
METALE
METALE
W SYNTEZIE
W SYNTEZIE
ORGANICZNEJ
ORGANICZNEJ
Tworzenie pojedynczego
wiązania węgiel-węgiel
część I

Lista podręczników
•
R. P. Houghton
„Kompleksy metali w chemii organicznej”
•
W. Carruthers, I. Coldham
„Modern Methods of Organic Chemistry”
•
M. Bochmann
„Organometalics 1” and „Organometallics 2”
•
J. Gawroński, K. Gawrońska, K. Kacprzak, M. Kwit
„Współczesna
synteza organiczna”
•
F. Pruchnik
„Chemia metaloorganiczna – pierwiastki przejściowe”
•
J. Skarżewski
„ Wprowadzenie do syntezy organicznej”

Pierwszy syntetyczny związek metaloorganiczny został
otrzymany ponad 200 lat temu w 1760 roku przez francuskiego
chemika L. C. Cadet de Gassicourt.
Podczas prac nad nowym atramentem sympatycznym uzyskał
on nieprawdopodobnie śmierdzącą ciecz, która - jak się potem
okazało – była związkiem arsenoorganicznym o wzorze
As
2
(CH
3
)
4
. Połączenie to nazwano kakodylem (po grecku
κακοδια oznacza śmierdzieć).

Pierwszy kompleks π otrzymał w 1827 roku duński chemik W. C.
Zeise (profesor Uniwersytetu w Kopenhadze, znany również jako
odkrywca ksantogenianów). Ogrzewając sól K
2
PtCl
4
w etanolu
uzyskał związek o sumarycznym wzorze KPtCl
3
C
2
H
4
. Sam
odkrywca nie zdawał sobie sprawy z wagi swojego odkrycia.
Wiele lat później uświadomiono sobie, że Zeise otrzymał
pierwszy organometaliczny związek metalu przejściowego,
etylenowy kompleks π platyny(II). Ten sam kompleks uzyskuje
się z dużo lepszą wydajnością działając bezpośrednio etylenem
na K
2
PtCl
4
w obecności katalizatora SnCl
2
. W eksperymencie
Zeisa etylen tworzył się in situ przez odwodnienie (dehydratację)
etanolu.
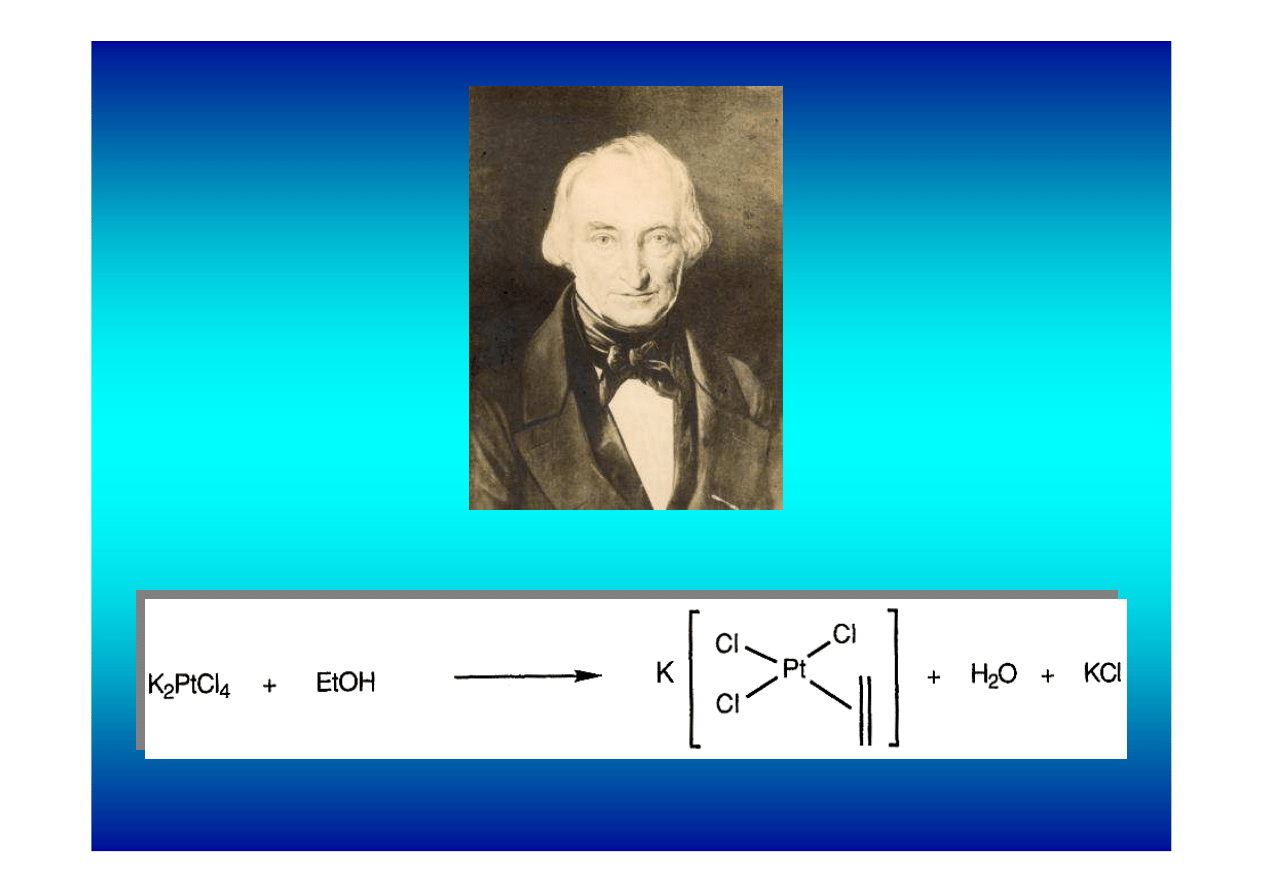
William Christopher Zeise (15.10.1789 – 12.11.1847)
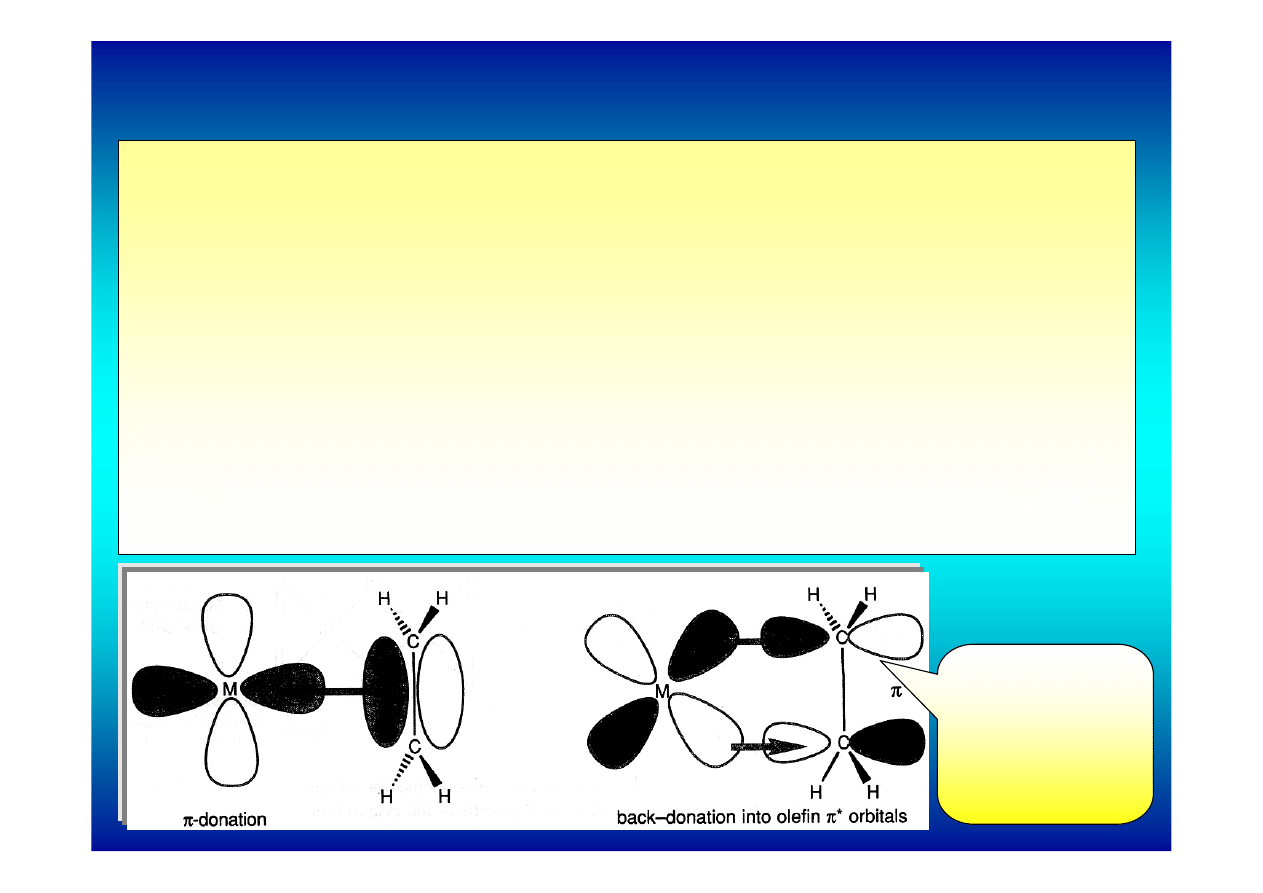
Wiązanie olefin z metalami przejściowymi
model
Dewara-Chatta-
Duncansona
Alkeny i alkiny kompleksują metale przejściowe za
pomocą wiążących orbitali π, które tworzą wiązania z
pustymi orbitalami ns, np, (n-1)d
z
2
, (n-1)d
x
2
-y
2
metalu lub
ich hybrydami.
Olefiny są słabymi zasadami więc wiązanie z metalem
musi być dodatkowo stabilizowane przez redonorowe
wiązanie metal-alken (alkin) polegające na przeniesieniu
elektronów z obsadzonych orbitali metalu na pusty orbital
π* olefiny. Jest to tak zwane wiązanie π-zwrotne.

