1
Czynniki warunkujące aktywność enzymów na przykładzie fosfatazy
kwaśnej (EC 3.1.3.2 – fosfohydrolaza monoestrów ortofosforanowych
kwaśne opimum).
Cel ćwiczenia
Celem ćwiczenia jest wykazanie, na przykładzie fosfatazy kwaśnej, że takie czynniki
jak stężenie enzymu, temperatura, stężenie jonów wodorowych (pH) oraz inhibitory wpływają
na szybkość reakcji enzymatycznych.
Wprowadzenie
Fosfatazy (fosfomonoesterazy) należą do enzymów klasy hydrolaz, podklasy esteraz
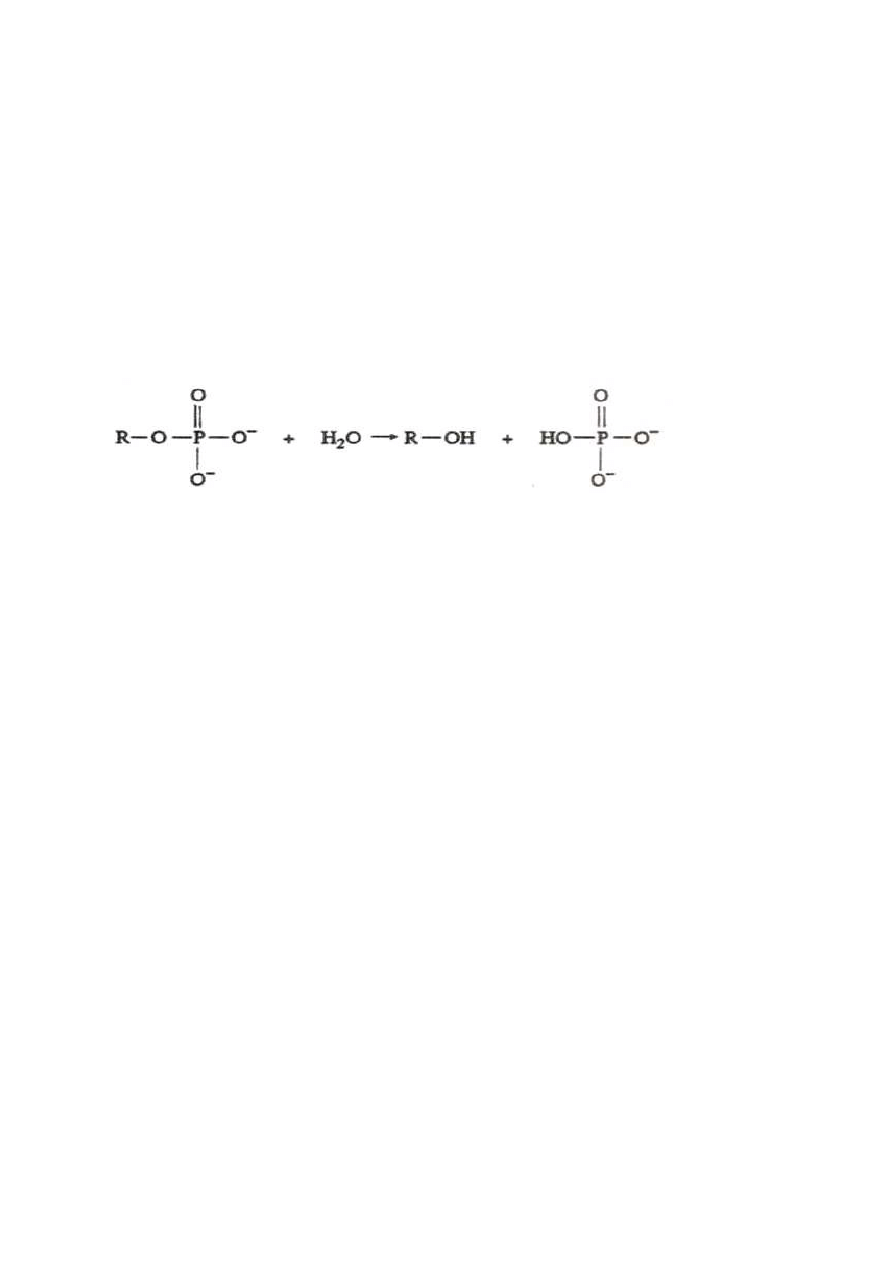
i katalizują hydrolizę różnych estrów fosforanowych, zgodnie z reakcją:
monoester fosforanowy alkohol monofosforan
Ze względu na na optimum pH działania fosfatazy zostały podzielone na dwie grupy:
• fosfatazy kwaśne aktywne w zakresie pH 4,0 do 7,0;
• fosfatazy alkaliczne aktywne w zakresie pH 9,0 do 11,0.
