
WYDZIAŁ LEKARSKI
AKADEMII MEDYCZNEJ W GDA
Ń
SKU
Joanna Balon
Kliniczne znaczenie wczesnego całkowitego
chimeryzmu hematopoetycznego po allogenicznej
transplantacji komórek macierzystych hematopoezy
Rozprawa na stopie
ń
doktora nauk medycznych
Praca wykonana w Klinice Hematologii
Instytutu Chorób Wewn
ę
trznych
Promotor: Prof. dr hab. Andrzej Hellmann
Gda
ń
sk, 2005

1
SPIS TRE
Ś
CI
1
WST
Ę
P ......................................................................................................................... 6
1.1
Allogeniczna transplantacja komórek hematopoezy – wprowadzenie ......................... 6
1.2
Przewlekła choroba przeszczep przeciwko gospodarzowi........................................... 7
1.2.1
Klasyfikacja cGvHD........................................................................................... 10
1.2.2
Leczenie cGvHD ............................................................................................... 13
1.3
Chimeryzm hematopoetyczny .................................................................................... 14
1.3.1
Metody okre
ś
lania i monitorowania chimeryzmu hematopoetycznego ............ 14
1.3.2
Kliniczne znaczenie chimeryzmu ...................................................................... 18
2
ZAŁO
ś
ENIA I CEL PRACY ........................................................................................ 20
3
MATERIAŁ I METODY ............................................................................................... 21
3.1
Badani chorzy ............................................................................................................. 21
3.1.1
Choroba przeszczep przeciwko gospodarzowi................................................. 23
3.2
Metody bada
ń
............................................................................................................. 24
3.2.1
Izolacja leukocytów krwi obwodowej................................................................. 24
3.2.2
Ocena chimeryzmu ........................................................................................... 26
3.3
Analiza statystyczna ................................................................................................... 29
4
WYNIKI ....................................................................................................................... 30
4.1
Chimeryzm hematopoetyczny w komórkach j
ą
drzastych krwi obwodowej ................ 30
4.1.1
Charakterystyka chorych w zale
ż
no
ś
ci od rodzaju chimeryzmu....................... 31
4.2
Pó
ź
ne powikłania poprzeszczepowe.......................................................................... 35
4.2.1
Przewlekła choroba przeszczep przeciwko gospodarzowi ............................... 35
4.2.1.1
cGvHD a rodzaj materiału przeszczepowego ................................................. 37
4.2.1.2
cGvHD a aGvHD ............................................................................................. 38
4.2.1.3
Charakterystyka stosowanego leczenia cGvHD ............................................. 41
4.2.1.4
Zgony w grupie z cGvHD ................................................................................ 42
4.2.2
Wznowa choroby zasadniczej........................................................................... 44
4.2.3
Zgony pó
ź
ne ..................................................................................................... 45
4.3
Zale
ż
no
ść
pomi
ę
dzy typem pełnego chimeryzmu a powikłaniami
poprzeszczepowymi ................................................................................................... 47
4.3.1
Typ pełnego chimeryzmu a cGvHD .................................................................. 47
4.3.1.1
Cz
ę
sto
ść
wyst
ę
powania cGvHD w zale
ż
no
ś
ci od typu CC............................. 47
4.3.1.2
cGvHD a aGvHD w zale
ż
no
ś
ci od typu pełnego chimeryzmu ........................ 48
4.3.1.3
Rodzaj CC a ci
ęż
ko
ść
przebiegu cGvHD........................................................ 51
4.3.1.4
Zale
ż
no
ść
pomi
ę
dzy czasem stwierdzenia pełnego chimeryzmu a
wyst
ą
pieniem objawów cGvHD ....................................................................... 53
4.3.2
Typ CC a wznowa choroby zasadniczej .......................................................... 54
4.3.3
Przyczyna zgonów pó
ź
nych w zale
ż
no
ś
ci od rodzaju CC ................................ 56
4.4
Chimeryzm w poszczególnych subpopulacjach leukocytów krwi obwodowej............ 58
5
OMÓWIENIE WYNIKÓW I DYSKUSJA ..................................................................... 61
5.1
Chimeryzm hematopoetyczny .................................................................................... 61
5.2
Choroba przeszczep przeciwko gospodarzowi .......................................................... 67
5.3
Wznowa choroby zasadniczej .................................................................................... 70
6
WNIOSKI .................................................................................................................... 74
7
STRESZCZENIE ........................................................................................................ 75

2
8
SUMMARY ................................................................................................................. 77
9
PI
Ś
MIENNICTWO ...................................................................................................... 79

3
SPIS TABEL
Tabela I Czynniki ryzyka cGvHD .................................................................................................. 9
Tabela II Oryginalna i zmodyfikowana klasyfikacja cGvHD........................................................ 11
Tabela III Najcz
ęś
ciej stosowany model prognostyczny dla chorych z cGvHD.......................... 12
Tabela IV Charakterystyka dawców i biorców ............................................................................ 22
Tabela V Stopniowanie ostrej choroby przeszczep przeciwko gospodarzowi............................ 23
Tabela VI Charakterystyka przeciwciał u
ż
ytych w badaniu ........................................................ 25
Tabela VII. Charakterystyka pacjentów oraz przeszczepu w zale
ż
no
ś
ci od czasu
uzyskania CC.............................................................................................................. 34
Tabela VIII Charakterystyka chorych bez i z cGvHD .................................................................. 36
Tabela IX. Charakterystyka cGvHD w grupie chorych z wczesnym i pó
ź
nym chimeryzmem .... 53
Tabela X Charakterystyka wznowionych chorych w grupie z pó
ź
nym CC ................................. 55
Tabela XI Charakterystyka wznowionych chorych w grupie chorych z wczesnym
chimeryzmem.............................................................................................................. 55

4
SPIS RYCIN
Rysunek 1 Grupy chorych w zale
ż
no
ś
ci od rodzaju chimeryzmu............................................. 30
Rysunek 2 Zmiana odsetkowego udziału CC w kolejnych dobach kontrolnych ....................... 31
Rysunek 3 Zmienna wieku biorcy w zale
ż
no
ś
ci od rodzaju osi
ą
gni
ę
tego chimeryzmu............ 32
Rysunek 4 Zmienna wieku dawcy w zale
ż
no
ś
ci od rodzaju osi
ą
gni
ę
tego chimeryzmu ............. 33
Rysunek 5 Cz
ę
sto
ść
wyst
ę
powania cGvHD ............................................................................. 35
Rysunek 6 cGvHD w zale
ż
no
ś
ci od rodzaju materiału przeszczepowego ............................... 37
Rysunek 7 Wyst
ę
powanie ostrej choroby przeszczep przeciwko gospodarzowi w badanej
grupie chorych ......................................................................................................... 38
Rysunek 8 Wyst
ę
powanie aGvHD u chorych z rozległa postaci
ą
cGvHD w porównaniu do
chorych bez cGvHD................................................................................................. 39
Rysunek 9 Wyst
ę
powanie cGvHD w zale
ż
no
ś
ci od aGvHD..................................................... 40
Rysunek 10 Ostra choroba przeszczep przeciwko gospodarzowi w zale
ż
no
ś
ci od rozległo
ś
ci
cGvHD ..................................................................................................................... 40
Rysunek 11 Typ cGvHD w zale
ż
no
ś
ci od rodzaju pocz
ą
tku..................................................... 41
Rysunek 12 Ilo
ść
leków stosowanych w terapii immunosupresyjnej ........................................ 42
Rysunek 13 Przyczyny zgonu w zale
ż
no
ś
ci od wyst
ę
powania cGvHD .................................... 43
Rysunek 14 Prawdopodobie
ń
stwo prze
ż
ycia w grupie chorych z i bez cGvHD....................... 43
Rysunek 15 Wznowa choroby zasadniczej w zale
ż
no
ś
ci od diagnozy .................................... 44
Rysunek 16 Przyczyny zgonów pó
ź
nych.................................................................................. 45
Rysunek 17 Krzywa prze
ż
ycia w badanej grupie 51 chorych................................................... 46
Rysunek 18 cGvHD w grupach chorych z wczesnym i pó
ź
nym chimeryzmem ....................... 47
Rysunek 19 Ostra choroba przeszczep przeciwko gospodarzowi w zale
ż
no
ś
ci od rodzaju
pełnego chimeryzmu ............................................................................................... 48
Rysunek 20 Cz
ę
sto
ść
wyst
ę
powania cGvHD w zale
ż
no
ś
ci od aGvHD w grupie chorych
z wczesnym chimeryzmem...................................................................................... 49
Rysunek 21 Cz
ę
sto
ść
wyst
ę
powanie cGvHD w zale
ż
no
ś
ci od stopnia aGvHD w grupie chorych
z pó
ź
nym CC ........................................................................................................... 50
Rysunek 22 Typ pocz
ą
tku cGvHD w zale
ż
no
ś
ci od czasu uzyskania CC................................ 51
Rysunek 23 Rodzaj pełnego chimeryzmu a terapia cGvHD..................................................... 52
Rysunek 24 Rodzaj pełnego chimeryzmu a zgony z powodu cGvHD....................................... 52
Rysunek 25 Korelacja mi
ę
dzy czasem uzyskania CC a ró
ż
nic
ą
czasu pomi
ę
dzy czasem do
uzyskania CC a czasem wyst
ą
pienia cGvHD ......................................................... 54

5
Rysunek 26 Wznowa choroby zasadniczej w grupie chorych z wczesnym i pó
ź
nym
chimeryzmem .......................................................................................................... 56
Rysunek 27 Przyczyny zgonów pó
ź
nych w zale
ż
no
ś
ci od czasu uzyskania pełnego
chimeryzmu ............................................................................................................. 57
Rysunek 28 Prawdopodobie
ń
stwo prze
ż
ycia w zale
ż
no
ś
ci od czasu uzyskania pełnego
chimeryzmu ............................................................................................................. 57
Rysunek 29 Przykładowy przebieg chimeryzmu w poszczególnych populacjach krwi
obwodowej u jednego z chorych ............................................................................. 59
Rysunek 30 Przykład mieszanego chimeryzmu w wysortowanych limfocytach CD3 wykazany
metod
ą
multiplex PCR w 2 loci (strzałki pionowe)................................................... 60

6
1
WST
Ę
P
1.1 Allogeniczna transplantacja komórek hematopoezy – wprowadzenie
Transplantacja komórek hematopoetycznych (Hematopoietic Stem Cell
Transplantation HSCT) jest obecnie uznan
ą
metod
ą
leczenia chorób rozrostowych
układu krwiotwórczego, wrodzonych zaburze
ń
metabolicznych i immunologicznych. Jej
wprowadzaniu w latach 60tych towarzyszył du
ż
y sceptycyzm, obecnie na całym
ś
wiecie zabiegowi przeszczepienia szpiku ka
ż
dego roku poddawanych jest tysi
ą
ce
chorych. Znaczny post
ę
p zarówno w post
ę
powaniu przedprzeszczepowym: kwalifikacji
chorych, oznaczaniu antygenów HLA, a tak
ż
e wprowadzenie profilaktyki choroby
przeszczep przeciwko gospodarzowi i post
ę
py w terapii powikła
ń
infekcyjnych w istotny
sposób poprawiły wyniki transplantacji. Nadal ogromnym wyzwaniem pozostaje
zmniejszenie
ś
miertelno
ś
ci zwi
ą
zanej z przewlekł
ą
chorob
ą
przeszczep przeciwko
gospodarzowi, a u chorych przeszczepionych z powodu chorób rozrostowych
- wznow
ą
choroby zasadniczej (19;21;63;65;100;106).
Allogeniczne przeszczepianie szpiku jest skomplikowan
ą
procedur
ą
, w której
komórki hematopoetyczne biorcy zostaj
ą
zast
ą
pione komórkami dawcy. W przypadku
chorób wrodzonych lub aplazji szpiku stanowi to istot
ę
leczenia, natomiast
w chorobach nowotworowych zabieg ten umo
ż
liwia eskalacj
ę
dawek chemioterapii
i napromieniania z pomini
ę
ciem ogranicze
ń
wynikaj
ą
cych z mielotoksyczno
ś
ci.
Dodatkowo, korzystny efekt przeciwnowotworowy wynika z reakcji immunologicznej
powodowanej przez przeszczepiane limfocyty T dawcy (Graft versus Leukemia - GvL;
Graft versus Tumor GvT)(18;19;22;31;96;105;115;130).
Tradycyjnie post
ę
powanie przygotowawcze do przeszczepu (kondycjonowanie)
polegaj
ą
ce na podaniu wysokodawkowanej (mieloablacyjnej) chemioterapii lub
radiochemioterapii (w celu zniszczenia komórek nowotworowych, wytworzenia wolnej
przestrzeni w szpiku oraz zapobie
ż
enia odrzuceniu przeszczepu) było uznawane za
warunek konieczny do uzyskania wszczepienia. Obecnie przeprowadza si
ę
równie
ż
procedury
przeszczepienia
poprzedzone
zredukowanym,
niemieloablacyjnym
kondycjonowaniem. Post
ę
powanie to nie prowadzi do nieodwracalnej aplazji szpiku
-
działanie
mieloablacyjne
zostało
zast
ą
pione
uniemo
ż
liwiaj
ą
c
ą
odrzucenie
przeszczepu terapi
ą
immunoablacyjn
ą
. Proces zamiany hematopoezy biorcy na

7
komórki dawcy (pełny chimeryzm) uzyskiwany jest przez etap mieszanego chimeryzmu
i niekiedy wymaga wspomo
ż
enia infuzj
ą
limfocytów dawcy (Donor Lymphocytes
Infusion - DLI). Efekt przeciwnowotworowy wynika w tej sytuacji głownie z reakcji GvL.
Zalet
ą
post
ę
powania niemieloablacyjnego jest unikni
ę
cie ci
ęż
kich powikła
ń
zwi
ą
zanych
z wysokodawkow
ą
chemioterapi
ą
, co umo
ż
liwia kwalifikowanie do przeszczepu
chorych w coraz starszym wieku, a tak
ż
e obci
ąż
onych chorobami współistniej
ą
cymi
(20;58;64;116;119;121;122;122;123).
Głównym
ź
ródłem pozyskiwania komórek progenitorowych jest krew szpikowa
oraz komórki progenitorowe pobrane z krwi obwodowej. Komórki progenitorowe krwi
p
ę
powinowej w przypadku przeszczepu osób dorosłych maj
ą
marginalne znaczenie
(25;61;106).
Zasadnicz
ą
przyczyn
ą
niepowodze
ń
we wczesnym okresie poprzeszczepowym
(do 100 doby po przeszczepie) s
ą
powikłania wynikaj
ą
ce z aplazji szpiku (krwawienia,
powikłania infekcyjne), toksyczno
ś
ci wysokodawkowanej terapii oraz ostra choroba
przeszczep przeciwko gospodarzowi. Do najwa
ż
niejszych powikła
ń
pó
ź
nych (powy
ż
ej
100 doby po przeszczepie) nale
żą
przewlekła choroba przeszczep przeciwko
gospodarzowi, infekcje oportunistyczne, wznowa choroby zasadniczej, oraz wtórne
nowotwory (5;65;101;126).
1.2 Przewlekła choroba przeszczep przeciwko gospodarzowi
Przewlekła choroba przeszczep przeciwko gospodarzowi (chronic Graft versus
Host Disease - cGvHD) jest najpowa
ż
niejszym i najcz
ę
stszym powikłaniem pó
ź
nym
transplantacji allogenicznej. Wyst
ę
puje u 30 - 70% pacjentów, którzy prze
ż
yli pierwsze
100 dni po transplantacji, a jej cz
ę
sto
ść
w ostatnich latach znacz
ą
co si
ę
zwi
ę
ksza
(9;78;98). Spowodowane jest to wzrostem ilo
ś
ci transplantacji niemieloablacyjnych,
wzrostem cz
ę
sto
ś
ci stosowania infuzji limfocytów dawcy (leczenie wznowy choroby
zasadniczej, uzupełnienie terapii niemieloablacyjnej), zwi
ę
kszeniem ilo
ś
ci HSCT
wykonywanych od dawców alternatywnych (w pełni zgodnych lub niezgodnych
w układzie
HLA
niespokrewnionych,
oraz
rodzinnych
przeszczepów
haploidentycznych) (104). Nie bez znaczenia pozostaje równie
ż
coraz cz
ę
stsze
wykorzystanie jako
ź
ródła materiału przeszczepowego komórek progenitorowych
hematopoezy pobranych z krwi obwodowej (Peripheral Blood Stem Cell

8
Transplantation - PBSCT), gdzie zwi
ę
kszone ryzyko cGvHD zostało potwierdzone
w wielu badaniach (26;27;36;46;47;53;95;124;129).
cGvHD obok wznowy choroby zasadniczej jest obecnie główn
ą
przyczyna
ś
miertelno
ś
ci pó
ź
nej u chorych poddanych allogenicznemu przeszczepieniu szpiku
(powoduje 25% zgonów pó
ź
nych u chorych poddanych HSCT z powodu ostrej
białaczki i ok. 60% u chorych przeszczepionych z powodu anemii aplastycznej).
Przyczyn
ą
ś
mierci w przebiegu cGvHD mo
ż
e by
ć
niewydolno
ść
zaj
ę
tego narz
ą
du, ale
przede wszystkim powikłania infekcyjne spowodowane zarówno eskalacj
ą
leczenia
immunosupresyjnego jak i wyst
ę
puj
ą
cymi w tej chorobie zaburzeniami rekonstytucji
immunologicznej. Pojawiaj
ą
si
ę
równie
ż
doniesienia,
ż
e u chorych z cGvHD cz
ęś
ciej
obserwuje si
ę
nowotwory wtórne, b
ę
d
ą
ce efektem przewlekłego procesu zapalnego,
terapii immunosupresyjnej i zaburze
ń
immunologicznych (43;44). Wreszcie
konsekwencj
ą
zaj
ę
cia narz
ą
dowego cGvHD jest znacz
ą
ce pogorszenie jako
ś
ci
ż
ycia
chorego lub nawet kalectwo (26;27;46;47;53;66;77;78;85;101;103;126;129;151).
Z drugiej strony jednak u chorych, u których obserwuje si
ę
cGvHD rzadziej dochodzi do
wznowy choroby zasadniczej, co jest spowodowane faktem,
ż
e efekt przeszczep
przeciwko białaczce cz
ęś
ciowo wynika z reakcji przeszczep przeciwko gospodarzowi
(78;107;141).
Tradycyjnie chorob
ę
przewlekł
ą
rozpoznaje si
ę
je
ż
eli jej objawy wyst
ę
puj
ą
powy
ż
ej doby 100, jednak u niektórych chorych objawy charakterystyczne dla cGvHD
pojawiaj
ą
si
ę
znacznie wcze
ś
niej i to typ morfologiczny zmian – wyst
ą
pienie objawów
charakterystycznych dla choroby przewlekłej powinno by
ć
podstaw
ą
rozpoznania, a nie
arbitralnie wprowadzony podział czasowy (52). Zwykle pierwsze objawy pojawiaj
ą
si
ę
pomi
ę
dzy 3 – 24 miesi
ą
cem po przeszczepieniu (mediana 4 - 5 miesi
ę
cy), bardzo
rzadko przed doba 80 a tylko u 5% powy
ż
ej 12 miesi
ę
cy (66;151).
Patogeneza cGvHD pozostaje nadal niewyja
ś
niona. Istot
ą
procesu wydaje si
ę
defekt zarówno centralnych jak i obwodowych mechanizmów tolerancji, co prowadzi do
ekspansji alloreaktywnych limfocytów T. Nowopowstaj
ą
ce limfocyty T niszcz
ą
tkanki
docelowe w bezpo
ś
rednim ataku cytolitycznym, poprzez wydzielanie cytokin zapalnych
powoduj
ą
cych włóknienie narz
ą
dów. U chorych z cGvHD obserwuje si
ę
zaburzenia
prowadz
ą
ce do przewagi komórek Th2 co manifestuje si
ę
podwy
ż
szonym poziomem
cytokin jak IL4 czy IL5. Efektem tego jest aktywacja komórek B z produkcj
ą
autoprzeciwciał. Mimo
ż
e cGvHD przypomina nakładanie si
ę
w ró
ż
nym stopniu chorób
tkanki ł
ą
cznej, w których główn
ą
rol
ę
odgrywa obecno
ść
autoprzeciwciał, nie udało si
ę
do tej pory jednoznacznie udowodni
ć
ich udziału w patogenezie tej choroby. Wykazano

9
natomiast,
ż
e wydzielane cytokiny stymuluj
ą
fibroblasty do produkcji kolagenu i jego
odkładania w tkankach, co jest jednym z patomorfologicznych wykładników choroby
(49;66;77;103;151).
Brak zrozumienia patofizjologii cGvHD jest niew
ą
tpliwie jednym z czynników
uniemo
ż
liwiaj
ą
cych znacz
ą
cy post
ę
p w leczeniu tej choroby. Tym bardziej istotne
wydaj
ą
si
ę
działania prowadz
ą
ce do zmniejszenia ryzyka wyst
ą
pienia klinicznie istotnej
choroby poprzez celowe unikanie modyfikowalnych czynników wpływaj
ą
cych na rozwój
tego powikłania. Do działa
ń
takich nale
ż
y dobór najbardziej optymalnego dawcy, pod
wzgl
ę
dem płci i wieku, wybór materiału przeszczepowego czy ograniczenie ilo
ś
ci
komórek CD3 w przetaczanym materiale (66;77;103).Tabela I
Tabela I Czynniki ryzyka cGvHD
Biorca
Dawca oraz
charakterystyka przeszczepu
Przebieg
poprzeszczepowy
Powszechnie uznane
Starszy wiek
Dawca kobieta dla m
ęż
czyzny
aGvHD
CML
Przeszczep niezgodny w układzie HLA
Anemia aplastyczna
Przeszczep od dawcy
niespokrewnionego
PBSCT
Infuzja limfocytów dawcy
Du
ż
a ilo
ść
komórek T w materiale
przeszczepowym
Przypuszczalne (dyskusyjne)
Biorca CMV (+)
Ró
ż
nice etniczne mi
ę
dzy dawc
ą
i biorc
ą
Glikokortykosteroidy
w profilaktyce
aGvHD
Reaktywacja zaka
ż
enia
CMV
Rzadziej u chorych po przeszczepie
z krwi p
ę
powinowej
Du
ż
a ilo
ść
komórek
CD34 w materiale
przeszczepowym
(PBSCT)
Splenektomia
Brak metotreksatu
w profilaktyce
aGvHD (PBSCT)
Klinicznie cGvHD przebiega jako pleomorficzny zespół i mo
ż
e dotyczy
ć
praktycznie ka
ż
dego układu. Do najcz
ęś
ciej zaj
ę
tych narz
ą
dów nale
żą
: skóra (dwie
postacie: z obecno
ś
ci
ą
zmian lichenoidalnych lub sclerodermii) 65-80%, jama ustna
48-72%, w
ą
troba 40-73%, oczy 18-47%. Rzadziej zmiany dotycz
ą
przewodu

10
pokarmowego / utraty masy ciała 16-26%, płuc 10-15%, przełyku 6-8% i stawów 2-12%
(66).
1.2.1 Klasyfikacja cGvHD
Obecna klasyfikacja cGvHD obejmuje okre
ś
lenie typu rozwoju choroby w zale
ż
no
ś
ci od
wcze
ś
niejszego wyst
ą
pienia ostrej choroby przeszczep przeciwko gospodarzowi,
ocen
ę
rozległo
ś
ci pod k
ą
tem wskaza
ń
do terapii immunosupresyjnej oraz ryzyka
niekorzystnego przebiegu choroby.
U wi
ę
kszo
ś
ci pacjentów rozwój choroby przewlekłej poprzedzony jest
wyst
ą
pieniem ostrej choroby przeszczep przeciwko gospodarzowi (acute Graft versus
Host Disease aGvHD). Je
ż
eli objawy ostrej choroby płynnie przechodz
ą
w chorob
ę
przewlekł
ą
typ ten nazywamy post
ę
puj
ą
cym (progressive), je
ż
eli pojawienie si
ę
choroby przewlekłej oddzielone jest okresem ust
ą
pienia objawów aGvHD typ ten
nazywamy wyciszonym (quiscent lub interrrupted). Natomiast wyst
ą
pienie cGvHD bez
poprzedzenia chorob
ą
ostr
ą
- de novo. Według IBMTR (International Bone Marrow
Transplant Registry) typ post
ę
puj
ą
cy stanowi 20-30%, wyciszony 30 - 40%, a de novo
35% cGvHD u chorych poddanych transplantacji od HLA zgodnego rodze
ń
stwa (66)
Okre
ś
lenia typu rozwoju choroby ma ogromne znaczenie prognostyczne. W najgorzej
rokuj
ą
cym typie o rozwoju post
ę
puj
ą
cym wg niektórych autorów
ś
miertelno
ść
mo
ż
e
si
ę
ga
ć
nawet 50 - 60% i typ ten wyodr
ę
bnia grup
ę
chorych wymagaj
ą
cych szczególnie
intensywnej terapii immunosupresyjnej (85).
Pierwotna klasyfikacja Seattle definiuj
ą
ca rozległo
ść
w oparciu o podział
choroby na posta
ć
ograniczon
ą
lub rozległ
ą
ustalona została w latach 80-tych na
podstawie badania histopatologicznego narz
ą
dów 20 pacjentów (120). Ograniczone
zaj
ę
cie skóry lub/i łagodne zaj
ę
cie w
ą
troby zostało okre
ś
lone jako choroba
ograniczona niewymagaj
ą
ca systemowego leczenia immunosupresyjnego. Natomiast
uogólnione zmiany skórne lub zaj
ę
cie innych narz
ą
dów oraz dysfunkcja w
ą
troby było
traktowane jako posta
ć
rozległa, wymagaj
ą
ca terapii systemowej. Obecnie
obowi
ą
zuj
ą
ca, zmodyfikowana klasyfikacja w bardziej precyzyjny sposób okre
ś
la
stopie
ń
rozległo
ś
ci, wprowadzaj
ą
c mi
ę
dzy innymi
ś
cisłe kryteria ilo
ś
ciowe rozpoznania
(79) (
Tabela
II.) Minusem tej klasyfikacji pozostaje jej niskie znaczenie prognostyczne .

11
Tabela II Oryginalna i zmodyfikowana klasyfikacja cGvHD
Oryginalna klasyfikacja Seattle
(Shulman et al.,1980)
Zmodyfikowana klasyfikacja Seattle (Lee et al., 2003)
Posta
ć
ograniczona
1. ograniczone zaj
ę
cie skóry
1. Zmiany na
ś
luzówkach jamy ustnej w przebiegu
cGvHD, dodatnia biopsja skóry lub wargi bez innych
objawów cGvHD
2.dysfunkcja w
ą
troby spowodowana
cGvHD
2. Zwi
ę
kszona aktywno
ść
fosfatazy alkalicznej (FALK
≤
2xnorma, AST, ALT
≤
3xnorma, bilirubina całkowita
≤
27,3
µ
mol/l) z dodatni
ą
biopsj
ą
skóry lub wargi bez
innych objawów cGvHD
3. Niewielkie zmiany skórne: mniej ni
ż
6 zmian
grudkowo - łuskowych, rumie
ń
<20%powierzchni ciała
(BSA), lub rumie
ń
<50% BSA, dodatnia biopsja skóry
bez innych objawów cGvHD
4. Dodatni test Schirmera
≤
5 mm bez innych zmian w
narz
ą
dzie wzroku, dodatnia biopsja skóry lub wargi
bez innych objawów cGvHD
5. Zmiany w narz
ą
dach płciowych potwierdzone
biopsj
ą
bez innych objawów cGvHD
Posta
ć
rozległa
1. uogólnione zaj
ę
cie skóry
1. Zaj
ę
cie 2 lub wi
ę
cej narz
ą
dów w przebiegu cGvHD,
potwierdzone biopsj
ą
jednego z nich
2. Karnofsky<60%, utrata masy ciała
≥
15%,
nawracaj
ą
ce infekcje bez innej przyczyny,
potwierdzona biopsj
ą
cGvHD w jakimkolwiek innym
narz
ą
dzie
3. Potwierdzone biopsj
ą
zaj
ę
cie skóry o powierzchni
wi
ę
kszej ni
ż
w postaci ograniczonej
4. Twardzina
5. Onycholysis(oddzielanie si
ę
ło
ż
yska od paznokcia)
lub onychodystrofia, potwierdzona biopsj
ą
cGvHD w
jakimkolwiek narz
ą
dzie
2. Ograniczone zmiany skórne lub
dysfunkcja w
ą
troby w przebiegu
cGvHD plus:
a. w biopsji przewlekłe post
ę
puj
ę
ce
zapalenie w
ą
troby, martwica
mostowa, marsko
ść
w
ą
troby
b. zmiany oczne (dodatni test
Schirmera
≤
5 mm) lub
c. potwierdzone biopsj
ą
zmiany
ś
luzówek jamy ustnej lub gruczołów
ś
linowych
d. zaj
ę
cie jakiegokolwiek innego
organu
6. Zapalenie obturacyjne oskrzelików nie
spowodowane inn
ą
przyczyn
ą
7. Dodatnia biopsja w
ą
troby lub (FALK) >2xnorma,
AST, ALT > 3xnorma, bilirubina całkowita > 27,3
µ
mol/l)
z potwierdzeniem cGvHD w innym narz
ą
dzie
8. Potwierdzone biopsj
ą
zmiany górnego lub dolnego
odcinaka przewodu pokarmowego
9. Zapalenie powi
ę
zi lub błon surowiczych bez innej
przyczyny; Przykurcze spowodowane przez cGvHD

12
W ostatnim okresie czasu coraz wi
ę
ksze znaczenie w ocenie cGvHD maj
ą
ró
ż
ne modele prognostyczne oparte na obecno
ś
ci niekorzystnych klinicznych
czynników ryzyka. Do potwierdzanych przez wszystkich badaczy złych czynników
prognostycznych nale
ż
y zaj
ę
cie skóry, poziom płytek krwi poni
ż
ej 100G/l i typ
post
ę
puj
ą
cy (2;66;85). Niektórzy autorzy zaliczaj
ą
do tych czynników równie
ż
zaj
ę
cie
przewodu pokarmowego i niski wska
ź
nik oceny stanu ogólnego (Karnofsky). Jeden
z cz
ęś
ciej proponowanych modeli prognostycznych stworzony przez grup
ę
z Baltimore
przedstawia
Tabela III
(2).
Tabela III Najcz
ęś
ciej stosowany model prognostyczny dla chorych z cGvHD
Czynnik ryzyka:
A Zaj
ę
cie >50% powierzchni skóry
B poziom płytek krwi <100G/l
C post
ę
puj
ą
cy typ choroby
Mimo wprowadzonych zmian w klasyfikacji i w ocenie zaawansowania cGvHD
nadal podkre
ś
la si
ę
ich ograniczenia. Chorzy z rozpoznan
ą
postaci
ą
rozległa choroby
stanowi
ą
bardzo heterogenn
ą
grup
ę
, co w du
ż
ej mierze utrudnia ocen
ę
skuteczno
ś
ci
leczenia i rokowania. Próby wprowadzenia podziału choroby rozległej na łagodn
ą
,
umiarkowan
ą
i ci
ęż
k
ą
nie zyskały powszechnego uznania, ze wzgl
ę
du na du
ż
y
subiektywizm oceny wynikaj
ą
cy z do
ś
wiadcze
ń
o
ś
rodka i braku jednoznacznych
kryteriów oceny. W ostatnim czasie wysuni
ę
to propozycj
ę
standaryzacji kryteriów
rozpoznania oraz oceny zaawansowania cGvHD, poprzez wprowadzenie systemu
oceny ka
ż
dego zaj
ę
tego narz
ą
du punktacj
ą
od 0-3 (oceny ci
ęż
ko
ś
ci oraz zaburzenia
funkcji narz
ą
du)(52).
Grupa ryzyka
Kryteria
Niskie
Bez czynników ryzyka
Umiarkowane
1 czynnik ryzyka
Wysokie
2 czynniki ryzyka
Bardzo wysokie
3 czynniki ryzyka

13
1.2.2 Leczenie cGvHD
Podstaw
ą
wł
ą
czenia systemowej terapii immunosupresyjnej jest rozpoznanie
rozległej cGvHD. Najskuteczniejszym lekiem jest prednizon stosowany w dawce
pocz
ą
tkowej 1mg/kg masy ciała. Nie ma jednoznacznych opinii oceniaj
ą
cych rol
ę
inhibitorów kalcineuryny (cyklosporyna, tacrolimus) w leczeniu cGvHD. Wi
ę
kszo
ść
badaczy uznaje,
ż
e w przypadku braku przeciwwskaza
ń
, inhibitory kalcineuryny
powinny by
ć
podawane u chorych z nowo rozpoznan
ą
cGvHD, aby zmniejszy
ć
dawk
ę
sterydów niezb
ę
dnych do kontroli objawów choroby. Je
ż
eli cGvHD zostanie
rozpoznana w trakcie podawania cyklosporyny utrzymuje si
ę
podawanie tego leku po
wprowadzeniu prednizonu. U chorych, u których objawy cGvHD pojawiaj
ą
si
ę
w okresie, kiedy chory nie otrzymuje ju
ż
inhibitorów kalcineuryny mo
ż
e by
ć
podejmowana próba monoterapii cyklosporyn
ą
po rozwa
ż
eniu czynników ryzyka
cGvHD, jej rozległo
ś
ci oraz typu zmian narz
ą
dowych (32;68;76;85;131;152).
Wi
ę
kszo
ść
chorych wymaga leczenia immunosupresyjnego przez okres 1 – 3 lat.
Progresja cGvHD wyra
ż
aj
ą
ca si
ę
nasileniem objawów, zaj
ę
ciem nowego narz
ą
du, lub
niemo
ż
no
ś
ci
ą
rozpocz
ę
cia redukcji dawki prednizonu w ci
ą
gu 2 miesi
ę
cy od
rozpocz
ę
cia leczenia jest wskazaniem do wdro
ż
enia terapii drugiego rzutu. Nie ma
obecnie
schematu,
który
mo
ż
na
rekomendowa
ć
jako
standard
leczenia
drugorzutowego przy stwierdzeniu sterydooporno
ś
ci. Najbardziej korzystne wydaje si
ę
stosowanie mykofenolatu mofetilu, szczególnie w postaci w
ą
trobowej cGvHD
(23;24;30;94). Mniejsze znaczenie ze wzgl
ę
du na nasilone działania niepo
żą
dane ma
rapamycyna i talidomid (45;103). Zmiany skórne mo
ż
na skutecznie leczy
ć
fotochemioterapi
ą
(doustny 8- metoksy-psoralen z nast
ę
powym promieniowaniem
ultrafioletowym A ang.: psoralen plus ultraviolet irradiation – PUVA) lub pozaustrojow
ą
fotochemioterapi
ą
(extracoporeal photochemotherapy – ECP) która wykazuje równie
ż
skuteczno
ść
w przypadku zaj
ę
cia w
ą
troby (118). Z innych stosowanych sporadycznie
metod nale
ż
y wymieni
ć
napromienianie w
ę
złów chłonnych, stosowanie Rituximabu,
Etanerceptu – rozpuszczalnego receptora TNF-
α
i pentostatyny (77;103).
Ze wzgl
ę
du na zwi
ę
kszone ryzyko powikła
ń
infekcyjnych zalecane jest równoczesne
stosowanie profilaktyki infekcji oportunistycznych (85;152).

