mogą asymilować kwas octowy,
rozkładając go do CO2 i H2O. Dobrym, dodatkowym czynnikiem utrwalającym zwiększającym
trwałość marynat, zwłaszcza łagodnych, jest pasteryzacja. Niekiedy dodaje się kwas benzoesowy
lub benzoesan sodu, zwłaszcza w przypadku produktów w dużych opakowaniach (dotyczy to
głównie ogórków).
Marynaty grzybowe
Jakkolwiek ocet, czasem także sok z cytryn i kwas mlekowy, służą przede wszystkim do
przyrządzania marynat z surowców roślinnych (grzybów, ogórków) to w pewnych okolicznościach

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
są wykorzystywane również do warunkowego utrwalania produktów mięsnych. Marynaty
grzybowe mogą być ostre i łagodne. Marynaty ostre zawierają 2-3% kwasu octowego i przy
opakowaniu naczyniach niehermetycznych znoszą dłuższe przechowywanie w temperaturze niższej
od 10°C. Łagodne marynaty grzybowe (0,5-1% kwasu octowego) do utrwalenia wymagają
opakowań hermetycznych (słojów, puszek) oraz dodatkowo zabiegu pasteryzacji.
Grzyby wymagają bardzo starannego uprzedniego spreparowania (przebierania, czyszczenia
nożem, płukania, przycinania i 5-20-minutowego, zależnie od gatunku grzybów, obgotowywania w
wodzie z ewentualnym jej zakwaszeniem kwasem cytrynowym). Impregnowanie grzybów octem (z
dodatkiem kilku procent soli kuchennej i przypraw korzennych) odbywa się na zimno w ciągu 2-3
dni, przy stopniowym zwiększaniu stężenia kwasu Pasteryzację prowadzi się w temp. 75-80°C w
ciągu 15-20 min. Normy ustalają m.in. maksymalną ilość zalewy w stosunku do samych grzybów
(np. 15-18% zalewy).
Marynaty ogórkowe
Rozróżnia się kilka kategorii marynat:
Łagodne marynaty ogórkowe w puszkach i słojach, czyli tzw. ogórki konserwowane, mają duże
znaczenie w przetwórstwie krajowym, jak również stanowią ważny przedmiot eksportu. Zawierają
zwykle tylko 0,3-0,6% kwasu octowego, przy czym część kwasu octowego może być zastąpiona
przez spożywczy kwas mlekowy. Ważne jest staranne wyjałowienie, możliwe wskutek znacznego
zakwaszenia ogórków, z wykluczeniem zbytniego ich rozgotowania, powodującego m.in. utratę
pożądanej „chrupkości" i „świeżości” smakowej.
Ogórki-korniszony stanowią przykład ostrych marynat o zawartości 2,5-3% kwasu octowego,
soli, przypraw, niekiedy marchwi. Ogórki są małe, długości 3-7 cm i grubości do 2,5 cm. Produkt
ten w opakowaniu szklanym jest poddawany pasteryzacji.
Marynaty z innych warzyw
Mixed pickles - ostra marynata (3% i więcej kwasu octowego) z pokrajanych warzyw: selera,
jarmużu, cebuli i małych ogórków.
Ć
wikła buraczana - krajane, uprzednio ugotowane i obrane buraki ćwikłowe z dodatkiem
tartego chrzanu, soli, ok. 5% cukru, przypraw i octu, po opakowaniu w słojach poddawane
pasteryzacji.
Buraczki młode i cebula w zalewie kwasu octowego.
Marynaty owocowe
Odgrywają one rolę raczej w przetwórstwie domowym. Sporządza się je z takich owoców, jak
śliwki, gruszki, brzoskwinie, melony i in. Owoce impregnuje się zalewą octową z dużym dodatkiem
cukru (10-50%) i przypraw korzennych.
Marynaty rybne
Od dawna jest znane i praktykowane utrwalanie całych, oczyszczonych, mniejszych ryb lub
płatków mięsa w zalewie octowej, przy prowadzeniu impregnacji mięsa ryb octem na zimno lub na
gorąco. Również w przypadku ryb część octu może być zastąpiona kwasem mlekowym. Z uwagi na
dużą zawartość białka w marynowanym materiale, dość silnie występuje działanie buforujące ze
strony wolnych grup aminowych białek i stąd kwasowość aktywna przy określonej dawce, np.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
12 luty 2010
88
kwasu octowego, jest znacznie mniejsza, niż w przypadku surowców roślinnych. Wskutek
przekraczania w kierunku kwaśnym punktu izoelektrycznego białek, część kwasu octowego lub
mlekowego tworzy białczany. Większa kwasowość, zwłaszcza w połączeniu z działaniem cieplnym
(np. przy pasteryzacji marynat w opakowaniu puszkowym), powoduje zmiękczenie - rozluźnienie
mięsa i znaczne zmiękczenie ości. Oprócz octu, czy ogólnie kwasów, do marynat tych dodaje się
jeszcze sól kuchenną i przyprawy.
Najbardziej znane marynaty rybne to rolmopsy (śledzie zawijane) oraz moskaliki (małe
oczyszczone śledzie lub szproty) przygotowywane wg różnorodnych receptur na zimno, a ponadto
różnorodne marynaty rybne w galarecie, otrzymywane z ryb gotowanych, niekiedy także
wędzonych, impregnowanych octem z przyprawami i zalewanych roztworem żelatyny z dodatkiem
octu i soli kuchennej. Marynaty rybne niepasteryzowane powinny być przechowywane a
temperaturze ok. 0°C.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
9.2.
W
YKONANIE ĆWICZENIA
Cel ćwiczenia
Celem ćwiczenia jest zapoznanie się z procesem utrwalania żywności za pomocą zakwaszania przy
zastosowaniu kwasów organicznych (mlekowy, octowy i cytrynowy) oraz ocena różnic
sensorycznych produktów tak zakwaszonych.
Zakres ćwiczenia
Na ćwiczeniach zostaną wykonane marynaty warzywne, w których będzie należało zmierzyć
pH (produktu i zalewy) oraz ocenić cechy sensoryczne.
Przed przystąpieniem do zadania należy przygotować 3 słoiki typu twist-off, myjąc je dokładnie
(wraz z pokrywkami) i wyparzając gorącą wodą (operacja taka wyjaławia opakowania i hartuje
zabezpieczając je przed pękaniem podczas sterylizacji).
Należy wykonać 3 słoiki marynaty warzywnej według przepisu podanego przez osobę
prowadzącą ćwiczenia. Przed sporządzaniem marynat należy poddać ocenie surowiec
wykorzystywany do procesu oraz zalewę wykonaną według przepisu.
Ocena surowców przed procesem
Surowiec należy ocenić, mierząc jego pH i określając jego cechy sensoryczne.
Pomiar pH surowca
Surowiec przed pomiarem pH musi zostać dokładnie rozdrobniony blenderem. Rozdrobniona
ilość surowca powinna wynosić co najmniej 5 g.
pH próbki surowca powinno się mierzyć w naczyniu, w którym przeprowadzono rozdrabnianie,
gdyż przemieszczanie próbki wiąże się z utratą soku komórkowego, który jest podstawowym
materiałem do pomiaru.
Oznaczenia pH dokonuje się przy użyciu pehametru, zwracając szczególną uwagę na zasady
pomiaru zawarte w instrukcji obsługi aparatu. Istotnymi czynnościami, o których należy
bezwzględnie pamiętać, są przemywanie elektrody wodą destylowaną i jej osuszanie po
każdorazowym pomiarze oraz przetrzymywanie jej w roztworze KCl w okresie, w którym nie jest
wykorzystywana. Przed pomiarem należy upewnić się, czy temperatura nastawiona na suwaku
pehametru jest odpowiednia i dopiero wówczas można dokonać kalibracji aparatu.
Ocena sensoryczno surowców
Badane wyróżniki sensoryczne surowca należy ocenić w skali pięciopunktowej uwzględniając
określenia odpowiadające poszczególnym stopniom jakości (5 – nota najwyższa, 1 – nota
najniższa).
Zgodnie ze schematem podanym w tabeli 1 należy ocenić zarówno owoce, jak i warzywa,
wybierając odpowiednie dla nich określenia.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
12 luty 2010
90
Tabela 1. Punkty i odpowiadające im określenia słowne charakteryzujące poszczególne wyróżniki
sensoryczne surowca.
Liczba
punktów
Wygląd ogólny
1
Zwrócić uwagę na
wygląd produktu
dokonując oględzin
zewnętrznych, włączając
kształt i barwę
Zapach
2
Ocenić intensywność i
typowość wrażeń węcho-
wych produktu
Konsystencja
3
Określić soczystość,
miękkość lub twardość,
jędrność i spoistość -
przez dotyk oraz rozgry-
zienie i miażdżenie w
jamie ustnej
Smakowitość
4
Ocenić intensywność i
typowość wrażeń
doustnych dostarczanych
przez produkt
5
typowy dla danego
rodzaju surowca; barwa,
wygląd zewnętrzny i
kształt; właściwe,
atrakcyjne, zachęcające
do spożycia
bardzo aromatyczny,
typowy dla danego su-
rowca, zachęcający do
spożycia
prawidłowa, soczysta,
odpowiednio miękka lub
odpowiednio twardawa
(w zależności od rodzaju
surowca), jędrna, spręży-
sta, spoista
właściwa dla danego
produktu, z wyczu-
walnym, przyjemnym
posmakiem
charakterystycznym dla
surowca
4
typowy dla danego
surowca z niewielkimi
odchyleniami od
kształtu; barwa i wygląd
zewnętrzny nieco
zmienione
aromatyczny,
charakterystyczny dla
tego typu surowca
soczysta, miękka lub
twarda, dość jędrna, dość
sprężysta, dość spoista
właściwa dla danego
rodzaju produktu
3
typowy dla danego
produktu z widocznymi
odchyleniami od
kształtu; barwa i wygląd
zewnętrzny zmienione
mniej aromatyczny
trochę zbyt miękka lub
zbyt twarda, mało soczy-
sta, mało jędrna, mało
spoista
smakowitość nieco
zmieniona
2
wygląd zewnętrzny,
barwa i kształt
nietypowe, wyraźnie
zmienione
zmieniony, mniej
atrakcyjny
wyraźnie zbyt miękka
lub zbyt twarda,
niesoczysta, niespoista
smakowitość wyraźnie
zmieniona
1
nietypowy, nie-
atrakcyjny, o wyraźnie
zdeformowanym
kształcie, barwa
całkowicie zmieniona
nieswoisty, obcy
nietypowa, wyraźnie
zmieniona, niewłaściwa
nietypowa, z obcym
nieprzyjemnym
posmakiem
1
Wygląd ogólny – zespół wszystkich istotnych dla danego produktu cech decydujących o jego wartości użytkowej dla
konsumenta.
2 Zapach – wrażenie odbierane przez zmysł powonienia lub wąchania określonych substancji lotnych.
3 Konsystencja – tekstura – spójność pomiędzy cząsteczkami oraz budowa produktu oceniana za pomocą czucia
głębokiego lub czucia doustnego, w niektórych przypadkach również wzrokowo; na teksturę produktu
składa się wiele cech jednostkowych, np. twardość, elastyczność, soczystość, kruchość, chrupkość,
miałkość, włóknistość, smarowność, lepkość.
4 Smakowitość – kompleksowe wrażenia doustne odczuwane przez zmysł powonienia, smaku i powierzchni jamy
ustnej, które może zawierać również ból, temperaturę i wrażenia dotykowe.
Przygotowanie surowca do marynat
Surowiec (pietruszkę, ziemniaki, buraki, marchew itp.) należy obrać i odpowiednio rozdrobnić,
np. marchew umyć w strumieniu bieżącej wody, oczyścić, obrać i pokroić w kostkę o boku ok. 0,5

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
cm. Marchew przenieść do sita i przeprowadzić blanszowanie, zanurzając w garnku z wodą o
temperaturze 90˚C przez 5 min. Po zakończeniu blanszowania marchew ostudzić do temperatury
ok. 20˚C przez zanurzenie w naczyniu z zimną wodą. Próbki warzyw umieścić po 40 g w słoikach.
Sporządzenie zalewy i przygotowanie surowca do utrwalenia
Zalewę do sporządzenia marynaty oraz odpowiednie przygotowanie surowca należy
przeprowadzić zgodnie z instrukcją otrzymaną na ćwiczeniach.
Wybraną przez prowadzącego ćwiczenia marynatę wykonać następujących składników
(dokładne ilości składników poda prowadzący):
•
marynata łagodna: 0,45-0,8% kwasu, 0,5-2% cukru oraz 0,5-1,5 % soli;
•
marynata średnio kwaśna: 1-3% kwasu, 0,7-2% cukru oraz sól;
•
marynata ostra: 3% i więcej kwasu, 3% cukru i 2% soli.
Należy wykonać po 100 cm3 marynat zawierających każdy z kwasów.
W obliczeniu niezbędnej ilości zalewy mogą pomóc poniżej podane wzory, z których można
wyliczyć dokładną ilość octu (lub innego kwasu) i wody potrzebnych do jej sporządzenia.
Obliczenie ilości octu do przygotowania zalewy:
P
p
Z
W
O
)
(
+
=
gdzie:
O - masa octu potrzebnego do sporządzenia zalewy [g],
W- masa warzyw przeznaczonych do marynowania [g],
Z - masa zalewy [g],
p - końcowa (żądana) zawartość kwasu octowego w produkcie [%],
P - procentowa zawartość kwasu octowego w occie [%].
Ilość wody potrzebnej do sporządzenia zalewy:
W = Z-(O + C)
gdzie:
W- ilość wody [g],
Z - masa zalewy [g],
O - masa octu [g],
C-masa cukru [g].
50 cm3 przygotowanej zalewy należy pozostawić przelewając do niewielkiej zlewki, a
następnie dokonać jej oceny. Ocenę zalewy wykonuje się po obniżeniu jej temperatury do
temperatury otoczenia, mierząc pH i cechy sensoryczne w skali trój-punktowej.
Jednocześnie powinno się pamiętać, że zalewa wykorzystywana do napełniania opakowań musi
być gorąca!
Zalewy zawierające kwas mlekowy i cytrynowy przygotować w analogiczny sposób.
Ocena zalewy przed procesem
Ocena pH
Przy ocenie pH istotne jest przestrzeganie odpowiedniej temperatury próbki (zalewa musi być
schłodzona do temperatury pokojowej, o czym wspomniano powyżej). Ocenę pH wykonuje się przy
U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
12 luty 2010
92
użyciu niewielkiej zlewki o objętości 50 cm3, zachowując wszelkie zasady pomiaru, o których
wspomniano już wcześniej.
Ocena sensoryczna zalewy
Ocenę sensoryczna należy przeprowadzić zgodnie z tabelą 2, posługując się skalą trójpunktową:
3 punkty - najwyższa nota, 1 punkt - najniższa. Skala 5-punktowa w przypadku zalewy byłaby zbyt
rozbudowana.
Tabela 2. Punktacja i odpowiadające poszczególnym punktom określenia słowne do oceny jakości zalewy
Liczba
punktów
Barwa
Ocenić barwą zalewy
Zapach
Ocenić intensywność i
typowość
wrażeń
wę-
chowych zalewy
Smakowitość
Ocenić intensywność i typo-
wość wrażeń doustnych dos-
tarczanych przez zalewę
3
typowa, prawie bezbarw-
na, nieco zmieniona przez
dodanie przypraw
przyjemny, aromatyczny,
typowy dla danej zalewy,
atrakcyjny, odpowiednio
kwaśny
właściwa, dobrze doprawio-
na, nie za słodka, nie za
słona, odpowiednio kwaśna
itp.
2
widocznie zmieniona
przez dodanie przypraw,
mniej atrakcyjna
mniej atrakcyjny, lekko
drażniący, trochę zbyt
kwaśny
gorzej doprawiona, nieco za
słodka, nieco za słona, trochę
za kwaśna itp.
1
nietypowa, zbyt mocno
zmieniona przez dodanie
przypraw, nieatrakcyjna
zmieniony, nieatrakcyjny,
mocno drażniący, zbyt
kwaśny
niewłaściwie doprawiona: za
słodka, za słona, zbyt mocno
kwaśna itp.
Przygotowane marynaty powinno się poddać pasteryzacji zgodnie z instrukcją. Po jednym ze
słoików przygotowanych produktów, czyli jedną konserwę warzywną i jedną owocową należy
ocenić pod koniec zajęć (po ich ostudzeniu do temperatury pokojowej).
Schładzanie marynaty musi zostać przeprowadzone bardzo ostrożnie, trzeba uważać, żeby słoiki
nie pękły - przede wszystkim nie należy ich wstawiać do zimnej wody bezpośrednio po
pasteryzacji!
Oceny skuteczności procesu dokonuje się mierząc pH zalewy i produktów, badając
instrumentalnie barwę utrwalonej żywności, a także zwracając uwagę na cechy sensoryczne, tj.
barwę, zapach oraz smak zarówno zalewy, jak i utrwalonych produktów. Cechy sensoryczne ocenia
się w skali 5-punktowej, przyjmując skalę od 1 do 5 (1 - najniższa nota, 5 - wartość najwyższa).
Schemat postępowania podano poniżej.
Ocena produktu po utrwaleniu i ostudzeniu
Pomiar pH produktu po procesie
Oznaczenie pH musi być przeprowadzone tak jak opisano powyżej, należy pamiętać, że produkt
przed pomiarem pH powinno się dokładnie rozdrobnić i ostudzić.
Ocena sensoryczna produktów po procesie
Ocenę sensoryczną przeprowadza się zgodnie z tabelą 3, biorąc pod uwagę zawarte w niej
wyróżniki sensoryczne.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
Tabela 3. Wyróżniki sensoryczne i określenia odpowiadające poszczególnym wartościom punktowym dla
produktu bezpośrednio po utrwaleniu i po próbie trwałościowej
Liczba
punktów
Wygląd ogólny
Zwrócić uwagę na wy-
gląd produktu
dokonując oględzin
zewnętrznych,
włączając kształt i
barwę
Zapach
Ocenić intensywność i
typowość wrażeń
węchowych produktu
Konsystencja
Określić soczystość,
miękkość, jędrność i
spoistość -przez dotyk
oraz rozgryzienie i
miażdżenie w jamie
ustnej
Smakowitość
Ocenić intensywność i
typowość wrażeń doustnych
dostarczanych przez produkt
5
typowy dla danego ro-
dzaju produktu; barwa,
wygląd zewnętrzny i
kształt zbliżone do su-
rowca, niezmienione w
wyniku przeprowa-
dzonego procesu (lub po
próbie trwałościowej)
bardzo aromatyczny,
świeży, typowy dla da-
nego produktu,
odpowiednio
(przyjemnie) kwaśny
prawidłowa, soczysta,
odpowiednio miękka,
jędrna, sprężysta,
spoista, zbliżona do
surowca
właściwa dla danego pro-
duktu, z wyczuwalnym,
przyjemnym posmakiem
kwaśnym
4
typowy dla danego pro-
duktu z niewielkimi
odchyleniami od kształ-
tu; barwa i wygląd zew-
nętrzny nieco zmienione
w stosunku do surowca
przed procesem
bardzo aromatyczny,
charakterystyczny dla
produktu mary-
nowanego
soczysta, miękka, dość
jędrna, dość sprężysta,
dość spoista, nieco
zmieniona w porównaniu
z surowcem przed
procesem
właściwa dla danego pro-
duktu, smakowitość cha-
rakterystyczna, nieco
zmieniona po utrwaleniu
(trochę zbyt lub za mało
kwaśna)
3
typowy dla danego pro-
duktu z widocznymi
odchyleniami od kształ-
tu; barwa i wygląd ze-
wnętrzny zmienione w
stosunku do surowca
przed procesem
mniej aromatyczny,
charakterystyczny dla
produktu mary-
nowanego, lekko
drażniący (trochę zbyt
kwaśny lub za mało
kwaśny)
trochę zbyt miękka, mało
soczysta, mało jędrna,
nieco za rzadka, mało
spoista
wyczuwalny posmak
octowy, produkt zbyt
kwaśny lub za mało kwaśny
2
wygląd zewnętrzny,
barwa i kształt nietypo-
we, wyraźnie zmienione
w stosunku do surowca
zmieniony w wyniku
przeprowadzonego
procesu, wyraźnie zbyt
kwaśny lub w inny
sposób nietypowy
wyraźnie zbyt miękka,
za rzadka, mazista
smakowitość wyraźnie
zmieniona,
nieprzypominająca smaku
surowca; dominujący smak
zalewy octowej
1
nietypowy, nieatrakcyj-
ny, o wyraźnie zdefor-
mowanym kształcie,
barwa całkowicie zmie-
niona w porównaniu z
surowcem
nieswoisty, obcy,
drażniący, nieprzyjemny
nietypowa, bardzo
mazista, cieknąca
nietypowa, z nieprzyjemnym
posmakiem
Ocena zalewy po utrwaleniu
Zalewę należy ocenić, mierząc jej pH i określając cechy sensoryczne, tak jak przed procesem.
Pomiar pH zalewy przy użyciu pehametru
Przed pomiarem pH zalewę należy schłodzić do temperatury pokojowej.
Ocena sensoryczna zalewy
Ocenę sensoryczna zalewy wykonuje się zgodnie z określeniami zawartymi w tabeli 2.

