
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
MINISTERSTWO EDUKACJI
NARODOWEJ
Jolanta Wąsikowska
Bartosz Wąsikowski
Wykonywanie pomiarów parametrów procesowych
311[02].Z1.04
Poradnik dla ucznia
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy
Radom 2007

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
1
Recenzenci:
mgr Urszula Ciosk-Rawluk
mgr Barbara Przedlacka
Opracowanie redakcyjne:
mgr Jolanta Łagan
Konsultacja:
mgr inż. Gabriela Poloczek
Poradnik stanowi obudowę dydaktyczn
ą programu jednostki modułowej 311[02].Z1.04,
„Wykonywanie pomiarów parametrów procesowych”, zawartego w modułowym programie
nauczania dla zawodu technik analityk.
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy, Radom 2007

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
2
SPIS TREŚCI
1. Wprowadzenie
3
2. Wymagania wstępne
5
3. Cele kształcenia
6
4. Materiał nauczania
7
4.1. Podstawy metrologii
7
4.1.1. Materiał nauczania
7
4.1.2. Pytania sprawdzające
10
4.1.3. Ćwiczenia
11
4.1.4. Sprawdzian postępów
12
4.2. Wybrane pomiary w procesach technologicznych
13
4.2.1. Materiał nauczania
13
4.2.2. Pytania sprawdzające
20
4.2.3. Ćwiczenia
21
4.2.4. Sprawdzian postępów
22
4.3.Wybrane zagadnienia analizy instrumentalnej
23
4.3.1. Materiał nauczania
23
4.3.2. Pytania sprawdzające
31
4.3.3. Ćwiczenia
31
4.3.4. Sprawdzian postępów
34
4.4. Wybrane zagadnienia z kontroli procesów technologicznych
35
4.4.1 Materiał nauczania
35
4.4.2. Pytania sprawdzające
51
4.4.3. Ćwiczenia
51
4.4.4. Sprawdzian postępów
55
5. Sprawdzian osiągnięć
56
6. Literatura
61

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
3
1. WPROWADZENIE
Poradnik będzie Tobie pomocny w przyswajaniu wiedzy i umiejętności z zakresu
wykonywania pomiarów parametrów procesowych.
W poradniku zamieszczono:
−
wymagania wstępne – wykaz umiejętności, jakie powinieneś mieć już ukształtowane abyś
bez problemu mógł korzystać z poradnika,
−
cele kształcenia – wykaz umiejętności, jakie ukształtujesz podczas pracy z poradnikiem,
−
materiał nauczania zawierający niezbędne wiadomości teoretyczne, umożliwiający
opanowanie treści jednostki modułowej,
−
pytania sprawdzające wiedzę potrzebną do wykonania ćwiczenia,
−
ćwiczenia, które pomogą ci zweryfikować wiadomości teoretyczne oraz ukształtować
umiejętności praktyczne,
−
sprawdzian osiągnięć – przykładowy zestaw zadań i pytań sprawdzających Twoje
opanowanie wiedzy i umiejętności z zakresu całej jednostki modułowej,
−
literaturę.
Bezpieczeństwo i higiena pracy
W czasie pobytu w pracowni zobowiązany jesteś przestrzegać przepisów bezpieczeństwa
i higieny pracy, instrukcji przeciwpożarowych oraz zasad ochrony środowiska wynikających
z rodzaju wykonywanych ćwiczeń. Przepisy te poznasz w trakcie nauki.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
4
Schemat układu jednostek modułowych
311[02].Z1.01
Stosowanie zasad bezpiecznej pracy
w laboratorium
311[02].Z1
Podstawowe czynności preparatywne oraz pomiary
parametrów procesowych
311[02].Z1.02
Zastosowanie technik komputerowych
do obliczeń chemicznych
311[02].Z1.03
Zastosowanie technik laboratoryjnych do sporządzania
preparatów chemicznych
311[02].Z1.04
Wykonywanie pomiarów parametrów
procesowych

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
5
2. WYMAGANIA WSTĘPNE
Przystępując do realizacji programu jednostki modułowej powinieneś umieć:
−
przestrzegać przepisów bezpieczeństwa i higieny pracy, ochrony przeciwpożarowej oraz
ochrony środowiska,
−
przestrzegać zasad dobrej techniki laboratoryjnej,
−
przestrzegać zasad bezpieczeństwa podczas badania analitycznego,
−
posługiwać się nomenklaturą związków nieorganicznych i organicznych,
−
określać właściwości fizyko-chemiczne substancji,
−
stosować stechiometrię do obliczeń chemicznych,
−
stosować obowiązujące jednostki układu SI,
−
sporządzać wykresy i interpretować wyniki,
−
sporządzać roztwory o określonym stężeniu,
−
przygotowywać próbki materiału do analizy,
−
przygotowywać sprzęt laboratoryjny, aparaturę, odczynniki,
−
korzystać z norm, przepisów, procedur i dostępnych instrukcji,
−
określać
sposoby
kontroli
procesów
technologicznych,
zastosować
kontrolę
międzyoperacyjną i finalną,
−
pobierać próbki analityczne w warunkach ciągłego procesu technologicznego.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
6
3. CELE KSZTAŁCENIA
W wyniku realizacji programu jednostki modułowej powinieneś umieć:
−
scharakteryzować parametry procesowe oraz wielkości fizyczne stosowane w technologii
chemicznej i analitycznej kontroli procesów technologicznych,
−
odczytać i sporządzić schematy aparatury pomiarowej stosowanej w technologii
chemicznej,
−
wyjaśnić budowę i działanie aparatury pomiarowej,
−
obsłużyć aparaturę pomiarową oraz dokonać jej konserwacji,
−
określić zastosowanie aparatury pomiarowej w analityce i procesach technologicznych,
−
dokonać
kontroli
analitycznej
charakterystycznej
dla
technologii
zakładów
przemysłowych,
−
dokonać pomiaru wartości wielkości fizycznych charakteryzujących materiały,
−
określić użytkowe właściwości substancji, jak: odporność na temperaturę i czynniki
chemiczne,
−
sporządzić dokumentację pomiarów i badań oraz zinterpretować uzyskane wyniki,
−
przedstawić wyniki w formie tabelarycznej i graficznej,
−
zinterpretować wyniki analiz ilościowych z zastosowaniem metod statystycznych,
−
zorganizować stanowisko pracy z uwzględnieniem przepisów bezpieczeństwa i higieny
pracy oraz ochrony przeciwpożarowej,
−
dokonać obserwacji przebiegu ćwiczeń, zapisać wartości mierzonych wielkości
i parametrów oraz istotne zjawiska i przemiany,
−
scharakteryzować procesy jednostkowe realizowane w węźle technologicznym oraz określić
poziom zagrożenia powodowanego ewentualną awarią,
−
dokonać analizy graficznego przedstawienia koncepcji procesowej i określić położenie
węzłów analitycznych,
−
zastosować zasady technologiczne do oceny organizacji systemu kontroli analitycznej,
−
określić
sposoby
kontroli
procesów
technologicznych,
zastosować
kontrolę
międzyoperacyjną i finalną,
−
pobrać próbki analityczne w warunkach ciągłego procesu technologicznego,
−
scharakteryzować proces technologiczny, maszyny i urządzenia techniczne oraz budowę
i działanie aparatury pomiarowej,
−
wykonać pomiary parametrów procesowych i wielkości stosowanych w analityce,
−
dokonać kontroli analitycznej charakterystycznej dla technologii zakładów regionalnych.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
7
4. MATERIAŁ NAUCZANIA
4.1. Podstawy metrologii
4.1.1. Materiał nauczania
Regulamin laboratorium zawiera przepisy będące częścią przepisów regulaminu
szkolnego, który uczeń ma obowiązek przestrzegać, lecz ponadto uwzględnia specyfikę zajęć.
W laboratorium uczeń styka się z substancjami szkodliwymi dla zdrowia, z kruchymi
naczyniami szklanymi, z instalacją gazową, elektryczną i wodną. Przy nieumiejętnym
posługiwaniu się tymi substancjami, naczyniami i instalacjami uczeń może być narażony na
różne niebezpieczeństwa (zatrucie, poparzenie, skaleczenie, porażenie prądem elektrycznym),
a ponadto może spowodować uszkodzenie sprzętu laboratoryjnego.
Uczeń powinien dbać o oszczędne używanie odczynników, oszczędne gospodarowanie
wodą, gazem i prądem elektrycznym. Zasady pracy w laboratorium powinny być
ogólnodostępne.
Metrologia zajmuje się nauką o pomiarach. Procesy chemiczne charakteryzuje się podając
parametry procesowe.
Wielkość to właściwość, którą można określić ilościowo. Pomiar wielkości polega na
porównaniu jej z wielkością tego samego rodzaju przyjętą umownie za równą jednostce.
Umowna wartość dająca informacje, ile razy mierzona wielkość jest większa lub mniejsza od
tej umownej jednostki, nazywamy jednostką miary:
wielkość = liczbowa wartość wielkości · jednostka miary
Międzynarodowy Układ Jednostek Miar, zwany układem SI, ma siedem jednostek
podstawowych: metr, kilogram, sekunda, amper, kelwin, mol, kandela.
Pozostałe jednostki są pochodnymi wynikają z równań definicyjnych. W celu zapisu
bardzo dużych i bardzo małych miar, wprowadzono jednostki wtórne, których nazwy tworzy
się z odpowiedniego przedrostka.
Rozróżnia się:
−
pomiary bezpośrednie- poszukiwaną wartość uzyskuje się wprost z pomiaru;
−
pomiary pośrednie – poszukiwaną wartość oblicza się z pomiarów innych wielkości
odpowiednio z nią związanych.
Pomiary można prowadzić metodą bezpośredniego porównania z wzorcem lub metodą
kompensacyjna zwaną zerową.
Wynik pomiaru na ogół odbiega od wartości wielkości mierzonej. Różnica ta nosi nazwę
błędu pomiaru. Dla użytkownika wynik pomiaru jest przydatny tylko wtedy, gdy potrafi on
określić błąd popełniony w trakcie pomiaru. Rozróżniamy pojęcie błędu bezwzględnego
i błędu względnego.
Błąd bezwzględny jest to różnica algebraiczna pomiędzy wynikiem pomiaru
X
i
a wartością wielkości mierzonej X
X
X
e
i
−
=
Wartość wielkości mierzonej X użyta w tym wyrażeniu jest wartością porównawczą.
W zasadzie powinna to być wartość mierzonej wielkości, w praktyce używa się jednak
wartości poprawnej (tzn. na tyle przybliżonej do wartości rzeczywistej, że różnicę pomiędzy
nimi można pominąć z punktu widzenia celu, do którego wartość ta ma służyć) lub średniej
arytmetycznej wyników serii pomiarów. Błąd bezwzględny wyraża się w tych samych
jednostkach co wielkość mierzoną.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
8
Stosunek błędu bezwzględnego do wartości wielkości mierzonej zastosowanej do
obliczania tego błędu nosi nazwę błędu względnego ε
X
e
=
ε
Najczęściej błąd względny określamy w procentach
%
100
X
e
⋅
=
ε
Niezale
ż
nie od przyczyn powstawania bł
ę
dów mo
ż
na podzieli
ć
je na dwie grupy: bł
ę
dy
systematyczne i bł
ę
dy losowe. Bł
ę
dy systematyczne s
ą
to bł
ę
dy powtarzaj
ą
ce si
ę
przy
kolejnych pomiarach. Przyczyn
ą
powstawania bł
ę
dów systematycznych mo
ż
e by
ć
zarówno
niedoskonało
ść
wykonania przyrz
ą
du pomiarowego, jak i daj
ą
cy si
ę
okre
ś
li
ć
wpływ
czynników zewn
ę
trznych, takich jak np. temperatury, wilgotno
ś
ci powietrza, ci
ś
nienia
atmosferycznego. Bł
ą
d ten jest stały dla danego przyrz
ą
du albo zmienia si
ę
według jakiego
ś
znanego prawa. Bł
ę
dy losowe s
ą
to bł
ę
dy zmieniaj
ą
ce si
ę
zarówno co do wielko
ś
ci, jak
i znaku przy powtarzaniu pomiarów tej samej wielko
ś
ci w praktycznie jednakowych
warunkach. Bł
ę
dy te powstaj
ą
w wyniku działania na przyrz
ą
d nieuchwytnych zmian
warunków
zewn
ę
trznych,
niedoskonało
ś
ci
samego
przyrz
ą
du
pomiarowego
oraz
indywidualnych wła
ś
ciwo
ś
ci obserwatora. Bł
ę
du losowego nie mo
ż
na wyeliminowa
ć
przez
wprowadzenie odpowiedniej poprawki. Mo
ż
na jedynie na podstawie serii pomiarów
(wykonywanych przez tego samego obserwatora, za pomoc
ą
tego samego narz
ę
dzia
pomiarowego i w tych samych warunkach zewn
ę
trznych) okre
ś
li
ć
z danym
prawdopodobie
ń
stwem granice, w których znajduje si
ę
ten bł
ą
d.
Wyniki pomiarów na ogół układa si
ę
w tablice. Nale
ż
y pami
ę
ta
ć
o zanotowaniu na
tablicy daty i miejsca wykonywania pomiaru oraz oznaczenia jednostek, w których s
ą
podawane wyniki.
Wad
ą
tabelarycznego uj
ę
cia wyników s
ą
trudno
ś
ci w okre
ś
laniu warto
ś
ci po
ś
rednich oraz
mała przejrzysto
ść
nieobrazuj
ą
ca jasno charakteru zale
ż
no
ś
ci pomi
ę
dzy wyst
ę
puj
ą
cymi
wielko
ś
ciami. Dlatego d
ąż
ymy do przedstawienia wyników w postaci graficznej, je
ś
li
mierzona wielko
ść
A jest funkcj
ą
jednej zmiennej B, zale
ż
no
ść
A = f(B) mo
ż
na łatwo
przedstawi
ć
w układzie współrz
ę
dnych prostok
ą
tnych. Stosownie do charakteru tej zale
ż
no
ś
ci
odpowiednio dobiera si
ę
skal
ę
. Przy sporz
ą
dzaniu wykresów nale
ż
y na osi odci
ę
tych
odkłada
ć
zmienn
ą
niezale
ż
n
ą
. Skale wykresu mog
ą
nie zaczyna
ć
si
ę
od zera, lecz ich punkty
graniczne musz
ą
by
ć
tak dobrane,
ż
eby obejmowały najni
ż
sze i najwy
ż
sze warto
ś
ci
zmiennych. Skal
ę
dobiera si
ę
tak,
ż
eby mo
ż
na było łatwo odczyta
ć
współrz
ę
dne ka
ż
dego
punktu. Przy doborze skali nale
ż
y pami
ę
ta
ć
,
ż
e powinna ona zapewni
ć
tak
ą
dokładno
ść
odczytu przedstawionych graficznie wielko
ś
ci, z jak
ą
dokonano ich pomiaru.
Krzywa obrazuj
ą
ca zale
ż
no
ść
mi
ę
dzy zmiennymi powinna by
ć
ci
ą
gła i bez załama
ń
.
Na skutek bł
ę
dów losowych nie wszystkie punkty pomiarowe le
żą
na takiej krzywej.
W zasadzie dokładnie wykre
ś
lona krzywa powinna przebiega
ć
tak,
ż
eby suma kwadratów
odległo
ś
ci punktów od krzywej była najmniejsza. Przy pewnej wprawie krzyw
ą
mo
ż
na
wykre
ś
li
ć
bez tych oblicze
ń
, nale
ż
y j
ą
prowadzi
ć
tak,
ż
eby w przybli
ż
eniu połowa punktów
do
ś
wiadczalnych le
ż
ała po jednej stronie krzywej, a połowa po drugiej. Przebieg krzywej jest
tym pewniejszy, im wi
ę
cej mamy punktów pomiarowych. Jako minimum dla wykre
ś
lenia
krzywej w sposób pewny przyjmuje si
ę
5 punktów.
Urz
ą
dzenia techniczne stosowane w technice pomiarowej dziel
ą
si
ę
na wzorce miar
i przyrz
ą
dy pomiarowe.
Przyrz
ą
d pomiarowy mo
ż
na zdefiniowa
ć
jako urz
ą
dzenie słu
żą
ce do przetwarzania
wielko
ś
ci mierzonej lub innej wielko
ś
ci zwi
ą
zanej z ni
ą
znan
ą
funkcj
ą
na wskazania lub inn
ą
równorz
ę
dn
ą
informacj
ę
. Przyrz
ą
dy te s
ą
niezb
ę
dne do kontrolowania procesów

