TTTTTTTT
AAAAA
TTTTTTT
C
TTTT
AAA
#2 Metody
dystansowe
C
D
E
F
G
H
I
J
K
L
M
N
O
A
B
Przykładowe rodzaje cech stosowanych w rekonstrukcji filogenii
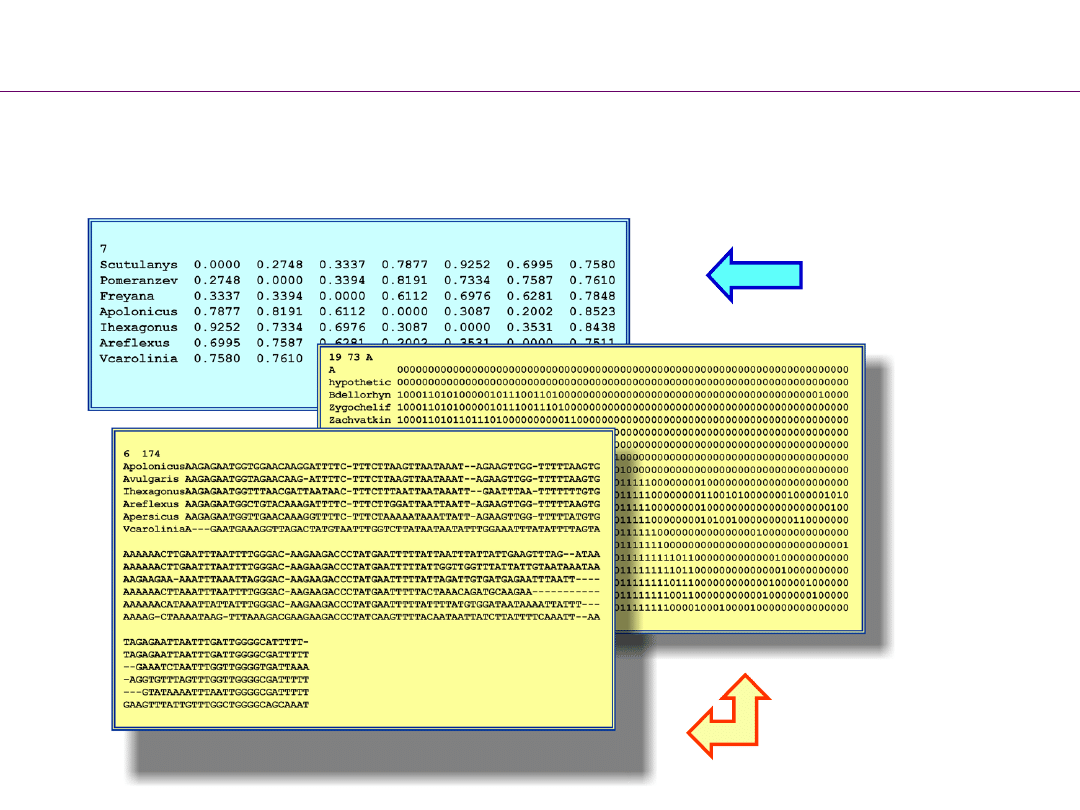
Typy matryc danych
matryca
cech
matryca
dystansów

Czy cechy morfologiczne są nadal istotne?
ukierunkowane
konwergencje
ograniczona liczba cech
trudności w znalezieniu cech
homologicznych między
odległymi taksonami
Zalety
Wady
tanie
możliwość korzystania ze
źródeł muzealnych
taksony wymarłe mogą być
analizowane głównie na
podstawie danych
morfologicznych
dane morfologiczne mogą
być testowalne na wszystkich
etapach analizy filogenetycznej

Metody konstruowania drzew filogenetycznych
Metoda obliczeniowa
optymalizacja
analiza klastrów
• Parsymonia
• Maximum Likelihood
• wnioskowanie
Bayesowskie
Cechy
• Minimum Evolution
• UPGMA
• Neighbor-Joining
Dystanse
Zasada działania
1.