Wiązanie π-zwrotne polegające na obsadzeniu
antywiążącego orbitalu π* alkenu obniża krotność
wiązania, wydłuża je i osłabia. Można to
zaobserwować przy pomocy metod spektroskopii IR
gdzie zjawisko wydłużenia wiązania C=C wyraża się
obniżeniem częstości drgań rozciągających w regionie
1600-1500 cm
-1
.
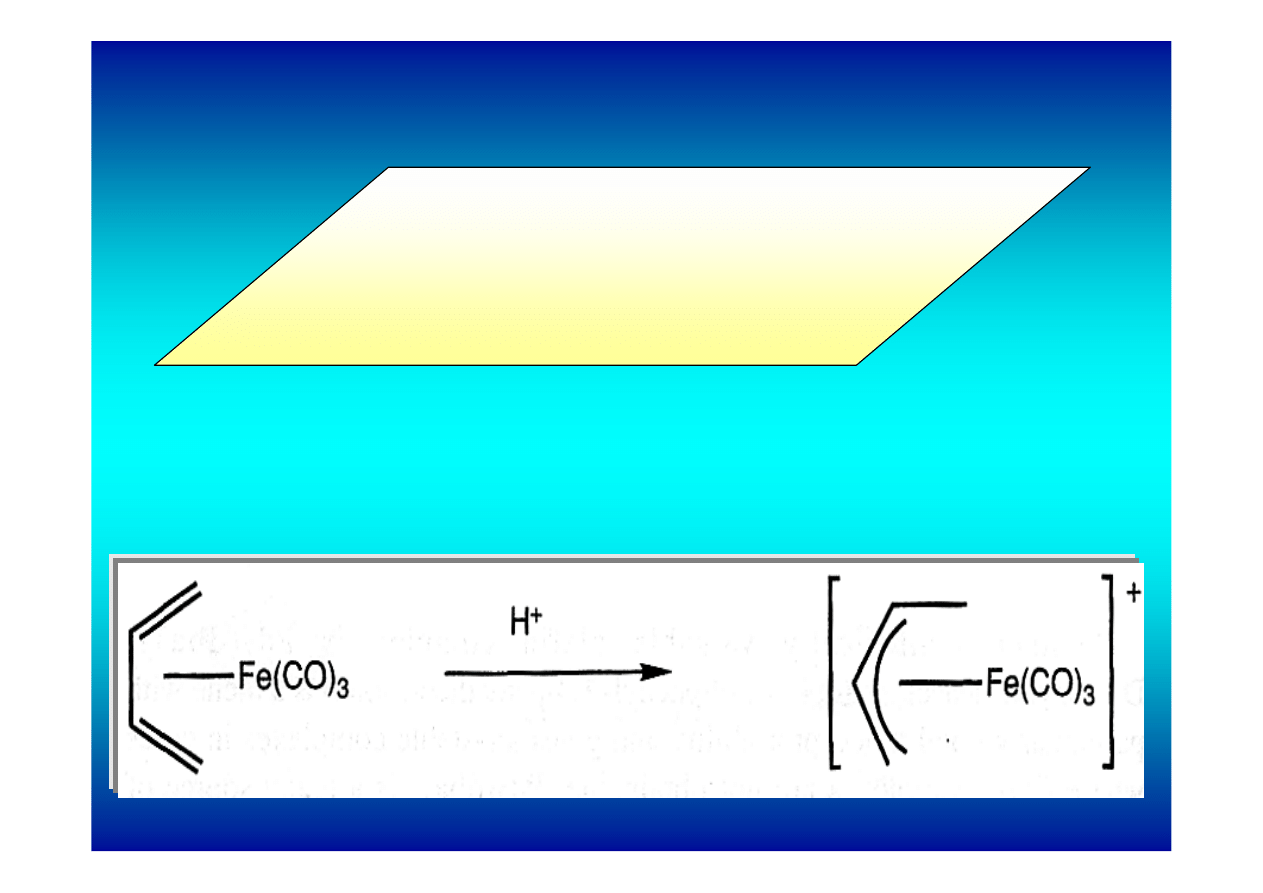
Reakcje kompleksów π z elektrofilami
Reakcje kompleksów π alkenów z elektrofilami,
takimi jak HX, zazwyczaj prowadzą do reduktywnej
eliminacji alkenu i utworzenia kompleksu typu
L
n
MX
y
. Tylko w wyjątkowych sytuacjach udało się
wyizolować pochodne alkilowe.
Reakcje kompleksów π z elektrofilami
Protonowanie kompleksów π
allenowych i dienowych prowadzi
do izolowalnych pochodnych
allilowych.
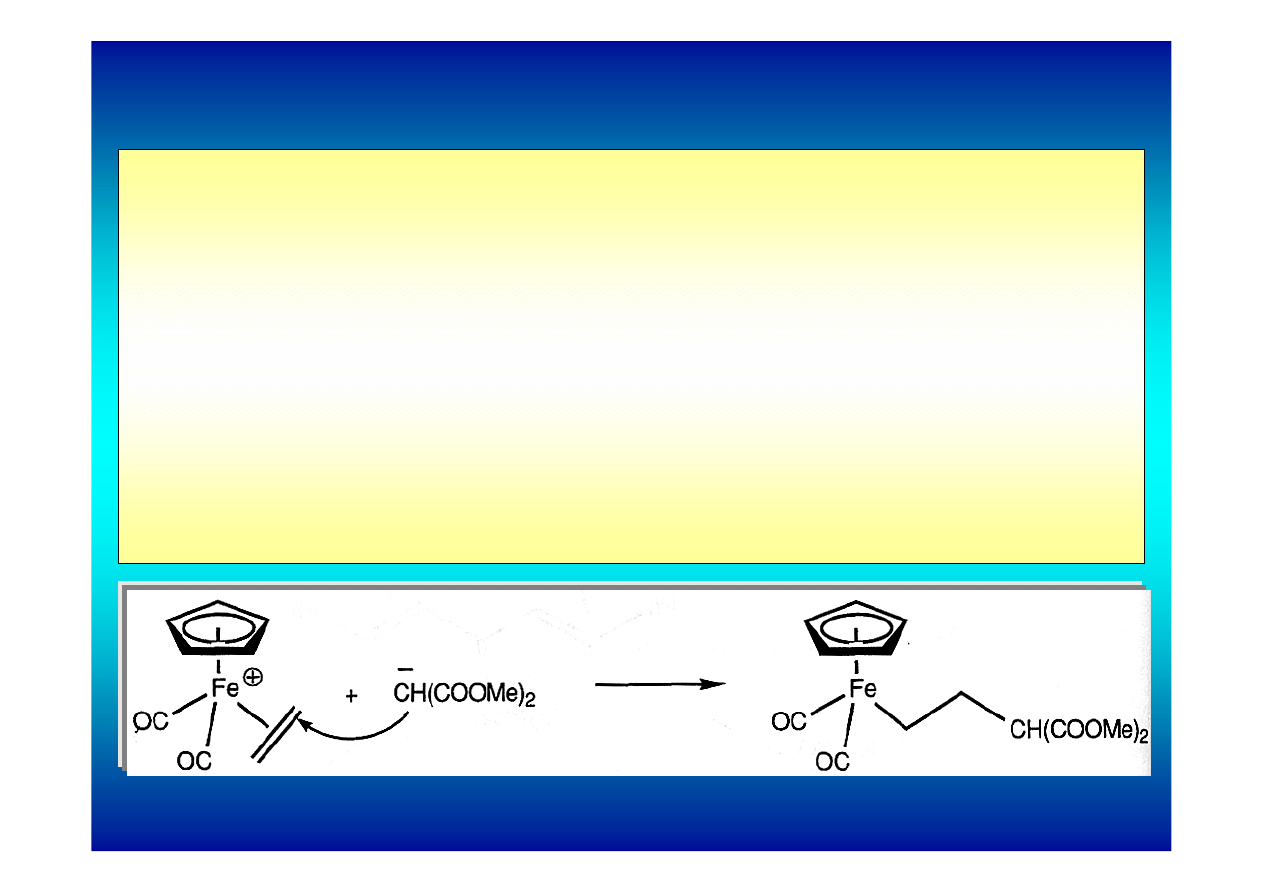
Reakcje kompleksów π z nukleofilami
Najważniejsze zastosowanie olefinowych kompleksów π to
reakcje addycji nukleofilowej. Są one możliwe dzięki
wysokiej podatności skoordynowanych olefin na atak
nukleofilowy.
Reaktywność etylenu w stosunku do nukleofilów i
elektrofilów jest bardzo mała. Po skoordynowaniu etylenu
przez metal przejściowy jego reaktywność gwałtownie
rośnie – ulega on łatwo atakowi nukleofilowemu, zwłaszcza
gdy kompleks jest kationowy.
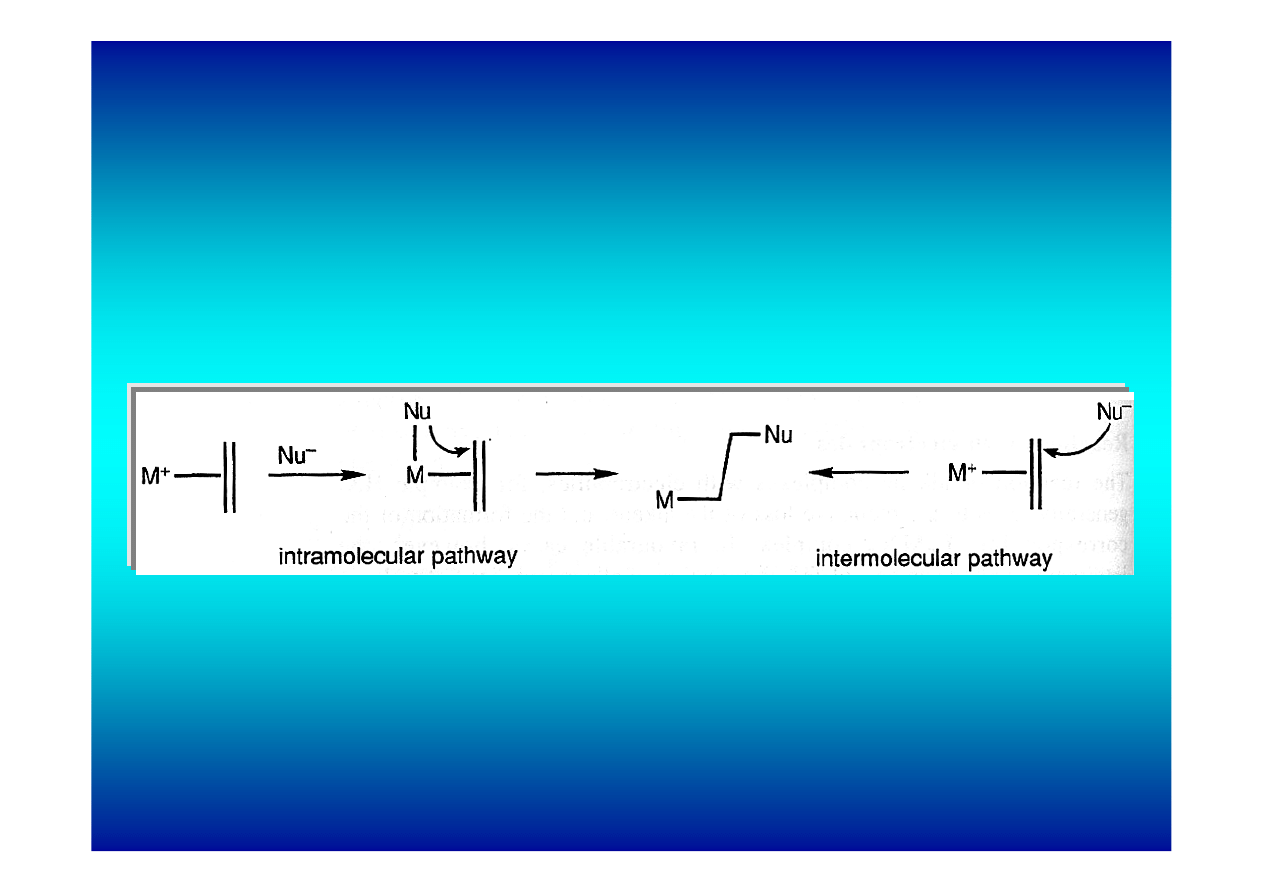
Teoria międzycząsteczkowego i wewnątrzcząsteczkowego
mechanizmu reakcji skoordynowanych olefin
z nukleofilami

Wiązanie
alkanów
z metalami
Pierwszy związek metaloorganiczny – kompleks σ – został
otrzymany przypadkowo przez Edwarda Franklanda w 1848
roku. Frankland chciał uzyskać stabilne rodniki etylowe
działając cynkem na jodoetan a wyizolował dwa produkty
metaloorganiczne: EtZnI oraz ZnEt
2
. Było to wyjątkowe
osiągnięcie naukowe w tamtym czasie – zwłaszcza gdy weźmie
się pod uwagę piroforyczną naturę związków
cynkoorganicznych i ich ekstremalną podatność na hydrolizę.
Swoje reakcje Frankland prowadził w atmosferze wodoru!!!
kompleksy σ

Pierwsza synteza związków alkilocynku
zapoczątkowała szybki rozwój chemii
kompleksów σ. Wkrótce po odkryciu Falklanda
otrzymano kolejne kompleksy σ: związki glino-,
magnezo-, tręcio-, krzemo-, cyno- i
ołowioorganiczne.
Pierwszy metaloorganiczny kompleks σ metalu
przejściowego otrzymał William Jackson Pope
(1870-1939) w 1907 roku. Jednak struktura
tetramerycznego kompleksu Pt(IV) przez 50 lat
pozostawała niewyjaśniona. W następnych
latach podejmowano liczne i nieudane próby
syntezy nowych kompleksów σ metali
przejściowych dowodząc w końcu, że wiązanie σ
C-M (węgiel-metal_przejściowy) jest z natury
niestabilne, chyba że obecne są stabilizujące
ligandy π.

Sytuacja zmieniła się w latach 1960. Okazało się, że obecność
ligandów π nie jest konieczna aby kompleksy metali
przejściowych typu metal-alkil były trwałe.
Uzyskano szereg stabilnych kompleksów a nawet dla wielu z
nich wyznaczono wartości entalpii wiązania M-C.
Stabilność związku chemicznego oznacza jego trwałość w
standartowych warunkach w obecności różnych reagentów.
Istnieją związki, które mogą być ogrzewane bez rozkładu do
wysokiej temperatury w atmosferze gazu obojętnego ale gwałtownie
reagują w obecności wilgoci lub tlenu atmosferycznego.
Są to związki niestabilne.
Istnieją także związki, dla których dane termodynamiczne sugerują
labilność ale są trwałe w obecności tlenu i nie reagują z wodą.
Są to związki stabilne.

