Postępy Biochemii 59 (1) 2013
45
Paulina Pergoł
1,
Agata Nowak-Stępniowska
1
Katarzyna Drela
2
Alfreda Padzik-Graczyk
1
1
Pracowania Biochemii, Instytut Optoelektro-
niki, Wojskowa Akademia Techniczna, War-
szawa
2
Zakład Neurobiologii Naprawczej, Instytut
Medycyny Doświadczalnej i Klinicznej im. M.
Mossakowskiego PAN, Warszawa
Pracowania Biochemii, Instytut Optoelektro-
niki, Wojskowa Akademia Techniczna, ul. Ka-
liskiego 2, 00-908 Warszawa; tel.: (22) 683 70 17;
e-mail: paulinapergol@wp.pl
Artykuł otrzymano 11 sierpnia 2012 r.
Artykuł zaakceptowano 15 stycznia 2013 r.
Słowa kluczowe: komórki macierzyste, nowo-
tworowe komórki macierzyste, nowotwory
Wykaz skrótów: APC (ang. adenomatous po-
lyposis coli) — białko supresyjne szlaku Wnt;
ASC (ang. adult stem cells) — somatyczne (doj-
rzałe) komórki macierzyste; BCC (ang. basal cell
carcinoma) — nowotwór podstawnokomórko-
wy skóry; CSC (ang. cancer stem cells) — nowo-
tworowe komórki macierzyste; Dvl (ang. dishe-
velled) — białko cytoplazmatyczne szlaku Wnt;
ESC (ang. embryonic stem cells) — embrionalne
komórki macierzyste; Fzd (ang. frizzled) —
białko transbłonowe szlaku Wnt; LRP (ang. li-
poprotein receptor related protein) — białko zwią-
zane z receptorem LDL; MMP (ang. matrix me-
talloproteinase) — metaloproteazy macierzowe;
N
EC
(ang. notch extracellular domain) — domena
zewnątrzbłonowa białka Notch; N
IC
(ang. notch
intracellular domain) — domena cytoplazma-
tyczna białka Notch; N
TM
(ang. notch transmem-
brane domain) — domena transbłonowa białka
Notch; Oct-3/4 (ang. octamer3/4) — czynnik
transkrypcyjny aktywujący proliferację komór-
kową; PDGF (ang. platelet derived growth factor)
— pochodzący z płytek czynnik wzrostu; POU
(ang. pit-oct-unc) — czynniki transkrypcyjne
wiążące się z DNA; SC (ang. stem cells) — ko-
mórki macierzyste; Shh (ang. sonic hedgehog) —
białko aktywujące szlak sygnałowy Shh; SMO
(ang. smothened) — białka transbłonowe szlaku
Shh, ang. Patched (Ptch) — rodzina białek re-
ceptorowych szlaku Shh; TGFβ (ang. transfor-
ming growth factor) — transformujący czynnik
wzrostu; VSEL (ang. very small embryonic-like)
— bardzo małe komórki macierzyste o cechach
komórek embrionalnych; Wnt (ang. wingless/
integration signaling) — białko aktywujące szlak
sygnałowy Wnt
Znaczenie komórek macierzystych w inicjacji i rozwoju nowotworów
STRESZCZENIE
I
nicjacja nowotworu może być wynikiem nagromadzenia się mutacji zachodzących w
prawidłowych komórkach macierzystych, powodujących zablokowanie różnicowania
się tych komórek. Wiele wspólnych cech, jakie posiadają komórki macierzyste z niektóry-
mi komórkami nowotworowymi sugeruje, że za inicjację i progresję nowotworu mogą być
odpowiedzialne nowotworowe komórki macierzyste. Szczególne znaczenie ma zdolność do
samoodnowy i proliferacji komórkowej, które są głównym powodem wznowy choroby no-
wotworowej i tworzenia przerzutów. Za proliferację komórkową odpowiedzialne są przede
wszystkim szlaki sygnałowe Wnt, Notch, Shh i czynniki transkrypcyjne Oct-4, Nanog. Po-
znanie przyczyn inicjacji i rozwoju nowotworów jest kluczowe dla udoskonalenia leczenia
tych groźnych dla życia chorób.
WPROWADZENIE
Komórki macierzyste (SC, ang. stem cells) ze względu na swoje wyjątkowe
właściwości i wiążące się z nimi perspektywy terapeutyczne są przedmiotem
wielu badań biomedycznych. Komórki te odpowiedzialne są za wzrost prawi-
dłowych tkanek oraz naprawę tkanek uszkodzonych w organizmach [1]. Pod-
stawowymi właściwościami umożliwiającymi pełnienie powyższych funkcji są:
i) zdolność do samoodnowy — odtwarzania w wyniku symetrycznych bądź
niesymetrycznych podziałów oraz ii) zdolność do wielokierunkowego różnico-
wania.
Za regenerację uszkodzonych tkanek w dorosłych organizmach odpowie-
dzialne są somatyczne komórki macierzyste ASC (ang. adult stem cells). Znajdują
się one w tzw. niszach w określonych miejscach w organizmie m.in. w szpiku
kostnym, tkance tłuszczowej, trzustce, wątrobie, naskórku oraz innych tkankach
i narządach. ASC głównie posiadają ograniczoną zdolność do różnicowania i
mogą się przekształcić w komórki potomne w obrębie tkanki z której pochodzą.
Można jednak znaleźć publikacje wykazujące, że wyizolowane ze szpiku me-
zynchymalne komórki macierzyste, mogą miedzy innymi zróżnicować się w
kierunku neuralnym czy epitelialnym. Somatyczne komórki macierzyste róż-
nią się przede wszystkim stopniem zróżnicowania oraz pełnionymi funkcjami
od embrionalnych komórek macierzystych ESC (ang. embryonic stem cells), naj-
słabiej zróżnicowanych, z których powstają wszystkie komórki [2-4]. Ostatnio
zidentyfikowano w szpiku kostnym dorosłego organizmu populacje komórek
o morfologii i markerach powierzchniowych charakterystycznych dla embrio-
nalnych komórek macierzystych, które nazwano VSEL (ang. very small embry-
onic-like). Stanowią one nieliczną grupę, małych komórek o średnicy 2-4 µm o
bardzo szerokim spektrum różnicowania. VSEL są zdolne różnicować się we
wszystkie trzy listki zarodkowe i mogą być źródłem pluripotencjalnych komó-
rek macierzystych potrzebnych do regeneracji uszkodzonych tkanek. Analiza z
zastosowaniem mikroskopu elektronowego wykazała, że komórki te posiadają
cechy budowy morfologicznej charakterystyczne również dla embrionalnych
komórek macierzystych. Mianowicie, posiadają one stosunkowo duże jądra oto-
czone wąskim pasmem cytoplazmy, w których znajduje się rozluźniona forma
chromatyny, euchromatyna[5].
Komórki macierzyste można również sklasyfikować pod względem zdolno-
ści do różnicowania na następujące grupy:
• komórki totipotencjalne — występują w pierwszych 2-3 dniach po zapłodnie-
niu i mogą różnicować się we wszystkie komórki organizmu, w tym komórki
popłodu (łożysko, sznur pępowinowy, błony płodowe);
• komórki pluripotencjalne — są zdolne do przekształcenia się w każdy typ
komórki czy tkanki, oprócz komórek popłodu;
• komórki multipotencjalne — różnicują się w obrębie jednego z trzech listków
zarodkowych: ektodermy, endodermy, mezodermy;
• komórki unipotencjalne — zdolne jedynie do różnicowania się w określony
typ komórek [6].
46
www.postepybiochemii.pl
Wyżej wynmienione właściwości komórek macierzy-
stych skłoniły wielu badaczy do podejmowania prób zasto-
sowania tych komórek w medycynie regeneracyjnej oraz w
leczeniu nowotworów. Terapie z zastosowaniem komórek
macierzystych mogą być powiązane z bezpośrednim zastę-
powaniem przez nie komórek uszkodzonych lub oddzia-
ływaniem pośrednim, poprzez korzystny wpływ na endo-
genne procesy regeneracyjne organizmu. Najnowsze dane
ukazują potencjalne możliwości wykorzystania komórek
macierzystych nie tylko w hematologii, ale również w te-
rapii przeciwnowotworowej oraz w leczeniu chorób prze-
wlekłych i genetycznych, chorób neurologicznych zarówno
tych o charakterze neurodegeneracyjnym, jak i w ostrych
uszkodzeniach układu nerwowego oraz wielu innych [4,7].