Metody dystansowe zakładają stochastyczny
model ewolucji (np. sekwencji)
2.
Stosują ten model do określenia prawdziwej liczby
różnic (np. substytucji) z obserwowanych różnic
3.
Budują drzewo, które jest dopasowane najlepiej
do oszacowanych dystansów ewolucyjnych
Zasada działania
•
Liczona jest całkowita liczba substytucji, które występują
w parze porównywanych sekwencji (=taksonów) od
momentu dywergencji od wspólnego przodka.
•
Liczba ta jest dzielona przez długość sekwencji.
•
Dystans ten wyrażany jest w liczbie substytucji/miejsce
(dystans p)
przodek
sekwencja 1
sekwencja 2
substytucja
substytucja
substytucja
substytucja
substytucja

UPGMA
•
UPGMA (unweighted pair group method with arithmetic mean)
to najprostsza metoda grupująca taksony według ogólnego
podobieństwa lub odległości.
•
Pracuje wyłącznie na matrycach dystansowych np.
hybrydyzacja DNA-DNA lub konstruowanych z danych
sekwencyjnych na podstawie ilości substytucji.
•
UPGMA
umożliwia określenie długości gałęzi (odlegości
ewolucyjnej) jak i uporządkowania gałęzi.
•
Zakłada stały zegar molekularny – możliwe jest teoretycznie
oszacowanie czasu dywergencji na podstawie różnic w
sekwencjach.
