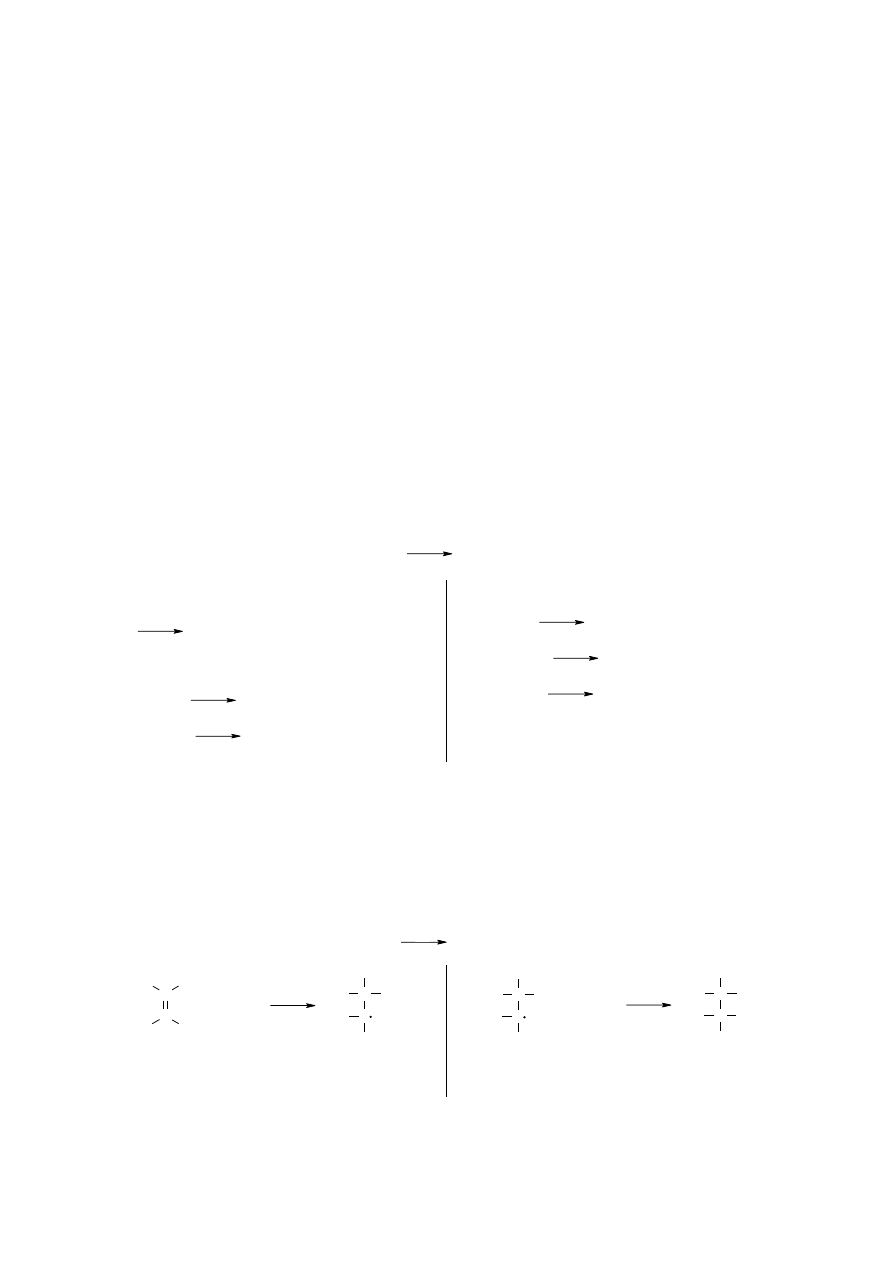
1
1. Mechanizm halogenowania jonowego i wolnorodnikowego.
W przypadku alkanów halogenowanie (Cl
2
, Br
2
) przebiega jako reakcja substytucji
wolnorodnikowej, a do wytworzenia wolnych rodników zastosowanie ma światło widzialne
lub nadfioletowe. W wyniku reakcji substytucji rodnikowej otrzymuje się zazwyczaj
mieszaninę izomerów, w ogólności, najłatwiej podstawione zostają wodory przy węglach
trzeciorzędowych (CH), później drugorzędowych (CH
2
) a najmniej reaktywne są wodory grup
węgli pierwszorzędowych (CH
3
). Reakcja ta jest mało użyteczna dla celów preparatywnych
dlatego, ponieważ podstawienie zachodzi właściwie na każdym atomie węgla, a ponadto
zawsze powstają produkty dwu- i wielohalogenowe, nawet gdy zastosuje się duży nadmiar
substratu w stosunku do halogenu. Dlatego w wyniku halogenowania metanu otrzymujemy
zawsze chloro-, dichloro- trichloro- i tetrachlorometan w ilościach, które zależą od stosunku
molowego substratów, ale powstają zawsze wszystkie cztery produkty.
Substytucja wolnorodnikowa na przykładzie halogenowania metanu
Cl
2
CH
4
CH
3
Cl
+
+
HCl
hv
Etap I: inicjowanie reakcji łaocuchowej:
Cl
2
Cl*
hv
2
Etap II: propagacja reakcji łaocuchowej:
Cl*
CH
4
CH
3
*
Cl
H
+
+
Cl
2
CH
3
*
CH
3
Cl
Cl*
+
+
Etap 4: terminacja reakcji łaocuchowej
Cl*
Cl*
Cl
2
+
CH
3
*
CH
3
*
C
2
H
6
+
Cl*
CH
3
*
CH
3
Cl
+
Halogenowanie rodnikowe w obecności hv zachodzi również w przypadku addycji do
wielokrotnego wiązania, przy czym przyłączenie zachodzi niezgodnie z regułą
Markownikowa.