U
TRWALANIE
ś
YWNO
Ś
CI PRZEZ ZAKWASZENIE
12 luty 2010
94
9.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń umieścić w sprawozdaniu w formie oddzielnych punktów wraz z
przeliczeniami i przedłożyć osobie prowadzącej ćwiczenie w celu sprawdzenia (wzór, załącznik 1
na stronie 121). Należy także zaznaczyć wszelkie odstępstwa od metod podanych w opracowaniu
ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski w formie zwięzłych punktów.
Przykładowe zestawienie wyników badań
Surowiec
Marynata z kw.
mlekowym
Marynata z kw.
octowym
Marynata z kw.
cytrynowym
P
rz
ed
st
er
y
li
za
cj
ą
P
o
st
er
y
li
za
cj
i
P
rz
ed
st
er
y
li
za
cj
ą
P
o
st
er
y
li
za
cj
i
P
rz
ed
st
er
y
li
za
cj
ą
P
o
st
er
y
li
za
cj
i
pH zalewy
Ocena sensoryczna
zalewy
•
barwa
•
zapach
•
smak
pH produktu/surowca
Ocena sensoryczna
produktu/surowca
•
wygląd ogólny
•
zapach
•
konsystencja
•
smakowitość
9.4.
S
PRZĘT I ODCZYNNIKI
Aparatura i odczynniki:
•
autoklaw
•
słoiki
•
duża zlewka
•
duża kolba
•
pipeta
•
maszynka elektryczna
•
garnek + sitko
•
pH-metr laboratoryjny
•
waga analityczna + łyżeczka + szkiełko zegarkowe
•
sól
•
sacharoza
•
kwas octowy
•
kwas cytrynowy
•
kwas mlekowy
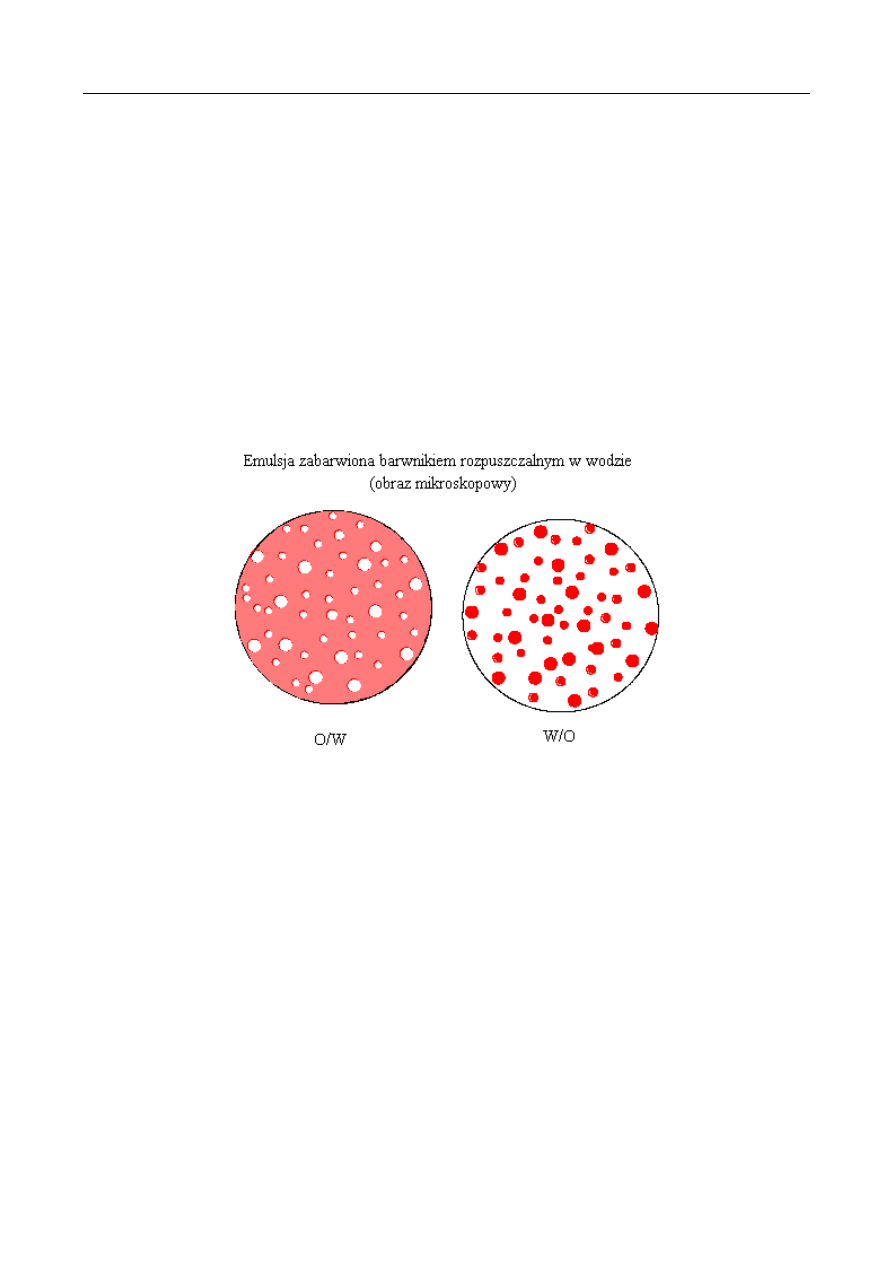
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
10.
EMULSJE
10.1.
WPROWADZENIE
Emulsjami nazywamy dwufazowe, ciekłe układy dyspersyjne, czyli mówiąc bardziej
przystępnym językiem układy, w których w jednej cieczy, zwanej fazą ciągłą lub zewnętrzną,
zawieszone są subtelne kropelki drugiej cieczy, zwanej fazą rozproszoną lub wewnętrzną. Emulsją
jest mleko, majonez, kremy, maści, tzw. mleczka itp.
Układ emulsyjny dwóch nie mieszających się cieczy określamy jako olej w wodzie (O/W) jeżeli
hydrofobowa faza, zwana "olejem" jest zawieszona w postaci rozproszonej w fazie hydrofilowej,
zwanej "wodą". Dla przypadku, gdy fazą ciągłą, zewnętrzną jest ciecz hydrofobowa ("olej") a
cieczą zdyspergowaną jest ciecz hydrofilowa ("woda") mówimy o emulsji woda w oleju (W/O).
Określenia typu emulsji można dokonać pod mikroskopem, oglądając warstwę emulsji
zabarwiona barwnikiem rozpuszczalnym w olejach (np. Sudan IV) lub w wodzie (np. oranż
metylowy). Zabarwieniu ulegnie oczywiście ta faza, która rozpuszcza barwnik.
Drugi sposób na określenie typu emulsji polega na sprawdzeniu przewodności elektrycznej emulsji.
Emulsje typu O/W, gdzie fazą zewnętrzna, ciągłą, jest rozpuszczalnik polarny (najczęstszy
przypadek to wodny roztwór elektrolitów) będzie przewodzić prąd elektryczny. Emulsja W/O
będzie wykazywać dużą oporność.
Emulgatory
Ponieważ w emulsjach powierzchnia styku dwóch faz (niemieszajacych się składników) jest
bardzo duża, układ może być trwały tyko w przypadku, gdy siły napięcia powierzchniowego
między fazami będą bliskie zeru. W przypadku przeciwnym faza rozproszona w bardzo krótkim
czasie łączy się w fazę ciągłą i emulsja rozdziela się na dwie fazy ciągłe (zjawisko zwane
koalescencją). Tak w przyrodzie, jak i w emulsjach tworzonych przez człowieka w celu obniżenia
napięcia powierzchniowego między fazami wykorzystuje się substancje trzecie, zwane
emulgatorami, tenzydami, środkami powierzchniowoczynnymi, których budowa powoduje, że
umieszczają się one na granicy faz, jedną częścią "zanurzone" w fazie hydrofilowej a drugą w
hydrofobowej. Tworząc taką monomolekularną warstewkę przyjmują na siebie "bycie granicą faz",
a ponieważ ich powinowactwo do obu faz jest podobne, powodują praktycznie zanik napięcia
powierzchniowego (dobre emulgatory) lub przynajmniej jego poważne obniżenie (środki
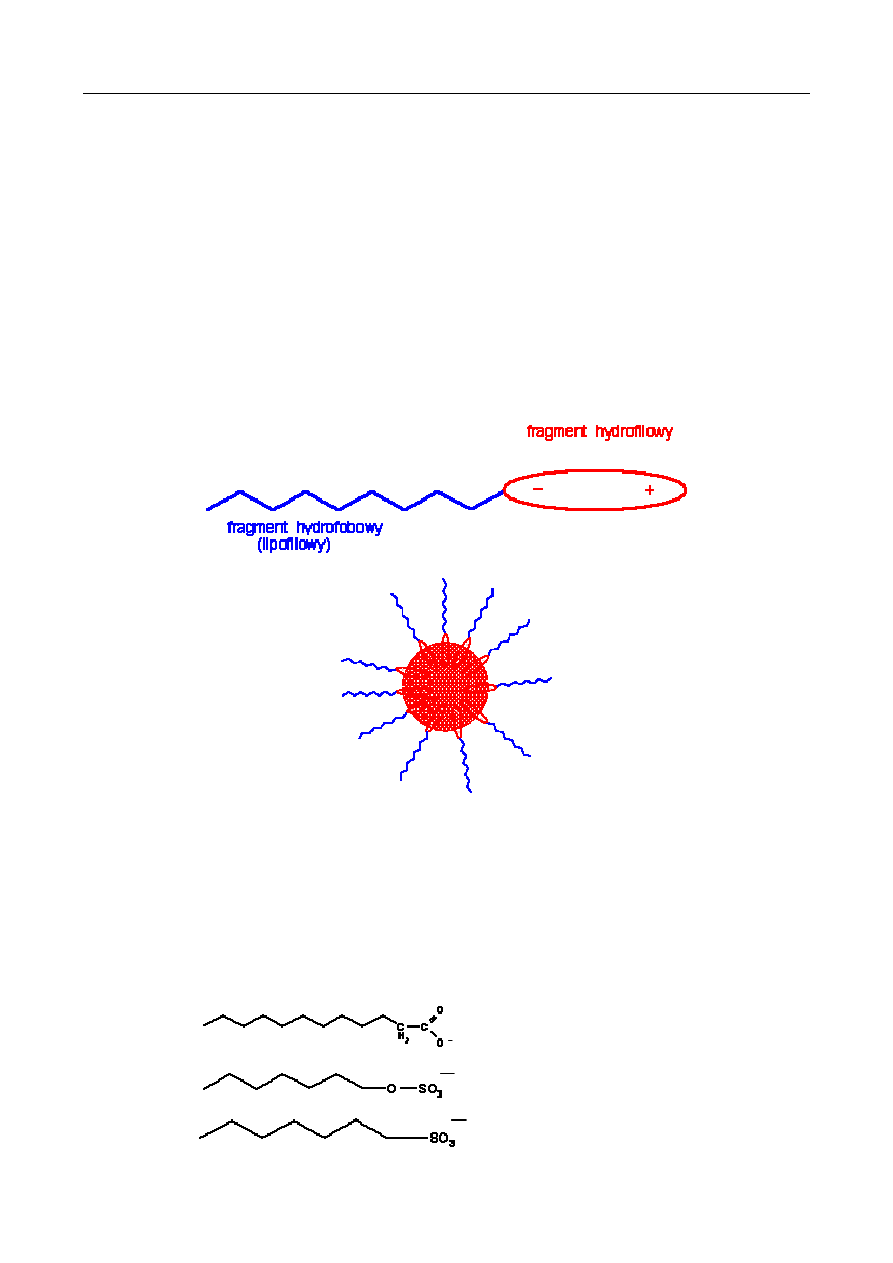
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
96
powierzchniowe czynne) i tym sposobem stabilizują emulsje. Powinowactwo emulgatora do fazy
olejowej i fazy wodnej określa parametr HLB (Hydrophilic-Liophilic Balance). Wartość HLB,
zależna głównie od budowy cząsteczki, a dokładniej od stosunku części hydrofilowej do
hydrofobowej, określa, czy dany środek powierzchniowo czynny stabilizuje lepiej emulsje O/W czy
W/O. Umownie przyjęto skale HLB w zakresie 1 - 40. Wartość 1 odnosi się do kwasu olejowego,
wartość 40 do laurylosiarczanu sodowego. Emulgatory o HLB <10 pozwalają wytwarzać emulsje
typu W/O (są lepiej rozpuszczalne w fazie niepolarnej). HLB powyżej 10 predysponuje emulgator
do użycia w emulsji O/W. Liczba HLB charakteryzuje się addytywnością , tzn. dla mieszaniny
emulgatorów można ją obliczyć na podstawie wartości HLB poszczególnych składników i ich
względnego udziału w mieszaninie.
Schemat budowy cząsteczki emulgatora
emulsja W/O
Ponieważ mechanizm obniżania napięcia powierzchniowego polega na tworzeniu cienkiej,
najczęściej monomolekularnej warstewki na granicy faz, emulgator dodany do emulsji tylko do
pewnego stężenia wspomaga tworzenie i utrzymanie emulsji, dodany w nadmiernej ilości nie
powoduje już obniżenia napięcia powierzchniowego i nie gromadzi się więcej na granicy faz tylko
tworzy skupiska (micele) w fazie ciągłej. Część hydrofilowa emulgatora zawsze jest „zanurzona” w
wodzie, bez względu na to, czy woda stanowi substancję rozproszoną, czy ośrodek dyspersyjny.
Podział emulgatorów ze względu na charakter chemiczny
Anionowe
Mydła (aniony kwasów tłuszczowych)
oraz aniony monoestrów kwasu
siarkowego
(np. laurylosiarczan Na) i aniony
alkilosulfonowe
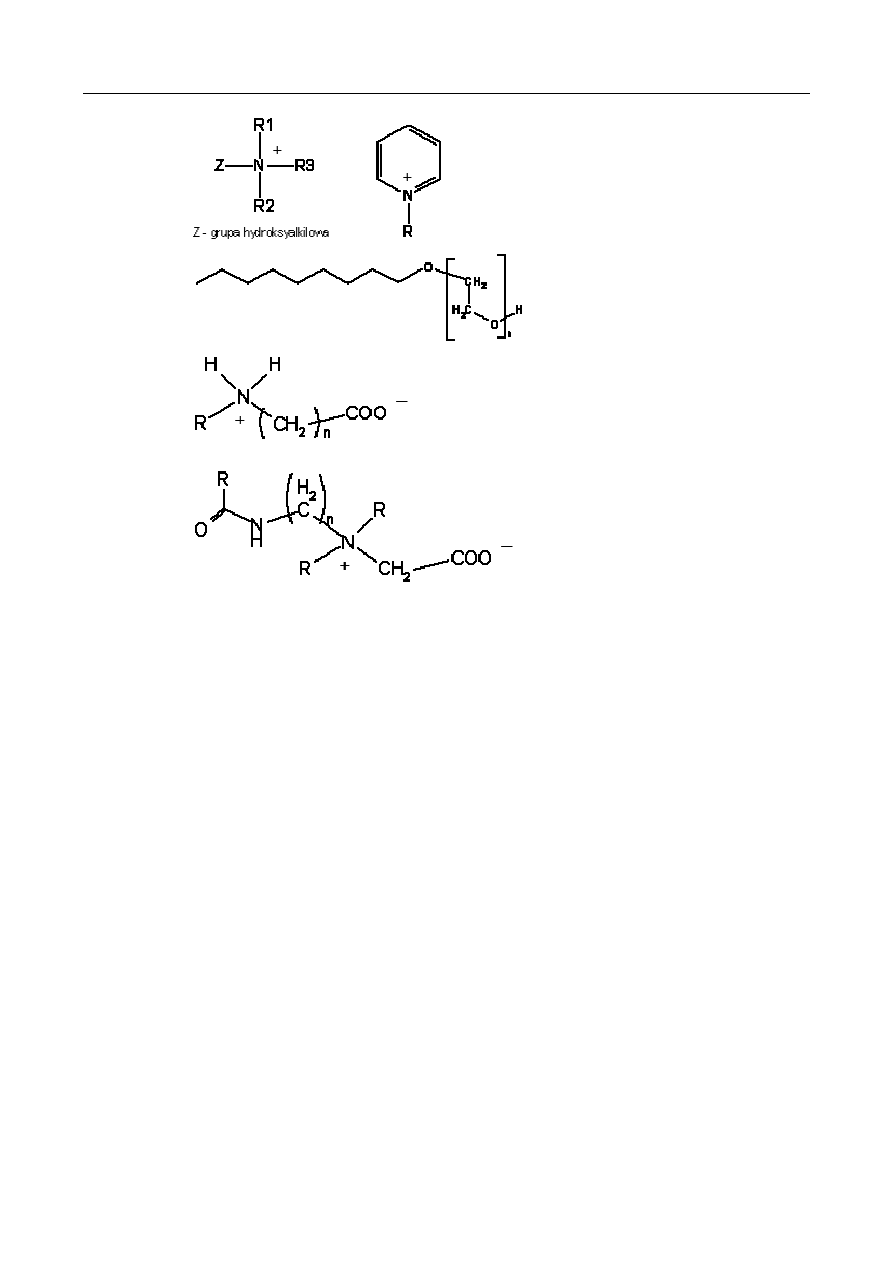
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
Kationowe
Czwartorzędowe sole amoniowe lub
czwartorzędowe heterocykliczne
związki amoniowe (pochodne
pirydyny)
Niejonowe
Fragment hydrofilowy cząsteczki to
najczęściej ugrupowanie alkoholu
wielowodorotlenowego lub łańcuch
poli(oksymetylenowy)
Amfolityczne
Wykazują charakter anionowy lub
kationowy w zależności od pH
roztworu. Sole wewnętrzne
Trwałość układów emulsyjnych
Jeżeli emulsja została wytworzona świadomie, jako postać np. leku czy kosmetyku to zależy
nam na długim okresie trwałości. Bywa jednak i tak, że emulsja powstała samorzutnie w trakcie
jakiegoś procesu technologicznego (częsty przypadek w czasie pracy z surowcami naturalnymi
zawierającymi często substancje spełniające role niepożądanego w tym przypadku emulgatora) i
jest poważnym utrudnieniem w procesie produkcyjnym. Dość często obserwujemy takie zjawisko
podczas procesu ekstrakcji, dokonywanego metodą wytrząsania. W tych przypadkach zależy nam
na szybkim zniszczeniu emulsji i doprowadzeniu do rozdzielenia faz. Pamiętając, że głównym
czynnikiem wpływającym na trwałość emulsji jest napięcie międzyfazowe (powierzchniowe),
zarówno przy tworzeniu, jak i niszczeniu emulsji będziemy się starać wpływać na wielkość tego
parametru. Drugim czynnikiem warunkującym trwałość emulsji jest wielkość kulek fazy
rozproszonej. Emulsja o większej dyspersji będzie trwalsza, ze względu na mniejsza masę
pojedynczej kuleczki i związaną z tym większą ich ruchliwość.
Szybkość rozkładu emulsji jest proporcjonalna do kwadratu promienia kuleczek fazy
rozproszonej, różnicy gęstości obu faz i przyspieszenia działającego na emulsję (zazwyczaj
przyspieszenia ziemskiego, chyba że wirujemy emulsje, wtedy jest to przyspieszenie nadawane
przez wirówkę) a odwrotnie proporcjonalna do lepkości fazy rozpraszającej.
Jeżeli zależy nam na szybkim zniszczeniu niepożądanej emulsji możemy starać się zniszczyć
ochronną warstewkę emulgatora wokół kropel fazy zdyspergowanej. Możemy też starać się
zmieniać pozostałe parametry warunkujące trwałość (lepkość, przyspieszenie), niestety jednak
najczęściej zdarza się tak, że emulsje, na trwałości których nam zależy rozwarstwiają się zbyt
szybko (wie to niejedna gospodyni, która samodzielnie próbowała ukręcić majonez, wie to też
niejeden student farmacji), zaś niepożądane emulsje opierają się najczęściej bardzo skutecznie
naszym staraniom, mającym doprowadzić do ich złamania.