Roślinne fosfatazy kwaśne, w większości są dimerami zbudowanymi z podjednostek o
charakterze glikoprotein i masie 50-60 kD. Występują w różnych organach roślin: w bulwach,
nasionach,
warstwie
aleuronowej
ziarniaków,
owocach,
liścieniach,
brodawkach
korzeniowych i liściach. Na terenie komórki fosfatazy kwaśne zlokalizowane są w cytosolu,
wakuolach lub ścianie komórkowej. Fosfatazy kwaśne charakteryzują się niską
specyficznością substratową. Mogą katalizować defosforylację różnych naturalnych
substratów, takich jak 3-fosfoglicerynian, fosfoenolopirogronian (PEP), fityna, ATP, czy
ufosforylowane na tyrozynie białka. Funkcje biochemiczne fosfataz kwaśych w komórkach
roślin nie zostały dokładnie poznane. Przypuszcza się, że uwalniają fosforan ze związków
organicznych w warunkach niedoboru fosforanów, w warunkach stresu zasolenia lub deficytu
wody, bądź w warunkach stresu wywołanego atakiem patogenów, w ontogenezie. Do
najbardziej poznanych enzymów roślinnych należą: fitaza jak również fosfatazy należące do
nadrodziny fosfataz tyrozynowych.
U zwierząt, fosfataza kwaśna jest enzymem obecnym głównie w lizosomach komórek
kości, gruczołu krokowego, jelit, trzustki i nerek, w płytkach krwi i krwinkach czerwonych.
Zwiększony poziom fosfatazy kwaśnej jest obserwowany w niektórych chorobach
nowotworowych.
In vitro,
aktywność fosfataz oznacza się zazwyczaj używając sztucznych substratów,
takich jak: 2-glicerofosforan, fenylofosforan, fosforan fenoloftaleiny, p-nitrofenylofosforan
pNPP). Aktywność enzymu mierzy się ilością uwolnionego fosforanu lub części organicznej
estru po reakcji enzymu z substratem prowadzonej w ściśle określonych warunkach.
Szczególnie dogodnym substratem jest p-nitrofenylofosforan, gdyż jeden z produktów reakcji
po zalkalizowaniu środowiska przechodzi w formę barwną.
Na ćwiczeniu będzie wykorzystany wodny wyciąg fosfatazy kwaśnej z siewek pszenżyta.
Jako substrat będzie zastosowany p-nitrofenylofosforan (pNPP). Reakcję hydrolizy tego
związku przedstawia rysunek1. Powstający produkt reakcji, p-nitrofenol (pNP), przyjmuje w

2
ś
rodowisku zasadowym barwę żółtozieloną, a jego ilość jest miarą aktywności fosfatazy.
Rys. 1. Reakcja hydrolizy p-nitrofenylofosforanu z udziałem fosfatazy kwaśnej.
Na szybkość reakcji enzymatycznych wpływa szereg czynników, przede wszystkim
stężenie substratu, stężenie enzymu, stężenie jonów wodorowych w środowisku reakcji,
temperatura a także obecność inhibitorów.
Badanie aktywności enzymów
Miarą aktywności enzymu jest szybkość reakcji enzymatycznej (V) mierzona ilością substratu
przekształconego w jednostce czasu lub ilością produktu wytworzonego w jednostce czasu.
Aktywność enzymu wyraża się w jednostkach aktywności.
Podstawową jednostką jest jednostka uniwersalna ( standardowa. Za taką jednostkę
przyjęto ilość enzymu, która katalizuje przemianę 1 µmola substratu w ciągu 1 min., w
temperaturze 30°C i optymalnych warunkach pH oraz stężenia substratu.
Odczynniki:
1.
Ekstrakt enzymu: do150 mg liści 10 – dniowych siewek pszenżyta dodać 100 ml wody
destylowanej i homogenizować przez 5 min. Homogenat przesączyć przez sączek z
waty i przechowywać w lodówce.
2.
Roztwór substratu: 0.0075 M p - nitrofenylofosforan disodowy (pNPP).
3.
0.05 M bufor cytrynianowy o pH 5,2
4.
0.05 M bufor cytrynianowy o pH 3,0
5.
0.05 M bufor cytrynianowy o pH 4,4
6.
0.05 M bufor cytrynianowy o pH 6,0
7.
10% Na
2
CO
3
(węglan sodowy).
8.