Mechanizm:
Br
H
H*
Br*
+
hv
C
C
H
H
C
H
3
H
Br*
C
C
H
Br
H
H
CH
3
+
3
o
C
C
H
H
H
H
CH
3
H*
C
C
H
Br
H
H
CH
3
H
+
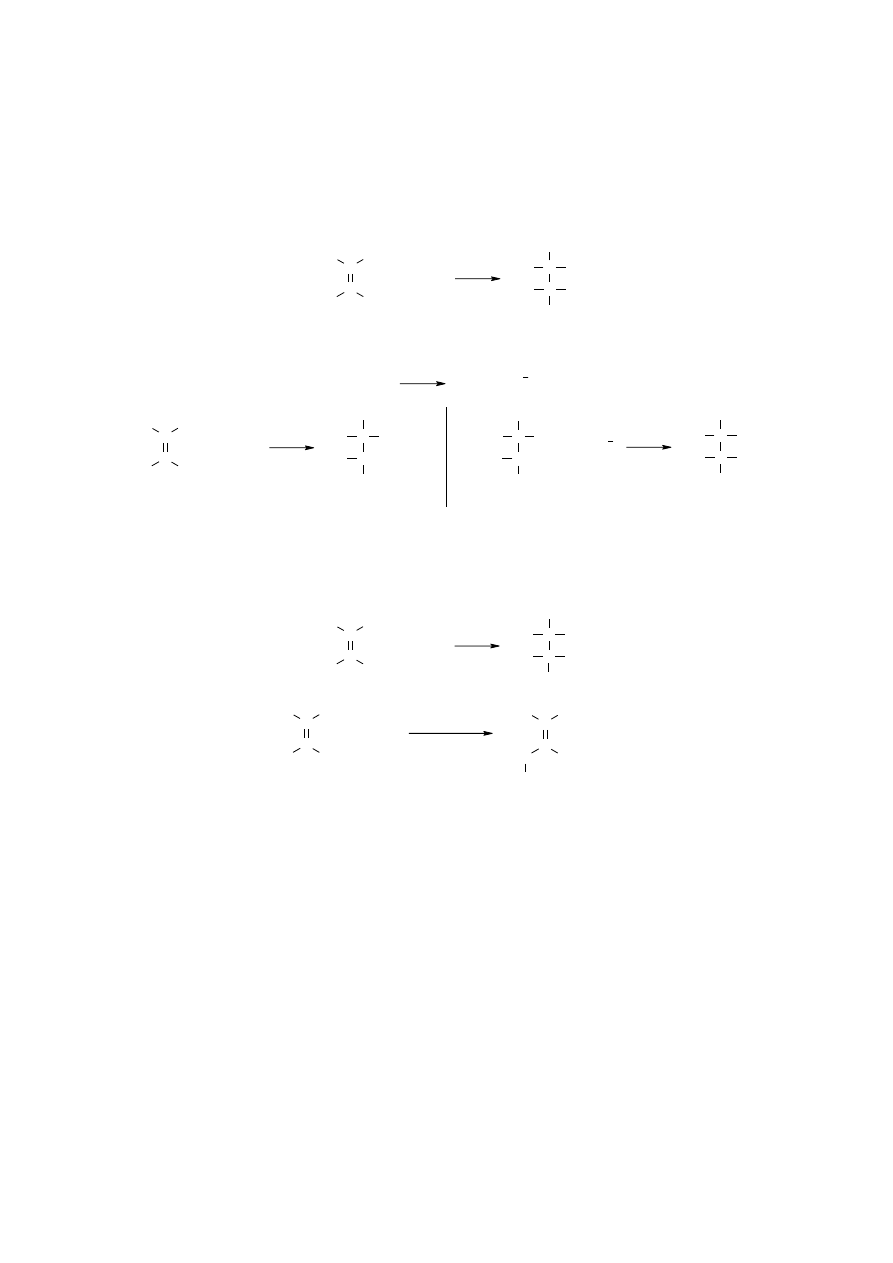
2
Halogenowanie jonowe zachodzi w przypadku addycji do wielokrotnego wiązania.
Addycja halogenowodorów do podwójnego wiązania zachodzi dośd łatwo, a jako produkt
powstaje odpowiedni halogenek alkilu. Reakcja zachodzi zazwyczaj zgodnie z regułą
C
C
H
H
C
H
3
H
Br
H
C
C
H
H
H
H
Br
CH
3
+
Mechanizm:
Br
H
H
+
Br
+
C
C
H
H
C
H
3
H
H
+
C
C
+
H
H
H
H
CH
3
+
3
o
C
C
+
H
H
H
H
CH
3
Br
C
C
H
H
H
H
CH
3
Br
+
Reakcja z chlorowcem prowadzona w wysokiej temperaturze w fazie gazowej całkowicie zmienia
swój mechanizm i zamiast reakcji addycji (przyłączenia) mamy reakcje substytucji (podstawienia).
C
C
H
H
C
H
3
H
Br
2
C
C
H
Br
H
H
Br
CH
3
+
C
C
H
H
C
H
3
H
Br
2
C
C
H
H
C
H
2
H
Br
Br
H
+
+
t=500
o
C
faza gazowa
2. Katalizatory elektrofilowego podstawienia chlorowca do pierścienia aromatycznego –
pojęcie kwasów i zasad wg Lewisa
Wg definicji Lewisa, atom, cząsteczkę lub jon dostarczający parę elektronową (donor)
nazywamy zasadą, zaś atom, cząsteczkę lub jon przyjmujący parę elektronową (akceptor)
nazywamy kwasem. Zgodnie z definicją Lewisa katalizatory stosowane w halogenowaniu – AlCl
3
,
FeCl
3
, są kwasami Lewisa posiadającymi deficyt elektronów.
3. Środki chlorowcujące
Jak już wspomniano halogenowanie można przeprowadzid za pomocą chloru i bromu.
Ponieważ brom jest mniej reaktywny niż chlor w przypadku bromowania można spodziewad się
większej selektywności i jest to zgodne z doświadczeniem. Fluor stosuje się rzadko, ponieważ jest
on zbyt reaktywny i reakcje są trudne do kontrolowania. Fluor często rozrywa łaocuchy węglowe
3
na mniejsze jednostki. Do halogenowania można zastosowad również jod. Reakcję tę stosuje się
rzadko, przede wszystkim dlatego, że wytwarzający się HI redukuje jodki alkilowe.
Najpopularniejszym środkiem chlorującym jest chlorek sulfurylu. SO
2
Cl
2
. Jest dogodniejszy
w użyciu i reaguje szybciej niż sam chlor. Inne stosowane reagenty to: N-bromosukcynimid (NBS),
CCl
4
, chlorek oksalilu, BrCCl
3
, PCl
5
, fosgen, podbromin t-butylu, podchloryn t-butylu /(CH
3
)
3
COCl/.
We wszystkich przypadkach do zapoczątkowania reakcji łaocuchowej wymagany jest katalizator,
zwykle nadtlenek lub światło nadfioletowe.
Źródłem bromu jest szybka reakcja jonowa pomiędzy NBS a HBr.
N
Br
O
O
Br
H
N
H
O
O
Br
2
NBS
+
+
chlorek oksalilu
WODA BROMOWA
Nasycony roztwór wodny bromu; ma kolor jasnobrązowy (ze względu na barwę bromu –
brązowobrunatnej cieczy). Wytrząsana z ciekłym węglowodorem nienasyconym odbarwia się,
gdyż brom zostaje przyłączony (addycja) do wiązao wielokrotnych.
