R E A K T Y W N O Ś Ć A R E N Ó W
Aleksander Kołodziejczyk listopad 2006
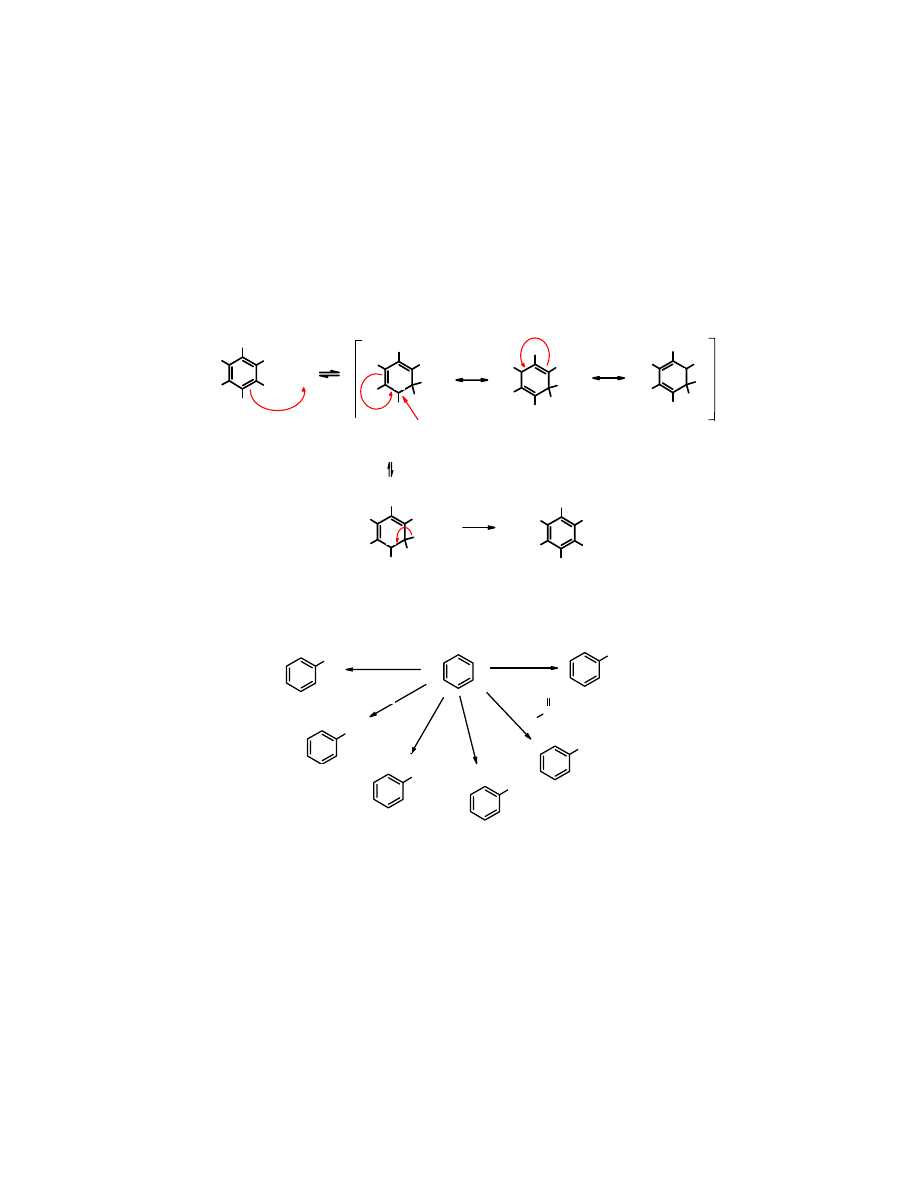
1. Substytucja elektrofilowa S
E
Charakterystyczną reakcją związków aromatycznych jest
substytucja elektrofilowa
(S
E
), zwana
również
reakcją aromatycznej substytucji elektrofilowej
. Polega ona najczęściej na tym, że
elektrofil (
E
+
) podstawia proton związany z pierścieniem aromatycznym.
Elektrofil może
podstawić także inny atom lub grupę związaną z pierścieniem aromatycznym.
W pierwszym etapie dochodzi do
addycji
elektrofila
E
+
do pierścienia aromatycznego, w wyniku
czego powstaje stabilizowany mezomerycznie karbokation, po czym po odszczepieniu protonu
odtwarza się układ aromatyczny i powstaje pochodna zawierająca elektrofil w miejscu protonu.
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
+
+
elektrofil E
+
przyłącza się
do atomu C o hybrydyzacji sp
2
tworzy się stabilizowany mezomerycznie karbokation, który
w wyniku odszczepienia protonu ulega dalszej stabilizacji
E
E
E
E
E
E
+
+
+
+
+
H
+
W ten sposób zachodzą reakcje typu
halogenowania
,
alkilowania
,
acylowania
,
nitrowania
,
sulfonowania
,
diazowania
i inne.
X
NO
2
SO
3
H
R
COR
NO
R
O
C+
X
+
+
NO
2
SO
3
R
+
+
NO
halogenowanie
nitrowanie
sulfonowanie
alkilowanie
acylowanie
nitrozowanie
Rys. 10.1 Przykłady głównych reakcji aromatycznej substytucji elektrofilowej
1.1 Halogenowanie
W arenach, pod wpływem halogenów (Cl
2
i Br
2
), w obecności kwasów Lewisa (np. FeX
3
)
dochodzi do wymiany atomu wodoru na atom halogenu. Pomimo dużego stopnia nienasycenia
pierścienia benzenowego (3 C=C) dochodzi do reakcji
substytucji
, a nie
addycji!
Obie te
reakcje łatwo rozróżnić po zewnętrznych efektach – w obu zanika barwa bromu, jednak w trakcie
substytucji wydziela się gazowy bromowodór
(jest to widoczną oznaką reakcji), a
w
addycji
cały brom zostaje przyłączony do wielokrotnego wiązania
.
1
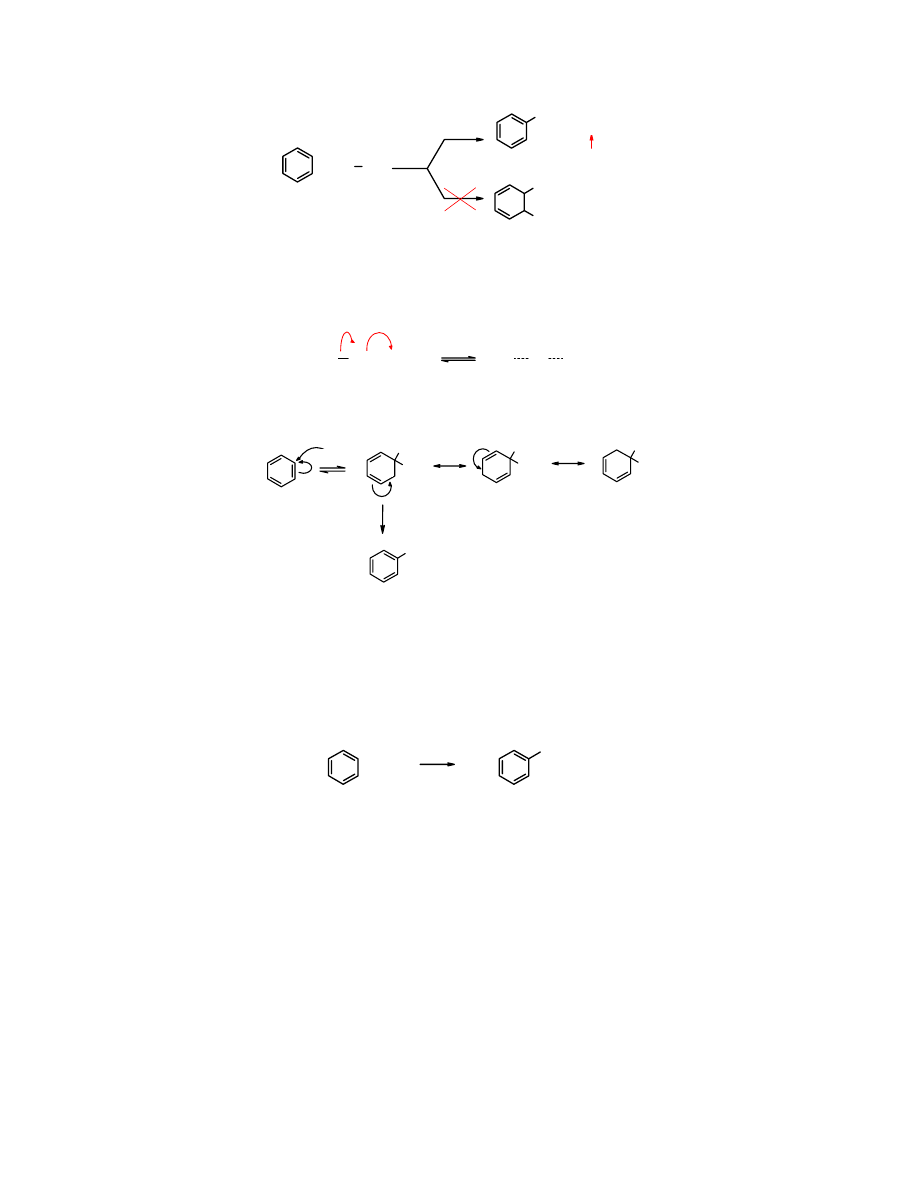
Br
Br Br
Br
Br
1,2-dibromo-
cykloheksa-
3,5-dien
+
+ HBr
FeBr
3
benzen
bromobenzen
gaz
Reakcja biegnie wg mechanizmu S
E
. Cząsteczka bromu w kontakcie z kwasem Lewisa ulega
polaryzacji i dodatnio naładowany atom bromu staje się silnym elektrofilem. Br
+
przyłącza się do
pierścienia
benzenu
i powstaje mezomerycznie stabilizowany karbokation, który odszczepiając
proton przekształca się w
bromobenzen
(
bromek fenylu
).
Br
Br
δ
+
Br
+ FeBr
3
..
..
..
..
:
:
Br
FeBr
3
..
..
..
..
:
δ
-
brom - słaby elektrofil brom spolaryzowany, z ładunkiem dodatnim
staje się silnym elektrofilem
H
Br
H
Br
H
Br
Br
Br
+
+
+
+
benzen
bromobenzen
(72%)
- H
+
Duża energia sprzężenia 6 elektronów
π (energia rezonansu) jest przyczyną powrotu produktu
addycji kationu bromkowego do układu aromatycznego poprzez odszczepienie protonu.
Podobnie jak
bromowanie
biegnie reakcja
chlorowania
arenów.
Jodowanie
w takich samych
warunkach jest reakcją odwracalną i do przesunięcia równowagi na prawo potrzebny jest
utleniacz utleniający wydzielający się jodowodór. Utleniaczami stosowanymi do tego celu są
nadtlenek wodoru, kwas azotowy (V) lub sole miedzi (II).
I
+ I
2
2
HNO
3
2
+ HOH
benzen
jodobenzen
(87%)
1.2 Nitrowanie
Grupę nitrową do pierścienia aromatycznego wprowadza się najczęściej za pomocą mieszaniny
nitrującej, czyli kwasu azotowego i siarkowego o różnych stężeniach, zależnych od reaktywności
arenu. W mieszaninie tych kwasów wytwarza się
kation nitroniowy
+
NO
2
, który jako silny
elektrofil wchodzi na miejsce jednego z aromatycznych atomów wodoru.
2
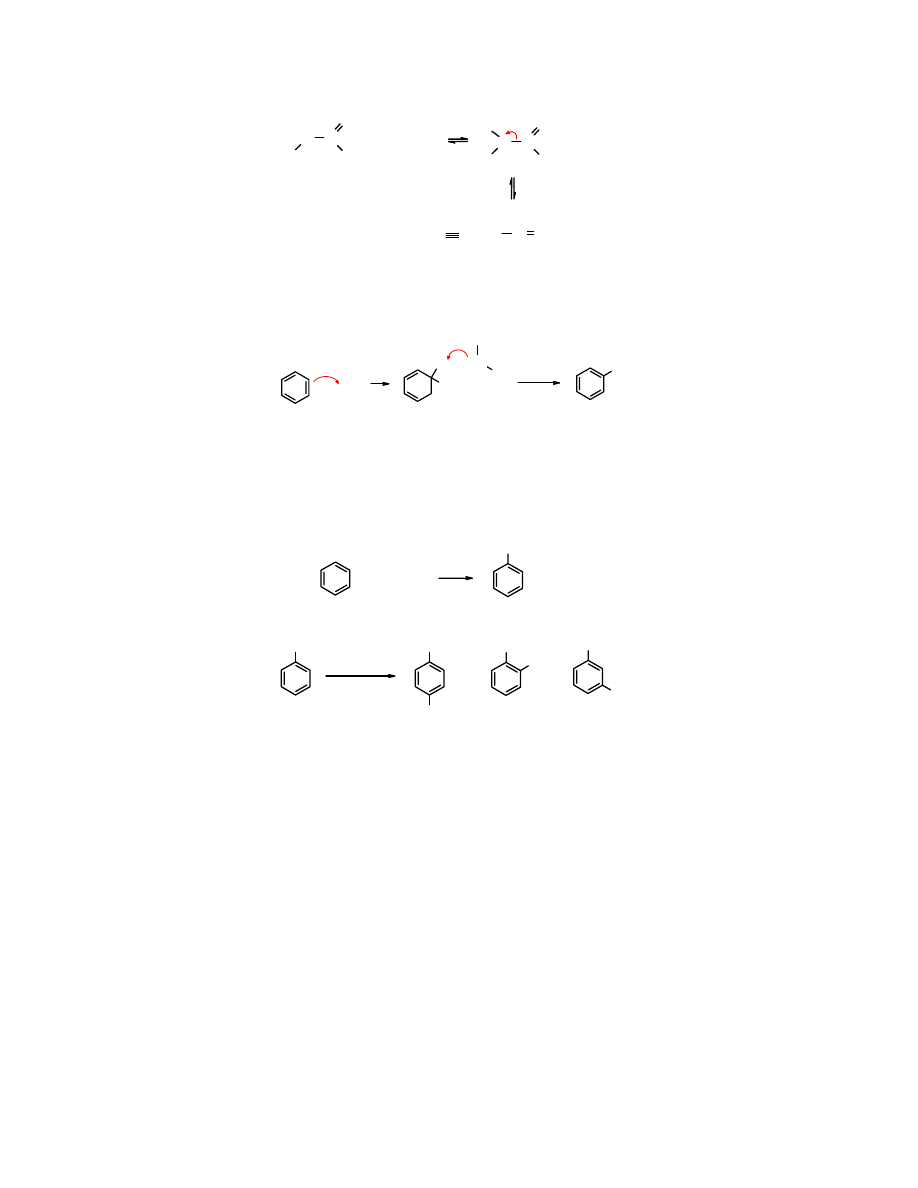
O N
H
O
O
O N
H
O
O
H
O
O
..