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
9
technologicznych. Stosuje się je zarówno w kontroli procesów podstawowych, całego
procesu, kontroli między operacyjnej i finalnej.
W każdym przyrządzie pomiarowym można wyodrębnić podstawowe części składowe,
rozpatrywane od strony spełnianej funkcji. Są one połączone szeregowo i przekazują
jednokierunkowo informacje za pośrednictwem linii łączących, zwanych torem pomiarowym.
Czujnik jest częścią przyrządu, na którą oddziałuje wielkość mierzona. Rozróżnia się
czujniki stykowe, stykające się bezpośrednio ze środowiskiem mierzonym oraz czujniki
bezstykowe, spotykane rzadziej. Czujnik przetwarza wielkość mierzoną na inną wielkość,
łatwiejszą do wykorzystania praktycznego w elemencie wskazującym wartość zmierzoną,
w mierniku. Przetworzenie następuje według znanej zależności, sygnału pomiarowego, jest
wprost proporcjonalna do wartości wejściowego sygnału czujnika, to znaczy stanu mierzonej
wielkości.
Czujnik i miernik, występują nie tylko w złożonych układach pomiarowych, ale również
w przyrządach prostych.
Tor pomiarowy może mieć długość od kilku centymetrów do setek kilometrów.
W praktyce przemysłowej tory pomiarowe mają najczęściej długości do kilkudziesięciu
metrów, a to w celu umożliwienia centralizacji pomiarów przez umieszczenie wszystkich
mierników danego agregatu, oddziału produkcyjnego lub całej linii produkcyjnej na jednej
tablicy pomiarowej, umieszczonej w sterowni.
Mierniki są tylko jednym z przykładów odbiornika sygnałów pomiarowych. Odróżnia się
następujące odbiorniki:
−
wskaźnik, stwierdzający jedynie występowanie lub niewystępowanie danej wielkości;
−
miernik, wskazujący zmierzoną wartość;
−
rejestrator inaczej samopis, wskazujący i zapisujący zmierzoną wartość jako funkcję
czasu;
−
rejestrator XY, zapisujący mierzoną wartość wielkości X jako funkcję innej jednocześnie
mierzonej wielkości Y;
−
miernik sumujący zwany licznikiem;
−
sygnalizator, który wizualnie lub akustycznie sygnalizuje przekroczenie określonej
wartości;
−
drukarka, która wynik pomiaru zapisuje w postaci cyfrowej;
−
regulator automatyczny, służący do automatycznego sterowania przebiegiem procesu.
Odbiornik w układzie pomiarowym musi być dopasowany do postaci fizycznej
dochodzącego sygnału pomiarowego. Dowolny układ pomiarowy składa się zawsze
z czujnika i z odbiornika, natomiast przetwornik stosuje się jedynie w razie potrzeby.
Duża część przyrządów pomiarowych jest wyposażona w mierniki wskazówkowe.
Położenie ruchomej wskazówki jest zależne od wartości sygnału pomiarowego i odczytuje się
je na podziałce składającej się ze zbioru wskazów. Każde wskazanie odpowiada określonej
mierze, wyrażonej w jednostkach danej wielkości. Najczęściej stosowanym rodzajem są
tu kreski. Jeżeli ostrze wskazówki zajmuje położenie pośrednie między sąsiednimi kreskami,
wówczas odczyt wartości mierzonej uściśla się przez interpolację.
Odróżnia się następujące rodzaje podziałek:
−
jednostajna, jeżeli podziałka ma jednakową długość wszystkich działek,
−
równomierna, jeżeli wszystkie działki mają równą wartość,
−
regularna, jeżeli podziałka jest jednocześnie jednostajna i równomierna.
Podziałki są naniesione na część przyrządu pomiarowego zwaną podzielnią. Często na
podzielni umieszcza się informacje o właściwościach przyrządu, (dokładność, pozycję pracy,
metodę pomiaru, napięcie zasilania itp.). Przyrządy pomiarowe czułe na pozycję pracy są
często wyposażone w poziomnice, umożliwiającą ich prawidłowe ustalenie.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
10
Zakres pomiarowy przyrządu określa wartości, które można mierzyć z określoną
dokładnością. Zakres pomiarowy jest określony dolną granicą, zazwyczaj równą zeru i górną
granicą.
Dokładność przyrządu pomiarowego określa się często terminem klasy. Klasa przyrządu,
podana przeważnie na podzielni w postaci liczby oderwanej. Jest równa największemu
błędowi bezwzględnemu podanemu w stosunku do różnicy między górną i dolną granicą
zakresu pomiarowego, np. klasa 1 amperomierza o zakresie pomiarowym 0-50A oznacza, że
jego błąd względny wynosi 1% w stosunku do górnej granicy zakresu pomiarowego,
co odpowiada błędowi bezwzględnemu 0,5A na całym zakresie pomiarowym.
Przyrząd pomiarowy można scharakteryzować jego czułością. Miarą czułości jest
najmniejsza zmiana wartości mierzonej powodująca zmianę wskazania przyrządu. Pojęcie
czułości jest związane z konstrukcją i starannością wykonania danego przyrządu.
Inną właściwością jest przełożenie pomiarowe przyrządu, które można zdefiniować jako
stosunek przesuwu wskazówki wyrażony miarą długości lub kąta do jednostkowego
przyrostu wartości mierzonej.
Wzorcowanie przyrządów pomiarowych polega na ustaleniu położenia wskazów
odpowiednio do różnych wartości wielkości mierzonej. Na podstawie wzorcowania
przeprowadza się skalowanie przyrządu. Polega ona na wykonaniu podziałki na podstawie
wskazów określonych wzorcowaniem, jeżeli wzorcowanie objęło tylko główne wskazy,
resztę wskazów nanosi się przez interpolację.
Obie te czynności są wykonywane pod nadzorem wyspecjalizowanej jednostki
organizacyjnej, spełniającej funkcję państwowej służby metrologii prawnej. W Polsce funkcje
te spełnia Polski Komitet Normalizacji i Miar (PKNiM), uprawniony do sprawowania opieki
nad całą gospodarką przyrządami pomiarowymi.
PKNiM ma uprawnienia ustalania obowiązujących wzorców miar, opracowywania metod
ich odtwarzania, sposobów ich przechowywania oraz metod porównywania z wzorcami
praktycznymi, również ustalania pochodnych jednostek miar. PKNiM ma więc decydujący
wpływ na ustalanie podstaw metrologii stosowanej.
Legalizacja jest to stwierdzenie i zaświadczenie, że dany przyrząd pomiarowy spełnia
całkowicie wymagania przepisów legalizacyjnych.
Przyrządy pomiarowe z upływem czasu zmieniają swoją dokładność i dlatego muszą być
okresowo sprawdzane. Jeżeli sprawdzona dokładność przyrządu nie mieści się już w jego
klasie, wówczas przyrząd należy naprawić, wyregulować, zmienić klasę podaną na podzielni
lub zlikwidować. Własności metrologiczne przyrządów muszą być zgodne z ich danymi
technicznymi. Wszystkie obowiązki użytkowników, mające na celu zabezpieczenie
rzetelności pomiarów, są ujęte w przepisach państwowych.
W każdym zakładzie przemysłowym jest laboratorium analityczne, które odpowiedzialne
jest za pobór oraz analizę substratów, półproduktów i produktów. Pracownik laboratorium
pobiera próbki analityczne w warunkach ciągłego procesu, co pozwala kontrolować
poprawność przebiegu produkcji.
4.1.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Ile i jakie jednostki podstawowe obowiązują w układzie SI?
2.
Jakie są rodzaje błędów pomiaru?
3.
Jaka jest różnica pomiędzy błędem bezwzględnym a błędem względnym?
4.
Jak należy wyeliminować błędy losowe (systematyczne)?
5.
Jak należy zapisać wynik końcowy pomiaru, który jest rezultatem działań stopnia
wyższego niż pierwszy?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
11
6.
Jak należy wykreślić krzywą obrazującą zależność między zmiennymi?
7.
Jakie są rodzaje podziałek przyrządów pomiarowych?
8. Jak określa się dokładność przyrządu pomiarowego?
4.1.3. Ćwiczenia
Ćwiczenie 1
Określ
niedokładność
termostatu,
wykorzystywanego
w procesie
fermentacji
beztlenowej.
Pomiar niedokładności termostatu prowadzi poprzez pomiar temperatury dodatkowym
termometrem rtęciowym umieszczonym w termostacie.
Związki chemiczne: woda.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
napełnić termostat wodą, tak aby pokrywała ona pompę,
2)
ustawić za pomocą pokrętła na termometrze kontaktowym, umieszczonym w termostacie,
temperaturę 50ºC, w termostacie powinien być także termometr rtęciowy z podziałką
w skali 0,1% za pomocą którego należy kontrolować działanie procesu,
3)
włączyć termostat, mierzyć temperaturę i czas w momencie włączania się i wyłączania
grzałki (kontrola za pomocą żarówki i charakterystycznego odgłosu),
4)
prowadzić obserwację do momentu ustabilizowania się temperatury przez 45 minut,
5)
zestawić zebrane wyniki w tabeli,
6)
sporządzić wykres zależności temperatury od czasu czyli T(K)=f[t(s)],
7)
zinterpretować otrzymany wykres, obliczyć wariancję i odchylenie standardowe od
nastawionej temperatury w termostacie,
8)
określić niedokładność termostatu.
Wyposażenie stanowiska pracy:
–
termostat z termometrem kontaktowym i rtęciowym,
–
stoper,
–
papier milimetrowy i przyrządy kreślarskie.
Ćwiczenie 2
Przeprowadź wzorcowanie rotametru, na przepływ powietrza przy użyciu anemometru,
wykorzystywanego w procesie spalania węgla w kotle pyłowym.
Związki chemiczne: powietrze.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
podłączyć rotametr do sprężarki,
2)
skierować wylot powietrza rotametru na skrzydełka anemometru,
3)
przepuszczać sprężone powietrze tak, aby w rotametrze pływak utrzymywał się na
wybranym poziomie,
4)
zmierzyć anemometrem prędkość liniową powietrza oraz wysokość, na jakiej oscyluje
pływak,
5)
pomiar wykonać 3-krotnie dla różnych położeń pływaka,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
12
6)
zanotować wyniki w tabeli,
7)
obliczyć natężenie przepływu powietrza,
Wzór tabeli do ćwiczenia 2
Lp.
Ilość podziałek
Prędkość liniowa
[m/min]
Natężenie przepływu
[dm
3
/h]
1
2
3
8)
wykonać wykres: natężenie przepływu powietrza V(m3/s)w funkcji położenia pływaka
h(mm) czyli V(m3/s)=f[h(mm)].
Wyposażenie stanowiska pracy:
–
rotametr zamontowany w statywie,
–
anemometr skrzydełkowy,
–
sprężarka do powietrza,
–
przewody gumowe (węże) z zaworami,
–
papier milimetrowy i przyrządy kreślarskie.
4.1.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
wymienić zadania metrologii?
2)
wymienić czynności, jakie należy wykonywać przy każdym
pomiarze?
3)
scharakteryzować rodzaje błędów pomiaru?
4)
wymienić odbiorniki sygnałów pomiarowych?
5)
scharakteryzować właściwości przyrządów pomiarowych: klasę,
czułość, przełożenie pomiarowe?
6)
sklasyfikować przyrządy pomiarowe?
7)
wymienić informacje, jakie powinna zawierać instrukcja eksploatacji
przyrządów pomiarowych?
8)
dokonać wyboru przyrządu do określonego pomiaru?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
13
4.2. Wybrane pomiary w procesach technologicznych
4.2.1.
Materiał nauczania
Ciśnienie (p) to wielkość skalarna określona jako wartość siły (F
n
) działającej prostopadle
do powierzchni podzielona przez powierzchnię (S) na jaką ona działa, co przedstawia
zależność:
Jednostką ciśnienia jest paskal (Pa) z definicji -niuton na metr kwadratowy (N/m
2
).
Ciśnienie hydrostatyczne p, wyrażone zależnością p = hρg (g – przyspieszenie ziemskie),
jest wprost proporcjonalne do wysokości słupa cieczy h i jej gęstości ρ. Ciśnienie wyrażone
wysokością słupa cieczy, najczęściej wody lub rtęci, stosuje się w laboratoriach. Jednostka
1milimetr słupa wody (1 mm H
2
O) jest to ciśnienie, które na swoją podstawę wywiera słup
wody destylowanej o temperaturze 277,15 K=4°C i wysokości 1mm przy przyspieszeniu
ziemskim g = 9,80665 N/kg. Analogicznie jednostka 1milimetr słupa rtęci (1 mm Hg) określa
ciśnienie, które ma swoją podstawę wywiera w temperaturze 273,15 K=0°C i przy
g = 9,80665 N/kg
słup rtęci wysokości 1 mm. Ciśnienie 760 mm Hg jest to atmosfera
fizyczna (symbol atm.).
Zależnie od wielkości ciśnienia przyjętego za podstawę (umowne zero), od którego
zaczynamy mierzyć ciśnienie, rozróżniamy:
1.
ciśnienie absolutne p
a
(bezwzględne), dla którego ciśnieniem zerowym jest próżnia
absolutna.
2.
ciśnienie względne mierzone od ciśnienia zerowego różnego od próżni absolutnej. Jako
ciśnienie zerowe najczęściej przyjmuje się ciśnienie barometryczne (p
b
) tj. ciśnienie
otaczającej atmosfery. Różnica między ciśnieniem absolutnym większym od
barometrycznego nazywana jest nadciśnieniem (p
n
), a różnica między ciśnieniem
barometrycznym a mniejszym od niego ciśnieniem absolutnym nazywana jest
podciśnieniem (p
p
).
Przyrządy do pomiaru ciśnienia można klasyfikować według różnych kryteriów, takich
jak zasada działania, dokładność przyrządu, rodzaj mierzonego ciśnienia.
Biorąc pod uwagę rodzaj ciśnienia, przyrządy do jego mierzenia można podzielić na:
1. Barometry – do pomiaru ciśnienia atmosferycznego w otwartej przestrzeni.
2. Manometry bezwzględne (manobarometry) – do pomiaru ciśnienia bezwzględnego
w zamkniętej przestrzeni – zwane potoczni próżniowymi.
3.
Manometry – do pomiaru nadciśnienia w zamkniętej przestrzeni.
4.
Wakuometry – do pomiaru podciśnienia w zamkniętej przestrzeni.
5.
Manowakuometry – do pomiaru nadciśnienia i podciśnienia.
6.
Manometry różnicowe – do pomiaru różnicy ciśnień w dwóch zamkniętych
przestrzeniach.
Podział z uwagi na zasadę działania przyrządu:
1.
Cieczowe (hydrostatyczne) – oparte na równoważeniu mierzonego ciśnienia ciśnieniem
hydrostatycznym słupa cieczy.
2.
Hydrauliczne (obciążeniowe) – oparte na zasadzie równowagi hydraulicznej pomiędzy
ciśnieniem mierzonym, działającym na jedną stronę przegrody ruchomej, a siłą
zewnętrzną działającą na drugą stronę tej przegrody.
3.
Sprężynowe, w którym miarą ciśnienia jest odkształcenie elementu sprężystego.
4. Elektryczne oparte na zmianie właściwości elektrycznych pewnych materiałów pod
wpływem zmian ciśnienia.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
14
W technice (szczególnie w przypadku pomiaru wysokich ciśnień) mamy przeważnie do
czynienia z pomiarem różnicy ciśnień. Pomiary ciśnienia bezwzględnego to najczęściej
pomiar ciśnienia atmosferycznego, ciśnień niższych od atmosferycznego albo ciśnień
niewiele wyższych od atmosferycznego.
Temperatura jest wielkością określającą stopień ogrzania jakiegoś ciała, zależną od
średniej energii kinetycznej cząstek tego ciała. Temperatury nie można zmierzyć
bezpośrednio, dokonuje się tego przez pomiar innych wielkości fizycznych ciała
jednoznacznie zależnych od jego temperatury i dających się łatwo zmierzyć. Takimi
wielkościami są rozszerzalność objętościowa, ciśnienie pary nasyconej, opór elektryczny,
natężenie promieniowania itp.
Temperatura, podobnie jak ciśnienie, jest jedną z podstawowych wielkości
warunkujących przebieg procesów technologicznych w przemyśle chemicznym. Jej wartość
wywiera znaczny wpływ na szybkość i wydajność reakcji.
Ze względu na sposób wykonywaniu, pomiary temperatury można podzielić na:
1.
pomiary stykowe, w których następuje wymiana ciepła pomiędzy badanym obiektem
i dotykającym do niego bezpośrednio czujnikiem,
2.
pomiary bezstykowe, w których do oceny temperatury służy jakaś wielkość dająca się
zmierzyć bez bezpośredniego dotyku czujnika do badanego obiektu.
Przyrządy służące do pomiaru temperatury (termometry) najczęściej klasyfikujemy
według zasady działania:
1.
Termometry działające na zasadzie rozszerzalności cieplnej cieczy, ciał stałych i gazów.
W termometrach
cieczowych
szklanych,
wypełnionych
odpowiednią
cieczą
termometryczną (rtęć, alkohol inne), pod wpływem zmian temperatury zmienia się
objętość cieczy. Na zasadzie rozszerzalności cieplnej ciał stałych oparte są dwa rodzaje
termometrów: prętowe (dylatacyjne) i bimetaliczne. W termometrach manometrycznych,
ogrzewaniu w stałej objętości cieczy lub gazu towarzyszy wzrost ciśnienia.
2.
Termometry rezystancyjne (oporowe) działające na zasadzie zmian rezystancji (oporu
elektrycznego) pod wpływem zmian temperatury.
3.
W termometrach termoelektrycznych wykorzystuje się występowanie różnicy
potencjałów elektrycznych pomiędzy dwoma spoinami dwóch różnych metali,
znajdującymi się w różnej temperaturze. Pomiar temperatury sprowadza się do pomiaru
siły elektromotorycznej, zależnej od różnicy temperatury obu złączy i rodzaju użytych
metali.
4.
Termometry optyczne, w których wykorzystuje się występowanie zależności pomiędzy
temperaturą rozżarzonego ciała a wydzielanym przez nie promieniowaniem.
Idealną skalą temperatury niezależną od właściwości fizycznych ciał jest
termodynamiczna skala temperatury wyprowadzona przez Kelwina w oparciu o II zasadę
termodynamiki. Jednostką temperatury termodynamicznej jest kelwin (symbol K),
definiowany jako
15
,
273
1
część temperatury termodynamicznej punktu potrójnego
wody,. dla którego przyjęto wartość 273,15 K. Woda występuje jednocześnie w trzech
stanach skupienia w temperaturze 0,1°C i pod ciśnieniem 0,00623 at. Powiązanie
pomiędzy powszechnie używaną temperaturą Celsjusza t (wyrażaną w stopniach
Celsjusza – symbol °C) a temperaturą termodynamiczną ma postać:
15
,
273
−
=
T
t
Stopie
ń
Celsjusza równy jest kelwinowi. Punkt zerowy termodynamicznej skali
temperatury odpowiada najni
ż
szej temperaturze, jak
ą
mo
ż
na by uzyska
ć
, w której zanika ju
ż
wszelki ruch post
ę
powy i drgaj
ą
cy atomów i cz
ą
steczek.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
15
Poziom cieczy w zbiorniku mierzy się w celu określenia jej ilości, bądź w celu utrzymania
jej poziomu na określonej wysokości. Pomiar poziomu cieczy dla określenia jej ilości
w zbiorniku różni się od pomiaru poziomu w celu regulowania i utrzymania go na określonej
wysokości. W pierwszym przypadku zakres pomiaru jest określony wymiarami zbiornika
i przyrząd pomiarowy powinien umożliwiać mierzenie poziomu od poziomu najniższego,
tj. od dna, do górnej krawędzi zbiornika. W drugim przypadku pomiar poziomu waha się
w wąskich granicach, zwykle 100–200 mm. Odpowiednio do celu, któremu ma służyć pomiar
poziomu cieczy, stosujemy różne metody pomiaru, różne typy poziomowskazów o różnej
czułości i dokładności pomiaru.
Rozróżniamy dwa podstawowe typy poziomowskazów:
1.
Poziomowskazy pływakowe. Zasadniczą częścią urządzenia jest pływak unoszący się na
powierzchni cieczy i wznoszący się lub opadający wraz z nią. Ruchy pływaka są
przekazywane mechanicznie urządzeniu wskazującemu.
2.
Poziomowskazy
zbudowane
na
zasadzie
naczyń
połączonych.
Najprostszym
poziomowskazem tego typu jest zwykłe szkło wodowskazowe.
Przyrządy o małym zakresie pomiarowym mają punkt zerowy, odpowiadający
optymalnemu poziomowi cieczy, w środku podzielni i podziałkę określającą odchylenia od
normalnego poziomu w obie strony od punktu zerowego.
Przyrządy o dużym zakresie pomiarowym mają punkt zerowy na początku podzielni,
podziałkę położoną z jednej tylko strony, liczoną od zera do maksimum, i wskazują
wysokość poziomu cieczy nad dnem.
Pomiaru poziomu cieczy w zbiorniku można dokonać bądź określając bezpośrednio
odległość zwierciadła cieczy od jakiegoś poziomu odniesienia, np. dna zbiornika,
lub w sposób pośredni mierząc wielkości zależne od wysokości słupa cieczy w zbiorniku.
Jeden z najprostszych sposobów pomiaru poziomu cieczy w otwartym zbiorniku polega na
umieszczeniu podziałki na wewnętrznej ścianie zbiornika lub po zanurzeniu w zbiorniku
listwy z podziałką, która może wyrażać bądź wysokość cieczy w zbiorniku w metrach, bądź
jej objętość w m
3
.
Pomiar ilości cieczy i gazów, czyli ogólnie płynów, w procesach technologicznych jest
czynnością bardzo istotną, ważną i często spotykaną. Ilość płynu mierzymy bądź
w jednostkach masy (kg, t), bądź w jednostkach objętości (m
3
). Przeliczenia ilości płynu
wyrażonej w jednostkach objętości na jego masę, dokonuje się przy pomocy wzoru:
ρ
⋅
=
V
m
gdzie: m – masa płynu [kg]
V – objętość płynu [m
3
]
ρ – gęstość płynu [kg/m
3
]
Ilość cieczy lub gazu przepływająca w ciągu jednostki czasu nosi nazwę natężenia
przepływu masowego lub objętościowego zależnie od tego, czy jest mierzona
w jednostkach masy, czy w jednostkach objętości. Masowe natężenie przepływu wyrażamy
w kg/s, kg/h, a objętościowe natężenie prze pływu w m
3
/s, m
3
/h.
Ilość gazu w warunkach przemysłowych zwykle wyrażamy w jednostkach objętości.
Objętość tej samej, masy gazu jest różna w różnych warunkach temperatury i ciśnienia. Chcąc
jednoznacznie określić objętość gazu należy podawać zarówno temperaturę jak i ciśnienie
w warunkach pomiaru. Zmiana
objętości cieczy w zależności od zmian temperatury bądź
ciśnienia jest tak niewielka, że można jej w pomiarach przemysłowych nie uwzględniać.
Jednak w przypadkach wymagających wysokiej dokładności, gdy należy uwzględnić zmianę
gęstości cieczy wynikającą ze zmiany temperatury, gęstość tę w temperaturze t można
określić w sposób przybliżony z zależności:

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
16
)
20
(
1
20
t
t
−
−
=
β
ρ
ρ
gdzie: ρ
20
– gęstość cieczy w temperaturze 20°C
β – współczynnik rozszerzalności objętościowej [1/deg]
t – temperatura [°C]
Jeśli wprawimy w ruch jakąś warstwę cieczy lub gaz, to pociągnie ona za sobą
nieruchome warstwy sąsiednie. Siła, jaka będzie działać na kolejne warstwy sąsiednie, jest
proporcjonalna do powierzchni zetknięcia się warstw, ich odległości i prędkości przesuwania się
względem siebie Współczynnik proporcjonalności nosi nazwę współczynnika lepkości lub
krócej lepkości dynamicznej. W układzie SI wymiarem jednostki lepkości jest N·s/cm
2
.
Często można się spotkać z ilorazem lepkości i gęstości zwanej lepkością kinetyczną.
ρ
µ
ν
=
gdzie: ν – lepkość kinematyczna
µ – lepkość dynamiczna
ρ – gęstość
Jednostką lepkości kinematycznej w układzie SI jest m
2
/s. Mając daną lepkość
dynamiczną gazu w temperaturze 273°K (0°C) można przeliczyć ją na lepkość w dowolnej
temperaturze posługując się wzorem:
3
0
0
0
+
+
=
T
T
S
T
S
T
µ
µ
gdzie: µ – lepkość dynamiczna w temperaturze T
µ
0
– lepkość dynamiczna w temperaturze T
0
= 273°K
S – Stała Sutherlanda
T i T
0
– temperatura [°K
]
Lepkość gazu zależy od temperatur i od ciśnienia. W przypadku ciśnień wyższych od
5 MPa, ten wpływ należy uwzględniać a wzory znaleźć w literaturze monograficznej.
Przepływ płynu przez rurociąg lub kanał może mieć dwojaki charakter. Przy małych
prędkościach przepływu można stwierdzić, że poszczególne cząstki płynu poruszają się
równolegle do osi rurociągu, przy czym cząstki znajdujące się bliżej ścianek przewodu
poruszają się wolniej, a znajdujące się bliżej środka przewodu szybciej. Z największą
prędkością poruszają się cząstki płynące wzdłuż osi rurociągu. Przepływ z takim rozkładem
prędkości nosi nazwę przepływu uwarstwionego (laminarnego). Przy zwiększaniu prędkości
przepływu w pewnym momencie zauważymy występowanie poprzecznych do osi przewodu
ruchów cząstek płynu i tworzenie się wirów. W tym przypadku poza niewielką ilością
płynu w pobliżu ścianek przewodu pozostała masa będzie poruszała się jednakową
prędkością niezależnie od odległości od osi przewodu, taki rodzaj przepływu nosi nazwę
przepływu burzliwego.
W obliczeniach technicznych operujemy średnią prędkością płynu określaną zależnością:
F
V
u
śr
=
gdzie:
u
śr
– średnia prędkość płynu [m/s]
V
– objętościowe natężenie przepływu [m
3
/s];stosuje się również oznaczenie Q
r
F
– przekrój strumienia płynu [m
2
]
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
17
O tym czy ruch jest burzliwy czy laminarny przesądza nie tylko u – prędkość przepływu
płynu [m/s], ale również D – średnica przewodu [m], ρ – gęstość płynu [kg/m
3
], µ – lepkość
dynamiczna płynu [N·s/m
2
= kg/m·s].Wszystkie te parametry decydujące o rodzaju ruchu
ujęte są w jedną zależność zwaną kryterium (liczbą) Reynoldsa, którą oznaczamy symbolem
Re:
µ
ρ
uD
=
Re
Równanie Bernoulliego opisuje parametry płynu doskonałego płynącego w rurze
(niekoniecznie materialnie istniejącej) o zmiennym przekroju. Wynika ono wprost z faktu
zachowania objętości cieczy doskonałej (która jest nieściśliwa) i zasady zachowania energii
mechanicznej. Założenie, że ciecz jest nieściśliwa, nie jest lepka a przepływ stacjonarny
i bezwirowy pozwala sformułować uproszczoną zależność:
.
2
2
const
p
gh
v
e
m
=
+
+
=
ρ
gdzie: e
m
– energia jednostki masy płynu [J/kg]
ρ – gęstość cieczy [kg/m
3
]
v – prędkość cieczy w rozpatrywanym miejscu [m/s]
h – wysokość w układzie odniesienia, w którym liczona jest energia potencjalna [m]
g – przyspieszenie grawitacyjne [m/s
2
]
p – ciśnienie cieczy w rozpatrywanym miejscu [N/m
2
]
Poszczególne człony to: energia kinetyczna, energia potencjalna przyciągania ziemskiego,
energia ciśnienia. Energia jest stała tylko wówczas, kiedy element porusza się wzdłuż linii
prądu. Istnienie lepkości, przepływu wirowego rozprasza energię a ściśliwość zmienia
zależność prędkości przepływu od ciśnienia
Równanie Bernoulliego może być z pewną dokładnością stosowane także dla cieczy
ściśliwych. Opracowano wersję równania dla płynów uwzględniającą zmianę energii
wewnętrznej płynu w wyniku zmiany różnych czynników.
Uwzględniając właściwości gazów można przekształcić to równanie tak, by było
spełnione także dla gazów. Choć pierwotne równanie Bernoulliego nie jest spełnione dla
gazów, to ogólne wnioski płynące z niego mogą być stosowane również dla nich.
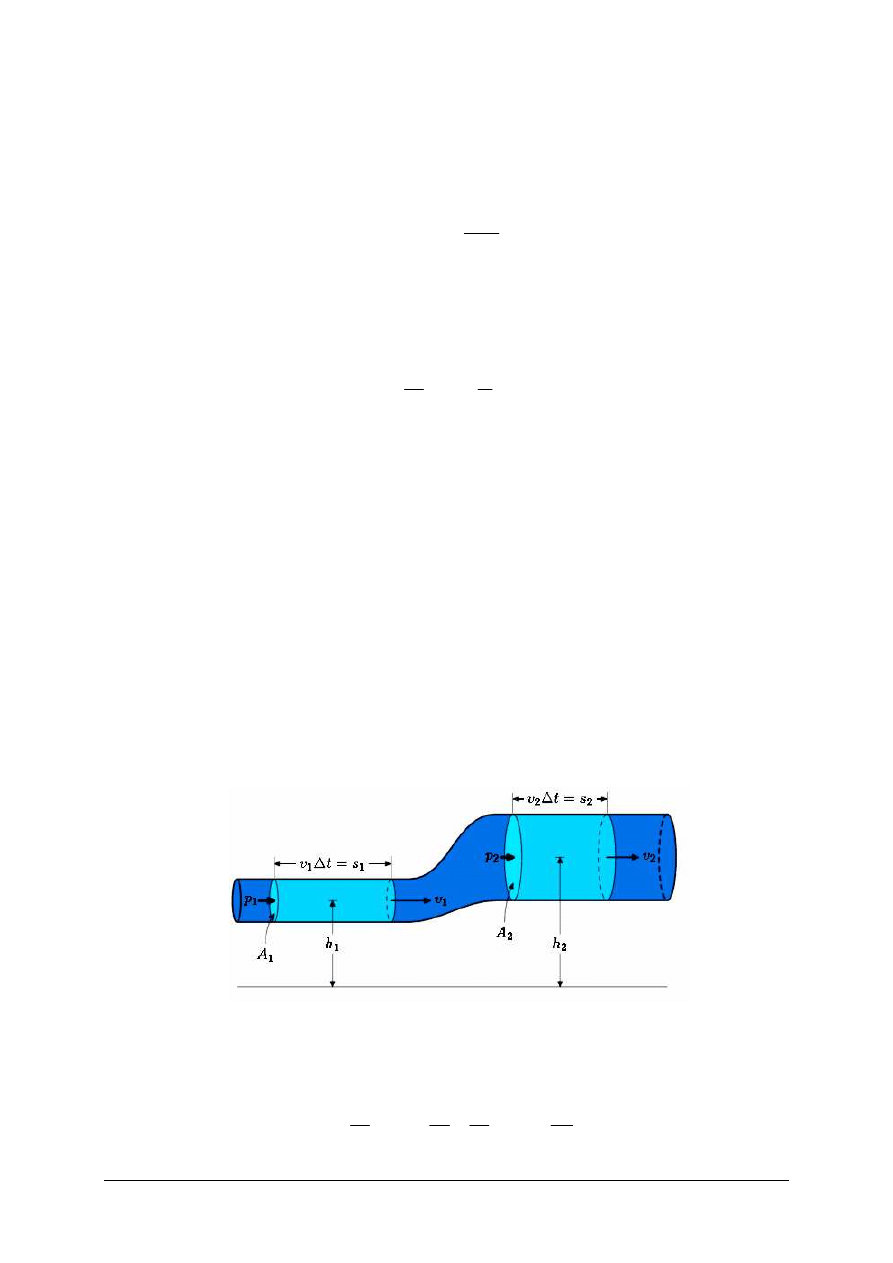
Rys. 1. Graficzne przedstawienie równania Bernoulliego [1]
Z równania Bernoulliego (Rys. 1) dla sytuacji przedstawionej na rysunku zachodzi
prawidłowość:
ρ
ρ
2
2
2
2
1
1
2
1
2
2
p
gh
v
p
gh
v
+
+
=
+
+
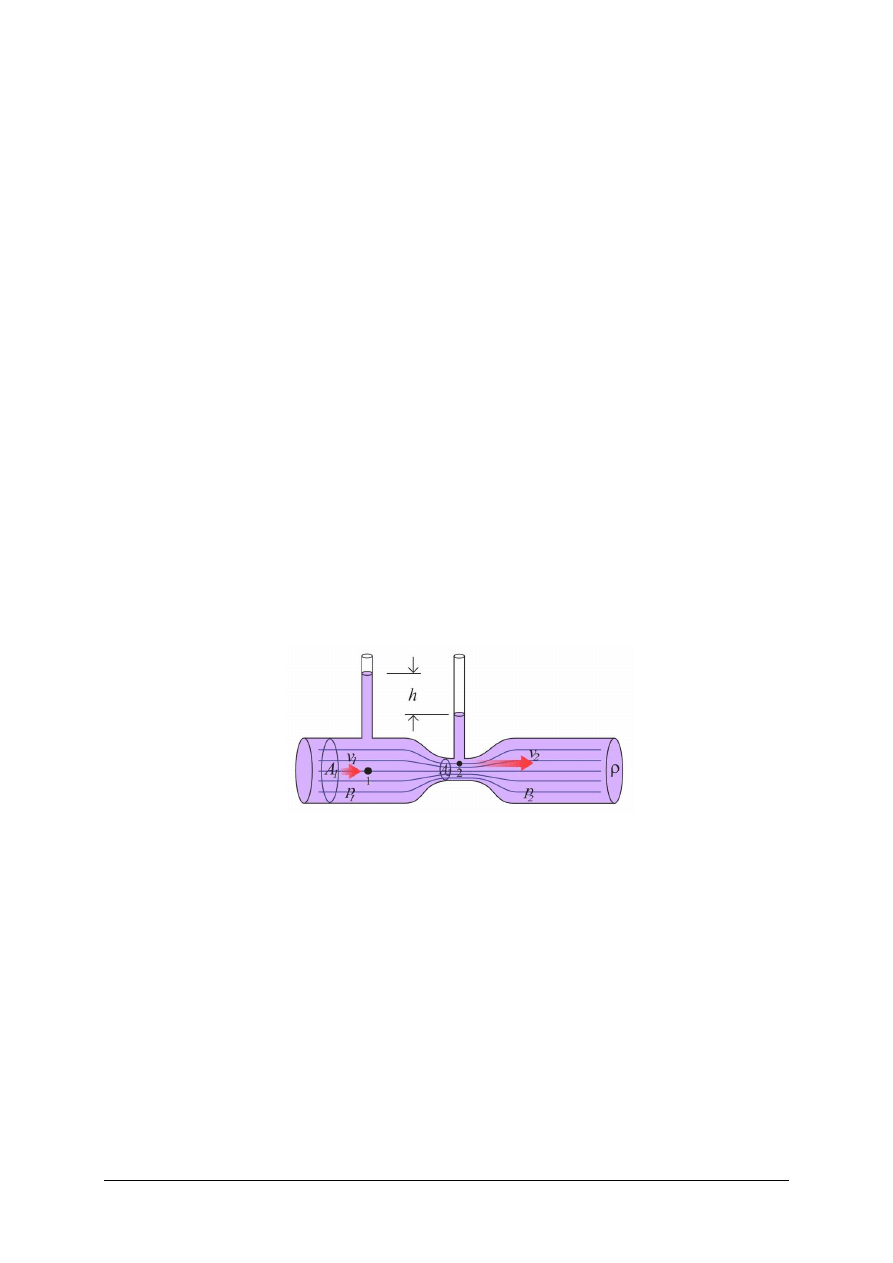
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
18
W rurze o mniejszym przekroju ciecz płynie szybciej (v
1
> v
2
), w związku z tym panuje
w niej mniejsze ciśnienie niż w rurze o większym przekroju. Ciecz płynąc w rurze
o zmieniającym się przekroju ma mniejsze ciśnienie na odcinku, gdzie przekrój jest mniejszy.
Przepływomierzami nazywamy przyrządy służące do pomiaru ilości przepływającego
płynu. Można je podzielić na przepływomierze silnikowe i przepływomierze zwężkowe.
W przyrządach grupy pierwszej przepływający płyn napędza bezpośrednio element
roboczy przepływomierza, który z kolei uruchamia mechanizm liczący. W przepływomierzach
zwężkowych ustawiony na drodze przepływającego płynu element spiętrzający wywołuje
różnicę ciśnień przed i za nim, zależną od natężenia przepływu. Z kolei przepływomierze
silnikowe dzielimy na wirnikowe i komorowe.
Przepływomierze służące do pomiaru ilości względnie natężenia przepływu wody noszą
nazwę wodomierzy. Przepływomierze budowane są na określone natężenia przepływu płynu.
Największą wartość natężenia przepływu, przy której przepływomierz może krótkotrwale
pracować w sieci w normalnych warunkach użytkowych, nazywamy przepuszczalnością
przepływomierza. Drugim czynnikiem charakteryzującym wielkość przepływomierza jest
jego średnica nominalna lub średnica montażowa (średnica przewodu, do którego
przepływomierz jest przeznaczony). Wskazania danego przepływomierza są poprawne tylko
w pewnych granicach natężenia przepływu i zakres ten nosi nazwę obszaru stosowalności
przepływomierza.Górną granicą tego obszaru jest przepuszczalność przepływomierza, zaś
dolną najmniejsze natężenie przepływu zwane dolną granicą dokładności, przy którym błędy
wskazań przepływomierza nie przekraczają granic uchybień dopuszczalnych.
Jeżeli do przewodu, przez który płynie ciecz lub gaz, wstawimy przewężenie, zwężkę
i będziemy mierzyli manometrem różnicowym ciśnienie przed przewężeniem i blisko za
przewężeniem, to manometr wykaże różnicę ciśnień. Ciśnienie przed zwężką będzie większe
niż ciśnienie za zwężką (Rys. 2).
Rys. 2. Zwężka z manometrem różnicowym [6]
Różnica ta (∆p) będzie tym większa, im większe będzie natężenie przepływu płynu
przez rurociąg. Średnica otworu zwężki jest mniejsza od średnicy rurociągu, jeśli zatem
przez otwór w zwężce ma przepłynąć ta sama ilość płynu, co przez rurociąg, to jego
prędkość w otworze zwężki musi być większa niż w przewodzie przed zwężką.
Zwiększenie prędkości płynu powoduje zwiększenie jego energii kinetycznej.
Ten wzrost prędkości i energii kinetycznej płynu w otworze zwężki odbywa się kosztem
jego energii potencjalnej. Zmiany energii potencjalnej płynu powodują zmiany ciśnienia
statycznego, zmniejszenie energii potencjalnej wywołuje zmniejszenie ciśnienia statycznego.
W wyniku tego manometr różnicowy wykazuje różnicę ciśnień statycznych przed i za
zwężką. Tę zależność różnicy ciśnień od natężenia przepływu wykorzystujemy do
pomiaru natężenia przepływu cieczy i gazów w rurociągach. Przyrządy pomiarowe oparte
na tej zasadzie są bardzo szeroko stosowane w praktyce przemysłowej. Poważną ich zaletą
jest prosta budowa. Stosowane są trzy zasadnicze typy zwężek: kryzy, dysze i dysze