4. Wpływ skierowujący chlorowca i grupy alkilowej w pierścieniu aromatycznym – efekt
indukcyjny i mezomeryczny
Podstawniki należące do grupy III – halogeny kierują elektrofil w położenie orto- lub para-.
Jest to wynik efektu +M, który zwiększa ładunek ujemny w tych pozycjach, czyniąc je podatne na
atak odczynnika elektrofilowego.
Natomiast efekt indukcyjny halogenów -I powoduje obniżenie szybkości reakcji S
E
halogenoarenów, wobec czego halogeny dezaktywują pierścieo na podstawienie elektrofilowe.
Warto zwrócid uwagę, iż największy dezaktywujący wpływ ma fluor i że ten wpływ słabnie wraz z
odległością – silniejszy jest w pozycji orto- niż w para-.
Podstawniki należące do grupy I – grupy alkilowe kierują elektrofil w położenie orto- lub
para-. Jest to wynik występowania dodatniego efektu mezomerycznego +M i indukcyjnego +I.
Podstawniki aktywujące powodują przesunięcie ładunku ujemnego na pierścieo aromatyczny, przy
czym bardziej ujemne bieguny elektryczne pojawiają się właśnie w położeniach orto- i para-.

4
Podstawniki elektronodonorowe (EDG): Podstawniki elektronoakceptorowe (EWG):
-C N
..
-NH
2
, -NHR, -NR
2
..
..
-OH, -OR, -O
..
..
..
..
..
.. :
-
-SH, -SR, -S
..
..
..
..
..
..:
-
-Alk, -Ar
podstawniki
grupy I
wykazują efekt
+I lub +M/+I
lub +M/-I
-NHCOR
..
+
+
-NO
2
,
-CHO, -COR, -COOH, -COOR
-SO
3
H, -SO
2
OR, -SO
2
R
-CF
3
, -CCl
3
, -NR
3
, -SR
2
podstawniki
grupy II
wykazują efekt
-I lub -M/-I
Podstawniki elektrodonorowo-elektroakceptorowe (EDG/EAG):
-F , -Cl , -Br , -I
..
..
..
..
..
..
..
..
:
:
:
:
podstawniki grupy III wykazują efekt -I i +M
Efekt indukcyjny − w chemii wpływ grup funkcyjnych i ogólnie wszelkich podstawników (w
cząsteczkach związków organicznych), bardziej lub mniej elektroujemnego od wodoru, na rozkład
gęstości
elektronowej
w
sąsiedztwie
tego
podstawnika
lub
nawet
w całej cząsteczce na skutek nakładania się ich orbitali σ lub s z orbitalami s lub σ atomów
do których są one przyłączone. Efekty indukcyjne kilku podstawników są kumulowane.
Efekt indukcyjny jest dobrze przenoszony przez układ pierścienia aromatycznego, szybko
natomiast słabnie w łaocuchu węglowym o wiązaniach pojedynczych.
Efekt mezomeryczny zwany też efektem rezonansowym to w chemii zdolnośd grup
funkcyjnych i ogólnie wszelkich podstawników do zmniejszania lub zwiększania reaktywności
związków chemicznych na skutek nakładania się ich orbitali π lub p z orbitalami p lub π atomów do
których są one przyłączone.
Zmniejszanie lub zwiększanie reaktywności związków, w których występuje efekt
mezomeryczny jest związane z wpływem na gęstośd elektronową występującą na atomie do
którego jest przyłączona pojedynczym wiązaniem chemicznym dana grupa funkcyjna.
Efekt mezomeryczny może byd dodatni (+M) lub ujemny (-M). Ujemny efekt ma miejsce
wtedy, gdy podstawnik powoduje "wyciąganie" elektronów z reszty cząsteczki, czyli powoduje
spadek gęstości elektronowej na atomie do którego jest przyłączony. Dodatni efekt ma miejsce
wtedy, gdy podstawnik "dostarcza" elektrony do reszty cząsteczki. Zarówno dodatni jak i ujemny
efekt mezomeryczny może powodowad zmniejszanie lub zwiększanie reaktywności związków
chemicznych.
5
5. Halogenowanie związków aromatycznych – wpływ katalizatorów na mechanizm i miejsce
wprowadzenia chlorowca do związku aromatycznego
W arenach, pod wpływem halogenów (Cl
2
i Br
2
), w obecności kwasów Lewisa (np. FeX
3
)
dochodzi do wymiany atomu wodoru na atom halogenu. W tym przypadku dochodzi do reakcji
substytucji, a nie addycji. Obie te reakcje łatwo rozróżnid po zewnętrznych efektach – w obu zanika
barwa bromu, jednak w trakcie substytucji wydziela się gazowy bromowodór (jest to widoczną
oznaką reakcji), a w addycji cały brom zostaje przyłączony do wielokrotnego wiązania.
Br
Br Br
Br
Br
1,2-dibromocykloheksa-3,5-dien
+
+ HBr
FeBr
3
benzen
bromobenzen
gaz
Reakcja biegnie wg mechanizmu S
E
. Cząsteczka bromu w kontakcie z kwasem Lewisa ulega
polaryzacji i dodatnio naładowany atom bromu staje się silnym elektrofilem. Br
+
przyłącza się do
pierścienia benzenu i powstaje mezomerycznie stabilizowany karbokation, który odszczepiając
proton przekształca się w bromobenzen (bromek fenylu).
Br
Br
+
Br
+ FeBr
3
..
..
..
..
:
:
Br
FeBr
3
..
..
..
..
:
-
brom - słaby elektrofil brom spolaryzowany, atom z ładunkiem dodatnim
staje się silnym elektrofilem
H
Br
H
Br
H
Br
Br
Br
+
+
+
+
benzen
bromobenzen
(72%)
- H
+
Duża energia sprzężenia elektronów π (energia rezonansu) jest przyczyną powrotu
produktu addycji kationu bromkowego do układu aromatycznego poprzez odszczepienie protonu.
Podobnie jak bromowanie biegnie reakcja chlorowania arenów. Jodowanie w takich
samych warunkach jest reakcją odwracalną i do przesunięcia równowagi na prawo potrzebny jest
utleniacz utleniający wydzielający się jodowodór. Utleniaczami stosowanymi do tego celu są
nadtlenek wodoru, kwas azotowy (V) lub sole miedzi (II).
