
______________________________________________
Oznaczanie wybranych węglowodorów aromatycznych
(WWA) przy zastosowaniu chromatografii gazowej z
detekcją mas (GC/MS) i wysokosprawnej chromatografii
cieczowej w układzie faz odwróconych z detekcją
fluorescencyjną (RP-HPLC-FLD)

Improved Analytical Assay for Selected PAHs Using GC/MS and RP-HPLC-FLD
Wykaz słów kluczowych:
wielopierścieniowe węglowodory aromatyczne (WWA),
benzo[a]piren,
wysokosprawna chromatografia cieczowa (HPLC),
chromatografia gazowa ze spektrometrem mas (GC/MS),
chromatografia wykluczania sterycznego (SEC).
3

1
SPIS TREŚCI
2.1. Ogólna Charakterystyka Wielopierścieniowych Węglowodorów Aromatycznych .........
2.3. Toksyczność WWA, absorpcja i metabolizm w organizmach żywych..........................10
Działanie kancerogenne i mutagenne WWA .........................................................11
Preparatywny chromatograf wykluczania sterycznego (SEC) ..............................20
Chromatograf gazowy ze spektrometrem mas (GC/MS) I.....................................20
Chromatograf gazowy ze spektrometrem mas (GC/MS) II ...................................20
Analityczny wysokosprawny chromatograf cieczowy (HPLC)..............................21
Przygotowanie próbek olei i wyodrębnienie frakcji WWA z matrycy tłuszczowej 24
Warunki analizy za pomocą chromatografii cieczowej.........................................25
Przeprowadzanie reakcji B[a]P z rodnikami hydroksylowymi .............................26

2

3
1. WSTĘP
Wielopierścieniowe węglowodory aromatyczne (WWA) to bardzo zróżnicowana i
wszechobecna grupa zanieczyszczeń występujących zarówno w środowisku jak i w artykułach
spożywczych. Zazwyczaj występują one w postaci wieloskładnikowych mieszanin różnych
WWA. Liczne badania sugerują ich kancero- i mutagenne własności.
Wielopierścieniowe węglowodory aromatyczne należą do grupy związków chemicznych
zanieczyszczających środowisko i artykuły spożywcze, które ze względu na powszechność ich
występowania i ich zdolność do wchłaniania są skażeniami trudnymi do uniknięcia.
Problematyka skażenia zarówno środowiska naturalnego jak i żywności w ostatnich
latach wzbudza duże zainteresowanie badaczy,
a także zwykłych konsumentów. Świadomość
występowania niekorzystnych zmian w organizmach, jakie powodują wielopierścieniowe
węglowodory aromatyczne powoduje poszukiwanie i udoskonalanie metod analitycznych ich
oznaczania. Zostało to w szczególności wymuszone przez przystąpienie Polski do Unii
Europejskiej. W Rozporządzeniu Komisji (UE) nr 208/2005 z dnia 4 lutego 2005r.
zamieniającym rozporządzenie (UE) nr 466/2001 w odniesieniu do wielopierścieniowych
węglowodorów aromatycznych ustalono dopuszczalne stężenia benzo[a]pirenu w żywności. Z
kolei w Rozporządzeniu nr 2065/2003 z dnia 10 listopada 2003r. określono dopuszczalne
stężenie benzo[a]antracenu w dymach wędzarniczych. Komisja Nauki UE zasugerowała, aby w
przyszłości oznaczać 15 WWA w żywności, zgodnie z podaną listą.
2. CZĘŚĆ LITERATUROWA
2.1. Ogólna Charakterystyka Wielopierścieniowych Węglowodorów
Aromatycznych (WWA)
Wielopierścieniowe Węglowodory Aromatyczne WWA (ang. Polycyclic Aromatic
Hydrocarbons - PAHs) stanowią grupę związków chemicznych zawierającą dwa lub więcej
skondensowane pierścienie aromatyczne. Cząsteczka WWA ma budowę płaską. Poznanych
zostało około 100 homocyklicznych węglowodorów występujących w środowisku, a ponadto
kilkaset ich pochodnych alkilowych, aminowych, nitrowych itp. Znane są również
heterocykliczne WWA z wbudowanymi atomami tlenu, siarki, lub azotu. W środowisku
występują WWA głównie w postaci mieszanin tych związków. Najbardziej poznanym i
najczęściej oznaczanym WWA jest benzo[a]piren.
Tabela 1.
Najczęściej oznaczane WWA - lista EPA-US (1-16) i KN UE (SCF-UE) (9-23) [1, 2]
EPA-US – Environmental Protecting Agency United States, KN UE- Komisja Nauki Unii Europejskiej, (SCF-UE –
Union European Commission Scientific Committee on Food )
L.p Nazwa
związku
Akronim
Masa
cząsteczkowa
Wzór
sumaryczny
Wzór strukturalny
1 Acenaften
ACF 154
C
12
H
12
2 Acenaftylen
ACN
152
C
12
H
10
3 Antracen
AT 178
C
14
H
10
4 Fluoranten
F 202
C
16
H
10
4
5 Fluoren
FL 166
C
13
H
10
6 Naftalen
NA 128
C
10
H
8
7 Fenantren
FA
178
C
14
H
10
8 Piren
P 202
C
16
H
10
9 Benzo[a]antracen B[a]A
228
C
18
H
12
10 Benzo[a]piren
B[a]P 252
C
20
H
12
11 Benzo[b]fluoranten B[b]F 252
C
20
H
12
12 Benzo[ghi]perylen B[g]P 276
C
22
H
12
13 Benzo[k]fluoranten B[k]F 252
C
20
H
12
5
14 Chryzen
Ch 228
C
18
H
12
15
Dibenzo[a,h]antracen D[h]A 278
C
22
H
14
16 Indeno[1,2,3-cd]piren
I[c]P 276
C
22
H
12
17 Benzo[j]fluoranten B[j]F 252
C
20
H
12
18 Cyklopenta[c,d]piren
CPP 226
C
18
H
10
19 Dibenzo[a,e]piren D[e]P 302
C
24
H
14
20 Dibenzo[a,h]piren D[h]P 302
C
24
H
14
6
21 Dibenzo[a,i]piren D[i]P 302
C
24
H
14
22 Dibenzo[a,l]piren D[l]P 302
C
24
H
14
23 5-Metylochryzen
5MC 242
C
19
H
14
C
H
3
2.1.1. Właściwości fizyczne WWA
WWA są to ciała stałe o krystalicznej budowie, które w stanie czystym występują jako
bezbarwne, jasno żółte lub zielone kryształy. Mają niską lotność (głównie cięższe WWA) i słabą
rozpuszczalnością w wodzie (brak grup polarnych). Im większa jest ich masa cząsteczkowa tym
mniejsza rozpuszczalność węglowodorów. Przykładowo, rozpuszczalność naftalenu (M=128) w
wodzie wynosi 37,2 [µg/l], benzo[a]antracenu (M=228) - 14 [µg/l], benzo[a]pirenu (M=252) -
3,8 [µg/l] natomiast perylenu (M=252) tylko 0,4 [µg/l]. Z tego powodu bardzo łatwo adsorbują
się one na cząstkach niepolarnych, takich jak np. pyły. Absorbują one światło w zakresie UV-
VIS, co wykorzystuje się to do oznaczeń zarówno ilościowych i jakościowych [3, 4].
2.1.2. Właściwości chemiczne WWA
Węglowodory aromatyczne pod wpływem światła oraz tlenu łatwo ulegają reakcjom
fotochemicznym tworząc epoksydy, chinony, diole, fenole i aldehydy (Schemat 1). W układach
biologicznych reakcje te są kontrolowane enzymatycznie (Schemat 2). Im większa liczba
7
skondensowanych pierścieni w cząsteczce WWA tym łatwiej się ona utlenia. Będąc związkami
aromatycznymi ulegają także reakcjom substytucji elektrofilowej.
[O]
[O]
[O]
[O]
O
O
O
OH
O
H
OH
epoksyd
dihydrodiol
fenol
chinon
Schemat 1. Utlenianie pierścienia aromatycznego WWA.
O
O
2
enzym
OH
O
H
enzym
OH
O
H
O
O
2
enzym
benzo[a]piren
7,8-epoksybenzo[a]piren
trans-7,8-dihydroksybenzo[a]piren
9,10-epoksy-trans-7,8-dihydroksybenzo[a]piren
8
Schemat 2. Utlenianie benzo[a]pirenu w układach biologicznych.

9
2.2. Odkrycie i występowanie WWA
Już w 1775 roku anglik sir Percival Pott wskazał na powiązania pomiędzy wzmożoną
zachorowalnością na raka moszny u londyńskich kominiarzy, a ekspozycją na WWA. Wysunął
hipotezę, że zachorowalność na nowotwory jest powiązana z ekspozycją na smołę i sadzę, z
którą mieli do czynienia podczas wykonywania swojej pracy. Pott nie potrafił jeszcze nazwać i
wyodrębnić związków chemicznych, które zawarte w sadzach powodowały nowotwory. Dopiero
po ponad 150 latach, w roku 1929 zidentyfikowano dibenzo[a,h]antracen występujący w
sadzach, który jest odpowiedzialny za powstawanie chorób nowotworowych [5].
WWA powstają podczas niepełnego spalania substancji organicznej (z syntezy
niskocząsteczkowych związków lub rozpadu wysokocząsteczkowych, głównie lignin). Jako
naturalne źródła emisji wielopierścieniowych węglowodorów aromatycznych do środowiska
należy wymienić: pożary, wybuchy wulkanów, wypalanie traw i nieużytków. W niewielkich
ilościach zawierają je także naturalne kopaliny, a mianowicie węgiel kamienny czy ropa
naftowa. Spośród źródeł generowanych przez człowieka najważniejszymi są emisja gazów i
dymów z zakładów przemysłowych (szczególnie z przemysłu ciężkiego) oraz procesy
wytwarzania energii w elektrowniach i elektrociepłowniach. Istotnymi źródłami uwalnianie
WWA do środowiska są także: motoryzacja (spaliny samochodowe, ścieranie się opon) oraz
dymy z kotłowni i pieców domowych [6]. Wielopierścieniowe węglowodory aromatyczne
występują także w surowcach przemysłowych takich jak pak węglowy, oleje mineralne oraz w
produktach ich obróbki: smoła pogazowa, sadze czy też olej kreozotowy. Skład i ilość mieszanin
WWA emitowanych do środowiska zależy od rodzaju substancji spalanej, metody spalania oraz
stosowania filtrów i innych urządzeń chroniących przed ich emisją [7].
Większość wielopierścieniowych węglowodorów w powietrzu występuje pod postacią
par i aerozoli zaadsorbowanych na pyłach o średnicy około 0,5 nm. Tak zaadsorbowane
cząsteczki WWA mogą być przenoszone za pomocą wiatru daleko od miejsc, w których
powstały, zanieczyszczając glebę, wodę i rośliny. Szczególnie dotyczy to węglowodorów
ciężkich.
W literaturze można znaleźć dużo prac odnośnie zależności charakterystyki emisji WWA
od źródeł ich powstawania. Jednym z ważniejszych czynników wpływających na immisję
węglowodorów do środowiska są ruchy powietrza [8]. Węglowodory skażające atmosferę to
głównie węglowodory alifatyczne zarówno nasycone jak i nienasycone
,
a także węglowodory
aromatyczne o małej masie cząsteczkowej [9]. Tylko cząstki o średnicy mniejszej niż 1 µm
wnikają drogą oddechową. W szczególności zagrożenie ekspozycją na WWA występuje w
takich gałęziach przemysłu jak hutnictwo, przemysł gumowy, produkcja sadzy czy
koksownictwo [7]. Odrębnym źródłem powstawania i emisji WWA jest palenie papierosów.
2.3. Toksyczność WWA, absorpcja i metabolizm w organizmach żywych
2.3.1. Aktywność biologiczna i absorpcja WWA
Wielopierścieniowe węglowodory aromatyczne mają różne struktury, w których
pierścienie benzenu przyjmują różne wzajemne położenia. W niektórych cząstkach WWA
występują charakterystyczne obszary, zwane regionem K (zewnętrzna krawędź pierścienia
fenantrenu) oraz region M (przeciwstawne atomy struktury antracenu). Ważne jest także
położenie regionu, zwanego regionem zatoki. To właśnie na te obszary i pozycje wskazują
badacze i przypisują im aktywność biologiczną [10]. Schemat budowy WWA na przykładzie
benzo[a]piranu wraz z zaznaczonymi regionami o biologicznej aktywności przedstawia
Rysunek 1.
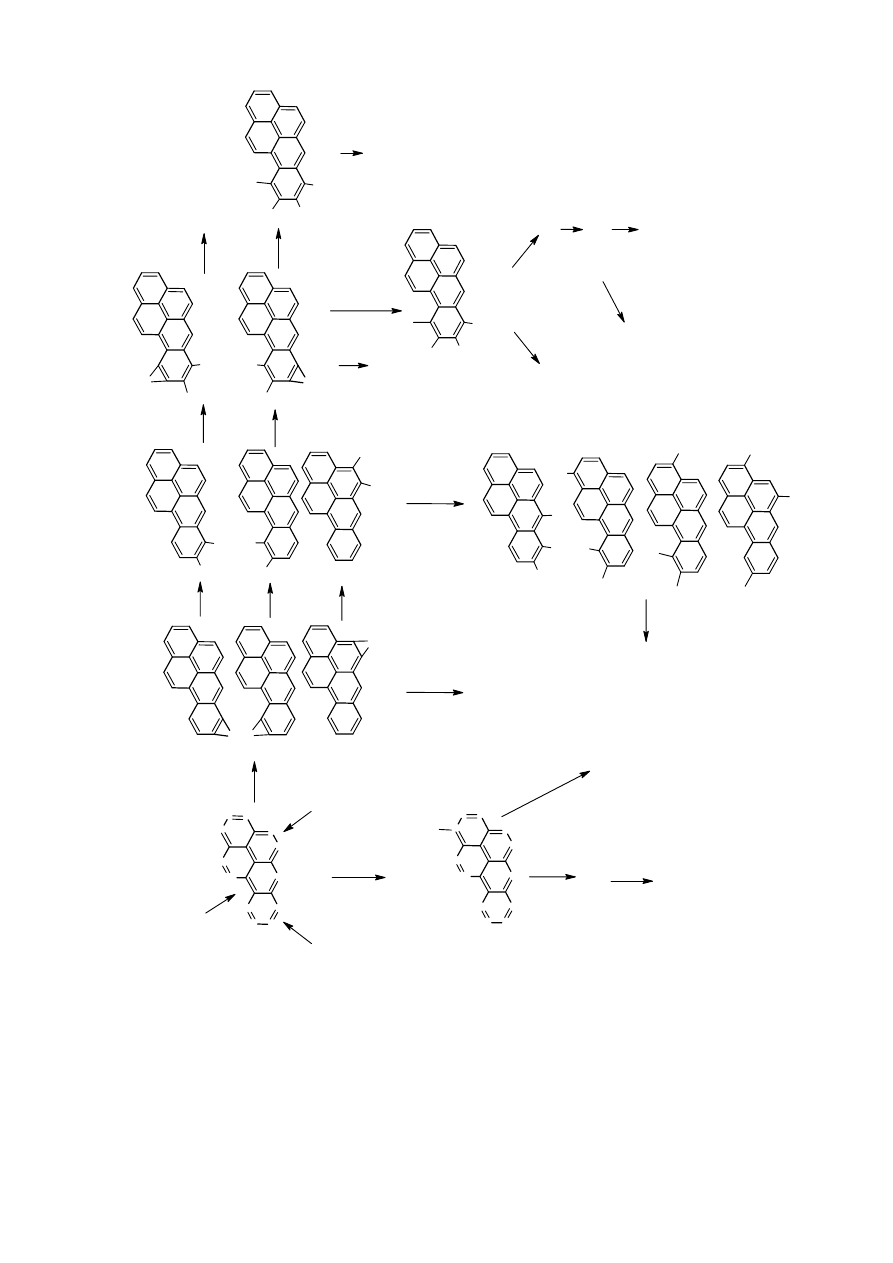
6
12
11
2
1
3
4
5
7
8
10
9
region zatoki
region M
region K
Rysunek 1. Wzór strukturalny B[a]P wraz z zaznaczonymi regionami o biologicznej aktywności.
Drogi przenikania węglowodorów do organizmów żywych [11]:
- wziewna, przez układ oddechowy,
- pokarmowa, wraz ze spożywanym pokarmem,
- poprzez skórę.
10
11
W procesie pobrania drogą pokarmową WWA stwierdzono, że największy udział mają
produkty pochodzenia zwierzęcego, następnie oleje i tłuszcze roślinne, produkty zbożowe, ryby i
produkty rybne. Udział warzyw i owoców w przeciętnym pobraniu WWA jest minimalny [12].
2.3.2. Działanie kancerogenne i mutagenne WWA
Wyniki wieloletnich badań wykazały, że dłuższa ekspozycja organizmów żywych na
WWA może powodować szereg niekorzystnych oddziaływań i zmian. Wiele WWA ma
właściwości kancero- i mutagenne, a także geno- i embriotoksyczne [13]. Nie zaobserwowano
natomiast ich ostrego działania toksycznego. Wielopierścieniowe węglowodory aromatyczne
wykazują toksyczność układową, co potwierdziły badania przeprowadzone na zwierzętach.
Celowe narażanie zwierząt, powodowało u nich uszkodzenie układu chłonnego, oddechowego i
krwiotwórczego[14].
Tabela 2.
Względne aktywności kancero- i mutagenna wybranych WWA [15,16]
Nazwa związku
Aktywność
kancerogenna
Aktywność
mutagenna
dibenzo[a,h]antracen
benzo[a]piren
indeno[1,2,3-cd]piren
benzo[a]antracen
benzo[a]fluoranten
benzo[k]fluoranten
benzo[j]fluoranten
piren
benzo[ghi]perylen
benzo[e]piren
chryzen
1,110
1,000
0,232
0,145
0,141
0,066
0,061
0,081
0,022
0,004
0,004
0,47
1,00
0,14
0,62
0,20
-
-
0,20
0,08
0,42
0,37
Następstwem długotrwałej ekspozycji na ciężkie WWA jest ich działanie muta- i
teratogenne. W 1981 roku Amerykańska Agencja Ochrony Środowiska (EPA) ustaliła listę 126
związków chemicznych jako szczególnie toksycznych zanieczyszczeń. W tej grupie związków
12
znalazło się 16 wielopierścieniowych węglowodorów aromatycznych. Eksperci z tejże agencji
podzielili wyselekcjonowane WWA na dwie podgrupy według ilości pierścieni i właściwości
toksycznych. W celu określenia ryzyka kancerogennego w odniesieniu do poszczególnych
WWA, przyjęto benzo[a]piren za związek wzorcowy ze względu na to, że jest on najbardziej
rozpowszechnionym i poznanym WWA, dla którego względny współczynnik kancerogenności
(WWK) wynosi 1. Siła działania rakotwórczego pozostałych WWA obliczana jest względem
niego. Wartości
WWK dla niektórych WWA przedstawia Tabela 2.
Tabela 3.
Klasyfikacja WWA ze względu na siłę działania kancerogennego według IARC
(Międzynarodowa Agencja Badań nad Rakiem) [17]
Prawdopodobnie kancerogenny Możliwie kancerogenny
benzo[a]antracen
benzo[a]piren
dibenzo[a,h]antracen
benzo[b]fluoranten
benzo[j]fluoranten
benzo[k]fluoranten
dibenzo[a,e]piren
dibenzo[a,h]piren
dibenzo[a,i]piren
dibenzo[a,l]piren
indeno[1,2,3-cd]piren
5-metylochryzen
dibeno[a,h]antracen
dibenzo[a,j]antracen
Badania Międzynarodowej Agencji Badań nad Rakiem (IARC) potwierdzają właściwości
kancerogenne, teratogenne oraz mutagenne wielu przebadanych WWA. Jednakże eksperci z
IARC sugerują pewne zastrzeżenia i konieczność rozpatrzenia następujących kwestii [17]:
badania były przeprowadzane na zwierzętach doświadczalnych, więc należy z pewną
ostrożnością interpretować wyniki względem ekspozycji oddziaływania WWA na
organizm ludzki,
w dużej mierze badania dotyczyły poszczególnych wybranych WWA, nie
uwzględniając, że występują one przeważnie w mieszaninach,
badania nie brały po uwagę WWA dostających się do organizmu drogą pokarmową oraz
poprzez układ oddechowy.

