
Clinical
complex clinical syndromes, such as
Wolfram and Behr’s. These forms of optic
atrophy are also linked to causal genes, but
the damage to the optic nerve may be
secondary to other pathological changes
that take place in the body.
General signs and symptoms
The primary optic atrophies are associated
with a number of general signs and
symptoms. In the majority of patients,
there is a light, flat optic disc with well-
defined margins. There is a reduction in
the numbers of the smaller diameter
blood vessels supplying the disc, as well as
attenuation of the peripapillary vessels. In
the primary optic atrophies, the pathology
largely affects the retinal ganglion cells and
loss of these cells leads to a reduction of
optic nerve tissue and thinning of the
nerve along its length
2
. Pupil function is
less affected than visual function,
suggesting that the retinotectal fibres,
responsible for the pupil light reflex, are
less susceptible to damage than the
retinogeniculate fibres
3
. Visual field
defects, including diffuse or sectorial
scotomas, can usually be observed but are
highly variable.
Although considerable overlap of the
signs and symptoms may occur in primary
and secondary forms of optic atrophy,
there are also some consistent differences.
Richard A. Armstrong DPhil and Stephen N. Smith PhD
32
|
July 30
|
2004 OT
Optic atrophy describes a pathological
state resulting from the degeneration of
optic nerve fibres and their supporting
vascular system. It can result from many
disease processes affecting the eye, orbit or
brain including neurological disorders,
metabolic disease, glaucoma, retinal
degeneration and optic nerve
compression. Hereditary factors, however,
are one of the most important causes,
inherited optic atrophy being a significant
cause of childhood and adult blindness
1
.
The mode of inheritance of the familial
forms of optic atrophy may be autosomal
dominant or recessive, X-linked recessive
or mitochondrial. As a result of genetic
heterogeneity, there are considerable
variations in clinical presentation of optic
atrophy and this makes the exact cause of
the condition difficult to diagnose in the
individual patient.
The hereditary optic atrophies can be
divided into two main groups. First, there
are the primary optic atrophies, such as
autosomal dominant optic atrophy (DOA)
and Leber’s hereditary optic neuropathy
(LHON). In primary optic atrophy, the
disease is linked to a specific genetic defect
which results in degeneration of the retina
and/or optic nerve fibres. Second, in
secondary optic atrophy, the pathological
changes affecting the optic disc are
accompanied by many other symptoms in
Secondary optic atrophy is usually
associated with swelling and/or
inflammation of the optic nerve head. The
appearance of the disc is variable,
depending on the associated disease, but
the presence of a pale, slightly elevated
disc with blurred margins is a common
feature. Within the disc there is a reduction
in the smaller diameter blood vessels and
a proliferation of reactive glial cells.
Mitochondrial DNA (mtDNA)
The majority of cases of primary optic
atrophy are either linked to nuclear genes
which control the function of
mitochondria, or are defects of
mitochondrial DNA (mtDNA) itself. In
common with other mammals and
primates, most human inheritable
genomic material in the form of DNA
resides within the nucleus. Because of the
peculiar inheritance patterns observed in
yeast during the late 1940s and early
1950s, however, it became apparent that
DNA also existed in complex eukaryote
cells. Research then isolated DNA from
such cellular organelles as chloroplasts and
mitochondria, giving rise to speculation
that these organelles were at some stage
free-living entities, which were captured
and became incorporated into eukaryote
cells. These organelles are particularly
important to life on earth, as chloroplasts
support photosynthesis while
mitochondria supply cells with the energy
required for metabolism and functions
such as muscle contraction and nerve
impulse transmission.
Throughout the plant kingdom, the
chloroplast genome is relatively similar in
size, but that of mitochondria varies
considerably depending on its origin. The
genetic material of mitochondria is located
inside the organelle in a region known as
the nucleoid. Human mitochondria
genomes, such as those of other mammals,
are relatively small (16.5kb). MtDNA,
which may occur in a loose association
with the inner mitochondrial membrane
(Figure 1), is double stranded in the form
of a closed loop and lacks complexed
proteins. Mitochondrial genes unlike their
nuclear counterparts have no introns and
the mitochondrial genome is so compact
that a pair of genes can share the first and
last nucleotides. Each mitochondrion may
have up to 10 near identical genome
copies and, as a human cell may have
hundreds of mitochondria to furnish its
energy requirements, 0.5-1.0% of human
cellular DNA is mitochondrial in origin.
The two thousand million years which
may have elapsed since mitochondria
became incorporated into eukaryote cells
T
his article considers the clinical symptoms associated with
hereditary optic atrophy and reviews recent progress in our
understanding the genetics of the disorder. The major genes
linked to optic atrophy are identified, and how defects in these
genes could lead to the optic disc pathology is discussed.
