
POLITECHNIKA GDAŃSKA
WYDZIAŁ CHEMICZNY
KATEDRA TECHNOLOGII CHEMICZNEJ
Technologia Chemiczna, VII sem.,
Technologia Organiczna
Tytuł ćwiczenia: Produkcja leków
Opracowała: mgr. inż. Agnieszka Pazik, p. 406 ChB
Gdańsk 2012
1.
Wiadomości ogólne
Lek jest to substancja, którą wprowadza się do organizmu w celu osiągnięcia efektu terapeutycznego lub w celu
zapobiegania chorobie. Lekiem może być substancja pochodzenie naturalnego lub syntetycznego, która oddziałuje z
wielocząsteczkowymi strukturami docelowymi, wywołując odpowiedź biologiczną – hamuje przyczyny lub objawy
choroby lub zapobiega jej rozwojowi. Większość leków stosuje się również w celach diagnostycznych oraz środkach
modyfikujących niezmienione chorobowo funkcje organizmu (np. leki antykoncepcyjne). W zbyt dużych dawkach
część substancji stosowanych w medycynie, jako leki mogą stać się truciznami. Nauką o produkcji leków i ich
działaniu zajmuje się farmacja, zaś farmakologia terapeutycznym zastosowaniem leku. Nauką zajmującą się
wykorzystaniem wiedzy o sposobach działania farmaceutyków na poziomie molekularnym oraz do projektowania i
syntezy nowych pochodnych jest chemia leków.
W Polsce podstawowym aktem prawnym regulującym zasady wprowadzania leków, ich dystrybucji i działania jest
ustawa Prawo Farmaceutyczne (Dz. U. z dnia 31 października 2001 roku). Prawo to nie zawiera pojęcia lek lecz
produkt leczniczy, który definiowany jest jako „substancja lub mieszanina substancji, przeznaczona do zapobiegania
lub leczenia chorób występujących u ludzi lub zwierząt, lub podawana człowiekowi lub zwierzęciu w celu postawienia
diagnozy lub w celu przywrócenia, poprawienia czy modyfikacji fizjologicznych funkcji organizmu ludzkiego lub
zwierzęcego”.
Prawo farmaceutyczne precyzuje również rodzaje produktów leczniczych (leków), jako produkty przeznaczone do
specjalnych celów żywieniowych, leczniczy homeopatyczny, immunologiczny, krwiopochodny i roślinny.
2.
Klasyfikacja leków
Podstawą klasyfikacji substancji leczniczych może być ich budowa chemiczna lub działanie farmakologiczne.
Różnorodność budowy chemicznej, wprowadzanie coraz to nowszych leków o niespotykanej budowie utrudnia ich
klasyfikacje pod względem chemicznym. Podział leków na podstawie klasyfikacji stosowanej w chemii organicznej
nie daje jednoznacznego podziału związków leczniczych, które często w swojej budowie posiadają różne struktury i
grup funkcyjnych. Powszechnie proponowany podział leków bazuje na ich farmakologicznym działaniu. Taki podział
daje możliwość wyjaśnienia zależności budowy między budową a działaniem leku, powiązania wchłaniania leku, jego
rozmieszczenia w organizmie i biotransformacji z jego właściwościami fizykochemicznymi.
S
NH
2
O
O
NHR
R'
"R
CH
3
H
H
CH
3
R
H
H
N
S
O
CH
3
CH
3
COOH
H
H
N
H
R
O
penicyliny
steroidy
sulfonamidy
COOH
O
C
CH
3
O
HOH
-CH
3
COOH
COOH
OH
OH
- CO
2
Na
+
N
-
N
N
N
O
O
CH
3
CH
3
CO
2
, H
2
O
N
N
N
N
O
O
CH
3
CH
3
H
NaHCO
3
3.
Wpływ czynników fizykochemicznych na trwałość i wchłanianie leków
Zasadniczą właściwością każdego leku jest jego trwałość, czyli odporność na działanie czynników fizycznych,
chemicznych lub biologicznych (np. światło, wilgoć, pH środowiska, enzymy). Jakakolwiek zmiana w budowie i
właściwościach leku, może osłabić lub obniżyć aktywność farmakologiczną leku, a nawet spowodować wzrost jego
toksyczności. Spośród reakcji chemicznych, będących przyczyną rozkładu leków, należy wyróżnić: hydrolizę,
utlenianie, redukcję, dekarboksylację i karboksylację oraz swoiste przemiany leków związane z ich budową
(racemizacja) i występowaniem pewnych grup funkcyjnych.
Najczęstszą przyczyną rozkładu leków jest reakcja hydrolizy. Mogą jej ulegać estry, amidy, cykliczne imidy, laktamy,
wiązania eterowe w glikozydach. Reakcje hydrolizy przyśpiesza: woda, podwyższona temperatura oraz środowisko
kwasowe lub zasadowe. Leki ulegają również rozkładowi pod wpływem czynników utleniających, tj. podwyższona
temperatura, obecność katalizatorów utleniania, enzymów typu oksydaz oraz środowiska kwasowego. Procesom
utleniania najczęściej ulegają grupy aldehydowe, fenolowe, pochodne fenotiazyny, niektóre alkaloidy, którym
towarzyszy zwykle zmiana cech zewnętrznych (zabarwienia) oraz zmiana w budowie. Zabezpieczeniem przed
szkodliwym wpływem czynników utleniających jest wprowadzenie tzw. przeciwutleniaczy - akceptory tlenu lub
donory wodoru. Rozkład leku na skutek reakcji redukcji jest rzadziej spotykane, dotyczą one głównie jonów srebra
lub jonów rtęci w niektórych formach leków. Zaś dekarboksylacja leku zachodzi zwykle jako jeden etap w cyklu
przemian chemicznych rozkładu środka leczniczego, głownie ulegają temu kwasy aromatyczne. W związkach o
charakterze estrów obserwuje się zjawisko dekarboksylacji, jako reakcję wtórną po wcześniejszej ich hydrolizie (np.
kwas acetylosalicylowy).
W solach zasadowych bardzo słabych kwasów zachodzi proces karboksylacji, które pod wpływem, CO
2
i powietrza
rozkładają się na wolne kwasy o znaczniej mniejszej rozpuszczalności. Jako przykład można przedstawić sole sodowe
pochodnych kwasów barbiturowego lub sole teobrominy:
Oprócz przemian prowadzących do rozkładu leku, występują również przemiany przestrzenne w grupach izomerów
optycznych, geometrycznych czy konformacyjnych prowadzące do obniżenia aktywności farmakologicznej.
Aktywność ta związana jest najczęściej z jedną tylko formą optycznie czynnych izomerów, które pod wpływem
wewnątrzcząsteczkowych przegrupowań podstawników przy węglu
asymetrycznym przekształcają się w mieszaninę
racemiczną. Pary izomerów geometrycznych lub konformacyjnych w normalnych warunkach przygotowania i
przechowywania leków są stabilne, a ich wzajemne przemiany są możliwe jedynie po dostarczeniu znacznych ilości
energii.
Złożone procesy przemiany leków rozpatrywane są w szczególności w warunkach in vivo, które zalezą nie tylko od
budowy i właściwości fizykochemicznych leku, ale i od wielu zmiennych czynników ustrojowych. Lek zanim dotrze

do miejsca działania (najczęściej receptora) w organizmie musi pokonać wiele przeszkód związanych z błonami
biologicznymi. Transport leku przez błony jest uzależniony od rozpuszczalności w lipidach i płynach ustrojowych,
wielkości cząsteczek związku, stopnia jonizacji oraz miejsca wprowadzenia (drogi podania), pH środowiska oraz
struktury i właściwości błon, przez które dany lek przechodzi. Większość leków jest transportowana w organizmie na
zasadzie dyfuzji biernej. Dyfuzja ta polega na przechodzeniu niezjonizowanych cząsteczek leku rozpuszczalnego w
fazie wodnej przez półprzepuszczalną błonę lipidową do fazy wodnej po drugiej stronie błony. Szybkość tego procesu
zależy od rozpuszczalności leku w lipidach błonowych oraz od masy cząsteczki. Związki niezjonizowane przechodzą
przez błony łatwiej niż zjonizowane. Transport bierny zachodzi bez zużywania energii.