:
-
:
..
..
..
..
..
+
+ H
2
SO
4
:
-
:
..
..
..
..
+
+
+ HSO
4
-
N
2
+
-
:
:
..
..
+ HOH
NO
2
+
kation nitroniowy
Kwas siarkowy pełni rolę kwasu (dawcy protonu), czynnika wiążącego wodę oraz ułatwia
rozpuszczanie arenów. Dwie pierwsze funkcje kwasu siarkowego sprzyjają powstawaniu kationu
nitroniowego, który jako elektrofil reaguje z arenem tworząc addukt, po czym następuje
stabilizacja przez odszczepienie protonu. Jest to typowy mechanizm
reakcji S
E
.
H
NO
2
H
H
NO
2
+ NO
2
+
+
O
..
:
-
H
+
/HOH
benzen nitrobenzen
(85%)
Nitrowanie arenów jest reakcją wysoko egzotermiczną.
Zadanie: napisz mechanizm
nitrowania
benzenu
z uwzględnieniem wszystkich stanów mezomerycznych adduktu
po przyłączniu kationu nitroniowego.
Przykłady:
NO
2
+ HNO
3
H
2
SO
4
+ HOH
benzen
nitrobenzen
(85%)
CH
3
CH
3
NO
2
CH
3
NO
2
CH
3
NO
2
HNO
3
/H
2
SO
4
40
o
C
+
+
toluen
60%
36%
4%
p-nitrotoluen o-nitrotoluen m-nitrotoluen
W 94% kwasie siarkowym stężony kwas azotowy jest prawie całkowicie zdysocjowany na jony
+
NO
2
. W roztworze wodnym HNO
3
i w rozcieńczonym kwasie siarkowym przeważają
utleniające jony NO
3
-
. W samym bezwodnym kwasie azotowym stężenie jonów
+
NO
2
dochodzi
do 1%.
Kation nitroniowy nie jest wymyślonym tworem. Tworzy on trwałe sole, np. znany jest
nadchloran nitroniowy – NO
2
ClO
4
czy fluoroboran – NO
2
BF
4
. Te sole też mają właściwości
nitrujące. Areny wrażliwe na działanie kwasów
nitruje
się kwasem azotowym wobec P
2
O
5
,
kwasem azotowym w mieszaninie z
kwasem octowym
lub
bezwodnikiem octowym
.
Aromatyczne nitrozwiązki mają duże znacznie praktyczne, dlatego też
nitrowaniu
na
przemysłową skalę poddawane jest wiele arenów, w tym
benzen
,
toluen
,
fenol
,
chlorobenzen
,
anilina
,
naftalen
,
antrachinon
i inne. Otrzymuje się z nich mono-, di- i polinitrozwiązki. Sam
nitrobenzen
jest cennym, wysokowrzącym, nie mieszającym się z wodą rozpuszczalnikiem (tw.
210
o
C), służy też do produkcji
aniliny
i izocyjanianów. Nitrozwiązki stanowią ważne substraty w
produkcji amin, barwników, leków, substancji zapachowych, materiałów wybuchowych (
trotyl
,
3
ksylit
,
heksanitrobenzen
,
kwas pikrynowy
,
tetryl
czy
heksyl
), tworzyw syntetycznych i wielu
innych produktów.
1.3 Sulfonowanie
Utworzenie wiązania pomiędzy atomem węgla związku organicznego a atomem siarki grupy
sulfonowej – -SO
3
H lub jej odpowiednikiem chlorosulfonowym – -SO
2
Cl nazywa się
sulfonowaniem
.
Sulfonowanie
arenów biegnie wg mechanizmu S
E
. Prowadzi się je za pomocą
SO
3
, jego kompleksów, np. z
pirydyną
oraz stężonym kwasem siarkowym lub dymiącym
kwasem siarkowym (oleum). Podczas
sulfonowania
kwasem siarkowym powstają duże ilości,
trudnych do zagospodarowania kwaśnych odpadów.
Sulfonowanie
tritlenkiem siarki jest w
zasadzie bezodpadowe. Trójtlenek siarki jest jednak substancją stałą, co utrudnia prowadzenie
reakcji i łatwo dochodzi do zestalenia całej mieszaniny reagującej. Często stosuje się procedurę
mieszaną –
sulfonowanie
rozpoczyna się w niewielkiej ilości kwasu siarkowego i w miarę
postępu reakcji dodaje się SO
3
. Ten sposób postępowania zapewnia nie tylko utrzymywanie
stałego stężenia środka sulfonującego, ale i wiązanie wody powstającej w reakcji; ponadto kwas
siarkowy dodatkowo pełni rolę rozpuszczalnika. Wiązanie lub usuwanie wody ze środowiska
reakcji jest koniecznie, gdyż reakcja
sulfonowania
jest odwracalna i tylko odpowiednie stężenie
kwasu (zależne od aktywności arenu) zapewnia przesunięcie równowagi na korzyść produktów.
Wodę powstającą w reakcji
sulfonowania
można usuwać azeotropowo. Tak często postępuje się
podczas sulfonowania
benzenu
lub
toluenu
.
Czynnikiem sulfonującym jest SO
3
lub tworzący się z kwasu siarkowego i SO
3
kation
+
SO
3
H.
H
2
SO
4
+ SO
3
→ HSO
4
-
+
+
SO
3
H
Mechanizm reakcji z SO
3
:
O
O
O
H
SO
3
H
SO
3
H
SO
3
SO
3
S
O
OH
O
+
S
-
+
-
+
-
+
:B
-
H
+
-
H
+
/HOH
kwas
benzenosulfonowy
benzen
Mechanizm reakcji z kationem
+
SO
3
H:
OH
O
O
H
SO
3
H
SO
3
H
+
+
S
+
:B
benzen kwas benzenosulfonowy
Zadanie: przedstaw mechanizm sulfonowania
benzenu
, w którym udział bierze kation
+
SO
3
H, uwzględniając
wszystkie stany mezomeryczne adduktu po przyłączeniu się kationu sulfoniowego
4
Areny zawierające grupy sulfonowe są zwykle rozpuszczalne w wodzie, wykazują także
powinowactwo do wielu materiałów (szczególnie wełny), stąd często barwniki wełny zawierają
grypy sulfonowe. Sole kwasów alkiloarenosulfonowych wykorzystywane były jako detergenty,
obecnie ich stosowanie ograniczono, bądź zakazano z uwagi na to, że są to związki trudno
biodegradowalne.
Aromatyczne kwasy sulfonowe znalazły zastosowanie w syntezie związków organicznych,
ponieważ grupę sulfonową wprowadzać można czasowo do cząsteczki arenu, tak żeby podczas
kolejnej operacji następny podstawnik zajął oczekiwaną pozycję, po czym funkcję -SO
3
H usuwa
się lub wymienia na inną grupę.
SO
3
H
H
2
SO
4
/98%
HOH, 150
o
C
benzen
kwas benzenosulfonowy
+ HOH
(80%)
Kwas benzenosulfonowy
jest dobrze rozpuszczalny w wodzie i w uwodnionym kwasie
siarkowym, dlatego w celu jego wyodrębnienia z mieszaniny reakcyjnej przeprowadza się go w
trudniej rozpuszczalną sól wapniową (proces wapniowania).
Łatwiejszy do wyodrębniania jest
kwas p-toluenosulfonowy
, bowiem krystalizuje on w postaci
monohydratu.
CH
3
CH
3
SO
3
H
H
2
SO
4
110
o
C
+ HOH
toluen
kwas p-tolueno-
sulfonowy
(83%)
Alklilobenzeny zawierające 10-14 węglowy łańcuch alifatyczny po
sulfonowaniu
i
zalkalizowaniu
służyły jako środki powierzchniowo czynne. Do ich otrzymywania stosowano
SO
3
. Obecnie tego typu surfaktanty zastępowane są przez lepiej biodegradowalne sole kwasów
alkanosulfonowych.
SO
3
H
C
n
H
2n+1
+ SO
3
40
o
C
C
n
H
2n+1
alkilobenzeny
kwasy alkilo-
arenosulfonowe
Sulfonowanie
kopolimeru styrenu sieciowanego diwinylobenzenem prowadzi do
nierozpuszczalnego w wodzie kwasu polisulfonowego. Służy on wyrobu żywic jonowymiennych
– silnych kationitów.
5
SO
3
H
SO
3
H
SO
3
H
SO
3
H
SO
3
H
-CH
2
-CH-CH
2
-CH-CH
2
-CH-CH
2
CH-CH
2
CH-
H
2
SO
4
-CH
2
-CH-CH
2
-CH-CH
2
-CH-CH
2
CH-CH
2
CH-
polistyren
Chlorosulfonowanie
przeprowadza się za pomocą kwasu chlorosulfonowego. Reakcja biegnie
dwuetapowo. W pierwszym etapie powstaje kwas arenosulfonowy i wydziela się chlorowodór.
Dopiero w drugim etapie kwas arenosulfonowy pod wpływem kwasu chlorosulfonowego zostaje
przekształcony w chlorek arenosulfonylowy. Pierwszy etap reakcji jest nieodwracalny, ponieważ
gazowy chlorowodór opuszcza środowisko reakcji. Drugi odwracalny etap wymaga nadmiaru
kwasu chlorosulfonowego, żeby przesunąć równowagę reakcji w stronę produktu. Trzykrotny
nadmiar kwasu chlorosulfonowego zapewnia dobrą wydajność. Konieczność stosowania
nadmiaru kwasu chlorosulfonowego nie tylko podnosi koszt reakcji, ale i zwiększa ilość
kłopotliwych kwaśnych ścieków.
Ar-H + ClSO
3
H
Ar-SO
3
H + HCl
Ar-SO
3
H + ClSO
3
H
Ar-SO
2
Cl + H
2
SO
4
SO
2
Cl
ClSO
3
H
benzen
chlorek
benzenosulfonylu
(76%)
Chlorosulfonowanie
jest wykorzystywane do produkcji sulfonoamidów – ważnych leków, a
także w produkcji
sacharyny
, znanego słodzika.
Surowcem w produkcji
sacharyny
jest
toluen
, z którego w reakcji
chlorosulfonowania
w niskiej
temperaturze powstają dwa izomery
chlorku toluenosulfonylu
, z przewagą izomeru orto-. Izomer
para- usuwa się przez krystalizację.
CH
3
CH
3
SO
2
Cl
CH
3
SO
2
Cl
ClSO
3
H
0
o
C
+
toluen
chlorek o-tolueno-
sulfonylu
chlorek p-tolueno-
sulfonylu
(58%)
(14%)
Następnie
chlorek o-toluenosulfonylu
przeprowadza się w
amid
w reakcji z węglanem amonu. Z
amidu
po utlenieniu go do
soli
kwasu o-sulfamoilobennzoesowego
i zakwaszeniu powstaje
sacharyna
.
6
CH
3
SO
2
Cl
CH
3
SO
2
NH
2
SO
2
NH
2
COONa
S
NH
O
O
O
(NH
4
)
2
CO
3
100
o
C
1.
KMnO
4
2.
NaOH
HCl
chlorek o-toluenosulfonylu o-toluenosulfamid sacharyna sól kwasu o-sulfamoilo-
benzoesowego
1.4 Reakcje Friedela-Craftsa
Tworzenie nowego wiązania C-C pomiędzy pierścieniem aromatycznym, a inną resztą
organiczną, tj. katalizowane chlorkiem glinu reakcje
alkilowania
i
acylowania
arenów,
nazywane są
reakcjami Friedela-Crafstsa
(F-C).
Charles Friedel (1832-1899); ur. w Strasburgu, Francja; studia na Sorbonie, prof. w École des Mines i w Paryżu.
James M. Crafts (1876-1917); ur. w Bostonie, USA; doktorat z prawa w Harvardzie (1898); prof. w Cornell
University i w Massachusetts Institute of Technology.
Elektrofilem w reakcji
alkilowania
F-C jest karbokation Alk
+
powstający w reakcji halogenków
alkilowych z AlCl
3
, lub z protonowanych alkenów albo też z alkoholi uaktywnionych BF
3
.