Genetics of optic atrophy
Recent progress in understanding
Ribosome
DNA
Cristae
Matrix
Intermembrane space
Inner membrane
Outer membrane
Nuclear coded
proteins and
sub-units
Mitochondrial structures
Figure 1
Structure of mitochondria showing mitochondrial DNA (mtDNA)
(by courtesy of G. Smith, Aston University)
Clinical
33
|
July 30
|
2004 OT
has seen the loss of mitochondrial genes to
the nucleus, where as many as a thousand
redundant mitochondrial pseudogenes
have been recognised. However, the
mammalian mitochondrial genome retains
transcription control function and codes
for 37 genes and is responsible for two
ribosomal RNAs, more than 20 transfer
RNAs and a selection of polypeptides – the
latter forming part of the vital
mitochondrial energy generating function.
All other mitochondrial coding
requirements are furnished by the nucleus,
suggesting great intergenomic regulation
and also presenting molecular biologists
with the interesting question as to why all
mitochondrial coding requirements have
not been entirely taken over by the
nucleus.
Although mitochondria draw heavily
on nuclear coding resources, their own
DNA and mode of inheritance differs
markedly to that of the nucleus. The vital
energy generating function of
mitochondria suggests that their genomes
should be stable and conserved to ensure
efficient biological function. However, the
human mitochondrial genome, possibly
due to the proximity of energy rich and
potentially destructive chemical
intermediates, poor repair facilities, and
compact nature has been shown to mutate
and evolve at a much greater rate than its
nuclear counterpart. Hence, the unique
inheritance pattern of mitochondrial
genomes, which precludes recombination
has simplified the study of human
pedigrees and played a major role in
determining the evolution of primates.
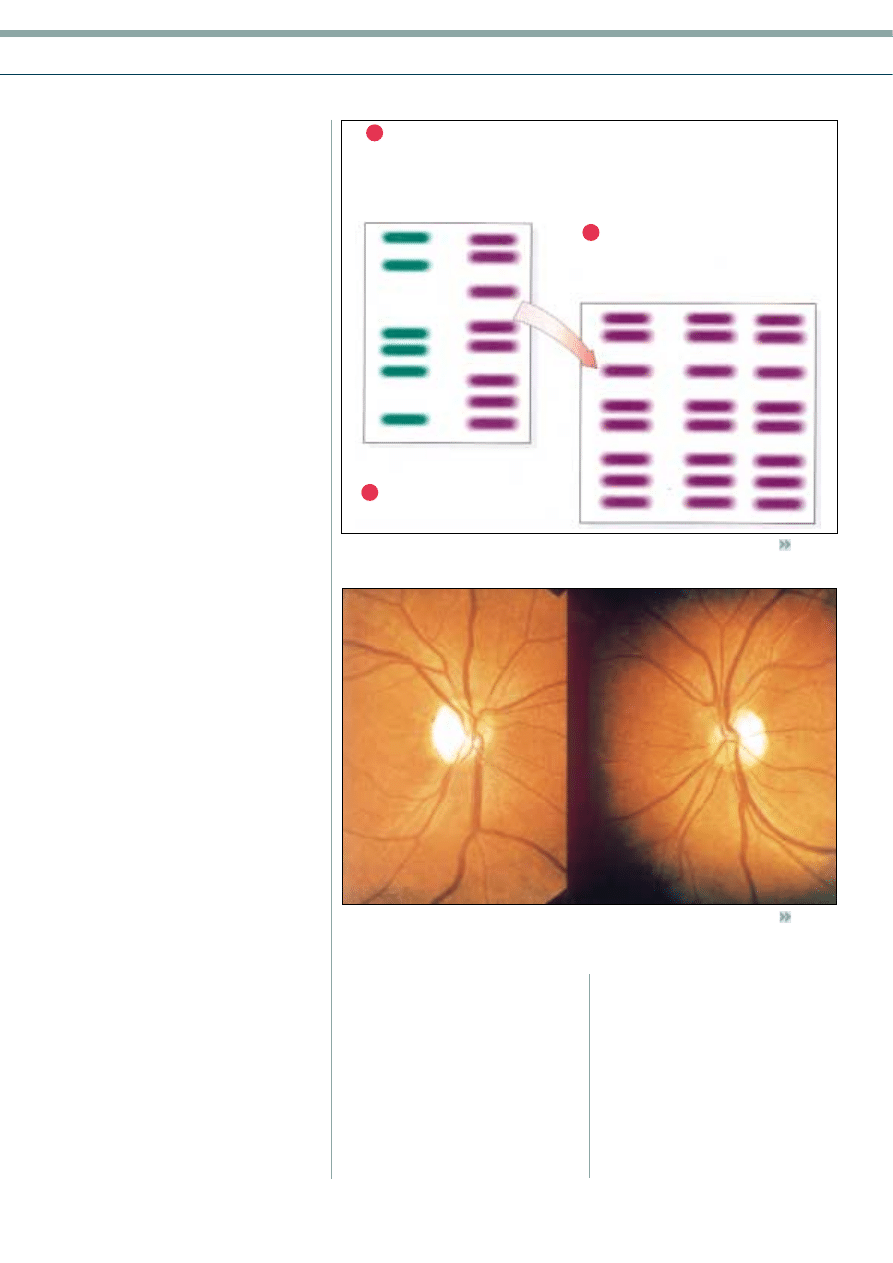
Many studies have attempted to
demonstrate that there is some paternal
inheritance of mtDNA. Analysis of parental
and offspring mtDNA, using restriction
enzymes which cut specific DNA
sequences (Figure 2), clearly demonstrates
that offspring only inherit the
mitochondrial genome of the mother.