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
19
Venturiego (zwane potocznie zwężką Venturiego). Każdy z tych typów zwężek inaczej
wpływa na przepływający strumień płynu i powoduje inny rozkład ciśnień w płynie.
Zasada działania zwężek jest idealną ilustracją równania Bernoulliego. W pewnym
miejscu rurociągu, w którym z prędkością v przemieszcza się płyn (gaz lub ciecz), znajduje
się przewężenie o znacznie mniejszym przekroju. Z prawa Bernoulliego, oraz warunku
ciągłości przepływu, wynika, że kwadrat prędkości płynu przed zwężką jest wprost
proporcjonalny do różnicy ciśnień przed zwężką i na niej.
Wody znajduje się w powietrzu, materiałach pochodzenia naturalnego, wielu produktach
wytwarzanych sztucznie, jeżeli w czasie produkcji czy przechowywania miały one kontakt
z wodą lub materiałami wilgotnymi. Woda w powietrzu, i w innych gazach, znajduje się
w postaci pary. Zawartość wody w powietrzu wpływa na zawartość wilgoci w materiałach
stykających się z powietrzem, co ma wpływ na właściwości tych materiałów. Zawartość
wody w gazach jest ograniczona zjawiskiem tzw. nasycania się gazu. Gaz jest nasycony parą
wodną, jeżeli jej ciśnienie cząstkowe jest równe ciśnieniu pary nasyconej nad czystą wodą.
Zawartość wilgoci podaje się w jednostkach tzw. wilgotności bezwzględnej w kg H
2
O/kg
substancji suchej. Wilgotność gazów można wyrażać w jednostkach tzw. wilgotności
względnej. Wilgotność względna jest to stosunek ciśnienia cząstkowego pary wodnej w gazie
do ciśnienia pary wodnej nasyconej w tej samej temperaturze:
%
100
n
P
P
=
ϕ
gdzie:
φ
– wilgotno
ść
wzgl
ę
dna [%]
P – ci
ś
nienie pary wodnej w gazie [Pa]
P
n
– ci
ś
nienie pary wodnej nasyconej w tej samej temperaturze [Pa]
Gdy ci
ś
nienie pary wodnej nasyconej ro
ś
nie ze wzrostem temperatury, wilgotno
ść
wzgl
ę
dna maleje przy niezmiennym ci
ś
nienie pary wodnej w gazie.
Zale
ż
no
ść
mi
ę
dzy wilgotno
ś
ci
ą
wzgl
ę
dn
ą
i bezwzgl
ę
dn
ą
podaje nast
ę
puj
ą
ca zale
ż
no
ść
:
M
P
P
P
M
P
P
P
x
c
n
c
n
02
,
18
02
,
18
⋅
−
=
⋅
−
=
ϕ
ϕ
gdzie: P
c
– ci
ś
nienie całkowite gazu [Pa]
18,02 – masa molowa pary wodnej [kg/kmol]
M – masa molowa gazu [kg/kmol]
Je
ż
eli gaz jest mieszanin
ą
, wówczas przyjmujemy
ś
redni
ą
mas
ę
molow
ą
obliczon
ą
z udziałów molowych poszczególnych składników.
Najprostszym sposobem pomiaru jest przepuszczenie okre
ś
lonej obj
ę
to
ś
ci wilgotnego
gazu przez rurk
ę
lub płuczk
ę
wypełnion
ą
substancj
ą
silnie pochłaniaj
ą
c
ą
wilgo
ć
. Znaj
ą
c mas
ę
naczynia z pochłaniaczem wilgoci przed przepuszczaniem gazu i po przepuszczeniu jego
okre
ś
lonej ilo
ś
ci mo
ż
na łatwo obliczy
ć
wilgotno
ść
ze wzoru:
%
100
0
1
ρ
V
m
m
x
−
=
gdzie: m
0
i m
1
– masa naczynia przed i po pochłanianiu [kg]
V – obj
ę
to
ść
gazu po pochłanianiu[m
3
]
ρ
– g
ę
sto
ść
gazu suchego[kg/m
3
]

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
20
Najczęściej stosowanymi przyrządami do pomiaru wilgotności gazów są: higrometr
włosowy; higrometr kondensacyjny; psychrometr.
Najprostszą metodą oznaczania wilgoci w ciałach stałych jest suszenie ich do stałej masy.
Dla najczęściej badanych materiałów warunki suszenia zostały znormalizowane. Ze zmiany
masy określa się wilgotność w procentach odniesionych do masy materiału suchego lub
wilgotnego. Warunki suszenia należy tak dobierać, aby substancja badana nie ulegała
przemianom z wydzieleniem innych składników niż woda, czy też pochłanianiem składników
atmosfery, w której przeprowadza się suszenie. Metoda suszenia jest dokładna, lecz
pracochłonna i długotrwała.
Dużo szybszymi, chociaż mniej dokładnymi, są metody oparte na pomiarach właściwości
elektrycznych materiałów zależnych od zawartości wilgoci. Najczęściej w tym celu
wykorzystuje się zmiany oporności oraz stałej dielektrycznej materiału. W metodach
mechanicznych mierzy się takie parametry, jak gęstość, lepkość, ciśnienie cząstkowe
i objętość mieszaniny gazów. W metodach akustycznych wykorzystuje się pomiar prędkości
rozchodzenia się ultradźwięków lub ich pochłanianie przez mieszaninę gazów. Inna grupa
metod jest oparta na pomiarze właściwości cieplnych gazu. Oprócz wymienionych metod
stosuje się jeszcze pomiar wielkości elektrochemicznych, optycznych, magnetycznych
i jonizacyjnych, związanych ze składem mieszaniny gazów.
Stosowane przyrządy można podzielić w zależności od rodzaju analizy wykorzystanej do
oznaczania składu mieszaniny. W przypadku przyrządów chemicznych wykorzystuje się
reakcje chemiczne, które powodują zmianę objętości, zabarwienia, temperatury itp.
Analizatory fizykochemiczne są oparte na reakcjach chemicznych, którym towarzyszą
zjawiska fizyczne, np. pomiar ciepła reakcji chemicznej.
Podczas pracy w laboratorium najczęściej stosowanymi metodami pomiaru składu
mieszanin gazowych są: analizy oparte na pomiarze gęstości gazu; analizy oparte na pomiarze
przewodnictwa cieplnego gazu i analizy chemiczne.
4.2.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Co to jest ciśnienie i jaka jest jego jednostka w układzie SI?
2.
Jaki jest podział i s zasady działania przyrządów do pomiaru ciśnienia?
3.
Jak zdefiniujesz pojęcie temperatury?
4.
Czym charakteryzują się podstawowe grupy termometrów?
5.
W jakim celu mierzy się poziom cieczy w zbiorniku?
6.
Co opisuje równanie Bernoulliego?
7.
Na jakie grupy dzielimy przepływomierze?
8.
Co rozumiesz pod pojęciem wilgotności względnej gazów?
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
21
4.2.3. Ćwiczenia
Ćwiczenie 1
Dokonaj pomiaru ciśnienia ciśnieniomierzem naczyniowym z rurką pochyłą.
Związki chemiczne: powietrze.
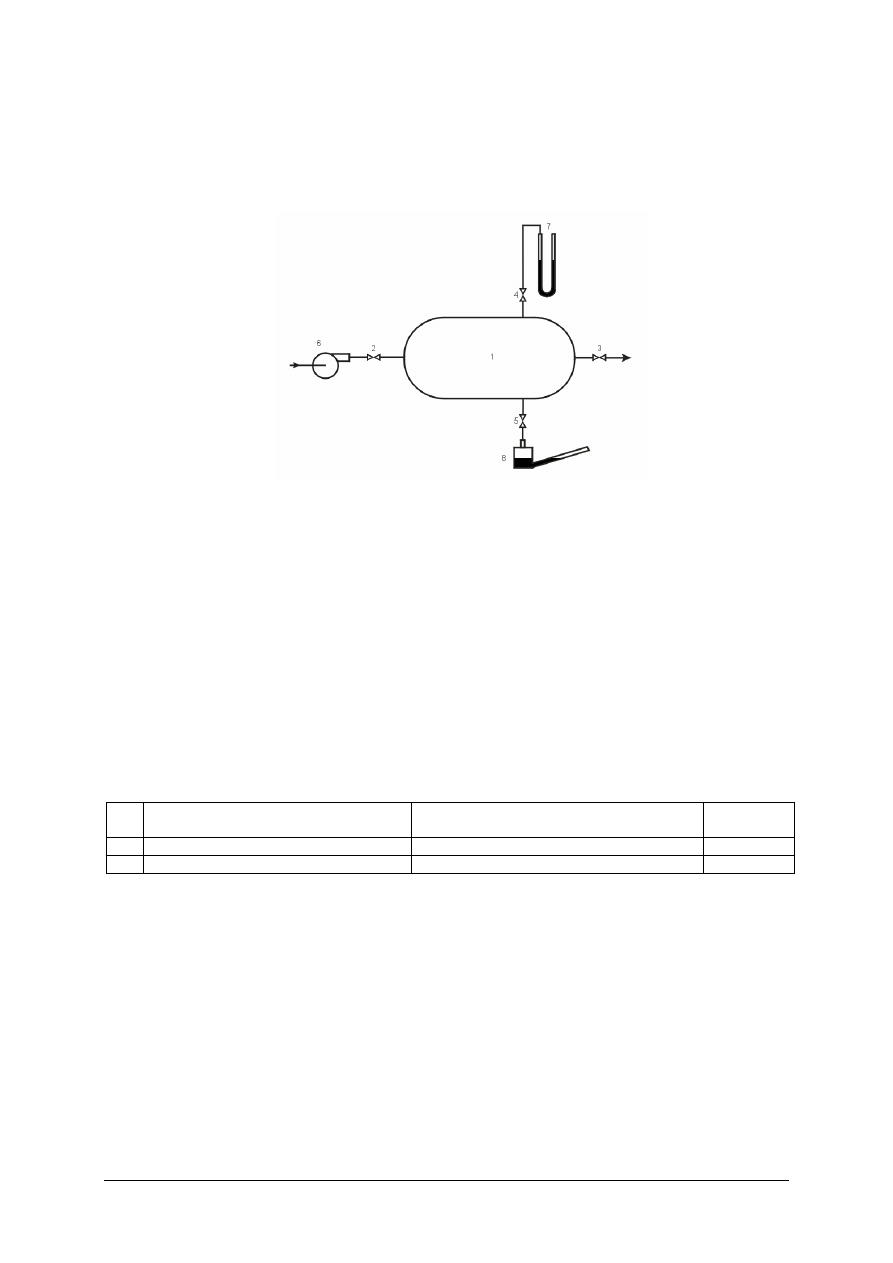
Rysunek do ćwiczenia 1. Schemat układu do pomiaru ciśnienia: 1 – naczynie, 2, 3, 4, 5 – zawory,
6 – wentylator, 7 – ciśnieniomierz U-rurka, 8 – ciśnieniomierz naczyniowy z pochyłą rurką.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
zestawić układ w sposób pokazany na rysunku (ciśnieniomierze połączyć z króćcami
naczynia za pomocą elastycznych przewodów),
2)
otworzyć zawory 2 i 3 i włączyć wentylator. Zawór 3 należy tak ustawić, żeby
wytworzone w naczyniu nadciśnienie nie przekraczało górnego zakresu pomiarowego
ciśnieniomierza z pochylą rurką (sprawdzić to za pomocą U-rurki),
3)
zmierzyć ciśnieniomierzami kilka wartości nadciśnienia. Nadciśnienie w zbiorniku
obniżyć otwierając zawór 3,
4)
uzupełnić tabelę. Porównać wskazania przyrządów. Zinterpretuj wyniki.
Tabela do ćwiczenia 1
Lp.
Ciśnienie odczytane z ciśnieniomierza z
U rurką [mm Hg]
Ciśnienie odczytane z ciśnieniomierza
z rurką pochyłą [mm Hg]
Różnica
wskazań
1.
2.
Wyposażenie stanowiska pracy:
–
naczynie (pojemność ok. 30 dm
3
) z czterema króćcami z zaworami odcinającymi,
–
wentylator,
–
ciśnieniomierze z U rurką i z pochyłą rurką.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
22
Ćwiczenie 2
Określ wilgotność powietrza psychrometrem Assmanna.
Związki chemiczne: woda, powietrze w dwóch pomieszczeniach o różnej wilgotności.
Pomiar wilgotności powietrza prowadzi się psychrometrem aspiracyjnym Assmanna.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
nasycić wodą gazę, którą owinięty jest termometr „mokry” przyrządu,
2)
nakręcić mechanizm sprężynowy, za pomocą klucza, który uruchamia wiatraczek
zasysający powietrze z przestrzeni wokół termometrów,
3)
odczytać temperatury z termometru „suchego” i „mokrego” w momencie, gdy wiatraczek
przestaje się obracać, obliczyć różnicę temperatur i z tabeli psychrometrycznej odczytać
wartość wilgotności względnej w procentach,
4)
wykonać 10 pomiarów wilgotności w różnych miejscach w laboratorium i innym
pomieszczeniu lub na powietrzu poza budynkiem, przedstawić wyniki w tabeli,
5)
podać średnią wilgotność względną w badanych miejscach.
Wyposażenie stanowiska pracy.
–
psychrometr aspiracyjny Assmanna,
–
tabela psychrometryczna,
–
zlewka,
–
kalendarz chemiczny.
4.2.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
wymienić i opisać przyrządy do mierzenia różnego rodzaju ciśnienia?
2)
wymienić
podstawowe
rodzaje
wodowskazów
stosowane
w przemyśle?
3)
opisać zasadę pomiaru przepływu płynu za pomocą zwężki?
4)
wymienić i opisać rodzaje przepływów płynu przez rurociąg?
5)
wykorzystać wykres suszarniczy do określenia wilgotności
powietrza?
6)
przedstawić zasadę pomiaru termometrem oporowym?
7)
opisać budowę i zasadę działania psychrometru?
8)
dokonać pomiaru ciśnienia ciśnieniomierzem naczyniowym z rurką
pochyłą?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
23
4.3. Wybrane zagadnienia analizy instrumentalnej
4.3.1. Materiał nauczania
W przemyśle chemicznym często stosuje się elektrolizę do ilościowego oznaczania
pierwiastków. Metoda ilościowego oznaczania pierwiastków metalicznych, które zostały
wydzielone na katodzie w postaci metalu lub na anodzie w postaci tlenku metalu przy
zastosowaniu elektrolizy, nazywa się analizą elektrograwimetryczną.
Najczęściej ilość pierwiastka wydzielonego na elektrodzie wyznacza się z różnicy między
masą katody przed i po elektrolizie.
Proces elektrolizy zachodzi po zanurzeniu do roztworu elektrolitu dwóch elektrod
i przyłożeniu do nich pewnego napięcia. Jeżeli przyłożone napięcie będzie stopniowo
wzrastało, to obserwuje się, że po przekroczeniu pewnego określonego napięcia zacznie
przepływać prąd. Napięcie to nazywa się napięciem rozkładowym. Niekiedy przy napięciach
mniejszych od napięcia rozkładowego obserwuje się nieznaczny przepływ prądu.
Na podstawie znajomości reakcji elektrodowych można, stosując równanie Nernsta,
wyrazić potencjały reakcji elektrodowych. Całkowite napięcie przyłożone do elektrod
wyrazić wzorem:
iR
E
E
E
anod
kat
+
−
=
.
.
gdzie: E
kat.
– potencjał katody [V]
E
anod.
– potencjał anody [V]
i – natężenie prądu [A]
R – rezystancja (opór) elektrolitu [Ω]
Iloczyn iR odpowiada napięciu potrzebnemu do spowodowania przepływu prądu
o natężeniu i przez elektrolit o rezystancji R (wynika to z prawa Ohma).
Wydzielanie metalu nie zachodzi przy potencjale obliczonym z równania Nernsta, lecz
przy napięciu większym. Różnicę między obliczonym napięciem a rzeczywistym nazywa się
nadnapięciem.
Ilość metalu m [g] wydzielona w czasie elektrolizy t [s] przez prąd o natężeniu i [A]
określona jest pierwszym prawem Faradaya:
kit
m
=
gdzie:
k
.
– równoważnik elektrochemiczny, określający ilość substancji wydzielonej przez
ładunek o wartości 1 kulomba [g/C].
Prawidłowe wykonanie elektrolizy wymaga dobrania odpowiednich warunków
zapewniających ilościowe wydzielenie oznaczanego składnika i odpowiednią jego strukturę.
Elektrolizę przeprowadza się przy takim napięciu, aby natężenie prądu było możliwie
duże. W ten sposób szybkość procesu jest duża. Jednakże zbyt duże napięcie może być
przyczyną wydzielania się drugiego metalu, jeżeli w roztworze jest ich kilka.
Wprowadzenie do roztworu dodatkowej elektrody porównawczej pozwala na mierzenie
potencjału elektrody, na której następuje wydzielanie oznaczanego składnika. Znając napięcie
rozkładowe składników roztworu, można tak regulować przyłożone napięcie, aby nie
przekroczyć napięcia rozkładowego drugiego metalu. Taki sposób przeprowadzania
elektrolizy nazywa się elektrolizą z kontrolowanym potencjałem.
Najczęściej stosuje się elektrolizę do wydzielania jednego składnika roztworu, który
oznacza się następnie wagowo. Niekiedy elektrolizę przeprowadza się w celu usunięcia

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
24
z roztworu wszystkich składników, które wydzielają się przy napięciu mniejszym od
przyłożonego. W ten sposób można rozdzielić metale alkaliczne i metale ziem alkalicznych
od większości metali ciężkich, które wydziela się np. na katodzie rtęciowej.
Natężenie prądu powinno być tak dobrane, aby zapewniało odpowiednią gęstość prądu.
Wzrost gęstości prądu powoduje wzrost szybkości elektrolizy, jednakże zbyt duża gęstość
prądu jest przyczyną powstawania osadu gąbczastego, który łatwo odpada od elektrody
i utlenia się tlenem z powietrza. Prąd o małej gęstości powoduje powstawanie osadów
krystalicznych niepokrywających całej elektrody. Jedynie prąd o średniej gęstości daje osad
ściśle przylegający do elektrody, o powierzchni gładkiej i błyszczącej. Optymalne gęstości
prądu są różne dla różnych metali.
Niekiedy w roztworze powinny znajdować się specjalnie dodane związki chemiczne
przyczyniające się do właściwej struktury osadu, np. miedź tworzy odpowiednie osady
jedynie w przypadku elektrolizy z roztworów zawierających kwas azotowy(V).
Natężenie prądu zależy od oporu wewnętrznego elektrolitu. W celu zmniejszenia tego
oporu stosuje się dodawanie specjalnych elektrolitów, ogrzewanie roztworu, dobieranie
elektrod o odpowiedniej powierzchni i regulowanie odległości pomiędzy elektrodami.
Czas elektrolizy zależy od kształtu elektrolizera i elektrody. Elektroliza może trwać od
kilku do kilkunastu godzin. Czynnikami skracającymi czas elektrolizy są: mieszanie
roztworu, jego ogrzewanie, stosowanie nierozcieńczonych roztworów i elektrod o dużej
powierzchni (tzw. elektroliza przyspieszona). Wymieniane czynniki powodują wzrost
natężenia, a więc i zwiększenie gęstości prądu, co powoduje szybszy przebieg elektrolizy
i zapewniają odpowiednie właściwości osadu.
Zależnie od rodzaju oznaczanego pierwiastka należy dobrać skład roztworu elektrolitu.
Najczęściej przeprowadza się elektrolizę z roztworów zawierających kwas azotowy(V) lub
siarkowy(VI). Jednakże zbyt duże stężenie kwasu może pod koniec elektrolizy spowodować
ponownie rozpuszczanie wydzielonego metalu. Obecność jonów chlorkowych jest
niekorzystna, ponieważ wydziela się wówczas na anodzie chlor, może on powodować
rozpuszczanie się anody lub metalu wydzielonego na katodzie.
W przemyśle chemicznym najczęściej stosuje się elektrody wykonane z platyny. Kształt
takich elektrod może być bardzo różny. Powszechnie stosuje się elektrody w kształcie walca
wykonane z siatki platynowej. Różna średnica walca umożliwia umieszczanie jednej
elektrody wewnątrz drugiej. Elektrodę o mniejszej średnicy można wykorzystywać jako
mieszadło, jednakże wygodniej jest stosować mieszadło magnetyczne. Wykonanie elektrod
z siatki ułatwia mieszanie roztworu.
Niekiedy wydzielany metal, np. cynk, bizmut, tworzy stopy z platyną i niszczy elektrodę.
Wówczas stosuje się elektrody z innego metalu, np. srebra. Można stosować elektrody
z mniej szlachetnych metali, np. do wydzielania miedzi można użyć elektrody miedzianej.
Polarografia jest jedną z elektrochemicznych metod analizy. Polega ona na badaniu zmian
natężenia prądu elektrycznego płynącego przez roztwór elektrolitu w zależności od potencjału
jednej z elektrod. W czasie procesu elektrolizy potencjał drugiej elektrody jest stały (nie ulega
ona polaryzacji). W analizie polarograficznej wyniki pomiarów podaje się w postaci
wykresów przedstawiających zależność natężenia prądu od napięcia (potencjału elektrody)
i znajduje się zależność między natężeniem prądu a składem roztworu.
Polarografia wykazuje wiele zalet. Badania polarograficzne wykonuje się szybko i często
można równocześnie oznaczać kilka substancji. Oznaczenia polarograficzne można powtarzać
kilkakrotnie w tym samym roztworze. Oznaczenia te są w dużym stopniu zautomatyzowane.
Metody polarograficzne są bardzo czułe i precyzyjne, mogą być stosowane do oznaczania
stężeń rządu 10
-2
- 10
-6
mol/1.
Metodami polarograficznymi można oznaczać większość metali, niektóre aniony
i związki organiczne, które ulegają reakcjom elektrodowym. Polarografię zastosowano do