14
1.3 Chimeryzm hematopoetyczny
Allogeniczna transplantacja szpiku prowadzi do powstania w organizmie biorcy
zjawiska zwanego chimeryzmem hematopoetycznym. Słowo chimeryzm wywodzi si
ę
od greckiego słowa chimera, którym w mitologii greckiej był stwór b
ę
d
ą
cy poł
ą
czeniem
lwa, kozy i w
ęż
a (134). W transplantologii poj
ę
cie to jest stosowane zarówno do
okre
ś
lenia faktu,
ż
e komórki hematopoezy jak i pozostałe komórki somatyczne ró
ż
ni
ą
si
ę
materiałem genetycznym jak i do okre
ś
lenia zjawiska wyst
ę
powania komórek
o ró
ż
nym genotypie w obr
ę
bie samej hematopoezy. Zale
ż
no
ś
ci te stały si
ę
podstaw
ą
do wyodr
ę
bnienia
pełnego
chimeryzmu
(Complete
Chimerism – CC),
charakteryzuj
ą
cego si
ę
obecno
ś
ci
ą
w obr
ę
bie hemopoezy jedynie komórek o genotypie
dawcy oraz mieszanego chimeryzmu (Mixed Chimerism – MC), który oznacza
obecno
ść
zarówno komórek o genotypie dawcy jak i biorcy (7;75;134). Jako warunki
uzyskania CC wymienia si
ę
zniszczenie hematopoezy biorcy przez zastosowanie
chemio- lub radioterapii w trakcie post
ę
powania przygotowawczego przed
przeszczepem (mieloablacja) oraz reakcje immunologiczne zachodz
ą
ce po uzyskaniu
wszczepienia mi
ę
dzy komórkami dawcy a przetrwałymi komórkami biorcy, w tym
równie
ż
nowotworowymi (GvL). Zmiany wymienionych warunków np. poprzez
zastosowanie materiału przeszczepowego po deplecji limfocytów T lub zmniejszenie
dawki chemioterapii, jak ma to miejsce w transplantacjach o zredukowanym
kondycjonowaniu, sprzyja przetrwaniu hematopoezy biorcy, a wi
ę
c mieszanego
chimeryzmu (7;17;89;110;111;113;114;133).
1.3.1 Metody okre
ś
lania i monitorowania chimeryzmu hematopoetycznego
Ocena chimeryzmu po allogenicznej transplantacji mo
ż
liwa jest dzi
ę
ki
wyst
ę
powaniu w populacji zjawiska polimorfizmu. Poniewa
ż
wszystkie odmiany
polimorfizmu s
ą
wynikiem ró
ż
nic w sekwencji DNA, mog
ą
by
ć
one wykrywane
metodami biologii molekularnej. Istnieje równie
ż
mo
ż
liwo
ść
identyfikacji produktów
polimorficznych genów np. dziedzicznych wariantów enzymów lub antygenów.

15
Poni
ż
ej przedstawiono mo
ż
liwo
ś
ci oceny chimeryzmu na podstawie metod wykrywania
polimorfizmu (55):
1. Identyfikacja przez bezpo
ś
rednie wykrycie sekwencji DNA
a. polimorfizm długo
ś
ci fragmentów restrykcyjnych /RFLP – Restriction Fragment
Length Polimorfizm/ - warianty sekwencji DNA które polegaj
ą
na braku lub
obecno
ś
ci miejsc rozpoznawanych przez endonukleazy restrykcyjne
b. Powtórzenia DNA
- VNTR (mikro i minisatelitarne DNA)
c. sekwencje charakterystyczne dla chromosomu Y
2. Wykorzystanie heteromorfizmu chromosomów
a. badaniem cytogenetycznym – rozró
ż
nienie chromosomów pochodz
ą
cych od dawcy
czy biorcy na podstawie:
- ró
ż
nic w budowie chromosomów np. zmienno
ść
wielko
ś
ci heterochromatyny
centromerowej czy zmienno
ść
wielko
ś
ci i struktury satelitów
- chromosomów płci
- aberracji chromosomowych zarówno u dawcy jak i biorcy
b. FISH /Fluorescence In Situ Hybridization/
- u
ż
ycie sond specyficznych dla chromosomów płci
- aberracje chromosomowe zwi
ą
zane z chorob
ą
podstawow
ą
np. bcr/abl
3. Identyfikacja produktów polimorficznych genów
a. antygeny grupowe krwi oraz izoaglutyniny
b. dziedziczne warianty enzymów czerwonokrwinkowych np. warianty elektroforetyczne
kwa
ś
nej fosfatazy, dehydrogenazy glukozo-6-fosforanowej, fosfoglukomutazy czy
glioksalazy I
c. immunoglobuliny
d. antygeny HLA
Pocz
ą
tkowo oceniano chimeryzm hematopoetyczny opieraj
ą
c si
ę
na analizie
specyficznych antygenów erytrocytarnych lub elektroforetycznie, analizuj
ą
c polimorfizm
enzymów erytrocytów i leukocytów. Długa
ż
ywotno
ść
erytrocytów, konieczno
ść
transfuzji masy erytrocytarnej, oraz niska czuło
ść
tych metod nie pozwalała jednak na

16
precyzyjn
ą
ocen
ę
(17;134). Badanie antygenów HLA mogło mie
ć
zastosowanie jedynie
w przypadku przeszczepu niezgodnego w układzie HLA. Inne metody jak badanie
izotypów immunoglobulinowych i konwencjonalna cytogenetyka cechuj
ą
si
ę
stosunkowo nisk
ą
czuło
ś
ci
ą
i ograniczonym stopniem polimorfizmu (72;75;134).
Do
ść
istotnym przełomem w ocenie chimeryzmu było wprowadzenie techniki
fluorescencyjnej hybrydyzacji in situ. W metodzie tej za pomoc
ą
odpowiednich sond
molekularnych wykrywa si
ę
charakterystyczne dla choroby aberracje cytogenetyczne
lub chromosom Y. Zalet
ą
tej metody jest do
ść
wysoka czuło
ść
, oraz mo
ż
liwo
ść
oceny
ilo
ś
ciowej Jej głównym ograniczeniem jest mo
ż
liwo
ść
stosowania jedynie u chorych,
u których mi
ę
dzy dawc
ą
a biorc
ą
wyst
ę
puje ró
ż
nica płci lub je
ż
eli u dawcy lub biorcy
wyst
ę
puje znana wcze
ś
niej aberracja cytogenetyczna wzgl
ę
dem której wyst
ę
puje
odpowiednia sonda (60;99).
Obecnie najwi
ę
ksze zastosowanie w monitorowaniu chimeryzmu maj
ą
metody
oparte na analizie minisatelitarnych i mikrosatelitarnych fragmentów DNA poddanych
amplifikacji w procesie PCR (Polymerase Chain Reaction - PCR) (35;75;117;137;153).
Około 20 % genomu człowieka stanowi
ą
sekwencje powtarzaj
ą
ce si
ę
(zmienna liczba
tandemowych powtórze
ń
- Variable Number of Tandem Repeats - VNTR). Wyró
ż
nia
si
ę
dwa główne rodzaje VNTR: sekwencje minisatelitarne, których powtarzaj
ą
cy si
ę
fragment zawiera zwykle 7-50 nukleotydów oraz sekwencje mikrosatelitarne.
Sekwencje mikrosatelitarne (Short Tandem Repeats - STR) to polimorficzne loci,
zawieraj
ą
ce sekwencje nukleotydowe (zazwyczaj 10-50 powtórze
ń
motywu, na który
składa si
ę
od 1-6 nukleotydów w danym locus). Do najcz
ęś
ciej wyst
ę
puj
ą
cych nale
żą
powtórzenia mono-, di-, tri- i pentanukleotydowe. Wysoki polimorfizm tych sekwencji
sprawił,
ż
e ich analiza została uznana za jedn
ą
z najbardziej u
ż
ytecznych technik
w badaniu zjawiska chimeryzmu komórkowego po HSCT (1;135). Produkt PCR mo
ż
e
by
ć
rozdzielony na
ż
elu agarozowym, hybrydyzowany z radioaktywnymi sondami
uwidacznianymi nast
ę
pnie przez autoradiografi
ę
. Obecnie metoda oparta na
wykorzystaniu znaczników fluorescencyjnych zdominowała wcze
ś
niejsze sposoby.
Wyznakowane
fluorescencyjnie
produkty
reakcji
PCR
analizowane
s
ą
w automatycznym sekwenatorze DNA, przy zastosowaniu systemu opartego na
elektroforezie kapilarnej (139). Metoda fluorescencyjnej PCR jest nie tylko bardziej
czuła, ale pozwala tak
ż
e na półilo
ś
ciowe monitorowanie dynamiki zmian chimeryzmu
mieszanego, co jest kluczowym warunkiem wykorzystania chimeryzmu do oceny
zagro
ż
enia wznow
ą
czy odrzuceniem przeszczepu (117;136). Zalet
ą
tej metody jest
równie
ż
mo
ż
liwo
ść
jednoczesnej analizy kilku loci mikrosatelitarnych dzi
ę
ki

17
wykorzystaniu starterów wyznakowanych ró
ż
nymi fluorochromami (kompleksowa
reakcja PCR - multiplex PCR). Równoczesna amplifikacja wielu loci umo
ż
liwia
w krótkim czasie zró
ż
nicowanie DNA biorcy i dawcy równie
ż
u tych chorych, u których
powszechnie stosowane w identyfikacji układy VNTR s
ą
identyczne. Zalet
ą
tego
zestawu jest równie
ż
mał
ą
ilo
ść
DNA konieczna do analizy (1-2ng/20
µ
l mieszaniny
reakcyjnej). Limit detekcyjny tego zestawu wynosi 1% - 3% dla komponenty
znajduj
ą
cej si
ę
w próbce w mniejszo
ś
ci. Metoda ta jest wystandaryzowana, oraz
cechuje si
ę
wysok
ą
powtarzalno
ś
ci
ą
, co umo
ż
liwia porównywanie wyników
uzyskiwanych w ró
ż
nych o
ś
rodkach (134). Obecnie metoda ta została uznana jako
najbardziej warto
ś
ciowa dla oceny chimeryzmu hematopoetycznego i powinna by
ć
u
ż
ywana standardowo (14;35;66;72;77;82;91;103;151).
Ogromny post
ę
p w ocenie chimeryzmu wynika nie tylko z wprowadzania
nowych metod oceny DNA. W ostatnich latach nowe mo
ż
liwo
ś
ci w diagnostyce
u chorych poddanych HSCT wniosła ocena chimeryzmu w poszczególnych
populacjach, zarówno krwi szpikowej jak i obwodowej. Chocia
ż
pierwsze próby takiej
oceny przeprowadzono ju
ż
w latach 70tych, dopiero rozwój metod umo
ż
liwiaj
ą
cych
pozyskanie odpowiedniej ilo
ś
ci materiału do bada
ń
, takich jak cytometria przepływowa
czy izolacja immunomagnetyczna, a tak
ż
e wprowadzenie techniki PCR ułatwiaj
ą
cej
analiz
ę
nawet niewielkich ilo
ś
ci DNA, spowodowały wykorzystanie tych oznacze
ń
na
szersz
ą
skal
ę
(59;143). Ocena chimeryzmu w populacjach z wykorzystaniem STR
PCR zwi
ę
ksza czuło
ść
metody nawet do 0,1 – 0,0001%. Wykrycie obecno
ś
ci DNA
biorcy w populacji komórek posiadaj
ą
cych na swojej powierzchni antygeny
charakterystyczne dla rozpoznanej przed transplantacj
ą
choroby nowotworowej stało
si
ę
jedn
ą
z najczulszych metod detekcji choroby resztkowej (Minimal Residual
Disease - MRD). Ocena chimeryzmu w populacji limfocytów T umo
ż
liwia identyfikacj
ę
chorych zagro
ż
onych odrzuceniem przeszczepu lub w przypadku transplantacji
niemieloablacyjnych jest istotn
ą
informacj
ą
w ocenie wskaza
ń
do stosowania DLI
(10;38;71;86;87). Ocena chimeryzmu w poszczególnych populacjach komórek nie jest
jednak badaniem przeprowadzonym rutynowo, ze wzgl
ę
du na czasochłonno
ść
,
wysokie koszty a tak
ż
e konieczno
ść
odpowiedniego wyposa
ż
enia laboratorium, mo
ż
e
by
ć
przeprowadzana jedynie w wybranej grupie chorych.

18
1.3.2 Kliniczne znaczenie chimeryzmu
Ocena chimeryzmu hematopoetycznego u chorych poddanych allogenicznej
transplantacji jest obecnie rutynowo wykorzystywanym badaniem diagnostycznym.
Umo
ż
liwia ona potwierdzenie przyj
ę
cia si
ę
przeszczepionych komórek w organizmie
biorcy, a monitorowanie w regularnych odst
ę
pach czasowych umo
ż
liwia wczesne
wykrycie odrzucania przeszczepu. Ocena chimeryzmu jest równie
ż
najwa
ż
niejszym
kryterium w ocenie wskaza
ń
do wykonania infuzji limfocytów dawcy u chorych
poddanych transplantacji o zredukowanym kondycjonowaniu (14). Kolejnym wa
ż
nym
aspektem jest wykorzystanie oceny chimeryzmu w prognozowaniu wznowy choroby
zasadniczej (11-13). Monitoruj
ą
c chimeryzm hematopoetyczny wyodr
ę
bniono dwie
zasadnicze grupy chorych. Pierwsza, u której poprzez zanik własnej hematopoezy
dochodzi do wytworzenia CC, oraz drug
ą
– z utrzymuj
ą
cym si
ę
przewlekle MC. Mimo
dotychczasowych bada
ń
i obserwacji du
ż
ych grup chorych po transplantacji szpiku nie
ma jednoznacznej opinii, czy utrzymuj
ą
cy si
ę
długotrwale MC ma wpływ na
powodzenie
terapii,
poprzez
zwi
ę
kszenie
ryzyka
wznowy.
Publikowane
w pi
ś
miennictwie doniesienia ró
ż
ni
ą
si
ę
skrajnie: od opinii,
ż
e w grupie chorych z MC
ryzyko wznowy choroby jest istotnie wi
ę
ksze, po stwierdzenie,
ż
e MC nie ma
znaczenia w ocenie zagro
ż
enia wznow
ą
(11;12;57;67;69;72;113;142;143).
Jednym z mo
ż
liwych tłumacze
ń
jest fakt,
ż
e wykrywana, przetrwała hematopoeza
biorcy mo
ż
e mie
ć
zarówno cechy hematopoezy prawidłowej jak i resztkowej,
nowotworowej. Ostatnio pojawiły si
ę
pojedyncze doniesienia,
ż
e MC mo
ż
e dotyczy
ć
jedynie okre
ś
lonej populacji komórek hematopoezy. Wykrycie MC jedynie w populacji,
z której wywodzi si
ę
klon nowotworowy mo
ż
e by
ć
czasami jedyn
ą
wskazówk
ą
dotycz
ą
c
ą
obecno
ś
ci choroby resztkowej, a mechanizm wznowy wydaje si
ę
by
ć
w tym
przypadku stosunkowo jasny (87).
Brak jest natomiast danych, jak
ą
rol
ę
mo
ż
e odgrywa
ć
obecno
ść
jedynie
wybranych populacji prawidłowej hematopoezy. Przetrwanie komórek biorcy mo
ż
e
wi
ą
za
ć
si
ę
z wytworzeniem stanu tolerancji immunologicznej, co znajduje
potwierdzenie w klinicznych obserwacjach, bowiem ci
ęż
ka posta
ć
GvHD rzadko
dotyczy chorych z MC (132). Obserwacje te nie tłumacz
ą
jednak wyst
ę
powania
wznowy u chorych z utrzymuj
ą
cym si
ę
CC. Kluczowe znaczenie mo
ż
e odgrywa
ć
metoda stosowana do oznaczenia chimeryzmu: na podstawie własnych do
ś
wiadcze
ń
wykazano,
ż
e znaczny odsetek chorych, u których metoda tradycyjn
ą
(elektroforezy na

19
ż
elu) stwierdzono CC, wykazuje MC w badaniach metod
ą
elektroforezy kapilarnej ze
znakowaniem starterów barwnikiem fluorescencyjnym.
Kolejnym aspektem w wykorzystaniu oceny chimeryzmu mo
ż
e by
ć
jego znaczenie
w ocenie ryzyka jednego z najwa
ż
niejszych powikła
ń
immunologicznych – chorobie
przeszczep przeciwko gospodarzowi. Oprócz nielicznych doniesie
ń
potwierdzaj
ą
cych
rzadsze wyst
ę
powanie cGvHD u chorych z przetrwałym MC, w ostatnim czasie
pojawiły si
ę
pojedyncze prace, w których sugeruje si
ę
,
ż
e szybkie osi
ą
gni
ę
cie CC
u chorych poddanych niemieloablacyjnemu przeszczepowi szpiku towarzyszy
zwi
ę
kszone ryzyko cGvHD. Nie ma natomiast doniesie
ń
potwierdzaj
ą
cych te
obserwacje w odniesieniu do chorych poddanych transplantacji mieloablacyjnej.

20
2
ZAŁO
ś
ENIA I CEL PRACY
Celem podj
ę
tych bada
ń
było:
1. Okre
ś
lenie
i
monitorowanie
rodzaju
chimeryzmu
hematopoetycznego
w komórkach j
ą
drzastych oraz w poszczególnych populacjach leukocytów krwi
obwodowej u chorych poddanych allogenicznej transplantacji komórek
progenitorowych hematopoezy od dawcy rodzinnego w pełni zgodnego
w układzie HLA.
2. Okre
ś
lenie czy istnieje korelacja mi
ę
dzy rodzajem chimeryzmu stwierdzonym
we
krwi
obwodowej
lub
poszczególnych
populacjach
leukocytów
a wyst
ą
pieniem powikła
ń
pó
ź
nych: przewlekłej choroby przeszczep przeciwko
gospodarzowi oraz wznowy choroby zasadniczej.

21
3
MATERIAŁ I METODY
Badania chimeryzmu przeprowadzono u 54 chorych poddanych allogenicznej
transplantacji szpiku od dawcy rodzinnego, w pełni zgodnego w układzie HLA.
Transplantacj
ę
u wszystkich chorych wykonano w Klinice Hematologii AMG mi
ę
dzy
wrze
ś
niem 1999 a sierpniem 2003. Z oceny wył
ą
czono chorych po allogenicznym
przeszczepie od dawcy spokrewnionego, którzy otrzymali kondycjonowanie
niemieloablacyjne, przeszczepianych wielokrotnie, oraz tych, którzy zmarli z powodu
powikła
ń
wczesnych przed 100 dob
ą
po przeszczepie (u 6 chorych zgon przed
uzyskaniem wszczepienia – chimeryzm nie został oceniony, u 2 chorych chimeryzm
oceniony w 30 dobie - MC).
3.1 Badani chorzy
Wszyscy chorzy otrzymali kondycjonowanie mieloablacyjne. U 46 chorych
ź
ródłem materiału przeszczepowego były komórki progenitorowe krwi obwodowej
(Peripheral Blood Stem Cell - PBSC), u 8 krew szpikowa (Bone Marrow – BM).
Pacjenci z ostr
ą
białaczk
ą
limfoblastyczn
ą
oraz 1 chory z ostr
ą
białaczk
ą
szpikow
ą
-
AML
(transformacja
zespołu
mielodysplastycznego
MDS)
otrzymali
jako
kondycjonowanie na
ś
wietlenie całego ciała (Total Body Irradiation – TBI) w dawce
12Gy (w 6 frakcjach) oraz cyklofosfamid w ł
ą
cznej dawce 120mg/kg masy ciała
Pozostali chorzy otrzymali kondycjonowanie wg schematu Bu/Cy 120 (busulfan
podawany doustnie w ł
ą
cznej dawce 16mg/kg masy ciała oraz cyklofosfamid
podawany do
ż
ylnie w ł
ą
cznej dawce 120mg/kg masy ciała; u chorych których waga
odbiega o ponad 25% od tzw. idealnej masy ciała, leki dawkowano wg tzw. idealnej
dostosowanej masy ciała)(8;62).
Wszyscy dawcy komórek progenitorowych byli mobilizowani granulocytarnym
czynnikiem wzrostu G-CSF w dawce 10
µ
g/kg masy ciała /dob
ę
. Kolekcj
ę
komórek
progenitorowych za pomoc
ą
separatora Baxter Fenwall CS 3000 plus wykonywano
w 12 godzin po 4 i 5 dawce G-CSF. Krew szpikow
ą
pobierano w warunkach sali
operacyjnej w znieczuleniu ogólnym.
Pacjenci poddani PBSCT otrzymali 6,3x10
6
CD34+ komórek / kg masy ciała (mediana);
zakres 2,8 - 8,6 komórek / kg masy ciała. Natomiast biorcy krwi szpikowej otrzymali
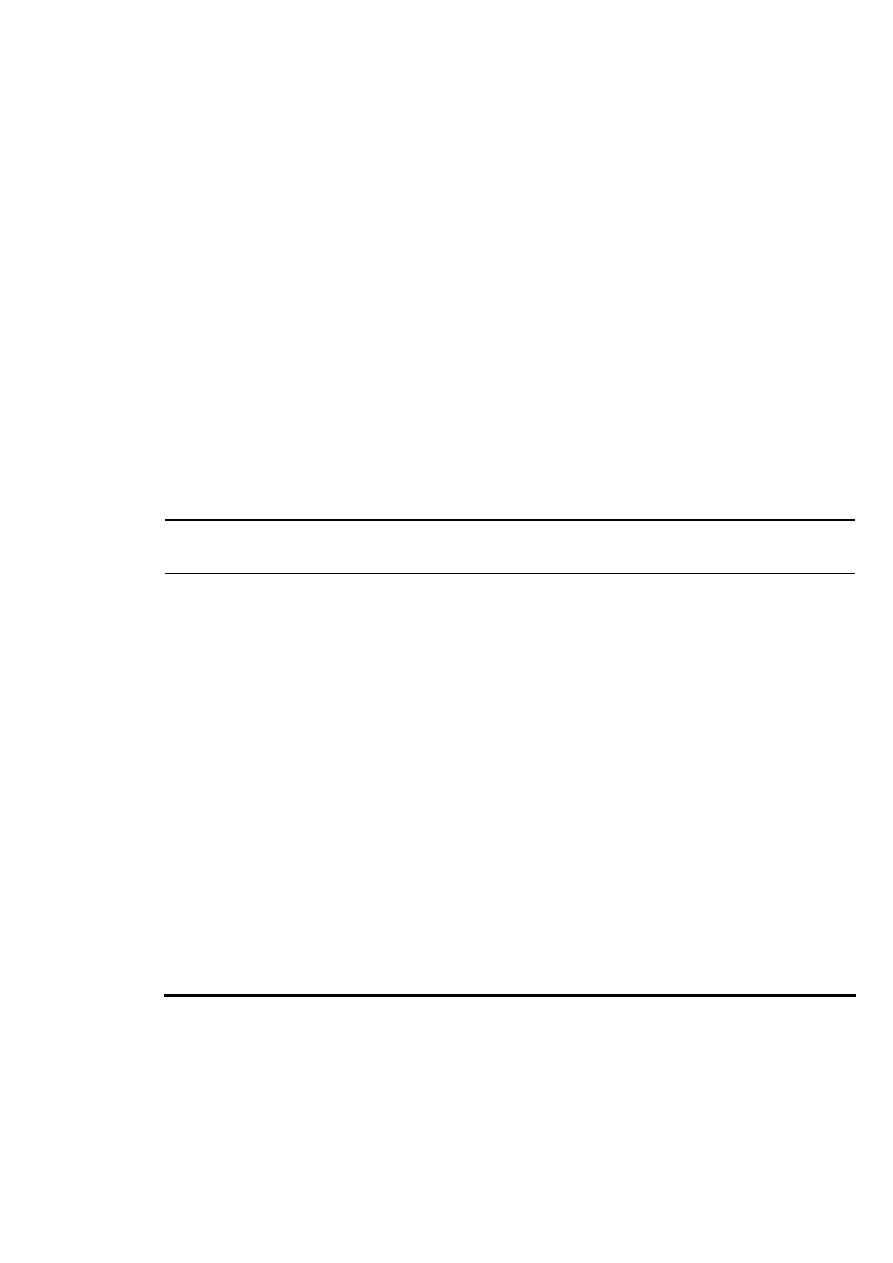
22
3,2x10
6
CD34+ komórek / kg masy ciała (mediana) zakres 1,1 - 5,2 komórek / kg masy
ciała. Charakterystyk
ę
dawców i biorców przedstawia Tabela IV.
Wszyscy chorzy otrzymywali profilaktyk
ę
choroby przeszczep przeciwko gospodarzowi
zgodnie z protokołem Seattle (109;127) (CsA/Mtx). Cyklosporyn
ę
A (CsA) pocz
ą
tkowo
stosowano w dawce 3mg/kg masy ciała na dob
ę
do
ż
ylnie, nast
ę
pnie 5 mg/kg masy
ciała doustnie; do doby 100 po przeszczepie dawki cyklosporyny były modyfikowane
tak, aby st
ęż
enie leku we krwi pełnej wynosiło co najmniej 150-200
µ
g/l. Je
ż
eli chory
nie miał cGvHD od doby 101 do 180 stopniowo odstawiano lek. Metotreksat (Mtx)
podawano do
ż
ylnie w dniach: 1 (15mg/m
2
powierzchni ciała), 3, 6 i 11 po przeszczepie
1 (10mg/m
2
powierzchni ciała).
Wszyscy chorzy spełnili kryteria wszczepienia, okre
ś
lone jako wzrost bezwzgl
ę
dnej
liczby neutrocytów >0,5G/l, przed dob
ą
30 po przeszczepie (mediana 17 dni zakres 10-
29).
Tabela IV Charakterystyka dawców i biorców
Charakterystyka dawców i biorców
Wiek w dniu transplantacji, mediana (zakres)
Pacjenci
37 (18-53)
Dawcy
36 (10-67)
Płe
ć
biorca/dawca
kobieta/m
ęż
czyzna
14
m
ęż
czyzna / m
ęż
czyzna
18
m
ęż
czyzna / kobieta
11
kobieta / kobieta
11
Choroba zasadnicza
AML*
18
ALL*
6
CML*
25
MDS*
3
Inne (CEL, PNH)
2
AML (Acute Myeloid Leukemia) - ostra białaczka szpikowa, ALL (Acute Lymphoblastic
leukemia) - ostra białaczka limfoblastyczna, CML (Chronic Myeloid Leukemia)
przewlekła
białaczka
szpikowa,
MDS
(Myelodysplastic
Syndrome) – zespól
mielodysplastyczny. CEL (Chronic Eosinophilic Leukemia) - białaczka eozynofilowa,
PNH (Paroxysmal Nocurnal Hemoglobinuria) - nocna napadowa hemoglobinuria
Jako cezur
ę
powikła
ń
pó
ź
nych przyj
ę
to wg ogólnie uznanych kryteriów dob
ę
100.

23
3.1.1 Choroba przeszczep przeciwko gospodarzowi
Ocen
ę
ostrej oraz przewlekłej choroby przeszczep przeciwko gospodarzowi
dokonywano zgodnie z kryteriami Seattle
(Tabela II, Tabela V)
(79;102;120) .
Tabela V Stopniowanie ostrej choroby przeszczep przeciwko gospodarzowi
Stopie
ń
Skóra
W
ą
troba
Jelito
+
Rumie
ń
<25% powierzchni
ciała
Bilirubina 2-3 mg/dL
Biegunka, 500-1000
ml/dob
ę
lub nudno
ś
ci *
++
Rumie
ń
25 – 50 %
powierzchni ciała
Bilirubina 3-6 mg/dL
Biegunka, 1000-1500
ml/dob
ę
+++
Uogólniona erytrodermia
Bilirubina 6-15 mg/dL
Biegunka >1500 ml/dob
ę
++++
Złuszczanie lub p
ę
cherze
Bilirubina >15 mg/dL
Ból +/- niedro
ż
no
ść
*Rozpoznanie aGvHD na podstawie utrzymuj
ą
cych si
ę
nudno
ś
ci wymaga
potwierdzenia biopsj
ą
ż
oł
ą
dka lub dwunastnicy
Stage
Skóra
W
ą
troba
Jelito
Zaburzenia funkcji
0
0
0
0
0
I
+ do ++
0
0
0
II
+ do +++
+
+
+
III
++ do +++
++ do +++
++ do +++
++
IV
++ do ++++
++ do ++++
++ do ++++
+++

24
3.2 Metody bada
ń
3.2.1 Izolacja leukocytów krwi obwodowej
Izolacj
ę
populacji leukocytów krwi obwodowej wykonywano w Pracowni
Cytometru Przepływowego Kliniki Hematologii AMG przy u
ż
yciu cytometru
przepływowego wyposa
ż
onego w przystawk
ę
sortuj
ą
c
ą
oraz koncentrator komórkowy
(FACS - Calibur; Becton Dickinson). Znakowanie CD66b oraz sortowanie granulocytów
przeprowadzano z krwi pełnej. W celu zwi
ę
kszenia czysto
ś
ci oraz odzysku barwienie
oraz sortowanie monocytów i poszczególnych populacji limfocytów wykonywano na
jednoj
ą
drzastych komórkach krwi obwodowej .
Izolacja
jednoj
ą
drzastych
komórek
krwi
obwodowej - (Peripheral
Blood
Mononuclear Cell - PBMC)
Jednoj
ą
drzaste komórki krwi obwodowej uzyskiwano drog
ą
wirowania pobranej
krwi
ż
ylnej w gradiencie st
ęż
e
ń
, przy u
ż
yciu Histopaq 1077 (Sigma). Krew pobran
ą
na
EDTA rozcie
ń
czano w płynie CellWash w stosunku 1:1, nast
ę
pnie nawarstwiano na
tak
ą
sam
ą
ilo
ść
Histopaq 1077 i wirowano przez 15 minut (RCF 1000). Po odwirowaniu
uzyskiwano widoczne rozdzielenie faz z wyra
ź
nie zaznaczonym pier
ś
cieniem komórek
jednoj
ą
drzastych, które przenoszono do innej probówki. Po dwukrotnym płukaniu
komórki zawieszano w roztworze CellWash.
Sortowanie w cytometrze przepływowym
W osobnych próbówkach 200
µ
l PBMC inkubowano z 20
µ
l monoklonalnego
przeciwciała anty CD3 sprz
ęż
onego z FITC, anty CD19 sprz
ęż
onego z PE,
antyCD3/CD16/CD56 FITC/PE, anty CD14 FITC
(Tabela VI).
Wszystkie przeciwciała
pochodziły z firmy Becton Dickinson San Jose, CA, USA,. Dla ka
ż
dej próbki stosowano
kontrol
ę
izotypow
ą
γ
1/
γ
2a. Komórki inkubowano przez 30 minut w ciemni, a nast
ę
pnie
płukano w płynie CellWash.

25
Tabela VI Charakterystyka przeciwciał u
ż
ytych w badaniu
Przeciwciało
Klon
Pochodzenie
Podklasa
Immunoglobulin
γ
1/
γ
2a
X39/X40
mysie
IgG1
CD3
SK7
mysie
IgG1,
κ
CD14
M5E2
mysie
IgG2a,
κ
CD 19
4G7
mysie
IgG1,
κ
CD3/CD16CD56
SK7/B73.1/MY31
mysie
IgG1,
κ
CD66b
G10F5
mysie
IgG1,
κ
Znakowanie granulocytów: do 100
µ
l krwi pełnej dodawano 2 ml roztworu
lizuj
ą
cego erytrocyty (FACS lysing solution, Becton Dickinson, San Jose, CA, USA), po
10 min inkubacji próbki odwirowywano (5 minut, RCF 1000) i dodawano 2ml roztworu
CellWash. Procedur
ę
płukania powtarzano, odwirowany i ods
ą
czony osad komórek
zawieszano w 200
µ
l CellWash a nast
ę
pnie dodawano 20
µ
l przeciwciała
monoklonalnego CD66b FITC. Komórki inkubowano przez 30 minut a nast
ę
pnie
powtarzano procedur
ę
płukania w CellWash. Do analizy cytometrycznej komórki
zawieszano w CellWash tak, aby uzyska
ć
st
ęż
enie 2x10
5
/leukocytów. Tak
przygotowane próbki poddawano dalszym procedurom.
Sortowan
ą
populacj
ę
wyznaczano na podstawie dodatniego sygnału
fluorescencji, powy
ż
ej fluorescencji kontroli izotypowej, w odpowiedniej bramce:
limfocytarnej dla komórek CD3, CD16/CD56 oraz CD19, monocytarnej dla komórek
CD14, oraz granulocytarnej dla komórek CD66b.
W celu uzyskania jak najwi
ę
kszej czysto
ś
ci uzyskanej populacji wszystkie
komórki sortowano w trybie „pojedynczej komórki (ang. mode „single cell”), co znaczy
ż
e komórki sortowane były tylko w takiej sytuacji, gdy w strumieniu znajdowały si
ę
pojedynczo rozdzielone komórki
żą
dane. Ka
ż
dorazowo sortowano 50 tysi
ę
cy komórek.
Komórki zostały zatrzymywane na jałowym filtrze komórkowym koncentratora.
Poniewa
ż
limfocyty B stanowi
ą
poni
ż
ej 1% populacji leukocytów a
ż
do doby 180 nie
udało uzyska
ć
odpowiedniej ilo
ś
ci tej populacji do dalszych analiz.

