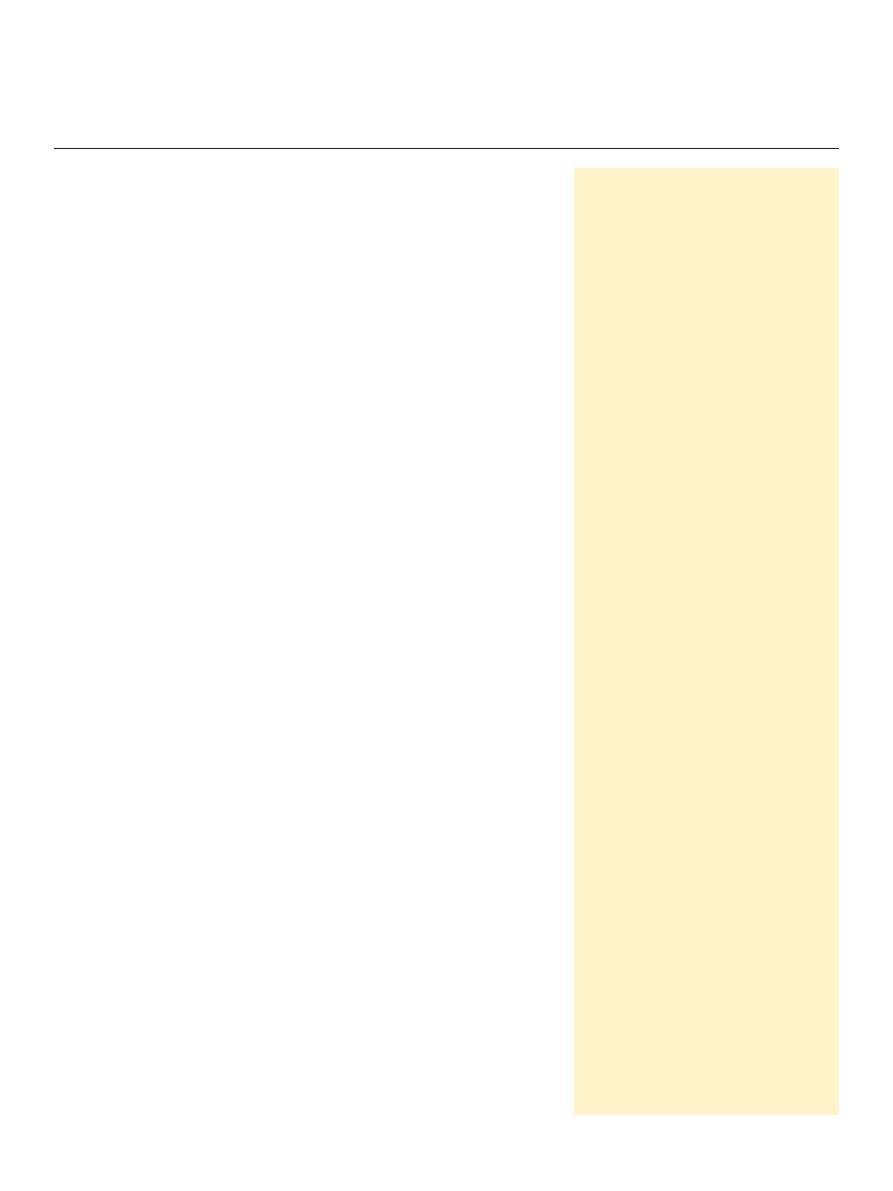
Postępy Biochemii 58 (4) 2012
403
Elżbieta Salińska
*
Jerzy W. Łazarewicz
Instytut Medycyny Doświadczalnej i Klinicz-
nej im. M. Mossakowskiego Polskiej Akademii
Nauk, Warszawa
*
Pracownia
Farmakoneurochemii,
Zakład Neurochemii, Instytut Medycyny
Doświadczalnej i Klinicznej im. M.
Mossakowskiego Polskiej Akademii Nauk, ul.
Pawińskiego 5, 02-106 Warszawa; tel.: (22) 608
65 28, e-mail: elas@cmdik.pan.pl
Artykuł otrzymano 11 września 2012 r.
Artykuł zaakceptowano 2 października 2012 r.
Słowa kluczowe: homeostaza wapnia, ische-
mia mózgu, plastyczność neuronalna, recepto-
ry glutaminianergiczne, transdukcja sygnału,
zwyrodnienie neuronów
Wykaz skrótów: CICR (ang. Ca
2+
-induced Ca
2+
release) — indukowany przez wapń wyrzut
wapnia; ER — siateczka śródplazmatyczna;
IP
3
— inozytolo-1,4,5-trifosforan; LTD (ang.
long term depression) — długotrwałe osłabienie
synaptyczne; LTP (ang. long term potentiation)
— długotrwałe wzmocnienie synaptyczne;
NCX — wymiennik Na
+
/Ca
2+
; PMCA (ang.
plasma membrane Ca
2+
-ATPase) — Ca
2+
-ATPaza
błony plazmatyczej; ROC (ang. receptor-opera-
ted channels) — receptory jonotropowe; SERCA
— Ca
2+
-ATPaza siateczki sarko/śródplazma-
tycznej (ang. sarcoplasmic/endoplasmic reticulum
Ca
2+
-ATPase); VOCC (ang. voltage-operated Ca
2+
channels) — kanały wapniowe zależne od po-
tencjału błony
Podziękowanie: Praca powstała w trakcie re-
alizacji projektu badawczego nr N N401 003935
finansowanego ze środków przyznanych
przez MNiSW.
Rola wapnia w fizjologii i patologii neuronów
STRESZCZENIE
W
artykule na wstępie przedstawiono ewolucyjne aspekty dwoistej roli jonów wapnia
jako cząsteczek sygnałowych oraz kationów o działaniu cytotoksycznym. Następnie
omówiono mechanizmy homeostazy wapnia w neuronach z uwzględnieniem specyficznych
funkcji tych komórek pobudliwych, oraz mechanizmy generacji i przekazywania sygnału
wapniowego. W oparciu o te informacje została przedstawiona w zarysie rola Ca
2+
w specy-
ficznych funkcjach komórek nerwowych, jak pobudliwość, przewodnictwo nerwowe, prze-
kaźnictwo synaptyczne, plastyczność i różne formy mobilności. Następnie omówiono rolę
zaburzeń homeostazy wapnia i jego funkcji sygnałowych w mechanizmach uszkodzenia i
śmierci neuronów w ostrych schorzeniach, ze szczególnym uwzględnieniem niedokrwienia
mózgu, oraz w przewlekłych chorobach neurozwyrodnieniowych.
WPROWADZENIE
Wapń jest dwuwartościowym kationem zewnątrzkomórkowym, a wysoki
gradient jego stężeń w przedziałach na zewnątrz i wewnątrz komórki prze-
kracza wartość 10
4
. We krwi i płynach tkankowych ssaków stężenie Ca
2+
jest
utrzymywane na stałym poziomie około 10
–3
M, podczas gdy jego spoczynkowe
stężenie w cytosolu wynosi poniżej 10
–7
M. Równie niskie stężenia jonów Ca
2+
obserwuje się zarówno w komórkach ssaków jak i u prokaryota, co świadczy
o tym, że mechanizmy usuwania wapnia z komórek są stare i ewolucyjnie za-
chowane. Wiadomo od dawna, że jony Ca
2+
odgrywają kluczową rolę w fizjo-
logii komórek, a zwłaszcza neuronów. Z drugiej strony masowy napływ Ca
2+
jest toksyczny dla komórek, a udział wapnia w mechanizmach uszkodzenia i
śmierci neuronów jest przedmiotem badań od wielu lat. Uważa się, że cyto- i
neurotoksyczność Ca
2+
wynika z jego właściwości chemicznych oraz szczególnej
roli w mechanizmach przekazywania sygnału, leżących u podstaw fizjologicz-
nych funkcji komórek, a szczególnie neuronów. Stąd też ich zaburzenie może
mieć skutki letalne. Uniwersalne mechanizmy komórkowej homeostazy Ca
2+
oraz sygnalnej roli wapnia, dotyczące także neuronów, zostały przedstawione
szczegółowo w innych rozdziałach bieżącego zeszytu Postępów Biochemii. W
tym rozdziale zwrócimy uwagę na wybrane szczegółowe aspekty, ważne dla
funkcji fizjologicznych lub patologii neuronów.
DWOISTA ROLA WAPNIA W KOMÓRKACH: ASPEKTY EWOLUCYJNE
Ewolucja życia na Ziemi potoczyła się w kierunku wykorzystania fosforanów
jako anionów wewnątrzkomórkowych kluczowych dla metabolizmu energe-
tycznego i przemian lipidów, regulacji aktywności białek i replikacji DNA. Prze-
ciw-jonem w tych procesach jest Mg
2+
, natomiast długotrwały wzrost stężenia
Ca
2+
wewnątrz komórki jest dla niej zabójczy, ponieważ jony Ca
2+
konkurują z
Mg
2+
, a fosforany wapnia są praktycznie nierozpuszczalne w wodzie. Dla unik-
nięcia wytrącania się tych soli, stężenie Ca
2+
wewnątrz komórki musi być utrzy-
mywane poniżej poziomu 10
–5
M [1].
Sugerowane są hipotezy dotyczące rozwoju mechanizmów homeostazy
Ca
2+
w komórkach, które stały się kluczowe dla pojawienia się funkcji tych jo-
nów w procesach przekazywania sygnałów, a których zaburzenie prowadzi do
śmierci komórek. Chociaż obecnie stężenie Ca
2+
w oceanach waha sie w grani-
cach 10
–2
-10
–3
M, zapewne ewolucja najwcześniejszych form życia, a być może
także prostych eukaryota, miała miejsce w wodach w których stężenie jonów
Ca
2+
początkowo nie przekraczało 10
–4
M. Późniejszy jego wzrost do obecnego
poziomu wymusił na komórkach ewolucję mechanizmów adaptacyjnych [1]. W
rezultacie zarówno współczesne prokaryota jak i eukaryota dysponują dwoma
podstawowymi mechanizmami usuwania Ca
2+
z komórek przez błonę plazma-
tyczną, to jest ATPazami PMCA zależnymi od dostępności ATP, oraz wymienni-
404
www.postepybiochemii.pl
kiem NCX napędzanym gradientem jonów sodu zależnym
od aktywności Na
+
-K
+
-ATPazy.
W przebiegu ewolucji eukaryota wzbogaciły mechani-
zmy homeostazy Ca
2+
w bardziej rozwinięte systemy bu-
forowania wapnia wewnątrz komórek. Należy do nich po-
bieranie Ca
2+
przez struktury wewnątrzkomórkowe: ER i
mitochondria, a także wiązanie przez liczne białka wiążące
wapń o znaczącej pojemności buforowej. Ponadto pojawiły
się kanały pojemnościowe w błonach plazmatycznych oraz
receptory/kanały wrażliwe na IP
3
i rianodynę, odgrywające
rolę w utrzymaniu zrównoważonego bilansu wapniowe-
go w komórkach, uzupełnianiu zasobów Ca
2+
i uwalnianiu
Ca
2+
z ER do cytosolu.
W neuronach, komórkach pobudliwych cechujących się
intensywnymi przepływami jonów wapnia, obserwujemy
różnorodne drogi kontrolowanego wzrostu wewnątrzko-
mórkowego stężenia Ca
2+
i usuwania go z cytosolu [2]. Roz-
budowany system homeostazy wapnia u eukaryota stał się
w przebiegu ewolucji sprawnym narzędziem dla wykorzy-
stania jonów wapnia w wewnątrzkomórkowych mechani-
zmach przekazywania sygnałów, szczególnie silnie rozwi-
niętych w neuronach [3]. Uważa się, że uniwersalna rola Ca
2+
jako cząsteczki służącej do przekazu sygnału wynika z uni-
kalnych właściwości fizykochemicznych tego kationu, ponie-
waż w swej sferze koordynacyjnej może on pomieścić od 4
do 12 atomów tlenu [4]. Dzięki obecności obfitych zasobów
jonów Ca
2+
w przestrzeniach na zewnątrz i wewnątrz komór-
ki indukcja sygnału wapniowego jest błyskawiczna, ponadto
jest możliwość jego szybkiego wygaszenia dzięki sprawnym
mechanizmom usuwania tego kationu z cytosolu.
GENEROWANIE SYGNAŁU
WAPNIOWEGO W NEURONACH
Wewnątrzkomórkowe sygnały wapniowe w neuronach
są elementem łańcucha przekazu informacji w układzie ner-
wowym, zapoczątkowanego przez sygnał zewnątrzkomór-
kowy, w postaci pobudzenia synaptycznego docierającego
do tych komórek w przebiegu neurotransmisji chemicznej
lub elektrycznej [3]. W efekcie depolaryzacji błony plazma-
tycznej i/lub aktywacji receptorów, dochodzi do napływu
jonów Ca
2+
do cytosolu ze środowiska zewnątrzkomórko-
wego lub do ich mobilizacji z zasobów wewnątrz komórki,
co prowadzi do wzrostu stężenia Ca
2+
wewnątrz neuronów.
Napływ Ca
2+
do neuronów przez błonę plazmatyczną za-
chodzi za pośrednictwem kanałów wapniowych VOCC lub
receptorów jonotropowych ROC, a zwłaszcza receptorów
dla glutaminianu specyficznie wrażliwych na N-metyl-
-D-asparaginian (NMDAR) [5]. Z kolei pobudzenie recep-
torów metabotropowych sprzężonych za pośrednictwem
białek G z fosfolipazą C, do których należą receptory me-
tabotropowe dla glutaminianu (mGluR) grupy I, prowadzi
do mobilizacji wapnia z zasobów w ER, zachodzącej za po-
średnictwem wtórnego przekaźnika IP
3
i specyficznych dla
niego receptorów [6]. Początkowy krótkotrwały wzrost stę-
żenia Ca
2+
w cytosolu spowodowany napływem z zewnątrz
może być przedłużony i wzmocniony w wyniku wtórnego
uwalniania Ca
2+
z ER, głównie za pośrednictwem recepto-
rów rianodynowych, w mechanizmie CICR [7]. Dla kodo-
wania sygnału przekazywanego za pośrednictwem jonów
Ca
2+
wykorzystywana jest charakterystyka wzrostu we-
wnątrzkomórkowego stężenia tych jonów: jego amplituda,
częstotliwość, czas trwania i lokalizacja wewnątrz komórki
Rycina 1. Komórkowa homeostaza Ca
2+
na przykładzie dendrytu neuronów glutaminianergicznych w warunkach komfortu i niedoboru energetycznego. (A) Warunki
prawidłowe. Ściśle kontrolowany napływ Ca
2+
przez VOCC i ROC (głównie NMDAR) jest równoważony przez wyrzut jonów wapnia z neuronów za pośrednictwem
pompy PMCA i wymiennika NCX. Buforowanie Ca
2+
wewnątrz komórki przez białka wiążące wapń (CaBP) oraz pobieranie przez mitochondria (na drodze uniportu
zgodnie z gradientem elektrochemicznym) i ER dzięki pompie SERCA. Mobilizacja Ca
2+
z ER w wyniku aktywacji receptorów metabotropowych grupy I (mGluR1/5) i
otwarcia kanałów receptorów IP
3
R lub w drodze CICR. (B) Warunki niedoboru energetycznego, np. w niedokrwieniu mózgu. Zahamowanie ATPaz, depolaryzacja neu-
ronów oraz pobudzenie VOCC i ROC, zwłaszcza NMDAR prowadzi do napływu Ca
2+
do neuronów, wspomaganego przez odwrócenie trybu pracy NCX. Pobudzenie
mGluR I i wzrost stężenia Ca
2+
w cytosolu indukuje wyrzut wapnia z zasobów w ER za pośrednictwem IP
3
R i RyR (CICR). Początkowe przeładowanie mitochondriów
wapniem owocuje ich deenergizacją, aktywacją megakanałów (MPT) z uwalnianiem Ca
2+
z mitochondriów.

