
Zespół Turnera
1
Zespół Turnera
Zespół Turnera
syndroma Turner
Q96
Q96.0 Kariotyp 45,X
Q96.1 Kariotyp 46,X izo (Xq)
Q96.2 Kariotyp 46,X, z anormalnym chromosomem płciowym z wyjątkiem izo(Xq)
Q96.3 Mozaicyzm 45,X/46,XX lub 46,XY
Q96.4 Mozaicyzm 45,X/inne linie komórkowe z nieprawidłowym chromosomem płciowym
Q96.8 Inne warianty zespołu Turnera
Q96.9 Zespół Turnera, nie określony
Kariotyp 45,X0
Kariotyp kobiety z zespołem Turnera; w 70%
komórek pacjentki stwierdzono kariotyp 45,X0,
w pozostałych 30% obecny był izochromosom X
zawierający podwójne długie ramię – 46,X
i(Xq)[1] .
Zespół Turnera (ang. Turner's syndrome, łac. syndroma Turner) –
uwarunkowany genetycznie zespół wad wrodzonych spowodowany
całkowitym lub częściowym brakiem jednego z chromosomów X we
wszystkich komórkach organizmu lub w pewnej ich części. Występuje
u 1 na 2500 urodzonych dziewczynek
[2]
. Najważniejsze cechy
występujące u osób z zespołem Turnera to: niski wzrost, słabo
zaznaczone cechy żeńskie i wrodzona dysgenezja gonad powodująca w
większości przypadków bezpłodność.
Nazewnictwo
Spotykane nazwy zespołu to: zespół Turnera, zespół Ullricha
(zwłaszcza w literaturze niemieckojęzycznej), zespół Ullricha-Turnera,
zespół Szereszewskiego-Turnera (w krajach byłego Związku
Radzieckiego), zespół szczątkowych jajników
[3]
, zespół
Morgagniego-Turnera-Albrighta, zespół Turnera-Albrighta, zespół
Morgagniego-Szereszewskiego-Turnera-Albrighta.
Zespół Bonnevie-Ullricha
[3]
jest zarzuconym, historycznym terminem,
stosowanym do określenia zespołu Turnera i zespołów wad
wrodzonych o zbliżonym fenotypie u niemowląt, zanim poznano
etiologię choroby.
Zespół Turnera u chłopców i mężczyzn
[3]
, zespół pseudoturnerowski,
rodzinny zespół Turnera, zespół Ullricha-Noonan, fenotyp turnerowski
z normalnym kariotypem, to zarzucone, nieprawidłowe nazwy
stosowane na określenie zespołu Noonan zanim ostatecznie nie
poznano różnic w etiologii tych dwóch chorób.
Postać eunuchoidalna zespołu Turnera to zarzucona, nieprawidłowa nazwa zespołu Swyera
[3]
.

Zespół Turnera
2
Historia
Klasyczny opis zespołu
[4]
przedstawił w 1938 roku amerykański endokrynolog z Oklahomy, Henry Hubert Turner
(1892-1970); od jego nazwiska jednostka chorobowa wzięła nazwę. Turner w swojej pracy opisał siedem młodych
pacjentek z typowymi cechami zespołu (niski wzrost, płetwiasta szyja, hipogonadyzm, opóźniony wiek kostny), ale
błędnie przyczynę choroby upatrywał w dysfunkcji przedniego płata przysadki. Jednak pojedyncze przypadki
przypadek pacjentki z monosomią chromosomu X już w 1768 roku
[5]
. U zmarłej niskorosłej kobiety stwierdził
malformację nerek i dysgenezję gonad. W 1902 roku Otto Funke opisał przypadek piętnastoletniej dziewczynki z
dysgenezją gonad, niskim wzrostem, niedojrzałością płciową, wrodzonym obrzękiem limfatycznym i płetwiastą
szyją
[6]
. W 1925 roku w na spotkaniu Rosyjskiego Towarzystwa Endokrynologicznego klasyczne objawy zespołu u
25-letniej kobiety opisał Nikołaj Adolfowicz Szereszewskij (1885-1961); w krajach byłego Związku Radzieckiego
używa się nazwy zespół Szereszewskiego-Turnera. W 1929 roku na sympozjum Monachijskiego Towarzystwa
Pediatrycznego inny opis choroby przedstawił Otto Ullrich (1894-1957)
[7]
; do dziś niekiedy spotyka się określenie
zespołu Ullricha-Turnera (głównie w Niemczech). Spotyka się również (zwłaszcza w starszej literaturze) określenie
zespół Bonnevie-Ullricha; Kristine Bonnevie, norweska biolog, pracowała przypuszczalnie nad mysim modelem
[8]
, a określenia zespołu Bonnevie-Ullricha (albo status Bonnevie-Ullrich) używano zwłaszcza w
odniesieniu do dzieci z dodatkowymi wadami układu kostnego, mięśniowego i naczyniowego
[3]
[9]
[10]
[11]
[12]
.
Pierwszeństwo w wyjaśnieniu genetycznych podstaw zespołu przypisuje się Charlesowi Fordowi i jego
współpracownikom, którzy w 1959 roku w pracy na łamach Lancet opisali brak chromosomu X u 14-letniej
dziewczynki z zespołem Turnera
[13]
.
Na górę strony
Epidemiologia
Zespół Turnera jest jedną z najczęstszych aberracji chromosomowych. Ocenia się, ze występuje u 3% wszystkich
ludzkich płodów. Jednakże tylko 1% spośród tych płodów dożywa porodu
[14]
. Tym samym, zespół Turnera
przeżywa pierwszego trymestru ciąży, co ilustrują wyniki badań prenatalnych: częstość zespołu Turnera wykrytego
w amniocentezie (16. tydzień życia płodowego) szacuje się na 176:100 000, a w CVS (średnio 11. tydzień) 392:100
000
[15]
.
Częstość zespołu Turnera szacuje się na 25-55:100 000 żywych urodzeń noworodków płci żeńskiej u rasy białej
[16]
[17]
[18]
; badanie na populacji japońskiej pozwoliło oszacować częstość zespołu Turnera w tej grupie na 70-210:100
000 urodzeń
[19]
. Przyjmując szacunkową częstość zespołu 1:2000 – 1:2500 urodzeń można obliczyć, że każdego
roku w Polsce rodzi się około 100 dziewczynek dotkniętych zespołem Turnera
[20]
, a liczbę chorych na tę chorobę w
Polsce można szacować na około 10 tysięcy osób
[21]
. Na całym świecie liczba pacjentów z zespołem Turnera
wynosi 1,5 miliona
[22]
. Istotny wpływ na częstość zespołu Turnera ma przerywanie ciąży po prenatalnym
zdiagnozowaniu zespołu Turnera; duże badanie przeprowadzone w jedenastu krajach europejskich wykazało, że
66% tych ciąż było przerywanych, przy najwyższym wskaźniku aborcji po rozpoznaniu postawionym na podstawie
badania USG (79%, w porównaniu z 42% przerwanych ciąż rozpoznanych kariotypowaniem)
[15]
Århus znajduje się jeden z czołowych ośrodków badań nad zespołem Turnera, upowszechnianie wiedzy o zespole i
możliwościach jego leczenia zmniejszyło częstość aborcji płodów zdiagnozowanych prenatalnie ze 100% w latach
1970-1980 do 60% w latach 1985-1988
[1]
.
Na górę strony
Zespół Turnera
3
Etiologia
Mapa chromosomu X; przedstawiono gęstość
genów, ilość powtórzeń GC i SNP.
Poza monosomią chromosomu X zespół Turnera może być też
spowodowany strukturalnymi zmianami jednego chromosomu X.
Możliwe są następujące aberracje chromosomalne skutkujące
fenotypem zespołu Turnera:
• monosomia chromosomu X (oznaczana 45,X albo 45,XO)
• częściowa lub całkowita delecja krótkiego ramienia chromosomu X
(delXp)
• całkowita delecja długiego ramienia chromosomu X (delXq)
• izochromosom długiego ramienia chromosomu X (i(Xq))
• chromosom pierścieniowy (kolisty, ring-chromosome, r(X))
• chromosom markerowy (46,X+m)
• mozaicyzm, czyli obecność więcej niż dwóch linii komórkowych u
jednej osoby, najczęściej 45,X/46,XX i 45,X/46,XY.
W zależności od wielkości zmian objawy kliniczne są zróżnicowane i
nie wszystkie występują. W wypadku mozaicyzmu też może nie
występować pełne spektrum objawów.
Niektórzy badacze sugerują, że "czysty" kariotyp 45,X warunkuje
letalny zespół i płody o takim kariotypie są ronione przez matki
[23]
[24]
[25]
. Hipotezę tę potwierdzają badania kariotypu komórek innych niż limfocyty na obecność mozaicyzmu
[15]
.