Obecnie podejmowane są próby zastosowania modyfiko-
wanych genetycznie komórek macierzystych jako specjal-
nie zaprojektowanych nośników dostarczających określoną
substancję do komórek lub tkanek [8]. W przyszłości ruty-
nowe wykorzystanie komórek macierzystych w medycynie
jest wielce prawdopodobne, jednak dalszy rozwój tej tech-
nologii wymaga przede wszystkim ustalenia i zoptyma-
lizowania standardów izolacji oraz ekspansji wraz z okre-
śleniem możliwości różnicowania poszczególnych typów
komórek w zależności od potrzeb, w różnych przypadkach
chorobowych. Mimo znacznego postępu w badaniach nad
terapią komórkową dotychczasowe efekty kliniczne wyda-
ją się nie do końca satysfakcjonujące, przede wszystkim z
powodu ryzyka powstania transformacji nowotworowej.
W wielu publikacjach z zastosowaniem modeli zwierzę-
cych zwraca się uwagę na negatywne skutki związane z
przeszczepem komórek macierzystych, możliwości zaini-
cjowania procesu nowotworowego i powstanie guza [9,10].
Ostatnio okazało się, że po kilku latach od przeszczepu neu-
ralnych komórek macierzystych, u pacjenta zidentyfikowa-
no nowotwór mózgu, którego komórki nie były zgodne pod
względem antygenów HLA z komórkami biorcy. Dalsze ba-
dania wykazały, że nowotwór składał się z dwóch populacji
komórek, które wcześniej zostały przeszczepione [11].
NOWOTWOROWE KOMóRKI MACIERZYSTE
PRZEGLąD HISTORYCZNY
Po raz pierwszy podobieństwo komórek nowotworowych
do komórek macierzystych zauważyli Rudolf Virchow i Ju-
lius Conhein już w XIX wieku, lecz hipotezy te nie zostały
potwierdzone badaniami eksperymentalnymi [12]. Przeło-
mowym odkryciem dla istnienia nowotworowych komórek
macierzystych CSC (ang. cancer stem cells) były testy wyko-
nane w 1985 roku przez Anne Hamburger i Sydney Salmon.
Zauważyli oni, że nie wszystkie komórki wyizolowane z li-
tych guzów posiadają zdolność do proliferacji. Okazało się,
że tylko 1 na 1000 do 1 na 5000 komórek wyizolowanych
były zdolne do tworzenia kolonii [13]. Ponadto w 1961 roku
doświadczenia przeprowadzone przez Chester Soutcham i
Aleksander Brunschwing wykazały, że komórki pobrane od
chorych z rozsianym nowotworem złośliwym wstrzyknięte
pod skórę tym samym pacjentom w małym stopniu powo-
dowały formowanie nowych ognisk nowotworowych. Do
zainicjowania guza niezbędne było przeszczepienie przy-
najmniej 1 000 000 komórek nowotworowych [14]. Wyniki
tych doświadczeń pozwoliły wnioskować, że istnieją małe
populacje komórek odpowiedzialne za inicjacje, wzrost i
proliferacje nowotworów. W toku długoletnich badań, do-
piero w 1997 roku udało się zespołowi John’a Dick wyizo-
lować pierwsze nowotworowe komórki macierzyste z całej
populacji w ostrej białaczce szpikowej (AML) [15]. Pierwsza
identyfikacja CSC z nowotworu piersi opublikowana zosta-
ła w 2003 roku, natomiast rok później opisano CSC central-
nego układu nerwowego [16-18]. Nowotworowe komórki
macierzyste są przedmiotem zainteresowania naukowców
różnych dziedzin, choć są tematem dużej liczby badań to
wiele aspektów dotyczących powstawania i rozwoju CSC
czeka jeszcze na wyjaśnienie.
HIPOTEZY POWSTAWANIA CSC
Nowotworowe komórki macierzyste mogą powstawać w
wyniku mutacji genetycznych zachodzących w komórkach
macierzystych, progenitorowych lub nawet w zróżnicowa-
nych komórkach somatycznych. Pochodzenie tych komó-
rek pozostaje dyskusyjne i może być różne, w zależności od
typu danej tkanki i nowotworu.
Hipoteza zakładająca, że nowotworowe komórki macie-
rzyste powstają w wyniku zmian epigenetycznych i nagro-
madzenia mutacji zachodzących w prawidłowych komór-
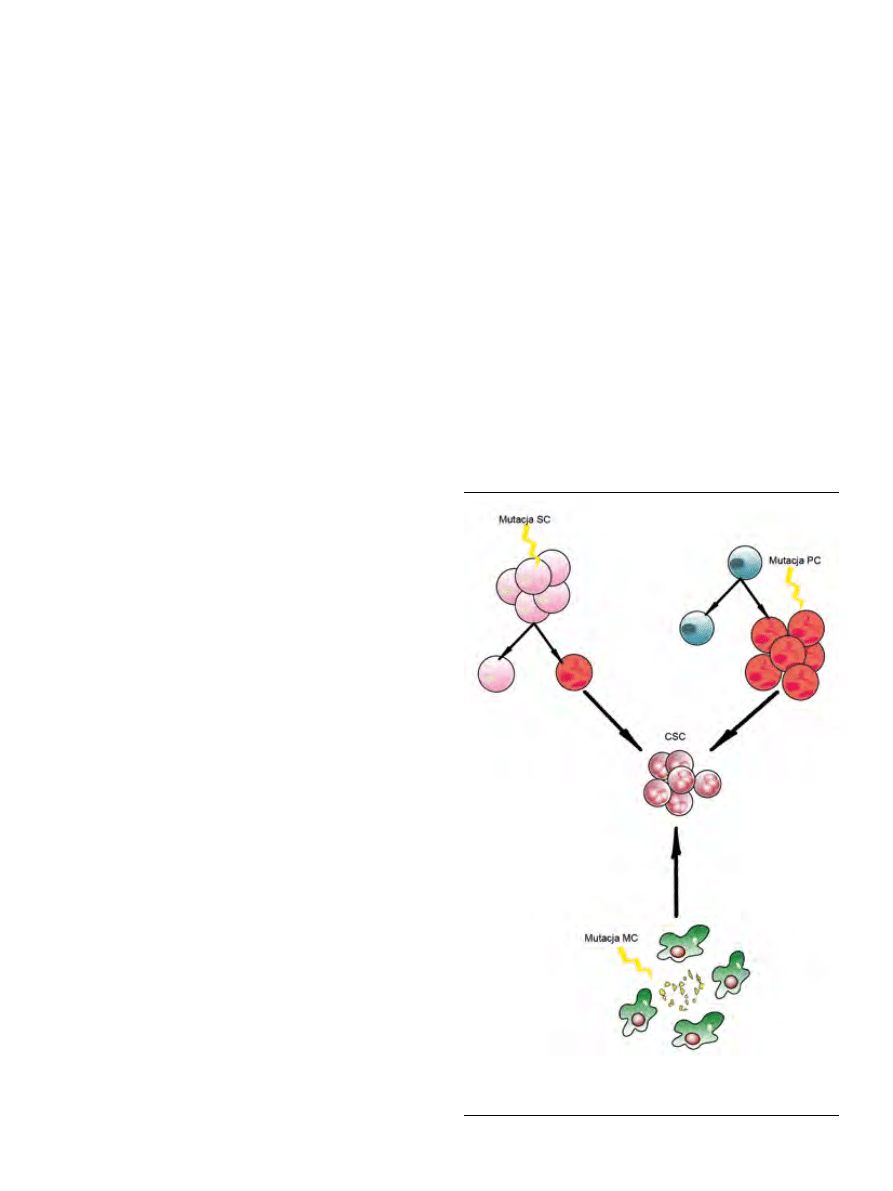
Rycina 1. Mechanizmy transformacji nowotworowej SC. Nowotworowe komórki
macierzyste mogą powstawać na skutek mutacji zachodzących w macierzystych
komórkach (SC), progenitorowych (PC) lub komórkach zróżnicowanych (MC).

Postępy Biochemii 59 (1) 2013
47
kach macierzystych potwierdza wiele wspólnych cech, któ-
re te dwa typy komórek posiadają. Transformacja prawidło-
wej komórki, w komórkę nowotworową nie jest jednoeta-
powym procesem, tylko nagromadzeniem znaczącej liczby
mutacji. Komórki macierzyste żyją dłużej od komórek zróż-
nicowanych i bardziej narażone są na kumulację zmian pro-
wadzących do inicjacji nowotworu. Przypuszcza się rów-
nież, że źródłem nowotworowych komórek macierzystych
mogą być komórki progenitorowe (prekursorowe), które są
komórkami pośrednimi pomiędzy macierzystymi komór-
kami, a zróżnicowanymi i posiadają częściową zdolność do
samoodnowy. Część badaczy sugeruje, że CSC pochodzą z
komórek już zróżnicowanych, które w wyniku nagroma-
dzonych mutacji zyskały umiejętność samoodnawiania się
oraz odróżnicowując, zdobyły inne cechy komórek macie-
rzystych (Ryc. 1). Model ten zakłada, że relatywnie duża
populacja komórek mogłaby być źródłem nowotworowych
komórek macierzystych, choć faktycznie tylko mała grupa
z nich jest zdolna do inicjacji nowotworu [19,20]. Jadnakże,
ostatnio przeprowadzone badania wykazały, że komórki
nowotworu piersi pod wpływem radioterapii, uległy trans-
formacji do nowotworowych komórek macierzystych [21].