I
benzen
jodobenzen
+ I
2
2
HNO
3
2
- HOH
(87%)
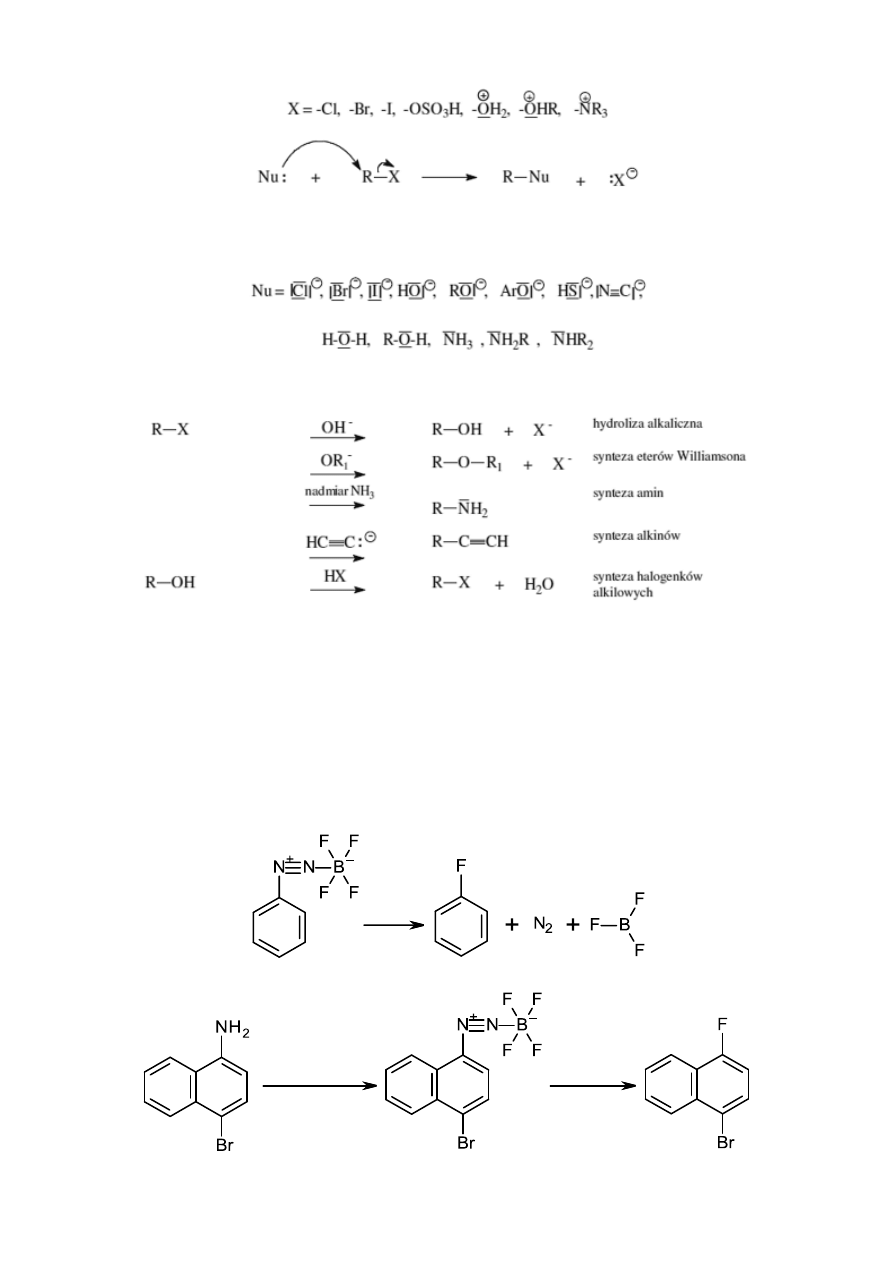
6. Nukleofilowe wprowadzenie chlorowca do związku aromatycznego
Reakcja podstawienia nukleofilowego polega na wymianie grupy X związanej
z atomem węgla na odczynnik nukleofilowy Nu. Podstawnikiem X jest przeważnie grupa
elektronoakceptorowa, która polaryzuje wiązanie C–X a następnie odchodzi z parą elektronową.
6
Odczynnikami nukleofilowymi są obojętne cząsteczki z wolnymi parami elektronowymi
parami elektronowymi lub aniony, dla przykładu:
Reakcje substytucji nukleofilowej:
7. Otrzymywanie związków fluoroaromatycznych
Fluorki arylowe można otrzymywad w reakcji Schiemanna /termiczny rozkład stałych
fluoroboranów diazoniowych/. Do roztworu soli diazoniowej wprowadza się kwas fluor oborowy,
w wyniku czego wytrąca się odpowiedni fluoroboran diazoniowy. Każdy fluoroboran ma
charakterystyczną temperaturę rozkładu, niewiele wyższą od jego temperatury topnienia i
niekontrolowany rozkład może doprowadzid do wybuchu. Reakcja przebiega wg mechanizmu S
N
1.