Reakcja
sulfonowania
zaczyna się od wytworzenia karbokationu.
CH
3
CH
2
-Cl + AlCl
3
CH
3
CH
2
....
AlCl
4
+
-
chlorek etylu
karbokation
Karbokation tworzy z arenem stabilizowany mezomerycznie kompleks, który powraca do
układu aromatycznego przez odszczepienie protonu pod wpływem zasady (w tym przypadku
anionu halogenkowego -Cl
-
).
H
CH
2
CH
3
CH
2
CH
3
H
CH
2
CH
3
H
CH
2
CH
3
CH
3
CH
2
+
AlCl
4
-
+
- AlCl
3
Cl
-
- HCl
+
+
benzen etylobenzen
Przykłady:
CH
3
CH=CH
2
+ HF
CH
3
CH-CH
3
F
+
-
CH(CH
3
)
2
+ CH
3
CH=CH
2
HF
benzen
(izopropylobenzen)
kumen
(84%)
+
0
o
C
HF
benzen
cykloheksen cykloheksylobenzen
(62%)
O
H
+
60
o
C
BF
3
benzen
cykloheksanol cykloheksylobenzen
(56%)
7
Problemy
alkilowania
reakcji F-C
1.4.1 Polialkilowanie
Wprowadzenie reszty alkilowej do pierścienia aromatycznego zwiększa jego podatność na
reakcje S
E
do tego stopnia, że di- i trialkiloareny tworzą się łatwiej niż monoalkiloareny, tak
więc powstający produkt jest mieszaniną mono-, di-, tri-, a nawet polialkilowych pochodnych.
CH
3
C
H
3
C
H
3
C
H
3
C
H
3
CH
3
+ CH
3
Cl
AlCl
3
+
+
+ ....
benzen
Jeżeli celem reakcji jest związek monopodstawiony należy użyć nadmiaru alkilowanego arenu,
w tym przypadku
benzenu
. Takie postępowanie zapewnia wysoką wydajność i zadawalającą
czystość oczekiwanego produktu.
CH
3
+ CH
3
Cl
AlCl
3
15
+
benzen
toluen
benzen
(85%)
Odzyskanie nadmiaru
benzenu
lub zawrócenie go do reakcji pozwala na lepsze wykorzystanie
surowca.
1.4.2. Przegupowanie karbokationu
W reakcjach z udziałem karbokationu dochodzi do jego przegrupowania, np. podczas alkilowania
benzenu chlorkiem n-butylu
otrzymuje się mieszaninę obu butylobenzenów.
CH
2
CH
2
CH
2
CH
3
CH
3
CH
2
CH
2
CH
2
Cl
AlCl
3
CH
3
CHCH
2
CH
3
+
(27%)
(52%)
benzen n-butylobenzen s-butylobenzen
W reakcji
chlorku n-butylu
z chlorkiem glinu tworzy się karbokation 1
o
, który przegrupowuje się
do trwalszego 2
o
karbokationu. Oba karbokationy biorą udział w
alkilowaniu
benzenu
.
H
-
CH
3
CH
2
CH
2
CH
2
-Cl + AlCl
3
CH
3
CH
2
CHCH
2
...
AlCl
4
CH
3
CH
2
CHCH
3
...
AlCl
4
+
+
-
karbokation
1
o
2
o
Najtrwalsze są karbokationy 3
o
, dlatego też w reakcji
alkilowania
benzenu 1-chloro-2,2-
dimetylopropanem
tworzy się jeden związek, którym jest
(1,1-dimetyloprobylo)benzen
; produkt
powstały w reakcji przegrupowania karbokationu 1
o
.
CH
3
CH
3
CCH
2
CH
3
C
H
3
CH
3
+ CH
3
CCH
2
Cl
AlCl
3
benzen
(1,1-dimetylo-
propylo)benzen
8
Problem, jaki podczas alkilowania arenów metodą F-C stwarza niepożądane
przegrupowanie
karbokationów można rozwiązać pośrednio zastępując
alkilowanie
kolejno reakcjami
acylowania
i
redukcji
otrzymanego ketonu alkilowo-arylowego do n-alkiloarenów.
CH
2
CH
2
CH
3
CCH
2
CH
3
O
CH
2
CH
2
CH
3
CH
3
CH
2
CH
2
Cl
AlCl
3
AlCl
3
CH
3
CH
2
COCl
CH
3
CHCH
3
+
n-propylobenzen izopropylobenzen
H
2
NNH
2
/-OH
200
o
C
benzen
1.4.3 Arenów nie można winylować ani arylować. Halogenki winylowe i arylowe są
niereaktywne w reakcja F-C.
Reakcje acylowania F-C
Acylowanie
arenów metodą Friedela-Craftsa polega na reakcji arenów z odczynnikami
acylującymi (halogenkami kwasowymi lub bezwodnikami) w obecności AlCl
3
. Elektrofilem w
tych reakcjach są kationy acyliowe tworzące się z odczynnika acylującego i AlCl
3
.
O
C
H
3
Cl
C
H
3
O
C
H
3
O
O
CH
3
H
O
CH
3
CH
3
O
C
AlCl
3
..
..
:
C
+
C
+
..
..
..
+ AlCl
4
+
C
+
C
Cl
..
..
:
- HCl
chlorek acetylu
kation acyliowy
-
-
benzen
acetofenon
(95%)
Podobnie biegną reakcje z użyciem
bezwodnika octowego
.
CCH
3
O
CH
3
COOCOCH
3
AlCl
3
naftalen
2-acetylonaftalen
(79%)
O
O
O
OH
O
O
+
C
AlCl
3
C
C
+
C
benzen bezwodnik benzoesowy benzofenon
(85%)
kwas benzoesowy
Problemy reakcji acylowania F-C
Wprowadzenie reszty acylowej do pierścienia aromatycznego zmniejsza podatność produktu na
podstawienie elektrofilowe (S
E
), wobec czego w trakcie reakcji
acylowania
F-C nie dochodzi do
diacylowania, co było obserwowane podczas
alkilowania
F-C.
Reakcjom
acylowania
nie ulegają areny zawierające podstawniki wyciągające elektrony
(EWG), ponieważ one dezaktywują pierścień na reakcje S
E
. Areny zawierające przy pierścieniu
9
aromatycznym grupę nitrową lub amoniową
nie ulegają acylowaniu
.
Acylowanie
acyloarenów
jest bardzo utrudnione.
NO
2
N(R)
3
+
te areny nie ulegają reakcjom
S
E
, są silnie zdezaaktywowane
R: H, alkil lub aryl
Również aminy aromatyczne są nieaktywne w reakcjach F-C, ponieważ dezaktywuje je sam
„katalizator” tej reakcji.
AlCl
3
:
AlCl
3
H
2
N
H
2
N
+
-
1.5. Efekt podstawnikowy w reakcjach S
E
Reaktywność podstawionych arenów reakcjach S
E
zależy od właściwości podstawnika
(podstawników) związanego z pierścieniem aromatycznym. Może on ułatwiać reakcję S
E
(zwiększać szybkość reakcji poprzez uaktywnienie pierścienia aromatycznego) lub ją utrudniać
(obniżać szybkość reakcji poprzez dezaktywację układu aromatycznego). Dodatkowo podstawnik
obecny w pierścieniu wpływa na miejsce przyłączenia grupy biorącej udział w S
E
.
Podstawniki związane z pierścieniem aromatycznym z uwagi na wpływ, jaki wywierają na
reakcje S
E
dzielą się na trzy grupy:
I
:
aktywujące
; kierują one nowy podstawnik w położenie orto- lub para-. Należą do nich grupy
elektronodonorowe (EDG), a więc -OH,
-OR, -NH
2
, -NHR, NR
2
, -alkil.
II
:
dezaktywujące, które kierują nowy podstawnik w położenie meta-
Należą do nich grupy elektroakceptowowe wykazujące równocześnie efekt -M i -I. Należą do
nich grupa -NO
2
, -COOH, -COOR, -CONR
2
, -CN, -COR, -COH i
+
-NR
3
(R: H, alkil lub
aryl).
III
:
dezaktywujące, które kierują nowy podstawnik w położenie orto- lub para-
. Należą do
nich grupy wykazujące efekt +M i –I; są to halogeny.
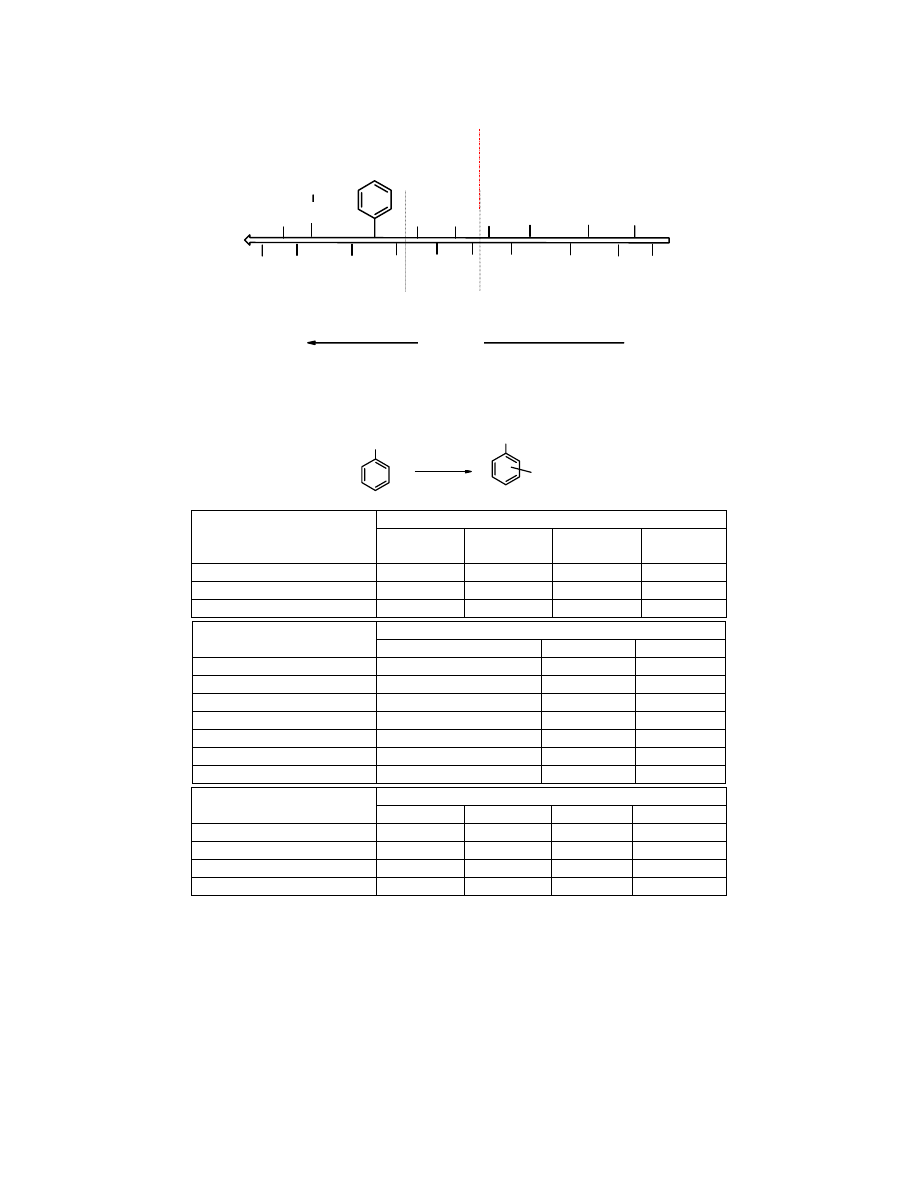
Jak silne jest takie oddziaływania świadczy fakt, że
benzen
jest 1000 razy mniej podatny na
reakcję
nitrowania
niż
fenol
(wpływ grupy -OH), a z kolei 2
.
10
7
bardziej aktywny na
nitrowanie
niż
nitrobenzen
.
10
NR
3
CN
COR
COOH
COOR
SO
3
H
CHO
I
Br
Cl
F
H
OR
OH
NR
2
alkil
COR
NO
2
+
NH
grupa I
grupa III
grupa II
reaktyność
efekt kierujący w położenia
orto- i para-
meta-
Tabela 10.1
Efekt oddziaływania poszczególnych podstawników można zilustrować na
przykładzie
nitrowania
podstawionego
benzenu
.
Y
Y
NO
2
HNO
3
/H
2
SO
4
25
o
C
produkt – izomery [%]
Y - podstawnik
grupy I
orto-
meta-
para- Razem
o- i
p-
-OH 50
0
50
100
-NHCOCH
3
19
2
79
98
-CH
3
63
3
34
97
produkt – izomery [%]
Y - podstawnik
grupy II
orto-
meta-
para-
-NO
2
7
91
2
+
-N(CH
3
)
3
2
87
11
-CN
17
81
2
-COOH
22
76
2
-COCH
3
26
72
2
-CHO
19
72
9
-COOEt
28
66
6
produkt – izomery [%]
Y - podstawnik
grupy III
orto-
meta-
para- Razem
o- i p-
-F
13
1
86
99
-Cl
35
1
64
99
-Br
43
1
56
99
-I
45
1
54
99
Jak widać z powyższej tabeli efekt skierowujący niektórych podstawników w określone pozycje
jest bardzo wyraźny, wynosi 99 a nawet 100% dla grupy -OH i halogenów), natomiast dla
innych podstawników jest mniej zdecydowany, np. 66% dla estru etylowego. Efekt skierowujący
wynika z rozkładu ładunków i stabilności adduktu, jaki powstaje po przyłączeniu elektrofila.