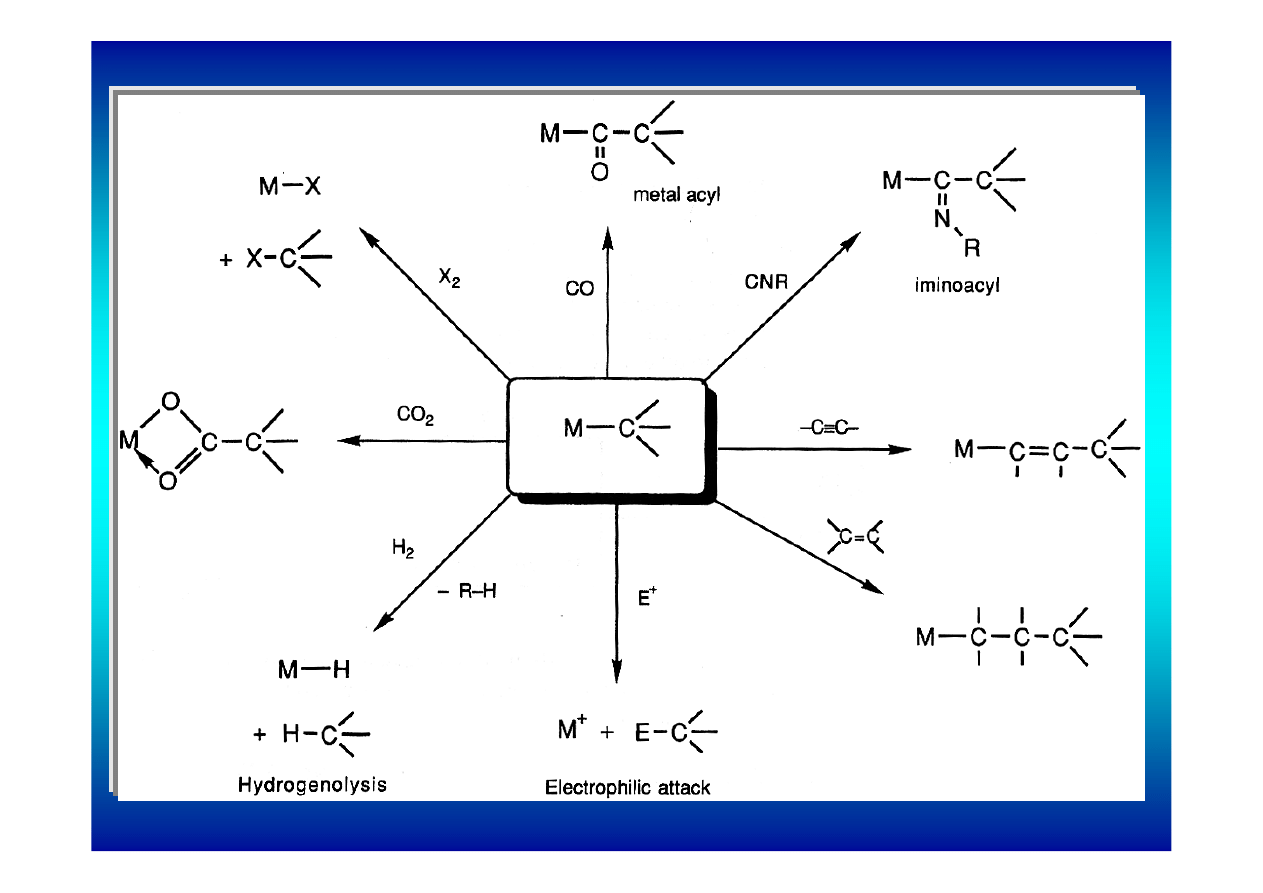
Reaktywność kompleksów σ metal-alkil
Większość kompleksów σ metal_przejściowy-alkil to związki
bardzo reaktywne. Najważniejsze typy reakcji, którym
ulegają to:
1.
Reakcje z rozerwaniem wiązania M-C:
a)
wodoroliza wiązania M-C
b) reakcje z elektrofilami (CO
2
, H
+
, I
2
)
2.
Reakcje wbudowania (insercji) nienasyconego reagenta
w wiązanie M-C. Najczęściej biorą udział ligandy CO,
izocyjanki, alkeny i alkiny.
3.
Reakcje reduktywnej eliminacji (jak łączenie ligandów
alkilowych z nukleofilami)
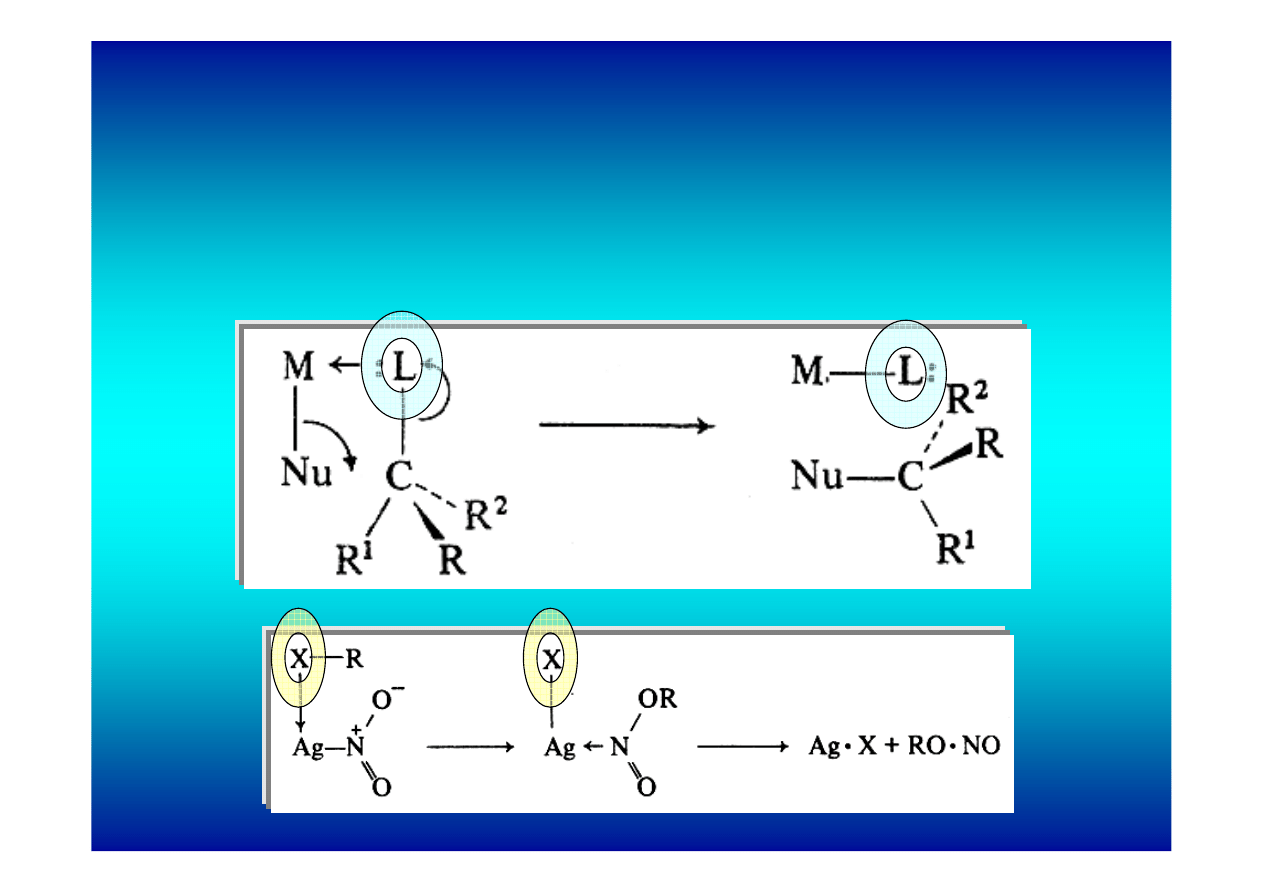
Efekty i zastosowanie kompleksowania w reakcjach
podstawienia nukleofilowego
1. Koordynowanie grupy odchodzącej
a) powstanie kompleksu przyspiesza podstawienie
b) ułatwia atak wewnątrzcząsteczkowy
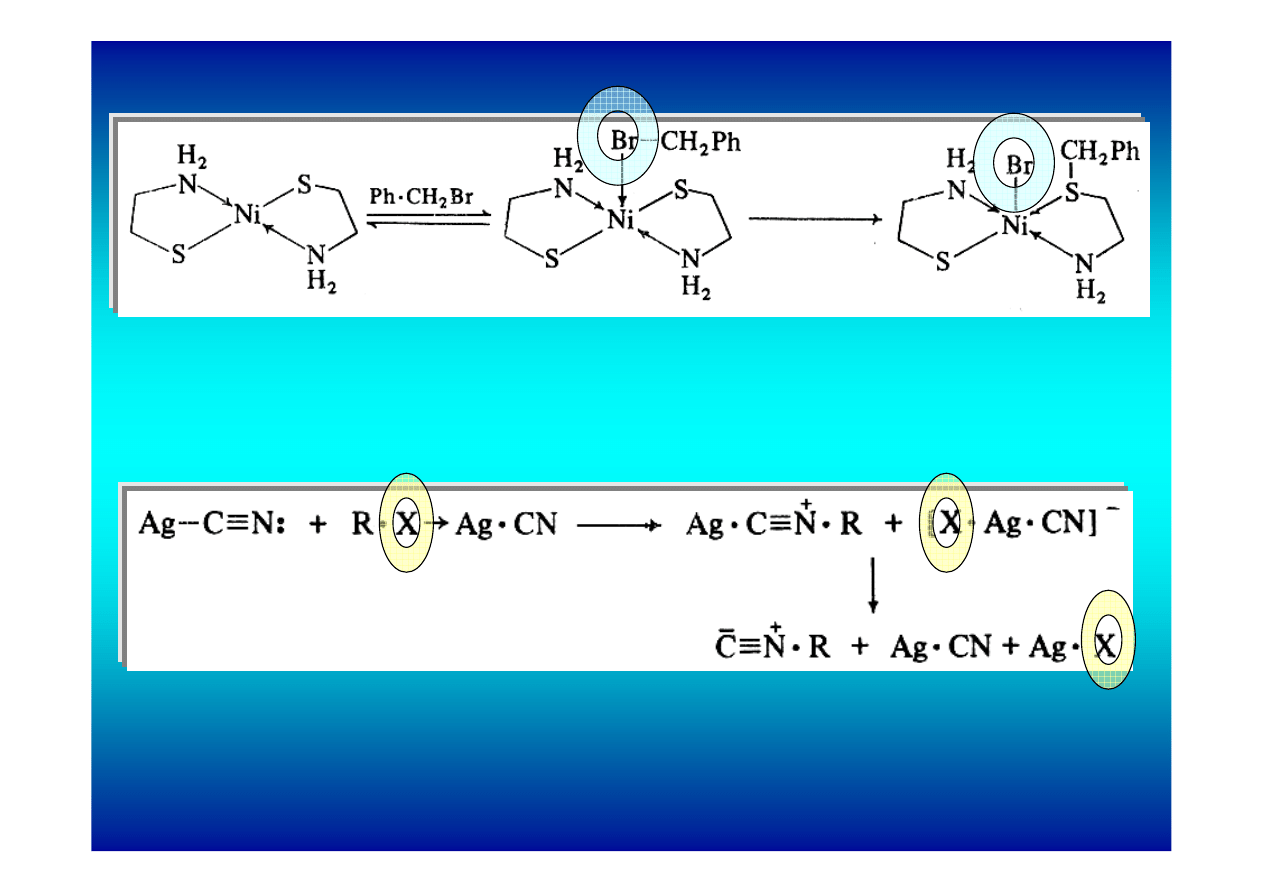
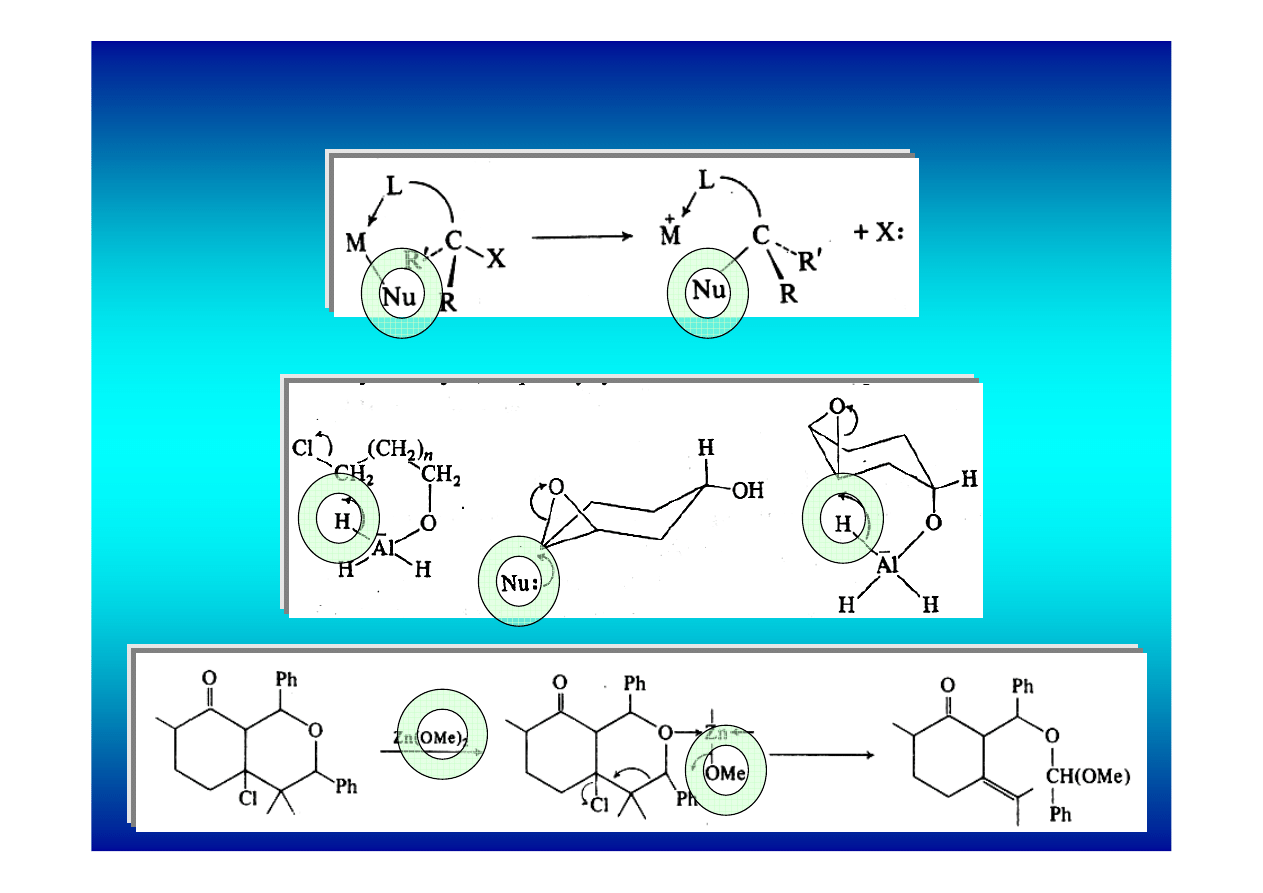
2. Koordynowanie nukleofila
Stan przejściowy sześcioczłonowy
(alkilowanie enolanów)
Stan przejściowy sześcioczłonowy
(alkilowanie allilu)
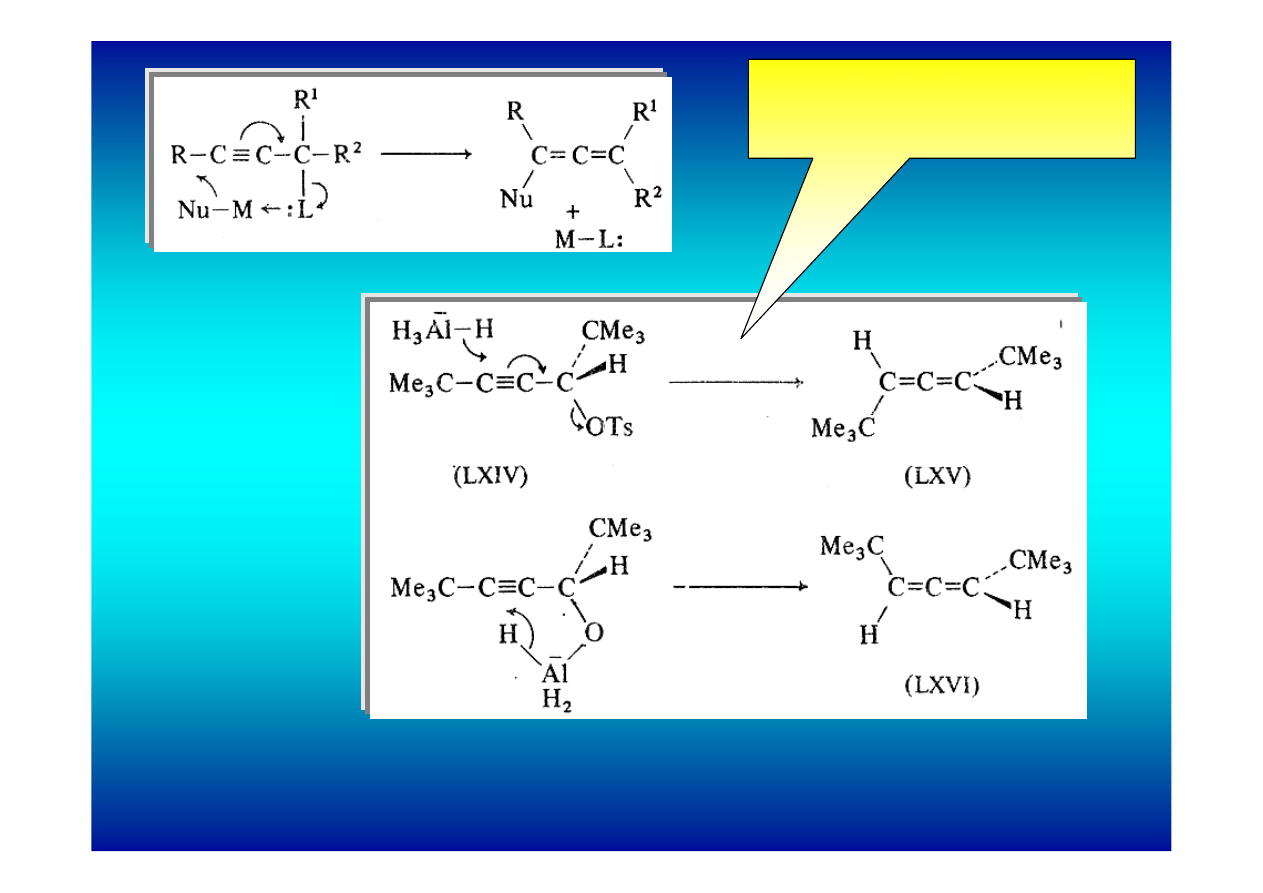
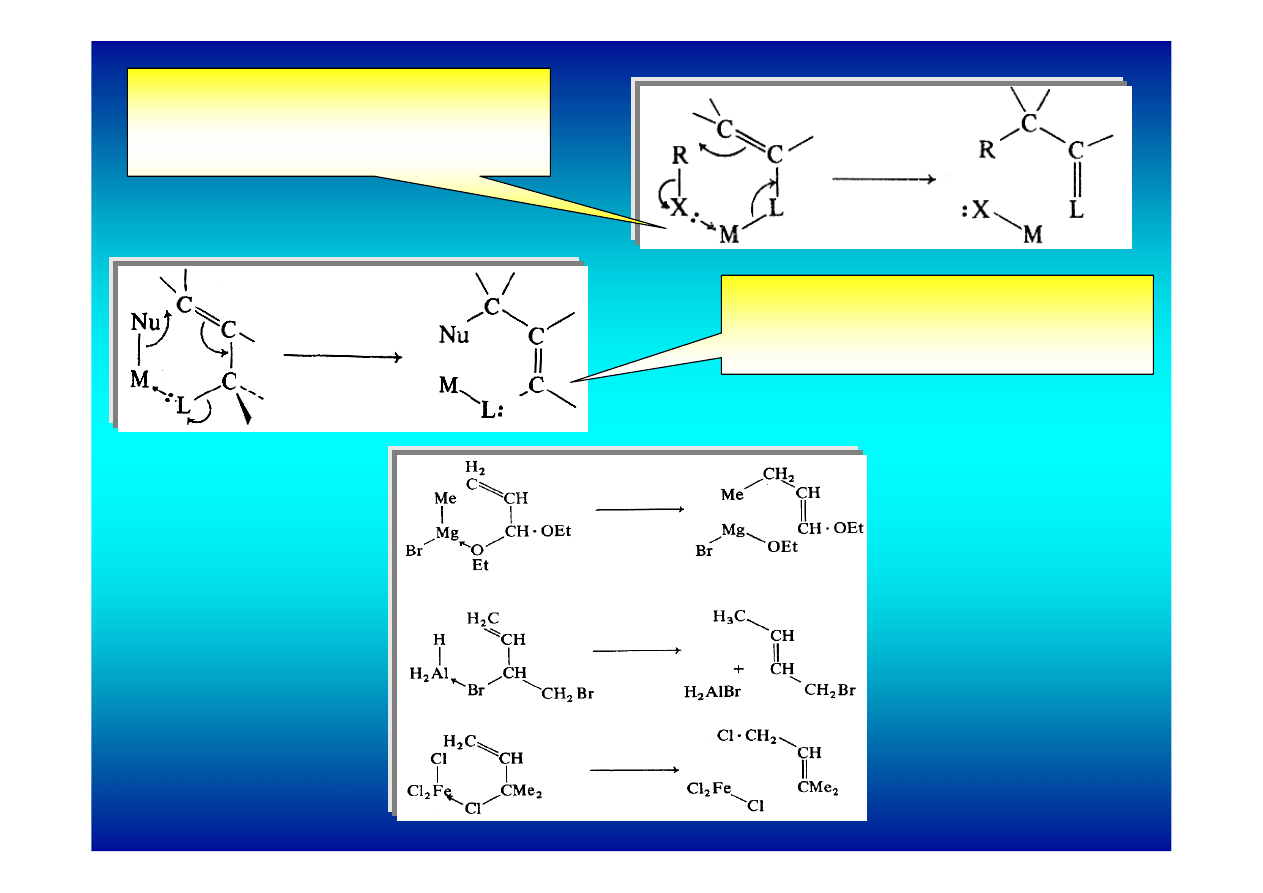
Synteza kumulenów

