
1 WYKŁADY Z INTERNY
2001/2002
WYKŁAD 1 Niedokrwistości
02.10.2001
Mianem niedokrwistości określa się stan chorobowy, w którym liczba erytrocytów i stężenie hemoglobiny
zmniejsza się poniżej wartości optymalnych dla organizmu, tzn. takich, które zapewniają prawidłowe utleno-
wanie tkanek. Zgodnie z wytycznymi WHO u ludzi niedokrwistość rozpoznajemy, gdy poziom Hb < 12 g/dl u
kobiet i < 14 g/dl u mężczyzn.
Klasyfikacja niedokrwistości na podstawie przyczyn:
I.
Niedokrwistości spowodowane utratą krwi (pokrwotoczne)
II.
Niedokrwistości powstałe wskutek zaburzeń w wytwarzaniu erytrocytów i hemoglobiny (zmiany
jakościowe erytropoezy) – niedoborowe
III.
Niedokrwistości towarzyszące różnym chorobom (objawowe)
IV.
Niedokrwistości wywołane nadmiernym rozpadem erytrocytów (hemolityczne)
V.
Niedokrwoistości powstałe wskutek niedostatecznego tworzenia erytrocytów (aplastyczne)
I.
Niedokrwistości pokrwotoczne
Jest to najczęściej spotykana grupa niedokrwistości. Występuje, gdy utrata krwi przekracza zdolności kompen-
sacyjne układu krwiotwórczego lub mechanizmy wyrównawcze. Podział:
•
ostre, zależne od nagłej utraty większej objętości krwi krążącej (pojawiają się po 24h)
•
przewlekłe, zależne od stopniowej utraty krwi w niewielkich ilościach (podwyższone retikulocyty)
II. Niedokrwistości niedoborowe
Są to stany, gdy poziom erytrocytów i hemoglobiny jest niższy od prawidłowego na skutek braku jednego lub
kilku składników niezbędnych do prawidłowej erytropoezy.
Rozpoznanie jest możliwe, gdy podanie brakującego czynnika przywraca prawidłowe wartości erytr. i Hb.
Wszystkie niedokrwistości, których przyczyną jest brak czynników krwiotwórczych w samych komórkach he-
mopoetycznych zalicza się do niedokrwistości wtórnych lub achrestycznych.
A)
Niedokrwistości z niedoboru żelaza (anaemia sideropenica)
Przyczyny:
1.
Niedobór Fe w pokarmach
2.
Zwiększone zużycie Fe
•
ciąża
•
okres karmienia piersią
•
okres dojrzewania
3.
Zmniejszone wchłanianie Fe w takich stanach, jak:
•
bezkwaśność soku żołądkowego (sok żołądkowy powoduje przejście Fe
2+
Fe
3+
)
•
zapalenie błony śluzowej przewodu pokarmowego
•
stan po resekcji żołądka
4.
Nadmierna utrata Fe na skutek:
•
krwawień
•
zespołu złego wchłaniania
Objawy:
•
osłabienie
•
łatwe męczenie się
•
bóle i zawroty gło-
wy
•
mroczki przed
oczami
•
szum w uszach
•
drażliwość
•
trudności w koncentracji
•
apatia
•
senność
•
łatwe marznięcie
•
niechęć do jedzenia
•
kołatanie serca
•
bóle wieńcowe
•
zajady
•
pieczenie języka
•
zapalenie dziąseł
WYKŁADY Z INTERNY 2001/2002
2
•
nieżyt błony śluzo-
wej nosa
•
matowe włosy
•
łamliwość włosów i
paznokci
•
rozdwajanie się paznokci
•
prążkowanie paznokci
•
częste infekcje
Badania dodatkowe:
•
morfologia
-
Hb – obniżone
-
RBC – obniżone
-
MCV – obniżone
-
MCHC – obniżone
-
MCH – obniżone
-
hipochromia
-
mikrocytoza
-
retikulocyty – obniżone
•
Fe w surowicy – obniżone
•
TIBC – podwyższony poziom
•
badanie szpiku – wyklucza inne jednostki chorobowe
Wskaźniki krwinek czerwonych:
MCV – średnia objętość krwinki czerwonej – norma: 80 – 100 μm
3
MCH – średnia zawartość Hb w krwince – norma: 27 – 33 pg
MCHC – średnie stężenie Hb w krwince – norma: 32 – 36 g%
Profilaktyka:
1.
Produkty roślinne zawierające dużo Fe
2+
:
⇒
fasola
⇒
bób
⇒
soczewica
⇒
szpinak
⇒
zielona pietruszka
⇒
sałata
⇒
orzeszki ziemne
⇒
migdały
⇒
kakao
2.
Produkty pochodzenia zwierzęcego bogate w Fe
3+
:
⇒
podroby – głównie wątróbka
⇒
kaszanka
⇒
tatar
⇒
żółtko jaja
Leczenie:
preparaty doustne
-
Ascofer (tabl. 24 mg Fe
2+
): 4 – 6 tabl./d
-
Hemofer – krople
-
Hemofer prolongatum – tabl. 105 mg Fe
2+
-
Ferro-Gradumet – tabl. 105 mg Fe
2+
-
Tardyferon – tabl. 80 mg
preparaty pozajelitowe
-
Ferrum - Hausmann i.v. amp. co 2-gi dzień
-
Ferrum - Lek i.v. amp. co 2-gi dzień
-
Jectofer i.m. amp.
•
czas leczenia: 2 – 3 miesiące, by wysycić ustrój
Wskaźniki krwinek czerwonych:
MCV – średnia objętość krwinki czerwonej – norma: 80 – 100 μm
3
MCH – średnia zawartość Hb w krwince – norma: 27 – 33 pg
MCHC –
średnie stężenie Hb w krwince – norma: 32 – 36 g%

3 WYKŁADY Z INTERNY
2001/2002
B)
Niedokrwistości megaloblastyczne
Przyczyny:
I.