1 mM molibdenian amonu
1. Wpływ stężenia enzymu na szybkość przebiegu reakcji.
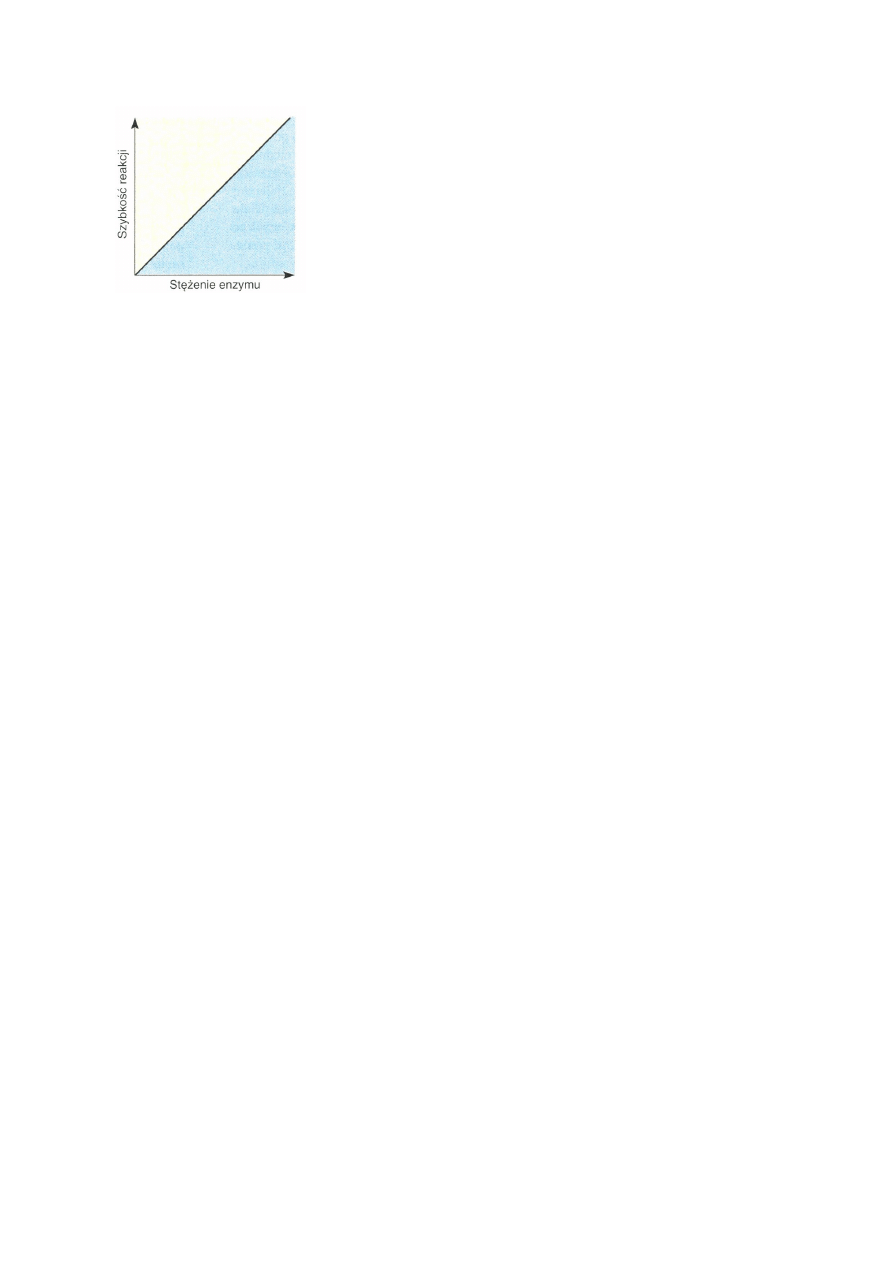
W warunkach optymalnych szybkość reakcji enzymatycznej zmienia się
proporcjonalnie do stężenia enzymu: im więcej jest cząsteczek enzymu, tym więcej
cząsteczek substratu zostanie przekształconych w jednostce czasu. Badanie zależność
szybkości reakcji od stężenia enzymu prowadzimy przy stałym wysokim stężeniu substratu,
bo wówczas szybkość reakcji jest proporcjonalna do stężenia enzymu. Wykresem zależności
szybkości reakcji od stężenia enzymu jest linia prosta.
3
Rys.2. Wpływ stężenia enzymu na szybkość reakcji.
Wykonanie
Do pięciu ponumerowanych probówek (próby właściwe) odmierzyć kolejno po 0,25; 0,5;
0,75; 1,0 i 1,25 ml roztworu enzymu (1), po 0,75 ml buforu cytrynianowego o pH 5,2 (3) a
następnie uzupełnić ich zawartość wodą destylowaną do objętości 2 ml. Do probówki numer
6 (próba kontrolna) odmierzyć 0,75 ml buforu cytrynianowego o pH 5,2 i 1,25 ml wody
destylowanej. Wszystkie probówki umieścić na 5 min (preinkubacja, czyli podgrzanie
mieszaniny reakcyjnej do temperatury, w której przebiega reakcja) w łaźni wodnej o
temperaturze 30
0
C. Po preinkubacji, nie wyjmując prób z łaźni ! dodać kolejno do
wszystkich po 0,5 ml roztworu substratu (2), wymieszać i inkubować w łaźni jeszcze przez 15
min., licząc czas reakcji od momentu dodania substratu. Następnie przerwać reakcję dodając
do wszystkich prób po 2,5 ml węglanu sodowego(4). Zmierzyć absorbancję prób właściwych
w fotometrze przy długości fali 420 nm ustawionym na zero wobec próby kontrolnej.
Obliczenia:
Obliczyć szybkość reakcji wyrażając ją jako ilość µmoli uwolnionego w reakcji
p-nitrofenolu w czasie 1 min., dzieląc absorbancję próby przez współczynnik absorbancji dla
1 µg p-nitrofenolu równy 0,002 oraz masę molową p-nitrofenolu równą 139 i czas reakcji
wynoszący10 min. ( A
420
/ 0,002 x 139 x 15 min.).
Przykładowe obliczenia:
A
420
= 0.34, to szybkość reakcji v = 0,34 / 0.002 x 139 x 15 = 0.082 µmola p-nitrofenolu/min.
Takie obliczenia wykonać dla każdej uzyskanej w doświadczeniu wartości absorbancji.
2. Wpływu pH na szybkość reakcji enzymatycznych.
Wpływ pH na aktywność enzymu związany jest z wartością stałych dysocjacji:
1.
grup czynnych w centrum aktywnym enzymu uczestniczących w wiązaniu substratu i
katalizie.
2.
grup funkcyjnych w cząsteczce substratu, którymi wiąże się on z enzymem.