Inna koncepcja zakłada, że istnieją w dorosłym organizmie
bardzo małe komórki macierzyste VSEL, które z powodu
ich słabego zróżnicowania i dużej zdolności do proliferacji
również mogą być potencjalnym źródłem nowotworu [22].
PORóWNANIE PRAWIDŁOWYCH I
NOWOTWOROWYCH KOMóREK MACIERZYSTYCH
W celu identyfikacji czynników regulujących funkcjono-
wanie nowotworowych komórek macierzystych, kluczo-
we zdaje się porównanie ich z prawidłowymi komórkami
macierzystymi. Obie populacje wykazują wiele wspólnych
cech, poniżej wymieniono najważniejsze z nich wraz z wy-
jaśnieniami [23].
1. Zdolność do samoodnowy
Samoodnawialność jest podstawową cechą komórek
macierzystych, umożliwiającą utrzymywanie pewnej puli
tych komórek przez całe życie organizmu. W zależności od
warunków i zapotrzebowania, komórka macierzysta może
ulegać podziałom symetrycznym, w wyniku których po-
wstają dwie komórki macierzyste bądź niesymetrycznym,
w których jedna komórka potomna jest macierzysta, a dru-
ga ulega różnicowaniu. Proces samoodnawialności cha-
rakterystyczny jest zarówno dla prawidłowych komórek
macierzystych, jak również nowotworowych komórek ma-
cierzystych. W kontrolowanie procesu samoodnowy zaan-
gażowane są szlaki przekazywania sygnału takie jak Wnt,
Shh, Notch. Właśnie rozregulowanie tych szlaków może
być przyczyną transformacji komórek macierzystych w no-
wotworowe i prowadzić do inicjacji nowotworów. W rezul-
tacie nadmierne namnażanie niezróżnicowanych komórek
nie pełniących swoich funkcji, może powodować progresję
nowotworu oraz inwazję do sąsiadującej tkanki [24,25].
2. Występowanie w podobnych regionach organizmu
Wykazano, iż komórki macierzyste zasiedlają miejsca
silnie ukrwione zwane niszami naczyniowymi, które pełnią
rolę mikrośrodowiska dla tych komórek. Podstawowym
składnikiem nisz są komórki śródbłonka, które zapewnia-
ją odpowiednią równowagę pomiędzy samoodnawianiem
się komórek macierzystych, a ich różnicowaniem. Ostatnie
badania wykazały, że nowotworowe komórki macierzyste
obecne in vivo w nowotworach mózgu, koncentrują się przy
naczyniach włosowatych. Natomiast hodowane in vitro
wraz z komórkami śródbłonka, wykazują selektywne powi-
nowactwo do tych komórek tworząc skupiska. Stwierdzo-
no również, że związanie się CSC z komórkami śródbłonka
powoduje wzmożenie procesów samoodnowy i zwiększa
populacje tych komórek.[23]
3. Wydzielanie czynników wzrostu oraz stymulacja angio-
genezy
Charakterystyczną cechą prawidłowych i nowotworo-
wych komórek macierzystych jest zdolność zapewnienia
sobie dopływu stałych czynników wzrostu. W tym celu an-
gażowane są różne czynniki między innymi cytokiny, któ-
re są autokrynnymi i parakrynnymi czynnikami wzrostu i
przeżycia komórek. Cytokiny proangiogenne są niezbęd-
ne do tworzenia nowych naczyń krwionośnych, które do-
starczają tlen i substancje odżywcze do komórek. Niektóre
nowotwory poprzez produkcje czynników wzrostu, takich
jak interleukiny promują angiogenezę, przerzuty i wzrost
nowotworu. Część badaczy sugeruje, że za procesy te od-
powiedzialne są nowotworowe komórki macierzyste, które
stymulują wydzielanie wyżej wymienionych czynników
[26,27]. Wykazano, że czynniki wzrostowe znajdujące się
w macierzy zewnątrzkomórkowej komórek macierzystych
tj. TGFβ, PDGF występują również w niektórych nowotwo-
rach. Podobnie podejrzewa się, że proliferację i migrację
komórek nowotworowych wspomagają metaloproteinazy
macierzowe MMP występujące w komórkach macierzy-
stych. Przykładem są metaloproteinazy MMP1, MMP2,
MMP7, MMP9, MMP11, których nadmierną aktywność wy-
kryto w nowotworach kości, jelita, trzustki, żołądka [28-30].
4. Odporność na chemioterapeutyki i radioterapię
Komórki nowotworowe nabywają odporności na che-
mioterapeutyki poprzez szereg mechanizmów. Często
można zaobserwować, że po pierwszej reakcji nowotworu
na leki dochodzi do wznowienia populacji nowotworowych
komórek odpornych na dany typ chemioterapii. Przypusz-
cza się, że za wznowę odpowiedzialne są CSC, które tak jak
SC mogą występować dłuższy czas w fazie G0 w tzw. sta-
nie „uśpionym” (ang. quiescence state) co czyni je niewraż-
liwymi na leki, działające jedynie na dzielące się komórki.
Dlatego wnioskuje się, że część nowotworowych komórek
macierzystych może przetrwać chemioterapię i powodo-
wać wznowienie nowotworu. Dodatkowo prawidłowe i
nowotworowe komórki macierzyste posiadają zdolności
naprawcze DNA. Cecha ta czyni je odpornymi na zmiany
genetyczne wywołane promieniowaniem [31,32]. Ważnym
czynnikiem chroniącym nowotworowe komórki macierzy-
ste przed związkami toksycznymi jest wysoka aktywność
białek błonowych — transporterów ABC występująca rów-
nież w SC. Zadaniem transporterów ABC jest usunięcie
czynników szkodliwych w tym leków przeciwnowotwo-
rowych poza komórkę przed osiągnięciem ich stężenia le-
48
www.postepybiochemii.pl
talnego, korzystając z energii pozyskanej z hydrolizy ATP.
Przykładem jest zidentyfikowana glikoproteina P, należąca
do rodziny transporterów ABC, której wzmożona synteza
była widoczna w wielu nowotworach [33].
5. Starzenie replikacyjne
Od dawna wiadomo, że komórki somatyczne zdolne są
do przejścia określonej liczby podziałów komórkowych, po
upływie których aktywowane zostają czynniki wywołują-
ce apoptozę. Proces ten jest kontrolowany przez telomery
— terminalne odcinki chromosomów. Po każdym podziale
długość telomerów ulega skróceniu, aż do pewnej długości,
która jest sygnałem dla komórki do zainicjowania procesu
apoptozy. Wykazano, że zarówno prawidłowe jak i nowo-
tworowe komórki macierzyste cechuje wysoka aktywność
telomerazy, enzymu powodującego wydłużanie się telo-
merów. Utrzymywanie stałej długości telomerów jest po-
wodem względnej „nieśmiertelności” obu typów komórek,
ponieważ nie podlegają one procesom starzenia komórko-
wego [34,35].
6. Synteza podobnych receptorów powierzchniowych
Każdy typ komórek charakteryzuje się swoistymi re-
ceptorami zgromadzonymi na powierzchni, co pozwala
na ich identyfikację. Prawidłowe i nowotworowe komór-
ki macierzyste posiadają wspólną gamę wielu markerów
powierzchniowych między innymi CXCR4, Sca-1, CD133,
CD24, CD34, CD44, c-kit, c-met, LIF-R, BMI1. Wykazano,
że specyficzny dla komórek macierzystych receptor CD133,
występuje również w niektórych komórkach nowotwo-
ru jelita grubego, wątroby, trzustki, prostaty, płuc czy w
raku mózgu. Udowodniono również obecność CD24 w no-
wotworze piersi wraz z CD44, który jest charakterystycz-
ny również dla nowotworowych komórek macierzystych
okrężnicy oraz prostaty. Zasługującym na szczególną uwa-
gę jest receptor CXCR4, który może być odpowiedzialny za
tworzenie przerzutów. Obecność CXCR4 na powierzchni
CSC jest pomocna w identyfikacji tej małej populacji komó-
rek w masie guza. [22,23,36-38].