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
25
analizy stopów metali wysokiej czystości, rud i innych materiałów nieorganicznych.
W analizie organicznej polarografia została zastosowana do oznaczania aminokwasów,
cukrów, alkaloidów, witamin, nitrozwiązków i innych substancji.
W polarografii stosuje się: kroplową elektrodę rtęciową i elektrodę porównawczą.
Kroplowa elektroda rtęciowa jest wykonana z grubościennej kapilary szklanej długości
kilkunastu centymetrów. Średnica wewnętrzna takiej kapilary wynosi 0,03–0,05 mm.
Kapilara jest połączona za pomocą węża polietylenowego ze szklanym zbiornikiem na rtęć.
W polarografii elektrodami porównawczymi są elektrody kalomelowe o dużej
powierzchni lub tak zwana elektroda rtęciowa, czyli warstwa rtęci umieszczona na dnie
naczynka polarograficznego połączona za pomocą przewodu elektrycznego z polarografem.
Do łączenia kapilary ze zbiornikiem można stosować wąż gumowy lub z tworzyw
sztucznych. Długość węża należy dobrać tak, aby maksymalna wysokość poziomu rtęci
w zbiorniku w stosunku do wylotu kapilary wynosiła około 50 cm. Z kapilarą polarograficzną
należy obchodzić się ostrożnie i nie dopuścić do jej zanieczyszczenia. Po użyciu kapilarę
należy wyjąć z roztworu i opłukać wodą destylowaną. Przy tych czynnościach z kapilary
powinny wypływać krople rtęci. Po opłukaniu kapilarę umieszcza się na statywie i opuszcza
się zbiornik z rtęcią do takiego poziomu, aby rtęć nie wypływała (kapilarę przechowuje się
w powietrzu, chroniąc ją przed kurzem). Ponowne uruchomienie kapilary następuje przez
podniesienie zbiornika rtęci na wyższy poziom, i umieszczenie kapilary z wypływającymi
kroplami rtęci w badanym roztworze. Należy sprawdzić, czy nie powstały przerwy w rtęci,
co uniemożliwia przepływ prądu przez kapilarę. Przerwy takie należy ostrożnie usunąć.
Elektroda kroplowa wykazuje następujące zalety w porównaniu z innymi elektrodami:
−
powierzchnia jej jest stale odnawiana i dlatego nie ma obawy o zmianę jej zachowania się
wskutek osadzania się na jej powierzchni metali, które mogą zmienić charakter elektrody,
−
nowe krople rtęci stykają się ze świeżym elektrolitem w wyniku mieszania roztworu przez
opadające krople,
−
wysokie nadnapięcie wodoru na rtęci powoduje, że praktycznie potencjał wydzielania
wodoru jest większy od potencjału innych jonów,
−
rtęć jest metalem szlachetnym i dlatego nie reaguje z roztworami,
−
elektrodę kroplową zazwyczaj stosuje się w zakresie potencjałów (+0,4)–(-0,7) V
w roztworach kwaśnych. W roztworach obojętnych lub zasadowych elektrodę można
stosować aż do -1,9V.
Do oznaczania małych zawartości substancji można wykorzystać fakt, te tworzą one
z odpowiednimi odczynnikami koloidalne zawiesiny. Jeżeli koloid jest nietrwały, można
zapobiec jego koagulacji i sedymentacji dodając koloidu ochronnego, np. żelatyny.
Trwałe zawiesiny wykazują specjalne właściwości optyczne. Wiązka promieniowania
padającego na mętny ośrodek zostaje tylko częściowo przepuszczona. Reszta ulega absorpcji
i rozproszeniu (zjawisko Tyndalla).
Turbidymetria zajmuje się pomiarami stosunku natężenia promieniowania padającego
I
0
do natężenia I
t
promieniowania, które przeszło przez mętny ośrodek, przy czym I
t
jest
odwrotnie proporcjonalne do ilości zawiesiny, a zatem do stężenia oznaczanego składnika.
Nefelometria opiera się na pomiarach natężenia promieniowania I
n
rozproszonego przez
cząstki ośrodka. Pomiar przeprowadza się na zasadzie porównania wielkości I
n
bądź
z natężeniem I
o
promieniowania padającego, bądź z natężeniem I'
n
promieniowania
rozproszonego przez odpowiedni wzorzec. Równania przedstawiające zależność pomiędzy
zdolnością rozpraszania promieniowania i pochłaniania go przez zawiesinę a stężeniem
substancji i grubością warstwy nie są w turbidymetrii tak proste, jak w przypadkach
opisanych prawami Bouguera-Lamberta-Beera.
Konieczne jest przy otrzymywaniu zawiesiny ścisłe zachowanie identyczności warunków,
które decydują o rozmiarach cząstek koloidalnych, a więc wpływają na wartość natężenia

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
26
promieniowania rozproszonego. Wielkość cząstek zawiesiny zależy nie tylko od składu
roztworu, temperatury, pH, sposobu i kolejności dodawania odczynników oraz od czasu,
który upłynął od ich wprowadzenia, ale także od stężenia substancji tworzącej koloidalny
osad.
Pomiary turbidymetryczne można wykonywać za pomocą wszystkich aparatów
stosowanych w spektrofotometrii (kolorymetrii). Należy tylko zaczernić lub osłonić boczne
ściany kiuwet, aby zniwelować ewentualne wtórne odbicie promieniowania rozproszonego.
Oznaczenia turbidymetryczne wykonuje się wizualnie w cylindrach Nesslera lub Hehnera,
albo z zastosowaniem kolorymetrów i spektrofotometrów na światło widzialne.
Pomiary nefelometryczne wymagają przyrządów lub aparatury zbudowanej nieco inaczej.
Najprostszą wizualną metodą nefelometryczną jest porównywanie ze skalą wzorców, można
w tym celu stosować cylindry kolorymetryczne, wstawione do zaczernionego wewnątrz
pudełka, z wyciętymi otworami w bocznej ścianie. Przez te otwory obserwuje się zawiesiny
umieszczone w cylindrach. W turbidymetrii i w nefelometrii stosuje się technikę
porównywania ze skalą wzorców i metodę krzywej wzorcowej.
Trudność otrzymania zawsze identycznych zawiesin sprawia, że metody turbidymetryczne
i nefelometryczne nie dają precyzyjnych wyników. Najczęściej stosuje się je w wersji
wizualnej do półilościowych oznaczeń lub do sprawdzenia, czy zawartość zanieczyszczającej
preparat substancji nie przekracza wartości dopuszczalnej według odpowiedniej normy.
Wykonuje się wówczas porównawczy pomiar turbidymetryczny lub nefelometryczny.
Wzorcem jest substancja zawierająca dopuszczalną ilość zanieczyszczenia.
Emisyjna analiza spektralna jest metodą polegającą na określeniu składu chemicznego
substancji na podstawie promieniowania, emitowanego przez wzbudzone atomy i cząsteczki.
Wzbudzenie atomów i cząsteczek zachodzi najczęściej w łuku lub iskrze elektrycznej.
Promieniowanie to rozszczepia się za pomocą pryzmatu lub siatki dyfrakcyjnej na
tzw. widmo. Uzyskane w ten sposób widma mogą być: liniowe (emitowane przez atomy
i jony gazów), pasmowe (emitowane przez cząsteczki w postaci pary) i ciągłe (emitowane
przez ciecze i ciała stałe).
W analizie znalazły zastosowanie jedynie widma liniowe, które są rejestrowane w sposób
fotograficzny za pomocą spektrografów. Analiza emisyjna jest metodą bardzo czułą, szybką
i uniwersalną, jednakże prawidłowe przeprowadzenie analizy nie jest łatwe.
Podczas analizy należy utrzymać jednakowe warunki wzbudzenia (często źródła
wzbudzenia są niestabilne) i rejestracji promieniowania (należy przestrzegać jednorodności
emulsji fotograficznej, jednakowych warunków wywoływania i utrwalania klisz). Istotne są:
wpływ struktury, zmiany składu chemicznego badanych próbek, oraz stosowane wzorce
(wzorce powinny mieć skład maksymalnie zbliżany do składu próbki, aby uniknąć wpływu
innych pierwiastków na wyniki oznaczenia).
Emisyjną analizę spektralną dzielimy na analizę jakościową i ilościową. Spektralna
analiza jakościowa pozwala określić skład jakościowy badanej substancji. Analiza spektralna
jest bardzo prosta w porównaniu z chemiczną analizą jakościową. Przeprowadzenie jej jest
możliwe z bardzo małą ilością substancji badanej (wystarczy 0,1–1 mg). Badana próbka nie
wymaga żadnej obróbki chemicznej. Jedynie w niektórych przypadkach skład chemiczny
próbki może być taki, że nie wszystkie pierwiastki ulegną wzbudzeniu. Spektralna analiza
ilościowa korzysta z zależności między stężeniem danego pierwiastka w materiale badanym
a natężeniem promieniowania emitowanego przez atomy tego pierwiastka po ich wzbudzeniu.
Fotometria płomieniowa jest metodą analizy spektralnej, polegającej na pomiarze
intensywności promieniowania atomów, które są wzbudzane w płomieniu. Na podstawie
intensywności promieniowania można wnioskować o ilości pierwiastka w badanej próbce.
Procesy zachodzące podczas wzbudzania atomów w płomieniu są takie same jak podczas
wzbudzania atomów w analizie spektralnej. Wielkość potencjału wzbudzenia linii ostatnich

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
27
jest podstawą podziału pierwiastków na trzy grupy: pierwiastki o niskich potencjałach
wzbudzania (od 1,4 do 3 eV), pierwiastki o średnich potencjałach wzbudzenia (od 3 do
10 eV), pierwiastki o wysokich potencjałach wzbudzenia (od 10 do 35 eV).Te trzy grupy
pierwiastków wymagają różnych źródeł wzbudzania. Grupą pierwszą można wzbudzać
w płomieniu gazowym. Do grupy tej należą głównie metale alkaliczne i metale ziem
alkalicznych. Po wzbudzeniu, emitują one promieniowanie widzialne i wywołują
charakterystyczne zabarwienie płomienia.
Absorpcyjna spektrometria atomowa wykorzystuje fakt, że atomy w stanie gazowym są
nie tylko źródłem promieniowania wzbudzonego, ale również absorbują promieniowanie
wysyłane przez atomy tego samego pierwiastka. Metoda polega na badaniu substancji
przeprowadzonej w stan gazowy (rozpylanie roztworu w płomieniu lub rozkład substancji
w wysokiej temperaturze w specjalnej kuwecie grafitowej) i poddaniu jej działaniu
promieniowania wysyłanego przez lampę z katodą wnękową wykonaną z oznaczanego
metalu. W widmie promieniowania emitowanego przez lampę występują jedynie linie metalu,
z którego jest ona wykonana. Po przejściu promieniowania przez płomień (lub kuwetę) ulega
ono osłabieniu (absorpcji), ponieważ jego część została zużyta na wzbudzenie atomów metalu
znajdujących się w płomienni w stanie podstawowym. Absorpcja promieniowani jest
proporcjonalna do stężenia metalu. Wynik analizy, zależy od. temperatury płomienia. W zbyt
niskiej temperaturze wzbudzeniu ulega tylko mała część atomów i intensywność
promieniowania jest za mała. W zbyt wysokiej temperaturze może w płomieniu nastąpić
jonizacja atomów oznaczanego pierwiastka. Powstające jony spowodują zmniejszenie
stężenia atomów i zmniejszenie natężenia promieniowania (widmo jonów jest inne niż widmo
atomów). Z tego powodu ważny jest odpowiedni dobór składu mieszaniny gazowej, którą jest
zasilany płomień. Najczęściej stosuje się mieszaninę gazu świetlnego z powietrzem, gazu
świetlnego z tlenem, mieszaninę acetylen-powietrze oraz acetylen-tlen.
Od temperatury płomienia zależą procesy zachodzące w płomieniu po wprowadzeniu do
niego badanego roztworu w postaci aerozolu. Drobne kropelki cieczy w płomieniu ulegają
odparowaniu i powstają z nich cząstki stałe, które parują, a powstające pary soli ulegają
dysocjacji termicznej na atomy, które mogą ulec wzbudzeniu, jonizacji lub utworzyć nowe
związki. Przebieg tego procesu zależy od szybkości odparowania roztworu, parowania
cząstek stałych i od stopnia rozpylenia cieczy wprowadzanej do płomienia. Również skład
chemiczny roztworu ma wpływ na wyniki. Jeżeli roztwór zawiera dużą ilość soli trudno
lotnych, to obserwowana intensywność promieniowania będzie mniejsza niż w przypadku
roztworu o tym samym stężeniu oznaczanego pierwiastka dla soli lotnych. Podobnie
wzbudzenie pierwiastków zależy od anionów zawartych w roztworze. Najłatwiej wzbudzeniu
ulegają siarczany metali alkalicznych. Chlorki tych metali wzbudzane są trudniej, a jeszcze
słabiej wzbudzane są fosforany. Efekt wpływu anionu obserwuje się w płomieniach
o niższych temperaturach.
W momencie kiedy temperatura płomienia jest odpowiednio wysoka, to efekt ten może
nie wystąpić. Wpływ anionów na intensywność promieniowania zależy od stężenia anionów.
Wpływu negatywnych czynników można uniknąć. W tym celu pomiary intensywności
promieniowania przeprowadza się dla roztworów o zbliżonym składzie chemicznym oraz
stosując stałą temperaturę płomienia (reguluje się ją w pewnym zakresie, zmieniając ciśnienie
gazu palnego i utleniacza doprowadzanych do palnika). W przypadku roztworów
o nieznanym składzie chemicznym najlepiej jest wykonywać oznaczenie fotometryczne
metodą dodawania wzorca.
Najistotniejszym parametrem opracowania metod absorpcyjnej spektrometrii atomowej
do oznaczania pierwiastków jest dobór linii analitycznych, na których opiera się metoda.
Wybór ten należy przeprowadzić pod kątem uzyskania największej możliwej absorpcji oraz
najmniejszych zakłóceń. W przypadku metali alkalicznych są to linie rezonansowe, te same,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
28
które dają najlepsze wyniki w emisji. W przypadku innych metali o bardziej
skomplikowanym widmie do pomiarów absorpcji niekoniecznie najlepsze są linie dające
najlepszą emisję. Oznaczalność metod absorpcji atomowej można poprawić stosując lampy
o większej intensywności, zmniejszając tło, które powoduje płomień, przez jego odpowiedni
dobór i zwiększając ilość roztworu podawaną do płomienia w jednostce czasu. Również
wybór właściwej strefy
w płomieniu i odpowiedni dobór szczeliny wpływa na nachylenie krzywej. Tło w stosunku do
mierzonego sygnału jest związane z określoną aparaturą, stałością pracy wzmacniacza
i zasilaczy oraz stałością pracy rozpylacza.
Metoda absorpcyjnej spektrometrii atomowej jest szeroko stosowana do oznaczania
śladowych zawartości wszystkich metali. Jest bardzo czułą metodą i służy do oznaczania
bardzo wielu pierwiastków.
Podstawą spektroskopii magnetycznej rezonansu jądrowego (NMR – z ang. Nuclear
Magnetic Resonance) jest zdolność absorbowania promieniowania o radiowej częstości drgań
(10
6
–10
8
Hz) przez jądra izotopów, mające moment magnetyczny, umieszczone w silnym
zewnętrznym polu magnetycznym (10000–25000 Gs).
Absorpcja promieniowania przez jądro daje sygnał NMR, którego natężenie jest
proporcjonalne do ilości zaabsorbowanego promieniowania, zależnej od całkowitej liczby
jąder. Prawdopodobieństwo zaabsorbowania promieniowania przez jądro jest jednak
stosunkowo małe, stąd metoda NMR jest mało czuła w porównaniu z innymi metodami
spektrofotometrii absorpcyjnej. Z analitycznego punktu widzenia ważne jest natężenie linii
NMR, czyli powierzchnia zawarta pod pikiem absorpcji. Częstość drgań promieniowania
absorbowanego w procesie NMR przy określonej wartość. H
0
zależy nie tylko od samego
jądra, lecz także od chemicznego otoczenia jądra oraz od tzw. sprzężenia spin-spin.
Pierwsza zmiana częstości drgań absorbowanych, zwana przesunięciem chemicznym,
wywołana jest ekranującym działaniem chmury elektronowej otaczającej jądro. Wirując
przeciwdziała ona zewnętrznemu polu magnetycznemu i powoduje, że jego natężenie się
zmniejsza. Przesunięcie chemiczne jest szczególnie silne, gdy w pobliżu protonów znajdują
się wiązania zawierające elektrony π, np. pierścień benzenowy, wiązania C=O, C=C, C≡C.
Przesunięcia chemiczne są charakterystyczne dla danej konfiguracji wokół jądra, są one
wykorzystywane w identyfikacjach substancji za pomocą metody NMR. Sprzężenie spin-spin
powoduje rozszczepienie linii NMR na dublety lub multiplety, a jest ono spowodowane
oddziaływaniem
poprzez
wiązanie
między
sąsiednimi
jądrowymi
momentami
magnetycznymi, co powoduje rozszczepienie poziomu energii jądra. Zamiast jednego
przejścia energetycznego, dającego jedną linię absorpcji, obserwuje się dwa lub więcej
przejść dających odpowiednio dublet lub multiplet. Zjawisko to jest również wykorzystywane
do identyfikacji określonych konfiguracji atomów. Przesunięcia chemiczne i rozszczepienia
spowodowane sprzężeniem spin-spin stanowią podstawę widma NMR cząsteczki.
Przesunięcia te obserwuje się w praktyce w stosunku do sygnału cząsteczki, dającej tylko
jedną linię, przyjętego umownie jako zerowy. Jedną z powszechnie w tym celu przyjętych
substancji jest czterometylosilan (TMS).
Analiza (spektroskopia) masowa wykorzystuje różnice w poruszaniu się zjonizowanych
fragmentów próbki w silnym polu magnetycznym. W polu tym jony są odchylane
proporcjonalnie do stosunku ich masy m do ładunku elektrycznego e. Jeśli na drodze jonów
ustawimy detektor, którym może być np. płyta fotograficzna, jony o różnych wartościach m/e
będą padać na różne miejsca płyty, powodując zaczernienie zależne od liczby jonów
o jednakowym stosunku m/e. Otrzymuje się w ten sposób widmo masowe.
Sprawne działanie spektrometru masowego wymaga pracy w wysokiej próżni, aby
wyeliminować obecność jonów tworzących się ze składników powietrza, dokładnej
stabilizacji procesu jonizacji próbki oraz dobrych detektorów. Nowoczesna aparatura do

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
29
spektroskopii masowej składa się z trzech zasadniczych bloków: urządzenia do jonizacji
(źródła jonów), analizatora mas i detektora. Dochodzą do tego urządzenia do wprowadzania
próbki i urządzenia do rejestracji wskazań detektora. Rozróżnia się cztery typy urządzeń do
jonizacji: termiczne, elektronowe, przez bombardowanie jonami i iskrowe próżniowe.
W przemyśle są głównie stosowane magnetyczne analizatory, (z pojedynczym lub
podwójnym ogniskowaniem), analizatory oparte na pomiarze czasu przelotu i analizatory
kwadrupolowe (omegatrony). Detektory stosowane w przemyśle to przede wszystkim
elektrometry, powielacze elektronowe i fotopowielacze oraz płyty fotograficzne.
Podstawą stosowania metod radiometrycznych w analizie jest pomiar promieniowania
α, β lub γ, emitowanego przez naturalne lub sztuczne izotopy promieniowaniotwórcze
pierwiastków. Izotopy znalazły różnorodne zastosowanie do kontroli procesów
technologicznych i analizy. Pod wpływem naświetlenia materiału badanego neutronami
powstają w nim izotopy promieniotwórcze pierwiastków, z których składa się dany materiał.
Zjawisko to wykorzystuje się w analizie aktywacyjnej. Ilość powstających atomów izotopu
promieniotwórczego jest wprost proporcjonalna do zawartości pierwiastka w badanym
materiale. Przy zachowaniu stałych warunków pomiaru można porównywać próbki o różnej
zawartości pierwiastka. Ilość powstałego izotopu określa się mierząc jego promieniowanie.
Najczęściej stosuje się pomiar za pomocą licznika scyntylacyjnego. Ponieważ w wyniku
naświetlenia powstają równocześnie izotopy różnych pierwiastków, to układ pomiarowy
powinien umożliwiać eliminację promieniowania innych izotopów. Przyrządem takim jest
spektrometr gamma, który pozwala na analizę wieloskładnikowej mieszaniny izotopów.
Oprócz badanej próbki równocześnie naświetla się wzorce o znanej zawartości oznaczanych
pierwiastków. Następnie mierzy się aktywność powstałych izotopów. Zawartość oznaczanego
pierwiastka oblicza się ze wzoru
I
I
m
m
x
x
=
gdzie: m
x
i m – zawartość oznaczanego pierwiastka w próbce badanej i wzorcu
I
x
i I – aktywność powstałego izotopu w próbce badanej i wzorcu
Oznaczanie tą metodą wymaga przestrzegania wielu warunków. Metodą tą można
oznaczyć wiele pierwiastków z dokładnością do kilku procent, jest to metoda bardzo czuła
i w niektórych przypadkach można nawet oznaczać 10
-9
–10
-12
g takich pierwiastków, jak glin,
arsen, ren, tantal.
Przyrządy do pomiarów radiometrycznych umożliwiają ilościowy pomiar pierwiastków
promieniotwórczych. Pomiar taki opiera się na rejestracji promieniowania α, β lub γ
wysyłanego
podczas
rozpadu
przez
atomy
pierwiastków
promieniotwórczych.
Promieniowanie to przechodząc przez materię wywołuje w niej zmiany. Pojawiają się cząstki
naładowane elektrycznie (jony i elektrony). Jeżeli proces zachodzi w gazie, to gaz przewodzi
prąd. Przepływ prądu jest rejestrowany w detektorze promieniowania. Przyrządy umożliwiają
rejestrację poszczególnych rozpadów. Jedna cząsteczka α, β lub γ jest rejestrowana przez
przyrząd jako jeden impuls. Liczba zarejestrowanych impulsów odpowiada liczbie
cząsteczek, które dotarły do przyrządu rejestrującego. W badanej próbce podaje się
zarejestrowaną liczbę impulsów w jednostce czasu (imp./min). Szybkość liczenia jest zawsze
mniejsza od liczby rozpadów atomów dlatego, że do przyrządu rejestrującego dociera tylko
część promieniowania wysyłanego przez próbkę. Przy pomiarze radiometrycznym, oprócz
warunku zachowania jednakowego położenia próbki względem przyrządu rejestrującego
(jest to tzw. geometria pomiaru), należy brać pod uwagę możliwość absorpcji
promieniowania w materiale próbki (samoabsorpcja) i na drodze od powierzchni próbki do
przyrządu rejestrującego (absorpcja promieniowania itp.).