Postępy Biochemii 58 (4) 2012
405
[8]. Postęp w badaniu z dużą rozdzielczością stężenia Ca
2+
w żywych komórkach pozwolił na ujawnienie, na tle global-
nych wzrostów stężenia Ca
2+
przyrównywanych do analo-
gowych sygnałów wapniowych, iskrzeń wapniowych (ang.
calcium sparks) będących krótkotrwałymi lokalnymi bardzo
znacznymi wzrostami stężeń Ca
2+
, którym przypisuje się
rolę cyfrowych sygnałów wapniowych [9]. Na dywersyfika-
cję sygnałów wapniowych generowanych w neuronach ma
także wpływ zróżnicowanie struktury molekularnej i skła-
du podjednostkowego białek zaangażowanych w regulację
przepływów Ca
2+
.
NAPŁYW JONÓW WAPNIA DO NEURONÓW
Z PRZESTRZENI ZEWNĄTRZKOMÓRKOWEJ
Sygnał elektryczny docierający do neuronu (tj. depolary-
zacja błony) jest przetwarzany przez VOCC w sygnał wap-
niowy, którego pierwszym elementem jest napływ Ca
2+
do
komórki i podwyższenie jego lokalnego stężenia (Ryc. 1A).
Ten efekt, zwykle krótkotrwały, ma jednak doniosłe skut-
ki regulacyjne. Heteromeryczne VOCC są pentamerami o
złożonej strukturze podjednostkowej. W ich skład wchodzą
podjednostki α1, kodowane przez 10 genów, zawierające
sensor napięcia, por kanału selektywnego dla Ca
2+
i miejsce
fosforylacji przez kinazę białkową zależną od cAMP, pod-
jednostki α2 i δ, kodowane przez 3 geny i regulujące am-
plitudę prądów jonowych, oraz podjednostki β i γ pełniące
funkcje regulatorowe. Skład podjednostkowy determinuje
właściwości czynnościowe VOCC. W oparciu o kryteria
elektrofizjologiczne i farmakologiczne wyodrębniono pięć
typów VOCC, kanały L, P/Q, N, R i T. Wykazano istotne
różnice w lokalizacji poszczególnych typów tych kanałów
w różnych częściach komórki nerwowej i w ich funkcjach.
Kanały N i P/Q są głównie zlokalizowane presynaptycznie
i regulują zależną od Ca
2+
egzocytozę pęcherzyków synap-
tycznych [10]. Kanały L są zlokalizowane zarówno w części
pre- jak i postsynaptycznej, głównie jednak w części prok-
symalnej dendrytów i ciele neuronów. Przypisuje się im
rolę w regulacji za pośrednictwem jonów Ca
2+
transkrypcji
genów, w aktywacji genów szybkiej odpowiedzi i w akty-
wacji wraz z kalmoduliną białka CREB [11]. Znane są zło-
żone oddziaływania i powiązania czynnościowe kanałów
L z różnorodnymi zależnymi od wapnia mechanizmami
przekazywania sygnałów w neuronach [12]. Obecnie dys-
ponujemy rozbudowanym zestawem narzędzi farmakolo-
gicznych służących do sterowania aktywnością VOCC [13].
ROC przetwarzają docierający z zewnątrz sygnał che-
miczny, jakim jest związanie się specyficznego neuroprze-
kaźnika z receptorem w błonie synaptycznej w sygnał wap-
niowy wewnątrz komórki. Pobudzenie receptorów jonotro-
powych prowadzi bowiem do aktywacji kanału przepusz-
czalnego dla Ca
2+
. Wśród ROC najbardziej rozpowszech-
nione w mózgu są jonotropowe receptory dla glutaminianu
trzech podtypów: NMDAR, AMPAR i kainianowe (KAR)
(Ryc. 1A) [14]. Szczególnie wysoką przepuszczalnością dla
jonów wapnia cechują się NMDAR. Są to tetramery, składa-
jące się z dwóch podjednostek NR1 i dwóch podjednostek
NR2. Występuje wiele wariantów poszczególnych podjed-
nostek (NR1
1-4a/b
oraz NR2
A-D
), co zapewnia ogromną różno-
rodność tych receptorów. Ich unikalną cechą jest detekcja
koincydencji zdarzeń kluczowych dla aktywacji receptora:
dostępności agonisty, którym jest neuroprzekaźnik - gluta-
minian, koagoniasty - glicyny lub D-seryny, oraz depolary-
zacji błony plazmatycznej neuronu, w czym mogą pośred-
niczyć AMPAR [15]. Aktywacja kanału jonowego NMDAR
zależy bowiem od depolaryzacji, która zwalnia jony Mg
2+
blokujące kanał. C-końcowe cytoplazmatyczne fragmen-
ty podjednostek NMDAR służą do połączenia tych recep-
torów z innymi elementami gęstości postsynaptycznych
(PSD, ang. post-synaptic density) w kolcach dendrytycznych
synaps, co ma zasadnicze znaczenie dla procesów przeka-
zywania sygnałów. AMPAR są heteromerami składającymi
się z podjednostek GluR1-4 kodowanych przez cztery geny.
Ich przepuszczalność dla jonów Ca
2+
jest normalnie bardzo
ograniczona, jednak wzrasta ona dramatycznie przy braku
w strukturze podjednostki GluR2, co ma istotne znaczenie
funkcjonalne i patologiczne [16]. KAR mają strukturę i wła-
ściwości zbliżone do AMPAR. Inne przepuszczalne dla Ca
2+
podtypy ROC, to receptory jonotropowe dla acetylocholiny
(nAch), ATP (P2X) i serotoniny (5-HT3) [17], które cechują
się mniej intensywnymi przepływami Ca
2+
i pozostają w cie-
niu receptorów dla glutaminianu.
Podobnie jak w komórkach niepobudliwych, w neuro-
nach zachodzi napływ wapnia za pośrednictwem kanałów
pojemnościowych SOCC (ang. store-operated calcium chan-
nels) z udziałem białka Orai1, których aktywność jest re-
gulowana przez wrażliwe na zawartość Ca
2+
w ER białka
sensorowe STIM [18], wykryte w neuronach licznych okolic
mózgu [19]. Napływowi pojemnościowemu wapnia przypi-
suje się udział w mechanizmach plastyczności neuronalnej
[20].
UWALNIANIE JONÓW WAPNIA
Z WEWNĘTRZNYCH ZASOBÓW NEURONÓW
W uwalnianiu Ca
2+
zmagazynowanego w ER do cytosolu
pośredniczą dwa typy kanałów wapniowych zlokalizowa-
nych w błonach ER (Ryc. 1A) [6,7]. Jeden z nich, to receptor
IP
3
(IP
3
R) o budowie tetrameru (monomeru), zbudowane-
go z trzech sklonownych izoform, a poszczególne podtypy
IP
3
R różnią się właściwościami funkcjonalnymi [21]. Ago-
nista tego receptora — IP
3
, to produkt sprzężonej z recepto-
rami metabotropowymi fosfolipazy C [3]. IP
3
pełni funkcję
globalnego przekaźnika sygnału w komórce, co wynika z
jego długiego, mierzonego w sekundach okresu półtrwa-
nia i rozległego zakresu dyfuzji w komórkach w granicach
20 μm. IP
3
Rs integrują ten sygnał i przekształcają go w sy-
gnał lokalny przekazywany przez jony Ca
2+
. Ten podział
ról wynika z faktu, że droga dyfuzji uwalnianych z ER jo-
nów Ca
2+
jest 200 razy krótsza niż IP
3
[22]. Aktywność IP
3
R
jest zwrotnie regulowana przez jony Ca
2+
, przy czym Ca
2+
w stężeniach do 0,1–0,3 μM potęgują aktywność kanału, a
przy wyższych stężeniach go inaktywują [23]. Z kolei białka
wiążące wapń oraz cytochrom c hamują inaktywację kanału
przez wyższe stężenia jonów Ca
2+
[24,25].
Drugi typ receptorów/kanałów wapniowych zaanga-
żowanych w proces wyrzutu Ca
2+
z ER w neuronach, to
receptor rianodynowy (RyR) (Ryc. 1A) [26]. RyR to homo-
tetramer o dużej masie cząsteczkowej ok. 500 kDa, z N-
-końcową domeną cytoplazmatyczną, domeną błonową w
pobliżu końca C cząsteczki oraz regionem pośrednim, za-
wierającym miejsca wiążące jony Ca
2+
, ATP, kalmodulinę

406
www.postepybiochemii.pl
(CaM) i inne modulatory [27]. Sklonowane trzy izoformy
RyR charakteryzują się różną lokalizacją w tkankach i nieco
innymi właściwościami. Izoforma RyR1 występuje głównie
w mięśniach szkieletowych, RyR2 w sercu i neuronach mó-
zgu, a RyR3, choć nazywana neuronalną lub mózgową, jest
wykrywana głównie w tkankach obwodowych. Aktywność
RyR jest regulowana przez wapń, przy czym Ca
2+
w niskich
stężeniach pobudza, a w wysokich hamuje aktywność RyRs
[28]. Kofeina i cykliczna ADP-ryboza są odpowiednio egzo-
i endogennymi aktywatorami RyR [29].
Uwalnianie jonów Ca
2+
z ER w neuronach, zachodzące
za pośrednictwem IP
3
R, jest wykorzystane w mechanizmie
przekazywania sygnałów z udziałem receptorów metabo-
tropowych, w tym w mGluR grupy I, obejmującej dwa typy
receptorów: mGluR1 i mGluR5 (Ryc. 1A) [30]. Jak wspo-
mniano powyżej, niskie stężenia jonów Ca
2+
aktywują oba
rodzaje receptorów/kanałów wapniowych występujących
w ER. Ta cecha jest podstawą dla CICR, czynnościowego
sprzężenia poprzez Ca
2+
kanałów i receptorów pośredniczą-
cych w napływie Ca
2+
ze środowiska zewnętrznego (zwłasz-
cza VOCC i NMDAR) z receptorami uwalniającymi Ca
2+
z
ER, a szczególnie RyR. Znaczenie czynnościowe CICR po-
lega na wzmacnianiu i przedłużaniu sygnału wapniowego
w neuronach [31]. Bez względu na drogę uwolnienia Ca
2+
z
ER, opróżnienie tych zasobów wapnia indukuje pojemno-
ściowy napływ Ca
2+
do komórki oraz uzupełnienie zasobów
w ER [18]. Niezależnie od znanych funkcji sekrecyjnych
związanych z tworzeniem amyloidu β, białko presenilina
odgrywa ważną rolę w integracji homeostazy wapnia w ko-
mórce, moduluje aktywność IP
3
R i RyR, pracę pompy wap-
niowej SERCA, wpływa na aktywację SOCC, a sama tworzy
w błonie ER kanał wycieku Ca
2+
(ang. leak channel) [32].
BUFOROWANIE Ca
2+
I WYGASZANIE SYGNAŁU
WAPNIOWEGO W NEURONACH
Kontrolowany wzrost wewnątrzkomórkowego stężenia
Ca
2+
w neuronach (sygnał wapniowy), dla zachowania swej
kluczowej roli w systemie przekazu informacji w komór-
kach, wymaga skutecznych mechanizmów wygaszania, co
sprowadza się do usuwania wolnych jonów Ca
2+
z cytosolu.
Do tych mechanizmów należą wyrzut wapnia na zewnątrz
komórki za pośrednictwem pompy PMCA i wymiennika
NCX oraz buforowanie wewnątrz neuronów, polegające
na pobieraniu Ca
2+
przez organelle komórkowe - ER i mito-
chondria oraz wiązaniu przez wyspecjalizowane białka [2].
USUWANIE Ca
2+
Z NEURONÓW
Dla pełnego i długoterminowego wyrównania bilansu
wapniowego komórki nerwowej oraz utrzymania niskiego
stężenia Ca
2+
w cytosolu decydujące są mechanizmy usu-
wania tych jonów na zewnątrz (Ryc. 1A). W warunkach
spoczynkowych tę rolę spełnia PMCA. Cząsteczka tego
enzymu ma złożoną strukturę, w tym 10 fragmentów prze-
szywających błonę i 4 pętle wewnątrzkomórkowe z krótkim
fragmentem N-końcowym, miejsce wiążące kwaśne fosfo-
lipidy, domenę katalityczną oraz fragment C-końcowy za-
wierający miejsca modulatorowe wiążące CaM i fosforylo-
wane przez kinazy białkowe C i A [33]. W mózgu występuje
ponad 30 izoform PMCA; białko to jest kodowane przez 4
geny i są cztery alternatywne warianty jego składania. Daw-
no poznany mechanizm działania PMCA polega na zmianie
konformacyjnej cząsteczki, zachodzącej dzięki hydrolizie
ATP. Wyrzutowi wapnia z cytoplazmy towarzyszy napływ
protonów do wnętrza komórki. Jako błonowa pompa wap-
niowa PMCA cechuje się względnie niską wydajnością, ale
wysokim powinowactwem do jonów Ca
2+
, dzięki czemu
utrzymuje ich stężenie na prawidłowym poziomie poniżej
10
–7
M.
W neuronach pobudzonych dochodzi do znacznego
wzrostu stężenia Ca
2+
w komórce. Do jego usuwania włącza
się wymiennik NCX, wykorzystujący energię elektroche-
micznego gradientu sodu (Ryc. 1A). Posiada on znacznie
niższe niż PMCA powinowactwo dla jonów Ca
2+
, ale dużą
wydajność transportu. NCX jest białkiem transportowym
niezależnym od potasu, kodowanym przez geny NCX1, 2 i
3 [34]. Ponadto występuje też transporter zależny od pota-
su, kodowany przez geny NCKX1 i 2. Białka te mają rozbu-
dowaną strukturę zawierającą 11 domen transbłonowych,
pętle cytoplazmatyczne z miejscami modulatorowymi, a po
stronach na zewnątrz i wewnątrz komórki posiadają liczne
miejsca wiążące jony [35]. Aktywność NCX jest modulowa-
na przez ATP, stopień ufosforylowania, fosfoargininę, jony
H
+
i Na
+
. Zależy ona też od stężenia jonów Ca
2+
w cytosolu,
jest bardzo niska przy wartościach spoczynkowych poniżej
10
-7
M, natomiast osiąga maksymalną wartość przy stężeniu
Ca
2+
10
-4
M [36]. Transport polega na wymianie 1 jonu Ca
2+
na 3 jony Na
+
, lub 1 jonu Ca
2+
i 1 jonu K
+
na 4 jony Na
+
, i
jest odwracalny, przy czym kierunek transportu jest regulo-
wany przez potencjał błonowy oraz gradienty stężeń jonów
Ca
2+
i Na
+
. Odwrócony tryb pracy prowadzący do napływu
wapnia, cechuje komórkę o niskim potencjale błonowym,
z podwyższonym stężeniem sodu. Tak więc, wymiennik
NCX pracujący w trybie odwróconym jest alternatywną
drogą wnikania wapnia do komórek.
POBIERANIE Ca
2+
PRZEZ MITOCHONDRIA I ER
Obok wytwarzania ATP i regulacji metabolizmu w ko-
mórce, inną uniwersalną funkcją mitochondriów jest pobie-
ranie jonów Ca
2+
. Fakt, że ulega on znaczącej aktywacji do-
piero przy stężeniu Ca
2+
> 0,5µM czyni z tego mechanizmu
bufor wapniowy o stosunkowo niskim powinowactwie, ale
dużej pojemności (Ryc. 1A). Ponieważ zewnętrzna błona
mitochondrialna jest przepuszczalna dla substancji drob-
nocząsteczkowych (o małej masie cząsteczkowej), napływ
Ca
2+
do mitochondriów polega na transporcie przez błonę
wewnętrzną na zasadzie uniportu napędzanego przez po-
tencjał mitochondrialny i gradient elektrochemiczny Ca
2+
[37]. Usuwanie Ca
2+
z mitochondriów do cytosolu zacho-
dzi ze znacznie mniejszą szybkością, głównie na zasadzie
wymiany z jonami Na
+
[38]. Zarówno uniporter jak i mito-
chondrialny wymiennik NCX są hamowane przez czerwień
rutenową i kationy dwuwartościowe, przy czym jony Mg
2+
hamują aktywność uniportera przez działanie na miejsce
regulatorowe. Za mechanizm mogący prowadzić do szyb-
kiego usuwania nadmiaru jonów Ca
2+
z mitochondiów
uważana jest aktywacja megakanałów (kanałów MPT, ang.
mitochondrial permeability transition), co przy przeładowa-
niu mitochondriów jonami wapnia może się stać zarazem
elementem dysfunkcji mitochondriów mogącej prowadzić

Postępy Biochemii 58 (4) 2012
407
do nekrotycznej lub apoptotycznej śmierci neuronów [39].