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
98
Procesy starzenia emulsji
Procesy starzenia emulsji to powolne, stopniowe przemiany jej struktury prowadzące do jej
rozdziału, zżelowania, koagulacji lub tylko subtelnych zmian rozmiarów tworzących ją miceli.
Procesy starzenia dzieli się na:
•
śmietankowanie i sedymentację
•
flokulację
•
koalescencję
•
inwersję faz
•
dojrzewanie Ostwaldowskie
Śmietankowanie i sedymentacja emulsji
Jeżeli dwie fazy tworzące emulsję różnią się znacznie ciężarem właściwym, to cięższa faza
przemieszcza się pod wpływem grawitacji do dolnej części naczynia. Śmietankowanie emulsji
można łatwo odwrócić przez zmieszanie warstwy górnej i dolnej. Śmietankowanie można
rozpatrywać jako pewien rodzaj sedymentacji.
Szybkość śmietankowania zwiększają elektrolity, których działanie zależy od ładunku kationu.
Zapobieganie śmietankowaniu polega na rozdrobnieniu emulsji do rozmiaru 0,1 µm, zwiększeniu
lepkości ośrodka dyspersyjnego i zwiększeniu gęstości fazy ciągłej.
Sedymentacja przebiega we wszystkich emulsjach, a czynnikiem determinującym ten proces
jest ziemskie pole grawitacyjne. Proces ten przebiega, gdy krople fazy rozproszonej zaczynają się
zbierać na dole lub górze układu emulsyjnego. Sedymentację zmniejsza się poprzez zmiany
niektórych parametrów emulsji, takich jak:
•
gęstość jednej lub obu faz,
•
rozmiar miceli,
•
ładunek miceli,
•
lepkość fazy zewnętrznej.
Flokulacja emulsji
Koagulacja emulsji polega na łączeniu się cząstek fazy zdyspergowanej, co prowadzi do
powstania dużych agregatów i przebiega ona w dwóch etapach. Pierwszym etapem jest flokulacja,
w której micele łączą się ze sobą i tworzą zgęstniałe bryłki. Czasami flokulacja powoduje
początkowe zwiększenie pozornej lepkości. Proces ten wpływa bardzo źle na strukturę, właściwości
użytkowe i stabilność emulsji. Flokulację można łatwo odwrócić poprzez wstrząsanie całego
układu, ale agregacja kropelek sprzyja sedymentacji. Dla uniknięcia tego zjawiska zwiększa się
stężenie emulgatora lub dobiera taki, który tworzy pełniejszą otoczkę solwatacyjną.
Koalescencja emulsji
Koalescencja emulsji wiąże się z procesem flokulacji, ponieważ jest to drugi etap koagulacji
emulsji. Jeżeli emulsja nie posiada wystarczającej ilości stabilizatora to występuje jego brak na

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
powierzchni micel. Dzięki ruchom Browna micele zderzają się ze sobą, a brak emulgatora
powoduje, że nie mają żadnych przeszkód w łączeniu się ze sobą. Jest to proces nieodwracalny,
który zmniejsza liczbę micel i w konsekwencji prowadzi do rozbicia emulsji. Koalescencję można
ograniczyć poprzez zmniejszenie sedymentacji, zwiększenie wzajemnego odpychania się micel czy
wybór lepszego stabilizatora. Czasami na proces koalescencji dobrze wpływa zwiększenie lepkości
fazy zewnętrznej.
Inwersja faz emulsji
Inwersja faz emulsji polega na nagłej zmianie typu emulsji. Wcześniejsza faza rozproszona staje
się rozpuszczalnikiem, a dawna faza ciągła grupuje się w micele. Inwersję wywołują takie czynniki,
jak: zbyt mała ilość surfaktanta lub nieodpowiedni jego rodzaj, duże zmiany temperatury, za
wysokie stężenie fazy wewnętrznej. Proces ten zachodzi ze wzrostem stężenia fazy rozproszonej,
kiedy micele zaczynają zbliżać się do siebie. Cząsteczki z miceli przenikają przez powierzchnię
międzyfazową i łączą się ze sobą tworząc fazę zewnętrzną. Po tym procesie typ emulsji zmienia się.
Zjawisko obserwuje się dzięki pomiarowi lepkości zmieniając jednocześnie objętość jednej z faz.
Podczas inwersji można wyróżnić trzy etapy:
•
zniszczenie filmu powierzchniowego na granicy dwóch faz,
•
rozwarstwienie się emulsji,
•
ponowne zemulgowanie faz.
Ostwaldowskie dojrzewanie emulsji
Ostwaldowskie dojrzewanie emulsji ma związek z ciśnieniem wewnątrz miceli, które jest
wyższe niż w fazie zewnętrznej. W związku z tym rozpuszczalność fazy wewnętrznej jest wyższa w
mniejszych micelach niż w większych. Podczas długiego przechowywania emulsji różnica ciśnień
prowadzi do dyfuzji składników emulsji fazy wewnętrznej z mniejszych miceli do większych.
Podczas tego procesu następuje wyrównanie ciśnień wewnątrz niejednorodnych miceli. Proces ten
prowadzi do zaniku małych miceli na rzecz dużych. Prowadzi to do sedymentacji lub tzw.
odstawania
się
emulsji.
Zapobieganie
temu
zjawisku
polega
na
wprowadzeniu
wielocząsteczkowego surfaktanta lub zamrażaniu emulsji.
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
100
10.2.
W
YKONANIE ĆWICZENIA
Cel ćwiczenia
Celem ćwiczenia jest zapoznanie się z operacją wytwarzania emulsji na przykładzie majonezu, z
zastosowaniem zmiennej ilości składników i różnego czasu mieszania.
Próba I
Składniki: 1/3 jaja, 83 cm
3
oleju, 1,7 g musztardy, 3,4 cm
3
octu, 1,7 cm
3
przegotowanej wody o
temperaturze pokojowej. Musztarda jest składnikiem stabilizującym układ.
Wykonanie: Jajo z musztardą wodą i octem dokładnie zmiksować do chwili uzyskania jednolitej,
gładkiej masy. Zanotować czas operacji. Uzyskany półprodukt przenieść dokładnie do naczynia
miarowego i zmierzyć jego objętość w cm
3
. Wynik umieścić w tabeli 1 jako V
w
.
Próbę ponownie przenieść do naczynia, w którym poprzednio była mieszana, a następnie, ciągle
mieszając, kroplami dozować olej, przy czym w pierwszych 5 minutach mieszania należy dodać 8,3
cm
3
oleju, w następnych 5 minutach dodać 33,4 cm
3
oleju, natomiast w ostatnich 5 minutach –
pozostały olej. Objętość oleju wykorzystaną do wytworzenia emulsji należy wpisać do tabeli 1
(V
o
). W taki sposób należy postąpić w przypadku każdej próby.
Dokładnie należy notować czas trwania poszczególnych operacji. Uzyskany wynik wpisać do tabeli
3.
Próbę odstawić na 30 minut, a następnie poddać ocenie.
Próba II
Składniki: 1/3 jaja, 83 cm
3
oleju, 1,7 g musztardy, 3,4 cm
3
octu, 1,7 cm
3
przegotowanej wody o
temperaturze pokojowej.
Jajo z musztardą, wodą i octem dokładnie zmiksować do chwili uzyskania jednolitej, gładkiej
masy. Następnie, ciągle mieszając, kroplami dozować olej, przy czym czas mieszania (podczas
dodawania oleju) nie może przekraczać 10 minut. W pierwszych 5 minutach mieszania należy
dodać 41,5 cm
3
oleju i w następnych 5 minutach również 41,5 cm
3
oleju.
Dokładnie notować czas trwania poszczególnych etapów procesu. Wynik wpisać do tabeli 3.
Informacja ta dotyczy również pozostałych prób.
Próbę, jak poprzednio, odstawić na 30 minut, a następnie poddać ocenie. Tak samo należy
postąpić z pozostałymi dwiema emulsjami.
Próba III
Składniki: 2/3 jaja, 83 cm
3
oleju, 1,7 g musztardy, 3,4 cm
3
octu, 1,7 cm
3
przegotowanej wody o
temperaturze pokojowej.
Jajo z musztardą, wodą i octem dokładnie zmiksować do chwili uzyskania jednolitej, gładkiej
masy. Następnie, ciągle mieszając, kroplami dozować olej. Przy czym w pierwszych 5 minutach
mieszania należy dodać 8,3 cm
3
oleju, w następnych 5 minutach dodać 3,4 cm
3
oleju, natomiast w
ostatnich 5 minutach - pozostały olej.
Dokładnie notować czas trwania poszczególnych etapów procesu.
Próba IV
Składniki: 2/3 jaja, 83 cm
3
oleju, 1,7 g musztardy, 3,4 cm
3
octu, 1,7 cm
3
przegotowanej wody o
temperaturze pokojowej.
Jajo z musztardą, wodą i octem dokładnie zmiksować do chwili uzyskania jednolitej, gładkiej masy.
Następnie, ciągle mieszając, kroplami dozować olej, przy czym czas mieszania (podczas dodawania
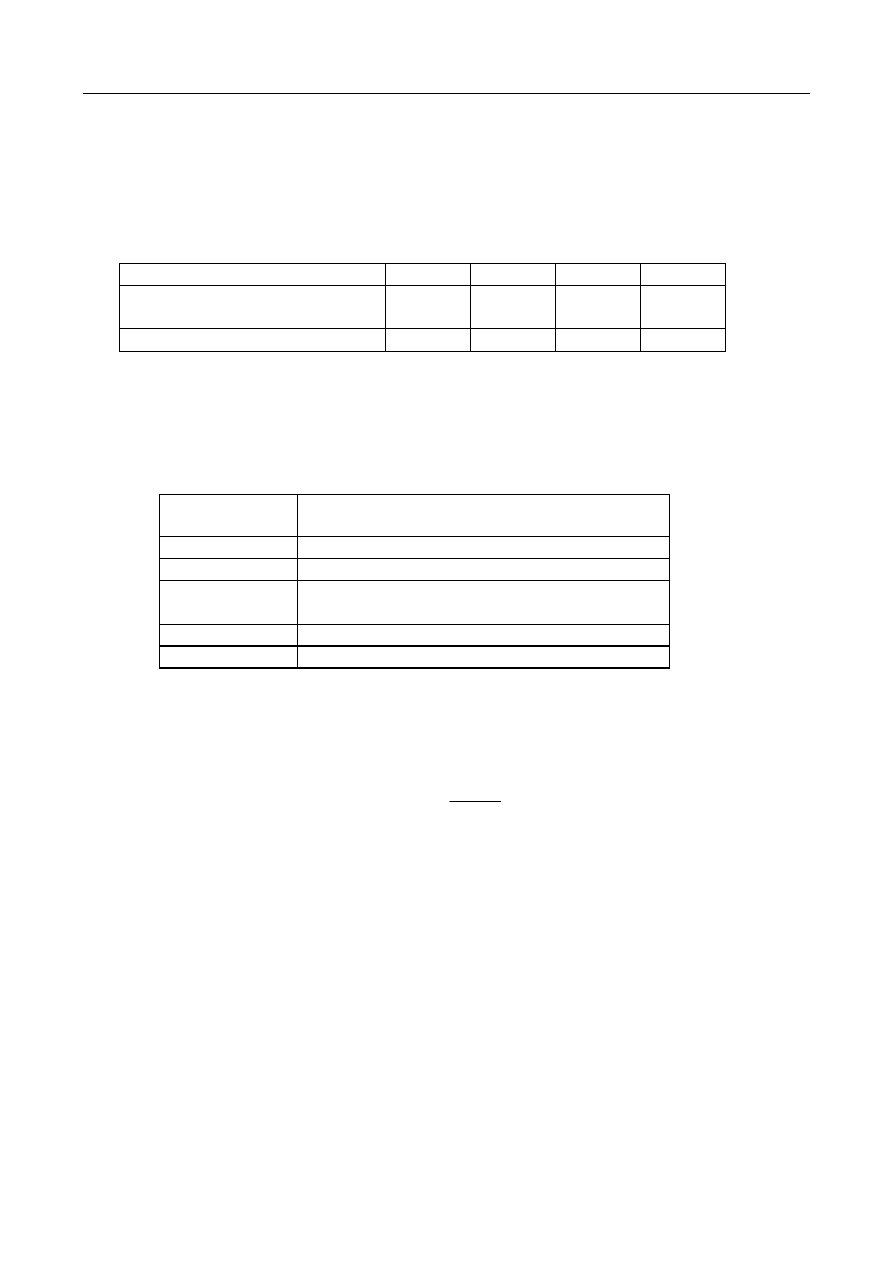
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
oleju) nie może przekraczać 10 minut. W pierwszych 5 minutach mieszania należy dodać 41,5 cm
3
oleju i w następnych 5 minutach również 41,5 cm
3
oleju.
Szczegółowo notować czas operacji.
Dokładny sposób przygotowania emulsji wyjaśni prowadzący ćwiczenia.
Tabela 1. Objętość fazy tłuszczowej i wodnej dla poszczególnych prób.
Składniki
Próba I
Próba II Próba III Próba IV
V
w
[cm
3
]
(jaja + musztarda + woda +ocet)
V
o
[cm
3
] (olej)
Ocena konsystencji emulsji
Ocenę konsystencji należy przeprowadzić zgodnie z określeniami zawartymi w tabeli 2, a
uzyskane wyniki (w punktach) wpisać do tabeli VII.5.
Tabela 2. Wyróżniki charakteryzujące konsystencję.
Liczba
punktów
Konsystencja
5
gładka, aksamitna, jednorodna
4
w miarę jednorodna
3
zauważalne, ale nieznaczne rozdzielanie się
składników - układ nie stanowi jednolitej całości
2
wyraźne rozdzielenie się składników
1
produkt „zwarzony"
Klasyfikacja emulsji
Na podstawie wzoru i danych zamieszczonych w tabeli 1 dokonać klasyfikacji wykonanych
emulsji na emulsje o małej ilości fazy rozproszonej, emulsje o średniej zawartości fazy
rozproszonej bądź emulsje o znacznej ilości fazy rozproszonej.
e
i
i
V
V
V
F
+
=
gdzie:
V
i
- objętość fazy rozproszonej,
V
e
- objętość fazy ciągłej.
Na podstawie wartości F można podzielić emulsje na trzy grupy:
•
F<0,3 – emulsje o małej ilości fazy rozproszonej; zalicza się do nich mleko, lody, sery,
masło i margarynę;
•
0,3<F<0,7 – emulsje o średniej zawartości fazy rozproszonej (kremy)
•
F>0,7 – emulsje o znacznej ilości fazy rozproszonej – np. majonezy i dressingi.
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
102
Uzyskaną wartość F dla każdej próby wpisać do tabeli 3.
Nr próby
Czas mieszania
(łącznie z dodatkiem
wody) [min]
Konsystencja [pkt]
F
Próba I
Próba II
Próba III
Próba IV
Test rozpuszczalności (Wygląd makroskopowy)
Umieścić bagietkę w próbce majonezu i mieszając dodawać powoli po 1 ml wody kranowej (max.
10 ml), obserwując wygląd majonezu. Emulsja W/O będzie unosić się na wodzie, zaś emulsja O/W
wymiesza się, powodując zmętnienie wody. Należy opisać wynik w sprawozdaniu.
Barwienie (Wygląd mikroskopowy)
Umieścić kroplę mieszaniny z poprzedniego testu na szkiełku podstawowym, dodać dwie krople
1% błękitu metylenowego (w wodzie), dokładnie wymieszać, przykryć szkiełkiem nakrywkowym.
Obserwować pod mikroskopem (450x). Wykonać rysunek spod mikroskopu (kształt, regularność i
kolor kropelek). Powtórzyć wszystko z barwnikiem Sudan III.
Opracowanie wyników
Na podstawie przeprowadzonego doświadczenia należy:
1.
Omówić, czy zaobserwowano zależności między czasem mieszania a konsystencją uzyskanej
emulsji.
2.
Ocenić, czy zmienna zawartość jaj zawierających środek emulgujący wpływała na konsystencję
produktu końcowego.
3.
Omówić wartości współczynnika F dla poszczególnego rodzaju majonezu.
4.
Ocenić, która z podanych receptur i technologii pozwoliła na uzyskanie produktu o najlepszej
jakości. Spróbować wyjaśnić, dlaczego.
5.
Podać uwagi (zastrzeżenia) do pozostałych procedur, wskazując na rozwiązanie zaistniałych
problemów.