13
Okazało się, że dwu-, trzy- oraz czteropierścieniowe węglowodory nie są kancerogenne.
Również 9,10-dwumetyloantracen, 1,2,3,4-terametylofenantren i benzo[a]antracen posiadają
niski potencjał kancerogenny. Natomiast dibenzo[a,h]antracen oraz benzo[a]piren są silnie
kancerogenne [17].
Korzystając z wyników badań przeprowadzonych na zwierzętach doświadczalnych
wyznaczono dawkę minimalnego poziomu ryzyka (z ang. minimal risk lewel – MRL). Dla ludzi
wynosi ona 0,01 mg B[a]P na kg mc na dzień oraz 3,65 mg na kg mc na rok. Należy zaznaczyć,
że sugerowana dawka nie oznacza konieczności wystąpienia choroby nowotworowej, a jedynie
prowizoryczną dawkę względnego ryzyka.
2.3.3. Metabolizm WWA w organizmie
Po wniknięciu do organizmu WWA ulegają szeregowi przemian metabolicznych,
koniecznych do ich detoksytacji i wydalenia z organizmu. Podobna struktura różnych
węglowodorów aromatycznych powoduje ich zbieżne przemiany metaboliczne. Najdokładniej
poznanym i opisywanym w literaturze jest metabolizm benzo[a]piranu. Mechanizm przemian
został przedstawiony na Schemacie 3 [18]. Wśród zachodzących przemian przeważają reakcje
epksydacji i hydroksylacji, stanowiące reakcje I fazy. Metabolity powstałe w I fazie zawierają
czynne grupy funkcyjne reagujące z glukuronianami i siarczanami w II fazie. Powstałe produkty
są łatwo wydalane z organizmu wraz z moczem [19, 20, 21]. W reakcjach I fazy tworzą się
tlenki arenowe, chinony, fenolodiole, fenole, będące metabolitami przejściowymi, z których
najważniejszym jest 7,8-diol-9,10-epoksybenzo[a]piren, o silnych właściwościach muta- i
kancerogennych. Zidentyfikowano go w organach zwierząt poddawanych eksperymentowi
narażania na B[a]P [19, 20, 21]. Metabolity benzo[a]pirenu wykazują bardzo silne
powinowactwo do struktur wewnątrzkomórkowych i mogą uszkadzać DNA. W wyniku
nieprawidłowej replikacji i błędnych procesów odnowy uszkodzonego DNA dochodzi do
mutacji, a to sprzyja propagacji procesu rakotwórczego.
Stwierdzono także, że wielopierścieniowe węglowodory aromatyczne wnikają do
wszystkich narządów, a ich zwartość we wszystkich organach jest niezależna od drogi
wchłaniania. Narządy bogate w tkankę tłuszczową mogą kumulować WWA. Uwalniają się one z
nich stopniowo, przez dłuższy okres czasu. Wydalane są one z organizmów przez mleko
zarówno jako metabolity jak i WWA pierwotne [22].
14
6
12
11
2
1
3
4
5
7
8
10
9
re
g
io
n
z
a
to
k
i
re
g
io
n
M
re
g
io
n
K
M
FO
O O
O
E
H
E
H
E
H
OH
OH
O
H
O
H
OH
OH
O
O
H
O
H
O
OH
OH
M
FO
M
FO
OH
OH
O
H
O
H
E
H
E
H
OH
OH
O
H
DNA
OH
OH
OH
O
H
O
H
O
H
O
H
O
H
OH
O
H
OH
OH
6
12
11
2
1
3
4
5
7
8
10
9
O
H
B
[a
]P
e
p
o
k
s
y
d
y
d
ih
y
d
ro
k
s
y
d
io
le
e
p
o
k
s
y
-d
ih
y
d
ro
k
s
y
d
io
le
te
tr
a
-o
le
a
d
d
u
k
t
D
N
A
fe
n
o
lo
d
io
le
fe
n
o
le
w
p
o
z
y
c
ja
c
h
1
-,
3
-,
6
-,
7
-
lu
b
9
-
W
y
d
a
la
n
ie
z
m
o
c
z
e
m
K
w
a
s
g
lu
k
u
ro
n
o
w
y
w
y
d
a
la
n
ie
z
m
o
c
z
e
m
M
u
ta
c
ja
D
N
A
T
ra
n
s
fo
rm
a
c
ja
N
O
W
O
T
W
O
R
Y
O
d
n
o
w
a
D
N
A
E
li
m
in
a
c
ja
N
a
p
ra
w
a
t
k
a
n
k
i
E
s
tr
y
g
lu
k
u
ro
n
o
w
e
i
s
ia
rk
o
w
e
w
y
d
a
la
n
ie
z
m
o
c
z
e
m
W
y
d
a
la
n
ie
z
m
o
c
z
e
m
C
h
in
o
n
y
W
y
d
a
la
n
ie
z
m
o
c
z
e
m
MFO – kompleks enzymów o różnych funkcjach oksydacyjnych
EH – hydrolazy epoksydowe
Schemat 3. Etapy przemian WWA, na przykładzie B[a]P, w organizmach żywych [19].

15
2.4. Charakterystyka
skażenia żywności WWA
Główną przyczyną skażenie żywności WWA są zanieczyszczenia środowiskowe.
Skażona woda, powietrze oraz gleba wpływają na zawartość WWA szczególnie w produktach
pochodzenia roślinnego. Jak wcześniej zostało już wspomniane WWA adsorbują się na
cząstkach pyłów, które osadzają się na liściach roślin i warzyw. Wnikają i kumulują się w
określonych warstwach roślin, unikając tkanek z wysoką zawartością wody. Liczne badania
wykazały także, że warzyw i owoce uprawiane w rejonach zurbanizowanych i
uprzemysłowionych zawierają od 5 do 10 razy więcej WWA niż te uprawiane w rejonach
czystych ekologicznie [23].
Występowanie wielopierścieniowych węglowodorów aromatycznych w żywności jest
spowodowane głównie jej obróbką. Do procesów obróbki i przetwarzania żywności, podczas
których tworzą się WWA, zaliczamy [24]:
-
smażenie,
-
wędzenie,
-
grillowanie,
-
suszenie,
-
pieczenie,
itp.
Szczególnie intensywnie były badane procesy wędzenia i grillowania. Wykazano, że na
zawartość WWA w produktach wędzonych i grillowanych ma wpływ wiele parametrów, takich
jak [25]:
- rodzaj użytego drewna,
- temperatura wędzenia,
- rodzaj wędzenie (wędzenie na zimno, wędzenie na gorąco, wędzenie za pomocą dymu
wędzarniczego),
- stosowanie mycia po wędzeniu,
-
używane przyprawy,
-
zawartość tłuszczu w wędzonym mięsie,
- czas trwania procesu.
Ważnym źródłem wielopierścieniowych węglowodorów aromatycznych w diecie są oleje
i tłuszcze roślinne. Używa się ich nie tylko do bezpośredniej konsumpcji, są one także
składnikami różnych produktów i półproduktów spożywczych. Ich występowanie w olejach jest
głównie uwarunkowane procedurą suszenia ziarna (szczególnie gazami grzewczymi).
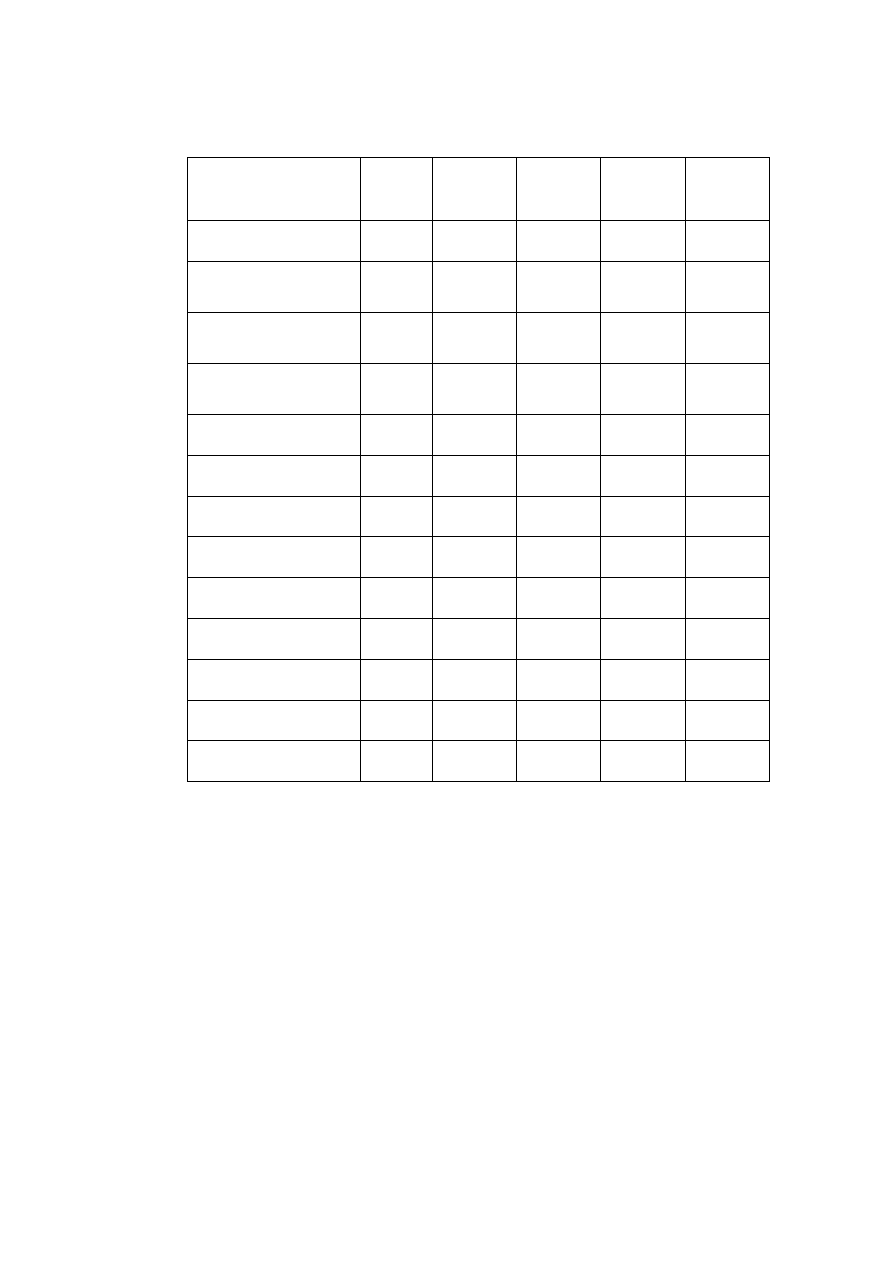
16
Tabela 4.
Zawartości oznaczanych WWA w różnych olejach roślinnych [26].
Próbka oleju
B[a]A
[µg/kg]
Ch
[µg/kg]
B[b]F
[µg/kg]
B[k]F
[µg/kg]
B[a]P
[µg/kg]
Surowy palmowy
62,6 105,3 55,1 12,1 40,6
Rafinowany
palmowy
ppw Ppw ppw ppw ppw
Rzepakowy tłoczony
na zimno
4,7
5,6 5,6 2,2 4,4
Oczyszczony
rzepakowy
ppw 0,6
0,5
ppw ppw
Oliwa
0,5
2,0 1,0 0,4 0,7
Słonecznikowy
ppw 0,4
ppw ppw 0,3
Sojowy
0,6
2,1 0,9 0,6 1,0
Winogronowy
0,4
1,7 0,4 0,3 0,5
Lniany
0,6 0,9 0,6 ppw 0,5
Z dyni
ppw 0,8
0,6
ppw 0,5
Arachidowy
5,1
5,2 4,0 1,6 3,1
Sezamowy
5,5
6,1 3,8 1,5 3,1
Tran
ppw Ppw ppw ppw 0,1
ppw – poniżej progu wykrywalności
Analizując wyniki zawartości poszczególnych WWA w tabeli zauważamy, że oleje
tłoczone na zimno zawierają znacznie więcej WWA od olejów poddanych procesowi rafinacji.
Takie zabiegi technologiczne jak bielenie (dodatek węgla aktywnego) oraz rafinacja wyraźnie
zmniejszają zawartość zanieczyszczeń, w tym WWA [27].
Obecnie przepisy prawne nie limitują dopuszczalnych zawartości poszczególnych WWA
w żywności poza B[a]P, którego zawartość w olejach jadalnych nie może być wyższa niż 2
µg/kg, w wyrobach wędzonych – 5 µg/kg, ostrygach – 10 µg/kg natomiast w produktach dla
dzieci – 1 µg/kg. Regulacje europejskie limitują również zawartości B[a]P i B[a]A w dymach
wędzarniczych, które nie mogą przekraczać odpowiednio 10 µg/kg dla B[a]P i 20 µg/kg dla
B[a]A [28].

17
2.5. Metody oznaczania WWA
Większość metod oznaczania WWA dotyczy próbek środowiskowych (próbki wody,
ścieków, gleby). Metody oznaczania wielopierścieniowych węglowodorów aromatycznych w
matrycach żywnościowych, charakteryzują się wysokim stopniem trudności. Matryce
żywnościowe są problematyczne z powodu tego, że lipofilne węglowodory przenikają do
wnętrza komórek. By wyodrębnić frakcję WWA z matryc stosuje się hydrolizę
w środowisku
zasadowym, wielokrotne ekstrakcje ciecz-ciecz lub ciecz-ciało stałe. Metody te są pracochłonne,
skomplikowane i kosztowne. Inną metodą wyodrębniania wielopierścieniowych węglowodorów
aromatycznych z matryc żywnościowych jest ekstrakcja płynem w stanie nadkrytycznym, lub
ekstrakcja w podwyższonej temperaturze. Głównie stosuje się je do wyodrębnienia WWA z
próbek o konsystencji stałej. Alternatywną metodą przygotowania próbek do analizy jest
preparatywna chromatografia wykluczania sterycznego (SEC). Jest to metoda względnie szybka
i prosta [26].
Rozdziały wyodrębnionych węglowodorów wykonuje się za pomocą chromatografii
gazowej połączonej ze spektrometrem mas (GC/MS) lub przy zastosowaniu wysokosprawnej
chromatografii cieczowej (HPLC). Istotną zaletą GC/MS jest wysoka rozdzielczość, co jest
bardzo korzystne przy rozdziale mieszanin oraz możliwość identyfikacji rozdzielonych
związków,. Wadą natomiast trudności w analizie „ciężkich” (sześcio-, siedmiopierścieniowych)
WWA (kolumna może ulegać rozkładowi).
HPLC z detektorem fluorescencyjnym charakteryzuje się wprawdzie gorszą sprawnością
rozdziału, ale niższym progiem wykrywalności (rzędu ułamków ppb)
WWA. Analizę
wielopierścieniowych węglowodorów aromatycznych za pomocą HPLC prowadzi się w układzie
faz odwróconych (faza stacjonarna jest niepolarna). Ta technika chromatografii jest najlepsza do
rozdziału mieszanin związków różniących się hydrofobowością. Fazę ruchomą stanowią zwykle
acetonitryl i woda lub metanol i woda podawane w odpowiednim gradiencie lub izokratycznie.