Ponieważ w warunkach spoczynkowych w neuronach
stężenie Ca
2+
jest znacznie niższe niż powinowactwo mi-
tochondrialnego uniportera, buforowanie przez mitochon-
dria wewnątrzkomórkowego stężenia Ca
2+
nabiera znacze-
nia regulacyjnego przy lokalizacji tych struktur w pobliżu
zewnątrzkomórkowych lub wewnątrzkomórkowych ka-
nałów wapniowych, gdzie lokalne stężenia jonów wapnia
mogą na krótko osiągać wysokie wartości i wymagają bu-
forowania [40].
Wysokie powinowactwo do Ca
2+
wykazuje system buforo-
wania wapnia w ER, w skład którego wchodzą wapniowa AT-
Paza SERCA, transportująca wapń do światła ER, białka wią-
żące Ca
2+
zlokalizowane wewnątrz kanałów ER, oraz recepto-
ry/kanały IP
3
i RyR uwalniające Ca
2+
(Ryc. 1A) [7,41]. Aktyw-
ności SERCA i pobieraniu Ca
2+
przez cysterny ER towarzyszy
przeciwprądowy przepływ H
+
. Wykryto dotąd trzy izoformy
i szereg wariantów SERCA: 1a i b, 2a i b oraz 3. Najlepiej po-
znano strukturę molekularną SERCA 2b zawierającą dziesięć
domen transbłonowych, trzy domeny cytoplazmatyczne, w
których zlokalizowane są miejsca wiązania ATP i ulegające
fosforylacji, oraz dwa miejsce wiążące Ca
2+
[42].
Wzrastającą uwagę przyciąga homeostaza wapnia w ją-
drze komórkowym. Podwójna błona jądrowa jest w ciągłym
kontakcie z ER, zachowując jej funkcje transportu Ca
2+
. Pory
w błonie jądrowej są przepuszczalne dla małych cząstek, w
tym dla jonów Ca
2+
. Wykazano, że transport Ca
2+
w błonie
jądrowej jest spolaryzowany i są różnice w zawartości białek
transportujących Ca
2+
w błonie zewnętrznej i wewnętrznej.
Błona wewnętrzna obfituje w receptory IP
3
i rianodynowe, a
błona zewnętrzna zawiera SERCA. Ułatwia to generowanie
i wygaszanie sygnału Ca
2+
w nukleoplazmie, obfitującej w
białka wiążące wapń. Te mechanizmy stanowią podłoże dla
bezpośredniej roli wapnia w mechanizmach przekazywania
sygnałów w jądrze komórkowym [43].
BIAŁKA WIĄŻĄCE Ca
2+
O znaczeniu mechanizmów wiązania Ca
2+
wewnątrz
komórki świadczy to, że wolne jony Ca
2+
w cytosolu sta-
nowią zaledwie 1% całkowitej zawartości wapnia, a resz-
ta przypada na wapń związany z cząsteczkami jak ATP, a
przede wszystkim z różnorodnymi białkami. Pomijając en-
zymy takie jak niektóre fosfolipazy A
2
, posiadające w swojej
strukturze miejsca wiążące Ca
2+
z niskim powinowactwem
i aktywowane w obecności tych kationów [44], należy pod-
kreślić znaczenie właściwych białek wiążących Ca
2+
[45].
Wśród nich wyróżniamy bufory wapniowe jak parwalbu-
mina, kalbindyna i kalretinina, którym przypisuje się m. in.
udział w wiązaniu i wewnątrzkomórkowym transporcie
tego kationu, oraz sensory wapnia jak CaM, kalcyneuryna,
białka S100 i troponina, a także neuronalne sensory wapnia
(NCS), spełniające omówione poniżej funkcje regulacyjne
ważne w mechanizmach przekazywania sygnałów. Właści-
we białka wiążące wapń posiadają w swej strukturze różną
liczbę domen zwanych „EF-hand”, umożliwiających wiąza-
nie Ca
2+
[2,46]. Białka wiążące wapń, zarówno o niskim jak i
o wysokim powinowactwie, występują w dużych ilościach
zarówno w cytosolu, jak i w świetle cystern ER, w jądrze
oraz w macierzy mitochondriów.
CZUJNIKI WAPNIA; NIEGENOMOWE
KONSEKWENCJE PRZEKAZYWANIA SYGNAŁU
WAPNIOWEGO W NEURONACH
Wynikiem wzrostu wewnątrzkomórkowego stężenia
Ca
2+
jest ich przyłączenie się do białek zawierających dome-
ny EF-hand, będących wewnątrzkomórkowymi czujnikami
Ca
2+
i
mediatorami zależnych od wapnia dalszych szlaków
przekazywania sygnału. Przykładem specyficznych dla
neuronów i wyspecjalizowanych białek będących sensora-
mi wapnia są synaptotagminy, zlokalizowane w części pre-
synaptycznej zakończeń synaptycznych i odgrywające klu-
czową rolę w zależnym od Ca
2+
wyrzucie neurotransmite-
rów nagromadzonych w pęcherzykach synaptycznych [47].
Kalmodulina (CaM) odgrywa szczególną rolę uniwersal-
nego czujnika wapnia integrującego sygnał wapniowy w
neuronach i zaangażowanego w regulację bardzo licznych
procesów komórkowych (Ryc. 2). W kompleksie z Ca
2+
wiąże się ona z szeregiem białek efektorowych, w tym en-
zymów takich jak kinazy i fosfatazy białkowe, cyklazy czy
syntazy NO i reguluje ich aktywność [48]. Ważne miejsce
wśród zależnych od wapnia i CaM mechanizmów przeka-
zywania sygnałów w neuronach zajmuje szlak aktywowany
przez pobudzenie receptorów NMDA lub VOCC i napływ
Ca
2+
, obejmujący aktywację syntazy NO, pobudzenie cy-
klazy guanylowej, produkcji cGMP i odpowiednich kinaz
białkowych, fosfataz i kanałów jonowych, co prowadzi
do modulacji aktywności synaptycznej [49]. Istotną rolę w
procesie przekazywania sygnału odgrywają cyklazy adeny-
lanowe wrażliwe na wapń i CaM. Natomiast wśród kinaz
białkowych zależnych od wapnia i CaM (CaM kinaz), nale-
ży wyróżnić CaM kinazę II (CaMKII) zlokalizowaną postsy-
naptycznie, która jest podstawowym zależnym od wapnia
regulatorem plastyczności synaptycznej. Przykłady mecha-
nizmów, ważnych dla krótko- i długotrwałych zmian pla-
stycznych w neuronach, a kontrolowanych przez Ca
2+
, CaM
i CaMKII to fosforylacja receptorów AMPA oraz aktywacja
jądrowego czynnika transkrypcyjnego CREB [50]. Fosfata-
za białkowa, kalcyneuryna jest enzymem aktywowanym
przez wapń i CaM, co powoduje, że CaM kontroluje także
defosforylację białek [51].
Ca
2+
A REGULACJA EKSPRESJI GENÓW
Szybkie odpowiedzi wewnątrzkomórkowe na wzrost
stężenia Ca
2+
angażują głównie potranslacyjną modyfika-
cję białek już obecnych w komórce. Jednakże długotrwałe
zmiany strukturalne i funkcjonalne komórki, będące od-
powiedzią na sygnał wapniowy, wymagają uruchomienia
ekspresji odpowiednich genów. Wykazano, że sygnał wap-
niowy jest niezbędny do uruchomienia ekspresji genów
wczesnej odpowiedzi takich jak np. c-fos czy zif/268, a w
konsekwencji również do aktywacji ekspresji genów róż-
nych białek docelowych. W neuronach można wyróżnić
dwa mechanizmy, dzięki którym elektryczny sygnał pobu-
dzenia komórki przekładany jest na transkrypcję. W pierw-
szym, jony Ca
2+
po wniknięciu do komórki aktywują znaj-
dujące się w pobliżu cytoplazmatyczne przekaźniki, które
następnie przekazują sygnał do jądra. W drugim jony Ca
2+
działają bezpośrednio w jądrze.
408
www.postepybiochemii.pl
Podstawowym szlakiem uruchamianym przez wzrost
stężenia Ca
2+
w cytoplazmie jest szlak Ras/MAP kinaza/
ERK1/2 (ang. extracellular signal-regulated kinase 1/2), który
prowadzi do aktywacji jądrowych kinaz Rsk 1-3, mających
zdolność fosforylacji związanych z DNA białek regulują-
cych transkrypcję (Ryc. 2). Szlak ten aktywowany jest głów-
nie napływem Ca
2+
przez kanały związane z NMDAR [52],
jednakże napływ Ca
2+
przez zależne od potencjału kanały
typu L (L-VOCC) również może uruchomić ten szlak [53].
Nie jest do końca wyjaśnione, w jaki sposób sygnał wap-
niowy uruchamia kaskadę Ras/MAPK/ERK1/2. Część ba-
daczy uważa, że kompleks Ca
2+
/CaM reguluje aktywację
Ras poprzez czynnik uwalniający nukleotyd guanylowy
Ras (Ras-GRF), inni z kolei opowiadają się za udziałem za-
leżnej od Ca
2+
/CaM kinazy kinazy (CaMKK) oraz zależnej
od Ca
2+
/CaM kinazy I (CaMKI) [54]. Przekaz sygnału przez
ERK1/2 w jądrze zachodzi albo bezpośrednio za pomocą
ufosforylowanego ERK1/2 (pERK1/2) przedostającego się
do jądra, albo pośrednio poprzez kinazy fosforyzowane
przez pERK1/2 w cytoplazmie przed ich przemieszczeniem
się do jądra. Do kinaz tych należą zlokalizowana w jądrze
i wymagająca obecności aktywnej MAPK — MKS1/2 oraz
RSK2, która jest importowana do jądra po jej ufosforylowa-
niu przez REK1/2 w cytoplazmie [54].
Wzrost stężenia Ca
2+
w cytoplazmie aktywuje również
przekazywanie sygnału przez kinazy zależne od Ca
2+
i CaM
(CaMKs) (Ryc. 2). CaMKs I, II i IV mogą fosforylować CREB
na reszcie seryny 133 (Ser133), co jest niezbędne do zapo-
czątkowania transkrypcji, jednakże największe znaczenie
wydają się mieć CaMKs II oraz IV. CaMK II zlokalizowa-
na jest głównie w cytoplazmie, a jej aktywna forma może
ulegać przemieszczeniu do jądra. W jądrze znajdują się
również izoformy CaMKII mogące być aktywowane przez
przedostające się do jądra jony Ca
2+
związane z CaM. Na-
tomiast CaMK IV jest enzymem zlokalizowanym głównie
w jądrze i jak wykazano jej aktywacja zbiega się w czasie
z aktywacją CREB [55]. CREB nie jest jedynym czynnikiem
transkrypcyjnym, który może aktywować jony Ca
2+
i CaM.
Wykazano bowiem, że działanie CaMKIV jest niezbędne
przy aktywowaniu C/EBPβ, ATF-1 oraz SRF [52].
Wiadomo, że chociaż napływ Ca
2+
przez L-VOCC silnie
aktywuje CREB, to równoważny napływ Ca
2+
przez pozo-
stałe VOCC nie skutkuje zainicjowaniem zależnej od CREB
ekspresji genów [55]. Napływ Ca
2+
przez kanały związane z
NMDAR jest dominującym elementem aktywacji ekspresji
genów w embrionalnym rozwoju neuronów, jednakże wraz
z dojrzewaniem tkanki mózgowej tę rolę przejmują również
inne mechanizmy powodujące wzrost wewnątrzkomórko-
wego stężenia Ca
2+
[55]. Wykazano, że w dojrzałej tkance
mózgowej napływ Ca
2+
przez VOCC typu L prowadzi do
długotrwałej fosforylacji CREB, podczas gdy wejście Ca
2+
kanałami związanymi z NMDAR skutkuje krótkotrwałą
fosforylacją [56].
Oprócz opisanych powyżej dwóch szlaków przekazywa-
nia sygnału wapniowego w cytoplazmie, transkrypcja może
być aktywowana bezpośrednio przez wzrost stężenia Ca
2+
w jądrze. Podwyższenie poziomu Ca
2+
w jądrze najczęściej
związane jest z uwalnianiem Ca
2+
z magazynów wewnątrz-
komórkowych, ale może też być wynikiem przemieszczenia
się kompleksu Ca
2+
/CaM do jądra. Bezpośredni napływ Ca
2+
do jądra wiąże się z silną lub powtarzającą się stymulacją
dendrytów, co powoduje masowy napływ Ca
2+
, przemiesz-
czającego się do ciała komórki. Ponieważ struktura błony
jądrowej pozwala na dyfuzję Ca
2+
do jądra, przy wzroście
stężenia Ca
2+
w pobliżu tej błony, część przemieszcza
się do nukleoplazmy, a ponadto jony Ca
2+
mogą być
uwalniane z zasobów wewnątrz podwójnej błony ją-
drowej [43,57]. Uważa się, że jony Ca
2+
w jądrze nie
angażują się w aktywację specyficznych czynników
transkrypcyjnych, ale raczej kontrolują, poprzez ak-
tywację jądrowej CaMKIV, aktywność białek wią-
żących CREB (CBP). CBP może oddziaływać z wie-
loma czynnikami transkrypcyjnymi, stanowiąc me-
chanizm, poprzez który sygnał wapniowy w jądrze
może modulować aktywność potencjalnie dużej licz-
by tych czynników, a co za tym idzie ekspresję dużej
liczby genów [58].