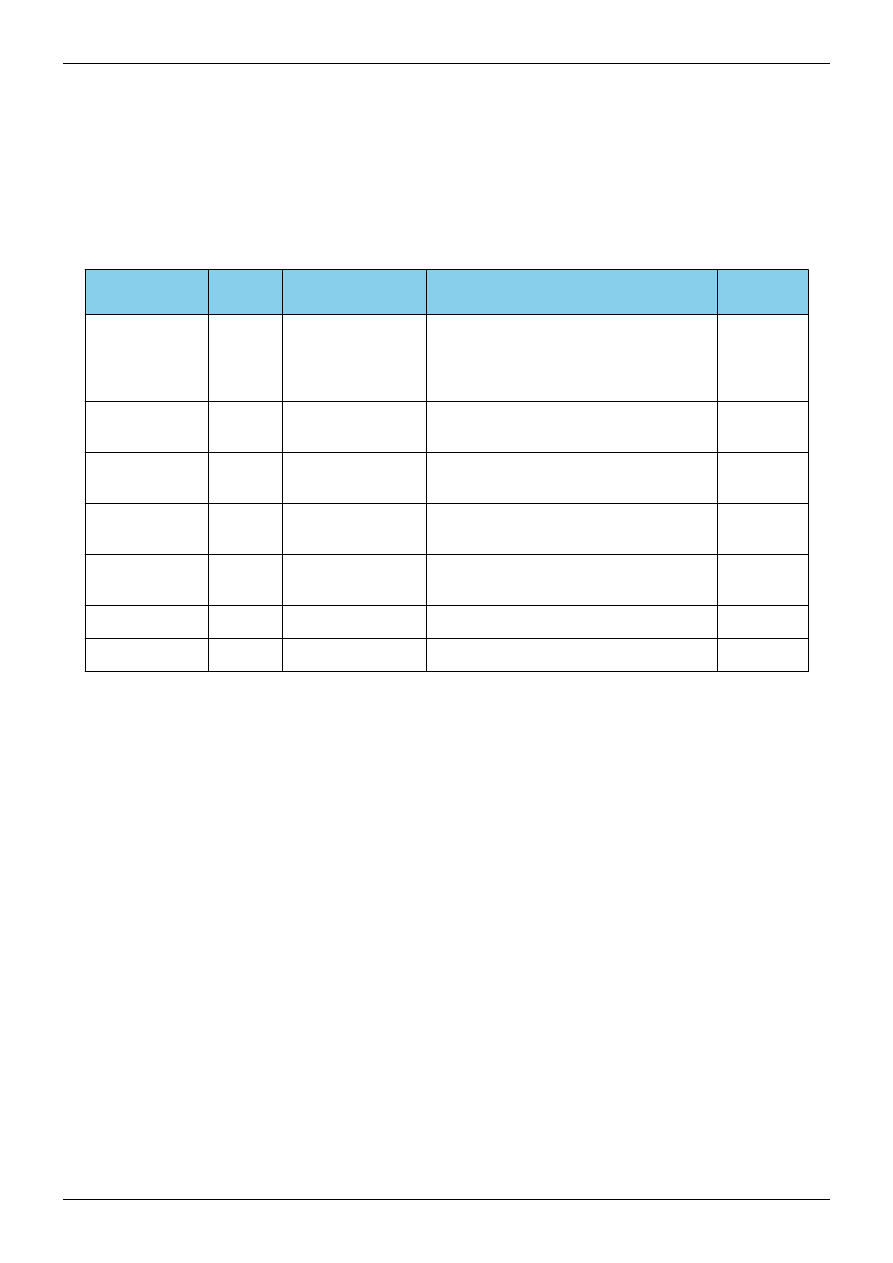
Częstość poszczególnych aberracji chromosomalnych u pacjentów z fenotypem zespołu
Turnera w populacji duńskiej według danych Danish Cytogenetic Central register (DCCR)
w latach 1970-2002
[15]
Kariotyp
Częstość w okresie prenatalnym Częstość w okresie postnatalnym
45,X
64%
47%
45,X/46,XX
22%
17%
45,X/46,XX, i(Xq);
46,X,i(Xq)
45,X/46,X,i(Xq)/47,X,i(Xq),i(Xq)
i inne warianty
4%
12%
45,X/46,X,del(X);
46,X,del(X)
7%
8%
45,X/46,XX/47,XXX;
45,X/47,XXX;
45,X/46,XX/47,XXX/48,XXXX
2%
5%
45,X/46,X,r(X)
<1%
6%
45,X/46,XY
3%
Inne warianty z
obecnością części chromosomu
Y
3%
Na górę strony
Zespół Turnera
4
Patofizjologia
Zmapowanie chromosomu X i poznanie części jego genów stworzyło możliwość powiązania niektórych cech
zespołu Turnera z zaburzeniem funkcji określonych genów. Okazało się również, że zmienność obrazu
cytogenetycznego przekłada się na zmienność fenotypu pacjentek z zespołem Turnera, co ma istotne znaczenie w
prognozowaniu przebiegu choroby.
Geny mające udział w patogenezie zespołu Turnera
Gen
Locus
Mechanizm
patogenetyczny
Cechy fenotypu
OMIM
SHOX (PHOG)
Xpter-p22.32
Niskorosłość i wady kończyn
(zniekształcenia kłykci przyśrodkowych, deformacja
Madelunga,
koślawość łokci)[27] [28]
OMIM 312865
ODG2 (BMP15)
Xp11.2
Haploinsuficjencja
Nieprawidłowy rozwój gonad
OMIM 300247
Putative lymphogenic
gene
Xp11.4-21.1
Haploinsuficjencja
Rozwój obrzęków limfatycznych i płetwistości szyi[31]
[32]
.
GBY
Y
?
Nowotworzenie w dysgenetycznych gonadach
(gonadoblastoma)[33]
OMIM 424500
VSPA
Xp22.33
Haploinsuficjencja
Ograniczenie zdolności neurokognitywnych[35]
OMIM 313000
STS
Xp22.3
Haploinsuficjencja
Ograniczenie zdolności neurokognitywnych[37]
NLGN4X
Xp22.3
Haploinsuficjencja
Ograniczenie zdolności neurokognitywnych[37]
ODG2 – ovarian dysgenesis 2
BMP 15 – bone morphogenetic protein 15
SHOX – short stature homeobox
VSPA – visuospatial/perceptual abilities gene
ODG2 – ovarian dysgenesis, hypergonadotropic, X linked
SOX3 – sex determining box 3 gene
GBY – gonadoblastoma locus on the Y chromosome
PHOG – pseudoautosomal homeobox-containing osteogenic gene
Na górę strony
Czynniki ryzyka
Czynniki ryzyka wystąpienia zespołu Turnera u dziecka są słabo poznane. Wiadomo, że częstość zespołu u dziecka
nie ma związku z wiekiem matki, jak w trisomii 21, 18 czy 13
[38]
. Niektórzy autorzy stwierdzili odwrotnie
proporcjonalną zależność między wiekiem matki a ryzykiem wystąpienia zespołu Turnera u dziecka
[39]
.
Sugerowano związek między alkoholizmem u ojca a wystąpieniem monosomii chromosmomu X u dziecka, ale
ostatnie badania nie potwierdziły tej hipotezy
[40]
.
Na górę strony

Zespół Turnera
5
Objawy i przebieg
Objawy w okresie prenatalnym
99% płodów z zespołem Turnera ulega spontanicznemu poronieniu. W badaniu USG w 13.-14. tygodniu ciąży i w
autopsji martwych płodów można stwierdzić obecność wodniaka w okolicy karkowej (hygroma nuchae); hipotezę
głoszącą, że przyczyną obrzęku jest zaburzenie rozwoju układu chłonnego tej okolicy
[41]
, potwierdziły badania
histologiczne
[42]
, immunologiczne
[43]
[44]
dowodzące hipoplazji naczyń chłonnych
odprowadzających chłonkę do żył szyjnych, a także do żył kończyn dolnych, miednicy i przestrzeni
zaotrzewnowej
[44]
.
Budowa ciała
Jedną z najważniejszych cech zespołu jest niski wzrost, występujący w blisko 100% przypadków zespołu
[15]
. Zespół
Turnera w krajach zachodnich jest najczęstszą, po konstytucyjnie niskim wzroście i rodzinnym niskim wzroście,
przyczyną niskorosłości u dziewczynek
[45]
. Średni ostateczny wzrost osiągany przez pacjentki z zespołem Turnera
wynosi 143 cm i jest o około 20 cm niższy niż przeciętna wzrostu kobiet w danej populacji
[20]
. Tempo wzrastania w
dzieciństwie jest wolniejsze i nie obserwuje się skoku pokwitaniowego; zwolnienie szybkości wzrostu można
stwierdzić już w 18. miesiącu życia
[15]
. Zakończenie wzrastania jest opóźnione i przypada na 20.-21. rok życia
[20]
.
Proporcje ciała chorych są nieprawidłowe: budowa ciała jest krępa, szyja jest krótka, a kończyny dolne skrócone w
stosunku do długości tułowia.
W zdecydowanej większości przypadków (80-90%)
[15]
pacjentki z zespołem Turnera mają dysgenetyczne gonady.
Mają one postać pasm łącznotkankowych długości 2-3 cm i szerokości około 0,5 cm. Nie pełnią one właściwej
jajnikom funkcji, stąd mówi się o pierwotnej niewydolności jajników (infantylizmie płciowym). Objawia się ona:
• opóźnionym dojrzewaniem płciowym, brakiem telarche i pubarche
• brakiem pierwszej miesiączki (menarche)
• pierwotną niepłodnością
• obniżonym stężeniem estrogenów
• podwyższonym poziomem gonadotropin: LH i FSH.
W części przypadków (10-20%)
[20]
; 40% wśród pacjentek z mozaicyzmem 45,X/46,XX
[45]
) czynność jajników jest
częściowo zachowana: występuje menarche, ale po kilku- kilkunastu miesiącach cykle zanikają. Rzadko kobiety z
zespołem Turnera mają zachowaną czynność jajników, prawidłowo wykształcają drugorzędowe cechy płciowe i są
płodne.
jest mała z drobną szyjką, a pochwa jest stosunkowo wąska. Może to, wspólnie ze zmianami atroficznymi śluzówki
wskutek niedoboru estrogenów, implikować dyspareunię
[45]
.
