BUDOWA CIAŁA STAŁEGO.
STRUKTURA KRYSZTAŁÓW
Podstawowe
rodzaje
materii,
pierwiastki
i związki chemiczne, występują w zależności od
ciśnienia i temperatury w trzech stanach skupienia.
Każdy
z
tych
stanów
jest
odrębną
fazą
o specyficznych cechach i właściwościach fizycznych.
Istotną cechą danego rodzaju materii w różnych
stanach skupienia jest stopień uporządkowania drobin
– tj. atomów, cząsteczek lub jonów.
3
Stany skupienia substancji czystej:
W
zr
o
st
u
p
o
rz
ąd
k
o
w
a
n
ia
d
ro
b
in
W
zr
o
st
te
m
p
er
a
tu
ry
(P
=
c
o
n
st
)
W
zr
o
st
ci
śn
ie
n
ia
(T
=
c
o
n
st
)
Gazowy
Ciekły
Stały
(alotropia, polimorfizm)
Szklisty
Ciekłokrystaliczny
Amorficzny
Rys. 1. Stany skupienia materii
W zależności od warunków zewnętrznych stany
skupienia, czyli fazy danego rodzaju materii, są
wzajemnie powiązane następującymi relacjami:
faza gazowa ↔ faza ciekła, skraplanie ↔ parowanie;
faza gazowa ↔ faza stała, kondensacja ↔ sublimacja;
faza ciekła ↔ faza stała, krzepnięcie ↔ topnienie.
Gazy i ciecze (płyny)
W stanie gazowym nie ma jakiekolwiek
uporządkowania, ponieważ wzajemne oddziaływania
cząsteczek siłami van der Waalsa są zbyt słabe.
Cząsteczki gazu mają swobodę rotacji i translacji,
ale w wyniku wzajemnych zderzeń poruszają się
chaotycznie po trajektoriach zygzakowatych (ruchy
Browna).
Gazy są ściśliwe i mają małą gęstość. Z powodu
braku własnego kształtu zawsze wypełniają
równomiernie całą objętość naczynia, w którym się
znajdują. Przy ustalonych warunkach zewnętrznych,
jednakowe objętości różnych gazów zawierają tę
samą liczbę atomów lub cząsteczek (prawo Avogadro
dla gazów). Gazy jako płyny mają zdolność do
przepływu w kierunku gradientu ciśnienia.
Właściwości
fizyczne
gazów,
np.
lepkość,
przewodnictwo cieplne, współczynnik załamania
światła, są jednakowe w całej objętości, tzn. nie

zależą od wyróżnionego kierunku w przestrzeni,
dlatego wszystkie gazy są ciałami izotropowymi.
Oddziaływania międzycząsteczkowe w cieczach są
silniejsze niż w fazie gazowej, wskutek czego w fazie
ciekłej występuje uporządkowanie bliskiego zasięgu.
Zasięg
tego
uporządkowania
jest
największy
w temperaturze bliskiej temperatury krzepnięcia
i zanika w temperaturze wrzenia. Ciecze są trudno
ściśliwe i mają znacznie większą gęstość niż gazy.
Każda ciecz, podobnie jak gazy, nie ma własnego
kształtu i wypełnia równomiernie naczynie pod
warunkiem, że jej własna objętość nie jest większa od
objętości naczynia. Ciecze jako płyny są zdolne do
przepływu w kierunku gradientu ciśnienia lub różnicy
wysokości. Powierzchnia swobodna cieczy jest
powierzchnią graniczną jej styku z fazą gazową (np.
powietrzem lub parą). Wielkość tej powierzchni zależy
od objętości cieczy oraz od wielkości i geometrii
zbiornika, w której się ona znajduje. Pomimo
uporządkowania bliskiego zasięgu, cząsteczki w fazie
ciekłej są równomiernie rozproszone w wyniku
przypadkowych ruchów translacyjnych, rotacyjnych
i wzajemnych zderzeń. Zatem ciecze są izotropowe,
ponieważ ich właściwości fizyczne nie zależą od
jakiekolwiek wyróżnionego kierunku. Wyjątkiem od
tej reguły są substancje ciekłokrystaliczne wykazujące
w fazie ciekłej anizotropię, czyli zależność niektórych
właściwości fizycznych od kierunku wyróżnionego w
przestrzeni. Cząsteczki tych związków mają wydłużony,
wrzecionowaty kształt i są usztywnione przez wiązania
wielokrotne, a ich kryształy po stopieniu przechodzą

w stan ciekłokrystaliczny o uporządkowaniu dalekiego
zasięgu.
Przykładem substancji ciekłokrystalicznej jest
p-azoksyanizol (PAA): CH
3
O-C
6
H
4
-N=NO-C
6
H
4
OCH
3
o temperaturze topnienia 391 K i temperaturze
przejścia w ciecz izotropową 408 K. Wzajemna
orientacja cząsteczek w fazie ciekłokrystalicznej jest
uporządkowaniem dalekiego zasięgu:
Takie uporządkowanie jest przyczyną anizotropii
właściwości optycznych
ciekłych kryształów, co
wykorzystuje się monitorach komputerowych typu
LCD, w czytnikach urządzeń elektronicznych, np.
kalkulatorów, telefonów komórkowych, itp.
Ciała stałe
Pierwiastki i związki chemiczne w stanie stałym
mają najczęściej budowę krystaliczną, polegającą na
regularnym
i
okresowo
powtarzającym
się
rozmieszczeniu
przestrzennym
określonych
konfiguracji atomów (pierwiastki) i cząsteczek lub
jonów (związki). Taki sposób rozmieszczenia atomów,
cząsteczek lub jonów jest uporządkowaniem dalekiego
zasięgu w sieci przestrzennej kryształu i może pociągać
za sobą anizotropię niektórych właściwości fizycznych
jednorodnej substancji krystalicznej, np. wytrzymałości
mechanicznej, rozszerzalności cieplnej, przewodnictwa
elektrycznego,
współczynnika
załamania
światła
i innych
.
Od ciał krystalicznych należy odróżnić ciała
stałe bezpostaciowe - amorficzne, takie jak szkła,
smoły, żywice. Ciała bezpostaciowe wykazujące co
najwyżej tylko dość przypadkowe uporządkowanie
bliskiego zasięgu, są izotropowe i można je
traktować
jak
przechłodzone
ciecze.
Ciała
krystaliczne
mają
ściśle
określoną,
„ostrą”
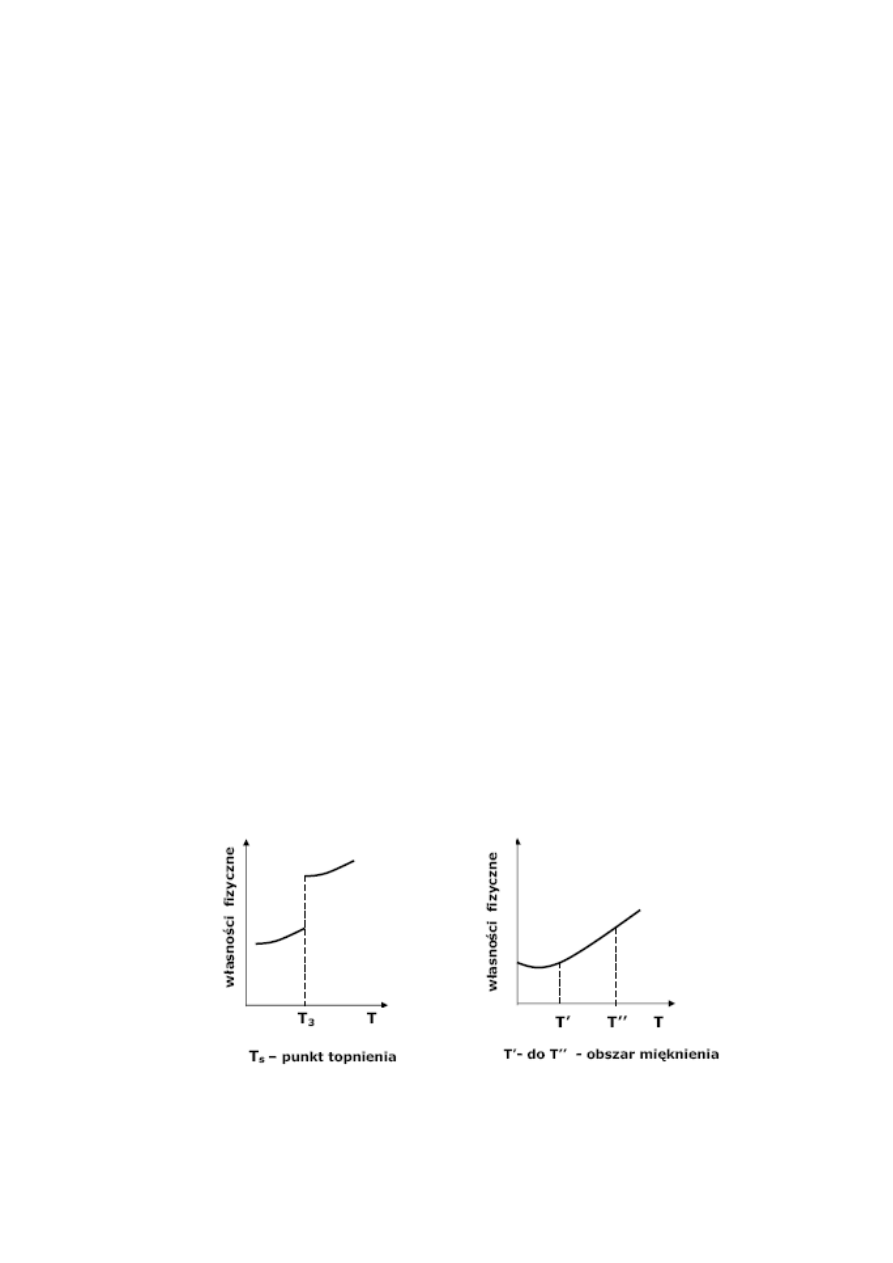
temperaturę topnienia, a ich właściwości po
przejściu w stan ciekły zmieniają się skokowo.
Inaczej wygląda topnienie ciał bezpostaciowych,
które nie mają „ostrej” temperatury topnienia.
Przejście ciał bezpostaciowych w stan ciekły jest
poprzedzone fazą mięknienia, dzięki czemu ich
właściwości fizyczne zmieniają się w sposób ciągły
(rys. 2).
Rys. 2. Topnienie ciał stałych: ciała krystalicznego (z lewej)
i ciała bezpostaciowego (z prawej)
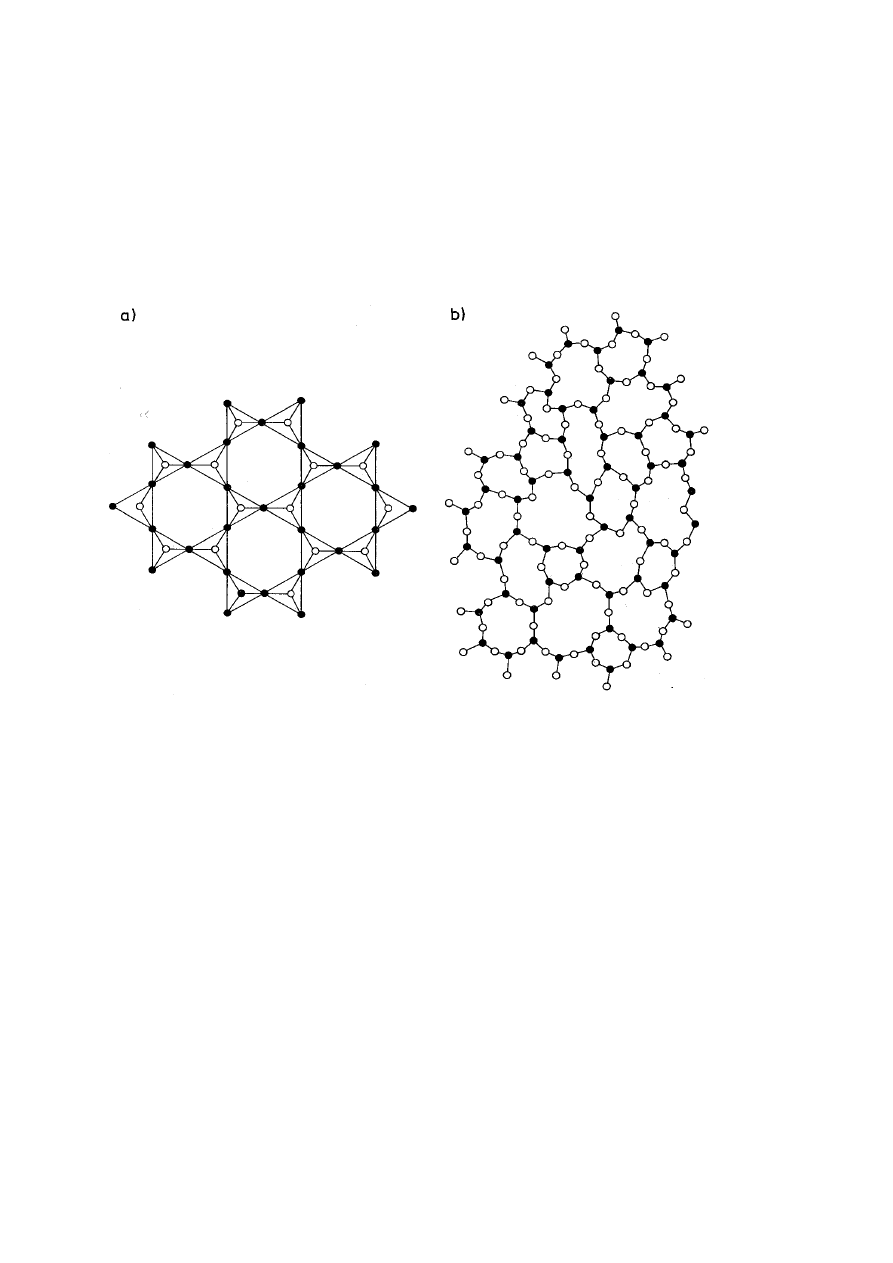
Porównanie uporządkowania dalekiego zasięgu
tetraedrycznych ugrupowań SiO
4
w kryształach
kwarcu (SiO
2
) i bardzo przypadkowego rozmieszczenia
tych ugrupowań w szkle kwarcowym przedstawia rys.