2.6. Wpływ rodników hydroksylowych na benzo[a]piren
Ważnym problemem związanym z ochroną środowiska jest usuwanie z niego związków
toksycznych. W literaturze opisane są metody ich rozkładu oparte na ich utlenianiu ozonem,
działaniem promieniowania UV czy ultradźwięków [31]. Ostatnio opisano również metody
utleniania fenoli za pomocą rodników hydroksylowych. Rodniki te generowane są w reakcji
Fentona [32, 33]:
H
O
OH
Fe
O
H
Fe
-
3
2
2
2
•
+
+
+
+
=
+
.
Rodniki te utleniają badane próbki. Dotychczas metoda nie była wykorzystywana w celu
dezaktywacji WWA.
18

19
3.
CEL PRACY
Celem pracy było opracowanie metody analizy benzo[a]pirenu i benzo[a]antracenu,
(które powinny być oznaczane w żywności zgodnie z wytycznymi KN UE) oraz WWA z listy 15
WWA rekomendowanych do badań przez KN UE, których maksymalne stężenia w żywności
podane zostaną w przyszłości, po opracowaniu odpowiednich metod analitycznych.
Porównane
zostaną dwie techniki analityczne: GC/MS i HPLC z detekcją
fluorescencyjną. Integralną częścią pracy będzie opracowanie metody przygotowania próbki.
Opracowane techniki analityczne zostaną praktycznie przetestowane w analizie
wybranych WWA w olejach jadalnych.
Zbadana zostanie następnie możliwość dezaktywacji benzo[a]pirenu przez rodniki
hydroksylowe.

4. CZEŚĆ EKSPERYMENTALNA
4.1. Aparatura
4.1.1. Preparatywny chromatograf wykluczania sterycznego (SEC)
Chromatograf preparatywny (Dionex, Germering, Niemcy)
składał się z:
- interfejsu UCI-100,
- autosamplera ASI-100,
- gradientowej pompy, zawierającej degazer, P-580,
- kolumny, wraz z prekolumną, do wykluczania sterycznego PLgel (5µm, 50 Å, 600 x 7,8 mm
I.D; złoże-PS/DVB) (Polymer Laboratories, Amherst, Stany Zjednoczone),
- detektora spektrofotometrycznego z matrycą diodową UVD-340S,
- kolektora frakcji Foxy Jr.fraction collector (Isco, Lincoln, Stany Zjednoczone),
- programu do naboru i obróbki danych Chromleon v. 6,20 (Dionex-Softron, Germering,
Niemcy) zainstalowanym komputerze IBM-PC kompatybilnym
4.1.2. Chromatograf gazowy ze spektrometrem mas (GC/MS) I
Część pomiarów GC/MS została wykonana na aparacie GC-17A z kwadrupolowym
spektrometrem mas MS - QP500 (Schimadzu, Tokio, Japonia). Kolumna chromatograficzna:
BPX-5, 30m x 0,25 mm I.D x 0,25
μm FT (Phenomenex, Torrance, CA, USA) o składzie złoża:
5% polisarylenu, 95% polidimetylosiloksanu. Gaz nośny – hel o czystości 99,99999%, przepływ
– 0,7 ml/min, temperatura dozownika – 180
o
C, temperatura interfejsu – 260
o
C, programowalna
temperatura kolumny: 80
o
C - 3 min, izoterma końcowa: 300
o
C - 7 min, zakres m/z -110 – 310.
4.1.3. Chromatograf gazowy ze spektrometrem mas (GC/MS) II
Część pomiarów wykonanych zostało na, znajdującym się na terenie Instytutu Przemysłu
Mięsnego i Tłuszczowego, aparacie GC/MS TSQ500 połączonym z spektrometrem mas
20

21
17A/QP5050 (Finnigan – Thermo Quest, Waltham, MA, USA) z pułapką jonową, wyposażonym
w kolumnę BPX-35, 30m x 0,025mm I.D x 0,25
μm FT (Phenomenex, Torrance, CA, USA).
Jako gaz nośny został użyty hel o czystości 99,99999%
i przepływie 0,8 ml/min. Temperatura
dozownika wynosiła 200
o
C, programowalna temperatura kolumny: izoterma 80
o
C – 2 min,
przyrost 5
o
C/min, izoterma końcowa 320
o
C - 15 min, temperatura linii transferowej – 310
o
C,
zakres m/z -110 – 310.
4.3.2. Analityczny wysokosprawny chromatograf cieczowy (HPLC)
Pomiary i analizy chromatograficzne HPLC wykonywane były na aparaturze firmy
Knauer (Berlin, Niemcy) składającej się z:
pompy dwutłokowej Smartline 1000 (zakres przepływu 0,001 - 50ml/min). Pompa
przystosowana jest do pracy w układzie czterokanałowego gradientu niskociśnieniowego,
uniwersalnego interfejsu Smartline Manager 500, zawierającego degazer z opatentowaną
technologią Teflon,
mieszadła magnetycznego,
kolumny analitycznej – Hypresil (5
μm, 250x3 mm I.D),
termostatu Smartline 400 (zakres temperatur 5 ÷ 85 ºC),
dozownika o objętość pętli -
20µl,
detektora spektrofotometrycznego z matrycą diodową (DAD) Smartline 2600, zakres
pracy lampy 190 – 510 nm,
detektora fluorescyjnego (Schimadzu, Tokio, Japonia),
programu do naboru i obróbki danych Eurochrom 2000, zainstalowanego na komputerze
kompatybilnym z IBM PC.
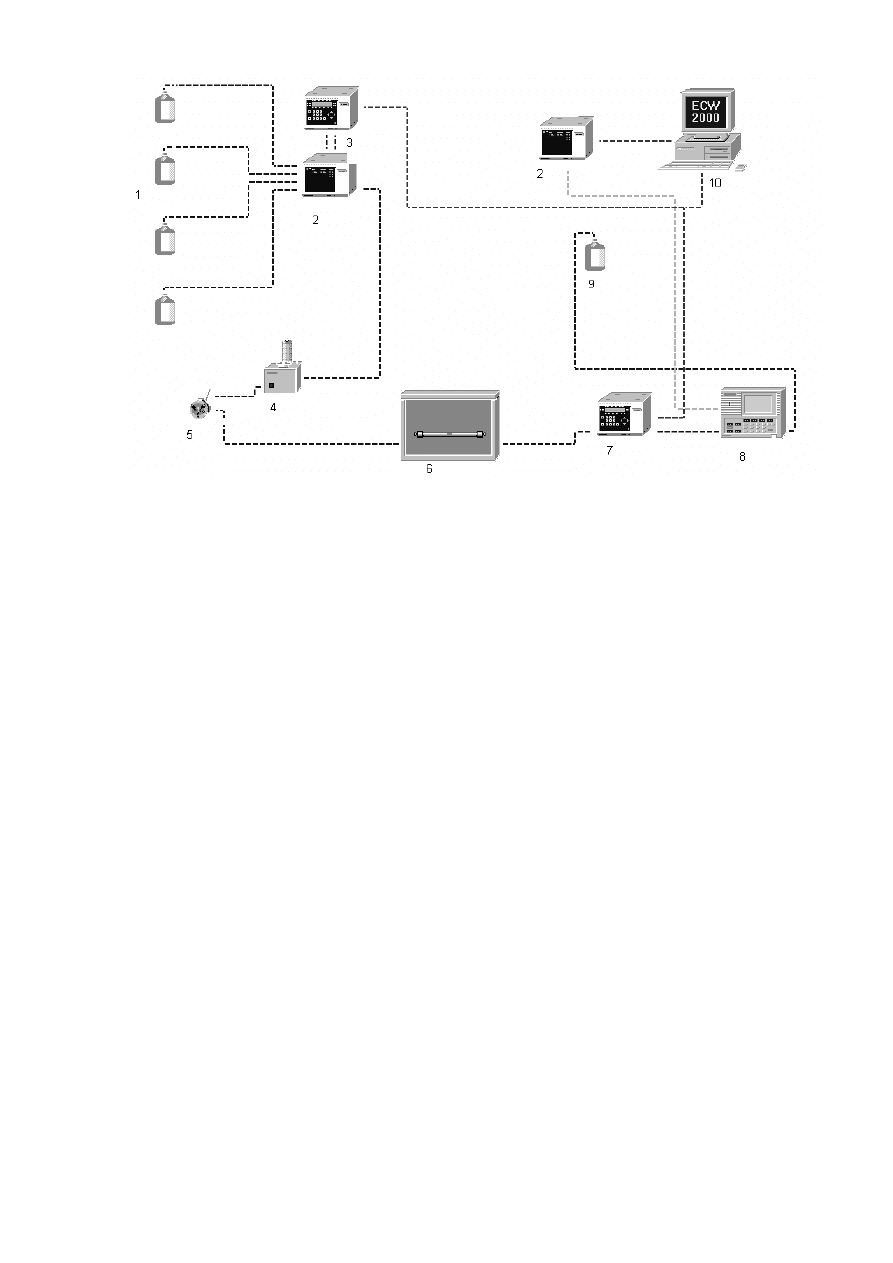
Rysunek 2. Schemat zestawu HPLC: 1 - butle ze składnikami fazy ruchomej, 2 - interfejs, 3 -
pompa, 4 - mieszadło, 5 - dozownik, 6 - termostat z kolumną, 7 - detektor spektrofotometryczny,
8 - detektor fluorescencyjny, 9 - butla na ścieki i 10 - komputer.
4.2. Odczynniki
W pracy stosowałam następujące odczynniki:
- acetonitryl, czysty do HPLC, 99,9 % (Sigma – Aldrich, Niemcy),
- metanol, czysty do HPLC, 99,9% (Sigma – Aldricht, Niemcy),
- dichlorometan, czysty do HPLC
(Labscan, Irlandia),
- certyfikowany roztwór cyklopenta[c,d]pirenu (50 ng/µl w acetonitrylu); dibenzo[a,i]pirenu (30
ng/µl w acetonitrylu); dibenzo[a,h]pirenu (30 ng/µl w acetonitrylu); dibenzo[a,l]pirenu (30
ng/µl w acetonitrylu); dibenzo[a,e]pirenu (30 ng/µl w acetonitrylu); 5-metylochryzenu (50
ng/µl w acetonitrylu); benzo[j]fluorantenu (10 ng/µl w acetonitrylu); benzo[a]pirenu (10 ng/µl
w acetonitrylu), dibenzo[a,h]antracenu (30 ng/µl w acetonitrylu), indeno [1,2,3-cd]pirenu (10
ng/µl w acetonitrylu), benzo[ghi]perylenu (10 ng/µl w acetonitrylu), benzo[k]fluorantenu (30
ng/µl w acetonitrylu), benzo[b]fluorantenu (10 ng/µl w acetonitrylu), benzo[a]antracenu (10
ng/µl w acetonitrylu), chryzenu (10 ng/µl w acetonitrylu), (Ehrenstorfer, Augsburg, Niemcy),
22
- certyfikowany roztwór węglowodorów aromatycznych SRM 1491 (NIST, Gaithersburg, USA),
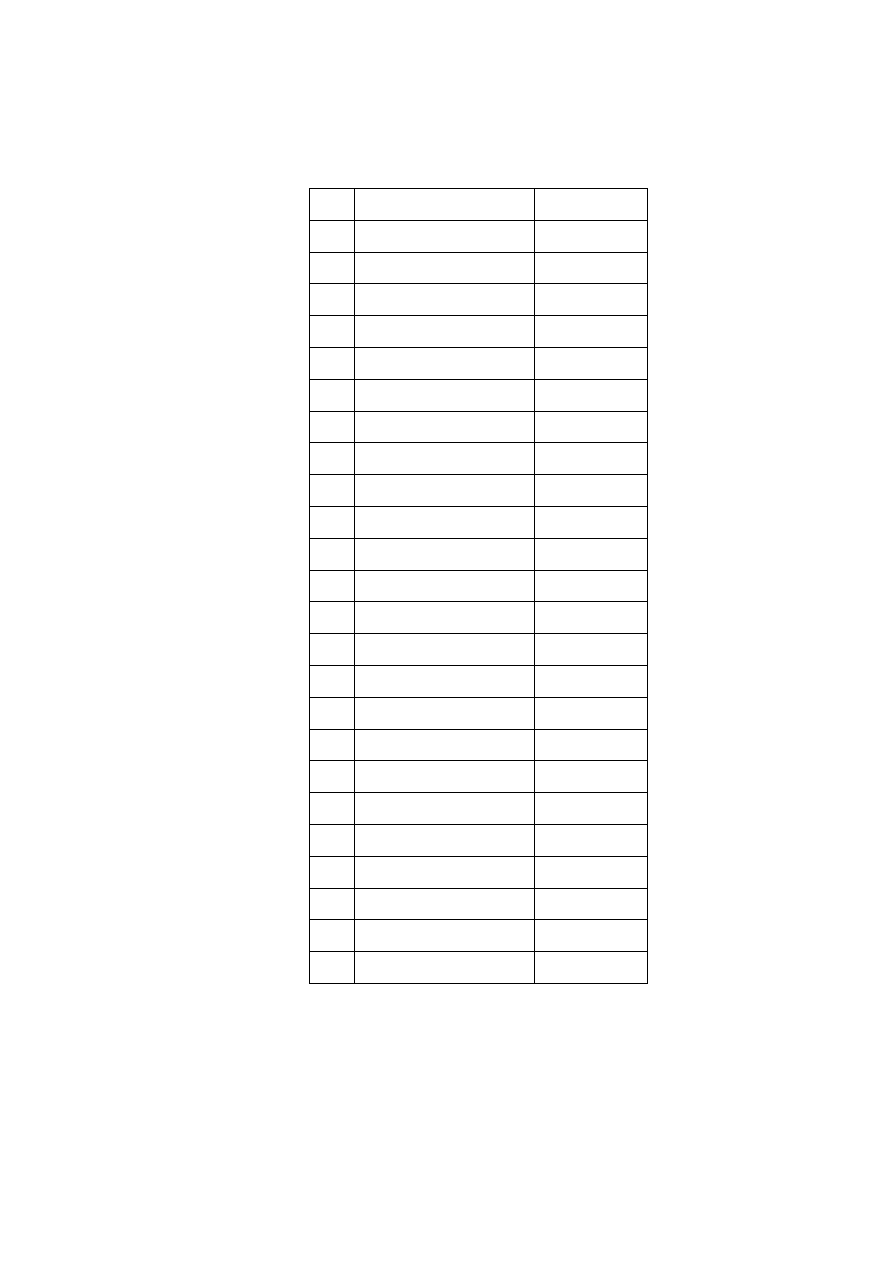
23
Tabela 5.
Stężenia poszczególnych WWA w SRM 1491.
L.p. WWA Stężenie [µg]
1 Naftalen
10,30±0,10
2 Acenaften
10,89±0,15
3 Acenaftylen
10,40±0,07
4 Fluoren
10,87±0,08
5 Feantren
10,48±0,07
6 Antracen
11,69±0,06
7 Fluorantenu
8,84±0,06
8 Piren
8,81±0,08
9 Benzo[a]antracen
5,37±0,04
10 Chryzen
10,50±0,06
11 Benzo[b]fluoranten 7,85±0,05
12 Benzo[k]fluoranten 8,33±0,12
13 Benzo[a]piren
10,14±0,09
14 Indeno[1,2,3-cd]piren 9,40±0,07
15 Dibenzo[a,h]antracen 7,74±0,18
16 Benzo[ghi]perylen
7,90±0,13
17 1-Metylonaftalen
12,4±0,5
18 Bifenyl
10,46±0,04
19 2,6-Dimetylonaftalen 10,8±0,4
20 2,3,5-Trimetylonftalen 9,9±0,4
21 1-Meteylofenantren 10,4±0,3
22 Perylen
10,65±0,06
23 2-Metylonaftalen
11,3±0,01
24 Benzo[e]piren
8,40±0,04
- certyfikowany roztwór benzo[b]chryzenu (10 ng/µl w acetonitrylu) (Ehrenstorfer, Augsburg,
Niemcy),
- materiał referencyjny: SRM 2978 (liofilizowane małże), CRM 458 (tłuszcz kokosowy) (NIST,
Gaithersburg, USA),