Niedobór witaminy B
12
1.
Niedobory pokarmowe
a)
wegetarianizm
b)
brak pokarmów pochodzenia zwierzęcego
2.
Zaburzenia wchłaniania
a)
choroba Addisona-Biermera
b)
Stan po gastrektomii
c)
Zapalenie żołądka z alkaliczną treścią
d)
Rak żołądka
e)
Choroby jelita cienkiego
3.
Niedostateczne wykorzystanie (zaburzenia polekowe)
a)
PAS
b)
pochodne fenforminy
c)
neomycyna
d)
kolchicyna
e)
cholestyramina
4.
Zwiększenie zapotrzebowania i/lub zużycia
a)
zużycie przez bakterie jelitowe lub pasożyty
-
bruzdogłowiec szeroki
-
zespół ślepej pętli
II.
Niedobór kwasu foliowego
WITAMINA B
12
Głównym źródłem witaminy B
12
dla człowieka jest mięso, mleko, jaja. Witamina B
12
uwalnia się w żołądku ze
związków z białkiem pod wpływem niskiego pH i działania pepsyny. Wolna cząsteczka witaminy B
12
zostaje
związana w żołądku przez czynnik wewnętrzny Castle’a (IF – Intrinsinc Factor) w stabilny kompleks umożli-
wiający jego wchłonięcie w alkalicznym środowisku jelita cienkiego.
NIEDOKRWISTOŚĆ ADDISONA-BIERMERA (N. ZŁOŚLIWA – Anaemia perniciosa)
Przyczyną choroby jest reakcja autoimmunologiczna skierowana przeciwko czynnikowi wewnętrznemu IF i
komórkom okładzinowym żołądka z wytworzeniem trzech rodzajów przeciwciał:
•
typ I – p/ciała skierowane przeciw IF blokują jego wiązanie z witaminą B
12
•
typ II – p/ciała przeciwko kompleksom IF-B
12
blokują jego wiązanie z receptorem jelitowym i
wchłanianie w jelicie cienkim
•
typ III – p/ciała skierowane przeciw komórkom okładzinowym żołądka prowadzą do nieżytu
zanikowego i ustania produkcji IF
Objawy kliniczne:
Choroba dotyczy przeważnie ludzi starszych (> 60 r.ż.) i rozwija się powoli i podstępnie. Objawy dotyczą 3
układów: pokarmowego, krwiotwórczego i nerwowego.
1.
Układ pokarmowy
zmiany zanikowe błony śluzowej:
-
języka – glossitis Hunteri (język gładki, lśniący, żywoczerwony)
-
żołądka – gastritis atrophicans
–
achlorhydia histaminooporna
OBJAWY:
pieczenie, palenie języka, brak łaknienia, uczucie pełności, wzdęcia, biegunki, zaparcia
2.
Układ krwiotwórczy

WYKŁADY Z INTERNY 2001/2002
4
niedokrwistość – często poniżej 1 mln /
µ
l
makrocytoza
leukopenia z hipersegmentacją jąder granulocytów
trombocytopenia
w ciężkich postaciach niewielka hemoliza – wzrost bilirubiny
szpik – hematopoeza megaloblastyczna
3.
Układ nerwowy
parestezje
uczucie drętwienia i mrowienia kończyn
zaburzenia psychiczne
niepokój
osłabienie pamięci
Leczenie:
-
witamina B
12
podawana pozajelitowo (i.m.)
-
witamina B
12
codziennie ok. 1000
µ
g i.m. – około 2 tyg.
-
do końca życia 100
µ
g i.m. 1 raz w miesiącu
PRZYCZYNY NIEDOBORU KWASU FOLIOWEGO
1.
Niedobory pokarmowe
a)
alkoholizm !!!
b)
brak świeżych, surowych pokarmów roślinnych
2.
Zaburzenia wchłaniania
3.
Niedostateczne wykorzystanie (zaburzenia polekowe)
a)
metotreksat
b)
izoniazyd
c)
sulfonamidy
d)
leki p/padaczkowe
e)
leki antykoncepcyjne
4.
Zwiększone zapotrzebowanie / zużycie
a)
ciąża i karmienie
b)
okres wzrostu
c)
niedoczynność tarczycy
IV. Niedokrwistości hemolityczne
Niedokrwistość hemolityczna jest stanem chorobowym powstałym w wyniku przyspieszonego rozpadu krwi-
nek czerwonych na skutek ich defektu strukturalnego lub wpływu różnorodnych czynników zewnętrznych. Po-
dejrzenie obecności hemolizy stwarzają skojarzenie podwyższonego odsetka retikulocytów z hiperbilirubine-
mią. Wskaźnikiem hemolizy jest obniżony poziom haptoglobiny.
A.
Nieprawidłowości pozakrwinkowe
1.
Czynniki immunologiczne
a)
niedokrwistości autoimmunohemolityczne
-
na tle p/ciał ciepłych
-
na tle p/ciał zimnych
b)
odczyny hematologiczne poprzetoczeniowe
c)
żółtaczka hemolityczna noworodków
2.
Czynniki nieimmunologiczne
a)
niektóre substancje chemiczne i leki
b)
bakterie, pasożyty, jad żmij
c)
czynniki fizyczne
d)
hipersplenizm
B.
Nieprawidłowoci wewnątrzkrwinkowe
1.
Dziedziczne (wrodzone)
a)
sferocytoza

5 WYKŁADY Z INTERNY
2001/2002
b)
eliptocytoza
c)
zespół hemolityczny na tle niedoborów enzymatycznych
-
niedobór enzymów procesów redukcyjnych
-
niedobór enzymów glikolitycznych
2.
Nabyte
a)
hemoglobinuria napadowa nocna (nie brać wtedy witaminy C !)