3.
Rys. 3. Uporządkowane (a) i nieregularne (b) rozmieszczenie
tetraedrów SiO
4
w krysztale kwarcu i szkle kwarcowym
Właściwości fizyczne substancji krystalicznych
zależą od rodzaju wiązań chemicznych działających
w ich sieciach przestrzennych. Z tego powodu
wygodnie jest wprowadzić podział kryształów na
następujące rodzaje:
a) kryształy molekularne,
b) kryształy kowalencyjne,
c) kryształy jonowe,
d) kryształy metaliczne.
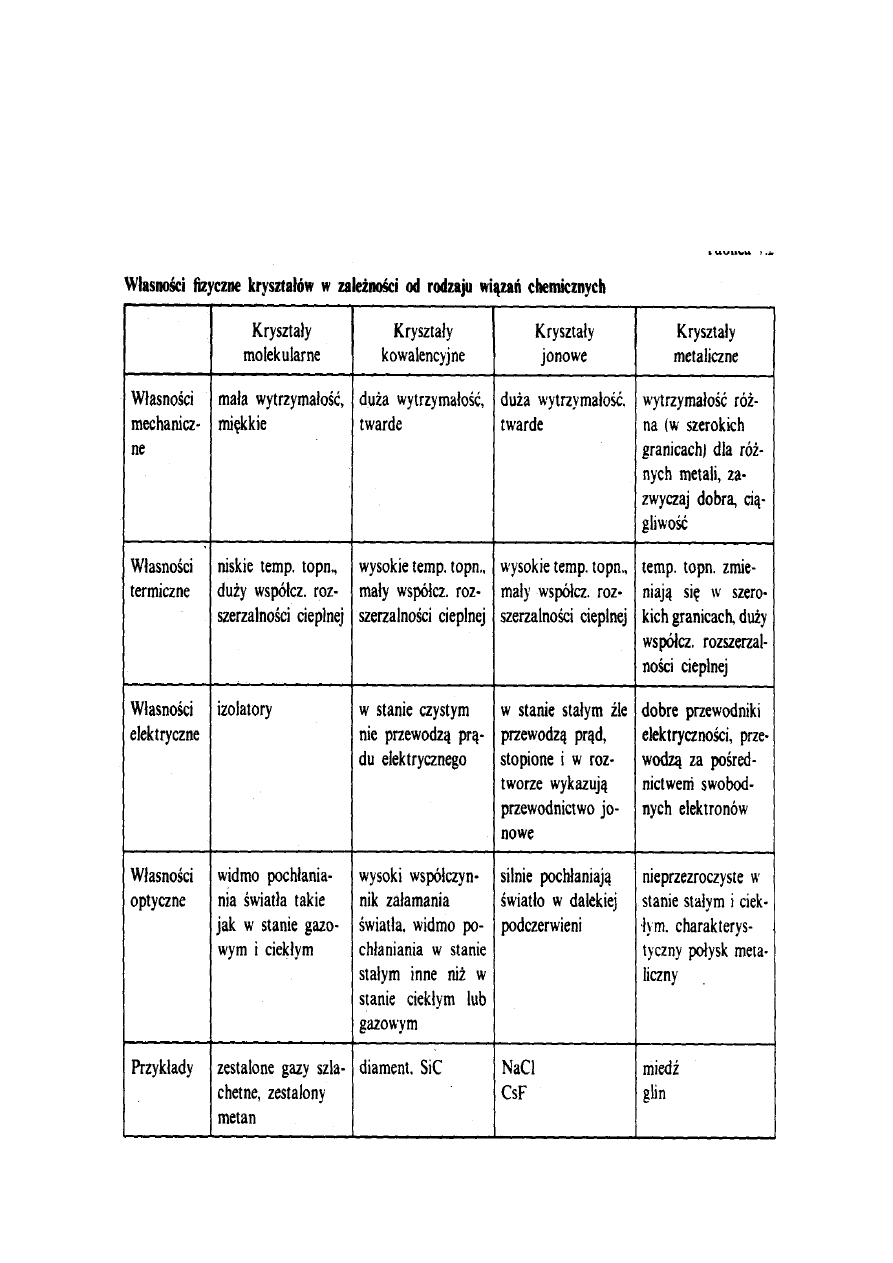
Charakterystykę
wybranych
właściwości
fizycznych różnych rodzajów kryształów podano
w tabeli 1.
Tabela 1.
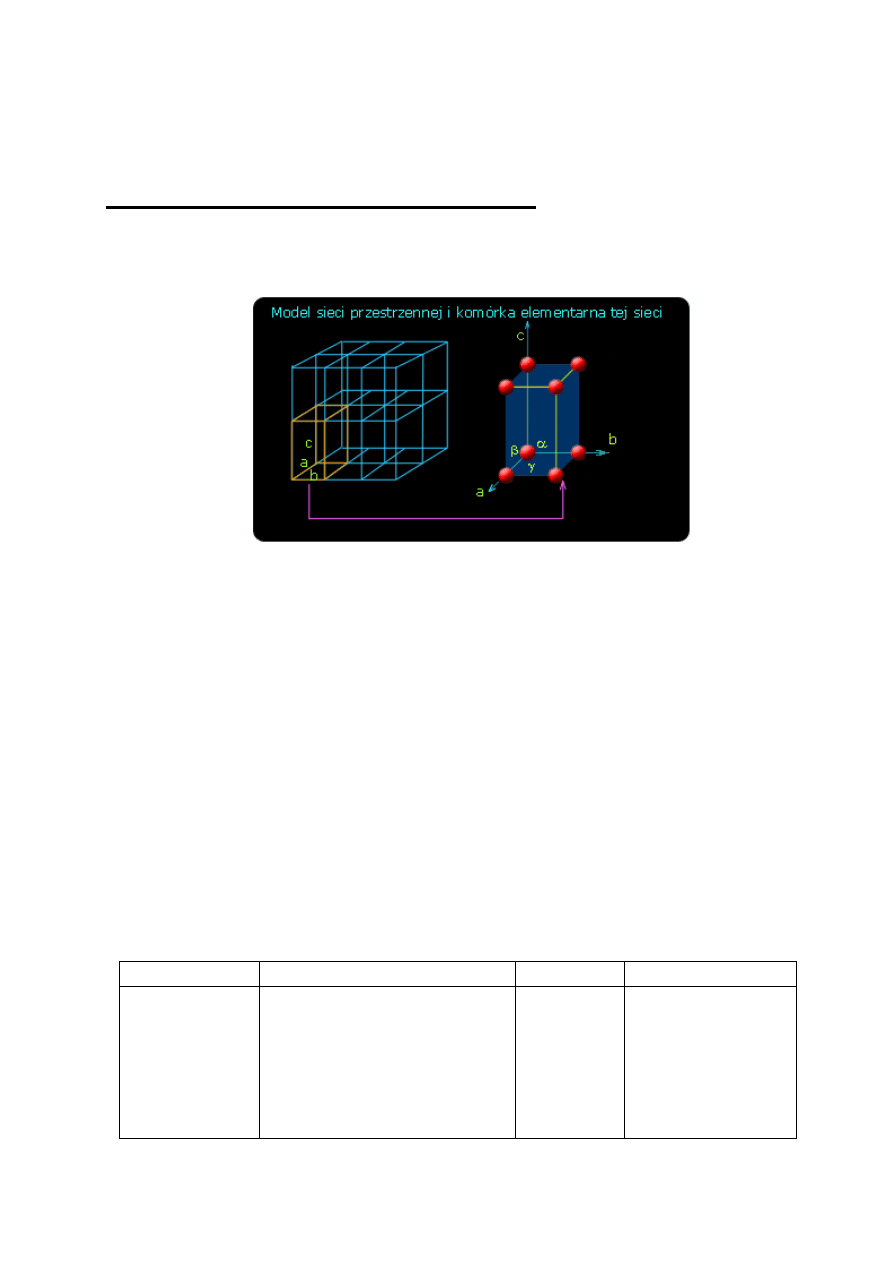
W
opisie
sieci
przestrzennej
substancji
krystalicznej wygodnie jest posłużyć się pojęciem
elementarnej komórki sieciowej, tj. najmniejszym
wycinkiem tej sieci, który jeszcze wykazuje
wszystkie jej cechy charakterystyczne (rys. 4).
Rys. 4. Komórka sieciowa jako najmniejszy element sieci
przestrzennej kryształu
Komórka elementarna jest równoległościanem
o ściśle określonych długościach krawędzi i kątach
między nimi. Przesuwanie (translacja) komórki
elementarnej w trzech kierunkach równoległych do jej
krawędzi pozwala odtworzyć całą sieć przestrzenną.
Wyróżniamy siedem typów komórek sieciowych
odpowiadających różnym układom krystalograficznym
(tabela 2).