24
- wtórne materiały odniesienia FAPAS oliwy: 0618, 0621,0615 (Central Science Laboratory,
York, UK),
- próbki olejów jadalnych (rzepakowy rafinowany , słonecznikowy, oliwa z oliwek) zakupione
zostały w najbliższym sklepie,
- butlę ze sprężonym azotem o czystości 5,0 (99,999%),
- bufor fosforowy w tabletkach PBS (0,02M) (Phosphate Buffered Saline),
- kwas tereftalowy (TFA) (10mM) (Sigma-Aldrich, Niemcy),
- 3% wodę utlenioną (Infarm, Polska),
- siarczan (VI) żelaza (II) (10 mM) (Sigma-Aldrich, Niemcy).
4.3. Procedura
analityczna
4.3.1. Przygotowanie roztworów wzorcowych
Standardowa mieszanina 23 WWA z listy US-EPA i UE-SCF została przygotowana
przez zrobienie naważek poszczególnych WWA, a następnie ich rozpuszczenie w acetonitrylu.
Jako standard wewnętrzny stosowany był B[b]Ch.
Roztwory przechowywane były w temp. 7-8°C w ciemnym pomieszczeniu.
Zachowywały swoją ważność przez okres ok. 3 tygodni.
4.3.2. Przygotowanie próbek olei i wyodrębnienie frakcji WWA z matrycy tłuszczowej
Próbki olei jadalnych zostały zakupione w sklepach spożywczych i przygotowane do
analizy GC/MS i HPLC za pomocą chromatografii wykluczania sterycznego (SEC) w Instytucie
Przemysłu Mięsnego i Tłuszczowego w Warszawie. Do próbek olei o masie 1g dodawany był
B[b]Ch (100µl, 50µg/L roztworu), następnie całość została rozpuszczona w dichlorometanie do
objętości końcowej 5 ml. Oddzielenie części tłuszczowej próbki od frakcji węglowodorów
wykonywano na preparatywnym chromatografie cieczowym połączonym z detektorem UV-VIS
z matrycą diodową DAD. Rozdział następował na kolumnie do chromatografii preparatywnej
Plgel column (5 µm, 50 Å, 600 x 7,8 mm), która była uprzednio stabilizowana jedną godzinę.
Frakcję WWA została odparowana na łaźni wodnej o temp. 40
o
C w atmosferze azotu i
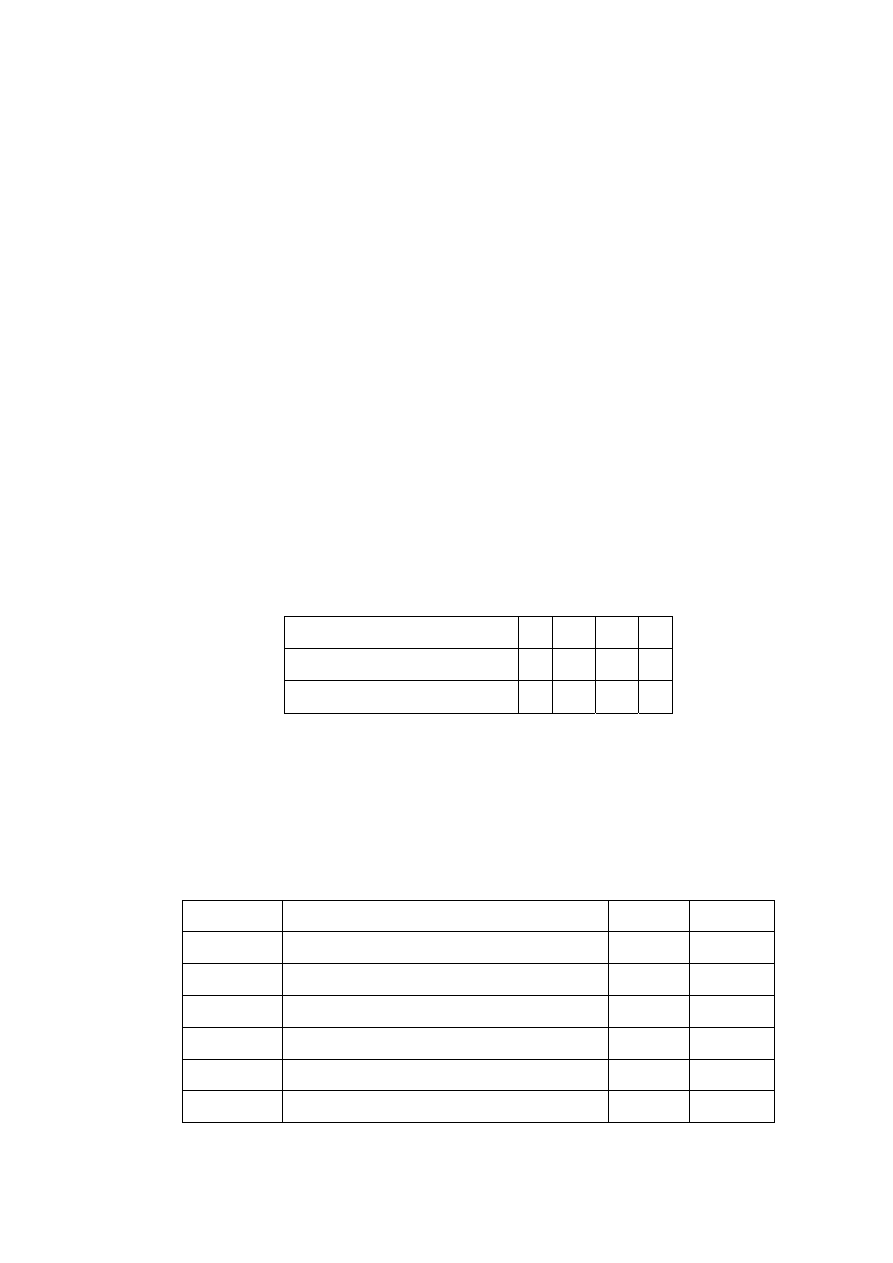
25
rozpuszczona w acetonitrylu (200 µl), a następnie poddana analizie na GC/MS i HPLC. Warunki
pracy preparatywnego chromatografu cieczowego:
- faza ruchoma - dichlorometan (temp. pokojowa),
- szybkość przepływu fazy ruchomej - 1 ml/min,
- objętość wstrzykiwanej próbki - 400 µl,
- długość fali detektora - 254 nm,
- czas zbierania frakcji WWA - 18-24 min.
4.3.3. Warunki analizy za pomocą chromatografii cieczowej
Rozdział WWA na HPLC był wykonywany w
układzie faz odwróconych (na niepolarnej
kolumnie). Warunki rozdziału zostały zoptymalizowane eksperymentalnie w oparciu o dostępne
dane literaturowe.
Warunki pracy analitycznego chromatografu cieczowego:
- Faza ruchoma:
- woda – acetonitryl lub woda – metanol o gradiencie:
Czas [min]
0 20 35 40
Woda [%]
50
0 0 50
Acetonitryl lub metanol [%] 50 100 100 50
- szybkość przepływu fazy ruchomej - 0,8 ml/min,
- objętość próbki
- 20 µl,
- pomiary były wykonywane w różnych temperaturach kolumny: 5, 10, 13, 15, 18 i 25 °C,
- długości fal wzbudzenia
(EX) i emisji (EM) detektora fluorescencyjnego:
Czas [min] Węglowodory Aromatyczne
λ
ex
[nm] λ
em
[nm ]
0 Nieanalizowane
niskocząsteczkowe WWA 248
375
13,9 B[a]A,
Ch
270
385
19,0 B[b]F
256
446
20,8
B[k]F, B[a]P, D[h]A
295
410
25,7 I[c]P
274
507
27,0 B[a]Ch
295
410

Stężenie, c
i
, węglowodoru w próbce obliczano ze wzoru (1):
(1)
m
A
V
c
A
c
S
c
S
c
s
st
st
b
S
b
b
s
i
=
gdzie:
S
b
– pole powierzchni węglowodoru w materiale certyfikowanym (SRM 1491),
S
S
– pole powierzchni benzo[b]chryzenu dodanego do materiału certyfikowanego (SRM 1491),
c
b
– stężenie węglowodoru w materiale certyfikowanym (SRM 1491) [
µ
g/kg],
c
s
– stężenie benzo[b]chryzenu dodanego do materiału certyfikowanego (SRM 1491) [
µ
g/kg],
A
b
– pole powierzchni badanego węglowodoru w próbce,
A
s
- pole powierzchni benzo[b]chryzenu dodanego do próbki,
c
st
– stężenie benzo[b]chryzenu dodanego do próbki,
V
st
– objętość roztworu benzo[b]chryzenu dodanego do próbki,
m – masa próbki [kg].
4.3.4. Przeprowadzanie reakcji B[a]P z rodnikami hydroksylowymi
W pracy zbadano możliwość usuwania benzo[a]piranu ze środowiska za pomocą
rodników hydroksylowych. Rodniki te generowane były w reakcji Fentona.
Do 500 µl buforu
fosforanowego pH = 7,4 o stężeniu c = 0,02M,
dodano 100 µl,
Fe (II) o stężeniu 10 mM, 100 µl,
3% H
2
O
2,
50 µl
10ng/µl B[a]P i uzupełniono wodą destylowaną do objętości 1000 µl. Tak
przygotowane próbki analizowano chromatograficznie. Próbki wstrzykiwano na kolumnę i
czynność tę powtarzano po upływie 2, 20 i 50 minut oraz 24 godzin.
Przygotowanie buforu fosforowego (pH=7,4).
Rozpuszczono:
27,598 g NaH
2
PO
4
,
34,838 g K
2
HPO
4
,
dopełniono wodą destylowaną do objętości 1 litra.
4.3.5. Statystyczna analiza danych
Pomiary
były wykonywane trzykrotnie. Wynikiem była średnia odniesiona do kontrolnej
próbki ślepej, niezawierającej oznaczanego węglowodoru. Wyniki były porównywane stosując t-
test Studenta dla zmiennych niezależnych przy p < 0,05.
26
5.
WYNIKI I DYSKUSJA
5.1. Przygotowanie próbki – chromatografia preparatywna
Próbki
były przygotowywane (usuwanie matrycy tłuszczowej i zatężanie) przy użyciu
preparatywnej chromatografii wykluczania sterycznego. Metoda ta wcześniej została
opracowana w Instytucie Przemysłu Mięsnego i Tłuszczowego [26]. Okazało się, że może być
ona zastosowana do bardzo różnorodnych próbek. W literaturze opisane zostały metody
przygotowania próbki oparte na kilkukrotnie powtarzanych ekstrakcjach ciecz-ciecz i/lub
ekstrakcjach na ciele stałym. Te trudności spowodowane są dużymi podobieństwami
fizykochemicznymi tłuszczy i WWA. Należy zaznaczyć, że w tym przypadku bardzo istotne jest
całkowite usunięcie lipidów gdyż uniemożliwiają one rozdział na fazach odwróconych (lipidy
nie są rozpuszczalne w rozpuszczalnikach polarnych) oraz wpływają na przesunięcie
fluorescencyjnych pasm wzbudzenia/emisji. Olbrzymią zaletą zastosowanej metody jest
możliwość przeprowadzenia oczyszczenia próbki w jednym kroku.
Przykładowy chromatogram SEC oliwy z oliwek pokazano na Rysunku 3. Okazało się,
że w przypadku oliwy zebrany eluat zawierał wprawdzie małe ilości tłuszczy, ale były one
rozpuszczalne w acetonitrylu, stosowanym jako faza ruchoma. Przesunięcie czasu zbierania
eluatu powodowało niecałkowite zbieranie WWA.
27
Rysunek 3. Chromatogram SEC oliwy z oliwek. Warunki chromatograficzne: Plgel column - 5
μm, 50 Å, 600x7,8 mm I.D.; faza ruchoma – dichlorometan, detektor spektrofotometryczny,
długość fali - 254 nm; temperatura – pokojowa, szybkość przepływu – 1 ml/min, objętość
próbki– 400
μl.
5.2. Analizy chromatograficzne GC/MS
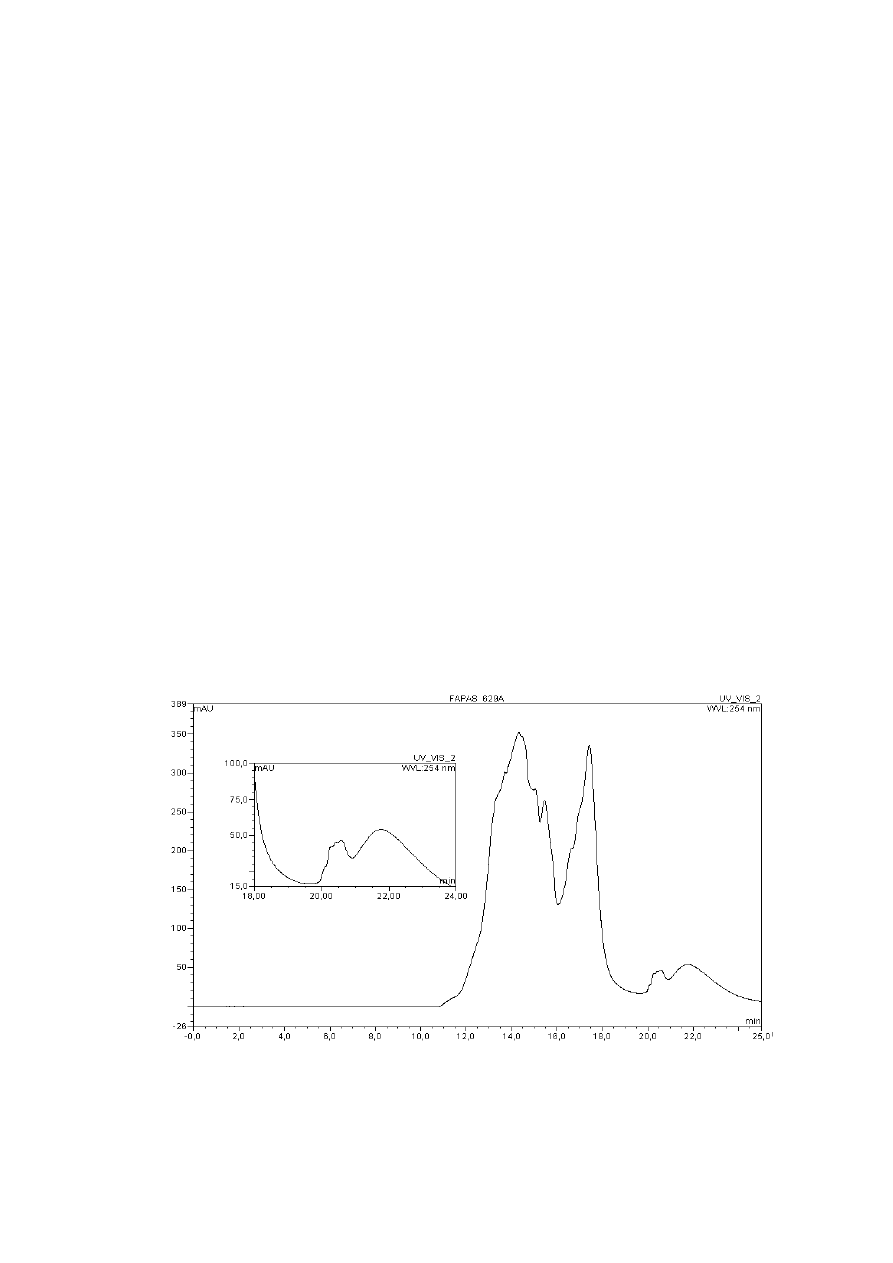
Rysunek 4. Chromatogram GC/MS (m/z=128, 140, 152,154, 166, 178, 192, 202, 226, 228, 242,
252, 276, 278) 24 WWA SRM 1491 (1 - naftalen, 2 - 2-metylonaftalen, 3 - 1-metylonaftalen, 4 -
bifenyl, 5 - 2,6-dimetylonaftalen, 6 - acenaftylen, 7 - acenaften, 8 - 2,3,5-trimetylonaftalen, 9 -
fluoren, 10 - fenantren, 11 - antracen, 12 - 1-metylofenantren, 13 - fluoranten, 14 - piren, 15 -
benzo[a]antracen, 16 - chryzen, 17 - benzo[b]fluoranten, 18 - benzo[k]fluoranten, 19 -
benzo[e]piren, 20 - benzo[a]piren, 21 - perylen, 22 - indeno[1,2,3-cd]piren, 23 -
dibenzo[a,h]antracen, 24 - benzo[ghi]perylen). Warunki chromatograficzne zostały
przedstawione w opisie aparatury (4.1.2.)
28
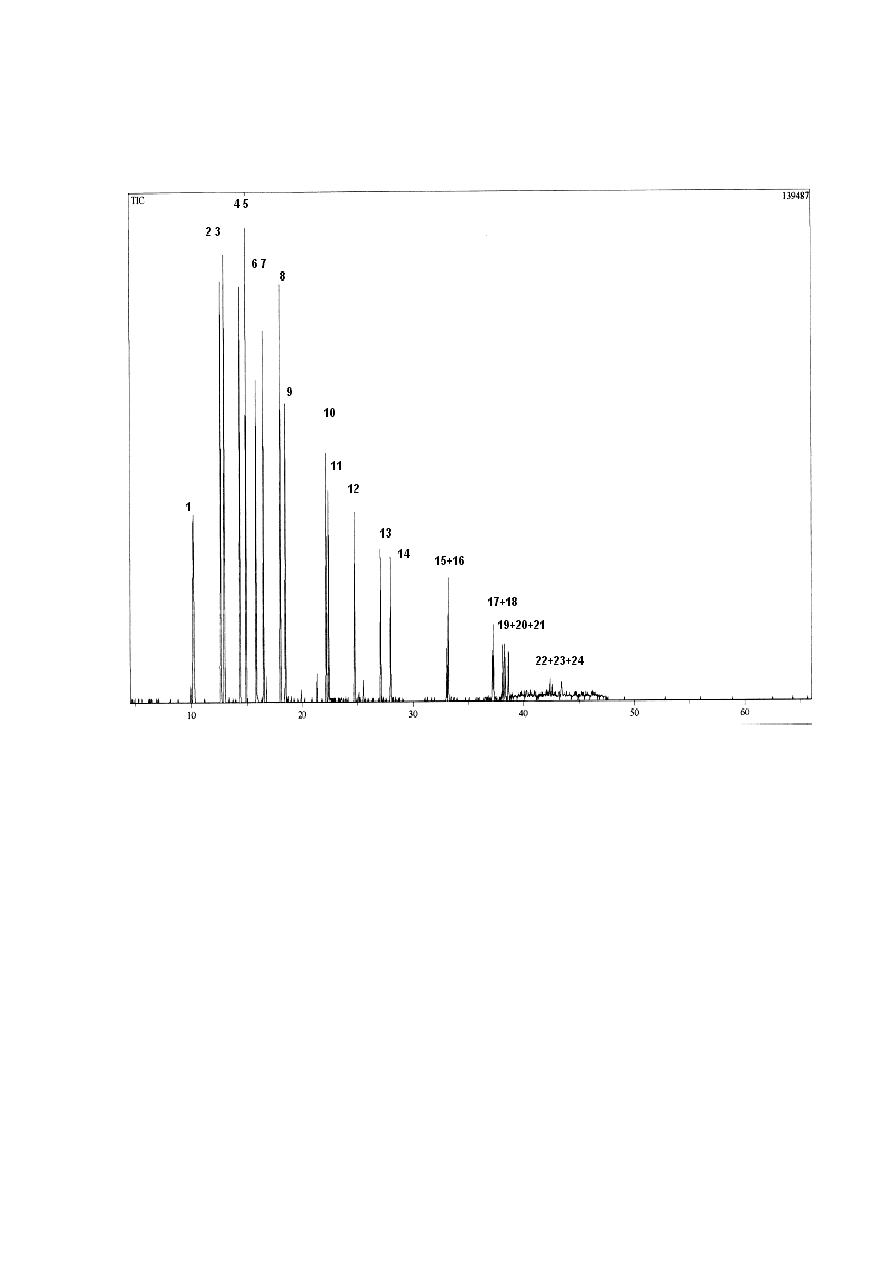
Rysunek 5. Chromatogram GC/MS (m/z=128, 152, 166, 178, 202, 226, 228, 242, 252, 276, 278,
302) 23 WWA z list US-EPA i KN UE (1 - naftalen, 2 - acenaftalen, 3 - acenaftylen, 4 -
fenantren, 5 - fluoren, 6 - antracen, 7 - fluoranten, 8 - piren, 9 - cyklopenta[cd]piren, 10 -
benzo[a]antracen, 11 - chryzen, 12 - 5-metylochryzen, 13 - benzo[b]fluoranten, 14 -
benzo[j]fluoranten, 15 - benzo[k]fluoranten, 16 - benzo[a]piren, 17 - indeno[1,2,3-cd]piren, 18 -
benzo[ghi]perylen, 19 - dibenzo[a,h]antracen, 20 - dibenzo[a,l]piren, 21 - dibenzo[a,e]piren, 22 -
dibenzo[a,i]piren, 23 - dibenzo[a,h]piren). Warunki chromatograficzne zostały przedstawione w
opisie aparatury (4.1.3.).
Rysunek 6. Chromatogram GC/MS (m/z=226, 242, 252, 302) 7 WWA z listy KN UE (9 -
cyklopenta[cd]piren, 12 - 5-metylochryzen, 14 - benzo[j]fluoranten, 20 -dibenzo[a,l]piren, 21 -
dibenzo[a,e]piren, 22 - dibenzo[a,i]piren, 23 - dibenzo[a,h]piren). Warunki chromatograficzne
zostały przedstawione w opisie aparatury (4.1.3.)
29