Sferocytoza wrodzona (choroba Minkowskiego-Chauffarda)
Dziedziczy się autosomalnie dominująco.
Etiopatogeneza
Zaburzenia budowy białek strukturalnych błony erocytów, głównie spektyny, powodują upośledzenie zdolności
do odkształcania i zmiany kształtu krwinki (sferocyt). Jest to przyczyną ich zalegania i nasilonego niszczenia w
śledzionie.
Objawy kliniczne
bladość skóry
inne zaburzenia wrodzone (wieżowata czaszka, podniebienie gotyckie)
powiększenie śledziony
żółtaczka
Badania dodatkowe
niedokrwistość
obecność sferocytów
zwiększona liczba retikulocytów
↑
poziom bilirubiny (pośredniej)
↓
oporność osmotyczna erytrocytów
Leczenie
splenektomia
w przełomach koncentrat krwinek czerwonych
sterydy
Niedokrwistości autoimmunohemolityczne
Spowodowane są reakcją pomiędzy antygenem krwinek czerwonych a skierowanym przeciwko nim p/ciałom.
Wyróżniamy p/ciała typu zimnego i ciepłego. Kompleks erytrocyt – p/ciało wiąże i aktywuje dopełniacz, co w
konsekwencji prowadzi do hemolizy wewnątrznaczyniowej.
Objawy kliniczne
niedokrwistość
żółtaczka
bóle w okolicy nerek, wątroby i śledziony
podwyższona temperatura
ciemne zabarwienie moczu
Badania laboratoryjne
niedokrwistość
podwyższona liczba erytrocytów
podwyższony poziom bilirubiny pośredniej
dodatni odczyn Coombsa
-
bezpośredni – p/ciała związane z krwinkami

WYKŁADY Z INTERNY 2001/2002
6
-
pośredni – p/ciała nie związane z krwinkami
czas przeżycia erytrocytów
stopień rozpadu w śledzionie
Leczenie
kortykoterapia
Encorton 1 mg/kg
immunoterapia – Imuran, Cyklofosfamid
splenektomia
plazmoforeza
przetaczanie masy erytrocytarnej
V. Niedokrwistości aplastyczne
Charakteryzują się ubogokomórkowym szpikiem i pancytopenią we krwi obwodowej. Przyczyną n.a. mogą być
zakażenia wirusowe, związki chemiczne, leki oraz promieniowanie jonizujące. W ponad 50% przypadków nie
udaje się ustalić przyczyny.
Niedokrwistość aplastyczna – stan głębokiego upośledzenia czynności krwiotwórczej szpiku, polegający na
zniknięciu ze szpiku komórek układu czerwonokrwinkowego, granulocytowego i płytkowego.
Przyczyny nabytych niedokrwistości aplastycznych:
I. Czynniki fizyczne – promieniowanie jonizujące, kosmiczne, UV
II. Środki chemiczne – leki, trucizny, przemysł, skażenie środowiska, detergenty, środki ochrony
roślin, a zwłaszcza owadobójcze
III. Wirusy (Parvowirusy, WZW)
IV.
Bakterie (np. prątki gruźlicy, Salmonella)
V.
Nowotwory złośliwe i choroby mielo- i limfoproliferacyjne, zespoły przedbiałaczkowe
VI.
Choroby immunologiczne
VII. Przyczyny immunologiczne bez jawnej choroby
VIII. Zwłóknienie szpiku
IX.
Niedokrwistość hemolityczna
X.
Nocna napadowa hemoglobinuria (?)
Leczenie
globuliny antymonocytarne lub antylimfocytarne
cyklosporyna A 5-10 mg/kg (średnio 400-600 mg)
sterydy
przeszczep szpiku
WYKŁAD 2
Białaczki ostre
09.10.2001
Terminem białaczki ostre określa się grupę złośliwych chorób nowotworowych wywodzących się z różnych li-
nii rozwojowych układu krwiotwórczego.
Białaczki ostre wywodzą się z wczesnych stadiów rozwoju ontogenetycznego, co obok złego naturalnego prze-
biegu, prowadzącego bez leczenia szybko do śmierci, jest zasadniczą cechą odróżniającą je od białaczek prze-
wlekłych.
W chwili rozpoznania są nowotworami rozsianymi z obecnością w organizmie ogromnej liczby komórek
białaczkowych (często przekraczającą 10
12
komórek, w przybliżeniu ok. 1 kg), naciekających szpik kostny,
śledzionę, wątrobę oraz węzły chłonne i inne narządy.
Etiopatogeneza
Etiologia białaczek, podobnie jak i innych nowotworów nie jest jeszcze wyjaśniona. U podstaw procesu
białaczkowego leży transformacja białaczkowa w obrębie określonej linii komórkowej prowadząca do za-
burzenia różnicowania i dojrzewania na różnym szczeblu rozwoju ontogenetycznego.

7 WYKŁADY Z INTERNY
2001/2002
Istota i mechanizm transformacji białaczkowej
Transformacja białaczkowa prawdopodobnie związana jest ze zmianami w genach, a ściślej ze zmianami
sekwencji kodujących etapy rozwoju osobniczego lub proliferacji. Istotą transformacji białaczkowej jest blok
w różnicowaniu i dojrzewaniu oraz niereagowanie na czynniki regulujące.