26
Ka
ż
dorazowo czysto
ść
uzyskanej populacji sprawdzano w cytometrze przepływowym.
Analizie DNA poddawano próbki o czysto
ś
ci powy
ż
ej 97%.
3.2.2 Ocena chimeryzmu
Badania wykonywano w Pracowni Genetyki S
ą
dowej Zakładu Medycyny
S
ą
dowej AMG (Kierownik Prof. R. Pawłowski).
Chimeryzm oceniano w 30, 60, 100 i 180 dobie po przeszczepie, a nast
ę
pnie co
3 miesi
ą
ce. Mediana okresu obserwacji wynosiła 32 miesi
ą
ce (zakres 24 - 70).
Izolacja DNA z krwi.
Ekstrakcj
ę
DNA przeprowadzono z wykorzystaniem gotowego zestawu Blood DNA
Prep Plus (A@A Biotechnology, Gda
ń
sk). Do 100
µ
l
ś
wie
ż
ej lub mro
ż
onej krwi
dodawano 200
µ
l uniwersalnego buforu lizuj
ą
cego LT i 20
µ
l proteinazy K (st
ęż
enie
20mg/ml). Cało
ść
mieszano i inkubowano w ła
ź
ni wodnej w temperaturze 37°C przez
minimum 20 minut. Nast
ę
pnie próbk
ę
intensywnie worteksowano przez 20s
i nanoszono na minikolumn
ę
do oczyszczania genomowego DNA. Po odwirowaniu
(1min przy 10-15 RPM) minikolumn
ę
wraz z probówk
ą
wyjmowano i dodawano 500
µ
l
roztworu płucz
ą
cego A1. Próbk
ę
ponownie wirowano (1min przy 10-15 RPM)
Nast
ę
pnie minikolumn
ę
przenoszono do nowej probówki 2ml i nanoszono na ni
ą
400
µ
l
roztworu płucz
ą
cego A1. Próbk
ę
wirowano przez 2 minuty przy 10-15tys RPM.
Osuszon
ą
minikolumn
ę
umieszczano w nowej probówce 1,5ml i dodawano do niej 90
µ
l
buforu TRIS.HCl o pH 8.5. Próbk
ę
inkubowano przez 5min w temperaturze pokojowej,
po czym wirowano przez 1minut
ę
przy 10-15 tys RPM. Po usuni
ę
ciu minikolumny
oczyszczone DNA poddawano dalszym analizom.
Izolacja DNA z filtra komórkowego
Filtr z wysortowanymi populacjami leukocytów umieszczano w probówce i dodawano
kolejno:
100
µ
l buforu Tris
200
µ
l uniwersalnego roztworu lizuj
ą
cego LT
20
µ
l Proteinazy K (20mg/ml)

27
Cało
ść
mieszano i inkubowano w temperaturze 37°C do czasu całkowitego wypłukania
materiału biologicznego z podło
ż
a (zwykle 2-3 godziny). Po inkubacji próbk
ę
intensywnie worteksowano przez 20 s, a nast
ę
pnie wirowano przez 3 minuty, przy 10-
15 tys. RPM, celem oddzielenia roztworu od podło
ż
a. Supernatant nanoszono na
minikolumn
ę
do oczyszczania DNA. Dalsza procedura przebiegała podobnie jak
podczas izolacji DNA z krwi.
St
ęż
enie DNA oznaczano metod
ą
fluorymetryczn
ą
. Otrzymane próbki DNA poddawano
amplifikacji metod
ą
multiplex PCR u
ż
ywaj
ą
c komercyjnie dost
ę
pnego zestawu Profiler
Plus firmy Perkin – Elmer (PE), zawieraj
ą
cego 9 loci typu STR (D3S1358, VWA, FGA,
D8S1179, D21S11, D18S51, D5S818, D13S317 i D7S820) i marker płci w postaci
locus amelogeniny. Amplifikacj
ę
DNA przeprowadzono zgodnie z warunkami podanymi
przez producenta wykorzystuj
ą
c w tym celu termocykler 2400 firmy Perkin – Elmer,
USA. Do amplifikacji w przypadku ka
ż
dej próby u
ż
ywano optymalnego st
ęż
enia DNA tj.
1,25ng/25
µ
L
mieszaniny
PCR.
Wielko
ść
produktów
PCR
znakowanych
fluorescencyjnie analizowano metod
ą
elektroforezy kapilarnej przy zastosowaniu
automatycznego sekwenatora DNA ABI310 (Perkin – Elmer, USA). Produkty
rozdzielano w kapilarze o długo
ś
ci 47cm wypełnionej denaturuj
ą
cym no
ś
nikiem POP4.
Rozdział prowadzono przez 24min. Przy 15kV (constans), 9mW i ok. 8mA.
Standardem wewn
ę
trznym wielko
ś
ci DNA był marker ILS 400 znakowany CXR
(Promega, USA). Analiz
ę
wielko
ś
ci fragmentów DNA przeprowadzono posługuj
ą
c si
ę
tzw. „drabin
ą
alleli” zawieraj
ą
c
ą
zsekwencjonowane allele wszystkich loci
wyst
ę
puj
ą
cych w zestawie. Po ustaleniu profilu genetycznego z krwi pobranej przed
przeszczepem dla ka
ż
dej pary dawca - biorca, okre
ś
lano locus lub loci, w których dla
dawcy i biorcy okre
ś
lono ró
ż
ne allele. Najbardziej optymalnym było ustalenie takiego
lotus, w którym zarówno dawca jak i biorca byli ró
ż
nymi od siebie heterozygotami
i
ż
aden z badanych alleli nie wyst
ę
pował w pozycji „stuttera” (n-4) wzgl
ę
dem kolejnego.
W takim przypadku oceny chimeryzmu, mierzonego odsetkiem DNA biorcy (%MC)
dokonywano stosuj
ą
c nast
ę
puj
ą
c
ą
formuł
ę
:
B1+ B2
%MC = ------------------------- x 100%
B1+ B2+ D1+D2

28
gdzie B1 i B2 to pola powierzchni pod szczytami poszczególnych alleli biorcy, a D1
i D2 – pola powierzchni pod pikami obu alleli pochodz
ą
cych od dawcy. W przypadku,
gdy w zakresie badanego locus heterozygotyczny dawca i biorca mieli jeden wspólny
allel, oceny chimeryzmu dokonywano stosuj
ą
c nast
ę
puj
ą
cy wzór:
B1
%MC = ---------- x 100%
D2
gdzie B1 to pole powierzchni pod pikiem biorcy a D2 – pod pikiem dawcy. Wytypowane
dla celów diagnostycznych loci lub locus poddawano badaniu przez cały czas
monitorowania chimeryzmu.
Jako CC okre
ś
lano jedynie sytuacj
ę
, w której nie wykrywano nawet
ś
ladowych ilo
ś
ci
DNA biorcy. Natomiast jako MC traktowano ka
ż
d
ą
ilo
ść
wykrywanego DNA biorcy
w badaniach kontrolnych.

29
3.3 Analiza statystyczna
We wnioskowaniu statystycznym przyj
ę
to próg istotno
ś
ci równy p= 0.05.
Porównanie rozkładów zmiennych jako
ś
ciowych przeprowadzono z zastosowaniem
testu
χ
2
. Hipotez
ę
o normalnym rozkładzie warto
ś
ci badanej zmiennej przeprowadzano
z wykorzystaniem testu Shapiro-Wilka. Korelacj
ę
pomi
ę
dzy zmiennymi wykazuj
ą
cymi
cechy rozkładu normalnego badano przy pomocy korelacji Pearsona, w przypadku, gdy
zało
ż
enie normalno
ś
ci rozkładu zmiennych nie było spełnione – posługiwano si
ę
korelacj
ą
Spearmana.
Warto
ś
ci
grupowe
zmiennych
ci
ą
głych
porównywano
za
pomoc
ą
testów
parametrycznych (analiza wariancji) w przypadku, gdy zmienne miały rozkład normalny
i odpowiednio testów nieparametrycznych (test Manna-Whitneya), gdy zmienne nie
wykazywały cech rozkładu normalnego.
Porównanie czasu prze
ż
ycia chorych w badanych podgrupach przeprowadzono przy
pomocy analizy Kaplana-Meyera.
Oblicze
ń
dokonywano przy pomocy programu STATISTICA (data analysis software
system), wersja 7.1., StatSoft, Inc. (2005).
30
4
WYNIKI
4.1 Chimeryzm hematopoetyczny w komórkach j
ą
drzastych krwi
obwodowej
Spo
ś
ród 54 chorych, u których oceniono chimeryzm w leukocytach krwi obwodowej 51
chorych w trakcie obserwacji osi
ą
gn
ę
ło chimeryzm pełny (Complete Chimerism - CC)
(Rysunek 1).
U 25 pacjentów osi
ą
gni
ę
cie pełnego chimeryzmu stwierdzono w czasie
100 dni po transplantacji (wczesny chimeryzm). Tylko u 9 chorych obserwowano pełny
chimeryzm ju
ż
w 30 dobie. U 10 chorych pełny chimeryzm wykazano w dobie 60, a u 6
w dobie 100. Mediana czasu do osi
ą
gni
ę
cia CC w tej grupie wynosiła 60 dni.
U 26 pacjentów obserwowano osi
ą
gni
ę
cie CC w trakcie redukcji lub po odstawieniu
terapii immunosupresyjnej (pó
ź
ny chimeryzm – osi
ą
gni
ę
ty po 100 dobie po
przeszczepie). Mediana czasu uzyskania CC wynosiła 270 dni (zakres 180 - 600).
U wszystkich chorych w tej grupie odsetek własnego DNA w okresie pomi
ę
dzy 100
dob
ą
a osi
ą
gni
ę
ciem CC nie przekraczał 2%.
U trzech chorych wykazano utrzymywanie si
ę
MC przez cały okres obserwacji. Ze
wzgl
ę
du na cel pracy chorych tych wył
ą
czono z dalszych analiz statystycznych
zaw
ęż
aj
ą
c badan
ą
grup
ę
do 51 chorych
.
Rysunek 1 Grupy chorych w zale
ż
no
ś
ci od rodzaju chimeryzmu
31
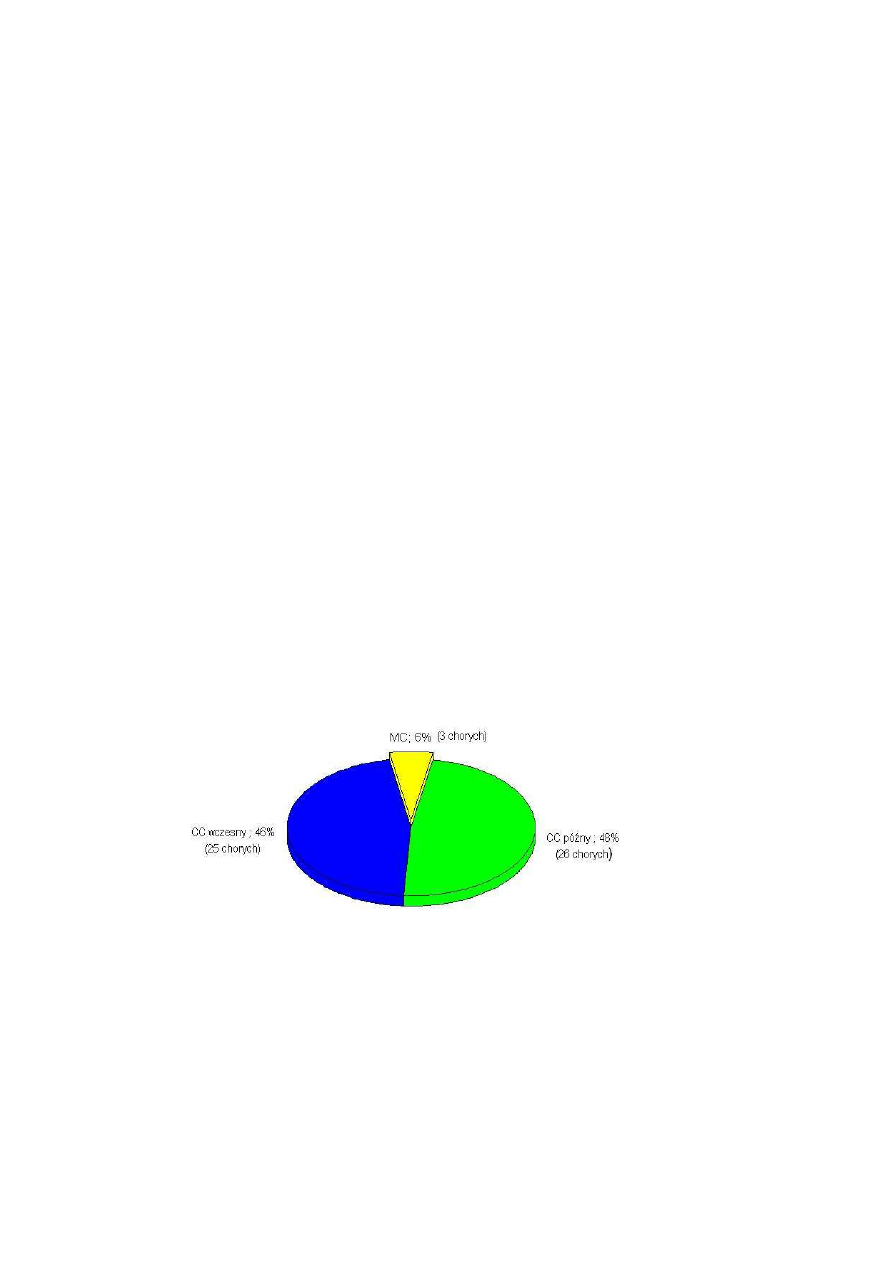
Zmian
ę
odsetkowego udziału CC w badanej grupie w poszczególnych dobach
kontrolnych przedstawia
(Rysunek 2).
Rysunek 2 Zmiana odsetkowego udziału CC w kolejnych dobach kontrolnych
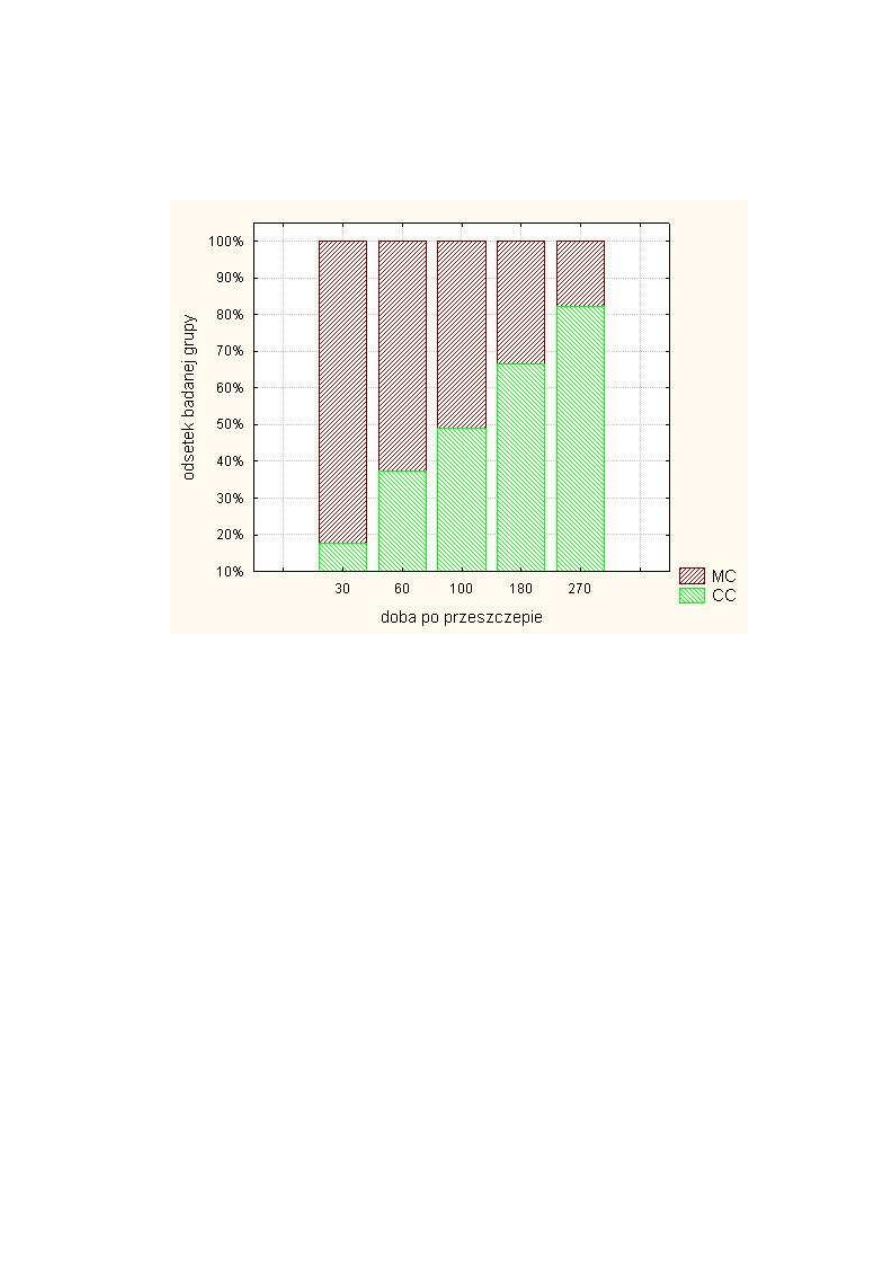
4.1.1 Charakterystyka chorych w zale
ż
no
ś
ci od rodzaju chimeryzmu
W grupie chorych z wczesnym chimeryzmem mediana wieku wynosiła 33 lata (zakres
wieku 20-53 lata), natomiast w grupie chorych z pó
ź
nym chimeryzmem mediana wieku
wynosiła 43 lata (zakres wieku 18-53 lata). W badanych grupach chorych,
wyró
ż
nionych na podstawie czasu uzyskania pełnego chimeryzmu wykazano
statystycznie istotn
ą
ró
ż
nic
ę
wieku biorcy (Test U Manna – Whitneya p=0,037)
(Rysunek 3).
Chorzy, u których obserwowano wyst
ą
pienie wczesnego CC byli istotnie
młodsi od chorych w grupie pó
ź
nego chimeryzmu.
Poniewa
ż
w grupie chorych z wczesnym CC znale
ź
li si
ę
wszyscy chorzy, u których
jako post
ę
powanie przygotowawcze stosowano TBI dodatkowe analizy wieku biorcy
przeprowadzono po wykluczeniu tych chorych z badanej grupy. Nie obserwowano
ró
ż
nic wieku biorcy w grupach chorych z wczesnym i pó
ź
nym chimeryzmem, u których
32
jako kondycjonowanie stosowano chemioterapi
ę
wg schematu BuCY120 (p=0,07).
(mediana wieku 33, zakres 20-53).
Chorzy kwalifikowani do kondycjonowania TBI byli młodsi (wszyscy chorzy w grupie
z wczesnym CC) mediana wieku 34 lata zakres 20-45).
Rysunek 3 Zmienna wieku biorcy w zale
ż
no
ś
ci od rodzaju osi
ą
gni
ę
tego chimeryzmu
Stwierdzono istotn
ą
statycznie ró
ż
nic
ę
w wyst
ę
powaniu wczesnego i pó
ź
nego
chimeryzmu w grupach wydzielonych w oparciu o cezur
ę
wieku równ
ą
45 lat. W grupie
z chimeryzmem wczesnym wiek 22 z 25 osób (88%) nie przekraczał 45 lat.
Analogiczny odsetek okre
ś
lony dla grupy chorych z chimeryzmem pó
ź
nym wynosił
61,5%. Przy pomocy test chi2 wykazano istotno
ść
statystyczn
ą
zale
ż
no
ś
ci
porównywanych zmiennych na poziomie p=0,03
Nie wykazano istotnej statystycznie ró
ż
nicy wieku dawcy w podgrupach chorych
z wczesnym i pó
ź
nym chimeryzmem
(Rysunek 4).
Nie wykazano ponadto istotnej
statystycznie zale
ż
no
ś
ci pomi
ę
dzy wyst
ę
powaniem wczesnego lub pó
ź
nego
chimeryzmu u biorcy a przynale
ż
no
ś
ci
ą
dawcy do grup wiekowych wyró
ż
nionych
w oparciu o cezur
ę
45 lat.
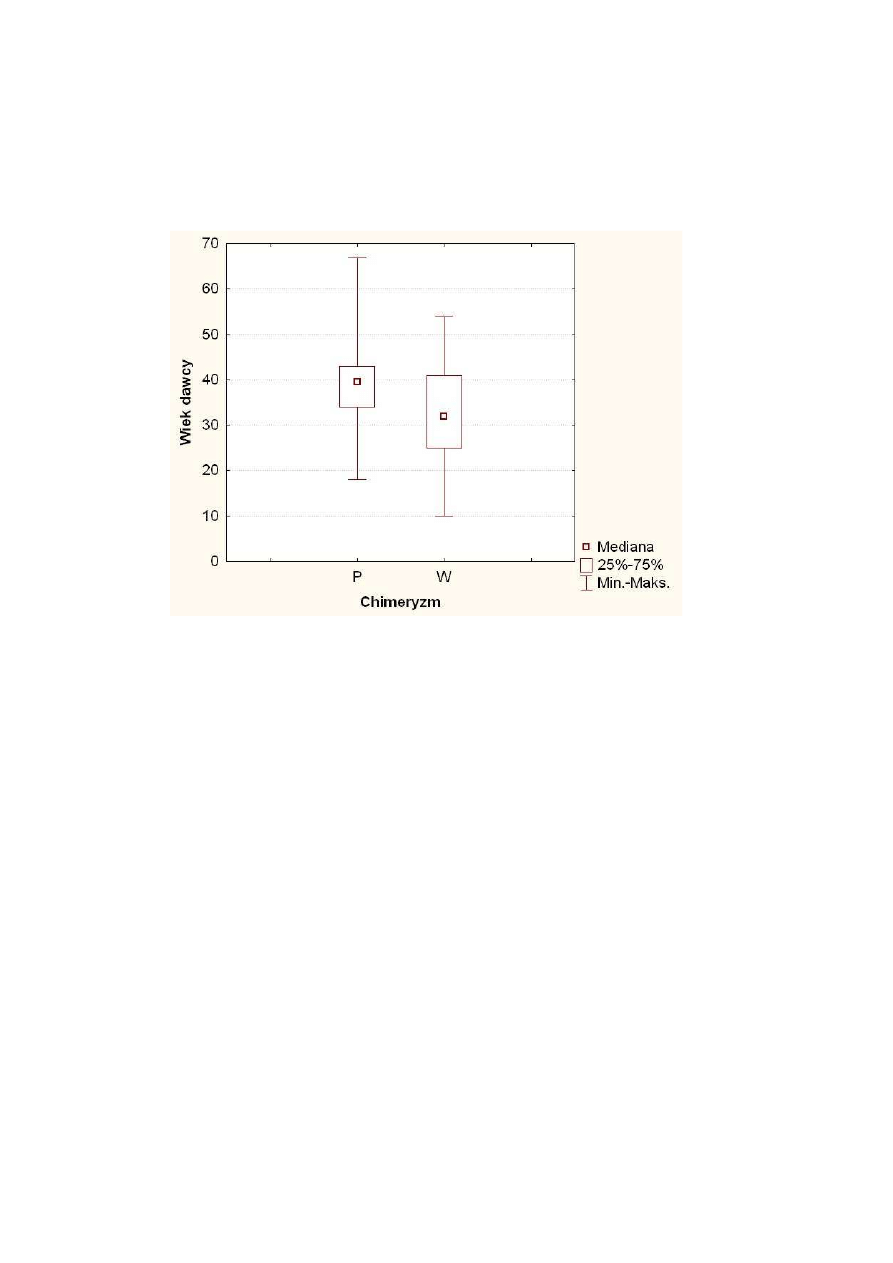
33
Nie stwierdzono statystycznie istotnej ró
ż
nicy pomi
ę
dzy płci
ą
biorców, rodzajem
materiału przeszczepowego, a rodzajem pełnego chimeryzmu w kolejnych punktach
oceny.
Rysunek 4 Zmienna wieku dawcy w zale
ż
no
ś
ci od rodzaju osi
ą
gni
ę
tego chimeryzmu
Wykazano natomiast wpływ rodzaju kondycjonowania na szybko
ść
eliminacji własnej
hematopoezy. Wszyscy 5 chorzy, którzy otrzymali w procesie przygotowawczym
na
ś
wietlenie całego ciała (TBI) uzyskali wczesny pełny chimeryzm: 3 chorych w dobie
30, dwóch w dobie 60 (p=0,02).
Dla cało
ś
ci rozpozna
ń
nie stwierdzono statystycznie istotnej zale
ż
no
ś
ci pomi
ę
dzy
rozpoznaniem choroby zasadniczej a typem osi
ą
gni
ę
tego chimeryzmu.
W grupie chorych poddanych transplantacji komórek hematopoetycznych z powodu
CML 17/25 chorych (68%) uzyskało CC po dobie 100. Ró
ż
nica w cz
ę
sto
ś
ci
wyst
ę
powania pó
ź
nego chimeryzmu u chorych z CML była statystycznie znamienna
(p<0,05).
Wszyscy chorzy przeszczepieni z powodu ALL osi
ą
gn
ę
li CC do doby 100. Zale
ż
no
ść
pomi
ę
dzy ALL a rodzajem chimeryzmu była statystycznie istotna (p<0,05).

34
Osi
ą
gni
ę
cie wczesnego CC cz
ęś
ciej obserwowano w przypadku, gdy dawc
ą
dla
m
ęż
czyzny była kobieta
.
Powy
ż
sze ró
ż
nice nie były jednak statystycznie znamienne
(p=0,14).
Szczegółow
ą
charakterystyk
ę
poszczególnych grup w zale
ż
no
ś
ci od rodzaju
osi
ą
gni
ę
tego chimeryzmu przedstawia Tabela VII
.
Tabela VII. Charakterystyka pacjentów oraz przeszczepu w zale
ż
no
ś
ci od czasu
uzyskania CC
Wczesny CC
(25 chorych)
Pó
ź
ny CC
(26 chorych)
≤
45 r.
ż
.
22
16
Wiek biorcy
>45 r.
ż
.
3
10
≤
45 r.
ż
.
22
21
Wiek dawcy
>45 r.
ż
.
3
5
ALL *
4
0
AML
10
7
CML*
8
17
MDS
2
1
Rozpoznanie
INNE
1
1
TBI
5
0
Kondycjonowanie
BuCY 120
20
26
BMT
4
4
Materiał
przeszczepowy
PBSCT
21
22
Kobieta
11
13
Płe
ć
biorcy
M
ęż
czyzna 14
13
TAK
7
3
Dawca kobieta dla
m
ęż
czyzny
NIE
18
23
* - p<0,05
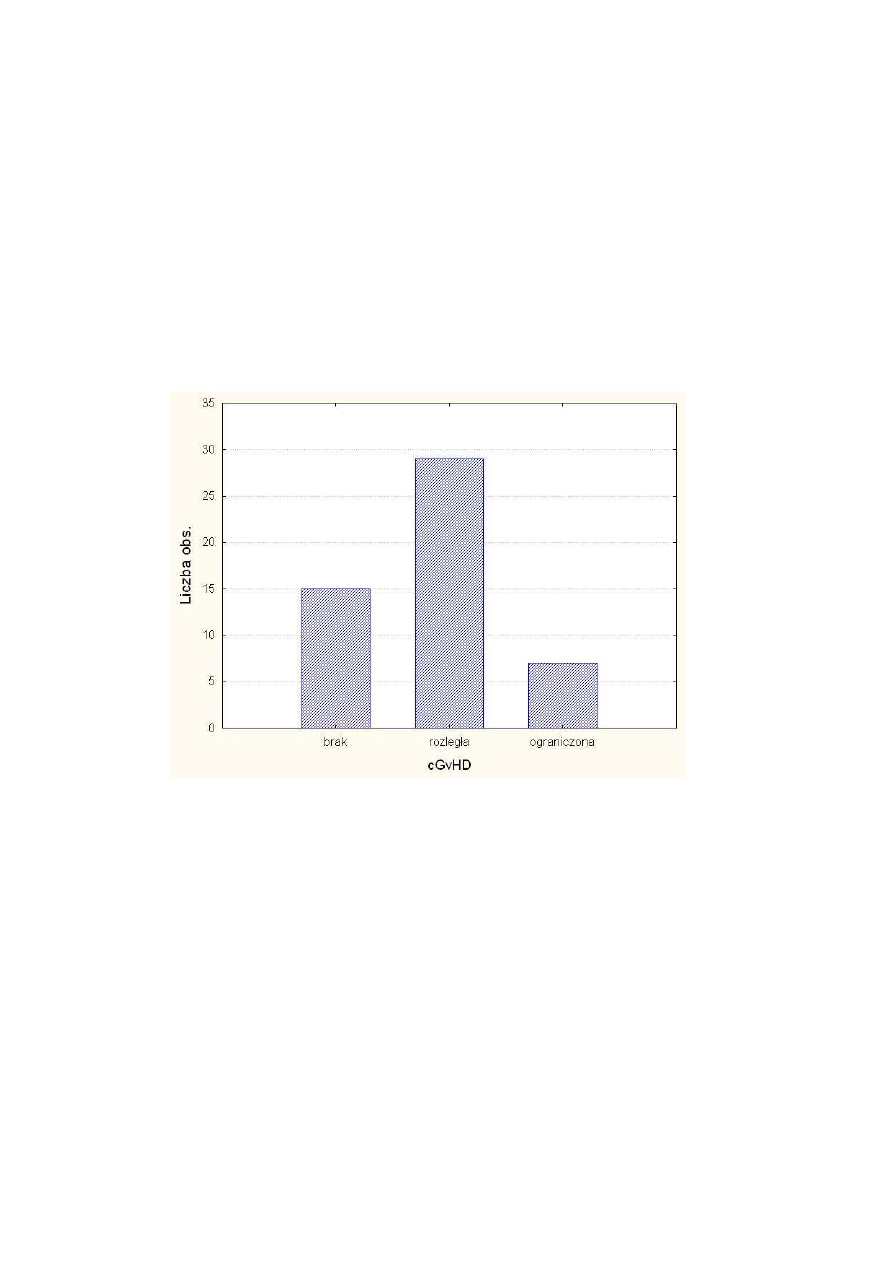
35
4.2 Pó
ź
ne powikłania poprzeszczepowe
4.2.1 Przewlekła choroba przeszczep przeciwko gospodarzowi
Przewlekł
ą
chorob
ę
przeszczep przeciwko gospodarzowi rozpoznano u 36
analizowanych chorych (71%) w tym posta
ć
rozległa u 29 (z 57%), a posta
ć
ograniczon
ą
u 7 chorych (14%).
(Rysunek 5)
Rysunek 5 Cz
ę
sto
ść
wyst
ę
powania cGvHD
Nie stwierdzono statystycznie istotnej zale
ż
no
ś
ci pomi
ę
dzy wiekiem dawcy i biorcy
rodzajem materiału przeszczepowego oraz płci
ą
biorcy, a cz
ę
sto
ś
ci
ą
wyst
ę
powania
cGvHD.
Przewlekła choroba przeszczep przeciwko gospodarzowi wyst
ą
piła u wszystkich
5 chorych, u których stosowano w post
ę
powaniu przygotowawczym napromieniowanie
całego ciała (TBI) oraz 31 z 46 chorych, u których w kondycjonowaniu stosowano
BuCY 120.
Wszyscy chorzy, u których dawc
ą
dla m
ęż
czyzny była kobieta rozwin
ę
li objawy
cGvHD. Zale
ż
no
ść
ta była statystycznie istotna (p=0,02).