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
30
Przyrząd do pomiarów radiometrycznych rejestruje promieniowanie spowodowane
zanieczyszczeniami promieniotwórczymi powietrza oraz promieniowaniem kosmicznym.
Zarejestrowane z tego powodu promieniowanie nazywa się tłem. Wartość tła odejmuje się od
wyniku pomiaru badanej próbki. W celu uzyskania porównywalnych wyników, należy:
przestrzegać tych samych warunków geometrycznych pomiarów, mierzyć próbki o takiej
samej samoabsorpcji, stosować tę samą aparaturę. Błąd uzyskanych wyników można ocenić
metodami statystycznymi. W odpowiednich warunkach można mierzyć aktywność próbek
gazowych i ciekłych. Najczęściej mierzy się aktywność preparatów stałych.
Chromatografia jest jedną z metod rozdzielania mieszanin substancji ciekłych i gazowych.
Chromatografię stosuje się do wydzielania zanieczyszczeń w celu ich oznaczenia.
W technologii chemicznej stosuje się chromatografię m.in. do otrzymywania bardzo czystej
wody, rozdzielania metali o podobnych właściwościach, np. ziem rzadkich.
W chromatografii wykorzystuje się różnice w podziale substancji między dwie różne
fazy: fazę ruchomą (ciecz lub gaz) i fazę nieruchomą (stałą).Niekiedy faza nieruchoma jest
ciałem stałym z trwale zaabsorbowaną cieczą. Wówczas faza ruchoma powinna być cieczą
niemieszającą się z cieczą zaadsorbowaną na powierzchni ciała stałego.
Chromatografia
adsorpcyjna
wykorzystuje
różnice
zdolności
do
adsorpcji
poszczególnych składników mieszaniny na odpowiednio dobranej fazie nieruchomej, zwanej
sorbentem.
Chromatografia rozdzielcza (podziałowa) polega na podziale składników mieszaniny
między dwie niemieszające się ciecze, z których jedna zatrzymana jest na specjalnym nośniku
(np. bibuła, żel krzemionkowy), a druga przepływa przez nośnik.
Chromatografia osadowa (chemichromatografia) wykorzystuje reakcje chemiczne, jakie
mogą zachodzić pomiędzy sorbentem a składnikami mieszaniny.
Chromatografia jonowymienna polega na wymianie jonów zawartych w roztworze
z jonami wchodzącymi w skład tzw. jonitów. W wyniku tego procesu następuje wydzielenie
jonów z roztworu i zastąpienie ich innymi jonami z jonitów.
Stosuje się dodatkowe czynniki zwiększające zdolności rozdzielcze poszczególnych
układów. Stosując przykładanie odpowiednio dobranego napięcia wywołuje się migrację
jonów, co znalazło zastosowanie w jonoforezie bibułowej i elektrochromatografii. Można
stosować zmiany temperatury wzdłuż kolumny chromatograficznej (termochromatografia).
Rozróżniamy różne techniki (wykonywania) rozdzielania metodą chromatograficzną.
Do tego celu najczęściej stosuje się metody kolumnowe i cienkowarstwowe. W metodzie
kolumnowej stosuje się kolumny napełnione sorbentem w postaci ziaren. Przez kolumnę
przepuszcza się ciecz (lub gaz) badaną, a następnie odpowiednie roztwory (lub gaz nośny),
które powodują rozdzielanie składników zatrzymanych przez sorbent i wymycie ich
z kolumny.
W wyniku tego procesu otrzymuje się barwne warstwy na sorbencie lub roztwory zawierające
poszczególne składniki mieszaniny rozdzielanej.
W metodach cienkowarstwowych stosuje się najczęściej bibułę lub sorbent naniesiony
np. na szkło w postaci cienkiej warstwy. W tym przypadku procesy rozdzielania zachodzą na
powierzchni
sorbentu
w
wyniku
poruszania
się
cieczy
siłami
kapilarnymi.
Po przeprowadzeniu rozdzielenia powierzchnię sorbenta najczęściej spryskuje się specjalnymi
roztworami, które z zaabsorbowanymi składnikami mieszaniny badanej tworzą związki
barwne. Możliwe są również inne sposoby wykrywania rozdzielonych substancji.
Do rozdzielania chromatograficznego mieszanin gazowych stosuje się specjalne kolumny.
Szczególnym przypadkiem chromatografii rozdzielczej jest chromatografia bibułowa. W tym
przypadku rolę nośnika spełnia bibuła. Unieruchomioną fazą jest roztwór wody zawierający
rozdzielane składniki. Woda unieruchamiana jest na bibule w wyniku adsorpcji na celulozie.
Rozpuszczalnikiem ruchomym są najczęściej rozpuszczalniki organiczne (niemieszające się

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
31
z wodą). Rozpuszczalnik taki wciągany jest siłami kapilarnymi na bibułę. Przesuwając się nad
unieruchomioną fazą wodną, rozpuszczalnik organiczny w wyniku wielokrotnie
powtarzającego się procesu ekstrakcji powoduje rozdzielenie składników zawartych
w roztworze wodnym.
Ważne jest nanoszenie próbki na bibułę. Linię startu- miejsce nanoszenia -zaznacza się
ołówkiem (odległe od brzegu arkusza o około 2 cm, punkty nanoszenia są oddalone od siebie
o około 2 cm). Na zaznaczone miejsca nanosi się kroplę badanego roztworu za pomocą
pipetki. Następną kroplę nanosi się po wysuszeniu poprzedniej. Potem wykonuje się
rozwinięcie chromatogramu w tzw. komorach chromatograficznych. Komory powinny być
szczelne, co zapewnia stałość atmosfery, nasyconej parami rozpuszczalników. Komora
chromatograficzna powinna być nasycona parami rozpuszczalników w ciągu kilku godzin
przed użyciem. Rozwijanie chromatogramu można podzielić na metodę zstępującą
i wstępującą.
4.3.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1.
Jakie warunki należy spełnić w pomiarach elektrograwimetrycznych, aby ilościowo
oznaczyć metal?
2.
Jakie elektrody stosuje się w polarografii i jakie mają zalety?
3.
Jakie zjawiska są podstawą analizy turbidymetrycznej (nefelometrycznej)?
4.
Czym różni się ilościowa analiza spektralna od jakościowej analizy spektralnej?
5.
Jakie czynniki wpływają na wynik badania absorpcyjnej spektrometrii atomowej?
6.
Jak prowadzi się analizę z wykorzystaniem NMR?
7.
Jakie detektory stosuje się w analizie masowej wykorzystywanej w przemyśle?
8.
Jakie rodzaje promieniowania wykorzystywane są w pomiarach radiometrycznych?
4.3.3. Ćwiczenia
Ćwiczenie 1
Oznacz elektrolityczne miedź, ołów i cynk w stopie.
Elektrolityczne oznaczanie miedzi i ołowiu może zachodzić równocześnie. Miedź wydziela
się z roztworu kwasu azotowego (V) na katodzie, a w tych samych warunkach ołów wydziela
się na anodzie w postaci dwutlenku ołowiu. Stężony kwas azotowy (V) wpływa korzystnie na
strukturę wydzielanego metalu, jednak zbyt duże stężenie kwasu może spowodować jej
rozpuszczenie. Pod koniec elektrolizy roztwór rozcieńcza się i częściowo zobojętnia
amoniakiem. Ołów oznacza się w ilościach 100–120
mg PbO
2,
gdyż
przy większych ilościach
osad może odpadać od elektrody. Cynk z roztworów kwaśnych nie wydziela się. Po
wydzieleniu miedzi i ołowiu można wydzielić cynk z roztworu zasadowego (azotany
i amoniak wpływają ujemnie na oznaczanie i dlatego usuwa się je). Wydzielony cynk może
spowodować zniszczenie elektrody platynowej, dlatego też stosuje się elektrodę platynową,
pokrytą elektrolitycznie miedzią lub srebrem.
Związki chemiczne; mosiądz, stężony kwas azotowy (V), stężony kwas siarkowy (VI),
granulowany wodorotlenek sodu, 25% wodny roztwór amoniaku, 10% roztwór siarczku
amonu, aceton, odczynniki do jakościowego sprawdzania oznaczanych kationów.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
odważyć 0,5–0,7 g mosiądzu (bez cyny), przenieść odważkę do zlewki, dodać 15 cm
3
wody i 5–7
cm
3
kwasu azotowego (V),

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
32
2)
stop roztworzyć, ogrzewając roztwór aż do odpędzenia tlenków azotu i rozcieńczyć wodą
do objętości 30 cm
3
.
3)
umieścić zważone elektrody w roztworze, ogrzać do temperatury około 70°C
i przepuszczać prąd o napięciu 3–4 V i gęstości 2 A/100 cm
2
.
4)
po upływie pewnego czasu (zależnie od zawartości ołowiu), dodać 10 cm
3
wody,
(na nowo pokrytej roztworem części anody nie powinien wydzielać się tlenek ołowiu),
5)
roztwór po zakończeniu wydzielania ołowiu rozcieńczyć wodą do 100 cm
3
, częściowo
zobojętnić kwas azotowy (V) wodą amoniakalną (przyspieszasz wydzielenie się miedzi),
6)
sprawdzić, czy miedź wydzieliła się ilościowo, wyjąć elektrody ze zlewki, przemyć je
wodą (wodą z przemycia dołączyć do roztworu po elektrolizie) i acetonem,
7)
wysuszyć i zważyć elektrody. Gdy wydzielona miedź jest zanieczyszczona (barwa
brunatna), musisz ją rozpuścić w kwasie azotowym (V) i ponownie wykonać oznaczanie.
8)
po wydzieleniu miedzi i ołowiu zadać 5 cm
3
kwasu siarkowego (VI) i odparować w celu
usunięcia kwasu azotowego (V),
9)
rozcieńczyć roztwór do 100 cm
3
wodą, dodać 4–6 g wodorotlenku sodu (roztwór
powinien mieć odczyn zasadowy) i ogrzać w celu odparowania amoniaku (powstał on
w wyniku redukcji kwasu azotowego (V),
10)
zanurzyć elektrody do roztworu (katoda powinna być pokryta miedzią tak, aby
wydzielony cynk nie stykał się z platyną, katoda powinna być zważona),
11)
ustalić napięcie prądu, aby przez roztwór przepływał prąd o natężeniu 1 A; po upływie 10
minut zwiększyć natężenie prądu do 2 A,
12)
po upływie 30 minut, pobrać pipetką, kilka kropel roztworu dodać kroplę roztworu
siarczku amon; jeżeli cynk wydzielił się ilościowo, nie wytrąca się biały siarczek cynku.
13)
po wydzieleniu cynku przemyć elektrody wodą i acetonem, ostrożnie wysuszyć i zważyć
14)
obliczyć procentową zawartość miedzi i cynku na podstawie przyrostu masy elektrody:
[%]
100
a
b
x
=
gdzie:
a
– odwa
ż
ka mosi
ą
dzu [g]
b
– masa wydzielonego metalu [g]
15)
obliczy
ć
procentow
ą
zawarto
ść
ołowiu na podstawie wzoru:
[%]
100
8662
,
0
a
b
x
⋅
=
gdzie:
a
– odwa
ż
ka mosi
ą
dzu [g]
b
– masa wydzielonego dwutlenku ołowiu [g]
0,8662
– mno
ż
nik analityczny do przeliczenia masy PbO
2
na Pb
Wyposa
ż
enie stanowiska pracy:
−
elektrolizer z dwoma elektrodami platynowymi,
−
waga analityczna,
−
szkło: zlewka, pipety, naczynko wagowe, bagietka, tryskawka,
−
maszynka do ogrzewania z płytk
ą
izolacyjn
ą
,
−
suszarka,
−
kalendarz chemiczny.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
33
Ćwiczenie 2
Rozdziel mieszaninę jonów Mn
2+
, Co
2+
i Ni
2+
Stosując technikę bibułową można rozdzielić mieszaninę kationów będącą półproduktem
w technologii otrzymywania odczynników chemicznych. Rozdzielenie jonów i położenie
plam na bibule zależy od składu eluentu. Jony manganu, kobaltu i niklu można po naniesieniu
na bibułę rozdzielić stosując alkohol n-butylowy. Otrzymane plamy są rozmyte.
Wprowadzenie kwasu solnego do alkoholu n-butylowego powoduje zmianę warunków
adsorpcji kationów na bibule. Powstają wówczas wyraźne i rozgraniczone plamy.
Związki chemiczne: roztwór manganu, niklu i kobaltu: odważyć 0,2025 g NiCl
2
·6H
2
O,
0,1801 g MnCl
2
·H
2
O i 0,2019 g CoCl
2
·H
2
O i rozpuścić w małej objętości wody, przenieść do
kolby pomiarowej o pojemności 100 cm
3
dodać 1 cm
3
kwasu solnego i dopełnić wodą
destylowaną do kreski; roztwór wymieszać; w 1 cm
3
tego roztworu znajduje się 0,5 mg
manganu, niklu i kobaltu, alkohol n-butylowy, stężony kwas solny, 1% roztwór alkoholowy
dimetyloglioksymu, 1% roztwór alkoholowy tiocyjanianu amonu, 1% roztwór azotanu (V)
srebra, 6 mol/dm
3
wodny roztwór amoniaku.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
wyciąć trzy paski bibuły chromatograficznej o wymiarach 3x30 cm,
2)
zaznaczyć linię startu w odległości 1,5 cm od brzegu i nanieść po kropli roztworu
manganu, niklu i kobaltu, a następnie wysuszyć,
3)
umieścić paski bibuły w cylindrach, w których na dnie znajduje się mieszanina alkoholu
n-butylowego i kwasu solnego w stosunku: a) 4:1, b) 4:1,7 i c) 4:2,5; paski zanurzyć
plamą do dołu tak, aby plama nie była zanurzona w rozpuszczalniku (linia startu musi
być powyżej poziomu rozpuszczalnika),
4)
przykryć cylindry szkłem (można zastosować wysoką zlewkę),
5)
przerwać wymywanie, gdy czoło rozpuszczalnika podniesie się na wysokość 25 cm;
wyjąć paski bibuły; wysuszyć je; spryskać je kolejno roztworem dimetyloglioksymu
(wywołuje plamę niklu), roztworem tiocyjanianu amonu (wywołuje plamę kobaltu),
roztworem azotanu (V) srebra, a następnie roztworem amoniaku (wywołuje plamę
manganu) spryskiwanie musisz wykonać pod wyciągiem.
Aby opracować wyniki, powinieneś:
1)
zmierzyć odległość od linii startu do środka plamy każdego jonu i wyznaczyć
współczynnik R
f,
2)
ocenić zarys plam i odległość między plamami, na tej podstawie wybrać najlepszy eluent
spośród stosowanych,
3)
zapisać spostrzeżenia i wnioski.
Wyposażenie stanowiska pracy.
–
bibuła chromatograficzna,
–
cylindry miarowe lub komora chromatograficzna z pokrywą,
–
szkło laboratoryjne: zlewki, bagietka, naczynka wagowe, kolby miarowe na 100 cm
3
,
tryskawka, naczynia do nanoszenia wywoływaczy, pipety i wkraplacze,
–
waga analityczna,
–
suszarka do włosów,
–
kalendarz chemiczny.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
34
4.3.4.
Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
skorzystać z prawa Faradaya w obliczeniach teoretycznej ilości
produktu przy podanych parametrach pomiarowych?
2)
przeprowadzić jakościową analizę polarograficzną mieszaniny
wybranych metali w roztworze?
3)
przeprowadzić
analizę
turbidymetryczną
lub
nefelometryczną
układów naturalnie mętnych np. ścieków przemysłowych?
4)
oznaczyć jakościowo mieszaninę wybranych kationów metodą
chromatografii cienkowarstwowej?
5)
omówić podstawowy analizy spektroskopii masowej?
6)
przeprowadzić pomiar tła w analizie radiometrycznej?
7)
scharakteryzować
metodę
badania
absorpcyjnej
spektrometrii
atomowej?
8)
scharakteryzować jakościową analizę spektralną?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
35
4.4.
Wybrane zagadnienia z kontroli procesów technologicznych
4.4.1 Materiał nauczania
Technologia jest nauką o przekształcaniu surowców w produkty o pożądanych
wartościach użytkowych. Głównym jej zadaniem jest wskazanie najwłaściwszego sposobu
przeprowadzenia procesów produkcyjnych. Podstawy procesowe technologii stosowanych
w przemyśle wynikają z praw fizyki i chemii. Istotny jest optymalny dobór technologii
wytwarzania lub przetwarzania uwzględniający warunki realizacji przy minimum kosztów
ogólnych. Materiały stosowane w technologii chemicznej i przetwórstwa to: surowce,
półprodukty, produkty będące substancjami prostymi lub związkami chemicznymi. W wyniku
przetwarzania surowców wytwarza się półprodukty i produkty stosowane we wszystkich
dziedzinach życia. Przetwarzanie polegać może jedynie na zmianie czyli modyfikacji cech
fizycznych. Przebieg procesów technologicznych wymaga przestrzegania zasad: najlepszej
dostępnej techniki, w tym najlepszego wykorzystania surowców, energii, aparatury
procesowej oraz zachowania umiaru technologicznego przy wytwarzaniu produktów
użytecznych niewywierających negatywnego oddziaływania na środowisko.
Opłacalność procesu technologicznego zależy od dobrego wykorzystania surowców, co
osiąga się dzięki przestrzeganiu zasad:
1.
zasada przeciwprądu materiałowego – stosuje się do układów, w których występuje
granica rozdziału faz, a fazy różnią się znacznie gęstością. Zapewnia uzyskanie dużej
szybkości przenikania masy,
2.
zasada maksymalnego wykorzystania produktów ubocznych – decyduje o wskaźnikach
ekonomicznych procesu, gdyż poprawia wykorzystanie surowców i nie komplikują
gospodarki odpadami. Produkty uboczne powstają również w wyniku niepożądanych
reakcji ubocznych. Dąży się do stosowania technologii bezodpadowych (z pełnym
wykorzystaniem produktów ubocznych) lub mało odpadowych,
3.
zasada indywidualnego regulowania szybkości procesów głównych i ubocznych –
stosowana, gdy zachodzi kilka procesów. Wymaga dobrania takich warunków, aby
przebiegał najszybciej główny proces, przy niezmienionej szybkości procesów
pozostałych,
4.
zasada regeneracji materiałów – w przypadku stosowania w procesie surowców
pomocniczych ważne jest ich odzyskanie i zawrócenie do procesu (regeneracja). Małe
straty w obiegu surowca pomocniczego obniżają jego zużycie i poprawiają opłacalność
procesu głównego.
Problem najlepszego wykorzystania energii rozwiązuje się w oparciu o bilans
energetyczny procesu, tak jak wykorzystanie surowców w oparciu o bilans materiałowy.
Podstawowe zasady:
1.
zasada odzysku ciepła – pozwala zmniejszyć koszty procesu. Klasycznym przykładem są
wymienniki ciepła, w których ciepło strumieni płynów chłodzonych wykorzystuje się do
jednoczesnego ogrzania innych płynów przepływających w drugiej przestrzeni
wymiennika.
2.
zasada przeciwprądu cieplnego – pozwala maksymalnie wykorzystać cenną energię np.
ogrzać płyn do temperatury wyższej lub ochłodzić do temperatury niższej niż przy
współprądzie. W wymiennikach przenikanie ciepła zachodzi przez ściankę oddzielającą
płyny, zatem nie występują ograniczenia powierzchni międzyfazowej.
3.
zasada wykonywania tylko prac niezbędnych – zapobiega marnowaniu energii na zbędne
operacje lub czynności, np. na rozdrabnianie materiału już dostatecznie rozkruszonego.
Procesy i operacje technologiczne powinny być tak prowadzone, aby wydajność produkcji
z jednostki objętości aparatu była możliwie duża. Dobre wykorzystanie aparatury osiąga się

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
36
zarówno dzięki odpowiedniemu poziomowi technicznemu produkcji, jak i prawidłowej
organizacji pracy. Podstawową zasadą jest więc zasada ciągłości pracy. W procesach ciągłych
nie ma potrzeby przerywania pracy aparatu na powtarzające się załadowanie surowcami
i opóźnianie z produktów. Proces przebiega aż do planowego remontu aparatu, instalacji lub
w przypadku awarii. O wyborze metody ciągłej lub nie ciągłej (periodycznej) decyduje
rachunek ekonomiczny i możliwości technologiczne. Niekiedy metody periodyczne są
bardziej opłacalne i stosowane w dużej skali (np. w koksowniach).
W wielu procesach występuje oddziaływanie sprzecznych czynników, co zmusza do
dobierania optymalnych parametrów. Np. niska temperatura sprzyja wydajnemu przebiegowi
reakcji, w której powstają kryształy ciała stałego, jednocześnie w niskiej temperaturze
wytrącający się osad jest drobnokrystaliczny i trudno go potem odsączyć. Poszukiwanie
optymalnych parametrów procesu opiera się na jego wnikliwej analizie. Przyjęte wartości
parametrów są zazwyczaj kompromisem pomiędzy wieloma czynnikami sprzecznie
oddziałującymi na proces.
Znaczna różnorodność produktów chemicznych narzuca dużą liczbę procesów
technologicznych, jednakże większość produktów przemysłu chemicznego i przetwórczego
otrzymuje się w wyniku prowadzenia stosunkowo niedużej liczby procesów podstawowych,
powtarzających się w różnych odmianach w wielu procesach technologicznych.
Podstawowe procesy, związane z przemianami fizyko-chemicznym, określa się jako
procesy jednostkowe. Wyróżnia się następujące procesy podstawowe i jednostkowe:
1.
mechaniczne: magazynowanie, transport, rozdrabnianie, klasyfikacja i przesiewanie,
flotacja, mieszanie, separacja (filtracja, wirowanie).
2.
cieplne: ogrzewanie, chłodzenie, kondensacja, odparowanie, zatężanie.
3.
dyfuzyjne: destylacja, rektyfikacja, absorpcja, adsorpcja, ekstrakcja i ługowanie,
suszenie, nawilżanie, krystalizacja sublimacja.
4.
procesy z udziałem reakcji chemicznej: związane z wyżej wymienionymi i dodatkowo
z przemianami chemicznymi np.: utlenianie i redukcja, nitrowanie, chlorowanie,
sulfonowanie.
Poniżej zostały scharakteryzowane najważniejsze procesy podstawowe i jednostkowe.
Do realizacji procesów mechanicznych konieczne jest doprowadzenie energii
mechanicznej z zewnątrz. Do przebiegu procesów cieplnych i dyfuzyjnych niezbędna jest
odpowiednia różnica temperatur i stężenia w środowisku przebiegu procesu.
Zmniejszenie rozmiarów ciała stałego połączone ze zniszczeniem jego struktury
nazywane jest rozdrabnianiem lub niekiedy kruszeniem. Celem rozdrabniania jest
wytworzenie produktu o pożądanych rozmiarach, zakresie rozmiarów lub docelowo, separacji
składników ciała stałego. Wymagania użytkowe i procesowe dotyczyć mogą surowca lub
produktu, który powinien mieć określone rozmiary lub zakres rozmiarów ziaren co łączy się
z klasyfikacją i przesiewaniem mieszaniny ziaren. W klasyfikacji maszyn do rozdrabniania
bierze się pod uwagę rodzaj przyłożonych sił i wielkość wywołanych naprężeń oraz stopień
rozdrobnienia. Rodzaje rozdrobnień przedstawiono w Tabeli 1.
Tabela 1. Stosowane nazwy cząstek ciała stałego w zależności od ich rozmiarów
Lp.
Rozdrobnienie
Rozmiar cząstek
[mm]
1
Grube
>100
2
Średnio grube
10
3
Średnio drobne
1
4
Drobne
0,1
5
Superdrobne
0,025
6
Ultradrobne
0,010
7
Koloidalne
≤0,005

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
37
Stopień rozdrobnienia n określony jest stosunkiem średniej średnicy ziaren przed
rozdrobnieniem do średniej średnicy ziaren po rozdrobnieniu. Wyróżnia się rozdrobnienie:
−
wstępne, n<5,
−
średnie, n = 5-10,
−
drobne, n = 10-50,
−
bardzo drobne, n>50.
Rozdrabnianie może być prowadzone w sposób ciągły lub okresowo. Do rozdrabniani
służą urządzenia zwane rozdrabniarkami (kruszarkami). Służą one do rozdrabniania
wstępnego i średniego przez ściskanie, rozłupywanie, uderzanie, ścieranie. Do kruszarek
zalicza się między innymi łamacze, gniotowniki, rozdrabniarki młynkowe, rozdrabniarki
udarowo-prętowe. Rozdrabnianie do najmniejszych rozmiarów prowadzone jest w młynach,
w których dominującymi mechanizmami jest ścieranie i uderzanie. Istnieje wiele klasyfikacji
młynów. Do najpowszechniejszych należy podział na: młyny wolnobieżne (kulowe oraz
prętowe) i młyny szybkobieżne (rolkowo-pierścieniowe, wibracyjne).
Rozdrabniarki (młyny) oznacza się symbolem:
Mieszanie to jeden z najistotniejszych procesów technologicznych. Bez względu na to,
czy mieszane są ciała stałe, ciekłe czy gazowe lub też mieszaniny tych ciał, istotą procesu jest
uzyskanie jednorodnej mieszaniny, w możliwie krótkim czasie, przy możliwie najmniejszym
zużyciu energii. Mieszanie pozwala przyspieszyć reakcję chemiczną (zwiększa szybkość
wymiany ciepła i masy). Mieszanie może być prowadzone jako samodzielna operacja
w
specjalnych
urządzeniach
lub
towarzyszyć
niektórym
procesom
fizycznym
(np. ogrzewaniu, chłodzeniu) czy chemicznym (np. nitrowaniu, sulfonowaniu). Proces
mieszania ułatwia rozpuszczanie cieczy w cieczy lub też ciał stałych w cieczy. Szybkość
reakcji chemicznych wzrasta przy zwiększeniu powierzchni zetknięcia się składników reakcji.
Z tego powodu większość aparatów czy urządzeń, w których prowadzona jest reakcja,
wyposażona jest w mieszadła różnego typu.
W przemyśle stosuje się różnorodne urządzenia do mieszania. Dobór typu zależy od
właściwości substancji (między innymi od stanu skupienia), zastosowanej metody,
konstrukcji urządzenia i mieszadeł. Urządzenia do mieszania można podzielić ogólnie na:
−
urządzenia do mieszania ciał sypkich – mieszarki,
−
urządzenia do mieszania past (ciał ciastowatych) i plastycznych – zagniatarki,
−
urządzenia do mieszania ciał ciekłych z ciekłymi albo ciekłych z niewielką ilością ciał
stałych lub gazowych – mieszalniki,
−
urządzenia do mieszania gazów – zwane również mieszalnikami.
Mieszalniki (ogólnie) oznacza się symbolem:
Proces odwrotny do procesy mieszania nazywany jest rozdzielaniem. Z operacjami
rozdzielania mamy do czynienia na różnych etapach produkcji np. usuwamy z surowców
niepożądane zanieczyszczenia, oddzielamy od sienie niejednorodne produkty itp. Filtrowanie
stanowi najważniejszy i bardzo szeroko rozpowszechniony proces oddzielania ciał stałych od
cieczy. Polega on na stosowaniu przegrody porowatej, nieprzepuszczającej cząstek ciała
stałego, znajdującego się w zawiesinie. Tworzą one na przegrodzie osad, a przechodząca przez
przegrodę klarowna ciecz stanowi przesącz. Proces ten prowadzi się w różnego rodzaju filtrach.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
38
Wyróżniamy filtry:
−
grawitacyjne – które pracują pod ciśnieniem hydrostatycznym słupa filtrowanej cieczy,
oznaczane symbolem:
−
próżniowe – w których wytwarza się próżnię pod przegrodą filtracyjną, oznaczane
symbolem:
−
ciśnieniowe – w których nad przegrodą panuje ciśnienie wyższe od atmosferycznego,
oznaczane symbolem:
Filtry podzielić można również na: powolne i pośpieszne.
Przegrody w filtrach wykonywane są z różnych materiałów porowatych. Rozróżnia się
przegrody: ziarniste, tkaninowe, ceramiczne.
Adsorpcja polega na wydzieleniu i zatrzymaniu składników płynu na powierzchni
zewnętrznej i wewnętrznej (w porach) ciała stałego zwanego adsorbentem (sorbentem).
Zatrzymanie cząsteczek zachodzi w wyniku działania sił fizycznych i chemicznych bliskiego
zasięgu. Proces adsorpcji jest egzotermiczny. Proces odwrotny zwany desorpcją wymaga
doprowadzenia energii. Liczba cząstek możliwa do zaadsorbowania na powierzchni
adsorbentu jest ograniczona stanem równowagi adsorpcyjnej i maleje wraz ze wzrostem
temperatury. Efektywnej adsorpcji sprzyja duża powierzchnia właściwa adsorbentu i niska
temperatura. Adsorpcja jest selektywna a największą zdolność do adsorpcji wykazują cząstki
gazów o dużej masie cząsteczki o mniejszej energii wiązania. Podczas adsorbowania cząstki
na powierzchni adsorbentu stosunkowo małymi siłami nazywany jest adsorpcją fizyczną. Gdy
siły wiązania cząstek na powierzchni adsorbentu są zbliżone do wiązania chemicznego,
proces jest określany jako adsorpcja chemiczna lub chemisorpcja. W momencie gdy
zaadsorbowana masa substancji, w danych warunkach, zbliżona jest do równowagowej, to
następnym etapem procesu adsorpcyjnego jest usunięcie tej substancji z powierzchni
adsorbentu, co nazywane jest regeneracją adsorbentu. Regeneracja (desorpcja) może być
przeprowadzana przez ogrzewanie adsorbentu, działanie parą wodną, przedmuchiwanie
gazem obojętnym, obniżenie ciśnienia oraz metodami mieszanymi.
Adsorbenty stanowią materiały porowate o dużej efektywnej powierzchni, otrzymywanie
z węgla i substancji organicznych, minerałów (tlenków glinu czy krzemionki) i innych.
Pożądane właściwości adsorbentów to: duża powierzchnia właściwa, odpowiednia wielkość
granulatu, twardość i sypkość, łatwość regeneracji i powtórnego stosowania. Wyróżnia się
trzy grupy adsorbentów: niepolarne, polarne i chemiczne.
Rodzaj i typ aparatury do prowadzenia adsorpcji jest związany z okresowym lub ciągłym
sposobem prowadzenia procesu, sposobem kontaktu stopniowym lub ciągłym, rodzajem
adsorbentu i parametrami procesu. Powszechnie stosowanym podziałem, związanym
z położeniem i stanem ruch adsorbentu w adsorberze, według którego wyróżnia się adsorbery
z warstwą nieruchomą, ruchomą i fluidalną.
Prowadzenie adsorpcji z fazy ciekłej związane jest z usuwaniem zanieczyszczeń ciekłych,
często substancji barwiących. Proces adsorpcji z cieczy jest mniej poznany w porównaniu
z fazą gazową. Adsorpcja okresowa z cieczy odbywa się zwykle w aparatach typu
zbiornikowego. Adsorpcję z cieczy w sposób ciągły prowadzi się w pionowych kolumnach,
w których adsorbent przepływając od góry, a ciecz od dołu kolumny kontaktują się