Większość występujących leków ma charakter słabych elektrolitów, które w roztworach wodnych ulegają częściowej
dysocjacji. Leki o charakterze słabych kwasów (np. pochodne fenoli, hydroksykwasy, pochodne kwasu
barbiturowego, hydantoiny, dimetyloksantyn) wchłaniane są z kwasowego środowiska żołądkowego na skutek
występowania w tym środowisku w formie niezjonizowanej. Natomiast leki o słabym charakterze zasadowym (np.
alkaloidy, pochodne pyrazolonu-5, prokaina, lidokaina) występują w środowisku kwasowym żołądka w formie
zjonizowanej na skutek protonizacji. Dlatego leki te wchłaniane są głównie w jelicie cienkim, gdyż w tym środowisku
występują w formie niezjonizowanej.
Leki o właściwościach hydrofilnych trudno przenikają przez błony lipidowe, dlatego przenoszone są na zasadzie
transportu czynnego, tj. przy udziale odpowiednich endogennych nośników i energii pochodzącej z rozkładu ATP.
Transport aktywny może przebiegać wbrew gradientowi stężeń leku po obu stronach błony. Modyfikując chemicznie
strukturę związku aktywnego farmakologicznie, można zmieniać właściwości fizykochemicznie w kierunku
zwiększenia lipofilności lub hydrofilności leku. Dobrym na to przykładem jest wzrost lipofilności oraz znaczna
poprawa wchłaniania leku po estryfikacji obecnych w cząsteczce macierzystej grup karboksylowych lub
hydroksylowych.
4.
Ogólne wiadomości dotyczące otrzymywania leków
Duża liczba leków stosowanych obecnie w terapii oraz różnorodność ich budowy chemicznej wymaga
zróżnicowanych metod ich wytwarzania. Metody można podzielić na dwie zasadnicze grupy: jedna z nich obejmuje
metody polegające na wyodrębnieniu substancji czynnych pochodzenia naturalnego, zarówno roślinnego, jak i
zwierzęcego, druga zaś obejmuje syntezę chemiczną oraz biosyntezę.
Wyodrębnianie związków biologicznie czynnych z produktów pochodzenia naturalnego polegają głównie na
ekstrakcji substancji czynnych przy użyciu odpowiednich rozpuszczalników. Dodatkowo należy przeprowadzić takie
procesy fizykochemiczne jak destylacja, krystalizacja oraz stosowanie wymieniaczy jonowych lub chromatografii.
Przykładem leków wyodrębnianych przez ekstrakcję z produktów pochodzenia roślinnego są przede wszystkim
alkaloidy i glikozydy. Alkaloidy makowca, głównie morfina, uzyskiwane są przez ekstrakcję z opium, tj.
wysuszonego soku mlecznego niedojrzałych makówek lub słomy makowej. Metyloksantyny otrzymuje się z łusek
kakaowych lub ziaren kawy, chininę z kory drzew chinowych. Leki uzyskiwane wyłącznie przez ekstrakcję z
surowców roślinnych mogą być glikozydy nasercowe, np. glikozydy naparstnicy purpurowej i wełnistej.
Przez ekstrakcję z produktów pochodzenia zwierzęcego otrzymuje się wiele preparatów organicznych, hormonów i
enzymów. Przykładem może być insulina (peptydowy hormon trzustki) stosowany w leczeniu cukrzycy, produkuje się
na skalę przemysłowa z trzustek zwierzęcych przez ekstrakcję alkoholem etylowym zakwaszonym do pH 2,5. Dalsze
SO
2
H
2
N
NHR
oczyszczanie i frakcjowanie prowadzi się metodami chromatograficznymi. Obecnie również insulinę otrzymuje się
przy użyciu inżynierii genetycznej. Inną metodą otrzymywania większości stosowanych (70%) obecnie leków jest
synteza chemiczna. Dzięki syntezie produkuje się głównie leki nasenne, przeciwbólowe, przeciwzapalne, antyseptyki,
chemioterapeutyki, leki psychotropowe, leki hipotensyjne i moczopędne, leki działające na układ autonomiczny oraz
leki przeciwhistaminowe. Współczesny rozwój nowych metod syntezy organicznej daje praktycznie nieograniczone
możliwości uzyskania różnorodnych struktur chemicznych, obdarzonych określonym działaniem farmakologicznym.
Metody syntezy chemicznej mogą być także wykorzystywane do modyfikacji substancji wyodrębnionych z surowców
naturalnych. Takie modyfikacje związków biologicznie czynnych pozwoliły na uzyskanie wielu nowych leków, o
lepszych właściwościach leczniczych w porównaniu do substancji macierzystych. Przykładem może być: modyfikacja
alkaloidów fenanterowych występujących w opium, alkaloidów tropanowych, naturalnych metyloksantyn oraz
antybiotyków. Dużą grupę leków otrzymuje się przez biosyntezę i biotransformację. Przykładem mogą być
antybiotyki, wprawdzie niektóre z nich otrzymywane są w wyniku pełnej syntezy chemicznej, większość jednak
stosowanych antybiotyków powstaje przez biosyntezę produktów naturalnych. Proces biosyntezy antybiotyków
obejmuje kilka etapów produkcyjnych. Hodowlę wstępną wyselekcjowanych i przygotowanych drobnoustrojów
prowadzi się w skali laboratoryjnej i półtechnicznej na odpowiednich podłożach. Wytwarzanie antybiotyków odbywa
się w fermentatorach produkcyjnych o bardzo dużej pojemności, po czym antybiotyk się izoluje i oczyszcza za
pomocą odpowiedniego procesu fizykochemicznego a następnie nadaje mu się odpowiednią postać farmaceutyczną.
Innym zastosowaniem biosyntezy w produkcji leków jest wykorzystanie jej przy otrzymywaniu hormonów
steroidowych. Oprócz klasycznych metod chemicznych przekształcających naturalne sterole, stosuje się również etapy
syntezy z udziałem drobnoustrojów lub odpowiednich enzymów. Przykładem może być hydroksylowanie układu
steroidowego, zwłaszcza w położeniach C
11
, C
17
, C
21
, utlenianie II-rzędowych grup alkoholowych do grup
ketonowych oraz odwodornienie pierścienia A układu steroidowego. Procesy te polegają na wprowadzeniu zawiesiny
odpowiedniego związku steroidowego do uprzednio wyhodowanego na właściwej pożywce szczepu grzyba lub
bakterii.
5.
Środki lecznicze działające na drobnoustroje chorobotwórcze na przykładzie sulfonamidów
Ś
rodki lecznicze działające na drobnoustroje chorobotwórcze dzielimy na substancje specyficzne odkażające żywe
tkanki (antyseptyki) oraz na niespecyficzne służące do niszczenia drobnoustrojów poza organizmem ludzkim
(dezynfekujące). Związki te charakteryzują się dużą aktywnością w działaniu na bakterie, grzyby i pierwotniaki.
Amidy kwasu 4-aminobenzenosulfonowego (nazywane potocznie sulfonamidami) są skutecznymi środkami
leczniczymi o działaniu bakteriostatycznym.
Amidy kwasu sulfanilowego w momencie wprowadzenia do lecznictwa wykazywały szeroki zakres działania na
liczne bakterie Gram-dodatnie i niektóre Gram-ujemne. Jednakże z czasem bakterie uodporniły się na dane leki. W
obecnych czasach sulfonamidy stosowane są wobec paciorkowców (Streptococcus), gronkowców (Staphylococcus),
promieniowców (Actinomyces), dwoinki zapalenia płuc (Diplococcus pneumoniae) i pałeczki okrężnicy (Escherichia
coli), a także niektórych pierwotniaków.