7. Szlaki sygnałowe zaangażowane w samoodnowę oraz
proliferację komórkową
Za progresję nowotworu i samoodnowę prawdopodob-
nie odpowiedzialne są nowotworowe komórki macierzyste.
W procesy te zaangażowane są te same szlaki sygnałowe,
które występują w prawidłowych komórkach macierzy-
stych. Szybkie tempo proliferacji komórek nowotworowych
i ich zdolność do samoodnowy to cechy powodujące naj-
większą destrukcję organizmu. Zahamowanie tych proce-
sów jest głównym celem terapii antynowotworowej, dla-
tego temat ten zasługuje na szczególną uwagę i wymaga
głębszego wyjaśnienia [39,40].
ZNACZENIE CSC W PROGRESJI NOWOTWORóW
Wiele badań wskazuje, że za kontrolę procesów samood-
nowy i proliferacji komórek odpowiedzialne są określone
czynniki oraz wewnątrzkomórkowe szlaki metaboliczne.
Mutacje zachodzące w genach kodujących białka wchodzą-
ce w cykl tych przemian tj. Wnt, Notch, Shh mają znaczący
wpływ na powstawanie nowotworów. Kluczową rolę peł-
nią również białka które są czynnikami transkrypcyjnymi,
w szczególności białka Oct-3/4, Nanog, Rex1, Sox2. [41].
SZLAKI SYGNAŁOWE
SZLAK Wnt
Szlak Wnt został zidentyfikowany jako jeden z głównych
szlaków sygnałowych występujących w nowotworach, re-
gulujący wzrost, ruchliwość komórek oraz różnicowanie.
Przekazywanie sygnału za pomocą ścieżek Wnt może być
różne i zależy ono od rodzaju liganda Wnt oraz warun-
ków panujących w komórce. Wyróżniamy dwie ścieżki
sygnałowe: kanoniczną (zależną od β-kateniny) oraz nie-
kanoniczną (zależny m.in. od stężenia jonów wapnia). Na
rycinie 2 przedstawiona została kanoniczna ścieżka sygna-
łowa Wnt, która ma znaczący wpływ na proces nowotwo-
rzenia i proliferację chorobowych komórek.
Przyczyny aktywacji ścieżki Wnt w procesie nowotwo-
rzenia są wynikiem mutacji poszczególnych białek tego
szlaku lub wyciszania ekspresji negatywnych regulatorów
Wnt/β-kateniny tj DKK lub WIF1. Najbardziej narażo-
ne na mutacje są: białko supresyjne APC, Axin lub sama
β-katenina. Wszystkie mechanizmy powodują ten sam
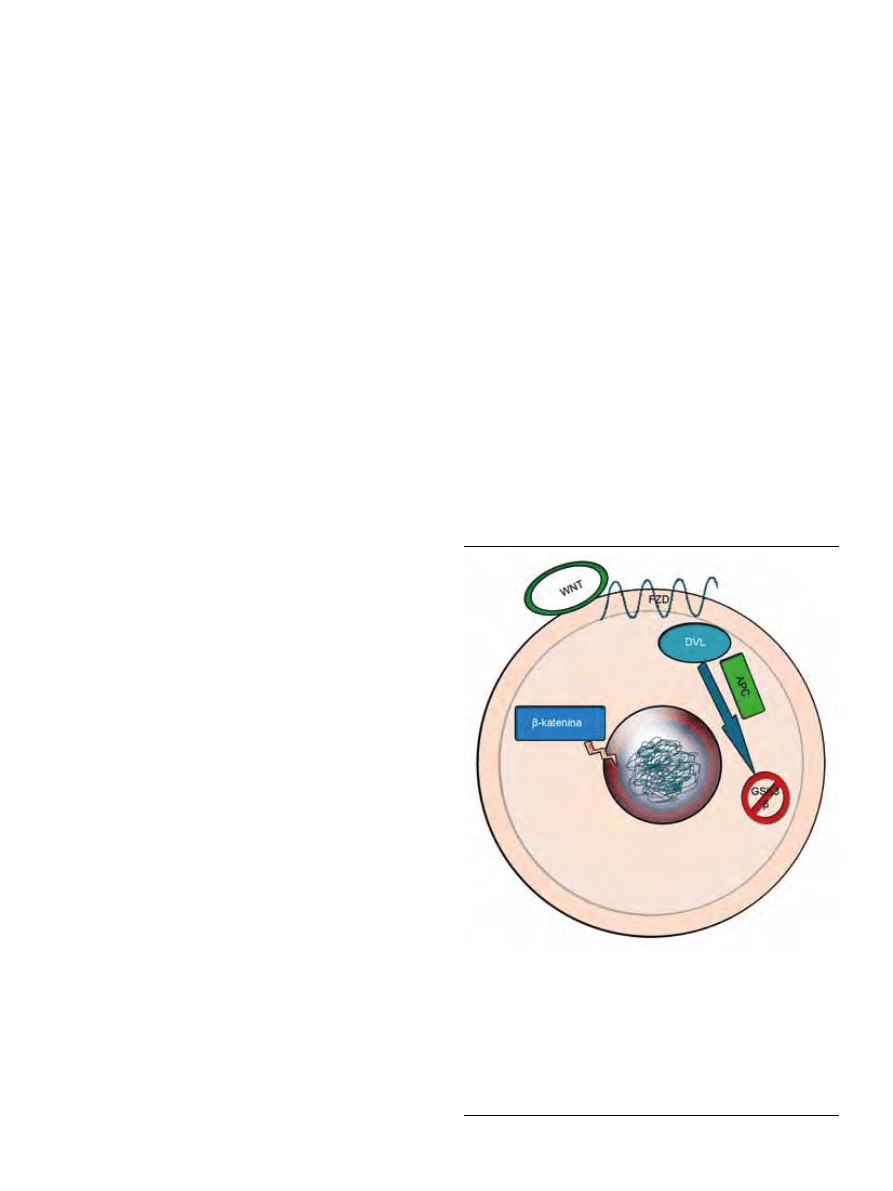
Rycina 2. Szlak sygnałowy Wnt. Sygnał na drodze tego szlaku przekazywany
jest w wyniku związania specyficznych białek Wnt z receptorami znajdującymi
się na powierzchni komórek. Jednym z nich jest receptor Fzd (ang. frizzled), który
po związaniu z białkiem Wnt powoduje fosforylację białka Dvl(ang. dishevelled).
Zaktywowane Dvl wiążą się z kompleksem APC-Acyna-Konduktyna hamując
działanie dwóch kinaz odpowiedzialnych za fosforylację β-kateniny: 3β syntazy
glikogenu oraz kazeiny 1α. Zablokowanie fosforylacji uniemożliwia degradację
β-kateniny w lizosomach ponieważ nie jest ona rozpoznawana przez składnik
ligazy ubikwitynowej: β-TrCP. Prowadzi to do stabilizacji i kumulacji β-kateniny,
która aktywuje czynniki transkrypcyjne Tcf/Lef indukując ekspresje genów od-
powiedzialnych za regulację cyklu komórkowego, proliferację, apoptozę oraz w
konsekwencji progresję nowotworową [42-44].
Postępy Biochemii 59 (1) 2013
49
efekt końcowy, mianowicie kumulację β-kateniny w jądrze
komórkowym, aktywującą czynniki transkrypcyjne Tcl/Lef
i w konsekwencji inicjującą ekspresję genów docelowych.
Niektóre z tych genów kodują białka o kluczowej roli dla
kancerogenezy. Należą do nich między innymi c-myc, cy-
klina D1, surwiwina [42-45].
Czynnik transkrypcyjny c-myc powoduje przejście ko-
mórki z fazy G1 do fazy S indukując wyjście komórek ze
stanu niezróżnicowanego. C-myc jest proonkogenem, który
ma znaczący wpływ na wzrost i proliferacje komórkową jak
również indukcje apoptozy. Wykazano, że podobny efekt
powodują cykliny D1, które poprzez tworzenie komplek-
sów z kinazami cdk4 i cdk6 oraz fosforylację białka pRb
również pobudzają proliferację komórkową i mają wpływ
na podział komórkowy [46]. Bardzo często w komórkach
nowotworowych i macierzystych można zaobserwować
zwiększoną ilość innego białka surwiwiny, która nie wy-
stępuje w komórkach już zróżnicowanych [47]. Surwiwiny
natomiast są inhibitorami apoptozy, a tym samym blokują
działanie chemioterapeutyczne [48,49].
Badania przeprowadzone na myszach udowodniły, że
zaburzenia Wnt1 prowadzą do nowotworów piersi oraz
nadmierna ekspresja tego genu czyni komórki odporne na
działanie leków. Natomiast nadekspresja Wnt5 powoduje
uzłośliwienie nowotworów, zwiększając ruchliwość i pro-
liferację zmienionych komórek [50].