3.
innych grup w cząsteczce enzymu, których stan zjonizowania może gwarantować
katalitycznie aktywną konformację.
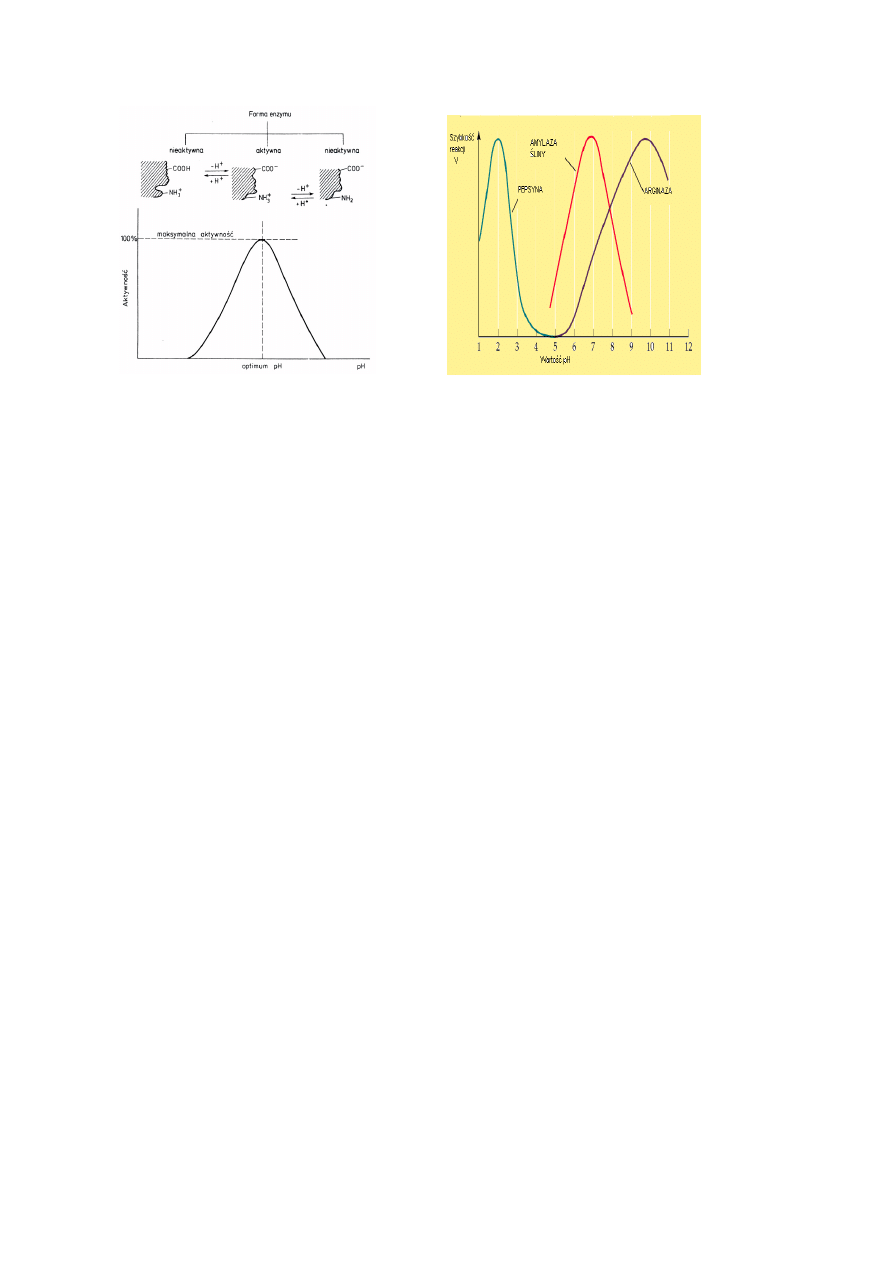
Wobec tego, każdy enzym ma optymalną wartość pH, przy której szybkość katalizowanej
reakcji jest maksymalna. Niewielkie odchylenia od optimum powodują zmniejszenie
szybkości reakcji, a duże odchylenia prowadzą do denaturacji białka enzymu, czego skutkiem
jest utrata aktywności..
4
Rys.3. Wpływ pH na aktywność enzymu. (Witwicki, Ardelt Elementy enzymologii PWN
1984).
Optimum pH może ulegać pewnym zmianom zależnie od stopnia czystości preparatu
enzymatycznego i temperatury środowiska. Wartość optymalnego pH działania enzymów
zależy organizmu oraz od lokalizacji enzymu w tkance, a nawet w przedziale komórkowym
(Rys. 3). Dlatego wartość pH wewnątrz komórki może stanowić ważny czynnik regulacji
metabolizmu.
Wykonanie
Do czterech kolejno ponumerowanych probówek (próby właściwe) odmierzyć po 0,5 ml
enzymu, dodać po 0,75 ml wody destylowanej oraz po 0,75 ml buforów: do pierwszej buforu
o pH 3,0; do drugiej o pH 4,4; do trzeciej o pH 5,2 i do czwartej o pH 6,0. Do probówki
numer 5 (próba kontrolna) odmierzyć 0,75 ml buforu cytrynianowego o pH 5,2 oraz 1,25 ml
wody destylowanej. Wszystkie probówki umieścić w łaźni wodnej o temperaturze 30
0
C na 5
min. (okres preinkubacji). Po preinkubacji, nie wyjmując prób z łażni dodać do wszystkich
po 0,5 ml roztworu substratu, wymieszać i inkubować jeszcze przez 15 min., licząc czas
reakcji od momentu dodania substratu. Następnie przerwać reakcję dodając do wszystkich
prób po 2,5 ml węglanu sodowego. Zmierzyć absorbancję prób właściwych w fotometrze,
przy długości fali 420 nm, ustawionym na zero wobec próby kontrolnej.