SO
2
H
2
N
NH
2
NH
CO
H
3
C
HOSO
2
Cl
NH
CO
H
3
C
SO
2
Cl
NH
CO
H
3
C
SO
2
NH
2
H
+
/ OH
-
NH
3
ASC
H
2
N
SO
2
NH
R
Sulfonamidy są podawane najczęściej doustnie. Pozajelitowo stosuje się jedynie ich sole sodowe. Sulfonamidy różnią
się miedzy sobą właściwościami farmakokinetycznymi. Leki podawane doustnie posiadają różny stopień wchłaniania
z przewodu pokarmowego, co jest uzależnione od stopnia zjonizowania i lipofilności cząsteczki, dlatego wyróżnia się:
1.
Sulfonamidy łatwo wchłaniające się z przewodu pokarmowego w krótkim czasie osiągające odpowiednie
stężenie w osoczu i tkankach. Należą do nich większość N
1
-podstawionych pochodnych sulfanilamidu.
2.
Sulfonamidy trudno wchłaniające się z przewodu pokarmowego, stosowane jedynie w bakteryjnych zakażeniach
przewodu pokarmowego. Należą do nich N
1
,N
4
-dipodstawione pochodne sulfanilamidu oraz sulfaguanidyna.
3.
Sulfonamidy stosowane jedynie miejscowo, zewnętrznie (sulfanilamid, sulfacetamid, sole srebrowe sulfadiazyny
i sulfatiazolu).
Sulfonamidy charakteryzuje duża rozpiętość czasu działania, co wiąże się z szybkością wydalania ich z organizmu.
Dlatego sulfonamidy dzielimy na leki krótko działające (okres półtrwania < 8 godzin), o średnio długim czasie
działania (okres półtrwania 8-20 godzin) i długo działające (okres półtrwania > 30 godzin).
Sulfonamidy otrzymuje się w reakcji acetanilidu z kwasem chlorosulfonowym, w wyniku, której powstaje chlorek
kwasu 4-acetyloaminobenzenosulfonowego (ASC). Związek ten amoniakiem tworzy amid kwasu 4-
acetyloaminobenzenosulfonowego, następnie w celu usunięcia grupy acetylowej, przeprowadza się hydrolizę
zasadową lub kwasową otrzymanego amidu. Schemat syntezy został przedstawiony na poniższym rysunku.
Zastępując amoniak aminowymi pochodnymi heterocyklicznymi otrzymuje się odpowiednie pochodne - sulfonamidy
zawierające podstawniki heterocykliczne pięcio- lub sześcioczłonowe.
Pierwszym sulfonamidem wprowadzonym do lecznictwa i zarazem najprostszym był sulfanilamid. Wszystkie