There are relatively few mitochondria in
male sperm compared to around 100,000
in each human female egg and this results
in almost a total loss of paternal mtDNA
at fertilisation. Such an occurrence
simplifies human pedigree analysis, but
also has marked implications for the
inheritance of those genetic diseases
associated with mtDNA.
Compromised mitochondrial function
is particularly manifest in those tissues and
entities demanding most energy, such as
the nervous system and muscle, and can
arise from mutations in either the nuclear
or mitochondrial genome. A considerable
variety of mutations have been found in
mitochondrial genomes including point
mutations, large deletions, and structural
abnormalities giving rise to a number of
human malignancies. However, the
defining characteristics of many
mitochondrial genetic diseases such as
Leber’s hereditary optic neuropathy
(LHON) are a maternal inheritance pattern
and have considerable variation in
symptom severity. The latter occurrence is
associated with the genetic heterogeneity
of an individual’s mitochondrial
population, in turn, resulting from the
proportion of compromised
mitochondrial genomes in the specific
maternal egg from which an offspring is
derived.
Autosomal dominant optic
atrophy (DOA, OPA1)
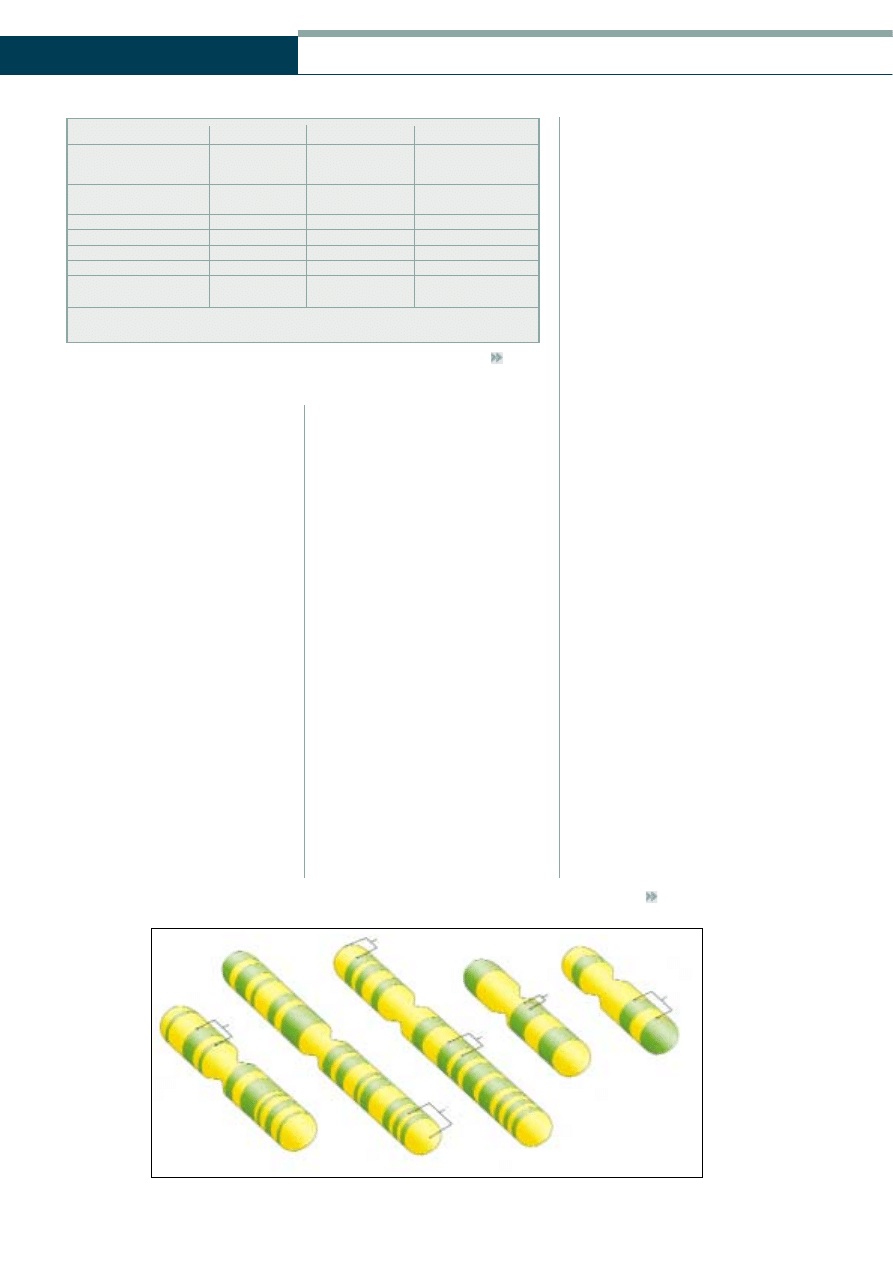
Autosomal dominant optic atrophy
(Figure 3) is the most common form of
non-glaucomatous optic atrophy and in
the general population, has an incidence
of 1/50,000
1
and a prevalence of
1/10,000
4
. The disorder has an insidious
onset and is most typically found in
children four to six years of age, but the
condition can also occur in infants as
young as one year. The first families with
this condition were described by Paul Kjer
and, hence, this disorder is also known as
‘Kjer-type optic atrophy’. The disease
phenotype is highly variable but the
presence of symmetrical bilateral visual
loss, temporal optic disc pallor,
Cut maternal and paternal
mitochondrial DNA with appropriate
restriction enzymes. Size separate
resulting DNA molecules and compare
maternal/paternal patterns
Maternal pattern repeated in all
progeny regardless of sex
Repeat analysis with motochondrial DNA
taken from progeny and compare size
patterns with parents
Father
Mother
Brother
Sisters
3
2
1
Figure 2
Maternal inheritance of mtDNA
(by courtesy of G. Smith, Aston University)
Figure 3
Autosomal dominant optic atrophy (reprinted with permission from: Hamilton AMP,
Gregson R, Fish GE (1998) Text Atlas of the Retina. Martin Dunitz, London)
Clinical
Richard A. Armstrong DPhil and Stephen N. Smith PhD
34
|
July 30
|
2004 OT