SZLAK Notch
Sygnalizacja Notch ma kluczowe znaczenie dla rozwoju i
utrzymania homeostazy tkanek. Reguluje procesy związane
z nabywaniem określonego fenotypu podczas ich różnico-
wania oraz kontroluje przeżywalność i interakcje między-
komórkowe [51,52]. Schemat przekazywania sygnału za po-
mocą tego szlaku został przedstawiony na rycinie 3.
Udowodniono, że ścieżka Notch aktywuje procesy zwią-
zane z przeżyciem komórek oraz inicjuje ich proliferację, ale
również zatrzymuje cykl komórkowy i decyduje o różnico-
waniu. Między innymi nadmierna aktywacja Notch pro-
wadzi do podwyższenia poziomu białek HES, HRT/HET
zaangażowanych w progresję czerniaka i do zahamowania
syntezy białka MAP-2 sprzyjającej proliferacji komórek.
Ponadto, zwiększona aktywacja szlaku Notch sprzyja
powstawaniu przerzutów poprzez polepszenie właściwo-
ści adhezyjnych i migracyjnych tych komórek. Przykładem
jest Notch1, który przez inicjację ekspresji m.in. kinazy ty-
rozynowej przyczyniającej się do tworzenia kompleksu
przylegania komórkowego sprzyja tworzeniu przerzutów
czerniaka.
Zaburzenia poziomu receptora Notch, zostały udoku-
mentowane w różnych typach nowotworów. Dużą zawar-
tość białka Notch1 i Notch2 zaobserwowano w nowotworze
szyjki macicy, jelita grubego, trzustki, skóry czy mózgu. Na-
tomiast wzmożona synteza Notch3 i Notch4 występuje w
nowotworze trzustki i w czerniaku. [53-56].
Przełomowym krokiem w terapii przeciwnowotworo-
wej wydaje się być ostatnio otrzymane, wysoce specyficz-
ne przeciwciało dla receptorów Notch1 i Notch2. Badania
wykonane na myszach wykazały, że podanie tego przeciw-
ciała hamuje wzrost komórek nowotworowych w różnych
typach nowotworów. Dodatkowo zaobserwowano zmniej-
szenie angiogenezy i redukcję masy guza. Otrzymane wy-
niki dowodzą, że ścieżka sygnałowa Notch pełni ważną
funkcję w inicjacji i progresji choroby nowotworowej [57].
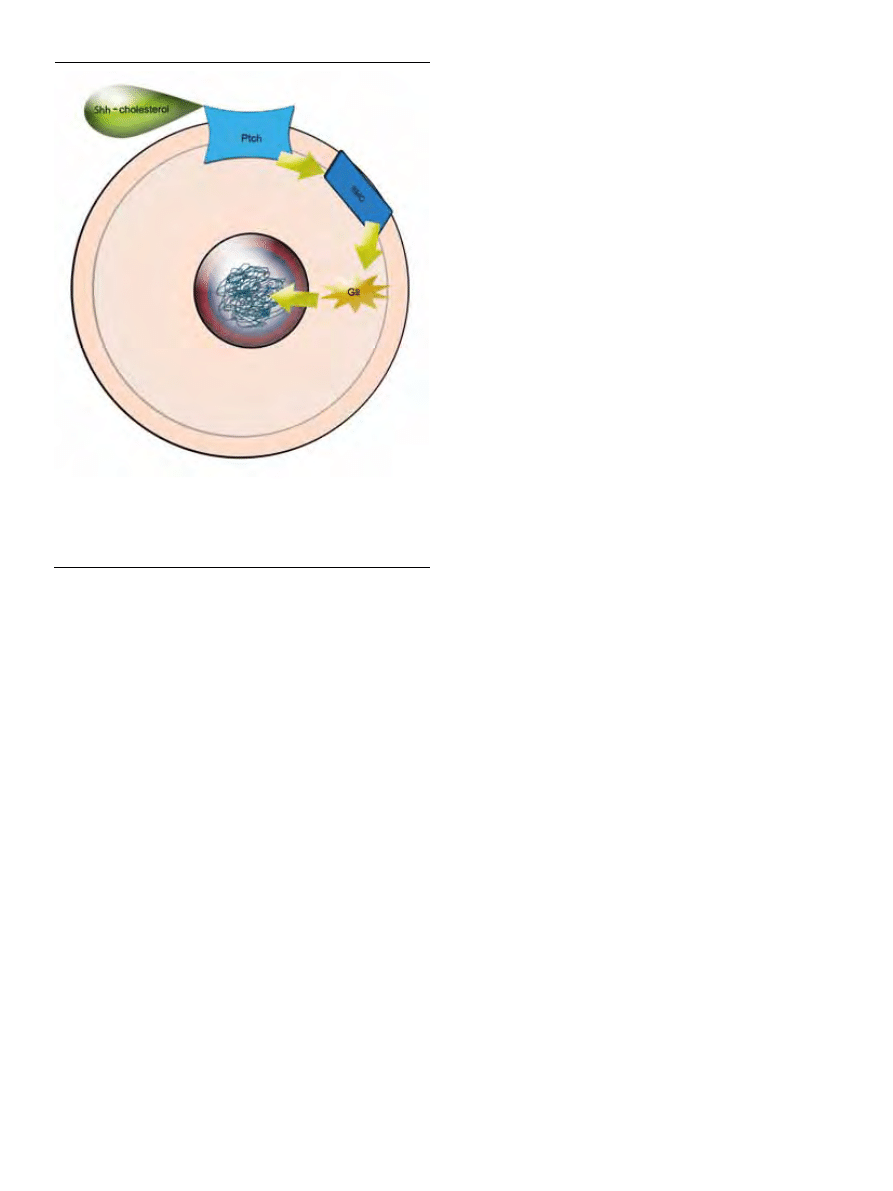
SZLAK Shh
Ścieżka sygnałowa Shh odgrywa istotną rolę w
embriogenezie i regulacji komórek macierzystych, a w
sytuacji patologicznej w indukcji procesu nowotworowego.
Nadekspresja białka Shh na błonie komórek nowotworo-
wych pobudza je autokrynnie do stałej proliferacji, powo-
dując progresję nowotworu.
Zapoczątkowanie szlaku sonic hedgehog następuje po-
przez połączenie aktywnego białka Shh z receptorem Ptch
i aktywację SMO, co powoduje rozszczepienie białek Gli
aktywujących geny docelowe (Ryc. 4). W prawidłowych
komórkach macierzystych aktywacja białka SMO zwięk-
sza transkrypcję Ptch1 i w wyniku sprzężenia zwrotnego
dochodzi do zahamowania procesu proliferacji. Jeśli doj-
dzie do mutacji któregoś ze składowych szlaku lub białko
Gli przedostanie się w całości do jądra, spowoduje to ciągłą
proliferację komórkową i może inicjować tworzenie nowo-
tworów.