pozostałe związki są traktowane, jako jego pochodne.
Nazwa międzynarodowa
Nazwa chemiczna
R
Sulfanilamid
Amid kwasu 4-aminobenzenosulfonowego
H
Sulfacetamid
Amid kwasu N’-acetylo-4-aminobenzenosulfonowego
CO
CH
3
Sulfacarbamid
4-Aminobenzenosulfonylomocznik
CO
NH
2
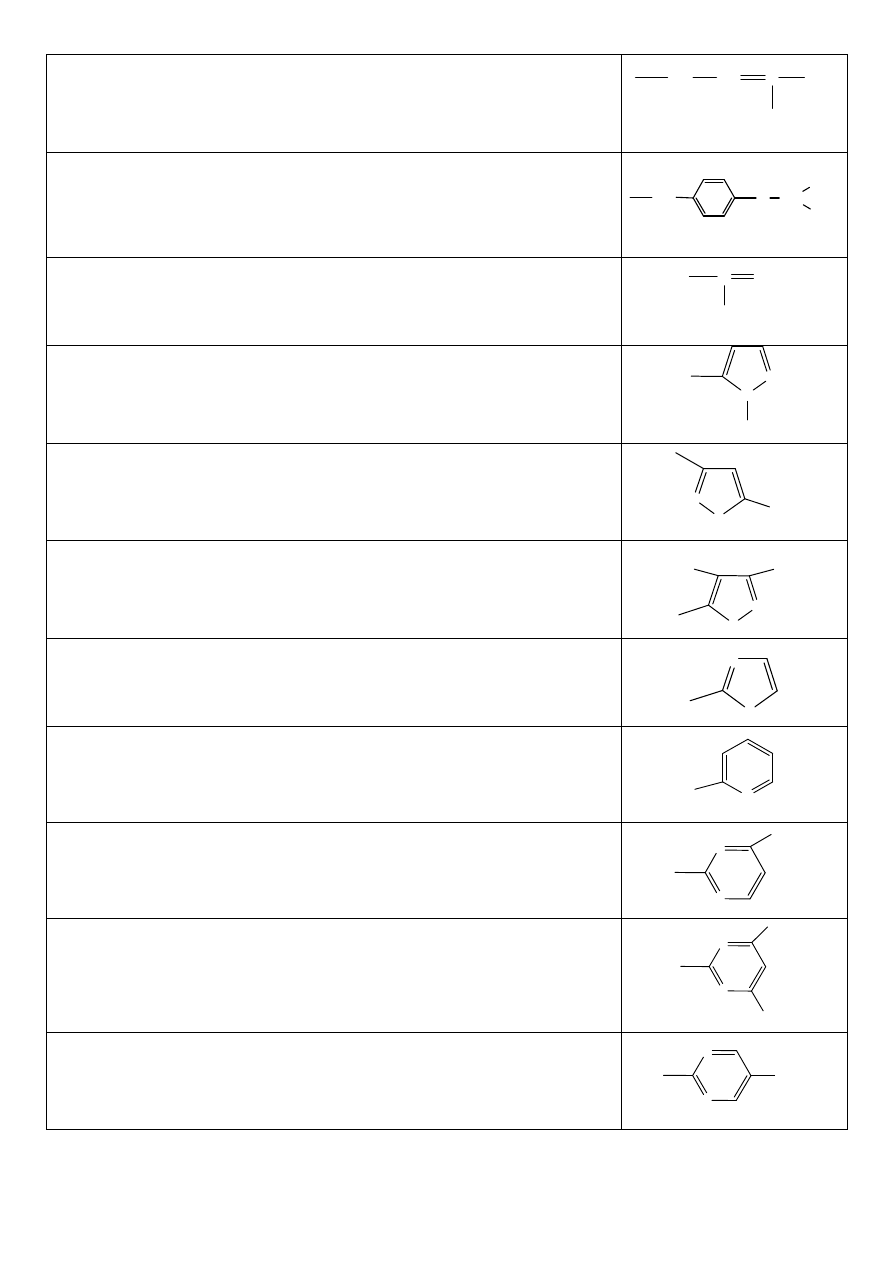
Sulfadikramid
Amid kwasu N’-(3,3-dimetyloakrylodo)-4-aminobenzeno-sulfonowego
CO
CH
C
CH
3
CH
3
Sulfaproksylina
Amid kwasu N’-(4-izopropoksybenzoilo)-4-amino-benzenosulfonowego
CO
O CH
CH
3
CH
3
Sulfaguanidyna
Amid kwasu N-guanidymo-4-aminobenzenosulfonowego
C
NH
NH
2
Sulfafenazol
Amid kwasu N’-(1-fenylopirazolo-5)-4-aminobenzeno-sulfonowego
N
N
C
6
H
5
Sulfametoksazol
Amid kwasu N’-(3,4-dimetyloizoksazolo-3)-4-amino-benzenosulfonowego
N
O
CH
3
Sulfafurazol
Amid kwasu N’-(3,4-dimetyloizoksazolo-5)-4-amino-benzenosulfonowego
O
N
H
3
C
CH
3
Sulfatiazol
Amid kwasu N’-(2-tiazolo)-4-aminobenzenosulfonowego
N
S
Sulfapirydyna
Amid kwasu N’-(2-pirydyno)-4-aminobenzenosulfo-nowego
N
Sulfamerazyna
Amid kwasu N’-(4-metylopirymidyno-2)-4-amino-benzenosulfonowego
N
N
CH
3
Sulfadimidyna
Amid kwasu N’-(4,6-dimetylopirymidyno-2)-4-amino-benzenosulfonowego
N
N
CH
3
CH
3
Sulfametoksydiazyna
Amid kwasu N’-(5-metoksypirymidyno-2)-4-amino-benzenosulfonowego
N
N
OCH
3
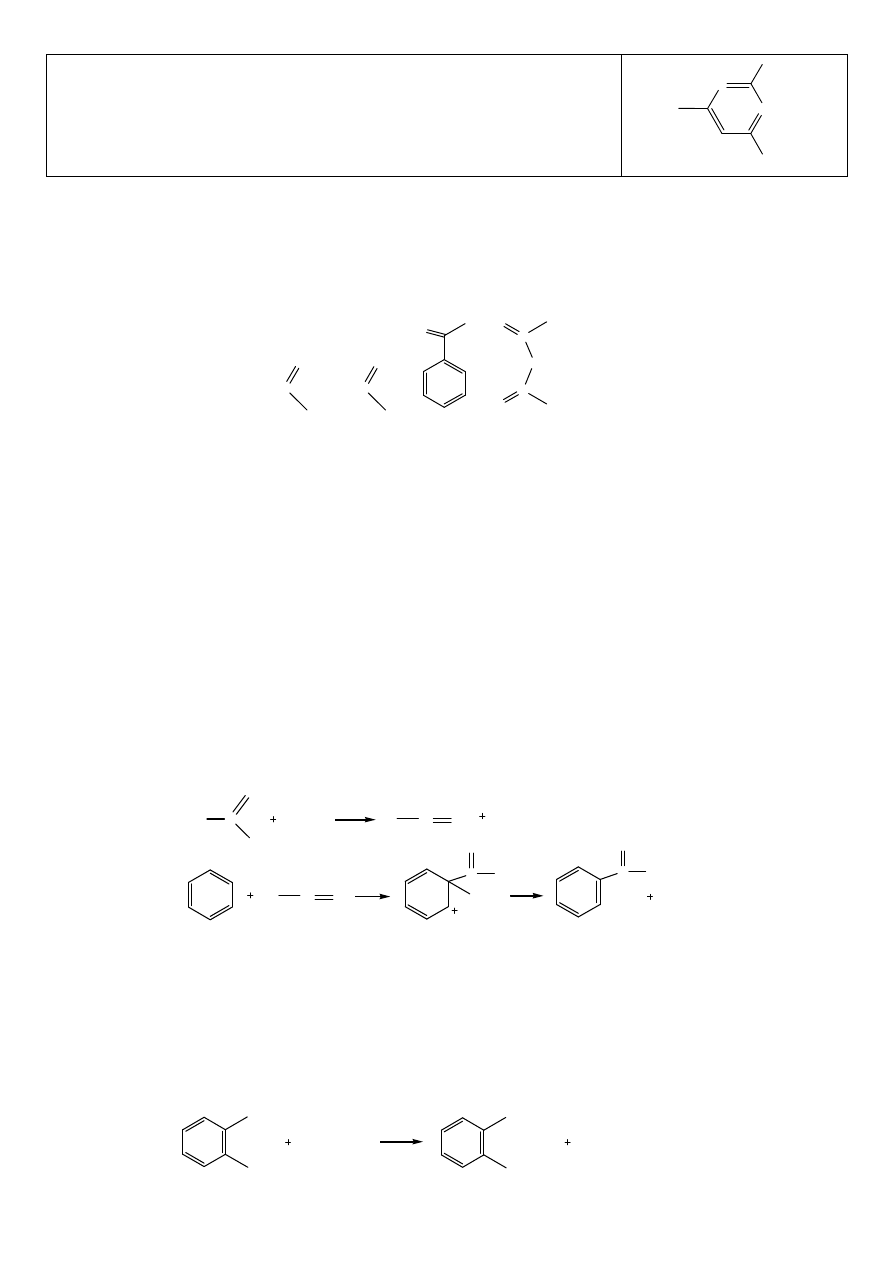
HC
O
formyl
CH
3
C
O
acetyl
O
benzoil
CH
2
C
C
O
O
malonyl
C
Cl
O
R
AlCl
3
R
C
+
O
AlCl
4
-
R
C
+
O
C
H
O
R
C
O
R
H
+
COOH
OH
(CH
3
CO)
2
O
COOH
OCOCH
3
CH
3
COOH
Sulfadimetoksyna
Amid kwasu N’-(2,6-dimetoksypirymidyno-4)-4-amino-benzenosulfonowego
N
N
OCH
3
OCH
3
6. Podstawowe procesy jednostkowe w produkcji leków (sulfonamidów)
6.1. Acylowanie
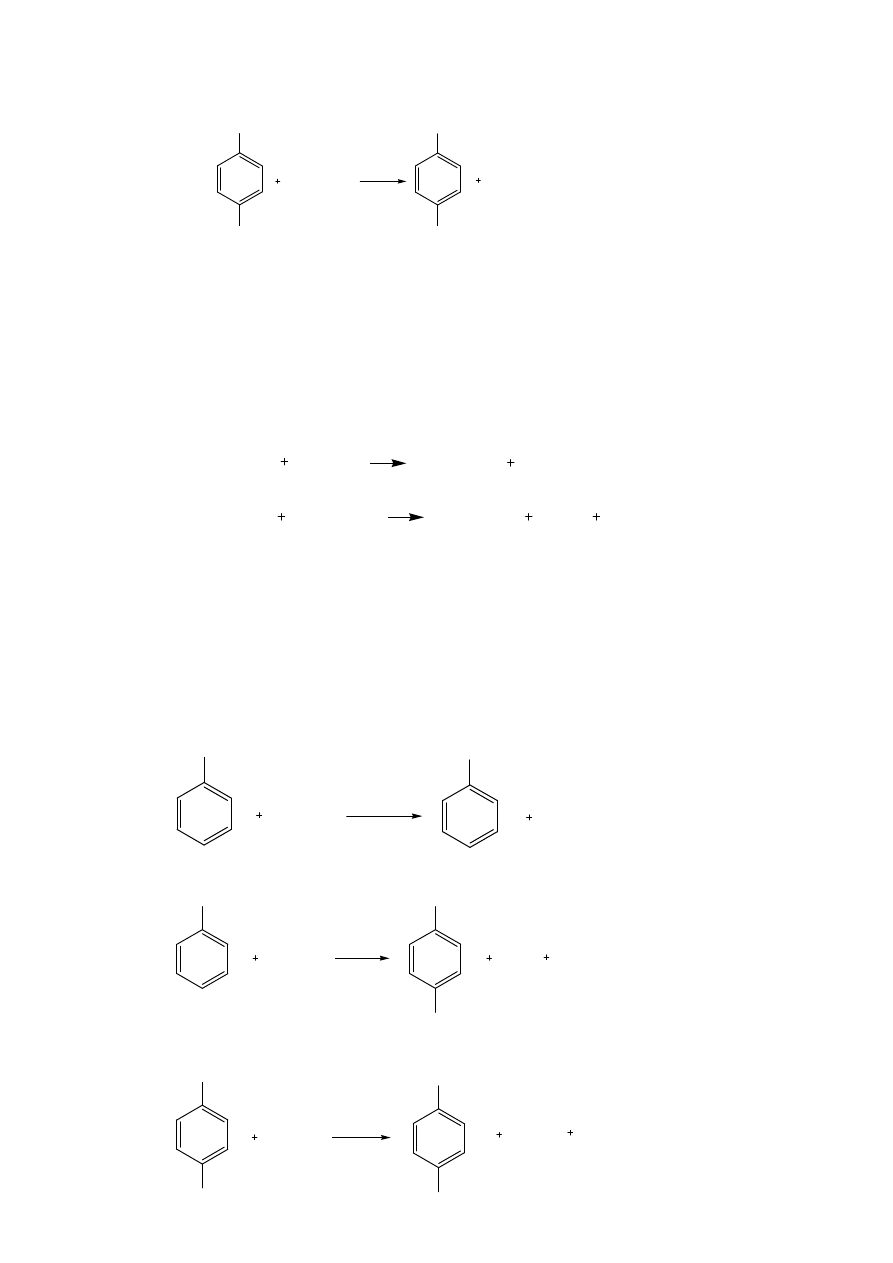
Acylowanie jest procesem polegającym na wprowadzeniu do cząsteczki związku organicznego grupy, acylowej
(reszty nieorganicznego lub organicznego kwasu tlenowego). Poniżej przedstawiono przykłady grup arylowych:
Największe znaczenie przemysłowe ma wprowadzenie do związków organicznych grup acetylowych i wtedy proces
nazywamy acetylowaniem.
