
INSULINOOPORNOŚĆ
OD PATOGENEZY
DO KLINIKI
Dr hab. n. med.
Magdalena Olszanecka-Glinianowicz
Definicja
Stan upośledzonej odpowiedzi biologicznej
tkanek na insulinę endogenną lub egzogenną
zarówno w zakresie
- metabolizmu węglowodanów, lipidów i białek,
jak też działania mitogennego insuliny -
wpływ na procesy:
- różnicowania i wzrostu komórkowego,
- syntezę DNA,
- ekspresję genów.
Rodzaje insulinooporności
• Przedreceptorowa
- zespół mutowanej insuliny, w którym wykazano
genetycznie uwarunkowaną nieprawidłową
budowę cząsteczki insuliny. W zespole tym
stwierdza się prawidłową reakcję na insulinę
egzogenną, natomiast występuje
insulinooporność w stosunku do endogennej,
zmienionej cząsteczki insuliny.
- Nadmiar hormonów działających
przeciwstawnie do insuliny:
a) zespół Cushinga;
b) Akromegalia;
c) Nadczynność tarczycy;
d) Guz chromochłonny
• Receptorowa
- Zaburzenia czynności;
- Zmiany w strukturze;
• Postreceptorowa
-
Zaburzenia procesów sygnalizujących
przyłączenie insuliny do receptora
insulinowego;
- Zaburzenia struktury i funkcji transporterów
glukozy do wnętrza komórki;
- Sytuacje kliniczne prowadzące do
zwiększenia lipolizy.
Metaboliczne skutki niedoboru
insuliny
• wzmożenie lipolizy,
• nasilenie ketogenezy,
• zahamowanie glikolizy,
• nadmierna glikogenoliza i glukoneogeneza

Geny kandydaci – odpowiedzialne
za rozwój insulinooporności
Monogenowe formy
insulinooporności

Poligenowa predyspozycja
do insulinooporności
• Wiek
• Płeć
• Niska aktywność fizyczna
• Dieta wysokokaloryczna
• Nadwaga i otyłość
• Stosowanie leków o działaniu diabetogennym
(glikokortykosteroidy, diuretyki tiazydowe, inhibitory
proteazy HIV, doustne leki antykoncepcyjne, diuretyki
pętlowe, blokery kanału wapniowego
• Alkohol
• Palenie tytoniu
• Ciąża
Czynniki środowiskowe
a insulinooporność
• Należy do rodziny receptorów
posiadających aktywność kinazy
tyrozynowej.
- Wyróżnia się 3 homologiczne receptory:
- insulinowy
- dla IGF-1
- związany z receptorem insulinowym–IRR
Receptor insulinowy
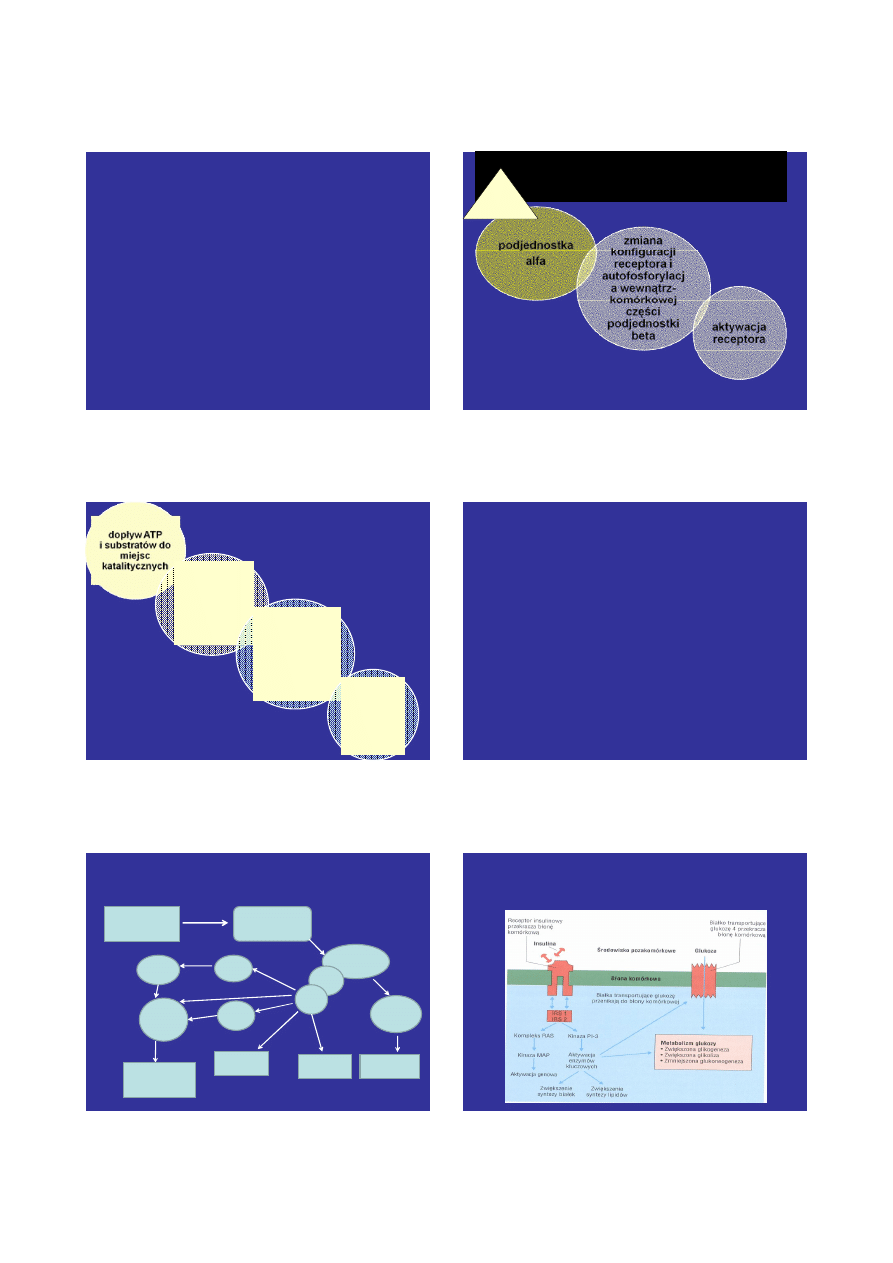
• Występują 2 izoformy receptora
insulinowego:
- A w OUN oraz tkankach płodowych
- B w wątrobie, mięśniach, tkance
tłuszczowej
insulina
Fosforylacja
reszt
tyrozynowych w
białkach
substratowych
(IRS 1-4, Gab-1,
p60dok, Cbl,
ASP, izoformy
białka Sbc
Ufosforylowane
reszty tyrozynowe
w białkach
substratowych
stanowią tzw.