30
W roku 1981 Amerykańska Agencja Ochrony Środowiska (EPA) ustaliła listę 16
wielopierścieniowych węglowodorów aromatycznych, które zostały wtedy uznane za
szczególnie toksyczne. Dopiero w roku 2005 Komitet Naukowy Unii Europejskiej (KN UE)
przedstawił zestawienie 15 WWA, które powinny być oznaczane zarówno w żywności jak i w
środowisku. Spowodowało to konieczność udoskonalania i opracowywania nowych metod ich
oznaczania.
Na Rysunku 4 przedstawiono chromatogram SRM 1491
WWA
.
Został on
zarejestrowany dla stosunków mas do ładunków (m/z) podanych pod rysunkami. Rysunek 5
przedstawia rozdział 23 WWA rekomendowanych do badań z list EPA i KN UE. Rysunek 6 to
chromatogram 7 WWA, które znajdują się na liście KN UE natomiast nie rekomenduje ich do
badań EPA, a prawdopodobnie są one silnie kancerogenne. Identyfikację rozdzielonych WWA
przeprowadziłam w oparciu o dostępne
dane literaturowe oraz
porównania czasów retencji
indywidualnych standardów WWA wstrzykiwanych oddzielnie. W oparciu o analizę
posiadanych standardów WWA stwierdzono, że widma masowe wielopierścieniowych
węglowodorów aromatycznych charakteryzują się intensywnym i stabilnym jonem
molekularnym (jon podstawowy) oraz wiązką jonów połówkowych o podwójnym ładunku
(m/2z=1/2m/z), co potwierdza liczna literatura dotycząca identyfikacji WWA techniką GC/MS
[29, 30]. Okazało się, że uzyskane progi wykrywalności (rzędu ppm) były za wysokie, aby
można tą techniką (mimo kilkakrotnego zatężania próbki) oznaczać WWA w próbkach
żywności. Szczególnie wysokie progi wykrywalności były obserwowane dla dibenzo
pochodnych pirenu. Wymywane one były z kolumny na końcu chromatogramu, tzn. przy
wysokiej temperaturze. W tych warunkach dochodziło do rozkładu fazy stacjonarnej kolumny.
Retencją WWA jest proporcjonalna do ich masy cząsteczkowej, a tak naprawdę do ich
temperatur wrzenia.
Okazało się, że znaczne obniżenie progu wykrywalności osiągnięto przy rejestracji
chromatogramu przy określonej wartości m/z w stosunku do chromatogramu rejestrującemu
całkowity prąd jonowy (TIC).
Pomiary wykonywane były na dwóch zestawach GC/MS, jak to zostało opisane w części
eksperymentalnej. Oba przyrządy różniły się analizatorami. W jednym z nich zastosowana była
pułapka jonowa, w drugim analizator kwadrupolowy. Niższy próg wykrywalności ok. rzędu
wielkości otrzymano na spektrometrze z pułapką jonową.
5.3. Analizy chromatograficzne HPLC
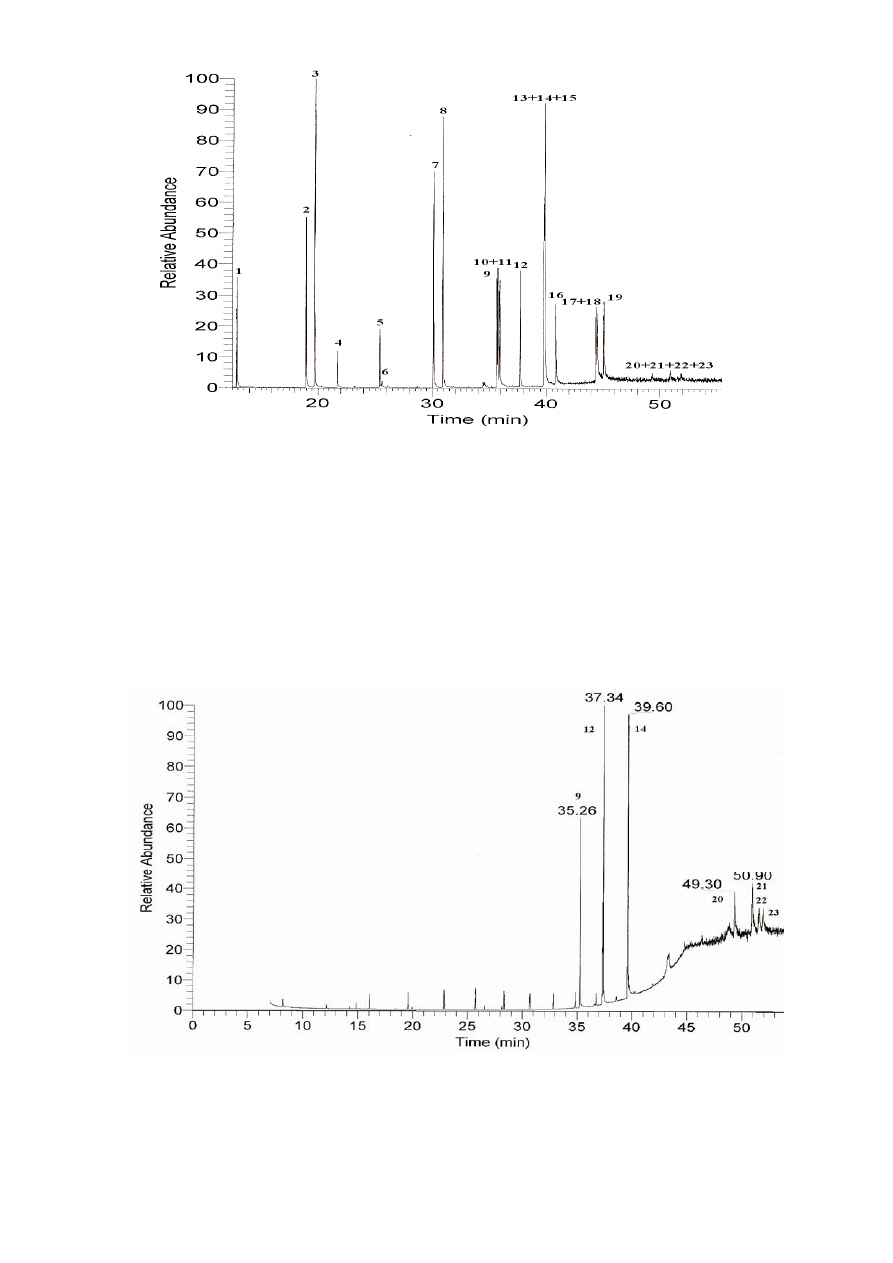
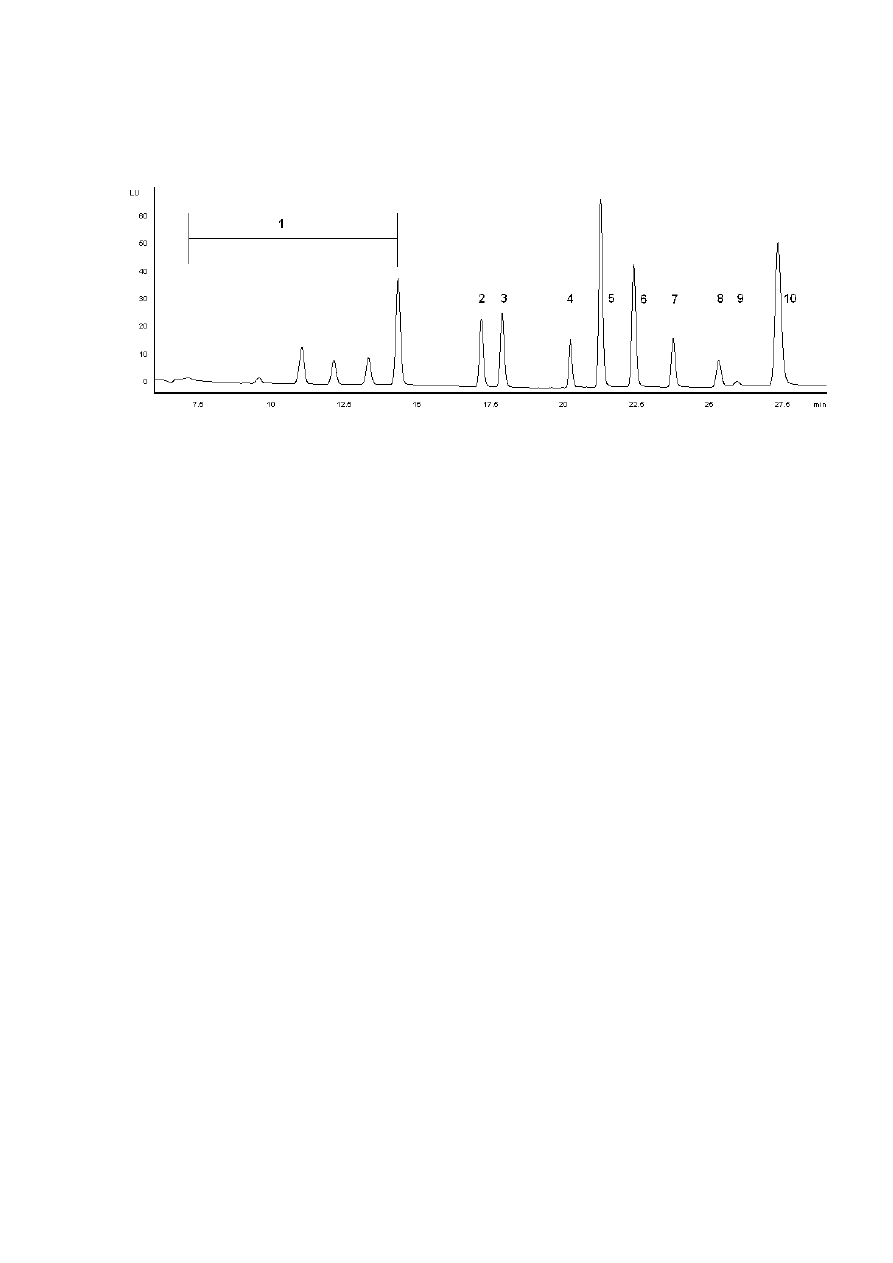
Rysunek 7. Chromatogram HPLC 16 WWA z listy EPA. SRM 1491 (1 – nie analizowane
niskocząsteczkowe WWA, 2 – benzo[a]antracen, 3 – chryzen, 4 – benzo[b]fluorantenu, 5 –
benzo[k]fluorantenu, 6 – benzo[a]piren, 7 – indeno[1,2,3-cd]piren, 8 – dibenzo[a,h]antracen, 9 –
benzo[ghi]perylen, i 10 – benzo[b]chryzen).Warunki chromatograficzne: kolumna Hypersil (5
μm, 250x3 mm I.D.), faza ruchoma : gradient acetonitrylu wody (zgodnie z tabelą podaną w
warunkach analizy za pomocą chromatografii cieczowej), detektor fluorescencyjny (długości fal
wzbudzenia i emisji podane w tabeli w warunkach analizy za pomocą chromatografii cieczowej),
temperatura - 25 ºC, przepływ – 0,8ml/min, objętość nastrzyku - 20
μl.
Ponieważ technika GC/MS charakteryzowała się zbyt wysokim progiem wykrywalności,
aby można ją zastosować do oznaczeń WWA w żywności, dlatego postanowiłam sprawdzić
możliwość zastosowania do tego celu wysokosprawną chromatografię cieczową. Z danych
literaturowych [26] wynika, że ich rozdział jest możliwy w układzie faz odwróconych.
Chromatogram 16 WWA z listy EPA (SRM 1491) został przedstawiony na Rysunku 7. Warunki
chromatograficzne (gradient elucji i parametry detektora fluorescyjnego) zostały oparte o dane
literaturowe [26]. Z rysunku tego wynika, że uzyskano dobry rozdział 8 tzw. ciężkich
węglowodorów, o prawdopodobnych właściwościach kancerogennych. Lżejsze węglowodory
były nierozdzielne w tej technice. Jednakże są one mniej interesujące z analitycznego punktu
widzenia, gdyż obecnie uważa się je za stosunkowo mało toksyczne. Ostatnim wymywanym
związkiem był B[b]Ch stosowany jako wzorzec wewnętrzny, zgodnie z równaniem (1).
Metoda wzorca wewnętrznego daje najbardziej wiarygodne wyniki, dlatego jest
najbardziej popularną metodą analizy ilościowej w chromatografii cieczowej i gazowej. Jeśli tok
31
analizy obejmuje szereg etapów wstępnych, na które składają się wielokrotne ekstrakcje oraz
oczyszczanie, wzorzec dodaje się na początku procesu analitycznego. Minimalizuje się w ten
sposób straty analitów, które mogą wystąpić na każdym etapie analizy. Zazwyczaj stosuje się
wzorzec jak najbardziej zbliżony budową strukturalną do związków znajdujących się w
analizowanej próbce. Wzorzec wewnętrzny nie jest jednym z analizowanych związków.
5.4. Oznaczanie WWA w próbkach olejów jadalnych
WWA
można oznaczać za pomocą najczęściej stosowanego w HPLC detektora -
fotometrycznego. Jednakże niższe progi wykrywalności uzyskiwane są na detektorze
fluorescencyjnym. Dla B[a]A i B[a]P wynosiły one odpowiednio 0,1 i 0,2
μg/kg są, więc
znacznie niższe niż maksymalne dopuszczalne stężenia w żywności [28]. Ich stężenia w olejach
jadalnych zostały przedstawione w Tabeli 6.
Tabela 6.
Stężenie B[a]A i B[a]P [µg/kg] w próbkach olejów jadalnych
Próbka oleju
B[a]A [µg/kg]
B[a]P [µg/kg]
Rzepakowy
ppw
ppw
Oliwa 0,5
0,7
Słonecznikowy ppw
0,3
ppw – poniżej progu wykrywalności
32
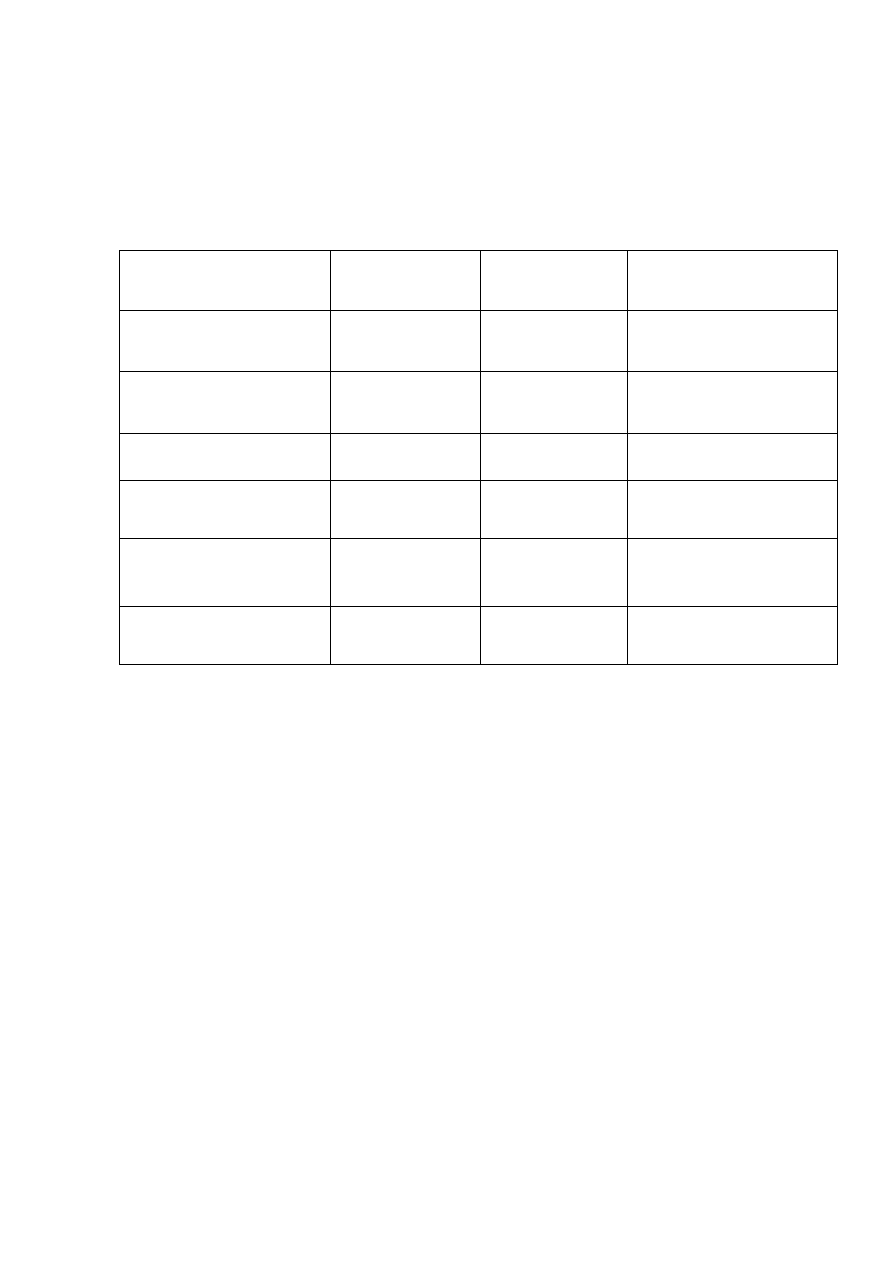
Tabela 7.
Porównanie wartości B[a]P i B[a]A oznaczonych w próbkach olejów z wartościami
rekomendowanymi przez UE.
Parametr
Wartości
wymagane
Wartości
wyznaczone
Odniesienie prawne
Granica wykrywalności
< 0,3 µg/kg
B[a]P – 0,1 µg/kg
B[a]A – 0,2 µg/kg
Zarządenie 2005/10/EC
Granica oznaczalności
< 0,9 µg/kg
B[a]P – 0,3 µg/kg
B[a]A – 0,5 µg/kg
Zarządenie 2005/10/EC
Odzysk 50 120%
÷
B[a]P 93 ÷ 106%
B[a]A 94 ÷ 104%
Zarządenie 2005/10/EC
Dopuszczalna zawartość
B[a]P w olejach i tłuszczach
2 µg/kg
D
L
= 0,1 µg/kg
Zarządzenie 208/2005/EC
Dopuszczalna zawartość
B[a]A w dymach
wędzarniczych
10 µg/kg
D
L
= 0,3 µg/kg
Zarządzenie 2065/2003/EC
Testy statystyczne wartości
odstających
Test: Cochrana,
Grubasa i Mandela
Zweryfikowano ISO
5725
Krzywe kalibracyjne charakteryzowały się dobrą liniowością dla wszystkich WWA w
zakresie stężeń 0,1÷100
µg/kg
. Precyzję metody wyznaczono poprzez sześciokrotne, w ciągu
jednego dnia oznaczanie mieszaniny związków i wyznaczenie względnego odchylenia
standardowego (RSD, n = 6). RSD dla 6 kolejnych dni nie przekraczał 12%. Uzyskane wartości
zawierały się w granicach 0,5 to 5%. Uzyskane wyniki umieszczono w tabelach danych
programu ProWL, w którym serie pomiarowe tworzą poszczególne dni wykonywania oznaczeń.
W programie ProWL uzupełniono dane z certyfikatów. Wartości odzysku od 80 do 109% dla
próbek wzmocnionych dodatkiem wzorców i materiałów odniesienia potwierdzają liniowość
stosowanej metody.
Próg wykrywalności, oznaczalności i odzysk dla benzo[a]pirenu wynosiły odpowiednio
0,1
µ
g/kg, 0,3
µ
g/kg, 93 ÷ 106%, a dla benzo[a]antracenu 0,2
µ
g/kg, 0,5
µ
g/kg, 94÷104%. Próg