Czynniki mogące wywołać transformację białaczkową
wirusy (HTLV-1)
promieniowanie jonizujące
czynniki chemiczne (papierosy, benzen, melfalan, chlorambucil, cyklofosfamid)
czynniki genetyczne (trisomia 21, XXY)
wtórne (zespoły mielodysplastyczne)
Obraz kliniczny ostrych białaczek
1.
początek choroby ostry lub podostry, osłabienie, łatwe męczenie, bóle głowy
2.
objawy zakażenia – afty, owrzodzenia j. ustnej, angina, zapalenia płuc
3.
objawy skazy krwotocznej – krwawienia z dziąseł, nosa, przewodu pokarmowego, dróg rodnych
4.
bóle kostno-stawowe
5.
zaburzenia gastryczne
6.
stan ogólny średnio- lub ciężki, chory cierpiący
7.
skóra – blada, sińce, wybroczyny, krwawienia z miejsc wkłucia, nacieki białaczkowe w skórze
(częściej w białaczkach limfoblastycznych o podostrym przebiegu)
8.
powiększenie węzłów chłonnych – głównie w białaczce limfoblastycznej
9.
zmiany w jamie ustnej i w gardle – w M
4
i M
5
znaczny przerost dziąseł, zmiany martwicze („fetor
ex ore”)
10. mierne powiększenie wątroby i śledziony (głównie w ostrej białaczce limfatycznej)
11. objawy oponowe – nacieki OUN (głównie w o.b.l.)
Badania dodatkowe
morfologia
-
ilość leukocytów – prawidłowa, podwyższona, obniżona
-
niedokrwistość
-
małopłytkowość
rozmaz krwi obwodowej
-
obecność komórek blastycznych
-
hiatus leucaemicus – dla ostrych białaczek – są komórki niedojrzałe (blasty) i krwinki
dojrzałe (brak form pośrednich)
rozmaz szpiku
-
komórki białaczkowe stanowią 30-100% elementów jądrzastych szpiku
W celu określenia linii komórkowej wykonujemy następujące badania:
1.
diagnostyka cytomorfologiczna
2.
diagnostyka cytochemiczna
3.
diagnostyka immunologiczna
4.
diagnostyka cytogenetyczna
Dokładne określenie podtypu białaczki uzyskuje się za pomocą oceny cytomorfologicznej, immunologicznej
oraz cytogenetycznej.
Diagnostyka cytomorfologiczna
Barwienie komórek krwi obwodowej i szpiku.
Diagnostyka cytochemiczna
Linie komórkowe zawierają charakterystyczne związki chemiczne i enzymy. 100% limfoblastów białacz-
kowych zawiera glikogen. Substancje lipidowe występują w mieloblastach, czasem w mielomonoblastach. W
mieloblastach, mielomonoblastach i monoblastach występuje mieloperoksydaza (POX), w monoblastach,
mielomonoblastach i limfoblastach – esterazy:
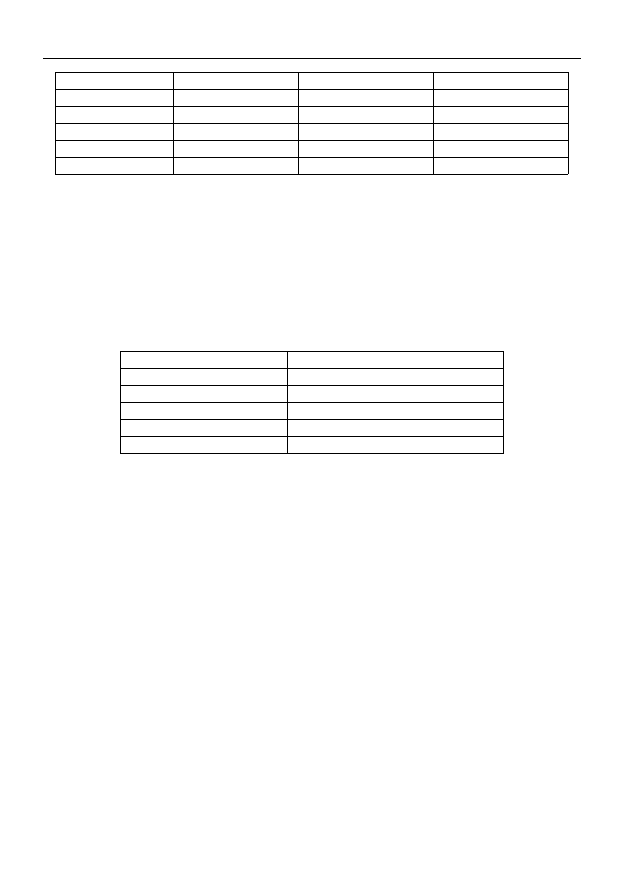
WYKŁADY Z INTERNY 2001/2002
8
reakcja
AMI
M1 – M5
M
6
ALL
POX
+
-
-
Sudan B
+
-
-
nieswoista esteraza
+ (
M
4
, M
5
, ham. NaF
*)
-
-
PAS
+
dyfuzyjna
+
blokowa
+
blokowa
fosfataza kwaśna
-
-
+
* reakcja hamowania fluorkiem sodu
Diagnostyka immunologiczna
Opiera się na technice fenotypowania białek przy pomocy markerów błon komórkowych i wewnątrzkomór-
kowych. Obecnie znamy ponad 130 antygenów różnicowania komórkowego wykrywanych na różnych etapach
dojrzewania komórek układu krwiotwórczego. Spośród tak dużej liczby p/ciał monoklonalnych tylko część jest
stosowana w praktyce codziennej do diagnostyki ostrych białaczek, a niektóre wykrywane przez nie antygeny
mogą mieć znaczenie prognostyczne. Immunotypizacja zmniejsza liczbę białaczek określanych jako „nieskla-
syfikowane” z 20% do ok. 3%.
Wykaz p/ciał monoklonalnych stosowanych do identyfikacji antygenów powierzchniowych blastów różnych li-
nii komórkowych:
markery linii komórkowej B
CD10, CD19, CD 20, CD22
markery linii komórkowej T
CD1, CD2, CD3, CD4, CD5, CD7, CD8
markery linii mielocytarnej
CD13, CD15, CD33, Cdw65
markery linii płytkotwórczej
Cdw41, Cdw42
markery linii erytroblastycznej
GpA-glikoforyna A
markery linii monocytarnej
CD11b, CD14
Diagnostyka cytogenetyczna
Współczesne badania cytogenetyczne obok oceny liczby chromosomów umożliwiają dokładniejsze określenie
ich struktury. Przyczyniło się to do identyfikacji określonych zmian u chorych na ostre białaczki (aneuploidii –
zmiany liczby chromosomów, anomalii chromosomów), które występują w sposób powtarzalny i często kore-
lują z ich cechami fenotypowymi oraz przebiegiem klinicznym.