miejsca
kotwiczące dla
białek
wykazujących
aktywność SH2
Pobudzenie
szlaków
sygnalizacji
insuliny
związanych
z
PI-3K
i MAPK
• MAPK (białkowa kinaza aktywowana
mitogenami) szlak ten pośredniczy w
przekazywaniu do jądra komórkowego
sygnału mitogennego – procesy
wzrostu, różnicowania i proliferacji
komórek pod wpływem insuliny.
• PI-3K (3-kinaza fosfatydyloinozytolu) –
szlak zaangażowany w metaboliczną
odpowiedź na insulinę.
Aktywacja szlaku PI-3K
insulina
Receptor
insuliny
Białka
IRS 1-2
MAPK
kinaza
Mitogeneza
p85
P
100
PI -3K
PDK
1
PKB/
Akt
PKC
GLUT4
Transport
glukozy
Synteza
glikogenu
Synteza
białek
Komórkowy tryb działania
insuliny na metabolizm glukozy
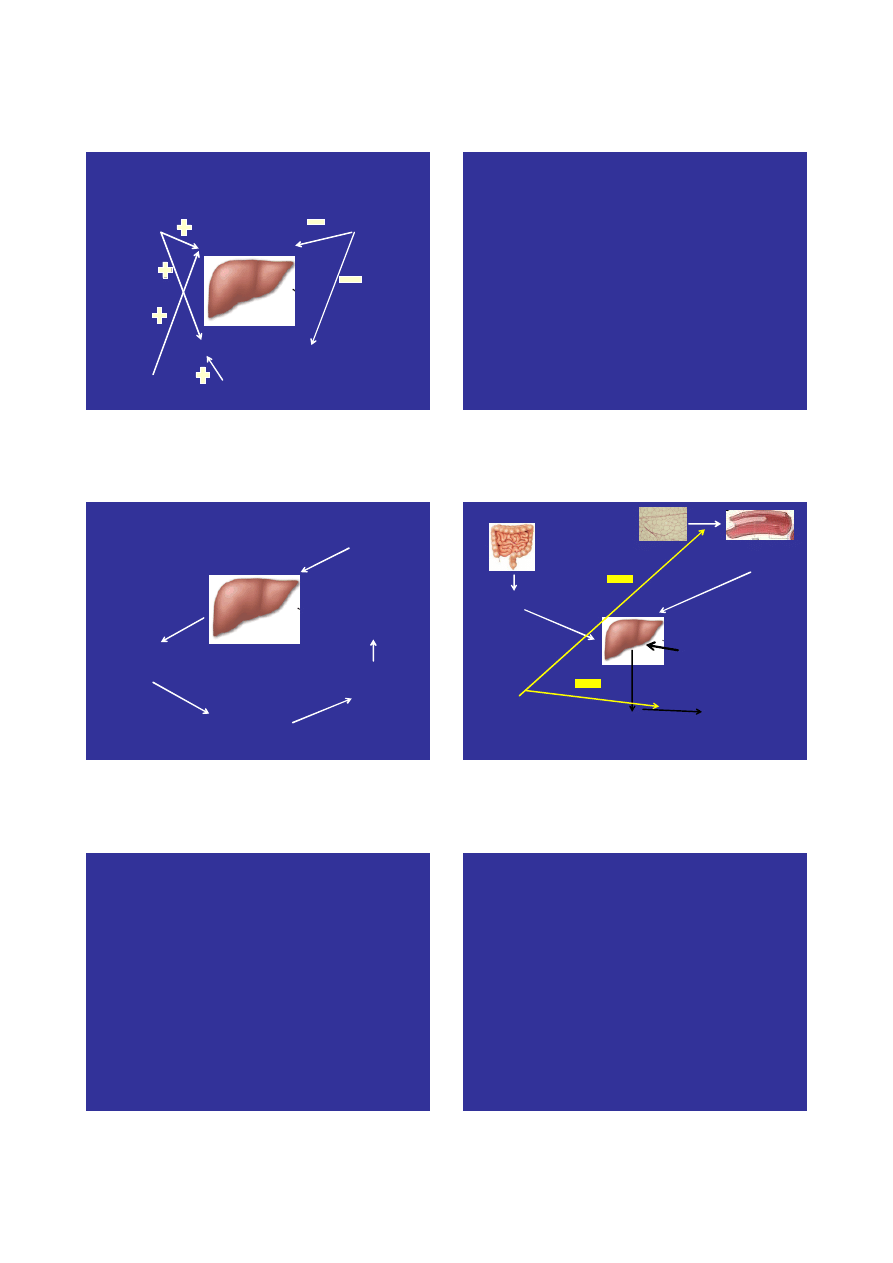
Rola wątroby w rozwoju
insulinooporności
glikogenoliza
glukoneogeneza
Insulina
Glukagon
Adrenalina
Kortyzol
Wątrobowa synteza glukoza na czczo
wynosi 1.8 – 2.2 mg/min/kg m.c.