Acylowaniu poddaje się:
•
Związki zawierające grupy –OH (alkohole i fenole)
•
Związki zawierające grupy – NH
2
(aminy alifatyczne, aromatyczne oraz amidy kwasowe)
•
Związki aromatyczne w pierścieniu benzenowym
Czynnikami acylującymi są kwasy organiczne oraz ich chlorki, bezwodniki i estry oraz keten. Reakcje prowadzi się w
umiarkowanie podwyższonych temperaturach (ok. 50 – 90 °C) i pod ciśnieniem atmosferycznym.
Acylowanie węglowodorów aromatycznych przebiega w reakcji Friedla-Craftsa przy udziale kwasów Lewisa, jako
katalizatorów. Początkowo powstaje jon acyliowy, który powstaje na skutek działania katalizatora na czynnik
acylujący. Jon ten atakuje pierścień benzenowy, zaś obecny w mieszaninie reakcyjnej chlorek glinowy z utworzonym
ketonem tworzy trwały kompleks.
Grupę acylową wprowadza się do związku na stałe lub w celu ochrony grup funkcyjnych wrażliwych np. –NH
2
, –OH.
W celu usunięcia grupy acylowej przeprowadza się deacylację poprzez hydrolizę, która zachodzi w wyniku
ogrzewania związku w wodnym roztworze kwasów lub zasad.
Proces acylowania ma szczególne znaczenie przy otrzymywaniu leków. Na przykład przez acetylowaniem kwasu
salicylowego bezwodnikiem octowym otrzymuje się kwas acetylosalicylowy będący substancją czynną takich leków
jak polopiryna (aspiryna), czy calcipiryna.
NHCOCH
3
2 HSO
3
Cl
NHCOCH
3
SO
2
Cl
HCl
H
2
SO
4
NHCOCH
3
SO
2
Cl
NH
4
OH
NHCOCH
3
SO
2
NH
2
NH
4
Cl
H
2
O
OC
2
H
5
NH
2
CH
3
COOH
OC
2
H
5
NHCOCH
3
H
2
O
Ar-H
2 ClSO
3
H
Ar-SO
2
Cl
HCl
H
2
SO
4
Ar-H
Ar-SO
3
H
HCl
ClSO
3
H
NH
2
CH
3
COCOCH
3
CH
3
COOH
NHCOCH
3
CH
3
COOH
Innym przykładem zastosowania acetylowania w przemyśle środków leczniczych jest otrzymywanie fenacetyny (lek
przeciwgorączkowy), synteza polega na acetylowaniu p-fenetydyny kwasem octowym lodowatym lub bezwodnikiem
octowym.
6.2. Sulfonowanie kwasem chlorosulfonowym
Zastosowanie kwasu chlorosulfonowego, jako odczynnika sulfonującego w zależności od użytych warunków reakcji i
ilości reagenta, prowadzi do otrzymania dwóch rodzajów produktów:
1.
Aromatycznych sulfochlorków Ar-SO
2
Cl
2.
Kwasów arylosulfonowych Ar-SO
3
H
W reakcji równomolowych ilości związku sulfonowanego i kwasu chlorosulfonowego lub przy małym nadmiarze tego
kwasu, powstają kwasu sulfonowe:
Stosowanie nadmiaru kwasu chlorosulfonowego prowadzi do otrzymania sulfochlorków wg poniższej reakcji:
Sulfochlorki ze względu na łatwość wymiany w ich grupie atomu chloru na inne podstawniki, stanowią produkty
pośrednie w syntezie aromatycznych sulfonamidów, estrów, anilidów, kwasów tiolowych i sulfinowych.
CZĘŚĆ DOŚWIADCZALNA
Ćwiczenie 1
Wieloetapowa synteza SULFANILAMIDU
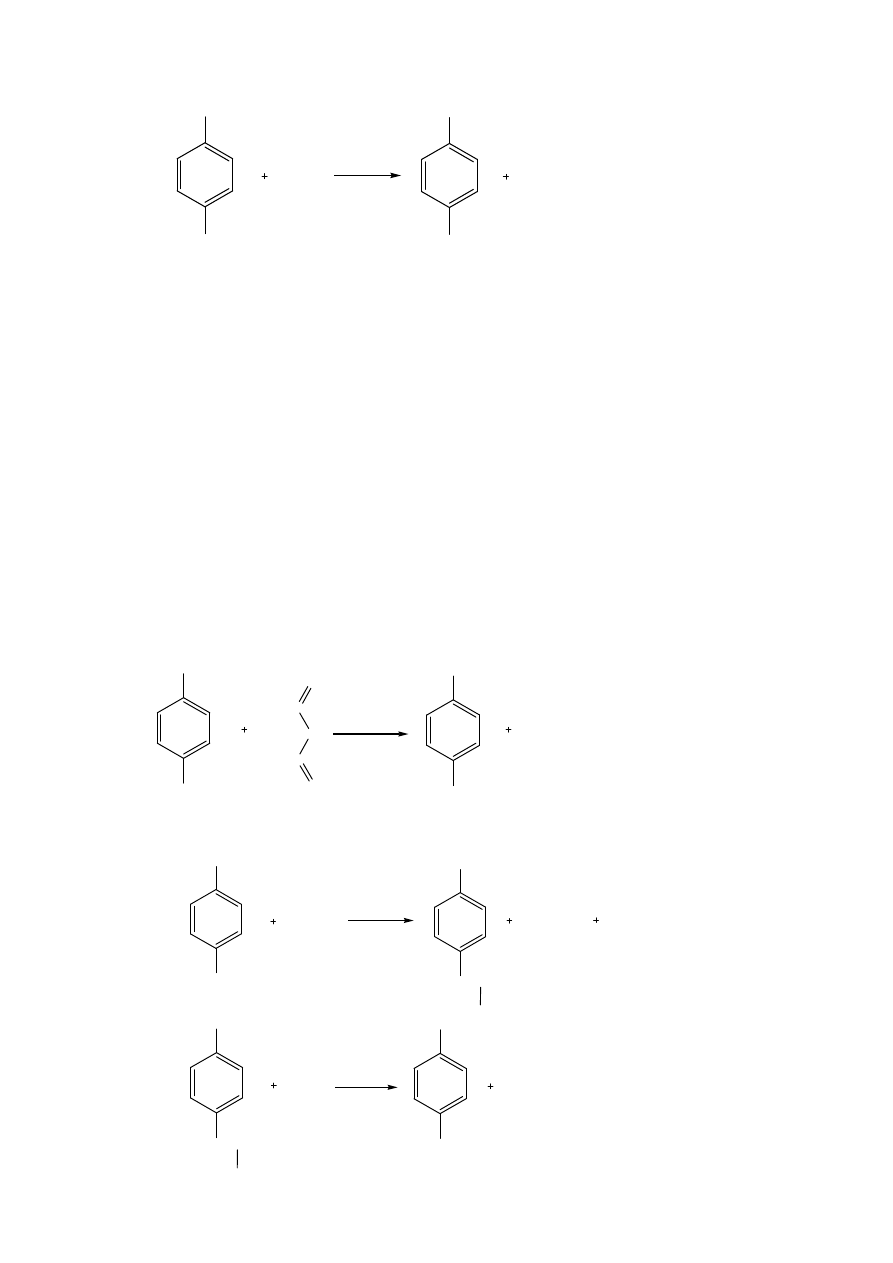
1etap. Otrzymywanie acetanilidu
2etap. Otrzymywanie p-acetyloaminobenzenosulfochlorku
3etap. Otrzymywanie acetylosulfanilamidu
NHCOCH
3
SO
2
NH
2
CH
3
C
O
O
CH
3
C
O
CH
3
COOH
NHCOCH
3
SO
2
NHCOCH
3
CH
3
COOH
NHCOCH
3
SO
2
NHCOCH
3
2 NaOH
NH
2
SO
2
NCOCH
3
Na
CH
3
COONa
H
2
O
NH
2
SO
2
NCOCH
3
Na
HCl
pH 4
NH
2
SO
2
NHCOCH
3
NaCl
NHCOCH
3
SO
2
NH
2
NaOH
NH
2
SO
2
NH
2
CH
3
COONa
4etap. Otrzymywanie sulfanilamidu
Acetanilid otrzymuje się poprzez acetylację aniliny mieszaniną kwasu octowego i bezwodnika octowego. p-
Acetyloaminobenzenosulfochlorek otrzymuje się w reakcji 20 mmol (1,32 ml) kwasu chlorosulfonowego z 10 mmol
(1,35 g ) acetanilidu, w temperaturze 53-55 °C w czasie 2 godzin. Mieszaninę reakcyjną po reakcji należy schłodzić a
następnie masę poreakcyjną wylać na wodę z lodem, w wyniku, czego wytrąca się osad p-
acetyloaminobenzenosulfochlorku. Dany osad oddziela się od ługów macierzystych i przemywa się wodą