Reakcja Wurtza
A. Wurtz, Ann. Chim. Phys. [3] 44, 275 (1855); Ann. 96, 364 (1855).
Połączenie dwóch rodników alkilowych w reakcji
dwóch moli halogenków alkilowych
z dwoma molami sodu.
2RX + 2Na
→
→
→
→
R-R + 2NaX
Sód
Na
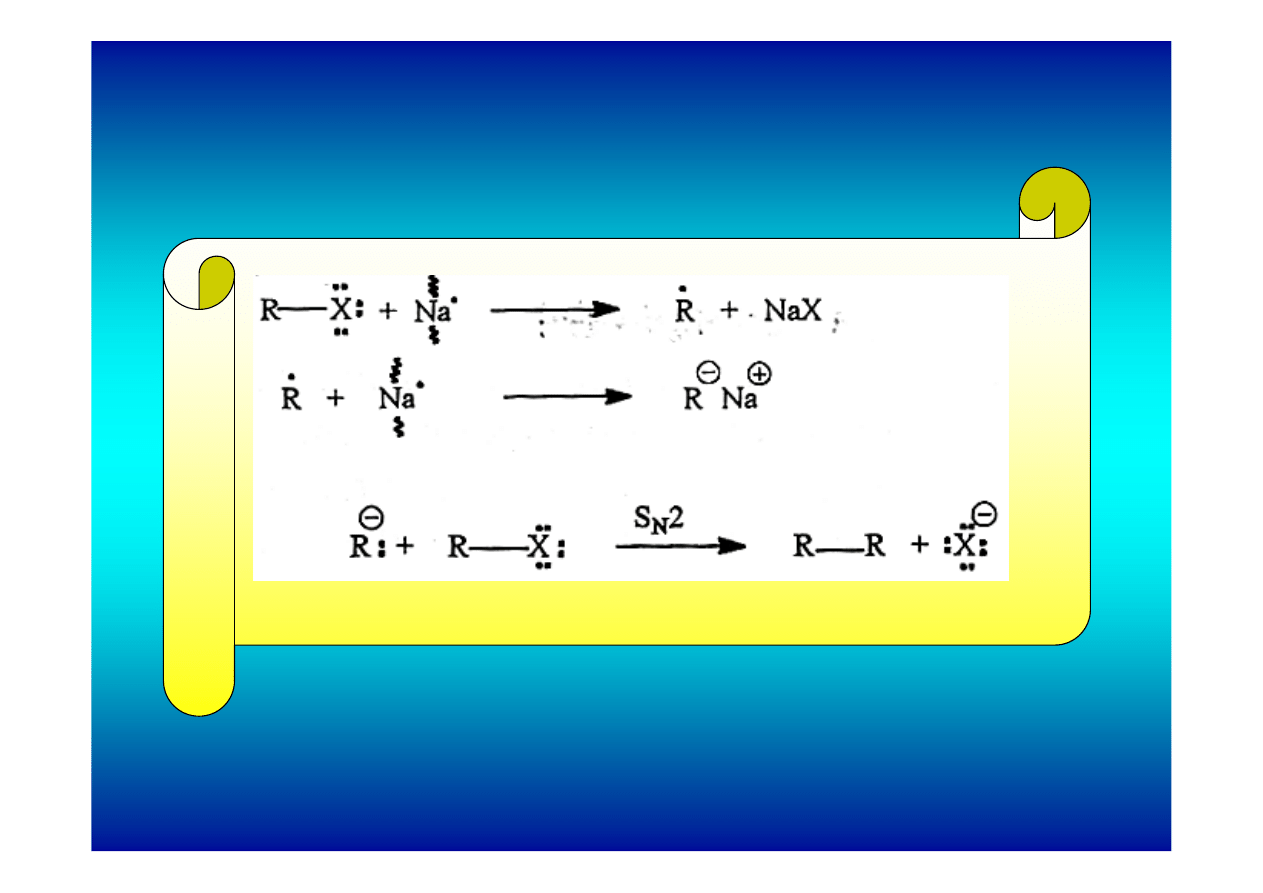
Mechanizm reakcji Wurtza