40-50% glukozy wydzielanej przez wątrobę
powstaje w procesie glikogenolizy
a reszta w procesie glukoneogenezy.
Wychwyt glukozy przez wątrobę zależy od
jej stężenia we krwi i od gradientu stężeń
glukozy przez błonę komórkową
hepatocyta.
Insulina
Aktywacja
glukokinazy
Fosforylacja
glukozy
Cykl kwasów
trójkarboksylowych
Synteza
glikogenu
estryfikacja kw.
tłuszczowych do TG
chylomikrony
20%
Kwasy
tłuszczowe
Synteza de novo
z acetylo –Co-A
TG
Lipoproteiny
głównie VLDL
INSULINA
• Wątroba jest głównym narządem
wydalającym z organizmu cholesterol.
Zasadniczą rolę odgrywają tu powstające
w wątrobie i jelicie HDL, które przejmują
cholesterol z powierzchni komórek i
posiadają zdolność jego estryfikacji.
Obładowane cholesterolem, jego estrami
i TG cząsteczki HDL trafiają do wątroby,
gdzie oddzielony wolny cholesterol zostaje
wydalony z żółcią.
• Duże stężenie VLDL wpływa również na
zwiększenie puli LDL. Komórki śródbłonka
zatok wątrobowych wydzielają lipazę
wątrobową, która powoduje hydrolizę TG
obecnych w LDL i HDL i powstanie
cząsteczek małych, gęstych LDL
i mniejszych HDL3.
• Insulina hamuje aktywność lipazy
wątrobowej.

• Stężenie WKT na czczo we krwi u ludzi
wynosi 0.4-1.0 mmol/l i zmniejsza się po
spożyciu węglowodanów.
• Brak antylipolitycznego działania insuliny
powoduje wzrost stężenia WKT w osoczu.
• Zwiększony dowóz WKT do wątroby
i mięśni szkieletowych powoduje
zmniejszenie metabolizmu glukozy w tych
tkankach.
- Obniżenie aktywności dehydrogenzy
pirogronianowej (oksydacja glukozy);
- Zmniejszenie aktywności fosfofruktokinazy
(glikoliza);
- Wzrost zawartości glukozo – 6 – fosforanu
w komórkach
Rola WKT w rozwoju insulinooporności
w wątrobie
Zwiększona kumulacja
TG w hepatocytach
• Mięśnie odpowiadają za około 80%
stymulowanego insuliną tkankowego wychwytu
glukozy.
• U ludzi wyróżnia się 3 typy mięśni szkieletowych
w zależności od potencjału oksydacyjnego:
- Typ 1 – mięśnie wolno kurczące się, oksydacyjne;
- Typ 2a – mięśnie szybko kurczące się,
oksydacyjne;
- Typ 2b – mięśnie szybko kurczące się,
glikolityczne;
Insulinooporność w mięśniach
szkieletowych
Metabolizm węglowodanów
• Insulina w mięśniach szkieletowych
stymuluje translokację GLUT4 z
cytoplazmy do błony komórkowej, co jest
kluczowe dla pobudzenia transportu
glukozy do wnętrza komórki.
• Mięśnie o największym potencjale
oksydacyjnym charakteryzujące się
największą wrażliwością na insulinę
charakteryzują się również największą
zawartością GLUT4.
• Po dostaniu się do wnętrza miocyta
glukoza jest fosforylowana przez
heksokinazę i powstaje glukozo – 6 –
fosforan , który może być użyty do syntezy
glukozy lub do glikolizy.
• Glikoliza z kolei prowadzi do powstawania
mleczanów (około 10%) lub do oksydacji
pirogronianów w cyklu Krebsa (około
90%). Insulina promuje tlenową przemianę
metabolitów glukozy.

• Insulina pobudza syntezę glikogenu –
aktywacja białkowej kinazy beta powoduje
fosforylację i inaktywację kinazy-3 syntazy
glikogenu (GSK-3). Fosforylacja syntazy
glikogenu powoduje jej inaktywację.
Mechanizm sygnalizacyjny insuliny
prowadzi w tym przypadku do
odblokowania aktywności syntazy
glikogenu poprzez zahamowanie GSK-3.
Metabolizm lipidów
• WKT pokrywają ponad 50%
zapotrzebowania energetycznego
mięśni szkieletowych w czasie
spoczynku.
• Ich źródłem są WKT krążące
w osoczu oraz TG zmagazynowane
w miocytach.
Metabolizm lipidów
• Śródbłonek naczyń jest
nieprzepuszczalny dla TG, WKT muszą
zostać uwolnione jeszcze w świetle
naczynia, uczestniczy w tym procesie
znajdująca się na powierzchni
śródbłonka LPL.
• Aktywność LPL zwiększają
katecholaminy i ACTH a zmniejsza
insulina.