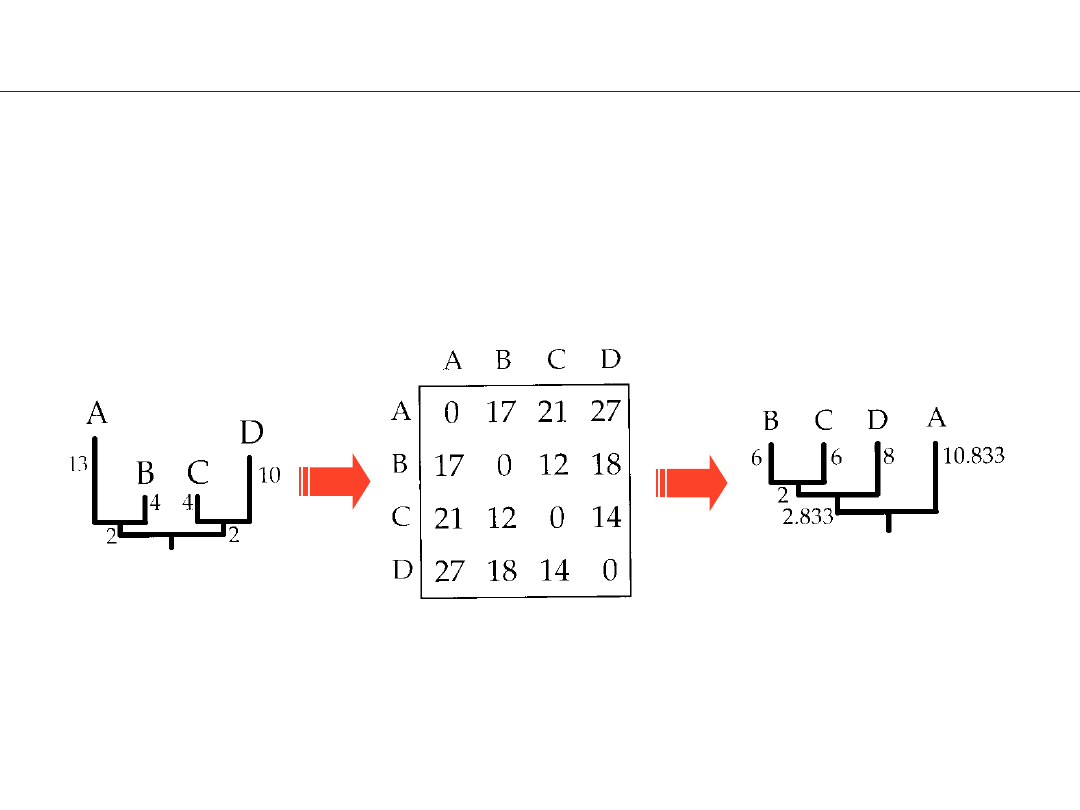
Jak działa UPGMA
OTU
A B C
D
A
-
8
7 12
B
-
9 14
C
- 11
D
-
Matryca dystansowa
– substytucje/100 nukleotydów
1. Znajdź najbliższą parę gatunków.
7
2. Połącz oba te gatunki w klaster.
3. Policz na nowo pozostałe dystanse jako średnią od A-C.
OTU A-C B
D
A-C
-
8,5 11,5
B
-
14
D
-
Matryca zredukowana
4. Idź do kroku 1 i powtórz procedurę, jeśli w tabeli pozostała tylko jedna
wartość to zakończ analizę.
D
B
C
A

Warunek „trzech punktów”
Aby analiza UPGMA mogła być przeprowadzona z sukcesem dane
muszą być zultrametryzowane. Oznacza to, że dla dowolnych trzech
taksonów (x, y, z) dystanse (d) pomiędzy nimi muszą spełniać
następujące wyrażenie:
Powyższą formułę nazywa się także
warunkiem „trzech punktów”
.
d(x,z)
≤ max (d(x,y), d(y,z))
Kiedy UPGMA zawodzi
Prawdziwe drzewo Matryca dystansów Drzewo UPGMA

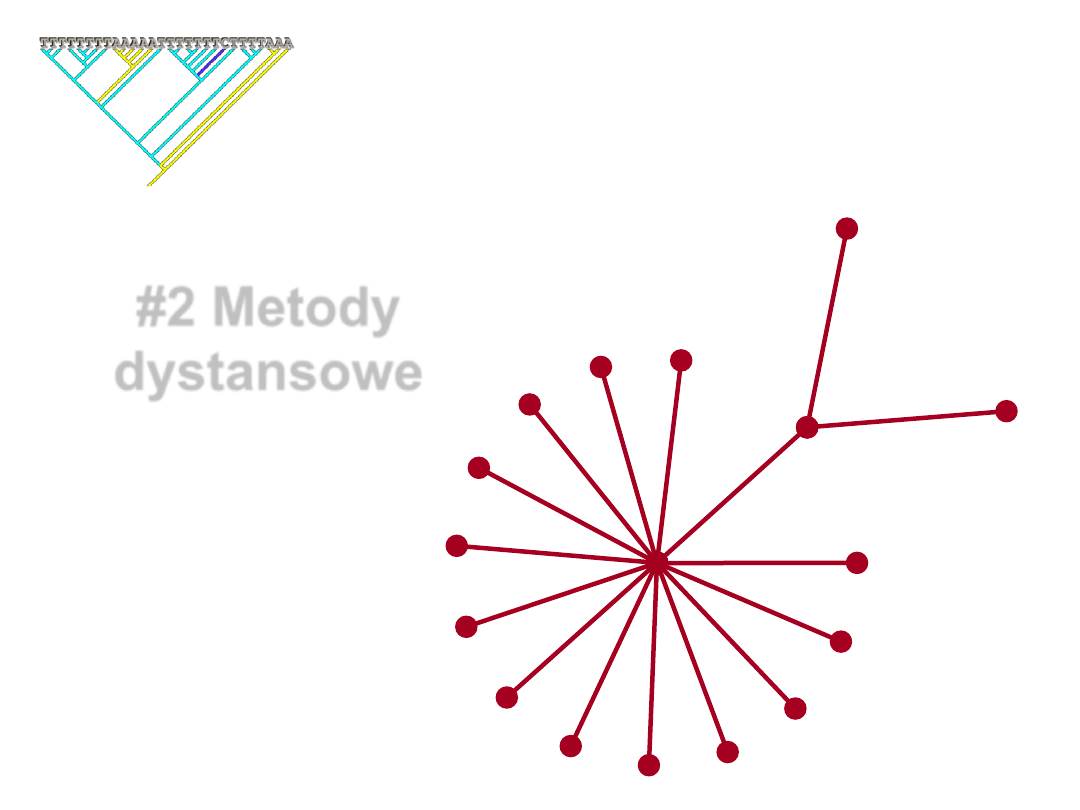
Neighbor-Joining (NJ)
•
Metoda koncepcyjnie zbliżona do analizy klastrów,
jednak dopuszcza niejednakowe tempo zmian
molekularnych wśród gałęzi.