Obliczenia:
Obliczyć szybkość reakcji wyrażając ją jako ilość µmoli uwolnionego w reakcji
p-nitrofenolu w czasie 1 min., dzieląc absorbancję próby przez współczynnik absorbancji dla
1 µg p-nitrofenolu równy 0,002 oraz masę molową p-nitrofenolu równą 139 i czas reakcji
wynoszący10 min. ( A
420
/ 0,002 x 139 x 15 min.).
Przykładowe obliczenia:
A
420
= 0.34, to szybkość reakcji v = 0,34 / 0.002 x 139 x 10 = 0.082 µmola p-nitrofenolu/min.
Takie obliczenia wykonać dla każdej uzyskanej w doświadczeniu wartości absorbancji.
3. Wpływ temperatury na aktywność enzymów, badanie termostabilności fosfatazy
kwaśnej.
Niska temperatura hamuje działanie enzymów ze względu na niską energię kinetyczną
reagujących cząsteczek. Szybkość reakcji enzymatycznych wzrasta wraz z podwyższeniem
temperatury. Wiąże się to ze wzrostem energii kinetycznej reagujących cząstek i większą
częstotliwością ich zderzeń. Zgodnie z regułą van’t Hoffa – podwyższenie temperatury o
10°C około dwukrotnie przyspiesza reakcje enzymatyczne. W temperaturze optymalnej –
szybkość reakcji jest największa. Zakres wartości temperatury optymalnej może się zmieniać
w zależności od czasu trwania reakcji, od pH, obecności innych substancji w mieszaninie
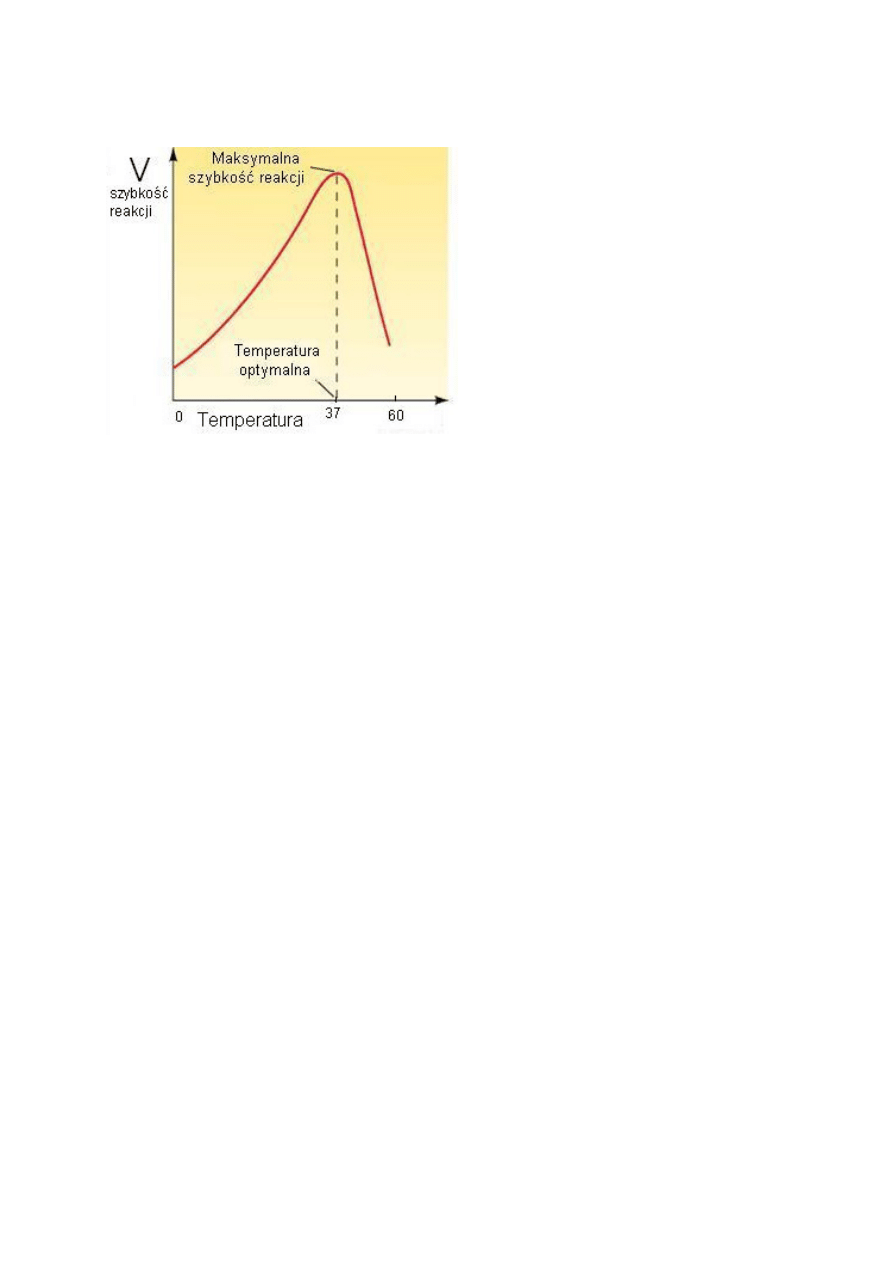
5
reakcyjnej. Przy wzroście temperatury powyżej optymalnej następuje gwałtowne obniżenie
szybkości reakcji, co jest związane z denaturacją cieplną białka enzymu.