wykrywalności (< 0,3
µ
g/kg), próg oznaczalności (< 0,9
µ
g/kg) i wartość odzysku (50 ÷ 120 %)
są zgodne z wytycznymi dyrektywy UE 2005/10 (02, 04, 2005).
33
5.5. Wpływ temperatury na rozdział WWA metodą HPLC
Ponieważ okazało się, że metoda GC/MS charakteryzowała się zbyt wysokim progiem
wykrywalności, dlatego dalsze pomiary przeprowadzałam z zastosowaniem HPLC w układzie
faz odwróconych z detekcją fluorescencyją. W tym przypadku progi wykrywalności były
wystarczające (poniżej ppb) do celów analitycznych. Okazało się jednakże, że nie mogłam
uzyskać rozdziału B[j]F i 5MCh poprzez zmianę składu i szybkości przepływu fazy ruchomej.
Rozdział chromatograficzny następuje wtedy, gdy
α
> 1. Współczynnik selektywności
jest wielkością termodynamiczną, co wynika ze wzoru (2).
(2)
-RTln
α
=
Δ
(
Δ
G).
Wynika z niego, że zmniejszenie temperatury zwiększa selektywność rozdziału.
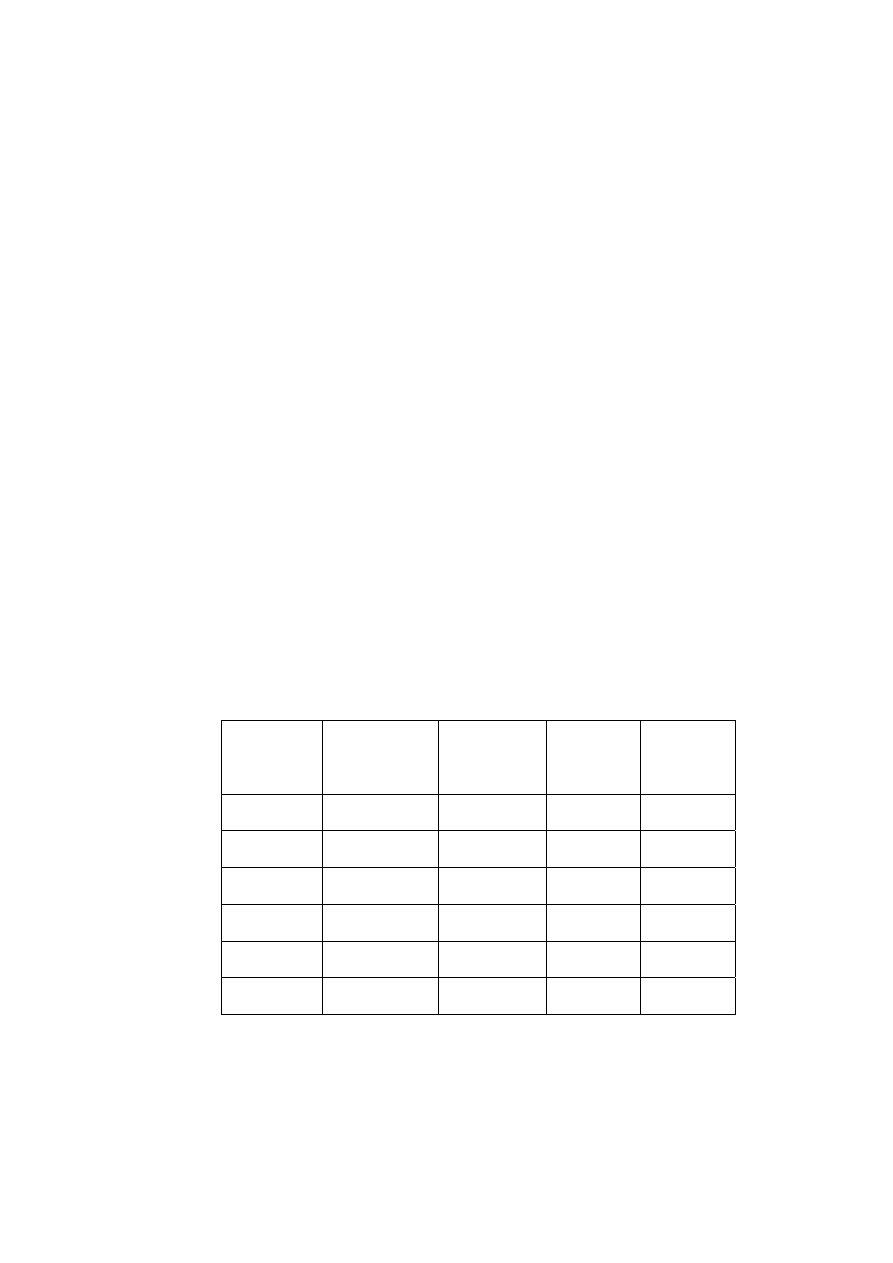
Tabela 8.
Czasy retencji 5-metylenochryzenu (5MCh) i benzo[j]fluorantenu (B[j]F) i wyliczone z nich
współczynniki rozdziału (Rs) i selektywności (α). Warunki chromatograficzne: kolumna –
Hypersil, 5µm, 250x3mm I.D; eluent - gradient acetonitrylu i wody (zgodnie z tabelą podaną w
warunkach analizy za pomocą chromatografii cieczowej); przepływ - 0,8 ml/min; objętość
wstrzykiwanej próbki - 20µl; detektor spektrofotometryczny, długość fali – 254 nm.
.
T [°C]
t
R
[min]
5MCh
t
R
[min]
B[j]F
R
S
α
5 26,70
27,62
2,10
1,038
8 27,17
27,82
2,25
1,026
13 26,15 26,54
1,49
1,016
18 25,37 26,61
1,12
1,010
23 24,47 24,56
0,42
1,004
25 23,90 23,90
0,00
1,000
34
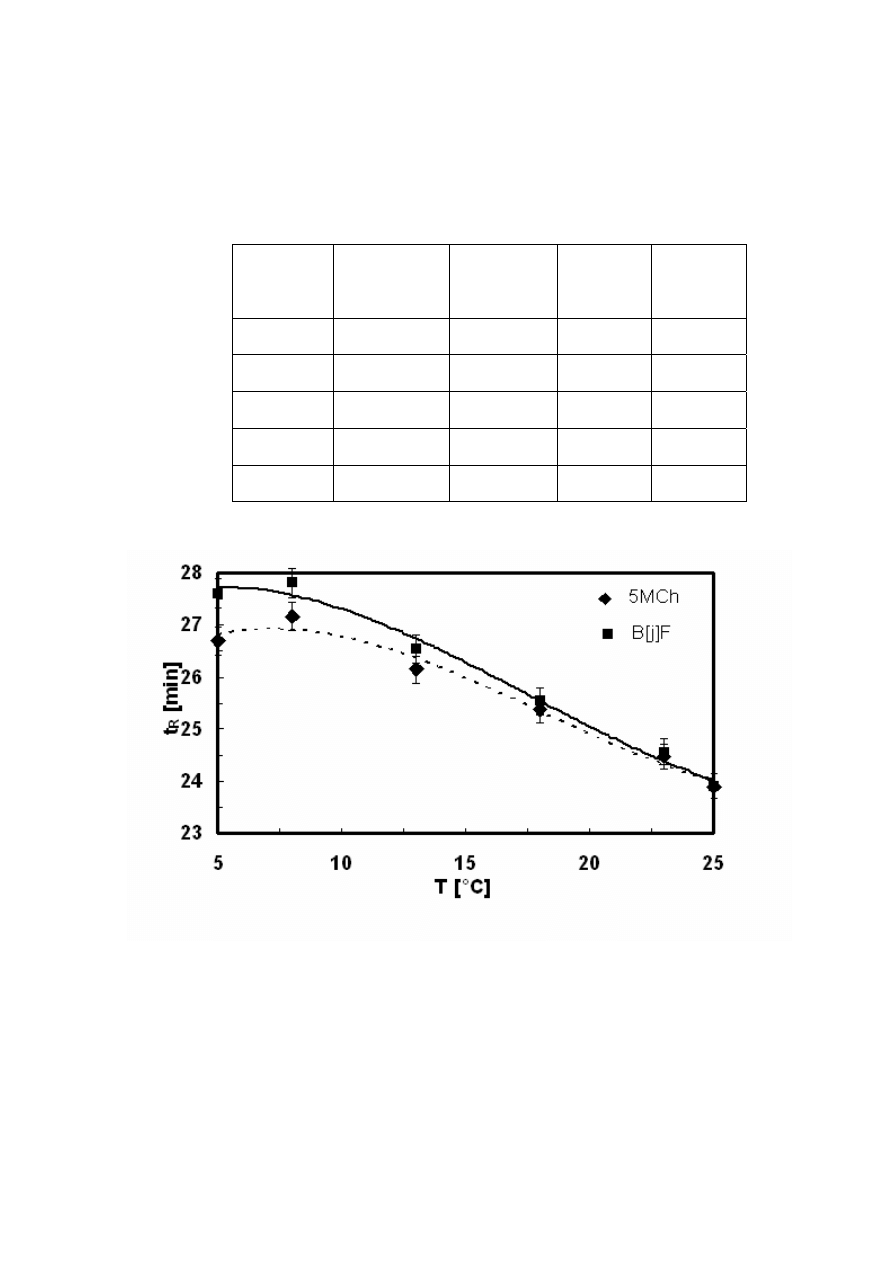
Tabela 9.
Czasy retencji 5-metylenochryzenu (5MCh) i benzo[j]fluorantenu (B[j]F) i wyliczone z nich
współczynniki rozdziału (Rs) i selektywności (α). Warunki chromatograficzne: kolumna –
Hypersil, 5µm, 250x3mm I.D; eluent - metanol; przepływ - 0,8 ml/min; objętość wstrzykiwanej
próbki - 20µl; detektor spektrofotometryczny, długość fali – 254 nm.
T [°C]
t
R
[min]
5MCh
t
R
[min]
B[j]F
R
S
α
8 11,36
12,06
1,51
1,080
10 10,40 10,89
1,20
1,061
15 9,79 10,10
0,96
1,040
18 9,40 9,54
0,45
1,020
23 9,00 9,00
0,00
1,000
Rysunek 8. Zależność czasu retencji (t
R
) od temperatury (T) dla 5-metylenochryzenu (5MCh) i
benzo[j]fluorantenu (B[j]F). Warunki chromatograficzne: kolumna – Hypersil, 5µm, 250x3mm
I.D; eluent - gradient acetonitrylu i wody (zgodnie z tabelą podaną w warunkach analizy za
pomocą chromatografii cieczowej); przepływ - 0,8 ml/min; objętość wstrzykiwanej próbki -
20µl; detektor spektrofotometryczny, długość fali – 254 nm.
35
Rysunek 9. Zależność czasu retencji (t
R
) od temperatury (T) dla 5-metylenochryzenu (5MCh) i
benzo[j]fluorantenu (B[j]F). Warunki chromatograficzne: kolumna – Hypersil, 5µm, 250x3mm
I.D; eluent - metanol; przepływ - 0,8 ml/min; objętość wstrzykiwanej próbki - 20µl; detektor
spektrofotometryczny, długość fali – 254 nm.
36
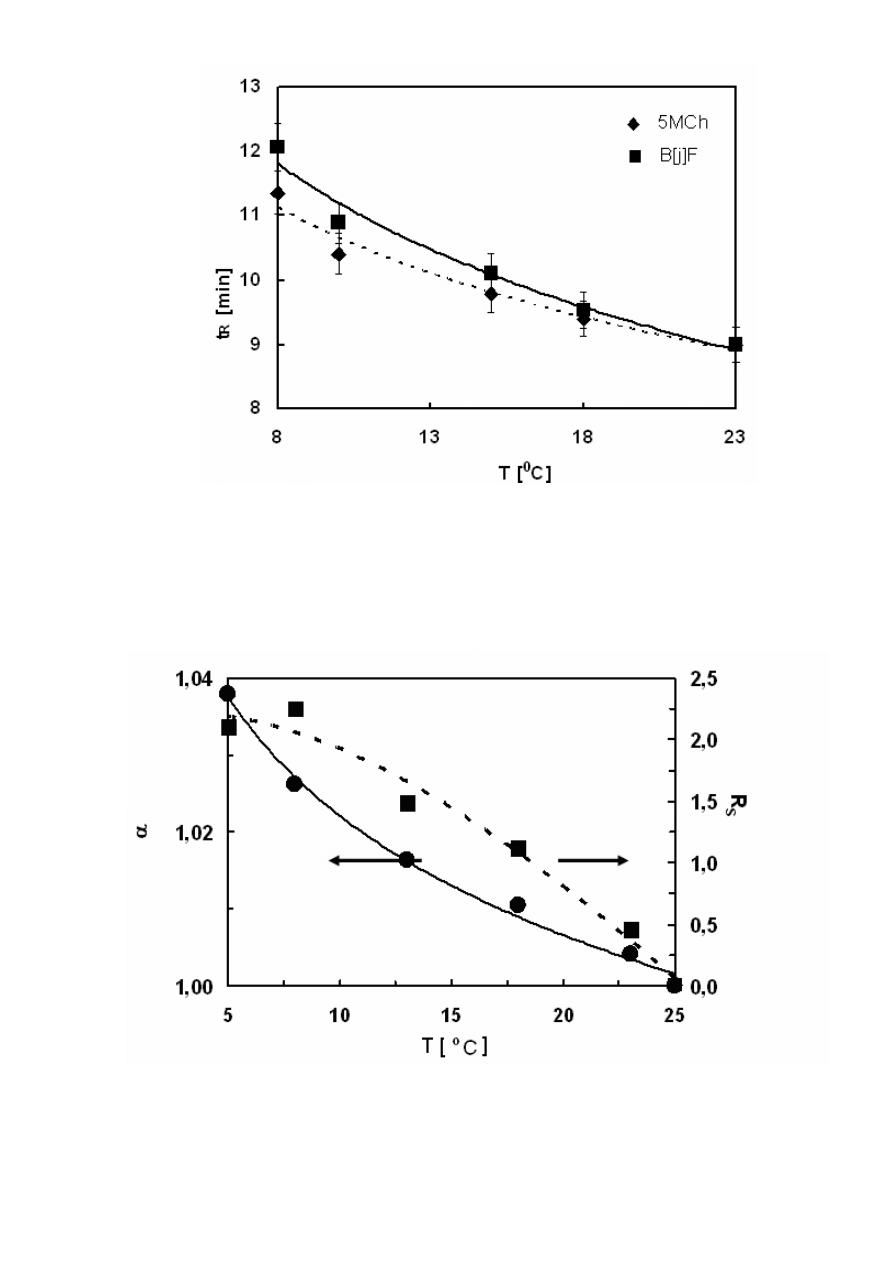
Rysunek10. Zależność selektywności (α) i współczynnika rozdziału (R
S
) 5-metylenochryzenu
(5MCh) i benzo[j]fluorantenu (B[j]F) od temperatury. Warunki chromatograficzne jak na
Rysunku 8.
1,00
1,05
1,10
1,15
8
10
12
14
16
18
20
22
T [
o
C]
α
0,00
0,40
0,80
1,20
1,60
R
S
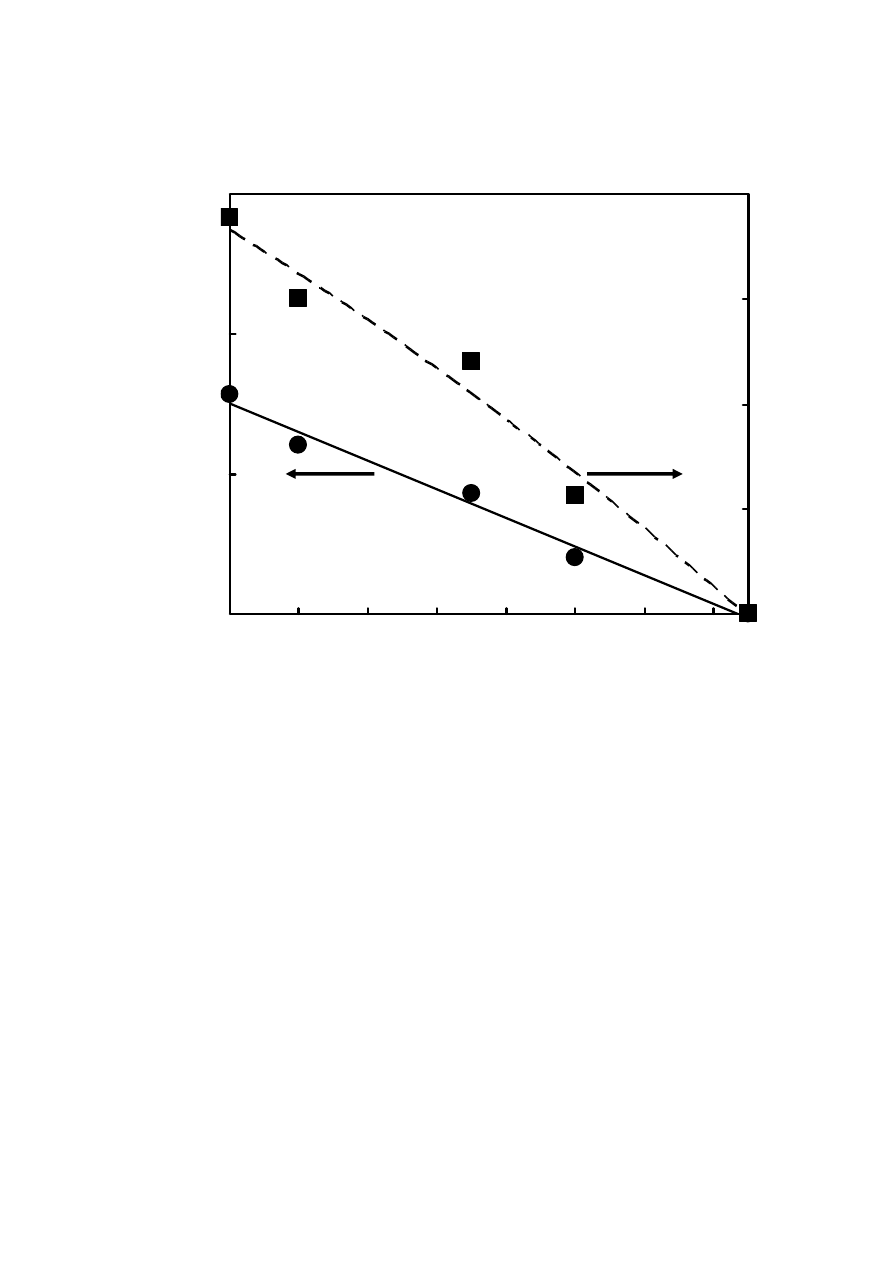
Rysunek11. Zależność selektywności (α) i współczynnika rozdziału (R
S
) 5-metylenochryzenu
(5MCh) i benzo[j]fluorantenu (B[j]F) od temperatury. Warunki chromatograficzne jak na
Rysunku 9.
37
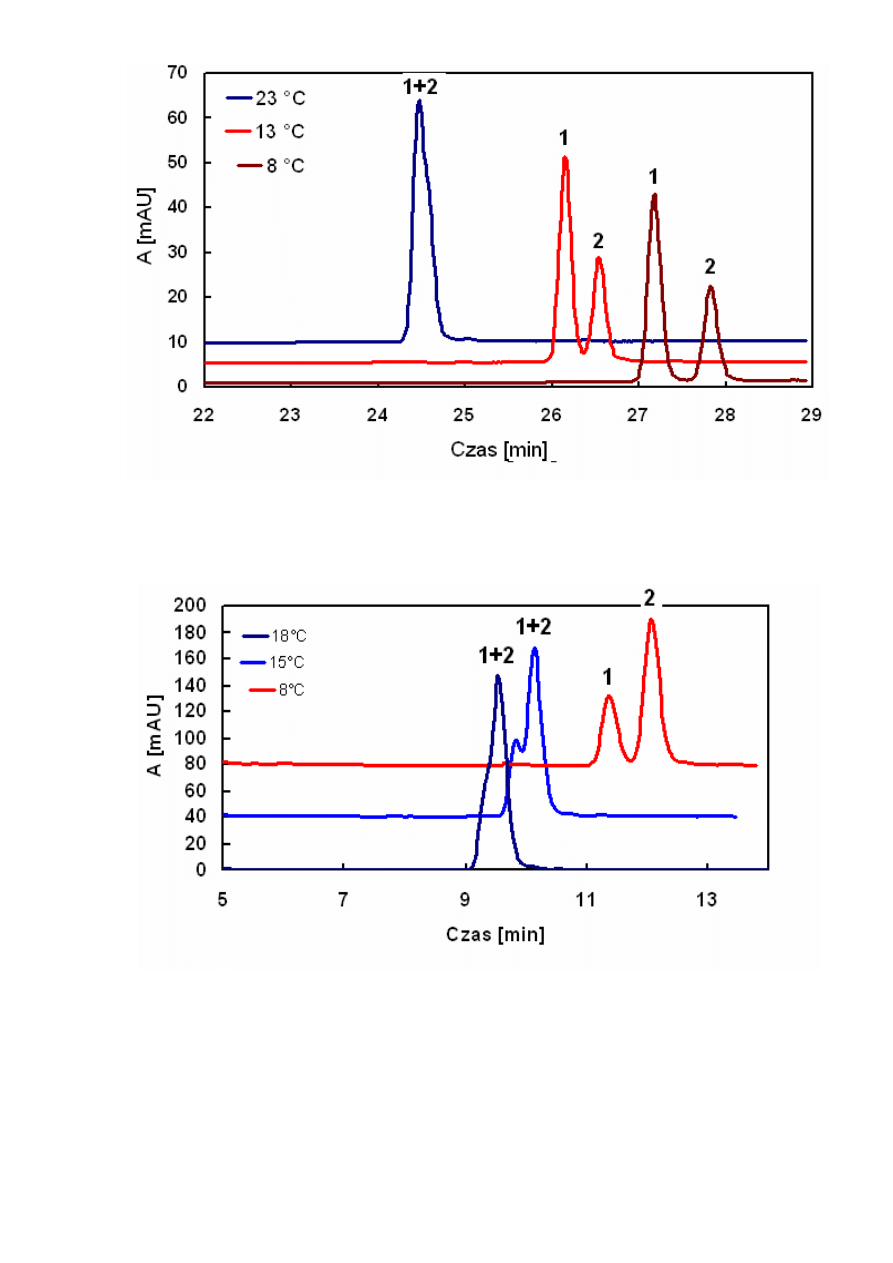
Rysunek 12. Chromatogramy HPLC 2 WWA zarejestrowane w różnych temperaturach (1 – 5-
metylochryzen, 2 –
benzo[j]fluoranten). Warunki chromatograficzne jak na Rysunku 8.
Rysunek 13. Chromatogramy HPLC 2 WWA zarejestrowane w różnych temperaturach (1 – 5-
metylochryzen, 2 – benzo[j]fluoranten).Warunki chromatograficzne jak na Rysunku 9.
38
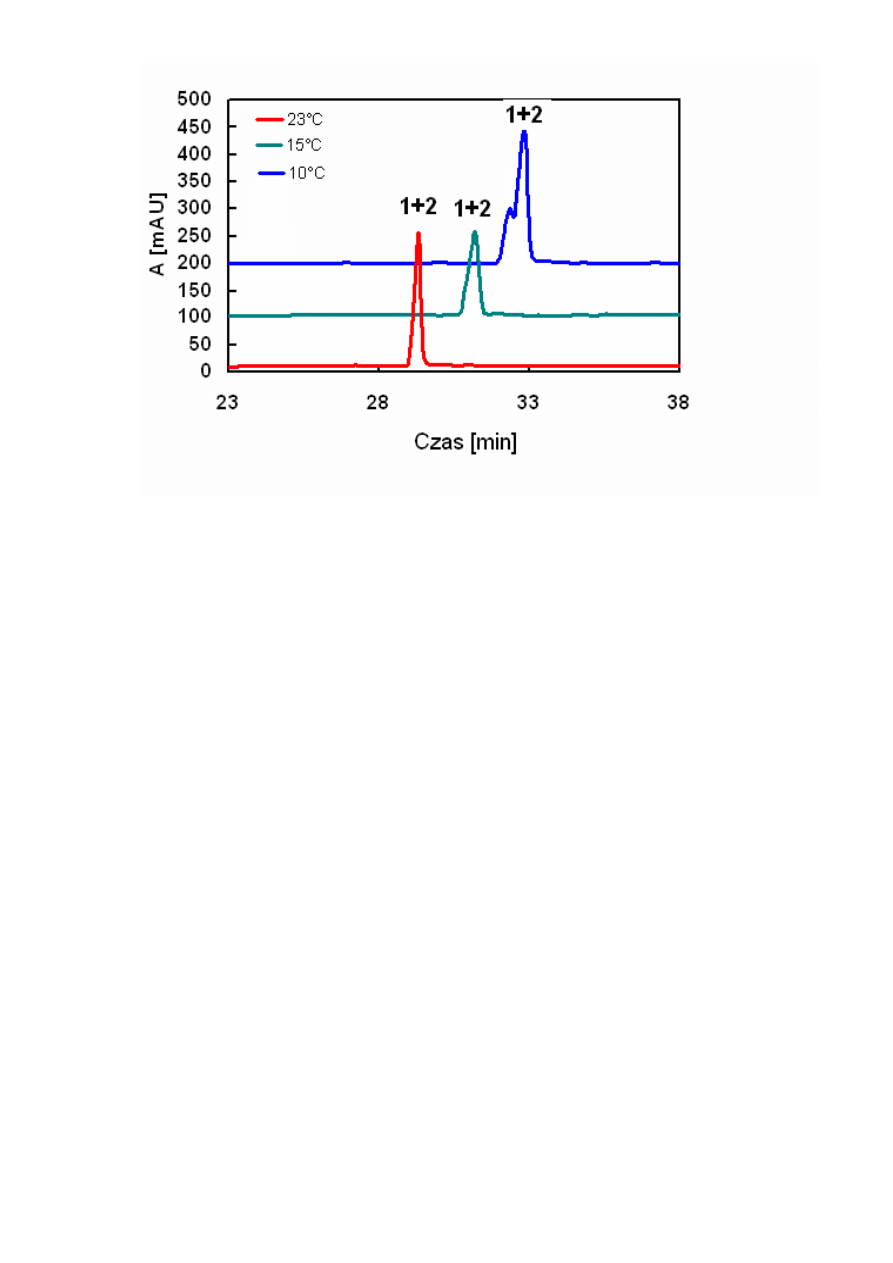
Rysunek 14. Chromatogramy HPLC 2 WWA zarejestrowane w różnych temperaturach (1 – 5-
metylochryzen, 2 –
benzo[j]fluoranten).Warunki chromatograficzne: kolumna – Hypersil, 5µm,
250x3mm I.D; eluent - gradient metanolu i wody (zgodnie z tabelą podaną w warunkach analizy
za pomocą chromatografii cieczowej); przepływ - 0,8ml/min; objętość wstrzykiwanej próbki -
20µl; detektor spektrofotometryczny, długość fali
-
254 nm.
Z Rysunków 12, 13 i 14 otrzymanych odpowiednio dla układów woda/acetonitryl,
metanol i woda/metanol wynika, że obniżenie temperatury zwiększało retencję. Ten wzrost był
różny dla obu badanych związków. Spowodowało to wzrost selektywności rozdziału, a więc i
współczynnika rozdziału (Rysunek 10 i 11). W konsekwencji umożliwiło to rozdział tych
związków, jak to zostało przedstawione na Rysunku 12 i 13. Krótsze czasy retencji otrzymano
dla fazy ruchomej acetonitryl/woda niż metanol/woda, co wiąże się z mniejszą polarnością
(mniejszymi wartościami współczynnika Hildebranda) acetonitrylu.
39
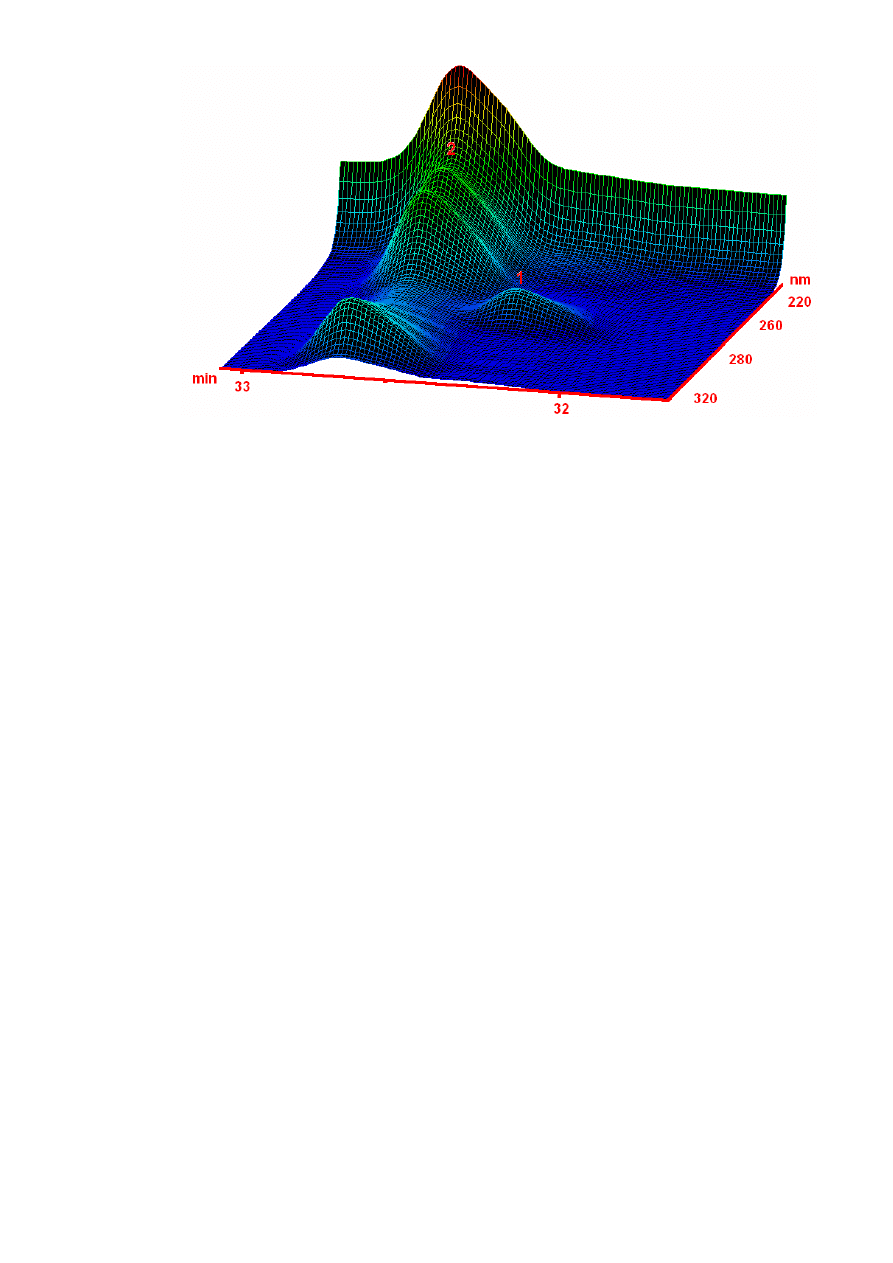
Rysunek 15. Trójwymiarowy chromatogram 5MCh (1) i B[j]F (2) zarejestrowany za pomocą
detektora z matrycą diodową.
Detektor z matrycą diodową pozwala dodatkowo na uzyskiwanie widma badanych
związków. Na Rysunku 15 został przedstawiony chromatogram zarejestrowany za pomocą
detektora z matrycą diodową. Widać na nim, że przy 254 nm (zwykle stosowanych do
oznaczania związków organicznych) pik chromatograficzny 5MCh zlewa się ze znacznie
wyższym pikiem B[j]F. Natomiast przy 273 nm pik chromatograficzny 5MCh jest wyższy niż
B[j]F i nadaje się do oznaczeń ilościowych.
5.6. Walidacja metody
W procesie walidacji metody wykorzystano dwa materiały odniesienia o różnych
zawartościach benzo[a]piranu i benzo[a]antracenu (liofilizowane małże SRM 2978, tłuszcz
kokosowy CRM 458), oraz trzy wtórne materiały odniesienia FAPAS 0615, 0618 i 0621. W
zastosowanych materiałach B[a]P występował w 5 różnych stężeniach (poziomach), a B[a]A w
3. Dla B[a]P próbki do analizy przygotowano w trzech seriach od 2 do 5 powtórzeń, natomiast
dla B[a]A przygotowano 4 serie od 2 do 5 powtórzeń. Uzyskane wyniki umieszczono w tabelach
danych programu ProWL, w którym serie pomiarowe tworzą poszczególne dni wykonywania
oznaczeń. W programie ProWL uzupełniono dane z certyfikatów.
40
41
Tabela 6.
Materiały odniesienia
Materiał odniesienia
B[a]A [µg/kg] B[a]P [µg/kg]
Wartość certyfikowana
-
0,97±0,07
CRM 458
tłuszcz kokosowy
Wynik pomiarów
-
0,96
Wartość certyfikowana
25±7
7±3
SRM 2978
liofilizowane małże
Wynik pomiarów
26,97
7,85
Tabela 7.
Wtórne materiały odniesienia
Wtórny materiał odniesienia
B[a]A [µg/kg] B[a]P [µg/kg]
Wartość certyfikowana
43,2±9,51
28,2±6,21
FAPAS 0621
oliwa
Wynik pomiarów
40,7
25,59
Wartość certyfikowana
-
2,36±0,52
FAPAS 0615
oliwa
Wynik pomiarów
-
2,23
Wartość certyfikowana
38,75±8,52
18,66±4,1
FAPAS 0618
oliwa
Wynik pomiarów
22,05
20,73
5.7. Utlenianie B[a]P przez rodniki hydroksylowe
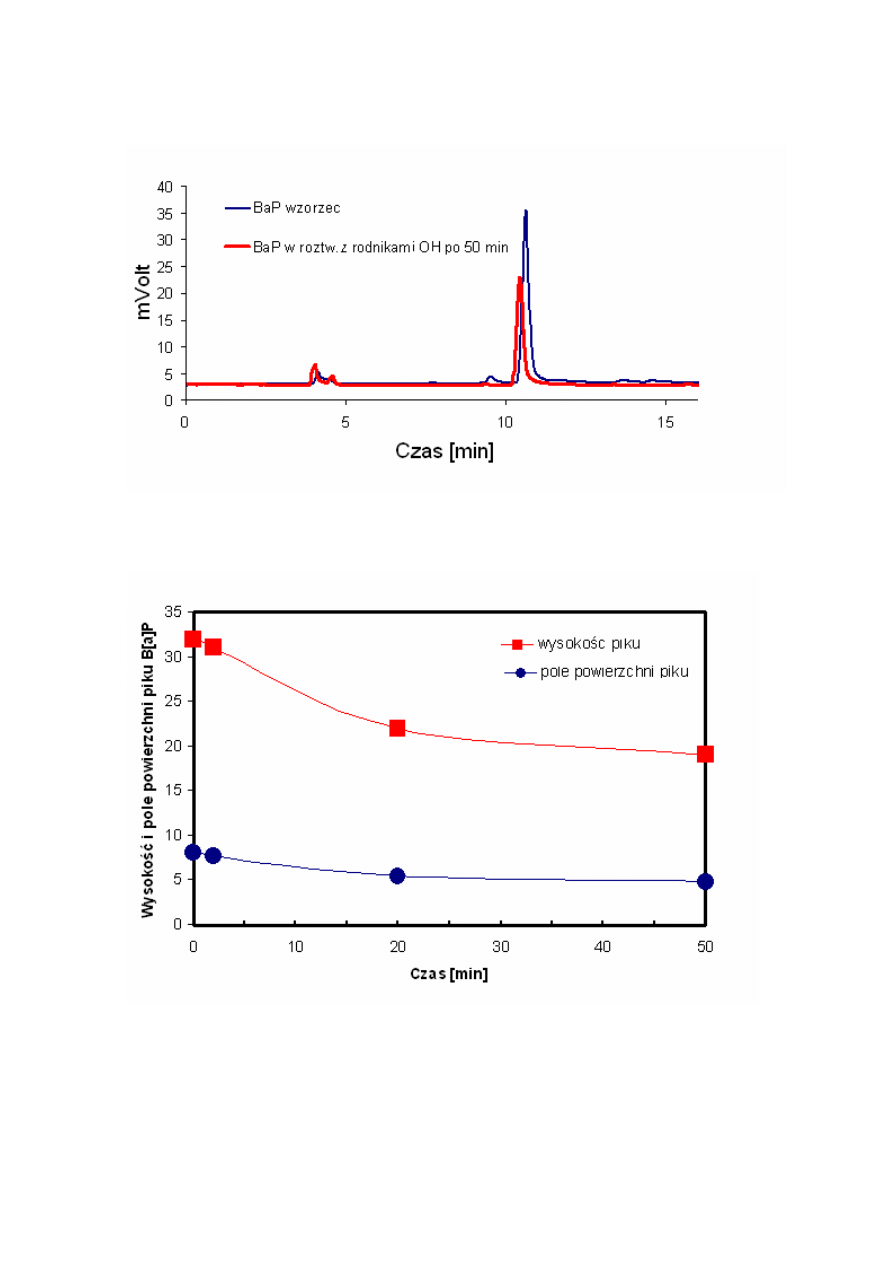
Rysunek 16. Obserwowane zmniejszenie wysokości piku roztworu B[a]P po upływie 50 minut.
Rysunek 17. Zmniejszenie
wysokości piku i pola powierzchni piku B[a]P po 3, 20 i 50 minutach
reakcji.
42