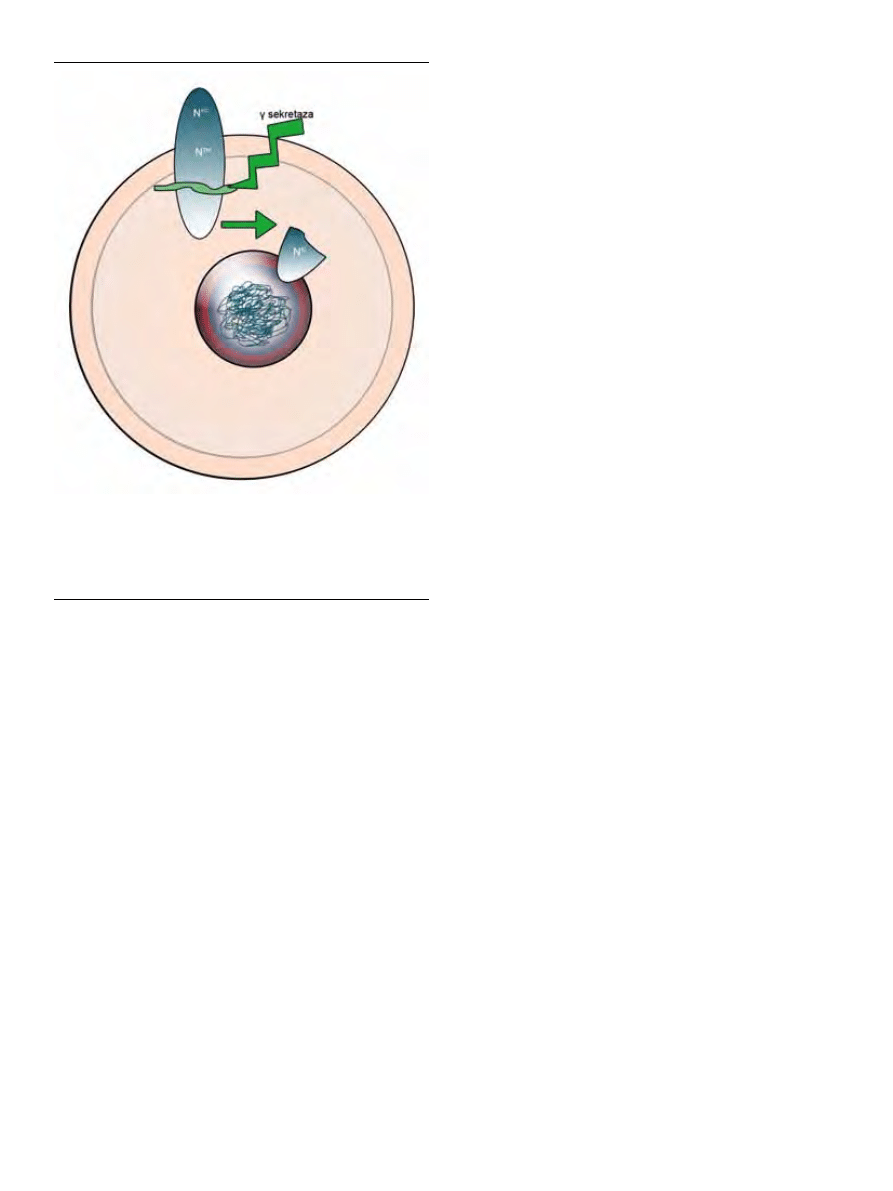
Rycina 3. Szlak sygnałowy Notch. Ligand znajdujący się na powierzchni błony
jednej komórki przyłączając się do zewnątrzkomórkowego fragmentu białka
Notch sąsiedniej komórki indukuje zmiany w jej transbłonowej części. Sygnał ten
powoduje zmiany konformacyjne w N
TM
odsłaniające domenę wrażliwą na cięcia
proteolityczne. Uwolniony fragment N
IC
przez enzymy proteolityczne (γ sekreta-
zę) wędruje do jądra komórkowego, gdzie przyłącza się do białka CBF-1/RBP-Jk
związanego z sekwencją genów docelowych [54].
50
www.postepybiochemii.pl
Wykazano, że szlak Shh wpływa również na cykl komór-
kowy, co ma kluczowe znaczenie dla kancerogenezy. Ostat-
nie badania wykazały, że uszkodzenie białka Ptch1 szlaku
Shh powoduje zaburzenia cyklu wynikające m.in. ze wzmo-
żonej syntezy białek regulatorowych takich jak cykliny B i
D. Przewiduje się, że proces ten przyczynia się do zwiększo-
nej proliferacji komórek nowotworowych oraz tworzenia
przerzutów. Mutacje w szlaku Shh występują ze wzmożoną
częstotliwością w nowotworze podstawnokomórkowym
skóry (BCC), kolczystokomórkowym (SCC) jak również w
glejaku [58-62].
CZYNNIKI TRANSKRYPCYJNE
Wiele obserwacji wskazuje, że za proliferację komórek
nowotworowych odpowiadają również niektóre czynniki
transkrypcyjne, które zmieniają ekspresję genów docelo-
wych. Komórka macierzysta poprzez sieć oddziaływań
białek oraz specyficznych czynników transkrypcyjnych tj.
Oct-3/4, Nanog, STAT3, Sox2, Bmi1, Rex1 może pozostać w
stanie pluripotencjalnym oraz jest zdolna do proliferacji, co
zostało udowodnione w badaniach in vitro. Podejrzewa się,
że te same mechanizmy działają w nowotworowych komór-
kach macierzystych [63,64].
Udowodniono, że czynnik transkrypcyjny Oct-3/4 (PO-
U5F1) jest niezbędny w utrzymaniu niezróżnicowanego
stanu komórek macierzystych. Białko to należy do rodzin-
ny czynników transkrypcyjnych POU (ang. pit-oct-unc) i in
vivo występuje w komórkach węzła zarodkowego blasto-
cysty. Wiadomo, że ekspresja genu Oct-3/4 zanika wraz ze
stopniem zróżnicowania komórki, natomiast w dojrzałych
komórkach somatycznych w ogóle nie występuje [65,66].
Ostatnio wykazano, że białko Oct-3/4 występuje w trzech
izoformach Oct-3/4A, Oct-3/4B i najsłabiej poznany Oct-
-3/4B1 oraz posiada kilka wariantów transkrypcyjnych i
pseudogenów. Wiadomo, że tylko Oct-3/4A jest odpowie-
dzialny za właściwości pluripotencjalne komórek, natomiast
funkcja pozostałych izoform nie jest w pełni poznana [67].
Badania kliniczne wykazały obecność Oct-3/4 w różnych
rodzajach nowotworów takich jak nowotwory piersi, jelita,
pęcherza [68]. Ponadto wykazano, że u pacjentów z nowo-
tworami charakteryzującymi się dużą zawartością tego biał-
ka, występuje szybsza progresja, częstsze przerzuty oraz
większa śmiertelność [69].
Za utrzymywanie komórek w stanie pluripotencji odpo-
wiada również białko Nanog, występujące między innymi
w embrionalnych komórkach macierzystych. Brak aktyw-
ności tego białka umożliwia różnicowanie się komórek, co
potwierdza jego znaczącą rolę w ich samoodnawialności
[65,70]. Wykazano, że transfekcja prawidłowych komórek
genem Nanog może powodować ich transformacje w CSC.
Ponadto najnowsze badania pokazują, że obniżenie zawar-
tości tego czynnika powoduje zmniejszenie tempa prolife-
racji linii komórkowych wywodzących się z nowotworu
piersi, jelita grubego, prostaty [71]. Sugeruje się, że Nanog
może służyć jako biomarker nowotworowych komórek ma-
cierzystych i być pomocny w identyfikacji tych komórek
w masie guza [72]. Dodatkowym aspektem, który podnosi
znaczenie Nanog w nowotworzeniu jest zdolność do two-
rzenia kompleksów z białkiem STAT-3 i aktywacji procesu
syntezy eksportera MDR1, należącego do rodziny białek
ABC, który odpowiedzialny jest za utrzymywanie leków
przeciwnowotworowych poza komórką [63]. W hodowli in
vitro obecność obu tych białek Oct-3/4 i Nanog w komórce
macierzystej jest wystarczająca do utrzymywania jej w sta-
nie niezróżnicowanym [73].
PODSUMOWANIE
Badania nad komórkami macierzystymi mają funda-
mentalne znaczenie w rozwoju leczenia chorób nowo-
tworowych. Szczególnie groźne dla organizmu są nowo-
twory złośliwe z tendencją do tworzenia przerzutów do
innych tkanek oraz zdolne do odnawiania się po zastoso-
wanym leczeniu. Z tego powodu, głównym celem terapii
onkologicznej powinny być nowotworowe komórki macie-
rzyste, dla których niewystarczająco skuteczne wydają się
być terapie konwencjonalne takie jak radioterapia czy che-
mioterapia i po zakończonym leczeniu komórki te mogą ini-
cjować powrót choroby. Istotnym aspektem jest wyjaśnienie
roli czynników regulujących szlaki proliferacji i różnicowa-
nia komórkowego, którego uszkodzenia mogą być przyczy-
ną inicjacji i progresji nowotworów, ale również tworzenia
przerzutów. Zablokowanie proliferacji tych komórek spo-
woduje zmniejszenie masy guza i zatrzymanie rozwoju
choroby nowotworowej. W wyniku badań okazało się, że
zablokowanie szlaku Notch spowodowało zredukowanie
do 1/5 komórek CSC, co zatrzymało proces formowania
guza [74]. Udowodniono również, że zastosowanie cyklo-
paminy, która wiąże białko SMO szlaku Shh powoduje
uwrażliwienie nowotworowych komórek macierzystych na
Rycina 4. Szlak sygnałowy Shh. Sygnałem dla komórki jest przyłączenie cząstecz-
ki cholesterolu do karboksylowego końca receptora Shh, a następnie połączenie
tego kompleksu z receptorem Ptch, co wywołuje aktywację białka SMO (ang.
smothened). Następnie SMO powoduje rozszczepienie białek Gli i przedostanie
się ich fragmentów do jądra komórkowego indukując geny odpowiedzialne za
proliferację[58].