36
Nie stwierdzono statystycznie istotnej zale
ż
no
ś
ci pomi
ę
dzy rozpoznaniem choroby
zasadniczej a cz
ę
sto
ś
ci
ą
wyst
ę
powania cGvHD.
Szczegółow
ą
charakterystyk
ę
poszczególnych grup przedstawia
Tabela VIII
.
Tabela VIII Charakterystyka chorych bez i z cGvHD
Bez cGvHD
(15 chorych)
cGvHD (36 chorych)
Wiek biorcy mediana (zakres)
37 (19-53)
37 (20-53)
Wiek dawcy mediana (zakres)
38 (18-67)
37 (10-54)
ALL
0
4
AML
4
13
CML
10
15
MDS
0
3
Rozpoznanie
INNE
1
1
TBI
0
5
Kondycjonowanie
BuCY 120
15
31
BMT
3
5
Materiał
Przeszczepowy
PBSCT
12
31
Kobieta
7
17
Płe
ć
biorcy
M
ęż
czyzna 8
19
TAK
0
10
Dawca kobieta dla
m
ęż
czyzny
NIE
15
26
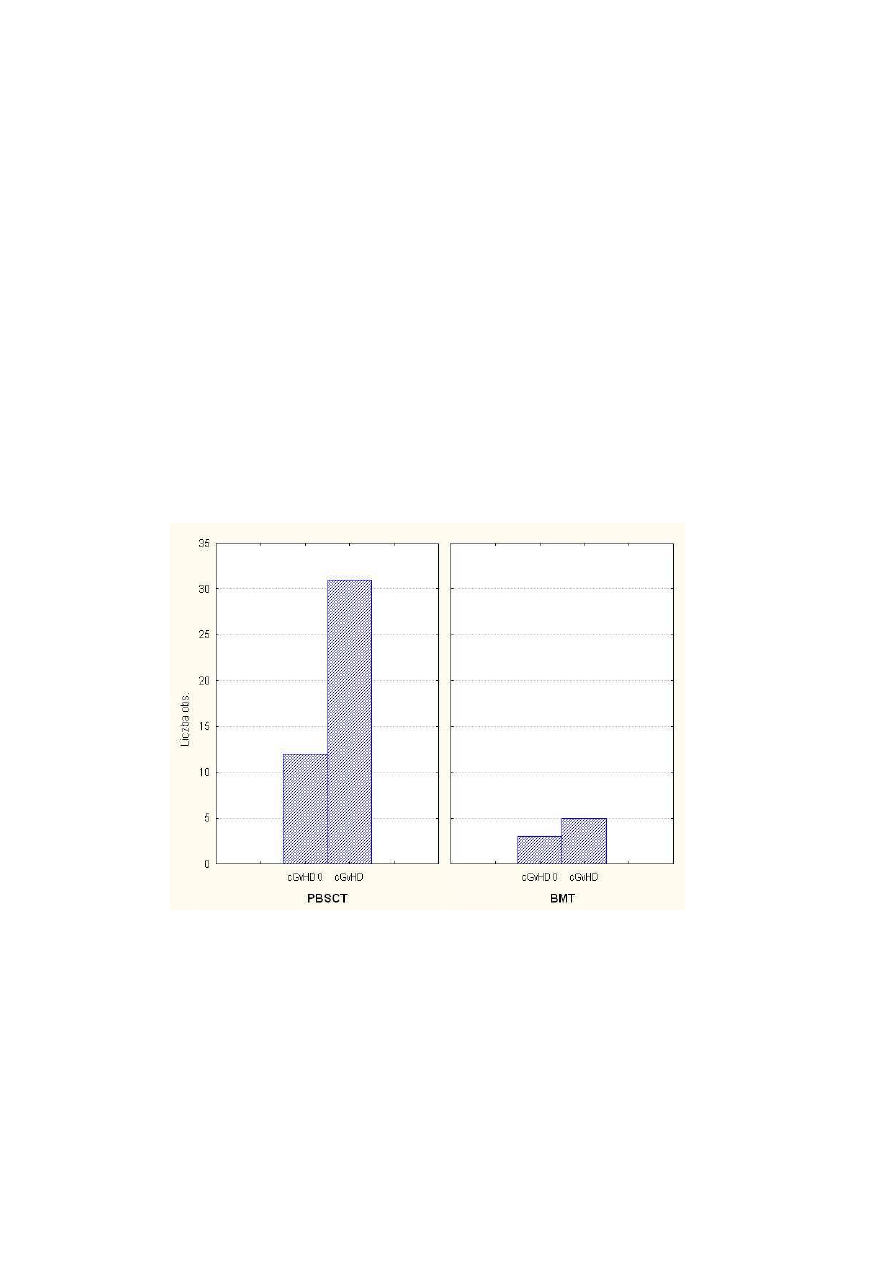
37
4.2.1.1 cGvHD a rodzaj materiału przeszczepowego
W analizowanej grupie transplantacj
ę
komórek progenitorowych krwi obwodowej
(PBSCT) wykonano u 43 chorych, natomiast u 8
ź
ródłem materiału przeszczepowego
była krew szpikowa (BMT)
Spo
ś
ród 43 chorych po PBSCT cGvHD obserwowano u 31 chorych (72%), z czego
u 25 chorych (58%) posta
ć
rozległ
ą
a u 6 ograniczon
ą
(14%).
Z 8 chorych po BMT, cGvHD rozwin
ę
ło 5 chorych (62%): 4 w postaci rozległej (50%),
1 w postaci ograniczonej (12,5%)
(Rysunek 6).
Ró
ż
nice w cz
ę
sto
ś
ci wyst
ę
powania cGvHD w zale
ż
no
ś
ci od rodzaju materiału
przeszczepowego nie były istotne statystycznie (p=0,58). Statystycznie istotnej
zale
ż
no
ś
ci nie wykazano równie
ż
rozpatruj
ą
c wyst
ę
powanie jedynie choroby rozległej
(p=0,66).
Rysunek 6 cGvHD w zale
ż
no
ś
ci od rodzaju materiału przeszczepowego
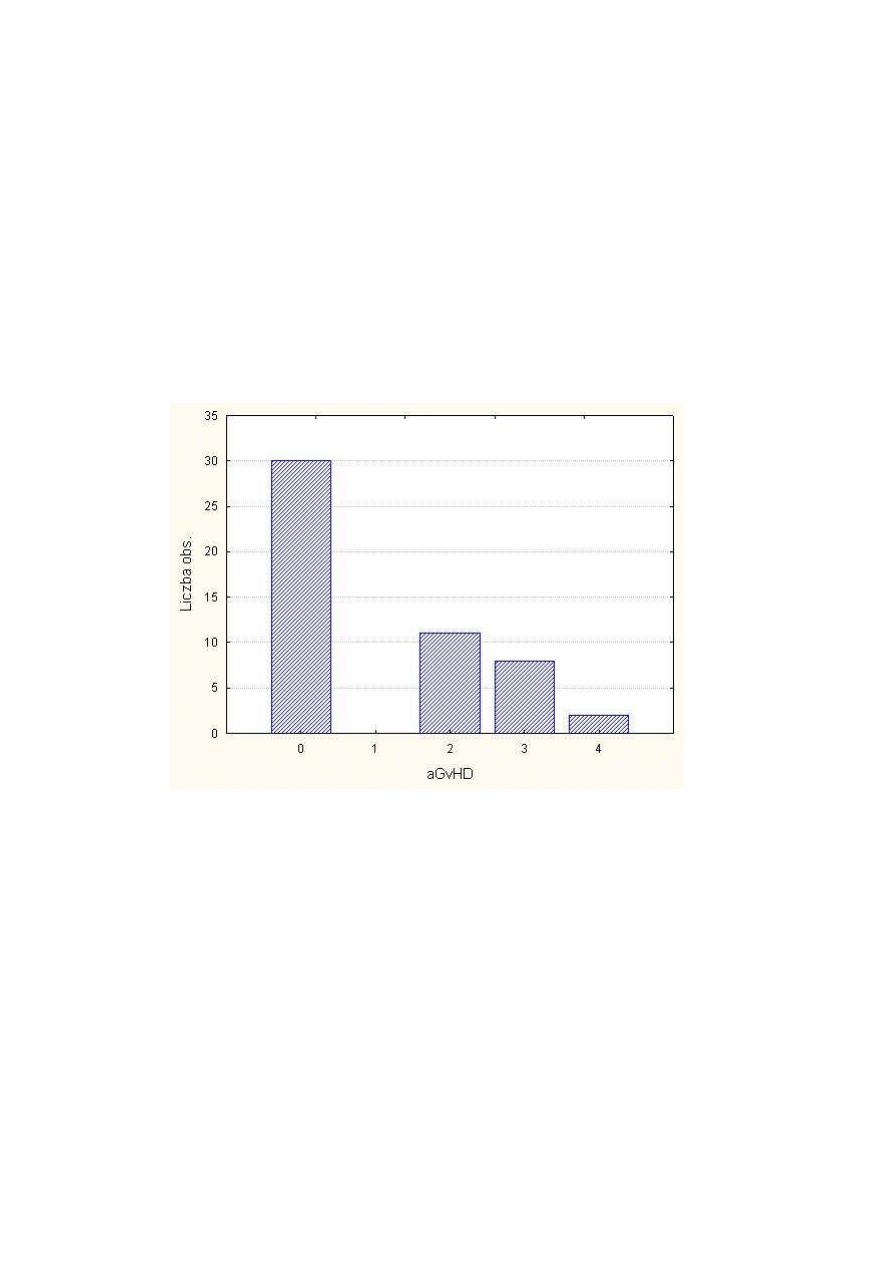
38
4.2.1.2 cGvHD a aGvHD
Wyst
ę
powanie aGvHD
W badanej grupie pacjentów ostr
ą
chorob
ę
przeszczep przeciwko gospodarzowi
rozpoznano w sumie u 21 chorych (41%), w tym u 11 w stopniu 2 (21%), u 8 w stopniu
3 (16%), a u 2 w stopniu 4 (4%)
(Rysunek 7).
Rysunek 7 Wyst
ę
powanie ostrej choroby przeszczep przeciwko gospodarzowi
w badanej grupie chorych
Cz
ę
sto
ść
wyst
ę
powania aGvHD u chorych z cGvHD
Spo
ś
ród 29 chorych z rozpoznan
ą
postaci
ą
rozległ
ą
cGvHD aGvHD poprzedzała
rozwój choroby przewlekłej u 15 (52%) chorych. U 7 chorych była to aGvHD 2 stopnia,
u 6 chorych w stopniu 3 i u 2 chorych w stopniu 4.
W grupie 7 osób z rozpoznan
ą
ograniczon
ą
postaci
ą
cGvHD, aGvHD rozpoznano
u 3 chorych (43%) u wszystkich w stopniu 2.
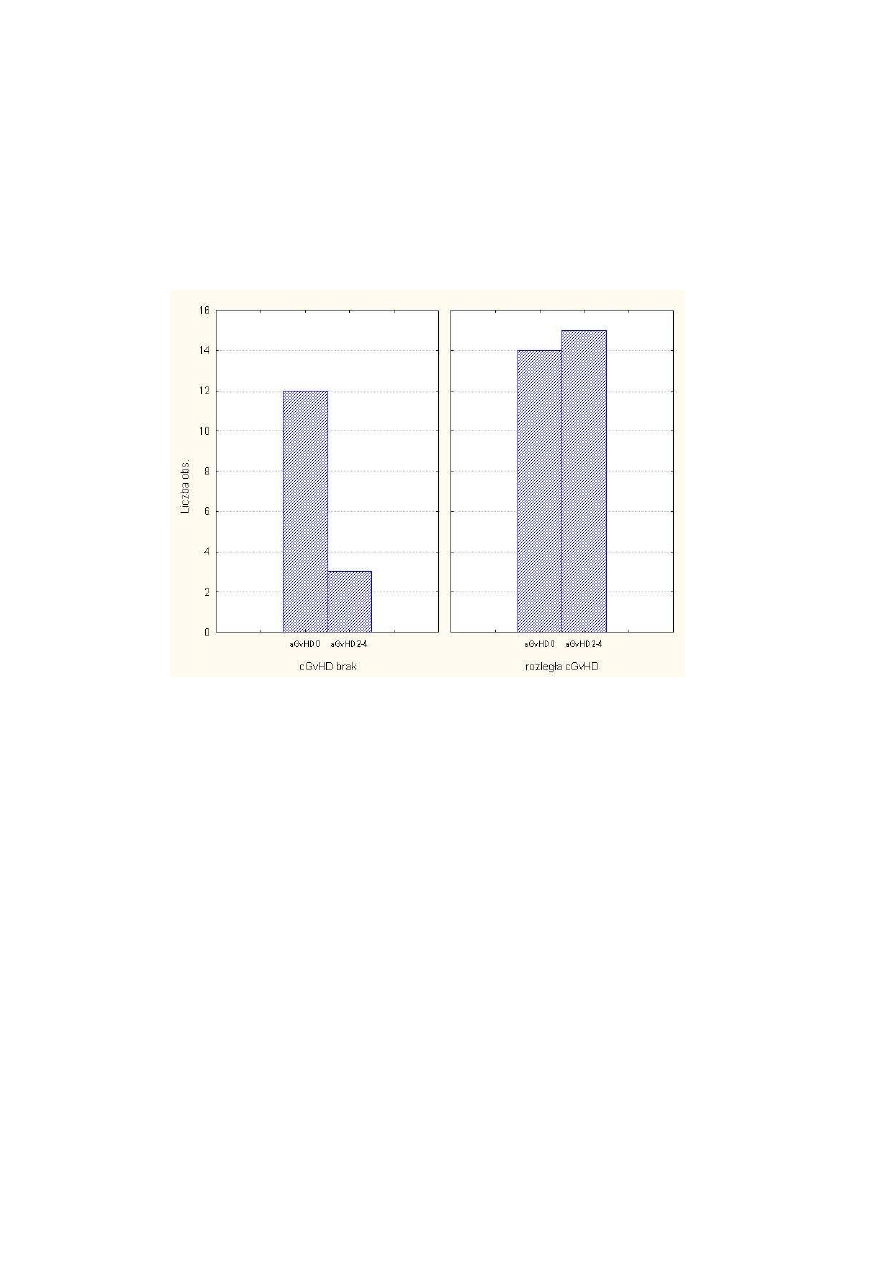
39
W
ś
ród 15 chorych, u których nie obserwowano objawów choroby przewlekłej, aGvHD
wyst
ą
piła u 3 osób (20%), w tym u 1 osoby w stopniu 2 (7%), a 2 w stopniu 3 (13%).
Zale
ż
no
ść
pomi
ę
dzy wyst
ę
powaniem aGvHD w grupie chorych z rozległ
ą
cGvHD
w porównaniu do grupy chorych bez cGvHD była statystycznie istotna (p=0,04)
(Rysunek 8).
Podobnych ró
ż
nic nie wykazano w grupie chorych z chorob
ą
ograniczon
ą
(p=0,24).
Rysunek 8 Wyst
ę
powanie aGvHD u chorych z rozległa postaci
ą
cGvHD w porównaniu
do chorych bez cGvHD
Nie stwierdzono statystycznie istotnych zale
ż
no
ś
ci pomi
ę
dzy wyst
ę
powaniem
poszczególnych stopni zaawansowania aGvHD a wyst
ę
powaniem cGvHD (p=0,24)
(Rysunek 9).
40
Rysunek 9 Wyst
ę
powanie cGvHD w zale
ż
no
ś
ci od aGvHD
Wyst
ę
powanie aGvHD w stopniu co najmniej III oraz rozległej postaci cGvHD
W
ś
ród 10 chorych, u których obserwowano aGvHD przynajmniej w stopniu 3 rozległ
ą
cGvHD rozpoznano u 8 chorych (80%). U pozostałych 41 chorych rozległ
ą
posta
ć
cGvHD obserwowano u 21 (51%). Ró
ż
nice wyst
ę
powania postaci rozległej cGvHD
w tak okre
ś
lonych podgrupach nie były statystycznie istotne (p=0,16).
(Rysunek 10).
Rysunek 10 Ostra choroba przeszczep przeciwko gospodarzowi w zale
ż
no
ś
ci od
rozległo
ś
ci cGvHD
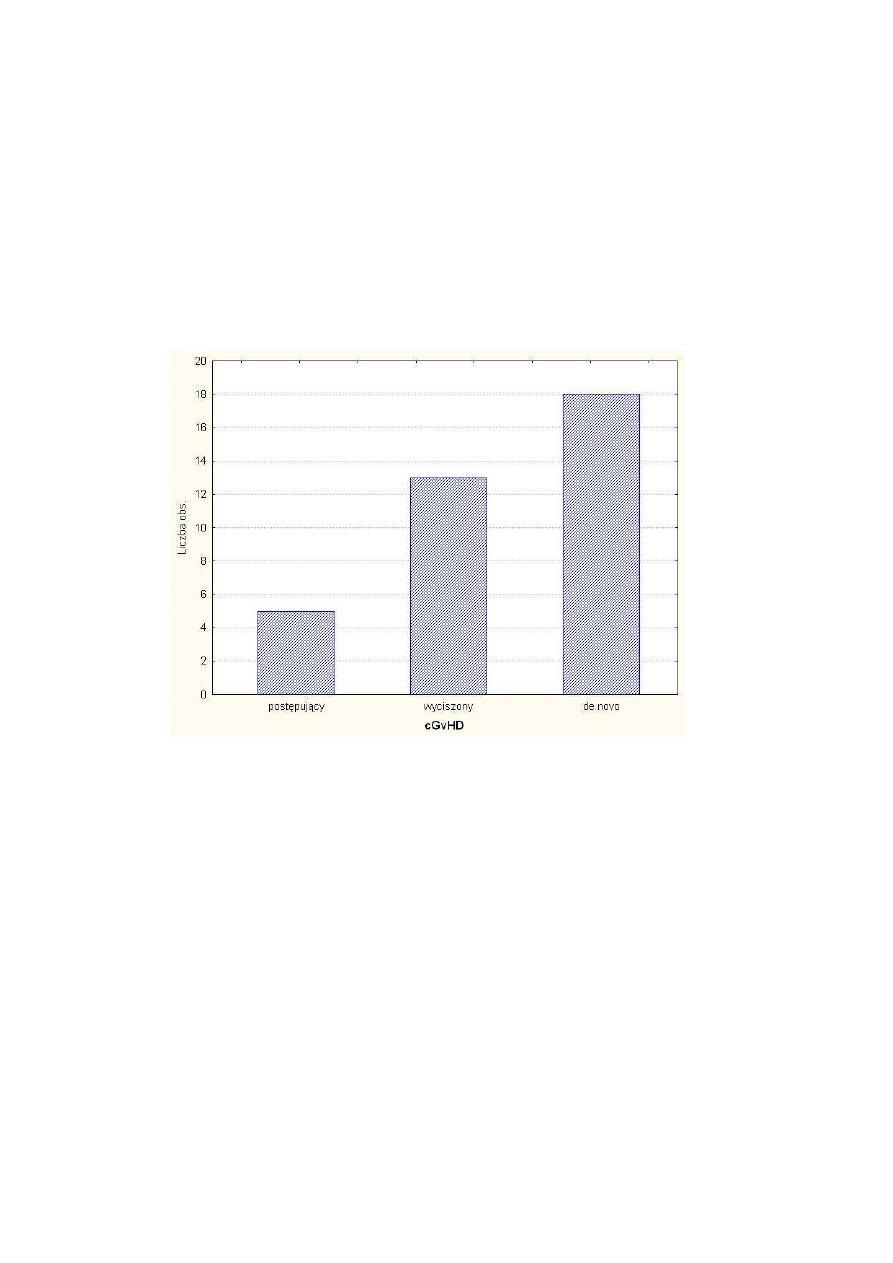
41
Typ rozwoju cGvHD stwierdzany w momencie rozpoznania choroby
U 5 chorych, u których mimo terapii immunosupresyjnej nie uzyskano ust
ą
pienia
objawów ostrej choroby przeszczep przeciwko gospodarzowi rozpoznano typ
post
ę
puj
ą
cy cGvHD. U 13 chorych objawy aGvHD całkowicie ust
ą
piły przed
rozpoznaniem cGvHD (typ wyciszony). Posta
ć
de novo wyst
ą
piła u połowy (18)
chorych
(Rysunek 11).
Rysunek 11 Typ cGvHD w zale
ż
no
ś
ci od rodzaju pocz
ą
tku
4.2.1.3 Charakterystyka stosowanego leczenia cGvHD
27 chorych, u których rozpoznano rozległ
ą
posta
ć
cGvHD, wymagało terapii
prednizonem i cyklosporyn
ą
A. U dwóch chorych z rozległ
ą
cGvHD o stosunkowo
łagodnym przebiegu (1 chory z zaj
ę
ciem w
ą
troby, 1 chory ze zmianami
ś
luzówkowymi
i zaj
ę
ciem w
ą
troby) prowadzono skuteczn
ą
monoterapi
ę
cyklosporyn
ą
A.
U 4 chorych, u których objawy ograniczonej postaci cGvHD pojawiły si
ę
po odstawieniu
cyklosporyny ponownie powrócono do dawki profilaktycznej CSA. Natomiast
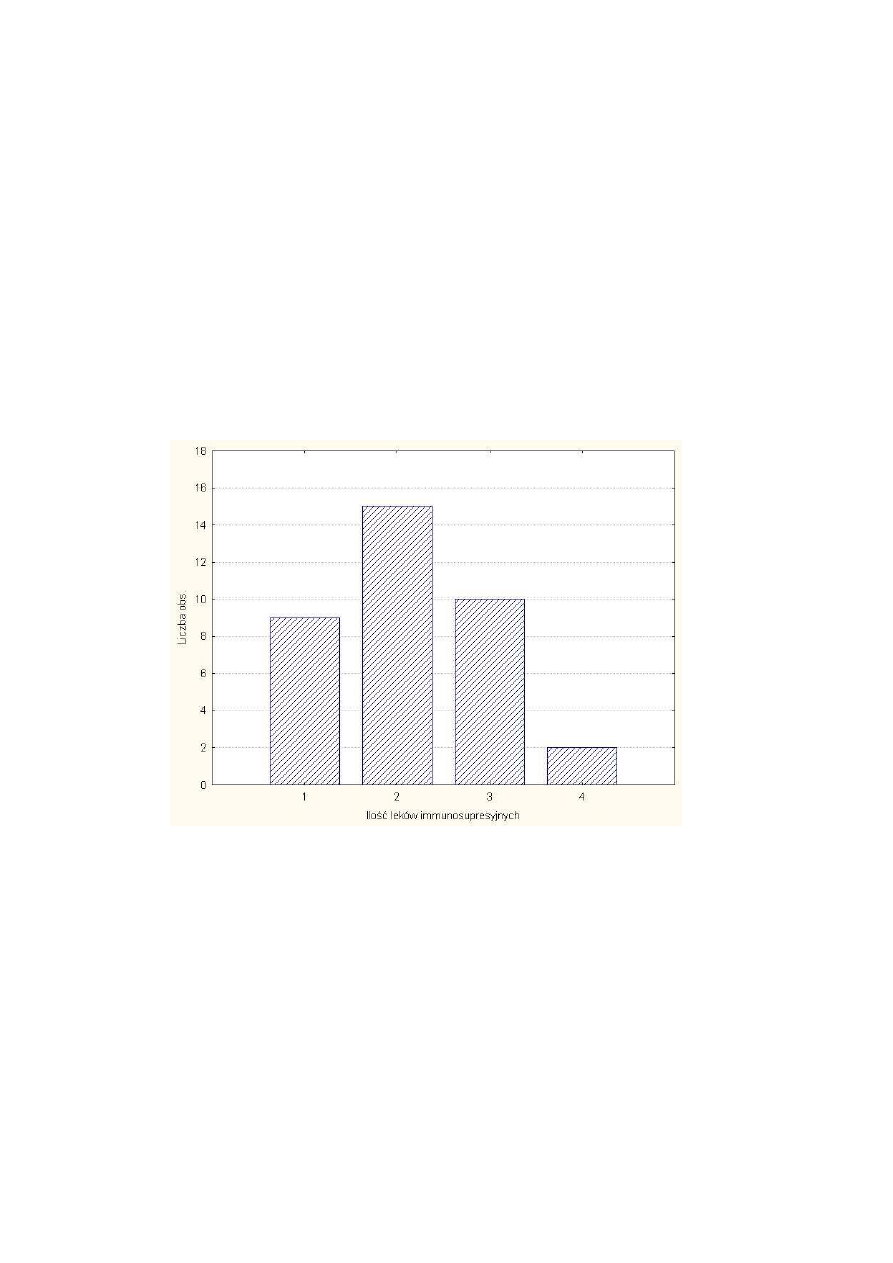
42
u kolejnych 3 pacjentów z powodu pojawienia si
ę
cGvHD czasowo przerwano redukcj
ę
dawki CSA.
Terapii drugiego rzutu z powodu progresji cGvHD wymagało 12 chorych, w tym dwóch
chorych – terapii czterolekowej. U wi
ę
kszo
ś
ci chorych (9) w leczeniu trzylekowym
stosowano dodatkowo mycofenolat mofetilu, jednej chorej oprócz standardowego
leczenia CSA i prednisonem podawano talidomid.
Jedna chora z 2 chorych, którzy wymagali stosowania czterolekowej terapii
immunosupresyjnej otrzymywała oprócz prednisonu i CSA, mycofenolat mofetilu
i pulsy cyklofosfamidu. Drugi chory otrzymywał dodatkowo talidomid oraz PUVA -
terapi
ę
z powodu rozległych zmian skórnych
(Rysunek 12)
Rysunek 12 Ilo
ść
leków stosowanych w terapii immunosupresyjnej
4.2.1.4 Zgony w grupie z cGvHD
W grupie 15 chorych bez cGvHD 2 chorych zmarło z powodu infekcji. Natomiast
w grupie 36 chorych z cGvHD obserwowano 9 zgonów: 4 z powodu wznowy choroby
zasadniczej, 2 z powodu infekcji oraz 3 z powodu cGvHD
(Rysunek 13) .
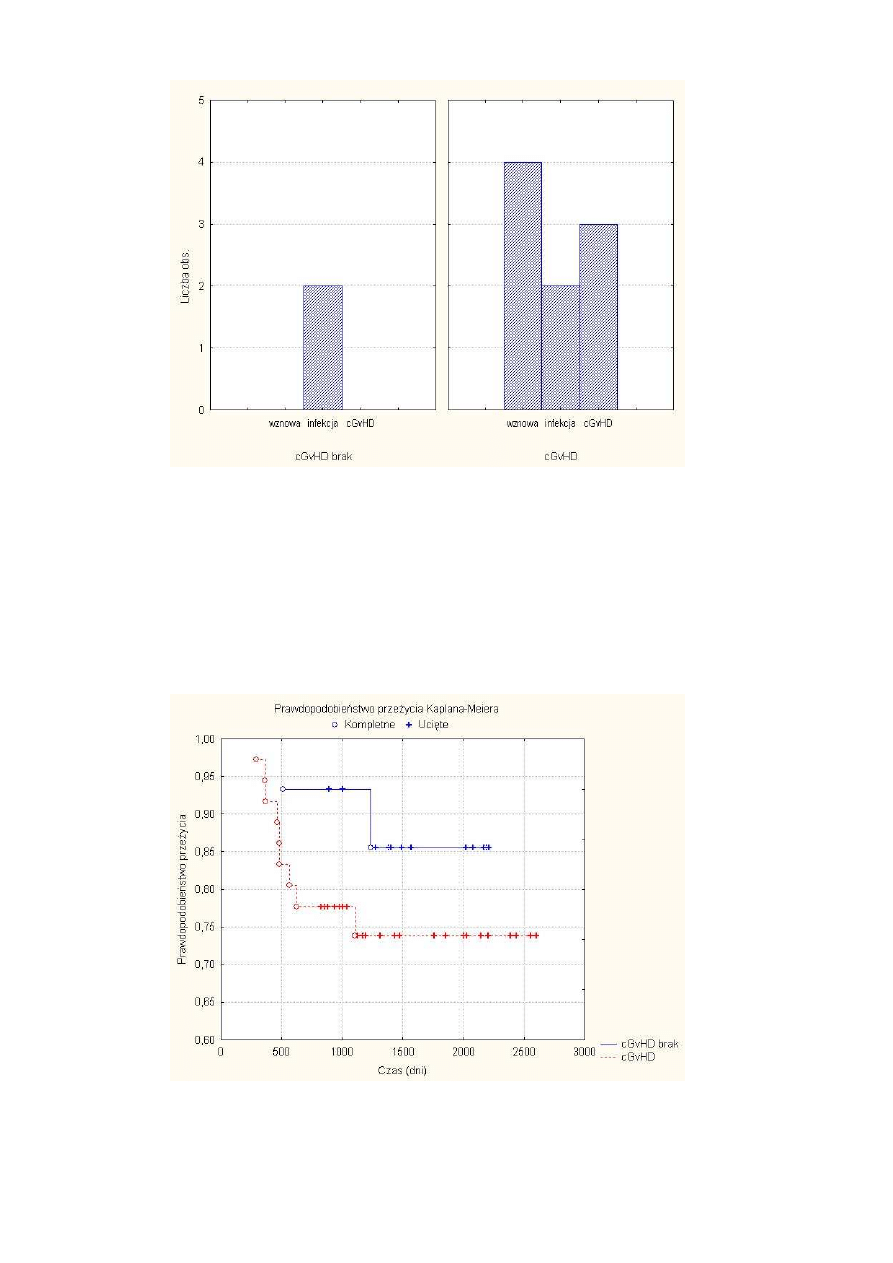
43
Rysunek 13 Przyczyny zgonu w zale
ż
no
ś
ci od wyst
ę
powania cGvHD
Prawdopodobie
ń
stwo prze
ż
ycia chorych z cGvHD było ni
ż
sze od prze
ż
ycia w grupie
bez cGvHD (odpowiednio 0,85 i 0,73 ). Ró
ż
nice te nie były jednak istotne statystycznie
p=0,28
(Rysunek 14).
Rysunek 14 Prawdopodobie
ń
stwo prze
ż
ycia w grupie chorych z i bez cGvHD
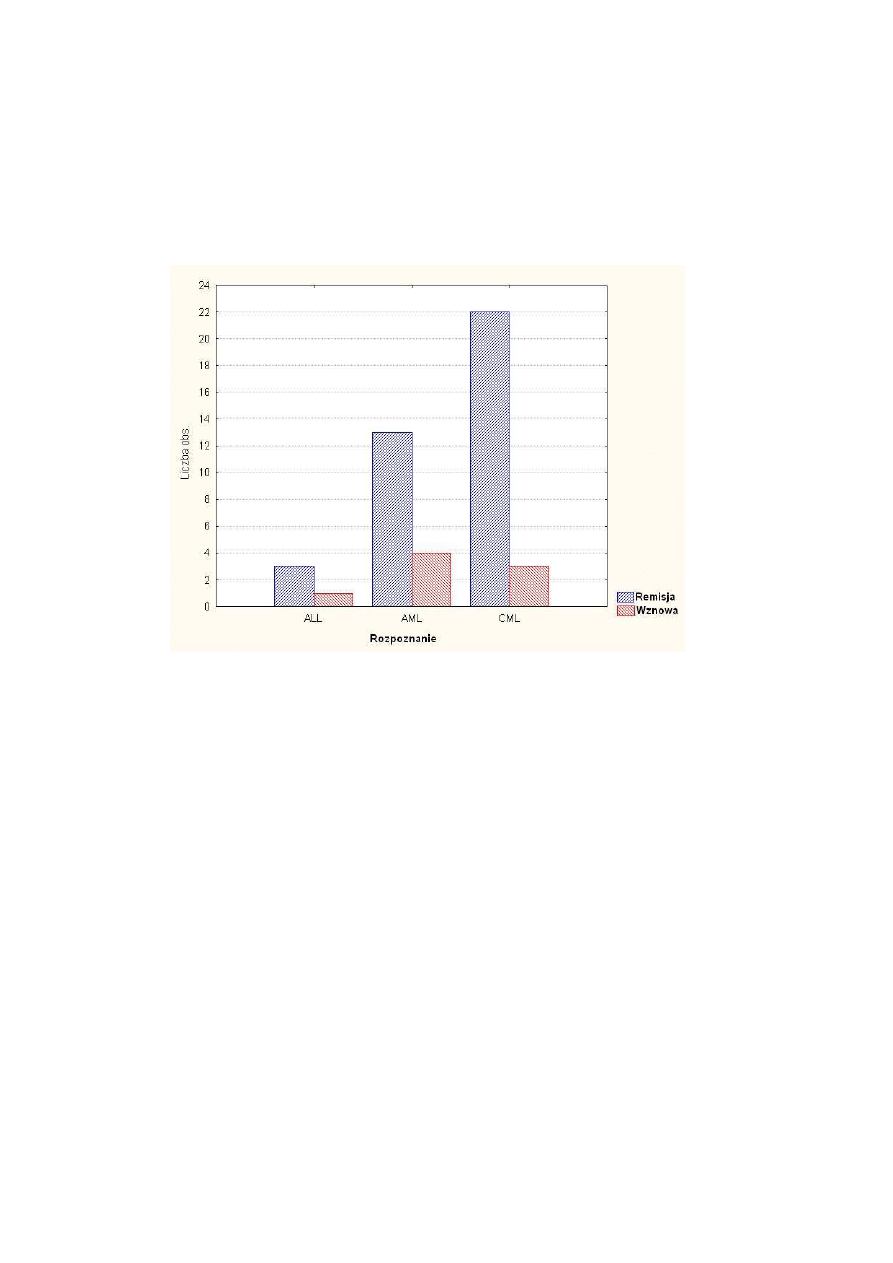
44
4.2.2 Wznowa choroby zasadniczej
Wznowa choroby zasadniczej wyst
ą
piła w sumie u 8 pacjentów: 3 chorych z CML,
4 chorych z AML, i u jednego chorego z ALL. Cz
ę
sto
ść
wyst
ę
powania nawrotu choroby
w poszczególnych rozpoznaniach nie była istotnie znamienna (p=0,91)
Rysunek 15
.
Rysunek 15 Wznowa choroby zasadniczej w zale
ż
no
ś
ci od diagnozy
W grupie chorych z CML wznow
ę
choroby zasadniczej obserwowano u 3 chorych
(u 2 chorych wznowa hematologiczna w fazie przewlekłej choroby, u jednego posta
ć
pozaszpikowa – guz klatki piersiowej, w badaniu histopatologicznym granulocytic
sarcoma)
U dwóch chorych, u których nawrót CML leczono preparatem Glivec (Imatinib)
uzyskano ponownie remisj
ę
cytogenetyczn
ą
. U jednego z tych chorych wykonano drugi
przeszczep od tego samego dawcy, uzyskuj
ą
c remisj
ę
molekularn
ą
(miesi
ę
cy po II
SCT).
Chory ze wznow
ą
pozaszpikow
ą
CML zmarł zanim rozpocz
ę
to terapi
ę
.
W grupie chorych z AML wznowa choroby zasadniczej wyst
ą
piła u 4 chorych.
U 2 chorych
rozpoznano
wznow
ę
hematologiczn
ą
,
u
jednego
chorego
przeszczepionego
z
powodu
AML
b
ę
d
ą
cej
transformacj
ą
zespołu
mielodysplastycznego wyst
ą
piła wznowa pozaszpikowa (guz
ś
ródpiersia w badaniu
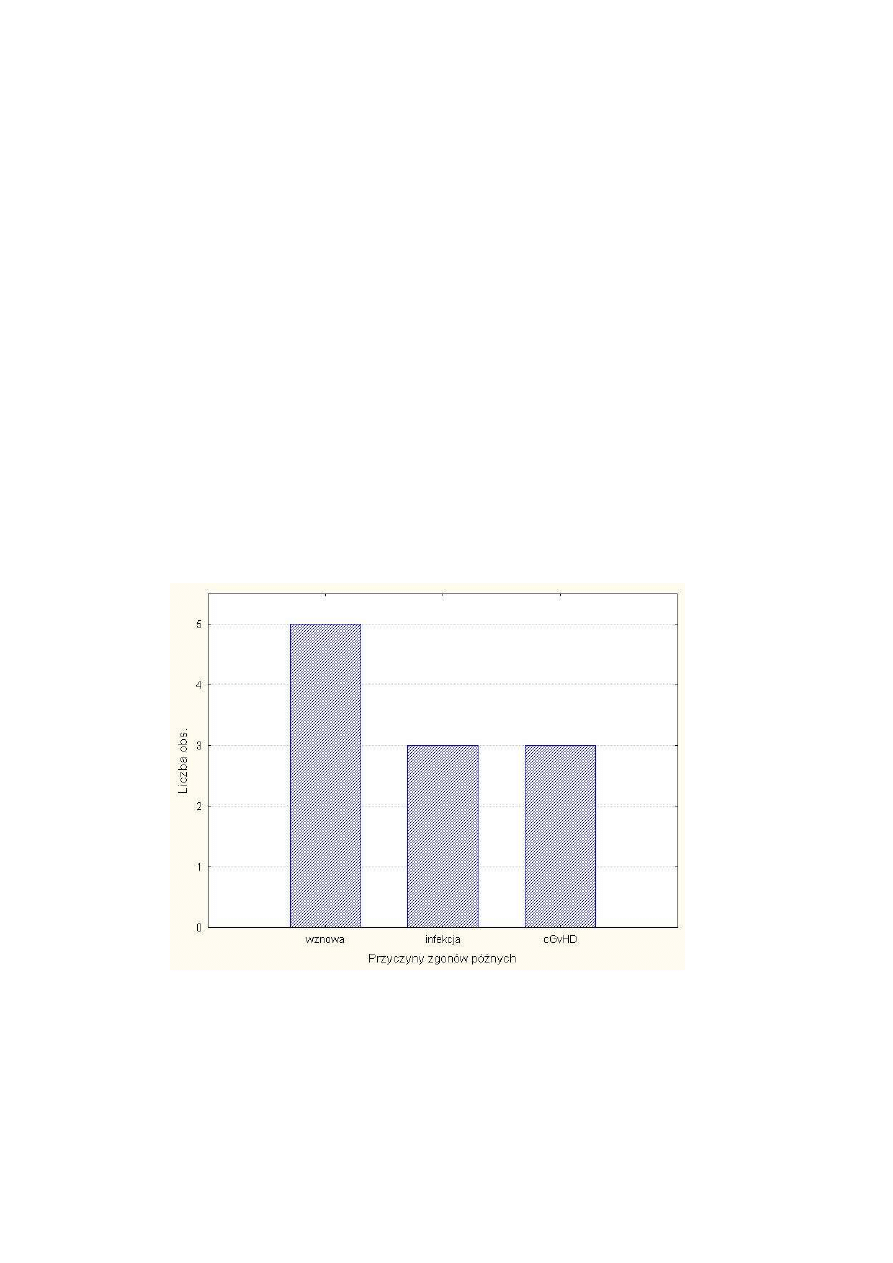
45
histopatologicznym – granulocytic sarcoma), u drugiej chorej z AML obserwowano
wznow
ę
choroby szpikow
ą
oraz zaj
ę
cie CUN.
2 chorych zmarło z powodu powikła
ń
terapii nawrotu lub progresji choroby zasadniczej
mimo leczenia. Natomiast u 1 chorej udało si
ę
uzyska
ć
całkowit
ą
remisj
ę
po leczeniu
reindukcyjnym i infuzji limfocytów dawcy. Chora zmarła około 12 miesi
ę
cy pó
ź
niej
w trakcie terapii trzeciej wznowy choroby. Jeden chory jest w trakcie terapii.
W grupie chorych z ALL wznow
ę
choroby zasadniczej (zarówno szpikow
ą
jak i zaj
ę
cie
CUN) obserwowano u 1 chorego. Chory zmarł z progresji choroby.
4.2.3 Zgony pó
ź
ne
Do chwili obecnej z badanej grupy 51 pacjentów zmarło 11 chorych (22%). U 5 chorych
przyczyn
ą
ś
mierci była wznowa choroby zasadniczej (46%), u 3 powikłania infekcyjne
(27%), u kolejnych 3 cGvHD (27%)
(Rysunek 16).
Rysunek 16 Przyczyny zgonów pó
ź
nych
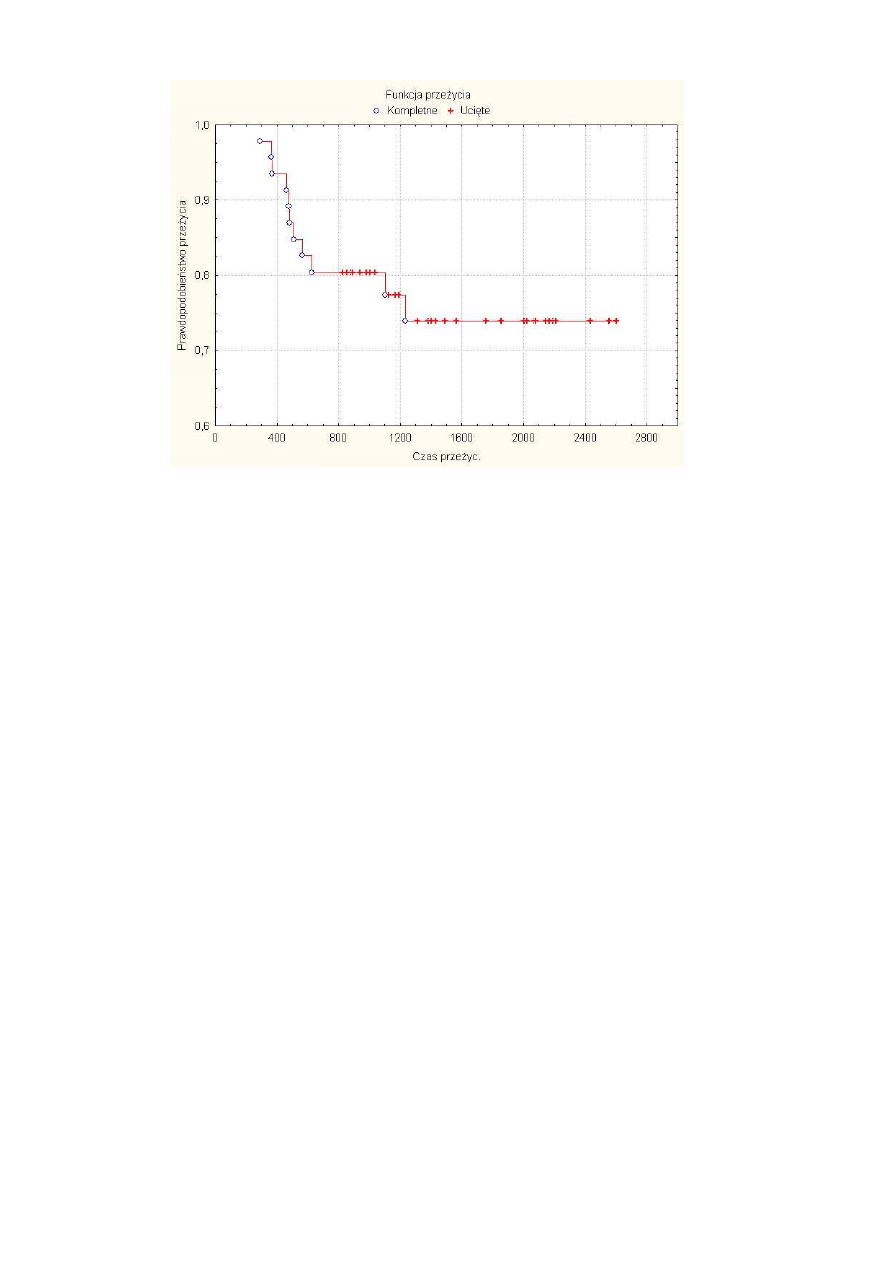
46
Rysunek 17 Krzywa prze
ż
ycia w badanej grupie 51 chorych
W badanej grupie chorych, którzy prze
ż
yli 100 dni po transplantacji 9 osób zmarło do
600 doby, a tylko 2 osoby do doby 1200 nast
ę
pnie krzywa osi
ą
gn
ę
ła plateau.
prawdopodobie
ń
stwo prze
ż
ycia wynosiło ok. 0,73
(Rysunek 17) .
47
4.3 Zale
ż
no
ść
pomi
ę
dzy typem pełnego chimeryzmu a powikłaniami
poprzeszczepowymi
4.3.1 Typ pełnego chimeryzmu a cGvHD
4.3.1.1 Cz
ę
sto
ść
wyst
ę
powania cGvHD w zale
ż
no
ś
ci od typu CC
W grupie 25 chorych, którzy osi
ą
gn
ę
li wczesny pełny chimeryzm, 23 (92%) chorych
rozwin
ę
ło cGvHD, tym posta
ć
rozległa dotyczyła 21 (84%) chorych.
W grupie 26 pacjentów, u których osi
ą
gni
ę
cie pełnego chimeryzmu obserwowano
pó
ź
niej ni
ż
w dobie 100, cGvHD rozwin
ę
ło 13 (50%) chorych, w tym posta
ć
rozległ
ą
rozpoznano jedynie u 8 (31%) chorych
(Rysunek 18).
Ró
ż
nica w cz
ę
sto
ś
ci
wyst
ę
powania rozległej postaci choroby cGvHD w grupie z wczesnym i pó
ź
nym
chimeryzmem była statystycznie znamienna (p
<
0.001).
Rysunek 18 cGvHD w grupach chorych z wczesnym i pó
ź
nym chimeryzmem
48
Statystycznie istotna zale
ż
no
ść
pomi
ę
dzy rodzajem chimeryzmu a wyst
ę
powaniem
cGvHD utrzymywała si
ę
równie
ż
po wykluczeniu z grupy wczesnego chimeryzmu
chorych, którzy otrzymali TBI (p<0,005).
4.3.1.2 cGvHD a aGvHD w zale
ż
no
ś
ci od typu pełnego chimeryzmu
Wyst
ę
powanie aGvHD w grupach chorych wyró
ż
nionych na podstawie czasu
uzyskania CC.
Klinicznie istotn
ą
(przynajmniej II
o
) ostr
ą
chorob
ę
przeszczep przeciwko gospodarzowi
rozpoznano u 11 (42%) i 10 (38%) pacjentów odpowiednio w grupie
z wczesnym i pó
ź
nym chimeryzmem.
W grupie chorych z wczesnym chimeryzmem cz
ęś
ciej obserwowano ostr
ą
chorob
ę
przeszczep przeciwko gospodarzowi w stopniu III i IV - u 7 chorych, w porównaniu
do grupy chorych z pó
ź
nym chimeryzmem - u 3 chorych
(Rysunek 19).
Ró
ż
nice
cz
ę
sto
ś
ci rozpoznania aGvHD w zale
ż
no
ś
ci od rodzaju chimeryzmu u chorych, którzy
prze
ż
yli pierwsze 100 dni po przeszczepie nie były statystycznie istotne (p=0,4).
Rysunek 19 Ostra choroba przeszczep przeciwko gospodarzowi w zale
ż
no
ś
ci od rodzaju
pełnego chimeryzmu
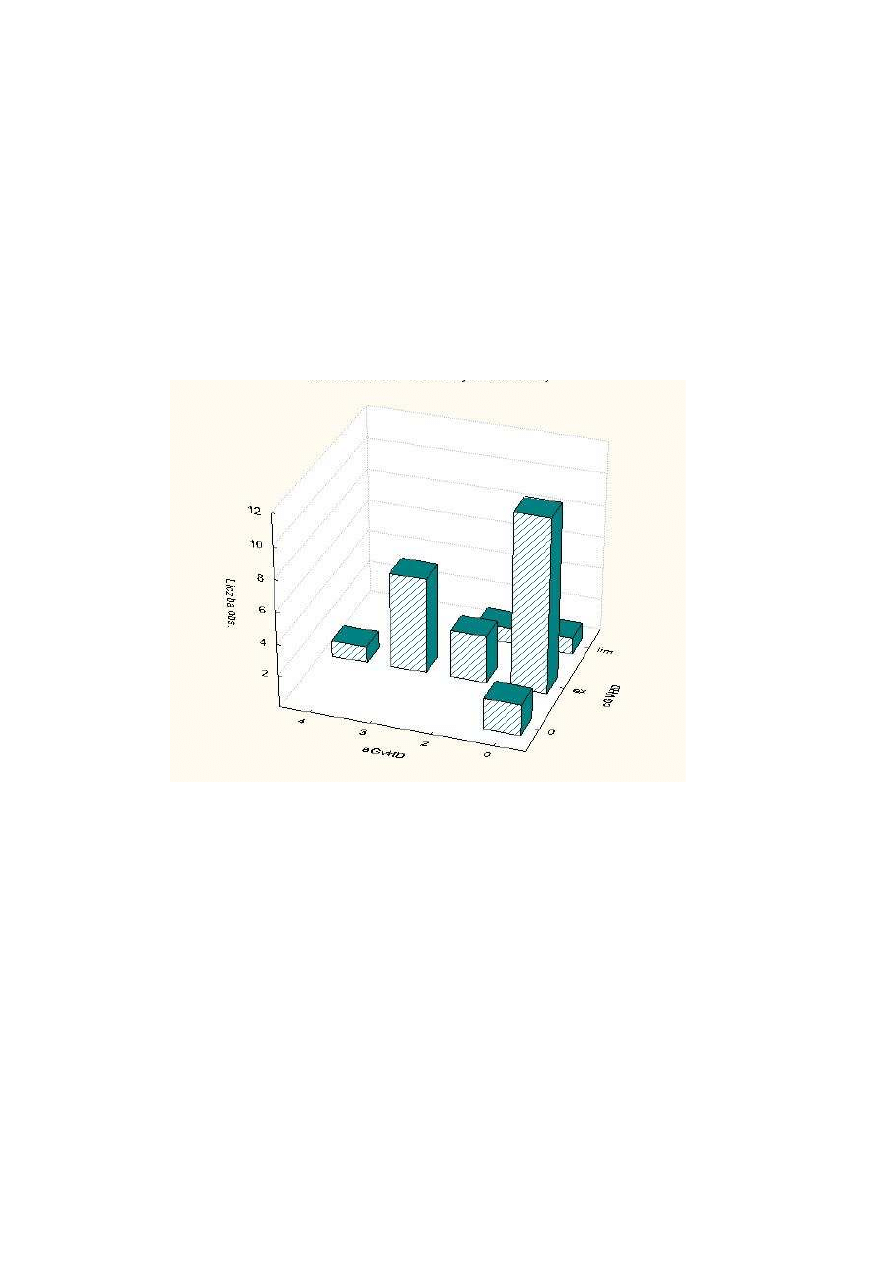
49
W grupie chorych z wczesnym chimeryzmem z 11 chorych, u których obserwowano
aGvHD wszyscy chorzy rozwin
ę
li chorob
ę
przewlekł
ą
, w tym 10 chorych posta
ć
rozległa.
U kolejnych 12 chorych, u których nie rozpoznano choroby ostrej równie
ż
wyst
ą
piły
objawy choroby przewlekłej: u 11 w postaci rozległej, u jednego w ograniczonej.
Tylko u 2 chorych nie rozpoznano
ż
adnej postaci GvHD
(Rysunek 20).
Ze wzgl
ę
du na niewielk
ą
liczebno
ść
poszczególnych grup powy
ż
szych zale
ż
no
ś
ci nie
poddawano analizie statystycznej.
Rysunek 20 Cz
ę
sto
ść
wyst
ę
powania cGvHD w zale
ż
no
ś
ci od aGvHD w grupie chorych
z wczesnym chimeryzmem
W grupie chorych z pó
ź
nym chimeryzmem u 10 pacjentów z rozpoznaniem aGvHD
chorob
ę
przewlekł
ą
rozpoznano u 7 chorych w tym tylko u 2 posta
ć
rozległ
ą
.
Natomiast u 16 chorych, u których nie obserwowano aGvHD u 3 wyst
ą
piły objawy
choroby rozległej i u 3 choroby ograniczonej.
W tej grupie u 10 chorych w ogóle nie prezentowało objawów GvHD
(Rysunek 21).
Ze wzgl
ę
du na niewielk
ą
liczebno
ść
poszczególnych grup powy
ż
szych zale
ż
no
ś
ci nie
poddawano analizie statystycznej.