Addukt taki jest stabilizowany mezomerycznie i im więcej uprzywilejowanych struktur
granicznych można wyszczególnić po przyłączeniu elektrofila w odpowiednią pozycję, tym
bardziej prawdopodobne jest utworzenie takiej struktury, a co za tym idzie powstanie
odpowiedniego produktu.
11
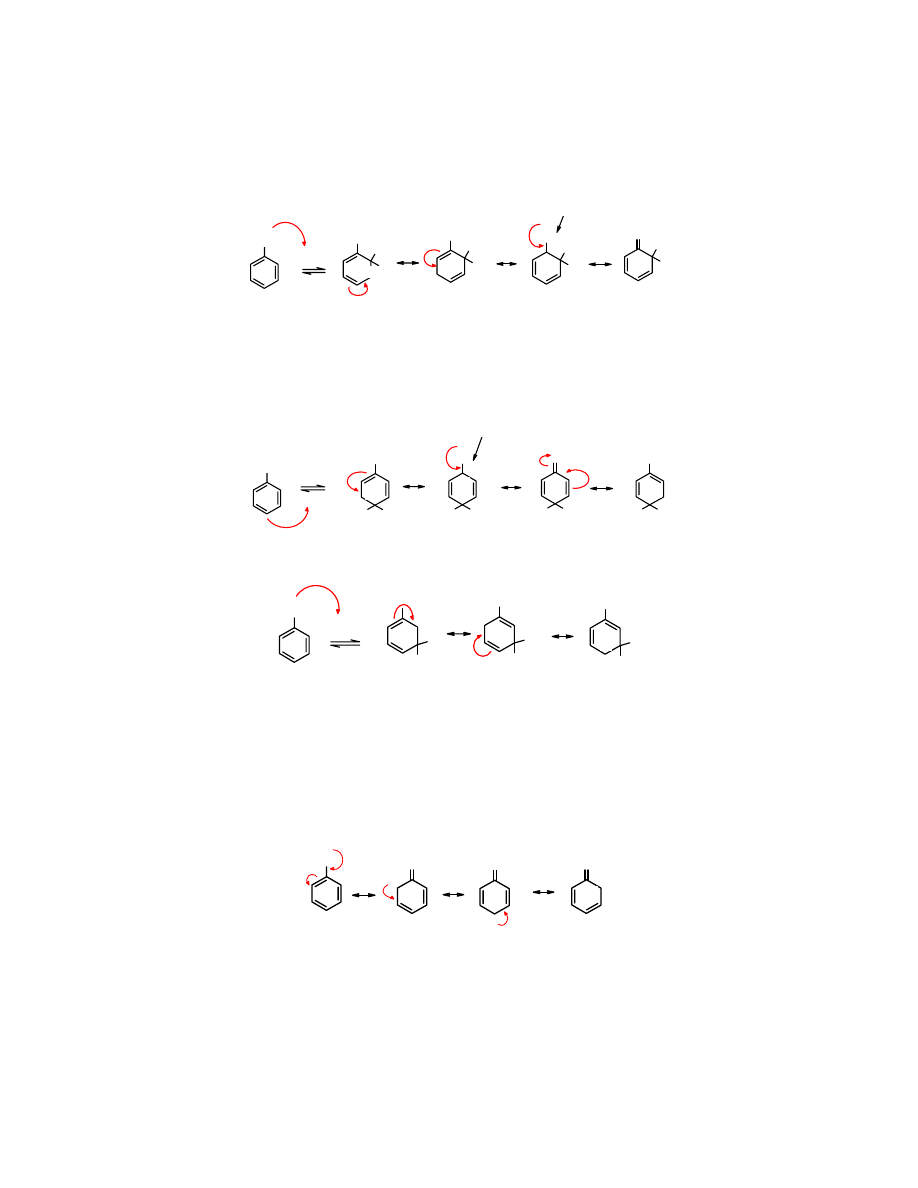
Rozpatrzmy reakcji S
E
podstawionego arenu zawierającego podstawnik Y o właściwościach
elektrodonorowych (EDG).
Jeżeli atak elektrofila jest skierowany w pozycje orto-,
Y
Y
H
E
Y
H
E
Y
H
E
Y
H
E
E
+
..
..
+
..
+
..
+
najtrwalsza
+
można wyszczególnić cztery struktury graniczne, przy czym jedna jest szczególnie
uprzywilejowana – ta w której podstawnik elektrodonorowy sąsiaduje z atomem C z deficytem
elektronów.
Podobnie jest w przypadku, kiedy atak elektrofila nastąpi w pozycję para-
Y
Y
H
E
Y
H
E
Y
H
E
Y
H
E
E
+
+
..
..
+
..
+
+
..
najtrwalsza
Natomiast po ataku w pozycje meta- jedynie trzy graniczne struktury mezomeryczne da się
wyróżnić:
Y
Y
E
H
Y
E
H
Y
E
H
E
+
+
..
..
+
..
+
..
Z analizy powyżej przedstawionych struktur mezomerycznych wynika, że najsilniej
stabilizowane addukty tworzą się po przyłączeniu elektrofila w pozycję
orto- i para-.
Dodatkowo należy uwzględnić fakt, iż w obecności podstawnika elektrodonorowego pozycje
orto- i para- są bardziej podatne na atak nukleofila, ponieważ w tych miejscach jest
zlokalizowany większy ładunek ujemny.
Wzrost ładunku ujemnego w pozycji
orto- i para-
uaktywnia pierścień aromatyczny na reakcje
S
E
,
co objawia się zwiększeniem szybkości
reakcji
.
..
Y
Y
Y
Y
..
..
..
+
-
+
-
+
-
Inna jest sytuacja, kiedy w reakcję S
E
wchodzi aren zawierający podstawnik elektroakceptorowy
(EWG). Podstawnik taki poprzez wyciąganie elektronów dezaktywuje pozycje orto- i para-.
12
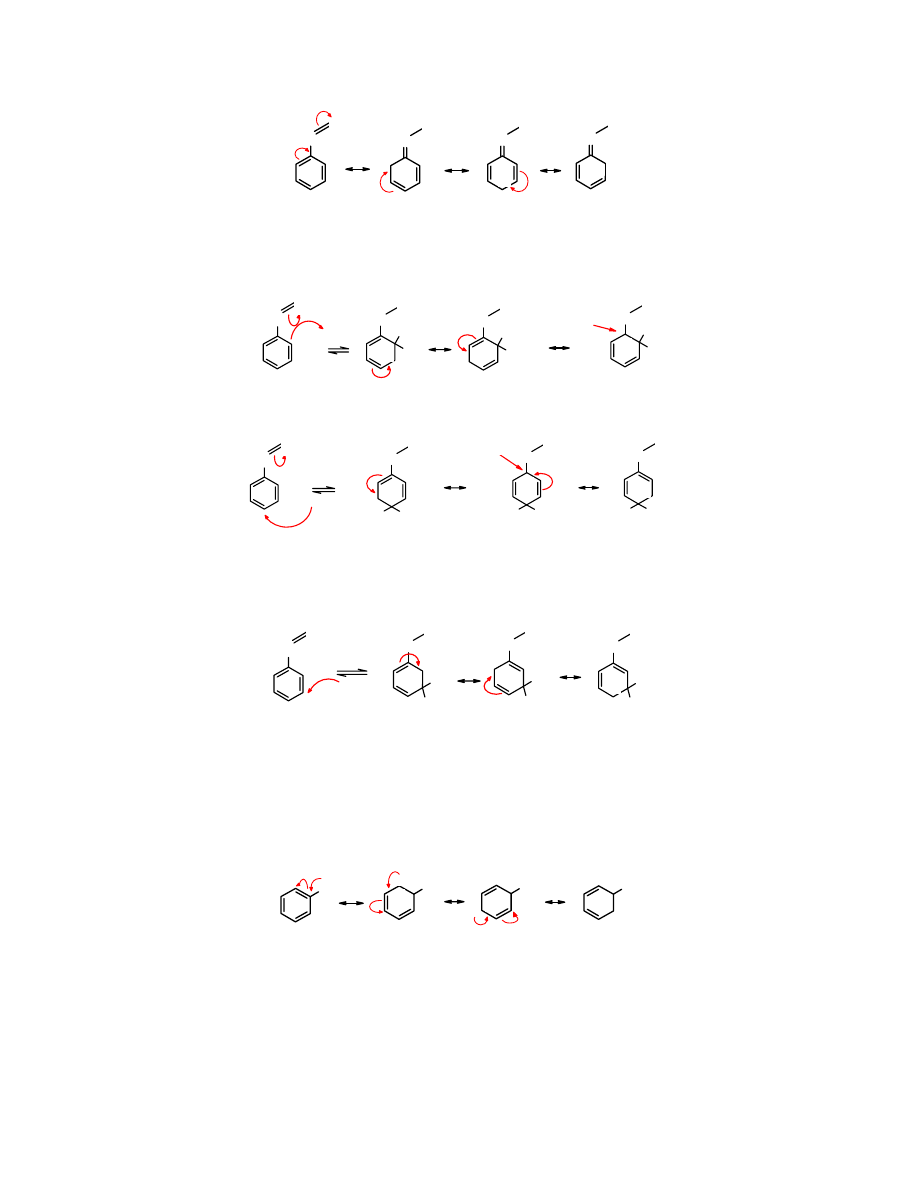
Y
Y
Y
Y
..
+
Z
Z
-
..
+
Z
-
..
+
Z
-
W addukcie powstałym po ataku elektrofila na pozycje orto- do podstawnika o właściwościach
EWG można wyodrębnić trzy graniczne struktury mezomeryczne, przy czym jedna ma charakter
destabilizujący, jako że ładunek
+
jest zlokalizowany w sąsiedztwie podstawnika
elektroakceptorowego:
Y
Y
H
E
Y
H
E
Y
H
E
E
+
+
Z
Z
+
Z
+
Z
+
+
+
-
-
-
destabilizacja
destabilizacja
Podobnie jest w przypadku ataku elektrofila na pozycję para-.
Y
Y
H
E
Y
H
E
Y
H
E
+
+
+
-
-
-
E
+
+
Z
Z
+
Z
+
Z
W jednej z trzech struktur granicznych dochodzi do destabilizacji.
Natomiast po ataku elektrofila w pozycji meta- wszystkie trzy struktury graniczne wpływają na
stabilizację układu.
Y
Y
E
H
Y
E
H
Y
E
H
+
+
+
-
-
-
E
+
+
Z
Z
+
Z
+
Z
Addycja
elektrofila w pozycję meta- w stosunku do podstawnika o właściwościach EWG nie
prowadzi do destabilizacji układu. Jednak jego obecność dezaktywuje aren na reakcję S
E
na
skutek wyciągania elektronów z pierścienia.
Podstawniki należące do
grupy III
– halogeny kierują elektrofil w położenie orto- lub para-. Jest
to wynik efektu +M, który zwiększa ładunek ujemny w tych pozycjach, czyniąc je podatne na
atak odczynnika elektrofilowego.
Cl
Cl
Cl
..
Cl
..
+
..
.. :
..
.. :
-
+
.. :
-
+
.. :
-
Natomiast efekt indukcyjny halogenów -I powoduje obniżenie szybkości reakcji S
E
halogenoarenów, wobec czego halogeny dezaktywują pierścień na
podstawienie elektrofilowe
.
Warto zwrócić uwagę (Tabela), iż największy dezaktywujący wpływ ma fluor i że ten wpływ
słabnie wraz z odległością – silniejszy jest w pozycji orto- niż w para-.
13
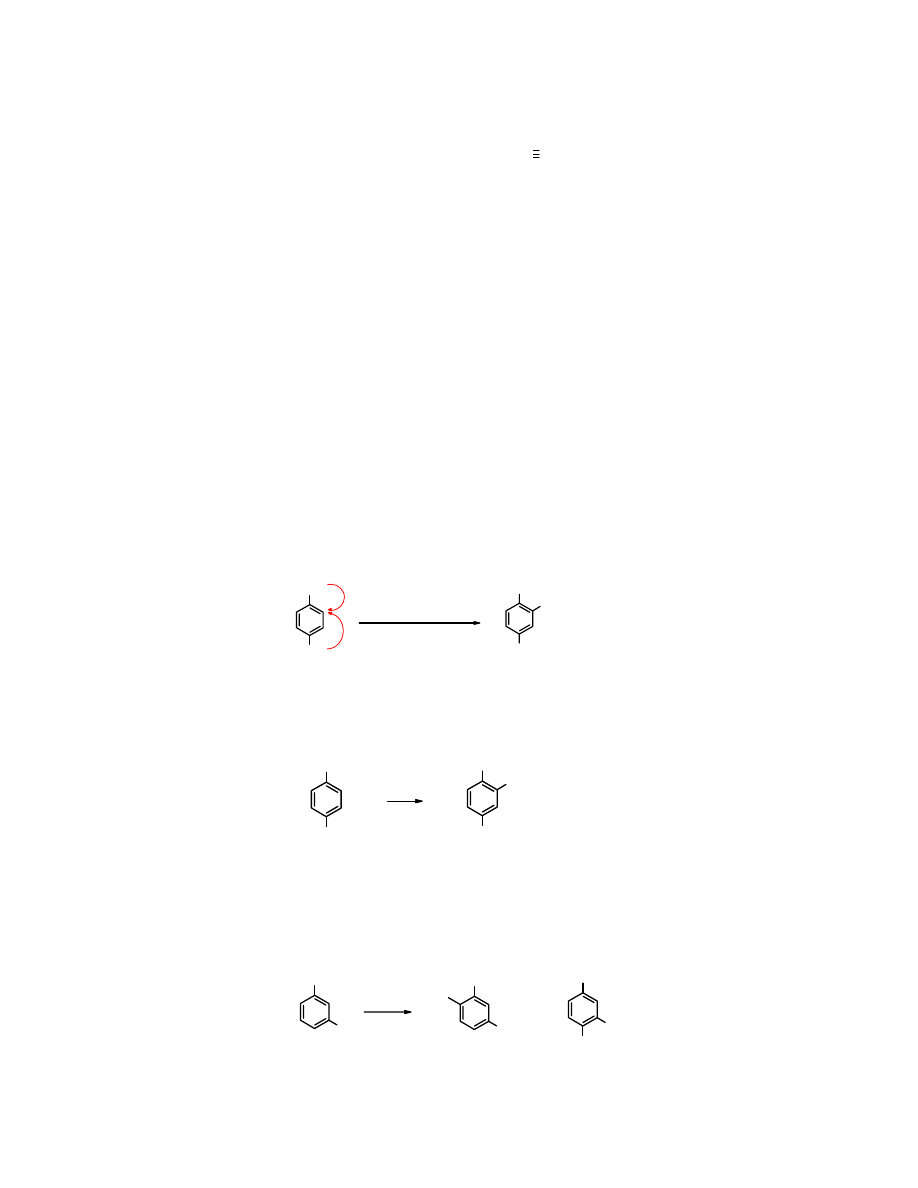
Podstawniki elektronodonorowe (EDG): Podstawniki elektronoakceptorowe (EWG):
-C N
..