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
11.
TERMICZNE UTRWALANIE śYWNOŚCI
11.1.
W
PROWADZENIE
Pasteryzacja i sterylizacja są operacjami jednostkowymi, w których żywność jest poddana
działaniu dostatecznie wysokiej temperatury i przez dostatecznie długi czas w celu zwiększenia jej
trwałości w czasie przechowywania. Podstawowym założeniem utrwalania żywności przez
ogrzewanie jest osiągnięcie jej mikrobiologicznej stabilności. Proces ten można podzielić na 3
kategorie: ogrzewanie do temperatur niższych niż 100°C, ogrzewanie w temperaturze 100°C i
powyżej 100°C. Proces ogrzewania poniżej 100°C jest nazywany pasteryzacją i powoduje
inaktywację wszystkich mikroorganizmów patogennych oraz niektórych ich gatunków
powodujących psucie żywności. Dobrym jej przykładem jest utrwalanie mleka. Drugi sposób,
apertyzacja, którego wynalazcą był Nicholas Appert, ciągle jest wykorzystywany w przetwórstwie
domowym, a na skalę przemysłową do produkcji konserw kwaśnych, np. owocowo-warzywnych.
Trzeci sposób, ogrzewanie powyżej 100°C, przeznaczony jest dla konserw o pH wyższym od 4,5 i
określany jest terminem sterylizacja. W czasie sterylizacji giną nie tylko wegetatywne, ale również
przetrwalnikujące formy mikroorganizmów. Dzięki bardziej surowym warunkom termicznym
sterylizacji utrwalona w ten sposób żywność zachowuje dobrą jakość, co najmniej 6 miesięcy.
Wpływ wysokiej temperatury na mikroorganizmy i niektóre składniki produktów
spożywczych
Najprostsze ujęcie kinetyczne procesu sterylizacji zakłada, że mszczenie mikroorganizmów
przebiega według kinetyki reakcji pierwszego rzędu. Jest to usprawiedliwione takim charakterem
reakcji termicznej denaturacji białek. Utrwalające działanie ciepła polega właśnie na denaturacji
białek, co pociąga za sobą inaktywację enzymów i mikroorganizmów występujących w żywności.
Jednakże złożoność budowy drobnoustrojów, możliwość występowania różnych form o różnej
termoodporności może spowodować, że przebieg rzeczywistego procesu odbiega czasem od
kinetyki pierwszego rzędu. W myśl tej kinetyki zmiana liczby drobnoustrojów w czasie jest
proporcjonalna do ich chwilowej liczby:
N
k
dt
dN
⋅
=
−
gdzie: -dN/dt - szybkość, z jaką zmniejsza się liczba drobnoustrojów,
N -liczba mikroorganizmów,
k - stała szybkość reakcji pierwszego rzędu.
Podobne równanie opisuje termiczną denaturację witamin i enzymów. Stała szybkości k zależna
jest od rodzaju i formy drobnoustrojów oraz od temperatury i innych czynników środowiskowych.
Zakładając, że proces sterylizacji przebiega okresowo i w stałej temperaturze, powyższe równanie
można całkować, przyjmując, że na początku sterylizacji liczba drobnoustrojów jest równa N.
Wówczas:
t
k
N
N
o
⋅
−
=
ln
Powyższe równanie wskazuje, że w danym czasie jest niszczony taki sam procent komórek
niezależnie od ich liczebności na początku procesu. Zjawisko to, zgodne z rzeczywistym
zachowaniem się ogrzewanych do odpowiednio wysokiej temperatury komórek, określa się jako
logarytmiczne tempo umierania, a opisuje je tzw. krzywa przeżycia drobnoustrojów. Przedstawia
ona zależność między liczbą komórek przeżywających proces termiczny w stałej temperaturze a
czasem jej działania (rys.1).
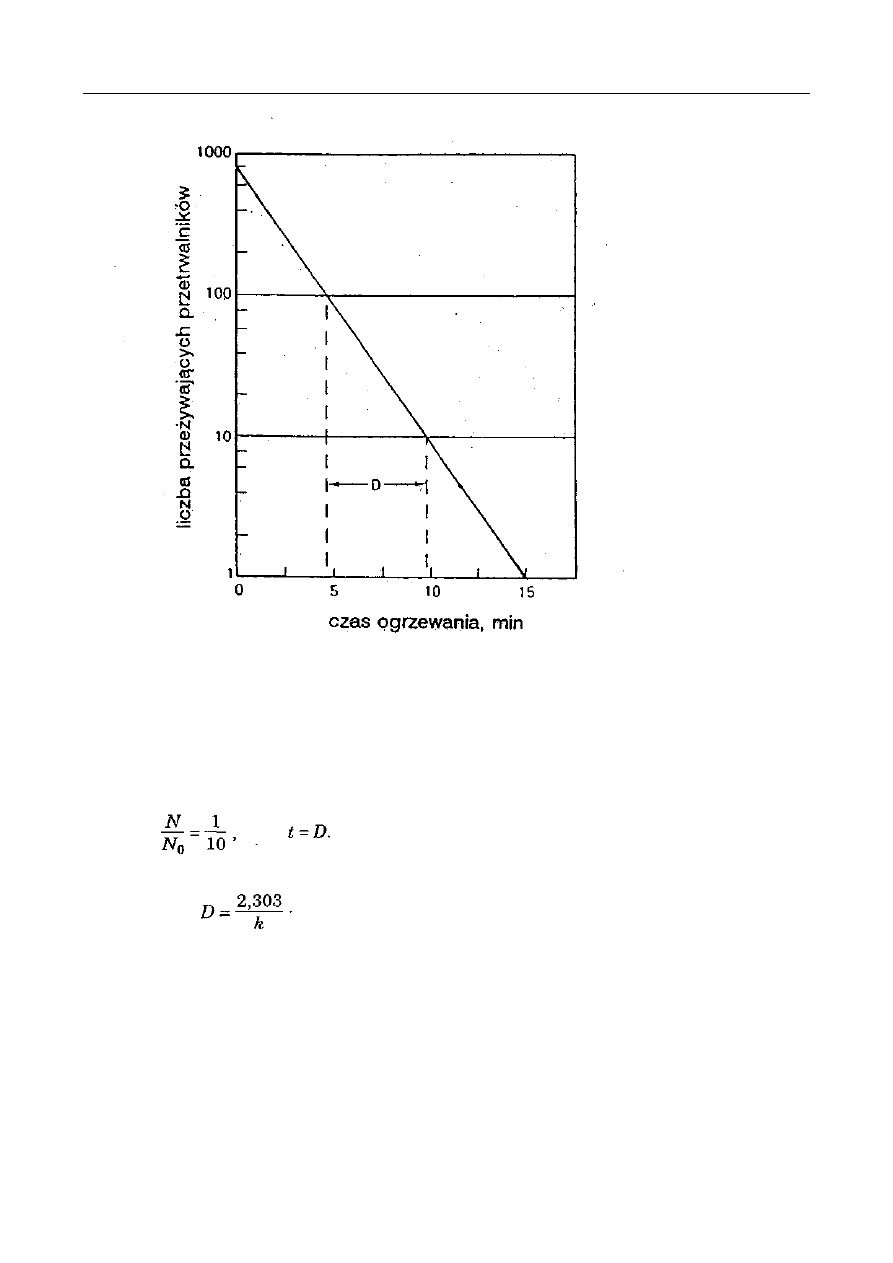
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
104
Rys. 1. Krzywa przeżycia drobnoustrojów.
Do opisu procesu sterylizacji - oprócz powyżej przedstawionych równań kinetycznych - stosuje
się pewne tradycyjne pojęcia. Związki pomiędzy nimi i ich definicje wynikają z przekształceń
równania kinetycznego.
Równanie definicyjne czasu dziesięciokrotnej redukcji liczby drobnoustrojów D otrzymuje się,
podstawiając
Parametr D (w warunkach pozwalających na założenie stałości współczynnika k) opisuje zatem
związki pomiędzy liczbą drobnoustrojów i czasem działania podwyższonej temperatury. Zależny
jest (podobnie jak stała szybkości k) od rodzaju i formy drobnoustrojów oraz od temperatury i
innych czynników środowiskowych. Jego wartość można odczytać z krzywej przeżycia drobno-
ustrojów. Jest to czas potrzebny do zniszczenia 90% mikroorganizmów lub, inaczej mówiąc, do
dziesięciokrotnego zredukowania ich liczby (na rys. 1 wynosi on 5 min). Wartość D jest niezależna
od stężenia początkowego drobnoustrojów.
Z zależności przedstawionej na rycinie 1 wynikają następujące konsekwencje. Po pierwsze, im
większa liczba mikroorganizmów jest obecna w surowcu, tym dłuższego czasu ogrzewania
potrzeba, aby zredukować ją do żądanego poziomu. Po drugie, ponieważ śmierć komórek następuje
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
w tempie logarytmicznym, praktycznie nie jest możliwe uzyskanie absolutnej sterylności, gdyż
jeżeli liczba komórek zbliża się do zera, to czas ogrzewania dąży do nieskończoności. Dlatego też
wprowadzono pojęcie sterylności handlowej albo sterylności technicznej, przez które rozumie się
zniszczenie wszystkich drobnoustrojów chorobotwórczych i zredukowanie mikroflory saprofitycz-
nej (łącznie z jej formami przetrwalnikowymi) do określonego, dostatecznie niskiego poziomu,
gwarantującego bardzo małe ryzyko zepsucia się zakonserwowanej żywności, np. l opakowanie na
10 000 opakowań.
Czas dziesięciokrotnej redukcji, czyli oporność cieplna D, jest także funkcją temperatury
ogrzewania D = φ(T). Jego zmienność temperaturowa charakteryzowana jest przez współczynnik
ciepłooporności z. Ponieważ na ogół przyjmuje się, że wartość D zmienia się w tempie
logarytmicznym wraz z temperaturą (zależność lgD= φ(T) jest prostoliniowa), zatem współczynnik
z definiowany jest jako przyrost temperatury odpowiadający dziesięciokrotnej zmianie
współczynnika D. Zależny jest od rodzaju drobnoustrojów. Jego graficznym obrazem jest krzywa
określana jako krzywa oporności cieplnej lub krzywa czasu śmierci cieplnej TDT (rys. 2). Wynika z
niej, że w wyższych temperaturach komórki giną szybciej (np. działanie temperaturą 102,5°C w
czasie 100 min daje ten sam letalny efekt, co temperatura 113°C w czasie 10 min). Nachylenie
krzywej, które można wyrazić wielkością tangensa kąta
α
, stanowi o wielkości wskaźnika z. Im
bardziej stromy jest przebieg krzywej, tym mniejsza jest wartość z. Wartość z łatwo można
odczytać z rysunku krzywej TDT (czasu śmierci cieplnej) w skali półlogarytmicznej jako zmianę
temperatury potrzebną, aby czas dziesięciokrotnej redukcji D zmniejszył się o jeden cykl
logarytmiczny, czyli dziesięciokrotnie (na rys. 5.2 wynosi on 10,5°C).
Rys. 2. Krzywa czasu śmierci cieplnej drobnoustrojów.
Wartości D i z są stosowane do charakterystyki ciepłooporności enzymów, mikroorganizmów
lub składników chemicznych żywności (tab. 1).
Do opisania zależności stałej szybkości k od temperatury najczęściej stosowane jest równanie
Arrheniusa:
⋅
−
=
T
R
E
k
k
exp
0
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
106
gdzie: E - energia aktywacji, R - stała gazowa i T - temperatura (K), k
o
-stała.
Wielkości energii aktywacji dla wegetatywnych form komórek oraz ich przetrwalników
mieszczą się w zakresie 210-420 kJ/mol i są większe od energii aktywacji dla enzymów lub
witamin (8,4-110 kJ/mol). Różnica między tymi wartościami tłumaczy, dlaczego możliwe jest
prowadzenie sterylizacji przy jednoczesnym częściowym zachowaniu właściwości odżywczych
żywności. Im wyższa jest temperatura procesu, tym szybciej przebiega ten z procesów, który ma
wyższą energię aktywacji, i dlatego proces sterylizacji powinien być prowadzony jak najkrócej oraz
w jak najwyższych temperaturach. Szybkość procesu o wyższej energii aktywacji szybciej się
zmienia przy podwyższeniu temperatury niż szybkość procesu o małej energii aktywacji (mniej
czułego na zmiany temperatury). Na rycinie 3 przedstawiono przykładową zależność pomiędzy 10-
12 krotnym czasem redukcji Cl. botulinum a czasem 90-procentowej denaturacji tiaminy (witaminy
B1).
Tabela.1. Wartości D i z dla niektórych mikroorganizmów i składników chemicznych żywności
Składnik
Źródło
pH
z
[°C]
D
121
[min]
Zakres
temperatur
[°C]
Tiamina
marchew
5,9
25
158
109-149
Lizyna
ziarno soi
—
21
786
100-127
Chlorofil a
szpinak
6,5
51
1.3,0
127-149
Chlorofil b
szpinak
5,5
79
14,7
127-149
Antocyjany
sok grejpfrutowy
nat.
23,2
17,8*
20-121
Betanina
sok buraczany
5.0
58,9
46,6*
50-100,
Karotenoidy
papryka
nat.
18,9
0,038
52-65
Peroksydaza
groszek
nat.
37,2
3,0
110-138
Clostridium botulinum
różne
>4,5
5,5-10
0,1-1,3
104
B. stearothermophilus
różne
>4,5
7-12
4,0-5,0
110
* Wskaźnik D dla temperatur innych niż 121°C
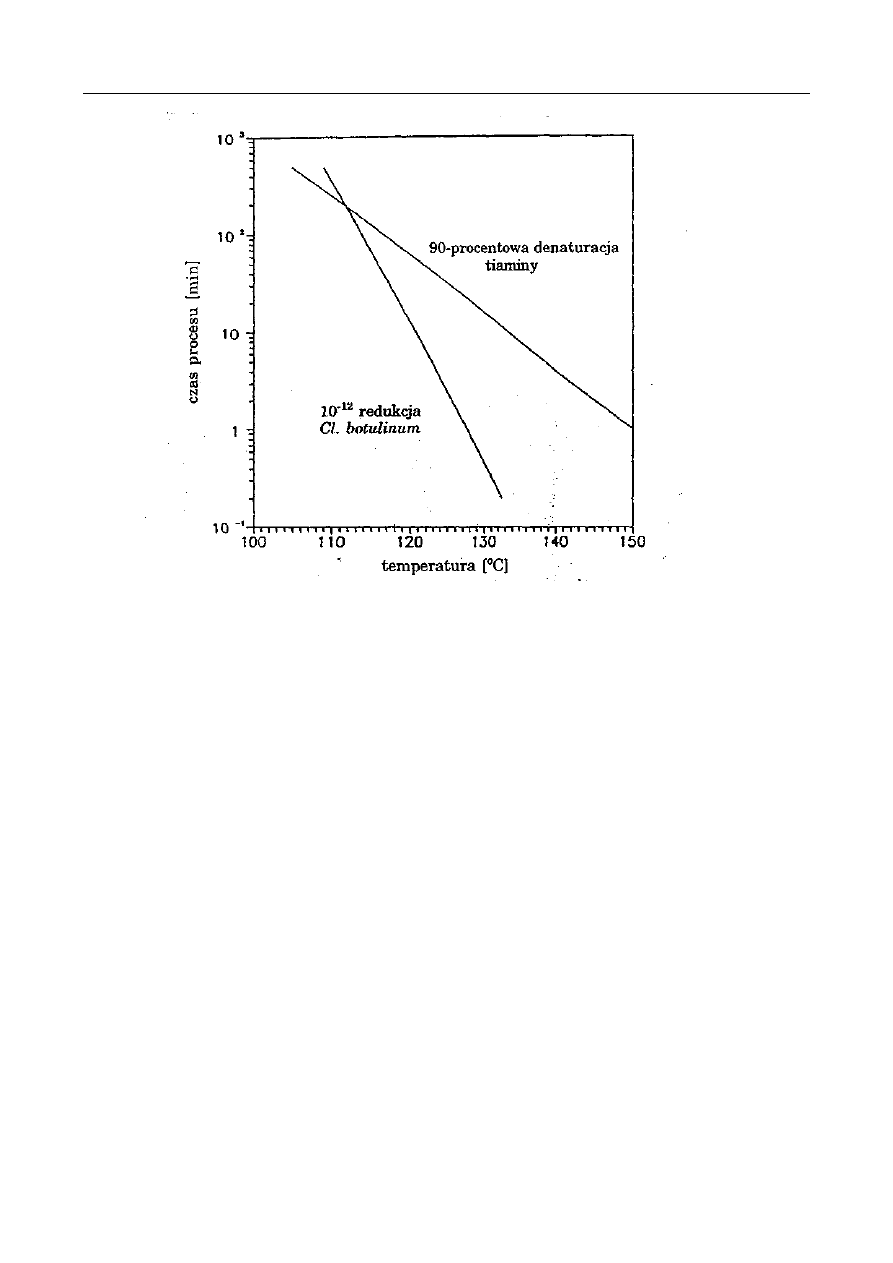
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
Rys. 3. Zależność między 10-12 krotnym czasem redukcji Cl. botulinum a czasem 90-% denaturacji
tiaminy
Na wykresie widać, że dla temperatur wyższych niż 115°C osiąga się wymagany stopień
sterylności przy minimalizacji strat witaminy B1. Straty innych witamin będą jeszcze mniejsze ze
względu na ich mniejsze energie aktywacji.
Dlatego też w nowoczesnych systemach pasteryzacji i sterylizacji dąży się do zastąpienia
ogrzewania w niższych temperaturach letalnych i w dłuższym czasie przez ogrzewanie w wysokich
temperaturach i w krótszym czasie przy takim samym efekcie biologicznym. Postępowanie takie
przyczynia się do ochrony składników odżywczych w procesie wyjaławiania cieplnego i jest znane
pod nazwą zasady wysokiej temperatury i krótkiego czasu albo HTST (High Temperaturę Short
Time), względnie systemu ultrawysokiej temperatury, czyli UHT (Ultra High Temperature)
Zasada UHT znalazła zastosowanie praktyczne w metodzie sterylizacji produktów płynnych
(np. mleka) przed ich zapakowaniem.
Na oporność cieplną mikroorganizmów mają wpływ następujące czynniki:
1.
Rodzaj drobnoustrojów i forma, w jakiej występują. Przetrwalniki bakteryjne są bardziej
ciepłooporne niż komórki wegetatywne. Spory pleśni mają niską ciepłooporność, zbliżoną do
cieplooporności komórek wegetatywnych bakterii mezofilnych. Najmniej oporne na ogrzewanie są
drożdże i to zarówno formy wegetatywne, jak i przetrwalnikowe.
2.
Warunki środowiska, w jakich przebywają podczas traktowania ciepłem, a są to:
•
Zawartość wody tak w środowisku, jak i w komórkach drobnoustrojów. Większą oporność
cieplną przetrwalników tłumaczy się między innymi tym, że zawierają one mniej wody niż
komórki wegetatywne. Podczas niszczenia przetrwalników ogrzewanie za pomocą pary wodnej
jest bardziej efektywne niż tzw. „ciepło suche";
•
Składniki żywności. Obecność tłuszczów, białek i wysoka koncentracja cukru zwiększa
oporność
cieplną
mikroorganizmów.
Szczególnie
tłuszcze
chronią
komórki
drobnoustrojów, tworząc dookoła nich otoczki pozbawione wody;
•
pH żywności. Bakterie patogenne i gnilne są bardziej ciepłooporne w środowisku o pH
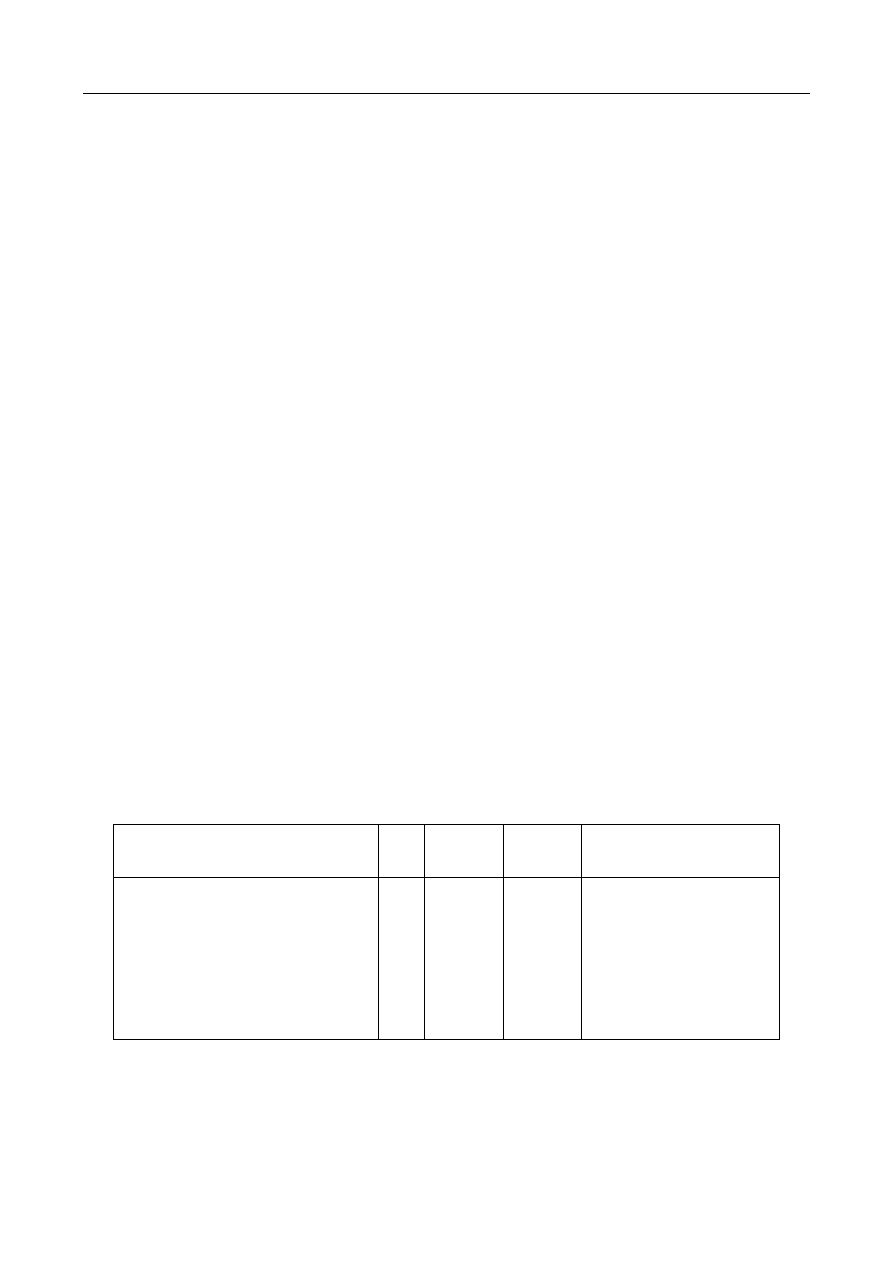
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
108
bliskim naturalnego, drożdże i grzyby tolerują bardziej kwaśne środowisko, ale są mniej
ciepłooporne niż przetrwalniki bakteryjne.
W zależności od pH żywność dzieli się na trzy zasadnicze grupy:
a)
żywność niekwaśna i mało kwaśna - o pH większym od 4,6, np. mleko, drób, ryby, groszek,
fasola, szpinak, buraki;
b)
żywność kwaśna - o pH od 3,7 do 4,6, do której zalicza się gruszki, morele, pomidory,
czerwoną kapustę;
c)
żywność bardzo kwaśna - o pH mniejszym od 3,7, np. kwaszona kapusta, kwaszone ogórki,
większość owoców.
śywność mało kwaśna do termicznego utrwalenia wymaga ogrzewania w temperaturze powyżej
100°C, natomiast w przypadku żywności kwaśnej ten sam efekt można osiągnąć przez ogrzewanie
w temperaturach nie przekraczających 100°C. Niektóre rodzaje bardzo kwaśnej żywności czy też
żywność o niskiej aktywności wodnej, jak słodzone zagęszczone mleko oraz suszone produkty, do
utrwalenia nie wymagają wcale lub też wymagają bardzo delikatnego ogrzewania głównie ze
względu na obecność drożdży, pleśni czy enzymów powodujących ich psucie.
Większość enzymów występujących w żywności ma wartości D i z zbliżone do tychże dla
mikroorganizmów i dlatego ulegają inaktywacji podczas normalnego procesu ogrzewania. Jednakże
niektóre z nich są bardziej ciepłooporne i szczególnie w kwaśnej żywności może zachodzić
niecałkowita ich denaturacja w związku ze stosunkowo krótkim czasem ogrzewania i niższą tempe-
raturą stosowaną do inaktywacji mikrobiologicznej.
Znajomość ciepłooporności drobnoustrojów lub enzymów znajdujących się w produktach
spożywczych jest wykorzystywana do ustalania warunków termicznych gwarantujących ich
zniszczenie. W praktyce problem ten sprowadza się do ustalenia takiego mikroorganizmu spośród
szkodliwej mikroflory występującej w danym asortymencie żywności, którego zniszczenie wymaga
stosunkowo największej dawki ciepła. Taki drobnoustrój uznaje się za krytyczny i dla niego jest
ustalana wielokrotność wartości D (czasu dziesięciokrotnej redukcji). W tabeli 2 przedstawiono
przykłady bakterii przyjmowanych za krytyczne przy sterylizacji cieplnej mało kwaśnych
produktów spożywczych.
Tabela 2. Bakterie przetrwalnikowe uznane za mikroflorę krytyczną w procesie termicznego
utrwalania mało kwaśnej żywności (w nawiasach podano optimum temperaturowe)
Rodzaj bakterii
n
z
[°C]
D121
[min]
Rodzaj
żywności
Bakterie termofilne (35-55°C):
Bacillus stearothermophilus
5
10,0
4,0
jarzyny, mleko
Cl. thermosaccharolyticum
5 7,2-10,0
3,0-4,0 jarzyny
Bakterie mezofilne (10-40°C):
Clostridium sporogenes
5 8,8-11,1
0,8-1,5 mięso
Bacillus subtilis
6
4,1-7,2
0,5-0,76 produkty mleczne
Clostridium botuiinum typ A i B
12 7,7-10,0
0,1-0,2 żywność mało kwaśna
W żywności mało kwaśnej za najbardziej niebezpieczny, ciepłooporny i wytwarzający
przetrwalniki mikroorganizm jest uznawany Clostridium botulinum, produkujący w warunkach
beztlenowych bardzo silne egzotoksyny. W praktyce międzynarodowej przyjęto, że wielokrotność
redukcji dziesiętnej w jego przypadku powinna wynosić 12, czyli l przetrwalnik na 10-12 tys.
opakowań, zakładając, że przed sterylizacją w opakowaniu znajduje się l przetrwalnik. Jednakże ze
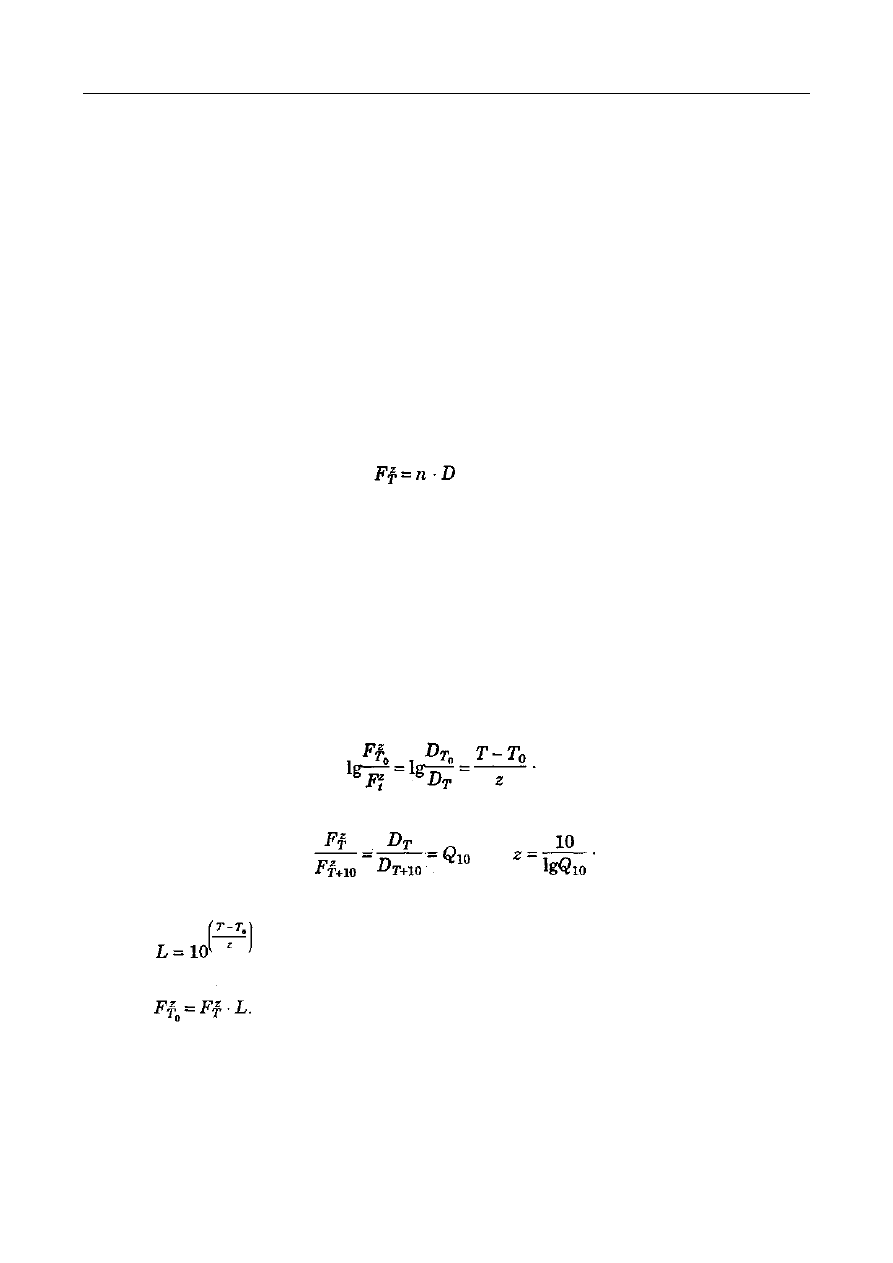
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
względu na prawdopodobieństwo występowania jeszcze bardziej ciepłoopornych bakterii, np.
Clostridium sporogenes, istnieje konieczność dodatkowego wydłużenia czasu ogrzewania.
W żywności kwaśnej i bardzo kwaśnej czynnikiem ograniczającym trwałość konserwy jest
działalność enzymów, stąd też warunki ogrzewania są łagodniejsze (wystarcza pasteryzacja).
Obliczanie czasu sterylizacji
W zależności od sposobu prowadzenia procesu sterylizacji (okresowo lub przepływowo)
podstawowym parametrem technologicznym jest odpowiednio czas trwania procesu oraz czas
przebywania w aparacie. W pewnym przybliżeniu czas przebywania można obliczać tak samo, jak
czas trwania procesu okresowego, zakładając, że przepływ produktu w sterylizatorze jest tłokowy.
Dokładniejsze obliczenia wymagają uwzględnienia rozkładu czasów przebywania (np. dla past lub
skoncentrowanych soków).
Czas trwania sterylizacji (dla danego poziomu redukcji liczby drobnoustrojów równego 10n)
zależy od temperatury procesu i ciepłooporności drobnoustrojów.
W warunkach zapewniających stałość współczynnika z i temperatury T czas sterylizacji można
obliczyć z zależności:
przy czym n = lg(N
0
/N),
gdzie: N
0
- początkowa liczba mikroorganizmów,
N - końcowa liczba mikroorganizmów. Początkowa liczba drobnoustrojów powinna
uwzględniać tylko spory, gdyż formy wegetatywne giną w niższych temperaturach.
Założony efekt sterylizacji zostanie osiągnięty tylko wówczas, gdy w całej masie
sterylizowanego produktu osiągnięta zostanie ta sama temperatura. Ponadto, jeśli produkt nie jest
jednorodny (np. zawiesina, ciało stałe), to spory mogą być izolowane od bezpośredniego działania
pary wodnej i łatwiej przeżyć sterylizację.
Proces sterylizacji prowadzony bywa czasem w innej temperaturze niż ta, dla której znana jest
wartość współczynnika D. Wymagany czas sterylizacji F
z
t
można przeliczyć z czasu F
z
To
, stosując
współczynnik z, za pomocą zależności
Powyższe wielkości wiążą się również z współczynnikiem Q
10
:
Wprowadza się kolejną wielkość zwaną szybkością śmierci termicznej i wówczas:
Jest ona zatem stosunkiem czasu trwania procesu w temperaturze T
0
(najczęściej 121°C) do
czasu jego trwania w temperaturze T. Jeżeli np. temperatura produktu wynosi 111°C, to oznacza, że
1,0 min sterylizacji przy temperaturze 111°C daje ten sam efekt letalny co 0,1 min przy 121°C.
Innymi słowy, jeżeli F
z
To
dla danego procesu równa się 1,0 min, wtedy L = 0,1 jest ułamkiem
jednostki sterylności spełnionym dla l min przy temperaturze T (tu 111°C). Natomiast jednostką
sterylności jest dawka ciepła powodująca śmierć mikroorganizmu w ciągu l min w
temperaturze121°C.
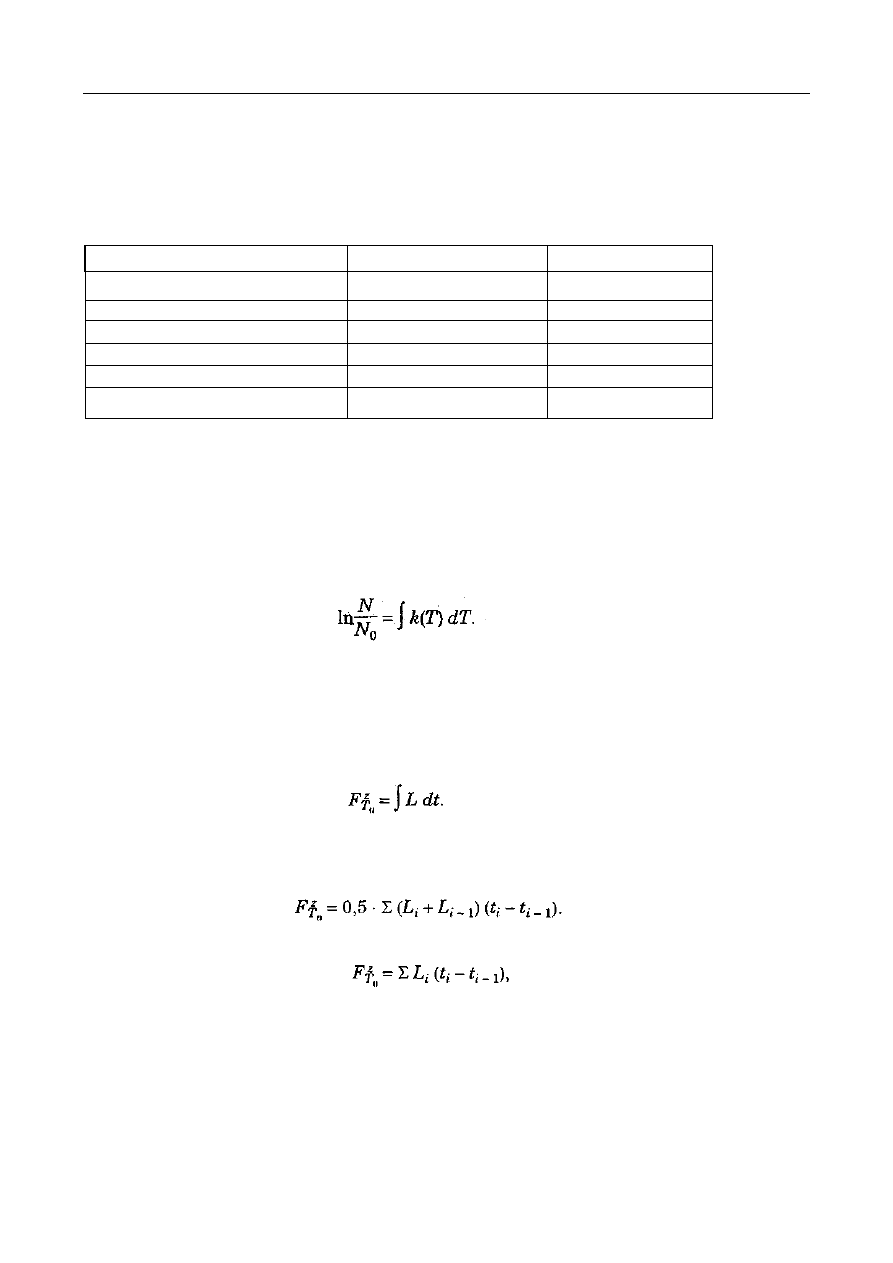
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
110
Przy obliczaniu dawki ciepła potrzebnej do uzyskania sterylności handlowej umownie przyjęto
symbol F
o
oznaczający liczbę minut potrzebną, aby w temperaturze 121°C zniszczyć
mikroorganizm, którego z=10. Przykładowe wielkości F
o
przedstawiono w tabeli 3. Przy produkcji
nowego asortymentu wartość F
o
należy ustalić doświadczalnie.
Tabela 3. Wartości Fo stosowane dla uzyskania słerylności handlowej niektórych konserw
Produkt
Wielkość puszki
F
o
[min]
Fasola w sosie pomidorowym
wszystkie
4-6
Groszek w zalewie
220 g
6
Marchewka
wszystkie
3-4
Grzyby w zalewie
A1
8-10
Siedź w pomidorach
owalne
6-8
Mięso w sosie
wszystkie
12-15
Rzeczywisty proces sterylizacji składa się z trzech etapów: ogrzania do zadanej temperatury,
przetrzymania w niej i następnie chłodzenia. Sumaryczny efekt działania temperatury podczas tych
trzech etapów można obliczyć na dwa sposoby, wykorzystując równanie pierwotne kinetyki
pierwszego rzędu lub podane wyżej zależności.
W pierwszej metodzie przyjmuje się, że stała szybkości reakcji k jest zmienna wraz z
temperaturą, zatem znając różne wartości k dla różnych temperatur T i wartość temperatury w
danym czasie t, oblicza się efekt sterylizacji z zależności:
Całka ta może być obliczona analitycznie, numerycznie lub graficznie.
Poniżej pokazany jest drugi sposób. Przeliczenie takie może być konieczne w przypadku, gdy
sterylizowany produkt jest czuły na wysoką temperaturę i nie powinien w niej przebywać dłużej niż
potrzeba.
Jeśli rozpatruje się chwilowe wartości L, to ekwiwalentny czas sterylizacji można obliczyć z
wzoru:
Przechodząc do skończonych wartości przyrostu czasu, można uznać, że w przedziałach ∆t
szybkość śmierci termicznej jest liniowa. Wartość F
z
To
można obliczyć, stosując zależność (wzór
trapezów):
Jeszcze prościej oblicza się całkę, stosując wzór prostokątów:
jednakże jest on wystarczająco dokładny przy dużej liczbie przedziałów czasu ∆t. Oczywiście
tak uproszczony sposób rozwiązania całki dt nie zawsze jest wystarczająco dokładny. Należy
wówczas skorzystać z innych metod numerycznych. Tych samych obliczeń można dokonać, rysując
krzywą szybkości śmierci termicznej w układzie współrzędnych: szybkość śmierci termicznej (L) –
czas (t). Powierzchnia poniżej krzywej równa jest co do wartości F
z
To
(rys. 4).
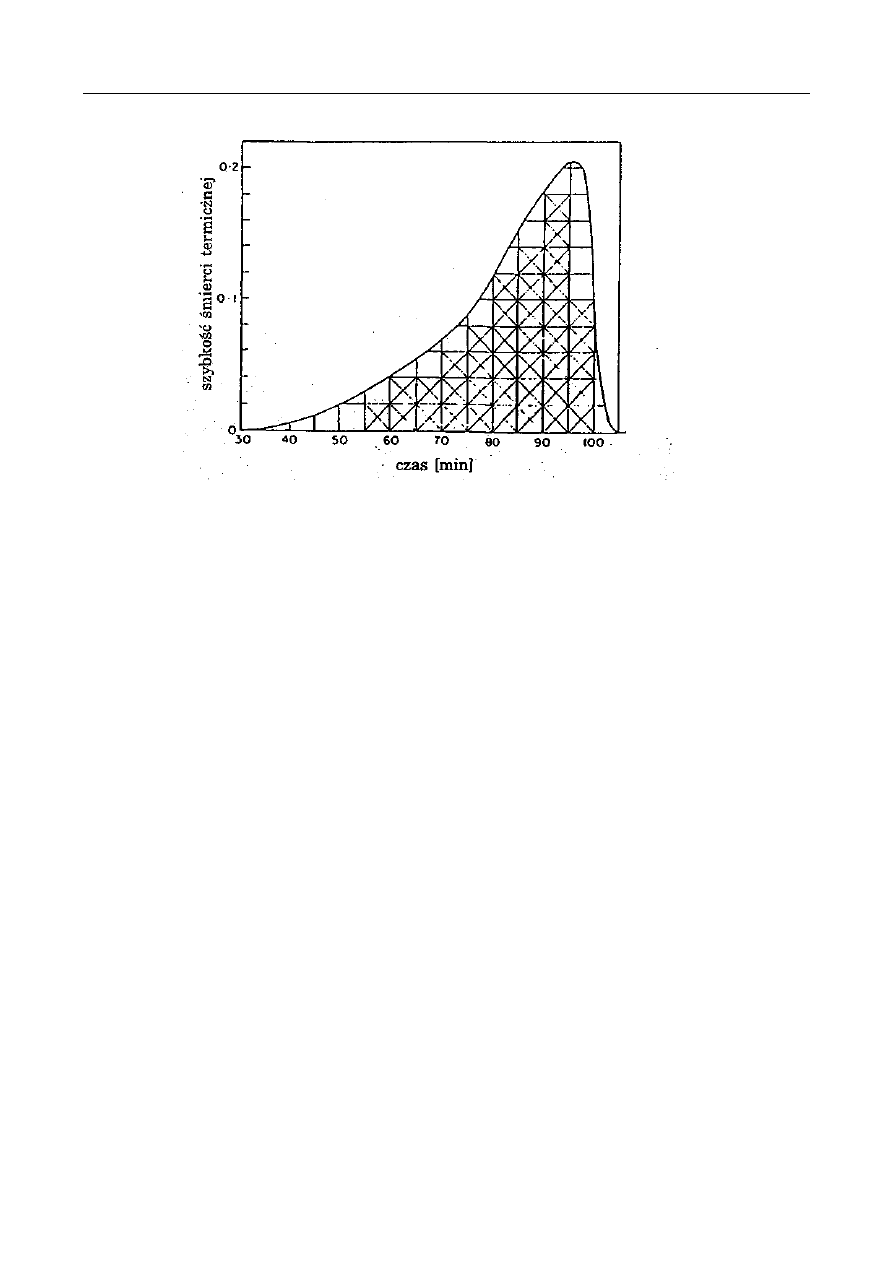
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
Rys .4. Graficzne wyznaczanie wartości F
z
To
Metody sterylizacji
Obecnie w przemyśle są stosowane dwie podstawowe metody sterylizacji cieplnej:
•
sterylizacja żywności w opakowaniach hermetycznych, czyli tzw. apertyzacja,
•
sterylizacja żywności przed zapakowaniem i aseptyczne pakowanie.
•
w niektórych przypadkach stosuje się tzw. sterylizację dwustopniową, będącą
kombinacją obu wyżej wymienionych metod.
Proces apertyzacji
Jako opakowania do konserw mogą służyć puszki metalowe, naczynia szklane i zgrzewalne
opakowania z tworzyw sztucznych. Puszki przeznaczone do pakowania produktów zawierających
barwniki antocjanowe są dodatkowo powlekane od wewnątrz różnymi odmianami emalii i
lakierów, dzięki czemu nie ulegają korozji, a produkty nie zmieniają barwy.
W przygotowaniu warzyw, a niekiedy owoców, mięsa i innych surowców przeznaczonych do
puszkowania bardzo ważnym procesem jednostkowym jest blanszowanie będące także wstępem do
mrożenia i suszenia. Głównym jego zadaniem jest inaktywowanie enzymów albo rozłożenie
substratów enzymatycznych, np. nadtlenków. Obecne w żywności czynne enzymy mogą spowodo-
wać niekorzystne zmiany barwy, zapachu i smaku podczas przerobu i przechowywania produktów.
Ogrzewanie we wrzącej wodzie inaktywuje np. oksydazę o-difenolową i przez to zapobiega
późniejszemu ciemnieniu surowców o jasnym miąższu (np. jabłka, ziemniaki, groszek) w wyniku
enzymatycznego utleniania związków fenolowych. Konsekwencją blanszowania jest także:
zmniejszenie zakażeń mikrobiologicznych, usuwanie gazów z komórek, zmiękczanie żywności i jej
kurczenie się, co ułatwia napełnianie opakowań, ale również straty rozpuszczalnych w wodzie
składników odżywczych.
W konserwach z zalewą w zamkniętej puszce powinna pozostać nieduża przestrzeń wolna
wynosząca ok. 0,25 cm. Jej brak może prowadzić do bombaży technicznych, natomiast zbyt duża
przestrzeń wolna może być przyczyną uszkodzeń stałych części konserwy podczas transportu.
Bardzo istotną operacją w procesie apertyzacji jest odpowietrzenie napełnionych opakowań. Ma
ono na celu:
•
ograniczenie szkodliwych procesów oksydacyjnych,
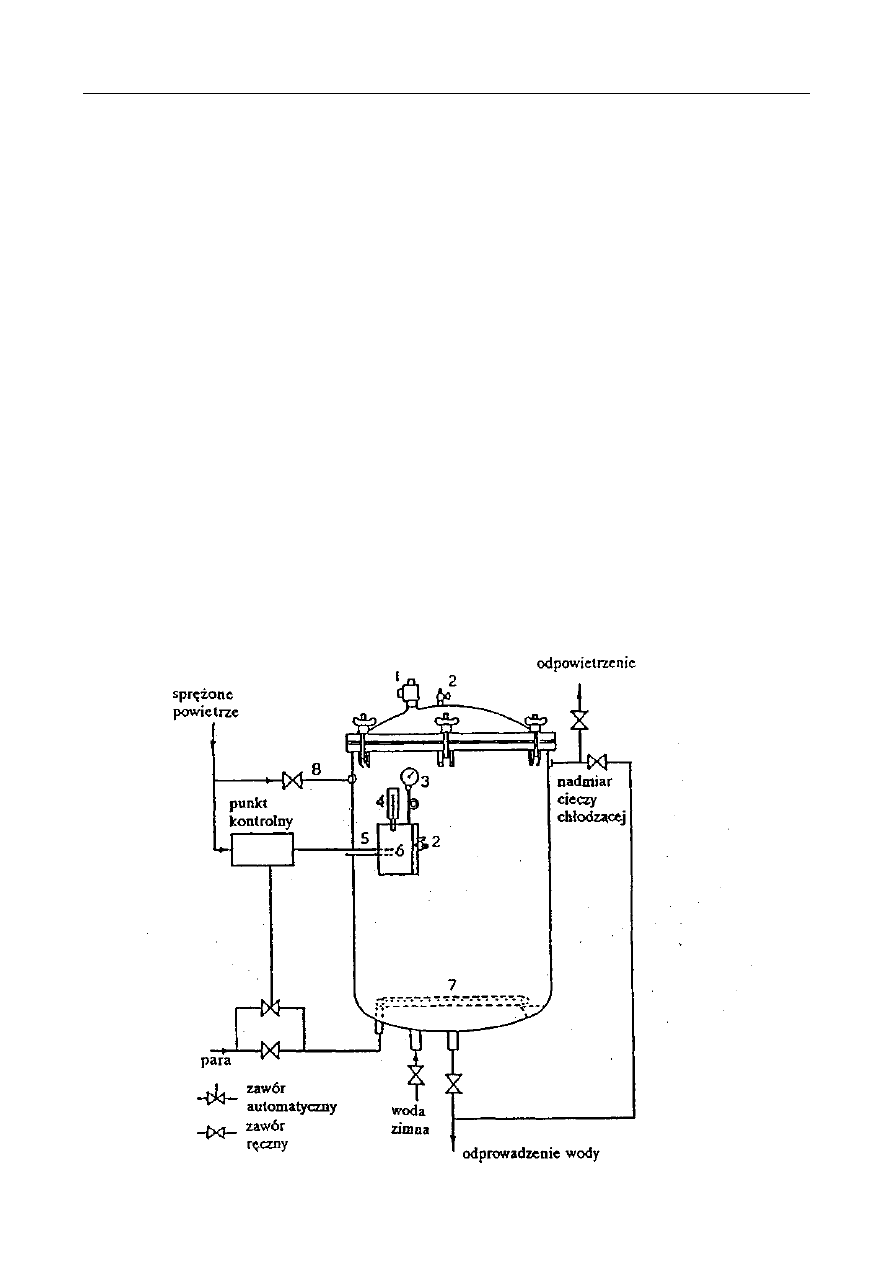
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
112
•
zachowanie większej wartości odżywczej,
•
ograniczenie procesów korozyjnych w puszkach oraz ograniczenie możliwości wykiełkowania
pojedynczych pozostałych przy życiu przetrwalników bakterii tlenowych,
•
zmniejszenie prawdopodobieństwa wystąpienia bombażu technicznego.
W wyniku prawidłowo przeprowadzonego procesu odpowietrzania w zamkniętym opakowaniu po
procesie sterylizacji panuje podciśnienie odpowiadające ok. 1/3 ciśnienia atmosferycznego.
Odpowietrzenie może być przeprowadzone jednym z trzech sposobów:
•
immersyjnie
•
za pomocą pary
•
mechanicznie
Odpowietrzanie immersyjne polega na przetrzymywaniu napełnionych naczyń w wodzie o
temperaturze 80-95°C i doprowadzeniu temperatury w materiale do 60-80°C. Ekshaustia
immersyjna jest stosowana przy produkcji kompotów, zwłaszcza z owoców zawierających duże
ilości powietrza (jabłka, truskawki, maliny).
Do odpowietrzania słoików z pastami i dżemami stosuje się system parowy. Powietrze z wolnej
przestrzeni nad produktem jest usuwane za pomocą strumienia pary, po czym naczynie zostaje
natychmiast zamknięte.
Odpowietrzanie mechaniczne stosuje się do konserw nie ogrzewanych po zamknięciu (jak
zgęszczone mleko) lub ogrzewanych łagodnie (konserwy szynkowe), a polega ono na usuwaniu
powietrza za pomocą pompy ssącej.
Najistotniejszą operacją w produkcji konserw jest sterylizacja, którą przeprowadza się w
sterylizatorach. Urządzenia te dzielą się na wsadowe (autoklawy) i do pracy ciągłej (rys. 5).
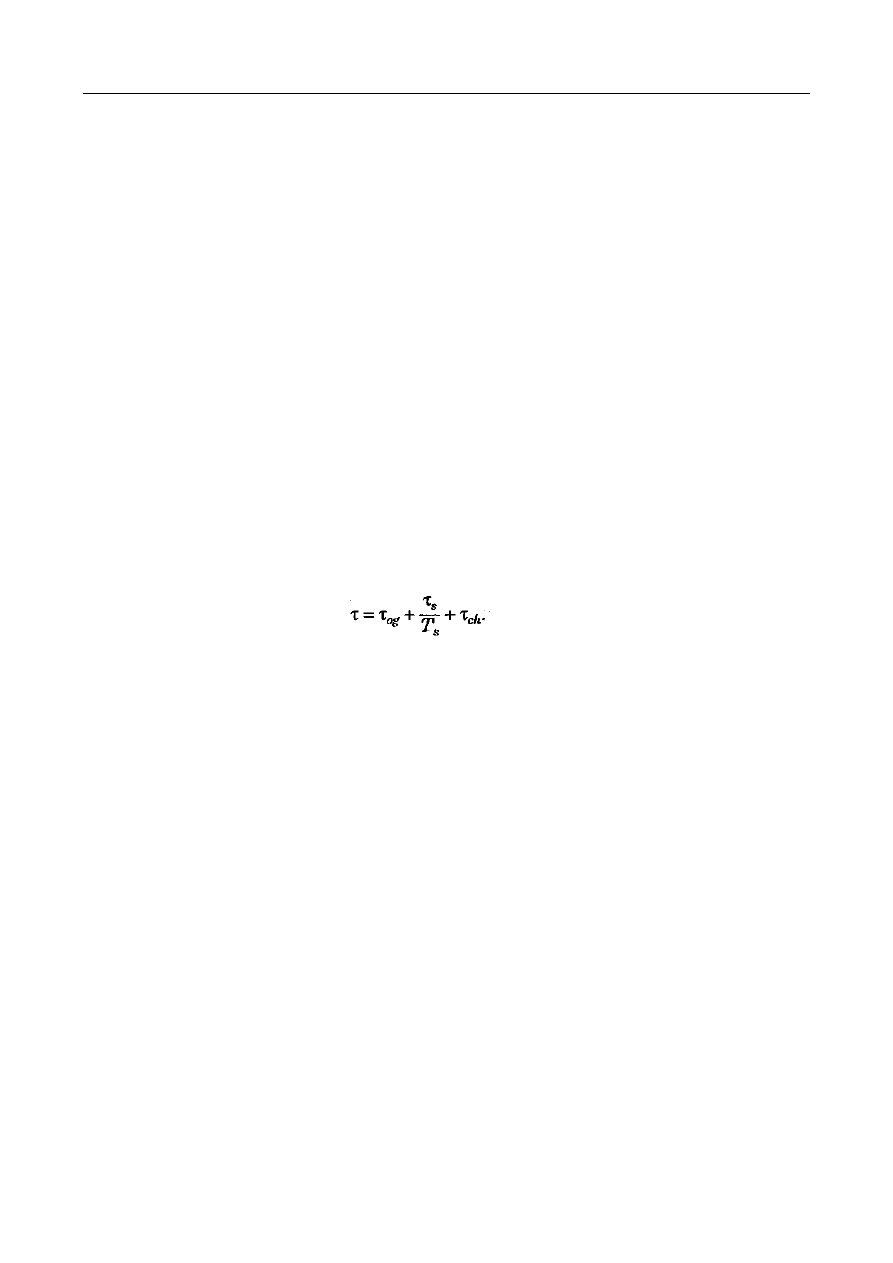
T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
Rys. 5. Sterylizator pionowy (autoklaw) przystosowany do chłodzenia pod ciśnieniem: 1-zawór
bezpieczeństwa, 2 -zawór parowy, 3- manometr, 4-termometr, 5-kontrolujący sensor, 6-
skrzynka z czujnikami temperaturowymi, 7-doprowadzenie pary, 8-wejście powietrza do
chłodzenia pod ciśnieniem.
Autoklawy mogą być pionowe lub poziome, z przystosowaniem do sterylizacji w wodzie, parze
lub homogennej mieszaninie powietrza i pary wodnej, z automatycznym załadowaniem i
wyładowaniem. Ich wadą jest większe zużycie pary i wody niż w nowoczesnych urządzeniach do
pracy ciągłej. Pionowe (rys. 5) zajmują mniej miejsca niż poziome, lecz te drugie są łatwiejsze do
załadowania i wyładowania. Urządzenia do pracy ciągłej pozwalają na ścisłą kontrolę warunków
procesu. Ciśnienie wewnątrz opakowań zmienia się w sposób łagodny, dzięki czemu
prawdopodobieństwo wystąpienia odkształceń czy też zniszczenia puszek jest mniejsze w
porównaniu z autoklawami wsadowymi.
Podstawowymi wadami urządzeń do pracy ciągłej są: znaczne załadowanie produktami, które w
razie awarii sterylizatora mogłyby ulec zniszczeniu, korozja metalu oraz zanieczyszczenie
bakteriami termofilnymi o ile nie są prowadzone właściwe pomiary. W praktyce sterylizatory do
pracy ciągłej są stosowane tam, gdzie nie zachodzi konieczność zmiany rozmiarów opakowań i
gdzie warunki procesu są stałe. Przykładem tego typu urządenia jest sterylizator hydrostatyczny
oraz śluzowy.
Czas sterylizacji w autoklawie τ dzieli się na trzy okresy:
•
czas ogrzewania konserw τ
og
do temperatury sterylizacji τ
s
•
czas utrzymywania konserw w temperaturze sterylizacji τ
s
•
czas chłodzenia τ
ch
co zapisuje się wzorem:
Szybkość przenikania ciepła do sterylizowanego produktu i tym samym czas nagrzewania
konserw zależy od następujących czynników:
•
Konsystencja konserwy. Im jej treść jest bardziej płynna, w tym większym stopniu może
zachodzić konwekcyjne przenoszenie ciepła, dzięki czemu czas nagrzewania jest krótszy niż
w przypadku konserw o stałej konsystencji, w których przenoszenie ciepła odbywa się na
drodze przewodzenia.
•
Wielkość opakowań. Im opakowanie jest większe, tym czas potrzebny do jego nagrzania jest
dłuższy.
•
Wytrząsanie opakowań podczas sterylizacji zwiększa szybkość przenoszenia ciepła na
drodze konwekcji w produktach półstałych i o dużej lepkości (np. fasola w sosie
pomidorowym), dzięki czemu można skrócić czas ogrzewania.
•
Różnica temperatur. Im wyższa jest temperatura w autoklawie, a niższa w puszce, tj. im
większa jest różnica temperatur, tym szybsze jest tempo nagrzewania konserwy.
•
Kształt opakowania. Wysokie opakowania wzmagają przewodzenie konwekcyjne w
żywności o konsystencji płynnej i półpłynnej.
•
Typ opakowania. Różnice w przewodności cieplnej i grubości ścianek opakowań sprawiają,
że czas sterylizacji dla konserw w opakowaniach metalowych jest o ok. 50% krótszy niż w
szklanych.
Po upływie czasu wymaganego do utrwalenia danej partii konserw rozpoczyna się proces
studzenia, który musi być prowadzony w warunkach uniemożliwiających powstawanie bombaży
technicznych czy też uszkodzenia podwójnej zakładki. Puszki o pojemności ponad l L, a
szczególnie duże opakowania 5-10 l, wymagają studzenia od temperatury 121-115°C najpierw do
ok. 100°C, a następnie dalszego chłodzenia do ok. 20-30°C przy utrzymywaniu nadciśnienia