• Transport WKT do wnętrza miocyta przez
sarkolemmę odbywa się m.in. przy udziale
białka FAT (transporter kwasów
tłuszczowych).
• Natomiast wewnątrzkomórki przy udziale
FABP (białko wiążące kwasy tłuszczowe).
Najwięcej FABP znajduje się w mięśniu
sercowym, następnie w mięśniach
o największym potencjale oksydacyjnym.
• Kwasy tłuszczowe w miocytach mogą
zostać wykorzystane jako:
- substrat energetyczny (proces beta –
oksydacji);
- uzupełnienia wewnątrzkomórkowej puli
lipidów;
- syntezy fosfolipidów błonowych.
• Beta – oksydacja kwasów tłuszczowych
odbywa się po ich aktywacji do
tłuszczowych acylo-CoA, które mogą
dyfundować przez błonę zewnętrzną
mitochondriów.
• Włókna oksydacyjne wykazują znacznie
większą aktywność
wewnątrzmitochondrialnych enzymów
beta – oksydacji w porównaniu do włókien
glikolitycznych.

• Transport przez błonę wewnętrzną jest
zależny od
palmitylotransferazy
karnitynowej 1.
• Silnym inhibitorem tego enzymu jest
malonylo – CoA,
• Jego zawartość wzrasta pod wpływem
hiperglikemii i hiperinsulinemii.
• KT wbudowane są do lipidów
mięśniowych lub do fosfolipidów
błonowych.
• TG mięśniowe mogą następnie ulegać
hydrolizie, dostarczając substratów
energetycznych. Źródło to ma największe
znaczenie w czasie przedłużonego
submaksymalnego wysiłku.
• Hydroliza zachodzi dzięki
lipazie
triacyloglicerolowej.
Rola lipidów wewnątrzmięśniowych
w indukowaniu insulinooporności
• Wysunięto hipotezę, że insulinooporność
jest stanem ektopowej akumulacji lipidów
w tkankach innych niż tkanka tłuszczowa
z powodu niewydolności tkanki
tłuszczowej jako „bufora energetycznego”
a więc mięśniowa akumulacja lipidów
może być przyczyną insulinooporności.
• WKT działają poprzez pierwotne
hamowanie transportu/fosforylacji glukozy
w komórkach mięśni, co powoduje
zmniejszenie oksydacji glukozy i syntezy
glikogenu.
Jest to efekt hamującego wpływu WKT na
szlak sygnałowy insuliny.
• TG same w sobie nie są szkodliwą frakcją
mięśniowej puli lipidów, ale raczej
markerem gromadzenia się innych
związków takich jak:
- diacyloglicerole (DAG)
- ceramidy.

Akumulacja DAG
w mięśniach szkieletowych
↑ PKC i jego
izoform
PCKβII i PKCδ
Aktywacja IKK
Aktywacja NF-κB
Sfingomielina
Seryna
+
Palmitylo-
CoA
CERAMID
hydroliza
Synteza de
novo
PKB/Akt
Translokacja GLUT4
do błony
komórkowej
Wychwyt
glukozy
Rola tkanki tłuszczowej w
patogenezie insulinooporności
Ekspresja niektórych receptorów
w tkance tłuszczowej
Receptory dla „tradycyjnych „
endokrynnych hormonów ;
- rec. insulinowy
- rec. glukagonowy
- rec. GH
- rec. TSH
- rec. Gatrynowy/CCK
- rec. GLP-1
- rec. Angiotensyny II t 1 i 2
Receptory dla katecholamin;
-
Jądrowe receptory;
- rec. glikosterydowy
- rec. witaminy D
- rec. hormonów tarczycy
- rec. Estrogenowy
- rec. Androgenowy
- rec. Progesteronowy
-- PPAR
Receptory cytokinowe:
- rec. leptynowy
- rec. IL-6
- rec. TNF
Rec. dla
lipoprotein
PPAR
Aktywacja
Heterodimer z RXR
Swoiste sekwencje DNA
Geny uczestniczące
w metabolizmie lipidów
i węglowodanów
wiązanie
Regulacja transkrypcji
Efekty aktywacji PPAR
• Zwiększenie aktywacji:
- białek transportujących i wiążących kwasy
tłuszczowe,
- syntazy kwasów tłuszczowych,
- glukokinazy.
• Zwiększenie:
- ekspresji GLUT4;
- sekrecji adiponektyny.

• zahamowanie sekrecji:
- TNFalfa;
- rezystyny.
• wpływ na różnicowanie adipocytów i powstawanie
małych adipocytów bardziej wrażliwych na działanie
insuliny.
- Czynniki aktywujące PPARgama to:
Niektóre kwasy tłuszczowe;
Prostanoidy;
Thiazolidinodiony
.
- Receptory PPARalfa znajdujące się
głównie w mięśniach szkieletowych
i wątrobie, zwiększają oksydację kwasów
tłuszczowych
• Transport glukozy do adipocytów zachodzi
przy udziale GLUT4.
• Najważniejszą konsekwencją udziału
tkanki tłuszczowej w rozwoju
insulinooporności jest brak hamowania
lipolizy i zwiększenie powstawania WKT i
glicerolu.