Tabela 2. Komórki elementarne sieci przestrzennych
w poszczególnych układach krystalograficznych
Układ
Kształt komórki
Krawędzie
Kąty
Regularny
Tetragonalny
Rombowy
Heksagonalny
Romboedryczny
Jednoskośny
Trójskośny
Sześcian
Prostopadłościan kwadratowy
Prostopadłościan prostokątny
Prostopadłościan rombowy
Równoległościan
Równoległościan
Równoległościan
a = b = c
a = b ≠ c
a ≠ b ≠ c
a = b ≠ c
a
a ≠ b ≠ c
a ≠ b ≠ c
α = β = γ = 90
o
α = β = γ = 90
o
α = β = γ = 90
o
α = β = 90
o
, γ = 120
o
ω ≠ 90
o
α = γ, β ≠ 90
o
α ≠ β, β ≠ γ ≠ 90
o
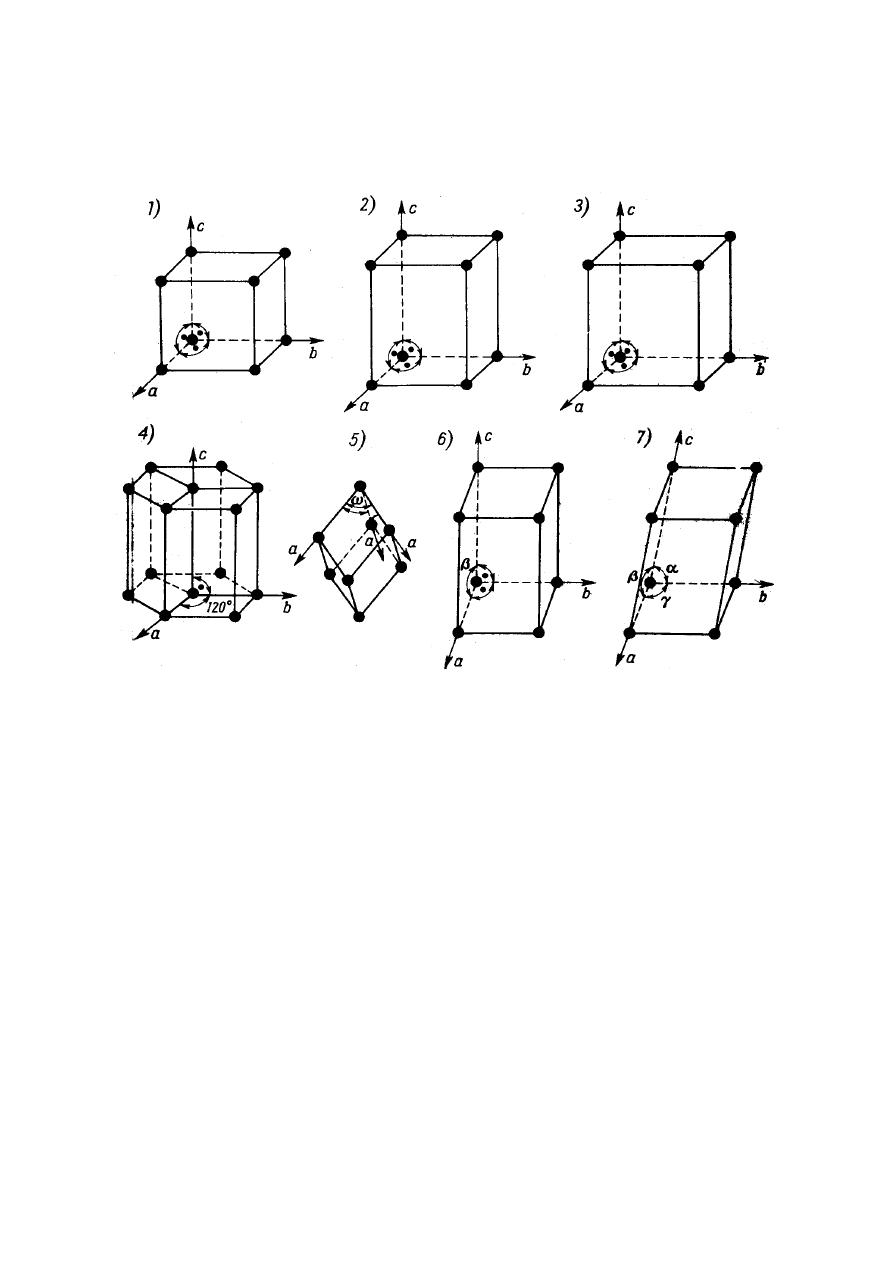
Typy elementarnych komórek sieciowych (według
kolejności w tabeli 2) pokazano na rys. 5.
Rys. 5. Typy elementarnych komórek sieciowych
1) komórka regularna; 2) komórka tetragonalna; 3) komórka rombowa;
4) komórka heksagonalna; 5) komórka romboedryczna, 6) komórka
jednoskośna, 7) komórka trójskośna
Atomy, cząsteczki lub jony mogą obsadzać w komórce
elementarnej naroża (komórka prymitywna), mogą
również zajmować środki podstaw i ścian bocznych
(komórka płasko centrowana) oraz pewne miejsca
wewnątrz komórki, np. jej środek geometryczny
(komórka przestrzennie centrowana). Wymienione
miejsca są węzłami sieci krystalicznej. Bravais
wyróżnił w siedmiu układach krystalograficznych
czternaście typów komórek elementarnych (rys. 6).
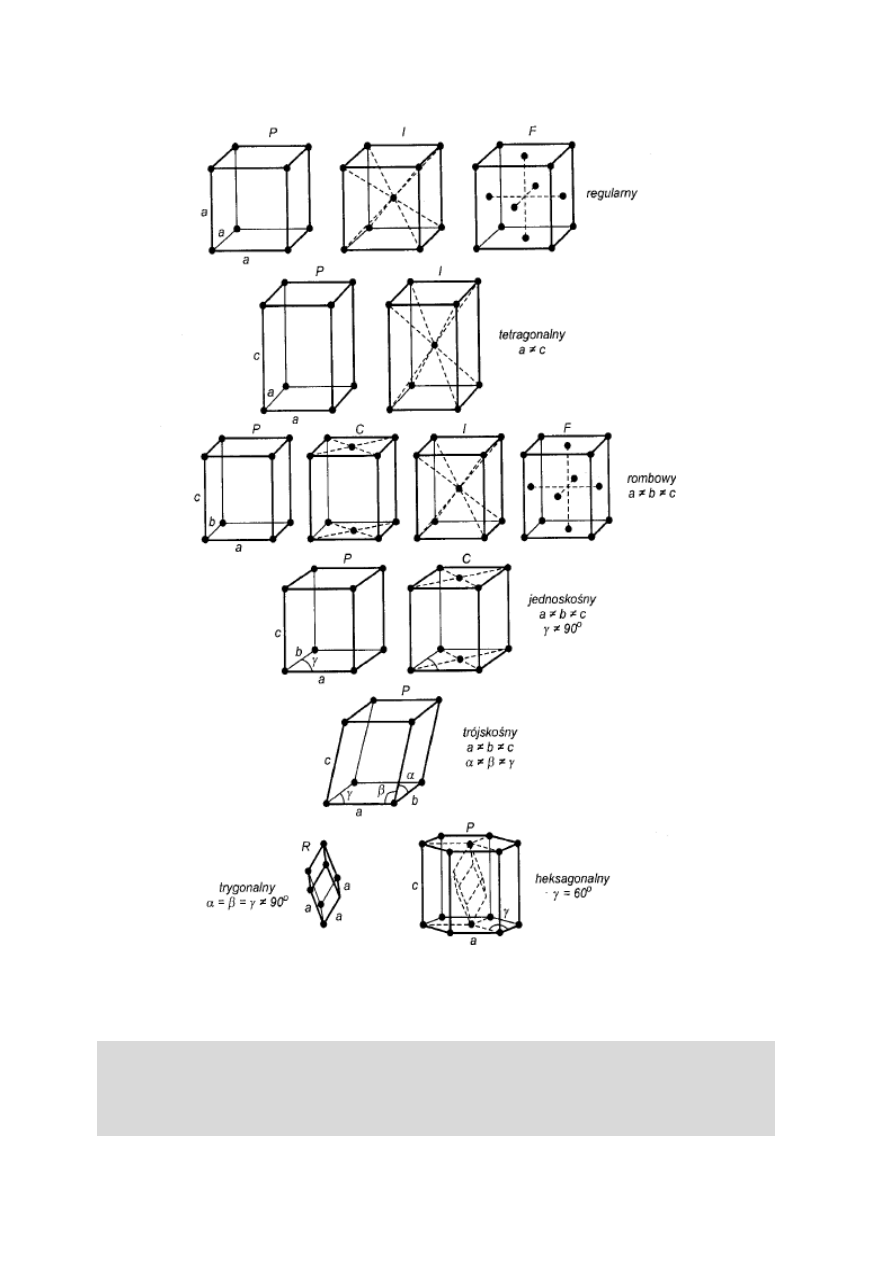
Rys. 6. Czternaście typów komórek elementarnych Bravais
w siedmiu układach krystalograficznych
Liczba najbliższych sąsiadów atomu, jonu lub cząsteczki
w danym węźle sieci przestrzennej nosi miano liczby
koordynacyjnej.
UWAGA:
Niekiedy cząsteczki lub jony obsadzają również
środki krawędzi komórki elementarnej.
STRUKTURA KRYSTALICZNA PIERWIASTKÓW
Pierwiastki metaliczne z reguły odznaczają się
wysoką symetrią struktury sieci przestrzennej.
Sieci te
są zbudowane z dodatnio naładowanych zrębów
atomowych,
natomiast
elektrony
walencyjne
wszystkich atomów metali są zdelokalizowane
tworząc tzw. gaz elektronowy, który neutralizuje
sumaryczny ładunek dodatni wszystkich zrębów
atomowych w sieci przestrzennej (rys. 7).
Rys. 7. Schemat struktury krystalicznej metalu:
●
–
zręby atomowe,
●
–
gaz elektronowy
Innymi słowy, mamy tu do czynienia
z
kooperatywnym
wiązaniem
metalicznym,
utworzonym
przez
delokalizację
elektronów
walencyjnych
wszystkich
atomów
metalu
w krysztale. Wiązanie to ma szczególnie dużą
trwałość, ponieważ metale z nielicznymi tylko
wyjątkami, np. Hg, Ga, mają na ogół wysokie lub
bardzo wysokie temperatury topnienia i wrzenia.
Trzeba podkreślić, że gaz elektronowy wykazuje
pewną mobilność, ponieważ metale pod wpływem
zewnętrznego pola elektrycznego przewodzą prąd
elektryczny.
Dla przeważającej liczby metali właściwy jest jeden
ze sposobów upakowania atomów w trzech typach
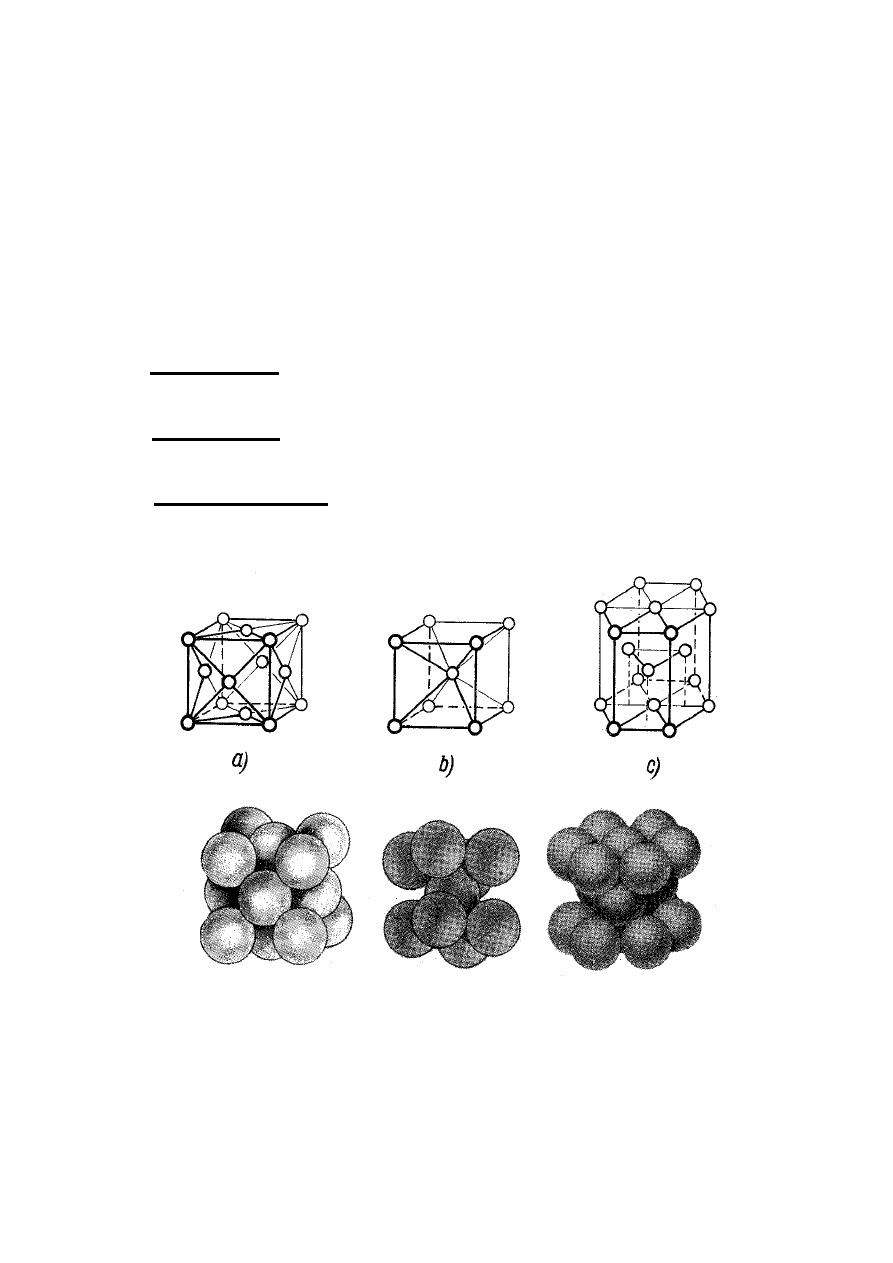
komórek elementarnych:
a) regularnej płasko centrowanej, A
1
, zawierającej
cztery atomy o liczbie koordynacji równej 12;
b) regularnej przestrzennie centrowanej, A
2
, która
zawiera dwa atomy o liczbie koordynacji równej 8;
c) heksagonalnej o najgęstszym upakowaniu, A
3
,
zawierającej sześć atomów o liczbie koordynacji
równej 12.