centrocaecal visual field defect, thinning of
the papillomacular bundle, and colour
vision defects (most often yellow-blue) are
fairly consistent features.
The first gene to be associated with
DOA was mapped in Danish families to
the long arm of chromosome 3 (3q27-
q29)
5
and subsequently designated as the
locus OPA1 (Table 1, Figure 4). The gene
has 30 exons (portions of DNA which
code for the amino acids of the gene) and
there are eight different isoforms of the
resulting protein due to alternative splicing
of the gene during transcription
6
. The
protein occurs within the inter-membrane
space, electron microscopy suggesting a
location close to the cristae of the
mitochondria
7
. The gene associated with
OPA1 is believed to code for a ‘dynamin-
related GTPase’ implicated in the
formation and maintenance of the
mitochondrial network
8
. Mutations of
OPA1, tend to cluster in the GTPase
domain of the gene
9
and lead to two
modifications, viz. an alteration of
GTPase activity or the loss of the last seven
amino acids of the protein
8
, a region
which is responsible for the interactions
between OPA1 and other proteins.
Subsequently, a second gene has been
found to be associated with DOA, located
to chromosome 18 (18q12.2-12.3)
(Table 1, Figure 4)
1
. Relatively little
clinical data is available at present on
patients expressing this gene, but the
symptoms appear to be similar to those of
OPA1. There is considerable variation in
visual symptoms from normal visual acuity
to legal blindness
10
but generally, the
prognosis for visual acuity is more
favourable with the 18q then the 3q
phenotype.
Autosomal recessive
optic atrophy
Autosomal recessive optic atrophy
(designated OAR1) is genetically distinct
from the dominant forms, but is difficult
to distinguish from them. The recessive
form, however, is generally more severe at
presentation and may also be accompanied
by nystagmus
1
. In addition, it should be
noted that optic atrophy is associated with
Behr’s and Wolfram syndromes, both of
which are autosomal recessive. At the time
of writing, research groups are close to
discovering the first genetic locus
associated with this disorder.
X-linked optic atrophy (OPA2)
Some families exhibit a clear pattern of X-
linked inheritance of optic atrophy, with
affected males showing very early onset
and slow progression of the disease.
Female carriers do not generally show
abnormalities. In one family with a four-
generation history of the disease, linkage
was demonstrated to a gene located at
Xp11.4-p11.21
11
.
Leber’s hereditary optic
neuropathy (LHON)
Leber’s hereditary optic neuropathy
(LHON) was described by Leber in 1871.