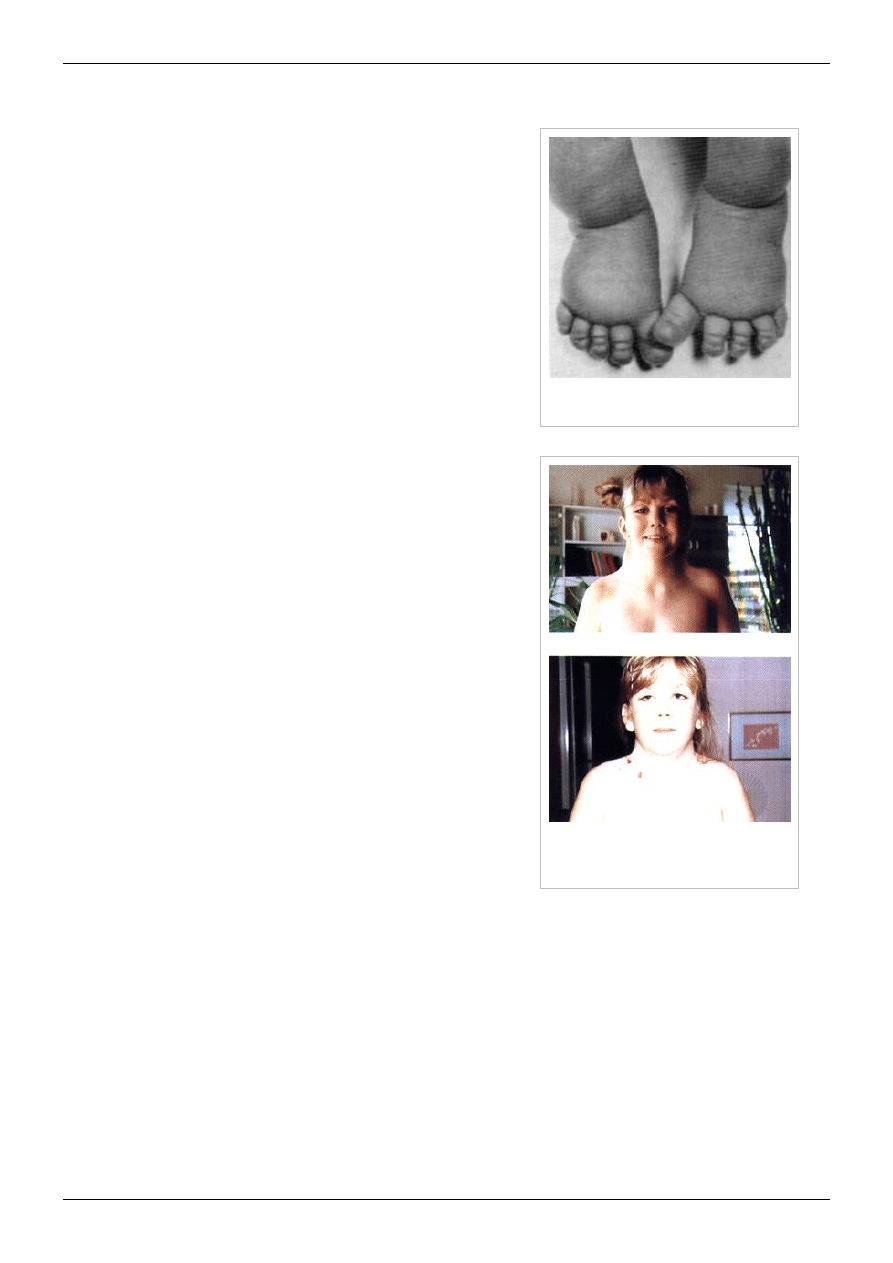
Zespół Turnera
6
Poduszeczkowaty obrzęk stóp u dziewczynki z
zespołem Turnera[1]
Pacjentka z zespołem Turnera przed operacją i
tuż po operacji plastycznej mającej na celu
skorygowanie płetwastości szyi[1]
W zespole Turnera spotyka się charakterystyczne cechy o charakterze
małych wad wrodzonych (anomalii, cech dysmorficznych); zwykle u
jednego pacjenta występuje kilka takich cech, nie spotyka się
natomiast wszystkich cech u jednej osoby. Te charakterystyczne cechy
fenotypowe to:
• poduszeczkowate obrzęki limfatyczne dłoni i stóp (ang. puffy feet)
w okresie noworodkowym i niemowlęcym (22%)
[46]
;
• nadmiar skóry na karku, płetwista szyja (25%)
[46]
[15]
;
• nisko osadzone, zrotowane ku tyłowi uszy, uszy dysplastyczne,
odstające (15%);
• uboga mimika;
• bogata oprawa oczu;
• opadanie powiek (ptoza) (10%)
[15]
;
• antymongoidalne ustawienie szpar powiekowych;
• zmarszczka nakątna (epicanthus) (20%)
[15]
;
• zez (15%
[15]
);
• wysokie, "gotyckie" podniebienie (38%)
[46]
;
• wady zgryzu;
[46]
[15]
;
• niska linia tylna włosów (42%)
[46]
;
• włosy "wełniste";
• nadmierne owłosienie (rzadko);
• krótka szyja (40%)
[15]
;
• szeroko rozstawione brodawki sutkowe (hiperteloryzm brodawek
sutkowych);
• wciągnięte brodawki sutkowe (łac. inversothelia) (5%)
[15]
;
• szeroka, puklerzowata klatka piersiowa (30%);
• dysplazja paznokci (13%)
[46]
;
• koślawość łokci (47%)
[46]
i kolan (35%)
[15]
;
• krótka czwarta kość śródręcza (37%)
[46]
;
[46]
;
• deformacja Madelunga (7%)
[46]
;
• objaw nadgarstkowy Kosowicza;
• objaw Kosowicza (pionowe wydłużenie wewnętrznego wyrostka kości udowej);
• objaw Archibalda (charakterystyczne zmiany w obrębie śródręcza);
• liczne znamiona barwnikowe (25%)
[46]
.
Wady narządów wewnętrznych
Wady układu krążenia
U około 23-40% chorych z zespołem Turnera występują wrodzone wady serca
[46]
. Większość z tych wad dotyczy
lewego serca; najczęstszą wadą jest dwupłatkowa zastawka aorty i koarktacja aorty. Stwierdzono ponadto trzykrotnie
większe ryzyko wystąpienie nadciśnienia tętniczego u chorych z zespołem Turnera
[47]
. Częstość poszczególnych
malformacji sercowo-naczyniowych określona na dużych grupach pacjentek z zespołem Turnera przedstawiona jest

Zespół Turnera
7
w tabeli.
Częstość malformacji sercowo-naczyniowych u pacjentek z zespołem Turnera
Wada
Częstość
6,9%[48]
10%[49]
12,5%[50]
14%[51]
Poszerzenie aorty wstępującej
2,9%[48]
6%[52]
12,5%[50]
Hipoplazja łuku aorty
2,5%[50]
Dwupłatkowa zastawka aorty
12,5%[48]
14%[49]
[51]
17,5%[50]
34%[52]
Wypadanie płatka zastawki mitralnej lub fala zwrotna przez zastawkę
0,6%[49]
2%[51]
5%[50]
6%[52]
8,9%[48]
Przerwanie ciągłości żyły głównej dolnej z jej kontynuacją drogą żyły nieparzystej
2,5%[50]
0,6%[49]
2,5%[50]
Stenoza i (lub) niewydolność zastawki aortalnej
3,2%[48]
6%[51]
11%[49]
Częściowy nieprawidłowy płucny spływ żylny
0,5%[51]
0,6%[49]
2,9%[48]
Ubytek przegrody międzykomorowej
0,5%[48]
Ubytek przegrody przedsionkowo-komorowej
0,2%[48]
5%[51]
Nieprawidłowości zastawki pnia płucnego (stenoza, fala zwrotna)
1%[49]
0,5%[51]
1%[49]

Zespół Turnera
8
Wady układu moczowego
W zespole Turnera częste (25-43% populacji) są wrodzone anomalie nerek: nerka podkowiasta (10-16%)
[46]
,
zdwojenie układu moczowego (5-11%)
[46]
, anomalie ułożenia nerek (6-8%)
[46]
, ektopia nerki (2,5-3,5%)
[46]
torbielowatość nerek, jednostronna agenezja nerki(2-5%
[46]
[53]
[54]
).
Rozwój umysłowy
Rozwój psychiczny jest zwykle prawidłowy. Opóźnienie umysłowe stwierdza się tylko u około 5% chorych
[20]
.
Nierzadkie są natomiast zaburzenia osobowości, instynktu poznawczego, rozwoju emocjonalnego. Jakość życia
chorych jest obniżona zwłaszcza w związku z ich niskorosłością; stwierdzono, że wprowadzenie leczenia hormonem
wzrostu znacznie poprawia komfort życia pacjentek
[38]
.
Współistniejące choroby
Genetyczny defekt związany z monosomią chromosomu X albo innymi aberracjami chromosomalnymi w zespole
Turnera przypuszczalnie sprzyja częstszemu występowaniu niektórych chorób.
Choroby tarczycy
W porównaniu ze zdrową populacją, pacjentki z zespołem Turnera częściej chorują na autoimmunologiczne choroby
tarczycy, zwłaszcza na zapalenie tarczycy Hashimoto. Niedoczynność tarczycy stwierdzono u 15-30% chorych
[45]
[55]
. Jeszcze większy odsetek chorych z zespołem Turnera ma podwyższone poziomy przeciwciał anty-TPO i
antytyreoglobulinowych w osoczu (35-75%). Stwierdzono korelację między występowaniem chorób z
autoimmunizacji a kariotypem z izochromosomem X
[56]
.