Istotnym problemem związanym z ochroną środowiska jest usuwanie z niego związków
toksycznych. W literaturze opisane są metody ich rozkładu oparte na ich utlenianiu ozonem,
działaniem promieniowania UV czy ultradźwięków [31].
Ostatnio opisano również metody
utleniania fenoli za pomocą rodników hydroksylowych. Rodniki te generowane są w reakcji
Fentona [32, 33]:
H
O
OH
Fe
O
H
Fe
-
3
2
2
2
•
+
+
+
+
=
+
Jako modelowy związek do zbadania reakcji wybrany został benzo[a]piren.. Okazało się,
że stężenie, (przedstawione w postaci wysokości piku) B[a]P po 50 min. reakcji zmniejszyło się
o połowę (Rysunek 16). Powyższe badania należy traktować jako pilotażowe. Dobór parametrów
reakcji może zwiększyć wydajność metody.
43

44
6. WNIOSKI
1. Metoda HPLC z detekcją fluorescencyjną okazała się znacznie bardziej czuła niż GC/MS.
2. Obniżenie temperatury kolumny poprawiło selektywność układu i umożliwiło rozdział B[j]F
i 5MCh.
3. Próg wykrywalności, oznaczalności i odzysk dla benzo[a]pirenu wynosiły odpowiednio 0,1
µ
g/kg, 0,3
µ
g/kg, 93÷106%, a dla benzo[a]antracenu 0,2
µ
g/kg, 0,5
µ
g/kg, 94÷104%. Próg
wykrywalności (< 0,3
µ
g/kg), próg oznaczalności (< 0,9
µ
g/kg) i wartość odzysku (50 ÷ 120
%) są zgodne z wytycznymi dyrektywy UE 10/2005 (02, 04, 2005).
4. Rodniki hydroksylowe mogą być stosowane do usuwania B[a]P ze środowiska, aczkolwiek
metoda ta wymaga jeszcze dopracowania.