Rys. 8. Komórki elementarne sieci przestrzennych metali
a) komórka płasko centrowana, A
1
; b) komórka przestrzennie
centrowana, A
2
; c) komórka heksagonalna, A
3
.
Komórki A
1
i A
3
charakteryzuje najwyższy stopień
upakowania atomów - 74%, natomiast w komórce A
2
wynosi on 68%. Znając parametry sieciowe komórki
elementarnej można obliczyć promień r atomu (zrębu
atomowego) metalu zakładając, że jest on sztywną kulą.
Komórka regularna A
1
o krawędzi a
:
a
r
4
2
Komórka regularna A
2
o krawędzi a
:
a
r
4
3
Komórka heksagonalna A
3
o krawędzi a i wysokości c:
2
a
r
c
r
2
4
3
Liczbę
wszystkich
atomów
w
komórce
elementarnej można łatwo obliczyć biorąc pod
uwagę, że:
- atomy położone w narożach przynależą do
sześciu komórek elementarnych w układzie
heksagonalnym
lub
ośmiu
komórek
w pozostałych układach;
- atomy położone na krawędziach należą do
czterech komórek elementarnych;
- atomy położone na ścianach bocznych
należą do dwu komórek elementarnych;
- atomy położone wewnątrz komórki są do
niej integralnie przynależne.

W heksagonalnej komórce A
3
dwanaście atomów
położone jest narożach, dwa na jej podstawach i trzy
w jej wnętrzu. Zatem komórka ta zawiera (12∙1/6 + 3
+ 2∙1/2) = 6 atomów metalu.
Przykłady:
A
1
– Cu, Ag, Au, Ca, Sr, Pd, Pt, γ-Fe, δ-Fe, β-Co, β-Ni;
A
2
– α-W, V, Nb, Ta, Mo, α-Fe, β-Zr;
A
3
– Mg, Zn, Cd, Ti, α-Zr, α-Co
.
Wymienione przykłady wskazują, że metale mogą
występować w różnych odmianach krystalicznych,
które są trwałe w ściśle określonych przedziałach
temperatury, np. Fe:
910
o
C (
α→γ), 1388
o
C (γ→δ), 1539
o
C (δ→faza ciekła).
α-Fe o właściwościach ferromagnetycznych oraz
paramagnetyczne
γ-Fe i δ-Fe są odmianami
alotropowymi żelaza.
Alotropia
Terminem
alotropia
określa
się
zjawisko
występowania danego pierwiastka chemicznego
w
kilku
różnych
postaciach,
najczęściej
krystalicznych, różniących się między sobą
właściwościami fizycznymi, a niekiedy różniących
się pod względem aktywności chemicznej.
Typowy pierwiastek niemetaliczny to węgiel, który
ma dwie podstawowe odmiany alotropowe, grafit
i diament, różniące się strukturą krystaliczną
i właściwościami fizycznymi.
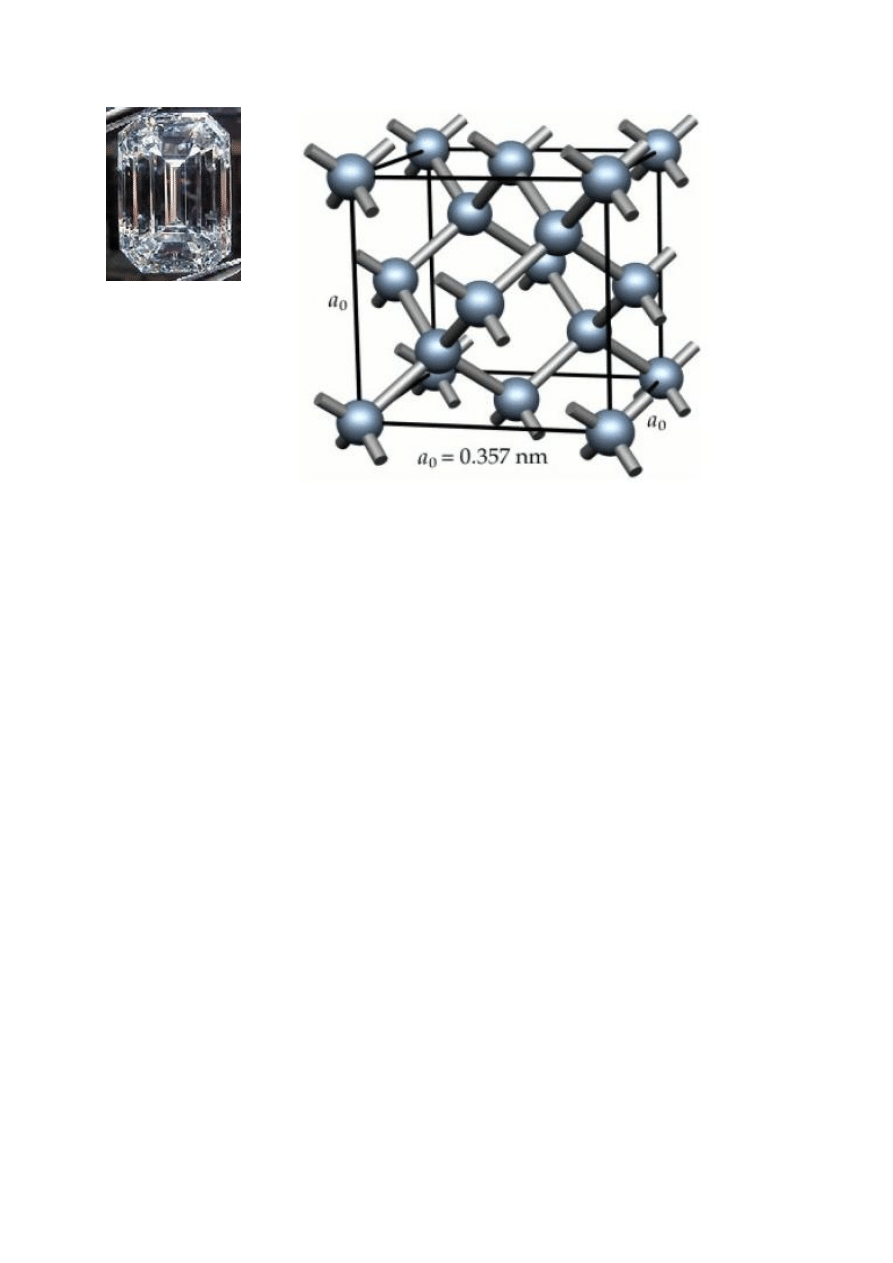
Rys. 9. Kryształ (z lewej) i komórka elementarna diamentu
Atomy węgla w regularnej komórce diamentu są
w stanie hybrydyzacji sp
3
, dlatego każdy z nich jest
związany kowalencyjnie czterema wiązaniami σ
o długości 154 pm z sąsiadami w sieci krystalicznej. Są
to wiązania mocne, ponieważ zerwanie ich wymaga
dużego nakładu energii, 348 kJ/mol, dlatego diament
jest bardzo twardy. Diament ma większą od grafitu
gęstość. Czysty diament jest bezbarwny, silnie załamuje
światło i nie przewodzi prądu elektrycznego. Niewielkie
domieszki nadają diamentom różne zabarwienie, np.
czerwone, żółte, fioletowe, niebieskie, a nawet czarne
(karbonado). Odpowiednio oszlifowane diamenty (tzw.
szlif brylantowy) są cenionymi przez jubilerów
kamieniami szlachetnymi. Diamenty syntetyczne są
używane jako ostrza wierteł, do cięcia szkła, do
produkcji
łożysk
osiowych
stosowanych
w instrumentach precyzyjnych.
Podobne do diamentu typy struktur mają krzem,
german i niskotemperaturowa odmiana cyny, α-Sn.
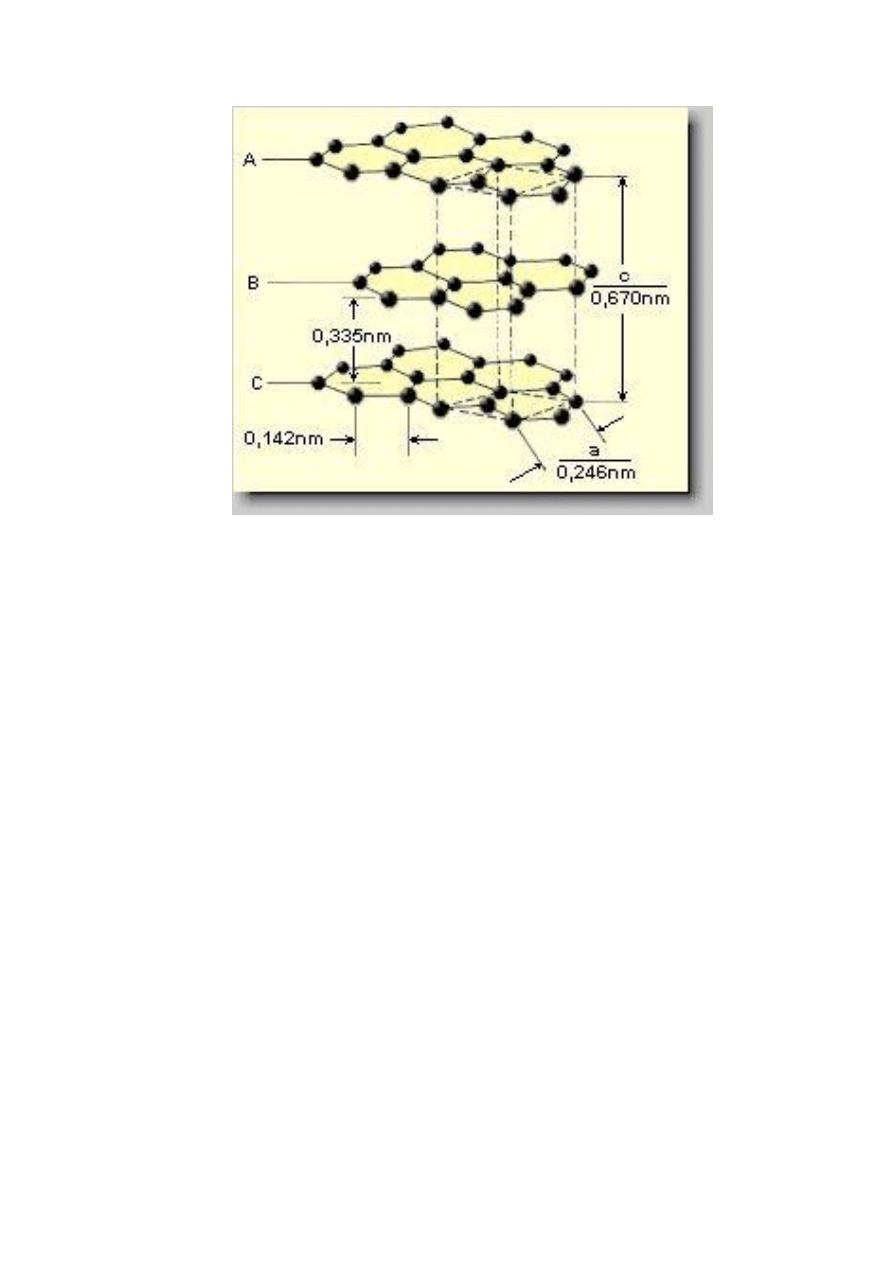
Rys. 10. Warstwowa, heksagonalna struktura grafitu
Grafit ma heksagonalną strukturę warstwową.
Atomy węgla w krysztale grafitu są w stanie
hybrydyzacji trygonalnej sp
2
, w wyniku czego każdy
z nich jest związany kowalencyjnie trzema
wiązaniami σ o długości 142 pm z sąsiadami w tej
samej
płaszczyźnie
sieciowej
(warstwie).