50
Rysunek 21 Cz
ę
sto
ść
wyst
ę
powanie cGvHD w zale
ż
no
ś
ci od stopnia aGvHD w grupie
chorych z pó
ź
nym CC
Rodzaj pocz
ą
tku cGvHD w zale
ż
no
ś
ci od typu CC
W grupie chorych z wczesnym CC najcz
ęś
ciej (12 chorych) obserwowano typ de novo
cGvHD (53%), typ wyciszony wyst
ą
pił u 7 chorych (30%), a typ post
ę
puj
ą
cy
u 4 pacjentów (17%).
W grupie chorych z pó
ź
nym chimeryzmem typ post
ę
puj
ą
cy wyst
ą
pił tylko u jednego
chorego (8%), a typ wyciszony i de novo z tak
ą
sam
ą
cz
ę
sto
ś
ci
ą
– 6 chorych (46%)
Ró
ż
nice w wyst
ę
powaniu okre
ś
lonego typu cGvHD w poszczególnych grupach
z wczesnym i pó
ź
nym chimeryzmem nie były statystycznie istotne (p=0,55).
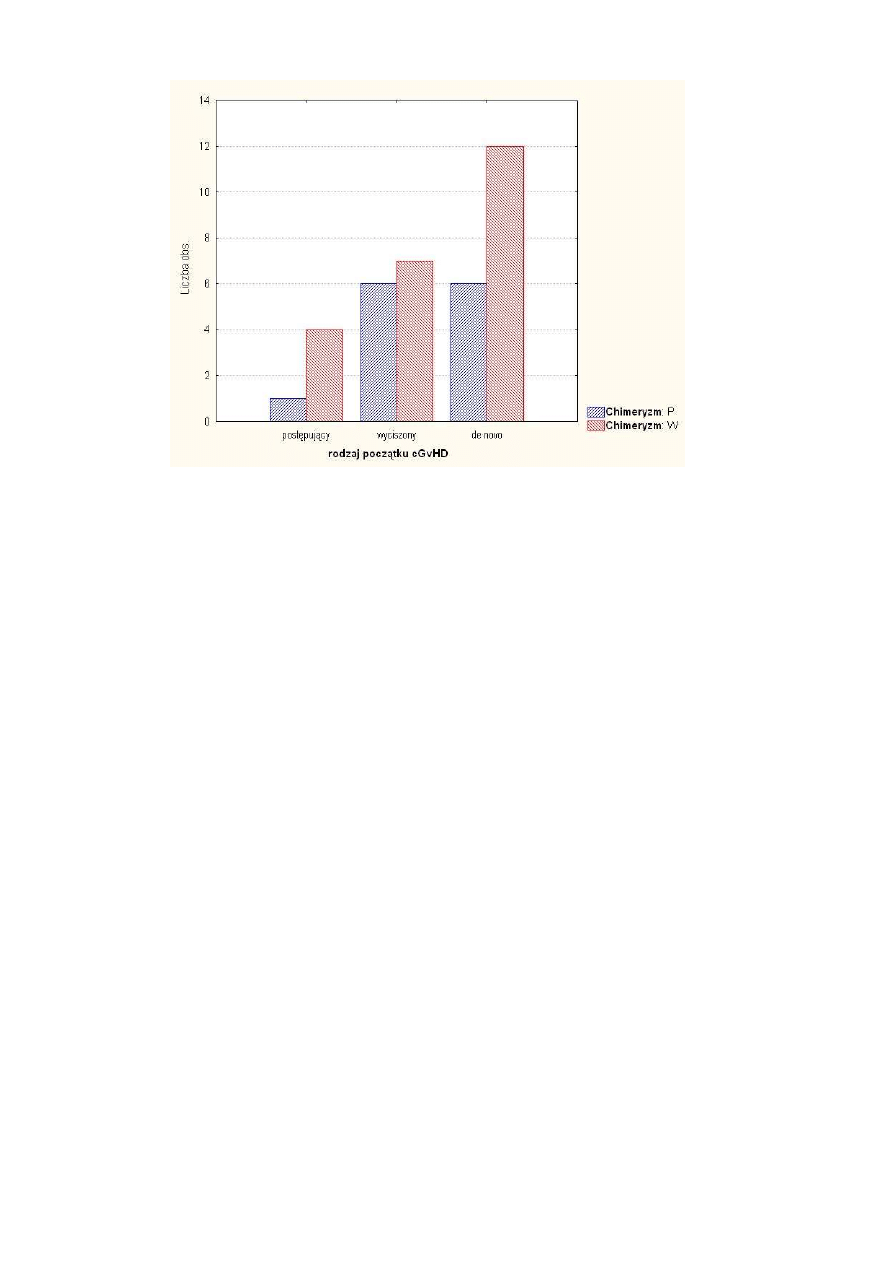
51
Rysunek 22 Typ pocz
ą
tku cGvHD w zale
ż
no
ś
ci od czasu uzyskania CC
4.3.1.3 Rodzaj CC a ci
ęż
ko
ść
przebiegu cGvHD
Ci
ęż
ko
ść
przebiegu cGvHD oceniana była ilo
ś
ci
ą
leków immunosupresyjnych
koniecznych do opanowania choroby. W grupie chorych z wczesnym CC obserwowano
wi
ę
kszy odsetek pacjentów, którzy do kontroli objawów choroby cGvHD wymagali
terapii przynajmniej trzylekowej (12 z 23) w porównaniu do grupy z pó
ź
nym CC gdzie
terapi
ę
trzylekow
ą
prowadzono tylko u 1 z 13
(Rysunek 23).
Powy
ż
sze wyniki były
statystycznie znamienne (P<0.005).
Innym
analizowanym
w
pracy
miernikiem
ci
ęż
ko
ś
ci
przebiegu
cGvHD
w poszczególnych grupach chorych było wyst
ą
pienie zgonu z powodu cGvHD.
W grupie chorych z wczesnym CC trzech pacjentów zmarło z powodu progresji
cGvHD, podczas gdy w grupie z pó
ź
nym CC nie obserwowano zgonów z tej przyczyny
(Rysunek 24).
Ró
ż
nice w cz
ę
sto
ś
ci wyst
ę
powania zgonu z powodu cGvHD
w poszczególnych grupach nie były statystycznie istotne (p=0,68).
Szczegółow
ą
charakterystyk
ę
cGvHD w obu badanych grupach przedstawia
Tabela IX
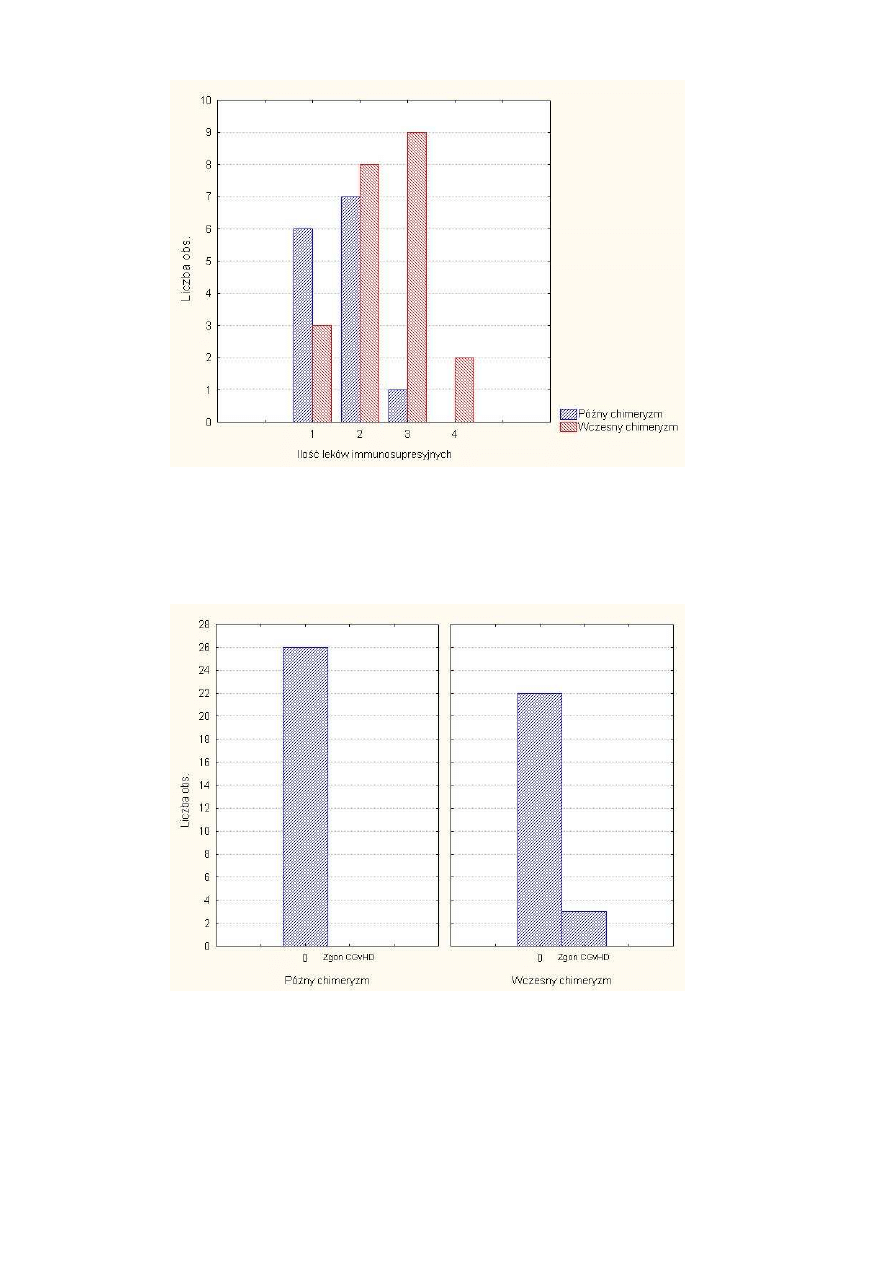
52
Rysunek 23 Rodzaj pełnego chimeryzmu a terapia cGvHD
Rysunek 24 Rodzaj pełnego chimeryzmu a zgony z powodu cGvHD

53
Tabela IX. Charakterystyka cGvHD w grupie chorych z wczesnym i pó
ź
nym chimeryzmem
Wczesny CC (25 chorych)
Pó
ź
ny CC (26 chorych)
CGvHD
23/25 (92%)
13/26 (48%)
ograniczona
2/25 (8%)
5/26 (18%)
rozległa
21/25 (84%)
8/26 (30%)
Typ rozwoju cGvHD
progressive
4/23 (17%)
1/13 (8%)
quiescent
7/23 (30%)
6/13 (46%)
de novo
12/23 (53%)
6/13 (46%)
Terapia immunosupresyjna
Jeden lek
3/23 (13%)
5/13 (38%)
Dwa leki
8/23 (34%)
7/13 (54%)
Trzy lub wi
ę
cej
11/23 (48%)
1/13 (8%)
Doba rozpoznanie cGvHD,
mediana, zakres
131 (88-178)
250 (98-306)
Zgony z powodu cGvHD
3
0
4.3.1.4 Zale
ż
no
ść
pomi
ę
dzy czasem stwierdzenia pełnego chimeryzmu a
wyst
ą
pieniem objawów cGvHD
U wszystkich chorych w grupie z wczesnym CC osi
ą
gni
ę
cie pełnego chimeryzmu
zawsze poprzedzało pojawienie si
ę
pierwszych objawów cGvHD (
ś
redni czas od
uzyskania CC do wyst
ą
pienia cGvHD wynosił 85 dni).
Z drugiej strony objawy cGvHD u pacjentów, którzy osi
ą
gn
ę
li pó
ź
ny CC zwykle
wyst
ę
powały przed osi
ą
gni
ę
ciem CC (
ś
redni czas od wyst
ą
pienia cGvHD
a pojawieniem si
ę
CC wynosił 100 dni). Jedynie w trzech przypadkach w tej grupie
wyst
ą
pienie pierwszych objawów cGvHD nieznacznie poprzedzało osi
ą
gni
ę
cie CC.
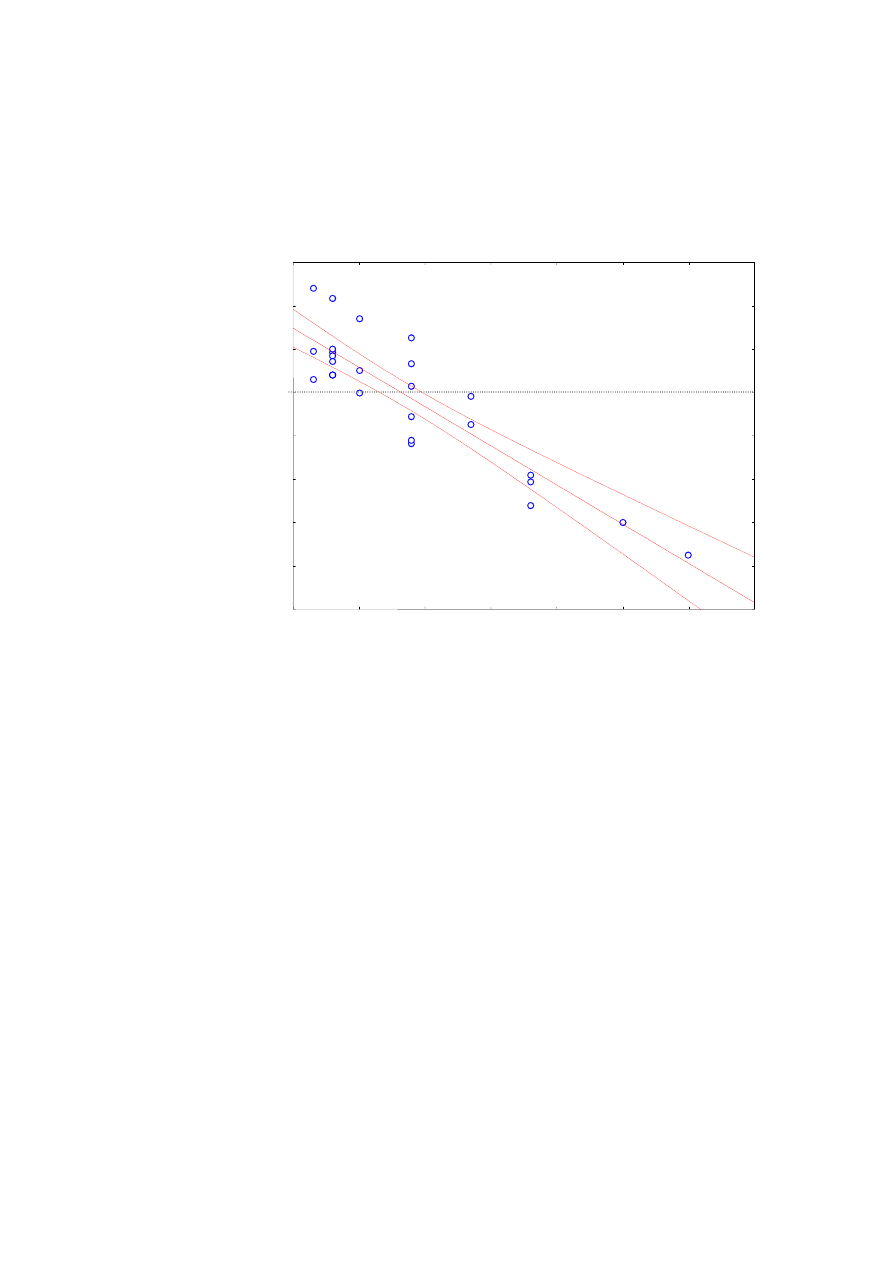
54
Korelacja pomi
ę
dzy czasem uzyskania CC a ró
ż
nic
ą
czasu pomi
ę
dzy czasem do CC
a czasem wyst
ą
pienia cGvHD była statystycznie znamienna (P<0.001)
(Rysunek 25).
Rysunek 25 Korelacja mi
ę
dzy czasem uzyskania CC a ró
ż
nic
ą
czasu pomi
ę
dzy czasem
do uzyskania CC a czasem wyst
ą
pienia cGvHD
4.3.2 Typ CC a wznowa choroby zasadniczej
Wznowa choroby zasadniczej wyst
ą
piła w sumie u 4 chorych w grupie z wczesnym CC
i 4 chorych w grupie z pó
ź
nym CC.
W grupie chorych z wczesnym chimeryzmem wznow
ę
choroby zasadniczej
obserwowano u 3 chorych z AML i u jednego chorego z ALL.
W grupie chorych z pó
ź
nym chimeryzmem do wznowy choroby zasadniczej doszło
u 3 pacjentów z CML i u jednego pacjenta z AML
(Rysunek 26).
Nie stwierdzono
statystycznie znamiennych ró
ż
nic w cz
ę
sto
ś
ci wyst
ę
powania wznowy choroby
zasadniczej w poszczególnych grupach (p=0,95).
0
100
200
300
400
500
600
700
Czas do uzyskania CC (dni)
-500
-400
-300
-200
-100
0
100
200
300
R
ó
z
n
ic
a
p
o
m
i
ę
d
z
y
w
y
s
t
ą
p
ie
n
ie
m
c
G
v
H
D
a
c
z
a
s
e
m
u
z
y
s
k
a
n
ia
C
C
a
(
d
n
i)

55
Tabela X Charakterystyka wznowionych chorych w grupie z pó
ź
nym CC
Chory Rozpoznanie Ryzyko choroby
zasadniczej przed
SCT
Najlepsza odpowied
ź
po SCT
cGvHD
Czas
wznowy
(miesi
ą
c)
TK
CML
I faza przewlekła
Remisja
cytogenetyczna
ograniczona 48
RU
CML
I faza przewlekła
Remisja
molekularna
0
43
GB
CML
I faza przewlekła
Remisja
cytogenetyczna
0
14
SS
AML
I całkowita remisja Całkowita remisja
0
26
Tabela XI Charakterystyka wznowionych chorych w grupie chorych z wczesnym
chimeryzmem
Chory Rozpoznanie
Ryzyko choroby
zasadniczej przed
SCT
Najlepsza odpowied
ź
po SCT
cGvHD
Czas
wznowy
(miesi
ą
c)
DS
AML
Transformacja
MDS
Całkowita remisja
Rozległa
10
DD
AML
I całkowita remisja Całkowita remisja
Rozległa
36
PP
ALL
II całkowita remisja Całkowita remisja
Rozległa
14
IA
AML
I całkowita remisja Całkowita remisja
0
8
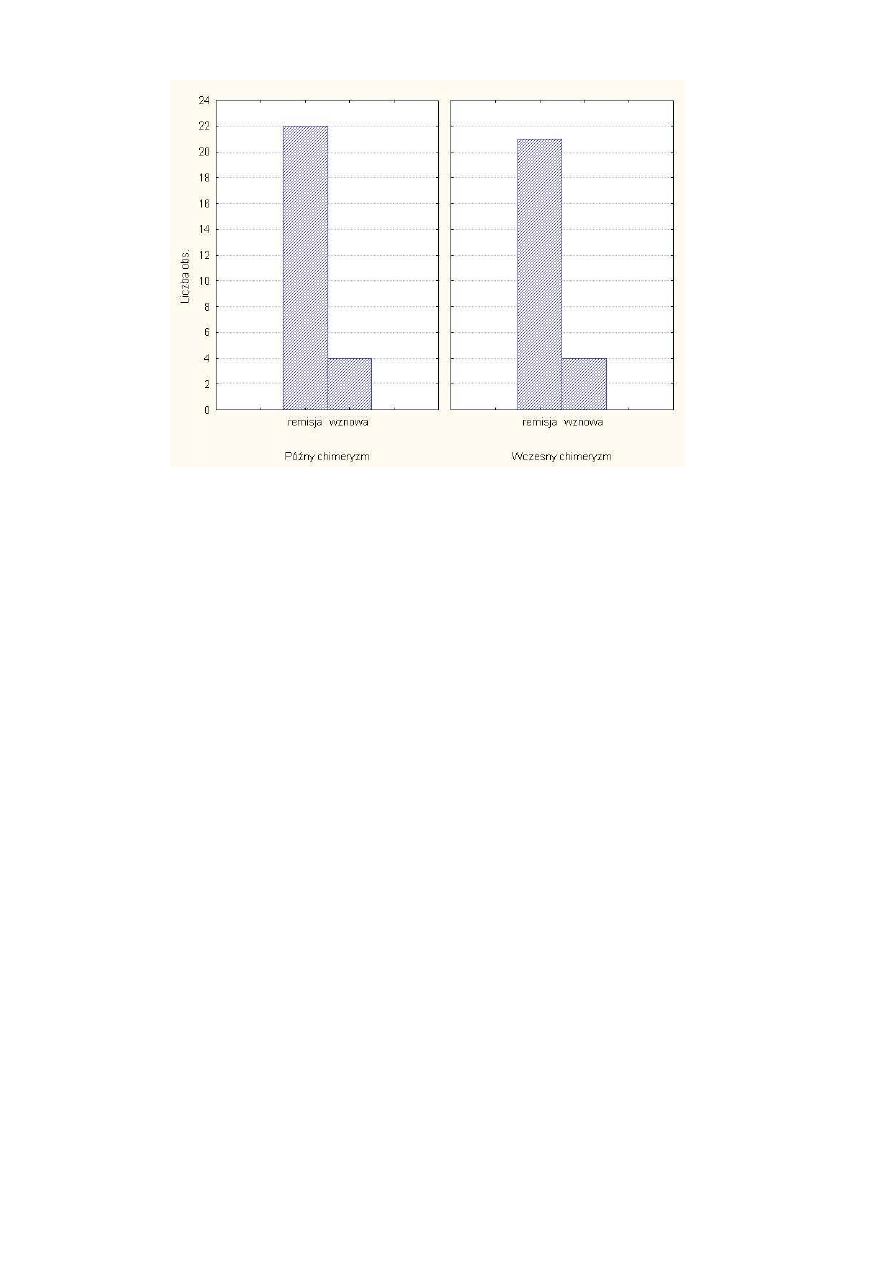
56
Rysunek 26 Wznowa choroby zasadniczej w grupie chorych z wczesnym i pó
ź
nym
chimeryzmem
4.3.3 Przyczyna zgonów pó
ź
nych w zale
ż
no
ś
ci od rodzaju CC
W grupie chorych z wczesnym chimeryzmem 3 chorych (43%) zmarło z powodu
przewlekłej choroby przeszczep przeciwko gospodarzowi, 3 (43%) z powodu wznowy
choroby zasadniczej i jedna chora (14%) z powodu powikła
ń
infekcyjnych. W grupie
chorych z pó
ź
nym chimeryzmem 2 chorych (50%) zmarło z powodu powikła
ń
infekcyjnych, a kolejnych 2 (50%) z powodu wznowy choroby zasadniczej. W tej grupie
nie obserwowano zgonów z powodu cGvHD
(Rysunek 27).
Ró
ż
nice w przyczynie
zgonów w poszczególnych grupach nie były istotne statystycznie (p=0,23).
Prawdopodobie
ń
stwo prze
ż
ycia w poszczególnych grupach przedstawia
(Rysunek 28).
Do około 1100 doby obserwowano ró
ż
nice w prze
ż
yciu chorych w grupach
wyró
ż
nionych na podstawie czasu uzyskania CC o ok. 20% na korzy
ść
grupy chorych
z pó
ź
nym CC. Na ko
ń
cu obserwacji ró
ż
nica pomi
ę
dzy prze
ż
yciem w obu grupach
zmniejszyła si
ę
12% i prawdopodobie
ń
stwo prze
ż
ycia wynosiło odpowiednio 72% dla
chorych z wczesnym CC i 84% dla chorych
z pó
ź
nym CC.
Ró
ż
nice nie były istotnie
statystycznie p=0,13.
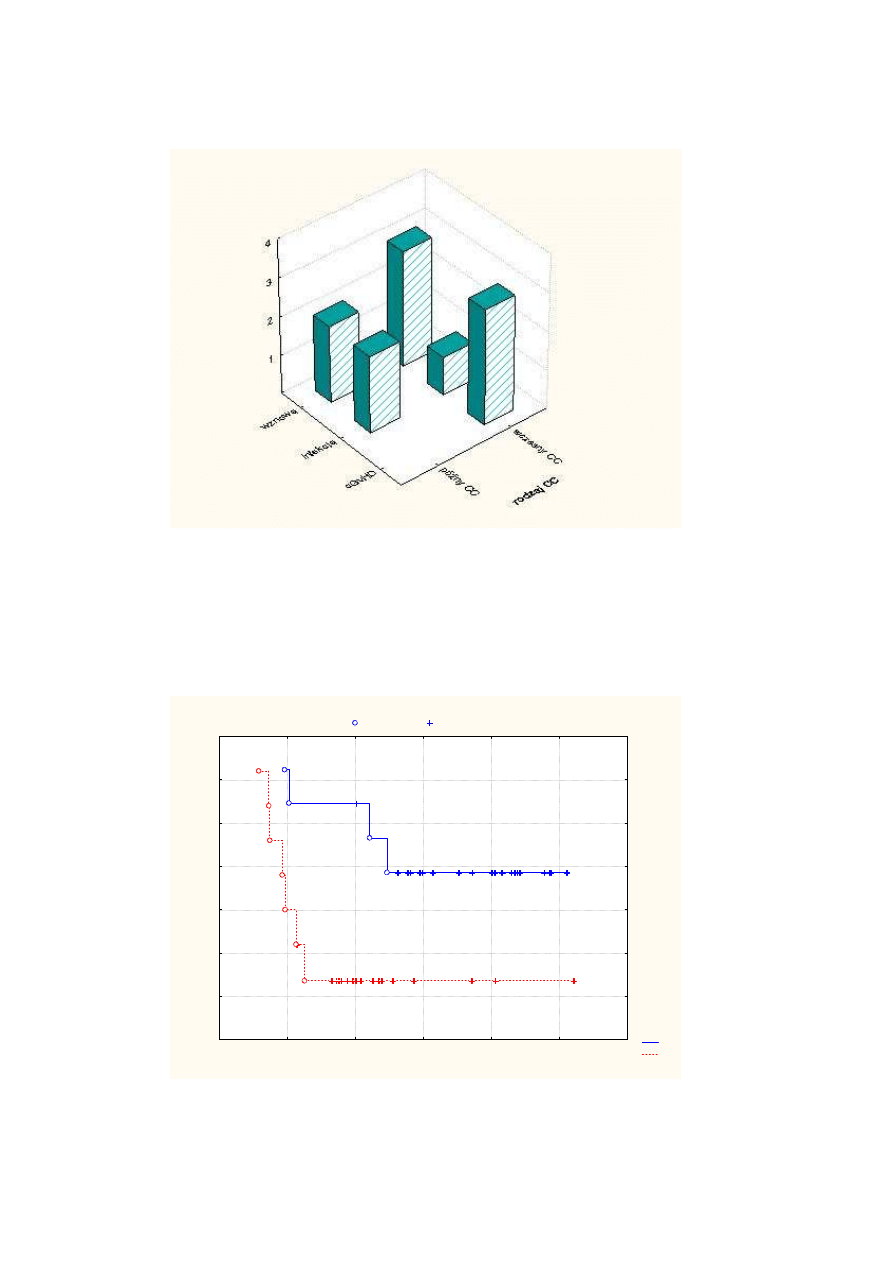
57
Rysunek 27 Przyczyny zgonów pó
ź
nych w zale
ż
no
ś
ci od czasu uzyskania pełnego
chimeryzmu
Prawdopodobie
ń
stwo prze
ż
ycia Kaplana-Meiera
Kompletne
Uci
ę
te
P
W
0
500
1000
1500
2000
2500
3000
Czas
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
P
ra
w
d
o
p
o
d
o
b
ie
ń
s
tw
o
p
rz
e
ż
y
c
ia
Rysunek 28 Prawdopodobie
ń
stwo prze
ż
ycia w zale
ż
no
ś
ci od czasu uzyskania pełnego
chimeryzmu