• Zasada analizy NJ:
Wyszukiwanie par taksonów (sąsiadów=neighbors),
które minimalizują totalną długość gałęzi na każdym
etapie grupowania taksonów początkowo zgrupowanych
w całkowicie politomicznym drzewie („gwiazda”).
Jak działa NJ
1. Inicjalne drzewo ma postać
w pełni politomicznej
gwiazdy.
3. Powtarzane jest to ze wszystkimi moż-
liwymi kombinacjami par, aż do znale-
zienia drzewa o najmniejszej całkowitej
długości gałęzi. Para sekwencji z tego
drzewa sąsiaduje ze sobą w finalnym
drzewie.
2. Losowo wybierana jest para sekwencji i
łączona gałęzią z centrum gwiazdy.
Liczona jest całkowita długość gałęzi
drzewa (=suma dystansów). Para jest
zwracana do gwiazdy.
4. Para ta jest tymczasowo kombinowana
w jednostkę, włączana do gwiazdy
krótszej o jedną gałąź i matryca
dystansów liczona jest na nowo.
5. Procedura jest powtarzana tak długo, aż wszyscy „sąsiedzi” zostaną
znalezieni i otrzymamy gotowe drzewo.
G
A
B
C
D
E
F
H
G
A
B
C
D
E
F
H
G
A
B
C
D
E
F
H
G
A
B
C
D
E
F
H
G
(A,B)
C
D
E
F
H
G
((A,B),H)
C
D
E
F
((A,B),H)
C
D
E
(G,F)
((A,B),H)
D
(G,F)
(C,E)
((A,B),H)
(G,F)
(D,(C,E))
(((A,B),H),(G,F))
(D,(C,E))
((((A,B),H),(G,F)),(D,(C,E)))
H
A
B
G
F
D
C
E

Minimum Evolution (ME)
•
Metoda ściśle „spokrewniona” z NJ.
• ME stosuje kryterium minimalnej ewolucji, czyli
wyszukiwania drzewa o totalnej najmniejszej długości gałęzi
(=sumie dystansów).
•
Testowane są wszystkie możliwe topologie i wybierana ta o
najmniejszej wartości – bardzo ekstensywna metoda.
• Inicjalne drzewo produkowane jest przez NJ, mierzona
totalna długość gałęzi dla tego drzewa oraz drzew
pochodnych podobnych topologicznie, różniących się od
inicjalnego drzewa o d
t
=2 i 4. Jest to powtarzane wielokrotnie -
Close-Neighbor-Interchange (CNI)

najprostszy
najbardziej
złożony
Modele ewolucji
Frekwencje nukleotydów i są równe i wszystkie
rodzaje substytucji są jednakowo prawdopodobne
(Jukes-Cantor)
Frekwencje nukleotydów i są równe, ale tranzycje i
transwersje występują w różnych proporcjach
(Kimura 2 parametrowy)
Frekwencje nukleotydów i są różne oraz tranzycje i
transwersje występują w różnych proporcjach
(HKY)
Frekwencje nukleotydów są różne oraz wszystkie
typy substytucji występują w różnych proporcjach
(GTR)
• stosuje kryterium optymalizacji
• jak NJ
• informacja z sekwencji jest
zredukowana (dystanse)
•
daje tylko jedno możliwe
drzewo
•
silnie zależy od rodzaju
zastosowanego modelu ewolucji
•
bardzo szybka (długie
sekwencje, bootstrap)
•
akceptuje linie wykazujące
różne tempo ewolucji
•
bardzo wolna i wymaga dużej
mocy komputera i/lub procedur
heurystycznych (CNI)
• jak NJ
ME
NJ
• bardzo
czuła na różne tempo
ewolucji
•
grupowanie możliwe jest
jedynie, jeśli dane są
ultrametryczne tzn. spełniają
warunek „trzech punktów”
• bardzo prosta i bardzo szybka
UPGMA
Wady
Zalety
Metoda
Wady i zalety metod dystansowych

Wady i zalety metod dystansowych
Generalnie wszystkie
metody dystansowe
są
fenetyczne -
konstruują drzewa poprzez
grupowanie OTU na podstawie ogólnego
podobieństwa (morfologicznego, sekwencji itp.).