The condition is inherited through the
female line by maternal transmission of
mtDNA. The majority of patients with
LHON are males in their 20s, but atypical
cases may be found in females who may
present at any age between 10 and 60
years. The male to female ratio is typically
5:1, but this varies with different mtDNA
mutations.
The condition appears suddenly and
usually affects both eyes. There is often an
acute, severe, painless loss of vision, both
eyes may either be affected simultaneously
or sequentially, with a gap of a few days or
weeks between the onset of symptoms.
Visual field defects usually affect central
vision, the peripheral field being spared.
The optic disc may appear normal in the
acute stage. Colour vision problems may
be present affecting the red-green axis and
there may be some pain during eye
movements. There is considerable
variation in symptoms between patients,
but the most typical cases show disc
hyperaemia, dilated capillaries on the disc
surface, deformed blood vessels, and
swelling of the peripapillary nerve fibre
layer. There is a modest degree of disc
elevation and no dye leakage on
fluorescein angiography. Visual prognosis
is poor although there is often some visual
recovery. The majority of patients will
exhibit permanent loss of vision, however,
with a final visual acuity of 6/60, although
this will vary with type of mutation.
There are three primary mutations of
mtDNA in LHON, viz. at base pair (bp)
11778, 3460 and 14484
12
. The mutation at
bp 11778 causes a change in the NADH
hydrogenase sub-unit four of complex one
of the respiratory chain, and is present in
Table 1
Genes associated with the hereditary forms of optic atrophy
D
Diisso
orrd
deerr
LLo
occuuss
IInnhheerriittaannccee
G
Geennee llo
occaattiio
onnss
Autosomal dominant
OPA1
AD
3q27-q29,
optic atrophy
18q12.2-12.3
Autosomal recessive
OAR1
AR
?
optic atrophy
X-linked optic atrophy
OPA2
XR
Xp11.4-p11.21
Wolfram syndrome
–
AR
4p16.1, 4q22-24
Behr’s syndrome
–
AR
?
Type III MGA
OPA3
?
19q13.2-q13.3
Leber’s optic neuropathy
–
mtDNA
Mutations at bp
11778, 3460, 14484
Abbreviations: AD = Autosomal dominant inheritance; AR = Autosomal recessive inheritance;
mtDNA = Mitochondrial inheritance; bp = base pair; MGA = 3-methylglutaconic aciduria
OPA3
DOA
WS
WS
OPA1
OPA2
X
3
4
18
19
Figure 4
Gene locations for the hereditary optic atrophies (by courtesy of G. Smith, Aston University)

Clinical
35
|
July 30
|
2004 OT
64% of cases of LHON. Mutation at bp
3460 is present in 10% of patients while
that at bp 14484 is found largely in the UK
and the Netherlands and accounts for 25%
of cases. Approximately 15 other changes
in mtDNA, including those at bp 5244,
9101, 9804; 14482 and 14498 have
also been identified but, at present,
linkage of these changes to LHON is
uncertain.
The majority of these mutations result
in partial defects of respiratory chain
function leading to deficits in ATP and an
increase in oxidative stress
13
. Optic nerve
axons may be particularly vulnerable to
oxidative stress because they have an
asymmetric pattern of myelination which
may require more energy to maintain than
other types of axon. The presence of
myelin pathology and a multiple sclerosis
like illness in some patients, especially
those with the 11778 mutation
12
, supports
the suggestion that oxidative stress is
important in LHON. The visual prognosis
of the condition appears to depend on
type of mutation, patients with the 11778
mutation having the worst prognosis.
In addition to uncomplicated LHON, a
condition termed ‘Leber’s plus’ has been
described in which clinical LHON occurs
accompanied by severe neurological or
systemic abnormalities. A variety of
accompanying syndromes may be present,
including a multiple sclerosis like
condition, dystonia, ataxia and a
peripheral neuropathy.
Behr’s syndrome
Optic atrophy can also be found in
association with particular combinations
of symptoms sufficiently frequently to
warrant a specific name. Of these, Behr’s
syndrome is a rare disorder with an
autosomal recessive pattern of inheritance,
and is found in infants and children up to
10 years of age. The syndrome is
characterised by a progressive visual loss
that leads, after a stationery period, to a
temporal field defect and horizontal
nystagmus. In addition, patients exhibit
marked neurological problems such as
ataxia, problems with the control of fine
movements, increased tendon reflexes,
dysarthria, and spastic paresis. Magnetic
resonance imaging (MRI) of these patients
often reveals marked atrophy of the
cerebellum, which may explain some of
the visual and motor symptoms.