Rys.4. Wpływ temperatury na aktywność enzymu.
Zazwyczaj obecność substratu i optymalne pH środowiska reakcji zmniejszają podatność
białka enzymu na denaturację.
Większość enzymów ulega powolnej denaturacji nawet w temperaturach optymalnych.
Podatność enzymu na denaturację cieplną, tak zwana termostabilność zależy od struktury
molekularnej oraz pochodzenia enzymu. Najwyższa temperatura, w której jeszcze nie
zachodzi termiczna inaktywacja enzymu w danych warunkach określa tak zwaną
termostabilność enzymu. Termostabilność enzymu badamy inkubując roztwór enzymu w
różnych temperaturach przez określony czas. Następnie doprowadzamy roztwór enzymu do
temperatury optymalnej, dodajemy substrat i pozostałe składniki reakcji, oznaczamy
aktywność i porównujęmy z aktywnością tego enzymu katalizującego reakcję w warunkach
optymalnych.
Wykonanie
Do czterech kolejno ponumerowanych probówek (próby właściwe) odmierzyć po 0,5
ml ekstraktu enzymatycznego, a następnie pierwszą umieścić w łaźni wodnej o temperaturze
30
0
C, drugą w łaźni o temperaturze 40
0
C, trzecią w łaźni o temperaturze 50
0
C i czwartą w
łaźni o temperaturze 60
0
C i inkubować przez 30 min. Następnie, wszystkie probówki po
schłodzeniu umieścić w łażni o temperaturze 30
0
C i po 5 min. preinkubacji przeprowadzić
reakcję dodając do każdej z nich po 0,75 ml buforu cytrynianowego o pH 5,2 i 0,75 ml H
2
O
dest., 0,5 ml substratu. Do probówki piątej (próba kontrolna) odmierzyć 0,75 ml buforu
cytrynianowego o pH 5,2 i 1,25ml H
2
O dest. oraz 0,5 ml substratu. Po 15 min. przerwać
reakcję dodając do wszystkich pięciu probówek po 2,5 ml 10% węglanu sodowego (4).
Zmierzyć absorbancję prób właściwych, przy długości fali 420 nm, ustawionym na zero
wobec próby kontrolnej.
Obliczenia
Na podstawie uzyskanych wyników obliczyć szybkość reakcji katalizowanej przez fosfatazę
kwaśną w poszczególnych temperaturach, wyrażając ją jako ilość µmoli uwolnionego w
reakcji p-nitrofenolu w czasie 1 min. (A
420
/ 0,002 x 139 x 15 min.).
4. Inhibicja aktywności enzymów.
Aktywność wielu enzymów może być hamowana przez związanie działających w
sposób specyficzny inhibitorów. Inhibitory hamują działanie enzymów wywołując zmiany w
obrębie centrum aktywnego. Inhibitorami mogą być metabolity komórkowe, leki, trucizny lub
inne substancje obce dla organizmu (ksenobiotyki). W zależności od sposobu działania