A ogólne podobieństwo nie koniecznie musi
odzwierciedlać prawdziwe pokrewieństwo
filogenetyczne.

Metody próbkowania (resampling)
•
Są to metody statystyczne służące do określenia stabilności
kladów.
•
Pobierane są wielokrotnie losowe próbki (pseudoreplikacje) z
danych.
•
Konstruowane są drzewka z wszystkich pseudoreplikacji i
procedura powtarzana jest wielokrotnie ( np. 1000 razy)
Następnie liczony jest 50% majority rule consensus.
•
Częstotliwość pojawiania się poszczególnych kladów w
drzewie konsensusowym stanowi miarę stabilności testowanej
topologii drzewa filogenetycznego.
•
Stosowane do wszelkich danych dyskretnych, także
dystansowych.
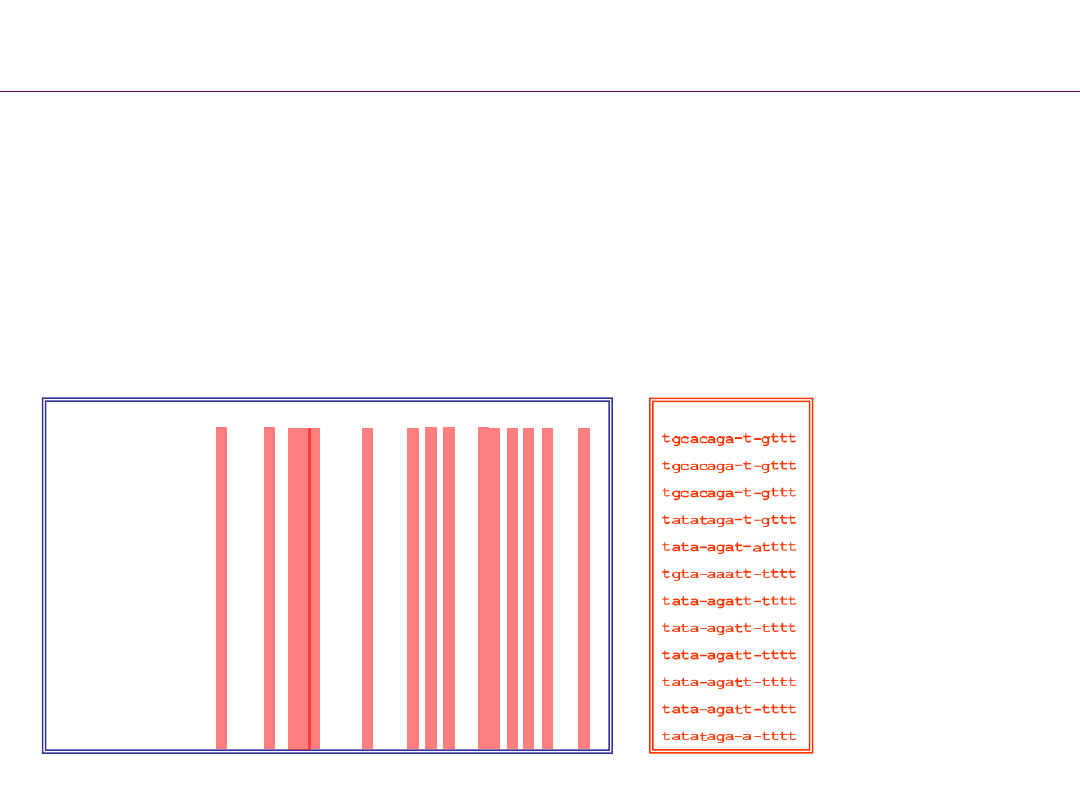
Jackknife
• Losowo pobierane do próbki cechy (dystanse) bez
zwracania danych do oryginalnej matrycy.