Molekularna diagnostyka hematologiczna służy do:
określenia genu markerowego dla określonego schorzenia
określenia ekspresji genów dla cytokin i mechanizmów ich regulacji
badania genów oporności wielolekowej
badania zgodności tkankowej w celu typizacji potencjalnych dawców szpiku kostnego
PODZIAŁ OSTRYCH BIAŁACZEK
klasyfikacja powinna określać z jakiego układu wywodzi się dana białaczka i na jakim szczeblu
określonej linii różnicowania nastąpiła transformacja nowotworowa
na podstawie kryteriów cytomorfologicznych oraz metod cytochemicznych powstała klasyfikacja
ostrych białaczek FAB (francusko-amerykańsko-brytyjska) używana głównie do ostrych białaczek
szpikowych
KLASYFIKACJA FAB
I
Ostra białaczka limfoblastyczna (o.b.l. – ALL)
II
Ostra białaczka szpikowa (o.b.sz. – AML)
M
1
Ostra białaczka mieloblastyczna bez dojrzewania
M
2
Ostra białaczka mieloblastyczna z dojrzewaniem
M
3
Ostra białaczka promielocytowa (przebiega z DIC)
M
4
Ostra białaczka mielomonocytowa
M
5
Ostra białaczka monocytowa
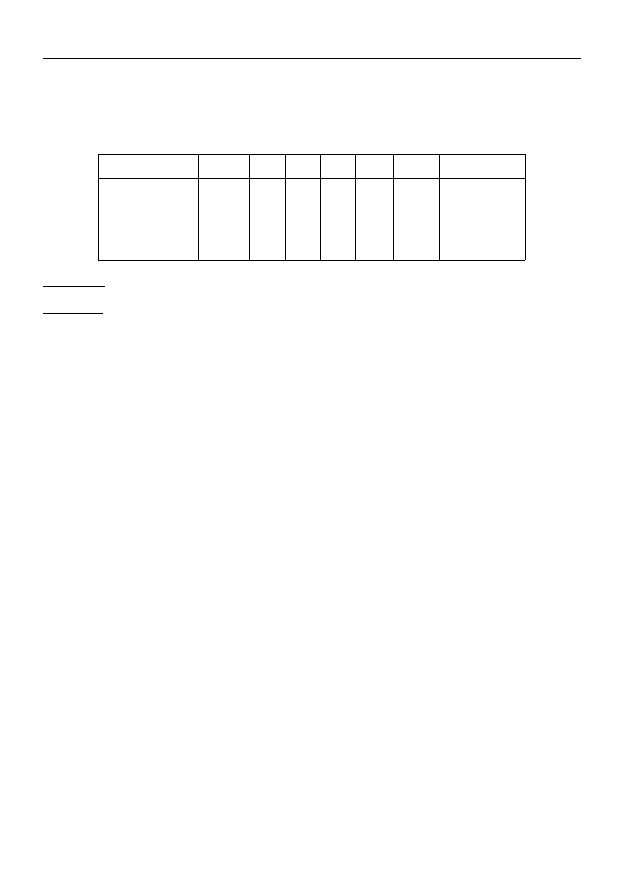
9 WYKŁADY Z INTERNY
2001/2002
M
6
Erytroleukemia
M
7
Ostra białaczka megakarioblastyczna
Czasem wyróżnia się też M
0
– białaczka bardzo nisko zróżnicowana
Markery immunologiczne podtypów ALL
podtypy ALL
HLA-DR
CD19 CD10 CytIg PowIg
CD7,1,
3
CD2 lub rozety E
null
common
pre-B komórkowa
B-komórkowa
pre-T komórkowa
T-komórkowa
+
+
+
+
+
+
+
+
+
+ / -
+ / -
+
+
+
+
+
Leczenie ostrych białaczek
Teoretycznie znając defekt genetyczny leżący u podstaw transformacji białaczkowej powinniśmy go naprawić
lub skompensować skutki defektu selektywnie działającym lekiem.
Praktycznie:
chemioterapia
zwalczanie zakażeń
zwalczanie zaburzeń krzepnięcia
leczenie niedokrwistości i leukopenii
zwalczanie hyperurykemii i zaburzeń wodno-elektrolitowych
leukostaza – wykonywanie leukaferezy
przeszczep szpiku
Chemioterapia
Podstawą chemioterapii jest stosowanie dużych dawek odpowiednio dobranych leków cytostatycznych.
Etapy:
indukcja remisji
konsolidacja
leczenie poemisyjne (pokonsolidacyjne)
Indukcja remisji – ma za zadanie zredukowanie masy guza do takiej ilości komórek białaczkowych, którą
zdrowy ustrój potrafi sam wyeliminować, czyli zmniejszenie około milion razy ilości komórek białaczkowych
– rzędu 10
5
– 10
6
.
Remisja kliniczna – brak objawów choroby.
Remisja cytogenetyczna – brak objawów w badaniach laboratoryjnych.
Remisja może też być częściowa lub całkowita.
Konsolidacja
-
ma miejsce po skutecznym leczeniu indukcyjnym
-
ma za zadanie zlikwidować tzw. chorobę resztkową – komórki, które przetrwały
-
ma na celu uzyskanie remisji
-
jest z reguły leczeniem silnym
Leczenie podtrzymujące (pokonsolidacyjne) – 3 opcje:
1.
Powtarzanie nieindukcyjne o rotacyjnym zmiennym składzie cytostatyków
2.