Nietolerancja glukozy
W grupie chorych jaką stanowią pacjentki z zespołem Turnera zapadalność na cukrzycę typu 2 jest 2 do 4 razy
większa niż w zdrowej populacji.
Choroby przewodu pokarmowego
W okresie noworodkowym i niemowlęcym u dziewczynek z zespołem Turnera częste są problemy z karmieniem,
refluks żołądkowo-przełykowy i niechęć do ssania, zarówno u dzieci karmionych piersią jak i butelką. Przypuszcza
się, że jest to związane z anatomicznymi różnicami w budowie części ustnej gardła oraz jej niedojrzałości
mięśniowej
[57]
. Kobiety z zespołem Turnera dwukrotnie częściej zapadają na wrzodziejące zapalenie jelita grubego
i chorobę Leśniowskiego-Crohna. Częstsze jest także występowanie teleangiektazji ściany jelita i celiakii. We
zdrowej populacji częstość choroby wynosi 0,55%
[58]
. Podwyższony poziom przeciwciał IgA skierowanych
przeciwko gliadynie i antygenom endomysium wystepują u większej grupy pacjentek
[45]
. Ryzyko wystąpienia
marskości wątroby u pacjentek z zespołem Turnera jest do 5 razy wyższe niż w ogóle populacji
[59]
. Przynajmniej w
części ma to związek z częstszym występowaniem pierwotnej marskości żółciowej u pacjentek z zespołem
Turnera
[60]
.
Dolegliwości otolaryngologiczne
Wrodzone anomalie twarzoczaszki i trąbki Eustachiusza upośledzają wentylację ucha środkowego i prawdopodobnie
ten mechanizm odpowiada za zwiększoną częstość zapaleń ucha środkowego u pacjentek z zespołem Turnera.
Istotne znaczenie może mieć opóźnienie wzrostu kości skroniowej u tych chorych
[61]
. Z wiekiem zwiększa się
częstość niedosłuchu i poważnego upośledzenia słuchu do głuchoty włącznie, sięgająca nawet 60%
[15]
. Ubytek
słuchu ma charakter czuciowy i manifestuje się obniżeniem progu czucia słuchowego; może wystąpić już w wieku 6
lat
[61]
. Często występują też problemy z mową, być może związane z malformacjami podniebienia i żuchwy.
Zespół Turnera
9
Osteoporoza
hormonem wzrostu. Nie stwierdzono zależności między kariotypem a częstością złamań kości, która niezależnie od
rodzaju aberracji jest stała i sięga nawet kilkudziesięciu procent
[20]
. Przyczyn redukcji masy kostnej i zmian
osteoporotycznych upatruje się w niskim stężeniu estrogenów, hormonu wzrostu i IGF1 w surowicy.
Otyłość
W zespole Turnera stwierdza się predyspozycję do otyłości, przypuszczalnie mającą związek z niedoborem
estrogenów.
Nowotwory
Nie wykazano częstszej zapadalności pacjentek z zespołem Turnera na raka sutka, jajnika i trzonu macicy. W
przypadku chorych z mozaicyzmem z obecnością chromosomu Y wyższe jest ryzyko zachorowania na nowotwory
złośliwe gonad (gonadoblastoma, dysgerminoma). Ryzyko szacuje się na 7 do 30%
[33]
. Sugerowano wprowadzenie
badań przesiewowych z użyciem cytometrii przepływowej lub metod hybrydyzacji DNA u wszystkich dziewczynek
z kariotypem 45,X w celu wykluczenia obecności linii komórkowych z obecnym chromosomem Y
[62]
. Spośród
innych nowotworów stwierdzono znacząco częstsze występowanie raka jelita grubego, co prawdopodobnie wynika z
niedoborów estrogenów; hormonalna terapia zastępcza redukuje ryzyko
[63]
.
Na górę strony
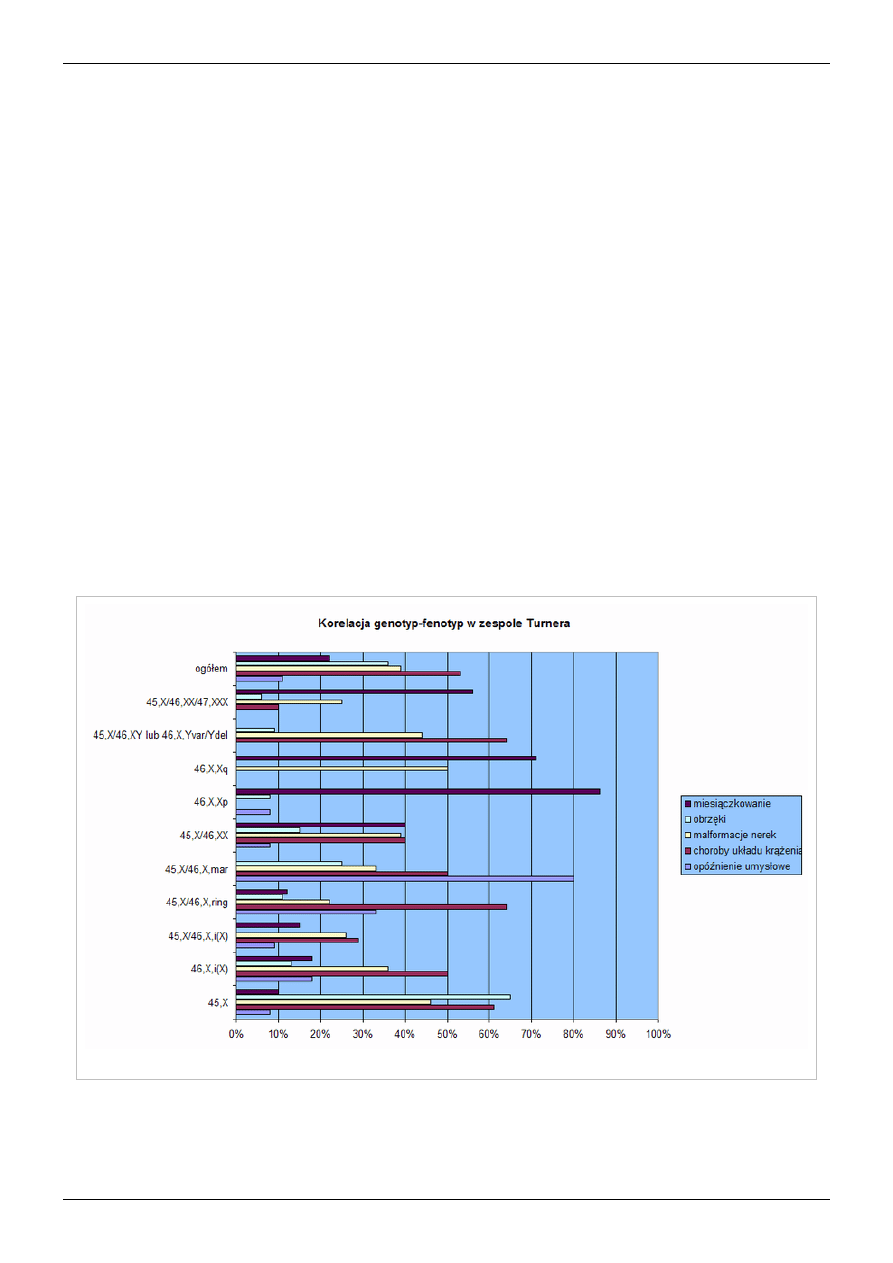
Korelacja genotyp-fenotyp
Częstość poszczególnych nieprawidłowości w spektrum zespołu Turnera; dane wg Sybert[45]
Kariotyp 45,X
Klinicznie u pacjentek z czystą monosomią chromosomu X cechy zespołu są nasilone. Stwierdza się niskorosłość,
nieprawidłowe proporcje ciała, deformacje układu kostnego, zaburzenia słuchu, dysgenezję gonad. Częste są wady

Zespół Turnera
10
serca, nerek i układu limfatycznego. Rozwój umysłowy jest zazwyczaj w normie
[45]
[15]
[20]
.
Kariotyp mozaikowy z linią komórkową zawierającą chromosom Y
W przypadku obecności chromosomu Y (kariotypy 45,X/46,XY, 45,X,Yvar lub 45,X,Ydel) znacznie zwiększone
jest ryzyko gonadoblastoma (10-30%) i nieco mniej dysgerminoma
[64]
. W przypadku stwierdzenia takiego
kariotypu w badaniu cytogenetycznym należy usunąć operacyjnie dysgenetyczne jajniki. Fenotyp jest zróżnicowany,
[15]
[20]
. Częste są malformacje sercowo-naczyniowe i nerek, rzadkie jest opóźnienie umysłowe.
Delecja krótkiego ramienia chromosomu X
W całkowitej delecji Xp fenotyp pacjentek jest zbliżony do fenotypu monosomii X. Stwierdza się niskorosłość,
wady układu kostnego, malformacje układu sercowo-naczyniowego, limfatycznego i nerek
[45]
. Może wystąpić
samoistne pokwitanie i miesiączkowanie, niekiedy kobiety z delecją Xp są płodne
[65]
.
Delecja długiego ramienia chromosomu X
U większości pacjentek stwierdza się pierwotny brak miesiączki, docelowy wzrost chorych w około połowie
przypadków jest w normie. Fenotyp tych pacjentek jest zmienny i często nie stwierdza się charakterystycznych dla
zespołu Turnera objawów.