Niezhybrydyzowane orbitale p
z
atomów węgla,
położonych w tej samej płaszczyźnie sieciowej,
tworzą zdelokalizowane orbitale π połowicznie
obsadzone elektronami. Elektrony π wykazują
zdolność do poruszania się i przenoszenia prądu
elektrycznego w obrębie każdej płaszczyzny
sieciowej.
Odległość
między
płaszczyznami
sieciowymi (warstwami) grafitu jest równa 335 pm.
Grafit jest łupliwy, ponieważ między poszczególnymi
warstwami działają słabe wiązania van der Waalsa.
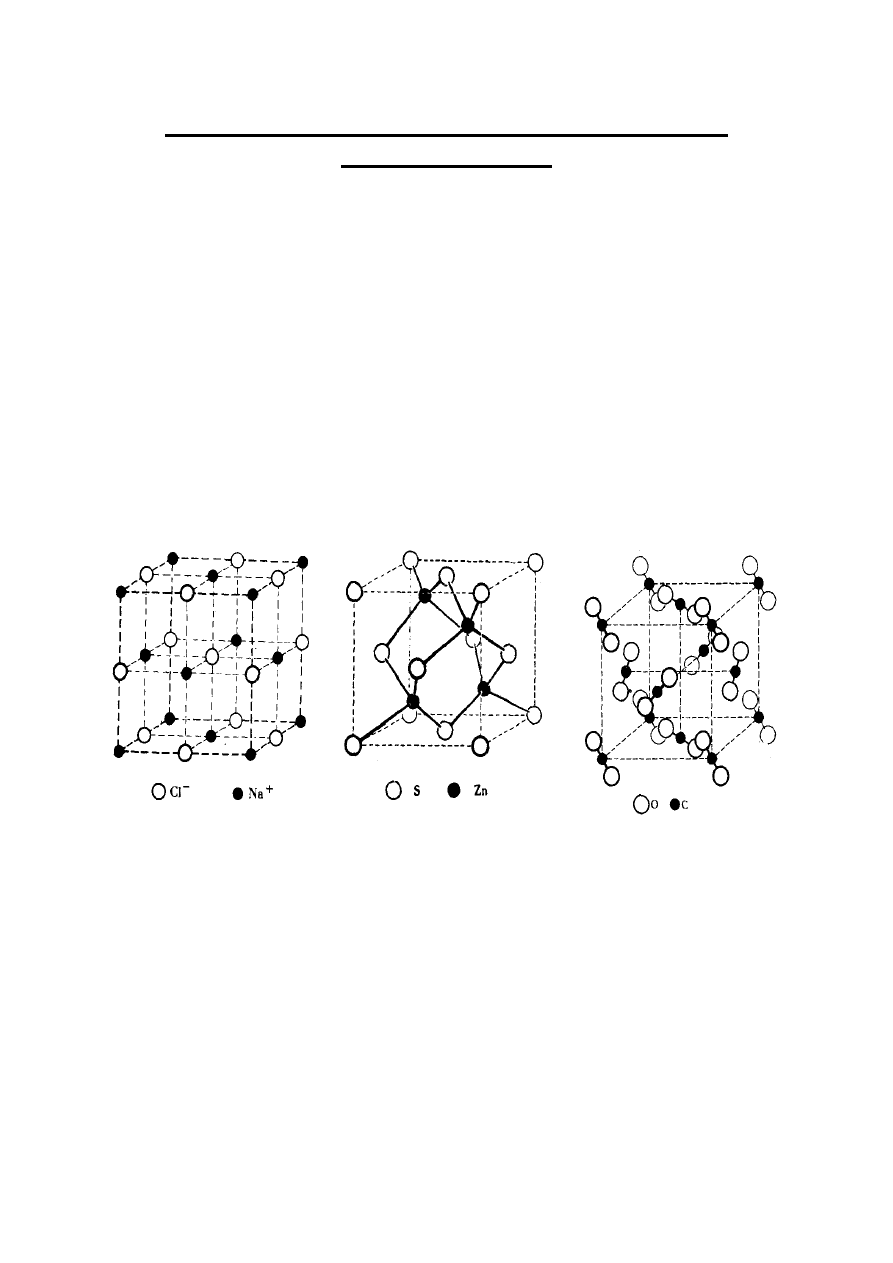
STRUKTURA KRYSTALICZNA ZWIĄZKÓW
CHEMICZNYCH
Kryształy związków chemicznych, w zależności
od natury wiązań działających w ich sieciach
przestrzennych, mogą mieć charakter jonowy,
kowalencyjny lub molekularny (Tabela 1).
Rysunek 11 przedstawia regularną komórkę
elementarnej
sieci
jonowej
chlorku
sodu,
regularną komórkę elementarną kowalencyjnej sieci
siarczku cynku (ZnS) oraz regularną komórkę
elementarną sieci molekularnej stałego ditlenku węgla,
w której działają jedynie wiązania van der Waalsa.
Rys. 11. Elementarne komórki sieciowe chlorku sodu NaCl,
siarczku cynku ZnS i stałego ditlenku węgla CO
2
Komórka sieciowa NaCl zawiera cztery kationy
Na
+
i cztery aniony Cl
–
, przy czym każdy z nich ma
liczbę koordynacji równą 6. Czysty, krystaliczny
chlorek sodu nie przewodzi prądu elektrycznego, zatem
jest dielektrykiem czyli izolatorem. Natomiast stopiony
NaCl jest cieczą jonową zdolną do przewodzenia prądu
elektrycznego. Roztwory wodne chlorku sodu również
przewodzą prąd elektryczny.
Siarczek
cynku,
ZnS,
ma
dwie
odmiany
krystalizujące w różnych układach, mianowicie:
sfaleryt (układ regularny, rys. 11b) oraz wurcyt (układ
heksagonalny, rys. 12).
Występowanie związku chemicznego w różnych
odmianach krystalicznych określa się mianem
polimorfizmu.
Regularna komórka sieciowa sfalerytu (rys. 11b)
zawiera cztery atomy cynku i cztery atomy siarki
połączone wiązaniami kowalencyjnymi, przy czym
każdy z nich ma liczbę koordynacji równą cztery.
Heksagonalna komórka sieciowa wurcytu zawiera sześć
atomów cynku i sześć atomów siarki związanych
kowalencyjnie, gdzie każdy z nich ma liczbę
koordynacji równą cztery. Czysty siarczek cynku jest
dielektrykiem.
●
S
●
Zn
Rys. 12. Komórka elementarna wurcytu

Płasko centrowana regularna komórka sieciowa
stałego ditlenku węgla zawiera cztery liniowe
cząsteczki CO
2
, przy czym każda z nich ma liczbę
koordynacji 6, ponieważ jest otoczona przez sześć
innych cząsteczek tego związku. Stały ditlenek węgla
jest oczywiście dielektrykiem. Ten sam typ płasko
centrowanej regularnej komórki sieciowej ma jod I
2
.
Sieci jonowe
Kryształy jonowe są elektrycznie obojętne,
ponieważ sumaryczny ładunek kationów jest
kompensowany przez sumaryczny ładunek anionów.
Trwałość sieci jonowych wynika z faktu, że
wypadkowa siła elektrostatycznego przyciągania
kationów i anionów przeważa nad wypadkową siłą
elektrostatycznego odpychania kationów i anionów.
Tworzenie trwałych, krystalicznych struktur
jonowych wiąże się z następującymi regułami:
● odległość międzyjonowa R jest sumą promieni
kationu i anionu, R = r
k
+ r
a
,
● stosunek promieni r
k
/r
a
określa liczbę koordynacji
danej sieci.
Tabela 3. Liczby koordynacji jonów w zależności od stosunku r
k
/r
a
r
k
/r
a
L.K.
Wielościan koordynacyjny
0,22-0,41
0,41-0,72
0,72-1
4
6
8
tetraedr
oktaedr
sześcian
Promienie jonowe kationu Na
+
i anionu Cl
-
są
równe odpowiednio 102 i 181 pm, a ich stosunek jest
równy 0,564. Stąd każdy z tych jonów w sieci
krystalicznej NaCl ma liczbę koordynacji równą 6
(rys. 11a). Regularna sieć NaCl składa się z dwóch
podsieci typu A
1
(pierwsza dla kationów, druga dla
anionów) nasuniętych na siebie tak, aby aniony
chlorkowe dzieliły krawędzie komórki elementarnej
na połowy. Sieć krystaliczną typu NaCl mają: AgCl,
AgBr, PbS.
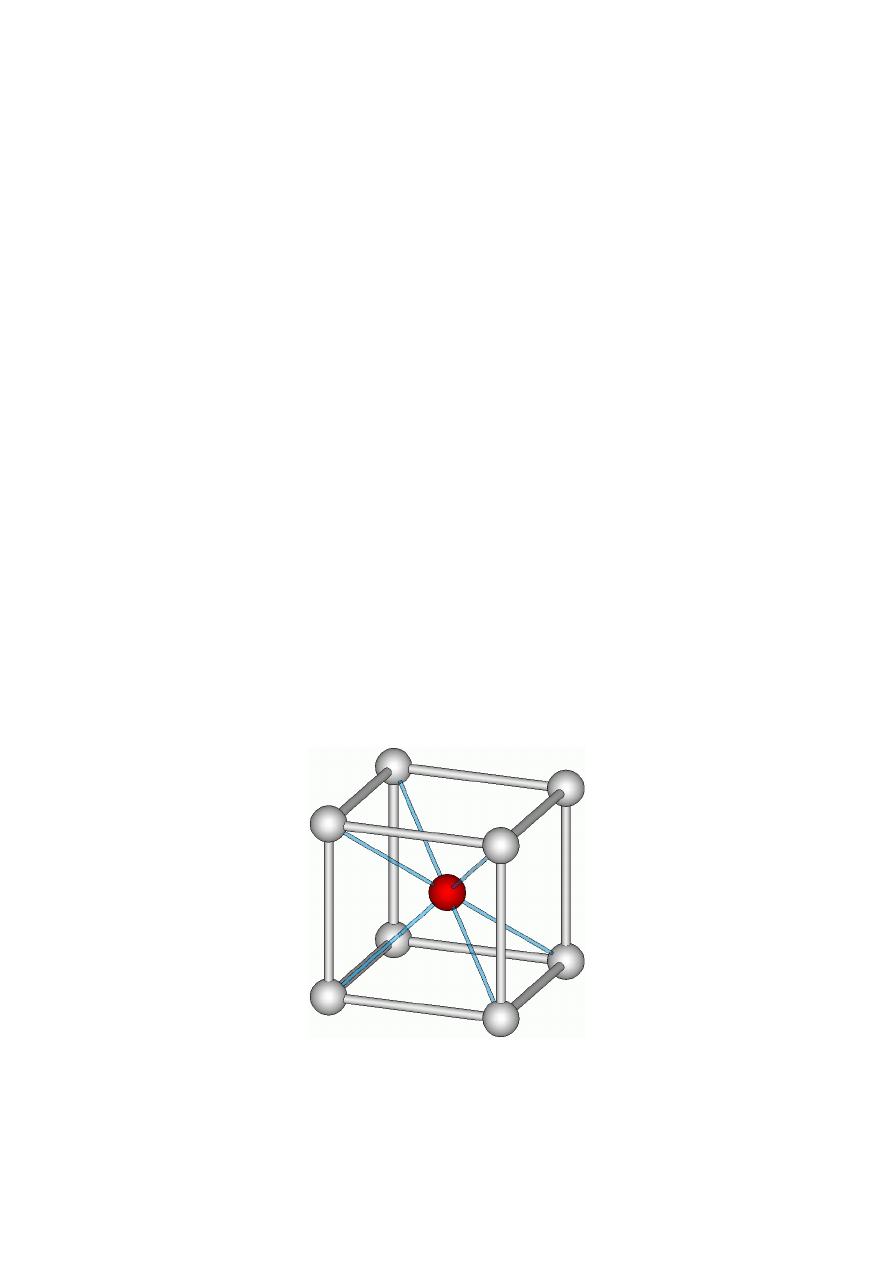
Inny typ struktury krystalicznej ma chlorek cezu,
CsCl, ponieważ promień jonowy kationu Cs
+
jest
równy 170 pm, a stosunek promieni r
k
/r
a
= 0,939.
Stąd każdy jon w regularnej strukturze tego związku
ma liczbę koordynacji równą 8. Regularną komórkę
elementarną CsCl można formalnie wyprowadzić
z komórki typu A
2
.