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
114
zewnętrznego. Studzenie pod ciśnieniem przeprowadza się w sposób ciągły w autoklawach
sprzężonych z urządzeniami do chłodzenia. W sterylizatorach do pracy okresowej studzenie
przeprowadza się przez stopniowe doprowadzenie chłodnej wody od spodu z jednoczesnym
wypieraniem górą pary lub gorącej wody, przy bardzo wolnym obniżaniu nadciśnienia.
W celu sprawdzenia trwałości konserw wykonuje się tzw. próbą termostatową, która polega na
przetrzymywaniu reprezentatywnie wybranych opakowań przez 3-8 dni w temperaturze 37-40°C.
W produkcji konserw przeznaczonych do krajów tropikalnych stosuje się temperaturę 55°C. Jeśli
proces sterylizacji nie został przeprowadzony właściwie, mogą się pojawić puszki z objawami
zepsuć. Próbę termostatową uważa się za pozytywną, jeżeli ilość pojawiających się wad konserw
nie przekracza 2%.
W żywności utrwalanej metodą apertyzacji mogą wystąpić trzy rodzaje zepsuć:
•
bombaże objawiające się wydęciem puszek;
•
zepsucia płasko-kwaśne;
•
zepsucia płaskie niekwaśne.
W zależności od przyczyny zepsucia wyróżnia się bombaż techniczny, chemiczny i
mikrobiologiczny.
Bombaże techniczne są spowodowane przeładowaniem puszek, brakiem odpowietrzenia, a więc
obecnością nadmiernej ilości tlenu lub zbyt szybkim obniżaniem ciśnienia w autoklawie i mogą
przyczyniać się do rozwoju bombaży chemicznych lub mikrobiologicznych.
Bombaże chemiczne lub inaczej wodorowe są spowodowane przez gromadzący się wodór, który
powstaje w wyniku reakcji między kwaśnymi związkami konserwy a metalowymi ściankami
opakowania. Tempo korozji zależy od porowatości blachy (miejsca nie pokryte cyną), od rodzaju
produktu w puszce, stopnia odpowietrzenia itp. Obecność tlenu, barwników antocyjanowych i
kwasów organicznych przyspiesza reakcje towarzyszące korozji.
Bombaże mikrobiologiczne spowodowane są przez drobnoustroje wytwarzające produkty gazowe.
W przypadku niedogrzania mogą to być drożdże wytwarzające C0
2
lub bakterie mlekowe.
Jednakże zepsucie konserw mało kwaśnych następuje najczęściej pod wpływem ciepłoopornych
bakterii beztlenowych z rodzaju Clostridium, wśród których mogą występować gatunki nie-
bezpieczne dla zdrowia, jak C. perfingens lub C. botulinum produkujące egzotoksyny o dużej
zjadliwości.
Innym rodzajem zepsuć, którym nie towarzyszą wydęcia puszek, są zepsucia płasko-kwaśne i
płaskie niekwaśne. Pierwsze z nich spowodowane są działalnością względnych beztlenowców
wytwarzających przetrwalniki i powodujących zakwaszenie konserw, natomiast drugie są skutkiem
działania bakterii tlenowych przetrwalnikujących powodujących rozluźnienie konsystencji
konserwy i powstanie nieprzyjemnych zapachów.
Sterylizacja żywności przed zapakowaniem i aseptyczne pakowanie
Charakterystyka procesu
Podstawowym problemem podczas apertyzacji produktów o stałej konsystencji lub dużej
lepkości jest mała szybkość przenikania ciepła do centrum termicznego konserwy, czego
następstwem jest utrata wartości odżywczych i sensorycznych żywności, zwłaszcza warstw
znajdujących się bezpośrednio przy ściankach naczynia, oraz długi czas procesu. Podwyższenie
temperatury w sterylizatorach pozwoliłoby na skrócenie czasu procesu i zmniejszyłoby straty
jakościowe żywności, ale wiązałoby się także ze zwiększeniem stosowanego ciśnienia, a w dalszej
konsekwencji ze wzrostem kosztów opakowań i wyposażenia. Problem ten został rozwiązany dzięki