Izochromosom długich ramion
Pacjentki z kariotypem 46,X,i(X) mają nietypowy dla zespołu Turnera fenotyp. Zdecydowanie większe jest u nich
ryzyko wystąpienia chorób autoimmunologicznych, przewlekłego limfocytarnego zapalenia tarczycy Hashimoto,
wrzodziejącego zapalenia jelita grubego lub choroby Crohna
. Stosunkowo rzadko mają malformacje układu
sercowo-naczyniowego i nerek, w większości przypadków rozwój umysłowy jest prawidłowy
[45]
.
Chromosom kolisty
W przypadku małych chromosomów kolistych (powstających z utratą znacznej części materiału genetycznego
chromosomu X; kariotyp 46,X,r(X)) rozwija się fenotyp podobny do tego u pacjentek z monosomią X, ale z bardzo
dużą częstością wad wrodzonych serca i nerek i częstym opóźnieniem umysłowym. Niekiedy występuje
spontaniczne menarche i płodność
[20]
.
Rozpoznanie
Dziewczynka z zespołem Turnera[1] . Dzieci z
zespołem Turnera rozwijają się początkowo
prawidłowo i często sprawiają wrażenie zupełnie
zdrowych, dopóki nie stwierdzi się u nich zbyt
wolnego tempa wzrastania. Dlatego właściwe
rozpoznanie może nastąpić późno.
W okresie prenatalnym
Rozpoznanie zespołu Turnera można postawić już w okresie
prenatalnym. W badaniu USG można stwierdzić obecność
uogólnionego obrzęku płodu, torbielowatego wodniaka szyi (cystic
hygroma), wrodzonych malformacji serca i (lub) układu moczowego.
Nieprawidłowy jest również wynik pomiaru przezierności fałdu
karkowego (ang. nuchal translucency, NT); wykazano, że wartość NT
jest wprost proporcjonalna do ryzyka wystąpienia zespołu Turnera u
dziecka
[40]
. Diagnostyczny jest także wynik tzw. testu potrójnego
(gonadotropina kosmówkowa, nieskoniugowany estriol,
alfa-fetoproteina). Testem potwierdzenia jest oznaczenie kariotypu
płodu. Prenatalne wykrycie zespołu Turnera u płodu nie zmienia
rokowania i dziecko zdiagnozowane po urodzeniu ma taką samą
.
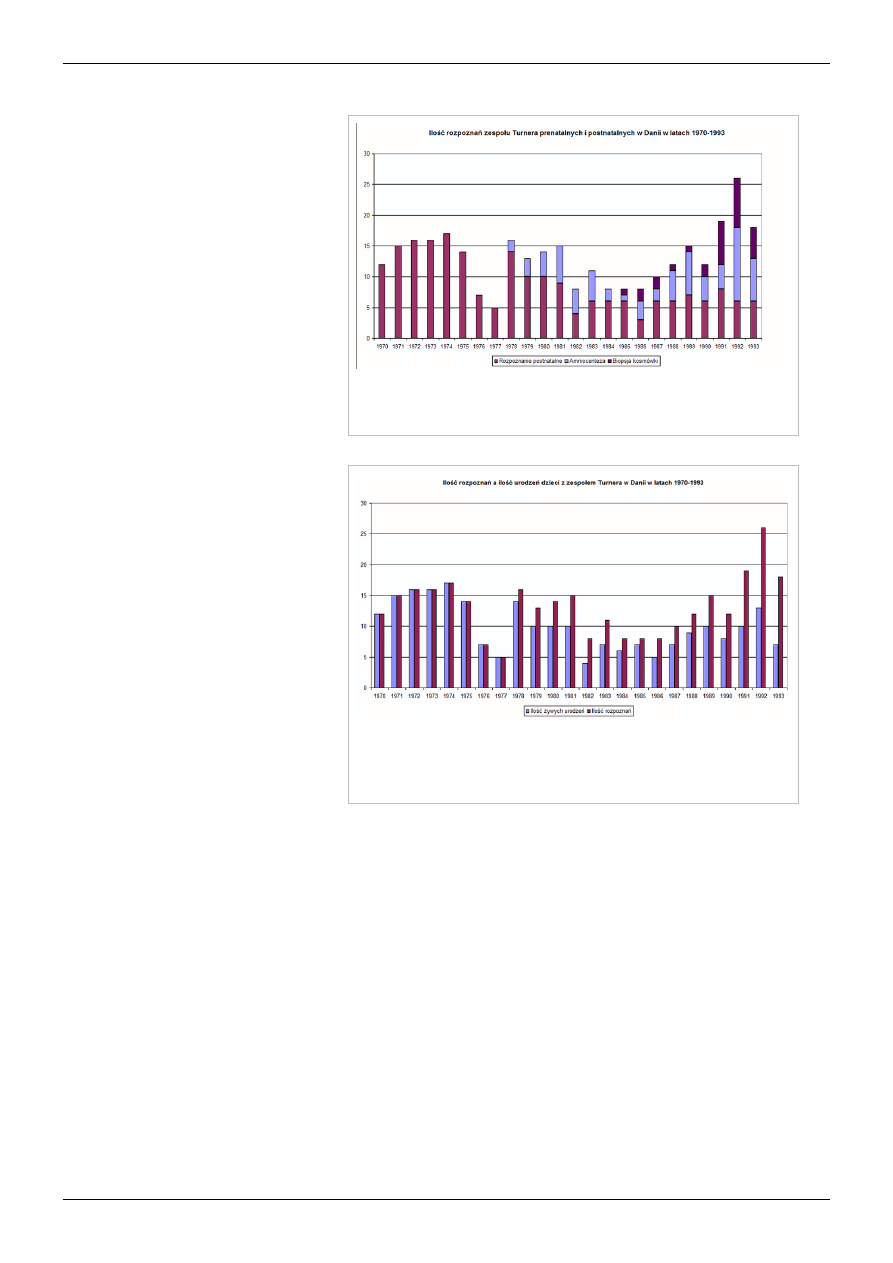
Zespół Turnera
11
Liczba rozpoznań zespołu Turnera prenatalnych i postnatalnych według danych Danish
Cytogenetic Central Register w latach 1970-1993. Widoczny wzrost ilości rozpoznań w
ostatnich latach, przypisywany upowszechnieniu badań prenatalnych[66] .
Liczba żywych urodzeń a liczba rozpoznań według danych Danish Cytogenetic Central
Register w latach 1970-1993. Rozbieżność wynika po części z faktu poronień części
płodów z zespołem Turnera, a także planowo wykonywanych zabiegów aborcji. W
ostatnich latach ilość zabiegów przerywania ciąży spadła[66]
W okresie postnatalnym
W okresie noworodkowym i
niemowlęcym rozpoznanie jest
stawiane najczęściej na podstawie
poduszeczkowatych obrzęków stóp i
dłoni oraz fałdu skóry karku,
pozostałości wodniaka szyi in utero.
Te charakterystyczne cechy pozwalają
na wykrycie zespołu Turnera w 1/5 do
1/3 przypadków
[45]
. Podkreśla się
fakt, że u każdej dziewczynki, u której
stwierdza się wrodzony obrzęk lub
koarktację aorty lub hipoplazję lewego
przedsionka serca powinno się
wykonać diagnostykę w kierunku
aberracji chromosomu X. Około 1/3
pacjentek diagnozuje się w
późniejszym dzieciństwie z powodu
niedoborów wzrostu. Pozostałe
zgłaszają się do lekarza z powodu
braku pokwitania lub niemożności
zajścia w ciążę. Każda kobieta,
szczególnie niska, z pierwotnym lub
wtórnym brakiem miesiączki, powinna
mieć wykonane badania w kierunku
zespołu Turnera
[45]
.
Wczesne rozpoznanie zespołu Turnera
ma znaczenie ze względu na
możliwość wczesnego wykrycia i
leczenia wad wrodzonych,
optymalizację czasu rozpoczęcia
terapii hormonalnej hormonem wzrostu i estrogenami, a także wczesne objęcie pacjentki i jej rodziny opieką lekarza
i psychologa. W Stanach Zjednoczonych średni wiek postawienia diagnozy u dzieci wynosił 7,7 roku ± 5,4 lat
[67]
.
W innym badaniu przeprowadzonym w Belgii mediana wieku pacjentek w momencie diagnozy wynosiła 6,6 lat;
pacjentki z kariotypem 45,X diagnozowano wcześniej
[68]
.
Na górę strony

Zespół Turnera
12
Leczenie
Leczenie przyczynowe w zespole Turnera jest niemożliwe, jednak odpowiednio wczesne wdrożenie leczenia
objawowego może jednak w znaczący sposób poprawić jakość życia chorych. Najważniejszymi kierunkami leczenia
pacjentek z rozpoznaną aberracją chromosomu X są:
• wykrycie i leczenie wad wrodzonych, zwłaszcza wad serca i układu moczowego;
• leczenie niskorosłości hormonem wzrostu;
• hormonalna terapia zastępcza indukująca dojrzewanie.
Terapię estrogenową u pacjentek z zespołem Turnera wprowadzono w USA już w latach 30. Badania nad
zastosowaniem rekombinowanego hormonu wzrostu w terapii niskorosłości w zespole Turnera zapoczątkowano w
1983 roku; FDA zaaprobowała tę terapię w 1997 roku
[69]
. Obecnie jest to standard leczenia i jako taki refundowany
jest przez państwo: obliczono, ze koszt terapii rekombinowanym hormonem wzrostu w przeliczeniu na 1 cm
oszacowanej różnicy wzrostu uzyskanej leczeniem wynosi 29 000 $
[70]
. W Polsce rozporządzeniem Ministra
Zdrowia wpisano leczenie zespołu Turnera hormonem wzrostu na listę wysokospecjalistycznych procedur
medycznych w 2000 roku
[71]
. Umożliwiło to objęcie sfinansowanym przez budżet państwa leczeniem w
wyspecjalizowanych placówkach większości pacjentek z zespołem Turnera.