Wolfram syndrome
Wolfram syndrome comprises a series of
clinically overlapping conditions first
described in 1938. The syndrome is also
known as ‘diabetes insipidus, diabetes
mellitus, optic atrophy and deafness’ and
is most commonly found in children less
than five years of age, but may also occur
in patients in their early 20s. Insulin-
dependent diabetes mellitus is usually the
first symptom to develop followed by
optic atrophy, and diabetes insipidus while
deafness develops later.
There is a severe visual loss, normally
6/60 or less, and there is a poor visual
prognosis for the patient. The optic disc
itself often exhibits a diffuse pallor with
some evidence of cupping. Colour vision
problems are usually present and the
visual field defect is most often concentric,
and is sometimes accompanied by a
peripheral scotoma. A pigmentary
retinopathy may be present in
approximately 30% of patients and
diabetic retinopathy in 20%. Abnormal
light reflexes and horizontal nystagmus
have also been reported. The visual evoked
potential (VEP) to flash and checkerboard
stimuli are abnormal with signals having
reduced amplitude. An MRI of patients
with Wolfram syndrome has revealed mild
to moderate atrophy of the optic nerve,
optic chiasm, cerebellum, basal ganglia
and the brainstem.
Wolfram syndrome is an autosomal
recessive disease linked to a gene on the
short arm of chromosome 4 (Table 1,
Figure 2) and subsequently refined to
location 4p16.1. The gene responsible for
Wolfram syndrome (locus WTS1) codes
for a protein called ‘Wolfranin’, the
function of which is uncertain but it may
play a homeostatic role within the inner
ear. Mutations of WTS1 have been
described including missense, frame-shift,
and splice site mutations. The clinical
symptoms of Wolfram syndrome have
some similarities with mitochondrial
disease but no defects in mDNA have
been found in the majority of patients. A
few patients with Wolfram syndrome,
however, also have the 11778 mDNA
mutation associated with LHON, a
condition which is believed to represent
the random overlap of the two disorders.
In addition, there is a variant of Wolfram
syndrome in which diabetes insipidus is
absent but gastrointestinal ulceration and
bleeding are common. There is some
indication of linkage of this specific
subtype of the disease to a gene located at
4q22-24.
Type III 3-methylglutaconic
aciduria (OPA3)
Type III 3-methylglutaconic aciduria
(MGA) is a syndrome reported in people
of Iraqi-Jewish origin comprising early
onset bilateral optic atrophy and later
spasticity, movement and cognitive
problems. There is excessive excretion of
3-methylglutaconic and 3-methylglutaric
acids in the urine. The gene responsible
for this condition has been mapped to the
long arm of chromosome 19 at location
19q13.2-q13.3, and has been designated
OPA3. OPA3 consists of two exons and
encodes a peptide of 179 amino acid
residues.
Discussion and conclusions
Optic atrophy is a complex clinical
symptom associated with both primary
hereditary disorders and in combination
with other symptoms. The secondary
disorders also form a heterogeneous group
and include the hereditary syndromes such
as Behr’s and Wolfram and also
neurological disorders, metabolic diseases,
glaucoma, retinal degenerations, and optic
nerve compression.
Diagnosing the cause of optic atrophy
is a particular problem for eye specialists.
Familial optic atrophy is most commonly
associated with an autosomal dominant
condition (OPA1) and occurs in families
affecting, on average, 50% of male and
female offspring. Simple clinical tests are
often useful in diagnosing DOA. Visual
acuity is highly variable but a mild degree
of temporal or diffuse pallor of the disc
and minimal colour vision defects in the
context of a familial pattern is highly
suggestive of DOA
14
.
DOA can be clinically difficult to
separate from normal tension glaucoma,
however, since both may be associated
with disc pallor cupping
15
. Nevertheless, in
DOA, the absence of a healthy
neuroretinal rim and a shallow degree of
shelving to the disc, in combination with
frequent peripapillary atrophy, should
allow the two conditions to be clinically
separated. The pattern of inheritance is
more difficult to predict in autosomal
recessive optic atrophy (OAR1) as carriers
of the recessive gene are phenotypically
similar to normal cases. Symptoms at
presentation are usually more severe in
autosomal recessive optic atrophy
compared with the dominant form. Of the
hereditary optic atrophies, LHON is the
most difficult to clinically diagnose as the
symptoms can be highly variable and there
may be significant numbers of cases which
fall outside the classic presentation.
Recent studies have emphasised that a
significant number of cases of optic
atrophy are genetic, and that genetically
based forms of the disease are
heterogeneous. There are at least six loci of
nuclear DNA associated with the
hereditary optic atrophies as well as several
mutations of mtDNA. There is also the
possibility that further genes associated
with optic atrophy will be identified. As in
other genetic ocular disorders that we have
discussed
16
, this genetic information is
likely to have a considerable impact on the
classification of the optic atrophies in
future. Individual genes and different gene
mutations are likely to be responsible for
distinct types of optic atrophy. In addition,
many of the gene defects which have been
identified code for proteins directly or
indirectly involved in respiratory chain
function and the generation of ATP. The
identification of specific defects in these
genes is therefore likely to lead to a better
understanding of the mechanisms
involved in optic atrophy and to
conventional treatments for the disease.