CREB jest najlepiej poznanym do tej pory czyn-
nikiem transkrypcyjnym regulowanym sygnałem
wapniowym (Ryc. 2). Wiąże się w postaci dimeru
do domeny DNA nazwanej elementem odpowiedzi
na cAMP (CRE) i jest kluczowym elementem w eks-
presji genów aktywowanej sygnałem wapniowym
w komórkach pobudliwych. Uruchomienie trans-
krypcji genów, w której pośredniczy CREB, wymaga
fosforylacji reszty Ser133 w tym białku. Fosforylacja
ta może być katalizowana przez szereg kinaz, takich
jak kinaza białkowa A (PKA), CaMK I, II i IV, regu-
lowana przez ERK kinaza RSK2 czy kinazy regulo-
wane przez p38 MAP [52]. Nie oznacza to jednak, że
każda fosforylacja reszty Ser133 skutkuje aktywacją
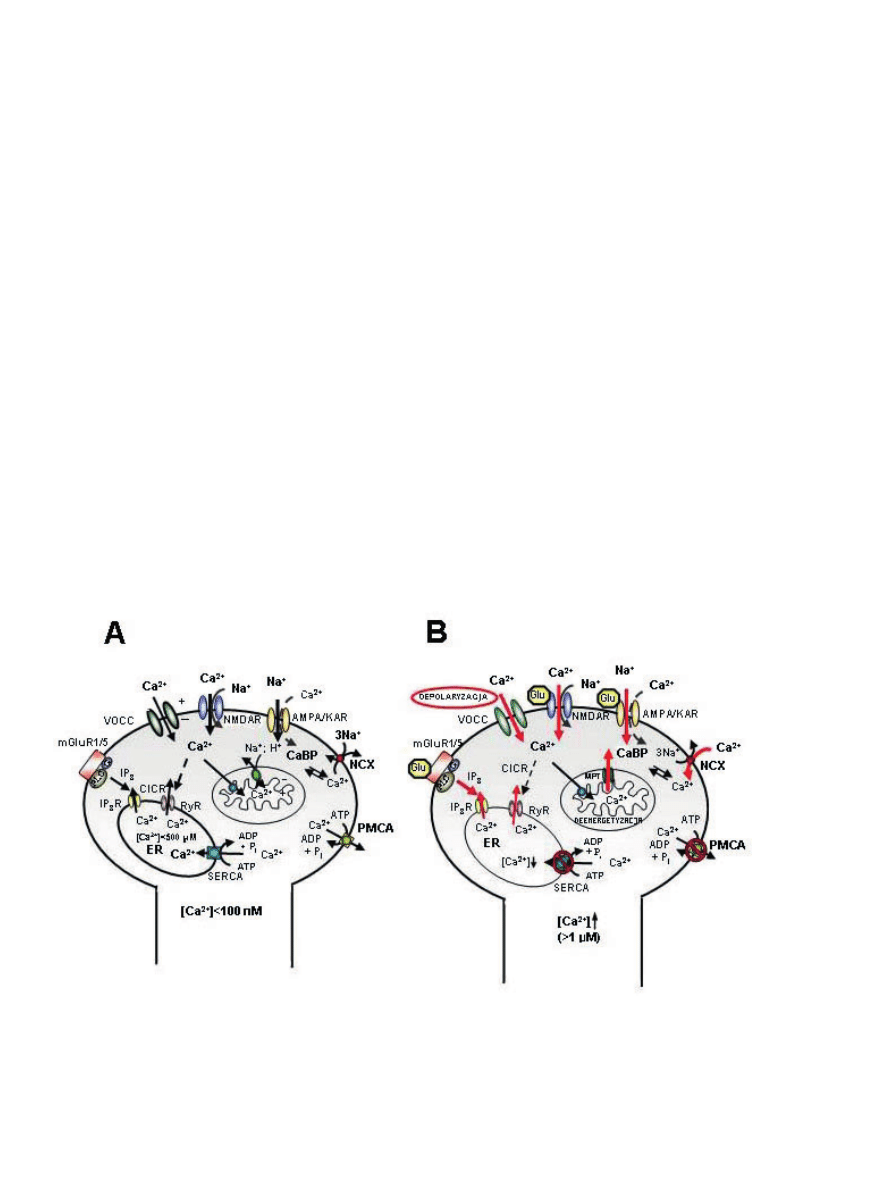
Rycina 2. Rola Ca
2+
w przekazywaniu sygnału uruchamiającego transkrypcję genów w neu-
ronach. Napływ Ca
2+
do wnętrza neuronów przez VOCC lub ROS powoduje aktywację kil-
ku szlaków sygnałowych jak cyklazy adenylanowej (AC), kinazy aktywowane kompleksem
Ca
2+
/kalmodulina przez Ras. Każda z tych dróg prowadzi do przekazania sygnału do jądra
i uruchomienia kinaz przemieszczonych do jądra (PKA, RSK1-3 i CaMKII) oraz aktywacji
jądrowych kinaz (CaMKII i IV), co w efekcie umożliwia fosforylację CREB i rozpoczęcie
transkrypcji genu.

Postępy Biochemii 58 (4) 2012
409
transkrypcji. Wykazano, że do zapoczątkowania transkryp-
cji niezbędny jest dodatkowy czynnik aktywowany jądrową
CaMKIV, którym jest białko wiążące CREB (CBP). CBP jest
białkowym koaktywatorem, który nie wiąże się bezpośred-
nio z DNA, może za to oddziaływać z wieloma czynnikami
transkrypcyjnymi [58]. Łączenie CBP z wybranym promoto-
rem jest inicjowane przez kaskady uruchamiane sygnałem
wapniowym, ale żaden z tych szlaków nie aktywuje samego
CBP. Aktywacja CBP kontrolowana jest przez jony Ca
2+
w
jądrze oraz jądrową CaMKIV, jednak sam mechanizm akty-
wacji jest do tej pory niejasny. CaMKII, która również może
fosforylować w CREB resztę Ser133, fosforyluje też resztę
Ser142, co skutkuje zahamowaniem transkrypcji. Fosforyla-
cja reszty Ser142 jest indukowana sygnałem pochodzącym z
depolaryzacji błony komórkowej, która ma miejsce w czasie
trwania transkrypcji aktywowanej CREB [55].
Oddziałujące z CBP czynniki transkrypcyjne inne niż
CREB, również mogą być zaliczone do regulowanych przez
sygnał wapniowy. Jednym z nich jest c-Jun, którego akty-
wacja przez sygnał wapniowy w komórkach hipokampa
nie wymaga, jak w innych przypadkach, fosforylacji reszt
seryny 63 i 73, za to jest kontrolowana przez CaMKIV i ak-
tywację CBP. To sugeruje, że CBP przyłączone do c-Jun jest
jednocześnie miejscem regulacji transkrypcji aktywowanej
przez Ca
2+
[52].
Innym elementem regulowanym przez jony Ca
2+
jest
czynnik SRF (ang. serum response factor), znaleziony na
promotorze c-fos i miejscach regulatorowych kilku in-
nych genów [59]. Łączy się on z domeną SRE (ang. serum
response element) w postaci dimeru, często w towarzy-
stwie jednego z koaktywatorów transkrypcji z rodziny
TCFs (ang. ternary complex factors), również regulowane-
go przez Ca
2+
lub MRTF (ang. myocardin-related transcrip-
tion factor) [59,60]. W odróżnieniu od CREB/CBP, ekspre-
sja genów regulowana przez SRF jest kontrolowana przez
mechanizm uruchamiany przez cytoplazmatyczny sy-
gnał wapniowy [57]. Uważa się, że szlakiem przekazywa-
nia sygnału zaangażowanym w aktywację SRF raczej nie
jest kaskada Ras/MAPK/ERK1/2 i że zaangażowane są
tu CaM kinazy [52]. CaMKII, która katalizuje fosforylację
reszty Ser103 w SRF wydaje się jednak nie być jedynym
aktywatorem SRF regulowanym Ca
2+
i prawdopodobnie
w aktywację transkrypcji zaangażowane są jeszcze inne
białka [61].
Wzrost stężenia jądrowego Ca
2+
może bezpośrednio re-
gulować proces transkrypcji poprzez DREAM (ang. down-
stream regulatory element antagonist modulator). Jest to biał-
ko zawierające wiążącą Ca
2+
domenę EF-hand, łączące się z
DRE (ang. downstream regulatory element). Kompleks DRE-
AM/DRE hamuje transkrypcję, prawdopodobnie przez
hamowanie elongacji transkrypcji. Gdy wzrasta stężenie
Ca
2+
w jądrze, Ca
2+
wiąże się do DREAM, zmniejszając
jednocześnie jego powinowactwo do DRE. Skutkiem tego
jest odłączenie się DREAM od DNA i zwolnienie hamulca
transkrypcji docelowego genu. DREAM jest białkiem sze-
roko rozpowszechnionym w układzie nerwowym, szcze-
gólnie w neuronach czuciowych, gdzie odpowiedzialny
jest za ekspresję prodynorfiny, genu związanego z modu-
lacją bólu [62].
FIZJOLOGICZNA ROLA JONÓW
WAPNIA W NEURONACH
Jony Ca
2+
są uniwersalnym przekaźnikiem informacji
we wszystkich komórkach, pełnią jednak szczególną rolę
w komórkach pobudliwych, gdzie sprzęgają pobudzenie z
ich specyficzną funkcją. W układzie nerwowym jony Ca
2+
są zaangażowane w neurotransmisję na wielu etapach tego
procesu. Regulują one pobudliwość neuronów, w części
presynaptycznej synaps aktywują sekrecję neuroprzekaź-
ników chemicznych, a w części postsynaptycznej pełnią
funkcję wtórnego przekaźnika sygnału [3]. Jak omówiono
powyżej, jony Ca
2+
i zależne od nich szlaki sygnałowe są w
neuronach zaangażowane w przekaz informacji od synapsy
do jądra. Zmiany stężenia jonów Ca
2+
wewnątrz komórki, a
więc sygnały wapniowe, są w neuronach wykorzystywane
do krótkotrwałej regulacji aktywności tych komórek lub w
procesach długo trwających. Wśród procesów długo trwa-
jących należy wymienić mechanizmy wzrostu neurytów,
tworzenia i przebudowy sieci neuronalnej, regulacji siły po-
łączeń synaptycznych oraz procesy warunkujące przeżycie
lub programowaną śmierć tych komórek. Te długotrwałe
mechanizmy wymagają zmiany ekspresji genów.
ROLA WAPNIA W NEUROTRANSMISJI
Od dawna znany jest udział Ca
2+
w stabilizacji poten-
cjału błony plazmatycznej i regulacji pobudliwości komó-
rek pobudliwych; wiadomo, że niedobory wapnia, któ-
re obserwuje się np. w tężyczce prowadzą do zaburzenia
pobudliwości. Szczegółowy mechanizm tego zjawiska jest
złożony [63], jego ważnym elementem jest aktywacja przez
Ca
2+
kanałów potasowych regulujących potencjał błonowy,
a zarazem biorących udział w mechanizmach plastyczności
neuronalnej [64].
Regulowany przez Ca
2+
proces sprzężenia depolaryzacji
z egzocytozą neuroprzekaźników chemicznych nagroma-
dzonych w pęcherzykach synaptycznych, zachodzi w czę-
ści presynaptycznej zakończenia nerwowego. W wyniku
depolaryzacji dochodzi do aktywacji VOCC typu N i P/Q
i do lokalnego wzrostu wewnątrzkomórkowego stężenia
Ca
2+
do poziomu ocenianego na 20-50 μM [65]. Jednak na-
tura i regulacja presynaptycznych sygnałów wapniowych
odpowiedzialnych za uwalnianie neuroprzekaźników jest
procesem bardziej złożonym, w którym poza wspomniany-
mi kanałami wapniowymi uczestniczą inne liczne elementy
regulacji homeostazy Ca
2+
. Jest to mechanizm przyczynia-
jący się do pewnej autonomii zakończeń synaptycznych
[66]. W regulacji aktywności kanałów wapniowych oraz
w dalszych etapach uwolnienia neuroprzekaźników z pę-
cherzyków synaptycznych do przestrzeni synaptycznej
uczestniczy szereg białek wiążących wapń, pełniących rolę
sensorów wapnia, zawierających motyw EF-hand, jak CaM,
kalcyklina i NCS-1 lub też, jak wspomniana wyżej synapto-
tagmina, domenę C2 wiążącą Ca
2+
i fosfolipidy, pierwotnie
odkrytą w strukturze kinazy białkowej C [47].
W części postsynaptycznej zakończenia nerwowego,
w wyniku pobudzenia receptorów i depolaryzacji, jony
Ca
2+
wnikają do komórki przez bramkowane ligandem ka-
nały wapniowe ROC, przez zależne od potencjału kanały

410
www.postepybiochemii.pl
wapniowe VOCC, a także są mobilizowane z ER za po-
średnictwem receptorów metabotropowych sprzężonych
przez białka G z fosfolipazą C i IP
3
R oraz za pośrednictwem
RyR w procesie CICR. W efekcie aktywowane są zależ-
ne od wapnia szlaki przekazywania sygnału. Jedne z nich
prowadzą poprzez potranslacyjną modyfikację (zwłasz-
cza fosforylację i nitrozylację) białek do wspomnianych
powyżej krótkotrwałych (mierzonych w minutach) zmian
aktywności neuronów. Istotnym aspektem wapniowej re-
gulacji transmisji synaptycznej jest zależna od Ca
2+
synteza
wstecznych neuroprzekaźników w synapsach. Wniknięcie
Ca
2+
do części postsynaptycznej aktywuje bowiem zależną
od Ca
2+
i CaM neuronalną syntazę tlenku azotu (nNOS), co
prowadzi do uwolnienia NO [49]. Z kolei efektem aktywacji
przez wapń fosfolipazy A
2
jest uwolnienie kwasu arachido-
nowego oraz jego metabolitów, eikozanoidów, a aktywacja
fosfolipazy Cβ w wyniku dalszych przemian doprowadza
do syntezy endokannabinoidów [67]. Od dawna sugerowa-
no, że NO, CO i eikozanoidy mogą pełnić rolę wstecznych
neuroprzekaźników transsynaptycznych i dyfundując do
części presynaptycznej zakończenia synaptycznego regulu-
ją uwalnianie glutaminianu, a przez to siłę neurotransmisji
pobudzającej [68,69]. Jednak ostatnio uwaga badaczy sku-
pia się głównie na endokannabinoidach [67].
Inną zależną od Ca
2+
specyficzną funkcją komórek ner-
wowych jest ruch w różnych formach. Takie fundamental-
ne procesy, jak migracja prekursorów komórek nerwowych,
wzrost aksonów, a w mniejszej skali zmiany morfologiczne
w aksonach i dendrytach związane z różnicowaniem, ucze-
niem się i pamięcią oraz regeneracją, są regulowane przez
zmiany stężenia Ca
2+
na zewnątrz i wewnątrz komórek i
modulację aktywności zależnych od Ca
2+
kinaz białkowych
[70].
UDZIAŁ SYGNAŁU WAPNIOWEGO
W LTP I FORMOWANIU PAMIĘCI
Jedną z najważniejszych funkcji jonów Ca
2+
w komór-
kach nerwowych jest udział w mechanizmach związanych
z uczeniem się i zapamiętywaniem. Większość badaczy jest
zgodna, że głównym elementem zapoczątkowującym pro-
cesy tworzenia się pamięci jest pobudzenie neurotransmisji
glutaminianergicznej, aktywacja NMDAR i napływ Ca
2+
do
komórek nerwowych.
Uczenie się i zapamiętywanie, procesy o podstawowym
życiowym znaczeniu, są od wielu lat obiektem intensyw-
nych badań, jednak ich dokładny mechanizm oraz bio-
chemiczne podstawy nie zostały jeszcze w wystarczający
sposób poznane. Wiadomo, że doświadczenia nabywane
w czasie nauki wywołują krótkotrwałe zmiany w przekaź-
nictwie synaptycznym w specyficznych rejonach mózgu,
a impulsy te zapoczątkowują kaskadę wewnątrzkomórko-
wych przemian, które prowadzą do trwałych zmian w po-
łączeniach synaptycznych, co wiązane jest z konsolidacją
pamięci. W związku z czasowym rozciągnięciem procesów
formowania pamięci, od lat funkcjonują pojęcia pamięci
krótkotrwałej (STM, ang. short term memory) oraz pamięci
długotrwałej (LTM, ang. long term memory). Uważa się, że
pamięć krótkotrwała tworzona jest w ciągu krótkiego cza-
su od nabycia informacji lub treningu i bazuje na przejścio-
wych modyfikacjach wcześniej istniejących białek, głównie
na fosforylacji i defosforylacji enzymów, receptorów lub
kanałów jonowych, mogących momentalnie zmieniać wy-
dajność przekaźnictwa synaptycznego [71]. Za rejon mózgu
odpowiedzialny za ten typ pamięci u ssaków uważa się hi-
pokamp oraz jego różne odpowiedniki w mózgach innych
zwierząt. Pamięć ta może funkcjonować od minut do go-
dzin. Natomiast zależna od syntezy nowych białek pamięć
długotrwała jest związana ze zmianami strukturalnymi
istniejących już oraz z generacją nowych połączeń synap-
tycznych [72]. Przechowywana jest ona przez godziny, dni,
tygodnie i lata od zarejestrowania informacji. Informacje za-
pisywane są w różnych rejonach mózgu, głównie w korze,
ale dokładny mechanizm tego zapisu oraz miejsca przecho-
wywania informacji nadal nie są w pełni poznane i pozosta-
ją przedmiotem badań.