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
wprowadzeniu sterylizacji produktu przed jego zapakowaniem, a następnie pakowaniu w sposób
aseptyczny. Tego rodzaju metoda pozwala na wykorzystanie zasady HTST (sterylizację polegającą
na błyskawicznym nagrzaniu, a potem schłodzeniu i aseptycznym pakowaniu określa się jako
fasteryzację). W przemyśle spożywczym ten sposób utrwalania znalazł szerokie zastosowanie.
Systemem UHT sterylizuje się produkty płynne, jak mleko, soki owocowe, wino, praż żywność
zawierającą cząstki o średnicy mniejszej od l cm, np. odżywki dziecięce, przetwory pomidorowe,
desery ryżowe. Wysoka jakość żywności utrwalanej systemem UHT współzawodniczy z żywnością
chłodzoną i utrwalaną przez napromienienie, ale ma dodatkową zaletę, a mianowicie zachowuje do-
brą jakość przez przynajmniej 6 miesięcy, nie wymagając chłodniczego zabezpieczenia w tym
czasie. Drugą ważną zaletą jest to, że warunki procesu nie są uzależnione od rozmiarów opakowań.
Wreszcie proces ten charakteryzuje wysoka wydajność ze względu na pełną automatyzację i
oszczędność energetyczną, np. ekonomiczność produkcji mleka systemem UHT wiąże się z tym, że
jego dystrybucja nie wymaga transportu chłodniczego w przeciwieństwie do mleka
pasteryzowanego. Wadami tej metody są wysoki koszt i złożoność urządzeń wynikające z
konieczności sterylizacji materiałów opakowaniowych, rurociągów i zbiorników, konieczność
utrzymywania powietrza i urządzeń napełniających w stanie sterylnym. Ponadto urządzenia te musi
obsługiwać wysoko wyspecjalizowany personel. Utrwalana żywność ogrzewana jest w stosunkowo
cienkich warstwach w wymiennikach ciepła z jednoczesną ścisłą kontrolą temperatury sterylizacji i
czasu jej utrzymywania. Ponieważ opakowania nie muszą być wytrzymałe na warunki sterylizacji,
dlatego też powszechnie są stosowane laminowane kartony. Kartony są wstępnie sterylizowane za
pomocą nadtlenku wodoru, a ich napełnianie odbywa się w warunkach sterylnych utrzymywanych
dzięki działaniu ultrafioletu i filtrowanego powietrza.
Urządzenia stosowane w procesie UHT dzielą się w zależności od sposobu doprowadzania
ciepła na:
•
bezpośrednie (ciepło doprowadzane przez iniekcję lub infuzję pary);
•
pośrednie (ciepło doprowadzane w wymiennikach ciepła, np. rurowych, płytowych);
•
inne (ciepło doprowadzane przez ogrzewanie dielektryczne, indukcyjne lub mikrofalowe).
Urządzenia do bezpośredniego ogrzewania produktów
Zasada działania urządzeń należących do tej grupy polega na dokładnym wymieszaniu produktu
z oczyszczoną parą wodną, co może odbywać się na dwa sposoby:
I. Metoda iniekcyjna. Para o ciśnieniu 965 kPa jest wprowadzana przez iniekcję do wstępnie
ogrzanego (do 76°C) płynnego produktu np. mleka, powodując jego natychmiastowe ogrzanie do
temperatury 150°C. Po odpowiednim czasie przetrzymywania w tej temperaturze (np. po 2,5 s)
produkt ze skroploną parą przechodzi do komory o zredukowanym ciśnieniu, zwanej komorą
ekspansyjną. Ciśnienie w komorze odpowiada temperaturze nieco niższej od temperatury mleka
przed iniekcją pary, co powoduje odparowanie takiej ilości wody, jaka została wprowadzona ze
skraplającej się pary. Odparowanie wody pod zredukowanym ciśnieniem powoduje
natychmiastowe oziębienie produktu do temperatury 76°C.
Zaletami tej metody są:
•
bardzo szybkie ogrzanie, a potem schłodzenie, dzięki czemu jest ona odpowiednia dla
większości produktów wrażliwych na wysoką temperaturę,
•
z produktu są usuwane gazy, co w przypadku niektórych produktów jest zaletą (np. z
mleka usuwany jest m.in. tlen, co hamuje proces utleniania). ,
Jednakże:
•
metoda ta jest odpowiednia tylko dla produktów o niskiej lepkości,
•
kontrola warunków procesu jest stosunkowo słaba,
•
mogą wystąpić trudności z utrzymaniem sterylności w niskociśnieniowych częściach
wyposażenia,
•
wymagania dotyczące czystości pary znacznie podrażają koszty procesu,