1. NaNO
2
, H
2
SO
4
2. HBF
4
150 - 155
o
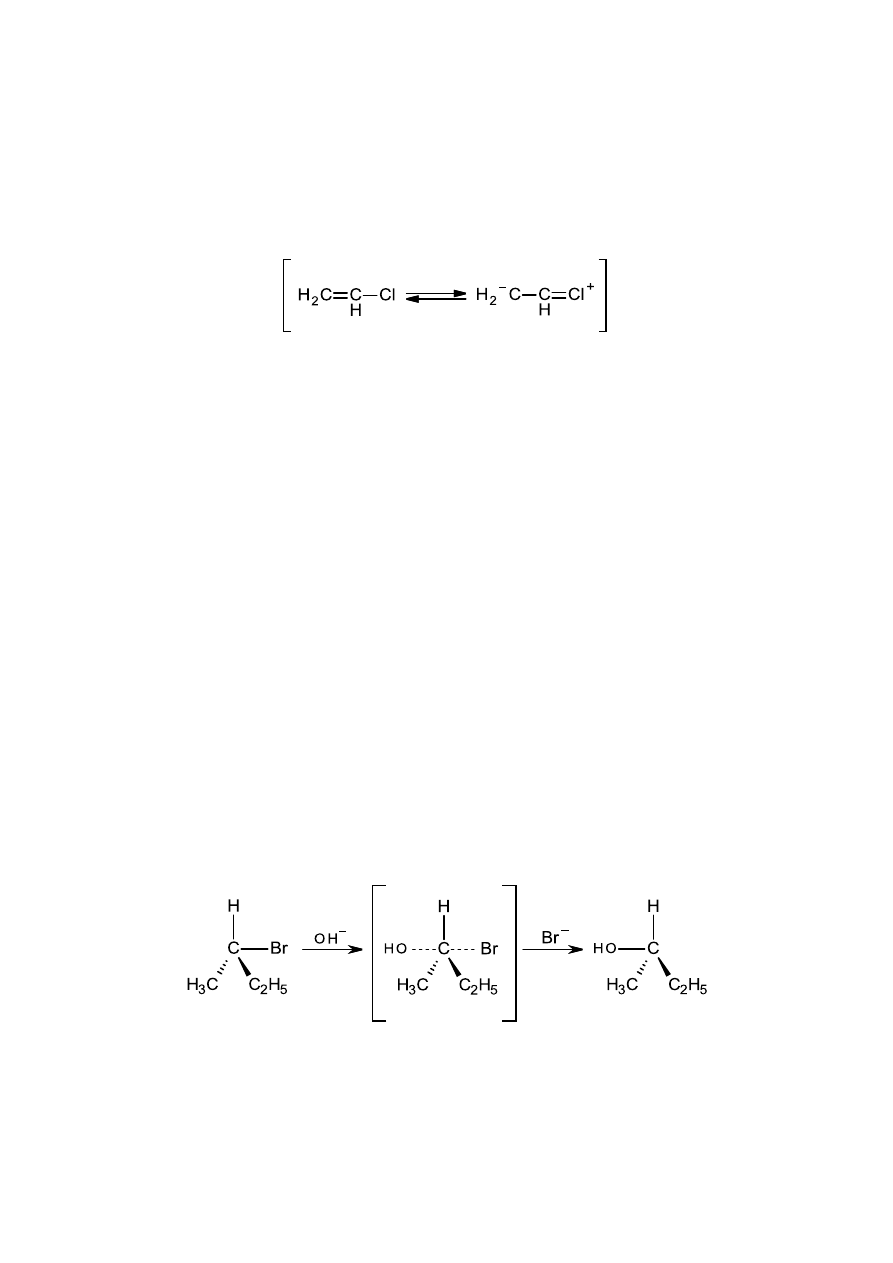
7
8. Reaktywnośd chloru w chlorku etylu, chlorku winylu i chlorku allilu.
Najmniej reaktywnym związkiem posiadającym atom chloru jest chlorek winylu – biernośd
w reakcjach SN1 i SN2. Powodem jego małej reaktywności jest stosunkowo silne wiązanie węgiel-
chlorowiec. Wiązanie węgiel-chlor nie wykazuje właściwości typowych dla wiązao pojedynczego
ani podwójnego. Przedstawiają to struktury rezonansowe:
W przeciwieostwie do chlorku winylu, chlorek allilu jest bardzo reaktywny zarówno
w reakcji SN1 i SN2. Chlorek allilu oraz inne pochodne allilowe wykazują niezwykłą reaktywnośd,
ponieważ obecnośd podwójnego wiązania wpływa aktywująco w kierunku rozerwania wiązania
z grupą funkcyjną.
9. Wymiana chlorowca w wyniku reakcji solwolizy – mechanizm reakcji SN1, SN2, E1, E2.
Substytucja nukleofilowa – reakcja chemiczna podstawienia, w której czynnikiem
atakującym jest nukleofil.
W zależności od mechanizmu przebiegu tej reakcji wyróżnia się dwa zasadnicze jej rodzaje:
a) substytucję jednocząsteczkową (S
N
1) - w etapie limitującym szybkośd reakcji reaguje jedna
cząsteczka: następuje odejście grupy odchodzącej i powstaje nietrwały produkt
przejściowy, zwykle kation; w następnym etapie (znacznie szybszym niż poprzedni) produkt
przejściowy łączy się z nukleofilem
b) substytucję dwucząsteczkową (S
N
2) - w etapie limitującym szybkośd reakcji następuje
jednoczesne przyłączenie nukleofila i odszczepienie grupy opuszczającej.
Substytucja nukleofilowa S
N
2 – mechanizm dwucząsteczkowy
Reakcja S
N
2 jest reakcją stereospecyficzną. W reakcji stereospecyficznej stereizomeryczne
substraty prowadzą do stereoizomerycznych produktów – inwersja konfiguracji absolutnej.
Reaktywnośd w reakcji S
N
2 zależy od rodzaju grupy opuszczającej, reaktywności nukleofila
oraz struktury substratu.
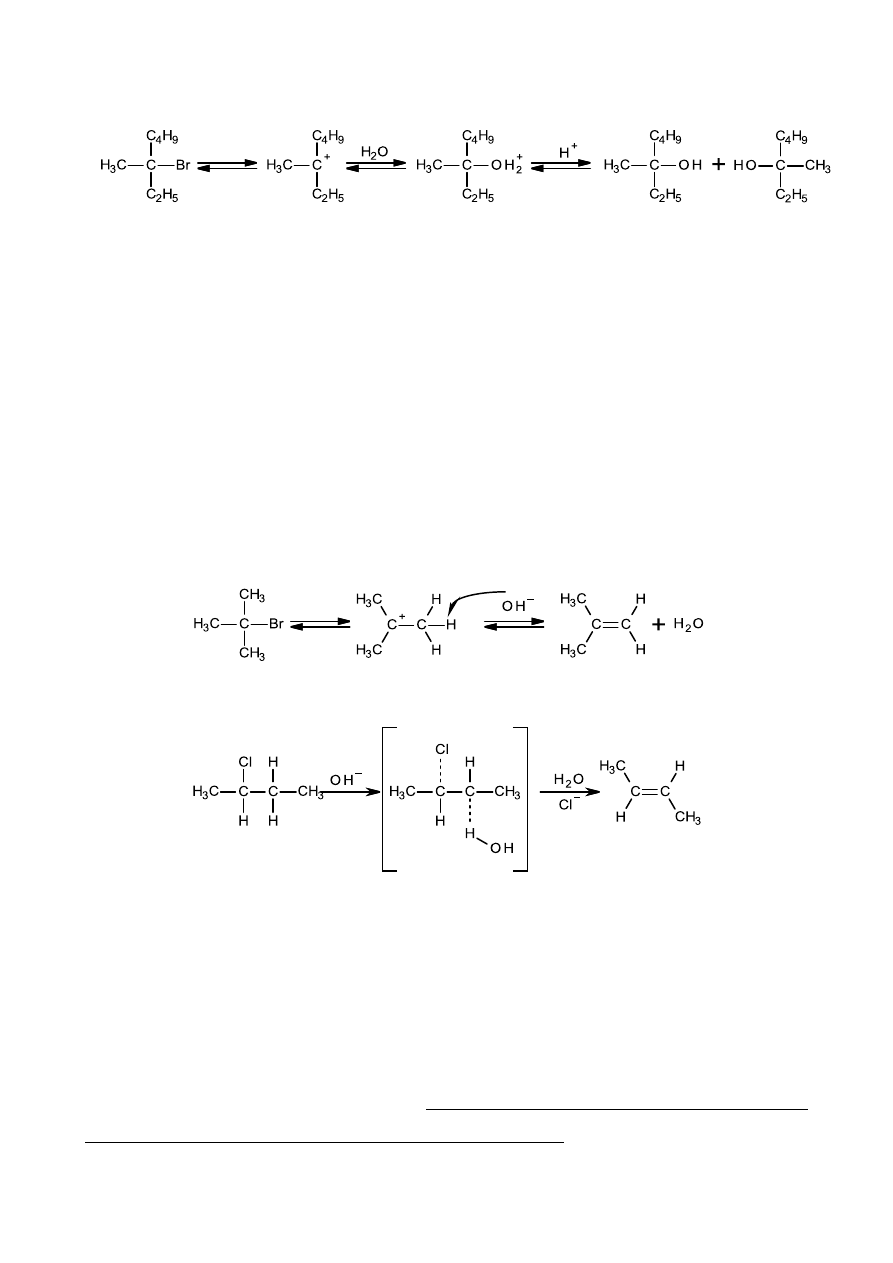
8
Substytucja Nukleofilowe S
N
1 – mechanizm jednocząsteczkowy
-
Wszystkie etapy reakcji są odwracalne – równowaga zależna od warunków reakcji; nadmiar
rozpuszczalnika elektrofilowego – 100% solwoliza.