Wydłużenie konsolidacji i następnie obserwacja chorego z nadzieją, że udało się wyleczyć chorobę
resztkową
3.
Transplantacja szpiku
Zwalczanie zakażeń
o
antybiotyki szerokowachlarzowe
o
leki przeciwgrzybicze
-
doustne: Nystatyna, Pimafucin, Ketokonazol
-
dożylne: Amfoterycyna B, Diflucan
o
leki przeciwwirusowe: Acyklowir (Zovirax), Groprinosin

WYKŁADY Z INTERNY 2001/2002
10
Zwalczanie zaburzeń krzepnięcia
o
leki uszczelniające (witamina C, cyklonamina, Ca)
o
glikokortykosteroidy (Encorton, Hydrokortyzon)
o
masa płytkowa
o
osocze mrożone, krioprecypitat
o
w DIC heparyna
Leczenie niedokrwistości i leukopenii
o
koncentrat krwinek czerwonych (KKCz)
o
masa leukocytarna
o
czynniki wzrostu
-
GM-CSF (Leucomax)
-
G-CSF (Neupogen)
Inne leczenie wspomagające
o
↑
poziomu kwasu moczowego – allopurinol (Allopurinol, Milurit)
o
nudności, wymioty –
-
metoklopramid i.v. (antagonista rec. DA)
-
ondansetron i.v. (Zofran – anty 5-HT
3
)
o
nawodnienie
o
niskie stężenie potasu w surowicy – Elkinton II i.v., Kalium Effervescens p.o.
o
kardioprotekcja – Kardioksan
Przeszczepy szpiku – rodzaje:
allogeniczny (BMT)
autogeniczny (ABMT)
syngeniczny
Allogeniczny - między osobnikami tego samego gatunku, ale genetycznie różnymi (rodzeństwo, bliźniaki 2-
jajowe, dawca spoza rodziny), o prawie identycznym układzie antygenów HLA.
Autogeniczny – pochodzi od tego samego biorcy szpiku, jest pobierany w okresie prawidłowej funkcji tkanki
szpikowej i przechowywany do czasu użycia.
Syngeniczny – dokonywany jest przy całkowitej zgodności antygenów HLA (wyłącznie bliźniaki 1-jajowe) –
najrzadziej wykonywana odmiana.
Komórki krwiotwórcze otrzymuje się z: szpiku, krwi obwodowej i krwi pępowinowej. Wieloletnie przeżycia u
chorych na ostre białaczki po transplantacji szpiku obserwuje się w 50-60% przypadków.
WYKŁAD 3
Przewlekła białaczka szpikowa
16.10.2001
Przewlekła białaczka szpikowa (PBS) = Myelosis leucaemia chronica (MLC) = Chronic myelogenes
leucaemia (CML)
PBS – jest to niepohamowana hiperplazja układu granulocytarnego z zaburzeniem dojrzewania i morfologicz-
nego różnicowania się komórek. Konsekwencją tego jest:
pojawienie się we krwi obwodowej nadmiernej liczby dojrzałych i młodych komórek szeregu
granulocytarnego
nacieczenie tkanki szpikowej i stłumienie pozostałych układów utkania szpikowego
wtórna niedokrwistość i trombocytopenia
metaplazja pozaszpikowa przede wszystkim do wątroby i śledziony
Epidemiologia
PBS stanowi 15-20% wszystkich białaczek
najczęściej chorują osoby w wieku średnim
w Polsce choruje ok. 500 osób

11 WYKŁADY Z INTERNY
2001/2002
u około 50% chorych PBS rozpoznawana jest przypadkowo podczas rutynowych badań krwi
ok. 80-90% chorych w chwili rozpoznania znajduje się w fazie przewlekłej choroby
Patogeneza
U ponad 90% chorych na PBS stwierdza się chromosom Philadelphia – zmieniony chromosom 22. Powstaje
on w wyniku wzajemnych translokacji części długich ramion chromosomów 9 i 22 pary –t(9,22). Fragment
BCR długiego ramienia jednego z chromosomów 22 łączy się z protoonkogenem abl pary 9. W wyniku tego
zjawiska powstaje onkogen hybrydowy BCR-abl, który koduje syntezę nieprawidłowego białka 210 kd.
Białko 210 kd:
-
zwiększa aktywność kinazy tyrozynowej
-
zaburza fizjologiczną regulację podziałów i dojrzewania w cyklu komórkowym
-
hamuje apoptozę komórek
Okresy kliniczne
1.
Okres przewlekły
Przebieg podstępny, często trwa wiele lat. Prognozowany czas przeżycia wynosi 3 – 6 lat. Objawy ogól-
ne: zmęczenie,
↓
sprawności, poty nocne.
2.
Okres przyspieszenia – akceleracji
Objawy ogólne: narastająca leukocytoza, niedokrwistość, małopłytkowość, powiększenie śledziony,
czasem gorączka. Prognozowany czas przeżycia wynosi 3 – 9 miesięcy.
3.
Przełom blastyczny (kryza blastyczna)
Do tego okresu dochodzi u wszystkich chorych, którzy nie zmarli z powodu wcześniejszych powikłań
choroby. Przebiega ze wzrostem odsetka mieloblastów i promielocytów we krwi obwodowej (przełom
mieloidalny) lub ze wzrostem odsetka komórek limfocytarncych (przełom limfoidalny). Przebieg jest po-
dobny do ostrej białaczki i z reguły kończy się zgonem. Prognozowany czas przeżycia wynosi 3 – 6 m-cy.
Badanie przedmiotowe
powiększenie węzłów chłonnych
splenomegalia (nawet do 11 kg !)
hepatomegalia
objawy niedokrwistości i skazy krwotocznej, głównie płytkowej
Badania dodatkowe
1.