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
39
w przepływie przeciwprądowym. Do tego samego rodzaju procesów sorpcyjnych zalicza się
również wymianę jonową prowadzoną w obecności sorbentów stałych zwanych jonitami,
kationitami i anionitami, stanowiących zwykle żywice syntetyczne.
Suszeniem nazywamy proces odparowywania to jest usuwania wilgoci lub innej
substancji lotnej z ciał stałych, szlamów i zawiesin. Suszenie jest etapem końcowym lub
przejściowym wielu technologii przemysłowych, w tym również wytwarzania wyrobów
gotowych. Suszenie na między innymi na celu: wytworzenie produktu lub półproduktu
o odpowiednich
właściwościach
użytkowych,
zwiększenie
trwałości
materiałów,
zmniejszenie kosztów transportu materiałów.
Do prowadzenia procesu suszenia używane są aparaty zwane suszarkami. Czynnikiem
suszącym jest zazwyczaj powietrze lub gazy spalinowe. Proces suszenia związany jest
z jednoczesnym przenikaniem masy i ciepła w czynniku suszącym i w ciele suszonym.
Duża różnorodność właściwości fizycznych i chemicznych suszonych materiałów
powoduje, że istnieje wiele sposobów prowadzenia procesu suszenia i konstrukcyjnych
rozwiązań suszarek. W klasyfikacji suszarek wynikającej z metody pracy i sposobu
dostarczenia ciepła wyróżnia się suszarki:
−
okresowe lub ciągłe,
−
z ogrzewaniem:
-
bezpośrednim – suszarki konwekcyjne,
-
pośrednim – suszarki przeponowe,
−
radiacyjne (promiennikowe),
−
mikrofalowe i dielektryczne.
Suszarki oznacza się symbolem:
Krystalizacja stanowi proces wydzielania substancji rozpuszczonej z roztworu
przesyconego lub z substancji macierzystej będącej w stanie ciekłym, bądź bezpośrednio
z fazy gazowej pominięciem fazy ciekłej – na skutek desublimacji (proces odwrotny do
sublimacji). W wyniku krystalizacji następuje wydzielanie produktu w postaci stałej lub
oczyszczanie roztworu macierzystego bądź gazu. Kryształ stanowi ciało stałe, jednorodne,
ograniczone płaszczyznami kryształu, które tworzą określoną strukturę geometryczną. Siłą
napędową procesów krystalizacji jest przesycanie roztworu uzyskiwane przez odparowanie
rozpuszczalnika (zatężanie roztworu), chłodzenie roztworu, reakcję chemiczną lub dodanie
trzeciego składnika. Po dojściu do stanu równowagi krystalicznej między procesem
rozpuszczania się ciała stałego o jego krystalizacją należy doprowadzić roztwór zwany ługiem
macierzystym do przesycenia. Zdolność do przesycenia roztworu zależy od rodzaju substancji
i określona jest jako stosunek stężenia aktualnego roztworu do stężenia równowagowego.
Przy dużym stopniu przesycenia powstaje znaczna liczba małych trudnych do filtracji
i mycia. Mały stopień przesycenia daje natomiast małą liczbę dużych kryształów,
w roztworze określonym jako ług pokrystaliczny. Krystalizacją frakcyjną nazywa się
krystalizację stosowaną do rozdzielania substancji obecnych w roztworze, różniących się
rozpuszczalnością lub szybkością wzrostu kryształów w zależności od temperatury.
Krystalizację prowadzi się w aparatach zwanych krystalizatorami, w których istnieją warunki
dla powstawania i wzrostu kryształów. Krystalizacja może być prowadzona w sposób ciągły
lub okresowy.
Wiele procesów technologicznych polega na wprowadzeniu do cząsteczki związku
organicznego określonych grup funkcyjnych poprzez ich podstawienie lub przyłączenie, jak
chlorowanie, nitrowanie, arylowanie, alkilowanie i sulfonowanie. Odrębną grupę stanowią
procesy związane z przekształcaniem grup funkcyjnych w inne, jak estryfikacja, redukcja czy

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
40
utlenianie. Zmniejszenie masy cząsteczkowej substancji i tworzenie nowych związków następuje
w procesach pirolizy, hydrolizy, alkoholizy. Zwiększenie uzyskuje się w procesach polimeryzacji,
kondensacji i polikondensacji. Procesy, w których następuje przebudowa struktury chemicznej
substratów, przy niezmienionej masie cząsteczkowej nazywa się izomeryzacją.
Redukcja (inna nazwa elektronacja) to proces, w trakcie którego atom lub ich grupa
przechodzi z wyższego na niższy stopień utlenienia. Każdej redukcji musi towarzyszyć
utlenienie. Łącznie takie procesy nazywa się reakcjami redoks. W praktyce, daną reakcję
nazywamy reakcją redukcji (zwłaszcza w chemii organicznej), gdy struktura głównego
substratu i głównego produktu różnią się tylko tym, że jedna niewielka grupa lub pojedynczy
atom zmniejszył w jej wyniku swój stopień utlenienia, kosztem utlenienia, zwykle
nieorganicznego, prostego związku zwanego w tym przypadku środkiem redukującym.
Procesy uwodorniania polegają na reakcji wodoru in statu nascendi ze związkami
organicznymi w obecności katalizatorów. W przemyśle katalitycznego uwodorniania węgla,
produktów jego pirolizy i pozostałości ropy naftowej, uzyskuje się węglowodory alifatyczne
i aromatyczne. Z tlenku węgla otrzymuje się węglowodory alifatyczne i alkohole,
z naturalnych tłuszczów utwardzone tłuszcze i alkohole tłuszczowe, z nienasyconych
węglowodorów nasycone, z nitryli i nitrozwiązków – odpowiednie aminy.
W procesach katalitycznego uwodorniania można rozróżnić kilka typów reakcji:
−
przyłączenie wodoru do związków nienasyconych,
−
redukcja grup karbonylowych, karboksylowych, nitrylowych i nitrowych,
−
rozpad cząsteczek z jednoczesnym przyłączeniem wodoru tzw. hydroliza,
−
aromatyzacja.
Destruktywne uwodornienie ma duże znaczenie technologiczne jako podstawowa reakcja
uwodorniania węgla, smół, pozostałości ropy, a także hydrorafinacji, powodując rozpad
wiązań nie tylko węglowych, ale również C-S i C-N.
Metody uwodorniania są bardzo różnorodne, a dobór warunków reakcji i katalizatorów
zależy od charakteru chemicznego surowca wyjściowego i produktów końcowych. Procesy
uwodorniania prowadzi się w fazie ciekłej i gazowej. Stosuje się katalizatory nieruchome,
ruchome i w zawiesinie. Dobór katalizatora ma wpływ na przebieg i kierunek reakcji.
Kontakty spełniają rolę aktywatorów wodoru i substratów. Wodór absorbuje się na
powierzchni katalizatora, przy czym siły działające pomiędzy wodorem i powierzchnią
katalizatora, osłabiają wiązania pomiędzy atomami wodoru umożliwiając wejście
uaktywnionej cząsteczki wodoru do reakcji. Podobnej aktywizacji na powierzchni katalizatora
ulega substancja uwodorniania.
Jako katalizatory do uwodorniania nadają się: rozdrobniona platyna, pallad tańsze od nich
nikiel i miedź.
Źródłami otrzymywania wodoru w przemyśle są: gaz wodny, gaz koksowniczy, gaz
zimny, elektroliza roztworów wodorotlenku sodu lub soli kuchennej.
Utlenienie to reakcja chemiczna, w której jakiś atom (lub ich grupa) przechodzi
z niższego na wyższy stopień utlenienia. W praktyce, daną reakcję nazywa się utlenieniem,
gdy struktura głównego substratu i głównego produktu różnią się tylko tym, że jedna
niewielka grupa lub pojedynczy atom zwiększył w jej wyniku swój stopień utlenienia,
kosztem redukcji prostego związku zwanego w tym przypadku środkiem utleniającym.
Zupełne utlenianie substancji organicznej, prowadzi do rozpadu cząsteczki
z wytwarzaniem dwutlenku węgla i wody, a jeśli w cząsteczce zawarte były azot i siarka,
tworzą się również tlenki azotu i siarki (proces spalania Reakcje niezupełnego utleniani
polegają na:
−
wprowadzeniu do cząsteczki związku organicznego atomu tlenu bez zasadniczej zmiany
w budowie cząsteczki, np.: utlenianie węglowodorów alifatycznych do alkoholi,
aldehydów lub kwasów, węglowodorów alkiloaromatycznych do fenoli,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
41
−
odwodornieniu związku organicznego z wytworzeniem wiązanie nienasyconego
pomiędzy atomami węgla,
−
wprowadzeniu atomów tlenu z jednoczesną zmianą podstawowego szkieletu cząsteczki.
Silnymi środkami utleniającymi są: tlen atomowy, nadtlenki, kwasy np. kwas
azotowy (V), ozon, gazowy fluor, chlor, brom i jod, manganiany (VII), chlorany. Najczęściej
stosowany jako środek utleniający jest tlen z powietrza lub czysty tlen. Utlenianie tlenem
cząsteczkowym
wymaga
stosowania
podwyższonej
temperatury
i
odpowiednich
katalizatorów. Procesy wykonywane są w fazie ciekłej i gazowej. Manganian (VII) potasu jest
bardzo silnym środkiem utleniających. Utlenianie prowadzi się w fazie ciekłej. Zależnie od
odczynu środowiska manganian rozkłada się oddając różne ilości tlenu. Dichromiany (VI)
sodu lub potasu są tanimi i energicznymi utleniaczami. Bezwodnik kwasu chromowego CrO
3
w roztworze lodowego kwasu octowego w szczególnych przypadkach używany jest jako
środek utleniający np. utlenia aromatyczne alkohole do aldehydów, antracen do antrachinonu.
Kwas chlorowy (I) i jego sole (NaOCl tzw. woda Javella) stosowane są powszechnie jako
łagodne środki utleniające np. bielące (włókna, celulozę). Tlenek manganu (IV) MnO
2
stosowany do utleniania, np. grup metylowych do aldehydowych (aldehyd benzoesowy).
Nadtlenek wodoru (H
2
O
2
) bardzo silny, ale kosztowny środek utleniający znajduje
przemysłowe zastosowanie w przyspieszonym procesie dojrzewania alkalicelulozy
i procesach bielenia. Kwas azotowy (V) HNO
3
zalicza się do energicznych środków
utleniających, stosuje się np. do otrzymywania kwasu adypinowego. Tlenki azotu (N
2
O
3
↔
NO + NO
2
) zaliczają się do silnych środków utleniających. Stosowane są w fazie ciekłej
i gazowej.
Celem nitrowania jest wprowadzenie grupy nitrowej (-NO
2
) do cząsteczki związku
organicznego:
RH + HONO
2
→ RNO
2
+ H
2
O
gdzie: R – rodnik.
Podstawowym środkiem nitrującym jest mieszanina składająca się z kwasu azotowego (V)
i środka odwadniającego (kwas siarkowy (VI), bezwodnik kwasu octowego). Nitrowanie
prowadzi się za pomocą mieszaniny nitrującej przygotowanej z kwasów azotowego (V)
i siarkowego(VI). Kwas siarkowy (VI) stosujemy do wiązania wody tworzącej się w reakcji
nitrowania., Obecność tego kwasu pozwala lepiej wykorzystać HNO
3
i umożliwia ponad to
przeprowadzenie nitrowania w nitratorach ze zwykłej stali. Dobór składu mieszaniny nitrującej
i warunków nitrowania uzależniony jest od właściwości związku poddawanego nitrowaniu i od
ilości grup nitrowanych, które zamierza się wprowadzić. Największe znaczenie przemysłowe
mają procesy nitrowania węglowodorów aromatycznych, a w szczególności nitrowanie benzenu
i toluenu. Zapotrzebowanie na nitro benzen jest duże ze względu jego zastosowanie do
produkcji aniliny oraz pochodnych chlorowych, sulfonowych i aminowych, które są surowcami
do syntezy lekarstw, barwników i materiałów wybuchowych.
Polimeryzację, polikondensację, poliaddycję stosuje się do syntezy związków wielko
cząsteczkowych wykorzystywanych do produkcji tworzyw sztucznych i syntetycznych.
W każdej dziedzinie życia wykorzystywane są nie tylko tworzywa naturalne ale przede
wszystkim tworzywa sztuczne i syntetyczne. Tworzywa sztuczne otrzymuje się
z wielkocząsteczkowych surowców naturalnych takich jak: celuloza, kauczuk naturalny,
kazeina.
Substancje, z których otrzymuje się syntetyczne związki wielkocząsteczkowe to: eten,
propylen, chlorek winylu oraz wiele innych.
Polimeryzację prowadzi się różnymi sposobami:
−
Polimeryzacja w masie, zwana blokową – prowadzona jest w czystym, ciekłym
monomerze i polega na ogrzaniu monomeru z dodatkiem inicjatora do temperatury
zapoczątkowania procesu.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
42
−
Polimeryzacja w roztworze – prowadzona jest w rozpuszczalniku, w którym rozpuszcza
się monomer i polimer.
−
Polimeryzacja roztworowo-strąceniowa – prowadzona jest w rozpuszczalniku, w którym
rozpuszcza się tylko monomer.
−
Polimeryzacja suspensyjna (perełkowa) – prowadzona jest w wodzie, gdzie poprzez
mieszanie, z inicjatorem procesu w postaci drobnych kropel, następuje rozproszenie
nierozpuszczalnego monomeru. Produkt ma postać twardych kulek (perełek).
−
Polimeryzacja emulsyjna – prowadzona jest w środowisku wodnym, w którym
z monomeru, środka powierzchniowo czynnego i inicjatora wytwarza się, poprzez
mieszanie, emulsję.
−
Polimeryzacja w fazie gazowej – prowadzona jest w reaktorach rurowych po uprzednim
sprężeniu monomeru i dodaniu inicjatorów (tlenków lub nadtlenków).
Polikondensacja polega na reakcji cząstek związków mających co najmniej dwie
odpowiednio aktywne grupy funkcyjne. Produktem jest związek wielkocząsteczkowy (żywica
polikondensacyjna) i produkt uboczny w postaci wody, chlorowodoru, amoniaku.
Polikondensacja jest reakcją odwracalną i stopniową. Przebieg reakcji zależy od temperatury,
kwasowości i szybkości usuwania produktów ubocznych ze środowiska procesu.
Polikondensację prowadzi się w reaktorach z mieszadłem kotwicowym, płaszczem grzejnym
oraz chłodnicą. Do typowych tworzyw polikondensacyjnych nalezą: fenoplasty i aminoplasty.
Metodą polikondensacji otrzymuje się związki krzemoorganiczne i polisiloksany.
Poliaddycja jest procesem otrzymywania produktów wielkocząsteczkowych w wyniku
rozerwania pierścienia heterocyklicznego lub wysycenia wiązań podwójnych w cząsteczkach
monomerów. Poliaddycja jest procesem stopniowym charakteryzującym się wzrostem masy
cząsteczkowej produktu w kolejnych etapach produkcji. Reakcja jest nieodwracalna i nie
wydziela produktów ubocznych. Poliaddycja dotyczy cząstek tego samego związku,
najczęściej heterocyklicznego lub cząstek dwóch różnych związków.
Wymienione powyżej procesy prowadzi się w różnego rodzaju aparatach, urządzeniach.
Aparat lub urządzenie stanowi zespół przedmiotów (części) skonstruowanych w celu
prowadzenia odpowiednich procesów z możliwością spełnienia wymagań procesowych.
Istnieje wiele podziałów aparatów na grupy związane z pełnioną funkcją w procesie
technologicznym i cechami konstrukcyjnymi.
Proces technologiczny jest jednym lub w szeregu aparatów i urządzeń tworzących tak
zwany ciąg technologiczny. Ze względu na charakter pracy i przetwarzania substancji
w wymienionym ciągu aparaty i urządzenia podzielić można na pracujące w sposób ciągły,
okresowy lub mieszany.
W procesie technologicznym istotny jest dobór reakcji chemicznych, które pozwolą na
optymalny przebieg przemiany od surowców do pożądanych produktów. Dobór właściwej
reakcji jest problemem złożonym.
Na stan równowagi chemicznej i wydajności reakcji chemicznej wpływają: temperatura,
ciśnienie, skład reagentów oraz dobór katalizatora. Warunkiem uzyskania wysokiego stopnia
przemiany jest duża wartość stałej równowagi reakcji i ujemna wartość potencjału
termodynamicznego układu reakcyjnego, w przeciwnym wypadku należy doprowadzać
energię z zewnątrz.
Szybkość procesu można zmienić stosując substancje zwane katalizatorami. Katalizator
zwykle nie ulega znaczącym zmianom chemicznym i ilościowym w trakcie procesu.
Katalizatory zmniejszające szybkość dojścia do stanu równowagi noszą nazwę inhibitorów.
Katalizatorami mogą być gazy, ciecze i ciała stałe. Katalizatory stałe zwane są również
kontaktami. W momencie, kiedy substraty lub produkty reakcji działają katalitycznie,
przyspieszając przebieg reakcji proces ten nazywamy autokataliza.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
43
Katalizatory stałe są to z reguły substancje pochodzenia nieorganicznego. Katalizatory
organiczne, biokatalizatory, są to enzymy stosowane powszechnie w procesach
biotechnologicznych. Kataliza homogeniczna, jednorodna, jednofazowa występuje wtedy,
gdy katalizator w środowisku procesu nie tworzy odrębnej fazy, lecz występuje w tej samej
postaci co reagenty. Natomiast, jeżeli katalizator występuje w postaci odrębnej fazy,
to kataliza nazywana jest mianem heterofazowej, heterogenicznej. W przemyśle chemicznym
i przetwórstwa w większości przypadków stosowana jest kataliza heterogeniczna a jako
katalizator stosuje się ciało stałe naniesione na nośnik o rozbudowanej powierzchni
zewnętrznej i często wewnętrznej. Substancje dodawane do katalizatora zwiększające jego
aktywność nazywa się promotorami.
W wielu procesach i urządzeniach wykorzystuje się działanie sił elektrycznych
w elektrofiltrach, elektrodializach, elektroforezie, odsalaniu wód, elektroeksykacji, filtracji
dielektrycznej i rozpylaniu elektrostatycznym. W elektrolizerach energię elektryczną
doprowadza się do środowiska reakcji chemicznej za pomocą elektrod, lub odwrotnie,
otrzymuje się energię elektryczną z reakcji chemicznych w ogniwach paliwowych.
Procesy elektrochemiczne (elektrolityczne) prowadzone w środowisku wodnym
elektrolitów stosowane są do wytwarzania produktów nieorganicznych i organicznych,
a także do otrzymywania metali. Pokrywanie powłokami metalicznymi realizowane jest
w procesach galwanicznych. Procesy elektrolityczne w środowisku niewodnym są stosowane
do otrzymywania i rafinacji metali, w tym aluminium. W procesach elektrotermicznych
energia elektryczna jest źródłem ciepła. W wyniku elektrorafinacji otrzymuje się metale
o wysokiej czystości, takie jak miedź, nikiel, kobalt. Procesy rozkładu elektrolitycznego służą
do otrzymywania wodoru i tlenu przez rozkład wody, a także chloru, glinu czy magnezu, gdy
elektrolizie zostaną poddane odpowiednie związki.
W procesach elektrotermicznych uzyskuje się o wiele wyższą temperaturę niż podczas
spalania klasycznych paliw. Reaktory termochemiczne, zwane piecami, są stosowane do
wytwarzania substancji, które wymagają temperatury do 3230
°
C i wyższych. W piecach
oporowych przepływ prądu odbywa się przez elementy grzejne. W piecach indukcyjnych
wykorzystuje się zjawisko indukcji do ogrzewania przetwarzanych materiałów prądami
wysokiej częstotliwości. W piecach łukowych ciepło do przetwarzanego materiału jest
dostarczane od łuku elektrycznego bezpośrednio lub przez promieniowanie.
Niektóre procesy technologiczne prowadzi się w plazmie. Doprowadzając do gazu
odpowiedni strumień energii, powoduje się dysocjację i jonizację cząsteczek, powstawanie
wolnych rodników, wzbudzanie atomów i emisję promieniowania. Mieszaninę gazową,
w której zachodzą wymienione stany nazywamy plazmą. W praktyce plazmę tworzy się przez
wyładowania elektryczne w gazach, gdy napięcie graniczne zostanie przekroczone powyżej
30 kV/cm i gaz wokół elektrody emisyjnej zacznie świecić. Wyładowanie może być iskrowe,
jarzeniowe lub łukowe. Najczęściej stosowane, w przemyśle, procesy plazmowe prowadzone
są wówczas, kiedy pozwalają uzyskać znaczącą szybkość procesu w temperaturze plazmy
oraz gdy trzeba wytworzyć odpowiednie rodniki do zainicjowania przebiegu procesu
otrzymywania produktów o wysokiej czystości, rozkładu niebezpiecznych substancji.
Procesy fotochemiczne przebiegają pod wpływem absorpcji promieniowania o długości
fali od ultrafioletu do podczerwieni. Procesy te wykorzystywane są w polimeryzacji, syntezie
związków światłoczułych, destylacji zanieczyszczeń z cieczy. Promieniowanie ultrafioletowe
jest stosowane do sterylizacji przy oczyszczaniu wody, ograniczenia wzrostu glonów,
farmaceutyków, kosmetyków, wyrobów żywnościowych, w procesach oczyszczania ścieków
i innych. Energia promieniowania świetlnego ma największe znaczenie w procesach takich
jak chlorowanie węglowodorów, otrzymywanie sulfochlorków, polimeryzacji olefin.
Fotokataliza ma duże znaczenie w procesach usuwania zanieczyszczeń z cieczy i gazów.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
44
W procesach radiacyjnych stosuje się promieniowanie gamma oraz promieniowanie typu
rentgenowskiego. Procesy radiacyjne umożliwiają przeprowadzenie procesów niemożliwych
do realizacji metodami standardowymi, jednak koszty energii są wyższe niż energii cieplnej
i chemicznej. W chemicznych procesach radiacyjnych energia jest wielokrotnie większa od
energii jonizacji.
Promieniowanie jonizujące może inicjować procesy chemiczne oraz stanowić siłę
napędową procesu. W reakcjach związków organicznych to promieniowanie stosuje się przy
polimeryzacji kopolimeryzacji oraz modyfikacji polimerów. Pod wpływem promieniowania
jonizującego następuje proces sterylizacji i utleniania żywności. W procesach ochrony
środowiska naturalnego, za pomocą aktywacji radiacyjnej usuwa się SO
2
i tlenki azotu NO
x
ze spalin odlotowych, unieszkodliwia osady czynne z biologicznych oczyszczalni ścieków,
neutralizuje odpady celulozowe i gnilne.
Wiele procesów technologicznych i przetwórstwa prowadzi się biochemicznie.
W procesach biochemicznych przebiegających w środowisku wodnym dużą rolę odgrywają
mikroorganizmy. Mikroorganizmy dzieli się na kilka grup: wirusy, bakterie (mikroorganizmy
jednokomórkowe), promieniowce, drożdże, pleśnie, glony.
Na wzrost i rozwój mikroorganizmów mają wpływ warunki środowiskowe, dostępność
pożywienia, temperatura, pH środowiska oraz jego zanieczyszczenie (obecność trucizn).
Dla mikroorganizmów w największym stopniu toksyczne są metale ciężkie. Procesy
biochemiczne prowadzi się w bioreaktorach z dodatkiem pożywek dla bakterii, z ciągłym
mieszaniem i napowietrzaniem, gdy jest to wymagane.
W warunkach utleniających przebiega właściwy proces biochemiczny:
substancja organiczna + O
2
→
bakterie
CO
2
+ H
2
O + przyrost biomasy
W środowisku bakterii beztlenowych w kolejnych etapach następuje przekształcenie
zanieczyszczeń w produkty przejściowe, kwasy organiczne i alkohole i na końcu w metan
i dwutlenek węgla:
substancja organiczna
→
bakterie
produkty przejściowe + CO
2
+ H
2
S + H
2
O
kwasy organiczne
→
bakterie
CH
4
+ CO
2
Procesy przemian biochemicznych określa się również mianem fermentacji.
Do prowadzenia procesów technologicznych konieczna jest energia w różnych postaciach,
w największym stopniu cieplna i elektryczna. Energia z paliw zostaje uwolniona podczas ich
spalania i jest używana bezpośrednio w procesach endotermicznych, służy do ogrzewania lub
jest zmieniana na energie elektryczną w generatorach prądu.
Wyróżniamy trzy podstawowe rodzaje paliw:
−
paliwa stałe naturalne to: drewno, torf, węgiel brunatny i kamienny, natomiast paliwa
stałe sztuczne to: koks, brykiety węglowe oraz drzewne.
−
paliwa ciekłe to: ropa naftowa i produkty jej przetwarzania w postaci różnego rodzaju
olejów, oleje pochodzenia roślinnego.
−
naturalne paliwa gazowe to: gaz ziemny, metan pochodzący z fermentacji, natomiast
paliwa gazowe sztuczne to: gazy koksownicze i generatorowe, acetylen, wodór i inne.
Najważniejszymi cechami charakteryzującymi paliwa są: wartość opałowa oraz ciepło
spalania. Wartość opałowa jest to ilość ciepła wydzielana przy spalaniu jednostki masy lub
jednostki objętości paliwa przy jego całkowitym i zupełnym spalaniu, przy założeniu, że para
wodna zawarta w spalinach nie ulega skropleniu, pomimo że spaliny osiągną temperaturę
początkową paliwa. Jednostką wartości opałowej jest kJ/kg. Ciepło spalania jest to ilość
ciepła, jaka powstaje przy spalaniu całkowitym i zupełnym jednostki masy lub jednostki
objętości analizowanej substancji w stałej objętości, przy czym produkty spalania oziębia się
do temperatury początkowej, a para wodna zawarta w spalinach skrapla się zupełnie.
Jednostką ciepła spalania jest J/kg.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
45
Spalanie jest procesem chemicznym przebiegającym w wielu stadiach i może być zupełne
(całkowite) lub niezupełne, gdy w spalinach jest obecny tlenek węgla lub inne substancje
niespalone całkowicie. W tym procesie dąży się do całkowitego spalenia wszystkich części
palnych paliwa przy minimalnej ilości powietrza (tlenu), zbliżonej do stechiometrycznej.
Warunki procesowe i aparatura do spalania paliw różnią się w zależności od postaci paliwa,
jego koncentracji i właściwości fizykochemicznych zawartych w nim składników palnych.
Surowce stosowane do wykorzystania produktów przemysłowych są przede wszystkim
materiałami pochodzenia naturalnego, które w zależności od pochodzenia dzieli się na
surowce mineralne (kopalne) oraz roślinne i zwierzęce (odtwarzalne). Surowce mineralne
występują w postaci rud, mniej lub bardzie czystych, zawierających obok składnika
zasadniczego tak zwany balast określony mianem skały płonnej. Wzrost ogólnych kosztów
pozyskiwania surowców o odpowiedniej czystości powoduje potrzebę ich odzysku
i powtórnego użycia lub tez stosowania w postaci bardziej stężonej.
Projektowanie procesu technologicznego stanowi złożony problem wymagający analizy
wielu różnorodnych zagadnień, wykonywania wielu symulacji i obliczeń. Projekt
wykonywany jest w wielu stadiach i wariantach zanim stanie się rozwiązaniem ostatecznym,
w których zastosowane zostaną właściwe rozwiązania konstrukcyjno-aparaturowe.
Schemat technologiczny jest kluczowym dokumentem w procesie projektowania
technologicznego. Stanowi on rysunek, na którym za pomocą symboli ilustrujących proste lub
złożone procesy jednostkowe oraz ich powiązanie przedstawia się przebieg produkcji. Schemat
taki musi być jasny, zwarty, dokładny i kompletny. Rozróżnia się różnego rodzaju schematy.
Schemat blokowy stanowi najprostszą formę. Procesy lub aparaty przedstawione są jako
kwadraty (prostokąty) lub koła. Sposób ten przydatny jest do pokazania procesu w sposób
poglądowy, w uproszczonej formie (w sprawozdaniach, raportach czy podręcznikach).
W wielostadiowym projektowaniu technologicznym zaleca się wykonywanie trzech
rodzajów schematów: ideowego, wstępnego i technicznego.
Schemat ideowy przedstawia proces w najprostszej postaci w kolejności wymaganej do
przeprowadzenia procesu technologicznego. Zalecane jest przy tym posługiwanie się
symbolami pokazanymi na schemacie:
a)
b)
bezciśnieniowe
ciśnieniowe
próżniowe
Rys. 3. Symbole schematu ideowego: procesy podstawowe, jednostkowe:
a) bez reakcji chemicznej; b) z reakcją chemiczną.
Posługując się wymienionymi symbolami, sporządza się schemat ideowy, w którym
symbole umieszcza się jeden pod drugim poczynając od góry. Jeżeli proces składa się z kilku
ciągów, wówczas wskazane jest rysowanie ciągów równolegle, obok siebie. Ciągi
pomocnicze rozmieszcza się obok głównego ciągu technologicznego. Poszczególne symbole
łączy się liniami obrazującymi drogę przepływu strumieni masowych, zaznaczając kierunek
strzałką. Strumienie pomocnicze oznaczane są linią o połowę cieńszą w wprowadzone do
prostokąta od góry z lewej strony, a odprowadzone od dołu z prawej strony. Dla czynników
pomocniczych zarezerwowana jest linia cienka przerywana lub punktowana. Wszystkie linie
na schemacie powinny być ponumerowane z wyjaśnieniem ich znaczenia lub opisane.
Schemat ideowy przedstawiono na Rys. 4.
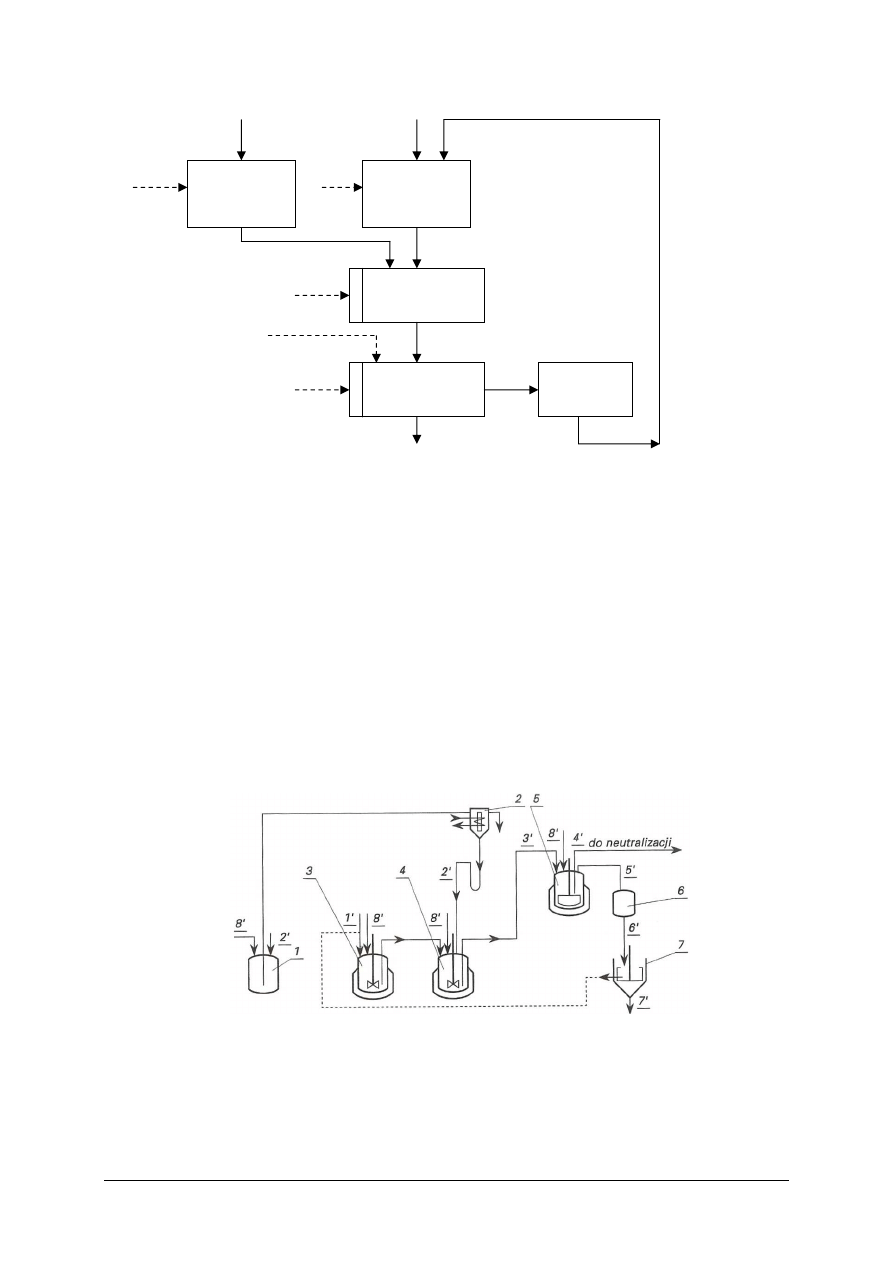
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
46
Rys. 4. Schemat ideowy sulfonowania naftalenu: 1 – naftalen, 2 i 3 – strumienie produktów sulfonowania,
4 – pary naftalenu, 5 – kwas siarkowy (VI), 6 – woda, 7 – para wodna [9]
Wstępny schemat technologiczny przedstawia proces za pomocą odpowiednich symboli –
kwadrat (prostokąt) oznaczał proces jednostkowy, co mogło odpowiadać nawet kilku
aparatom i urządzeniom, a na schemacie wstępnym są one wyodrębnione i przedstawione
oddzielnymi symbolami. Symbole schematu wstępnego podano w normie branżowej
BN-72/2200-01. Symbole graficzne i oznaczenia literowo-cyfrowe układów pomiarowych
i automatyki, stosowane przy projektowaniu i użytkowaniu schematów pomiarowych
i automatyzacji procesów technologicznych, podane są w normie PN – 70/M-42007.
Znormalizowane są również nazwy pojęć, symbole i określenia procesów podstawowych
inżynierii chemicznej, norma PN – 76/C-01350. Na wstępnym schemacie nie jest wymagane
określenie wielkości urządzeń i aparatów. Wykonując schemat wstępny, symbole szereguje
się w kolejności odpowiadającej przebiegowi procesu w kierunku od lewej ku prawej stronie
arkusza. Przykładowo wstępny schemat technologiczny pokazano na Rys. 5.
Rys. 5. Schemat wstępny sulfonowania naftalenu
Aparaty: 1 – przetłaczarka, 2 – miernik, dozownik, 3 – topielnik naftalenu,
4 – sulfonator, 5 – hydrolizatom, 6 – separator, łapacz naftalenu, 7 – wirówka.
Strumienie: 1’ – naftalenu, 2’ – kwasu siarkowego (VI), 3’ – strumień posulfonacyjny,
4’ – strumień pohydratacyjny, 5’ – par naftalenu i wody, 6’ – zawiesiny wodnej naftalenu,
7’ – wody, 8’ – powietrza sprężonego [9]
podgrzewanie
i dozowanie
H
2
SO
4
spalanie
naftalenu
395K
sulfonowanie
435K
hydroliza
410-420K
separacja
naftalenu
5
7
7
7
7
6
1
1
3
2
4