-NH
2
, -NHR, -NR
2
..
..
-OH, -OR, -O
..
..
..
..
..
.. :
-
-SH, -SR, -S
..
..
..
..
..
.. :
-
-Alk, -Ar
podstawniki
grupy I
wykazują efekt
+I lub +M/+I
lub +M/-I
-NHCOCH
3
..
+
+
-NO
2
,
-CHO, -COR, -COOH, -COOR
-SO
3
H, -SO
2
OR, -SO
2
R
-CF
3
, -CCl
3
, -NR
3
, -SR
2
podstawniki
grupy II
wykazują efekt
-I lub -M/-I
Zadanie: przypisz efekty poszczególnym podstawnikom
Podstawniki elektrodonorowo-elektroakceptorowe (EDG/EAG):
-F , -Cl , -Br , -I
..
..
..
..
..
..
..
..
:
:
:
:
podstawniki
grupy III
wykazują efekt -I i +M
Substytucja elektrofilowa dipodstawionego benzenu – sumowanie efektów kierujących
Efekty kierujące dwóch podstawników pierścienia aromatycznego sumują się lub odejmują w
zależności od wzajemnego położenia.
1.
Efekty kierujące podstawników
grupy I i II
znajdujących się względem siebie w
położeniach orto- lub para- sumują się. Przykładem może być nitrowanie
4-nitrotoluenu
– obie grupy kierują drugą grupę nitrową w położenie 2.
CH
3
NO
2
CH
3
NO
2
NO
2
produkt główny
efekt kierujący
grupy NO
2
efekt kierujący
grupy CH
3
HNO
3
/H
2
SO
4
4-nitrotoluen 2,4-dinitrotoluen
(69%)
2. Efekty
kierujące podstawników
grupy I
znajdujących się względem siebie w położeniach
orto- lub para- mają działanie przeciwne. Decyduje grupa o silniejszym efekcie.
OH
CH
3
OH
CH
3
Br
Br
2
produkt główny
4-metylofenol
(
p-krezol
)
2-bromo-4-metylofenol
Zadanie: wyjaśnij, co zadecydowało o powstaniu
2-bromo-4-metylotoluenu
.
3. Trzeci, nowo wchodzący podstawnik rzadko zajmuje położenie 2- pomiędzy
podstawnikami ulokowanymi w pozycjach 1- i 3-.
CH
3
Cl
CH
3
Cl
Cl
CH
3
Cl
Cl
+
Cl
2
/FeCl
3
3-chlorotoluen 3,6-dichlorotoluen 3,4-dichlorotoluen
14
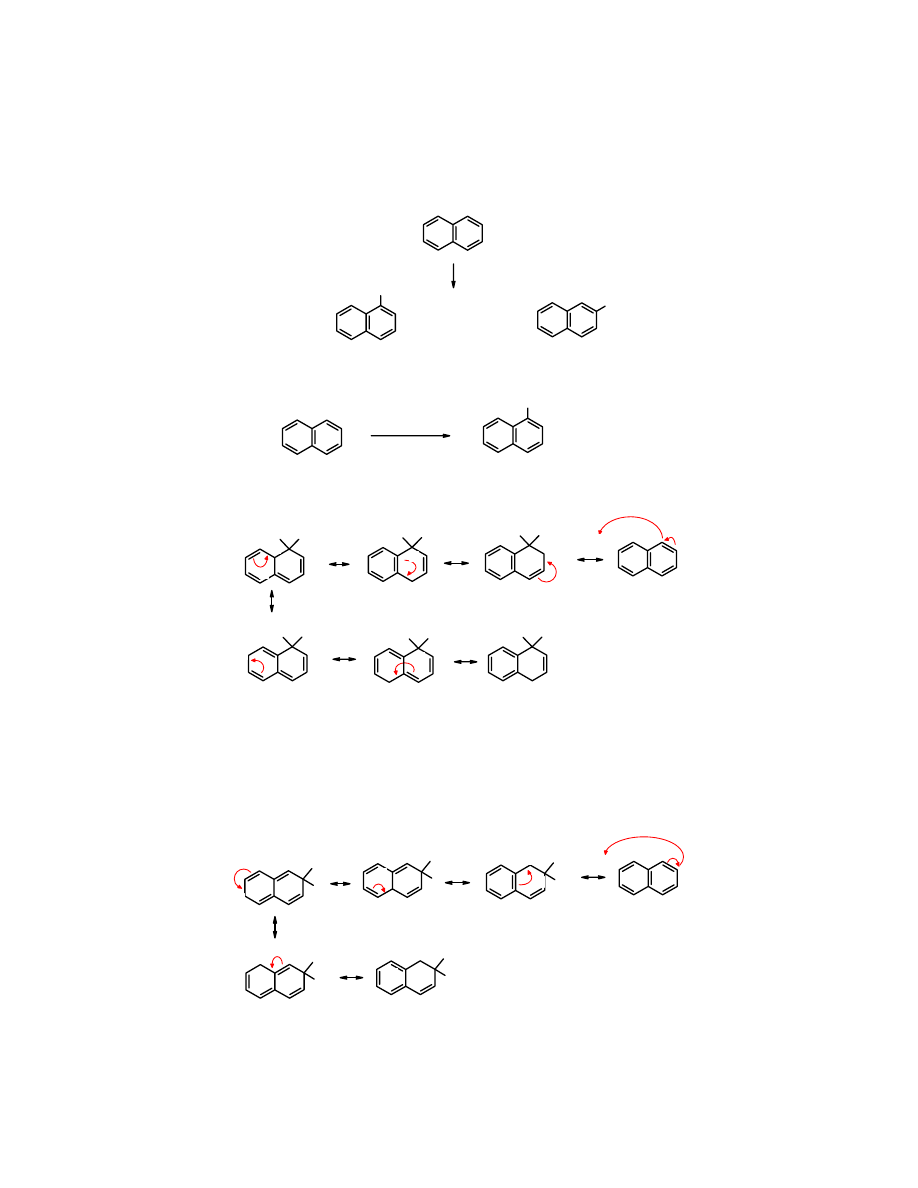
Reakcje S
E
naftalenu
Naftalen
jest bardziej podatny na reakcje substytucji S
E
niż
benzen
.
Naftalen
jest bardziej
podatny na reakcje substytucji S
E
niż
benzen
. W jego cząsteczce znajdują się dwie
nierównocenne pozycje, w których atom H – 1 lub 2 (
α lub β) może być podstawiony przez
elektrofil.
E
E
E
+
-
H
+
+
naftalen podstawiony
w pozycji 1 (
α)
naftalen podstawiony
w pozycji 2 (
β)
naftalen
W reakcjach S
E
łatwiej wymianie ulega atom H w pozycji 1.
NO
2
naftalen
HNO
3
/H
2
SO
4
1-nitronaftalen
(89%)
Większą reaktywność naftalenu w pozycji 1 tłumaczy się większą trwałością adduktu
powstającego po przyłączeniu elektrofila właśnie w pozycję 1 a nie 2.
H
E
H
E
H
E
H
E
H
E
H
E
E
+
+
+
+
+
+
1
2
3
4
5
6
naftalen
A
B
A
B
A
B
A
B
A
B
B
A
B
A
+
Po przyłączenie elektrofila w pozycję 1 w strukturach rezonansowych
1
,
2
i
6
w pierścieniu
B
zachowana zostaje aromatyczność (posiadają pełny sekstetet elektronów
π), jest to energetycznie
korzystne.
Natomiast po przyłączeniu elektrofila w pozycje 2 mniej jest mezomerycznych struktur
granicznych i tylko w dwóch zachowana zostaje pełna aromatyczność – w
1
i
5
.
H
E
H
E
H
E
H
E
H
E
E
+
1
2
3
4
5
naftalen
A
B
A
B
+
A
B
+
A
B
+
A
B
+
A
B
+
15
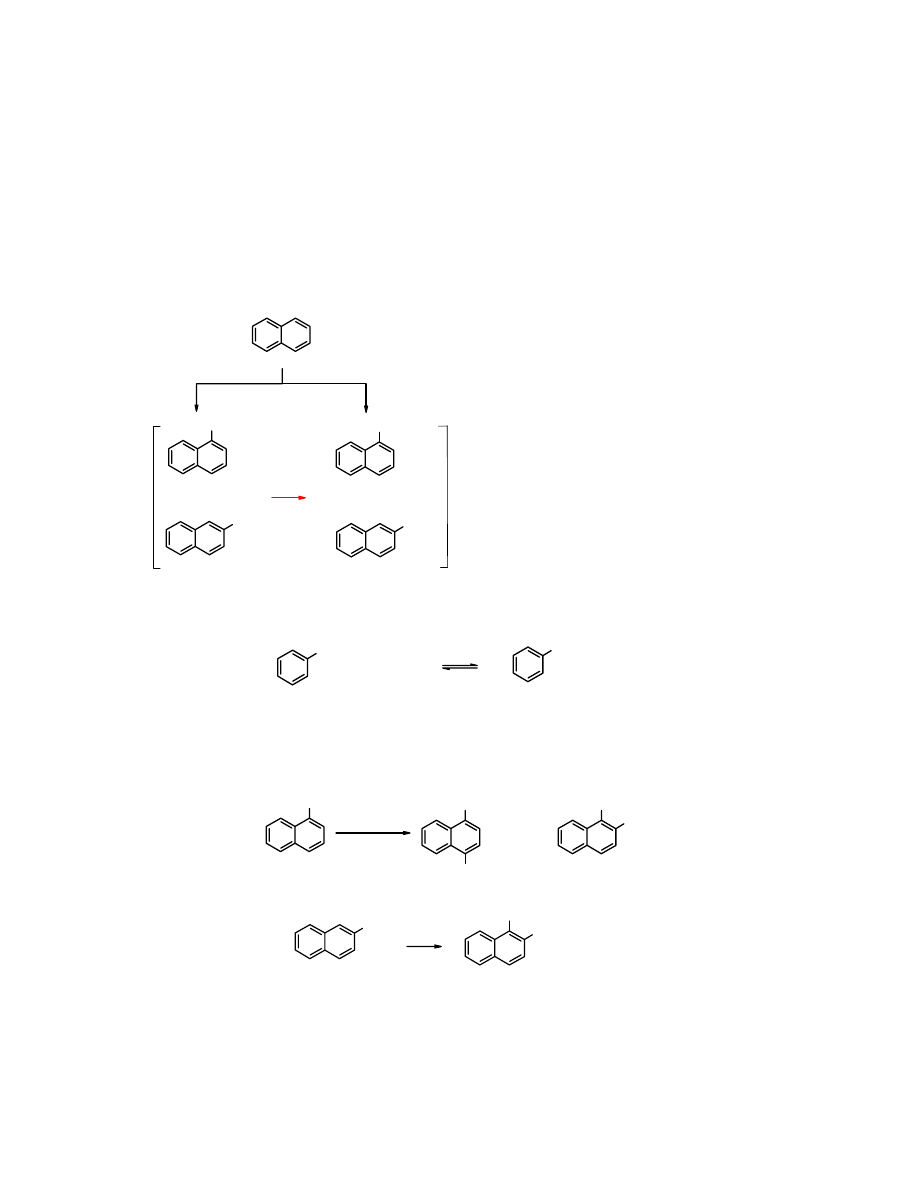
Sulfonowanie naftalenu
Produkt reakcji sulfonowania
naftalenu
zależy od temperatury procesu; w niskich temperaturach
w produktach przeważa
kwas 1-naftaleno sulfonowy
, a wraz ze wzrostem temperatury rośnie
udział
kwasu 2-naftalenosulfonowego
. Ponadto reakcja jest równowagowa – ogrzewanie izomeru
1- powoduje przekształcanie go w izomer 2-. Wytłumaczenie tego zjawiska jest proste. W niskich
temperaturach powstaje więcej tego produktu, który tworzy się z większą szybkością; mówimy
wówczas o
kinetycznej kontroli reakcji
. W przypadku
naftalenu
pozycja 1- jest aktywniejsza.