Eliminacja – to reakcja chemiczna powodująca usunięcie atomów lub ich grup z cząsteczki.
W zależności od mechanizmu, eliminacja może przebiegad jednocząsteczkowo (E1) lub
dwucząsteczkowo (E2), może też byd katalizowana kwasem lub zasadą, a także przebiegad
spontanicznie lub pod wpływem czynników środowiskowych (światła, ciepła itp.).
Eliminacja E1 – mechanizm jednocząsteczkowy
I) utworzenia karbokationu
II) deprotonacja przez zasadę Lewisa
III) utworzenie podwójnego wiązania.
Eliminacja E2 – mechanizm dwucząsteczkowy
-
-
10. Reguła Markownikowa i Zajcewa.
Ponieważ w większości przypadków podstawniki przy atomie węgla są bardziej
elektroujemne niż wodór, każde zastąpienie wodoru innym podstawnikiem prowadzi do
zwiększenia niedoboru ładunku ujemnego na węglu i tym samym zwiększenia polaryzacji wiązania
C–H, a w konsekwencji ułatwienia odłączenia wodoru.
Reguła Zajcewa jest regułą dotyczącą chemicznej reakcji eliminacji, w której powstają
nowe wiązania podwójne C=C. Reguła głosi, iż w reakcjach eliminacji, w których powstaje wiązanie
C=C, powstają zawsze w przewadze bardziej rozgałęzione izomery.
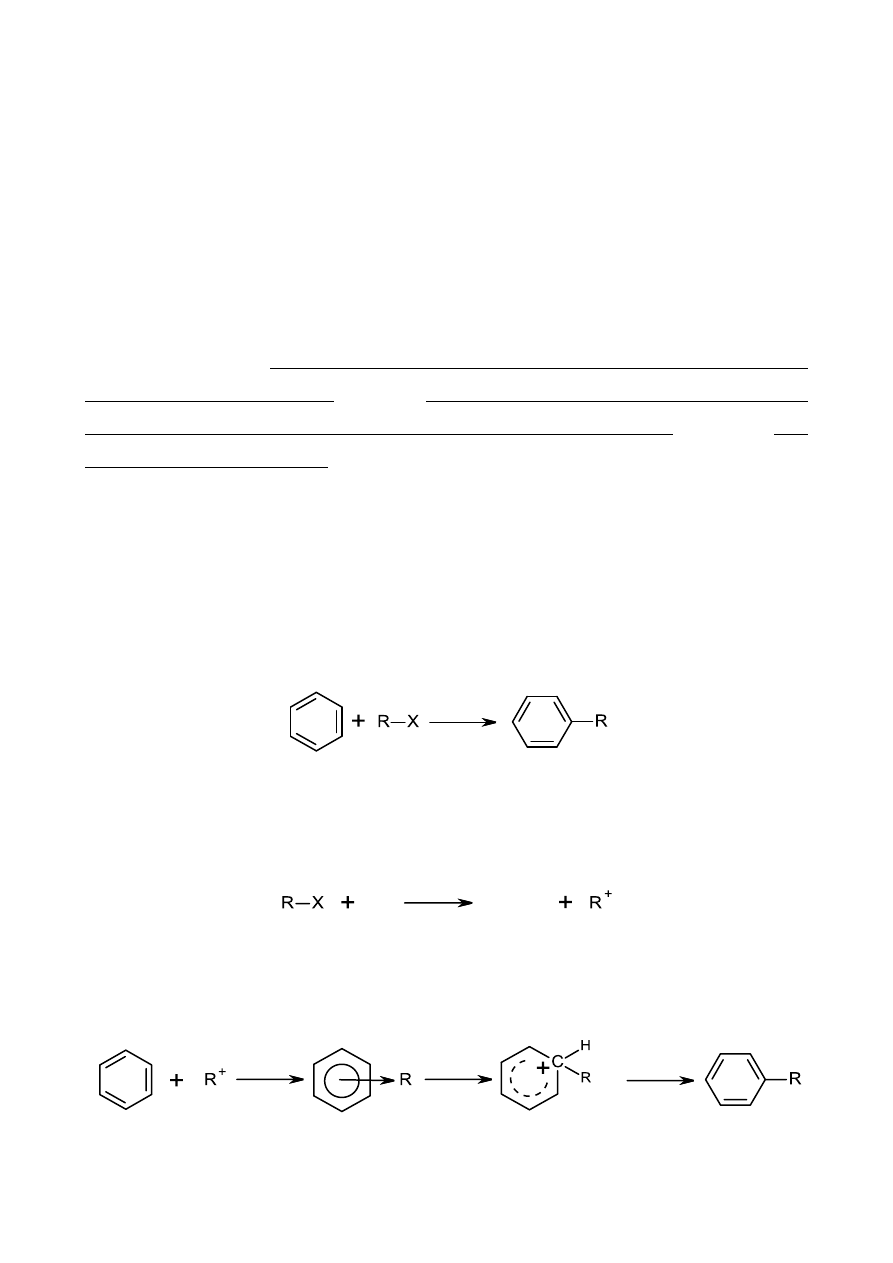
9
W przypadku reakcji eliminacji halogenowodorów (HX) z halogenków alkilowych z reguły
wynika, że jako produkt główny powstaje alken zawierający maksymalną liczbę grup alkilowych
przy atomach węgla posiadających wiązanie podwójne.