58
4.4 Chimeryzm w poszczególnych subpopulacjach leukocytów krwi
obwodowej
U 15 pacjentów chimeryzm został oceniony w poszczególnych populacjach
leukocytów: limfocytach T, komórkach NK, monocytach i granulocytach. Analizie
statystycznej poddano wyniki 12 chorych, którzy osi
ą
gn
ę
li CC w trakcie obserwacji.
Trzech pacjentów wykluczono ze wzgl
ę
du na przetrwały MC.
Z 12 ocenianych chorych, trzech osi
ą
gn
ę
ło CC w WBC przed doba 100 po
przeszczepie, natomiast 9 w dalszej obserwacji. U 8 z nich wykrywano MC
w przynajmniej 1 populacji leukocytów przy CC wykazanym w krwi pełnej. U wi
ę
kszo
ś
ci
chorych obserwowano wczesny CC w populacji monocytów (CD14), granulocytów
(CD66b) oraz komórek NK (CD3
-
CD16/56
+
). W dobie 30 po przeszczepie odpowiednio
92%(11 chorych), 85% (10 chorych) oraz 85% (10 chorych) wykazywało CC w tych
populacjach.
Natomiast w populacji limfocytów T pełny chimeryzm do doby 100 osi
ą
gn
ę
ło jedynie
trzech badanych chorych (23%). U tych chorych równie
ż
badania chimeryzmu
w leukocytach krwi obwodowej wykazywały CC. U wszystkich trzech chorych rozwin
ę
ła
si
ę
posta
ć
rozległa cGvHD (100%). U jednego chorego, u którego osi
ą
gni
ę
cie CC
zarówno w nie selekcjonowanych leukocytach jak i we wszystkich badanych
populacjach wykazano ju
ż
w dobie 30 doszło do rozwoju rozległej cGvHD o typie
post
ę
puj
ą
cym, poprzedzonej aGvHD w stopniu III. Chory ten wymagał trójlekowej
immunosupresji.
U 9 chorych którzy osi
ą
gn
ę
li CC w WBC po dobie 100, MC wykrywano najcz
ęś
ciej
w populacji limfocytów T. Dodatkowo jeden pacjent wykazywał MC w populacji
komórek NK i jeden zarówno w granulocytach jak i komórkach NK
(Rysunek 29).
W tej
grupie cGvHD obserwowano u 6 chorych, a trzech z nich rozwin
ę
ło chorob
ę
rozległ
ą
(33.3%). Dwóch pacjentów wykazywało przetrwały MC w populacji CD3 podczas gdy
we krwi obwodowej nie wykrywano ju
ż
własnego DNA
(Rysunek 29).
U obu pacjentów
nie obserwowano do tej pory objawów cGvHD. Byli to pacjenci z CML, którzy nigdy nie
osi
ą
gn
ę
li molekularnej remisji choroby zasadniczej po transplantacji: nadal utrzymuje
si
ę
obecno
ść
transkryptu bcr/abl natomiast nie wykryto obecno
ś
ci chromosomu Ph
zarówno w konwencjonalnej cytogenetyce jak i badaniu FISH (okres obserwacji u obu
chorych powy
ż
ej 30 msc.)
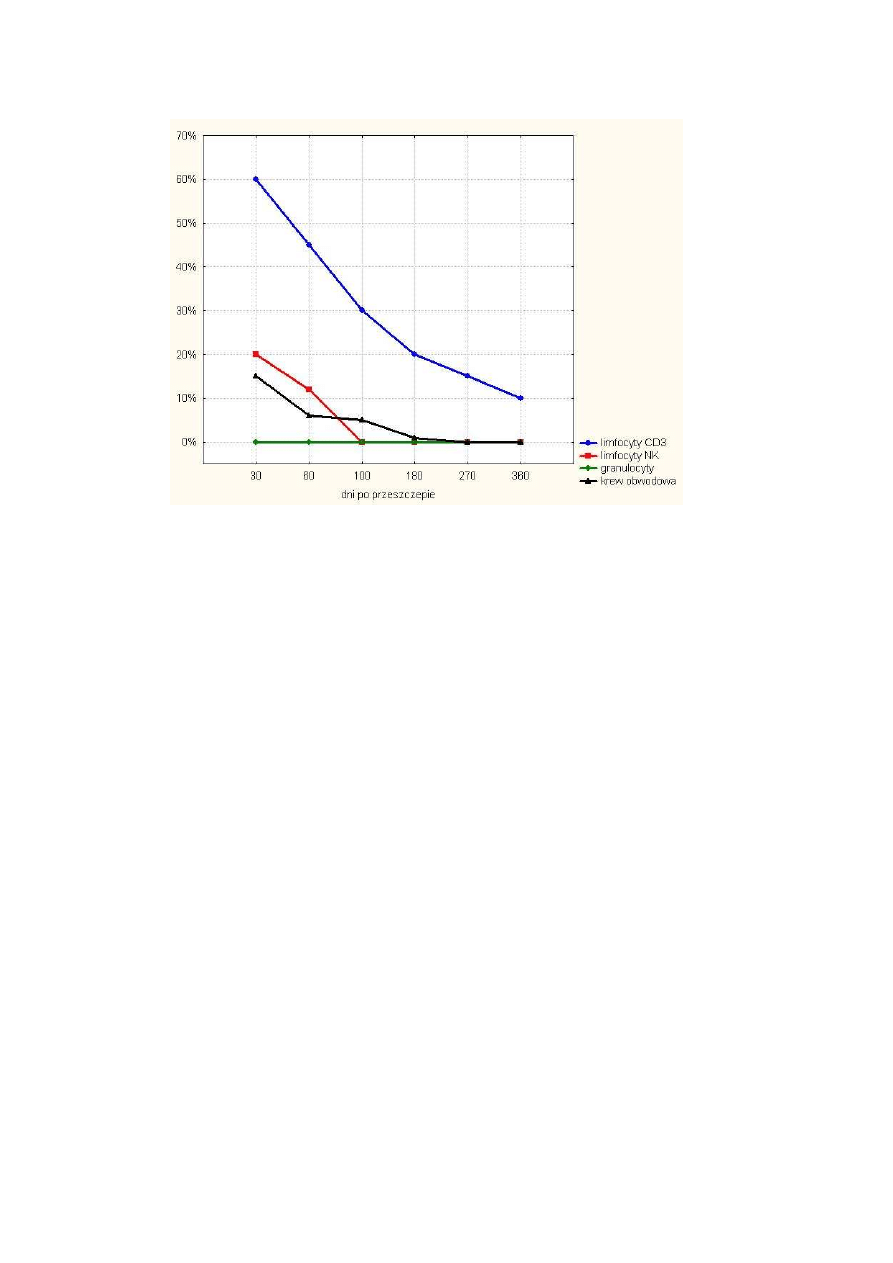
59
Rysunek 29 Przykładowy przebieg chimeryzmu w poszczególnych populacjach krwi
obwodowej u jednego z chorych
Pełny chimeryzm ju
ż
1 miesi
ą
c po transplantacji obserwowano w populacji
granulocytów, CC komórek NK w 3 miesi
ą
ce po przeszczepie. W populacji limfocytów T
(CD3) obserwowano stopniow
ą
redukcj
ę
odsetka DNA biorcy z 60% w 1 miesi
ą
c po
przeszczepie do 10% w ok. 1 rok po. Mimo utrzymywania si
ę
MC w populacji CD3 od
9 miesi
ą
ca po przeszczepie w krwi obwodowej nie wykrywano ju
ż
DNA biorcy.
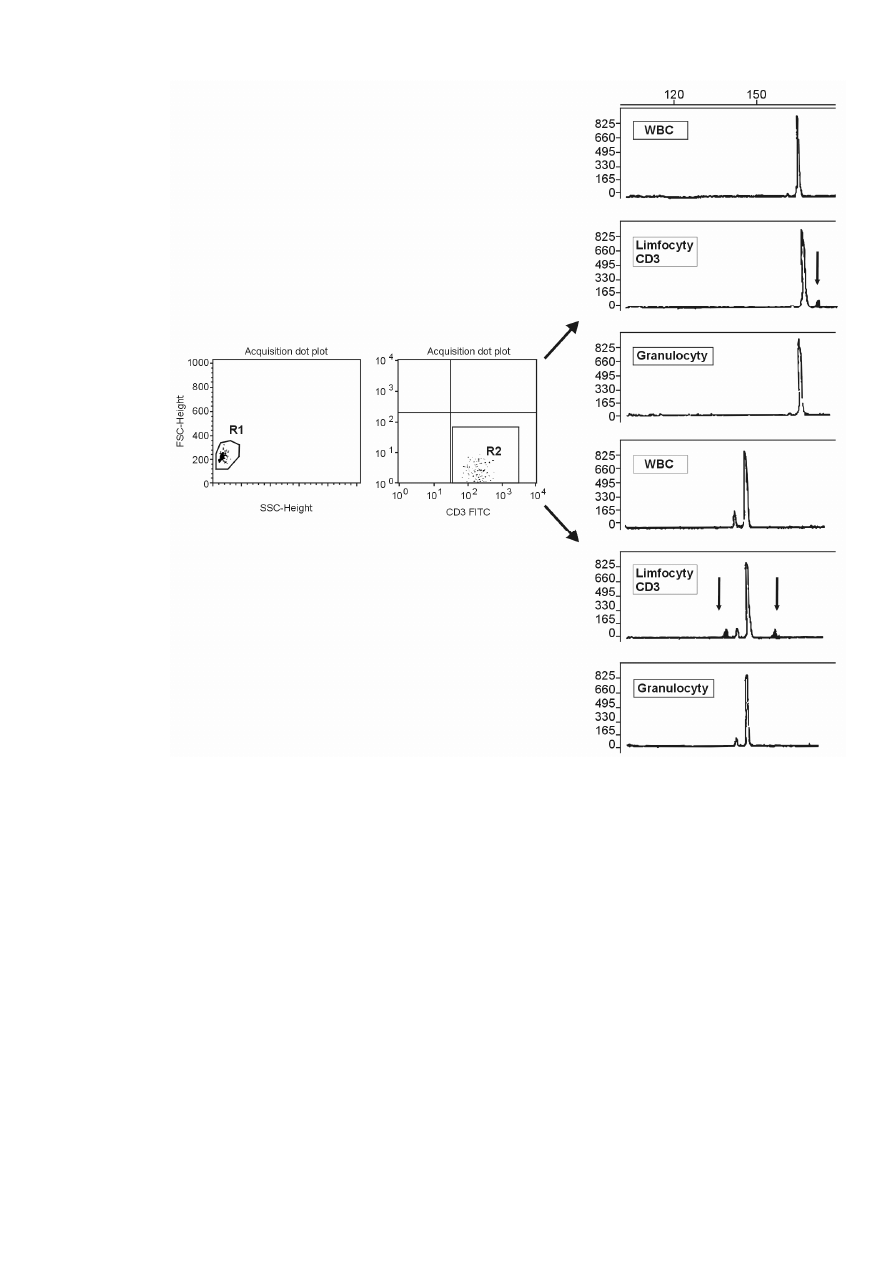
60
Rysunek 30 Przykład mieszanego chimeryzmu w wysortowanych limfocytach CD3
wykazany metod
ą
multiplex PCR w 2 loci (strzałki pionowe).

61
5
OMÓWIENIE WYNIKÓW I DYSKUSJA
5.1 Chimeryzm hematopoetyczny
Równocze
ś
nie z pierwszymi transplantacjami szpiku wykonanymi w naszym
o
ś
rodku w roku 1994 rozpocz
ę
to ocen
ę
chimeryzmu hematopoetycznego. Pocz
ą
tkowo
amplifikowane metod
ą
PCR allele VNTR rozdzielano na
ż
elu agarozowym (153).
W 1999
rozpocz
ę
to
ocen
ę
chimeryzmu
z
zastosowaniem
jednej
z najnowocze
ś
niejszych ówcze
ś
nie metod, opracowanej pierwotnie dla celów
kryminalistyki - identyfikacji
ś
ladów biologicznych w mieszaninie. Metoda ta polega na
rozdziale znakowanych fluorescencyjnie alleli STR metod
ą
elektroforezy kapilarnej na
automatycznym sekwenatorze DNA. Reakcj
ę
PCR przeprowadza si
ę
za pomoc
ą
komercyjnie dost
ę
pnego zestawu umo
ż
liwiaj
ą
cego równoczesn
ą
amplifikacj
ę
wielu loci
STR
z
jednoczesnym
wyznakowaniem
fluorescencyjnym
produktów
PCR
(kompleksowa reakcja PCR) (15). U wszystkich chorych przeszczepianych w naszym
o
ś
rodku powy
ż
sza metoda stosowana jest do rutynowej oceny chimeryzmu zarówno
we krwi obwodowej jak i szpikowej. Dodatkowo u chorych, u których wykonano
przeszczep od dawcy niezgodnego w układzie grup głównych krwi lub fenotypie Rh,
monitoruje si
ę
chimeryzm w układzie czerwonokrwinkowym metodami serologicznymi.
U niektórych chorych badania poszerza si
ę
równie
ż
o analiz
ę
antygenów grupowych
na krwinkach czerwonych metod
ą
cytometrii przepływowej.
Przedstawione w pracy wyniki dotyczyły oceny chimeryzmu komórek j
ą
drzastych
krwi obwodowej. Ze wzgl
ę
du na niepełne dane oceny krwi szpikowej zrezygnowano
z równoczesnej analizy tego materiału. Dodatkowo zalet
ą
oceny krwi obwodowej przy
tak sformułowanym celu pracy jest fakt,
ż
e limfocyty, które s
ą
komórkami efektorowymi
zarówno cGvHD jak i reakcji GvL, stanowi
ą
wi
ę
kszy odsetek we krwi obwodowej ni
ż
szpiku.
Wprowadzenie metody kompleksowej reakcji PCR do rutynowego oznaczania
chimeryzmu w naszym o
ś
rodku potwierdziło dokonane wst
ę
pnie obserwacje,
ż
e
stosuj
ą
c metod
ę
o wysokim stopniu czuło
ś
ci, eliminacj
ę
DNA biorcy obserwujemy
pó
ź
niej ni
ż
przypuszczano (28). W wi
ę
kszo
ś
ci prac badaj
ą
cych chimeryzm
hematopoetyczny u pacjentów po HSCT uzyskanie CC zarówno w krwi obwodowej jak
i szpiku stwierdzano ju
ż
w 30 dobie po przeszczepie (57;69;83;93;112). Wynika to
zarówno ze stosowania metod oceny chimeryzmu o znacznie mniejszej czuło
ś
ci,
a tak
ż
e ze sposobu definiowania pełnego chimeryzmu przez badaczy. Wiele prac

62
analizuj
ą
c znaczenie chimeryzmu wprowadza okre
ś
lenie CC w sytuacji gdy DNA
dawcy stanowi 95-98% zakładaj
ą
c,
ż
e ten niewielki odsetek nie ma znaczenia dla
funkcjonowania przeszczepu okre
ś
lanego jedynie kryteriami ilo
ś
ciowej regeneracji
hematopoezy (7;57;69;112). Jednak w
ś
ród pomijanych w tych 2 - 5% DNA biorcy
mog
ą
znajdowa
ć
si
ę
komórki maj
ą
ce kluczowe znaczenie w wielu reakcjach
immunologicznych jak kontrola choroby zasadniczej, indukcja procesu tolerancji
i ochrona przed rozwini
ę
ciem cGvHD (41;57;111;112;140;147). Z tego powodu bardziej
warto
ś
ciowe jest wprowadzenie
ś
cisłej definicji CC jako całkowitego braku komórek
biorcy. Potwierdzaj
ą
to równie
ż
wyniki naszych bada
ń
, gdy
ż
poziom DNA w grupie
chorych z pó
ź
nym CC nigdy nie przekroczył 2%.
U
ż
ycie bardziej czułych metod oraz zdefiniowanie chimeryzmu jako obecno
ś
ci
jedynie komórek o genotypie dawcy, umo
ż
liwiło wyodr
ę
bnienie dwóch grup chorych.
W pierwszej grupie CC został osi
ą
gni
ę
ty do 100 doby po przeszczepie (wczesny CC),
czyli do eliminacji własnych komórek hematopoetycznych doszło w okresie gdy
wszyscy pacjenci otrzymywali jeszcze profilaktyk
ę
GvHD w pełnej dawce
immunosupresyjnej. W drugiej grupie CC został osi
ą
gni
ę
ty pó
ź
niej ni
ż
w 100 dobie
– w okresie redukcji dawki leków immunosupresyjnych lub po ich całkowitym
odstawieniu (pó
ź
ny CC).
W tak wyró
ż
nionych podgrupach postanowiono oceni
ć
wpływ parametrów
charakteryzuj
ą
cych biorc
ę
, jak równie
ż
czynników zwi
ą
zanych z przebiegiem
transplantacji na czas uzyskania CC.
Nie stwierdzono statystycznie istotnej zale
ż
no
ś
ci pomi
ę
dzy płci
ą
biorców,
wiekiem dawcy, rodzajem materiału przeszczepowego, a rodzajem CC w kolejnych
punktach oceny.
Wykazano statystycznie istotn
ą
zale
ż
no
ść
pomi
ę
dzy czasem uzyskania CC
a wiekiem biorcy. CC wczesny osi
ą
gn
ę
li chorzy młodsi ni
ż
w grupie z pó
ź
nym CC,
a wi
ę
kszo
ść
chorych w grupie z wczesnym chimeryzmem wi
ę
kszo
ść
chorych była
młodsza ni
ż
45 lat. Wynikało to z osi
ą
gni
ę
cia wczesnego CC przez wszystkich chorych,
którzy otrzymali TBI.
Nie wykazano zale
ż
no
ś
ci pomi
ę
dzy diagnoz
ą
a czasem osi
ą
gni
ę
cia CC.
Natomiast w poszczególnych rozpoznaniach stwierdzono uzyskanie wczesnego
chimeryzmu przez wszystkich chorych z ALL, co było zwi
ą
zane z rodzajem
kondycjonowania. W grupie chorych z rozpoznaniem CML statystycznie cz
ęś
ciej
obserwowano pó
ź
ny pełny chimeryzm.

63
Wczesny CC został uzyskany przez wszystkich chorych, którzy w post
ę
powaniu
przygotowawczym otrzymali TBI.
Szybsze
osi
ą
gni
ę
cie
pełnego
chimeryzmu
u
chorych
poddawanych
napromienianiu całego ciała w porównaniu do chorych, którzy w post
ę
powaniu
przygotowawczym otrzymywali jedynie chemioterapi
ę
był obserwowany równie
ż
przez
innych badaczy. Spo
ś
ród innych badanych czynników wykazali oni równie
ż
wpływ
wzrostu wieku na szybsze tempo uzyskiwania CC, czego nie wykazano w badanej
grupie chorych (139).
W grupach wydzielonych na podstawie czasu uzyskania CC porównano
cz
ę
sto
ść
wyst
ę
powania dwóch zale
ż
nych od procesów immunologicznych powikła
ń
:
cGvHD oraz wznowy choroby zasadniczej. Wykazano
ż
e cz
ę
sto
ść
wyst
ę
powania
rozległej postaci cGvHD w grupie chorych z wczesnym CC była statystycznie istotnie
wy
ż
sza ni
ż
w grupie chorych którzy osi
ą
gn
ę
li CC powy
ż
ej doby 100 po transplantacji
(odpowiednio 84% do 31%). Powy
ż
sza zale
ż
no
ść
utrzymywała si
ę
równie
ż
w podgrupach wydzielonych na podstawie rodzaju kondycjonowania – po wykluczeniu
z badanej grupy chorych, którzy otrzymali TBI. Oceniono równie
ż
ci
ęż
ko
ść
przebiegu
cGvHD w poszczególnych grupach z wczesnym i pó
ź
nym CC traktuj
ą
c jako
podstawowy miernik oceny ci
ęż
ko
ś
ci choroby ilo
ść
leków immunosupresyjnych
koniecznych do kontroli objawów. Wi
ę
kszo
ść
pacjentów z grupy z wczesnym CC
wymagała terapii kolejnego /drugiego lub trzeciego/ rzutu i przynajmniej trzech leków
immunosupresyjnych do opanowania choroby. Ró
ż
nice były statystycznie znamienne.
Kolejnym wyznacznikiem oceny ci
ęż
ko
ś
ci przebiegu cGvHD w poszczególnych
podgrupach było wyst
ą
pienie zgonu z powodu cGvHD. Wykazano,
ż
e wszyscy chorzy,
których przyczyn
ą
zgonu była przewlekła choroba przeszczep przeciwko gospodarzowi
osi
ą
gn
ę
li wczesny CC.
Interesuj
ą
c
ą
z klinicznego punktu widzenia obserwacj
ą
było wykazanie
zale
ż
no
ś
ci pomi
ę
dzy czasem uzyskania CC a czasem wyst
ą
pienia pierwszych
objawów cGvHD. W podgrupie chorych z wczesnym CC objawy cGvHD wyst
ą
piły
wcze
ś
niej (mediana czasu rozpoznania cGvHD wynosiła 131 dni po przeszczepie).
Dodatkowo w tej grupie u wszystkich badanych osi
ą
gni
ę
cie CC (mediana 60 doba)
zawsze poprzedzało wyst
ą
pienie pierwszych objawów cGvHD, ze
ś
redni
ą
ró
ż
nic
ą
czasu pomi
ę
dzy dob
ą
, w której rozpoznano cGvHD, a czasem osi
ą
gni
ę
cia pełnego
chimeryzmu 85 dni. U prawie wszystkich chorych w tej grupie obserwowano objawy
choroby rozległej. Natomiast w podgrupie, okre
ś
lonej na podstawie czasu uzyskania
pełnego chimeryzmu jako pó
ź
ny CC, mediana czasu wyst
ą
pienia pierwszych objawów

64
cGvHD wynosiła 250 dni, podczas gdy mediana czasu uzyskania CC – 270 dni po
przeszczepie. Oprócz trzech chorych u wszystkich badanych objawy cGvHD
poprzedziły uzyskanie CC a
ś
redni czas mi
ę
dzy uzyskaniem CC a rozpoznaniem
cGvHD wynosił 100 dni.
Mechanizm, który mo
ż
e prowadzi
ć
do tak ró
ż
nego przebiegu cGvHD
w zale
ż
no
ś
ci od czasu osi
ą
gni
ę
cia CC pozostaje nadal niejasny. Mo
ż
na przypuszcza
ć
,
ż
e mechanizm prowadz
ą
cy do uzyskania CC i pojawienia si
ę
cGvHD ró
ż
ni si
ę
w obu
badanych grupach. W obu grupach cz
ę
sto
ść
wyst
ę
powania klinicznie istotnej ostrej
choroby przeszczep przeciwko gospodarzowi była podobna. W grupie chorych
z wczesnym CC u wszystkich oprócz 2 wyst
ą
piła posta
ć
rozległa (21/25 chorych) a
u wi
ę
kszo
ś
ci chorych rozpoznano typ cGvHD de novo. Mo
ż
e to sugerowa
ć
,
ż
e
uzyskanie CC nie jest bezpo
ś
rednio efektem ostrej GVHD. Jednak
ż
e nie mo
ż
na
wykluczy
ć
istnienia przebiegaj
ą
cej subklinicznie reakcji przeszczep przeciwko
gospodarzowi prowadz
ą
cej do eliminacji komórek gospodarza (84). W
ś
wietle ostatnich
doniesie
ń
immunologii transplantacyjnej, jak równie
ż
obserwacji prowadzonych
u chorych po przeszczepach narz
ą
dowych wyst
ą
pienie mikrochimeryzmu mo
ż
e
w pewnym stopniu tłumaczy
ć
ró
ż
ny przebieg uzyskania CC i cGvHD obserwowane
u naszych chorych. Współistnienie komórek gospodarza i dawcy mo
ż
e sprzyja
ć
rozwojowi tolerancji immunologicznej w mechanizmie obwodowym (34;132). U biorców
komórek hematopoetycznych mo
ż
e si
ę
to klinicznie przejawia
ć
brakiem lub
łagodniejszym przebiegiem cGvHD. Wydaje si
ę
,
ż
e najbardziej krytycznym okresem
poprzeszczepowym, wpływaj
ą
cym na cały jego przebieg, jest okres pierwszych trzech
miesi
ę
cy po przeszczepie. Eliminacja wykrywalnych komórek gospodarza w okresie,
gdy pacjent otrzymuje jeszcze profilaktyk
ę
immunosupresyjn
ą
w pełnej dawce, mo
ż
e
by
ć
po
ś
rednim dowodem wyj
ą
tkowo intensywnej reakcji immunologicznej skierowanej
przeciwko alloantygenom. Konsekwencj
ą
tej reakcji mo
ż
e by
ć
równie
ż
eliminacja
ochronnej, bior
ą
cej udział w procesie wytwarzania tolerancji, populacji komórek, co
z kolei prowadzi do ci
ęż
szego przebiegu cGvHD. Potwierdzaj
ą
to kliniczne obserwacje
u naszych chorych z wczesnym CC. Niektórzy autorzy wskazuj
ą
,
ż
e populacj
ą
leukocytów odpowiedzialn
ą
za udział w procesach tolerancji jest populacja limfocytów
CD3 (70;138;150). Zaobserwowano równie
ż
wpływ utrzymywania si
ę
mieszanego
chimeryzmu w populacji komórek CD3 na wznow
ę
choroby zasadniczej. Zale
ż
no
ść
tak
ą
wykazano tak
ż
e w przypadku chorób rozrostowych, w których wykrywane komórki
CD3 nie nale
żą
do klonu nowotworowego, a wi
ę
c przyczyn
ą
wznowy nie jest obecno
ść
choroby resztkowej (33;81;138;149;150). Jak wiadomo reakcja GvL przynajmniej

65
cz
ęś
ciowo jest zale
ż
na od rozpoznawania przez limfocyty T CD8 alloantygenów, np.
„słabych” antygenów zgodno
ś
ci tkankowej, a nie antygenów zwi
ą
zanych bezpo
ś
rednio
z klonem nowotworowym. Z tego powodu nabywanie tolerancji na alloantygeny wi
ąż
e
si
ę
równocze
ś
nie z osłabieniem reakcji GvL, a tym samym zwi
ę
ksza ryzyko wznowy.
W naszych obserwacjach u pacjentów, u których obserwowano utrzymywanie si
ę
MC
przez dłu
ż
szy okres czasu (przynajmniej do doby 100) objawy cGvHD pojawiły si
ę
pó
ź
niej, a choroba miała l
ż
ejszy przebieg. U wszystkich tych chorych ocena
chimeryzmu
w
subpopulacjach krwi
obwodowej
wykazała,
ż
e
komórkami
odpowiedzialnymi za utrzymywanie si
ę
MC były limfocyty CD3. W tej grupie eliminacja
spełniaj
ą
cego ochronn
ą
rol
ę
klonu mo
ż
e by
ć
wyja
ś
niona jako wynik cGvHD, która
rozwin
ę
ła si
ę
w trakcie odstawiania lub ju
ż
po odstawieniu immunosupresji. Nabyta
w okresie mieszanego chimeryzmu CD3 tolerancja, prowadzi do istotnie rzadszego
wyst
ę
powania cGvHD, a w razie jej wyst
ą
pienia do znacznie łagodniejszego przebiegu
choroby. Z drugiej strony eliminacja limfocytów w okresie pierwszych trzech miesi
ę
cy
po transplantacji obserwowana w grupie chorych z wczesnym CC mo
ż
e skutkowa
ć
statystycznie cz
ę
stszym wyst
ą
pieniem rozległej postaci cGvHD wymagaj
ą
cej bardziej
intensywnego leczenia immunosupresyjnego.
Identyfikacja ewentualnego, kolejnego czynnika ryzyka rozwoju rozległej
cGvHD, jakim mo
ż
e by
ć
tempo uzyskania CC, nasuwa pytanie, czy mo
ż
na praktycznie
wykorzysta
ć
t
ą
informacj
ę
w prowadzeniu pacjentów po transplantacji. Próby
zapobiegania cGvHD poprzez wydłu
ż
enie okresu profilaktyki immunosupresyjnej jak
dotychczas nie wykazały istotnego wpływu na cz
ę
sto
ść
cGvHD. Randomizowana
próba podj
ę
ta przez grup
ę
badaczy ze Seatlle nie wykazała zmniejszenia cz
ę
sto
ś
ci
cGvHD u chorych, u których wydłu
ż
ono okres profilaktycznej immunosupresji
cyklosporyn
ą
A z 6 miesi
ę
cy do 24 miesi
ę
cy. Do wydłu
ż
onej profilaktyki kwalifikowano
chorych, którzy mieli zwi
ę
kszone ryzyko cGvHD z powodu wcze
ś
niejszego
wyst
ę
powania choroby ostrej lub subklinicznych zmian charakterystycznych dla cGvHD
w biopsji skóry (73). Niepowodzeniem zako
ń
czyła si
ę
równie
ż
randomizowana,
podwójnie
ś
lepa próba polegaj
ą
ca na doł
ą
czeniu do profilaktyki cGvHD w dobie 80
talidomidu. W grupie chorych, którzy otrzymywali talidomid cz
ę
sto
ść
cGvHD oraz
ś
miertelno
ść
była wy
ż
sza (37). Ringen i wsp. utrzymywali sterydoterapi
ę
u chorych
z aGvHD a
ż
do 6 miesi
ę
cy po przeszczepie. Zaobserwowano w tej grupie wy
ż
sz
ą
cz
ę
sto
ść
cGVHD (108). Równie
ż
prowadzenie sterydoterapii w dobie 100 po
transplantacji, nawet ze wskaza
ń
innych ni
ż
cGvHD zwi
ę
ksza ryzyko rozległej GvHD
(4;145). W powy
ż
szym badaniu mo
ż
emy zaobserwowa
ć
,
ż
e u chorych szczególnie

66
nara
ż
onych na ci
ęż
ki przebieg cGvHD objawy choroby pojawiły si
ę
w czasie, gdy
chorzy otrzymywali jeszcze pełna dawk
ę
immunosupresji, a u trzech chorych
z post
ę
puj
ą
c
ą
chorob
ą
ostr
ą
– kilkulekowe leczenie immunosupresyjne. W tej grupie
chorych wyst
ą
pienie objawów klinicznych cGvHD nie zostało sprowokowane redukcj
ą
lub odstawieniem immunosupresji.
By
ć
mo
ż
e wi
ę
cej korzy
ś
ci z kontynuacji profilaktyki immunosupresyjnej
odnie
ś
liby chorzy, u których objawy cGvHD pojawiaj
ą
si
ę
w trakcie redukcji dawki
cyklosporyny A lub po jej odstawieniu. Potwierdzeniem tego mo
ż
e by
ć
zmniejszenie
cz
ę
sto
ś
ci wyst
ę
powania cGvHD u chorych, u których wydłu
ż
ono okresu redukcji dawki
cyklosporyny A do jednego roku (90). Nale
ż
y jednak pami
ę
ta
ć
o zachowaniu
równowagi pomi
ę
dzy kontrol
ą
cGvHD a hamowaniem GvL. Potencjalne korzy
ś
ci
wynikaj
ą
ce ze zmniejszenia cz
ę
sto
ś
ci GvHD, cz
ę
sto wi
ążą
si
ę
ze zwi
ę
kszeniem ryzyka
wznowy choroby zasadniczej oraz odrzucenia przeszczepu. Dlatego chorzy ci
wymagaj
ą
dłu
ż
szego okresu obserwacji w celu pełnej oceny ryzyka nawrotu, oraz
wtórnych nowotworów.
Kolejnym aspektem jest wykorzystanie informacji o potencjalnym czynniku
ryzyka w bardziej intensywnym leczeniu. Chorzy ci powinni by
ć
obj
ę
ci próbami
klinicznymi, a tak
ż
e próbami wykorzystania nowych leków immunosupresyjnych.
Ostatecznie, chocia
ż
jeszcze w sferze przyszło
ś
ci pozostaje celowana
manipulacja tempem uzyskania CC tak, aby zachowuj
ą
c korzy
ś
ci wynikaj
ą
ce
z procesów alloreaktywno
ś
ci, zmniejszy
ć
niekorzystne dla chorego powikłania
immunologiczne.
Kolejnym porównywanym w pracy parametrem była cz
ę
sto
ść
wyst
ę
powania
wznowy choroby zasadniczej w podgrupach chorych z wczesnym i pó
ź
nym
chimeryzmem. W niniejszej pracy nie oceniano warto
ś
ci oceny chimeryzmu jako
mo
ż
liwo
ś
ci przewidywania wznowy z odpowiednio wczesnym rozpocz
ę
ciem terapii.
W przypadku chorób rozrostowych układu krwiotwórczego bardziej warto
ś
ciowym
materiałem w mo
ż
liwo
ś
ci oceny obecno
ś
ci choroby resztkowej jest krew szpikowa (7).
Ponadto ogromne znaczenie, szczególnie w mo
ż
liwo
ś
ci oceny zagro
ż
enia wznow
ą
u chorych z ostr
ą
białaczk
ą
, ma wykonywanie bada
ń
w znacznie krótszych odst
ę
pach
czasowych – w okre
ś
lonych przypadkach nawet tygodniowych. Wi
ąż
e si
ę
to
z wykonywaniem procedury inwazyjnej, jak
ą
stanowi biopsja aspiracyjna szpiku.
Wykonywanie bada
ń
z krwi obwodowej w tym przypadku ma mniejsz
ą
warto
ść
diagnostyczn
ą
. W badanej grupie 51 chorych, u wszystkich osób stwierdzono pełny
chimeryzm, co oznacza,
ż
e dan
ą
metod
ą
nie stwierdzono obecno
ś
ci choroby