Tkanka tłuszczowa największy
„gruczoł” endokrynny
leptyna
rezystyna
RBP-4
apelina
wisfatyna
waspina
omentyna
TNF + sR
IL-6 + sR
NO
PAI-1
angiotensynogen
TGF
adipsyna
AdipoQ
ApoE
IGF-1
ASP
białko
Agouti
steroidy
Czynniki
komplementarne
adiponektyna
Mediatory insulinooporności –
wolne kwasy tłuszczowe ( WKT )
• Uwalniane w nadmiarze :
hamują wykorzystanie glukozy w wątrobie
i mięśniach szkieletowych
Pobudzają glukoneogenezę
Obniżają wątrobowy klirens insuliny co
doprowadza do hiperinsulinemi
Mediatory insulinooporności –
czynnik
martwicy nowotworów ( TNF - )
• Obniża autofosforylację kinazy tyrozynowej
w substracie insulinowym
• Hamuje sygnał insuliny na poziomie kinazy – 3
fosfatydyloinozytolu
• Hamuje ekspresję genu GLUT – 4
• W komórkach wysp trzustkowych hamuje
stymulowaną glukozą sekrecję insuliny

Mediatory insulinooporności -
leptyna
• Zmniejsza wrażliwość hepatocytów na
insulinę i nasila proces glukoneogenezy
• Hamuje pierwszą fazę sekrecji insuliny
przez komórki , co może wynikać z jej
działania na ATP – zależny kanał
potasowy
Mediatory insulinooporności –
IL 6
• Reguluje aktywność lipazy lipoproteinowej
i zwiększa ilość krążących WKT
Mediatory insulinooporności –
rezystyna
• Hamuje stymulowany insuliną wychwyt
glukozy w tkance tłuszczowej i mięśniach
szkieletowych
Adiponektyna
• Jej wydzielanie jest zmniejszone u otyłych
• Występująca w otyłości hipoadiponektynemia
sprzyja insulinooporności
• Działania adiponektyny :
Obniża stężenie glukozy w osoczu niezależnie od insuliny
Stymuluje zwiększony wychwyt WKT przez komórki mięśni
szkieletowych oraz zwiększa spalanie WKT w
mitochondriach tych komórek niezależnie od insuliny
Hamuje działanie TNF -
Hamuje wątrobową glukoneogenezę
Nie wyjaśniona rola
w rozwoju insulinooporności
• Wisfatyna
• Waspina
• Omentyna
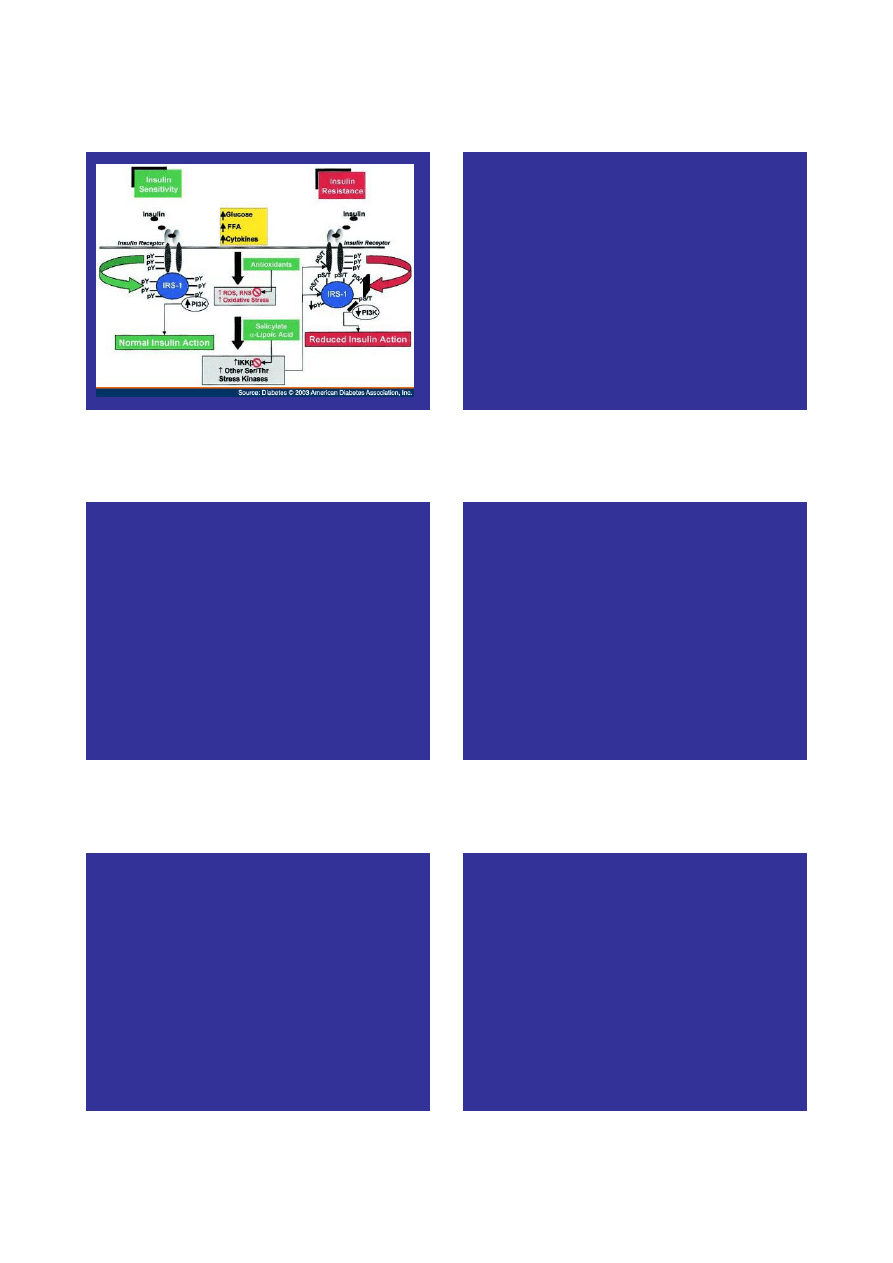
Metody pomiaru
insulinooporności in vivo
1. Stężenie insuliny na czczo;
2. Pośrednie wskaźniki insulinooporności oparte o
stężenia insuliny i glukozy na czczo;
3. Wskaźniki wykorzystujące stężenia glukozy i
insuliny podczas OGTT;
4. Dożylny test tolerancji glukozy oraz analiza
„minimal model”;
5. Test tolerancji insuliny;
6. Test supresji insuliny endogennej;
7. Klamra hiperinsulinemiczna normoglikemiczna
– „złoty standard”.