destylowaną. Mokry p-acetyloaminobenzenosulfochlorek poddaje się procesowi aminowania wodą amoniakalną
(stosunek 1:9). Mieszaninę reakcyjną ogrzewamy w temperaturze 75-80 °C przez 1,5 godziny. Następnie po
schłodzeniu próbki po procesie aminowania wydziela się osad acetylosulfanilamidu, który przesączamy pod próżnią
oraz przemywamy wodą destylowaną. Wysuszony osad kieruje się do syntezy sulfacetamidu lub poddaje zmydleniu w
celu usunięcia grupy ochronnej. Sulfanilamid suszy się w temperaturze 60-80 °C.
Ćwiczenie 2
Wieloetapowa synteza SULFACETAMIDU
1etap. Otrzymywanie acetylosulfacetamidu
2etap. Otrzymywanie sulfacetamidu
N
N
O
2
N
CH
3
N
N
O
2
N
CH
3
H
2
O
2
(NH
4
)
6
Mo
7
O
24
.
4 H
2
O
CH
2
CH
2
SCH
2
CH
3
CH
2
CH
2
SO
2
CH
2
CH
3
N
N
O
2
N
CH
3
N
N
O
2
N
CH
3
CH
2
CH
2
SCH
2
CH
3
CH
2
CH
2
SOCH
2
CH
3
CH
3
COOH
H
2
O
2
Acetylosulfacetamidu otrzymuje się przez acetylację acetylosulfanilamidu mieszaniną kwasu octowego i bezwodnika
kwasu octowego. Do reakcji używa się 10 mmol (2,14 g) acetylosulfanilamidu, 60 mmol (3,43 ml) kwasu octowego
oraz 30 mmol (2,83 ml). Mieszaninę reakcyjną ogrzewamy w temperaturze 118-122 °C przez 2,5 godziny. Następnie
chłodzimy mieszaninę i przesączamy powstały osad produktu, który po przemyciu wodą kierowany jest do procesu
otrzymywania sulfacetamidu farmakopealnego. W wyniku prowadzenia deacetylacji acetylosulfacetamidu roztworem
wodorotlenku sodowego powstaje sól sodowa sulfacetamidu. Do reakcji używa się 0,5 g NaOH przypadającego na 1 g
suchej masy acetylosulfacetamidu oraz 3 ml H
2
O na 1 g reagenta. Zawartość kolby ogrzewamy do temperatury 30-35
°C. W roztworze oprócz soli sodowej sulfacetamidu znajduje się w minimalnej ilości sól sodowa sulfanilamidu.
Roztwór po deacetylacji zobojętnia się kwasem solnym do pH=8, przy tym pH wytrąca się sulfanilamid. Do roztworu
po zobojętnieniu dodaje się węgiel aktywny i przeprowadza się filtrację. Na sączku pozostaje węgiel aktywny i
sulfanilamid. Filtrat oczyszcza się węglem aktywnym. Po oddzieleniu węgla aktywnego z roztworu kwasem solnym
do pH = 4,5 - 4,8 wytrąca się sulfacetamid farmakopealny. Sulfacetamid oddziela się od ługów macierzystych,
odmywa wodą od jonów chlorkowych i poddaje procesowi suszenia
.
Ćwiczenie 3
Otrzymywanie TINIDAZOLU w reakcji utlenienia 3-(2-etylotioetylo)-2-metylo-5-nitroimidazolu nadtlenkiem
wodoru w obecności katalizatora molibdenianu amonu lub lodowatego kwasu octowego.
6 mg katalizatora molibdenianu amonu rozpuszczamy w 5 ml wody destylowanej i dodajemy do 2,4 g 3-(2-
etylotioetylo)-2-metylo-5-nitroimidazolu. Całość ochładzamy do temperatury 15 °C i dodajemy 2,7 ml 33% roztworu
nadtlenku wodoru. Mieszaninę reakcyjną mieszamy i ogrzewamy w temperaturze ok. 65 °C przez około 2,5 godzin.
Powstały osad sączymy pod zmniejszonym ciśnieniem i krystalizujemy z 5 ml etanolu.
Do kolby zawierającej 1,5 g 3-(2-etylotioetylo)-2-metylo-5-nitroimidazolu dodajemy 0,4 ml lodowatego kwasu
octowego oraz 0,2 ml nadtlenku wodoru. Mieszaninę reakcyjną mieszamy przez 24 godziny w temperaturze
pokojowej. Następnie do mieszaniny dodajemy wodnego roztworu węglanu potasu w celu zobojętnienia i
przeprowadzamy ekstrakcję chloroformem. Fazę organiczną przemywamy małą ilością wody destylowanej, osuszmy
bezwodnym siarczanem magnezu i zagęszczamy pod zmniejszonym ciśnieniem. Produkt poddajemy krystalizacji z
małej ilości eteru etylowego.

N
N
N
N
O
O
CH
3
CH
3
H
O
Cl
N
N
N
N
O
O
CH
3
CH
3
O
K
2
CO
3
/ kat.
DMF
Badanie jakości otrzymanego leku
Masa C
8
H
13
N
3
O
4
S= 247,3 g/mol.
Temperatura topnienia: od 125ºC do 128ºC.
Wygląd: prawie biały lub jasnożółty, krystaliczny proszek.