W miarę podwyższania temperatury zaczyna przeważać udział izomeru termodynamicznie
trwalszego, w tym przypadku
kwasu 2-naftalenosulfonowego
. W podwyższonej temperaturze
dominuje
termodynamiczna kontrola reakcji
.
SO
3
H
SO
3
H
SO
3
H
SO
3
H
85%
15%
7%
93%
kwas 1-nafta-
lenosulfonowy
kwas 2-nafta-
lenosulfonowy
kontrola
kine-
tyczna
kontrola
termody-
namiczna
naftalen
40
o
C
160
o
C
+
+
160
o
C
Przemiana
kwasu 1-naftalenosulfonowego
w
2-naftalenosulfonowy
jest możliwa dzięki temu, że
reakcja sulfonowania jest reakcją odwracalną.
SO
3
H
H
+ HOH
∆
+ H
2
SO
4
Reakcje substytucji elektrofilowej podstawionego naftalenu
W cząsteczce
naftalenu
drugi podstawnik elektrofilowy zajmuje pozycję zgodnie z regułami
reakcji S
E
. Podstawnik
grupy I
aktywuje „swój” pierścień i kieruje nowy podstawnik w
położenie 2 i 4.
OMe
OMe
NO
2
OMe
NO
2
HNO
3
/AcOH
+
1-metoksynaftalen
1-metoksy-2-nitronaftalen
14%
CH
3
CH
3
Br
Br
2
CS
2
2-metylonaftalen
1-bromo-2-
-metylonaftalen
(91%)
Warto zwrócić uwagę, że
bromowanie
naftalenu
nie wymaga katalizatora, ponieważ jest on
bardziej podatny na reakcję S
E
niż
benzen
.
16
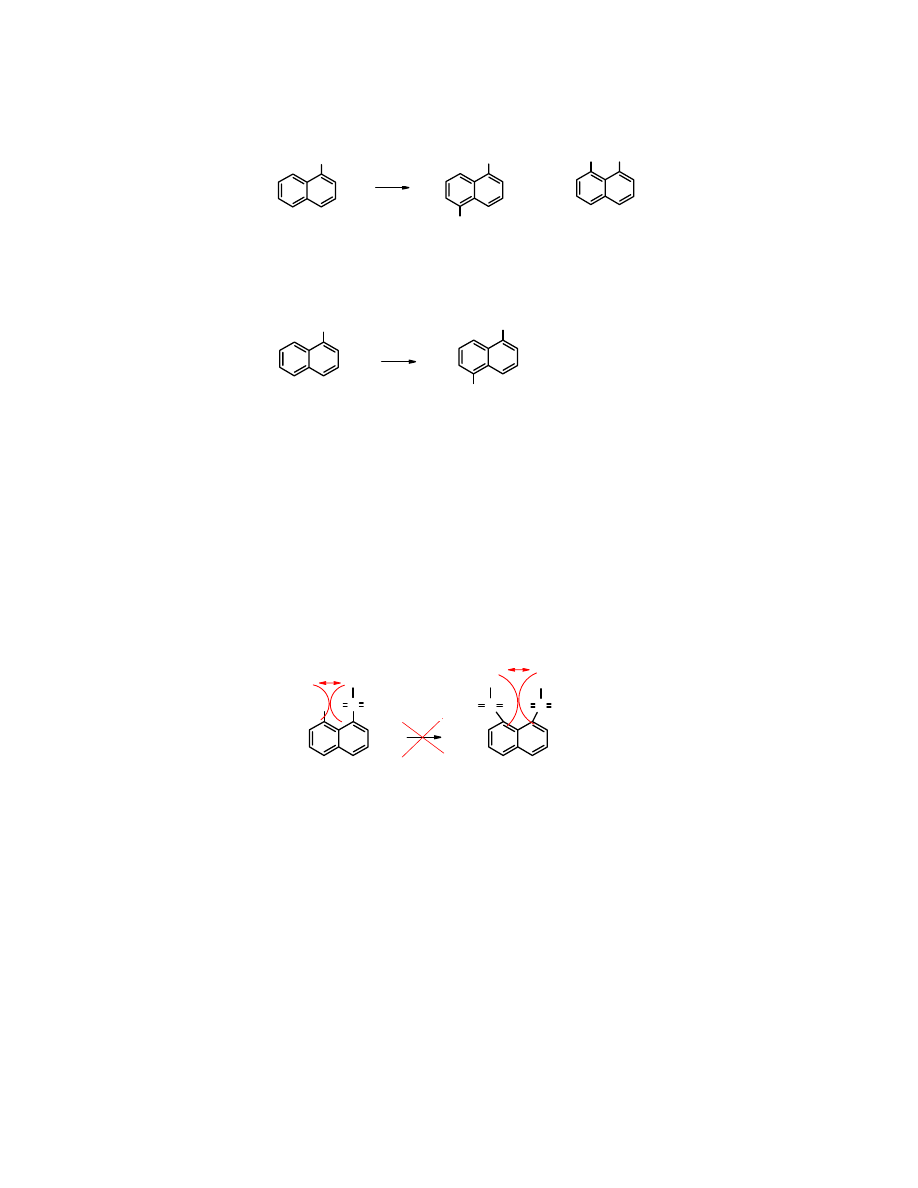
Podstawnik elektroakceptorowy (EWG) dezaktywuje „swój” pierścień, dlatego do
substytucji
dochodzi w drugim pierścieniu.
NO
2
NO
2
NO
2
NO
2
NO
2
1-nitronaftalen
HNO
3
H
2
SO
4
+
45% 31%
1,8-dinitronaftalen 1,5-dinitronaftalen
SO
3
H
SO
3
H
SO
3
H
H
2
SO
4
kwas 1-naftalenosulfonowy
kwas 1,5-naftaleno-
disulfonowy
72%
Polisulfonowanie naftalenu
Naftalen
w zależności od warunków sulfonowania, głównie temperatury i stężenia kwasu
siarkowego, można przeprowadzić z kwasy naftalenopolisulfonowe, aż do tetrasulfonowego.
Polisulfonowanie
biegnie wg określonych reguł.
Reguła 1
Druga grupa sulfonowa zostaje wprowadzona do drugiego pierścienia, ponieważ pierwszy jest
dezaktywowany obecnością grupy -SO
3
H, która należy do
grupy II podstawników
, tj.
dezaktywującej i kierującej nowy podstawnik w położenie meta-.
Reguła 2
Grupa -SO
3
H nie zajmuje pozycję peri-, tzn. 1,8 lub 4,5. W tych sąsiadujących z sobą pozycjach
jest za mało miejsca na dwie grupy sulfonowe obok siebie.
H
S
OH
O
O
S
S
OH
O
O
OH
O
O
H
2
SO
4
peri-
Reguła 3
Kolejna (trzecia) grupa sulfonowa zajmie pozycję meta- w stosunku do już obecnej, nie można,
więc wprowadzić drugiej grupę -SO
3
H do pierścienia, w którym znajduje się grupa sulfonowa w
pozycji 2.
17
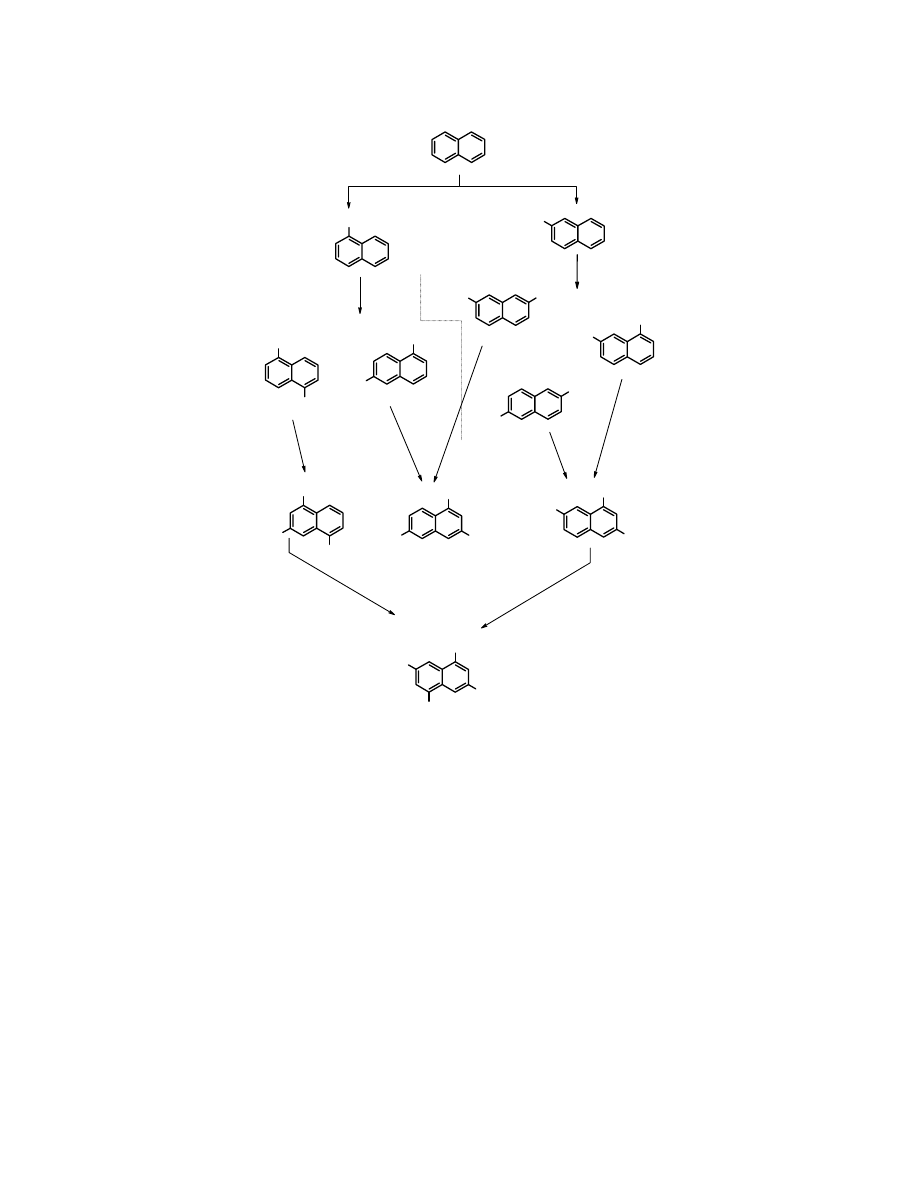
Możliwe produkty sulfonowania naftalenu
SO
3
H
HO
3
S
SO
3
H
SO
3
H
SO
3
H
HO
3
S
SO
3
H
HO
3
S
SO
3
H
HO
3
S
SO
3
H
HO
3
S
SO
3
H
SO
3
H
HO
3
S
SO
3
H
HO
3
S
SO
3
H
SO
3
H
SO
3
H
HO
3
S
SO
3
H
SO
3
H
SO
3
H
HO
3
S
40
o
C
oleum
H
2
SO
4
160
o
C
50
o
C
160
o
C
oleum
85
o
C
oleum
180
o
C
oleum
100
o
C
oleum
250
o
C, oleum
kwas naftaleno-1,3,5,7-
-tetrasulfonowy
naftalen
Zadanie: nazwij poszczególne kwasy naftalenosulfonowe
2. Reakcje substytucji nukleofilowej arenów - S
N
Charakterystyczną reakcją związków aromatycznych jest
substytucja elektrofilowa
S
E
. Zdarza
się jednak, że związki te ulegają
substytucji nukleofilowej
S
N
; jest to jednak sytuacja nietypowa.
Reakcji S
N
ulegają głównie halogenki alkilowe i inne pochodne o podobnych właściwościach,
np. estry – siarczany, fosforany czy tosylany alkilowe.
S
N
Alk-X + Nu
→ Alk-Nu + X
-
np.
HOH
CH
3
CH
2
-Br + NaCN
→ CH
3
CH
2
-CN + Na
+
Br
-
∆
bromek etylu cyjanek etylu
Takiej reakcji nie ulegają halogenki winylowe, podobnie jak arylowe.
18
CH
2
=CH-Br + NaOH
brak reakcji
100
o
C
Br
+ NaOH
brak reakcji
100
o
C
Hydroliza
halogenku fenylowego jest możliwa, ale dopiero w bardzo drastycznych warunkach.
Cl
OH
+ NaOH
350
o
C
HOH
H
+
/HOH
chlorek fenylu fenol
Reakcje S
N
podstawionych arenów biegną w łagodniejszych warunkach pod wpływem
silniejszych zasad:
Cl
NH
2
+ KNH
2
-33
o
C
+ KCl
NH
3
chlorek fenylu
amidek potasu
anilina
albo kiedy halogen „aromatyczny” jest uaktywniony grupą elektoakceptorową:
Cl
NO
2
OH
NO
2
NaOH/HOH
∆
H
+
/HOH
2-chloronitrobenzen 2-nitrofenol
Im więcej grup uaktywniających na
substytucję nukleofilowa
, tym łatwiej reakcja biegnie.
Cl
+ NaOH
brak reakcji
100
o
C
chlorek fenylu
Cl
NO
2
OH
NO
2
NaOH/HOH
H
+
/HOH
100
o
C
2-chloronitrobenzen 2-nitrofenol
2,4-Dinitrochlorobenzen
hydrolizuje już pod wpływem wrzącego roztworu węglanu sodu, a z
wodorotlenkiem sodu reakcja jest wydajna w temperaturze 80
o
C.