Terapia hormonem wzrostu
Uważa się, że leczenie niskorosłości podawaniem hormonu wzrostu należy rozpocząć, gdy wartości wzrostu
pacjentki znajdą się poniżej 5. percentyla na siatce centylowej dla zdrowej populacji
[61]
. Leczenie powinien
prowadzić endokrynolog dziecięcy; nie ustalono, od kiedy można zacząć leczenie, są doniesienia o rozpoczęciu
leczenia już od 2. roku życia. U dziewczynek poniżej 9-12 roku życia leczenie prowadzi się samym hormonem
wzrostu w zalecanej dawce 0,05 mg/dzień, u starszych pacjentek w wieku powyżej 9-12 lat lub u tych, u których
leczenie wdrożono gdy ich wzrost mieścił się dużo poniżej 5 percentyla na siatce centylowej, zaleca się podawanie
hormonu wzrostu wraz z oxandrolonem
[61]
. Najnowsze badania wskazują, że w terapii promocji wzrostu łączenie
hormonu wzrostu i estrogenów nie daje korzystnego efektu
[72]
.
Indukcja dojrzewania
Terapię estrogenową należy wprowadzić w odpowiednim czasie, biorąc pod uwagę wiek w którym kobiety w
rodzinie pacjentki zwykle rozpoczynały pokwitanie. W celu wykluczenia opóźnionego dojrzewania płciowego
należy oznaczyć poziom gonadotropin. Terapię estrogenową należy także skoordynować z terapią hormonem
wzrostu, by zminimalizować niekorzystne działanie estrogenów na wzrost kości długich. W celu wywołania i
ustalenia fizjologicznych cyklów miesięcznych, po 12-24 miesiącach terapii estrogenowej podaje się hormon ciałka
żółtego w postaci medroksyprogesteronu.
Na górę strony
Opieka nad chorymi
Złożoność i różnorodność problemów klinicznych związanych z fenotypem zespołu Turnera jest powodem, dla
którego pacjentki powinny zostać objęte wszechstronną opieką specjalistów i poddawane regularnym badaniom
przesiewowym w kierunku chorób, których ryzyko jest u nich zwiększone. Najważniejsze kierunki profilaktyki
chorób w zespole Turnera to
[45]
:
• dokładne badanie fizykalne po postawieniu rozpoznania, mające na celu wykrycie ewentualnych wad
wrodzonych;
• echokardiografia i badanie kardiologiczne w celu wykluczenia wad wrodzonych układu krwionośnego,
powtarzane co 3-5 lat od momentu postawienia diagnozy; pomiar ciśnienia tętniczego przynajmniej raz w roku;

Zespół Turnera
13
• badanie USG nerek jednorazowo po postawieniu rozpoznania, wykluczające ewentualne wrodzone malformacje
układu moczowego;
• testy czynnościowe tarczycy, po postawieniu rozpoznania i ponownie, w okresie pokwitania i po osiągnięciu
dorosłości: poziom TSH oraz całkowitego i wolnego T4;
okresie pokwitania opcjonalnie;
• badanie okulistyczne po postawieniu rozpoznania;
• badanie poziomu lipidów w osoczu w życiu dorosłym;
• testy czynnościowe wątroby w życiu dorosłym;
• skrining w kierunku cukrzycy, opcjonalnie w zależności od objawów klinicznych;
• testy czynnościowe jajników, od momentu rozpoznania;
• badanie wskaźników wzrostu, do osiągnięcia ostatecznej wysokości ciała;
• ocena rozwoju psychoruchowego od postawienia diagnozy i ocena zdrowia psychicznego w życiu dorosłym;
• kontrola masy ciała, o ile istnieje taka potrzeba;
• u chorych z kariotypem wykazującym obecność fragmentu chromosomu Y w celu uniknięcia ryzyka rozwoju
wyrażą zgody na interwencję chirurgiczną, zaleca się przezpochwowe USG powtarzane w regularnych odstępach
czasu.
Na górę strony
Płodność i problemy planowania rodziny
Według ostatnich badań nad medycznymi i psychosocjologicznymi problemami kobiet z zespołem Turnera,
wykorzystanie technik wspomaganego rozrodu, w tym możliwości krioprezerwacji tkanki jajników i niedojrzałych
oocytów, jest jednym z ich głównych problemów życiowych
[73]
[74]
[75]
[76]
[77]
.
Bibliografia
• Gravholt, CH. Epidemiological, endocrine and metabolic features in Turner syndrome
. „European Journal of
Endocrinology”. 151, ss. 657–687 (2004). PMID 16544046.
• Sybert, VP, McCauley, E. Medical Progress: Turner's Syndrome. „New England Journal of Medicine”. 351, ss.
1227-1238 (2004).
• Jerzy Stachura, Wenancjusz Domagała: Patologia znaczy słowo o chorobie. Kraków: Wydawnictwo PAU, 2003.
ISBN 83-88857-65-7.
• Łącka, K. Zespół Turnera – korelacja pomiędzy kariotypem a fenotypem
. „Endokrynologia Polska”. 56. 6, ss.
986-983 (2005). PMID 6821224.
• Andrzej Szczeklik (red.): Choroby wewnętrzne, t. I. Kraków: Medycyna Praktyczna, 2005, s. 1132. ISBN
83-7430-069-8.
• Walenty Hartwig (red.): Endokrynologia kliniczna, tom I. Warszawa: Państwowy Zakład Wydawnictw
Lekarskich, 1978, ss. 916-920.
• Łącka, K. Zespół Turnera – korelacja pomiędzy kariotypem a fenotypem. „Endokrynologia Polska”. 56. 6, ss.
986-983 (2005). PMID 16821224.

Zespół Turnera
14
Linki zewnętrzne
• Strona Turner Syndrome Society
• Artykuł z eMedicine
(
)
• Zespół Turnera
na stronie Geneva Foundation for Medical Education and Research
(
• Artykuł z TheFetus.net
• Morgagni-Turner-Albright syndrome
w bazie Who Named It
Linki do stron polskich
• Wielkopolskie Stowarzyszenie Wsparcia w Zespole Turnera
• Stowarzyszenie Pomocy Chorym z Zespołem Turnera
Przeczytaj zastrzeżenia dotyczące pojęć medycznych i pokrewnych w Wikipedii!
Przypisy
[1] Johannes Nielsen, Rune W. Naeraa, members of Turner contact groups in Denmark: Turner's syndrome and Turner contact groups (http:/
HTM#baby). 1991. [dostęp 2 sierpnia 2007].
[2] National Institutes of Health: Clinical Features of Turner syndrome (http:/
html). 2004. [dostęp
2006-07-17].
[3] Walenty Hartwig (red.): Endokrynologia kliniczna, tom I. Warszawa: Państwowy Zakład Wydawnictw Lekarskich, 1978.
[4] Turner, HH. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. „Endocrinology (Baltimore)”. 23, ss. 566-574 (1938).
[5] XLVII. W: Giovanni Battista Morgagni: Epistola anatomica medica. 1768, ss. art. 20.
[6] Funke, O. Pterygium colli. „Dtsch Z Chir”. 63, ss. 162-167 (1902).
[7] Ullrich, O. Über typische Kombinationsbilder multipler Abartungen. „Zeitschrift für Kinderheilkunde (Berlin)”. 49, ss. 271 (1930).
[8] Bonnevie, K. Die vererbbaren Kopf- und Fußanomalien der Little und Baggschen Mäuserasse in ihrer embryologischen Bedingtheit.
„Molecular and General Genetics”. 62. 1, ss. 73-84 (1932).
[9] Schönenberg, H. Zur Diagnose und Differentialdiagnose des Status Bonnevie-Ullrich (http:/
pdf). „European Journal of Pediatrics”. 70. 4 (1951).
[10] Petersen, GA. Zur Kasuistik des „Status Bonnevie-Ullrich” (http:/
„European Journal of Pediatrics”. 61. 1 (1939).
[11] Freund, J. Zur Phätogenese des Status Bonnevie-Ullrich (http:/
pdf).
„European Journal of Pediatrics”. 77. 5 (1956).
[12] Uhlig, H. Klinische Betrachtungen zum Status Bonnevie-Ullrich (http:/
pdf). „European Journal of Pediatrics”. 72. 1 (1952).
[13] Ford, CE, Jones, KW, Polani, P, de Almeida, JC, Briggs, JH. A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner's
syndrome). „Lancet”. 1, ss. 711-713 (1959). PMID 13642858.
[14] Stratakis, CA, Rennert, OM. Turner syndrome: molecular and cytogenetics, dysmorphology, endocrine, and other clinical manifestations
and their management. „Endocrinologist”. 4, ss. 442–453 (1994).
[15] Gravholt CH. Epidemiological, endocrine and metabolic features in Turner syndrome (http:/
pdf). „European Journal of Endocrinology”. 151, ss. 657–687 (2004). PMID 16544046.
[16] Jacobs PA, Melville M, Ratcliffe S, Keay AJ, Syme J. A cytogenetic survey of 11,680 newborn infants. „Ann Hum Genet”. 37, ss. 359-76
(1974). PMID 4277977.