Stężenie insuliny na czczo
• Im wyższy stopień insulinooporności, tym
wyższe stężenie insuliny.
• Przestaje być miarodajne w stanach takich
jak upośledzona tolerancja glukozy i
cukrzyca typu 2.
Pośrednie wskaźniki insulinooporności oparte
o stężenia insuliny i glukozy na czczo
• Homeostasis Model Assessment – Insulin
Resistance
HOMA-IR = glukoza (mmol/l)x insulina (IU/ml)/22,5
- Quantitative Insulin Sensitivity Check Index
QUICKI = 1/(log[glukoza] + log[insulina])
Ograniczenia:
- pulsacyjne wytwarzanie insuliny,
- regulacja stężenia glukozy przez inne hormony
Wskaźniki wykorzystujące stężenia glukozy
i insuliny podczas OGTT
• Wskaźnik insulinowrażliwości wg Matsuda i
DeFronzo = 1000/pierwiastek kwadratowy
(glukoza na czczo x insulina na czczo) x
(średnie stężenie glukozy x średnie stężenie
insuliny podczas OGTT)
• Wskaźnik insulinowraliwości wg Stumvolla i
wsp. = 0,226 – 0,0032 x BMI – 0,0000645 x
insulina w 120 min. OGTT – 0,00375 x
glukoza w 90 min. OGTT
Dożylny test tolerancji glukozy oraz
analiza „minimal model”;
• Podaje się iv glukozę w dawce 0,3 kg/kg m.c. w ciągu
60 s.
• W czasie 180 min 22 razy pobiera się krew w celu
oznaczeń glukozy i insuliny;
• W 20 minucie testu podaje się dożylnie tolbutamid,
aby zniwelować zaburzenia wydzielania insuliny.
• Analizy zależności między stężeniami insuliny
i glukozy dokonuje się za pomocą programu
komputerowego.

Test tolerancji insuliny
• Obserwuje się zmiany stężenia glukozy co 5-10 min
przez 40 min po dożylnym podaniu insuliny w dawce
0,1 j/kg m.c.
• Stężenie glukozy umieszcza się na skali
półlogarytmicznej, uzyskując liniowe obniżenie tych
stężeń;
• Stopień nachylenia tej linii (wartość k) odpowiada
procentowemu obniżeniu stężenia glukozy w ciągu
minuty i służy do oceny wrażliwości na insulinę – im
większa wartość k tym większa wrażliwość na
insulinę.
k = 0,693/t1/2glukozy) x 100%
Test supresji insuliny endogennej
• Stały wlew insuliny (0,48 nmol/min – stabilna
hiperinsulinemia rzędu 600 pmol/l w 3 godz testu) i
glukozy (33 mol/min/kg m.c.);
• Wartością badaną są stężenia glukozy uzyskane w
okresie stabilnych warunków metabolicznych (120-
180 min testu).
• Wyższe stężenia glukozy świadczą o mniejszej
wrażliwości na insulinę
• Aby zablokować wyrzut endogennej insuliny
dodatkowo podaje się somatostatynę w dawce 250
g/godz.
Klamra hiperinsulinemiczna
normoglikemiczna – „złoty standard”
• Insulinę podaje się w dawce 40
mU/m2/min, w ciągu pierwszych 10 min
badania najczęściej stosuje się 2-krotnie
większą dawkę aby wytworzyć stabilną
hiperinsulinemię
• Od 4 minuty badania rozpoczyna się wlew
glukozy, najpierw w dawce 2 mg/kg
m.c./min, a następnie od 10 min, w
zmiennym tempie, regulowanym co 5 min
w zależności od aktualnej glikemii.
• Wykorzystuje się analizator YSI 2300
STAT Plus do pomiarów stężeń glukozy.
• Klamra powinna trwać co najmniej 2
godziny.