Reguła ta obowiązuje dla reakcji zachodzących według mechanizmu dwucząsteczkowego
(E2) o ile w substratach nie ma blisko powstającego wiązania podwójnego podstawników
posiadających charakter silnie nukleofilowy oraz nie wchodzą w grę zjawiska zawady sterycznej.
Reguła Markownikowa jest zasadą określającą kierunek reakcji addycji do podwójnego
wiązania węgiel-węgiel (C=C).
Zasada głosi, że na ogół w reakcjach addycji do wiązania podwójnego występującego
w wielu związkach organicznych, atomy lub grupy o mniejszej elektroujemności przyłączają się
do tego z dwóch atomów węgla, do którego wcześniej było przyłączone więcej atomów lub grup
o własnościach elektrododatnich.
11. Reakcje alkilowania związków aromatycznych: metoda Friedla – Craftsa; metoda Wurza –
Fittiga
Alkilowanie pierścieni aromatycznych metodą Friedla – Craftsa jest reakcją o bardzo
szerokim zastosowaniu. Najważniejszymi odczynnikami są halogenki alkilowe, alkeny i alkohole.
Reaktywnośd halogenków alkilowych w reakcji Friedla – Craftsa maleje w szeregu F>Cl>Br>I.
Elektrofilem w reakcji alkilowania jest karbokation Alk
+
powstający w reakcji halogenków
alkilowych z AlCl
3
, lub z protonowanych alkenów albo też z alkoholi uaktywnionych BF
3
. Reakcja
alkilowania zaczyna się od wytworzenia karbokationu.
AlX
3
[AlX
4
]
-
Karbokation tworzy z pierścieniem aromatycznym stabilizowany mezomerycznie kompleks, który
powraca do układu aromatycznego przez odszczepienie protonu pod wpływem zasady (w tym
przypadku anionu halogenkowego –X
-
).
-H
+
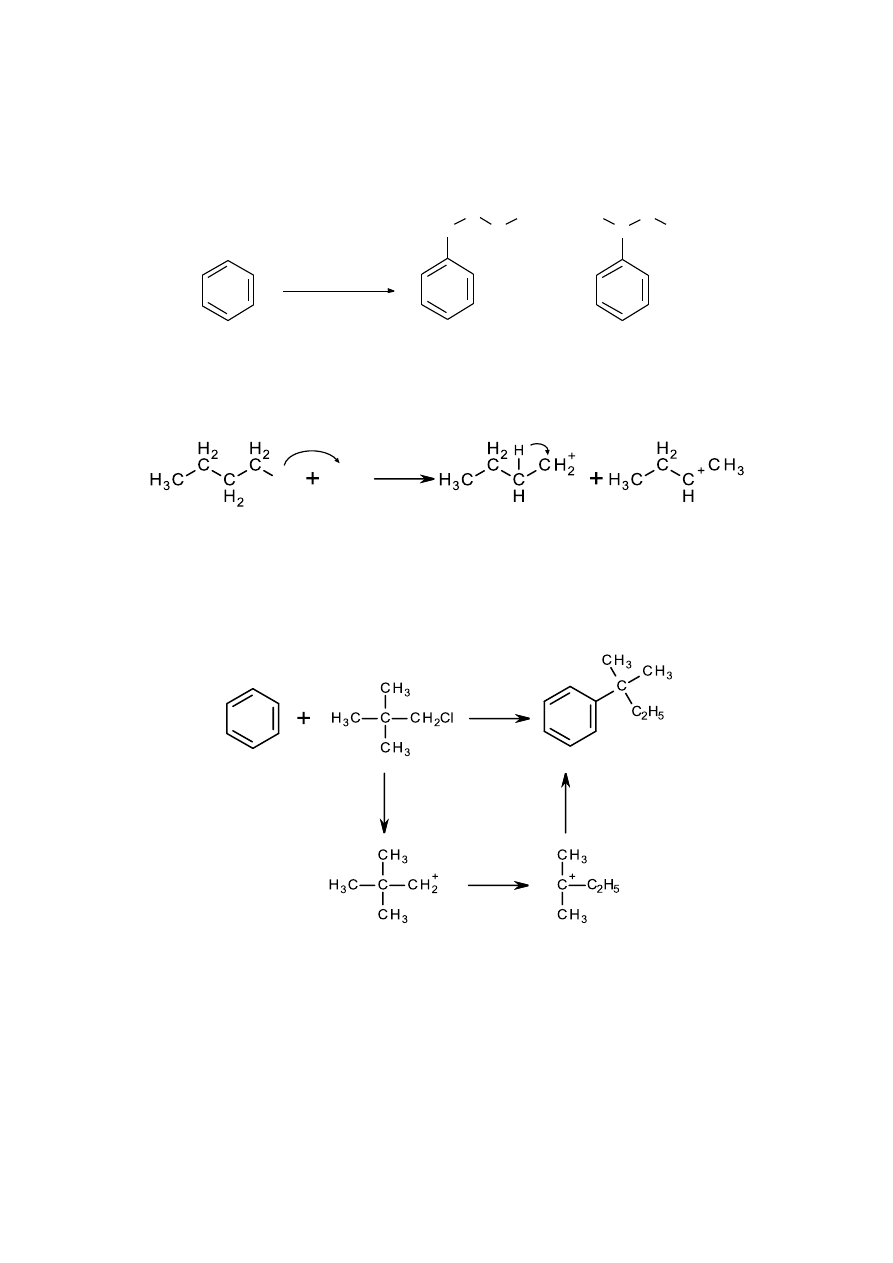
10
Przegupowanie karbokationu
W reakcjach z udziałem karbokationu alkilowego dochodzi do jego przegrupowania,
co prowadzi do mieszaniny produktów.
CH
3
CH
2
CH
2
CH
2
Cl
AlCl
3
C
H
3
C
H
C
H
2
C H
3
C H
3
C
H
2
C
H
2
C
H
2
+
W reakcji chlorku n-butylu z chlorkiem glinu tworzy się karbokation 1°, który
przegrupowuje się do trwalszego 2° karbokationu. Oba karbokationy biorą udział w alkilowaniu
benzenu.