[17] Hook EB, Warburton D. The distribution of chromosomal genotypes associated with Turner's syndrome: livebirth prevalence rates and
evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. „Hum Genet”. 64,
ss. 24-27 (1983). PMID 6683706.
[18] Nielsen J, Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus,
Denmark. „Hum Genet”. 87, ss. 81-83 (1991). PMID 2037286.
[19] Prevalence of Turner syndrome in Japan. W: Imaizumi K, Kuroki Y: Basic and clinical approach to Turner syndrome. Hibi I, Takano K
(red.). Amsterdam: Excerpta Medica, 1993, ss. 3-6.
[20] Łącka, K. Zespół Turnera – korelacja pomiędzy kariotypem a fenotypem (http:/
VSID=3f73c405e7c3f55ca81767352b7a456d). „Endokrynologia Polska”. 56. 6, ss. 986-983 (2005).
PMID 16821224.

Zespół Turnera
15
[21] Wiśniewski, A, Romer, TE. Zespół Turnera. „IMED Standardy Medyczne”.
[22] Tabor A, Starzyk J, Schlegel-Zawadzka M. Interdisciplinary nature of the integrated care model for children with Turner Syndrome (http:/
pdf). Probl Hig Epidemiol 2006; 87 (4), 372-381
[23] Kelly, TE, Ferguson, JE, Golden, W. Survival of fetuses with 45,X: an instructive case and an hypothesis. „Am J Med Genet”. 42. 6, ss.
825-826 (1992). PMID 1554022.
[24] Held, KR, Kerber, S, Kaminsky, E, Singh, S, Goetz, P, Seemanova, E, Goedde, HW. Mosaicism in 45,X Turner syndrome: does survival in
early pregnancy depend on the presence of two sex chromosomes?. „Human Genetics”. 88, ss. 288–294 (1992). PMID 1733830.
[25] Fernandez, R, Mendez, J, Pasaro, E. Turner syndrome: a study of chromosomal mosaicism. „Human Genetics”. 98, ss. 29–35 (1996). PMID
8682502.
[26] Ogata, T. SHOX haploinsufficiency and its modifying factors. „J Pediatr Endocrinol Metab”. Suppl. 5, ss. 1289-94 (2002). PMID 12510982.
[27] Ellison, JW, Wardak, Z, Young, MF, Gehron, Robey, P, Laig-Webster, M, Chiong, W. PHOG, a candidate gene for involvement in the short
stature of Turner syndrome (http:/
1341). „Human Molecular Genetics”. 6. 8, ss. 1341-1347
(1997). PMID 9259282.
[28] Clement-Jones, M, Schiller, S, Rao, E, Blaschke, RJ, Zuniga, A, Zeller, R, Robson, SC, Binder, G, Glass, I, Strachan, T, Lindsay, S,
Rappold, GA. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome (http:/
695). „Human Molecular Genetics”. 9. 5, ss. 695-702 (2000). PMID 10749976.
[29] http:/
[30] http:/
[31] Bouchera, CA, Sargenta, CA, Ogatab, T, Affara, NA. Breakpoint analysis of Turner patients with partial Xp deletions: implications for the
lymphoedema gene location (http:/
591?ijkey=04e4f03f18075e00c92fca6ee6de73b92e0092d5&
keytype2=tf_ipsecsha). „J Med Genet”. 38, ss. 591-598 (2001). PMID 11546827.
[32] Berdahl, LD, Wenstrom, KD, Hanson, JW. Web neck anomaly and its association with congenital heart disease. „Am J Med Genet”. 56. 3,
ss. 304-307 (1995). PMID 7778596.
[33] Gravholt, CH, Fedder, J, Naeraa, RW, Müller, J, Fisker, S, Christiansen, JS. Occurrence of gonadoblastoma in females with Turner
syndrome and Y chromosome material: a population study (http:/
3199). „J Clin Endocrinol
Metab”. 85, ss. 3199-3202 (2000). PMID 10999808.
[34] http:/
[35] Ross, JL, Roeltgen, D, Kushner, H, Wei, F, Zinn, AR. The Turner syndrome-associated neurocognitive phenotype maps to distal Xp (http:/
html). „Am J Hum Genet”. 67. 3, ss. 672-681 (2000). PMID
10931762.
[36] http:/
[37] Zinn AR, Roeltgen D, Stefanatos G, Ramos P, Elder FF, Kushner H, Kowal K, Ross JL. A Turner syndrome neurocognitive phenotype maps
to Xp22.3 (http:/
24). „Behav Brain Funct”. 3. 24 (2007). PMID 17517138.
[38] Romer TE, Walczak M, Lewiński A, Roszkowska-Blaim M i wsp. Ogólnopolski program leczenia niedoboru wzrostu u dzieci i młodzieży
w następstwie somatotropinowej niedoczynności przysadki, zespołu Turnera i przewlekłej niewydolności nerek, przez zastosowanie hormonu
ogolnopolski_program_niedoborwzrostu.
doc). Opracowanie
przygotowane dla potrzeb Ministerstwa Zdrowia 20-27
[39] Ljunger, E, Cnattingius, S, Lundin, C, Annerén, G. Chromosomal anomalies in first-trimester miscarriages. „Acta Obstetricia et
Gynecologica Scandinavica”. 84. 11, ss. 1103–1107 (2005). PMID 16232180.
[40] Kagan-Krieger, S, Selby, P, Vohra, S, Koren, G. Paternal Alcohol Exposure and Turner Syndrome (http:/
613). „Alcohol & Alcoholism”. 37. 6, ss. 613–617 (2002). PMID 12414557.
[41] Van der Putte, SCJ. Lymphatic malformation in human fetuses. A study of fetuses with Turner's syndrome or status Bonnevie-Ullrich.
„Virchows Arch A Pathol Anat Histol”. 376, ss. 233-246 (1977). PMID 145719.
[42] ,Chitayat, D, Kalousek, DK, Bamforth, JS. Lymphatic abnormalities in fetuses with posterior cervical cystic hygroma. „Am J Med Gen”. 33,
ss. 352-356 (1989). PMID 2801770.
[43] von Kaisenberg, CS, Nicolaides, KH, Brand-Saberi, B. Lymphatic vessel hypoplasia in fetuses with Turner syndrome (http:/
823). „Human Reproduction”. 14. 3, ss. 823-826 (1999). PMID 10221720.
[44] Vittay, P, Bosze, P, Gaal, M, Laszlo, J. Lymph vessel defects in patients with ovarian dysgenesis. „Clin Gen”. 18. 1980. Ss. 387-391. PMID
7460375.
[45] Sybert, VP, McCauley, E. Medical Progress: Turner's Syndrome. „New England Journal of Medicine”. 351, ss. 1227-1238 (2004).
[46] Elsheikh, M, Dunger, DB, Conway, GS, Wass, JAH. Turner’s Syndrome in Adulthood (http:/
120). „Endocrine Reviews”. 23. 1, ss. 120-140 (2002). PMID 11844747.
[47] Nathwani, NC, Unwin, R, Brook, CG, Hindmarsh, PC. Blood pressure and Turner syndrome (http:/
x). „Clin Endocr”. 52, ss. 363-370 (2000). PMID 10718835.
[48] Mazzanti L, Cacciari E. Congenital heart disease in patients with Turner’s syndrome. Italian Study Group for Turner Syndrome (ISGTS).
„Journal of Pediatrics”. 133. 5, ss. 688–692 (1998). PMID 9821430.
[49] Gøtzsche CO, Krag Olsen B, Nielsen J, Sorensen KE & Kristensen, BO. Prevalence of cardiovascular malformations and association with
karyotypes in Turner’s syndrome. „Archives of Diseases in Childhood”. 71. 5, ss. 433–436 (1994). PMID 7826114.

Zespół Turnera
16
[50] Dawson FKL, Wright AM, Bakker B, Pitlick PT, Rosenfeld RG. Cardiovascular evaluation in Turner syndrome: utility of MR imaging.
„Australasian Radiology”. 36. 3, ss. 204–209 (1992). PMID 1445102.
[51] Sybert VP. Cardiovascular malformations and complications in Turner syndrome (http:/
e11). „Pediatrics”. 101. 1, ss. E11 (1998). PMID 9417175.
[52] Miller MJ, Geffner ME, Lippe BM, Itami RM, Kaplan SA, DiSessa TG, Isabel-Jones JB, Friedman WF. Echocardiography reveals a high
incidence of bicuspid aortic valve in Turner syndrome. „Journal of Pediatrics”. 102. 1, ss. 47–50 (1983). PMID 6848727.
[53] Lippe, B, Geffner, ME, Dietrich, RB, Boechat, MI, Kangarloo, H. Renal malformations in patients with turner syndrome: imaging in 141
patients. „Pediatrics”. 82, ss. 852-856 (1988). PMID 3054787.
[54] Flynn, MT, Ekstrom, L, DeArce, M, Costigan, C, Hoey, HM. Prevalence of renal malformation in Turner syndrome. „Pediatr Nephrol”. 10.
4, ss. 498-500 (1996). PMID 8865252.
[55] El-Mansoury, M, Bryman, I, Berntorp, K, Hanson, C, Wilhelmsen, L, Landin-Wilmhelsen, K. Hypothyroidism is common in turner
syndrome: results of a five-year follow-up (http:/
2131). „J Clin Endocrinol Metab”. 90. 4,
ss. 2131-2135 (2005). PMID 15623818.
[56] Elsheikh, M,Wass, JA, Conway, GS. Autoimmune thyroid syndrome in women with Turner's syndrome – the association with karyotype.
x). „Clin Endocrinol”. 55, ss. 223-226 (2001). PMID
11531929.