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
116
•
możliwa jest regeneracja mniej niż 50% energii w porównaniu z ponad
90-procentowym odzyskiem w systemie pośrednim,
•
możliwości dokonywania zmian dla różnych typów produktów są małe.
II. Metoda infuzyjna. Produkt jest rozpylany w postaci swobodnie opadającej „błonki" wewnątrz
komory, w której znajduje się para wodna o ciśnieniu 450 kPa. W tych warunkach produkt
natychmiast (0,3 s) ogrzewa się do temperatury 142-146°C i utrzymywany jest w niej przez 3 s, po
czym przechodzi do komory ekspansyjnej, gdzie następuje błyskawiczne oziębienie go do
temperatury 65-70°C. Ciepło odzyskane z chłodzonego produktu jest wykorzystywane do
wstępnego ogrzania wchodzącego materiału.
Przewaga tej metody nad iniekcyjna polega na tym, że utrwalany płyn nie kontaktuje się z
gorącymi powierzchniami urządzenia, w związku z czym nie następuje jego przypalanie się.
Inne zalety tej metody to:
•
prawie natychmiastowe ogrzanie surowca do temperatury pary wodnej i bardzo
szybkie schłodzenie, dzięki czemu zostają zachowane jego właściwości sensoryczne i
odżywcze,
•
lepsza kontrola warunków procesu niż w metodzie iniekcyjnej,
•
mniejsze ryzyko miejscowego przegrzania produktu,
•
metoda ta jest bardziej odpowiednia dla żywności o wyższej lepkości niż iniekcyjna.
Podstawową wadą tej metody jest zatykanie się dyszy rozpyłowej urządzenia i rozdzielanie
składników w niektórych produktach.
Urządzenia do pośredniego ogrzewania produktów
1. Wymienniki płytowe
Stosowanie wymienników płytowych wiąże się z wieloma ograniczeniami wynikającymi ze
stosowania wyższej temperatury i ciśnienia. Ponadto mogą być one wykorzystywane tylko do
ogrzewania płynów o małej lepkości. Wymienniki płytowe obok tych wad mają wiele zalet, a
mianowicie:
•
są stosunkowo niedrogie,
•
są ekonomiczne pod względem zajmowanej powierzchni i zużywanej wody,
•
są oszczędne pod względem energetycznym (ponad 90% energii ulega regeneracji),
•
istnieją możliwości dopasowania wydajności produkcji dzięki zmiennej ilości płyt.
2. Wymienniki rurowe
Zastosowanie wymienników rurowych pozwala na pracę przy wyższym ciśnieniu (7 000-10 000
kPa), a to powoduje większą szybkość przepływu cieczy sterylizowanej, czego konsekwencją jest
bardziej równomierne przenoszenie ciepła i nieosadzanie się produktu na ściankach rur. Lecz w
tego rodzaju urządzeniach nie ma możliwości regulowania wydajności procesu, tak jak to ma
miejsce w przypadku wymienników płytowych (np. przez podłączenie dodatkowego pakietu płyt).
Chcąc zwiększyć wydajność sterylizacji wymiennika rurowego, należy zamontować drugie takie
samo urządzenie, gdyż zwiększenie średnicy rur wymagałoby wyższego ciśnienia do utrzymania
szybkości przepływu na tym samym poziomie, a przewody o zwiększonej średnicy są mniej
wytrzymałe na zwiększone ciśnienie.
Sterylizacja systemem dwustopniowym