Morfologia krwi obwodowej
-
leukocytoza (nawet do 500000 / mm
3
) z „przesunięciem leukogramu w lewo”
-
niedokrwistość
-
nadpłytkowość, małopłytkowość, zaburzenia czynności płytek
2.
Szpik
-
badania cytologiczne (
↑
promielocytów, mielocytów)
-
badania cytogenetyczne (obecność chromosomu Ph
1
)
-
badania molekularne (obecność genu fuzyjnego BCR-abl)
3.
Badania biochemiczne -
↑
LDH
4.
USG jamy brzusznej – ocena powiększenia wątroby i śledziony
5.
FAG (aktywność fosfatazy alkalicznej granulocytów) – znacznie zmniejszona
Leczenie
Zależy od wieku chorego i fazy klinicznej choroby

WYKŁADY Z INTERNY 2001/2002
12
1.
Chemioterapia
Hydroxycarbamid tabl. a 500 mg, dawka dzienna 0,5 – 4,0 g
Busulfan (Myleran) tabl. a 2 mg (długo stosowany daje zwłóknienie szpiku)
-
w okresie indukcji dawka dzienna 6-10 mg,
-
leczenie podtrzymujące 2 mg / dobę
2.
Leukoforeza – u chorych z dużą leukocytozą i objawami leukostazy
3.
Interferon
α
- Intron A
4.
Przeszczep allogeniczny
Długoletnie przeżycia (średnio 10 lat) obserwowane są u 55-80% chorych. Najlepsze wyniki
osiągane są przy przeszczepieniu szpiku w przewlekłej fazie choroby, w jak najkrótszym
czasie od rozpoznania PBS oraz u chorych młodszych i mających dawców o lepszej zgodności
w układzie HLA (może wystąpić reakcja przeszczep przeciwko gospodarzowi, czasem tak sil-
na, że niszczy ustrój biorcy i doprowadza do śmierci)
5.
Napromienianie śledziony
6.
Splenektomia – usunięcie śledziony
7.
Fazę blastyczną leczymy jak białaczkę ostrą
8.
STI 571 (Glivec)
inhibitor kinazy tyrozynowej
hamuje miejsce wiązania ATP w strukturze enzymu a tym samym upośledza fosforylację białek
BCR-abl zaangażowanych w przenoszeniu sygnałów do komórek (reguluje podział i dojrzewanie
komórek w cyklu komórkowym), co prowadzi do remisji hematologicznej i cytologicznej
jego zastosowanie prawdopodobnie będzie miało miejsce u chorych w fazie przewlekłej,
przejściowej jak i kryzy blastycznej
minusem jest to, że hamuje kinazy tylko w momencie podania – trzeba podawać cały czas
koszt kuracji rocznej – ok. 120 tys. zł.
Przewlekła białaczka limfatyczna (PBL)
Leucaemia lymphatica chronica (LLC) = Chronic lymphocytic leucaemia (CLL)
PBL jest nowotworem heterogennym cechującym się akumulacją limfocytów we krwi obwodowej, szpiku
kostnym oraz infiltracją przez te komórki węzłów chłonnych, śledziony i wątroby.
Choroba występuje częściej w wieku średnim i starszym (45-70 r.ż.) oraz u mężczyzn.
99% przypadków dotyczy limfocytów B.
Etiologia
Etiologia choroby jest nieznana, wymienia się jednak różne czynniki mogące mieć znaczenie patogenetyczne,
takie jak: predyspozycja genetyczna, ekspozycja na szkodliwe czynniki środowiskowe, promieniowanie jonizu-
jące oraz wirusy. W chorobie tej wykazano
↑
ekspresji onkogenu bel-2. Gen ten jest odpowiedzialny za ha-
mowanie apoptozy, co może tłumaczyć wzrastającą akumulację białaczkowych limfocytów.
Objawy kliniczne
początek choroby – wykrycie często przypadkowe
w wywiadzie:
-
częste infekcje (np. półpasiec)
-
narastające osłabienie
-
utrata masy ciała
-
brak apetytu
-
nocne poty
-
świąd skóry
Badanie przedmiotowe
uogólnione powiększenie węzłów chłonnych
powiększenie śledziony i wątroby

13 WYKŁADY Z INTERNY
2001/2002
cechy niedokrwistości często szybko narastającej – niedokrwistość immunohemolityczna
cechy skazy krwotocznej
infekcje
często półpasiec (Herpes zoster)
Badania laboratoryjne
krew obwodowa
-
leukocytoza 20 – 500 tys. / ml
-
w rozmazie gł. limfocyty (90 – 98% komórek)
-
obecne cienie Gumprechta (cienie rozpadłych komórek)
-
niedokrwistość późno (wyj: stany hemolityczne)
-
małopłytkowość późno
szpik
-
dominują komórki limfatyczne
biopsja węzłów chłonnych
Rozpoznanie PBL
Opiera się na wywiadzie, badaniu klinicznym, morfologii krwi obwodowej, badaniu szpiku oraz badaniach im-
munofenotypowych. Do oceny stopnia zaawansowania choroby należy wykonać badania RTG oraz USG lub
CT jamy brzusznej. Badanie histopatologiczne węzłów chłonnych jest potrzebne do różnicowania PBL z inny-
mi chorobami limfoproliferacyjnymi.
Leczenie
Ustalenie rozpoznania PBL B-komórkowej nie jest równoznaczne z rozpoczęciem leczenia. U chorych w do-
brym stanie ogólnym z niskim stopniem zaawansowania, niską lub umiarkowanie podwyższoną leukocytozą
należy przyjąć postawę wyczekującą – wait and watch.
Kiedy leczyć?