67
resztkowej we krwi obwodowej. Celem powy
ż
szej pracy była ocena wyst
ę
powania
wznowy jako jednego z klinicznych wyrazów aktywno
ś
ci reakcji GvL.
Rozpatruj
ą
c rodzaj chimeryzmu nie stwierdzono statystycznie istotnej ró
ż
nicy
w cz
ę
sto
ś
ci wyst
ę
powania wznowy w grupie z wczesnym i pó
ź
nym chimeryzmem.
Jednak
ż
e rozpatruj
ą
c wyst
ę
powanie wznowy w poszczególnych rozpoznaniach
zwrócono uwag
ę
,
ż
e w grupie chorych z CML nawrót choroby obserwowano jedynie
w grupie chorych z pó
ź
nym chimeryzmem. Powy
ż
sza obserwacja mo
ż
e
ś
wiadczy
ć
,
ż
e
nasilona
alloreaktywno
ść
klinicznie
wyra
ż
ona
obecno
ś
ci
ą
rozległej
cGvHD
zaowocowała równie
ż
efektywniejsz
ą
GvL. Zale
ż
no
ś
ci te obserwowano jedynie
u chorych z CML chorobie, w której skuteczno
ść
reakcji GvL jest szczególnie wysoka.
Nie obserwowano podobnych zale
ż
no
ś
ci w grupie chorych z ALL i AML, co mo
ż
e by
ć
zwi
ą
zane nie tylko z mniejszym nasileniem reakcji GvL w tych rozpoznaniach, ale
równie
ż
stosunkowo niewielk
ą
liczebno
ś
ci
ą
tych podgrup.
5.2 Choroba przeszczep przeciwko gospodarzowi
Przewlekła choroba przeszczep przeciwko gospodarzowi była obserwowana
u 71% chorych, u wi
ę
kszo
ś
ci (57%) rozpoznano posta
ć
rozległ
ą
, u 14% chorych posta
ć
ograniczon
ą
. W badanej grupie zbadano wpływ powszechnie uznawanych czynników
ryzyka (Tabela I) na wyst
ą
pienie przewlekłej choroby przeszczep przeciwko
gospodarzowi. Nie wykazano zale
ż
no
ś
ci pomi
ę
dzy wiekiem dawcy i biorcy,
a wyst
ę
powaniem cGvHD. Wi
ę
kszo
ść
autorów obserwuje wzrost cz
ę
sto
ś
ci
wyst
ę
powania cGvHD wraz z wiekiem biorcy, natomiast cz
ę
sto
ść
wyst
ę
powania
u dzieci jest niska, cho
ć
inne czynniki ryzyka, prezentacja kliniczna i wpływ
na prze
ż
ycie pozostaj
ą
podobne jak u chorych dorosłych (3;74;154). Brak tych
zale
ż
no
ś
ci w analizowanym materiale mo
ż
e wynika
ć
ze stosunkowo niskiej liczebno
ś
ci
badanej grupy.
W badanej grupie chorych cGvHD rozpoznano u wszystkich chorych, u których
jako post
ę
powanie przygotowawcze przed przeszczepem zastosowano na
ś
wietlenie
całego ciała (TBI) z cyklofosfamidem (4 ALL, 1 AML). Wpływ rodzaju post
ę
powania
przygotowawczego na cz
ę
sto
ść
wyst
ę
powania cGvHD pozostaje nadal kontrowersyjny.
Tylko w pojedynczych badaniach obserwowano zwi
ę
kszenie ryzyka cGvHD u chorych,
u których jako kondycjonowanie stosowano TBI. Wzrost ryzyka cGvHD mógłby
wynika
ć
z wi
ę
kszego ni
ż
w przypadku chemioterapii uszkodzenia grasicy przez
promieniowanie jonizuj
ą
ce. Uszkodzenie grasicy powoduje upo
ś
ledzenie centralnych

68
mechanizmów wytwarzania tolerancji, co w konsekwencji prowadzi do rozwoju cGvHD
(29;39;42;125).
Nie zaobserwowano statystycznie istotnej zale
ż
no
ś
ci pomi
ę
dzy, rozpoznaniem
choroby zasadniczej a wyst
ę
powaniem cGvHD. Wyst
ą
pienie cGvHD u wszystkich
chorych z ALL zwi
ą
zane było z zastosowanym rodzajem kondycjonowania:
u wszystkich chorych z ALL post
ę
powaniem przygotowawczym było TBI. Opisywane
w literaturze cz
ę
stsze wyst
ę
powanie cGvHD u chorych z CML równie
ż
nie zostało
potwierdzone w badanej grupie.
Nie stwierdzono statystycznej ró
ż
nicy w proporcji kobiet : m
ęż
czyzn w grupach
chorych z i bez cGvHD. Wykazano natomiast,
ż
e u wszystkich m
ęż
czyzn, u których
wykonano przeszczep komórek progenitorowych pobranych od siostry doszło do
rozwoju cGvHD. Zale
ż
no
ść
ta była statystycznie znamienna (p=0,02). Wielu autorów
potwierdza,
ż
e w przypadku przeszczepu, w którym płe
ć
dawcy ró
ż
ni si
ę
od płci biorcy
czynnikiem ryzyka dla cGvHD jest jedynie układ dawca kobieta dla biorcy m
ęż
czyzny
(66;77;103;151) .
Kolejn
ą
analizowan
ą
zale
ż
no
ś
ci
ą
była ocena wpływu rodzaju materiału
przeszczepowego na cz
ę
sto
ść
wyst
ę
powania cGvHD. W analizowanej grupie chorych
u wi
ę
kszo
ś
ci chorych wykonano przeszczep komórek progenitorowych pobranych
z krwi obwodowej (PBSCT - 43 chorych, co stanowi 84%) natomiast przeszczep
komórek progenitorowych pobranych ze szpiku (BMT) wykonano jedynie u 8 chorych.
Ró
ż
nice wynikały z preferencji stosowania PBSCT w naszym o
ś
rodku. Cz
ę
sto
ść
wyst
ę
powania cGvHD była wy
ż
sza u chorych poddanych PBSCT w stosunku do BMT
i wynosiła odpowiednio 72% i 62%. Powy
ż
sze wyniki s
ą
potwierdzeniem doniesie
ń
,
ż
e
przeszczep komórek progenitorowych krwi obwodowej istotnie zwi
ę
ksza ryzyko
przewlekłej choroby przeszczep przeciwko gospodarzowi. Jedn
ą
z prób wyja
ś
nienia
cz
ę
stszego wyst
ę
powania cGvHD u chorych po PBSCT jest stwierdzenie zwi
ę
kszenia
ilo
ś
ci limfocytów TH2 w materiale przeszczepowym uzyskanym z krwi obwodowej
zdrowych dawców stymulowanych granulocytarnym czynnikiem wzrostu. Stosunkowo
wysoki w badanej grupie odsetek chorych po PBSCT mógł mie
ć
wpływ na wi
ę
ksz
ą
ni
ż
opisywana w pi
ś
miennictwie cz
ę
sto
ść
wyst
ę
powania cGvHD - zwykle u chorych
poddanych transplantacji od zgodnego w układzie HLA dawcy rodzinnego nie
przekracza 50-60% BMT (36). Według wi
ę
kszo
ś
ci autorów nie zaobserwowano
zwi
ę
kszenia cz
ę
sto
ś
ci wyst
ę
powania aGvHD u chorych po PBSCT. Zwi
ę
kszenie
cz
ę
sto
ś
ci cGvHD bez wpływu na chorob
ę
ostr
ą
spowodowało wi
ę
kszy odsetek
w badanej grupie cGvHD o typie „de novo” (50% w porównaniu do danych

69
literaturowych – 35%). Ze wzgl
ę
dów na liczebno
ść
grup zale
ż
no
ś
ci te nie były istotne
statystycznie.
Oceniono równie
ż
zale
ż
no
ść
pomi
ę
dzy wyst
ę
powaniem aGvHD i cGvHD.
U 52% chorych, u których rozpoznano rozległ
ą
cGvHD we wczesnym okresie
poprzeszczepowym obserwowano aGvHD. W grupie chorych, u których nie
rozpoznano cGvHD, aGvHD obserwowano jedynie u 20% chorych. Cz
ę
sto
ść
wyst
ę
powania aGvHD w grupie chorych z rozległ
ą
postaci
ą
cGvHD w porównaniu do
chorych bez cGvHD była statystycznie znamienna (p=0,04). Nie wykazano podobnej
zale
ż
no
ś
ci u chorych, u których rozpoznano posta
ć
ograniczon
ą
cGvHD. Porównano
równie
ż
zale
ż
no
ść
pomi
ę
dzy stopniem zaawansowania aGvHD a wyst
ę
powaniem
cGvHD. Nie wykazano równie
ż
zale
ż
no
ś
ci pomi
ę
dzy poszczególnymi stopniami
zaawansowania, a wyst
ę
powaniem cGvHD, równie
ż
w wydzielonych podgrupach
z chorob
ą
rozległa b
ą
d
ź
ograniczon
ą
. 80% chorych, u których wyst
ą
piła aGvHD
przynajmniej III stopnia rozwin
ę
ło rozległ
ą
cGvHD (wyniki nie były istotnie
statystycznie). Wyst
ą
pienie aGvHD jest jednym z najwa
ż
niejszych czynników ryzyka
cGvHD. 70-80% chorych, u których rozpoznano chorob
ę
ostr
ą
rozwija cGvHD.
W analizach na du
ż
ych grupach wielu autorów wykazuje równie
ż
zale
ż
no
ść
pomi
ę
dzy
stopniem zaawansowania aGvHD a cz
ę
sto
ś
ci
ą
wyst
ę
powania cGvHD (77). Sugeruje
si
ę
cztery mechanizmy, by
ć
mo
ż
e współistniej
ą
ce ze sob
ą
, które mogłyby tłumaczy
ć
zale
ż
no
ść
wyst
ę
powania choroby ostrej i przewlekłej: 1) Objawy cGvHD mog
ą
by
ć
pó
ź
niejsz
ą
manifestacj
ą
alloreaktywacji manifestuj
ą
cej si
ę
aGvHD. 2) cGvHD mo
ż
e
wynika
ć
z uszkodzenia tkanek, w szczególno
ś
ci grasicy przez chorob
ę
ostr
ą
(146).
3) cGvHD jest wynikiem leczenia aGvHD, w szczególno
ś
ci stosowania steroidów.
Mog
ą
o tym
ś
wiadczy
ć
doniesienia,
ż
e u chorych, u których stosowano prednison jako
profilaktyk
ę
GvHD cz
ę
sto
ść
wyst
ę
powania cGvHD była istotnie wy
ż
sza, a tak
ż
e fakt
cz
ę
stszego wyst
ę
powania cGvHD u chorych, którzy otrzymywali steroidy
we wczesnym okresie po transplantacji z przyczyn innych ni
ż
GvHD (48;145).
4) Wreszcie aGvHD mo
ż
e by
ć
epifenomenem, którego wyst
ę
powanie jest zwi
ą
zane
z cGvHD, ale nie ma z ni
ą
zwi
ą
zku etiologicznego (77).
Nie wykazano statystycznie istotnych ró
ż
nic w prze
ż
yciu chorych w zale
ż
no
ś
ci
od wyst
ą
pienia cGvHD. W grupie 15 chorych bez cGvHD, 2 chorych zmarło z powodu
infekcji. Natomiast w grupie 36 chorych z cGvHD obserwowano 9 zgonów: 4 z powodu
wznowy choroby zasadniczej, 2 z powodu infekcji oraz 3 z powodu cGvHD.
Prawdopodobie
ń
stwo prze
ż
ycia w badanych grupach chorych wynosiło 0,85 dla
chorych bez rozpoznanej cGvHD i 0,73 dla chorych z cGvHD. W badaniach

70
przeprowadzonych na du
ż
ej ilo
ś
ci chorych wykazano,
ż
e prze
ż
ycie chorych z cGvHD
jest gorsze w porównaniu do chorych bez cGvHD. W szczególno
ś
ci dotyczy to
chorych, u których rozpoznano chorob
ę
rozległ
ą
. Obni
ż
enia krzywej prze
ż
ycia wynika
ze zwi
ę
kszenia
ś
miertelno
ś
ci z powodu powikła
ń
nie zwi
ą
zanych ze wznow
ą
choroby
zasadniczej (infekcja, cGvHD) mimo korzy
ś
ci wynikaj
ą
cej z rzadszego wyst
ę
powania
wznowy choroby zasadniczej (77;78;124;126). W badanym materiale nie analizowano
prze
ż
ycia w zale
ż
no
ś
ci od rozległo
ś
ci cGvHD, ze wzgl
ę
du na stosunkowo niskie
liczebno
ś
ci poszczególnych podgrup.
5.3 Wznowa choroby zasadniczej
Wznow
ę
choroby zasadniczej obserwowano u 8 chorych (3 chorych z CML,
4 chorych z AML oraz 1 chorego z ALL ).
W przedstawionej grupie chorych z rozpoznaniem przewlekłej białaczki szpikowej
cz
ę
sto
ść
wznowy choroby zasadniczej była ni
ż
sza ni
ż
w pi
ś
miennictwie i wynosiła
około 12% (3 chorych z 25). Rozpatruj
ą
c nawrót CML w zale
ż
no
ś
ci od
ź
ródła materiału
przeszczepowego mo
ż
emy zauwa
ż
y
ć
,
ż
e cz
ę
sto
ść
wznowy u chorych poddanych
przeszczepowi komórek z krwi szpikowej wynosiła ok. 20% (jeden chory
z 5 po BMT), natomiast u chorych którzy otrzymali przeszczep komórek z krwi
obwodowej ok. 10% (2 z 20 po PBSCT). Wyst
ę
powanie ni
ż
szej cz
ę
sto
ś
ci wznowy
u chorych poddanych PBSCT była obserwowana przez innych badaczy i przynajmniej
cz
ęś
ciowo mo
ż
e by
ć
efektem zwi
ę
kszenia cz
ę
sto
ś
ci wyst
ę
powania cGvHD (47).
Sukces terapeutyczny allogenicznej transplantacji komórek hematopoezy wynika nie
tylko z mo
ż
liwo
ś
ci eskalacji dawki chemio/radioterapii do poziomu toksyczno
ś
ci
„nie-szpikowych” organów. Wyniki transplantacji syngenicznych wykazały,
ż
e
skuteczno
ść
całkowitej eradykacji komórek nowotworowych, mimo stosowania
post
ę
powania mieloablacyjnego u chorych z AML w pierwszej remisji i CML w fazie
przewlekłej nie przekracza 40%. Znacz
ą
c
ą
popraw
ę
wyników transplantacji
zaobserwowano u chorych poddanych transplantacji od niemonozygotycznego
rodze
ń
stwa, gdzie cz
ę
sto
ść
wznowy wynosiła ju
ż
tylko 10-20% (50;56;128;144).
Dalsze badania potwierdziły znaczenie roli układu immunologicznego dawcy
w eliminacji komórek nowotworowych. Wykazano,
ż
e proces eliminacji komórek
nowotworowych prowadz
ą
cy ostatecznie do pełnej remisji molekularnej zachodzi
stopniowo przez wiele miesi
ę
cy, co oznacza,
ż
e nie jest on wynikiem bezpo
ś
redniego
działania mieloablacyjnego kondycjonowania (88). Przewlekła białaczka szpikowa jest

71
chorob
ą
, w której jako pierwszej udowodniono wyst
ę
powanie efektu przeszczep
przeciwko białaczce (GvL). Znaczenie tego zjawiska potwierdziły wyniki przeszczepów
o zredukowanym kondycjonowaniu. Wykazano równie
ż
mo
ż
liwo
ść
uzyskania pełnej
remisji stosuj
ą
c infuzj
ę
limfocytów dawcy w leczeniu wznowy choroby zasadniczej
nawet u chorych, u których wznowa wyst
ą
piła po przeszczepie mieloablacyjnym.
Ze szczególnie wysok
ą
efektywno
ś
ci
ą
GvL u chorych z CML wi
ąż
e si
ę
zmniejszenie
cz
ę
sto
ś
ci wyst
ę
powania wznowy u tych chorych, którzy rozwin
ę
li cGvHD, poniewa
ż
reakcja przeszczep przeciwko białaczce jest procesem cz
ęś
ciowo zale
ż
nym od reakcji
przeszczep przeciwko gospodarzowi (92;128). W badanej grupie spo
ś
ród 3 chorych
z CML, u których doszło do wznowy choroby zasadniczej, cGvHD w postaci
ograniczonej rozpoznano tylko u jednego chorego, natomiast nie obserwowano
wznowy u tych chorych z CML, którzy rozwin
ę
li chorob
ę
rozległ
ą
.
Znacznie cz
ęś
ciej, bo u około 25% wznow
ę
choroby zasadniczej obserwowano
w grupie chorych z ostr
ą
białaczka szpikow
ą
(4/17) oraz u chorych z ostr
ą
białaczk
ą
limfoblastyczn
ą
(1/4). W tej grupie do wznowy doszło mimo obecno
ś
ci objawów
rozległej postaci cGvHD (2 chorych) i postaci ograniczonej u 1 chorego, chocia
ż
w momencie rozpoznania wznowy chorzy nie mieli ju
ż
objawów aktywnej cGvHD.
Potwierdza to doniesienia o znacznie mniejszym nasileniu GvL w tych rozpoznaniach
ni
ż
w CML. Obecno
ść
efektu GvT wykazano ju
ż
w wielu chorobach rozrostowych
układu krwiotwórczego, a tak
ż
e niektórych guzach litych (rak nerki, rak piersi). Stopie
ń
jego nasilenia, a co si
ę
z tym wi
ąż
e skuteczno
ść
przeszczepu zale
ż
y od rodzaju
choroby zasadniczej. Najwi
ę
ksz
ą
skuteczno
ść
wykazano w CML i CLL, po
ś
redni
ą
w AML i szpiczaku mnogim a najni
ż
sz
ą
w ALL i chłoniakach o wysokim stopniu
zło
ś
liwo
ś
ci (42;54;97;128;141). Do zwi
ą
zanych z chorob
ą
zasadnicz
ą
czynników
maj
ą
cych wpływ na efektywno
ść
reakcji GvL nale
ż
y ekspresja immunogennych
antygenów białaczkowych, a tak
ż
e agresywno
ść
przebiegu choroby rozrostowej.
Reakcja
przeszczep
przeciwko
guzowi
(GvL,
GvT)
wymaga
czasu,
a szybkie narastanie ilo
ś
ci komórek nowotworowych obserwowane w przypadku
wznowy ostrej białaczki znacz
ą
co ogranicza skuteczno
ść
(6;40;141).
Wyst
ą
pienie cGvHD zmniejsza, ale nie wyklucza mo
ż
liwo
ś
ci wznowy choroby
zasadniczej. Wg niektórych autorów wznowa jest przyczyn
ą
10-20% zgonów u chorych
z rozpoznan
ą
cGvHD i zwykle dotyczy chorych z choroba o łagodniejszym przebiegu.
(2;51;144;148).
Oceniaj
ą
c
wyst
ę
powanie
wznowy
choroby
zasadniczej
u chorych z cGvHD wielu autorów zwraca uwag
ę
na niedoskonało
ść
klasyfikacji,
w której chorzy z rozpoznaniem cGvHD stanowi
ą
skrajnie heterogenna grup
ę
.

72
Dodatkowym problemem s
ą
bł
ę
dy wynikaj
ą
ce z nadrozpoznawania cGvHD, co zdarza
si
ę
cz
ęś
ciej u chorych z chorob
ą
łagodniejsz
ą
.Zarówno fałszywe rozpoznanie jak
i brak dostosowywania standardów leczenia do stopnia ci
ęż
ko
ś
ci stwarza mo
ż
liwo
ść
„przeleczenia” (80). Efektem tego jest zmniejszenie skuteczno
ś
ci GvL niepotrzebn
ą
terapi
ą
immnosupresyjn
ą
, co w konsekwencji mo
ż
e prowadzi
ć
do wznowy choroby.
Równie
ż
obserwacje prowadzone w naszym o
ś
rodku wykazały,
ż
e u cz
ęś
ci chorych
ponowne pojawienie si
ę
własnego DNA mo
ż
e pojawia
ć
si
ę
w trakcie terapii
immunosupresyjnej (16). W obserwowanej grupie chorych wznow
ę
choroby
zasadniczej obserwowano w okresie wyciszenia objawów cGvHD podczas gdy chory
otrzymywał jeszcze podtrzymuj
ą
c
ą
terapi
ę
immunosupresyjn
ą
. Powy
ż
sze wyniki
potwierdzaj
ą
konieczno
ść
dalszego dopracowania klasyfikacji cGvHD, szczególnie
w ocenie postaci rozległej, co stworzyłoby mo
ż
liwo
ść
dostosowania schematów
leczenia cGvHD do ci
ęż
ko
ś
ci przebiegu i mogłoby zmniejszy
ć
ś
miertelno
ść
wynikaj
ą
c
ą
z nawrotu choroby zasadniczej w tej grupie. Niew
ą
tpliwie opracowanie metod terapii
prowadz
ą
cej do kontroli objawów cGvHD bez zaburzania GvL stanowi jedno
z najwa
ż
niejszych wyzwa
ń
współczesnej transplantologii.
Wreszcie oceniaj
ą
c wyst
ę
powanie wznowy u chorych z cGvHD, nale
ż
y zwróci
ć
uwag
ę
nie tylko na heterogenno
ść
przebiegu cGvHD, ale równie
ż
znaczne ró
ż
nice
w zaawansowaniu choroby podstawowej b
ę
d
ą
cej wskazaniem do wykonania
transplantacji szpiku. U chorych z chorob
ą
zaawansowan
ą
– w kolejnej remisji, kryzie
blastycznej CML, oraz u chorych przeszczepianych bez remisji nie obserwuje si
ę
tak
korzystnego efektu cGvHD jak u chorych z mniej zaawansowan
ą
chorob
ą
(faz
ą
przewlekła CML lub w pierwszej remisji). Ogromne znaczenie ma równie
ż
rodzaj
choroby rozrostowej – poniewa
ż
udowodniono,
ż
e nasilenie efektu GvT wynika w du
ż
ej
mierze z samego rozpoznania.
Zwraca równie
ż
uwag
ę
du
ż
a cz
ę
sto
ść
wyst
ę
powania, w badanej grupie wznowy
pozaszpikowej, co mo
ż
na tłumaczy
ć
gorsz
ą
dost
ę
pno
ś
ci
ą
pewnych obszarów
organizmu dla komórek układu immunologicznego.
Do chwili obecnej z badanej grupy 51 pacjentów zmarło 11 chorych (22%).
U 5 (46%)
chorych
przyczyn
ą
ś
mierci
była
wznowa
choroby
zasadniczej,
u 3 (27%)chorych powikłania infekcyjne, u kolejnych 3 (27%) cGvHD. 9 chorych zmarło
w przedziale czasowym pomi
ę
dzy 100 a 600 dob
ą
po transplantacji, a tylko 2 kolejne
osoby do doby 1200. Nast
ę
pnie krzywa osi
ą
gn
ę
ła plateau z prawdopodobie
ń
stwem
prze
ż
ycia ok. 0,73. Nawrót choroby zasadniczej stanowi obecnie jedn
ą
z głównych
przyczyn zgonów pó
ź
nych, u chorych, którzy prze
ż
yli 24 miesi
ą
ce po przeszczepie jest

73
najcz
ę
stsz
ą
przyczyn
ą
ś
mierci (47 – 56%). Natomiast zgony z powodu cGvHD
obserwuje si
ę
głownie do 24 miesi
ę
cy po przeszczepie (65;126).
Podsumowanie
Wykorzystanie chimeryzmu hematopoetycznego do oceny zagro
ż
enia
wyst
ą
pieniem cGvHD mo
ż
e by
ć
kolejn
ą
cenn
ą
informacj
ą
ułatwiaj
ą
c
ą
prowadzenie
chorych po HSCT. W powy
ż
szej pracy wykazano zale
ż
no
ś
ci mi
ę
dzy czasem uzyskania
CC a cGvHD na stosunkowo niewielkiej liczbie (51) chorych. Ze wzgl
ę
du na
nowoczesno
ść
u
ż
ytej metody oraz stosunkowo długi okres obserwacji po przeszczepie
uzyskane wyniki mog
ą
by
ć
wskazówk
ą
do dalszych bada
ń
. Ostatecznie rola pełnego
chimeryzmu w zagro
ż
eniu rozwojem cGvHD wymaga dalszej obserwacji oraz
potwierdzenia na du
ż
ej grupie chorych.

74
6
WNIOSKI:
1a Oceniaj
ą
c chimeryzm metod
ą
STR multiplex PCR z zastosowaniem
elektroforezy kapilarnej na automatycznym sekwenatorze DNA wykazano,
ż
e
u wi
ę
kszo
ś
ci chorych eliminacj
ę
hematopoezy biorcy obserwuje si
ę
pó
ź
niej ni
ż
w 30 dobie.
1b Zastosowanie w kondycjonowaniu na
ś
wietlenia całego ciała (TBI) w dawce
12Gy powoduje szybsze osi
ą
gni
ę
cie pełnego chimeryzmu w porównaniu do
chorych poddanych jedynie chemioterapii.
1c Oznaczanie chimeryzmu w poszczególnych populacjach leukocytów krwi
obwodowej zwi
ę
ksza czuło
ść
metody umo
ż
liwiaj
ą
c wykrywanie przetrwałych
komórek biorcy nawet u chorych, u których stwierdzono CC w krwi obwodowej.
Przetrwał
ą
populacj
ą
biorcy manifestuj
ą
c
ą
si
ę
jako mieszany chimeryzm krwi
obwodowej u wi
ę
kszo
ś
ci chorych jest populacja limfocytów CD3.
2a W grupie chorych, u których obserwowano wczesny pełny chimeryzm
statystycznie znamiennie cz
ęś
ciej wyst
ę
puje rozległa posta
ć
cGvHD, jej
przebieg jest ci
ęż
szy: wi
ę
kszy odsetek chorych wymaga przynajmniej
trzylekowej terapii immunosupresyjnej, cz
ęś
ciej obserwuje si
ę
zgony z powodu
cGvHD.
2b Nie stwierdzono statystycznie istotnej ró
ż
nicy w cz
ę
sto
ś
ci wyst
ę
powania
wznowy w obu grupach. W grupie chorych z wczesnym chimeryzmem nie
obserwowano wznowy u chorych z CML, a u wi
ę
kszo
ś
ci chorych rozpoznano
wznow
ę
pozaszpikow
ą
(75%).
2c Prawdopodobie
ń
stwo prze
ż
ycia jest ni
ż
sze u chorych, którzy osi
ą
gn
ę
li wczesny
CC (0,72) w porównaniu do chorych z pó
ź
nym chimeryzmem (0,84) i wynika
z cz
ę
stszego wyst
ę
powania zgonów niezwi
ą
zanych ze wznow
ą
choroby
zasadniczej.
.

75
7
STRESZCZENIE
Allogeniczna transplantacja komórek układu krwiotwórczego jest dla wielu
schorze
ń
hematologicznych jedyn
ą
metod
ą
prowadz
ą
c
ą
do wyleczenia. Do
najwa
ż
niejszych przyczyn niepowodze
ń
zalicza si
ę
chorob
ę
przeszczep przeciwko
gospodarzowi i wznow
ę
choroby zasadniczej. W
ś
ród wielu wykonywanych rutynowo
bada
ń
diagnostycznych u chorych poddanych transplantacji szpiku na szczególn
ą
uwag
ę
zasługuje ocena chimeryzmu hematopoetycznego. Wykorzystywana jest ona do
potwierdzenia przyj
ę
cia lub odrzucenia przeszczepu, a tak
ż
e do oceny stopnia
zagro
ż
enia wznow
ą
choroby zasadniczej, co umo
ż
liwia podj
ę
cie wyprzedzaj
ą
cych
działa
ń
terapeutycznych. Po
ś
ród wielu metod wykorzystywanych do oceny chimeryzmu
hematopoetycznego szczególne uznanie zyskała metoda oparta na analizie alleli STR
amplifikowanych w reakcji multiplex PCR, a nast
ę
pnie rozdzielanych za pomoc
ą
elektroforezy kapilarnej na automatycznym sekwenatorze DNA.
Prowadz
ą
c ocen
ę
chimeryzmu krwi obwodowej t
ą
metod
ą
wykazano,
ż
e po
ś
ród
ocenionych 51 chorych, którzy osi
ą
gn
ę
li pełny chimeryzm, 25 pacjentów wyeliminowało
komórki własnej hematopoezy do doby 100 wł
ą
cznie (wczesny pełny chimeryzm),
u kolejnych 26 CC został uzyskany w dalszej obserwacji (pó
ź
ny chimeryzm). Mediana
czasu uzyskania CC w pierwszej grupie wynosiła 60 dni, w drugiej 270 dni.
W podgrupach chorych wydzielonych na podstawie czasu uzyskania CC oceniono
cz
ę
sto
ść
wyst
ę
powania cGvHD oraz wznowy choroby zasadniczej.
W grupie chorych z wczesnym chimeryzmem cGvHD w postaci rozległej
obserwowano u 21/25 chorych, w porównaniu do grupy chorych z pó
ź
nym CC, gdzie
rozległ
ą
cGvHD rozpoznano u 8 pacjentów. Ró
ż
nice w cz
ę
sto
ś
ci wyst
ę
powania
rozległej cGvHD w poszczególnych grupach były statystycznie istotnie (p<0,001).
Wykazano równie
ż
ci
ęż
szy przebieg cGvHD w grupie chorych z wczesnym CC: 52%
chorych do kontroli objawów choroby wymagała przynajmniej trzylekowej terapii
(p<0,005), cz
ęś
ciej równie
ż
obserwowano zgony z powodu cGvHD. Zaobserwowano
równie
ż
statystycznie istotn
ą
korelacj
ę
czasow
ą
pomi
ę
dzy czasem osi
ą
gni
ę
cia CC
a wyst
ą
pieniem cGvHD. W grupie chorych z wczesnym CC u wszystkich chorych
osi
ą
gni
ę
cie CC poprzedzało wyst
ą
pienie pierwszych objawów cGvHD,
ś
rednio o 85
dni. Natomiast u 10/13 chorych w grupie z pó
ź
nym chimeryzmem objawy cGvHD
poprzedziły osi
ą
gni
ę
cie CC,
ś
rednio o 100 dni.

76
Oceniono równie
ż
cz
ę
sto
ść
wyst
ę
powania wznowy w obu grupach, nie
stwierdzaj
ą
c statystycznie istotnej ró
ż
nic.
U 15 chorych wykonano bardziej szczegółowe analizy, oceniaj
ą
c chimeryzm
w wysortowanych populacjach krwi obwodowej. Wykazano,
ż
e oznaczanie chimeryzmu
w poszczególnych populacjach zwi
ę
ksza czuło
ść
metody, umo
ż
liwiaj
ą
c wykrywanie
przetrwałych komórek biorcy nawet u chorych, u których stwierdzono CC w krwi
obwodowej. Przetrwał
ą
populacj
ą
biorcy manifestuj
ą
c
ą
si
ę
jako mieszany chimeryzm
krwi obwodowej u wi
ę
kszo
ś
ci chorych jest populacja limfocytów CD3.

77
8
SUMMARY
Transplantation of allogeneic hematopoietic stem cells is the only curative method
of the treatment in several haematological diseases. The main reasons of failure in this
therapeutical approach is graft versus host disease and relapse of underlying disease.
Among many routine tests done in patients after transplantation haematopoietic
chimerism evaluation is particularly interesting. It is performed in order to confirm donor
engraftment or graft rejection as well as to evaluate risk of disease recurrence. In some
cases it allows employment of pre-emptive therapeutical measures to avoid relapse. Of
many methods used for chimerism evaluation one has gained particular importance. It
is analysis of STR allele amplification in multiplex PCR reaction after capillary
electrophoresis and automatic DNA sequencing.
Peripheral blood chimerism was evaluated with that method in patients after
allogeneic stem cell transplantations. Of 51 patients who achieved complete donor
chimerism (CC), 25 eliminated own cells up to 100 days after transplantation (early
CC). The remaining 26 patients achieved complete donor chimerism at a later time
during the course of follow-up (late CC). Median time to reach CC in the first group was
60 days, while in the latter it was 270 days. Chronic graft versus host disease (cGvHD)
and relapse was evaluated in the two groups of patients.
In early CC group extensive cGvHD was seen in 21 of 25 patients, while in late CC
group it was noted only in 8 of 26 patients (p<0,001). Clinical course of cGvHD was
more severe in early CC than in late CC group: 52% of patients in the first one required
three-drug immunosuppression (p<0,005) and deaths resulting from that complication
were more frequent. A statistically important time correlation between time to reach CC
and emergence of cGvHD symptoms was also seen. In early CC group achievement of
CC invariably preceded appearance of cGvHD symtoms by a mean time of 85 days. In
late CC group symptoms of cGvHD preceded achievement ot CC by a mean time of
100 days in 10 of 13 patients.
Incidence of relapse in the two groups was not statistically different.
15 patients had chimerism evaluation performed in a more detailed way in isolated
peripheral blood cell populations. It was proved that it enhances the method’s
sensitivity an allows to detect residual recipient cells in patients who show CC in total

78
white blood cell assesment. Most ferquently residual recipient cells were found within
CD3 lymphocytes.

79
9
PI
Ś
MIENNICTWO
(1) Agrawal S, Khan F, Talwar S, Nityanand S. Short tandem repeat technology has
diverse applications: individual identification, phylogenetic reconstruction and
chimerism based post haematopoietic stem cell transplantation graft monitoring.
Indian J Med Sci 2004; 58:297-304.
(2) Akpek G, Zahurak ML, Piantadosi S, Margolis J, Doherty J, Davidson R et al.
Development of a prognostic model for grading chronic graft-versus-host disease.
Blood 2001; 97:1219-1226.
(3) Alyea EP, Kim HT, Ho V, Cutler C, Gribben J, DeAngelo DJ et al. Comparative
outcome of nonmyeloablative and myeloablative allogeneic hematopoietic cell
transplantation for patients older than 50 years of age. Blood 2005; 105:1810-1814.
(4) Ancin I, Ferra C, Gallardo D, Peris J, Berlanga J, Gonzalez JR et al. Do
corticosteroids add any benefit to standard GVHD prophylaxis in allogeneic BMT?
Bone Marrow Transplant 2001; 28:39-45.
(5) Antin JH. Clinical practice. Long-term care after hematopoietic-cell transplantation in
adults. N Engl J Med 2002; 347:36-42.
(6) Antin JH. Stem cell transplantation-harnessing of graft-versus-malignancy. Curr Opin
Hematol 2003; 10:440-444.
(7) Antin JH, Childs R, Filipovich AH, Giralt S, Mackinnon S, Spitzer T et al.
Establishment of complete and mixed donor chimerism after allogeneic
lymphohematopoietic transplantation: recommendations from a workshop at the 2001
tandem meetings. Biol Blood Marrow Transplant 2001; 7:473-485.
(8) Apperley JF, Girinsky T, Friedrich W. Conditioning Regiments. The EBMT Handbook.
2004: 99-101.
(9) Atkinson K, Horowitz MM, Gale RP, van Bekkum DW, Gluckman E, Good RA et al.
Risk factors for chronic graft-versus-host disease after HLA-identical sibling bone
marrow transplantation. Blood 1990; 75:2459-2464.
(10) Auffermann-Gretzinger S, Lossos IS, Vayntrub TA, Leong W, Grumet FC, Blume KG
et al. Rapid establishment of dendritic cell chimerism in allogeneic hematopoietic cell
transplant recipients. Blood 2002; 99:1442-1448.
(11) Bader P, Duckers G, Kreyenberg H, Hoelle W, Kerst G, Lang P et al. Monitoring of
donor cell chimerism for the detection of relapse and early immunotherapeutic
intervention in acute lymphoblastic leukemias. Ann Hematol 2002; 81 Suppl 2:S25-
S27.

80
(12) Bader P, Kreyenberg H, Hoelle W, Dueckers G, Handgretinger R, Lang P et al.
Increasing mixed chimerism is an important prognostic factor for unfavorable outcome
in children with acute lymphoblastic leukemia after allogeneic stem-cell
transplantation: possible role for pre-emptive immunotherapy? J Clin Oncol 2004;
22:1696-1705.
(13) Bader P, Kreyenberg H, Hoelle W, Dueckers G, Kremens B, Dilloo D et al. Increasing
mixed chimerism defines a high-risk group of childhood acute myelogenous leukemia
patients after allogeneic stem cell transplantation where pre-emptive immunotherapy
may be effective. Bone Marrow Transplant 2004; 33:815-821.
(14) Bader P, Niethammer D, Willasch A, Kreyenberg H, Klingebiel T. How and when
should we monitor chimerism after allogeneic stem cell transplantation? Bone Marrow
Transplant 2005; 35:107-119.
(15) Balon J, Bieniaszewska M, Reichert M, Pawłowski R, Hellmann A. Monitorowanie
chimeryzmu
hemopoetycznego
po
allogenicznej
transplantacji
komórek
progenitorowych
półilo
ś
ciow
ą
metod
ą
elektroforezy
kapilarnej
przy
u
ż
yciu
automatycznego sekwenatora DNA . Acta Haematol.Pol. 30[Supplement 2], 429.
1999.
(16) Balon J, Bieniaszewska M, Hałaburda K, Reichert M, Pawłowski RHA. Wpływ terapii
immunosupresyjnej na chimeryzm hemopoetyczny w 180 i 270 dobie po
allotransplantacji komórek progenitorowych hemopoezy. Hematologia 2000 .
(17) Bar BM, Schattenberg A, de Man AJ, Hoogenhout MJ, Boezeman J, De Witte T.
Influence of the conditioning regimen on erythrocyte chimerism, graft-versus-host
disease and relapse after allogeneic transplantation with lymphocyte depleted
marrow. Bone Marrow Transplant 1992; 10:45-52.
(18) Barkholt L, Hentschke P, Zetterquist H, Mattsson J, Uzunel M, Wersall P et al. An
allogeneic anti-cancer effect after hematopoietic stem cell transplantation. Transplant
Proc 2001; 33:1862-1864.
(19) Baron F, Storb R. Allogeneic hematopoietic cell transplantation as treatment for
hematological malignancies: a review. Springer Semin Immunopathol 2004; 26:71-94.
(20) Baron F, Storb R. Current roles for allogeneic hematopoietic cell transplantation
following nonmyeloablative or reduced-intensity conditioning. Clin Adv Hematol Oncol
2005; 3:799-819.
(21) Baron F, Storb R, Little MT. Hematopoietic cell transplantation: five decades of
progress. Arch Med Res 2003; 34:528-544.
(22) Barrett AJ, van Rhee F. Graft-versus-leukaemia. Baillieres Clin Haematol 1997;
10:337-355.
(23) Basara N, Fauser AA. Safety profile of mycophenolate mofetil. Bone Marrow
Transplant 2000; 26:1362-1363.
(24) Basara N, Kiehl MG, Blau W, Romer E, Bischoff M, Schmetzer B et al.
Mycophenolate Mofetil in the treatment of acute and chronic GVHD in hematopoietic
stem cell transplant patients: four years of experience. Transplant Proc 2001;
33:2121-2123.