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
47
Schemat techniczny (techniczno-technologiczny) jest dalszym etapem graficznego, już
ścisłego, pokazania przebiegu procesu. Przebieg produkcji jest przedstawiony za pomocą
uproszczonych rysunków lub wyżej wymienionych symboli. Rysunki wykonywane są
w skali. W skali podane są również poziomy, na których rozmieszczone są aparaty
i urządzenia. Sposób rysowania jest analogiczny do schematu wstępnego z tym, że na
przewodach (rurociągach) podaje się średnice i ewentualnie długości. Zaznaczone są miejsca,
w których odbywa się pomiar ciśnienia, temperatury, stężenia itp. Przy sporządzaniu
schematu technicznego powinny być już określone: liczba, rodzaje, typy i wymiary aparatów
i urządzeń.
Woda w odpowiedniej ilości i odpowiedniej jakości jest potrzebna w wielu dziedzinach
bytowo-gospodarczych. Rozróżniamy kilka typów wód: wody gruntowe, wody
powierzchniowe, wody opadowe (deszczowe), wody morskie. Jakość oraz ilość wód określają
warunki geograficzne, klimatyczne i działalność człowieka.
W różnych gałęziach przemysłu woda stanowi: surowiec, reagent, rozpuszczalnik
substancji stałych, ciekłych i gazowych, absorbent, nośnik ciepła do chłodzenia i ogrzewania.
W przemyśle główne źródła zaopatrzenia w wodę stanowią wody powierzchniowe
i gruntowe. Stosuje się nazwy wód określając ich pochodzenie i przeznaczenie. Woda
nieuzdatniona to woda surowa, woda technologiczna (obiegowa) to woda krążąca
w instalacjach grzejnych i przemysłowych, woda kotłowa znajduje się w kotłach, wody
zrzutowe (kondensat lub skropliny) to wody usuwane na zewnątrz z instalacji
technologicznej. W technologiach przemysłowych szczególnie zalecanym rozwiązaniem jest
stosowanie obiegu zamkniętego (cyrkulacji).
Woda zawiera wiele substancji (zanieczyszczeń), które mogą stanowić cząstki
zawieszone lub rozpuszczone. Wody, zależnie od pochodzenia, mogą być zanieczyszczone
mechanicznie bądź chemicznie.
Oczyszczanie wody, zwane uzdatnianiem wody, obejmuje wszystkie procesy
mechaniczne i fizykochemiczne usuwania zanieczyszczeń. Rodzaj i stopień zanieczyszczeń
powodują, że oczyszczanie wód powierzchniowych różni się od oczyszczania wód
gruntowych. Oczyszczanie mechaniczne, czyli usuwanie zanieczyszczeń stałych, polega na
ich zatrzymaniu na kratach oraz sitach, koagulacji (flokulacji), sedymentacji lub filtracji.
Oczyszczanie chemiczne polega na wytrącaniu z wody, w postaci soli nierozpuszczalnych,
substancji powodujących jej twardość lub reakcji wymiany na sole rozpuszczalne,
niepowodujące tworzenia się kamienia kotłowego.
Twardość wody wynika z obecności różnego rodzaju soli. Usuwanie twardości wody
(zmiękczanie) metodą fizyczną prowadzi się poprzez podgrzanie wody do temperatury
65–90°C, w wyniku czego następuje rozkład wodorowęglanów, lub odparowaniu wody
w wyparkach oraz kondensacji i skropleniu pary wodnej. Zmiękczanie wody metodą
chemiczną prowadzi się z wykorzystaniem mleka wapiennego, sody kalcynowanej,
mieszaniny wapnia i sody, fosforanów (V) sodu oraz z wykorzystaniem jonitów. Twardość
przemijająca wynika z obecności w wodzie wodorowęglanów wapnia i magnezu, natomiast
twardość niewęglanowa wynika z obecności w wodzie siarczanów i chlorków wapnia
i magnezu.
Aeracja (napowietrzanie) jest stosowana do usuwania nadmiaru jonów żelaza i manganu
oraz usunięcia z wody twardości węglanowej. Aerację prowadzi się poprzez barbotaż
powietrza przez warstwę wody lub kontakt rozpylonej wody z powietrzem.
Chemiczne metody oczyszczania wody polegające na wytrącaniu soli wapnia i magnezu
mają obecnie coraz mniejsze znaczenie. Szerokie zastosowanie znalazły chemiczne metody
polegające na wymianie jonowej na jonitach. Rozróżniamy dwa rodzaje jonitów, kationity
i anionity. Kationity stosowane w przemyśle do zmiękczania wody stanowią najczęściej sieć
polistyrenową powiązaną poprzecznie, do której przyłączone są grupy sulfonowe. Ziarna

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
48
kationitu w formie sodowej umieszczone w cylindrycznym zbiorniku, po nasyceniu wodą
zdolne są do absorpcji i wymiany kationitów powodujących twardość wody.
2RSO
3
Na + Ca(HCO
3
)
2
→ (RSO
3
)
2
Ca + 2NaHCO
3
2RSO
3
Na + MgSO
4
→ (RSO
3
)
2
Mg + Na
2
SO
4
gdzie: R – rodnik.
Po wyczerpaniu zdolności jonowymiennych regenerację kationitu prowadzi się roztworem
chlorku sodu, w wyniku czego następuje wyparcie z kationitu jonów wapnia i magnezu.
W wyniku zastosowania jonitów do zmiękczania, stężenie jonów wapnia i magnezu osiąga
wartości poniżej 1mg/dm
3
w wodzie oczyszczonej.
Demineralizacja wody (dejonizacja) pozwala na usunięcie obok kationów wapnia
i magnezu również innych kationów i anionów. Zastosowanie kationitów z grupami
sulfonowymi powoduje konwersję wszystkich metali z soli zawartych w wodzie,
z wytrąceniem wolnych kwasów.
RSO
3
H + NaCl ↔ RSO
3
Na + HCl
W drugim etapie przepływ wody przez warstwę anionitu mającego grupy aminowe
o charakterze zasadowym, zdolnych do wymiany anionów, powoduje absorpcję wolnych
kwasów:
na anionitach słabo zasadowych
R–N(CH
3
)
2
+ HCl ↔ R–N(CH
3
)
2
HCl
na anionitach silnie zasadowych
R–N(CH
3
)
2
OH + HCl ↔ R–N(CH
3
)
2
Cl + H
2
O
Regeneracja anionitów odbywa się za pomocą roztworu wodorotlenku sodu. Anionity
silnie zasadowe stosowane są również do oczyszczania wody z rozpuszczonej krzemionki
i CO
2
.
Woda stosowana w przemyśle musi spełniać odpowiednie wymagania charakterystyczne
dla danej technologii, np. woda stosowana do chłodzenia powinna charakteryzować się jak
najmniejszą twardością aby unikać powstawania kamienia kotłowego, który może
spowodować uszkodzenie lub nieprawidłowe działanie instalacji.
Powietrze można rozpatrywać jako źródło surowców do otrzymywania gazów
przemysłowych. Czystość powietrza dla wielu technologii przemysłowych i dziedzin
gospodarki nie obejmuje usuwania zanieczyszczeń aerozolowych i gazowych. Powietrze
w wielu procesach stanowi substancję pomocniczą lub substrat, reagent, a także jest
surowcem do otrzymywania gazów przemysłowych, takich jak azot, tlen, argon, neon, ksenon
oraz krypton. Rozdzielenie powietrza na składniki wykonywane jest różnymi metodami
fizycznymi i chemicznymi. Na skalę przemysłową, najczęściej stosuje się rektyfikacje
powietrza ciekłego, skroplonego. Skraplanie powietrza metodą Claude’a znalazło największe
zastosowanie. Powietrze oczyszcza się w filtrze i chłodzi się bezprzeponowo strumieniem
wody w szeregu wymienników – reagentów, gdzie jest chłodzone gazami odlotowymi –
tlenem i azotem – do temperatury – 172˚C, co powoduje usunięcie w dużym stopniu wilgoci
i dwutlenku węgla. Ciekłe powietrze kierowane jest następnie do dolnej części kolumny
rektyfikacyjnej. Destylacja frakcyjna ciekłego powietrza odbywa się z wykorzystaniem różnic
temperatur wrzenia, przy bardzo niskiej temperaturze powoduje konieczność stosowania
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
49
specjalnych materiałów konstrukcyjnych do wyrobu aparatury procesowej i metod
chłodzenia. W wyniku destylacji otrzymuje się neon, argon, krypton i ksenon.
Związki azotowe w postaci kwasu azotowego (V) i amoniaku są jednymi z podstawowych
półproduktów stosowanymi we współczesnym przemyśle chemicznym. Związki azotanu (V)
amonu, mocznika, fosforanu (V) amonu są stosowane jako nawozy sztuczne, a także są
stosowane do wyrobu barwników, środków wybuchowych, żywic i innych.
Amoniak jest wytwarzany z węglowodorów, głównie metanu, lecz standardowym
procesem jest synteza amoniaku metodą Habera w zakresie temperatury 400–500ºC
i ciśnienia 8–35 MPa, zgodnie z reakcją:
N
2
+ 3H
2
2NH
3
Na szybkość reakcji tworzenia amoniaku ma wpływ aktywność stosowanego katalizatora.
Głównym surowcem tej metody jest gaz ziemny, konieczne jest wielostopniowe oczyszczenie
tego gazu. Z gazów syntetycznych muszą być usunięte duże ilości CO
2
, CO i H
2
O oraz
mniejsze ilości COS i H
2
S, a także popiół lotny. Przy wytwarzaniu gazów syntetycznych
z ropy naftowej lub gazu ziemnego, poprzez działanie pary wodnej w procesie reformingu
stopień konwersji węglowodoru do wodoru i tlenku węgla jest ograniczony równowagą
reakcji:
C
n
H
2n+2
+ nH
2
O nCO + (2n + 1)H
2
Proces ten jest prowadzony w reaktorach (piecach) kontaktowych rurowych. W rurkach
znajduje się katalizator niklowy na nośniku tlenku glinu lub tlenku magnezu. Dodatkowa
ilość ciepła do reaktora wprowadza się poprzez spalenie gazu ziemnego w przestrzeni miedzy
rurkami. W reaktorze, który pracuje w warunkach zbliżonych do adiabatycznych, powietrze
podawane jest w nadmiarze tak aby uzyskać porządny stosunek wodoru do azotu
w mieszaninie gazu kierowanej do syntezy amoniaku. W wyniku częściowego spalenia gazów
kierowany do reaktora tlen z powietrza jest używany na powstanie CO i CO
2
. w gazach po
konwersji metanu znajduje się 20 do 40% objętości CO. Konwersję tlenku węgla niezbędną
do uzyskania dodatkowej ilości wodoru prowadzi się w konwertorze półkowym
z wykorzystaniem katalizatora cynkowo-chromowo-miedzianego w zakresie temperatur
250 do 300ºC i pod nieco podniesionym ciśnieniem. Gaz syntetyczny po konwersji zawiera
pewną ilość nieprzereagowanego metanu, argonu a także tlenku węgla, dwutlenku węgla
i siarkowodoru, które są szkodliwe dla katalizatora syntezy amoniaku. System oczyszczania
gazów oparty jest na absorpcji zanieczyszczeń gazowych w roztworach alkaicznych,
w absorberach półkowych. Końcowe oczyszczenie gazów syntetycznych, polega na
dodatkowym usunięciu CO do stężenia poniżej 0,1 ppm poprzez metalizację na katalizatorze
niklowym osadzonym na nośniku tlenku glinu w temperaturze 300–350ºC. Metan i argon są
gazami obojętnymi w procesie syntezy amoniaku. Po ewentualnym uzupełnieniu gazów
syntetycznych wodorem i azotem pochodzącymi z innych źródeł są one sprężane. Mieszanina
azotowo-wodorowa kierowana jest do górnej części reaktora, gdzie na katalizatorze
żelazowym ulega przemianie w amoniak w stosunku do około 20%. Mieszanina gazów po
opuszczeniu reaktora zostaje ochłodzona w kondensatorze do skroplenia się amoniaku.
Po oddzieleniu amoniaku w separatorze gaz nieprzereagowany zawraca się ponownie do
reaktora. W celu zapewnienia stabilnych warunków pracy reaktora co pewien czas z gazów
obiegowych usuwane są gazy inertne (metan i argon) przez co ich stężenie utrzymuje się na
stałym niskim poziomie.
Kwas azotowy (V) można otrzymać, łącząc tlenek kwasowy z wodą:
H
2
O + N
2
O
5
→ 2HNO
3