Cl
NO
2
O
2
N
OH
NO
2
O
2
N
NaOH/HOH
H
+
/HOH
80
o
C
2,4-dinitrochlorobenzen 2,4-dinitrofenol
(92%)
Jeszcze łatwiej
hydrolizuje
2,4,6-trinitrochlorobenzen
.
Cl
O
2
N
NO
2
NO
2
OH
NO
2
O
2
N
NO
2
NaOH/HOH
H
+
/HOH
30
o
C
2,4,6-trinitrochlorobenzen kwas pikrynowy
(94%) (
2,4,6-trinitrofenol
)
19
Spośród halogenoarenów najłatwiej wymianie ulega fluor.
F
NO
2
NO
2
OMe
CH
3
OK/CH
3
OH
60
o
C, 10 min
4-fluoro-
nitrobenzen
4-nitroanizol
(97%)
Ta obserwacja może dziwić, ponieważ chlorki alkilowe są znacznie bardziej reaktywne w
reakcjach S
N
niż odpowiednie fluorki.
Szereg szybkości w reakcjach S
N
halogenków alkilowych:
Alk-I >> Alk-Br > Alk-Cl >> Alk-F
Natomiast reaktywność halogenków arylowych przedstawia się odwrotnie – w reakcjach S
N
najaktywniejsze są fluorki arylowe:
Ar-F >> Ar-Cl > Ar-Br Ar-I
Ta odmienna podatność obu grup związków na działanie nukleofili świadczy o odmiennym
mechanizmie reakcji. Za innym mechanizmem reakcji przemawia również to, że w warunkach
reakcji S
N
1 i S
N
2 halogenki arylowe są nieaktywne. Ich brak reakcji wg mechanizmu S
N
1 łatwo
wytłumaczyć tym, że halogenki arylowe nie dysocjują na jony Ar
+
i X
-
, co jest warunkiem
koniecznym dla reakcji typu S
N
1. Niemożliwy jest również atak nukleofila na atom węgla z
przeciwnej strony do halogenu, co jest wymagane w reakcjach biegnących wg mechanizmu S
N
2,
ponieważ dostępu od tej strony broni pierścień aromatyczny.
X
Nu:
S
N
2
brak reakcji
Mechanizm reakcji S
N
– addycja/eliminacja
Halogenki arylowe uaktywnione obecnością elektroakceptorowych podstawników reagują z
nukleofilami wg mechanizmu
adycja/eliminacja
, co oznacza, że pierwszym etapem reakcji jest
addycja
nukleofila do atomu węgla związanego z halogenem, zmiana hybrydyzacji na tym
atomie z sp
2
na sp
3
, po czym następuje odtworzenie aromatyczności poprzez odszczepienie
anionu halogenkowego (
eliminacja
). Atak nukleofila na atom węgla związany z halogenem jest
ułatwiony, ponieważ w tym miejscu występuje deficyt elektronów
δ+
.
..
F
NO
2
F
NO
2
NO
2
: ..
+
δ
-
δ
+
OCH
3
..
..
-
OCH
3
-
OCH
3
- F
addycja
eliminacja
4-fluoronitrobenzen
kompleks Meisenheimera
4-metoksynitrobenzen
Addukt nukleofila z halogenkiem arylowym nazywany jest kompleksem Meisenheimera; jest on
stabilizowany mezomerycznie.
Jacob Meisenheimer (1876-1934); ur. w Greisheim, Niemcy; doktorat w monachium; prof. w Berlinie,
Greifswaldzie i Tybindze.
Zadanie: przedstaw graniczne wzory mezomeryczne kompleksu Meisenheimera.
20
Reakcjom
S
E
związków aromatycznych sprzyjają podstawniki typu
EDG
ulokowane w
pozycjach orto- i para-, natomiast reakcje
S
N
są uaktywniane przez podstawniki
EWG
w
położeniach orto- i para- w stosunku do grupy odchodzącej
.
Mechanizm reakcji S
N
– eliminacja/addycja
Inny jest mechanizm reakcji S
N
nieuaktywnionych halogenków arylowych, które jak zostało
pokazane powyżej też ulegają pewnym reakcjom, np.
hydrolizie
. Reakcje te wymagające bardzo
drastycznych warunków biegną wg mechanizmu nazwanego eliminacja/addycja. Wysoka
temperatura i silna zasada powodują oderwanie (
eliminację
) protonu, w wyniku czego tworzy się
benzyn
– węglowodór cykliczny zawierający jedno potrójne i dwa wiązania podwójne. W
następnej kolejności
benzyn
przyłącza odczynnik nukleofilowy (
addycja
).
Benzyn
Benzyn
jest nietrwałym, bardzo reaktywnym produktem, który powstaje z odpowiednio
podstawionych arenów pod wpływem silnych zasad w wysokiej temperaturze. Tworzenie się
benzynu
stanowi pierwszy etap reakcji S
N
arenów (
eliminację
), w drugim następuje
addycja
, np.
cząsteczki HOH.
Cl
H
OH
H
..
:
OH
:
..
..
-
- HCl
HOH
eliminacja
addycja
..
..
350
o
C
chlorek fenylu benzyn fenol
Benzyn
powstaje w znacznie łagodniejszych warunkach w reakcji z bardzo silnymi zasadami, np.
amidkami.
Cl
H
NH
2
..
NH
3
/-33
o
C
+ KNH
2
-
KCl, - NH
3
HNH
2
..
..
:
..
chlorek fenylu
amidek potasu
benzyn anilina
Temperatura reakcji -33
o
C jest determinowana temperaturą wrzenia amoniaku.
Reakcja
addycji
wody czy amoniaku do
benzynu
też zachodzi etapowo.
..
NH
2
NH
2
H
+ NH
2
..
-
-
H-NH
2
Zadanie: przedstaw etapy
addycji
cząsteczki wody do
benzynu
.
Benzynu
nie udało się wyizolować, jednak jego istnienie potwierdzono dowodami pośrednimi.
Jednym z nich jest reakcja
amonolizy
bromku fenylu zawierającego izotop
14
C w pierścieniu
benzenu w miejscu przyłączenia bromu.
Br
NH
2
NH
2
*
KNH
2
/NH
3
- KBr
*
NH
3
*
*
50%
50%
bromobenzen
benzyn
anilina
21
Gdyby reakcja biegła mechanizmem S
N
1 lub S
N
2 , a nawet addycji/eliminacji powstawałby
produkt zawierający grupę aminową wyłącznie przy C
*
.
Zadanie: przedstaw powyższą reakcję biegnąca hipotetycznie mechanizm S
N
1 lub addycji/eliminacji.
Innym pośrednim dowodem na istnienie
benzynu
jest jego reakcja z
furanem
. Jeżeli w warunkach
tworzenia się
benzynu
do środowiska reakcji zostanie dodany
furan
to izoluje się produkt, który
mógł powstać jedynie w reakcji Dielsa-Adlera i tylko
furanu
z
benzynem
.
Br
O
O
KNH
2
furan
(dien)
3. Halogenowanie alkiloarenów w łańcuchu bocznym
W alkiloarenach, pod wpływem chloru lub bromu w warunkach reakcji rodnikowej albo w
wyniku działania
N-bromoimidu kwasu bursztynowego
(NBS) dochodzi do
substytucji
atomów
wodoru
α.
CH
3
CCl
3
toluen
1,1,1-trichloro-
metylobenzen
Cl
2
, nadmiar
h
ν, - HCl
CH
3
NO
2
CH
2
Br
NO
2
+ Br
2
h
ν
- HBr
p-nitrotoluen
bromek
p-nitrobenzylu
(70%)
CH
2
CH
2
CH
2
Br
CHCH
2
CH
2
Br
Br
NBS
CCl
4
, 60
o
C
(99%)
(
3-bromopropylo)benzen (1,3-dibromopropylo)benzen
Atom wodoru w położeniu
α do pierścienia aromatycznego ma takie same właściwości jak
allilowy atom wodoru, łatwo więc ulega halogenowaniu pod wpływem chloru czy bromu na
świetle lub znanego odczynnika bromującego jakim jest NBS. Mechanizm tej reakcji jest
rodnikowy. Karborodnik allilowy (
α-arenowy) tworzy się łatwo, ponieważ jest stabilizowany
mezomerycznie.
Pierwszym etapem reakcji jest utworzenie rodnika halogenkowego.
Br
Br
h
ν
2 :Br
.
..
..
Następnie rodnik halogenkowy odrywa rodnik wodorowy z pozycji
α alkiloarenu i powstaje
stabilizowany mezomerycznie rodnik benzylowy. On z kolei w reakcji z cząsteczką halogenu
przekształca się odpowiednią pochodną, przy czy generuje się nowy rodnik halogenkowy.
Reakcja biegnie mechanizmem łańcuchowym.
22
CH
2
H
CH
2
CH
2
CH
2
CH
2
CH
2
Br
+ :Br:
..
.
.
.
.
.
Br
2
- Br
.
toluen
bromotoluen
3. Reakcje addycji do pierścienia aromatycznego – chlorowanie benzenu
Areny jak wiadomo trudno ulegają reakcji addycji, powoduje ona utratę aromatyczności, jest
więc niekorzystna energetycznie. W pewnych warunkach można ją jednak wymusić.
Benzen
w
reakcji z chlorem w podwyższonej temperaturze, pod wysokim ciśnieniem lub naświetlany
światłem ultrafioletowym przyłącza 3 mole chloru przekształcając się w
1,2,3,4,5,6-
heksachloroheksan
, dawniej zwany sześciochlorkiem benzenu (BHC).
H Cl
H
Cl
H
Cl
Cl
H
H
Cl
H
Cl
+ 3 Cl
2
∆ , ciś.
h
ν
benzen
1,2,3,4,5,6-heksa-
chlorocykloheksan
W reakcji tej powstaje mieszanina wielu stereoizomerów, a pośród nich znajduje się izomer
owodobójczy
γ, zwany
lindanem
–
1(a),2(a),3(a),4(e),5(e)6(e)-heksachlorocykloheksen
. Produkt
handlowy
lindanu
(znany również jako
gammeksan
) nie powinien zawierać izomeru
δ, który jest
szkodliwy dla roślin.
Cl
Cl
Cl
Cl
Cl
Cl
lindan
Lindan
wykazuje nie tylko działanie owadobójcze, jest szkodliwy również dla zwierząt i ludzi. Z
tego powodu jego stosowanie w krajach rozwiniętych jest zakazane. Nadal stosuje się go w wielu
innych krajach, szczególnie w ubogich krajach afrykańskich. Poprzez ruchy powietrza i w
eksportowanych produktach żywnościowych dociera jednak do wszystkich zakątków globu.
4. Utlenianie alkiloarenów
Utleniane
monoalkiloarenów, niezależnie od długości łańcucha alkilowego, prowadzi do
kwasu
benzoesowego
. Niepodatne na
utlenienie
są reszty alkilowe przyłączone do pierścienia poprzez
3
o
atom węgla, np.
t-butylobenzen
jest odporny na działanie utleniaczy.
CH
3
COOH
KMnO
4
/HOH
toluen
kwas
benzoesowy
(75%)
tw.
23
CHCH
2
CH
3
CH
3
COOH
[O]
sec-butylo-
benzen
kwas
benzoesowy
W ten sposób otrzymuje się wiele podstawionych kwasów aromatycznych.
CH
3
NO
2
NO
2
COOH
Na
2
Cr
2
O
7
/H
2
SO
4
HOH, tw.
p-nitrotoluen
kwas p-nitro-
benzoesowy
(86%)
Utlenianie
p-ksylenu
jest przemysłową metodą otrzymywania
kwasu tereftalowego
.
Jego
światowa produkcja sięga milionów ton rocznie. Służy głównie do wyrobu włókien
syntetycznych.
CH
3
CH
3
COOH
COOH
powietrze/AcOH
kat. - Co (III), 200
o
C
p-ksylen
kwas tereftalowy
Alkilobenzeny ulegają też
utlenieniu
do wodoronadtlenków. Z
kumenu
powstaje
wodoronadtlenek służący do otrzymywania
fenolu
i
acetonu
.
CH(CH
3
)
2
C(CH
3
)
2
OH
O
powietrze, 100
o
C
Cu
2+
HOO
H
2
SO
4
60
o
C
CH
3
CCH
3
+
kumen
fenol
aceton
wodoronad-
tlenek kumenu
5. Redukcja arenów
Katalityczna redukcja
benzenu
jest możliwa tylko w drastycznych warunkach – w
podwyższonej temperaturze, pod wysokim ciśnieniu i wobec katalizatorów (Pt, Pd, Ni, Ru lub
Rh). Przyłączenie pierwszego mola wodoru niszczy układ aromatyczny i redukcja biegnie dalej
łatwiej.
+ 3 H
2
200
o
C, 2-4 MPa
Ni
Ra
benzen
cykloheksan
(100%)
Stosuje się ją w przemysłowej metodzie produkcji czystego
cykloheksanu
, bowiem
otrzymywanie go poprzez destylację frakcyjną węglowodorów z ropy naftowej nie daje
jednorodnego produktu.
Podczas redukcji dipodstawionego
benzenu
powstaje mieszanina steroizomerów (cis- i trans-).