Reakcja Wurtza-Fittiga
B. Tollens, R. Fittig, Ann. 131, 303 (1864);
R. Fittig, J. König, ibid. 144, 277 (1867).
Powstawanie aromatycznych
alkilowanych węglowodorów w wyniku
połączenia halogenków alkilowych i
arylowych w obecności sodu:
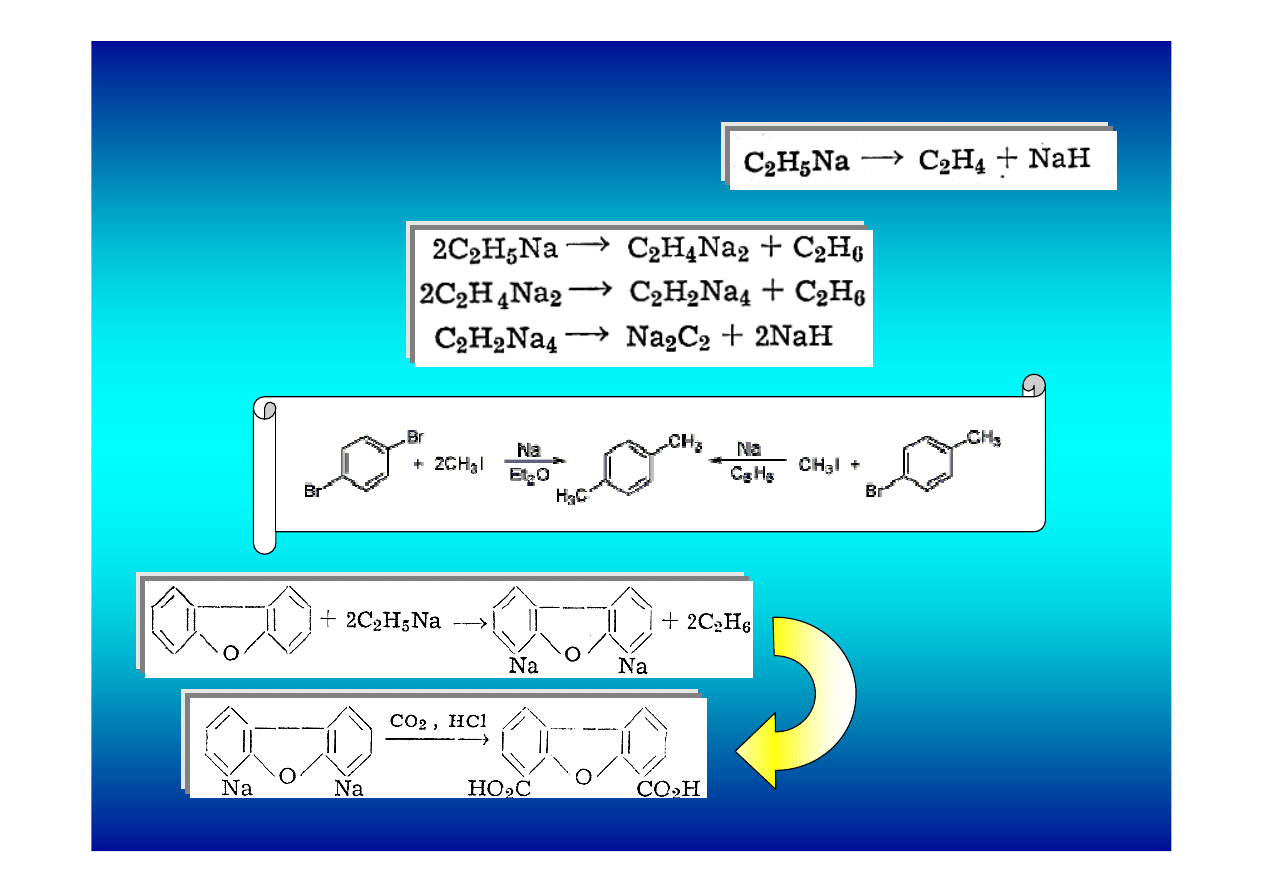
Reaktywność związków alkilosodowych:

Synteza pojedynczego wiązania C-C
Zastosowanie pochodnych alkilolitu
Lit
Li

Związki litoorganiczne mają szerokie zastosowanie w
syntezie organicznej jako zasady oraz jako nukleofile.
Reagują z wieloma elektrofilami a zakres tych reakcji od
oderwania protonu po atak nukleofilowy zależy:
od budowy związku litoorganicznego,
od budowy elektrofila,
od zastosowanych warunków.

Związki litoorganiczne zapisujemy ogólnie jako Li-R,
jednakże bardzo często tworzą one w roztworach złożone
struktury a wiązanie lit-węgiel uzyskuje w dużym stopniu
charakter kowalencyjny.
Rozpuszczalniki koordynujące THF, Et
2
O oraz silne σ-
donory jak TMEDA redukują stopień asocjacji związków
litoorganicznych zwiększając ich reaktywność.
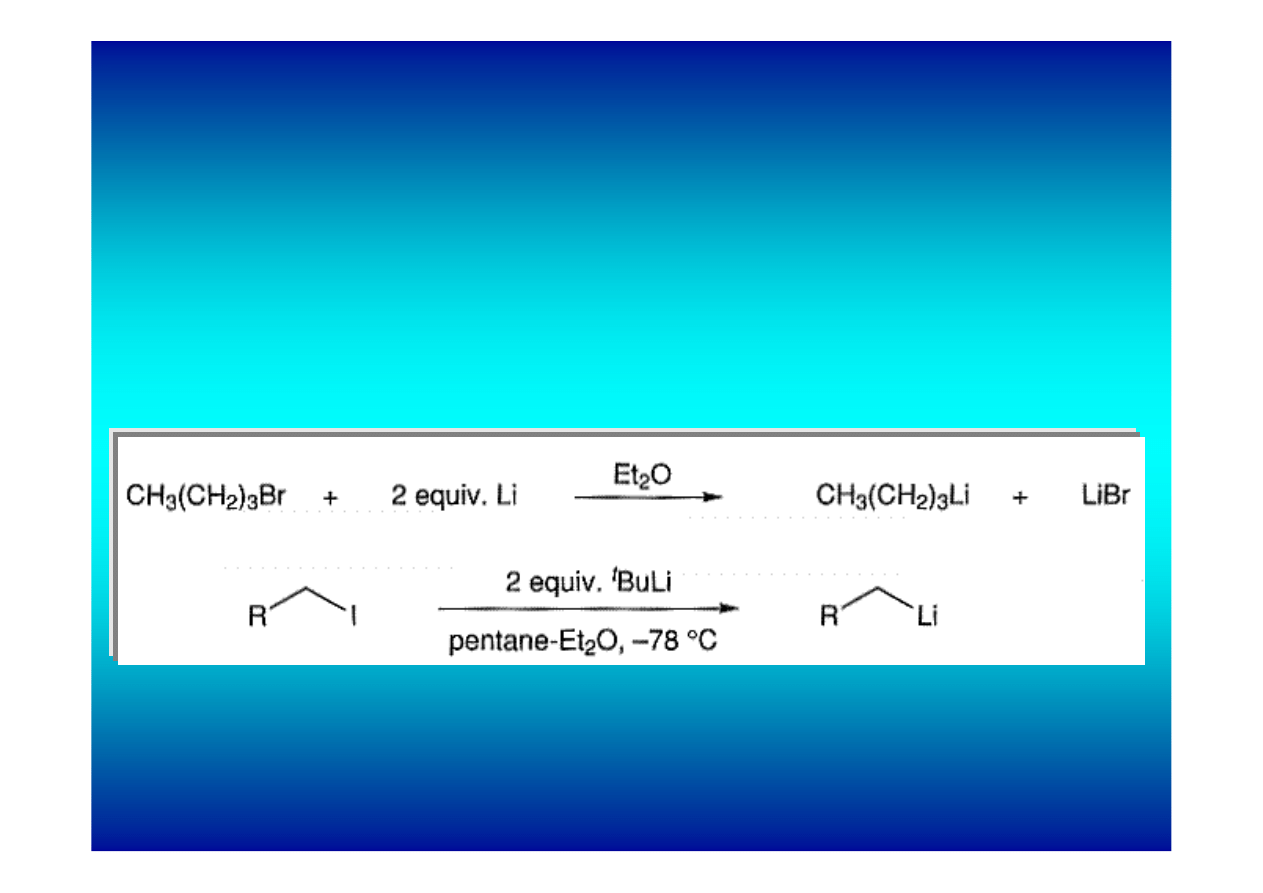
Synteza związków litoorganicznych
1. Bezpośrednia reakcja halogenków alkilowych z litem