Postępy Biochemii 59 (1) 2013
51
radioterapie [73]. Przypuszcza się również, że nieprawidło-
wa sygnalizacja Wnt powoduje transformacje normalnych
komórek macierzystych w nowotworowe i inicjuje powsta-
wanie nowotworu [64,75].
Wiele poruszanych tu aspektów dotyczących nowotwo-
rowych komórek macierzystych to jeszcze hipotezy, lecz są
one obecnie przedmiotem licznych badań, które pozwolą w
niedalekiej przyszłości na ich pełniejsze wyjaśnienie.
PIŚMIENNICTWO
1. Bach SP, Renehan AG, Potten CS (2000) Stem cells: the intestinal stem
cell as a paradigm. Carcinogenesis 3: 469-476
2. Gil J, Stembalska A, Pesz KA, Sąsiadek MM (2008) Cancer stem cells:
the theory and perspectives in cancer therapy. J App Genet 49: 193-199
3. Jiang J, Lv Z, Gu Y, Li J, Xu L, Xu W, Lu J, Xu J (2010) Adult rat me-
senchymal stem cells differentiate into neuronal-like phenotype and
express a variety of neuro-regulatory molecules in vitro. Neurosci Res
66: 46-52
4. Si YL, ZhaoYL, Hao HJ, Fu XB, Han WD (2011) MSCs: Biological cha-
racteristics, clinical applications and their outstanding concerns. Age-
ing Res Rev 10: 93-103
5. Kucia M, Reca R, Cambell FR (2006) A population of very smallembry-
onic-like (VESEL) CXCR4(+) SSEA-1(+) Oct4(+) stem cells identified in
adult bone marrow. Leucemia 20: 857-869
6. Jaenisch R, Young R (2008) Stem cells, the molecular circuitry of pluri-
potency and nuclear reprogramming. Cell 13: 567-582
7. Dai LJ, Moniri MR, Zeng ZR, Zhou JX, Rayat J, Warnock GL (2011)
Potential implications of mesenchymal stem cells in cancer therapy.
Cancer Lett 305: 8-20
8. Sun XY, Nong J, Qin K, Warnock GL, Dai LJ (2011) Mesenchymal stem
cell-mediated cancer therapy: A dual-targeted strategy of personalized
medicine. World J Stem Cells 3: 96-103
9. Sharpe ME, Morton D, Rossi A (2012) Nonclinical safety strategies for
stem cell therapies. Toxicol Appl Pharmacol 262: 223-231
10. Wong RS (2011) Mesenchymal Stem Cells: Angles or Demons? J Bio-
med Biotechnol doi:10.1155/2011/459510
11. Amariglio N, Hirshberg A, Scheithauer BW, Cohen Y, Loewenthal R,
Trakhtenbrot L, Paz N, Koren-Michowitz M, Waldman D, Leider-Tre-
jo L, Toren A, Constantini S, Rechavi G (2009) Donor-derived brain
tumor following neural stem cell transplantation in an ataxia telan-
giectasia patient. PLoS Med 6: 221-231
12. Virchow R (1855) Editorial Archive fuer pathologische Anatomie und
Physiologie fuer klinishe Medizin. 8: 23-54
13. Hamburger AW, Salmon SE (1977) Primary bioassay of human tumor
stem cells. Science 197: 461-463
14. Southam C, Brunschwig A (1961) Quantitative studies of autotrans-
plantation of human cancer. Cancer 14: 461-463
15. Bonnet D, Dick JE (1997) Human acute myeloid leukemiais organized
as a hierarchy that originates from a primitive hematopoietic cell. Na-
ture Med 3: 730-737
16. Al-Hajj M, Wicha MS, Benito-Hernandez, A, Morrison SJ, Clarke MF
(2003) Prospective identification of tumorigenic breast cancer cells.
Proc Natl Acad Sci USA 100: 3983-3988
17. Singh SK, Hawkins C, Clarke ID, Squire JA (2004) Identification of hu-
man brain tumour initiating cells. Nature 432: 396-401
18. Huntly BJ, Gilliland DG (2005) Leukaemia stem cells and the evolution
of cancer-stem-cell research. Nat Rev Cancer 5: 311-321
19. Xiong-Zhi Wu (2007) Origin of Cancer Stem Cells: The role of Self-Re-
newal and Differentiation. Ann Surg Oncol 15: 407-414
20. Shipitsin M, Polyak K (2008) The cancer stem cell hypothesis: in search
of definitions, markers, and relevance. Lab Invest 88: 459-463
21. Santostefano KE, Hamazaki T, Pardo CE, Kladde MP, Terada N (2012)
Fibroblast Growth Factor Receptor 2 homodimerization rapidly redu-
ces transcription of the pluripotency gene Nanog without dissociation
of activating transcription factors. J Biol Chem 36: 30507-30517
22. Kucia M, Ratajczak MZ (2006) Stem cells as a two edgeg sword-from
regeneration to tumor formation. J Physiol Pharmacol 7: 5-16
23. Staniszewska M, Słuczanowska-Głębowska S (2008) Komórki naskór-
ka i ich znaczenie w procesach odnowy. Dermatol Klin 10: 100-104
24. Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer,
and cancer stem cells. Nature 414: 105-111
25. Mackenzie IC (2008) Cancer stem cells. Ann Onkol 5: 40-43
26. Korkaya H, Liu S, Wicha MS (2011) Breast cancer stem cells, cytokine
networks, and the tumor microenvironment. J of Clin Invest 121: 3804-
3809
27. Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE (2008)
Drug-Selected Human Lung Cancer Stem Cells: Cytokine Network,
Tumorigenic and Metastatic Properties. PLoS One 8: 1-16
28. Sacewicz I, Wiktorska M, Wysocki T, Niewiarowska J (2009) Mecha-
nizmy angiogenezy nowotworowej. Postępy Hig Med Dosw 63: 159-
168
29. Discher DE, Mooney DJ, Zandstra PW (2009) Growth Factors, Matri-
ces, and Forces Combine and Control Stem Cells. Science 5935: 1673-
1677
30. Haghighipour N, Heidarian S, Shokrgozar MA, Amirizadeh N (2012)
Differential effects of cyclic uniaxial stretch on human mesenchymal
stem cell into skeletal muscle cell. Cell Biol Int 36: 669-675
31. Dean M, Fojo T, Bates S (2005) Tumor stem cells and drug resistance.
Nat Rev Cancer 5: 275-284
32. Statkiewicz M, Małecki M (2009) Macierzyste komórki nowotworowe,
a oporność nowotworów na terapie. Nowotrory 6: 456-463
33. Jamroziak K, Kowalczyk M, Robak T (2002) Białko oporności raka
piersi ABCG2(BCRP/MXR/ABCP) — nowy transporter z nadrodziny
ABC związany z opornością wielolekową. Acta Haem Pol 33: 4
34. Wai LK (2004) Telomeres, Telomerase, and Tumorigenesis. Med Gen
Med 6: 19
35. Hiyama E, Hiyama K (2007) Telomere and telomerase in stem cells. Br
J Cancer 96: 1020-1024
36. Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-
Wieczorek A, Ratajczak J, Ratajczak MZ (2005) Trafficking of normal
stem cells and metastasis of cancer cells involve similar mechanisms:
pivotal role of the SDF-1-CXCR4 axis. Stem Cells 23: 879-894
37. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClana-
han T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Veraste-
qui E, Zlotnik A (2001) Involvement of chemokine receptors in breast
cancer metastasis. Nature 410: 50-56
38. Janik P (2008) Stemowe komórki nowotworowe. Nowotwory 3: 221-
224
39. Pikuła M, Trzonkowski P (2009) Biologia komórek macierzystych
naskórka oraz ich znaczenie w medycynie. Postępy Hig Med Dosw
65: 449-456
40. Reya T (2003) Regulation of Hematopoietic Stem Cell Self-Renewal.
Recent Prog Horm Res 58: 283-295
41. Behbod F, Rosen JM (2004) Will cancer stem cells provide new thera-
peutic targets? Carcinogenesis 4: 703-711
42. Behrens J, Lusting B (2004) The Wnt connection to tumorigenesis. Int
J Dev Biol 48: 477-487
43. Janssens N, Janicot M, Perera T (2006) The Wnt-dependent signaling
pathways as target in oncology drug discovery. Invest New Drugs 24:
263-280
44. Staal FJ, Noort MM, Strous GJ, Clevers HC (2002) Wnt signals are
transmitted through N-terminally dephosphorylated beta-catenin.