inhibitora wyróżniamy inhibicję nieodwracalną i odwracalną.
6
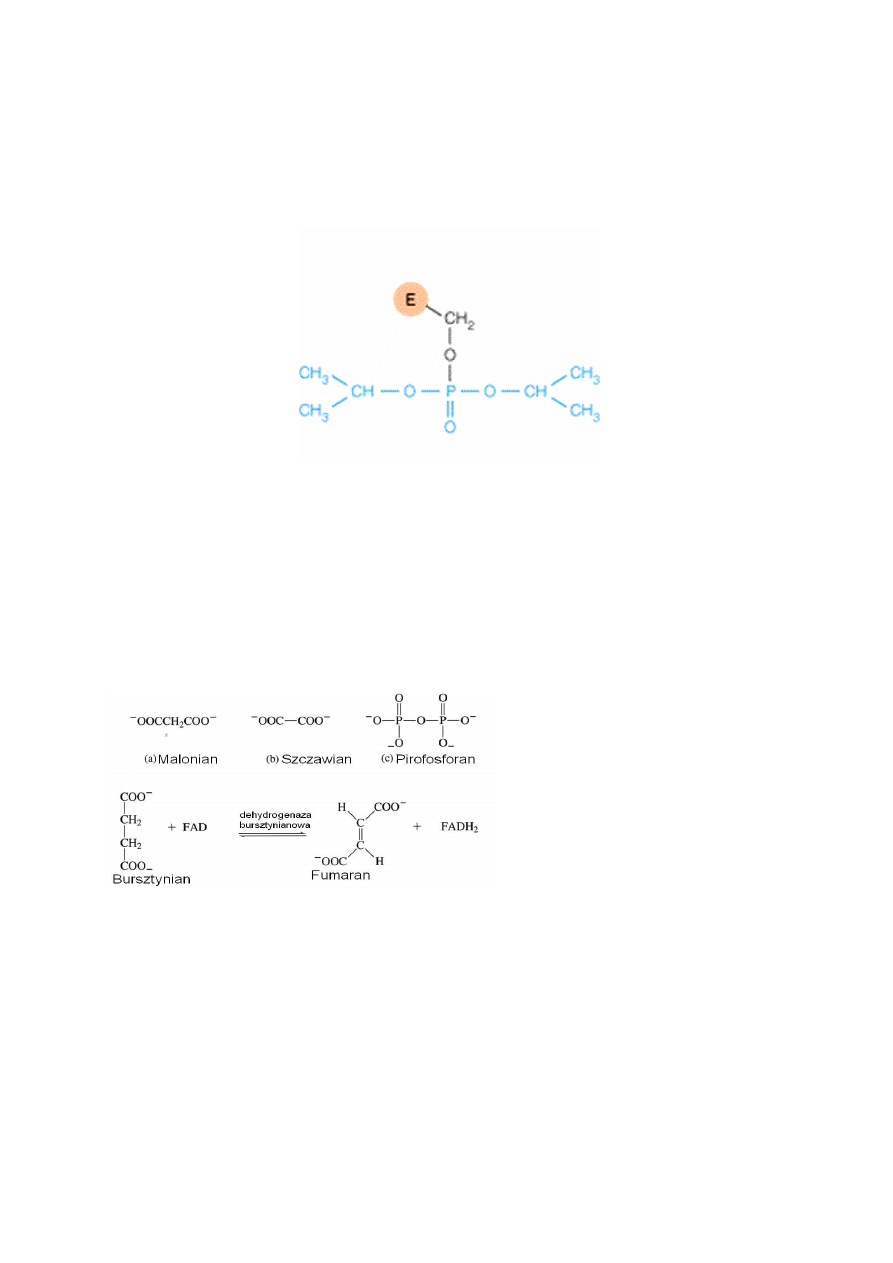
W inhibicji nieodwracalnej inhibitor wiąże się kowalencyjnie z enzymem. Przykładem
takiego inhibitora jest penicylina, która wytwarza wiązanie kowalencyjne z grupą –OH Ser w
centrum aktywnym transpeptydazy peptydoglikanu, enzymu uczestniczącego w syntezie
ś
ciany komórkowej bakterii i w rezultacie powoduje śmierć bakterii. Podobne działanie
wykazuje DFP – diizopropylofluorofosforan, który blokuje grupy –OH Ser w centrum
aktywnym esterazy acetylocholinowej. Stosowany jest jako pestycyd.
Rys. 5. Kompleks DFP (kolor niebieski) z grupą –OH Ser w centrum aktywnym enzymu.
Inhibicję odwracalną charakteryzuje szybka dysocjacja kompleksu enzym-inhibitor.
Wyróżniamy
dwa
typy
inhibicji
odwracalnej:
współzawodniczą
(kompetycyjną)
i niewspółzawodniczą (niekompetycyjną).
Inhibitor kompetycyjny przyłącza się tylko do wolnego enzymu (E) i blokuje jego
centrum aktywne, przez co zmniejsza liczbę cząsteczek enzymu wiążących substrat. Ten typ
inhibitora zwiększa wartość liczbową Km ale nie zmienia wartości Vmax. . Przy określonym
stężeniu inhibitora można cofnąć inhibicję kompetycyjną poprzez zwiększenie stężenia
substratu. Wiele leków działa jako inhibitory współzawodnicze kluczowych enzymów
przemian metabolicznych.
Rys. 6. Działanie inhibitora kompetycyjnego.
Stosowany na ćwiczeniach molibdenian amonu jest inhibitorem kompetycyjnym fosfatazy
kwaśnej.
Inhibitor niewspółzawodniczy (niekompetycyjny) może przyłączyć się zarówno do
wolnego enzymu (E) jak i do kompleksu enzym-substrat (ES). Inhibitor niekompetycyjny
obniża wartość Vmax ale nie zmienia wartości Km ponieważ nie zmniejsza liczby cząsteczek
enzymu wiążących substrat. Tego typu inhibicji nie można cofnąć poprzez zwiększenie
stężenia substratu, lecz należy zastosować związki reagujące z inhibitorem. Przykładem
inhibitora niekompetycyjnego jest pepstatyna A hamująca aktywność reniny, enzymu
uczestniczącego w mechanizmie regulacji ciśnienia tętniczego krwi lub jony metali, które
mogą reagować z grupami –SH w białku enzymu powodując zmiany konformacyjne.

7
A
B
Rys.6. Inhibitory niekompetycyjne : A -pepstatyna A inhibitor niekompetycyjny reniny,
B- działanie jonów metali jako inhibitorów niekompetycyjnych.