2. Metalowanie pozycji α w aminach związkami alkilolitowymi
Podstawienie protonu α w aminach litem jest podstawą
metody α-alkilowania amin drugorzędowych. Grupa
aminowa R
2
N-Me nie jest wystarczająco aktywująca aby
ugrupowanie metylenowe mogło być przekształcone w sól
litową R
2
N-CH
2
-M jednak niektóre pochodne amin
drugorzędowych mogą być już przekształcone w związki
litoorganiczne przy pomocy silnych zasad. Uzyskane w ten
sposób pochodne α-aminolitoorganiczne reagują łatwo z
elektrofilami (chlorkami alkilowymi i acylowymi,
aldehydami).
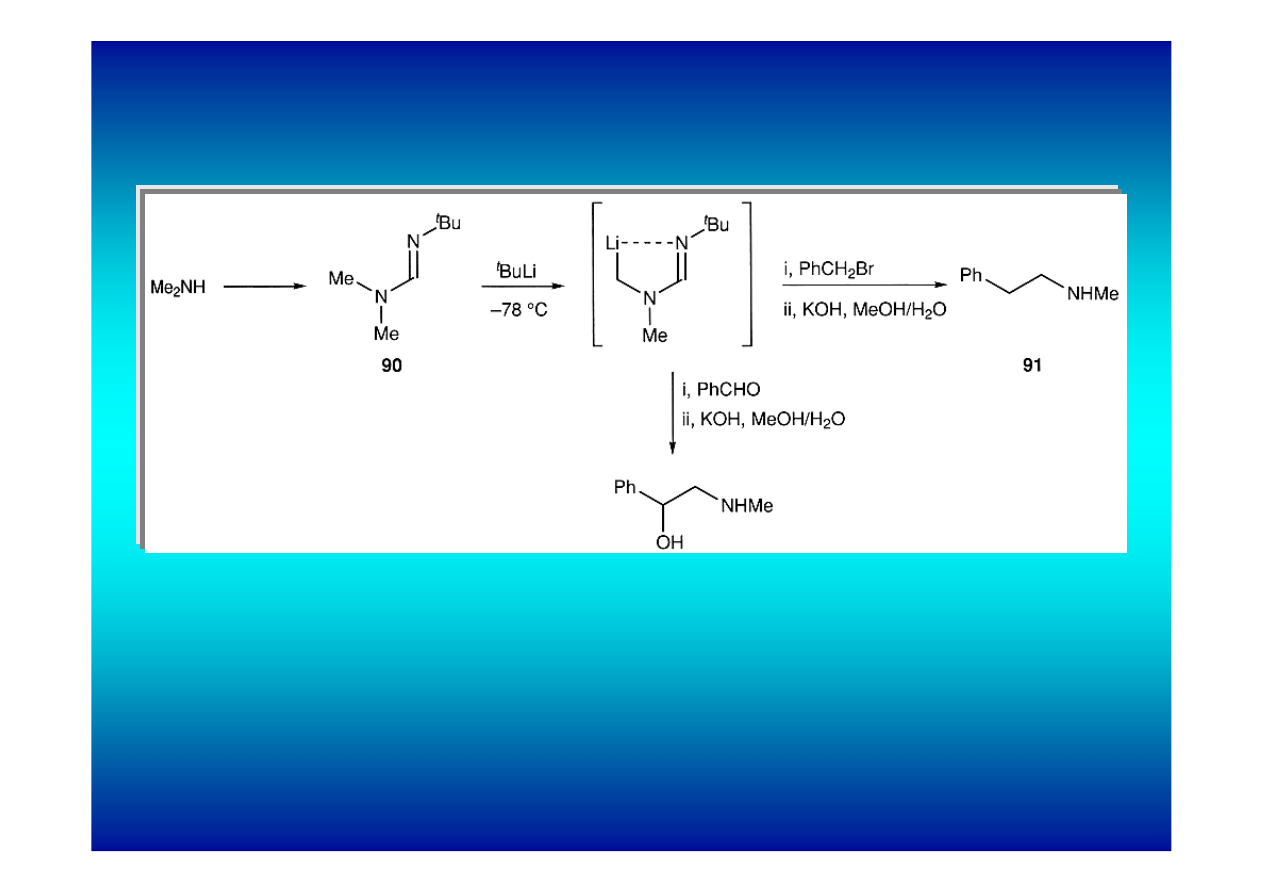
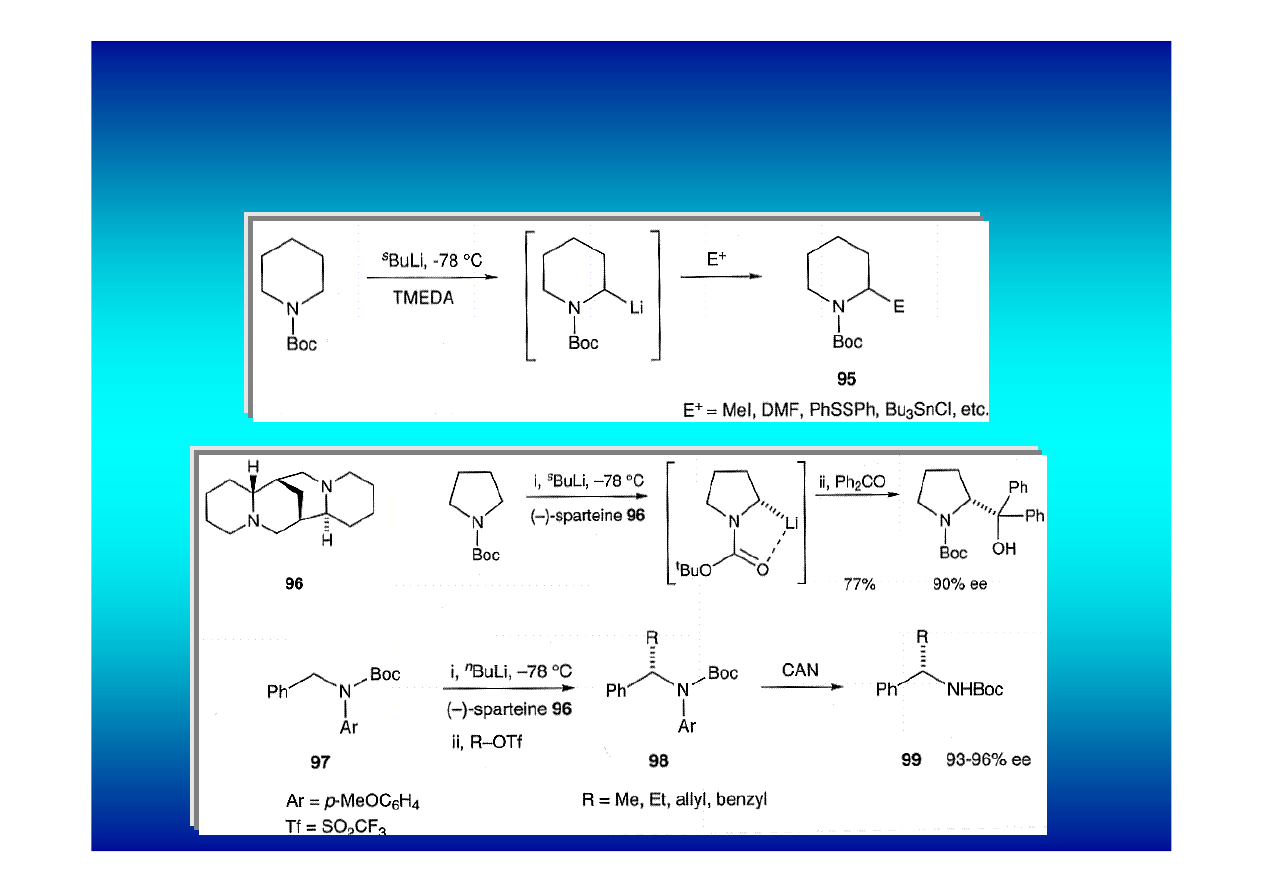
Jeszcze łatwiej przekształcane są w związki litoorganiczne
pochodne N-nitrozowe, sterycznie zatłoczone amidy i
formamidyny.
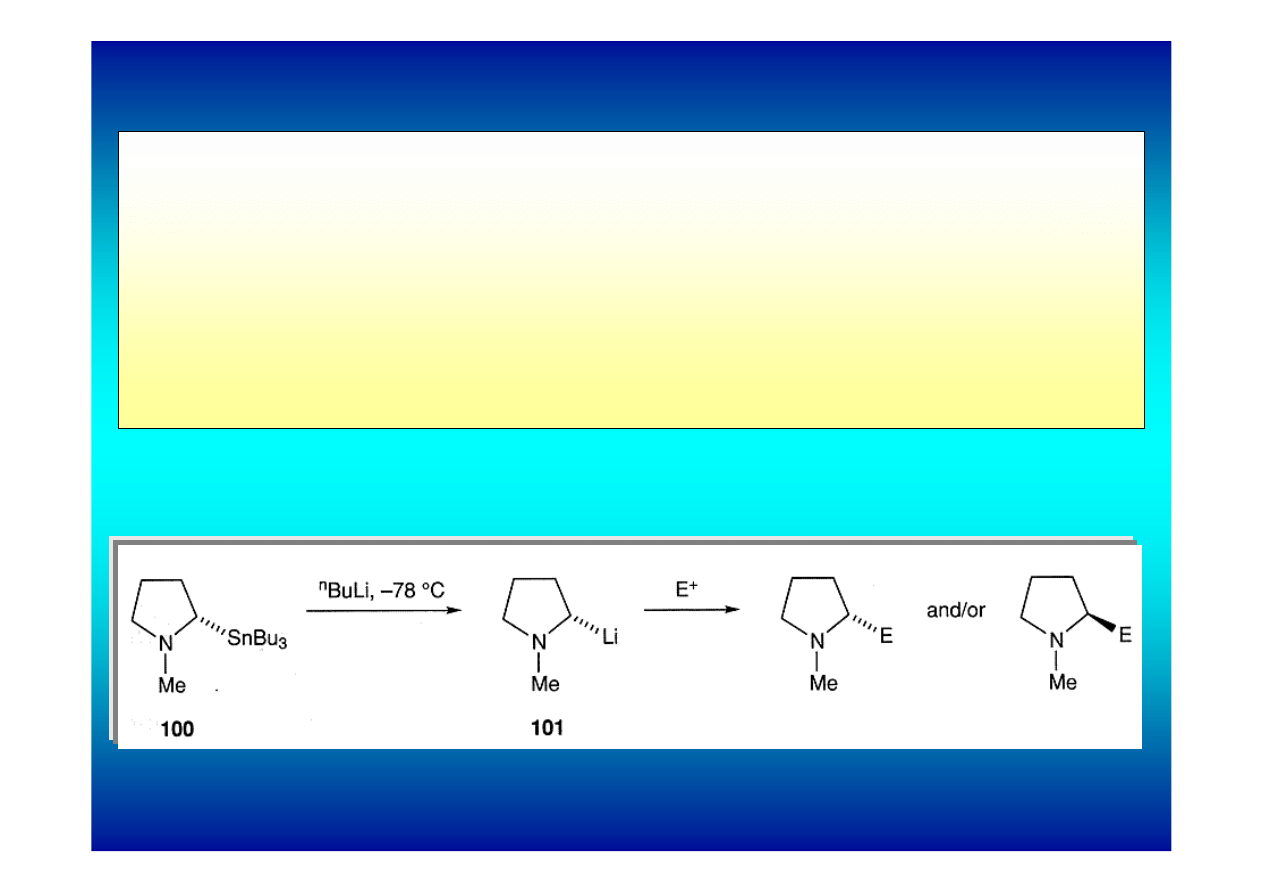
3. Transmetalowanie związków cynoorganicznych
Metoda ta pozwala łatwo uzyskać reaktywne związki α-
aminolitoorganiczne. Wymiana cyny na lit jest szczególnie
efektywna dla N-Boc zabezpieczonych drugorzędowych amin a
nawet dla zwykłych amin trzeciorzędowych.
Przykład: addycja n-butylolitu do związku cynoorganicznego 100 generuje
reaktywny związek α-aminolitoorganiczny 101, który reaguje z różnymi
elektrofilami.
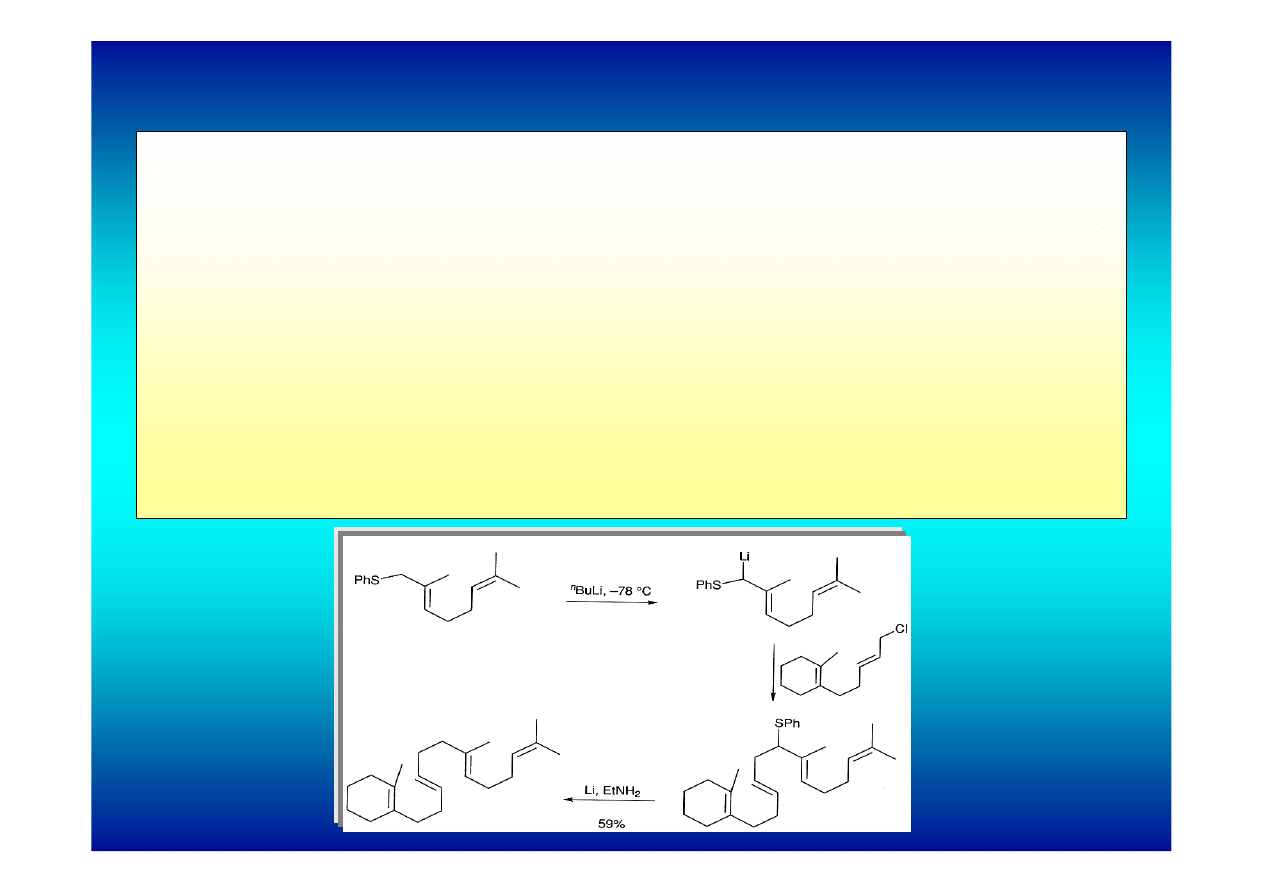
4. Bezpośrednie litowanie tioeterów w pozycji α
Alkilowe tioetery nie są szczególnie kwasowe ale mogą
ulegać litowaniu w pozycji α w przypadku obecności
dodatkowych grup aktywujących takich jak dodatkowy
atom siarki lub podwójne wiązanie.
Szczególnie ważną rolę pełnią siarczki allilowe ( zwłaszcza
tioetery allilowo-fenylowe), które łatwo ulegają reakcji z n-
butylolitem dając reaktywne związki litoorganiczne i mogą
być następnie alkilowane głównie w pozycji α.
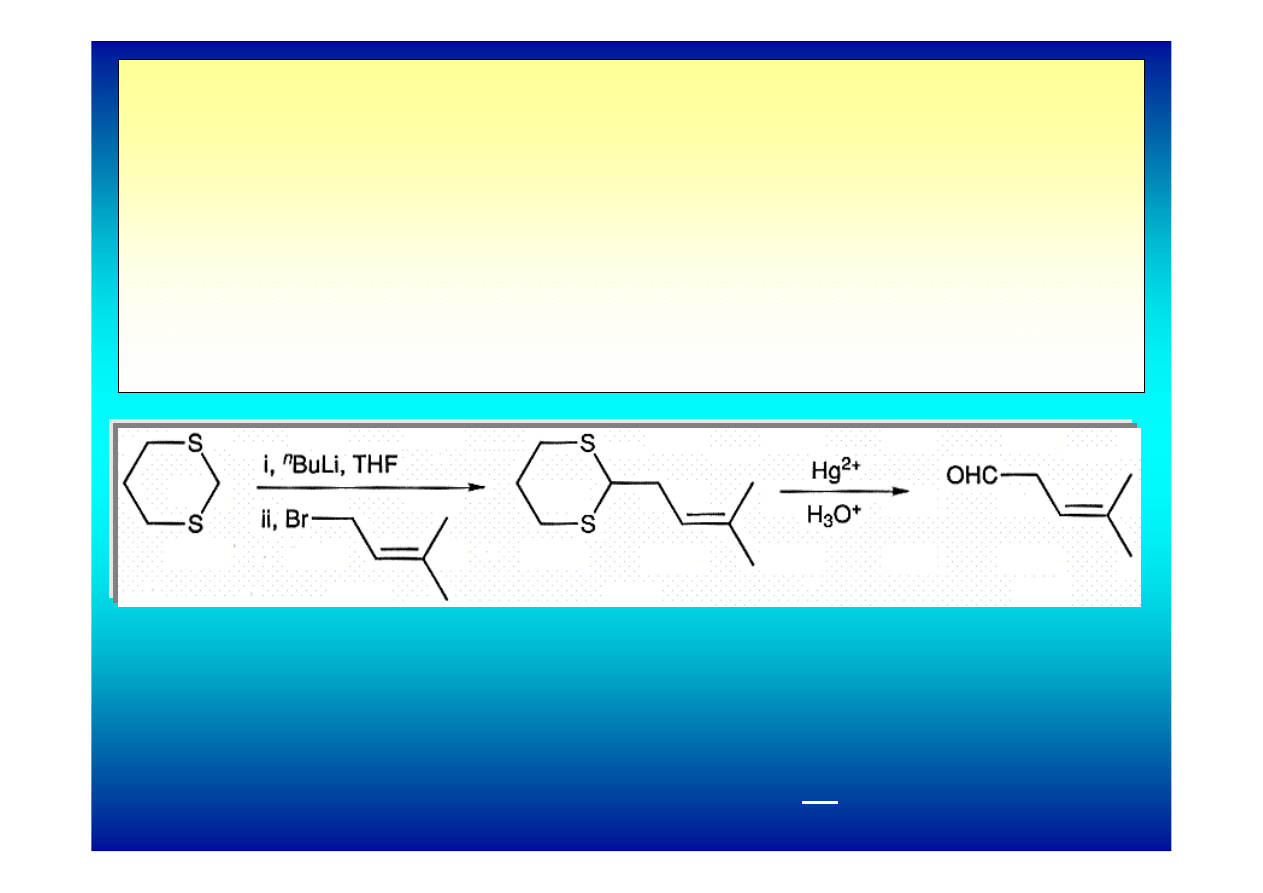
Niezwykle ważne są reakcje litowania i alkilowania atomów
węgla C-α w układach, gdzie 2 atomy siarki związane są z tym
samym atomem węgla. Oderwanie protonu jest tu bardzo
łatwe pod działaniem n-butylolitu.
Uzyskany 2-lito-1,3-ditian reaguje z licznymi elektrofilami:
chlorkami alkilowymi, epoksydami i związkami
karbonylowymi*.
* M.Yus, C.Najera, F.Foubelo, Tetrahedron, 59 (2003), 6147