Insulinooporność a zespół
metaboliczny
Otyłość trzewna
Wzrost stężenia FFA
Insulinooporność
zapalenie
Cukrzyca typu 2
Zaburzenia
fibrynolizy
NASH
Nadciśnienie
Zmiana reaktywności
naczyń
Dyslipidemia
Przyspieszony rozwój miażdżycy
Oporność na insulinę
i hiperglikemia w cukrzycy typu 2
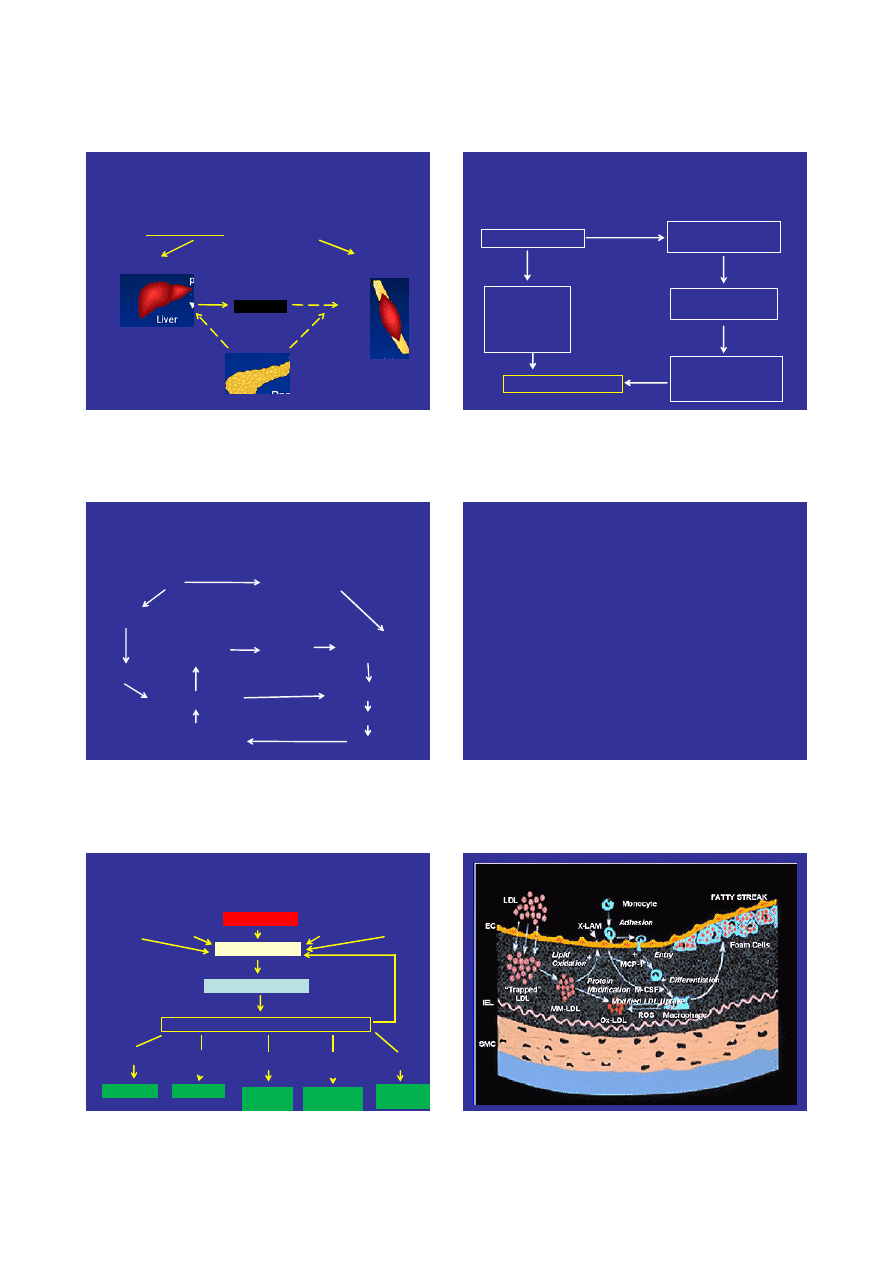
Przyczyny hiperglikemii
w cukrzycy typu 2
Insulinooporność: defekty receptorowe i post-receptorowe
Wzrost produkcji
glukozy
Upośledzony
wychwyt glukozy
X
Upośledzone wydzielanie insuliny
↑ Glukoza
Insulinooporność a nadciśnienie tętnicze
Hiperinsulinemia
Wzrost aktywności
SNS
Wzrost nerkowej
resorpcji Na
Wzrost objętości
krwi krążącej
i rzutu serca
Stabilizacja
receptorów dla
Ang II (ATR1)
przez wpływ na
mRNA i
wydłużenie jego
czasu półtrwania
NADCIŚNIENIE
Insulinooporność a funkcja
endotelium
↑ Lipolizy
↑ SNS
↑ skurcz naczyń
↓
wytwarzanie
endot. NO
↓ rozkurcz
naczyń
↓ obwodowego
przepływu krwi
↑
FFA
Insulinooporność
↓ sygnał insuliny
↓ wychwyt glukozy
↑ glukozy
↑ insuliny
Keulen L. et al. Journal of Clinical and Basic Cardiology 2001;4: 193-195
Inne mechanizmy patogenetyczne
nadciśnienia tętniczego w insulinooporności
• aktywacja układu renina-angiotensyna-
aldosteron ,
• hiperleptynemia,
• zmienionaczynność nerek i struktur
naczyniowych,
• zmniejszona aktywność ANP.