W latach 70-tych ubiegłego wieku opisano zjawisko dłu-
gotrwałego wzmocnienia połączeń synaptycznych w od-
powiedzi na krótką stymulację elektryczną, które nazwano
LTP, a w niedługi czas potem opisano zjawisko długotrwa-
łego osłabienia synaptycznego, LTD [73,74]. Do indukcji
LTP niezbędne jest zadziałanie impulsów elektrycznych o
wysokiej częstotliwości, natomiast przy wywołaniu LTD
działają impulsy o niskiej częstotliwości, ale czas ich trwa-
nia jest dłuższy [75]. Uważa się, że zarówno LTP jak i LTD
odzwierciedlają komórkowe i molekularne mechanizmy
leżące u podstaw plastyczności synaps zaangażowanej w
procesie uczenia się i zapamiętywania i do dziś opisano
wiele różnych form tych zjawisk [76]. Najlepiej poznanymi
formami LTP i LTD są te, które obserwuje się w rejonie CA1
hipokampa i które zapoczątkowane są aktywacją NMDAR.
Indukcję LTP zapoczątkowuje uwalnianie glutaminia-
nu, aktywacja NMDAR i napływ Ca
2+
do neuronów. Pod-
wyższony poziom wewnątrzkomórkowego Ca
2+
razem z
kalmoduliną aktywują CaMKII, która może fosforylować
podjednostki AMPAR, aktywując je w ten sposób i przy-
czyniając się do dalszego napływu Ca
2+
. Dochodzi do niego
albo bezpośrednio przez przepuszczalne dla Ca
2+
receptory
pozbawione podjednostki GluR2, lub pośrednio na skutek
depolaryzacji błon i aktywacji VOCC [77]. Utrzymanie LTP
wymaga przekazania komórkowego sygnału do jądra i uru-
chomienia syntezy białek. Wykazano, że w czasie LTP ma
miejsce zarówno aktywacja szlaku Ras/MAPK/ERK1/2 jak
i CaMKIV. Jak wiadomo, aktywacja tych szlaków prowadzi
do fosforylacji CREB i aktywacji ekspresji genów. Wzmożo-
ną fosforylację CREB obserwowano od 3 min do 60 min po
inicjacji LTP [73].
Dla zainicjowania LTP ważny jest nie tylko napływ Ca
2+
kanałami związanymi z NMDAR. Wykazano, że określone
formy LTP obserwowane w wielu rejonach mózgu wyma-
gają również aktywacji mGluR. Główną rolę odgrywają tutaj
mGluR grupy I — mGluR1 i mGluR5 [78]. LTP zależne od
mGluR w większości przypadków również wymaga wzro-
stu wewnątrzkomórkowego stężenia Ca
2+
, a źródła tego
wzrostu mogą różnić się w zależności od formy LTP. Może
on wynikać z napływu Ca
2+
przez aktywowane receptory
NMDA, AMPA, VSCCs typu L lub kanały TRP (ang. tran-
sient receptor potential) oraz uwalniania Ca
2+
z wewnątrzko-
mórkowych magazynów aktywowanych przez IP
3
[78,79].

Postępy Biochemii 58 (4) 2012
411
Według obecnej wiedzy, uruchomienie w neuronach
LTD związane jest z pamięcią przestrzenną, zapamiętywa-
niem informacji o nowych obiektach oraz o zmianach ich
rozmieszczenia, a także odpowiedzią na stres [74]. Spośród
wielu form LTD najważniejszymi wydają się być LTD zależ-
ne od NMDAR oraz od mGluR. Podobnie jak w przypadku
LTP zależnego od aktywacji NMDAR, zainicjowanie LTD
wiąże się z napływem Ca
2+
do neuronów postsynaptycz-
nych. O tym czy po napływie Ca
2+
przez kanały związane
z NMDAR zainicjowane zostanie LTP czy LTD decyduje
wielkość przepływu tego jonu. LTD wymaga niewielkiego
wzrostu stężenia Ca
2+
, podczas gdy do inicjacji LTP koniecz-
ny jest znaczny wzrost, przekraczający określoną krytycz-
ną wartość [75]. Czas utrzymywania się podwyższonego
stężenia Ca
2+
również jest czynnikiem decydującym, gdyż
niewielkie zmiany w długości trwania pobudzenia mogą
spowodować odwrócenie modyfikacji synaptycznej [80].
Sposób w jaki buforowany jest wapń w przedziale post-
synaptycznym też może stać się punktem wytyczającym
dalszy bieg procesów wewnątrzkomórkowych w kierunku
LTD lub LTP [76].
W odróżnieniu od LTP, LTD zależne od NMDAR nie
uruchamia aktywacji CaMKII, za to wzrost stężenia Ca
2+
ak-
tywuje kaskadę fosfatazy białkowej, w skład której wchodzi
kalcyneuryna, zależna od kompleksu wapń/CaM fosfataza
serynowo-treoninowa, fosfataza białkowa 1 (PP1) i inhibi-
tor-1, ufosforylowane białko odpowiedzialne za aktywność
PP1. Defosforylacja w czasie LTD specyficznych białek jak
również defosforylacja podjednostki GluR1 AMPAR w
miejscu reszty Ser831, może prowadzić do endocytozy i
internalizacji tych receptorów, co dodatkowo podtrzymuje
LTD [76].
Jony Ca
2+
są również ważnym elementem LTD zależne-
go od aktywacji mGluR. Najlepiej zbadany jest proces LTD
zależnego od mGluR w móżdżku, rejonie, gdzie nie zaob-
serwowano występowania LTP, ale jego występowanie wy-
kazano również w hipokampie, prążkowiu, korze i rdzeniu
kręgowym [81]. W móżdżku do aktywacji LTD niezbędna
jest równoczesna stymulacja znajdującej się na dendrycie
synapsy łączącej komórkę Purkiniego z włóknem pnącym,
będącym aksonem neuronu doprowadzającego sygnał do
móżdżku oraz synapsy łączącej ją z włóknem równoległym
sąsiadującej komórki. Wywołana sygnałem z włókna pnące-
go depolaryzacja części dendrytycznej powoduje otwarcie
VOCC i napływ Ca
2+
do wnętrza neuronu. Z kolei glutami-
nian uwolniony z zakończenia we włóknie równoległym
pobudza mGluR grupy I, uruchamiając wewnątrzkomórko-
wą kaskadę prowadzącą do mobilizacji Ca
2+
z otwieranych
przez receptory IP
3
magazynów wewnątrzkomórkowych.
W móżdżku dominującą rolę w tym procesie odgrywają re-
ceptory mGluR1. Podwyższenie poziomu Ca
2+
oraz diacyl-
glicerol (DC) powstający obok IP
3
po pobudzeniu mGluR1,
aktywują kinazę białkową C (PKC), która z kolei fosforyluje
resztę Ser880 podjednostki GluR2 AMPAR, co powoduje
odłączenie receptora od błony komórkowej, jego endocyto-
zę i internalizację [81].
Hipokampalna forma LTD aktywowanego przez mGluR
grupy I wydaje się przebiegać niezależnie od wzrostu we-
wnątrzkomórkowego stężenia Ca
2+
i uruchamiania uwal-
niania Ca
2+
z magazynów komórkowych oraz aktywacji
kinaz białkowych, chociaż wymaga udziału białka G zwią-
zanego z receptorami mGluR1 i mGluR5. Jednak dokład-
ny mechanizm hipokampalnej formy LTD uruchamianego
przez aktywację mGluR grupy I jest wciąż przedmiotem
intensywnych badań [81].
OSTRE ZABURZENIA HOMEOSTAZY WAPNIA
PROWADZĄCE DO USZKODZENIA I ŚMIERCI
NEURONÓW: ROLA W ISCHEMII MÓZGU
Utrzymanie wewnątrzkomórkowego stężenia jonów Ca
2+
na niskim poziomie jest warunkiem prawidłowego przeka-
zu sygnału wapniowego. Jest to proces wymagający dużych
nakładów energii. Neurony, z powodu masowych przepły-
wów jonów Ca
2+
związanych z aktywnością elektryczną,
są szczególnie podatne na zaburzenia metabolizmu ener-
getycznego prowadzące do nieprawidłowości homeostazy
tych jonów. Przyczyną ostrych zaburzeń homeostazy Ca
2+
może być także nadmierne pobudzenie neuronów, jak to
ma miejsce przy uszkodzeniu ekscytotoksycznym, w któ-
rym pośredniczą receptory dla pobudzających aminokwa-
sów [82,83]. W tych warunkach dochodzi do intensywnego
napływu jonów Ca
2+
do neuronów przez receptory jonotro-
powe, zwłaszcza NMDAR, przez VOCC, a także w wyni-
ku odwrócenia kierunku transportu Ca
2+
przez wymiennik
NCX [83]. Skutki nieprawidłowości homeostazy Ca
2+
i w
konsekwencji niewłaściwego przekazu sygnału wapniowe-
go, mogą się ograniczać do krótkotrwałych lub utrwalonych
(np. w formie ognisk padaczkorodnych) zaburzeń czynno-
ściowych komórek nerwowych, lub mogą się przejawić w
postaci nekrotycznej lub apoptotycznej śmierci neuronów.
W ostro przebiegającym niedokrwieniu mózgu pierwot-
nym czynnikiem zakłócającym homeostazę Ca
2+
jest deficyt
energetyczny, mogący doprowadzić do wyłączenia pomp
(ATPaz) sodowych i wapniowych oraz odwrócenia trybu
pracy transporterów błonowych, przemieszczenia glutami-
nianu do przestrzeni synaptycznej, pobudzenia NMDAR
i niekontrolowanego napływu jonów Ca
2+
do neuronów
zgodnie z mechanizmami ekscytotoksyczności (Ryc. 1B).
Wg obecnych poglądów zaburzenia homeostazy wapnia i
toksyczność pobudzeniowa, to nierozdzielnie ze sobą wią-
zane elementy ogólnej patogenezy uszkodzenia neuronów
w ostrych stanach patologicznych mózgu [83-85], obejmują-
cej ponadto stres oksydacyjny, dysfunkcję mitochondriów
i ER oraz inne procesy związne z nekrotyczną lub apop-
totyczną śmiercią komórek. Aspekt historyczny, a także
szczegóły klasycznej hipotezy o roli wapnia w ischemii
mózgu na tle innych mechanizmów patogennych, zostały
obszernie omówione poprzednio [85].
W wyniku niedokrwienia mózgu i rozwijających się zabu-
rzeń energetycznych oraz nadmiernego pobudzenia recepto-
rów dla glutaminianu, w neuronach obserwuje się zróżnico-
wany obraz zmian w homeostazie wapnia (Ryc. 1B). Ta różno-
rodność jest odbiciem istnienia różnych form niedokrwienia
mózgu (ogniskowego i globalnego, z odwracalnym lub nie-
odwracalnym upośledzeniem krążenia krwi) i różnorodnych
dodatkowych uwarunkowań patofizjologicznych jak kwasi-
ca, hiperglikemia, przewlekła hipoksja. Udokumentowany w
412
www.postepybiochemii.pl
licznych publikacjach [85] wzrost wewnątrzkomórkowego
stężenia Ca
2+
dotyczy zarówno okresu niedokrwienia, jak
i po niedokrwieniu (ten ostatni jest określany jako wtórny
wzrost stężenia wapnia). Wynika on zarówno z napływu jo-
nów wapnia z przestrzeni zewnątrzkomórkowej przez ROC
(zwłaszcza NMDAR), przez VOCC i na skutek odwrócenia
trybu pracy wymiennika NCX, jak i mobilizacji kationu z
zasobów wewnątrzko-
mórkowych (Ryc. 1B).
Wykazano jednak, że
neurotoksyczność
jest
skorelowana nie tyle z
poziomem wzrostu stę-
żenia jonów wapnia w
neuronach, co raczej z
całkowitym obciążeniem
komórek napływający-
mi jonami wapnia, przy
czym istotne znaczenie
patogenne ma napływ
Ca
2+
za pośrednictwem
NMDAR, podczas gdy
aktywacja VOCC mało
uszkadza neurony [83].
Istnieją jednak dane tok-
sykologiczne, wskazują-
ce na patogenną rolę nad-
miernej mobilizacji Ca
2+
z
ER. Wykazano silne tok-
syczne działanie na neu-
rony wywołanego przez
tapsygarginę niekontro-
lowanego wyrzutu Ca
2+
z
ER [86] oraz rolę tego mechanizmu w działaniu neurotoksyn
środowiskowych, takich jak polichlorowane bisfenole [87].
Kluczowe znaczenie w tych zjawiskach może odgrywać stres
ER (Ryc. 3 i 4). Udział opróżnienia zasobów Ca
2+
w ER i zwią-
zanych z nim mechanizmów molekularnych w uszkodzeniu
neuronów mózgu był już od dawna sugerowany [88].
Zaburzenia homeosta-
zy wapnia, prowadzące
do wzrostu stężenia Ca
2+
w cytosolu, aktywują
wrażliwe na Ca
2+
enzy-
my kataboliczne, prote-
azy i lipazy (Ryc. 3), co
wywołuje uszkodzenie
błon oraz zmiany w cy-
toszkielecie komórki, a
aktywacja endonukleaz
prowadzi do uszkodze-
nia DNA [83,84]. Stres
oksydacyjny jest niezwy-
kle ważnym elementem
patogenezy wywołane-
go przez niedokrwienie
uszkodzenia komórek
nerwowych, w którym
pośredniczy wapń [89].
Zaburzenia homeostazy
wapnia i niekontrolowa-
ny wzrost stężenia Ca
2+
w komórkach aktywują
produkcję reaktywnych
form tlenu, co z kolei
potęguje zaburzenia ho-
meostazy wapnia [85].
Ponadto istnieją wspólne
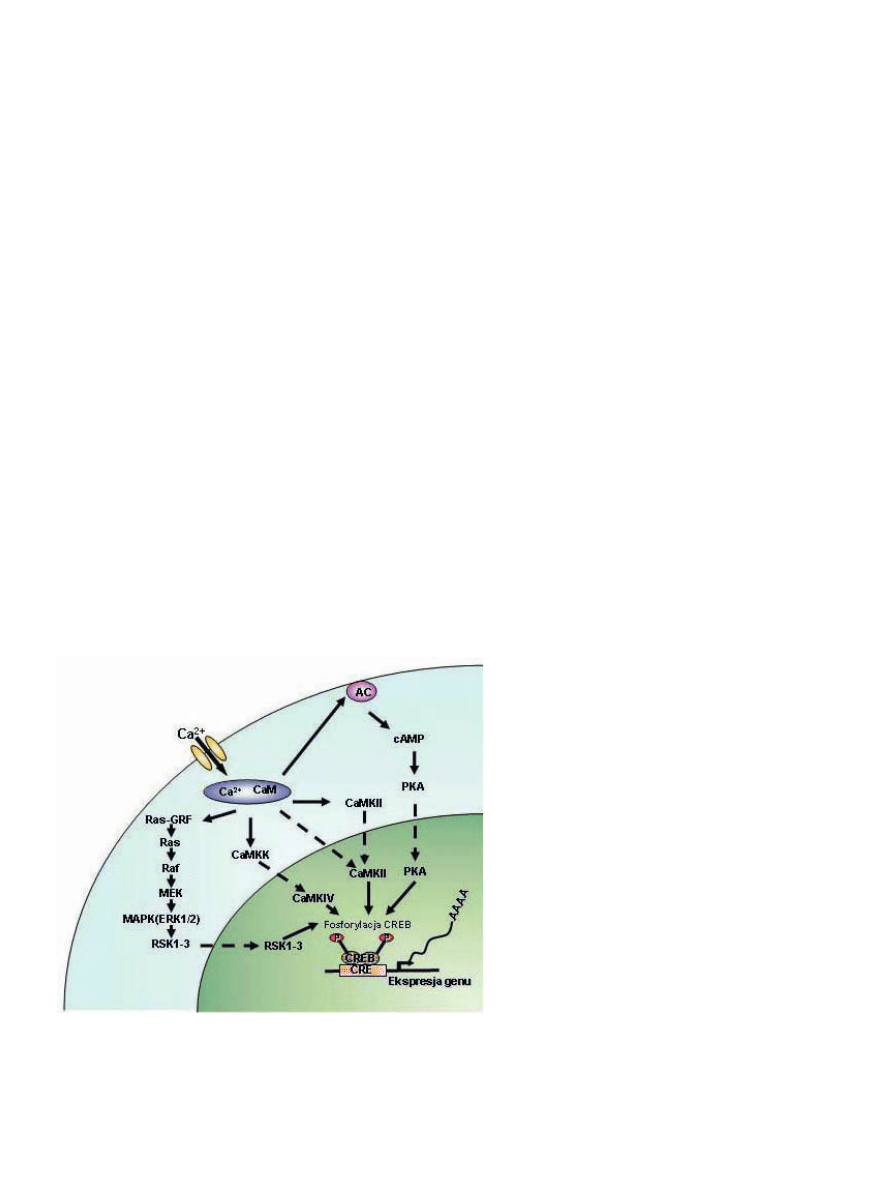
Rycina 3. Wybrane mechanizmy uszkodzenia neuronów zależne od Ca
2+
, rozwijające się w wyniku ostrych zaburzeń homeostazy
Ca
2+
. Niekontrolowany wzrost wewnątrzkomórkowego stężenia Ca
2+
zaburza przekazywanie sygnału w neuronach, prowadzi
do aktywacji wrażliwych na wapń enzymów katabolicznych jak protezy, lipazy i endonukleazy, uszkadza strukturę błon i DNA,
indukuje stres oksydacyjny, zaburza funkcję mitochondriów i ER. W efekcie dochodzi do uszkodzenia i śmierci neuronów. Szcze-
gółowe wyjaśnienia w tekście.