[57] Mathisen, B, Reilly, S, Skuse, D. Oral-motor dysfunction and feeding disorders of infants with Turner syndrome. „Dev Med Child Neurol”.
34, ss. 141-149 (1992). PMID 1733819.
[58] Bonamico, M, et al. Prevalence and Clinical Picture of Celiac Disease in Turner Syndrome (http:/
5495). „The Journal of Clinical Endocrinology & Metabolism”. 87. 12, ss. 5495-5498 (2002). PMID 12466343.
[59] Idilman R, De Maria N, Colantoni A, Kugelmas M, Van Thiel DH. Cirrhosis in Turner's syndrome: case report and literature review. „Eur J
Gastroenterol Hepatol”. 12. 6, ss. 707-709 (2000). PMID 10912494.
[60] Milkiewicz P, Heathcote J. Primary biliary cirrhosis in a patient with Turner syndrome. „Can J Gastroenterol”. 19. 10, ss. 631-3 (2005).
PMID 16247527.
[61] Saenger, P, Albertsson Wikland, K, Conway, GS, Davenport, M, Gravholt, CH, Hintz, R, Hovatta, O, Hultcrantz, M, Ladin-Wilhelmsen, K,
Lin, A, Lippe, B, Pasquino, AM, Ranke, RB, Rosenfeld, R, Silberbach, M. Recommendations for the Diagnosis and Management of Turner
Syndrome (http:/
3061). „The Journal of Clinical Endocrinology & Metabolism”. 86. 7, ss.
3061-3069 (2001). PMID 11443168.
[62] Alvarez-Nava, F, Soto, M, Sanchez, MA, Fernandez, E, Lanes, R. Molecular analysis in Turner syndrome (http:/
md5=91f5d9f50363bc896ec323c65e000b33). „J
Pediatr”. 142, ss. 336-340 (2003). PMID 12640385.
[63] Women’s Health Initiative. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the
Women’s Health Initiative randomized controlled trial. „JAMA”. 288, ss. 321–333 (2002). PMID 12117397.
[64] Page DCY. Chromosome sequences in Turner’s syndrome and risk of gonadoblastoma or virilisation. „Lancet”. 334, ss. 240-248 (1994).
[65] Fryns JP, Kleczkowska A, Petit P, van der Berghe H. Fertility in patients with X chromosome deletions. „Clin Genet”. 22, ss. 76-79 (1982).
[66] Gravholt CH, Juul S, Naeraa RW, Hansen J. Prenatal and postnatal prevalence of Turner's syndrome: a registry study (http:/
16). „BMJ”. 312. 7022, ss. 16-21 (1996). PMID 8555850.
[67] Savendahl, L, Davenport, ML. Delayed diagnoses of Turner’s syndrome: proposed guidelines for change. „Journal of Pediatrics”. 137, ss.
455–459 (2000). PMID 11035820.
[68] Massa, G et al. Trends in age at diagnosis of Turner syndrome (http:/
267?ijkey=592d9eb01b8f84f09925e86fce6c88f451fc91e6). „Archives of Disease in Childhood”. 90, ss. 267-268 (2005). PMID 15723912.
[69] Rosenfeld, FG, et al. Growth hormone therapy of Turner’s syndrome: beneficial effect on adult height. „J Pediatr”. 132, ss. 319-324 (1998).
PMID 9506648.
[70] Cave, CB, Bryant, J, Milne, R. Recombinant growth hormone in children and adolescents with Turner syndrome. „Cochrane Database Syst
Rev”. 3. CD003887 (2003).
[71] Rozporządzenie Ministra Zdrowia z dnia 21 grudnia 1999 roku zmieniające rozporządzenie w sprawie wykazu wysokospecjalistycznych
procedur medycznych finansowanych z budżetu państwa oraz zasad i trybu udzielania tych świadczeń. „Dziennik Ustaw”. Nr 106. Pozycja
1214.
[72] Johnston, DI, Betts, P, Dunger, D, Barnes, N, Swift, PG, Buckler, JM. A multicentre trial of recombinant growth hormone and low dose
oestrogen in Turner syndrome: near final height analysis (http:/
76?ijkey=f815fb739d94c5c64df068da77c7f80061de1d1a&
keytype2=tf_ipsecsha). „Arch Dis Child”. 84, ss. 76-81 (2001). PMID 11124794.
[73] Press F, Shapiro HM, Cowell CA, et al. Outcome of ovum donation in Turner`s syndrome patients. „Fertil Steril”. 64. 5, ss. 995-8 (1995).
PMID 7589649.
[74] Khastgir G, Abdalla H, Thomas A, Korea L, Latarche L, Studd J. Oocyte donation in Turner's syndrome: an analysis of the factors affecting
the outcome (http:/
279). „Hum Reprod”. 12. 2, ss. 279-85 (1997). PMID 9070711.
[75] Tarani L, Lampariello S, Raguso G, et al. Pregnancy in patients with Turner's syndrome: six new cases and review of literature. „Gynecol
Endocrinol”. 12. 2, ss. 83-7 (1998).

Zespół Turnera
17
[76] Foudila T, Soderstrom-Anttila V, Hovatta O. Turner's syndrome and pregnancies after oocyte donation (http:/
532). „Hum Reprod”. 14. 2, ss. 532-35 (1999). PMID 10100005.
[77] Abir R, Fisch B, Nahum R, et al. Turner's syndrome and fertility: current status and possible putative prospects. „Hum Reprod Update”. 7.
6, ss. 603-10 (2001).
[78] http:/
[79] http:/
VSID=3f73c405e7c3f55ca81767352b7a456d
[80] http:/
[81] http:/
[82] http:/
[83] http:/
[84] http:/
[85] http:/
[86] http:/
[87] http:/

Źródła i autorzy artykułu
18
Źródła i autorzy artykułu
Zespół Turnera Źródło: http://pl.wikipedia.org/w/index.php?oldid=20802134 Autorzy: ABX, BaQu, Bukaj, Buldożer, Chrumps, CiaPan, CommonsDelinker, Delimata, Dobromila, Drops44,
Filip em, Fraximus, Gluth, Gładka, Iksnigo, Kasei-jin, Kauczuk, Kiełek, Kocio, Kpjas, Krzysiek10, Leopold, Ludmiła Pilecka, Mario58, Masur, MatthiasGor, Miggawka, Monopol, Mpn,
Mroman, Mzopw, Ohtnim, PMG, Patrol110, Paweł ze Szczecina, Picus viridis, Polimerek, Pstan, Red 81, Seval, Slaweks, Staszek99, Stepa, Stok, Szwedzki, ToSter, Vuvar1, Woytekmm, 30
anonimowych edycji
Źródła, licencje i autorzy grafik
Plik:Caduceus.svg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Caduceus.svg Licencja: Public Domain Autorzy: User:Fadookie, User:Rama
Plik:45,X.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:45,X.jpg Licencja: GNU Free Documentation License Autorzy: Hämbörger, The cat
Plik:Karyotype isochromosomeX.JPG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Karyotype_isochromosomeX.JPG Licencja: Creative Commons Attribution 2.0 Autorzy:
Deadstar, Filip em
Plik:Chromosome X Etude Inactivation X.PNG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Chromosome_X_Etude_Inactivation_X.PNG Licencja: Public Domain Autorzy:
Original uploader was Mirmillon at fr.wikipedia
Plik:Puffy feet.JPG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Puffy_feet.JPG Licencja: Creative Commons Attribution 2.0 Autorzy: Filip em
Plik:Neck Turner.JPG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Neck_Turner.JPG Licencja: Creative Commons Attribution 2.0 Autorzy: Johannes Nielsen
Plik:Korelacja.PNG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Korelacja.PNG Licencja: Creative Commons Attribution 3.0 Autorzy: User:Filip em
Plik:Baby Turner.JPG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Baby_Turner.JPG Licencja: Creative Commons Attribution 2.0 Autorzy: Johannes Nielsen
Plik:Turner2.PNG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Turner2.PNG Licencja: Creative Commons Attribution-Sharealike 3.0 Autorzy: User:Filip em
Plik:Turner1.PNG Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Turner1.PNG Licencja: Creative Commons Attribution-Sharealike 3.0 Autorzy: User:Filip em
Plik:Star of life2.svg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Star_of_life2.svg Licencja: Attribution Autorzy: User:Verdy p
Licencja
Creative Commons Attribution-Share Alike 3.0 Unported
http:/
Document Outline
- Zespół Turnera
- Nazewnictwo
- Historia
- Epidemiologia
- Etiologia
- Patofizjologia
- Czynniki ryzyka
- Objawy i przebieg
- Korelacja genotyp-fenotyp
- Rozpoznanie
- Leczenie
- Opieka nad chorymi
- Płodność i problemy planowania rodziny
- Bibliografia
- Linki zewnętrzne
- Linki do stron polskich
- Licencja
Wyszukiwarka
Podobne podstrony:
Zespół Turnera, pliki zamawiane, edukacja
Biologia część V, Zespół Turnera choroba genetyczna
cw4 Zespół Turnera
Zespół Turnera, VI rok, Genetyka, Genetyka, Egzamin
Zespół Turnera
Zapalenia stawów i anomalie kostne w przebiegu zespołu Turnera, Ratownictwo medyczne, Ortopedia
ZESPOL TURNERA szkola
Zespol Turnera i Edukacja id 58 Nieznany
ZESPOL TURNERA
Zespół Turnera
Zespół Turnera i XXX
zespol Parsonagea Turnera, zespół Parsonage'a - Turnera
Zespół Downa i turnera(1)
Zespół Downa i Turnera
więcej podobnych podstron