Miedź
Cu
Reagenty miedzioorganiczne
Zastosowanie w tworzeniu wiązań C-C

Do najważniejszych związków miedzioorganicznych zalicza
się miedziany Gilmana R
2
CuLi otrzymywane w reakcji
jodku miedzi(I) z dwoma równoważnikami związku
litoorganicznego.
Gilman, Henry (1893-1986)
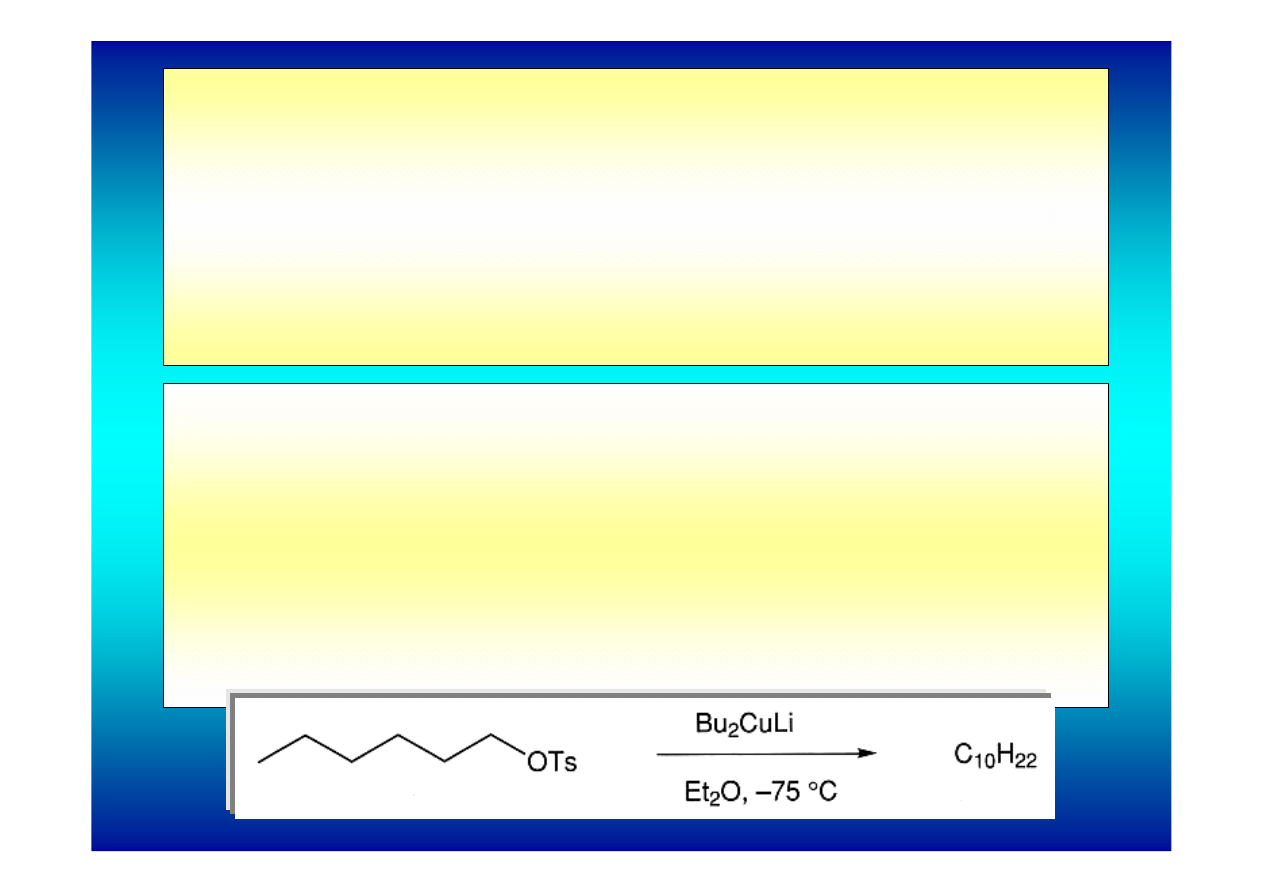
Znamy obecnie kilka typów miedzianów: R
2
CuLi
(klasyczne miedziany Gilmana), RCu(CN)Li oraz
R
2
Cu(CN)Li
2
. Miedziany te różnią się naturalnie
reaktywnością i wybór któregoś z nich jest uzależniony
od typu reakcji, którą chcemy przeprowadzić.
Na ogół generuje się je in situ i nie izoluje
Bardzo użyteczną reakcją tworzenia pojedynczego
wiązania C-C wykorzystującą miedziany jest reakcja
sprzęgania grup alkilowych miedzianu z halogenkami
alkilowymi lub tosylanami.
Reakcja zachodzi w temperaturze pokojowej dając
wysokie wydajności produktów. Miedziany tolerują
obecność wielu grup funkcyjnych.