Finally, the identification of specific
genetic defects leads to the possibility of
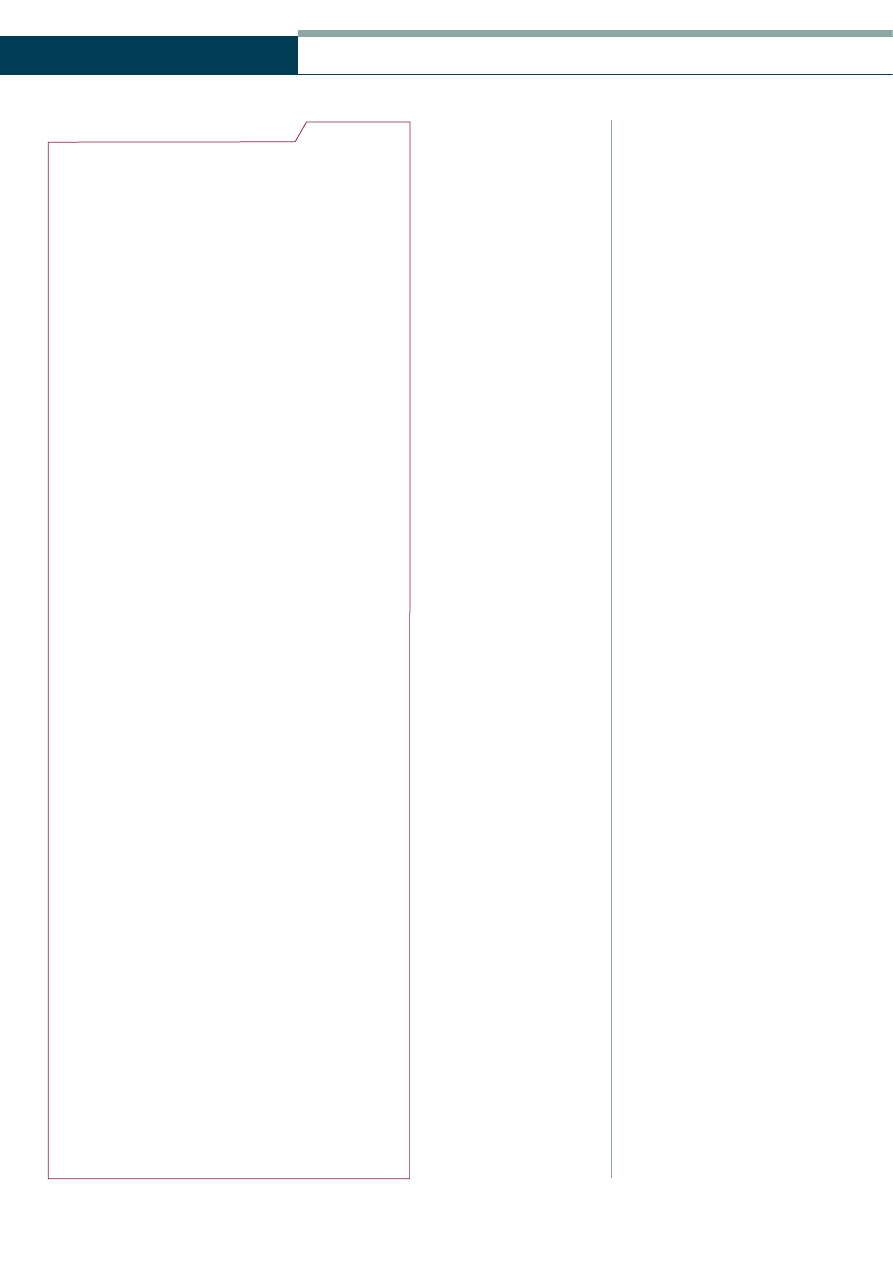
screening individuals in affected
families and hence, in recessive
forms of the disease, to the
identification of asymptomatic
carriers of the disease.
About the authors
Richard Armstrong is a Lecturer
in the Department of Optometry
and Vision Sciences at Aston
University. Stephen Smith is a
Lecturer in Pharmacological and
Biological Sciences at the
university.
References
1. Votruba M, Moore AT and
Bhattacharya SS (1998)
Clinical features, molecular
genetics and
pathophysiology of
dominant optic atrophy.
J. Med. Genet. 35: 793-800.
2. Votruba M, Leary S, Losseff
N, Bhattacharya SS, Moore
AT, Miller DH and Mosely IF
(2000) MRI of the
intraorbital optic nerve in
patients with autosomal
dominant optic atrophy.