81
(25) Benito AI, Diaz MA, Gonzalez-Vicent M, Sevilla J, Madero L. Hematopoietic stem cell
transplantation using umbilical cord blood progenitors: review of current clinical
results. Bone Marrow Transplant 2004; 33:675-690.
(26) Bensinger WI, Clift R, Martin P, Appelbaum FR, Demirer T, Gooley T et al. Allogeneic
peripheral blood stem cell transplantation in patients with advanced hematologic
malignancies: a retrospective comparison with marrow transplantation. Blood 1996;
88:2794-2800.
(27) Bensinger WI, Storb R. Allogeneic peripheral blood stem cell transplantation. Rev Clin
Exp Hematol 2001; 5:67-86.
(28) Bieniaszewska M, Balon J, Hałaburda K, Reichert M, Pawłowski R, Hellmann A.
Comparison of CR based methods for monitoring of chimerism after allogeneic stem
cell transplantation (alloSCT). Gel vs capillary electrophoresis of short tandem
repeats (STR) markers. Bone Marrow Transplant 2000. 25[Supplement 1], 277.
(29) Blaise D, Maraninchi D, Michallet M, Reiffers J, Jouet JP, Milpied N et al. Long-term
follow-up of a randomized trial comparing the combination of cyclophosphamide with
total body irradiation or busulfan as conditioning regimen for patients receiving HLA-
identical marrow grafts for acute myeloblastic leukemia in first complete remission.
Blood 2001; 97:3669-3671.
(30) Busca A, Saroglia EM, Lanino E, Manfredini L, Uderzo C, Nicolini B et al.
Mycophenolate mofetil (MMF) as therapy for refractory chronic GVHD (cGVHD) in
children receiving bone marrow transplantation. Bone Marrow Transplant 2000;
25:1067-1071.
(31) Butcher BW, Collins RH, Jr. The graft-versus-lymphoma effect: clinical review and
future opportunities. Bone Marrow Transplant 2005; 36:1-17.
(32) Carnevale-Schianca F, Martin P, Sullivan K, Flowers M, Gooley T, Anasetti C et al.
Changing from cyclosporine to tacrolimus as salvage therapy for chronic graft-versus-
host disease. Biol Blood Marrow Transplant 2000; 6:613-620.
(33) Chabannon C, Lafage M, Mozziconacci MJ, Faucher C, Maraninchi D, Blaise D. Early
establishment of chimerism in the B and T lymphoid lineages after transplantation of
allogeneic mobilized blood cells in leukemic patients. Transplantation 1997; 63:1646-
1652.
(34) Chakrabarti S, Handa SK, Bryon RJ, Griffiths MJ, Milligan DW. Will mixed chimerism
cure autoimmune diseases after a nonmyeloablative stem cell transplant?
Transplantation 2001; 72:340-342.
(35) Chalandon Y, Vischer S, Helg C, Chapuis B, Roosnek E. Quantitative analysis of
chimerism after allogeneic stem cell transplantation by PCR amplification of
microsatellite markers and capillary electrophoresis with fluorescence detection: the
Geneva experience. Leukemia 2003; 17:228-231.

82
(36) Champlin RE, Schmitz N, Horowitz MM, Chapuis B, Chopra R, Cornelissen JJ et al.
Blood stem cells compared with bone marrow as a source of hematopoietic cells for
allogeneic transplantation. IBMTR Histocompatibility and Stem Cell Sources Working
Committee and the European Group for Blood and Marrow Transplantation (EBMT).
Blood 2000; 95:3702-3709.
(37) Chao NJ, Parker PM, Niland JC, Wong RM, Dagis A, Long GD et al. Paradoxical
effect of thalidomide prophylaxis on chronic graft-vs.-host disease. Biol Blood Marrow
Transplant 1996; 2:86-92.
(38) Childs R, Clave E, Contentin N, Jayasekera D, Hensel N, Leitman S et al.
Engraftment kinetics after nonmyeloablative allogeneic peripheral blood stem cell
transplantation: full donor T-cell chimerism precedes alloimmune responses. Blood
1999; 94:3234-3241.
(39) Clift RA, Buckner CD, Thomas ED, Bensinger WI, Bowden R, Bryant E et al. Marrow
transplantation for chronic myeloid leukemia: a randomized study comparing
cyclophosphamide and total body irradiation with busulfan and cyclophosphamide.
Blood 1994; 84:2036-2043.
(40) Collins RH, Jr., Goldstein S, Giralt S, Levine J, Porter D, Drobyski W et al. Donor
leukocyte infusions in acute lymphocytic leukemia. Bone Marrow Transplant 2000;
26:511-516.
(41) Colson YL, Li H, Boggs SS, Patrene KD, Johnson PC, Ildstad ST. Durable mixed
allogeneic chimerism and tolerance by a nonlethal radiation-based cytoreductive
approach. J Immunol 1996; 157:2820-2829.
(42) Corvo R, Paoli G, Barra S, Bacigalupo A, Van Lint MT, Franzone P et al. Total body
irradiation correlates with chronic graft versus host disease and affects prognosis of
patients with acute lymphoblastic leukemia receiving an HLA identical allogeneic
bone marrow transplant. Int J Radiat Oncol Biol Phys 1999; 43:497-503.
(43) Curtis RE, Metayer C, Rizzo JD, Socie G, Sobocinski KA, Flowers ME et al. Impact of
chronic GVHD therapy on the development of squamous-cell cancers after
hematopoietic stem-cell transplantation: an international case-control study. Blood
2005; 105:3802-3811.
(44) Curtis RE, Rowlings PA, Deeg HJ, Shriner DA, Socie G, Travis LB et al. Solid cancers
after bone marrow transplantation. N Engl J Med 1997; 336:897-904.
(45) Cutler C, Antin JH. Sirolimus for GVHD prophylaxis in allogeneic stem cell
transplantation. Bone Marrow Transplant 2004; 34:471-476.
(46) Cutler C, Antin JH. An overview of hematopoietic stem cell transplantation. Clin Chest
Med 2005; 26:517-527.
(47) Cutler C, Giri S, Jeyapalan S, Paniagua D, Viswanathan A, Antin JH. Acute and
chronic graft-versus-host disease after allogeneic peripheral-blood stem-cell and
bone marrow transplantation: a meta-analysis. J Clin Oncol 2001; 19:3685-3691.

83
(48) Deeg HJ, Lin D, Leisenring W, Boeckh M, Anasetti C, Appelbaum FR et al.
Cyclosporine or cyclosporine plus methylprednisolone for prophylaxis of graft-versus-
host disease: a prospective, randomized trial. Blood 1997; 89:3880-3887.
(49) Deeg HJ, Storb R. Graft-versus-host disease: pathophysiological and clinical aspects.
Annu Rev Med 1984; 35:11-24.
(50) Enright H, Daniels K, Arthur DC, Dusenbery KE, Kersey JH, Kim T et al. Related
donor marrow transplant for chronic myeloid leukemia: patient characteristics
predictive of outcome. Bone Marrow Transplant 1996; 17:537-542.
(51) Enright H, Davies SM, Defor T, Shu X, Weisdorf D, Miller W et al. Relapse after non-
T-cell-depleted allogeneic bone marrow transplantation for chronic myelogenous
leukemia: early transplantation, use of an unrelated donor, and chronic graft-versus-
host disease are protective. Blood 1996; 88:714-720.
(52) Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ et al. National
Institutes of Health Consensus Development Project on Criteria for Clinical Trials in
Chronic Graft-versus-Host Disease: I. Diagnosis and Staging Working Group Report.
Biol Blood Marrow Transplant 2005; 11:945-956.
(53) Flowers ME, Parker PM, Johnston LJ, Matos AV, Storer B, Bensinger WI et al.
Comparison of chronic graft-versus-host disease after transplantation of peripheral
blood stem cells versus bone marrow in allogeneic recipients: long-term follow-up of a
randomized trial. Blood 2002; 100:415-419.
(54) Frassoni F, Scarpati D, Bacigalupo A, Vitale V, Corvo R, Miceli S et al. The effect of
total body irradiation dose and chronic graft-versus-host disease on leukaemic
relapse after allogeneic bone marrow transplantation. Br J Haematol 1989; 73:211-
216.
(55) Friedman J, McGillivray B. Polimorfizm genetyczny. Genetyka. 2005: 81-89.
(56) Gale RP, Horowitz MM, Ash RC, Champlin RE, Goldman JM, Rimm AA et al.
Identical-twin bone marrow transplants for leukemia. Ann Intern Med 1994; 120:646-
652.
(57) Gardiner N, Lawler M, O'Riordan J, DeArce M, Humphries P, McCann SR. Persistent
donor chimaerism is consistent with disease-free survival following BMT for chronic
myeloid leukaemia. Bone Marrow Transplant 1997; 20:235-241.
(58) Georges GE, Storb R. Review of "minitransplantation": nonmyeloablative allogeneic
hematopoietic stem cell transplantation. Int J Hematol 2003; 77:3-14.
(59) Ginsburg D, Antin JH, Smith BR, Orkin SH, Rappeport JM. Origin of cell populations
after bone marrow transplantation. Analysis using DNA sequence polymorphisms. J
Clin Invest 1985; 75:596-603.
(60) Gleissner B, Blau IW, Sindram A, Reinhardt R, Knauf W, Thiel E. Analysis of
chimerism during the early period after allogeneic peripheral stem cell transplantation.
Clin Lab Haematol 2001; 23:401-406.

84
(61) Gluckman E, Rocha V, Boyer-Chammard A, Locatelli F, Arcese W, Pasquini R et al.
Outcome of cord-blood transplantation from related and unrelated donors. Eurocord
Transplant Group and the European Blood and Marrow Transplantation Group. N
Engl J Med 1997; 337:373-381.
(62) Granados E, de la CR, Madero L, Diaz MA, Martin-Regueira P, Steegmann JL et al.
Hematopoietic cell transplantation in acute lymphoblastic leukemia: better long term
event-free survival with conditioning regimens containing total body irradiation.
Haematologica 2000; 85:1060-1067.
(63) Gratwohl A, Baldomero H, Passweg J, Frassoni F, Niederwieser D, Schmitz N et al.
Hematopoietic stem cell transplantation for hematological malignancies in Europe.
Leukemia 2003; 17:941-959.
(64) Gratwohl A, Baldomero H, Passweg J, Urbano-Ispizua A. Increasing use of reduced
intensity conditioning transplants: report of the 2001 EBMT activity survey. Bone
Marrow Transplant 2002; 30:813-831.
(65) Gratwohl A, Brand R, Frassoni F, Rocha V, Niederwieser D, Reusser P et al. Cause
of death after allogeneic haematopoietic stem cell transplantation (HSCT) in early
leukaemias: an EBMT analysis of lethal infectious complications and changes over
calendar time. Bone Marrow Transplant 2005; 36:757-769.
(66) Higman M, Vogelsang G. Chronic graft versus host disease. Br J Haematol 2004;
125:435-454.
(67) Hill RS, Petersen FB, Storb R, Appelbaum FR, Doney K, Dahlberg S et al. Mixed
hematologic chimerism after allogeneic marrow transplantation for severe aplastic
anemia is associated with a higher risk of graft rejection and a lessened incidence of
acute graft-versus-host disease. Blood 1986; 67:811-816.
(68) Hogan WJ, Storb R. Use of cyclosporine in hematopoietic cell transplantation.
Transplant Proc 2004; 36:367S-371S.
(69) Huss R, Deeg HJ, Gooley T, Bryant E, Leisenring W, Clift R et al. Effect of mixed
chimerism on graft-versus-host disease, disease recurrence and survival after HLA-
identical marrow transplantation for aplastic anemia or chronic myelogenous
leukemia. Bone Marrow Transplant 1996; 18:767-776.
(70) Imamura M, Tsutsumi Y, Miura Y, Toubai T, Tanaka J. Immune reconstitution and
tolerance after allogeneic hematopoietic stem cell transplantation. Hematology 2003;
8:19-26.
(71) Jaksch M, Uzunel M, Remberger M, Sundberg B, Mattsson J. Molecular monitoring of
T-cell chimerism early after allogeneic stem cell transplantation may predict the
occurrence of acute GVHD grades II-IV. Clin Transplant 2005; 19:346-349.
(72) Jolkowska JWM. Chimeryzm komórkowy po transplantacji szpiku kostnego. Acta
Haematol Pol 2004; 35:321-328.

85
(73) Kansu E, Gooley T, Flowers ME, Anasetti C, Deeg HJ, Nash RA et al. Administration
of cyclosporine for 24 months compared with 6 months for prevention of chronic graft-
versus-host disease: a prospective randomized clinical trial. Blood 2001; 98:3868-
3870.
(74) Keever-Taylor CA, Bredeson C, Loberiza FR, Casper JT, Lawton C, Rizzo D et al.
Analysis of risk factors for the development of GVHD after T cell-depleted allogeneic
BMT: effect of HLA disparity, ABO incompatibility, and method of T-cell depletion. Biol
Blood Marrow Transplant 2001; 7:620-630.
(75) Khan F, Agarwal A, Agrawal S. Significance of chimerism in hematopoietic stem cell
transplantation: new variations on an old theme. Bone Marrow Transplant 2004; 34:1-
12.
(76) Koc S, Leisenring W, Flowers ME, Anasetti C, Deeg HJ, Nash RA et al. Therapy for
chronic graft-versus-host disease: a randomized trial comparing cyclosporine plus
prednisone versus prednisone alone. Blood 2002; 100:48-51.
(77) Lee SJ. New approaches for preventing and treating chronic graft versus host
disease. Blood 2005; 105:4200-4206.
(78) Lee SJ, Klein JP, Barrett AJ, Ringden O, Antin JH, Cahn JY et al. Severity of chronic
graft-versus-host disease: association with treatment-related mortality and relapse.
Blood 2002; 100:406-414.
(79) Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood
Marrow Transplant 2003; 9:215-233.
(80) Lee SJ, Vogelsang G, Gilman A, Weisdorf DJ, Pavletic S, Antin JH et al. A survey of
diagnosis, management, and grading of chronic GVHD. Biol Blood Marrow Transplant
2002; 8:32-39.
(81) Lion T, Daxberger H, Dubovsky J, Filipcik P, Fritsch G, Printz D et al. Analysis of
chimerism within specific leukocyte subsets for detection of residual or recurrent
leukemia in pediatric patients after allogeneic stem cell transplantation. Leukemia
2001; 15:307-310.
(82) Maas F, Schaap N, Kolen S, Zoetbrood A, Buno I, Dolstra H et al. Quantification of
donor and recipient hemopoietic cells by real-time PCR of single nucleotide
polymorphisms. Leukemia 2003; 17:630-633.
(83) Mangioni S, Balduzzi A, Rivolta A, Rovelli A, Nesi F, Rossi V et al. Long-term
persistence of hemopoietic chimerism following sex-mismatched bone marrow
transplantation. Bone Marrow Transplant 1997; 20:969-973.
(84) Mapara MY, Kim YM, Wang SP, Bronson R, Sachs DH, Sykes M. Donor lymphocyte
infusions mediate superior graft-versus-leukemia effects in mixed compared to fully
allogeneic chimeras: a critical role for host antigen-presenting cells. Blood 2002;
100:1903-1909.
(85) Martin P, Carpenter PA, Sanders J, Flowers M. Clinical management of chronic Graft
versus host disease. Acta Haematol Pol 2003; 34:290-301.

86
(86) Matthes-Martin S, Lion T, Haas OA, Frommlet F, Daxberger H, Konig M et al.
Lineage-specific chimaerism after stem cell transplantation in children following
reduced intensity conditioning: potential predictive value of NK cell chimaerism for late
graft rejection. Leukemia 2003; 17:1934-1942.
(87) Mattsson J, Uzunel M, Tammik L, Aschan J, Ringden O. Leukemia lineage-specific
chimerism analysis is a sensitive predictor of relapse in patients with acute myeloid
leukemia and myelodysplastic syndrome after allogeneic stem cell transplantation.
Leukemia 2001; 15:1976-1985.
(88) McSweeney PA, Niederwieser D, Shizuru JA, Sandmaier BM, Molina AJ, Maloney
DG et al. Hematopoietic cell transplantation in older patients with hematologic
malignancies: replacing high-dose cytotoxic therapy with graft-versus-tumor effects.
Blood 2001; 97:3390-3400.
(89) McSweeney PA, Storb R. Mixed chimerism: preclinical studies and clinical
applications. Biol Blood Marrow Transplant 1999; 5:192-203.
(90) Mengarelli A, Iori AP, Romano A, Cerretti R, Cerilli L, De Propris MS et al. One-year
cyclosporine prophylaxis reduces the risk of developing extensive chronic graft-
versus-host disease after allogeneic peripheral blood stem cell transplantation.
Haematologica 2003; 88:315-323.
(91) Miflin G, Stainer CJ, Carter GI, Byrne JL, Haynes AP, Russell NH. Comparative serial
quantitative measurements of chimaerism following unmanipulated allogeneic
transplantation of peripheral blood stem cells and bone marrow. Br J Haematol 1999;
107:429-440.
(92) Mohty M, Kuentz M, Michallet M, Bourhis JH, Milpied N, Sutton L et al. Chronic graft-
versus-host disease after allogeneic blood stem cell transplantation: long-term results
of a randomized study. Blood 2002; 100:3128-3134.
(93) Molloy K, Goulden N, Lawler M, Cornish J, Oakhill A, Pamphilon D et al. Patterns of
hematopoietic chimerism following bone marrow transplantation for childhood acute
lymphoblastic leukemia from volunteer unrelated donors. Blood 1996; 87:3027-3031.
(94) Mookerjee B, Altomonte V, Vogelsang G. Salvage therapy for refractory chronic graft-
versus-host disease with mycophenolate mofetil and tacrolimus. Bone Marrow
Transplant 1999; 24:517-520.
(95) Morton J, Hutchins C, Durrant S. Granulocyte-colony-stimulating factor (G-CSF)-
primed allogeneic bone marrow: significantly less graft-versus-host disease and
comparable engraftment to G-CSF-mobilized peripheral blood stem cells. Blood 2001;
98:3186-3191.
(96) Nash RA, Storb R. Graft-versus-host effect after allogeneic hematopoietic stem cell
transplantation: GVHD and GVL. Curr Opin Immunol 1996; 8:674-680.
(97) Nordlander A, Mattsson J, Ringden O, Leblanc K, Gustafsson B, Ljungman P et al.
Graft-versus-host disease is associated with a lower relapse incidence after
hematopoietic stem cell transplantation in patients with acute lymphoblastic leukemia.
Biol Blood Marrow Transplant 2004; 10:195-203.

87
(98) Ochs LA, Miller WJ, Filipovich AH, Haake RJ, McGlave PB, Blazar BR et al.
Predictive factors for chronic graft-versus-host disease after histocompatible sibling
donor bone marrow transplantation. Bone Marrow Transplant 1994; 13:455-460.
(99) Palka G, Stuppia L, Di Bartolomeo P, Morizio E, Peila R, Franchi PG et al. FISH
detection of mixed chimerism in 33 patients submitted to bone marrow
transplantation. Bone Marrow Transplant 1996; 17:231-236.
(100) Pegg, D.E. Bone Marrow transplantation: Lloyd - Luke Medical Books. 2005: 77-101.
(101) Przepiorka D, Anderlini P, Saliba R, Cleary K, Mehra R, Khouri I et al. Chronic graft-
versus-host disease after allogeneic blood stem cell transplantation. Blood 2001;
98:1695-1700.
(102) Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J et al. 1994
Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant 1995;
15:825-828.
(103) Ratanatharathorn V, Ayash L, Lazarus HM, Fu J, Uberti JP. Chronic graft-versus-host
disease: clinical manifestation and therapy. Bone Marrow Transplant 2001; 28:121-
129.
(104) Remberger M, Beelen DW, Fauser A, Basara N, Basu O, Ringden O. Increased risk
of extensive chronic graft-versus-host disease after allogeneic peripheral blood stem
cell transplantation using unrelated donors. Blood 2005; 105:548-551.
(105) Riley RS, Idowu M, Chesney A, Zhao S, McCarty J, Lamb LS et al. Hematologic
aspects of myeloablative therapy and bone marrow transplantation. J Clin Lab Anal
2005; 19:47-79.
(106) Ringden O. Allogeneic bone marrow transplantation for hematological malignancies--
controversies and recent advances. Acta Oncol 1997; 36:549-564.
(107) Ringden O, Labopin M, Gorin NC, Schmitz N, Schaefer UW, Prentice HG et al. Is
there a graft-versus-leukaemia effect in the absence of graft-versus-host disease in
patients undergoing bone marrow transplantation for acute leukaemia? Br J Haematol
2000; 111:1130-1137.
(108) Ringden O, Paulin T, Lonnqvist B, Nilsson B. An analysis of factors predisposing to
chronic graft-versus-host disease. Exp Hematol 1985; 13:1062-1067.
(109) Ringden O, Remberger M, Runde V, Bornhauser M, Blau IW, Basara N et al.
Peripheral blood stem cell transplantation from unrelated donors: a comparison with
marrow transplantation. Blood 1999; 94:455-464.
(110) Schaap N, Schattenberg A, Bar B, Preijers F, van de Wiel van Kemenade, De Witte
T. Induction of graft-versus-leukemia to prevent relapse after partially lymphocyte-
depleted allogeneic bone marrow transplantation by pre-emptive donor leukocyte
infusions. Leukemia 2001; 15:1339-1346.
(111) Schaap N, Schattenberg A, Mensink E, Preijers F, Hillegers M, Knops R et al. Long-
term follow-up of persisting mixed chimerism after partially T cell-depleted allogeneic
stem cell transplantation. Leukemia 2002; 16:13-21.

88
(112) Schattenberg A, De Witte T, Salden M, Vet J, Van Dijk B, Smeets D et al. Mixed
hematopoietic chimerism after allogeneic transplantation with lymphocyte-depleted
bone marrow is not associated with a higher incidence of relapse. Blood 1989;
73:1367-1372.
(113) Schattenberg A, De Witte T, Salden M, Vet J, Van Dijk B, Smeets D et al. Mixed
hematopoietic chimerism after allogeneic transplantation with lymphocyte-depleted
bone marrow is not associated with a higher incidence of relapse. Blood 1989;
73:1367-1372.
(114) Schattenberg A, Schaap N, van de Wiel-van Kemenade, Bar B, Preijers F, van der
MR et al. In relapsed patients after lymphocyte depleted bone marrow transplantation
the percentage of donor T lymphocytes correlates well with the outcome of donor
leukocyte infusion. Leuk Lymphoma 1999; 32:317-325.
(115) Schattenberg AV, Dolstra H. Cellular adoptive immunotherapy after allogeneic stem
cell transplantation. Curr Opin Oncol 2005; 17:617-621.
(116) Schleuning M. Adoptive allogeneic immunotherapy--history and future perspectives.
Transfus Sci 2000; 23:133-150.
(117) Schraml E, Daxberger H, Watzinger F, Lion T. Quantitative analysis of chimerism
after allogeneic stem cell transplantation by PCR amplification of microsatellite
markers and capillary electrophoresis with fluorescence detection: the Vienna
experience. Leukemia 2003; 17:224-227.
(118) Seaton ED, Szydlo RM, Kanfer E, Apperley JF, Russell-Jones R. Influence of
extracorporeal photopheresis on clinical and laboratory parameters in chronic graft-
versus-host disease and analysis of predictors of response. Blood 2003; 102:1217-
1223.
(119) Shapira MY, Resnick IB, Bitan M, Ackerstein A, Samuel S, Elad S et al. Low
transplant-related mortality with allogeneic stem cell transplantation in elderly
patients. Bone Marrow Transplant 2004; 34:155-159.
(120) Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE et al.
Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20
Seattle patients. Am J Med 1980; 69:204-217.
(121) Slavin S. Non-myeloablative stem cell transplantation for induction of host-versus-
graft tolerance for adoptive immunotherapy of malignant and nonmalignant diseases
and towards transplantation of organ allografts. Transplant Proc 2002; 34:3371-3373.
(122) Slavin S. Graft-versus-host disease, the graft-versus-leukemia effect, and mixed
chimerism following nonmyeloablative stem cell transplantation. Int J Hematol 2003;
78:195-207.
(123) Slavin
S.
Reduced-intensity
conditioning
or
nonmyeloablative
stem
cell
transplantation: introduction, rationale, and historic background. Semin Oncol 2004;
31:1-3.

89
(124) Snowden JA, Nivison-Smith I, Atkinson K, Fay K, Concannon A, Dodds A et al.
Allogeneic PBPC transplantation: an effect on incidence and distribution of chronic
graft-versus-host disease without long-term survival benefit? Bone Marrow Transplant
2000; 25:119-120.
(125) Socie G, Clift RA, Blaise D, Devergie A, Ringden O, Martin PJ et al. Busulfan plus
cyclophosphamide compared with total-body irradiation plus cyclophosphamide
before marrow transplantation for myeloid leukemia: long-term follow-up of 4
randomized studies. Blood 2001; 98:3569-3574.
(126) Socie G, Stone JV, Wingard JR, Weisdorf D, Henslee-Downey PJ, Bredeson C et al.
Long-term survival and late deaths after allogeneic bone marrow transplantation. Late
Effects Working Committee of the International Bone Marrow Transplant Registry. N
Engl J Med 1999; 341:14-21.
(127) Sorror ML, Leisenring W, Deeg HJ, Martin PJ, Storb R. Twenty-year follow-up of a
controlled trial comparing a combination of methotrexate plus cyclosporine with
cyclosporine alone for prophylaxis of graft-versus-host disease in patients
administered HLA-identical marrow grafts for leukemia. Biol Blood Marrow Transplant
2005; 11:814-815.
(128) Storb RF, Champlin R, Riddell SR, Murata M, Bryant S, Warren EH. Non-
myeloablative transplants for malignant disease. Hematology (Am Soc Hematol Educ
Program ) 2001;375-391.
(129) Storek J, Gooley T, Siadak M, Bensinger WI, Maloney DG, Chauncey TR et al.
Allogeneic peripheral blood stem cell transplantation may be associated with a high
risk of chronic graft-versus-host disease. Blood 1997; 90:4705-4709.
(130) Sullivan KM, Fefer A, Witherspoon R, Storb R, Buckner CD, Weiden P et al. Graft-
versus-leukemia in man: relationship of acute and chronic graft-versus-host disease
to relapse of acute leukemia following allogeneic bone marrow transplantation. Prog
Clin Biol Res 1987; 244:391-399.
(131) Sullivan KM, Witherspoon RP, Storb R, Deeg HJ, Dahlberg S, Sanders JE et al.
Alternating-day cyclosporine and prednisone for treatment of high-risk chronic graft-v-
host disease. Blood 1988; 72:555-561.
(132) Sykes M, Sachs DH. Mixed chimerism. Philos Trans R Soc Lond B Biol Sci 2001;
356:707-726.
(133) Sykes M, Spitzer TR. Non-myeloblative induction of mixed hematopoietic chimerism:
application to transplantation tolerance and hematologic malignancies in experimental
and clinical studies. Cancer Treat Res 2002; 110:79-99.
(134) Thiede C. Diagnostic chimerism analysis after allogeneic stem cell transplantation:
new methods and markers. Am J Pharmacogenomics 2004; 4:177-187.
(135) Thiede C, Bornhauser M, Ehninger G. Evaluation of STR informativity for chimerism
testing--comparative analysis of 27 STR systems in 203 matched related donor
recipient pairs. Leukemia 2004; 18:248-254.

90
(136) Thiede C, Bornhauser M, Oelschlagel U, Brendel C, Leo R, Daxberger H et al.
Sequential monitoring of chimerism and detection of minimal residual disease after
allogeneic blood stem cell transplantation (BSCT) using multiplex PCR amplification
of short tandem repeat-markers. Leukemia 2001; 15:293-302.
(137) Thiede C, Florek M, Bornhauser M, Ritter M, Mohr B, Brendel C et al. Rapid
quantification of mixed chimerism using multiplex amplification of short tandem repeat
markers and fluorescence detection. Bone Marrow Transplant 1999; 23:1055-1060.
(138) Toh HC, Spitzer TR, Preffer F, Alexander SI, McAfee S, Dombkowski D et al.
Fluctuating lymphocyte chimerism, tolerance and anti-tumor response in a patient
with refractory lymphoma receiving nonmyeloablative conditioning and a
haploidentical related allogeneic bone marrow transplant. Cytokines Cell Mol Ther
2002; 7:43-47.
(139) Tsutsumi Y, Tanaka J, Kato N, Zhang L, Mori A, Kobayasi R et al. Analysis of mixed
chimerism in patients after allogeneic stem cell transplantation using a capillary
electrophoresis system. Acta Haematol 2002; 107:195-202.
(140) Umemura A, Morita H, Li XC, Tahan S, Monaco AP, Maki T. Dissociation of
hemopoietic chimerism and allograft tolerance after allogeneic bone marrow
transplantation. J Immunol 2001; 167:3043-3048.
(141) Uzunel M, Mattsson J, Jaksch M, Remberger M, Ringden O. The significance of graft-
versus-host disease and pretransplantation minimal residual disease status to
outcome after allogeneic stem cell transplantation in patients with acute lymphoblastic
leukemia. Blood 2001; 98:1982-1984.
(142) van Leeuwen JE, van Tol MJ, Joosten AM, Wijnen JT, Khan PM, Vossen JM. Mixed
T-lymphoid chimerism after allogeneic bone marrow transplantation for hematologic
malignancies of children is not correlated with relapse. Blood 1993; 82:1921-1928.
(143) van Leeuwen JE, van Tol MJ, Joosten AM, Wijnen JT, Verweij PJ, Khan PM et al.
Persistence of host-type hematopoiesis after allogeneic bone marrow transplantation
for leukemia is significantly related to the recipient's age and/or the conditioning
regimen, but it is not associated with an increased risk of relapse. Blood 1994;
83:3059-3067.
(144) van Rhee F, Szydlo RM, Hermans J, Devergie A, Frassoni F, Arcese W et al. Long-
term results after allogeneic bone marrow transplantation for chronic myelogenous
leukemia in chronic phase: a report from the Chronic Leukemia Working Party of the
European Group for Blood and Marrow Transplantation. Bone Marrow Transplant
1997; 20:553-560.
(145) Wagner JL, Flowers ME, Longton G, Storb R, Schubert M, Sullivan KM. The
development of chronic graft-versus-host disease: an analysis of screening studies
and the impact of corticosteroid use at 100 days after transplantation. Bone Marrow
Transplant 1998; 22:139-146.
(146) Weinberg K, Blazar BR, Wagner JE, Agura E, Hill BJ, Smogorzewska M et al. Factors
affecting thymic function after allogeneic hematopoietic stem cell transplantation.
Blood 2001; 97:1458-1466.

91
(147) Wekerle T. Transplantation tolerance induced by mixed chimerism. J Heart Lung
Transplant 2001; 20:816-823.
(148) Wingard JR, Piantadosi S, Vogelsang GB, Farmer ER, Jabs DA, Levin LS et al.
Predictors of death from chronic graft-versus-host disease after bone marrow
transplantation. Blood 1989; 74:1428-1435.
(149) Winiarski J, Gustafsson A, Wester D, Dalianis T. Follow-up of chimerism, including T-
and B-lymphocytes and granulocytes in children more than one year after allogeneic
bone marrow transplantation. Pediatr Transplant 2000; 4:132-139.
(150) Wren SM, Hronakes ML, Ildstad ST. CD4+ T cells, but not CD8+ T cells, mediate the
breaking of tolerance in mixed allogeneic chimeras (B10 + B10.BR-->B10).
Transplantation 1993; 55:1382-1389.
(151) Zaucha J, Gozdzik J, Wylegała E, Wojnar J, Balon J, Gil L et al. Rozpoznanie i
klasyfikacja przewlekłej choroby przeszczep przeciw gospodarzowi - raport z
Warsztatów Transplantologicznych Falenty 2003 cz
ęść
I. Acta Haematol Pol 2004;
35:417-429.
(152) Zaucha J, Gozdzik J, Wylegała E, Wojnar J, Balon J, Gil L et al. Rozpoznanie i
klasyfikacja przewlekłej choroby przeszczep przeciw gospodarzowi - raport z
Warsztatów Transplantologicznych Falenty 2003 cz
ęść
II. Acta Haematol Pol 2004;
35:431-443.
(153) Zaucha JM, Pawlowski R, Welz A, Prejzner W, Hauser R, Hellman A. [Evaluation of
bone marrow grafts and hemopoietic chimerism using PCR hypervariable sequencing
with variable number tandem repeat sequences]. Pol Tyg Lek 1995; 50:73-74.
(154) Zecca M, Prete A, Rondelli R, Lanino E, Balduzzi A, Messina C et al. Chronic graft-
versus-host disease in children: incidence, risk factors, and impact on outcome. Blood
2002; 100:1192-1200.
Wyszukiwarka
Podobne podstrony:
Oporność wielolekowa i kliniczne znaczenie polimorfizmów
Kliniczne znaczenie dziedziczenia
15 Znaczenie wczesnych wiezi MC Nieznany (2)
Rozdz.10-Znaczenie wczesnych doświadczeń lękowych w rozwoju, Klein-Psychoanaliza dziecka (fragmenty)
Kliniczne+znaczenie+diagnostyki+obrazowej, Prezentacje ELEKTRO
przedszkole znaczenie wczesnej edukacji
Rola i znaczenie wczesnej edukacji, studia pedagogika
ZIEMIA OBIECANA ZNACZENIE WCZEŚNIEJ I DZIŚ
Znaczenie wczesnych doświadczeń w rozwoju psychospołecznym
Psychologia rozwojowa Magdalena Czub Znaczenie wczesnych więzi społecznych dla rozwoju emocjonalne
Znaczenie wczesnego operacyjnego odbarczenia rdzenia kręgowego po urazach szyjnego odc kręgosłupa
Rola i znaczenie wapnia fosforu i magnezu w organizmach przeżuwaczy z uwzględnieniem zmian kliniczny
Cierpiałkowska Koncepcje interakcyjne i systemowe oraz ich znaczenie dla psychologii klinicznej
Rozpoznawanie raka piersi, Dla wczesnego wykrywania raka piersi bardzo ważne znaczenie mają systemat
Rola i znaczenie wapnia fosforu i magnezu w organizmach przeżuwaczy z uwzględnieniem zmian kliniczny
Biochemia kliniczna W VII0 03 2011 Uracyl w DNA – znaczenie biologiczne
więcej podobnych podstron