•
Symulacje wykazały, że najodpowiedniejszą wielkością
próbki jest 1/e (ok. 36,8%) danych.
Av.calidridis1 tatgaatgaattttctgagaactgttttttctg--ttttt-c
Av.calidridis2 tatgaatgaattttctgagaactgttttttctg--ttttt-c
Av.calidridis3 tatgaatgaattttctgagaactgttttttctg--ttttt-c
Av.calidridisalp tatgaatgaattttctaagggttggtttttttg--ttttt-c
Av.phalaropi tatgaatgaatttactatgaattttttct---gaattttttc
Av.philomachi tatgaatgaattttctgaaaattttttttt--a--tttattc
Av.totanigla1 tatgaatgaatgttctaaaaattttttttt--g--ttttttc
Av.totanigla2 tatgaatgaatgttctaaaaattttttttt--g--ttttttc
Av.totanitot1 tatgaatgaattttctaaaaattttttttt--g--ttttttc
Av.totanitot2 tatgaatgaattttctaaaaattttttttt--g--ttttttc
Av.tretekiae tatgaatgaattttctaataattttttttt--g--ttttttc
Av.tringae tatgaatgaattttctaataatttttattattg--ttttt--
oryginalna matryca
N=42
próbka
N=15
Procedura ta jest
powtarzana
wielokrotnie (np.
1000 razy) i za
każdym razem z
próbki budowane
jest drzewo (-a)
filogenetyczne.
Następnie
konstruowany
jest konsensus.

Bootstrap
•
Losowo pobierane są pseudoreplikacje i, w odróżnieniu do
jackknife
, dane zwracane są do oryginalnej matrycy.
•
Wielkość próbki jest taka sama jak matrycy oryginalnej.
Oznacza to, że pewne pseudoreplikacje są pobierane
więcej niż jeden raz.
Av.calidridis1 tatgaatgaattttctgagaactgttttttctg--ttttt-c
Av.calidridis2 tatgaatgaattttctgagaactgttttttctg--ttttt-c
Av.calidridis3 tatgaatgaattttctgagaactgttttttctg--ttttt-c
Av.calidridisalp tatgaatgaattttctaagggttggtttttttg--ttttt-c
Av.phalaropi tatgaatgaatttactatgaattttttct---gaattttttc
Av.philomachi tatgaatgaattttctgaaaattttttttt--a--tttattc
Av.totanigla1 tatgaatgaatgttctaaaaattttttttt--g--ttttttc