CH
3
CH
3
H
CH
3
CH
3
H
+ 3 H
2
100
o
C, 2-4 MPa
Ru lub Rh
m-ksylen
(E,Z)-1,3-dime-
tylocykloheksan
(100%)
24
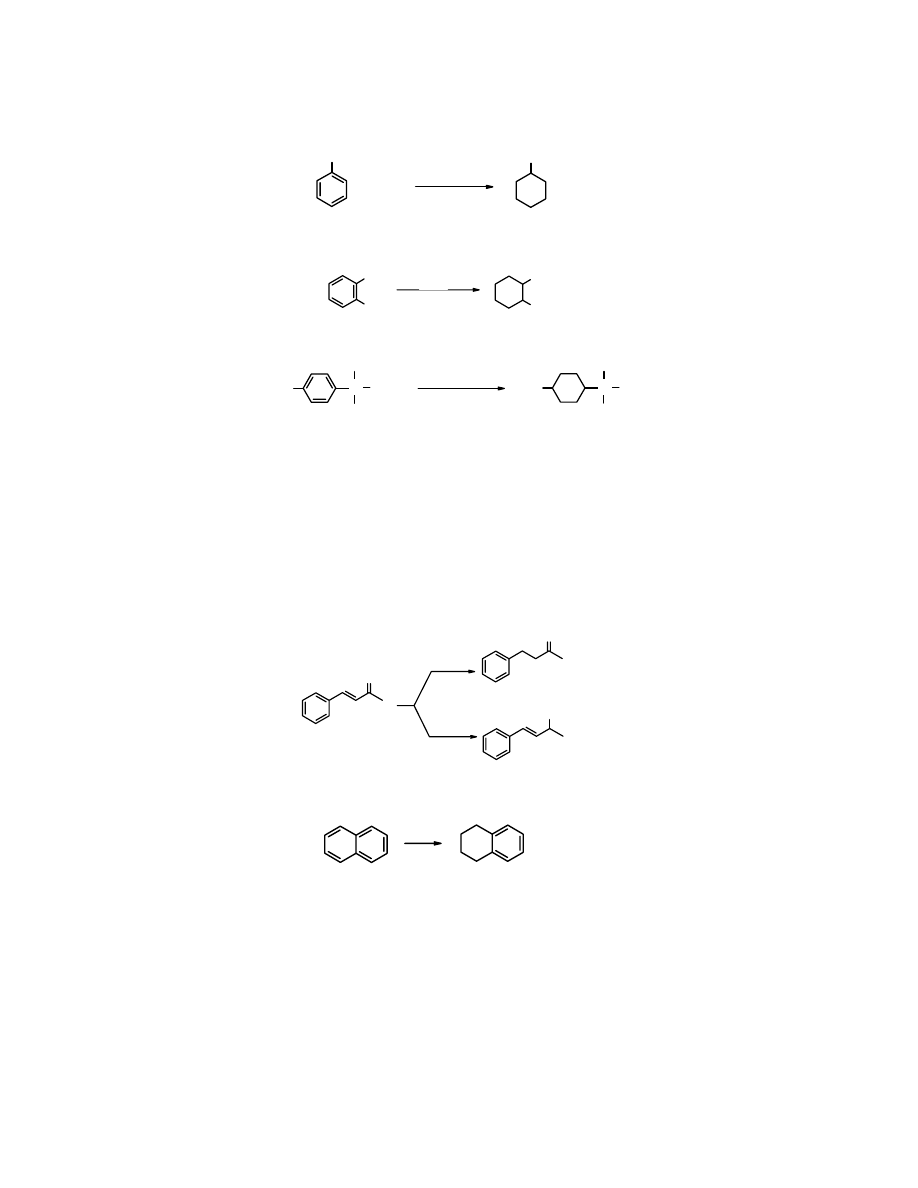
Katalityczne
uwodornienie
izolowanego pierścienia aromatycznego stosowane jest w przemyśle
do otrzymywania, np.
cykloheksanolu
z
fenolu
.
OH
OH
+ 3 H
2
150
o
C
1 MPa, Ni/Al
2
O
3
fenol
cykloheksanol
(96%)
Zastosowanie platyna jako katalizator pozwala na obniżenie ciśnienia i temperatury reakcji
uwodornienia.
CH
3
CH
3
CH
3
CH
3
H
2
/Pt
0,012 MPa, 25
o
C
o-ksylen
1,2-dimetylo-
cykloheksan
(100%)
Jeszcze łatwiej związki aromatyczne
redukuje
się wodorem w obecności katalizatora rodowego.
O
H
CH
3
CH
3
CH
3
O
H
CH
3
CH
3
CH
3
C
H
2
/Rh/C, EtOH
25
o
C
C
4-t-butylofenol 4-t-butylocykloheksanol
(100%)
Uwodornienie selektywne
Pierścień aromatyczny ulega trudno
redukcji katalitycznej
, bowiem jego uwodornienie
powoduje zanik aromatyczności, a tym samym utratę energii rezonansu. Z tego powodu można
selektywnie przeprowadzić
redukcję
w łańcuchach bocznych arenów. W
4-fenylobut-3-en-2-
onie
najłatwiej, a więc selektywnie
redukuje
się wiązanie podwójne za pomocą wodoru w
obecności palladu. Nienaruszona zostaje grupa karbonylowa i układ aromatyczny, zaś za pomocą
wodorku udaje się przekształcić nienasycony keton aromatyczny w nienasycony alkohol
aromatyczny.
O
O
OH
H
2
/Pd
NaBH
4
4-fenylobut-3-en-2-on
4-fenylobutan-2-on
4-fenylobutan-2-ol
(100%)
(100%)
W
naftalenie
łatwiej jest
uwodornić
jeden pierścień niż oba równocześnie. Jest to przemysłowa
metoda otrzymywania
tetraliny
.
H
2
/Ni
naftalen
tetralina
(tetrahydronaftalen)
Redukcja metalami
Areny podobnie jak alkiny reagują z metalami alkalicznymi w amoniaku, a produktami tej
reakcji są dihydroareny, tzn. związki częściowo zredukowane. Tego efektu nie da się osiągnąć za
pomocą
redukcji
wodorem wobec katalizatorów, ponieważ w pierwszym etapie takiego
uwodornienia tworzy się sprzężony dien, który uwodornia się łatwiej niż aren, wobec czego
redukcja biegnie aż do całkowitego wysycenia pierścienia wodorem.
25
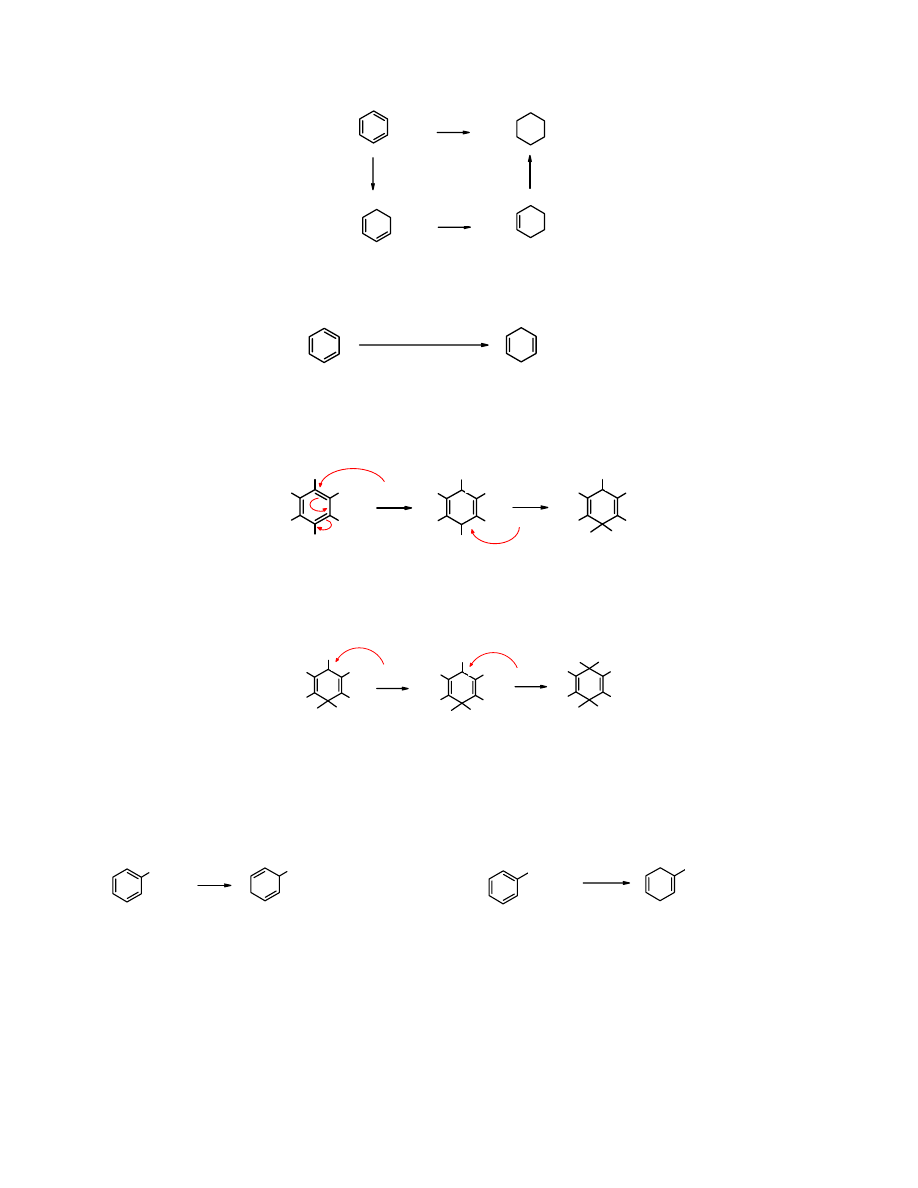
H
2
/Pt
H
2
/Pt
H
2
/Pt
H
2
/Pt
benzen
cykloheksan
cykloheksa-
-1,3-dien
cykloheksen
Natomiast redukcja
benzenu
litem lub sodem w amoniaku zatrzymuje się na etapie
dihydropochodnej. Reakcja ta nosi nazwę jej odkrywcy – redukcji Bircha (1944 r.).
Alkohol
pełni rolę donora protonów.
1.
Li/NH
3
lub Na/NH
3
2.
ROH
benzen
cykloheksa-
-1,4-dien
(90%)
Mechanizm tej
redukcji
jest identyczny jak w reakcji alkinów sodem lub litem w amoniaku.
Zaczyna się od przeniesienia elektronu (SET) z atomu metalu do pierścienia aromatycznego i
utworzenia anionorodnika, który po pobraniu protonu z rozpuszczalnika przekształca się w
rodnik.
..
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
Na
.
SET
.
-
H-OR
.
benzen
anionorodnik rodnik
Następnie rodnik po kolejnym przeniesieniu elektronu staje się anionem, który stabilizuje się w
wyniku protonowania. Reakcja zatrzymuje się na tym etapie, ponieważ utworzony dien nie
wykazuje powinowactwa do przyjęcia następnego elektronu.
H
H
H
H
H
H
H
H
H
H
H
H
H
H
..
H
H
H
H
H
H
H
H
.
Na
.
SET
-
H-OR
rodnik
karboanion
cykloheksa-1,4-dien
Podczas redukcji Bircha przejściowo powstają karboaniony, wobec tego podstawniki przy
pierścieniu aromatycznym będą miały wpływ zarówno na szybkość reakcji, jak i jej kierunek.
Podstawniki typu EDG utrudniają reakcję (poprzez destabilizację karboanionu), a podstawniki
EWG sprzyjają tworzeniu karboanionu. Jak widać ich wpływ jest przeciwny do obserwowanego
w reakcjach
aromatycznej substytucji elektrofilowej
.
CHO
CHO
Na/NH
3
EtOH
(90%)
OCH
3
Li/NH3
t-BuOH
OCH
3
(85%)
benzaldehyd 3-formylocyklohesa-1,4-dien metoksybenzen 1-metoksycykloheksa-1,4-dien
Lit i
t-butanol
używane są do
redukcji
związków zawierających silnie dezaktywujące
podstawniki (EWG), a więc arenów mniej podatnych na redukcję Bircha.
26
Document Outline
- R E A K T Y W N O Ś Ć A R E N Ó W
- Problemy reakcji acylowania F-C
- produkt – izomery [%]
- produkt – izomery [%]
- produkt – izomery [%]
- można wyszczególnić cztery struktury graniczne, przy czym jedn
- Sulfonowanie naftalenu
- Polisulfonowanie naftalenu
- Kolejna \(trzecia\) grupa sulfonowa zajmie pozycję meta- w sto
- Możliwe produkty sulfonowania naftalenu
- Benzyn
- 3. Halogenowanie alkiloarenów w łańcuchu bocznym
- 4. Utlenianie alkiloarenów
- 5. Redukcja arenów
- Uwodornienie selektywne
Wyszukiwarka
Podobne podstrony:
13 Reaktywno arenow
13 Reaktywne formy tlenu
Wyznaczanie stężenia nasycenia substancji reaktywnych (8), 1. 13 WYZNACZANIE ST??ENIA NASYCENIA SUBS
13 ZMIANY WSTECZNE (2)id 14517 ppt
13 zakrzepowo zatorowa
Zatrucia 13
pz wyklad 13
13 ALUid 14602 ppt
pz wyklad 13
ZARZ SRODOWISKIEM wyklad 13
Biotechnologia zamkniete użycie (2012 13)
Prezentacja 13 Dojrzewanie 2
SEM odcinek szyjny kregoslupa gr 13 pdg 1
w 13 III rok VI sem
więcej podobnych podstron