Rozpuszczalność: substancja praktycznie nierozpuszczalna w wodzie, rozpuszczalna w acetonie i chlorku metylenu,
dość trudno rozpuszczalna w metanolu.
Absorpcyjna spektrofotometria:
-zakres widma:220-350 nm (metanol),
-maksimum absorpcji: przy 310 nm.
Chromatografia cienkowarstwowa:
- faza ruchoma: butanol: octan etylu (25:75).
Chromatografia cieczowa:
- roztwór badany: rozpuścić 10 mg substancji badanej w 10 ml metanolu i uzupełnić fazą ruchomą do 100 ml.
- roztwór porównawczy (a): uzupełnić 1 ml roztworu badanego fazą ruchomą do 100 ml. Uzupełnić 1 ml tego
roztworu fazą ruchomą do 10 ml.
- roztwór porównawczy (b): rozpuścić 10 mg tynidazolu w 10 ml metanolu i uzupełnić fazą ruchomą do 100 ml.
Uzupełnić 2 ml tego roztworu fazą ruchomą do 10 ml.
- roztwór porównawczy (c): uzupełnić 1 ml roztworu porównawczego (b) fazą ruchomą do 50 ml.
Kolumna:
- wymiary: długość 0,25 m, średnica wewnętrzna 3 mm,
- faza nieruchoma: żel krzemionkowy do chromatografii z grupami oktylosilylowymi.
Kondycjonowanie kolumny wykonuje się poprzez przemywanie jej kolejno 50 ml wody, 100 ml metanolu, 25 ml
wody i 100 ml fazy ruchomej.
Faza ruchoma: acetonitryl : metanol : woda (10:20:70).
Szybkość przepływu: 0,5 ml/min
Detekcja: spektrofotometr przy 320 nm.
Ćwiczenie 4
Otrzymywanie PENTOKSYFILINY w sposób tradycyjny oraz przy użyciu reaktora mikrofalowego.
Otrzymywanie tradycyjnie: W kolbie reakcyjnej rozpuszczamy 0,45 g (2,5 mmol) teobrominy wraz z 0,09 g (10
mmol %) jodkiem tetrabutyloamoniowym, 0,69 g (5 mmol) bezwodnym K
2
CO
3
w 5 ml DMF. Następnie wkraplamy
powoli 0,32 ml (2,5 mmol) chloroheksanonu. Mieszaninę reakcyjną ogrzewamy w temperaturze 100
ο
C przez 8
N
N
S
O
O
CH
3
CH
3
O
2
N
godzin. Po ochłodzeniu mieszaniny rozpuszczalnik odparowujemy, a pozostałość ekstrahujemy chlorkiem metylenu.
Z frakcji organicznych odparowujemy rozpuszczalnik pod zmniejszonym ciśnieniem. Otrzymane związki
oczyszczamy za pomocą krystalizacji z alkoholu izopropylowego.
Otrzymywanie z użyciem reaktora mikrofalowego: W naczyniu reakcyjnym umieszczamy 0,45 g teobrominy, 0,69 g
bezwodnego K
2
CO
3
, 0,09 g jodku tetrabutyloamoniowego, 0,32 ml chloroheksanonu w 5 ml DMF. Mieszaninę
reagentów ogrzewamy w reaktorze mikrofalowym przez 20 min w przedziale temperaturze od 70 do 80
ο
C oraz mocy
reaktora w 100 W (10% maksymalnej mocy). Postęp reakcji sprawdzamy za pomocą płytek TLC. Po zakończeniu
reakcji mieszaninę przenosimy do kolby okrągłodennej i odparowujemy rozpuszczalnik pod zmniejszonym
ciśnieniem, a pozostałość ekstrahujemy chlorkiem metylenu. Z frakcji organicznej odparowujemy rozpuszczalnik na
wyparce próżniowej, a surowy produkt oczyszczamy za pomocą krystalizacji z alkoholu izopropylowego.
Badanie jakości otrzymanego leku
Masa C
13
H
18
N
4
O
3
= 278,3 g/mol.
Temperatura topnienia: od 103ºC do 107ºC.
Wygląd: biały lub prawie biały, krystaliczny proszek.
Rozpuszczalność: substancja rozpuszczalna w wodzie, łatwo rozpuszczalna w chlorku metylenu, dość trudno
rozpuszczalna w etanolu.
Chromatografia cienkowarstwowa:
- faza ruchoma: metanol: octan etylu (15:85).
Chromatografia cieczowa:
- roztwór badany: rozpuścić 50 mg substancji badanej w fazie ruchomej i uzupełnić do 25 ml.
- roztwór porównawczy (a): uzupełnić 2 ml roztworu badanego mieszaniną rozpuszczalników do 100 ml. Uzupełnić 5
ml tego roztworu mieszaniną do 100 ml.
- roztwór porównawczy (b): uzupełnić 10 ml roztworu porównawczego (a) mieszaniną rozpuszczalników do 50 ml.
- roztwór porównawczy (c): rozpuścić 5 mg kofeiny, 5 mg teobrominy w mieszaninie rozpuszczalników i uzupełnić
mieszaniną do 100 ml. Uzupełnić 1 ml roztworu mieszaniną rozpuszczalników do 25 ml.
Kolumna:
- wymiary: długość 0,25 m, średnica wewnętrzna 4,0 mm.
- faza nieruchoma: żel krzemionkowy do chromatografii z grupami oktadecylosililowymi.
Kondycjonowanie kolumny wykonuje się poprzez przemywanie jej kolejno 50 ml wody, 100 ml metanolu, 25 ml
wody i 100 ml fazy ruchomej.
Faza ruchoma: a). zmieszać 30 objętości metanolu i 70 objętości roztworu diwodorofosforanu potasu (5,44g/l);
b). zmieszać 30 objętości roztworu diwodorofosforanu potasu i 70 objętości metanolu.
Szybkość przepływu: 1 ml/min
Detekcja: spektrofotometr przy 272 nm
N
N
N
N
O
O
CH
3
CH
3
O

O
OH
O
CH
3
O
Ćwiczenie 4
Otrzymywanie kwasu acetylosalicylowego (ASPIRYNY)
W małej kolbie stożkowej umieszcza się 3 g bezwodnego kwasu salicylowego, 4,2 ml bezwodnika octowego i 2
krople stężonego kwasu siarkowego(VI). Po zamontowaniu chłodnicy zwrotnej z płaszczem wodnym, kolbę
ogrzewamy przez 15 minut w temperaturze 50-60ºC. Następnie mieszaninę pozostawia się do ostygnięcia, wstrząsając
co pewien czas, dodaje się 45 ml zimnej wody. Po wytrąceniu kryształów kwasu acetylosalicylowego odsącza się je
pod zmniejszonym ciśnieniem i pozostawia do wysuszenia.
Surowy produkt oczyszcza się poprzez krystalizację z etanolu. Gorący roztwór etanolowy wylewa się do ok. 30 ml
gorącej wody. Wytrącone kryształy kwasu acetylosalicylowego odsącza się pod zmniejszonym ciśnieniem i suszy.
Badanie jakości otrzymanego leku
Masa C
9
H
8
O
4
= 180,2 g/mol.
Temperatura topnienia: ok. 143ºC.
Wygląd: biały lub prawie biały, krystaliczny proszek lub bezbarwne kryształy.
Rozpuszczalność: substancja trudno rozpuszczalna w wodzie, łatwo rozpuszczalna w etanolu.
Chromatografia cieczowa:
- roztwór badany: rozpuścić 0,10 g substancji badanej w acetonitrylu i uzupełnić nim do 10 ml.
- roztwór porównawczy (a): rozpuścić 50 mg kwasu salicylowego w fazie ruchomej i uzupełnić nią do 50 ml.
Uzupełnić 1 ml tego roztworu fazą ruchomą do 100 ml.