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
Obok metody apertyzacji i aseptycznego wyjaławiania na uwagę zasługuje również sterylizacja
systemem dwustopniowym. Obejmuje ona:
•
sterylizację
wstępną
produktu
przed
jego
zapakowaniem
zachodzącą
w wymiennikach płytowych bądź rurowych w temperaturze 135-141°C,
•
rozlew do opakowań i ich zamknięcie,
•
sterylizację końcową produktu zamkniętego hermetycznie, która odbywa się w
autoklawach.
Dzięki zastosowaniu sterylizacji wstępnej warunki ogrzewania w autoklawach są łagodniejsze,
a to z kolei sprawia, że zmiany organoleptyczne produktu są niniejsze, a składniki pokarmowe
wrażliwe na wysokie temperatury lepiej zachowane.
Zmiany w żywności spowodowane działaniem wysokiej temperatury
Efektem cieplnego utrwalania żywności są zmiany jej koloru, smaku, zapachu i konsystencji,
przy czym stosowanie metod UHT pozwala na lepsze zachowanie wielu tych wskaźników, które
decydują o walorach sensorycznych i odżywczych. W mniejszym stopniu ulegają zmianie barwniki,
np. karoteny i betanina, które pozostają prawie nie zmienione, a chlorofil i antocjany są lepiej
zachowane. Sterylizowane aseptycznie mleko, soki owocowe i warzywa zachowują swój naturalny
smak. Również straty witamin są znacznie mniejsze.
Pasteryzacja
Pasteryzacja jest łagodniejszym sposobem termicznego utrwalania żywności, lecz okres
przydatności do spożycia produktów pasteryzowanych jest kilkakrotnie krótszy niż
sterylizowanych, a ponadto wymagają one dodatkowo zabezpieczenia chłodniczego. Jedynie
surowiec o pH niniejszym od 4,5 uzyskuje tą drogą trwałość równą produktom sterylizowanym.
Pasteryzacji poddaje się mleko, piwo, soki owocowe, ogórki konserwowe.
W technologii mleczarstwa rozróżnia się następujące jej rodzaje:
•
pasteryzacja niska lub długotrwała, która polega na przetrzymywaniu mleka w
temperaturze 63-65°C przez 30 min,
•
pasteryzacja krótkotrwała to działanie temperaturą 72°C przez 15-40 s,
•
pasteryzacja wysoka przebiegająca w temperaturze 80-85°C w czasie od 15-20s do
kilkunastu min,
•
pasteryzacja momentalna, czyli działanie temperaturą 85-90°C bez przetrzymywania.
Pasteryzacja powoduje znaczne zmniejszenie aktywności enzymów występujących w mleku,
np. termiczne warunki zniszczenia fosfatazy alkalicznej pokrywają się z warunkami pasteryzacji
krótkotrwałej, a zupełna inaktywacja laktoperoksydazy występuje po 13-sekundowym działaniu
temperatury 85°C. Właściwości tych enzymów zostały wykorzystane w przemyśle mleczarskim do
określania skuteczności-w pierwszym przypadku pasteryzacji niskiej i krótkotrwałej, a w drugim-
wysokiej.
Urządzenia, w których przeprowadza się ten proces, noszą nazwę pasteryzatorów i dzielą się
na:
•
pasteryzatory płytowe i rurowe,
•
pasteryzatory tunelowe,
•
pasteryzatory wannowe.
Zasadniczą częścią składową pasteryzatorów płytowych są płyty o charakterystycznym profilu,
dzięki któremu po zestawieniu ich w pakiet (zestaw) tworzą się kanały, którymi przepływają

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
118
przemiennie (co drugą płytą) i w przeciwprądzie mleko i woda. W obrębie zestawu płyt można
wyróżnić następujące działy:
•
regeneracji,
•
pasteryzacji,
•
przetrzymywania,
•
chłodzenia (wodą bieżącą, a następnie lodową).
Dział regeneracji składa się z 2-3 sekcji i służy do wstępnego ogrzewania mleka zimnego
ciepłem mleka spasteryzowanego.
W pasteryzatorach tunelowych opakowany produkt (butelki lub puszki) przesuwa się pod
natryskiem gorącej wody lub też jest zanurzany w wodzie o określonej temperaturze. Pasteryzatory
wannowe to zbiorniki, do których nalewa się produkt albo też napełnia się je wodą i wstawia do
nich zapakowany materiał.

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
11.2.
W
YKONANIE ĆWICZENIA
Celem ćwiczenia jest zapoznanie się z prędkością ogrzewania się najwolniej ogrzewającego się
miejsca w produkcie spożywczym umieszczonym w słoju.
Wykonanie oznaczenia
1.
Należy napełnić słoik w 90% produktem spożywczym (sok owocowy, mleko, kostki marchwi w
zalewie NaCl, buraczki w zalewie octowej itp.) w środku geometrycznym czujnika temperatury.
Podłączyć czujnik temperatury do układu pomiarowego.
2.
Zmierzyć i zapisać wymiary i masę słoja wg wzoru:
Wymiary słoja: Wartość . cm. średnio ......
Masa słoja: Napełniony, ....g, pusty.........g.
Masa żywności w słoju:..............g.
Skład produktu spożywczego:
Inne parametry produkty takie jak pH, lepkość itp.
Temperatura łaźni wodnej
o
C,
temperatura początkowa produktu .................................................... °C.
Czas ogrzewania w łaźni wodnej....... min.
3.
Umieścić słój w łaźni o temperaturze ok. 90°C. Początkowo temperaturę produktu wewnątrz
słoja zapisywać w odstępach czasu 0,5-1 minuty, później co 2 minuty. Temperaturę należy
notować do momentu uzyskania temperatury pasteryzacji.
UWAGA!!
N
ALEśY STALE KONTROLOWAĆ POZIOM WODY W ŁAŹNI WODNEJ
.
E
WENTUALNE JEJ BRAKI NALEśY UZUPEŁNIĆ WODĄ DESTYLOWANĄ
.
4.
Po odpowiednim okresie ogrzewania w łaźni słój należy chłodzić do temperatury ok. 20°C. W
czasie schładzania także należy co minutę zapisywać temperaturę (tabelka)
Nr
Czas, min.
Temperatura produktu
1
2
3
…
9
10 itd...
5.
Należy wykreślić krzywą zależności czasu od temperatury (T=
o
C, czas w minutach).
Otrzymany wykres należy zinterpretować.
11.3.
P
RZEDSTAWIENIE WYNIKÓW
Wyniki oznaczeń umieścić w sprawozdaniu [wzór, Załącznik 1. Załącznik 1. s.121] w formie
oddzielnych punktów wraz z przeliczeniami i przedłożyć osobie prowadzącej ćwiczenia w celu
sprawdzenia. Należy także zaznaczyć wszelkie odstępstwa od metod podanych w opracowaniu
ćwiczeń. Na końcu sprawozdania z ćwiczeń należy podać wnioski w formie zwięzłych punktów.

T
ERMICZNE UTRWALANIE
ś
YWNO
Ś
CI
12 luty 2010
120
11.4.
S
PRZĘT I ODCZYNNIKI
•
2 łaźnie wodne o temperaturach 90°C (łaźnie do ogrzewania słoi) i 20°C (łaźnia do chłodzenia
słoi)
•
słoiki typu „Twist” o pojemności 0,45l z zakrętkami
•
czujnik temperatury z układem pomiarowym.

Z
AŁ
Ą
CZNIK
ZAŁĄCZNIKI
Załącznik 1.
W
ZÓR SPRAWOZDANIA Z ĆWICZEŃ
Nazwisko i Imię:
Symbol podgrupy: 1A
Malinowska Agnieszka
Data(-y) wykonywania ćwiczenia: 28.02.2009
Kowalik Piotr
Osoba prowadząca ćwiczenia:
stopień naukowy, imię i nazwisko
Temat i numer ćwiczenia:
Wpływ pH i temperatury na aktywność glukoamylazy (ćwiczenie nr 6)
1. Cel ćwiczenia (nie więcej niż 1-3 zdania)
Nie należy zamieszczać teoretycznego opisu dotyczącego ćwiczenia!
2. Zadania do realizacji (w formie podpunktów)
np.:
a)
przygotowanie próbek piw do analiz
b)
oznaczenie gęstości piwa
c)
oznaczenie zawartości ekstraktu w piwie
d)
oznaczenie zawartości etanolu w piwie
e)
oznaczenie barwy piwa
itd.
3. Wprowadzone zmiany w metodykach
Należy w wyraźnie wydzielonych podpunktach wyszczególnić wprowadzone
zmiany w metodykach w stosunku do metod dostarczonych w tekście ćwiczenia.
4. Spostrzeżenia i obliczenia
Spostrzeżenia takie jak np. zmiana barwy, wygląd hodowli jak również pełne
przeliczenia wyników, rysunki, wykresy, tabele należy przedstawić w wyraźnie
wydzielonych podpunktach, adekwatnie do punktu 2 „Zadania do realizacji”.
Uwaga: Należy zamieszczać pełne obliczenia rachunkowe bez stosowania
skrótów myślowych.
5. Wnioski
Wnioski należy przedstawić w formie zwięzłych punktów.
W przypadku braku możliwości poprawnego wnioskowania np. na skutek
uzyskania błędnych lub niepewnych wyników eksperymentu należy przedstawić
na podstawie dogłębnej analizy przebiegu eksperymentu prawdopodobne
przyczyny uzyskania nieprawidłowego wyniku. Analizę błędów popełnionych w
czasie eksperymentu należy wyraźnie oddzielić od wniosków.

L
ITERATURA
12 luty 2010
122
LITERATURA
1.
Ogólna technologia żywności. Skrypt do ćwiczeń pod red. Ewy Hajduk. Wyd. AR w Krakowie.
Kraków 1998.
2.
Ogólna technologia żywności. Część II. Pod red. Włodzimierza Bednarskiego. Skrypt Akademii
Rolniczo-Technicznej w Olsztynie, Olsztyn 1991.
3.
Higiena produkcji żywności. Pod red. Danuty Kołożyn Krajewsiej. Wyd. SGGW. Warszawa
2001.
4.
Biotechnologia żywności. Praca zbiorowa pod red. Włodzimierza Bednarskiego i Arnolda
Repsa. Wyd. Naukowo-Techniczne. Warszawa 2001.
5.
Mikrobiologia i higiena w przemyśle spożywczym. Pod red. Zofii Zakowskiej i heleny
Stobińskiej. Wyd. Politechniki Łódzkiej, Łódź 2000.
6.
Przewodnik do ćwiczeń z ogólnej technologii żywności. Praca zbiorowa pod red. Gustawa
Sobkowicza. Wyd. AR we Wrocławiu. Wrocław 1998.
7.
Substancje dodatkowe i składniki funkcjonalne żywności. Rutkowski A., Gwiazda S., Dąbrowski
K., Czapski J., Kamiński E., Pluta A. Agro & Food Technology, Warszawa 1997.
8.
Handbook of food preservation. Edited by Shafiur Rahman. Marcel Dekker, Inc. New York-
Basel, 1999.
9.
Microbiological risk assessment in food processing. Ed. by Martyn Brown i Mike Stringer.
Woodhead Publishing Ltd.Cambridge England 2002.
10.
Handbook of Fermented Functional Foods. Edited by Edward R. Farnworth. CRC Press. Boca
Raton- London-N.Y.-Washington D.C. 2003.
11.
Modern Food Microbiology. James M. Jay. Sixth edition. Aspen Publishers Gaithers burg
Maryland 2000.
12.
The nutrition handbook for ford processors. C.J.K. Henry, C. Chapman. CRC Press. Boca
Raton- London-N.Y.-Washington D.C. 2000.
13.
Technologia żywności – wybrane zagadnienia. E. Biller. Wyd SGGW, Warszawa 2005
14.
Ogólna technologia żywności. E. Pijanowski, M. Dłużewski, A. Dłużewska, A. Jarczyk WNT,
Warszawa, 1996

S
PIS AUTORÓW
SPIS AUTORÓW
Prof. dr hab. Zdzisław Targoński
Dr inż. Jacek Pielecki
Dr Piotr Janas
Dr inż. Dominik Szwagier
Dr inż. Adam Waśko
Dr inż. Tomasz Czernecki
Dr inż. Bożena Sosnowska
Wyszukiwarka
Podobne podstrony:
2010.02.12 Roczny sprawdzian sprawności fizycznej żołnierzy, 002-05 WOJSKO POLSKIE OD 01.01.1990
2010 02 12 Co feministka ma kupić córce
2010 02 12 Rz, Czy tylko Żydzi mają wrażliwość K Jasiewicz & P Zychowicz
2010 02 12 Przemoc seksualna
Wprowadzenie do Pedagogiki 02.12.2010, RESOCJALIZACJA, wprow. do pedagogiki wykłady
02-12-2010, rosliny alternatywne
02 - 12. 10. 2010, Filozofia, Notatki FO, III Semestr, Semantyka logiczna
02 12 2010
2015 02 12 Popularny dziennikarz te
Jak Rosjanie pozbawili mjr Protasiuka licencji Nasz Dziennik, 2011 02 12
Pogoda załamała się już po lądowaniu Nasz Dziennik, 2011 02 12
02 2 1b KARO Interwencja (2010 10 12)
Smoleńskie żniwa Tuska Nasz Dziennik, 2011 02 12
Major Protasiuk nigdy nie ulegał panice Nasz Dziennik, 2011 02 12
MB ćwiczenia 24 04 2010 (02)
Prawo cywilne wyk.13 2010-02-16, Prawo Cywilne
fizyka na 2 02 12
Wykład 2# 02 12
2010 02 05 09;33;36
więcej podobnych podstron