45
7. BIBLIOGRAFIA
1.
European Union Commission Recomendation 2005/108/EC. L 34, 43, 2005.
2. Environmental Protection Agency (EPA) of the United States of America, Compedium
Method TO-13, EPA, Cincinnati, OH, USA, (http://www.epa.gov/epahome/index), 1981.
3. P. Welling, B. Kaandrop,
Determination of polycyclic aromatic hydrocarbons (PAH) in
edible vegetable oils by liquid chromatography and programmed fluorescence detection.
Comparison off caffeine complexation and XAD – 2 chromatography sample clean-up, Z
Lebensm. Unters. Forsch, 183, 111-15, 1986.
4.
S. Brzeźnicki, M. Jakubowski, B. Czerski,, Elimination of 1-hydroxypyrene After Human
Volunteer Exposure to Polycyclic Aromatic Hydrocarbons, J. Environ. Health., 70, 257-260,
1997.
5. P. Simko,
Determination of polycyclic aromatic hydrocarbons in smoked meat products and
smoke flavoring food additives. J. Chromatogr. A, 770, 3-8, 2002.
6.
M. Obiedziński, Wybrane zagadnienia zanieczyszczenia żywności wielopierścieniowymi
węglowodorami aromatycznymi (WWA). Post. Hig. Dośw., 39: 660-676, 1985.
7. S. F. Zakrzewski,
Podstawy toksykologii środowiska, PWN, Warszawa, 114,
1997.
8. D. M. Wagrowski, R. A. Hitem ,
Policyclic aromatic hydrocarbons accumulation In Urban,
suburban, and rural vegetation, Environ. Sci. Techn., 31, 1, 279-282, 1997.
9. S. Mitra, B. Ray,
Paterns and sources of polycyclic aromatic hydrocarbons and their
derivatives in idoor air. Atm. Environ., 22, 3345-335, 1995.
10. M. D. Guillen, P. Sopelana,
Polycyclic aromatic hydrocarbons in diverse foods reviews on, J.
Environ. Health, 12, 133-145, 1997.
11. A. Barranco, R. M. Alozo-Salces, A. Bakkali, L. A. Berruta, B. Gallo, F. Vonocenta,
M.Sarobe,
Solid-phase clean up in the liqid chromatography determination of polycyclic
aromatic hydrocarbons, J. Chromatogr. A, 67, 33-40, 2003.
12. P. S. Jankowski,
Analiza zanieczyszczenia wielopierścieniowymi węglowodorami
aromatycznymi (WWA) wybranych grup artykułów rolno spożywczych, praca doktorska,
Instytut Przemysłu Mięsnego i Tłuszczowego, Warszawa, 2004.
13. Agence for Toxic Substances And Disease Registry (ATSDR),
Polycyclic aromatic
hydrocarbons, Public Health Statement, 1990.
14.
R. K. Wolff i in., Effects of Repeated Inhalation exposures to 1-nitropyrene, Benzo(a)pyrene,
Ga203 Particles and SO2 Alone and in Combinations on Particle Clearance,

46
Bronchoalveolar Lavage Fluid Composition, and Histopathology, J. Toxicol. Environ.
Health, 27, 123-138, 1989.
15.
I. C. T. Nisbet, P. K. LaGoy, Toxic (TEFs) for polycyclic aromatic hydrocarbons (PAHs),
Toxicol. Pharmacol., 16, 290-300, 1992.
16. IARC,
Monographs on the Carcinogenic Risk of Chemicals to Humans, 32, 1983.
17. IARC, International Agency for Research on Cancer,
Monographs on the evaluation of
carcinogenic risk of the chemical to humans. Polynuclear aromatic compounds, Chem.
Environ. Exp. , Lyon France, 32, 1983.
18. M. D. Guilen, P. Sopelana,
Polycyclic aromatic hydrocarbons in diverse foods. Food Safety
Cont. Toxins, 17, 175-198, 1999.
19. B. Schoket.
DNA damage in humans exposed to environmental and dietary polycyclic
aromatic hydrocarbons. Mut. Res., 424, 143-153, 1999.
20. K Hemminki,
DNA adducts in humans related to occupational and environmental
exposureto aromatic compounds, IARC, 181-191, 1990.
21. K Hemminki., K Randerath., M Reddy,
Postlabelling and immunoassay analysis of
polycyclic aromatic hydrocarbons – adducts of deoxyribonucleic acid in white blood cells of
foundry workers.Scand. J. Environ. Health, 16, 158-162, 201, 1990.
22. N. Kishikawa, M Wada, N. Kuorda., S. Akiyama, K. Nakashima,
Determination of
polycyclic aromatic hydrocarbons In milk samples by high-performance liquid
chromatography witch fluorescence detection. J. Chromatogr. A, 789,
256-264, 2003.
23. S.Y.N. Yang, D. W. Conell, D. W. Havker, S. I. Kayal,
Policyclic aromatic hydrocarbons in
air, soil and vegetation in the vincinty of urban roadway, Sci. Total Environ., 102, 229 -
240, 1991.
24. W. Lijnsky,
The formation and occurrence of polynuclear aromatic hydrocarbons associated
witch food, Mut. Res., 259, 252-261, 1991.
25. M. E. Doremire, G. E. Harmon, D. E. Pratt,
3, 4-benzopyrene in charcoal grilled meats, J.
Food Sci., 622-623, 1979.
26. E. Węgrzyn, S. Grześkiewicz, W. Popławska, B. K. Głód, Udoskonalona metoda oznaczania
ośmiu WWA w olejach jadalnych przy zastosowaniu RP-HPLC-FLD oraz preparatywnej
SEC, Acta Chromatogr.,11, 2005.
27. K. Speer, A. Montag,
Polycyclishe aromatische kohlenwasserstoffe In nativen pflanzlichen
olen, Fat Sci. Techn., 163-167, 1988.
28. Regulation (EC) No. 2065/2003 of the Europen Parliament and of the Council of November
2003 on smoke flavourings used or intendent for use in foods. J. Eur. Comm. L 309

47
,http://europa.eu.int/eurlex/lex/LexUriServ/LexUriServ.do?uri=CELEX:32003R2065:EN:HT
Ml, 2003.
29. A. S. Płaziak,
Spektrometria masowa związków organicznych, Wydawnictwo Naukowe
UAM, Poznań 1997.
30. R. Simon, S. Palme, E. Anklam,
Single-laboratory validation of a gas chromatography-mass
spectrometry method for quantitation of 15 Europen priority polycyclic aromatic
hydrocarbons in spiked smoke flavourings, J. Chromatogr. A, 1103, 307-313, 2005.
31. M. Eberius, A. Berns, J. Schuphan,
Ozonation of pyrene and benzo[a]pyrene in silica and
soil -
14
C mass balances and chemical analisys of oxidation products as a first step to
exotoxicological evolution, J. Anal. Chem., 359, 274-279, 1997.
32. V.Kavitha, K. Palanivelu,
Destruction of cresolsby Fenton oxidation process, Wat. Res., 30,
62-3072, 2005.
33. F. J. Rivas, F. J. Beltron, J. F. And P. Buxeda,
Oxidation of p-hydroxybenzoic acid by
Fenton's reagent, Wat. Res., 35, 387-396, 2001.
Document Outline
- 1. WSTĘP 3
- 2. CZĘŚĆ LITERATUROWA
- 2.1. Ogólna Charakterystyka Wielopierścieniowych Węglowodorów Aromatycznych (WWA)
- 2.2. Odkrycie i występowanie WWA
- 2.3. Toksyczność WWA, absorpcja i metabolizm w organizmach żywych
- 2.4. Charakterystyka skażenia żywności WWA
- 2.5. Metody oznaczania WWA
- 2.6. Wpływ rodników hydroksylowych na benzo[a]piren
- 3. CEL PRACY
- 4. CZEŚĆ EKSPERYMENTALNA
- 5. WYNIKI I DYSKUSJA
- 6. WNIOSKI
- 7. BIBLIOGRAFIA
Wyszukiwarka
Podobne podstrony:
Oznaczanie wybranych węglowodorów aromatycznych przy zastosowaniu chromatografii gazowej(1)
Oznaczanie zawartości węglowodorów aromatycznych w paliwach dieslowych
Oznaczanie składu metodami chromatografii gazowej 1
Oznaczanie składu metodami chromatografii gazowej, AKADEMIA GÓRNICZO - HUTNICZA
wyk 4 węglow aromat
Przykłady Zmiany Oznaczeń Wybranych Gatunków Stali Niestopowych, Normy Polskie
Wycena nieruchomości ćwiczenie 2 Budowa modelu wartości nieruchomości przy zastosowaniu regresji wie
praca dyplomowa obróbka drewna przy zastosowaniu obrabiarek sterowanych numerycznie 4JPHWM2CIJ4QMD
WĘGLOWODORY AROMATYCZNE
aromaterapia, działanie i zastosowanie w kompozycji zapachowej olejku szałwiowego oraz spósób otrzym
Zabezpieczenie przy zastosowaniu kontraktu futures i koncepc
PKM9UZAS, Spo˙r˙d obliczonych wariant˙w przek˙adni pasowej dla pi˙y tarczowej wybrano wariant, w kt˙
Biznes plan, Biznes plan - hotel Hellena, III Analiz? strategiczn? firmy przeprowadzono przy zastoso
więcej podobnych podstron