Drugorzędowe bromki i jodki alkilowe nie reagują z dobrymi
wydajnościami z klasycznymi miedzianami Gilmana. Stosuje
się więc miedziany wyższych rzędów R
2
Cu(CN)Li
2
. Te
reagenty są bardziej uniwersalne – mogą reagować też z
halogenkami pierwszorzędowymi.
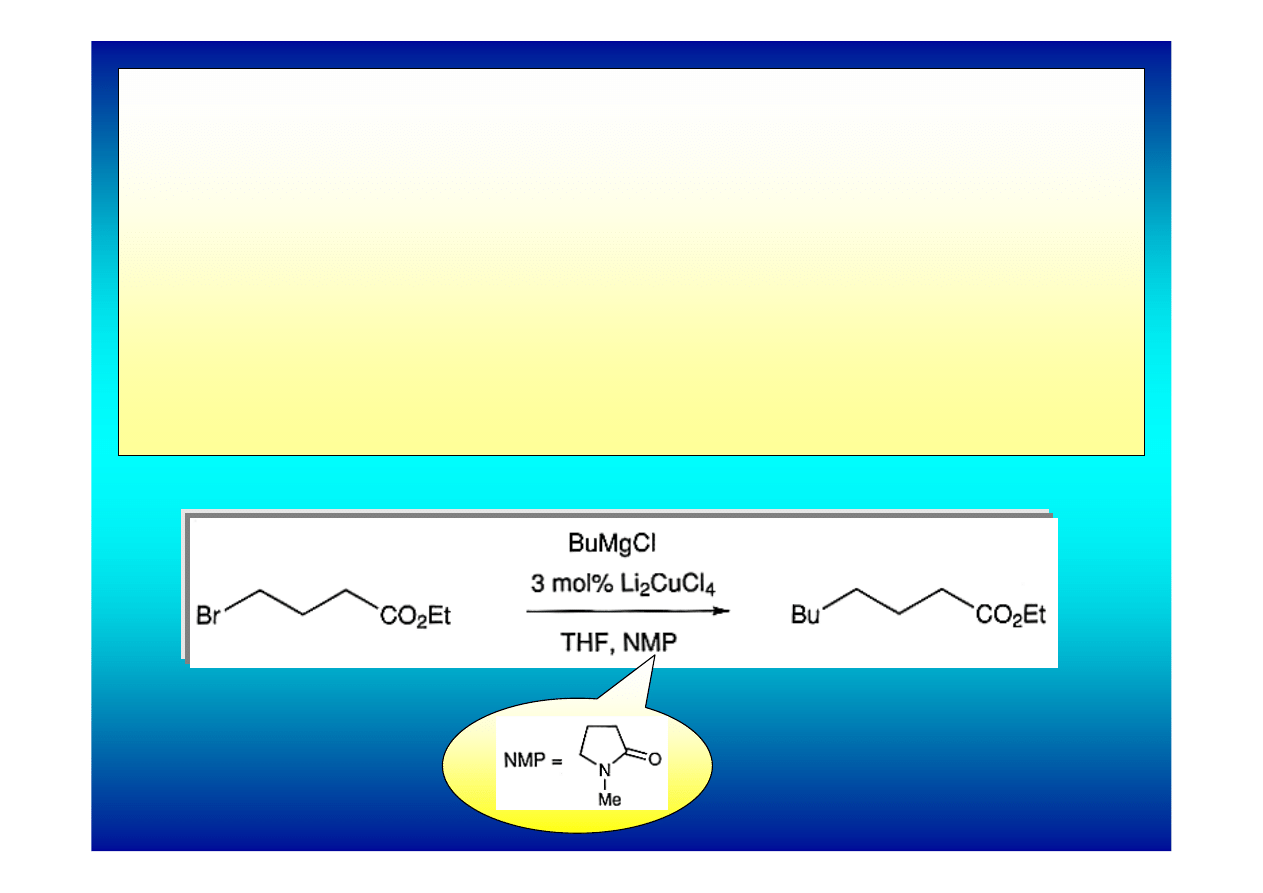
Odrębnym zagadnieniem są tetrachloromiedziany(II) litu,
Li
2
CuCl
4
, otrzymywane bezpośrednio z chlorku litu i chlorku
miedzi(II). Stosuje się je w ilościach katalitycznych w obecności
związków Grignarda. Przypuszcza się, że czynnikiem
aktywnym są w reakcjach alkilowania kompleksy
miedzio(I)organiczne o nie zbadanej jeszcze strukturze,
tworzące się w wyniku reakcji tetrachloromiedzianu z
halogenkiem alkilomagnezowym.
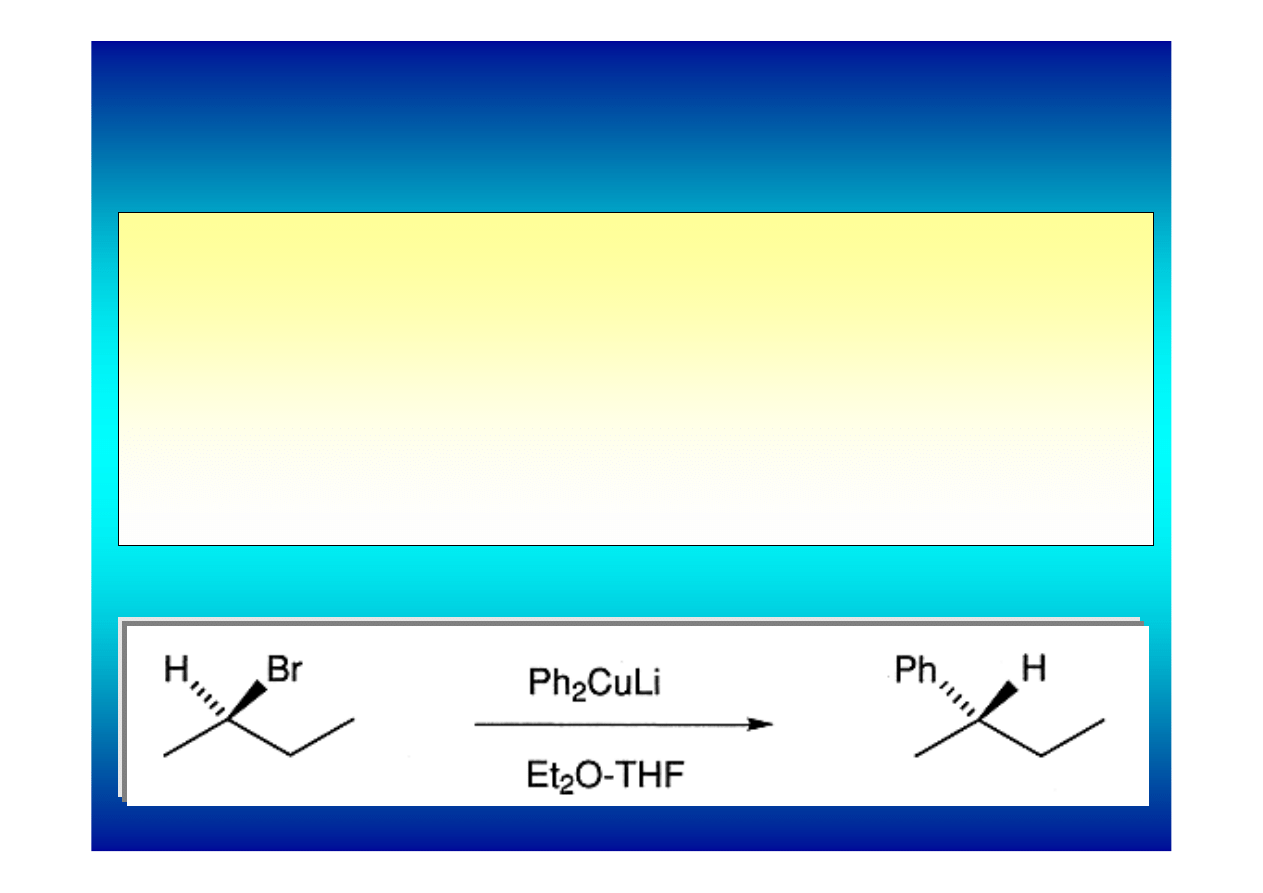
Reakcja difenylomiedzianu(I) litu z (-)-(R)-2-bromobutanem
zachodzi z inwersją konfiguracji – sugeruje to mechanizm S
N
2.
Jednakże podobny eksperyment wykonany z optycznie
czynnym jodkiem alkilowym daje produkt racemiczny co
oznacza, że reakcje miedzianów Gilmana z jodkami alkilowymi
przebiegają według mechanizmu określanego raczej jako one-
electron transfer niż według S
N
2.
Stereochemia alkilowania miedzianami Gilmana
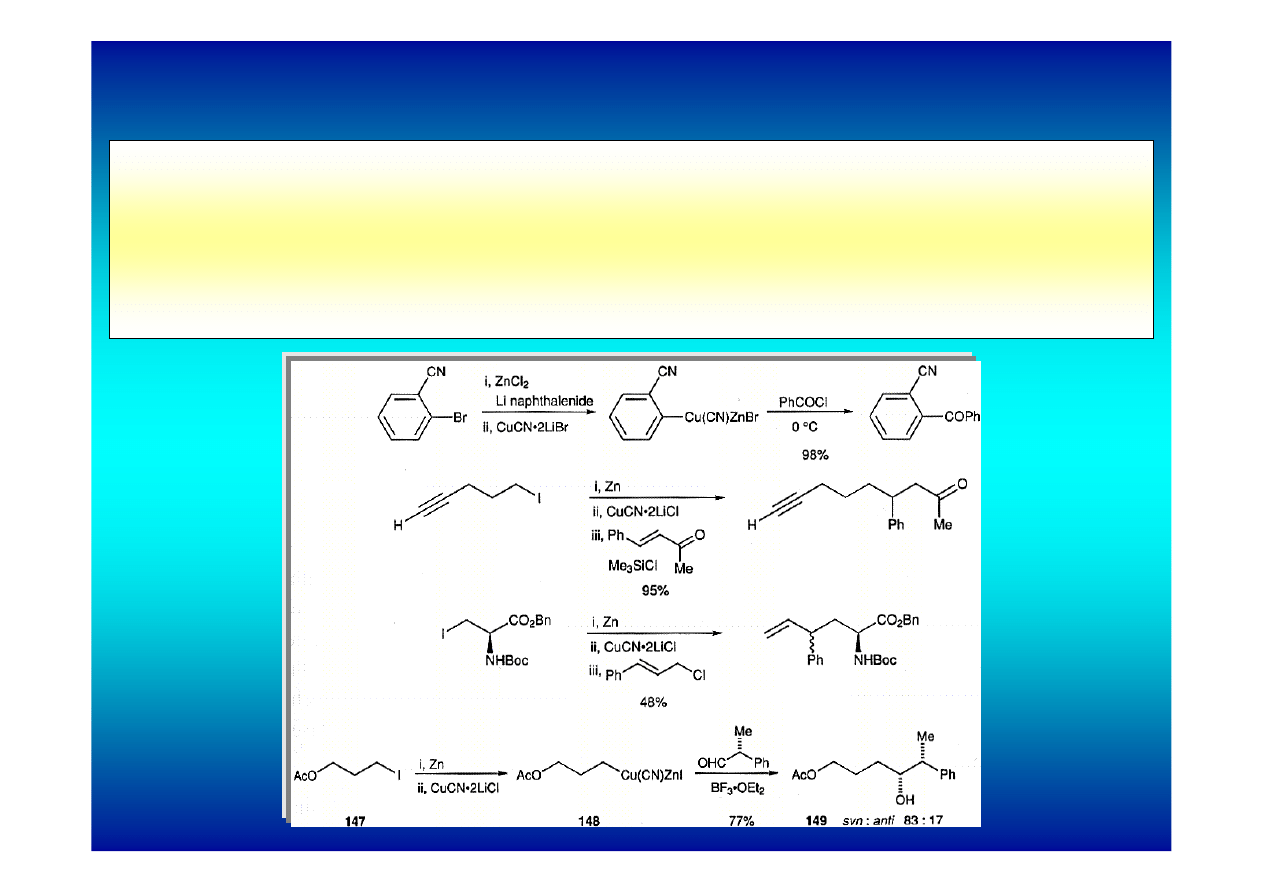
Kompleksy cynko-miedzioorganiczne
Reagenty te generowane są in situ w reakcji chlorku cynku z
kompleksem CuCN*2LiCl (rozpuszczalnym w THF).
Są bardziej reaktywne od związków cynkoorganicznych. Nie
izoluje się ich w trakcie reakcji.
Wyszukiwarka
Podobne podstrony:
ST14 20010 Met ppt
CC
met PCD
KM W 25 lekkie konst met stud
Met sta korekta ocen do e learningu
REGULACJA PID , Energetyka, sem5, sem5, met.ZN
Met. izol. oczysz.DNA dla studentów, Biologia molekularna
met.bad.ped.program, Studia, Semestry, semestr IV, Metody badań pedagogicznych
Cc, LITERKI
met
A dynamiki (przyklady 2 met klasyczna)
analityka światło i met opt 2012 2013
BAT met niez r6
2 przesylanie argumentow do met Nieznany
Proba statyczna roz met id 3926 Nieznany
metodyka met ind przyp, pr grup
egz CC 2011 06 25(SdS)
więcej podobnych podstron