EMBO Rep 3: 63-68
45. Gehrke I, Gandhirajan RK, Kreuzer KA (2009) Targeting the WNT/b-
catenin/TCF/LEF1 axis in solid and haematological cancers: Multi-
plicity of therapeutic options. Eur J Cancer 45: 2759-2767
46. Frye M, Gardner C, Li ER, Arnold I, Watt FM (2003) Evidence that Myc
activation depletes the epidermal stem cell compartment by modulat-
ing adhesive interactions with the local microenvironment. Develop-
ment 130: 2793-2808
52
www.postepybiochemii.pl
47. Dworakowska D (2005) Rola białka p53, pRb, p21
WAF1/CIP1
, PCNA,
MDM2 oraz cykliny D1 w regulacji cyklu komórkowego oraz apop-
tozy. Onkol Pol 4: 223-228
48. Marconi A, Dallaglio K, Lotti R, Vaschieri C (2007) Survivin identifi es
keratinocyte stem cells and is downregulated by anti-b1 integrin dur-
ing anoikis. Stem Cells 25: 149-155
49. Ambrosini G, Adida C, Altieri DC (1997) A novel anti-apoptosis gene,
survivin, expressed in cancer and lymphoma. Nat Med 3: 917-921
50. Weeraratna AT, Jiang Y, Hostetter
G, Rosenblatt K, Duray P, Bittner
M, Trent JM (2002) Wnt5a signaling directly affects cell motility and
invasion of metastatic melanoma. Cancer Cell 1: 279-288
51. Tataria MD, Perryman SV (2006) Stem cells: Tissue regeneration and
cancer. Semin Pediatr Surg 15: 284-292
52. Tsai RY (2004) A molecular view of stem cell and cancer cell self-re-
newal. Int J Biochem Cell Biol 36: 684-694
53. Balint K, Xiao M, Pinnix CC, Soma A, Veres I (2005) Activation of
Notch1 signaling is required for β-catenin-madiated human primary
melanoma progression. J Clin Invest 115: 3166-3176
54. Nickoloff BJ, Osborne BA, Miele L (2003) Notch signaling as a thera-
peutic target in cancer: a new approach to the development of cell fate
modifying agents. Oncogene 22: 6598-6608
55. Lai EC (2004) Notch signaling: control of cell communication and cell
fate. Development 131: 965-973
56. Liu ZJ, Xiao M, Balint K, Smalley KS (2006) Notch1 signaling promotes
primary melanoma progression by activating mitogen-activated pro-
tein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-reg-
ulating N-cadherin expression. Cancer Res 66: 4182-4190
57. Wu
Y, Cain-Hom
C, Choy
L, Hagenbeek
TJ, Leon GP (2010) Thera-
peutic antibody targeting of individual Notch receptors. Nature 464:
1052-1057
58. Daya-Grosjean L, Couve-Privat S (2005) Sonic hedgehog signaling in
basal cell carcinomas. Cancer Lett 225:181-192
59. Jia-xi Z, Li-Wei J, Wei-min L, Cheng-lin M (2006) Role of sonic hedge-
hog In maintaining a pool of proliferating stem cells In the human fetal
epidermie. Hum Reprod 21: 1698-1704
60. Lesiak A, Sysa-Jędrzejowska A, Narbutt J (2010) Rola ścieżki przeka-
zywania sygnału sonic hedgehog w procesie skórnej kancerogenezy.
Pol Merk Lec 29: 141-143
61. Frank-Kamentsky M, Zhang XM, Bottega S (2002) Small-molecule
modulators of Hedgehoge signaling: identification and characteriza-
tion of Smoothened agonists and antagonists. J Biol 1: 10-17
62. Clement
V, Sanchez P, Tribolet
N, Radovanovic
I (2007) HEDGEHOG-
GLI1 Signaling regulates human glioma growth, cancer stem cell self-
renewal, and tumorigenicity. Curr Biol 17: 165-172
63. Bourguignon LY, Peyrollier K, Xia W (2008) Hyaluronan-CD44 inter-
action activates stem cell marker Nanog, Stat-3-mediated MDR1 gene
expression, and ankyrin-regulated multidrug efflux in breast andovar-
ian tumor cells. J Biol Chem 283: 17635-17651
64. Wang J, Guo LP, Chen LZ (2007) Identification of cancer stem cell-like
side population cells in human nasopharyngeal carcinoma cell line.
Cancer Res 67: 3716-3724
65. Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G,
George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu
KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan
Y, Lim B, Ng HH (2006) The Oct-4 and Nanog transcription network
regulates pluripotency in mouse embryonic stem cells. Nat Genet 38:
431-440
66. Jungwoon Lee, Yeorim Go, Inyoung Kang, Yong-Man Han, Jungho
Kim (2010) Oct-4 controls cell-cycle progression of embryonic stem
cells. Biochem J 426: 171-181
67. Atlasi Y, Mowla SJ, Zieaee SA, Gokhale PJ, Andrews PW (2008) Oct4
Spliced variants are differentially expressed in human pluripotent and
nonpluripotent cells. Stem Cells 26: 3068-3074
68. Chang CC, Shieh GS, Wu P, Lin CC, Shiau AL, Wu CL (2008) Oct3/4
expression reflects tumor progression and regulates motility of blad-
der cancer cells. Cancer Res 68: 6281-6291
69. Kim RJ, Nam JS (2011) OCT4 Expression enhances features of cancer
stem cells in a mouse model of breast cancer. Lab Anim Res 27: 147-152
70. Rodda DJ, Chew JL, Lim LH, Loh YH (2005) Transcriptional regulation
of Nanog by Oct4 and Sox2. J Biochem 280: 24731-2473
71. Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Za-
ehres H, Shen JJ, Tang DG (2011) Nanog promotes cancer stem cell
characteristics and prostate cancer resistance to androgen deprivation.
Oncogene 30: 3833-3845
72. Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, Xu Y, Ma Q, Yang Z, Zhang
Q (2012) Nanog regulates self-renewal of cancer stem cells through the
insulin-like growth factor pathway in human hepatocellular carcino-
ma. Hepatology 56: 1004-1014
73. Pan G, Thomson JA (2007) Nanog and transcriptional networks in em-
bryonic stem cell pluripotency. Cell Res 17: 42-49
74. Fan X, Matsui W, Khaki L (2006) Notch pathway inhibition depletes
stemlike cells and blocks engraftment in embryonal brain tumors.
Cancer Res 66: 7445-7452
75. Hombach-Klonisch S, Paranjothy T, Wiechec E (2008) Cancer stem
cellsas targets for cancer therapy: selected cancers as examples. Arch
Immunol Ther Exp 56: 165-180
The importance of stem cells in the initiation and development of cancer
Paulina Pergoł
1,
, Agata Nowak-Stępniowska
1
, Katarzyna Drela
2
Alfreda Padzik-Graczyk
1
1
Biochemistry Laboratory, Institute of Optoelectronics, Military University of Technology, 2 Kaliskiego St., 00-908 Warsaw, Poland
2
NeuroRepair Department, Mossakowski Medical Research Centre, Polish Academy of Sciences, 5 Pawińskiego St., 02-106 Warsaw, Poland
e-mail: paulinapergol@wp.pl
Key words: stem cells, cancer stem cells, cancer
ABSTRACT
Initiation of cancer may be the result of mutations occurring in stem cells, which causes blocking the differentiation of these cells. Many
common properties of the stem cells and some tumor cells suggests that cancer stem cells may be responsible for the initiation and progres-
sion of cancer. The special properties of CSC is the ability to self-renewal and cell proliferation, which are the major cause of cancer recur-
rence and metastasis. Signaling pathways (Wnt, Notch, Shh) and pluripotency- connected transcription factors (Oct-4, Nanog) are primarily
responsible for cell proliferation. Understanding the causes of initiation and progression of cancer is crucial for improving treatment of these
life-threatening diseases.
Wyszukiwarka
Podobne podstrony:
03 PO Geometria 2013id 4609 Nieznany (2)
Konserwacja 2014 03 id 245321 Nieznany
03 Kinematykaid 4394 Nieznany
713[05] Z1 03 Wykonywanie izola Nieznany (2)
03 5id 4121 Nieznany
ais 03 id 53431 Nieznany (2)
712[06] S1 03 Montowanie system Nieznany (2)
03 4id 4118 Nieznany (2)
Chemia 03 id 557778 Nieznany
2014 Matura 01 03 2014id 28469 Nieznany (2)
Biul Moni Przyr 1(4)03 Aves id Nieznany
05 med dosw 4 2013id 5960 Nieznany (2)
03 a, l, o, m, t, i, eid 4311 Nieznany
03 12id 4271 Nieznany (2)
1 rok mgr22,02,2013id 9707 Nieznany
03 Rozdz I (B J 2012) Nieznany (2)
03 ulozeniaid 4513 Nieznany (2)
03 Organizowanie i prowadzenie Nieznany
PRZEKRA J TEOWY 2012 03 23 id 3 Nieznany
więcej podobnych podstron