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
50
Jest to jednak tylko teoretyczny proces, niemający praktycznego sensu. Na skalę
przemysłową kwas azotowy otrzymuje się w reakcji katalitycznego utleniania amoniaku
(w temperaturze 900°C w obecności platyny i rodu) według równania:
4NH
3
+ 5O
2
→ 4NO + 6H
2
O
Tak uzyskany tlenek azotu (II) utlenia się do tlenku azotu (IV):
2NO + O
2
→ 2NO
2
Następnie tlenek azotu (IV) rozpuszczony w wodzie ulega dysproporcjonowaniu
wg. równania:
2NO
2
+ H
2
O → HNO
3
+ HNO
2
W reakcji powstaje kwas azotowy (V) i kwas azotowy (III). Ten ostatni jest nietrwały
i ulega dalszemu dysproporcjonowaniu według równania:
3HNO
2
→ HNO
3
+ 2NO + H
2
O
Powstały tlenek azotu (II) jest ponownie utleniany i wykorzystywany w procesie. Metodę
otrzymywania kwasu azotowego z tlenku azotu opracował Ignacy Mościcki.
Katalizator charakteryzuje się wysoką selektywnością, co zapewnia stopień konwersji
ponad 95%, pod ciśnieniem wyższym od 1 MPa. Proces ten jest egzotermiczny. Chłodzenie
kwasu azotowego (V) do temperatury -2ºC jest konieczne, jeżeli chce się wytwarzać kwas
o stężeniu powyżej 65%.
Przetwórstwo surowców naturalnych obejmuje dużą grupę substancji, materiałów,
w których znajdują się surowce odnawialne i nieodnawialne.
Surowce zwierzęce to przede wszystkim tłuszcze, woski i białka. Tłuszcze zwierzęce
obejmują różnorodne produkty zależne od źródła pozyskania a także od rodzaju i pochodzenia
tłuszczu. Tłuszcze dzielą się na jadalne (spożywcze) oraz na tłuszcze techniczne. Tłuszcze
techniczne używane są do produkcji mydeł, gliceryny, stearyny, smarów, pokostów. Tłuszcze
ulegają hydrolizie i w reakcji z wodorotlenkiem sodu przechodzą w sól sodową kwasu
tłuszczowego, co określa się mianem mydła oraz w glicerol. Aby oddzielić mydło od
glicerolu do mieszaniny reakcyjnej dodaje się sól kamienną. Produkcja mydła może być
prowadzona przez działanie na tłuszcz parą wodną w autoklawie pod wysokim ciśnieniem
0,8 MPa i w temperaturze 170ºC. Otrzymuje się wówczas kwas tłuszczowy wysokiej
czystości oraz glicerol. Działając następnie na kwas tłuszczowy węglanem sodu lub potasu
wytwarza się mydło.
2C
15
H
31
−COOH + Na
2
CO
3
→ 2C
15
H
31
−COONa + CO
2
+ H
2
O
Mydło o odpowiedniej jakości (wyrób handlowy) otrzymuje się w wyniku dalszych
operacji, w dużej mierze mechanicznych.
Ze zwierząt morskich produkuje się oleje rybne i tran. Charakteryzują się one wysoka
zawartością specyficznych, nienasyconych kwasów tłuszczowych. Oleje rybne stanowią
doskonały surowiec do otrzymywania olejów specjalnych (sulfatowanych), używanych
w przemyśle włókienniczym i skórzanym. Tran,otrzymywany przez ekstrakcję z wątroby oraz
z tłuszczu ryb i ssaków morskich, jest wykorzystywany w celach leczniczych, a także jako
surowiec w przemyśle spożywczym i garbarskim.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
51
Surowce roślinne stanowią rośliny zbożowe, oleiste, włókniste, okopowe, a także
produkty roślinne takie jak drewno, kauczuk naturalny, wosk, tłuszcze i białka roślinne
rośliny do produkcji substancji zapachowych, garbników ziół i lekarstw.
Surowce roślinne wykorzystywane są w wielu gałęziach przemysłu, takich jak przemysł
gorzelniczy, spożywczy, paliwowy oraz wiele innych.
4.4.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonywania ćwiczeń.
1.
Jakie czynniki wpływają na stan równowagi i wydajność reakcji chemicznych?
2.
Jak nazywa się materiały biorące udział w procesie technologicznym?
3.
Jakie procesy najczęściej stosowane są w przemyśle chemicznym i przetwórczym?
4.
Co nazywa się ciągiem technicznym?
5.
Jakie wyróżnia się procesy podstawowe i jednostkowe?
6.
Jakie są zasady najlepszego wykorzystania energii?
7.
Na czym polega uzdatnianie wody przemysłowej?
8.
Jak przebiega proces skraplania powietrza metodą Claude’a?
4.4.3. Ćwiczenia
Ćwiczenie 1
Porównaj efektywność metod zmiękczania wody stosowanej jako chłodziwo
w technologii produkcji styrenu. Określ poziom zagrożenia spowodowany ewentualną awarią
podgrzewacza pary wynikającą z zastosowania wody twardej.
Celem ćwiczenia jest zbadanie skuteczności zmiękczania wody wodociągowej metodą
termiczną (gotowanie i destylowanie) i metodą wymiany jonitowej.oraz określenie poziom
zagrożenia spowodowany ewentualną awarią podgrzewacza pary wynikającą z braku kontroli
stanu technicznego urządzenia.
Związki chemiczne: próbki wody do uzdatniania, 0,05 mol/dm
3
roztwór EDTA (wersenian
sodu), bufor amoniakalny pH = 10, czerń eriochromowa.
Styren produkuje się na skalę przemysłową, gdyż przerabiany jest na polistyren oraz
kauczuk butadienowo-styrenowy. Styren otrzymuje się przez odwodornienie etylobenzenu,
który produkuje się przez alkilowanie benzenu etylenem. Należy rygorystycznie przestrzegać
zasad kontrolowania stanu technicznego stosowanych urządzeń.
Przebieg procesu (Rys. 6): etylobenzen odparowuje się w odparowalniku 2 ogrzewanych
ciepłem gazów spalinowych z pieca1.W wymiennikach ciepła 4 zostaje podgrzany do 700°C
i miesza z parami etylobenzenu przed aparatem kontaktowym 3, w którym następnie zachodzi
katalityczne uwodornienie etylobenzenu. Otrzymany styren schładza się w odbieralnikach 4
i 5. W oddzielaczu 6 skraplają się produkty smołowe. Po kolejnym schłodzeniu skropliny
styrenu oddziela się i gromadzi w zbiorniku 7. Przed rektyfikacją dodaje się siarkę, aby
zahamować proces polimeryzacji. Po rektyfikacji czysty styren przekazuje się do dalszego
przetwarzania, a zanieczyszczony zawraca do procesu.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
52
.
Rysunek do ćwiczenia 1. Schemat produkcji styrenu: 1 – podgrzewacz pary, 2 – odparowalnik etylobenzenu,
3 – aparat kontaktowy, 4 – wymienniki ciepła, 5 – chłodnice, 6 – oddzielacze, 7 – zbiornik styrenu surowego,
8 – pompa, 9 – kolumna rektyfikacyjna [5]
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
pobrać 50 cm
3
badanej wody do kolbki stożkowej,
2)
dodać 5 cm
3
buforu o pH = 10,0 oraz szczyptę czerni eriochromowej,
3)
zmiareczkować próbkę 0,05 molowym roztworem wersenianu do momentu, gdy
fiołkowo-czerwona barwa zmieni się na niebieską,
Zawarte w wodzie jony Mg
+2
oraz Ca
+2
reagują z wersenianem sodu (EDTA) dając
połączenia kompleksowe. Wprowadzona we wstępnej fazie do wody czerń eriochromowa
daje również połączenia kompleksowe z jonami Mg
+2
, przy czym połączenie to jest
barwne, ale słabsze od połączenia z EDTA; na początku analizy roztwór jest więc
barwny. Podczas miareczkowania (dodawania roztworu EDTA) najpierw jony Ca
+2
,
a później jony Mg
+2
zostają związane przy pomocy EDTA. Gdy już cała ilość Mg
+2
zostanie związana z EDTA, zabarwienie roztworu zmieni się z powodu zniknięcia
połączenia czerni eriochromowej z Mg
+2
. Moment zmiany zabarwienia oznacza koniec
miareczkowania,
4)
obliczyć stężenie jonów Mg
+2
oraz Ca
+2
w badanym roztworze wiedząc, że na każdy jon
Mg
+2
lub Ca
+2
zużyto jedną cząsteczkę EDTA:
a.
V
M
V
M
M
EDTA
EDTA
⋅
⋅
=
⋅
⋅
=
20
50
1000
gdzie:
M
– szukana molowo
ść
roztworu (twardo
ść
wody) [mol/dm
3
]
M
EDTA
– molowo
ść
roztworu EDTA [mol/dm
3
]
V
– obj
ę
to
ść
(w cm
3
) roztworu EDTA zu
ż
ytego na zmiareczkowania próbki
5)
oznaczenie powiniene
ś
wykona
ć
trzykrotnie, za wynik przyj
ąć
ś
redni
ą
arytmetyczn
ą
pomiarów,
6)
doprowadzi
ć
do wrzenia 200 cm
3
wody, stan wrzenia utrzyma
ć
przez 10 min, ostudzi
ć
do
temperatury pokojowej, przefiltrowa
ć
przez s
ą
czek, ponownie oznaczy
ć
twardo
ść
wody
metod
ą
miareczkowania EDTA w sposób opisany powy
ż
ej,
para
1
2
etylobenzen
3
4
4
5
5
6
6
7
8
9
styren
siarka
smoła
woda
woda
para
gazy odlotowe
4

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
53
7)
przesączyć 200 cm
3
wody surowej przez kolumny z anionitem, a następnie z kationitem
(poziom wody w kolumnach z jonitami powinien być zawsze wyższy, niż poziom
jonitów), powtórnie oznaczyć twardość wody teraz tej zdemineralizowanej przez
miareczkowanie EDTA (trzykrotne miareczkowanie, wynik średni),
8)
oznaczyć twardość wody wodociągowej i destylowanej metodą miareczkowania EDTA,
9)
porównać skuteczność stosowanych metod oczyszczania (zmiękczania i demineralizacji)
wody opierając się na wynikach miareczkowania,
10)
określić poziom zagrożenia ewentualną awarią podgrzewacza pary wynikającą z jego
zużycia a spowodowany brakiem okresowej kontroli stanu technicznego urządzenia.
Wyposażenie stanowiska pracy:
–
kolumny z jonitem,
–
maszynka elektryczna, płytka izolacyjna,
–
szkło laboratoryjne: kolbka stożkowa 250 cm
3
(do grzania wody), szkiełko zegarkowe,
kolbki do miareczkowania 125 cm
3
(3 szt.), zlewki 100 cm
3
(2 szt.), zlewka 25 cm
3
,
pipeta 50 cm
3
, pipeta miarowa 10 cm
3
, biureta ze statywem, lejek, cylinder 10 cm
3
lub
25 cm
3
,
–
papierki uniwersalne, sączki,
–
statyw do sączenia,
–
łopatka metalowa,
–
kalendarz chemiczny.
Ćwiczenie 2
Wykonaj schemat wstępny otrzymywania bezwodnika ftalowego metodą utleniania
naftalenu na podstawie schematu ideowego i opisu przebiegu. Scharakteryzuj procesy
i operacje. Podaj stosowane podstawowe zasady technologiczne.
Bezwodnik kwasu ftalowego jest jednym z ważniejszych półproduktów w produkcji
barwników i tworzyw sztucznych. Otrzymuje się go przez katalityczne utlenianie naftalenu.
Czynnikiem utleniającym jest powietrze pod normalnym ciśnieniem w temperaturze 625–675 K.
Naftalen w stanie pary o temperaturze ok. 352 K jest mieszany ze strumieniem gorącego
powietrza w mieszalniku 7. Oczyszczone i sprężone w sprężarce 9 powietrze podgrzewane
jest w wymienniku ciepła 6 strumieniem produktów opuszczających reaktor 8. Reaktor,
aparat kontaktowy stanowi zespół pionowych rurek wypełnionych nieruchomym
katalizatorem, tlenkiem wanadu (V) V
2
O
5
osadzonym na nośniku krzemionkowym. Ziarna
katalizatora mogą stanowić złoże fluidalne w reaktorze fluidalnym. Proces prowadzony jest w
temperaturze 625–675 K pod normalnym ciśnieniem. Gazy poreakcyjne, oddając część ciepła
w wymienniku 2, kierowane są do komór kondensacyjnych 6, gdzie następuje kondensacja
i krystalizacja bezwodnika ftalowego w temperaturze około 405 K. Oczyszczanie surowego
bezwodnika odbywa się zwykle metodą destylacji próżniowej (temperatura wrzenia 557 K)
(Rys. 7)
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
54
Rysunek do ćwiczenia 2. Schemat ideowy otrzymywania bezwodnika ftalowego metodą utleniania naftalenu.
Strumienie: 1 – para, 2 – naftalen,3 – powietrze, 4 – bezwodnik kwasu ftalowego.
Aparaty: 5 – topielnik, 6 – wymiennik ciepła, podgrzewacz, 7 – mieszalnik gazów, par,
8 – reaktor katalityczny, 9 – sprężarka, 10 – zespół komór kondensacyjnych [10, s. 113,114]
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1)
zapoznać się zasadą prowadzenia procesu technologicznego otrzymywania bezwodnika
ftalowego metodą utleniania naftalenu,
2)
zapoznać się ze schematem ideowy otrzymywania bezwodnika ftalowego metodą
utleniania naftalenu,
3)
skorzystać z norm: branżowej BN-72/2200-01 podającej symbole schematu wstępnego,
normy PN-70/M-420007 podającej symbole graficzne i oznaczenia literowo-cyfrowe
układów pomiarowych i automatyki, normy PN-76/C01350 podającej nazwy pojęć,
symbole i określenia procesów podstawowych inżynierii chemicznej,
4)
sporządzić wstępny schemat technologiczny, przedstawiając proces za pomocą
odpowiednich symboli aparatów i urządzeń,
5)
scharakteryzować procesy i operacje przedstawione na schemacie oraz podstawowe
zasady prowadzenia procesu technologicznego,
6)
zaproponować sposób jakościowego oznaczenia czystości otrzymanych kryształów
bezwodnika ftalowego,
7)
scharakteryzować zasady bezpiecznego prowadzenia procesu i przewidzieć skutki
ewentualnego wybuchu spowodowanego zastosowaniem źle dobranego składu czynnika
utleniającego.
Wyposażenie stanowiska pracy.
–
normy: PN-70/M-420007, PN-76/C01350, branżowa BN-72/2200-01,
–
instrukcja do ćwiczenia wraz ze schematem ideowym,
–
Internet, literatura techniczna,
–
arkusze papieru, przyrządy kreślarskie.
para
para
naftalen
powietrze
bezwodnik kwasu ftalowego

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
55
4.4.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1)
wymienić i scharakteryzować podstawowe zasady technologiczne?
2)
scharakteryzować zasadę najlepszego wykorzystania surowców?
3)
wymienić przyczyny zanieczyszczenia wód przeznaczonych dla
przemysłu i omówić metody usuwania obecnych w wodach
zanieczyszczeń?
4)
wymienić składniki powietrza i przedstawić metody rozdziału tej
mieszaniny?
5)
na podstawie schematu produkcji styrenu przedstawić przebieg
procesu?
6)
wymienić i scharakteryzować sposoby uzdatniania wody?
7)
przedstawić schematu ideowego sulfonowania naftalenu?
8)
przedstawić
symbole
filtrów
stosowane
na
schematach
technologicznych?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
56
5. SPRAWDZIAN OSIĄGNIĘĆ
INSTRUKCJA DLA UCZNIA
1.
Przeczytaj uważnie instrukcję.
2.
Podpisz imieniem i nazwiskiem kartę odpowiedzi.
3.
Zapoznaj się z zestawem zadań testowych.
4.
Udzielaj odpowiedzi na załączonej karcie odpowiedzi, wstawiając w odpowiedniej
rubryce znak X. W przypadku pomyłki należy błędną odpowiedź zaznaczyć kółkiem,
a następnie ponownie zaznaczyć odpowiedź prawidłową.
5.
Test zawiera 20 zadań, w tym: 15 z poziomu podstawowego i 5 z poziomu
ponadpodstawowego.
6.
Do każdego zadania dołączone są 4 możliwe odpowiedzi. Tylko jedna jest prawidłowa.
7.
Pracuj samodzielnie, bo tylko wtedy będziesz miał satysfakcję z wykonanego zadania.
8.
Kiedy udzielanie odpowiedzi będzie Ci sprawiało trudność, wtedy odłóż jego rozwiązanie
na później i wróć do niego, gdy zostanie Ci wolny czas.
9.
Na rozwiązanie testu masz 40 minut.
Powodzenia
ZESTAW ZADAŃ TESTOWYCH
1.
Legalizacja przyrządu pomiarowego polega na
a)
sprawdzeniu go za pośrednictwem wzorca.
b)
sprawdzeniu jednej czynności przyrządem kontrolnym o znanej i większej
dokładności od dokładności przyrządu legalizowanego.
c)
znalezieniu jego właściwych cech w Dzienniku Urzędowym lub Polskich Normach.
d)
sprawdzeniu właściwości metrologicznych przyrządu i jego ocechowaniu lub wydaniu
świadectwa legalizacyjnego przez wyspecjalizowane instytucje.
2.
Błąd bezwzględny to
a)
różnica algebraiczna pomiędzy wynikiem pomiaru a wartością wielkości mierzonej.
b)
błąd powtarzający się przy kolejnych pomiarach pod wpływem zmieniających się
czynników zewnętrznych np. temperatury, ciśnienia, wilgotności itp.
c)
błąd powstający w wyniku działania na przyrząd nieuchwytnych zmian warunków
zewnętrznych.
d)
błąd wynikający z indywidualnych właściwości obserwatora i jego niedoskonałości.
3.
Obliczony błąd względny ważenia, na wadze analitycznej, wykonanego z dokładnością do
0,0001 g dla próbki o masie 0,1549 wynosi
a)
– 0,060%.
b)
± 0,072%.
c)
± 0,065%.
d)
+ 0,060%.
4.
Do pomiaru ciśnienia atmosferycznego służą
a)
wakuometry.
b)
manometry.
c)
barometry.
d)
manowakuometry.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
57
5.
W przyrządach hydrostatycznych, np. „U” rurce lub manometrze rtęciowym, do pomiaru
ciśnienia wykorzystywana jest zasada
a)
odkształcenia elementu sprężystego.
b)
zmiany ciśnienia powodującej przesunięcie tłoka.
c)
zmiany właściwości materiału pod wpływem zmiany ciśnienia.
d)
równoważenia mierzonego ciśnienia ciśnieniem hydrostatycznym słupa cieczy.
6.
Do grupy podstawowych procesów dyfuzyjnych, związanych z przemianami fizycznymi
i chemicznymi nie należy
a)
rektyfikacja.
b)
ekstrakcja
c)
przesiewanie
d)
sublimacja.
7.
Temperatura wrzenia wody, w warunkach normalnych, w skali Kelwina wynosi
a)
273 K.
b)
373 K.
c)
223 K.
d)
323 K.
8.
W termometrach oporowych metalowe uzwojenie czujnika powinno odpowiadać
warunkowi
a)
zmianom temperatury powinny towarzyszyć nieznaczne zmiany oporu elektrycznego.
b)
w zakresie stosowania powinny zmieniać się właściwości fizyko–chemiczne.
c)
zmiany oporu elektrycznego powinny być proporcjonalne do zmian temperatury.
d)
temperatura topnienia lub przemian alotropowych nie wpływa na zakres stosowalności
termometru.
9.
Do pomiaru ilości przepływającej cieczy przez rurociąg nie stosuje się
a)
przepływomierzy komorowych.
b)
przepływomierzy skrzydełkowych.
c)
przepływomierzy śrubowych.
d)
przepływomierzy wirnikowych.
10.
W analizie polarograficznej nie można oznaczyć
a)
stopów metali wysokiej czystości.
b)
związków organicznych, które ulegają reakcjom elektrodowych.
c)
węglowodorów.
d)
rud metali.
11.
Potencjał półfali nie zależy od
a)
rodzaju badanego jonu.
b)
stężenia badanego jonu.
c)
pH roztworu, w którym prowadzone jest badanie.
d)
obecności substancji kompleksujących.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
58
12.
Promieniowanie α to
a)
strumień szybkich elektronów emitowanych z jądra atomu w wyniku zachodzących
w nim przemian.
b)
emisja promieniowania w postaci fali elektromagnetycznej.
c)
dodatnio naładowana cząstka, wyrzucona z jądra atomowego składająca się z dwóch
protonów i dwóch neutronów.
d)
emisja nowych neutronów wyrzuconych podczas tzw. rozszczepienia jądra przez
neutron pierwotny, znajdujący się w pobliżu tego jądra izotopu.
13.
Plateau licznika do pomiarów radiometrycznych to
a)
inna nazwa popularnego licznika Geigera-Müllera.
b)
zakres napięć, w którym liczba rejestrowanych impulsów zależy od przyłożonego
napięcia.
c)
wartość napięcia, przy którym układ pomiarowy rejestruje pierwszy impuls.
d)
zakres napięcia, przy którym liczba rejestrowanych impulsów nie zależy od
przyłożonego napięcia.
14.
Przyrządem do pomiaru natężenia przepływu nie jest
a)
pływakowy wskaźnik poziomu.
b)
zwężka znormalizowana.
c)
rotametr.
d)
przepływomierz komorowy.
15.
Podczas elektrolizy wodnego roztworu siarczanu (VI) miedzi (II) wydziela się
a)
na katodzie miedź na anodzie trójtlenek siarki.
b)
na katodzie wodór na anodzie tlen.
c)
na katodzie miedź na anodzie tlen.
d)
na katodzie miedź na anodzie siarka.
16.
Wskaż zdanie fałszywe
a)
na katodzie zachodzi proces redukcji np. wydzielanie się wodoru.
b)
elektrodę, która pobiera elektrony nazywamy katodą.
c)
w procesie elektrolizy na anodzie następuje utlenianie związków organicznych.
d)
w procesie elektrolizy po przyłożeniu źródła prądu stałego następuje wędrówka
kationów do katody a anionów do anody.
17.
Elektrolizę prowadzono prądem o natężeniu 10 amperów. Podczas elektrolizy stopionego
chlorku ołowiu (II) w czasie 20 minut, przyjmując M
Pb
= 207,2 u, ołowiu wydzieliło się na
katodzie
a)
12,882 g.
b)
25,76 g.
c)
0,215 g.
d)
0,043 g.
18.
Urządzenia do mieszania ciał ciastowatych i plastycznych to
a)
mieszarki.
b)
zgniatarki.
c)
mieszalniki.
d)
separatory.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
59
19.
W chemicznym procesie przetwarzania węgla (koksowania) nie otrzymuje się
a)
smoły.
b)
wody pogazowej.
c)
benzenu surowego.
d)
gazoliny.
20.W procesie polikondensacji nie otrzymuje się
a)
z kaprolaktamu – poliamidu.
b)
z związków krzemoorganicznych – polisiloksanów.
c)
z amin, mocznika i melaminy – aminoplastów.
d)
z formaldehydu i fenolu – fenoplastów.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
60
KARTA ODPOWIEDZI
Imię i nazwisko...........................................................................................................................
Wykonywanie pomiarów parametrów procesowych
Zakreśl poprawną odpowiedź.
Nr
zadania
Odpowiedź
Punkty
1.
a
b
c
d
2.
a
b
c
d
3.
a
b
c
d
4.
a
b
c
d
5.
a
b
c
d
6.
a
b
c
d
7.
a
b
c
d
8.
a
b
c
d
9.
a
b
c
d
10.
a
b
c
d
11.
a
b
c
d
12.
a
b
c
d
13.
a
b
c
d
14.
a
b
c
d
15.
a
b
c
d
16.
a
b
c
d
17.
a
b
c
d
18.
a
b
c
d
19.
a
b
c
d
20.
a
b
c
d
Razem:

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
61
6. LITERATURA
1.
Dramińska K., Różycki C., Tymiński B.: Pracownia pomiarów technicznych w przemyśle
chemicznym. WSiP, Warszawa 1987
2.
Jarosz M., Malinowska E.: Pracownia chemiczna. Analiza instrumentalna. WSiP,
Warszawa 1994
3.
Klepaczko-Filipiak B., Łoin J.: Pracownia chemiczna. Analiza techniczna, WSiP,
Warszawa 1994
4.
Łada Z. Różycki C.: Pracownia chemii analitycznej. Analiza techniczna i instrumentalna.
WSiP, Warszawa 1990
5.
Molenda J.: Technologia chemiczna. WSiP, Warszawa 1996
6.
Praca zbiorowa: Aparatura kontrolno-pomiarowa w przemyśle chemicznym. WSiP,
Warszawa 1993
7.
Szczepaniak W.: Metody instrumentalne w analizie chemicznej. PWN, Warszawa 2005
8.
Tuszyński K.: Pomiary i automatyka w przemyśle chemicznym. WSiP, Warszawa 1995
9.
Warych J.: Aparaty i urządzenia przemysłu chemicznego i przetwórczego. WSiP,
Warszawa 1996
10.
Warych J.: Podstawowe procesy przemysłu chemicznego i przetwórczego. WSiP,
Warszawa 1996
11.
Witkiewicz Z.: Podstawy chromatografii. WNT, Warszawa 1995
Wyszukiwarka
Podobne podstrony:
11 Wykonywanie pomiarow paramet Nieznany (2)
04 Wykonywanie pomiarów parametrów procesowych
04 Wykonywanie pomiarów parametrów procesowych
04 Wykonywanie izolacji termicz Nieznany (2)
10 Wykonywanie pomiarow krawiec Nieznany
04 Wykonywanie podstawowych for Nieznany (2)
04 Wykonywanie analiz ilosciowy Nieznany (2)
04 Wykonywanie zabiegow agrotec Nieznany (2)
08 Wykonywanie pomiarow warszta Nieznany (2)
19 Wykonywanie pomiarow realiza Nieznany (2)
04 Wykonywanie obliczen w uklad Nieznany
04 Wykonywanie pomiarów
04 Wykonywanie izolacji termicz Nieznany (2)
więcej podobnych podstron