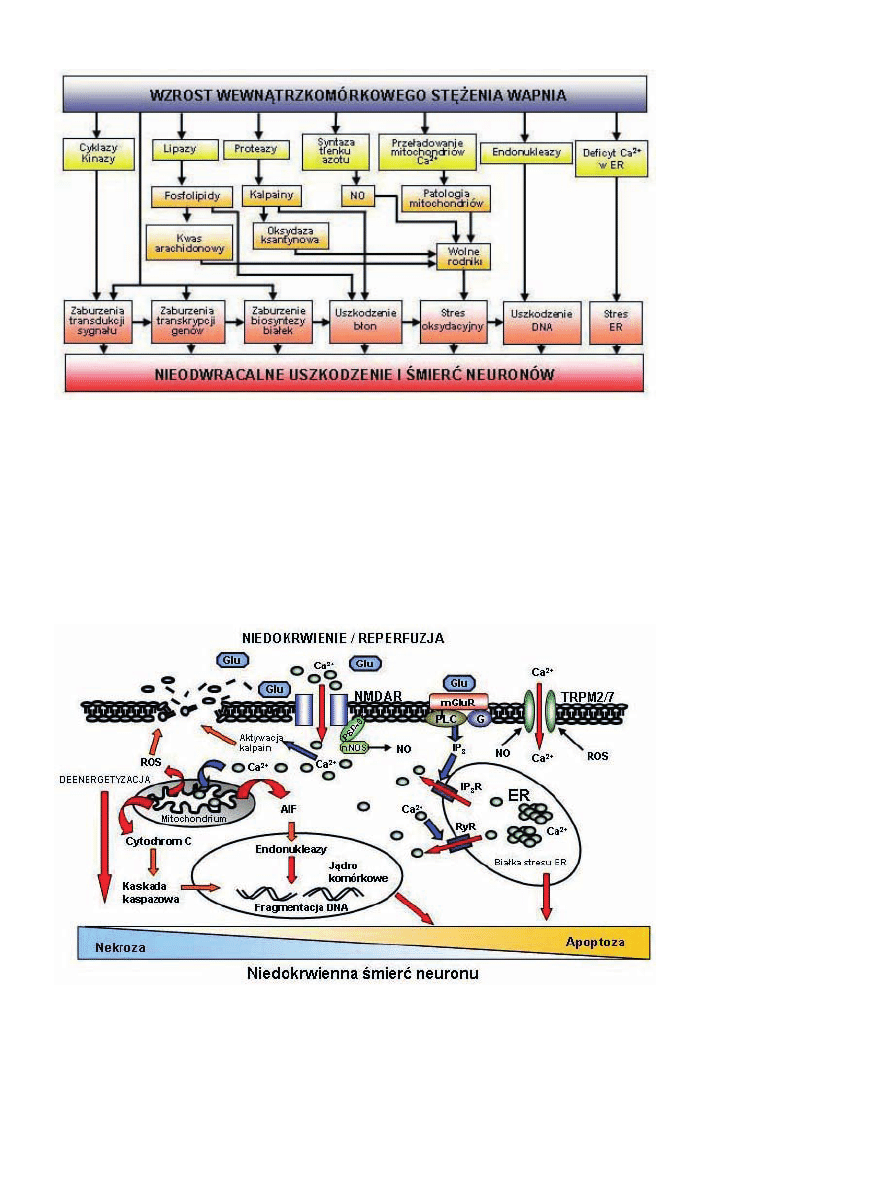
Rycina 4. Zaburzenia homeostazy Ca
2+
wywołane przez niedokrwienie mózgu indukują dysfunkcję mitochondriów i ER, co pro-
wadzi do śmierci neuronów o charakterze kontinuum nekrotyczno-apoptotycznego. Pobudzenie NMDAR i mGluR grupy I przez
uwolniony z komórek glutaminian (Glu) prowadzi do pierwotnego wzrostu stężenia Ca
2+
, co aktywuje neuronalną syntazę NO
(nNOS) i indukuje kaskady zaburzeń uszkadzających neurony, wymienione na Ryc. 3. Nagromadzenie Ca
2+
w mitochondriach
aktywuje megakanały, co wywołuje uwolnienie z nich białek proapoptotycznych, cytochromu c i AIF oraz zwiększoną produkcją
reaktywnych form tlenu (ROS). Zwiększona produkcja NO i ROS aktywuje kanały TRPM 2/7 i powoduje wtórny wzrost stężenia
Ca
2+
. Opróżnienie zasobów Ca
2+
w ER, w którym pośredniczą receptory IP
3
(IP
3
R) i rianodynowe (RyR) indukuje stres ER. O prze-
wadze nekrozy lub apoptozy decyduje stan energetyczny neuronu.

Postępy Biochemii 58 (4) 2012
413
dla jonów Ca
2+
i wolnych rodników mechanizmy cytotok-
syczne, do których należy aktywacja megakanałów mito-
chondrialnych, dysfunkcja i uszkodzenie mitochondriów,
oraz uwolnienie z nich białek proapoptotycznych [39].
W zależności od innych okoliczności towarzyszących
danej patologii, a zwłaszcza od zachowanego potencjału
energetycznego neuronów, zaburzenia homeostazy wapnia
mogą prowadzić do nekrotycznej lub apoptotycznej śmier-
ci neuronów (Ryc. 4) [82]. Towarzyszące niedokrwieniu
mózgu nieodwracalne zaburzenia energetyczne w rejonie
ogniska udarowego, w połączeniu z uszkodzeniem błon i
cytoszkieletu neuronów, prowadzą do nekrotycznej śmierci
neuronów. W przypadku odwracalnej globalnej ischemii w
obszarach mózgu szczególnie wrażliwych na ischemię do-
minuje apoptotyczna śmierć neuronów. W praktyce, akty-
wowane w wyniku niedokrwienia i urazu oraz stanu epi-
leptycznego złożone mechanizmy patologiczne związane z
ekscytotoksycznością i zaburzoną homeostazą jonów Ca
2+
prowadzą do śmierci neuronów, zawierającej w sobie ele-
ment zarówno nekrozy jak i apoptozy (Ryc. 4).
Drogi wnikania Ca
2+
do neuronów, a zwłaszcza NMDAR
wydawały się być obiecującymi celami terapeutycznymi,
zwłaszcza w udarze mózgu. Jednak stosowanie antagoni-
stów receptorów NMDA, wiążące sie z ryzykiem licznych
efektów niepożądanych, podobnie jak użycie blokerów
VOCC, nie przyniosło oczekiwanego przełomu terapeu-
tycznego, co skłania do dalszych poszukiwań. Nowsze
dane wskazują, że w patogennych efektach nadmiernej
aktywacji NMDAR istotną rolę odgrywają ścisłe fizyczne
połączenia pomiędzy tymi receptorami a innymi białkami
gęstości postsynaptycznych (PSD, ang. post-synaptic densi-
ty), a zwłaszcza NOS i PSD-95, przy czym wykazano, że ro-
zerwanie tych połączeń może zmniejszać neurotoksyczność
bez obniżania napływu Ca
2+
za pośrednictwem NMDAR
(Ryc. 4) [83]. Inne badania wykazały, że szlakiem wtórne-
go napływu Ca
2+
do neuronów, do którego dochodzi już po
przywróceniu krążenia, o dużym znaczeniu patogennym i
obiecującym potencjale terapeutycznym, mogą być aktywo-
wane przez wzrost wewnątrzkomórkowego stężenia jonów
wapnia kanały jonowe z rodziny TRPM (ang.
transient
recep-
tor
potential melastatin) typu 2 i 7
(Ryc. 4) [83].
ROLA Ca
2+
W PRZEWLEKŁYCH SCHORZENIACH
NEURODEGENERACYJNYCH
Rozregulowanie homeostazy Ca
2+
jest ważnym czynni-
kiem w wielu wolno rozwijających się chorobach neurodege-
neracyjnych. Zaburzenia homeostazy Ca
2+
w tych chorobach
są często spowodowane zmianami genetycznymi, ale są rów-
nież choroby, w których przyczyn takich zaburzeń jeszcze
nie zidentyfikowano. Wśród bardziej znanych przewlekłych
chorób neurodegeneracyjnych związanych z zaburzeniami
homeostazy Ca
2+
należy wymienić chorobę Alzheimera, Par-
kinsona czy Huntingtona, jednak zaburzenia te odgrywają
również ważną rolę w epilepsji i schizofrenii [2].
CHOROBA ALZHEIMERA
Choroba Alzheimera (AD, ang. Alzheimer’s disease) jest
jedną z najczęstszych chorób neurodegeneracyjnych zwią-
zanych z podeszłym wiekiem, objawiającą się postępującą
demencją. Choroba ta rozwija się powoli, a ponieważ brak
jest skutecznej terapii przyczynowej AD, najczęściej po oko-
ło 10 latach kończy się śmiercią. Charakteryzuje się stop-
niową, prowadzącą do śmierci komórki, degeneracją neu-
ronów w przodomózgowiu, a szczególnie w obrębie hipo-
kampa. Ponad 95% zachorowań ma charakter sporadyczny
(SAD, ang. sporadic Alzheimer’s disesase) i choroba ujawnia
się po przekroczeniu 65 roku życia. Występują też rzadkie
przypadki dziedzicznego, genetycznego uwarunkowania w
rodzinach (FAD, ang. family Alzheimer’s disease), w których
występuje dominująca mutacja w genie kodującym białko
prekursora amyloidu (APP) lub jednego z białek presynili-
ny (PS1 lub PS2), powodujące produkcję neurotoksycznego
białka amyloidu β (Aβ) z białka prekursorowego APP oraz
akumulację zmutowanych presynilin. W tych przypadkach
choroba objawia się znacznie wcześniej. W mózgach osób
chorych na AD stwierdzono podwyższony poziom Ca
2+
oraz wzmożoną aktywację enzymów zależnych od Ca
2+
.
Oddziaływanie Ca
2+
z Aβ i nadmiernie ufosforylowanym
białkiem tau oraz zmutowaną presyniliną są elementami
patogenezy obu typów AD [2].
Uważa się, że do rozwinięcia się SAD mogą przyczyniać
się niewielkie zaburzenia homeostazy Ca
2+
związane z pro-
cesem starzenia, podobnie jak obniżona aktywność inte-
lektualna i fizyczna oraz dieta bogata w kalorie, ale uboga
w kwas foliowy i antyoksydanty. Wirusowe infekcje oraz
urazy mózgu również mogą przyczynić się do rozwoju AD.
Wśród innych czynników sprzyjających rozwojowi tej cho-
roby wymieniane są też spowodowane przez Ca
2+
zaburze-
nia w funkcjonowaniu ER i mitochondriów oraz zmiany w
ekspresji genów [90]. Podwyższenie stężenia Ca
2+
w neuro-
nach może wpływać zarówno na fosforylację białka tau jak i
na enzymatyczną obróbkę APP, powodując nagromadzanie
się Aβ. Wykazano, że cząsteczki Aβ mogą tworzyć w błonie
komórkowej pory przepuszczalne dla Ca
2+
i w ten sposób
potęgować zaburzenia homeostazy Ca
2+
w neuronach, po-
wodując produkcję wolnych rodników tlenowych prowa-
dzącą do stresu oksydacyjnego oraz zaburzając aktywność
ATPaz wapniowych i receptorów glutaminianergicznych
[91]. Aβ powoduje osłabienie działania oraz zmniejszenie
zawartości NMDAR białka w błonach synaptycznych, a
stres oksydacyjny wywołany przez nagromadzone we-
wnątrz neuronów cząsteczek Aβ może prowadzić do pogłę-
bienia spowodowanego przez nadmiar wapnia nieprawi-
dłowego działania mitochondriów. Ponadto uwalnianie w
czasie enzymatycznej obróbki APP wewnątrzkomórkowej
domeny tego białka może przyczyniać się do wzrostu we-
wnątrzkomórkowego stężenia Ca
2+
, ponieważ ma ona wła-
ściwości regulatora zależnego od aktywacji receptorów IP
3
uwalniania Ca
2+
z ER [2]. Aβ może też być nagromadzany
w mitochondriach, zaburzając działanie łańcucha oddecho-
wego w obrębie kompleksu III i IV, a tym samym obniżając
produkcję ATP i pogłębiając stres oksydacyjny i zaburzenia
homeostazy Ca
2+
[4].
W przypadku FAD produkcja Aβ ze zmutowanej czą-
steczki APP wywołuje zaburzenia homeostazy Ca
2+
w spo-
sób podobny do opisanego powyżej. Presyniliny obecne w
błonach ER bezpośrednio lub pośrednio wpływają na RyR
i receptory IP
3
, w związku z tym mutacje w genach PS1 i

414
www.postepybiochemii.pl
PS2 powodują podwyższone uwalnianie Ca
2+
z magazynów
wewnątrzkomórkowych umiejscowionych w ER i funkcjo-
nowanie SOCC. Mutacje presynilin wiązane są również z
upośledzeniem mechanizmu magazynowania Ca
2+
w ER.
Prowadzi to do zaburzeń przekazywania wewnątrzkomór-
kowych sygnałów, wzmożonej produkcji Aβ i w konse-
kwencji do śmierci neuronów [2,4,89].
CHOROBA PARKINSONA
Choroba Parkinsona (PD, ang. Parkinson’s disease) jest często
występującą, wolno rozwijającą się chorobą neurodegenera-
cyjną wieku późnego, objawiającą się zaburzeniami zdolności
motorycznych. Związana jest ona z postępującą utratą neuro-
nów dopaminergicznych w istocie czarnej mózgu i zmniejsze-
niem wydzielania dopaminy w prążkowiu. Patogeneza proce-
sów neurodegeneracyjnych zachodzących w PD jest złożona
i uważa się, że składają się na nią zarówno zaburzenia gene-
tyczne jak i prawdopodobna ekspozycja na toksyny środowi-
skowe [92]. Kilka mutacji określonych symbolami PARK 1-13
w genach kodujących między innymi α-synukleinę (PARK1),
parkinę (PARK2), mitochondrialną kinazę PINK1 (PARK6)
czy sensor stresu oksydoredukcyjnego DJ-1 (PARK7), uważa
się obecnie za genetyczne przyczyny PD [4]. W przypadkach
spontanicznego, nie uwarunkowanego zmianami genetycz-
nymi występowania PD, związane z wiekiem zaburzenia ho-
meostazy Ca
2+
w mózgu uważa się obecnie za jedną z głów-
nych przyczyn neurodgeneracji. Związane z wiekiem zmiany
w funkcjonowaniu VOCC typu L wydają się być kluczowe.