Neuroradiology 42: 180-183.
3. Bremner FD, Tomlin EA,
Shallo-Hoffmann J, Votruba
M and Smith SE (2001) The
pupil in dominant optic
atrophy. Inv. Ophthalmol.
Vis. Sci. 42: 675-678.
4. Kjer B, Eiberg H, Kjer P and
Rosenburg T (1996)
Dominant optic atrophy
mapped to chromosome 3q
region II: clinical and
epidemiological aspects.
Acta. Ophthal. Scand. 74: 3-7.
5. Eiberg H, Kjer B, Kjer P and
Rosenburg T (1994)
Dominant optic atrophy
(OPA1) mapped to
chromosome 3q region.
Hum. Mol. Genetics 3:
977-980.
6. Delettre C, Griffoin JM,
Kaplan J, Dollfus H, Lorenz
B, Faivre L, Lenaers G,
Belenguer P and Hamel CP
(2001) Mutation spectrum
and splicing variants in the
OPA1 gene. Human Genetics
109: 584-591.
7. Olichon A, Emorine LJ,
Descoins E, Pelloquin L,
Brichese L, Gas N, Guillon E,
Delettre C, Valette A, Hamel
CP, Ducommun B, Lenaers
G and Belenguer P (2002)
The human dynamin-related
protein OPA1 is anchored to
the mitochondrial inner
membrane facing the
intermembrane space. Febs
Clinical
Richard A. Armstrong DPhil and Stephen N. Smith PhD
36
|
July 30
|
2004 OT
Letters 523: 171-176.
8. Delettre C, Lenaers G,
Pelloquin L, Belenguer P and
Hamel (2002) OPA1
(Kjer-type) dominant optic
atrophy: A novel
mitochondrial disease. Mole
Genet. and Met. 75: 97-107.
9. Pesch UEA, Leo-Kottler B,
Major S, Jurklies B, Kellner U,
Appelstedt-Sylla E, Zrenner E,
Alexander C and Wissenger B
(2001) OPA1 mutations in
patients with autosomal
dominant optic atrophy and
evidence for semi-dominant
inheritance. Human Mole
Genetics 10: 1359-1368.
10. Kerrison JB, Arnould VJ,
Sallum JMF, Vagefi MR,
Barmada MM, Li YY, Zhu DP
and Maumenee IH (1999)
Genetic heterogeneity of
dominant optic atrophy, Kjer
type: identification of a
second locus on
chromosome 18q12.2-12.3.
Arch. Ophthalmol. 117:
805-810.
11. Assink JJM, Tijmes NT,
tenBrink JB, Oostra RJ,
Riemslag FC, deJOng PTVM
and Bergen AAB (1997) A
gene for X-linked optic
atrophy is closely linked to
the Xp11.4-Xp11.2 region of
the X chromosome.
Am. J. Hum. Genet. 61:
934-939.
12. Sapey E, Burdon MA and
Nightingale S (2001)
Evidence of active
demyelination in a man with
Leber’s hereditary optic
neuropathy mtDNA 14484
genotype. Neuro-ophthalmol.
26: 119-126.
13. Carelli V, Ross-Cisneros FN
and Sadun AA (2002) Optic
nerve degeneration and
mitochondrial dysfunction:
genetic and acquired optic
neuropathies. Neurochem.
Int. 40: 573-584.
14. Johnston RL, Seller MJ,
Behnam JT, Burdon MA and
Spatton DJ (1999) Dominant
optic atrophy: refining the
clinical diagnostic criteria in
the light of genetic linkage
studies. Ophthalmol. 106:
123-128.
15. Votruba M, Thiselton D and
Bhattacharya SS (2003) Optic
disc morphology of patients
with OPA1 autosomal
dominant optic atrophy.
Brit. J. Ophthalmol. 87: 48-53.
16. Armstrong RA and Smith SN
(2001) The genetics of
glaucoma. OT 41; 22: 30-33.
Wyszukiwarka
Podobne podstrony:
Neuropatia n II 2
NEUROPSYCHOLOGIA W II Neuroanatomia zarys ogólny
NEUROPSYCHOLOGIA W II Neuroanatomia zarys ogólny
neuropatia n II
Neuropatia n II 2
Neuropeptydy kości, Weterynaria UP lublin, II rok, Materiały, Fizjologia
neuropatie cukrzycowe, Pielęgniarstwo licencjat cm umk, II rok, Patofizjologia
Prel II 7 szyny stałe i ruchome
Produkty przeciwwskazane w chorobach jelit II
9 Sieci komputerowe II
W wiatecznym nastroju II
W01(Patomorfologia) II Lek
Mała chirurgia II Sem IV MOD
Analiza czynnikowa II
więcej podobnych podstron