- roztwór porównawczy (b): rozpuścić 10 mg kwasu salicylowego w fazie ruchomej i uzupełnić fazą ruchomą do 10
ml. Do 1 ml tego roztworu dodać 0,2 ml roztworu badanego i uzupełnić fazą ruchomą do 100 ml.
Kolumna:
- wymiary: długość 0,25 m, średnica wewnętrzna 4,6 mm,
- faza nieruchoma: żel krzemionkowy do chromatografii z grupami oktadecylosililowymi.
Kondycjonowanie kolumny wykonuje się poprzez przemywanie jej kolejno 50 ml wody, 100 ml metanolu, 25 ml
wody i 100 ml fazy ruchomej.
Faza ruchoma: kwas fosforowy : acetonitryl : woda (2:400:600).
Szybkość przepływu: 1 ml/min
Detekcja: spektrofotometr przy 237 nm.
Ćwiczenie 5
Kontrolowane uwalnianie substancji z hydrożelu
1.
Ć
wiczenie rozpoczyna się od przygotowania 31 ml 5%, 10%, 15% roztworu żelatyny z badanym barwnikiem
spożywczym (tartrazyną lub indygo). Należy umieścić 31 ml roztworu barwnika w zlewce i podgrzać do
wrzenia a następnie dodać żelatynę i mieszać aż do jej rozpuszczenia. Następnie gorący roztwór żelatyny
wylewamy na szalkę Petriego i pozostawiamy do żelowania przez noc w zamrażarce.
2.
Przygotowujemy 100 ml roztworów papainy o stężeniu: 1mg/ml, 5mg/ml.
3.
Podstawowy roztwór barwnika przygotowujemy rozpuszczając 25 mg w 500 ml wody.

4.
Do 5 próbek o pojemności 10 ml odmierzamy pipetą 0.2, 0.4, 0.8, 1.2, 1.6 i 2.0 ml roztworu podstawowego
indygo lub tartazyny i uzupełnić wodą destylowaną do 10 ml. Obliczyć stężenia przygotowanych roztworów.
5.
Gotowe próbki barwników wlewamy do kuwet i mierzymy absorbancję A roztworów wzorcowych barwnika
dla odpowiedniej długości fali. Sporządzamy krzywą kalibracyjną.
6.
Do 3 kuwet do spektrofotometru UV nalewamy 4 ml wody destylowanej, do 3 następnych po 4 ml roztworu
papainy o stężeniu 1 mg/ml, a do ostatnich roztworu papainy o stężeniu 5 mg/ml.
7.
Z hydrożeli wycinamy po 3 sześciany 5x5x5 mm dla każdego stężenia żelatyny. Umieszczamy je w kuwetach
z wodą po sześcianie żelatyny o określonym stężeniu. To samo wykonujemy w roztworach z papainą.
8.
Kuwety przykrywamy i wykonujemy pomiary absorbancji przy wybranej długości fali po czasie 10 min, 30
min, 1h, 1,5h i 2h. Zależności zmiany absorbancji w czasie umieszczamy w tabeli i sporządzamy w formie
wykresu. Na podstawie pomiarów wyznaczyć stężenia barwników w różnym czasie dla wybranej próbki.
Skrócona instrukcja obsługi reaktora mikrofalowego PLAZMATRONIKA
Po umieszczeniu naczynia reakcyjnego wraz z chłodnicą w reaktorze, należy:
1.
Włączyć napięcie zasilające (czerwony włącznik z prawej strony)
2.
Uruchomić mieszadło oraz ustawić pożądaną szybkość mieszania
3.
Wcisnąć przycisk TIME – wpisać 1 i wcisnąć ENTER
4.
Wprowadzić zadany czas grzania (HEAT) i wcisnąć ENTER
5.
Wprowadzić czas chłodzenia (COOL) i wcisnąć ENTER
6.
Wprowadzić zadaną moc (%P) i wcisnąć ENTER
7.
Wprowadzić minimalną temperaturę grzania (TEMP MIN), wcisnąć ENTER
8.
Wprowadzić maksymalną temperaturę grzania (TEMP MAX), wcisnąć ENTER
9.
Wprowadzić zadany czas oczekiwania (SET WAIT TIME), wcisną ENTER
10.
Jeśli parametry zostały wprowadzone poprawnie wcisnąć przycisk START STOP
Literatura:
1.
A. Zejc, M. Gorczyca „Chemia Leków”, PZWL, Warszawa 1999.
2.
G. Patrick „Chemia Leków – krótkie wykłady”, PWN, Warszawa 2004.
3.
R. B. Silverman „Chemia organiczna w projektowaniu leków”, WNT, 2004.
4.
T. Tkaczyński, D. Tkaczyńska „Synteza i technologia chemiczna leków”, PZWL, Warszawa 1984.
5.
R. Bogoczek, E. Kociołek-Balawejder „Technologia chemiczna: surowce i półprodukty”, Akademia
Ekonomiczna, 1992.
6.
A. I. Vogel „Preparatyka Organiczna”, Wydawnictwo Naukowo-Techniczne, Warszawa 1984.
7.
Ustawa Prawo Farmaceutyczne (Dz. U. z dnia 31 października 2001 roku).
W sprawozdaniu musi być zawarty:
1.
Przebieg wykonywanego ćwiczenia, wydajność produktu i poszczególnych etapów syntezy. Określenie czystości
leku i ewentualnych produktów ubocznych.
2.
Krótki opis zsyntetyzowanego leku, jego właściwości farmakologicznych i zastosowanie.
3.
W ćwiczeniu 3 dodatkowo porównujemy wpływ odpowiednich katalizatorów na syntezę Tinidazolu.

4.
W przypadku wykonywania ćwiczenia 4 należy porównać metody otrzymywania Pentoksyfiliny w sposób
tradycyjny i przy zastosowaniu reaktora mikrofalowego. Zalety i wady tych metod. Przykładowe zastosowanie
reaktora mikrofalowego w przemyśle (3 przykłady).
5.
Poniżej zamieszczony jest wzór pierwszej strony sprawozdania. Sprawozdanie wysyłamy elektronicznie do
prowadzącego ćwiczenie laboratoryjne przed wykonywanym drugim ćwiczeniem.
POLITECHNIKA GDAŃSKA
WYDZIAŁ CHEMICZNY
KATEDRA TECHNOLOGII CHEMICZNEJ
SPRAWOZDANIE Z ĆWICZEŃ LABORATORYJNYCH
PRODUKCJI LEKÓW
PROWADZĄCY:
NAZWISKA OSÓB WYKONUJĄCYCH ĆWICZENIE:
1.
2.
3.
4.
KIERUNEK STUDIÓW:
GRUPA:
DATA WYKONANIA ĆWICZENIA:
DATA ODDANIA SPRAWOZDANIA:
GDAŃSK 2012
Wyszukiwarka
Podobne podstrony:
higiena produkcji 12 id 201601 Nieznany
PRODUKTY UOR id 394132 Nieznany
drDubas produkcja biomasy id 14 Nieznany
Produkcja wewnatrzzakl id 39404 Nieznany
Produkcja biomasy id 394025 Nieznany
Analiza Lekow id 60756 Nieznany
Notatki 03 PRODUKT id 322319 Nieznany
higiena produkcji 8 id 201603 Nieznany
4 Ksztaltowanie produktu id 377 Nieznany
KUPUJ TE PRODUKTY id 253983 Nieznany
Bezpieczenstwo produktu id 8355 Nieznany
Bezpieczenstwo produktu 2 id 83 Nieznany (2)
cykle produkcyjne id 126546 Nieznany
higiena produkcji 1 id 201600 Nieznany
II Sytem produkcyjny id 210031 Nieznany
bezpieczenstwo produktow id 320 Nieznany (2)
Notatki 03 PRODUKT id 322319 Nieznany
Abolicja podatkowa id 50334 Nieznany (2)
więcej podobnych podstron