Av.totanigla2 tatgaatgaatgttctaaaaattttttttt--g--ttttttc
Av.totanitot1 tatgaatgaattttctaaaaattttttttt--g--ttttttc
Av.totanitot2 tatgaatgaattttctaaaaattttttttt--g--ttttttc
Av.tretekiae tatgaatgaattttctaataattttttttt--g--ttttttc
Av.tringae tatgaatgaattttctaataatttttattattg--ttttt--
oryginalna matryca
N=42
próbka
N=42
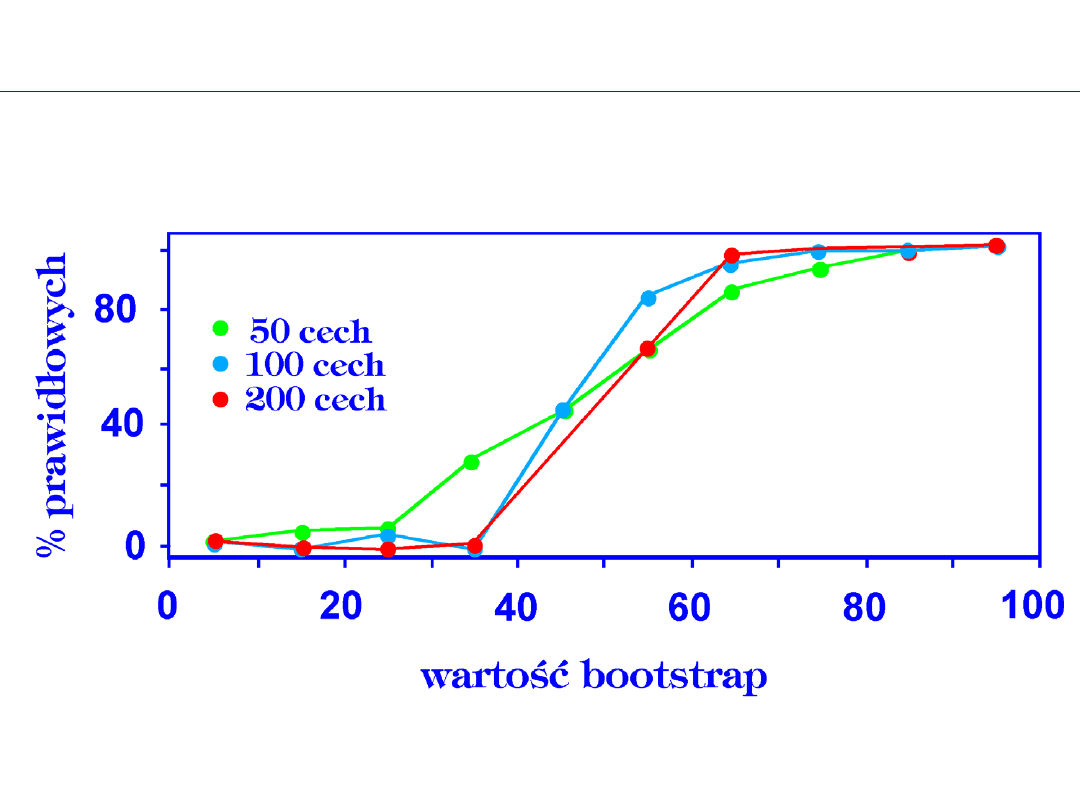
Bootstrap –
wartości krytyczne
Document Outline
- Slajd numer 1
- Przykładowe rodzaje cech stosowanych w rekonstrukcji filogenii
- Typy matryc danych
- Czy cechy morfologiczne są nadal istotne?
- Metody konstruowania drzew filogenetycznych
- Zasada działania
- Zasada działania
- UPGMA
- Jak działa UPGMA
- Warunek „trzech punktów”
- Kiedy UPGMA zawodzi
- Neighbor-Joining (NJ)
- Jak działa NJ
- Minimum Evolution (ME)
- Modele ewolucji
- Wady i zalety metod dystansowych
- Wady i zalety metod dystansowych
- Metody próbkowania (resampling)
- Jackknife
- Bootstrap
- Bootstrap – wartości krytyczne
Wyszukiwarka
Podobne podstrony:
02 Metody syntezy organicznej VI s1id 3675
17 02 Metodyka oceny ryzykaid 17384
02 metody pomiaru zdrowia
02 metody pomiaru zdrowia
02 metody badań neurobiologiczne podłoże pamięciid 3420 ppt
Metodyka wychowania przedszkolnego 19.02, metodyka wychowania przedszkolnego(1)
01 Wprowadzenie 02 metody geofizyczne 2id 3079 ppt
02 Metody syntezy organicznej VI s1id 3675
Zajęcia 02 Metody Badania Emocji
Rozdział 02 Metody wytwarzania materiałów i struktur półprzewodnikowych
02 Metody badan pedagogicznychid 3672 pptx
egzamin z metodyki 3 02 2009r 4rok
02 Wybrane metody numeryczne (aproksymacja funkcji, rozwiazy
07 02 2016 Metody obliczeniowe
więcej podobnych podstron