Wskazania do rozpoczęcia leczenia
1.
obecność objawów ogólnych
-
utrata masy ciała > niż 10% w ciągu ostatnich 6 miesięcy
-
bardzo duże osłabienie
-
gorączka > 38
°
C utrzymująca się min. 2 tygodnie i nocne poty bez cech infekcji
2.
narastająca niedokrwistość lub małopłytkowość
3.
masywne postępujące powiększenie węzłów chłonnych
4.
masywna lub postępująca splenomegalia
5.
cytopenia autoimmunologiczna
6.
gwałtownie narastająca liczba limfocytów we krwi obwodowej (czas podwojenia liczby < 6 m-cy)
Leczenie – chemioterapia:
1.
chlorambucil (Leukeran) p.o. tabl. 2 mg – 12 mg/m
2
przez 5-7 dni co 4-6 tygodni
2.
COP (cyklofosfamid + Oncovin (winkrystyna) + prednizon); CHOP (COP + doksorubicyna);
CHOP + bleomycyna
3.
nowe analogi puryn – stosowane we wlewach i.v.
-
2 CDa (Biodribin)
-
fludarabina (Fludara)
również w skojarzeniu z innymi cytostatykami
-
2 CDa + cyklofosfamid
-
2 CDa + cyklofosfamid + Mitoxantrone
cykle powtarzać co 28 dni
4.
monowalentne ludzkie p/ciała anty CD20 np. Rituksymeb
5.
allotransplantacja

WYKŁADY Z INTERNY 2001/2002
14
Leczenie wspomagające
glikokortykosteroidy (Encorton, Prednison)
leukoforeza
napromienianie miejscowe
splenektomia
preparaty immunoglobulin
koncentraty krwinek czerwonych i płytek
erytropoetyna
leczenie zakażeń – antybiotyki szerokowachlarzowe, leki p/grzybicze i p/wirusowe
BIAŁACZKA KOSMATOKOMÓRKOWA
Jest ona najbardziej znaną postacią przewlekłej białaczki limfatycznej B-komórkowej. Występuje 4-5 x
częściej u mężczyzn. Typowy obraz przedstawia pacjenta ze splenomegalią, której towarzyszy pancytopenia.
Limfadenopatia jest rzadkim objawem w momencie rozpoznania. Leukopenia jest bardzo charakterystycznym
znacznikiem tej białaczki różnicującym ją od innych białaczek B-komórkowych, ale należy pamiętać, że 10%
chorych ma leukocytozę > 10 x 10
9
/l. Charakterystyczną cechą rozpoznawczą jest reakcja cytochemiczna
wykazująca aktywność kwaśnej fosfatazy opornej na winian (TRAP).
SZPICZAK PLAZMOCYTOWY
(Plasmocytoma, Multiple myelome)
Choroba nowotworowa wywodząca się z limfocytów B, charakteryzująca się rozsianymi, wieloogniskowymi
naciekami szpiku kostnego. Prowadzi to do niszczenia kości i uniemożliwia prawidłowe wytwarzanie
elementów morfotycznych krwi.
Klon uzłośliwionych komórek plazmatycznych wytwarza:
cząsteczkę białka monoklonalnego (komponentę M) lub jej fragmenty (łańcuchy lekkie i ciężkie)
interleukiny (1 i 6) oraz TNF (
α
i
β
) – substancje pobudzające aktywność osteoklastów (OAF)
Etiologia
Przyjmuje się udział:
czynników genetycznych
promieniowania jonizującego
środowiskowych czynników agrotechnicznych
zakażeń wirusowych i bakteryjnych
Epidemiologia
częstość występowania – 3 / 100000 osób rocznie
choroba dotyczy osób po 40 rż., szczyt 60 rż.
najczęstszy nowotwór szpiku i kości
Objawy kliniczne
1.
Bóle kości – ogniskowa destrukcja z powodu rozplemu plazmocytów i działania czynników ak-
tywujących osteoklasty
2.
Krwawienia
3.
Narastające osłabienie
4.
Częste zakażenia – niedobory immunologiczne
5.
Zespół nadlepkości, zmiany zakrzepowo-zatorowe (dużo produkowanych białek)
6.
Nefropatia szpiczakowa
7.
Zaburzenia neurologiczne, obwodowe i mózgowe
8.
Zaburzenia widzenia

15 WYKŁADY Z INTERNY
2001/2002
Badania laboratoryjne
1.
Morfologia krwi
-
niedokrwistość z rulonizacją erytrocytów, małopłytkowość, leukopenia
2.
Szpik – dominacja plazmocytów w mielogramie
3.
Surowica krwi – obecność paraproteiny M (IgG, IgA, IgD) – proteinogram
-
wzrost LDH, kreatyniny, białka całkowitego, Ca
4.
Przyspieszone OB.
5.
W moczu – białko Bence-Jonesa (łańcuchy lekkie, łańcuchy ciężkie)
6.
RTG kości płaskich i długich – ogniska osteolityczne
Wyszukiwarka
Podobne podstrony:
LOGIKA wyklad 5 id 272234 Nieznany
ciagi liczbowe, wyklad id 11661 Nieznany
AF wyklad1 id 52504 Nieznany (2)
Neurologia wyklady id 317505 Nieznany
ZP wyklad1 id 592604 Nieznany
CHEMIA SA,,DOWA WYKLAD 7 id 11 Nieznany
or wyklad 1 id 339025 Nieznany
II Wyklad id 210139 Nieznany
cwiczenia wyklad 1 id 124781 Nieznany
BP SSEP wyklad6 id 92513 Nieznany (2)
MiBM semestr 3 wyklad 2 id 2985 Nieznany
algebra 2006 wyklad id 57189 Nieznany (2)
olczyk wyklad 9 id 335029 Nieznany
Kinezyterapia Wyklad 2 id 23528 Nieznany
AMB ME 2011 wyklad01 id 58945 Nieznany (2)
AWP wyklad 6 id 74557 Nieznany
PRAWO SPORTOWE Wyklady(1) id 38 Nieznany
AGH Wyklad 4 id 52883 Nieznany (2)
więcej podobnych podstron