Wykonanie
Do dwu ponumerowanych probówek (próby właściwe) odmierzyć po 0,5 ml enzymu,
po 0,75 ml buforu cytrynianowego. Następnie do probówki pierwszej dodać 0,25 ml 1 mM
molibdenianu amonu. Zawartość probówki pierwszej i drugiej uzupełnić wodą destylowaną
do objętości 2,0 ml. Do probówki numer 3 (próba kontrolna) odmierzyć 0,75 ml buforu
cytrynianowego o pH 5,2. i 1,25 ml wody. Próby wstawić do łaźni wodnej o temperaturze
30
0
C na 5 min. (preinkubacja), a następnie nie wyjmując prób z łaźni do wszystkich dodać
po 0,5 ml substratu, czyli p-nitrofenylofosforanu sodu (2), wymieszać i inkubować w łaźni
jeszcze przez 15 min, licząc czas reakcji od momentu dodania substratu. Następnie przerwać
reakcję dodając do wszystkich prób po 2,5 ml węglanu sodowego (4). Zmierzyć absorbancję
prób właściwych w fotometrze przy długości fali 420 nm ustawionym na zero wobec próby
kontrolnej.
Podobnie jak w przypadku wcześniejszych doświadczeń obliczyć aktywność fosfatazy
kwaśnej w reakcji bez inhibitora i w obecności inhibitora.
Opracowanie wyników
1. Obliczyć szybkość reakcji wyrażając ją jako ilość µmoli uwolnionego w reakcji p-
nitrofenolu w czasie 1 min., dzieląc absorbancję próby przez współczynnik absorbancji dla
1 µg p-nitrofenolu równy 0,002 oraz masę molową p-nitrofenolu równą 139 i czas reakcji
wynoszący10 min. ( A
420
/ 0,002 x 139 x 10 min.).
2. Sporządzić wykresy zależności szybkości reakcji od stężenia enzymu, wartości pH,
temperatury, odkładając na osi OY (rzędnych) µmole 4-nitrofenolu, a na osi OX ( odciętych)
odpowiednio ml enzymu, wartości pH.
3. Traktując jako 100% wynik aktywności fosfatazy kwaśnej uzyskany w temperaturze
30
0
C, wyrazić w procentach zmiany aktywności enzymu uzyskane w pozostałych
temperaturach i sformułować odpowiedni wniosek.
3. Porównując aktywność fosfatazy kwaśnej w reakcji bez inhibitora z aktywnością enzymu w
obecności inhibitorów oblicz stopień zahamowania reakcji przez zastosowane inhibitory.
Pytania
1.
Podaj ogólną reakcję katalizowaną przez fosfatazy oraz klasę enzymów do której
należą.
2.
Jak dzielimy fosfatazy ze względu na optimum działania.
3.
Podaj krótką charakterystykę oraz lokalizację fosfataz roślinnych.
4.
Wymień czynniki wpływające na aktywność enzymów. Omów działanie jednego z
nich.
5.
Podaj przykłady naturalnych substratów fosfatazy kwaśnej. Co może być miarą
aktywności enzymu. Podaj definicję jednostki uniwersalnej.
6.
Napisz reakcję zachodzącą podczas oznaczania fosfatazy kwaśnej na ćwiczeniach.
Podaj nazwy substratu i produktów reakcji.

8
7.
Podczas oznaczania aktywności fosfatazy kwaśnej w mieszaninie reakcyjnej wykryto
200 µmoli p-nitrofenolu. Oblicz ile µg substratu uległo rozłożeniu.
8.
Oblicz ile µmoli p-nitrofenylofosforanu uległo hydrolizie z udziałem fosfatazy
kwaśnej, jeżeli po 15 min. reakcji wartość absorbancji wynosiła 0,35, a 1 µg p-
nitrofenolu daje absorbancję równą 0,002.
Literatura
1.
Berg, J.M., Tymoczko J.L., Stryer L. Biochemia. Wydawnictwo Naukowe PWN 2009
2.
Witwicki J. Ardelt W. Elementy enzymologii PWN 1984
3.
Senna R., Simonin V., Silva-Neto M.A.C., Fialho E.. 2006. Induction of acid
phosphatase activity during germination of maize (Zea mays) seeds. Plant Phys.
Bioch. 44, 467-473.
Wyszukiwarka
Podobne podstrony:
Czynniki warunkujące aktywność enzymów na przykładzie fosfatazy kwaśnej
biochemia III, Czynniki warunkujące aktywność enzymów
biochemia III;], Czynniki warunkujące aktywność enzymów
Czynniki i warunki szkodliwe działające na pilotów, Szkolenie Szybowcowe
Czynniki wpływającego na aktywność enzymów
Czynniki wpływające na aktywność enzymów
6) Wyznaczanie stałej Michaelisa Menten (Km), Vmax oraz określanie typu inhibicji aktywności fosfata
Czynniki warunkujące wystąpienie i czas trwania częściowej remisji u chorych na cukrzycę typu 1
(), biochemia L, Wpływ temperatury na aktywność enzymów (ćw E)
Każdy dorosły człowiek posiada większą lub mniejszą wiedzę na temat czynników warunkujących prawidło
Czynniki warunkujące odporność na stres., Pedagogika
Oznaczanie fosfatazy kwaśnej enzymol, [ ARCHIWA ], [ 2008-2009 ], Enzymologia, instrukcje
ROLA CZYNNIKÓW MOTYWACYJNYCH W ZARZĄDZANIU ZASOBAMI LUDZKIMI NA PRZYKŁADZIE FUT ALCON SP praca ma
Przystosowanie roslin do gorskich warunkow srodowiskowych na przykladzie Tatr, Geografia Nauczyciels
badania marketingowe rynku wykład, badania - ściąga, Ekonomiczny Rynek oznacza zespół warunków i czy
więcej podobnych podstron