Rys. 13. Komórka elementarna chlorku cezu
●
Cl,
●
Cs

Sieć krystaliczną typu CsCl mają: TlCl, NH
4
Cl,
NH
4
Br.
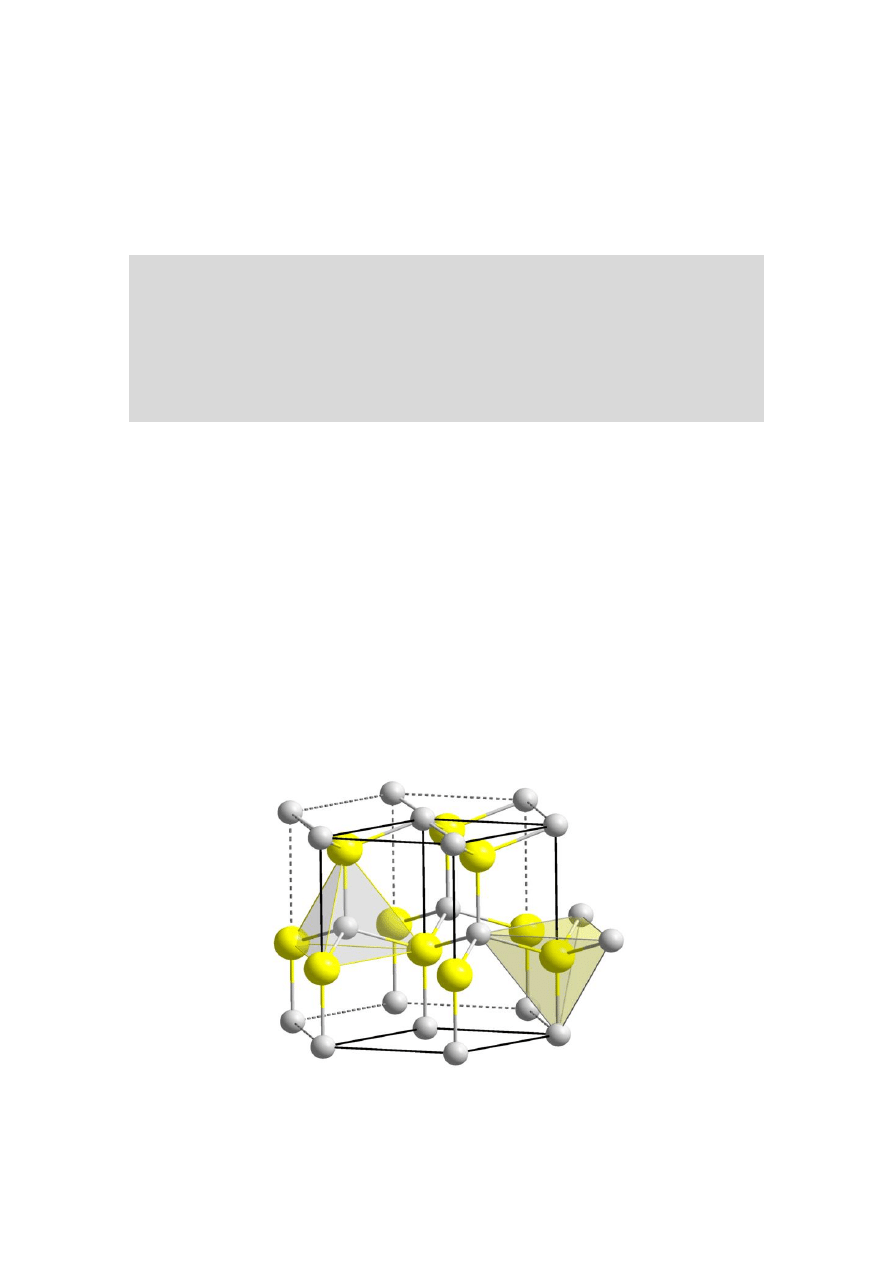
Związki typu AX
2
mają bardziej skomplikowane
struktury krystaliczne. Typowym przykładem jest
fluoryt, CaF
2
. W regularnej komórce tego związku
(rys.14) kationy Ca
2+
obsadzają naroża i środki
ścian, natomiast osiem anionów F
-
zajmuje jej
wnętrze.
●-
Ca
●
- F
Rys. 14. Komórka elementarna fluorytu
W komórce elementarnej CaF
2
jest cztery kationy
Ca
2+
o liczbie koordynacji 8 oraz osiem anionów F
-
o liczbie koordynacji 4. Podobny typ struktury mają
również SrF
2
, BaF
2
, PbF
2
.
Znana
jest również sieć odwrotna typu
antyfluorytu, powstała przez zamianę położeń
kationów i anionów w sieciach typu fluorytu.
Komórkę
elementarną
typu
antyfluorytu
przedstawia rys. 15. Należy pamiętać, że termin
antyfluoryt
nie oznacza tutaj polimorficznej odmiany
CaF
2
!!!
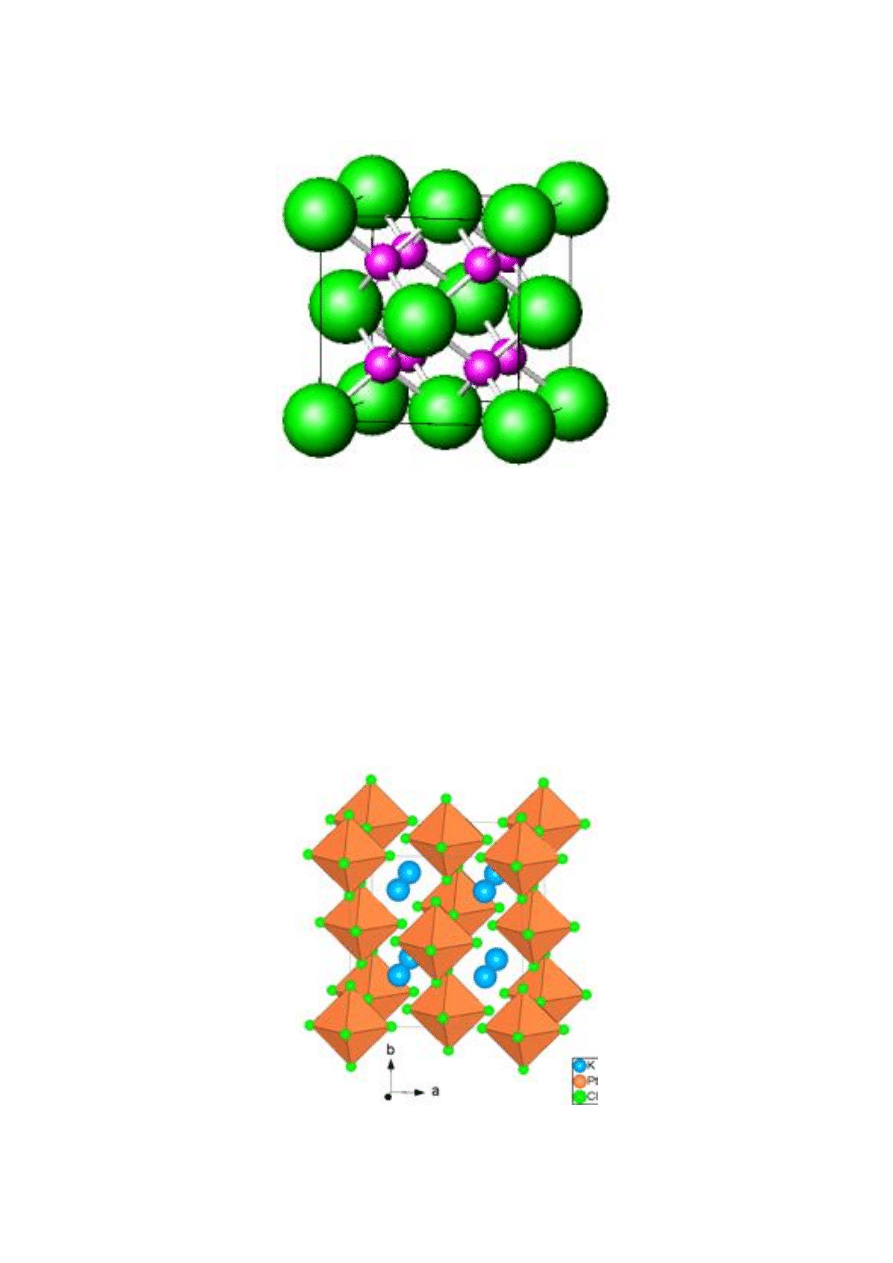
Rys. 15. Komórka elementarna typu antyfluorytu
Struktury typu antyfluorytu spotykamy u Li
2
S,
Na
2
S, Cu
2
S, Ag
2
Te, CeO
2
. Strukturę tę ma również
heksachloroplatynian(IV) potasu, K
2
[PtCl
6
]. Aniony
okteadryczne [PtCl
6
]
2-
obsadzają naroża i środki
ścian komórki regularnej, a kationy K
+
zajmują
położenia w jej wnętrzu (rys. 16).
e
Rys. 16. Struktura K
2
[PtCl
6
]

Z przedstawionych przykładów wynika, że różne
związki chemiczne często krystalizują w tym samym
układzie krystalograficznym i mają ten sam typ sieci
przestrzennej – innymi słowy są izostrukturalne.
Izostrukturalność jest nader często równoznaczna
z izomorfizmem.
Izomorfizm
to
równopostaciowość
różnych
substancji krystalicznych, wynikająca ze ścisłej
analogii ich struktur sieciowych. Do wystąpienia
izomorfizmu muszą być spełnione następujące
warunki:
1) identyczny typ wzoru chemicznego związków
izomorficznych, np. AB
2
, ABX
4
;
2) składniki związków izomorficznych nie mogą
wykazywać zbyt dużych różnic pod względem
promieni atomowych lub jonowych, przy
czym różnice te nie mogą przekraczać 15%;
Jeżeli wymienione warunki są spełnione, to
izomorfizm może, ale nie musi zaistnieć, np.
promienie jonowe kationów Tl
+
i Rb
+
są równe 149
pm, ale TlCl ma sieć typu CsCl, natomiast RbCl ma
sieć typu NaCl. Zatem dodatkowym warunkiem są
takie same lub zbliżone parametry komórek
elementarnych.
Izomorficzne
ałuny,
czyli
podwójne
siarczany
M
I
M
III
(SO
4
)
2
·12H
2
O, gdzie:
M
I
= K
+
, NH
4
+
, Rb
+
, Cs
+
, Tl
+
,
M
III
= Al
3+
, Fe
3+
, Ga
3+
, Cr
3+
, V
3+
,
krystalizują w układzie regularnym o typie sieci NaCl.