Zmiany te powodują łatwiejsze i częstsze otwieranie się tych
kanałów, co powoduje nadmierny napływ Ca
2+
do neuronów
[92]. Jednakże samo zaburzenie homeostazy Ca
2+
nie powo-
duje PD i dodatkowymi czynnikami są tu wpływy środowi-
skowe, takie jak obecność metali ciężkich, pestycydów, neuro-
toksyn czy stany zapalne w układzie nerwowym. Genetyczne
uwarunkowania, które mogą przyśpieszyć zmiany prowadzą-
ce do PD związane są z mutacjami białek mitochondrialnych
PINK1, DJ-1 i ligazą ubikwityny, parkiny. Białka te w odpo-
wiedzi na stres oksydacyjny, mitochondrialny czy ER, eskalują
zaburzenia homeostazy Ca
2+
i nieprawidłowe funkcjonowanie
neuronów [93]. Ostatnie badania wykazały zmniejszoną za-
wartość izoformy 1 kanałów TRP u chorych na PD, co również
jest wiązane z degeneracją neuronów dopaminergicznych [4].
CHOROBA HUNTINGTONA
Choroba Huntingtona jest autosomalnie dominującym
dziedzicznym schorzeniem neurozwyrodnieniowym, w któ-
rym występuje mutacja w genie kodującym białko hunting-
tynę. W wyniku tej mutacji białko zawiera długi ciąg reszt
glutaminy w rejonie N-końca. Choroba objawia się pomiędzy
20 a 50 rokiem życia i charakteryzuje się zaburzeniami rucho-
wymi (tzw. pląsaniem, stąd inna nazwa pląsawica Hunting-
tona) oraz postępującą demencją związaną z utratą średnich
neuronów kolczystych w obrębie prążkowia, ale zmiany do-
tyczą także innych rejonów mózgu. Huntingtonina związana
jest z mitochondriami i jak wykazano, zmutowana forma tego
białka rozregulowuje homeostazę Ca
2+
w mitochondriach [94].
Ponadto zmutowana huntingtonina wiąże się bezpośrednio z
receptorami IP
3
w ER i zwiększa ich wrażliwość na działanie
IP
3
co prowadzi do nadmiernego uwalniania Ca
2+
. Taki efekt
działania zmutowanej huntingtoniny wydaje się szczególnie
szkodliwy dla średnich neuronów kolczystych bogatych w
mGluR5, w których przekazywanie sygnału zachodzi za po-
średnictwem Ca
2+
uwalnianego z ER po pobudzeniu recepto-
rów IP
3
[95]. Wykazano również, że zmutowana huntingtoni-
na może wpływać na wiązanie się NMDAR, zwłaszcza tych
zawierających podjednostkę NR2B, do białka gęstości postsy-
naptycznej PSD-95, prowadząc do ich nadaktywności i napły-
wu Ca
2+
do neuronów [2]. Wzrastające stężenie Ca
2+
może w
konsekwencji doprowadzić do indukcji megakanałów mito-
chondrialnych i do apoptotycznej śmierci komórek.
AUTOSOMALNIE DOMINUJĄCA ATAKSJA MÓŻDŻKOWA
Ataksje móżdżkowe (SCA, ang. spinocerebellar ataxia) two-
rzą grupę autosomalnie dominujących neurodegeneracyj-
nych chorób genetycznych, charakteryzujących się zanikiem
móżdżku i postępującym upośledzeniem koordynacji ruchu
zwanym ataksją. Do tej pory opisano ponad 30 różnych mu-
tacji związanych z różnymi formami SCA, które zgodnie z
kolejnością odkrywania otrzymywały nazwy od SCA1 do
SCA30 [96]. Patogeneza SCA nie jest do końca poznana, ale
wiele wskazuje na udział w procesach neurodegeneracyjnych
zaburzeń w przekazywaniu sygnału wapniowego. W móżdż-
ku chorych dotkniętych mutacją SCA1 wykazano zmniejszoną
syntezę białek regulujących homeostazę Ca
2+
lub związanych
z przekazywaniem wewnątrzkomórkowego sygnału wap-
niowego. Mowa tu o kabindynie-D28k, parwalbuminie oraz
ATPazie wapniowej SERCA w ER [95]. Z kolei w SCA6 dege-
neracja komórek Purkiniego związana jest z ekspresją poliglu-
taminowego odcinka w genie CACNA1A, kodującym białko
podjednostki tworzącej kanał P/Q VSCC [2].
JASKRA (GLAUKOMA)
Jaskra, druga co do liczby przypadków przyczyna ślepo-
ty na świecie, jest chorobą zaliczaną do chorób neurodege-
neracyjnych. Przyczyną utraty wzroku jest postępujące wy-
mieranie komórek zwojowych siatkówki i uszkodzenia ner-
wu wzrokowego. Podtypy glaukomy charakteryzowane są
najczęściej na podstawie zmian w ciśnieniu wewnątrz gałki
ocznej, które są z kolei skutkiem zablokowania swobodnego
przepływu płynu wewnątrzgałkowego [97]. Patogeneza ja-
skry nie jest w pełni poznana, ale wiadomo, że w większości
przypadków na skutek nadciśnienia w gałce ocznej dochodzi
do znacznego obniżenia przepływu krwi w obrębie siatkówki
i nerwu wzrokowego, co prowadzi do powstania uszkodzeń
typu hipoksyjnego i ischemicznego. Podwyższony poziom
glutaminianu, nadmierna aktywacja NMDAR i AMPA/KAR
oraz VSCC uważane są za główne przyczyny obumierania
komórek zwojowych siatkówki. Uszkodzenia nerwu wzro-
kowego wiązane są natomiast z odwróconym działaniem
wymiennika NCX i napływem tą drogą Ca
2+
do aksonów
[97]. Podwyższony poziom Ca
2+
aktywuje kalpainy, powo-
dując szereg patologicznych zmian w komórkach. Wykazano
również, że podwyższone ciśnienie w gałce ocznej aktywuje
kalcyneurynę, co może uruchomić szlak apoptotyczny.
PADACZKA (EPILEPSJA)
Jest wiele uwarunkowanych genetycznie lub nabytych
przyczyn występowania epilepsji. Ocenia się, że ok. 50%
przypadków epilepsji wynika z przebytych schorzeń neu-

Postępy Biochemii 58 (4) 2012
415
rologicznych, takich jak udary czy urazy mózgu, po których
rozwija się chroniczne zaburzenie w funkcjonowaniu neu-
ronów. Badania wykazały, że w neuronach, które ocalały
po urazie, dochodzi do zaburzenia homeostazy Ca
2+
i me-
chanizmów regulowanych przez Ca
2+
, co przyczynia się do
epileptogenezy i rozwoju choroby. U podstaw pozostałych
przypadków leżą przyczyny genetyczne, przede wszystkim
uszkodzenia budowy kanału VOCC typu T i P/Q [98].
Typ epilepsji najczęściej spotykany u dzieci pomiędzy 4
i 12 rokiem życia to gwałtowne, niskoprogowe wapniowe
wyładowania neuronów wzgórza, spowodowane napływem
Ca
2+
przez kanały typu T, kiedy kanały te są aktywowane
przez hiperpolaryzację błony komórkowej pod wpływem
sygnału hamującego transmisję, napływającego z neuronów
GABAergicznych. U pacjentów, u których występują ataki
padaczkowe z utratą świadomości zazwyczaj stwierdza się
punktową mutację (E147K) w genie CACNA1A, kodującym
białko podjednostki kanału VOCC typu P/Q. Podobnie w
przypadku pojedynczego polimorfizmu nukleotydu w genie
kodującym białka VOCC typu T ujawniają się objawy epilep-
sji [98]. Wykazano, że nadmierna aktywacja receptorów glu-
taminianergicznych może sprzyjać zmianom prowadzącym
do epilepsji. Nadmierny napływ Ca
2+
do neuronów aktywuje
enzymy wiązane z epileptogenezą, takie jak CaMKII, kalcy-
neuryna czy białkowe kinazy tyrozynowe Scr i Fyn.
SCHIZOFRENIA
Schizofrenia jest chorobą psychiczną o nie do końca pozna-
nej etiologii, jednakże większość badań wskazuje na jej ge-
netyczne podłoże. Do tej pory zidentyfikowano prawie 1700
genów (białek), których zaburzona ekspresja wiązana jest z
występowaniem schizofrenii. Wiele z tych białek pełni ważną
rolę w homeostazie Ca
2+
oraz przekazywaniu sygnału wapnio-
wego [99]. U chorych na schizofrenię wykazano w korze skro-
niowej obniżoną aktywność białka regulującego sygnał białka
G -RGS4 (ang. regulator of G protein signaling 4), które hamuje
uwalnianie Ca
2+
z magazynów wewnątrzkomórkowych. W
korze mózgowej i hipokampie zaobserwowano wzrost synte-
zy białka GAP 43 (ang. growth associated protein 43), które kon-
troluje sygnał wapniowy poprzez przyłączanie wolnej CaM
i uniemożliwianie wiązania do niej Ca
2+
[2]. Zaobserwowano
również obniżoną produkcję białek buforujących Ca
2+
, takich
jak parwalbumina i kalbindyna D28k, a także białka Bcl-2,
które obniża uwalnianie Ca
2+
z magazynów komórkowych w
odpowiedzi na fizjologiczne i patologiczne sygnały. Obserwo-
wany wzrost poziomu białka oddziałującego z receptorem D1
(ang. calcyon) wpływającego na sygnał wapniowy, oraz NCS-1,
białka wiążącego Ca
2+
i hamującego desensytyzację receptora
D2, wskazują na powiązanie homeostazy Ca
2+
i receptorów
dopaminergicznych z patofizjologią schizofrenii [99]. Zabu-
rzenia w działaniu szlaków wewnątrzkomórkowych inicjowa-
nych aktywacją NMDAR również mają swój udział w rozwoju
objawów schizofrenii. Ostatnio dyskutowany jest też udział
zaburzeń ekspresji genu CACNA1C, kodującego białko będące
podjednostką VOCC typu L [100].
PODSUMOWANIE
Od początku swego istnienia żywe komórki, aby prze-
trwać, musiały przeciwstawiać się napływowi Ca
2+
z ze-
wnątrz. Rozwinięte w drodze ewolucji mechanizmy trans-
portu i kontroli homeostazy Ca
2+
zostały wykorzystane w
procesach przekazywania sygnałów. W neuronach są one
szczególnie rozwinięte i biorą udział w regulacji podstawo-
wych funkcji fizjologicznych układu nerwowego, łącznie z
mechanizmami uczenia się i pamięci. Homeostaza Ca
2+
w
neuronach jest procesem bardzo złożonym, w sposób kry-
tyczny zależnym od stałej dostawy energii oraz podatnym
na zaburzenia, np. w warunkach deficytu energii lub nad-
miernego pobudzenia komórek nerwowych. Może to dopro-
wadzić do dysfunkcji oraz nekrotycznej lub apoptotycznej
śmierci komórek. Ponieważ w zależnym od Ca
2+
uszkodze-
niu i śmierci neuronów biorą udział mechanizmy normal-
nie służące fizjologicznym funkcjom układu nerwowego,
próby neuroprotekcji poprzez ingerencję w homeostazę lub
procesy przekazywania sygnałów z udziałem jonów wap-
nia są zwykle obciążone efektami niepożądanymi, łącznie
z zaburzeniami procesów poznawczych. Istnieje więc pilne
zapotrzebowanie na nowe strategie terapeutyczne skutecz-
ne w zapobieganiu neurodegeneracji, które by hamowały
lub odwracały neurotoksyczne efekty Ca
2+
bez ingerencji w
fizjologiczne procesy przekazywania sygnałów.
PIŚMIENNICTWO
1. Kretsinger RH (1990) Why cells must export calcium, W: Bronner F
(red) Intracellular calcium regulation. Wiley-Liss, New York, str. 439-
457
2. Wojda U, Salinska E, Kuznicki J (2008) Calcium ions in neuronal dege-
neration. IUBMB Life 60: 575-590
3. Berridge MJ (2012) Calcium signalling remodelling and disease. Bio-
chem Soc Trans 40: 297-309
4. Zündorf G, Reiser G (2011) Calcium dysregulation and homeostasis
of neural calcium in the molecular mechanisms of neurodegenerative
diseases provide multiple targets for neuroprotection. Antioxid Redox
Signal 14: 1275-1288
5. Nakamura T, Lipton SA (2010) Preventing Ca
2+
-mediated nitrosative
stress in neurodegenerative diseases: possible pharmacological strate-
gies. Cell Calcium 47: 190-197
6. Carrasco MA, Jaimovich E, Kemmerling U, Hidalgo C (2004) Signal
transduction and gene expression regulated by calcium release from
internal stores in excitable cells. Biol Res 37: 701-712
7. Berridge MJ (2002) The endoplasmic reticulum: a multifunctional sig-
naling organelle. Cell Calcium 32: 235-249
8. Berridge MJ, Lipp P, Bootman MD (2000) The versatility and univer-
sality of calcium signalling. Nat Rev Mol Cell Biol 1: 11-21
9. Cheng H, Lederer WJ (2008) Calcium sparks. Physiol Rev 88: 1491-
1545
10. Matveev V, Bertram R, Sherman A (2011) Calcium cooperativity of
exocytosis as a measure of Ca²
+
channel domain overlap. Brain Res
1398: 126-138
11. Calin-Jageman I, Lee A (2008) Ca
v
1 L-type Ca
2+
channel signaling com-
plexes in neurons. J Neurochem 105: 573-583
12. Dai S, Hall DD, Hell JW (2009) Supramolecular assemblies and local-
ized regulation of voltage-gated ion channels. Physiol Rev 89: 411-452
13. Lacinová L (2005) Voltage-dependent calcium channels. Gen Physiol
Biophys 24 (Suppl 1): 1-78
14. Danysz W, Frankiewicz T, Sopala M (2004). Receptory glutaminia-
nergiczne, W: Nowak JZ, Zawilska JB (red) Receptory i mechanizmy
przekazywania sygnału. PWN, Warszawa, str. 382-413
15. Seeburg PH, Burnashev N, Köhr G, Kuner T, Sprengel R, Monyer H
(1995) The NMDA receptor channel: molecular design of a coincidence
detector. Recent Prog Horm Res 50: 19-34
16. Liu SJ, Zukin RS (2007) Ca
2+
-permeable AMPA receptors in synaptic
plasticity and neuronal death. Trends Neurosci 30: 126-134

416
www.postepybiochemii.pl
17. Kawamoto EM, Vivar C, Camandola S (2012) Physiology and pathol-
ogy of calcium signaling in the brain. Front Pharmacol 3: 1-17
18. Putney JW (2009) Capacitative calcium entry: from concept to mol-
ecules. Immunol Rev 231: 10-22
19. Skibinska-Kijek A, Wisniewska MB, Gruszczynska-Biegala J, Methner
A, Kuznicki J (2009) Immunolocalization of STIM1 in the mouse brain.