AlCl
3
-AlCl
4
-
-
-
1
o
2
o
Najtrwalsze są karbokationy 3°, dlatego też w reakcji alkilowania benzenu 1-chloro-2,2-
dimetylopropanem tworzy się jeden związek, którym jest (1,1-dimetylopropylo)benzen; produkt
powstały w reakcji przegrupowania karbokationu 1°.
1
o
AlCl
3
-AlCl
4
-
3
o
Problem, jaki stwarza niepożądane przegrupowanie karbokationów podczas alkilowania
arenów metodą Friedla - Craftsa można rozwiązad pośrednio zastępując alkilowanie reakcją
acylowania i następnie redukcją otrzymanego ketonu alkilowo-arylowego do n-alkiloarenu.
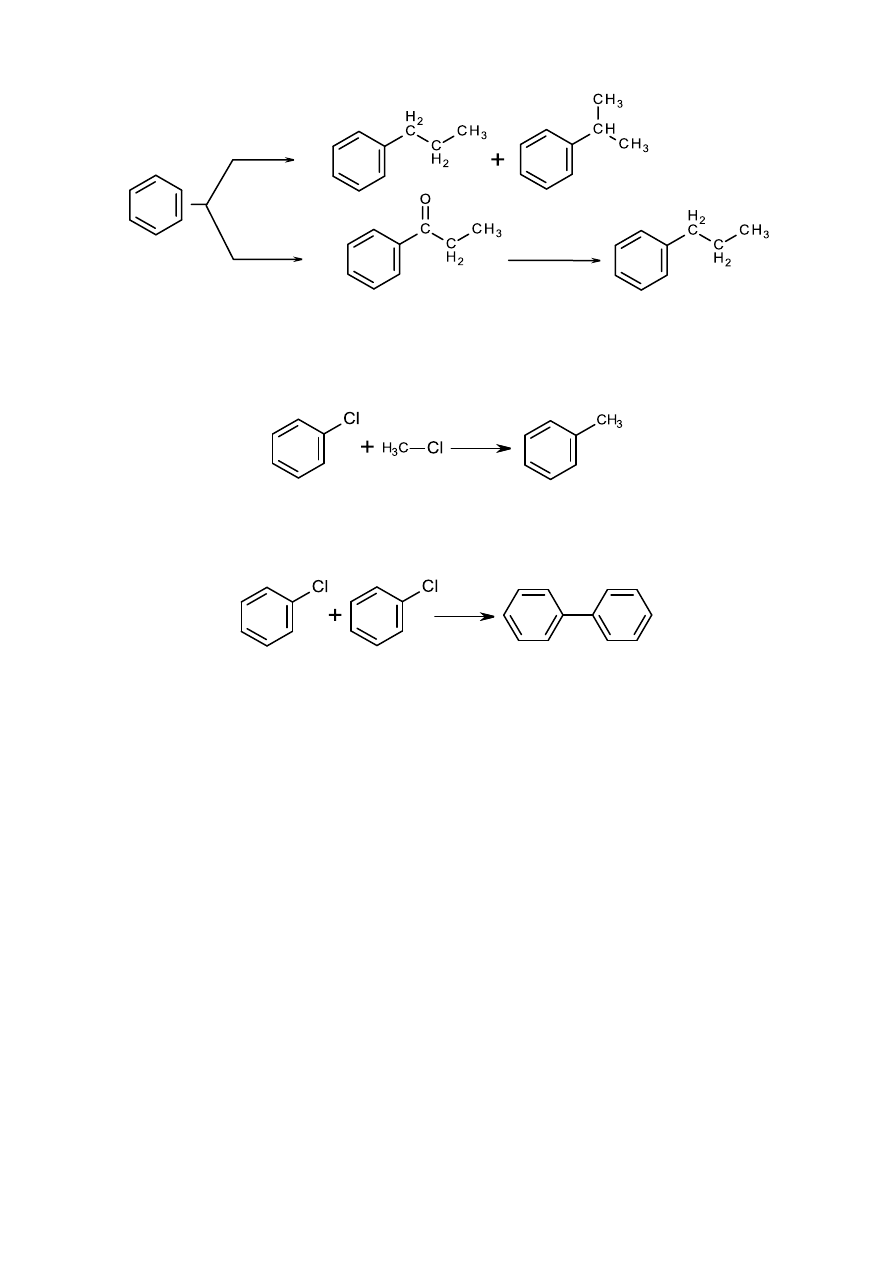
11
H
2
NNH
2
/
-
OH
CH
3
CH
2
CH
2
Cl
200
o
C
AlCl
3
AlCl
3
CH
3
CH
2
COCl
Proces Wurtza – Fittiga to reakcja pozwalająca na wprowadzanie podstawników alkilowych
do związków aromatycznych oraz syntezę symetrycznych związków biarylowych:
2Na
-2NaCl
2Ar-X + 2Na → Ar-Ar + 2NaX↓
2Na
-2NaCl
Otrzymywanie niesymetrycznych związków arylowo-alkilowych możliwe jest dzięki
różnicom w reaktywności halogenków arylowych i alkilowych. Prawdopodobny mechanizm
dwuetapowy składa się z reakcji halogenku alkilowego z sodem metalicznym oraz następczej
aromatycznej substytucji elektrofilowej.
Wyszukiwarka
Podobne podstrony:
halogenki alkilowe
Zadania z reakcji substytucji nukleofilowej i eliminacji w halogenkach alkilowych
4 halogenki alkilowe
Halogenki alkilowe – substytucja nukleofilowa – grupa opuszczająca – kwasy i zasady
09 Halogenki alkilowe substytucja eliminacja 26 05 2011 zadania
Halogenki alkilów, studia
09 Halogenki alkilowe substytucja eliminacja materialy dodatkowe
Ściąga ALKOHOLE,?NOLE, ETERY, HALOGENKI ALKILOWE
4 halogenki alkilowe SN materiały [tryb zgodności]
halogenki alkilowe
halogenki alkilowe
ZADANIE 25 halogenki alkilowe
12 halogenki alkiloweid 13245 pptx
Ściemniacz lampy halogenowej
Hydroksykwasy i halogenokwasy
REAKCJA ALKILOWANIA IV-RZĘDOWYCH SOLI AMONIOWYCH, Uczelnia PWR Technologia Chemiczna, Semestr 5,
więcej podobnych podstron