Dysfunkcja endotelium
Czynniki ryzyka
cukrzyca
palenie
↑
RR
↑ LDL-C
Stres oksydacyjny
Dysfunkcja endotelium
↓ NO ↑ lokalnych mediatorów ↑ tkankowej ACE-AII
PAI-1
VCAM
ICAM cytokiny
Endotelina
Czynniki
wzrostu
Proteoliza
zakrzepica
zapalenie
Skurcz
naczyń
Uszkodzenie
remodeling
Oderwanie
blaszki
Dzau and Gibbons 1997
Zapalenie łącznik między
insulinoopornością a miażdżycą
• ↑ FFA
• ↑TNF
• ↑CRP
• ↑ utlenowanych LDL
• ↑ wrażliwości na angiotensynę II
Hiperglikemia a dysfunkcja śródbłonka
Hierglikemia
eNOS
NO
Wolne rodniki
tlenowe
Jon
nadtlenoazotynowy
(ONOO
_
)
Ponadto przewlekła hiperglikemia
powoduje
• Aktywację PCK i fosfolipazy A2 – wzrost produkcji
metabolitów kw. arachidynowego;
• Zwiększenie ekspresji czynników wzrostowych
(angiotensyna II, endotelina) – remodeling;
• Wzrost stężenia końcowych produktów glikacji –
nasilenie stresu oksydacyjnego;
• Stymulacja glikacji i oksydacji LDL;
• Wzrost ekspresji NF-κB;
• Wzrost stężenia molekuł adhezyjnych, nasilenie migracji
i apoptozy komórek mięśni gładkich naczyń i dalsze
zmniejszenie dostępności NO;
• Wzrost stężenia IL-1 i TNF-α oraz
niektórych chemokin- nasilenie napływu
monocytów i limfocytów do ściany naczyń;
• Wzrost aktywności metaloproteinaz i
destabilizację blaszki miażdżycowej;
• Aktywacja PKC
• Wzrost aktywności PAI-1 i agregacji płytek
krwi.
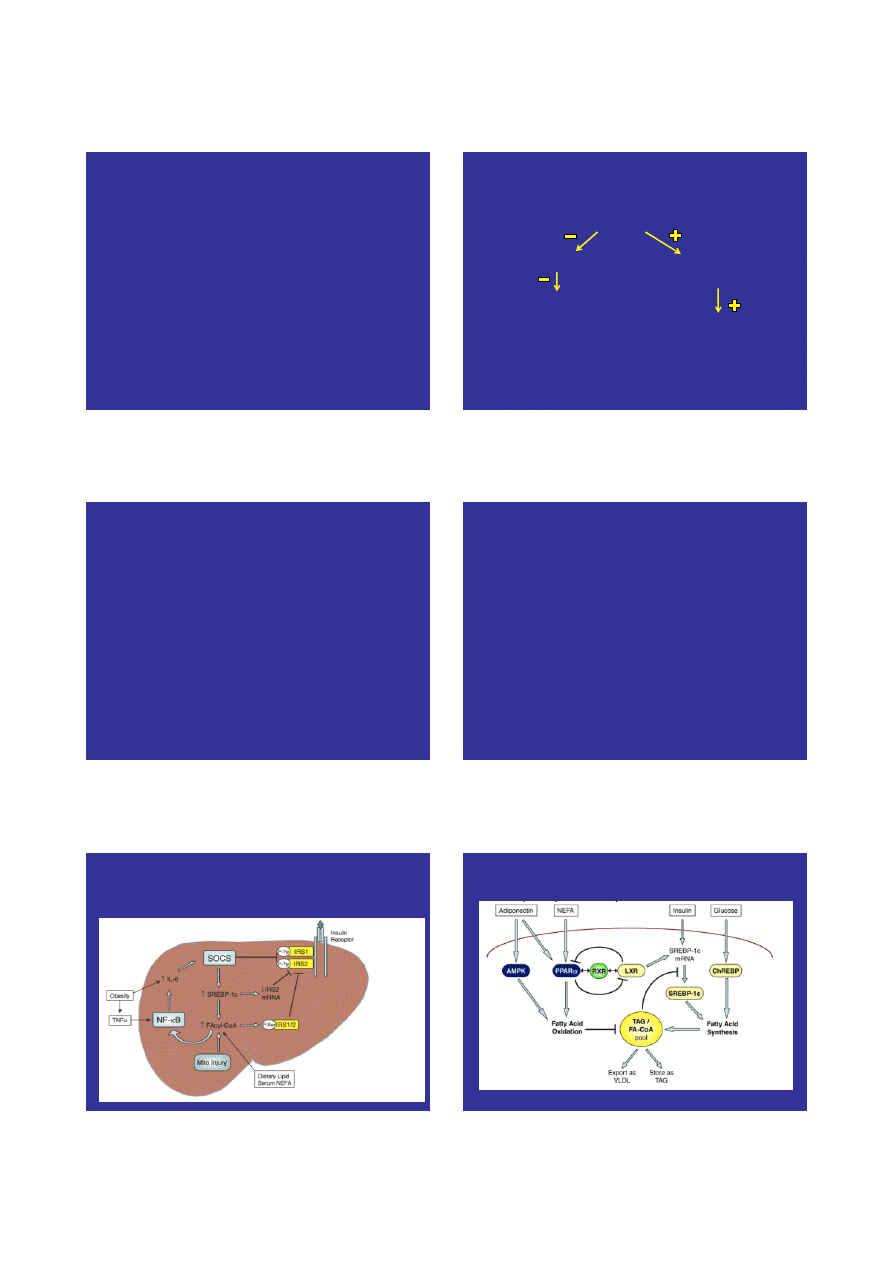
Związek między insulinoopornością
a NASH
Wyszukiwarka
Podobne podstrony:
spis lab I sem 2010
2010 ZMP studenci
W4 2010
LECZENIE STANÓW NAGLĄCYCH W DIABETOLOGII WYNIKAJĄCYCH Z NIEDOBORU INSULINY
wyklad 14 15 2010
W 8 Hormony 2010 2011
Insulinoterapia funkcjonalna
RI 12 2010 wspolczesne koncepcje
2009 2010 Autorytet
wyklad 2 2010
Wykład 3 powtórzenie 2010 studenci (1)
PD W1 Wprowadzenie do PD(2010 10 02) 1 1
BIOMATERIALY IV 2010
spis wykład I sem 2010
Wykład 5 2010 studenci
więcej podobnych podstron