Acta Neurobiol Exp 69: 413-428
20. Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N,
Matsuki N, Ikegaya Y (2003) Activity-evoked capacitative Ca
2+
entry:
implications in synaptic plasticity. J Neurosci 23: 7737–7741
21. Zhang S, Fritz N, Ibarra C, Uhlén P (2011). Inositol 1,4,5-trisphosphate
receptor subtype-specific regulation of calcium oscillations. Neuro-
chem Res 36: 1175-1185
22. Patterson RL, Boehning D, Snyder SH (2004). Inositol 1,4,5-trisphos-
phate receptors as signal integrators. Annu Rev Biochem 73: 437-465
23. Bezprozvanny I, Watras J, Ehrlich BE (1991) Bell-shaped calcium-re-
sponse curves of Ins(1,4,5)P3- and calcium-gated channels from endo-
plasmic reticulum of cerebellum. Nature 351: 751-754
24. Bootman MD, Berridge MJ, Roderick HL (2002) Activating calcium
release through inositol 1,4,5-trisphosphate receptors without inositol
1,4,5-trisphosphate. Proc Natl Acad Sci USA 99: 7320-7322
25. Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T,
Snyder SH (2003) Cytochrome c binds to inositol (1,4,5) trisphosphate
receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5:
1051-1061
26. Kimlicka L, Van Petegem F (2011) The structural biology of ryanodine
receptors. Sci China Life Sci 54: 712-724
27. Hamilton SL (2005) Ryanodine receptors. Cell Calcium 38: 253-260
28. Laver DR (2007) Ca
2+
stores regulate ryanodine receptor Ca
2+
release
channels via luminal and cytosolic Ca
2+
sites. Clin Exp Pharmacol Phy-
siol 34: 889-896
29. Fliegert R, Gasser A, Guse AH (2007) Regulation of calcium signalling
by adenine-based second messengers. Biochem Soc Trans 35: 109-114
30. Byrnes KR, Loane DJ, Faden AI (2009) Metabotropic glutamate recep-
tors as targets for multipotential treatment of neurological disorders.
Neurotherapeutics 6: 94-107
31. Verkhratsky A, Toescu EC (2003) Endoplasmic reticulum Ca
2+
home-
ostasis and neuronal death. J Cell Mol Med 7: 351-361
32. Honarnejad K, Herms J (2012) Presenilins: Role in calcium homeostasis.
Int J Biochem Cell Biol, http://dx.doi.org/10.1016/j.biocel.2012.07.019
33. Blaustein MP, Juhaszova M, Golovina VA, Church PJ, Stanley EF
(2002) Na/Ca exchanger and PMCA localization in neurons and astro-
cytes: functional implications. Ann N Y Acad Sci 976: 356-366
34. Lytton J (2007) Na
+
/Ca
2+
exchangers: three mammalian gene families
control Ca
2+
transport. Biochem J 406: 365-382
35. Breukels V, Touw WG, Vuister GW (2012) Structural and dynamic
aspects of Ca
2+
and Mg
2+
binding of the regulatory domains of the
Na
+
/Ca
2+
exchanger. Biochem Soc Trans 40: 409-414
36. Kimura J, Ono T, Sakamoto K, Ito E, Watanabe S, Maeda S, Shikama Y,
Yatabe MS, Matsuoka I (2009) Na
+
-Ca
2+
exchanger expression and its
modulation. Biol Pharm Bull 32: 325-331
37. Nicholls DG (2009) Mitochondrial calcium function and dysfunction
in the central nervous system. Biochim Biophys Acta 1787: 1416-1424
38. Castaldo P, Cataldi M, Magi S, Lariccia V, Arcangeli S, Amoroso S
(2009) Role of the mitochondrial sodium/calcium exchanger in neuro-
nal physiology and in the pathogenesis of neurological diseases. Prog
Neurobiol 87: 58-79
39. Pivovarova NB, Andrews SB (2010) Calcium-dependent mitochon-
drial function and dysfunction in neurons. FEBS J 277: 3622-3636
40. Pizzo P, Pozzan T (2007) Mitochondria-endoplasmic reticulum chore-
ography: structure and signaling dynamics. Trends Cell Biol 17: 511-
517
41. Guerrero-Hernandez A, Dagnino-Acosta A, Verkhratsky A (2010) An
intelligent sarco-endoplasmic reticulum Ca
2+
store: release and leak
channels have differential access to a concealed Ca
2+
pool. Cell Cal-
cium 48: 143-149
42. Vandecaetsbeek I, Trekels M, De Maeyer M, Ceulemans H, Lescrinier
E, Raeymaekers L, Wuytack F, Vangheluwe P (2009) Structural basis
for the high Ca
2+
affinity of the ubiquitous SERCA2b Ca
2+
pump. Proc
Natl Acad Sci USA 106: 18533-18538
43. Bengtson CP, Bading H (2012) Nuclear calcium signaling. Adv Exp
Med Biol 970: 377-405
44. Sun GY, Shelat PB, Jensen MB, He Y, Sun AY, Simonyi A (2010) Pho-
spholipases A2 and inflammatory responses in the central nervous
system. Neuromolecular Med 12: 133-148
45. Alpár A, Attems J, Mulder J, Hökfelt T, Harkany T (2012) The rena-
issance of Ca
2+
-binding proteins in the nervous system: secretagogin
takes center stage. Cell Signal 24: 378-387
46. Mikhaylova M, Hradsky J, Kreutz MR (2011) Between promiscuity
and specificity: novel roles of EF-hand calcium sensors in neuronal
Ca
2+
signalling. J Neurochem 118: 695-713
47. Chapman ER (2008) How does synaptotagmin trigger neurotransmit-
ter release? Annu Rev Biochem 77: 615-641
48. Xia Z, Storm DR (2005) The role of calmodulin as a signal integrator for
synaptic plasticity. Nat Rev Neurosci 6: 267-276
49. Vincent SR (2010) Nitric oxide neurons and neurotransmission. Prog
Neurobiol 90: 246-255
50. Miyamoto E (2006) Molecular mechanism of neuronal plasticity: in-
duction and maintenance of long-term potentiation in the hippocam-
pus. J Pharmacol Sci 100: 433-442
51. Groth RD, Dunbar RL, Mermelstein PG (2003) Calcineurin regulation
of neuronal plasticity. Biochem Biophys Res Commun 311: 1159-1171
52. Cruzalegui FH, Bading H (2000) Calcium-regulated protein kinase
cascades and their transcription factor targets. Cell Mol Life Sci. 57:
402-410
53. Barbado M, Fablet K, Ronjat M, De Waard M (2009) Gene regulation
by voltage-dependent calcium channels. Biochim Biophys Acta 1793:
1096-1104
54. Wiegert JS, Bading H (2011) Activity-dependent calcium signaling and
ERK-MAO kinases in neurons: A link to structural plasticity of the
nucleus and gene transcription regulation. Cell Calcium 49: 296-305
55. West AE, Chen WG, Davala MB, Dolmetch RE, Kornhauser JM, Shay-
witz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation
of neuronal gene expression. Proc Natal Acad Sciu USA 98: 11024-
11031
56. Sala C, Rudolph-Correia S, Sheng M (2000) Developmentally regula-
ted NMDA receptor-dependent dephosphorylation of cAMP respon-
se element-binding protein (CREB) in hippocampal neurons. J Neuro-
sci 20: 3529-3536
57. Bading H (2000) Transcription-dependent neuronal plasticity. The nu-
celar calcium hypothesis. Eur J Biochem 267: 5280-5283
58. Goldman PS, Tran VK, Goodman HR (1997) The multifunctional role
of the co-activator CBP in transcriptional regulation. Recent Prog
Horm Res 52: 103-120
59. Kalita K, Kuzniewska B, Kaczmarek L (2012) MKLs: Co-factors of se-
rum response factor (SRF) in neuronal responses. Int J Biochem Cell
Biol 44: 1444-1447
60. Besnard A, Galan-Rodriguez B, Vanhoutte P, Caboche J (2011) Elk-1
a transcription factor with multiple facets in the brain. Front Neurosci
5: art. 35
61. Gineitis D, Treisman R (2001) Differential usage of signal transduction
pathways defines two types of serum response factor target gene. J
Biol Chem 276: 24531-24539
62. Alonso MT, Garcia-Sancho J (2011) Nuclear Ca
2+
signalling. Cell Cal-
cium 49: 280-289
63. Gomez RS, Guatimosim C (2003) Mechanism of action of volatile ane-
sthetics: involvement of intracellular calcium signaling. Curr Drug
Targets CNS Neurol Disord 2: 123-129
64. Pedarzani P, Stocker M (2008) Molecular and cellular basis of small--
-and intermediate-conductance, calcium-activated potassium channel
function in the brain. Cell Mol Life Sci 65: 3196-3217

Postępy Biochemii 58 (4) 2012
417
Role of calcium in physiology and pathology of neurons
Elżbieta Salińska
*
, Jerzy W. Łazarewicz
Mossakowski Medical Research Centre, Polish Academy of Sciences, 5 Pawińskiego St., 02-106 Warsaw, Poland
*
e-mail: elas@cmdik.pan.pl
Key words: calcium homeostasis, brain ischemia, calcium channels, neuronal plasticity, glutamatergic receptors, signal transduction,
neurodegeneration
ABSTRACT
In this article we present evolutionary aspects of the dual role of Ca
2+
as signaling molecules and cytotoxic cations. We discuss the mechanisms
of calcium homeostasis in neurons, taking into account the specific features of excitable cells and the mechanisms of generation and transduc-
tion of calcium signals. Based on this information we outline the role of Ca
2+
ions in specific functions of the nerve cell, such as excitability,
propagation of the action potential, synaptic transmission, neuronal plasticity and various forms of mobility. Then we discuss the role of
disturbances of calcium homeostasis and signaling function in the mechanisms of injury and death of neurons in acute diseases with special
regard to cerebral ischemia, and in chronic neurodegenerative disorders.
65. von Gersdorff H, Matthews G (1994) Dynamics of synaptic vesicle fu-
sion and membrane retrieval in synaptic terminals. Nature 367: 735-
739
66. Scott R (2007) Use-dependent control of presynaptic calcium signal-
ling at central synapses. J Anat 210: 642-650
67. Ohno-Shosaku T, Hashimotodani Y, Maejima T, Kano M (2005) Cal-
cium signaling and synaptic modulation: regulation of endocannab-
inoid-mediated synaptic modulation by calcium. Cell Calcium 38:
369-374
68. Hawkins RD, Zhuo M, Arancio O (1994) Nitric oxide and carbon mon-
oxide as possible retrograde messengers in hippocampal long-term
potentiation. J Neurobiol 25: 652-665
69. Bazan NG, Packard MG, Teather L, Allan G (1997) Bioactive lipids in
excitatory neurotransmission and neuronal plasticity. Neurochem Int
30: 225-231
70. Zheng JQ, Poo MM (2007). Calcium signaling in neuronal motility.
Annu Rev Cell Dev Biol 23: 375-404
71. Wang H, Hu Y, Tsien JZ (2006) Molecular and systems mechanisms of
memory consolidation and storage. Prog Neurobiol 79: 123-135
72. Cowan N (2008) What are the differences between long-term, short-
term, and working memory. Prog Brain Res 169: 323-338
73. Miyamoto E (2006) Molecular mechanism of neuronal plasticity: In-
duction and maintenance of long-term potentiation in the hippocam-
pus. J Pharmacol Sci 1000: 433-442
74. Massey PV, Bashir ZI (2007) Long-term depression: multiple forms
and implications for brain function. Trends Neurosci 30: 176-184
75. Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of
riches. Neuron 44: 5-21
76. Citri A, Malenka RC (2008) Stnaptic plasticity: multiple forms, func-
tions and mechanisms. Neuropsychopharmacology 33: 18-41
77. Lamont MG, Weber JT (2012) The role of calcium in synaptic plasticity
and motor learning in the cerebellar cortex. Neurosci Biobehav Rev
36: 1153-1162
78. Anwyl R (2009) Metabotropic glutamate receptor-dependent long-
term potentiation. Neuropharmacology 56: 735-740
79. Reboreda A, Jimenez-Diaz L, Navarro-Lopez JD (2011) TRP channels
and neural persistent activity. Adv Exp Med Biol 704: 595-613
80. Dan Y, Poo MM (2006) Spike timing-dependent plasticity: from syn-
apse to perception. Physiol Rev 86: 1033-1048
81. Lüscher C, Huber KM (2010) Group 1 mGluR-dependent synaptic
long-term depression: mechanisms and implications for circuity and
disease. Neuron 65: 445-459
82. Salińska E, Łazarewicz JW (2003). Ekscytotoksyczność: uniwersalny
niespecyficzny mechanizm uszkodzenia neuronów, W: Liberski P,
Mossakowski MJ (red) Neurodegeneracje, Tom II. CUP PAN, Warsza-
wa, str. 10-32
83. Szydlowska K, Tymianski M (2010) Calcium, ischemia and excitotox-
icity. Cell Calcium 47: 122-129
84. Salińska E, Danysz W, Łazarewicz JW (2005) The role of excitotoxicity
in neurodegeneration. Folia Neuropathol 43: 322-339
85. Łazarewicz JW, Salińska E (2005) Udział jonów wapnia w patologii
niedokrwiennej mózgu, W: Strosznajder JB, Czernicki Z (red) Mózg a
niedokrwienie. Platan, Kraków, str. 16-46
86. Silverstein FS, Nelson C (1992) The microsomal calcium-ATPase in-
hibitor thapsigargin is a neurotoxin in perinatal rodent brain. Neurosci
Lett 145: 157-160
87. Pessah IN, Cherednichenko G, Lein PJ (2010) Minding the calcium
store: Ryanodine receptor activation as a convergent mechanism of
PCB toxicity. Pharmacol Ther 125: 260-285
88. Paschen W, Doutheil J (1999) Disturbances of the functioning of endo-
plasmic reticulum: a key mechanism underlying neuronal cell injury?
J Cereb Blood Flow Metab 19: 1-18
89. Gleichmann M, Mattson MP (2011) Neuronal calcium homeostasis
and dysregulation. Antioxid Redox Signal 14: 1261-1273
90. Bojarski L, Herms J, Kuznicki J (2008) Calcium disregulation in Alzhe-
imer’s disease. Neurochem Int 52: 621-633
91. Kawahara M (2010) Neurotoxicity of β-amyloid protein: oligomeri-
zation, channel formation, and calcium dyshomeostasis. Curr Pharm
Des 16: 2779-2789
92. Hurley MJ, Dexter DT (2012) Voltage-gated calcium channels and Par-
kinson’s disease. Pharmacol Ther 133: 324-333
93. Chan CS, Gertler TS, Surmeier DJ (2009) Calcium homeostasis, selecti-
vite vulnerability and Parkinson’s disease. Trends Neurosci 32: 249-256
94. Giacomello M, Hudec R, Lopreiato R (2011) Huntington’s disease, cal-
cium, and mitochondria. Biofactors 37: 206-218
95. Bezprozvanny I (2011) Role od inositol 1, 4, 5-trisphosphate receptors
in pathogenesis of Huntington’s disease and spinocerebellar ataxias.
Neurochem Res
36: 1186-1197
96. Verbeek DS, van de Warrenburg BP (2011) Genetics of the dominant
ataxias
. Semin Neurol 31: 461-469
97. Crish SD, Calkins DJ (2011) Neurodegeneration in galucoma: Progres-
sion and calcium-dependent intracellular mechanisms. Neuroscience
176: 1-11
98. Zamponi GW, Lory P (2010) Role of voltage-gated calcium channels in
epilepsy. Pfluges Arch 460: 395-403
99. Lidow MS (2003) Calcium signaling dysfunction in schizophrenia: a
unifying approach. Brain Res Brain Res Rev 43: 70-84
100. Giegling I, Genius J, Benninghoff J, Rujescu D (2010) Genetic findings
in schizophrenia patients related to alterations in the intracellular Ca-
-homeostasis. Prog Neuropsychopharmacol Biol Psychiatry 34: 1375-1380
Wyszukiwarka
Podobne podstrony:
403
403 ac
403
cziomer78 403
F LAB305, Fizyka, Laborki, cw. 417
Zobowiązania, ART 417 KC, Wyrok z dnia 5 sierpnia 2005 r
403
417 1
417, 417MISIE, Sprawozdanie z wykonanego ˙wiczenia nr 417
lanos manual cz III 1 403
403
medycyna wet 2009 nr 6 strony 399 403
417, 417(1), Temat : Wyznaczanie stosunku Cp/Cv metod˙ Clementa - Desormesa
kpk, ART 417 KPK, 1970
417
417 ac
417, #417
Księga 1. Proces, ART 403 KPC, 2005
więcej podobnych podstron