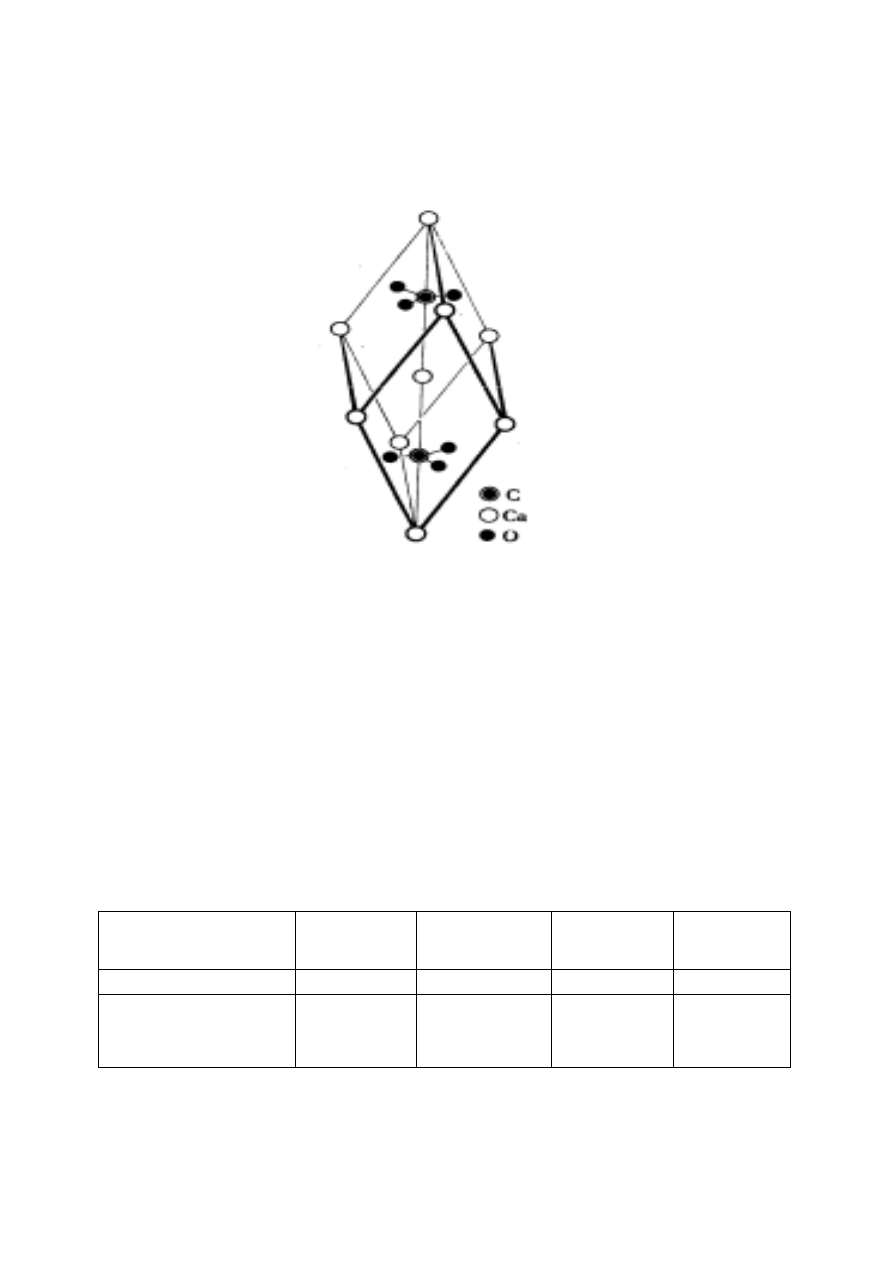
Węglan wapnia CaCO
3
(kalcyt)
krystalizuje
w układzie romboedrycznym (rys. 17).
Rys. 17. Struktura kalcytu
Jony Ca
2+
obsadzają naroża i centrum komórki
elementarnej, a płaskie aniony węglanowe znajdują się
w jej wnętrzu. Komórka elementarna kalcytu zawiera
dwa kationy i dwa aniony. Taki sam typ sieci
przestrzennej mają inne węglany M
II
CO
3
, M
II
= Mg
2+
,
Zn
2+
, Fe
2+
, Mn
2+
, Cd
2+
. Porównanie krystalicznych
węglanów MCO
3
o sieci typu kalcytu przedstawia
tabela 4.
Tabela 4. Porównanie wybranych węglanów MCO
3
MgCO
3
magnezyt
ZnCO
3
smitsonit
FeCO
3
syderyt
CaCO
3
kalcyt
r
kat
, pm
72
73
78
100
Parametry komórki
a, pm
c, pm
463
1501
465
1503
469
1538
499
1706
W świetle przedstawionych wyżej kryteriów magnezyt,
smitsonit i syderyt są izomorficzne.

Najdoskonalsza forma izomorfizmu przejawia się
w zdolności do tworzenia roztworów stałych, czyli
dwuskładnikowych kryształów mieszanych w całym
zakresie składów obydwu składników izomorficznych.
Jako typowe przykłady można wymienić ałuny
chromowo-potasowy i potasowo-glinowy lub chlorek
i
bromek
potasu.
Kryształami mieszanymi są
występujące w litosferze minerały, np. oliwin
(Mg,Fe)
2
SiO
4
oraz apatyt Ca
3
(PO
4
)
2
·Ca(F,Cl)
2
. Należy
podkreślić,
że
częściej
mamy
do
czynienia
z ograniczoną zdolnością do powstawania kryształów
mieszanych, np. NaCl – KCl, tylko w pewnym zakresie
składów. Kryształy podwójne obydwu tych związków,
krystalizowane w temperaturze 0, 25 i 100
o
C
z roztworów wodnych nasyconych względem NaCl
i KCl, zawierają odpowiednio 75,1, 64,5 i 43,8%
masowych NaCl. Ponadto, znane są przypadki
izomorfizmu, gdy nie wszystkie wymienione wyżej
warunki są spełnione, np. roztwory stałe tworzą CaF
2
i YF
3
. W innych układach dwuskładnikowych, np. MgO
– NaCl, PbS – NaCl, składniki mają ten sam typ sieci
przestrzennej, ale nie tworzą roztworów stałych
i wykazują jedynie zdolność do warstwicowego
narastania jednej substancji na drugiej.
Defekty sieci krystalicznej
W przyrodzie nic niej doskonałe, a kryształy nie
są wyjątkiem od tej reguły. Każdy kryształ
rzeczywisty zawiera niedoskonałości strukturalne,
czyli defekty sieci krystalicznej polegające na
zakłóceniu jej cech.
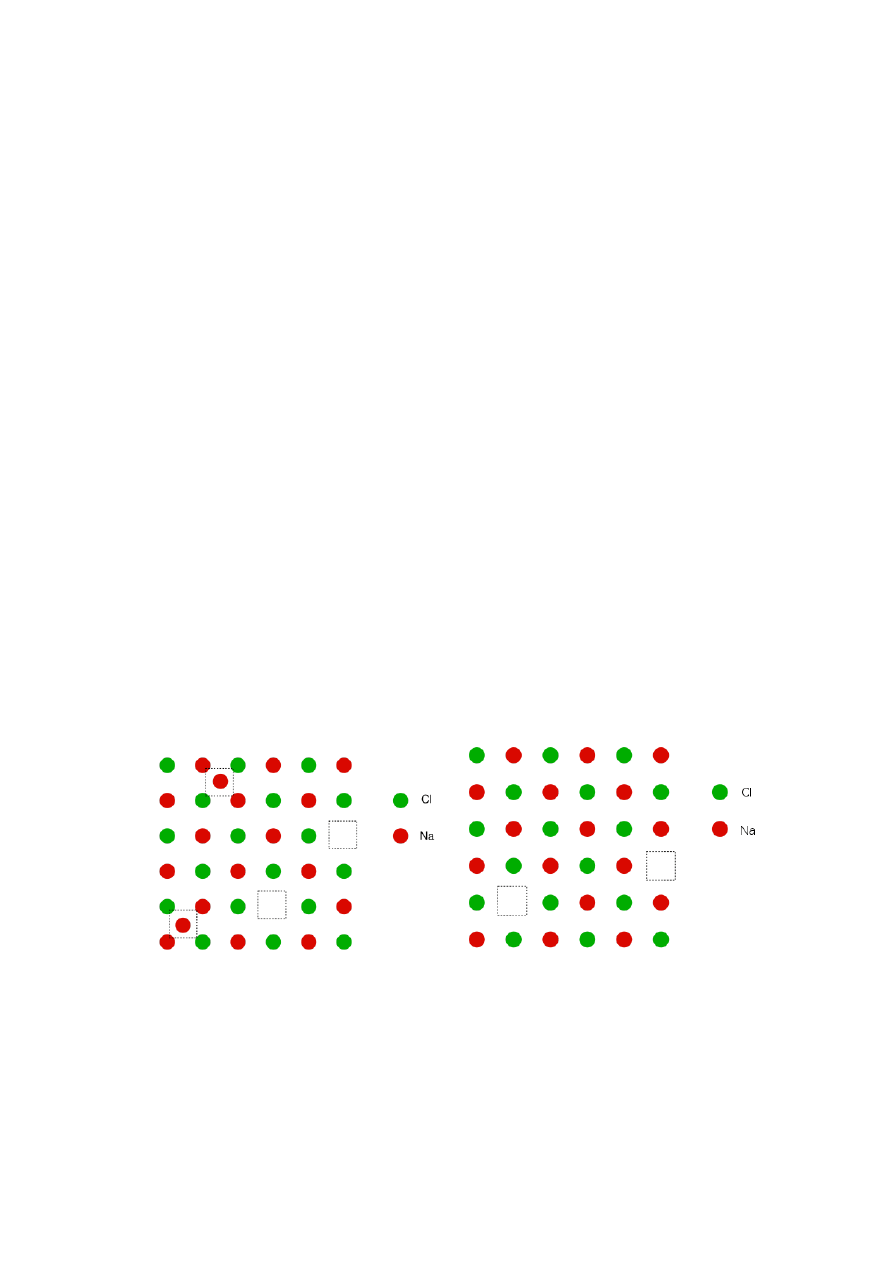
Defekty te w różny sposób wpływają na właściwości
fizyczne kryształów, np.:
- defekty w sieci diamentu zmieniają jego barwę;
- defekty sieciach metali zmieniają ich właściwości
mechaniczne, przewodnictwo elektryczne i cieplne.
Defekty w kryształach dzielimy na: a) punktowe
(defekty Frenkla i Schottky’ego); b) liniowe
(dyslokacje krawędziowe i śrubowe); c) defekty
powierzchniowe.
Defekty
Frenkla są spowodowane przez
przemieszczanie się atomów, jonów lub cząsteczek
w położenia międzywęzłowe i pojawienie się nie
obsadzonych węzłów czyli luk, zwanych również
wakansjami,
natomiast
defekty
Schottky’ego
polegają
na
występowaniu
luk
(wakansji)
kationowych i anionowych (rys. 18).
Rys. 18. Defekty Frenkla (z lewej) i Schottky’ego (z prawej)
w sieci NaCl

Luki sieciowe (wakansje) mogą być obsadzone
przez elektrony, co może prowadzić do powstawania
centrów barwnych, np. w halogenkach litowców.
Czyste kryształy NaCl, KCl, RbCl, LiCl, CsCl są
przeźroczyste w całym widzialnym obszarze widma.
Można je zabarwić, np. NaCl na niebiesko, przez
napromieniowanie (X, n, e, ...).
Defekty punktowe występują także w związkach,
których skład odbiega od stechiometrii wyrażonej za
pomocą prostych liczb całkowitych. Przykładowo,
zwykły tlenek żelaza(II) zaliczamy do klasy
daltonidów
, ponieważ jego skład dany jest prostym
wzorem stechiometrycznym FeO. Natomiast tlenek
żelaza, którego skład odpowiada formule Fe
0,93
O
1,00
zaliczany do klasy
bertolidów
. Związek ten wykazuje
defekty kationowe, ponieważ w jego sieci
krystalicznej niektóre węzły zajmują jony Fe
3+
.
W sieci krystalicznej tlenku cynku o składzie
Zn
1,05
O
1,00
nadmiarowe jony Zn
2+
zajmują pozycje
międzywęzłowe.
Dyslokacja krawędziowa ma miejsce wtedy, gdy
płaszczyzna sieciowa wewnątrz struktury kryształu
urywa się, tzn. jest zakończona krawędzią i działa
jak klin rozpychając pozostałe płaszczyzny sieciowe.
Dyslokacja śrubowa jest defektem powodującym, że
podczas krystalizacji wzrost kryształu w wybranym
kierunku zachodzi po linii śrubowej. Obydwa typy
dyslokacji przedstawia rys. 19.
Rys. 19. Dyslokacja krawędziowa i śrubowa
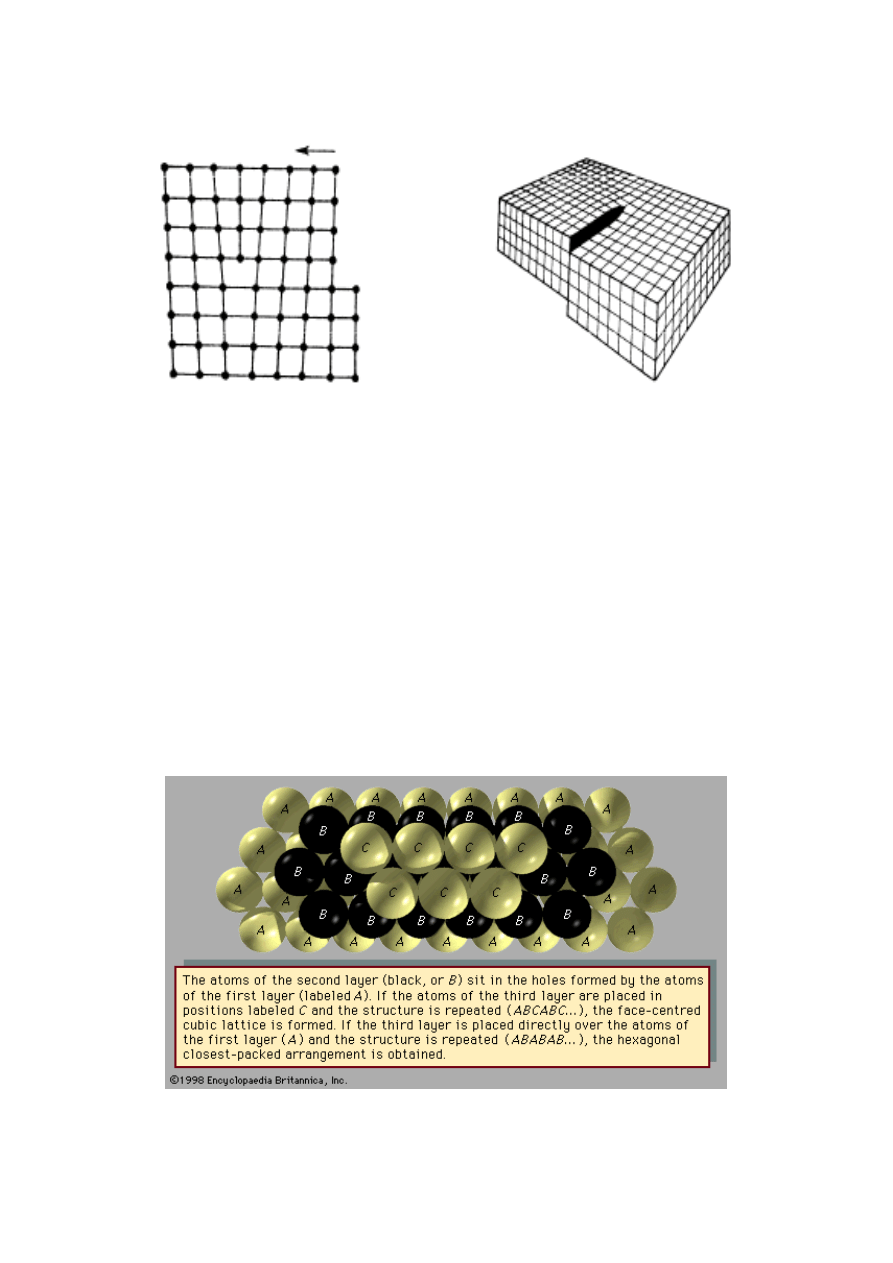
Defektami powierzchniowymi są między innymi
zaburzenia sieci o najgęstszym upakowaniu. W sieci
regularnej występują trzy typy warstw elementów
o symetrii sferycznej, oznaczone symbolami A, B i C.
W sieci heksagonalnej mamy tylko warstwy typu A
i B. Sekwencje uporządkowanych warstw w sieciach
nie wykazujących zaburzeń (rys. 20) są następujące:
sieć regularna - ABCABCABCABC… ,
sieć heksagonalna - ABABABAB …. .
Rys. 20. Struktury o najgęstszym upakowaniu atomów
traktowanych jako sztywne elementy o symetrii sferycznej
Defekty warstwowe polegają na zaburzeniu
uporządkowania warstw, które w sieci regularnej
mogą
mieć
przypadkową
kolejność,
np.
ABCABABCBA,
lub
w
sieci
heksagonalnej.
ABABBAABA.
Rentgenograficzne metody badań struktury kryształów
W
badaniach
wykorzystuje
się
dyfrakcję
promieniowania rentgenowskiego na kryształach,
ponieważ ich płaszczyzny sieciowe spełniają rolę siatki
dyfrakcyjnej (rys. 21).
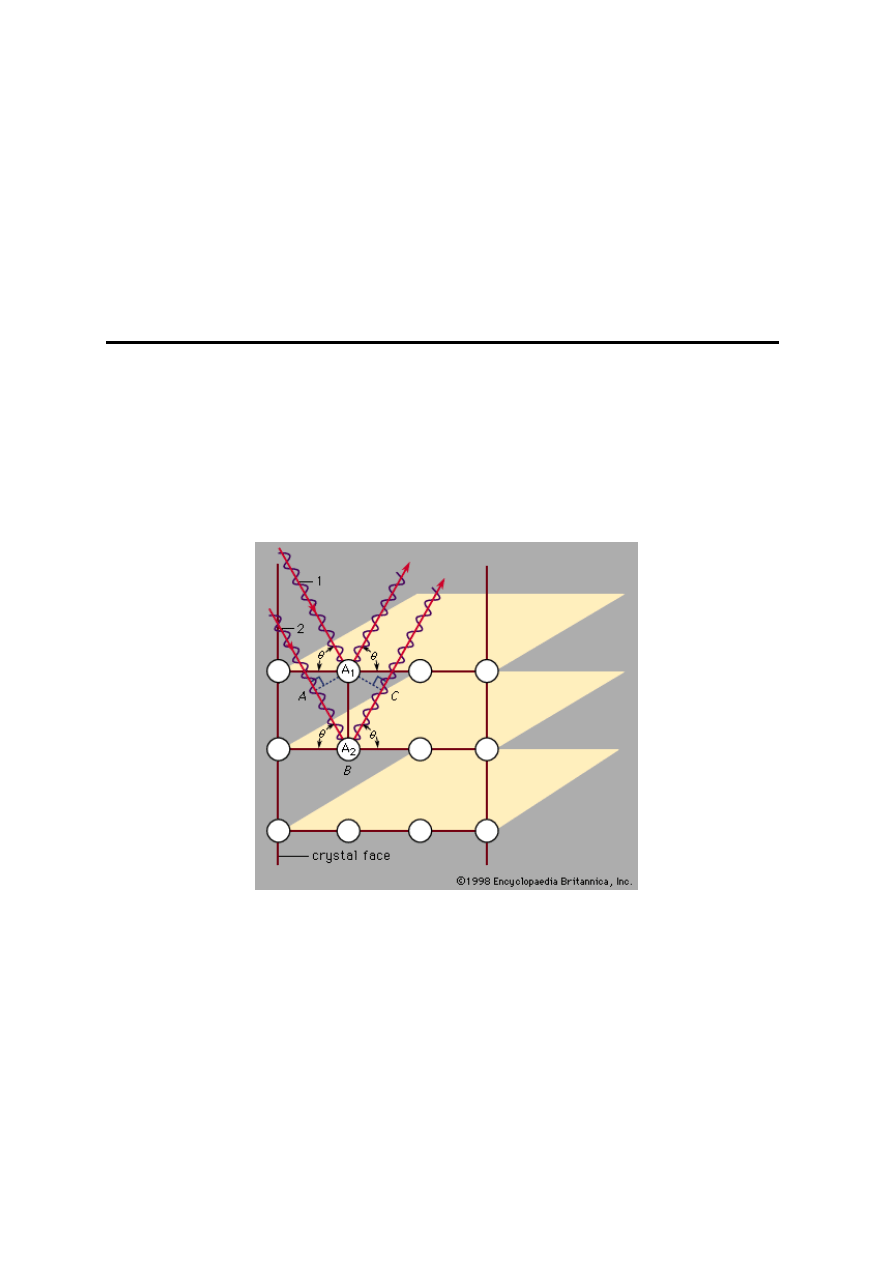
Rys. 21. Dyfrakcja promieni Roentgena na krysztale
Odległości międzypłaszczyznowe w krysztale można
obliczyć na podstawie równania Braggów:
nλ = 2d sin θ, n = 1, 2, 3
gdzie: d – odległość między płaszczyznami, λ – długość
fali promieniowania rentgenowskiego, θ – kąt pomiędzy
promieniowaniem padającym a płaszczyzną sieciową.
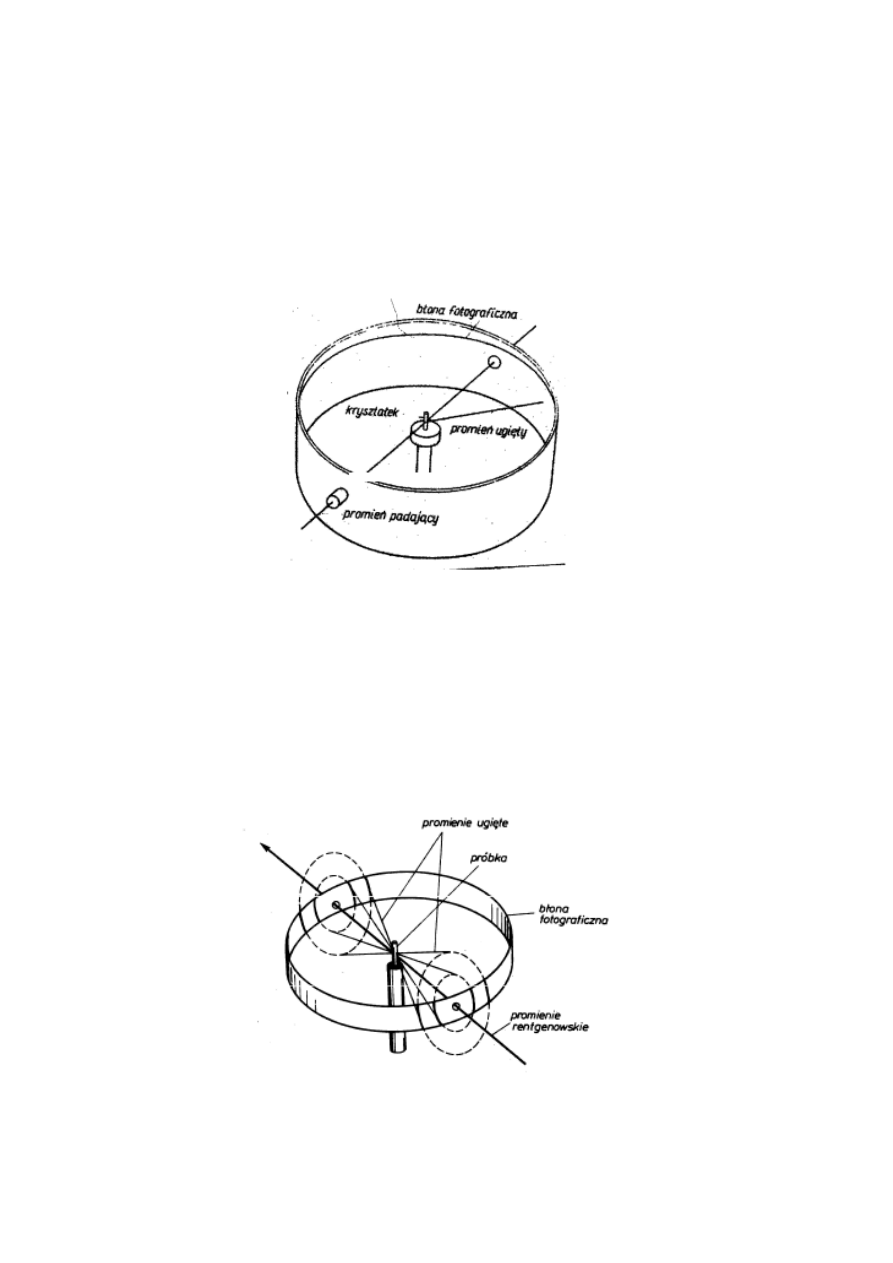
Metoda obracanego kryształu
W tej metodzie do badań potrzebny jest dobrze
wykształcony monokryształ, zwykle o wymiarach kilku
milimetrów.
Rys. 22. Zasada metody obracanego kryształu
Metoda proszkowa Debye’a – Scherrera
W tej metodzie badań stosuje się substancje
polikrystaliczne (proszki).
Rys. 22. Zasada metody proszkowej
Wyszukiwarka
Podobne podstrony:
04b BUDOWA CIALA STALEGOid 53 Nieznany (2)
budowa i dzialanie FDD id 94136 Nieznany (2)
Budowa osrodka sportowo1 id 943 Nieznany
budowa wyrazow i zdan id 94443 Nieznany (2)
Budowa ciała stałego
Budowanie systemu 11 id 94500 Nieznany (2)
budowa malej sieci id 94283 Nieznany (2)
Budowa Lampy Elektronowej id 94 Nieznany (2)
Budowa i sklad atm id 94182 Nieznany (2)
wykład 8 budowa ciała stałego
WYK-4c.Budowa ciala stalego
maszyny stalego2 id 282082 Nieznany
Budowa ciała stałego 3
Budowa monitora LCD id 94314 Nieznany (2)
więcej podobnych podstron